Область техники
Настоящее изобретение относится к новым лигандам меланокортиновых (MC) рецепторов. Эти лиганды предпочтительно демонстрируют селективность по отношению к рецепторам MC-4 и/или MC-3 по сравнению с другими меланокортиновыми рецепторами (в частности, рецептором MC-1).
Предшествующий уровень техники
Меланокортиновые пептиды (меланокортины) являются природными пептидными гормонами животных и человека, которые связываются с рецепторами MC и стимулируют их. Примерами меланокортинов являются α -MSH (меланоцит-стимулирующий гормон), β -MSH, γ -MSH, ACTH (адренокортикотропный гормон) и их пептидные фрагменты. MSH, главным образом, известен благодаря своей способности регулировать периферическую пигментацию (Eberle 1988), тогда как ACTH известен как индуцирующий стероидонеогенез (Simpson and Waterman, 1988). Кроме того, меланокортиновые пептиды являются посредниками ряда других физиологических действий. Сообщается, что они воздействуют на мотивацию, обучение, память, поведение, воспаление, температуру тела, ощущение боли, кровяное давление, частоту сердечных сокращений, сосудистый тонус, натрийурез, кровоснабжение мозга, рост и регенерацию нервов, развитие плаценты, синтез и высвобождение альдостерона, высвобождение тироксина, сперматогенез, овариальную массу, секрецию пролактина и FSH (фолликулостимулирующий гормон), маточное кровотечение у женщин,
выделение секрета сальных желез и феромона, половую активность, эрекцию полового члена, уровни глюкозы в крови, внутриматочное развитие плода; поведение, связанное с регуляцией аппетита, а также другие физиологические проявления, связанные с родами.
Пептидные последовательности ACTH и различных MSH имеют общий тетрапептидный скелет His-Phe-Arg-Trp. Все данные пептиды получаются в результате протеолитического процессинга пропептида преопиомеланокортина (POMC). За последние несколько лет было идентифицировано пять различных подтипов меланокортинового рецептора. Такие рецепторы MC относятся к классу 7 трансмембранных рецепторов, связанных с доменом G-белка. Пять рецепторов MC, называемые MC-1, MC-2, MC-3, MC-4 и MC-5, все связываются стимулирующим образом в отношении cAMP. Из указанных выше рецепторов, рецептор MC-2 представляет рецептор ACTH, тогда как другие представляют подтипы рецепторов MSH. Рецептор MC-1 присутствует на меланоцитах и меланоме. Рецептор MC-2 присутствует преимущественно в надпочечнике. мРНК (mRNA) для рецептора MC-3 обнаружена в головном мозге, а также в плацентарной и кишечной тканях (Gantz et al. 1993a, Desarnaud et al. 1994, Roselli Rehfuss et al. 1993). Рецептор MC-4 первоначально был обнаружен в головном мозге (Gantz et al. 1993b; Mountjoy et al 1994). Рецептор MC-5 экспрессируется в головном мозге, а также в отдельных периферических тканях (Chhajlani et al 1993; Gantz et al 1994; Griffon et al 1994; Labbu et al. 1994; Barrett et al. 1994; Fathi et al. 1995). Более современные данные, полученные от людей, указывают на то, что все клонированные MC-рецепторы имеют более широкое
распределение в тканях (Chhajlani, 1996), чем считали первоначально.
Как обсуждалось выше, представители семейства меланокортиновых рецепторов могут быть дифференцированы, исходя из их распределения в тканях. Рецепторы MC-4, так же как и рецепторы MC-3, были обнаружены в гипоталамусе, области головного мозга, которая, как полагают, вовлечена в модуляцию пищевого поведения. Было показано, что соединения, демонстрирующие селективность в отношении рецепторов MC-4/MC-3, изменяют отношение к потребности приема пищи после интрацеребровентрикулярной и периферической инъекции у грызунов. В частности, было показано, что агонисты регулируют питание в направлении снижения аппетита, в то время как антагонисты, как было показано, регулируют питание в направлении повышения аппетита. См., Fan, W. et al., “Role of Melanocortinergic Neurons in Feeding and the Agouti Obesity Syndrome”, Nature, 385(6612), pp. 165-8 (Jan. 9,1997).
Роль подтипа рецептора MC-4 в регулировании потребности приема пищи и регуляции массы тела была отчетливо подтверждена на млекопитающих. См., например, Huszer, D. et al., “Targeted Disruption of the Melanocortin-4 Receptor Results in Obesity in Mice”, Cell, Vol. 88, pp. 131-141 (1997); Klebig, M.L. et al., “Ectopic Expression of the Agouti Gene in Transgenic Mice Causes Obesity, Features of Type II Diabetes, and Yellow Fur”, Proc. Natl Acad Sci., Vol. 92, pp. 4728-32 (1995); Karbon, W. et al., “Expression and Function of Argt, a Novel Gene Related to Agouti”, Abstract from the Nineteenth Annual Winter Neuropeptide
Conference (1998); Fan, W. et al., “Role of Melanocortinergic Neurons in Feeding and the Agouti Obesity Syndrome”, Nature, Vol. 385, pp. 165-168 (1997); Seely, R.J., “Melanocortin Receptors in Leptin Effects”, Nature, Vol. 390, p. 349 (1997); Comuzzie, A.G., “A Major Quantitative Trait Locus Determining Serum Leptin Levels and Fat Mass is Located on Human Chromosome 2”, Nat. Gen., Vol. 15, pp. 273-276 (1997); Chagnon, Y.C. et al., “Linkage and Association Studies Between the Melanocortin Receptors 4 and 5 Genes and Obesity-Related Phenotypes in the Quebec Family Study”, Mol. Med., Vol. 3(10), pp. 663-673 (1997); Lee, F. and Huszar, D, “Screening Methods for Compounds Useful in the Regulation of Body Weight”, World Patent Publication WO 97/47316 (1997); and Shutter, J.R. et al., “Hypothalamic Expression of ART, a Novel Gene Related to Agouti, is Up-Regulated in Obese and Diabetic Mutant Mice”, Gen. & Dev. Vol. 11, pp. 593-602 (1997). Стимуляция рецептора MC-4 его эндогенным лигандом, α MSH, продуцирует сигнал насыщения и может быть посредником в снижении сигнала насыщения, регулируемого лептином. Полагают, что, обеспечивая сильнодействующие агонисты рецептора MC-4, можно подавить аппетит и достичь полезных результатов в потере массы тела.
Недавно была определена роль подтипа рецептора MC-3 в регулировании массы тела и распределении энергии. См., например, Chen, A.S. et al., “Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass”, Nature Genetics, Vol. 26, pp. 97-102 (2000); Butler, A.A. et al., “A Unique Metabolic Syndrome Causes Obesity in the
Melanocortin-3 Receptor -Deficient Mouse”, Endocrinology, Vol 141, pp 3518-3521 (2000). Полагают, что агонисты рецептора MC-3 могут модулировать распределение энергии и могут обеспечить полезные результаты в потере массы тела.
Указанные выше исследования подразумевают не-дублирующую роль рецептора MC-3, в сравнении с рецептором MC-4, в энергетическом гомеостазе. Поэтому соединения, которые стимулируют как рецепторы MC-3, так и рецепторы MC-4, могут оказывать более сильное воздействие на потерю массы тела по сравнению с соединениями, которые являются селективными в отношении либо подтипа рецептора MC-3, либо подтипа рецептора MC-4.
Заявителями был обнаружен класс соединений, которые неожиданно имели высокое сродство в отношении подтипов рецептора MC-4 и/или MC-3, и которые обычно селективны именно к этим подтипам MC рецепторов относительно других подтипов меланокортиновых рецепторов, в частности, подтипу MC-1. В соответствии с этим целью настоящего изобретения является разработка соединений, которые имеют сродство к подтипам рецепторов MC-4 и/или MC-3. Другой целью изобретения является способ введения указанных выше соединений животным или человеку. Последующие цели изобретения будут очевидны из следующего ниже раскрытия сущности изобретения.
Сущность изобретения
Изобретение относится к классу соединений, которые являются лигандами для рецепторов MC-4 и/или подтипа MC-3. В частности, изобретение относится к соединению, имеющему структуру,
соответствующую формуле (I):
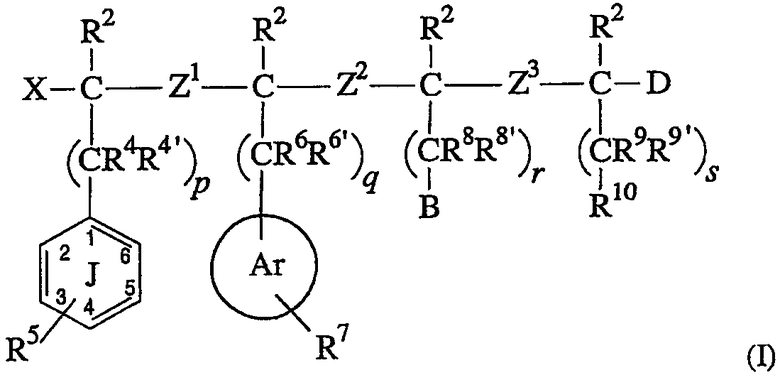
где:
(A) X выбран из водорода, фтора, арилокси, ацилокси, OR1, SR1, -NR1R1’ и –CHR1R1’, где R1 и R1’ независимо выбраны из группы, состоящей из водорода, алкила и ацила;
(B) (1) каждый R2 независимо выбран из группы, состоящей из водорода, алкила, галогена и гетероалкила; или
(2)(a) два соседних заместителя R2, или соседние заместители R2 и R3, могут соединяться с образованием (3-8)-членного карбоциклического или гетероциклического кольца; или
(b) R2, связанный с атомом углерода, который связан с X и Z1, и заместитель R5 могут необязательно соединяться, образуя карбоциклическое или гетероциклическое кольцо, которое конденсировано с фенильным кольцом J; или;
(c) R2, связанный с атомом углерода, который связан с кольцом Ar, может соединяться с R7 с образованием кольца, конденсированного с кольцом Ar; или
(d) R2, связанный с атомом углерода, который связан с Z2 и Z3, может необязательно соединяться с R8, образуя карбоциклическое или гетероциклическое
кольцо; или
(e) R2, связанный с атомом углерода, который связан с Z3 и D, может необязательно соединяться с R10, образуя карбоциклическое или гетероциклическое кольцо;
(C) каждый из Z1, Z2 и Z3 независимо выбран из -OC(R3)(R3a)-; -C(R3)(R3a)O-; -S(O)aC(R3)(R3a)-, где a равно 0, 1 или 2; -C(R3)(R3a)S(O)b-, где b равно 0, 1 или 2; -N(R3e)C(R3)(R3a)-; -C(R3)(R3a)N(R3e)-; -C(O)N(R3d)-; -N(R3d)C(O)-; -C(O)C(R3)(R3a)-; -C(R3)(R3a)C(О)-; -C(R3)(R3a)C(R3b)(R3c)-; -C(R3)=C(R3a)-; -С≡ C-; -SO2N(R3d)-; -N(R3d)SO2-; -C(R3)(R3a)P(=O)(OR3f)-; -P(=O)(OR3f)C(R3)(R3a)-; -N(R3d)P(=O)(OR3f)-; -P(=O)(ОR3f)N(R3d)-; -P(=О)(OR3f)О-; -O-P(=O)(OR3f)-; циклоалкила, имеющего от 3 до 8 кольцевых атомов, и гетероциклоалкила, имеющего от 4 до 8 кольцевых атомов; где
(1) каждый из R3, R3a, R3b и R3с, когда присутствует, независимо выбран из водорода, гидрокси, алкокси, арилокси, ацилокси, тиола, алкилтио, ацилтио, арилтио, амино, алкиламино, ациламино и алкила;
(2) R3d, когда присутствует, выбран из водорода, алкила и арила;
(3) R3e, когда присутствует, выбран из водорода, алкила, арила и ацила; и
(4) R3f, когда присутствует, выбран из водорода и алкила;
(D) p равно 0, 1, 2, 3, 4 или 5; где
(1) когда p больше, чем 0, каждый R4 и R4’ независимо выбран из водорода, алкила, арила, галогена, гидрокси, алкокси, амино и
ациламино;
(2) когда p больше, чем 1, два заместителя R4, вместе с атомами углерода, с которыми они связаны, могут соединяться, образуя гетероциклоалкильное, циклоалкильное или арильное кольцо; и
(3) когда p больше, чем 1, заместители R4 при двух смежных углеродных атомах могут быть оба равны нулю, так что образуется двойная связь между двумя смежными атомами углерода, или как заместители R4, так и заместители R4’ при двух смежных атомах углерода все равны нулю, так что образуется тройная связь между двумя смежными углеродными атомами;
(E) R5 представляет 5 заместителей (т.е. положения 2-6) в фенильном кольце J, где каждый R5 независимо выбран из водорода, гидрокси, галогена, тиола, -OR12, -SR12, -SO2N(R1Z)(R12’), -N(R12)(R12’), алкила, ацила, алкена, алкина, циано, нитро, арила, гетероарила, циклоалкила и гетероциклоалкила; где каждый R12 и R12’ независимо выбран из водорода, алкила, ацила, гетероалкила, арила, гетероарила, циклоалкила и гетероциклоалкила; или два заместителя R5 могут необязательно соединяться, образуя карбоциклическое или гетероциклическое кольцо, которое конденсировано с фенильным кольцом J;
(F) q равно 0, 1, 2, 3, 4 или 5; где
(1) когда q больше, чем 0, каждый R6 и R6’ независимо выбран из водорода, алкила, арила, галогена, гидрокси, алкокси, амино и ациламино;
(2) когда q больше, чем 1, два заместителя R6, вместе с атомами углерода, с которыми они связаны, могут соединяться с
образованием гетероциклоалкильного, циклоалкильного или арильного кольца; и
(3) когда q больше, чем 1, заместители R6 при двух смежных атомах углерода могут быть равны нулю, так что образуется двойная связь между двумя смежными атомами углерода, или оба заместители R6 и R6’ при двух смежных атомах углерода могут быть все равны нулю, так что образуется тройная связь между двумя смежными атомами углерода;
(G) Ar представляет арильное или гетероарильное кольцо, выбранное из группы, состоящей из фенила, тиофена, фурана, оксазола, тиазола, пиррола и пиридина;
(H) R7 обозначает все заместители в кольце Ar, где каждый R7 выбран из водорода, галогена, -NR13R13’, алкила, ацила, алкена, алкина, циано, нитро, арила, гетероарила, циклоалкила и гетероциклоалкила; где каждый R13 и R13’ независимо выбран из водорода, алкила, ацила, гетероалкила, арила, гетероарила, циклоалкила и гетероциклоалкила; или два заместителя R7 могут необязательно соединяться с образованием карбоциклического или гетероциклического кольца, конденсированного с кольцом Ar;
(I) r равно 0, 1, 2, 3, 4, 5, 6 или 7; где
(1) каждый R8 и R8’ независимо выбран из водорода, алкила, галогена, гидрокси, алкокси и амино;
(2) когда r больше, чем 1, два заместителя R8, вместе с атомами углерода, с которыми они связаны, могут соединяться, образуя гетероциклоалкильное, циклоалкильное или арильное кольцо; и
(3) когда r больше, чем 1, заместители R8 при двух смежных
атомах углерода могут быть равны нулю, так что образуется двойная связь между двумя смежными атомами углерода, или как оба заместители R8, так и оба заместители R8’ при двух смежных атомах углерода могут быть все равны нулю, так что образуется тройная связь между двумя смежными атомами углерода;
(J) B выбран из -N(R14)C(=NR15, =O или =S)NR16R17, -NR20R21, циано (-CN), гетероарильного кольца, например, тиофена, алкил- или диалкиламина, гетероарильного кольца, содержащего, по крайней мере, один атом азота в кольце, и гетероциклоалкильного кольца, содержащего, по крайней мере, один атом азота в кольце, где R14, R15 R16, R17, R20 и R21 независимо выбраны из водорода, алкила, алкена и алкина; где, кроме того, комбинация из двух или более R14, R15, R16 и R17 может необязательно объединяться с атомами, с которыми они связаны, с образованием моноциклического или бициклического кольца; предпочтительными являются -N(R14)C(=NR15)NR16R17, циано, N(R14)C(=O)NR16R17, гетероарильное кольцо, содержащее, по крайней мере, один атом азота в кольце, и гетероциклоалкильное кольцо, содержащее, по крайней мере, один атом азота в кольце. Более предпочтительными являются -N(R14)C(=NR15)NR16R17, N(R14)C(=O)NR16R17, циано и триазол и имидазол;
(K) s равно 0, 1, 2, 3, 4 или 5; где
(1) когда s больше, чем 0, каждый R9 и R9’ независимо выбран из водорода, алкила, арила, галогена, гидрокси, алкокси, амино и ациламино;
(2) когда s больше, чем 1, два заместителя R9, вместе с атомами углерода, с которыми они связаны, могут соединяться,
образуя гетероциклоалкильное, циклоалкильное или арильное кольцо; и
(3) когда s больше, чем 1, заместители R9 при двух смежных атомах углерода могут быть равны нулю, так что образуется двойная связь между двумя смежными атомами углерода, или как оба заместители R9, так и оба заместители R9’ при двух смежных атомах углерода могут быть все равны нулю, так что образуется тройная связь между двумя смежными атомами углерода;
(L) R10 выбран из группы, состоящей из необязательно замещенного бициклического арильного кольца и необязательно замещенного бициклического гетероарильного кольца; и
(M) D независимо выбран из водорода, фтора, гидрокси, тиола, ацилтио, алкокси, арилокси, алкилтио, ацилокси, циано, амино, ациламино, -C(O)R11 и -C(S)R11; где R11 выбран из группы, состоящей из гидрокси; алкокси; амино; алкиламино; -NHOR18, где R18 выбран из водорода и алкила; -N(R19)CH2C(O)NH2, где R19 представляет алкил; -NHCH2CH2OH; -N(CH3)CH2CH2OH; и -NHNHC(=Y)NH2, где Y выбран из О, S и NH; и
(N) где, если, по крайней мере, один из Z1, Z2 или Z3 является иным, чем -C(O)N(R3d)- или -N(R3d)C(O)-, тогда X и D могут необязательно соединяться вместе через связующую группу (линкер), L, которая содержит все ковалентные связи или ковалентные связи и ионную связь, так, чтобы образовать циклический аналог пептида;
или его оптическому изомеру, диастереомеру или энантиомеру; его фармацевтически приемлемой соли, гидрату или его биогидролизуемому сложному эфиру, амиду или имиду.
Кроме того, изобретение относится к фармацевтическим композициям, содержащим указанные выше соединения, и к способам лечения расстройств, опосредованных рецептором MC-3 или MC-4, путем введения указанных соединений.
Подробное описание изобретения
I.Определения:
“Аминокислота” относится к аланину (Ala; A), аргинину (Arg; R), аспарагину (Asn; N), аспарагиновой кислоте (Asp; D), цистеину (Cys; C), глутаминовой кислоте (Glu; Q), глутамину (Gln; E), глицину (Gly; G), гистидину (His; H), изолейцину (Ile; I), лейцину (Leu; L), лизину (Lys; K), метионину (Met; M), фенилаланину (Phe; F), пролину (Pro; P), серину (Ser; S), треонину (Thr; T), триптофану (Trp; W), тирозину (Tyr; Y) и валину (Val; V). Общие 3-буквенные и 1-буквенные аббревиатуры указаны в скобках. Ниже перечислены используемые в данном описании модифицированные аминокислоты (3-буквенная аббревиатура для каждой заместители указана в скобках): п-бензоилфенилаланин (Bpa); β -(1-нафтил)аланин (1-Nal); β -(2-нафтил)аланин (2-Nal); β -циклогексилаланин (Cha), 3,4-дихлорфенилаланин (3,4-Dcp); 4-фторфенилаланин (4-Fpa); 4-нитрофенилаланин (4-Npa); 2-тиенилаланин (Tha); 1,2,3,4-тетрагидроизохинолин-3-карбоновая кислота (Tic); 3-бензотиенилаланин (3-Bal); 4-цианофенилаланин (4-Ypa); 4-иодфенилаланин (4-Ipa); 4-бромфенилаланин (4-Rpa); 4,4’-бифенилаланин (Bip); орнитин (Orn); саркозин (Sar); пентафторфенилаланин (Pfp) и β ,β -дифенилаланин (Dip). Что касается заместителей, представленных в структурных формулах (I) и (А), то заместители, указанные в них с использованием
однобуквенного обозначения, являются такими, как они определены, и не относятся к однобуквенным аминокислотам, соответствующим тем же буквенным обозначениям.
Символ “D”, предшествующий указанным выше трехбуквенным аббревиатурам, например, как в “D-Nal” или “D-Phe”, обозначает D-форму аминокислоты. Символ “L”, предшествующий трехбуквенной аббревиатуре аминокислоты, обозначает природную L-форму аминокислоты. В настоящем описании, если не оговорено особо, отсутствие “D” или “L” обозначения указывает на то, что аббревиатура относится как к D-, так и к L-формам. Когда используют обычную однобуквенную аббревиатуру, обозначение заглавной буквой относится к L-форме и обозначение маленькой (строчной) буквой относится к D-форме, если не оговорено иначе.
“Ac” относится к ацетилу (т.е. CH3C(=O)-).
“Ациламино” относится к R-C(=O)N-.
“Ацилокси” относится к R-C(=O)O-.
“Ацилтио” относится к R-C(=O)S-.
“Алкокси” представляет собой кислородсодержащий заместитель имеющий углеводородную цепь, где углеводородная цепь представляет алкил или алкен (т.е., -O-алкил или -O-алкен). Предпочтительные алкоксигруппы включают (например) метокси (MeO), этокси, пропокси и аллилокси.
“Алкил” представляет собой насыщенную углеводородную цепь, имеющую 1-15 углеродных атомов, предпочтительно 1-10, более предпочтительно 1-4 углеродных атома. “Алкен” представляет углеводородную цепь, имеющую, по крайней мере, одну (предпочтительно только одну) углерод-углеродную двойную связь и
имеющую 2-15 углеродных атомов, предпочтительно 2-10, более предпочтительно 2-4 углеродных атома. “Алкин” представляет углеводородную цепь, имеющую, по крайней мере, одну (предпочтительно только одну) углерод-углеродную тройную связь и имеющую 2-15 углеродных атомов, предпочтительно 2-10, более предпочтительно 2-4 углеродных атома. Алкильные, алкеновые и алкиновые цепи (объединенные общим названием “углеводородные цепи”) могут быть прямыми или разветвленными и могут быть незамещенными или замещенными. Предпочтительные разветвленные алкильные, алкеновые и алкиновые цепи имеют одно или два разветвления, предпочтительно одно разветвление. Предпочтительными цепями являются алкильные. Алкильные, алкеновые и алкиновые углеводородные цепи, каждая, могут быть незамещенными или замещены от 1 до 4 заместителями; в случае замещения, предпочтительными цепями являются моно-, ди- или тризамещенные. Алкильные, алкеновые и алкиновые углеводородные цепи, каждая, могут быть замещены галогеном, гидрокси, арилокси (например, фенокси), гетероарилокси, ацилокси (например, ацетокси), карбокси, арилом (например, фенилом), гетероарилом, циклоалкилом, гетероциклоалкилом, спироциклом, амино, амидо, ациламино, кето, тиокето, циано или любой их комбинацией. Предпочтительные углеводородные группы включают метил (Ме), этил, пропил, изопропил, бутил, винил, аллил и бутенил.
Кроме того, указанный в данном описании “низший” алкил, алкен или алкин (например, “низший алкил”) представляет цепь, состоящую из 1-6, предпочтительно от 1 до 4, углеродных атомов в случае алкила и 2-6, предпочтительно 2-4, углеродных атома в
случае алкена и алкина.
“Алкилтио” представляет собой серусодержащий заместитель, имеющий углеводородную цепь, где углеводородная цепь представляет алкил или алкен (т.е. -S-алкил или -S-алкен). Предпочтительные алкилтиогруппы включают (например) метилтио (MeS) и этилтио.
“Арил” представляет собой ароматическое углеводородное кольцо. Арильные кольца являются моноциклическими или конденсированными бициклическими кольцевыми системами. Моноциклические арильные кольца содержат 6 углеродных атомов в кольце. Моноциклические арильные кольца также называют фенильными кольцами. Бициклические арильные кольца содержат от около 8 до около 17 углеродных атомов, предпочтительно от около 9 до около 12 углеродных атомов в кольце. Бициклические арильные кольца включают кольцевые системы, где одно кольцо представляет арил, а другое кольцо представляет арил, циклоалкил или гетероциклоалкил. Предпочтительные бициклические арильные кольца включают 5-, 6- или 7-членные кольца, конденсированные с 5-, 6- или 7-членными кольцами. Арильные кольца могут быть незамещенными или замещенными от 1 до 4 заместителями в кольце. Арил может быть замещен галогеном, циано, нитро, гидрокси, карбокси, амино, ациламино, алкилом, гетероалкилом, галогеналкилом, фенилом, арилокси, гетероарилокси или их любой комбинацией. Предпочтительные арильные кольца включают нафтил, толил, ксилил и фенил. Наиболее предпочтительным арильным кольцевым радикалом является фенил.
“Арилокси” представляет собой кислородсодержащий арильный
заместитель (т.е. -O-арил). Предпочтительные арилоксигруппы включают (например) фенокси, нафтилокси, метоксифенокси и метилендиоксифенокси.
Используемый в данном описании термин “основные аминокислоты” относится к His, Lys и Arg.
“Bс” относится к бутаноилу (т.е. CH3CH2CH2C(=O)-).
“Циклоалкил” представляет собой насыщенное или ненасыщенное углеводородное кольцо. Циклоалкильные кольца являются неароматическими кольцами. Циклоалкильные кольца являются моноциклическими или конденсированными, спиро или соединенными мостиковой связью бициклическими кольцевыми системами. Моноциклические циклоалкильные кольца содержат от около 3 до около 9 углеродных атомов, предпочтительно от 3 до 7 углеродных атомов в кольце. Бициклические циклоалкильные кольца содержат от 7 до 17 углеродных атомов, предпочтительно от около 7 до около 12 углеродных атомов в кольце. Предпочтительные бициклические циклоалкильные кольца включают 4-, 5-, 6- или 7-членные кольца, конденсированные с 5-, 6- или 7-членными кольцами. Циклоалкильные кольца могут быть незамещенными или замещенными от 1 до 4 заместителями в кольце. Циклоалкил может быть замещен галогеном, циано, алкилом, гетероалкилом, галогеналкилом, фенилом, кето, гидрокси, карбокси, амино, ациламино, арилокси, гетероарилокси или их любой комбинацией. Предпочтительные циклоалкильные кольца включают циклопропил, циклопентил и циклогексил.
Термин “конденсированный” относится к циклическим фрагментам, имеющим, по крайней мере, два общих кольцевых атома,
причем предпочтительное число конденсированных циклов равно трем.
“Галоген” представляет (F), хлор (Cl), бром (Br) или иод (I).
“Гетероатом” представляет атом азотa, серы или кислорода, с которым в соответствии с валентностью гетероатома могут быть связаны одна или несколько частей; в случае азота, один атом кислорода может быть необязательно связан с ним ковалентной координационной связью, такой как образующая N-оксид. Группы, содержащие больше, чем один гетероатом, могут содержать различные гетероатомы.
“Гетероалкил” представляет насыщенную или ненасыщенную цепь, содержащую углерод и, по крайней мере, один гетероатом, где нет расположенных рядом двух гетероатомов. Гетероалкильные цепи содержат от 2 до около 15 членов-атомов (углерод и гетероатомы) в цепи, предпочтительно от 2 до около 10, более предпочтительно от 2 до около 5. Например, алкокси (т.е. -О-алкил или -О-гетероалкил) радикалы включены в гетероалкил. Гетероалкильные цепи могут быть прямыми или разветвленными. Предпочтительные разветвленные гетероалкилы имеют одно или два разветвления, предпочтительно одно разветвление. Предпочтительные гетероалкилы являются насыщенными. Ненасыщенные гетероалкилы имеют одну или более двойных связей (также называемые как “гетероалкенил”) и/или одну или более тройных связей (также называемые как “гетероалкинил”). Предпочтительные ненасыщенные гетероалкилы имеют одну или две двойных связей или одну тройную связь, более предпочтительно одну двойную связь.
Гетероалкильные цепи могут быть незамещенными или замещены от 1 до 4 заместителями. Предпочтительные замещенные гетероалкилы являются моно-, ди- или тризамещенными. Гетероалкил может быть замещен низшим алкилом, галогеном, гидрокси, арилокси, гетероарилокси, ацилокси, карбокси, моноциклическим арилом, гетероарилом, циклоалкилом, гетероциклоалкилом, спироциклом, амино, ациламино, амидо, кето, тиокето, циано или их любой комбинацией.
“Гетероциклоалкил” представляет собой насыщенное или ненасыщенное, неароматическое кольцо, содержащее углерод и от 1 до около 4 (предпочтительно 1-3) гетероатомов в кольце, где в кольце нет расположенных рядом двух гетероатомов и нет углерода, который, имея связанный с ним гетероатом, имел бы также связанный с ним гидроксильный, амино или тиольный радикал. Гетероциклоалкильные кольца являются моноциклическими или конденсированными, соединенными мостиковой связью или спиро бициклическими кольцевыми системами. Моноциклические гетероциклоалкильные кольца содержат от около 4 до около 9 членов-атомов (углерод и гетероатомы), предпочтительно от 5 до 7 членов-атомов в кольце. Бициклические гетероциклоалкильные кольца содержат от около 7 до около 17 атомов, предпочтительно от 7 до 12 атомов. Бициклические гетероциклоалкильные кольца могут быть конденсированными, спиро или соединенными мостиковой связью кольцевыми системами. Предпочтительные бициклические гетероциклоалкильные кольца включают 5-, 6- или 7-членные кольца, конденсированные с 5-, 6- или 7-членными кольцами. Гетероциклоалкильные кольца могут быть незамещенными или
замещены от 1 до 4 заместителями в кольце. Гетероциклоалкил может быть замещен галогеном, циано, гидрокси, карбокси, кето, тиокето, амино, ациламино, ацилом, амидо, алкилом, гетероалкилом, галогеналкилом, фенилом, фенокси или их любой комбинацией. Предпочтительные заместители в гетероциклоалкиле включают фтор и алкил.
“Гетероарил” представляет ароматическое кольцо, содержащее углерод и от 1 до около 4 гетероатомов в кольце. Гетероарильные кольца являются моноциклическими или конденсированными бициклическими кольцевыми системами. Моноциклические гетероарильные кольца содержат от около 5 до около 9 членов-атомов (углерод и гетероатомы), предпочтительно 5 или 6 членов-атомов в кольце. Бициклические гетероарильные кольца содержат от около 8 до около 17 членов-атомов, предпочтительно около 8 до около 12 членов-атомов в кольце. Бициклические гетероарильные кольца включают кольцевые системы, где одно кольцо является гетероарилом, а другое кольцо представляет арил, гетероарил, циклоалкил или гетероциклоалкил. Предпочтительные бициклические гетероарильные кольцевые системы включают 5-, 6- или 7-членные кольца, конденсированные с 5-, 6- или 7-членными кольцами. Гетероарильные кольца могут быть незамещенными или замещены от 1 до 4 заместителями в кольце. Гетероарил может быть замещен галогеном, циано, нитро, гидрокси, карбокси, амино, ациламино, алкилом, гетероалкилом, галогеналкилом, фенилом, арилокси, гетероарилокси или их любой комбинацией. Предпочтительные гетероарильные кольца включают тиенил, тиазоло, имидазил, пуринил, пиримидил, пиридил и фуранил.
Используемый в данном описании термин “агонист MC-4” и “агонист MC-3” относится к соединению, обладающему сродством по отношению к рецептору MC-4 или рецептору MC-3, соответственно, воздействие которого может привести к проявлению измеримой биологической активности в клетках, тканях или организмах, которые содержат рецептор MC-4 или MC-3. Используемый в данном описании термин “агонист MC-4/MC-3” относится к соединению, которое является как агонистом MC-4, так и агонистом MC-3, в соответствии с определенной выше терминологией. Испытания, которые позволяют выявить агонистическую активность соединений по отношению к MC-4 и/или MC-3, хорошо известны в данной области. Одним таким обычно используемым тестом является иммуноферментная (EIA) система BioTrak TM, производимая Amersham Pharmacia Biotech (Piscataway, NJ), для прямого определения cAMP, которая позволяет количественно определить ответную реакцию cAMP клеток на лиганды MC. Другим используемым тестом является скрининг cAMP, Tropix cAMP Screen™ , выпускаемый Tropix. Такие системы позволяют провести количественную оценку суммарного клеточного cAMP в клетках, подвергнутых воздействию лигандов. В кратком изложении: клетки HEK, устойчиво трансфецированные рецепторами MC-1, MC-3 или MC-4, помещают в 96-луночные микротитрационные планшеты и культивируют на протяжении ночи. В клетки дозируют соответствующий лиганд MC в течение 1 часа и затем лизируют. Фракцию экстракта лизированных клеток переносят в аналитический планшет. ELISA анализ осуществляют в соответствии с инструкциями, прилагаемыми к набору. Каждый планшет содержит ряд стандартов cAMP для
построения калибровочной кривой, а также полный комплекс агонистов MC в качестве положительного контроля для каждого рецептора MC. Активность cAMP рассчитывают как% максимальной активности cAMP полного комплекса агонистов MC контроля.
Используемые в данном описании термины “антагонист MC-4” и “антагонист MC-3” относятся к соединению, обладающему сродством по отношению к рецептору MC-4 или рецептору MC-3, соответственно, и которое блокирует стимуляцию известным агонистом MC. Используемый в данном описании термин “антагонист MC-4/MC-3” относится к соединению, которое является как антагонистом MC-4, так и антагонистом MC-3, в соответствии с определенной выше терминологией. Тесты, которые позволяют выявить соединения с антагонизмом к MC-4 и/или MC-3, хорошо известны в данной области. Одним из особенно используемых испытаний является конкурентное связывание с использованием NDP-MSH, меченного европием. В кратком изложении: клетки HEK, устойчиво трансфецированные рецепторами MC-1, MC-3, MC-4 или MC-5, помещают в 96-луночные микротитрационные планшеты и культивируют на протяжении ночи. Клетки дозируют соответствующим лигандом MC в присутствии европилированного NDP-MSH в течение 60 мин, клетки промывают несколько раз, добавляют раствор для усиления и измеряют флуоресценцию. Значения IC50 и Ki с каждым рецептором для каждого лиганда MC можно рассчитать, используя стандартные графические программы, такие как GraphPad Prism, (GraphPad Software Inc., San Diego, CA).
Используемые в данном описании термины “рецептор MC-3” и “рецептор MC-4” означают известные рецепторы MC-3 и MC-4 и их
варианты, образованные в результате сплайсинга, и неописанные рецепторы. Рецепторы MC-3 описаны Gantz et at, выше (MC-3 человека); Desarnaud et al., выше (мышиный MC-3) and L. Reyfuss et al., “Identification of a Receptor for Gamma Melanotropin and Other Proopiomelanocortin Peptides in the Hypothalamus and Limbic System.”, Proc. Natl. Acad. Sci USA, vol. 90, pp. 8856-8860 (1993) (крысиный MC-3). Рецепторы MC-4 описаны Gantz et al., выше (MC-4 человека), J.D. Alvaro et al., “Morphine Down-Regulates Melanocortin-4 Receptor Expression in Brain Regions that Mediate Opiate Addiction”, Mol-Pharmacol Sep, vol. 50(3), pp. 583-91 (1996) (крысиный MC-4) and Takeuchi, S. and Takahashi, S., “Melanocortin Receptor Genes in the Chicken-Tissue Distributions”, Gen-Comp-Endocrinol, vol. 112(2), pp 220-31 (Nov. 1998) (куриный MC-4).
Используемый в данном описании термин “измеримый” означает, что биологический эффект является как воспроизводимым, так и существенно отличным от базового разброса данных анализа.
Термин “фармацевтически приемлемая соль” означает катионный противоион, образованный с любой кислотной (карбоновая кислота) группой, или анионный противоион, образованный с любой основной (например, амино) группой. Множество таких солей известно в данной области, как описано в международной патентной публикации 87/05297, Johnston et al., опубликованной 11 сентября 1987, включенной в данное описание в виде ссылки. Предпочтительные катионные соли включают соли щелочных металлов (таких как натрий и калий), соли щелочноземельных металлов (таких как магний и кальций) и органические соли. Предпочтительные анионные соли
включают галогениды (такие как хлоридные соли), сульфонаты, карбоксилаты, фосфаты, трифторацетат (TFA) и т.п. Очевидно, что среди таких солей предусматриваются аддитивные соли, которые могут обеспечивать оптический центр в тех случаях, где его не было. Например, из соединений изобретения можно получить хиральную тартратную соль, и данное определение включает такие хиральные соли.
Наличие таких солей очевидно для специалиста в данной области, и такой специалист способен получить любое число солей при условии, что они известны в данной области. Кроме того, очевидно, что специалиств в данной области может предпочесть одну из солей на основании ее растворимости, стабильности, легкости приготовления и т.п. Определение и оптимизация таких солей находится в пределах практической компетенции специалиста в данной области.
Используемый в данном описании термин “селективный” означает наличие предпочтения в активации конкретного рецептора по сравнению с другими рецепторами, которая может быть количественно определена, исходя из испытаний целостной клетки, ткани или организма, которые позволяют выявить активность рецептора, таких как иммуноферментная (EIA) система для прямого определения cAMP, которая была обсуждена выше. Селективность соединения определяют из сравнения значений EC50 для рассматриваемых рецепторов, подлежащих сравнению. Если не оговорено особо, то использование термина “селективный по сравнению с другими рецепторами MC” подразумевает селективность относительно других меланокортиновых рецепторов, включая
рецепторы MC-1, MC-2 и MC-5. Например, соединение, имеющее EC50 8 нМ для рецептора MC-4 и EC5O ≥ 80 нМ для рецепторов MC-1, MC-2 и MC-5, имеет отношение селективности для рецептора MC-4 по сравнению с другими MC рецепторами, по крайней мере, 1:10. Кроме того, очевидно, что селективность может быть также отнесена к одному из рецепторов MC-1, MC-2 или MC-5, индивидуально. Например, соединение, имеющее EC50 8 нМ для рецептора MC-4 и EC50 80 нМ для рецептора MC-1, имеет отношение селективности для рецептора MC-4 по сравнению с рецептором MC-1 1:10. Такое соединение является селективным по сравнению с рецептором MC-1, независимо от его значения EC50 для MC-2 или MC-5. Селективность описывается более подробно ниже и может быть определена, используя, например, программное обеспечение Prism v 2.0, которое доступно от GraphPad, Inc.
“Сольват” представляет комплекс, образованный комбинацией растворенного вещества (например, заявляемый лиганд рецептора MC-4/MC-3) и растворителя (например, вода). См. J. Honig et al., The Van Nostrand Chemist's Dictionary, p. 650 (1953). Фармацевтически приемлемые растворители, используемые в соответствии с данным изобретением, включают растворители, которые не мешают проявлению биологической активности соединения (например, вода, этанол, простые эфиры, уксусная кислота, N,N-диметилформамид и другие известные или без труда определяемые специалистом в данной области.
“Спироцикл” представляет алкильный или гетероалкильный бирадикальный заместитель алкила или гетероалкила, где указанный бирадикальный заместитель имеет общий углерод и где указанный
бирадикальный заместитель образует кольцо, причем указанное кольцо содержит от около 4 до около 8 членов-атомов (углерод или гетероатомы), предпочтительно 5 или 6 членов-атомов.
II. Соединения
Соединения настоящего изобретения являются лигандами рецептора MC-4 и/или MC-3, имеющими структуру, соответствующую формуле (I):
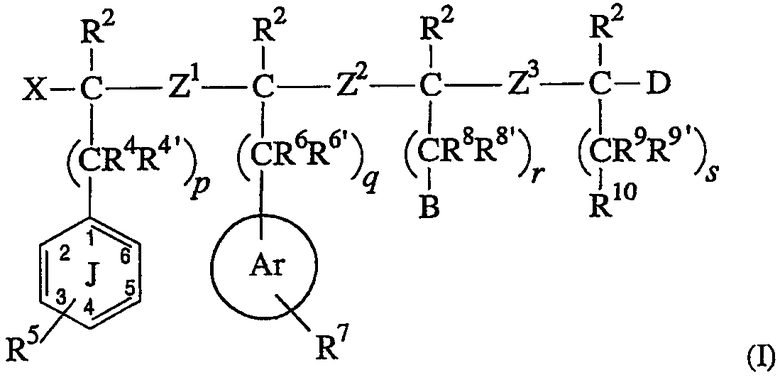
где R2, R4, R4’, R5, R6, R6’, R7, R8, R8’, R9, R9’, R10, Ar, Z1, Z2, Z3, X, B, D, p, q, r и s такие, как описано в разделе “Сущность изобретения”, представленном выше.
Помимо соединений, описанных формулой (I), предполагается, что пептидные остатки скелета могут быть модифицированы ПЭГ для того, чтобы повысить терапевтическую ценность, например, повысить эффективность действия путем пролонгирования полупериода существования лекарственного средства in vivo. Способы модифицирования пептидов ПЭГ хорошо известны в литературе. Например, это описано в представленных ниже источниках, описание каждого из которых включено в данное описание в виде ссылки: Lu, Y.A. et al., “Pegylated peptides. II. Solid-phase synthesis of amino-, carboxy- and side-chain pegylated peptides”, Int. J. Pept. Protein Res., Vol. 43(2), pp. 127-38 (1994); Lu, Y.A. et al., “Pegylated peptides. I. Solid-
phase synthesis of N alpha-pegylated peptides using Fmoc strategy”, Pept. Res., Vol. 6(3), pp. 140-6 (1993); Felix, A.M. et al., “Pegylated peptides. IV. Enhanced biological activity of site-directed pegylated GRF analogs.”, Int. J. Pept. Protein Res., Vol. 46(3-4), pp. 253-64 (1995); Gaertner, H.F. et al., “Site-specific attachment of functionalized poly(ethylene glycol) to the amino terminus of proteins”, Bioconjug Chem., Vol. 7(1), pp. 38-44 (1996); Tsutsumi, Y. et al., “PEGylation of interleukin-6 effectively increases its thrombopoietic potency”, Thromb Haemost, Vol. 77(1), pp. 168-73 (1997); Francis, G.E. et al., “PEGylation of cytokines and other therapeutic proteins and peptides: the importance of biological optimisation of coupling techniques”, Int. J. Hematol., Vol. 68(1), pp. 1-18 (1998); Roberts, M.J. et al., “Attachment of degradable poly(ethylene glycol) to proteins has the potential to increase therapeutic efficacy”, J. Pharm. Sci, Vol 87(11), pp. 1440-45 (1998); and Tan, Y. et al., “Polyethylene glycol conjugation of recombinant methioninase for cancer therapy”, Protein Expr. Purif, Vol. 12(1), pp. 45-52 (1998). Соединения формулы (I) могут быть модифицированы ПЭГ непосредственно, либо в соединения для облегчения операции модифицирования ПЭГ может быть введен “линкер (ветвь связывания)”.
Что касается формулы (I), ниже представлен неограничивающий перечень предпочтительных заместителей:
X выбран из водорода, фтора, арилокси, ацилокси, OR1, SR1, -NR1R1’ и -CHR1R1’. Предпочтительны водород (когда D не является также водородом), -NR1R1’ и -CHR1R1’. Более предпочтительны -NR1R1’
и -CHR1R1’. Еще более предпочтителен -NR1R1’.
R1 и R1’ независимо выбраны из группы, состоящей из водорода, алкила и ацила. Предпочтителен случай, когда R1 представляет водород или алкил и R1’ представляет ацил.
R2 независимо выбран из группы, состоящей из водорода, алкилгалогена и гетероалкила. Предпочтителен водород. Альтернативно, два следующих друг за другом заместителя R2 или следующие друг за другом заместители R2 и R3 могут соединяться с образованием (3-8)-членного карбоциклического или гетероциклического кольца. В другой альтернативе, R2, связанный с углеродным атомом, который связан с X и Z1, и заместитель R5 могут необязательно соединяться, образуя карбоциклическое или гетероциклическое кольцо, которое конденсировано с фенильным кольцом J. В другой альтернативе, R2, связанный с атомом углерода, который связан с кольцом Ar, может соединяться с R7 с образованием кольца, конденсированного с кольцом Ar. В другой альтернативе, R2, связанный с атомом углерода, который связан с Z2 и Z3, может необязательно соединяться с R8 с образованием карбоциклического или гетероциклического кольца. В следующем альтернативном варианте, R2, связанный с атомом углерода, который связан с Z3 и D, может необязательно соединяться с R10, образуя карбоциклическое или гетероциклическое кольцо. Что касается сказанного выше, то предпочтительно, когда R2 не образует кольца с другим R2, и когда R2 не образует кольца с R3, R7 или R8. Более предпочтительно, когда R2 не образует кольца с R10. Предпочтительно, когда кольца, образуемые между R2 и другим заместителем, имеют 5-8 кольцевых атомов.
Каждый из Z1, Z2 и Z3 независимо выбран из -OC(R3)(R3a)-; -C(R3)(R3a)O-; -S(O)aC(R3)(R3a)-, где a равно 0, 1 или 2; -C(R3)(R3a)S(O)b-, где b равно 0, 1 или 2; -N(R3e)C(R3)(R3a)-; -C(R3)(R3a)N(R3e); -C(O)N(R3d)-; -N(R3d)C(O)-; -C(O)C(R3)(R3a)-; -C(R3)(R3a)C(O)-; -C(R3)(R3a)C(R3b)(R3c)-; -C(R3)=C(R3a)-; -C≡ C-; -SO2N(R3d)-; -N(R3d)SO2-; -C(R3)(R3a)P(=O)(OR3f)-; -P(=O)(OR3f)C(R3)(R3a)-; -N(R3d)P(=O)(OR3f)-; -P(=O)(OR3f)N(R3d)-; -P(=O)(OR3f)O-; -O-P(=O)(OR3f)-; циклоалкила, имеющего от 3 до 8 кольцевых атомов, и гетероциклоалкила, имеющего от 4 до 8 кольцевых атомов. Предпочтительны -OC(R3)(R3a)-; -C(R3)(R3a)O-; -S(O)aC(R3)(R3a)-, где a равно 2; -C(R3)(R3a)S(O)b-, где b равно 2; -N(R3e)C(R3)(R3a)-; -C(R3)(R3a)N(R3e)-; -C(O)N(R3d)-; -N(R3d)C(O)-; -C(R3)(R3a)C(R3b)(R3c)-; -C(R3)=C(R3a)-; -C≡ C-; -SО2N(R3d)-; -N(R3d)SО2-; -C(R3)(R3a)P(=O)(OR3f)-; и -P(=O)(OR3f)C(R3)(R3a)-. Более предпочтительны -OC(R3)(R3a)-; -C(R3)(R3a)O-; -C(R3)(R3a)N(R3e)-; -C(O)N(R3d)-; -C(R3)(R3a)C(R3b)(R3c)-; -C(R3)=C(R3a)-; -SO2N(R3d)- и -P(=O)(OR3f)C(R3)(R3a)-. Наиболее предпочтительными являются -C(R3)(R3a)O-; -C(O)N(R3d)- и -C(R3)(R3a)C(R3b)(R3c)-. В другом аспекте, предпочтительными соединениями являются соединения, где, по крайней мере, один из Z1, Z2 или Z3 является иным, чем -C(O)N(R3d)-. Более предпочтительны соединения, где, по крайней мере, два из Z1, Z2 или Z3 являются иными, чем -C(O)N(R3d)-. Кроме того, более предпочтительны соединения, где (i), по крайней мере, один из Z1, Z2 или Z3 иной, чем -C(O)N(R3d)-, и (ii)(a) X является иным, чем -NR1R1’, где R1’ представляет ацил, и/или (b) D является иным, чем -C(O)R11.
Каждый из R3, R3a R3b и R3c, если присутствует, независимо выбран из водорода, гидрокси, алкокси, арилокси, ацилокси, тиола, алкилтио, ацилтио, арилтио, амино, алкиламино, ациламино и алкила. Предпочтительными являются водород, гидрокси, алкокси, арилокси и алкил. Наиболее предпочтительно, когда каждый из R3, R3a,R3b и R3c представляет водород.
R3d, если имеется, выбран из водорода, алкила и арила. Предпочтительно, когда R3d выбран из водорода и алкила.
R3e, если имеется, выбран из водорода, алкила, арила и ацила. Предпочтительно, когда R3e выбран из водорода и алкила.
R3f выбран из водорода и алкила. Когда R3f представляет алкил, предпочтительным является разветвленный алкил, предпочтительно изопропил.
p равно 0, 1, 2, 3, 4 или 5. Предпочтительно p равно 1 или 2, более предпочтительно 1.
Когда p больше, чем 0, каждый R4 и R4’ независимо выбран из водорода, алкила, арила, галогена (предпочтительно фтор), гидрокси, алкокси, амино и ациламино. Когда p больше, чем 1, два заместителя R4, вместе с атомами углерода, с которыми они связаны, могут соединяться, образуя гетероциклоалкильное, циклоалкильное или арильное кольцо. Когда p больше, чем 1, заместители R4 при двух смежных атомах углерода могут оба быть равными нулю, так что образуется двойная связь между двумя смежными атомами углерода, или оба заместители R4 и R4’ при двух смежных атомах углерода могут быть все равны нулю, так что образуется тройная связь между двумя смежными атомами углерода. Предпочтительно каждый R4, если присутствует, является водородом
и каждый R4’, если присутствует, представляет водород или алкил. Наиболее предпочтительно, когда нет ненасыщенности в цепи, связывающей кольцо J с Х-содержащим атомом углерода формулы (I).
R5 представляет 5 заместителей (т.е. положения 2-6) в фенильном кольце J, где каждый R5 независимо выбран из водорода, гидрокси, галогена, тиола, -OR12, -SR12, -SO2N(R12)(R12’), -N(R12)(R12’), алкила, ацила, алкена, алкина, циано, нитро, арила, гетероарила, циклоалкила и гетероциклоалкила. Каждый R12 и R12’ независимо выбран из водорода, алкила, ацила, гетероалкила, арила, гетероарила, циклоалкила и гетероциклоалкила; или два заместителя R5 могут необязательно соединяться с образованием карбоциклического или гетероциклического кольца, которое конденсировано с фенильным кольцом J.
Предпочтительными заместителями R5 являются водород, гидрокси, галоген, тиол, -OR12, где R12 представляет низший алкил или ацил, -SR12, где R12 представляет низший алкил или ацил, -SО2N(R12)(R12’), -N(R12)(R12’), алкил, циано, нитро, арил, гетероарил, циклоалкил и гетероциклоалкил. Более предпочтительными заместителями R5 являются водород, гидрокси, галоген, тиол, -SО2N(R12)(R12’), где R12 и R12’ оба представляют водород, -N(R12)(R12’), где R12 и R12’, каждый, представляют водород или алкил. Еще более предпочтительными заместителями R5 являются водород, гидрокси, хлор, фтор, -N(R12)(R12’), где R12 и R12’, каждый, представляют водород или алкил. Наиболее предпочтительными заместителями R5 являются водород, гидрокси, хлор, фтор и нитро.
Относительно кольца J, предпочтительно, когда четыре из
заместителей R5 представляют водород. Кроме того, предпочтительно, когда в положении 4 находится не водород. Наиболее предпочтительно, когда в положении 4 находится не водород, а остальные 4 заместителя представляют водород.
q равно 0, 1, 2, 3, 4 или 5, предпочтительно, q равно 0, 1 или 2, более предпочтительно q равно 1.
Когда q больше, чем 0, каждый R6 и R6’ независимо выбран из водорода, алкила, арила, галогена (предпочтительно фтор), гидрокси, алкокси, амино и ациламино. Когда q больше, чем 1, два заместителя R6, вместе с атомами углерода, с которыми они связаны, могут соединяться, образуя гетероциклоалкильное, циклоалкильное или арильное кольцо. Когда q больше, чем 1, заместители R6 при двух смежных атомах углерода могут быть равны нулю, так что образуется двойная связь между двумя смежными атомами углерода, или как заместители R6, так и заместители R6’ при двух смежных атомах углерода могут быть все равны нулю, так что образуется тройная связь между двумя смежными атомами углерода. Предпочтительно каждый R6, если присутствует, является водородом и каждый R6’, если присутствует, представляет водород или алкил. Наиболее предпочтительно, когда нет ненасыщенности в цепи, связывающей кольцо Ar с атомом углерода формулы (I), который связан с Z1 и Z2.
Ar представляет арильное или гетероарильное кольцо, выбранное из группы, состоящей из фенила, тиофена, фурана, оксазола, тиазола, пиррола и пиридина. Ar представляет предпочтительно фенил, тиофен или фуран. Наиболее предпочтительно Ar является фенилом.
R7 представляет заместители в кольце Ar, где каждый R7 независимо выбран из водорода; галогена; -NR13R13’, где R13 и R13’, каждый, представляет водород или алкил; алкила; ацила; алкена; алкина; циано; нитро; арила; гетероарила; циклоалкила и гетероциклоалкила. Необязательно, два заместителя R7 могут соединяться, образуя карбоциклическое или гетероциклическое кольцо, конденсированное с кольцом Ar. Когда Ar представляет фенил, предпочтительно, когда четыре из пяти заместителей R7 представляют водород или все пять заместителей R7 представляют водород. Также предпочтительно, когда два заместителя R7 выбраны из фтора, хлора, циано, брома, иода, нитро, алкокси и алкила; или два заместителя R7 соединяются с образованием карбоциклического или гетероциклического кольца, конденсированного с фенильным кольцом. Более предпочтительно, когда в положении 4 фенильного кольца находится водород, фтор, хлор, циано, бром, иод, нитро и алкил, а остальные четыре заместителя представляют водород. Наиболее предпочтительно, когда в положении 4 фенильного кольца находится водород или фтор, а остальные четыре заместителя представляют водород.
Когда Z1 и Z2 оба представляют -C(O)N(R3d)-, предпочтительными являются соединения, где атом углерода, который связан с Z1 и Z2, находится в R конфигурации в соответствии с нормами номенклатуры Кана-Ингольда-Прелога.
r равно 0, 1, 2, 3, 4, 5, 6 или 7. Предпочтительно r равно 2, 3, 4 или 5. Более предпочтительно r равно 2, 3 или 5. Наиболее предпочтительно r равно 3.
Каждый R8 и R8’ независимо выбран из водорода, алкила,
галогена (предпочтительно фтор), гидрокси, алкокси и амино. Необязательно, когда r больше, чем 1, два заместителя R8, вместе с атомами углерода, с которыми они связаны, соединяются с образованием гетероциклоалкильного, циклоалкильного или арильного кольца. Предпочтительно, каждый R8 и R8’ независимо выбран из водорода и алкила. Наиболее предпочтительно, каждый R8 и R8’ представляет водород. Необязательно, когда r больше, чем 1, заместители R8 при двух смежных атомах углерода могут быть равны нулю, так что образуется двойная связь между двумя смежными атомами углерода, или как заместители R8, так и заместители R8’ при двух смежных атомах углерода могут быть все равны нулю, так что образуется тройная связь между двумя смежными атомами углерода.
B выбран из -N(R14)C(=NR15, =O или =S)NR16R17, -NR20R21, циано (-CN), гетероарильного кольца, например, тиофена; алкил- или диалкиламина, гетероарильного кольца, содержащего, по крайней мере, один атом азота в кольце, и гетероциклоалкильного кольца, содержащего, по крайней мере, один атом азота в кольце. Предпочтительными являются -N(R14)C(=NR15)NR16R17, гетероарильное кольцо, содержащее, по крайней мере, один атом азота в кольце, и гетероциклоалкильное кольцо, содержащее, по крайней мере, один атом азота в кольце. Более предпочтительными являются -N(R14)C(=NR15)R16R17, циано, N(R14)C(=О)NR16R17, гетероарильное кольцо, содержащее, по крайней мере, один атом азота в кольце, и гетероциклоалкильное кольцо, содержащее, по крайней мере, один атом азота в кольце. Более предпочтительны -N(R14)C(=NR15)NR16R17, N(R14)C(=O)NR16R17, циано, триазол и имидазол.
R14 и R15 независимо выбраны из водорода, алкила, алкена и алкина. Предпочтительными являются водород и алкил. R16 и R17 независимо выбраны из водорода, алкила, алкена и алкина. Предпочтительны водород и алкил. R20 и R21 независимо выбраны из водорода, алкила, алкена и алкина. Предпочтительны водород и алкил.
Альтернативно, комбинация из двух или нескольких из R14, R15, R16 и R17 объединяется с образованием моноциклического или бициклического кольца. Например, R14 и R15, вместе с атомами, с которыми они связаны, могут соединяться, образуя гетероциклоалкил или гетероарил. Кроме того, R14 и R16, вместе с атомами, с которыми они связаны, могут соединяться с образованием гетероциклоалкила или гетероарила. Кроме того, R15 и R16, вместе с атомами, с которыми они связаны, могут соединяться, образуя гетероциклоалкил или гетероарил. Кроме того, R16 и R17 могут необязательно соединяться с образованием гетероарильного или гетероциклоалкильного кольца. Предпочтительно, когда R15 и R16 соединяются с образованием кольца.
s равно 0, 1, 2, 3, 4 или 5. Предпочтительно s равно 1 или 2, более предпочтительно 1.
Когда s больше, чем 0, каждый R9 и R9’ независимо выбран из водорода, алкила, арила, галогена (предпочтительно фтор), гидрокси, алкокси, амино и ациламино. Предпочтительно каждый R9, если присутствует, является водородом и каждый R9’, если присутствует, является водородом или алкилом. Необязательно, когда s больше, чем 1, два заместителя R9, вместе с атомами углерода, с которыми они связаны, соединяются, образуя
гетероциклоалкильное, циклоалкильное или арильное кольцо. Кроме того, когда s больше, чем 1, заместители R9 при двух смежных атомах углерода могут оба быть равны нулю, так что образуется двойная связь между двумя смежными атомами углерода. Кроме того, когда s больше, чем 1, оба заместители R9 и оба заместители R9’ при двух смежных атомах углерода могут быть все равны нулю, так что образуется тройная связь между двумя смежными атомами углерода. Наиболее предпочтительно, когда нет ненасыщенности в цепи, связывающей R10 c D-содержащим атомом углерода формулы (I).
R10 выбран из группы, состоящей из необязательно замещенного бициклического арильного кольца и необязательно замещенного бициклического гетероарильного кольца. Предпочтительные бициклические арильные кольца включают 1-нафтил, 2-нафтил, индан, 1H-инден, бензоциклобутан и бензоциклобутен. Предпочтительные бициклические гетероарильные кольца включают индол, индолин, пиридин, дигидропиридин, октагидропиридин, бензотиофен, бензофуран, бензимидазол, бензопиран, хинолин, хинолон и изохинолин. Более предпочтительно, когда R10 представляет 1-нафтил, 2-нафтил, индол, индан, 1H-инден, бензотиофен, бензофуран и бензопиран. Наиболее предпочтительно, когда R10 представляет 1-нафтил, 2-нафтил или индол (в частности, 3-индол).
D выбран из водорода, фтора, гидрокси, тиола, алкокси, арилокси, алкилтио, ацилокси, циано, амино, ациламино, -C(O)R11 и -C(S)R11. Предпочтительными являются фтор, гидрокси, тиол, алкокси, арилокси, алкилтио, ацилокси, циано, амино, ациламино, -C(O)R11 и -C(S)R11. Более предпочтительными являются алкокси,
циано, амино, ациламино, -C(O)R11 и -C(S)R11. Еще более предпочтительными являются -C(O)R11 и -C(S)R11. Наиболее предпочтителен -C(O)R11.
R11 выбран из группы, состоящей из амино; алкиламино; -NHOR18, где R18 выбран из водорода и алкила; -N(R19)CH2C(O)NH2, где R19 представляет алкил (предпочтительно низший алкил); -NHCH2CH2OH; -N(CH3)CH2CH2OH; и -NHNHC(=Y)NH2, где Y выбран из O, S и NH. Предпочтительными R11 являются амино; алкиламино; -NHOR18, где R18 выбран из водорода и алкила (предпочтительно водорода); -N(R19)CH2C(O)NH2, где R19 представляет алкил (предпочтительно низший алкил); -NHCH2CH2OH; и -N(CH3)CH2CH2OH. Более предпочтительными R11 являются амино; алкиламино; -NHOR18, где R18 выбран из водорода и алкила (предпочтительно водорода); и -N(R19)CH2C(O)NH2, где R19 представляет алкил. Наиболее предпочтительны амино и алкиламино.
Как указано в отношении формулы (I), если, по крайней мере, один из Z1, Z2 или Z3 является иным, чем -C(O)N(R3d)- или -N(R3d)C(O)-, тогда X и D могут быть необязательно связаны через линкер L, который содержит все ковалентные связи или ковалентные связи и ионную связь, образуя тем самым циклический аналог пептида. Такие циклические пептиды имеют структуру, соответствующую приведенной ниже формуле (II):
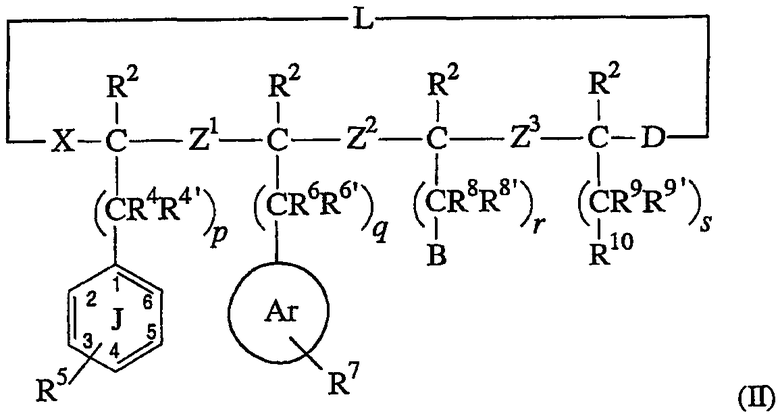
Что касается циклических соединений, содержащих линкер L, мостик, соединяющий X и D, может быть в форме связей ковалентной природы или, альтернативно, может включать солевой мостик, являющийся результатом образования ионных связей. Линкер может быть целиком пептидным по своей природе (т.е. содержащий только аминокислоты), не-пептидным по природе (т.е. не содержащий аминокислот) или он может включать как заместители пептидной природы, так и не-пептидной природы, вводимые с использованием хорошо известных способов химического синтеза. Линкер может включать алифатические остатки, ароматические остатки или гетероароматические остатки, или их любую комбинацию. В одном варианте осуществления, линкер может включать длинноцепочечные омега-аминокислоты, в которых амино- и карбоксильные группы разделены цепными участками, состоящими из от около 4 до около 24 метиленовых групп, или комбинацию указанных омега-аминокислот и аминобензойных кислот.
В другом варианте воплощения, который является предпочтительным вариантом осуществления, линкер может содержать все ковалентные связи, такие как амидные связи. Например, линкер может включать амид, образованный путем химического сочетания аминогруппы боковых цепей аминокислот, таких как Lys или Orn, и
карбоксильной группы боковой цепи аминокислотного остатка, такого как Asp или Glu. Альтернативно, линкер может включать амид, образованный между амино- и карбоксилатными группами, связанными с α -углеродом аминокислот, образующих мостиковую группу (в дальнейшем называемым “α -амино” группой аминокислоты или “α -карбоксильной” группой аминокислоты). В другой альтернативе, линкер может включать амид, образованный между любой комбинацией аминогруппы боковой цепи или карбоксильной группы боковой цепи (аминокислоты) и α -амино и α -карбоксильными группами. Связывающие остатки могут представлять амин- или карбоксилсодержащие структуры иные, чем природные аминокислоты, включая, например, 6-аминогексановую кислоту в качестве аминсодержащего остатка и янтарную кислоту в качестве карбоксилсодержащего остатка. Кроме того, изобретение учитывает связывание, используя другие типы химических функциональных групп. В этом случае, такие связывающие остатки могут содержать различные группы и заместители, включая алифатические, гетероалкильные, ароматические и гетероциклические. При ковалентном связывании, линкер может включать, но, не ограничиваясь ими, амид, сложный эфир, простой эфир, тиоэфир, аминоалкил, аминоарил, алкил, другой гетероалкил, алкен, алкин, гетероциклоалкил, арил и гетероарил. Предпочтительно, линкер может включать простой эфир, аминоалкил, аминоарил, алкил, другой гетероалкил, алкен, алкин, гетероциклоалкил, арил и гетероарил. Более предпочтительно, линкер может включать простой эфир, аминоалкил, алкил, алкен и алкин. Когда L содержит только
ковалентные связи, предпочтительными являются соединения, имеющие от около 12 до около 32 кольцевых атомов, более предпочтительными являются соединения, имеющие от около 22 до около 28 кольцевых атомов.
Линкер может альтернативно включать ионную связь/ассоциацию, которая благоприятствует образованию циклической структуры. Такой “ионный” мостик включает солеобразующую основную или кислотную функциональности. Например, связь может включать ионную связь, образованную между аминогруппой боковой цепи аминокислот, таких как Lys или Orn, и карбоксильной группой боковой цепи аминокислотного остатка, такого как Asp или Glu. Альтернативно, линкер может включать ионную связь, образованную между амино и карбоксилатными группами, связанными с α -углеродом аминокислот, образующих линкер. В другой альтернативе, линкер может включать амид, образованный между любой комбинацией аминогруппы боковой цепи или карбоксилом боковой цепи (аминокислоты) и α -амино и α -карбоксильной группами. Когда L содержит ионную связь, образованное кольцо предпочтительно может содержать от около 22 до около 28 кольцевых атомов.
Следует иметь в виду, что любые свободные пептидные α -карбокси и α -аминогруппы (т.е. α -карбокси и α -аминогруппы аминокислот), не участвующие в образовании кольца, могут необязательно быть в виде карбоксиамидной или ациламиногруппы, соответственно. Наиболее предпочтительными L-содержащими соединениями являются аналоги, где Х и D образуют ковалентно
связанные циклические структуры.
В отношении соединений настоящего изобретения, в общем, хотя алкильные, гетероалкильные, циклоалкильные и гетероциклоалкильные группы могут быть замещены гидрокси, амино и амидогруппами, как установлено выше, в изобретении не предусматривается наличие нижеследующего:
1. Енолы (OH, присоединенный к углероду, несущему двойную связь).
2. Аминогруппы, присоединенные к углероду, несущему двойную связь (за исключением винилогических амидов).
3. Более чем одна гидрокси, амино или амидогруппа, присоединенная к одному углероду (за исключением, когда два атома азота связаны с одним атомом углерода и все три атома являются атомами-членами гетероциклоалкильного кольца).
4. Гидрокси, амино или амидогруппа, присоединенная к sp3-гибридизированному углероду, который также имеет связанный с ним гетероатом.
5. Гидрокси, амино или амидогруппа, присоединенная к углероду, который также имеет связанный с ним галоген.
Предпочтительным подклассом соединений формулы (I), где нет линкера L, необходимого для образования макроциклического кольца, является подкласс соединений, имеющих структуру формулы (А), как следует ниже:
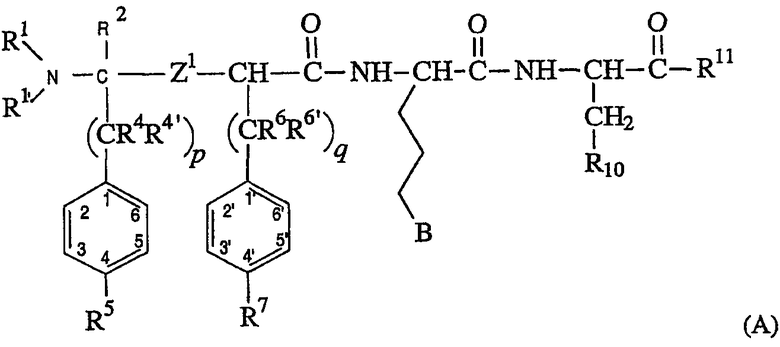
В указанном выше предпочтительном подроде соединений, заместители R1, R1’, Z1, R4, R4’, R5, R6, R6’, R7, B, R10 и R11 являются такими, как определено в отношении формулы (I). Ссылаясь на формулу (I), соединения формулы (A) представляют соединения, где кольцо J формулы является фенильным кольцом, где все из положений 2, 3, 5 и 6 представляют водород, так что кольцо замещено только в положении 4 заместителем R5, который является таким, как определено в отношении формулы (I). Подобно описанию формулы (I), заместитель R5 кольца и заместитель R2 могут необязательно соединяться с образованием кольца, конденсированного с изображенным фенильным кольцом. В таком варианте воплощения конденсированное кольцо может соединять фенильное кольцо в другом положении, чем положение 4. В формуле (А) кольцо Ar формулы (I) является фенильным кольцом, где все положения 2’, 3’, 5’ и 6’ представляют водород, а положение 4’ представляет R7, который является таким, как определено выше. В этом отношении, предпочтительно, когда R7 выбран из водорода и фтора.
В отношении формулы (A), p и q равны независимо 1 или 2, предпочтительно q равно 1. Кроме того, предпочтительно, когда R4, R4’, R6 и R6’ представляют все водород. Кроме того, предпочтительны соединения, где В представляет
-N(R14)C(=NR15)NR16R17 или -NR20R21.
Предпочтительным подклассом соединений формулы (II) являются соединения, имеющие структуру, соответствующую формуле (B), как следует ниже:
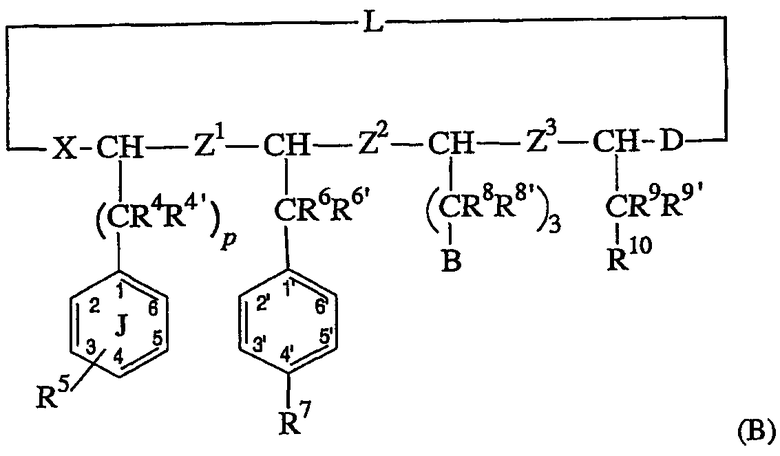
где X, Z1, Z2, Z3, D, R4, R4’, R5, R6, R6’, R7, R8, R8’, B, R9, R9’ и R10 являются такими, как определено выше, и p равно 1 или 2. Предпочтительны соединения, где R6 и R6’ представляют оба водород. Также предпочтительны соединения, где B представляет -N(R14)C(=NR15)NR16R17 или –NR20R21. Также предпочтительно, когда R8, R8’, R9 и R9’ представляют все водород. Предпочтительно, когда R7 выбран из водорода и фтора. Предпочтительными являются -N(R14)C(=NR15)NR16R17, циано, N(R14)C(=О)NR16R17, гетероарильное кольцо, содержащее, по крайней мере, один атом азота в кольце, и гетероциклоалкильное кольцо, содержащее, по крайней мере, один атом азота в кольце. Более предпочтительными являются N(R14)C(=NR15)NR16R17, N(R14)C(=O)NR16R17, циано и триазол и имидазол.
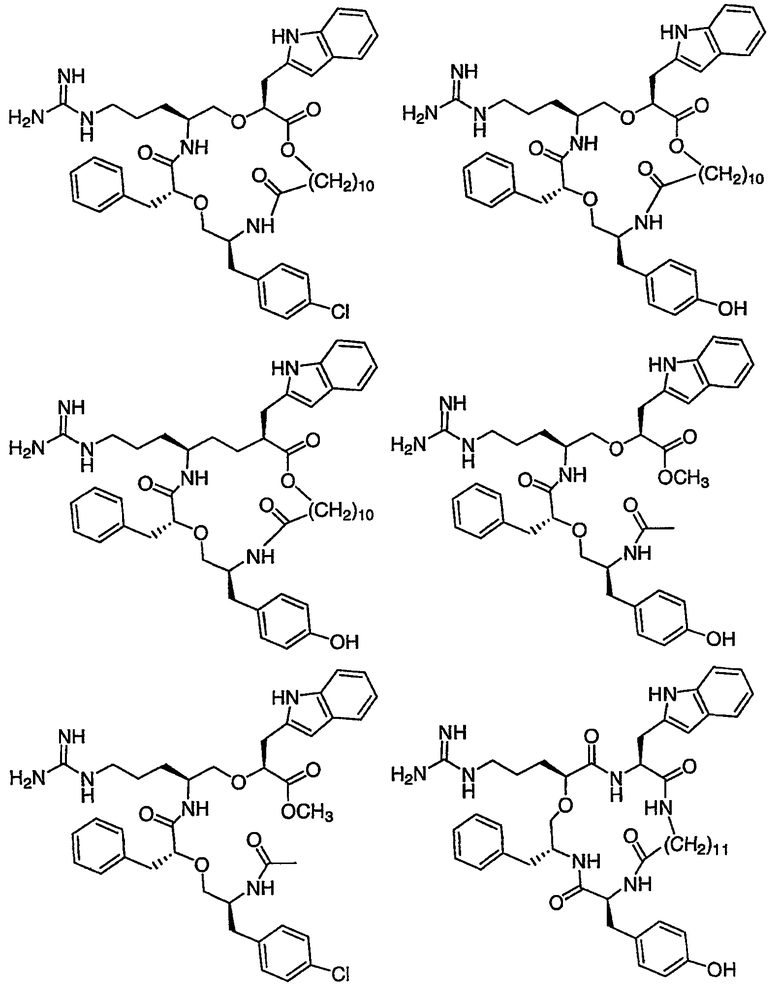
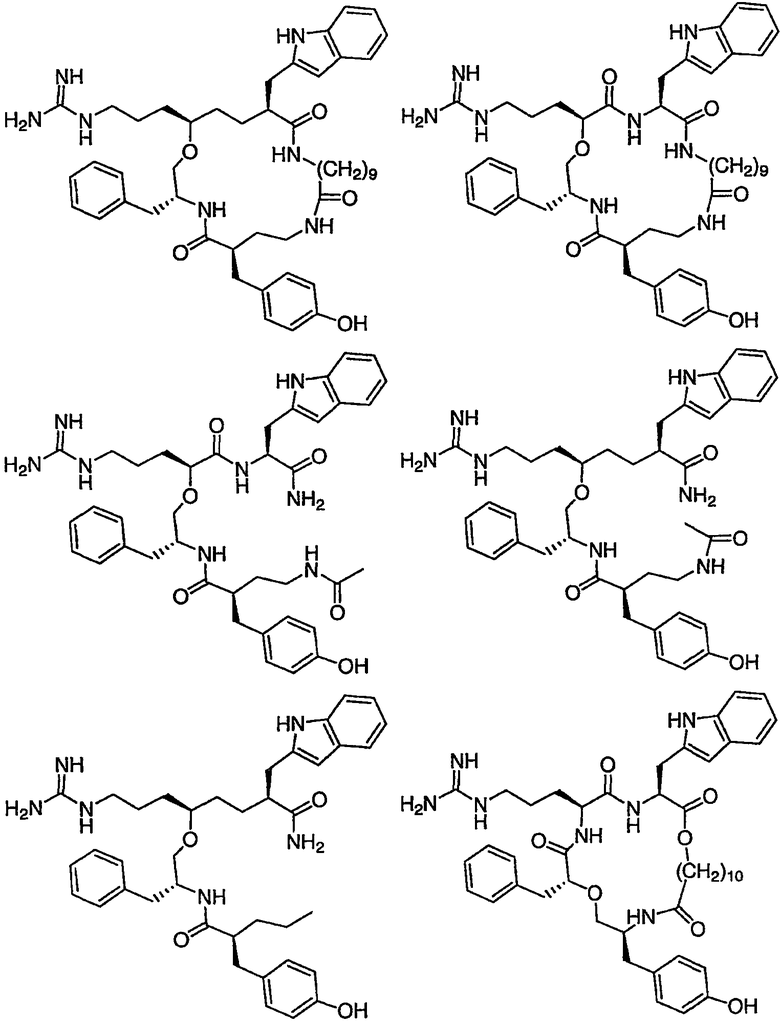
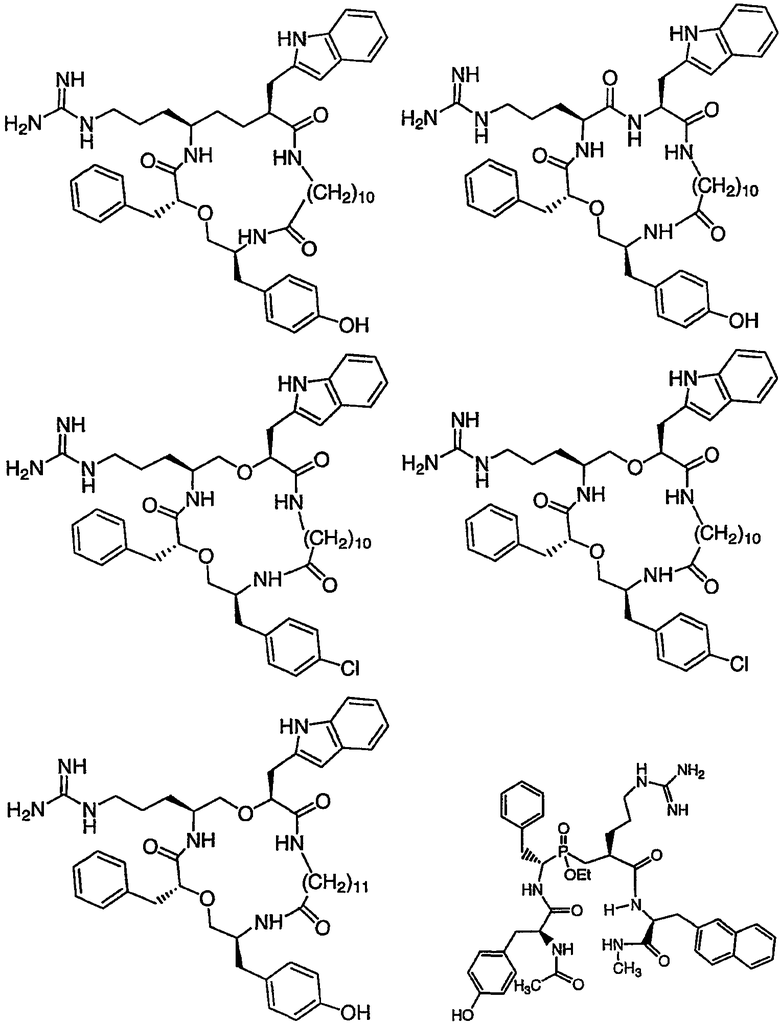
Ниже представлен неограничивающий перечень предпочтительных соединений формулы (I). В отношении соединений, изображенных указанными выше химическими структурами, в объем изобретения включены как “линейные” соединения (т.е. соединения, где
отсутствует линкер L, который обеспечивает макроциклическую молекулу), так и макроциклические соединения формулы (II).
Следующий ниже перечень использует одно- и/или трехбуквенные аббревиатуры аминокислот, которые обсуждались выше.
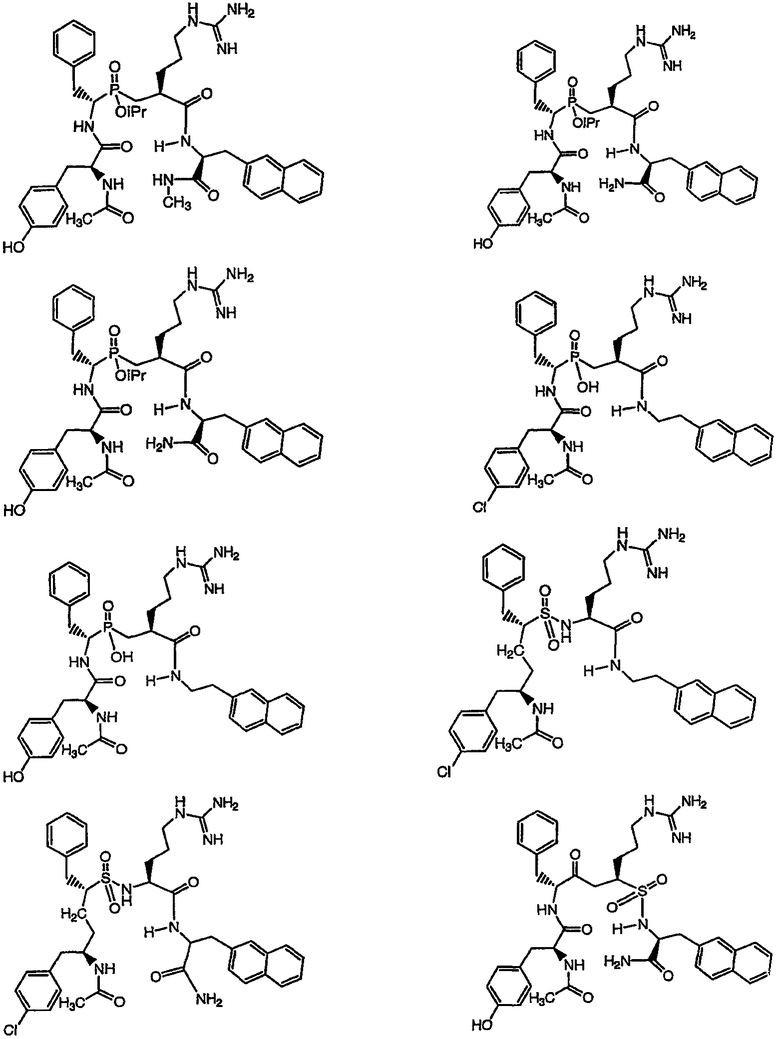
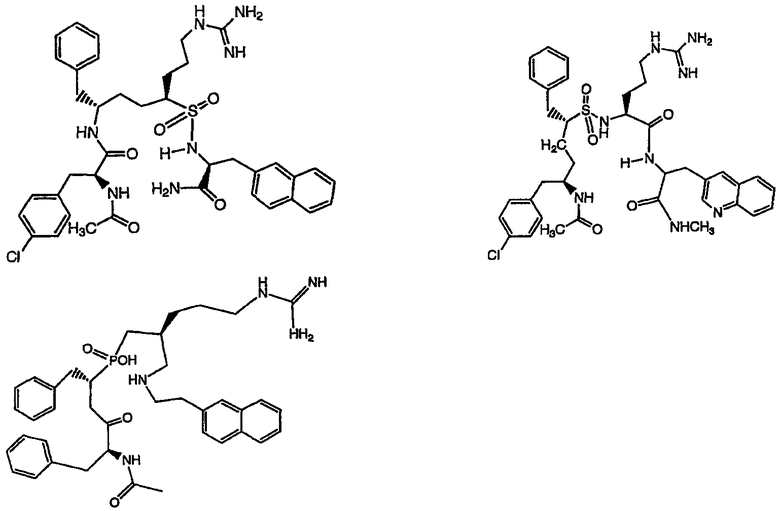
III. Синтез соединений
Соединения настоящего изобретения можно получить, используя ряд способов, включая методику твердофазного синтеза и методику жидкофазного синтеза в растворе. Ниже представлено общее описание методик как твердофазного, так и жидкофазного синтеза в растворе. В разделе VII приводятся несколько характерных примеров для каждой из указанных выше методик синтеза (пептидов).
A. Твердофазный синтез
Синтез линейных пептидов: Соединения синтезируют вручную (краткое описание представлено в разделе VII-C ниже) или автоматически либо на автоматизированном синтезаторе Perkin-Elmer Applied Biosystem Division (PE-ABD) Model 433, либо на SyntraPrep реакционной установке (производимой SyntraChem, Charlottesville, VA). Все реагенты, используемые для синтеза пептидов, могут быть поставлены PE-ABD. В случае PE-ABD автоматического синтезатора используют стандартную химическую
программу 0,25 ммоль FastMoc с контролем каждой ступени присоединения измерением проводимости. В общем случае (ступенчатый) синтез пептида с применением Fmoc-защитных групп в случае использования техники SPPS (твердофазный синтез пептидов) включает следующие стадии: 1) отщепление Fmoc-защитных групп пиперидином; 2) активация карбоксильной группы аминокислот; и 3) взаимодействие активированных аминокислот с аминоконцом пептидной цепи, связанной с полимером, с образованием пептидных связей. FastMoc-циклы, в которых аминокислоты активируют гексафторфосфатом 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU). 1,0 ммоль сухой защищенной аминокислоты в картридже растворяют в растворе HBTU, N,N-диизопропилэтиламина (DIEA) и добавляют 1-гидроксибензотриазол (HOBt) в N,N-диметилформамиде (DMF) с дополнительным N-метилпирролидоном (NMP). Активированная Fmoc аминокислота образуется почти мгновенно, и раствор переносят непосредственно в реакционный сосуд. Стадию снятия защиты Fmoc (стадия удаления защитной группы Fmoc) контролируют и регулируют измерением проводимости. Пептидная цепь строится на Rink Амидной смоле, так как требуется С-концевой амид. Ацетильную или бутильную группу вводят по N-концу пептидной цепи, после того как получают полную длину цепи пептида. Это осуществляется реакцией уксусного ангидрида или масляного ангидрида, (4,75% об.:об. уксусного ангидрида или масляного ангидрида, 0,2% HOBt масс.:об., 2,25% DIEA в NMP с α -аминогруппой N-концевого аминокислотного остатка. Конечный продукт синтеза в течение достаточного времени промывают NMP и дихлорметаном (DCM). Когда для пептидного синтеза используют
SyntraChem реакционную установку, в качестве реагента для активации, который заменяет HBTU, используют гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU).
Снятие защиты: Полимеры, содержащие синтезированные пептиды, выгружают из синтезатора и недолговременно сушат на воздухе. Используя 4,0-10,0 мл коктейля для расщепления (91% трифторуксусной кислоты (TFA), 2,3% этанодитиола, 2,3% тиоанизола и 2,3% фенола (мас.:об.) в воде) в течение 1,5-3,0 часов при комнатной температуре, отщепляют пептиды от смолы и в то же самое время удаляют защитные группы боковых цепей [O-трет-бутил (OtBu) для Asp, Glu, Tyr и Ser, пентаметилдигидробензофуран-5-сульфонил (Pbf) для Arg, трет-бутоксикарбонил (Boc) для Trp, Orn, Lys] в условиях снятия защиты. Раствор, содержащий продукты расщепления, отделяют от смолы фильтрованием. Затем осаждают пептид в фильтрате, добавляя 40 мл охлажденного эфира. Осадок пептида отфильтровывают и промывают охлажденным эфиром (4 x 40 мл). Для пептидов, которые не осаждаются в эфирном растворе из-за их высокой гидрофобности, эфир выпаривают в токе азота. Затем пептиды замораживают и лиофилизуют в течение более чем 24 часов. Пептиды регенерируют в раствор добавлением уксусной кислоты.
Очистка и характеристика: Порошок пептида вместе с другими побочными продуктами повторно растворяют в 50% растворе уксусной кислоты и подвергают очистке, инъецируя в колонку Vydac С-8, размером: ВД 1,0 см, длина 25 см, с размером частиц 5 мкм и размером пор 300 А. Используют систему ВЭЖХ Beckman System Gold с УФ-детектором с детектированием при двух длинах волн. Для
отделения пептидного продукта от других веществ устанавливают программируемый режим элюирования с линейным градиентом ацетонитрила и вводят в колонку. Элюат собирают в Pharmacia коллектор для сбора фракций и индивидуальные фракции после разделения подвергают анализу как методом аналитической ВЭЖХ, так и методом электрораспылительной масс-спектрометрии (MS) c целью идентификации продуктов, чтобы гарантировать их идентичность и чистоту.
B. Методика жидкофазного синтеза пептидов в растворе
Коммерчески доступные растворители и реагенты используют без очистки. Реакционные смеси перемешивают при помощи магнитной мешалки и их состав контролируют либо аналитической высокоэффективной жидкостной хроматографией (ВЭЖХ), либо тонкослойной хроматографией (ТСХ). Растворы концентрируют общепринятым способом, используя роторный испаритель Buchi при 15-25 мм рт. ст. ТСХ осуществляют, используя пластины, предварительно покрытые силикагелем 60 F254 с флуоресцентным индикатором. Визуализацию осуществляют стандартным способом с помощью УФ-излучения (254 нм). Флэш-хроматографию осуществляют на E. Merck силикагеле 60 (230-400 меш), используя требуемые элюенты; хроматографическое разделение контролировали анализами ТСХ. Аналитическую ВЭЖХ осуществляли либо на колонках MetaChem Kromasil C4 размером 4,6 x 250 мм, либо Polaris C18 с обращенной фазой (размер частиц 3,5 мк или 3,0 мк для С4 или С18, соответственно), используя градиент смеси 0,1% фосфорной кислоты в воде (A)/ацетонитрил (B) (5% B для C4 или 20% B для С18 до 100% на протяжении 20 мин, удерживание 5 мин) при объемном расходе
1,0 мл/мин; детектирование УФ-излучением как при 214 нм, так и при 254 нм. Препаративную ВЭЖХ осуществляли либо на колонке Polaris C18 с обращенной фазой размером 50 x 250 мм (размер частиц 10 мк, размер пор 100 Е), либо на колонке Rainin Dynamax C4 с обращенной фазой размером 41,4 x 250 мм (размер частиц 8 мк, размер пор 300 Е), используя градиент смеси 0,1% трифторуксусной кислоты в воде (А)/ацетонитрил (B) (5%→ 100% B на протяжении 55 мин, удерживание 10 мин); детектирование при помощи УФ-излучения при 214 нм.
C. Общие замечания
Признано, что предпочтительно использовать защитную группу для любой реакционноспособной функциональной группы, такой как карбоксил, гидроксил и т.п. Это является обычной практикой, использование которой находится в пределах экспериментального опыта специалиста в данной области.
Указанные стадии могут варьироваться, чтобы увеличить выход требуемого продукта. Для специалиста очевидно, что разумный подбор реактантов, растворителей и температур является важным фактором для успешного осуществления любого синтеза. Определение оптимальных условий и т.п. является общепринятой практикой. В соответствии с этим, специалист может получить целый ряд соединений, руководствуясь указанными выше общими описаниями, наряду с рекомендациями примеров Раздела VII.
Очевидно, что специалист в данной области органической химии может без труда выполнить обычные манипуляции с органическими соединениями без дальнейшего руководства; т.е. осуществление таких манипуляций находится в пределах
квалификации и практического опыта специалиста в данной области. Указанные выше манипуляции включают, но не ограничиваются ими, восстановление карбонильных соединений в их соответствующие спирты, окисление гидроксильных групп и т.п., ацилирование, ароматические замещения, как электрофильные, так и нуклеофильные, этерификация для получения простых и сложных эфиров и омыление и т.п. Примеры указанных выше манипуляций обсуждены в обычных публикациях, таких как March, Advanced Organic Chemistry (Wiley), Carey and Sundberg, Advanced Organic Chemistry (Vol. 2) и других публикациях, которые известны специалисту в данной области.
Для специалиста очевидно, что некоторые реакции лучше всего осуществлять, когда потенциально реакционноспособные функциональные группы в молекуле замаскированы или защищены, что дает возможность предотвратить любые нежелательные побочные реакции и/или увеличить выход целевой реакции. Зачастую специалистом в данной области используются защитные группы для того, чтобы повысить выход целевой реакции или избежать побочных реакций. Такие реакции имеются в литературе, и они также хорошо известны специалистам в данной области. Примеры многих таких манипуляций могут быть найдены, например, в T. Greene, Protecting Groups in Organic Synthesis. Конечно, аминокислоты с реакционноспособными боковыми цепями, используемые в качестве исходных веществ, предпочтительно блокируют с целью предотвращения протекания нежелательных побочных реакций.
IV. Функциональная активность и селективность в отношении меланокортиновых рецепторов
Функциональную активность можно оценить, используя различные способы, известные в данной области. Примерами таких способов являются измерение ответных реакций вторичного мессенджера, в частности cAMP, использование модифицированных клеточных систем, которые могут давать цветную реакцию после аккумулирования элементов вторичного мессенджера, таких как cAMP, например, как Chen et al. 1995 (Anal Biochem. 1995, 226, 349-54), Cytosensor Microphysiometer techniques (см. Boyfield et al. 1996), или можно воспользоваться исследованием физиологических эффектов, вызываемых соединениями изобретения, с использованием соединений изобретения, как таковых, или в сочетании с природными или синтетическими MSH-пептидами.
Соединения настоящего изобретения могут взаимодействовать предпочтительно (т.е. селективно) с MC-4 и/или MC-3, по сравнению c другими меланокортиновыми рецепторами. Селективность особенно важна, когда соединения вводят людям или другим животным, чтобы минимизировать ряд побочных эффектов, ассоциируемых с их введением. Селективность соединения в отношении MC-3/MC-4 определяется как отношение EС50 соединения для MC-1 рецептора (“EC50-MC-1”) к EC50 соединения для рецептора MC-3 (EC50-MC-3)/MC-4 (EC50-MC-4), при этом значения EC50 определяют, как описано выше. Формулы выглядят следующим образом:
MC-3-селективность = [EC50-MC-1]/[EC50-MC-3]
MC-4-селективность = [EC50-MC-1]/[EC50-MC-4]
Считают, что соединение проявляет “селективность в отношении MC-3 рецептора” в том случае, когда указанное выше
отношение “MC-3-селективность” равно, по крайней мере, приблизительно 10, предпочтительно, по крайней мере, приблизительно 100 и более предпочтительно, по крайней мере, приблизительно 500.
Соединение определяют, как имеющее “селективность в отношении MC-4 рецептора” в том случае, когда указанное выше отношение “MC-4-селективность” равно, по крайней мере, приблизительно 10, предпочтительно, по крайней мере, приблизительно 100 и более предпочтительно, по крайней мере, приблизительно 500.
V. Способы применения и композиции:
Исходя из способности заявляемых соединений проявлять агонизм или антагонизм в отношении рецептора МС-4 и/или МС-3, настоящее изобретение также относится к применению описанных в данном описании лигандов в способах лечения ожирения и других расстройств, связанных с отклонениями в массе тела, включая, например, анорексию и кахексию. Кроме того, соединения могут быть использованы в способах лечения нарушений, которые являются следствием нарушений, связанных с массой тела, включая, но, не ограничиваясь ими, инсулинорезистентность, интолерантность к глюкозе, сахарный диабет II Типа, коронарную болезнь сердца, повышенное кровяное давление, гипертензию, дислипидемию, рак (например, рак эндометрия, рак мозга, рак яичников, рак молочной железы, рак простаты, рак желчного пузыря, рак толстой кишки), менструальные отклонения, гирсутизм, бесплодие, болезнь желчного пузыря, ограниченное легочное поражение, приступы апноэ во сне, подагру, остеоартрит и тромбоэмболическую болезнь. Кроме того,
изобретение относится к лечению нарушений, имеющих отношение к физиологическому поведению, памяти (включая обучение), сердечно-сосудистой функции, воспалению, сепсису, кардиогенному и гиповолемическому шоку, половой дисфункции, эрекции полового члена, мышечной атрофии, росту и регенерации нерва, внутриматочному развитию плода и т.п.
Используемый в данном описании термин “лечение” означает, что, по меньшей мере, введение соединения настоящего изобретения ослабляет нарушение, действуя через рецептор МС-3 или МС-4. Таким образом, термины включают: предотвращение развития состояния болезни у млекопитающего, в частности, в том случае, когда оно имеет предрасположенность к приобретению такого заболевания, однако еще пока у него не диагностированного; ингибирование развития указанного состояния болезни; и/или облегчение или реверсию такого состояния болезни.
Соединения настоящего изобретения могут быть составлены в фармацевтические композиции, предназначенные для использования в лечении или профилактики указанных выше состояний болезни. Для этого используют стандартные методики приготовления лекарственных средств, такие как описаны в Remington’s Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., latest edition and Peptide and Protein Drug Delivery, Marcel Dekker, NY, 1991.
Композиции настоящего изобретения включают:
a. безопасное и эффективное количество соединения формулы (I); и
b. фармацевтически приемлемый наполнитель.
“Безопасное и эффективное количество” соединения формулы (I) представляет количество, которое эффективно для взаимодействия с рецептором МС-4 и/или МС-3, для животного, предпочтительно млекопитающего, более предпочтительно для человека, без чрезмерного проявления нежелательных побочных эффектов (таких как токсичность, раздражение или аллергическая реакция), соизмеримого с допустимым отношением польза/риск, при использовании способа данного изобретения. Очевидно, что конкретное “безопасное и эффективное количество” может варьироваться в зависимости от таких факторов, как конкретное состояние, подлежащее лечению; физическое состояние пациента, продолжительность лечения, характер сопутствующей терапии (если она вообще имеется), конкретная лекарственная форма, подлежащая использованию; применяемый эксципиент, растворимость соединения формулы (I) и необходимая для данной композиции схема приема лекарственного средства.
Помимо предлагаемого соединения, композиции настоящего изобретения содержат один или несколько фармацевтически приемлемых эксципиентов. Используемый в данном описании термин “фармацевтически приемлемый эксципиент” означает один или несколько совместимых твердых или жидких компонентов, которые являются подходящими для введения животному, предпочтительно млекопитающему, более предпочтительно человеку. Используемый в данном описании термин “совместимый” означает, что компоненты композиции могут быть смешаны с предлагаемым соединением и друг с другом до некоторой степени, так что при этом не будет иметь место взаимодействие, которое могло бы уменьшить
фармацевтическую эффективность композиции при обычных ситуациях ее применения. Фармацевтически приемлемые эксципиенты должны, конечно, иметь достаточно высокую чистоту и достаточно низкую токсичность, чтобы быть подходящими для введения животному, предпочтительно млекопитающему, более предпочтительно человеку, подлежащему лечению.
Некоторыми примерами веществ, которые могут быть полезными в качестве фармацевтически приемлемых эксципиентов или компонентов для композиции, являются сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и метилцеллюлоза; порошкообразный трагакант; солод; желатин; тальк; твердые смазывающие агенты, такие как стеариновая кислота и стеарат магния; растительные масла, такие как арахисовое масло, хлопковое масло, кунжутное масло, оливковое масло, кукурузное масло и масло теобромина; полиолы, такие как пропиленгликоль, глицерин, сорбит, маннит и полиэтиленгликоль; агар; альгиновая кислота, увлажняющие средства и смазывающие агенты, такие как лаурилсульфат натрия; красители; ароматизаторы; средства, способствующие таблетированию; стабилизаторы; антиоксиданты; консерванты; вода, свободная от пирогена; изотонический физиологический раствор; и буферы, такие как фосфатный, цитратный и ацетатный.
Выбор фармацевтически приемлемых эксципиентов, подлежащих использованию в сочетании с предлагаемым соединением, в основном определяется путем, при помощи которого соединение подлежит
введению. Если данное соединение подлежит введению путем инъекции, то предпочтительным фармацевтически приемлемым эксципиентом является стерильная вода, физиологический (солевой) раствор или их смеси, рН которых предпочтительно доводят до около 4-10 при помощи фармацевтического буфера; возможно также использование совместимого суспендирующего средства.
В частности, фармацевтически приемлемые эксципиенты для системного введения включают сахара, крахмалы, целлюлозу и ее производные, солод, желатин, тальк, сульфат кальция, лактозу, растительные масла, синтетические масла, полиолы, альгиновую кислоту, фосфатные, ацетатные и цитратные буферные растворы, эмульгаторы, изотонический физиологический раствор и свободную от пирогена воду. Предпочтительные фармацевтически приемлемые эксципиенты для парентерального введения включают пропиленгликоль, этилолеат, пирролидон, этанол и кунжутное масло. Предпочтительно, чтобы фармацевтически приемлемый эксципиент, в композициях для парентерального введения, составлял, по крайней мере, около 90% масс. от общей массы композиции.
Композиции настоящего изобретения предпочтительно формулируются в виде единичной дозированной формы. Используемый в данном описании термин “единичная дозированная форма” представляет композицию настоящего изобретения, содержащую количество соединения формулы (I), которое является подходящим для введения животному, предпочтительно млекопитающему, более предпочтительно человеку, в единичной дозе, согласно установленной медицинской практике. Указанные выше композиции
предпочтительно содержат от около 1 мг до около 750 мг, более предпочтительно от около 3 мг до около 500 мг, еще более предпочтительно от около 5 мг до около 300 мг соединения формулы (I).
Композиции настоящего изобретения могут быть представлены в любой из множества форм, подходящих для перорального, ректального, местного, назального, глазного, трансдермального, легочного или парентерального введения. В зависимости от желаемого конкретного пути введения, может быть использован целый ряд известных в данной области фармацевтически приемлемых эксципиентов. Такие эксципиенты включают твердые или жидкие наполнители, разбавители, гидротропы, поверхностно-активные вещества и инкапсулирующие вещества. Могут быть включены необязательные фармацевтически активные вещества, которые существенно не препятствуют ингибирующей активности соединения формулы (I). Количество эксципиента, используемого в сочетании с соединением формулы (I), должно быть достаточным для обеспечения введения на практике количества вещества, отвечающего единичной дозе соединения. Методики и композиции для приготовления лекарственных форм, используемых в способах настоящего изобретения, описаны в следующих ниже публикациях, которые включены в данное описание в виде ссылки: Modern Pharmaceutics, Chapters 9 and 10 (Banker & Rhodes, editors, 1979); Lieberman et al., Pharmaceutical Dosage Forms: Tablets (1981); and Ansel, Introduction to Pharmaceutical Dosage Forms 2d Edition (1976).
Могут быть использованы различные пероральные лекарственные формы, включая такие твердые формы, как таблетки, капсулы,
гранулы и сыпучие порошки. Указанные пероральные формы включают безопасное и эффективное количество, обычно, по крайней мере, около 5%, и предпочтительно от около 25% до около 50%, соединения формулы (I). Таблетки могут быть спрессованными, предназначенными для получения порошка, покрытыми энтеросолюбильным покрытием, покрытыми сахаром, покрытыми пленкообразующим веществом или спрессованными из нескольких слоев, могут содержать подходящие связующие, смазывающие агенты, разбавители, дезинтеграторы, красители, ароматизаторы, стимулирующие текучесть и способствующие плавлению агенты. Жидкие пероральные лекарственные формы включают водные растворы, эмульсии, суспензии, растворы и/или суспензии, восстановленные из нешипучих гранул, и шипучие препараты, восстановленные из шипучих гранул, содержащие подходящие растворители, консерванты, эмульгаторы, суспендирующие агенты, разбавители, подсластители, способствующие плавлению агенты, красители и ароматизаторы.
Фармацевтически приемлемые эксципиенты, подходящие для приготовления дозированных лекарственных форм для перорального введения, хорошо известны в данной области. Таблетки обычно включают обычные фармацевтически совместимые адъюванты в качестве инертных разбавителей, такие как карбонат кальция, карбонат натрия, маннит, лактоза и целлюлоза; связующие, такие как крахмал, желатин, поливинилпирролидон и сахароза; дезинтеграторы, такие как крахмал, альгиновая кислота и кроскармелоза; смазывающие агенты, такие как стеарат магния, стеариновая кислота и тальк. Для повышения характеристик текучести порошкообразной смеси могут быть использованы
глиданты, такие как диоксид кремния. Для придания внешнего вида могут быть использованы красители, такие как красители FD&C. Подсластители и ароматизаторы, такие как аспартам, сахарин, ментол, перечная мята и фруктовые отдушки, являются полезными адъювантами для разжевываемых таблеток. Капсулы обычно включают один или несколько твердых разбавителей, описанных выше. Выбор компонентов эксципиентов зависит от предусматриваемых вторичных характеристик, подобных таким, как вкус, стоимость и стабильность при хранении, которые не являются существенными для целей настоящего изобретения, подбор соответствующих эксципиентов может быть без труда сделан специалистом в данной области.
Пероральные композиции также включают жидкие растворы, эмульсии, суспензии и т.п. Фармацевтически приемлемые эксципиенты, подходящие для приготовления таких композиций, хорошо известны в данной области. Типичные компоненты эксципиентов для сиропов, эликсиров, эмульсий и суспензий включают этанол, глицерин, пропиленгликоль, полиэтиленгликоль, жидкую сахарозу, сорбит и воду. Для суспензии типичные суспендирующие агенты включают метилцеллюлозу, натрийкарбоксиметилцеллюлозу, Avicel® RC-591, трагакант и альгинат натрия; типичные увлажнители включают лецитин и полисорбат 80; и типичные консерванты включают метилпарабен, пропилпарабен и бензоат натрия. Пероральные жидкие композиции могут также содержать один или несколько компонентов, таких как подсластители, ароматизаторы и красители, описанные выше.
Кроме того, такие композиции могут быть покрыты
пленкообразующим покрытием обычными способами, обычно растворимость таких покрытий носит рН-зависимый или время-зависимый характер, так что заявляемое соединение высвобождается в желудочно-кишечном тракте вблизи желаемого места применения, или в различное время, пролонгируя желаемое действие. Такие лекарственные формы обычно включают в качестве одного или более пленкообразующих, но не ограничиваются ими, ацетат-фталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы, этилцеллюлозу, покрытия Eudragit® , воски и шеллак.
Так как соединения настоящего изобретения имеют пептидную природу, предпочтительным способом их введения является парентеральное (более предпочтительно внутривенная инъекция) или назальное введение, в виде единичной дозированной формы. Предпочтительные единичные дозированные формы включают суспензии и растворы, содержащие безопасное и эффективное количество соединения формулы (I). При введении парентеральным путем единичная дозированная форма обычно включает от около 1 мг до около 3 г, более предпочтительно от около 10 мг до около 1 г, соединения формулы (I), хотя количество вводимого соединения может зависеть, например, от его относительного сродства к подтипам рецепторов МС-4/МС-3, его селективности в сравнении с другими рецепторами, включая другие меланокортиновые рецепторы, и т.д.
Композиции настоящего изобретения необязательно могут включать другие активные лекарственные вещества.
Другие композиции, используемые для достижения системной
доставки заявляемых соединений, включают подъязычные и трансбуккальные лекарственные формы. Такие композиции обычно включают одно или несколько растворимых веществ-наполнителей, таких как сахароза, сорбит и маннит; и связующие, такие как акация, микрокристаллическая целлюлоза, карбоксиметилцеллюлоза и гидроксипропилметилцеллюлоза. Также могут быть включены указанные выше глиданты, смазывающие агенты, подсластители, красители, антиоксиданты и ароматизаторы.
VI. Способы введения:
Как отмечалось, композиции настоящего изобретения можно вводить местно или системно. Системное применение включает любой способ введения соединения формулы (I) в ткани организма, например, интраартикулярное, внутриоболочечное, эпидермальное, внутримышечное, трансдермальное, внутривенное, интраперитонеальное, подкожное, подъязычное, ректальное, назальное, легочное и пероральное введение. Соединения формулы (I) настоящего изобретения предпочтительно вводят системно, более предпочтительно парентерально и наиболее предпочтительно посредством внутривенной инъекции.
Индивидуальная доза соединения, подлежащего введению, а также продолжительность лечения, независимо от того, является ли лечение местным или системным, являются взаимозависимыми. Доза и схема лечения также будут зависеть от таких факторов, как конкретное используемое соединение формулы (I), показание к лечению, персональные отличительные признаки субъекта (как например, масса тела), соблюдение схемы лечения и присутствие и серьезность любых побочных эффектов лечения.
Обычно, для взрослого человека, массой приблизительно 70 килограмм, вводят в день при системном введении от около 1 мг до около 6 г, более типично от около 100 мг до около 3 г, соединения формулы (I). Очевидно, что эти границы дозы представлены только в качестве примера, и что суточное введение можно регулировать в зависимости от факторов, перечисленных выше.
Как известно и осуществляется на практике в данной области, все составы для парентерального введения должны быть стерильными. Для млекопитающих, особенно людей, (принимая приблизительную массу тела субъекта 70 килограмм) предпочтительны индивидуальные дозы от около 0,001 мг до около 100 мг.
Предпочтительным способом системного введения является внутривенная доставка. При использовании указанного способа доставки предпочтительны индивидуальные дозы от около 0,01 мг до около 100 мг, предпочтительно от около 0,1 мг до около 100 мг.
Во всех приведенных выше способах введения, несомненно, соединения настоящего изобретения могут быть введены, как таковые, или в виде смесей, и, кроме того, композиции могут содержать дополнительные лекарственные средства или наполнители, если это целесообразно по показанию.
Соединение настоящего изобретения может быть доставлено в предпочтительное место организма (человека или животного), используя подходящую систему доставки лекарственного средства. Системы доставки лекарственных средств хорошо известны в данной области. Например, техника доставки лекарственного средства,
используемая для соединений настоящего изобретения, состоит в конъюгировании соединения с активной молекулой, способной транспортироваться через биологический барьер (см., например, Zlokovic, B.V., Pharmaceutical Research, Vol. 12, pp. 1395-1406 (1995). Конкретный пример представляет связывание соединения настоящего изобретения с фрагментами инсулина с целью обеспечения транспорта лекарственного средства через гематоэнцефалический барьер (Fukuta, M., et al. Pharmaceutical Res., Vol. 11, pp. 1681-1688 (1994)). В качестве общих обзоров технологий для доставки лекарственных средств, подходящих для соединений изобретения, см. Zlokovic, B.V., Pharmaceutical Research, Vol. 12, pp. 1395-1406 (1995) and Pardridge, WM, Pharmacol. Toxicol, Vol. 71, pp. 3-10 (1992).
VII. Типичные примеры синтеза
В следующих ниже примерах изобретение описано более подробно со ссылками на ряд предпочтительных вариантов его осуществления, которые приводятся только в целях иллюстрации, и следует иметь в виду, что данные примеры никоим образом не ограничивают данное изобретение.
В примерах используют следующие аббревиатуры:
Ac: ацетил[-C(O)CH3]
Atc: (D,L)-2-аминотетралин-2-карбоновая кислота
Aun: аминоундекановая
Bc: бутаноил [-C(O)(CH2)2CH3]
Boc: трет-бутилоксикарбонил
DCM: дихлорметан
DEA: диэтиламин
DMF: N,N-диметилформамид
DMAP: 4-диметиламинопиридин
DME: 1,2-диметоксиэтан
DIEA: диизопропилэтиламин
DPPA: дифенилфосфорилазид
EtOAc: этилацетат
EDCI: гидрохлорид 1-этил-3-(3’-диметиламинопропил)карбодиимида
Fmoc: 9-флуоренилметоксикарбонил
HOBt: N-гидроксибензотриазол, моногидрат
HOAt: 1-гидрокси-7-азабензотриазол
i-PrOH: 2-пропанол
MeOH: метанол
NMM: N-метилморфолин
OtBu: трет-бутокси [-O-C(CH3)3]
Pbf: 2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонилPmc: 2,2,5,7,8-пентаметил-6-хромансульфонилp-TSA: п-толуолсульфонат
PyBOP: гексафторфосфат бензотриазол-1-илокси-трис-пирролидинофосфония
PyBroP: гексафторфосфат бром-трис-пирролидинофосфония
tBu: трет-бутил [-C(CH3)3]
TEA: триэтиламин
TFA: трифторуксусная кислота
THF: тетрагидрофуран
A. Автоматический твердофазный синтез пептидов
Пример 1
Синтез Ac-YfRW-NH2
Исходя из степени замещения Rink Амидной смолы 0,55 ммоль/г, взвешивают 0,45 г смолы для 0,25 ммольного масштаба синтеза. Функционирование пептидного синтезатора PE-ABD 433 проверяют до его использования различными поточными тестами, чтобы гарантировать точную подачу реагента. Fmoc аминокислоты: Tyr-OtBu, Arg-Pmc и Trp-Boc поставлялись коммерчески в 1 ммоль-картриджах. Fmoc-phe (387 мг, 1 ммоль) определяют и добавляют в картриджи для синтеза. Свежеприготовленный раствор уксусного ангидрида загружают в колбу #4 установки. Другие реагенты и растворители для синтеза поставляются коммерчески и загружаются в установку согласно прилагаемой инструкции. Для синтеза пептида используют химическую программу, называемую NAc-0,25ммоль MonPrePk. Снятие защиты Fmoc контролируют и регулируют измерением проводимости, при этом установленные критерии проводимости составляют 5% или менее, по сравнению с предыдущим циклом снятия защиты.
Смолу сушат на воздухе и переносят в стеклянный сосуд и добавляют свежеприготовленный реагент для расщепления (10 мл). Реакцию снятия защиты осуществляют в течение 2 часов при комнатной температуре при постоянном перемешивании. Затем супернатант отделяют от смолы фильтрованием. Затем синтезированный пептид осаждают в эфирном слое добавлением 40 мл охлажденного эфира. Осадки пептида центрифугируют (Heraeus Labofuge 400, Rotor #8179) при 3500 об/мин в течение четырех
минут. Эфир сливают и добавляют 40 мл свежего охлажденного эфира, чтобы промыть осадок пептида. Стадии промывки повторяют три раза, чтобы удалить побочные продукты реакции снятия защиты. Осадки конечного пептида сушат вымораживанием на протяжении ночи. Идентификацию и чистоту пептида определяют как методом МС, так и методом ВЭЖХ. Определяют ожидаемую молекулярную массу пептида.
Пептид повторно растворяют в 50% уксусной кислоте и очищают ВЭЖХ с обращенной фазой (С8), используя линейный градиент 0-70% растворителя В с растворителем А в течение 70 минут при объемном расходе 3 мл/мин. Состав растворителей А и В следующий: A: 0,1% TFA, 2% ацетонитрил в воде; B: 0,1% TFA в 95% ацетонитриле. Фракции собирают каждые 0,25 мин. Аликвоты каждой фракции анализируют как методом МС, так и методом ВЭЖХ с обращенной фазой (ОФ-ВЭЖХ). Фракции, которые содержат один пик поглощения при 220 нм с ожидаемой массой пептида, объединяют и лиофилизуют. Конечную чистоту пептида определяют аналитической ОФ-ВЭЖХ объединенных фракций.
Пептиды, описанные в примерах 2-54, представленных ниже, без труда синтезируют в соответствии с методикой примера 1, но с внесенными изменениями, которые особо оговариваются.
Пример 2
Синтез Ac-YFRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-Phe вместо Fmoc-D-Phe.
Пример 3
Синтез Ac-FfRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 4
Синтез Ac-PFRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-Pro вместо Fmoc-L-Tyr(OtBu).
Пример 5
Синтез Ac-AfRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-Ala вместо Fmoc-L-Tyr(OtBu).
Пример 6
Синтез Ac-(2-Nal)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-(2-Nal) вместо Fmoc-L-Tyr(OtBu).
Пример 7
Синтез Ac-YfR(2-Nal)-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-(2-Nal) вместо Fmoc-L-Trp(Boc).
Пример 8
Синтез Ac-YfR(1-Nal)-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-(1-Nal) вместо Fmoc-L-Trp(Boc).
Пример 9
Синтез Ac-YfHW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-His(Trt) вместо Fmoc-L-Arg(Pmc).
Пример 10
Синтез Ac-Y(D-2-Nal)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-(2-Nal) вместо Fmoc-D-Phe.
Пример 11
Синтез Ac-Y(L-N-Me-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-N-Me-Phe вместо Fmoc-D-Phe.
Пример 12
Синтез Ac-A(D-N-Me-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-N-Me-Phe вместо Fmoc-L-Phe, и Fmoc-L-Ala используют вместо Fmoc-L-Tyr(OtBu).
Пример 13
Синтез Ac-YF(L-N-Me-Arg)W-NH
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-N-Me-Arg(Mtr) вместо Fmoc-L-Arg(Pmc), и Fmoc-L-Phe используют вместо Fmoc-D-Phe.
Пример 14
Синтез Ac-Yf(L-N-Me-Arg)W-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-N-Me-Arg(Mtr) вместо Fmoc-L-Arg(Pmc).
Пример 15
Синтез Ac-(L-N-Me-Tyr)FRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-N-Me-Tyr(Bzl) вместо Fmoc-L-Tyr(OtBu) и Fmoc-L-Phe используют вместо Fmoc-D-Phe.
Пример 16
Синтез Ac-(L-N-Me-Tyr)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-N-Me-Tyr(Bzl) вместо Fmoc-L-Tyr(OtBu).
Пример 17
Синтез Ac-Y(D-4-хлор-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-4-хлор-Phe вместо Fmoc-D-Phe.
Пример 18
Синтез Ac-Y(D-4-фтор-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-4-фтор-Phe вместо Fmoc-D-Phe.
Пример 19
Синтез Ac-Y(D-3,4-дихлор-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-3,4-дихлор-Phe вместо Fmoc-D-Phe.
Пример 20
Синтез Ac-Y(D-4-Me-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-4-Me-Phe вместо Fmoc-D-Phe.
Пример 21
Синтез Ac-Y(D-4-нитро-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-4-нитро-Phe вместо Fmoc-D-Phe.
Пример 22
Синтез Ac-Y(D-фенилглицин)RW-NH2
Получают согласно примеру 1, за исключением того, что
используют Fmoc-D-фенилглицин вместо Fmoc-D-Phe.
Пример 23
Синтез Ac-Y(D-4-гомо-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-4-гомо-Phe вместо Fmoc-D-Phe.
Пример 24
Синтез Ac-Y(D-стирилаланин)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-стирилаланин вместо Fmoc-D-Phe.
Пример 25
Синтез Ac-Y(D-4-тиенилаланин)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-4-тиенилаланин вместо Fmoc-D-Phe.
Пример 26
Синтез Ac-Y(D-3-фтор-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-3-фтор-Phe вместо Fmoc-D-Phe.
Пример 27
Синтез Ac-(L-4-фтор-Phe)(D-4-фтор-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-фтор-Phe вместо Fmoc-L-Tyr(OtBu) и Fmoc-D-4-фтор-Phe используют вместо Fmoc-D-Phe.
Пример 28
Синтез Ac-Y(D-2-фтор-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-2-фтор-Phe вместо Fmoc-D-Phe.
Пример 29
Синтез Ac-(L-4-хлор-Phe)(D-4-фтор-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-хлор-Phe вместо Fmoc-L-Tyr(OtBu) и Fmoc-D-4-фтор-Phe используют вместо Fmoc-D-Phe.
Пример 30
Синтез Ac-(L-4-хлор-Phe)(D-4-фтор-Phe)RW(N-Me-Gly)NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-хлор-Phe вместо Fmoc-L-Tyr(OtBu), Fmoc-D-4-фтор-Phe используют вместо Fmoc-D-Phe и используют дополнительно Fmoc-N-Me-Gly.
Пример 31
Синтез Ac-(L-4-хлор-Phe)fRW(N-Me-Gly)-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-хлор-Phe вместо Fmoc-L-Tyr(OtBu) и используют дополнительно Fmoc-N-Me-Gly.
Пример 32
Синтез Ac-(L-4-хлор-Phe)(D-4-фтор-Phe)R-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-хлор-Phe вместо Fmoc-L-Tyr(OtBu), Fmoc-D-4-фтор-Phe используют вместо Fmoc-D-Phe и не используют Fmoc-L-Trp(Boc).
Пример 33
Синтез Ac-(L-4-хлор-Phe)(D-4-фтор-Phe)RG-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-хлор-Phe вместо Fmoc-L-Tyr(OtBu), Fmoc-D-4-фтор-Phe используют вместо Fmoc-D-Phe и Fmoc-L-Gly используют
вместо Fmoc-L-Trp(Boc).
Пример 34
Синтез Ac-(L-3,4-дифтор-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-3,4-дифтор-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 35
Синтез Ac-Y(D-2-Me-Phe)RW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-D-2-Me-Phe вместо Fmoc-D-Phe.
Пример 36
Синтез Ac-(L-4-бром-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-бром-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 37
Синтез Ac-(L-4-иод-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-иод-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 38
Синтез Ac-(L-пентафтор-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-пентафтор-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 39
Синтез Ac-(L-4-нитро-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-нитро-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 40
Синтез Ac-(L-аминометил-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-аминометил-Phe(Boc) вместо Fmoc-L-Tyr(OtBu).
Пример 41
Синтез Ac-(L-тетраизохинолин-3-карбоновая кислота)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-тетраизохинолин-3-карбоновую кислоту вместо Fmoc-L-Tyr(OtBu).
Пример 42
Синтез Ac-(L-гомо-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-гомо-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 43
Синтез Ac-(L-бифенилаланин)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-бифенилаланин вместо Fmoc-L-Tyr(OtBu).
Пример 44
Синтез Ac-(L-4-SO3-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-SO3-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 45
Синтез Ac-(L-2,6-диметил-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-2,6-диметил-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 46
Синтез Ac-(L-4-метил-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-метил-Phe вместо Fmoc-L-Tyr(OtBu).
Пример 47
Синтез Ac-(L-4-NH-Phe)fRW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-4-NH-Phe(Boc) вместо Fmoc-L-Tyr(OtBu).
Пример 48
Синтез Ac-YfKW-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-Lys(Boc) вместо Fmoc-L-Arg(Pmc).
Пример 49
Синтез Ac-Yf(Orn)W-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-Orn(Boc) вместо Fmoc-L-Arg(Pmc).
Пример 50
Синтез Bc-HFRW(N-Me-Gly)-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-His(Trt) вместо Fmoc-L-Tyr(OtBu), Fmoc-L-Phe используют вместо Fmoc-D-Phe, масляный ангидрид используют вместо уксусного ангидрида и используют дополнительно Fmoc-L-N-Me-Gly.
Пример 51
Синтез Bc-HfRW(N-Me-Gly)-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-His(Trt) вместо Fmoc-L-Tyr(OtBu), масляный ангидрид используют вместо уксусного ангидрида и используют дополнительно Fmoc-L-N-Me-Gly.
Пример 52
Синтез Bc-YfRW(N-Me-Gly)-NH2
Получают согласно примеру 1, за исключением того, что масляный ангидрид используют вместо уксусного ангидрида и используют дополнительно Fmoc-L-N-Me-Gly.
Пример 53
Синтез Bc-YFRW(N-Me-Gly)-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-Phe вместо Fmoc-D-Phe, масляный ангидрид используют вместо уксусного ангидрида и используют дополнительно Fmoc-L-N-Me-Gly.
Пример 54
Синтез Bc-FfRW(N-Me-Gly)-NH2
Получают согласно примеру 1, за исключением того, что используют Fmoc-L-Phe вместо Fmoc-L-Tyr(OtBu), масляный ангидрид используют вместо уксусного ангидрида и используют дополнительно Fmoc-L-N-Me-Gly.
B. Жидкофазный синтез в растворе
Пример 55
Синтез (нафталин-1-илметил)амида 5-гуанидино-2-[3-фенил-2-(3-фенилпропиламино)пропиониламино]пентановой кислоты
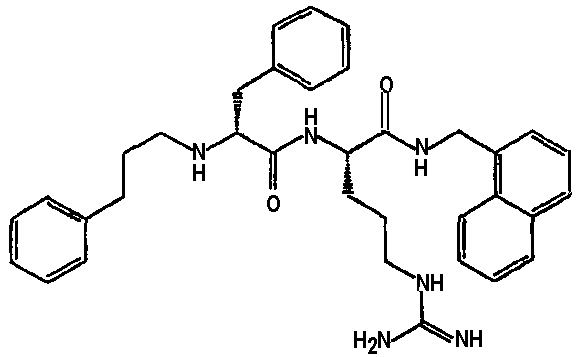
Метиловый эфир 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты
К раствору 2-(R)-(2-трет-бутоксикарбониламино-3-фенилпропионовой кислоты (2,0 г, 7,54 ммоль) и метилового эфира
2-(S)-амино-5-нитрогуанидинопентановой кислоты (2,0 г, 1,1 экв.) в 100 мл безводного DMF добавляют гидрохлорид 1-[3-(диметиламино)пропил)-3-этилкарбодиимида (1,5 г, 1,5 экв.), 1-гидроксибензотриазол (2,16 г, 1,4 экв.) и триэтиламин (3,0 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение 20 часов и затем растворители удаляют при пониженном давлении. Полученный остаток распределяют между 10% карбонатом натрия (100 мл) и метиленхлоридом (100 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворитель удаляют при пониженном давлении. Полученное неочищенное вещество очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
2-(S)-(2-(R)-трет-Бутоксикарбониламино-3-фенилпропиониламино)-5-нитрогуанидинопентановая кислота
К раствору метилового эфира 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (3,6 г, 7,5 ммоль) в тетрагидрофуране (100 мл) добавляют моногидрат гидроксида лития (387 мг, 1,2 экв.) и воду (10 мл). После перемешивания при комнатной температуре в течение одного часа реакционную смесь нейтрализуют трифторуксусной кислотой (0,7 мл) и растворители удаляют при пониженном давлении. Полученный остаток распределяют между водой (200 мл) и этилацетатом (200 мл). Водный слой экстрагируют этилацетатом (3× 250 мл), органическую фазу объединяют и сушат над безводным сульфатом магния, фильтруют и растворитель удаляют при пониженном давлении. Неочищенное
вещество используют без дальнейшей очистки.
трет-Бутиловый эфир (1-(S)-{4-нитрогуанидино-1-[нафталин-1-илметил)карбамоил]бутилкарбамоил}-2-(R)-фенилэтил)карбаминовой кислоты
К раствору 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-гуанидинопентановой кислоты (1,0 г, 2,15 ммоль) и C-нафталин-1-илметиламина (0,377 мл, 1,2 экв.) в 50 мл безводного DMF добавляют гидрохлорид 1-[3-(диметиламино)пропил)-3-этилкарбодиимида (532 мг, 1,5 экв.), 1-гидроксибензотриазол (376, 1,4 экв.) и триэтиламин (0,9 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение 20 часов и затем растворители удаляют при пониженном давлении. Полученный остаток распределяют между 10% карбонатом натрия (75 мл) и хлороформом (75 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворитель удаляют при пониженном давлении. Полученное неочищенное вещество очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония, получая указанное в заголовке соединение.
Соль трифторуксусной кислоты (нафталин-1-илметил)амида 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты
К раствору трет-бутилового эфира (1-(S)-{4-нитрогуанидино-[1-нафталин-1-илметилкарбамоил]бутилкарбамоил}-2-(R)-фенилэтил)карбаминовой кислоты (1,1 экв., 1,82 ммоль) в метиленхлориде (100 мл) добавляют трифторуксусную кислоту (50 мл). Полученный раствор перемешивают при комнатной температуре в
течение трех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают препаративной ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
(Нафталин-1-илметил)амид 5-нитрогуанидино-2-[3-фенил-2-(3-фенилпропиламино)пропиониламино]пентановой кислоты
К суспензии (нафталин-1-илметил)амида 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-гуанидинопентановой кислоты (750 мг, 1,21 ммоль), 3-фенилпропиональдегида (0,159 мл, 1,0 экв.) и активированных молекулярных сит (4 ангстрем, измельченные) добавляют триэтиламин (0,247 мл, 1,5 экв.). Полученную суспензию перемешивают при комнатной температуре в течение 24 часов и затем доводят рН до 5 уксусной кислотой. Затем добавляют 1,0М раствор цианоборгидрида натрия в тетрагидрофуране (1,44 мл, 1,2 экв.) со скоростью 0,2 мл/мин. с использованием шприца-насоса. Полученную суспензию перемешивают при комнатной температуре в течение 24 часов, фильтруют через целит и растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
(Нафталин-1-илметил)амид 5-гуанидино-2-[3-фенил-2-(3-фенилпропиламино)пропиониламино]пентановой кислоты
К раствору (нафталин-1-илметил)амида 5-нитрогуанидино-2-[3-фенил-2-(3-фенилпропиламино)пропиониламино]пентановой кислоты (140 мг, 0,165 ммоль) в метаноле (30 мл) добавляют уксусную кислоту (3 мл) и 5% палладий на сульфате бария (100 мг). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов и затем фильтруют через целит. Растворители удаляют при пониженном давлении и неочищенный
продукт очищают ВЭЖХ с обращенной фазой.
Пример 56
Синтез (2-нафталин-2-илэтил)амида 5-гуанидино-2-(S)-[3-фенил-2-(R)-пропиламино)пропиониламино]пентановой кислоты
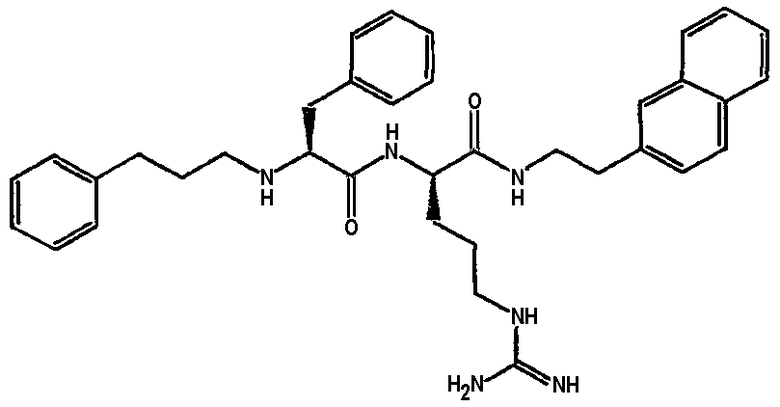
2-Нафталин-2-илэтиловый эфир толуол-4-сульфокислоты
К раствору 2-нафталин-2-илэтанола (3,0 г, 17,4 ммоль) в тетрагидрофуране (50 мл) добавляют ангидрид пара-толуолсульфокислоты (6,8 г, 1,2 экв.) и триэтиламин (7,1 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение одного часа и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают флэш-хроматографией на силикагеле (20% этилацетат/гексан), получая указанное в заголовке соединение.
2-(2-Азидоэтил)нафталин
К раствору 2-нафталин-2-илэтиловому эфиру толуол-4-сульфокислоты (5,0 г, 15,3 ммоль) в DMF (100 мл) добавляют азид натрия (1,3 г, 1,3 экв.). Полученную суспензию нагревают до 80° С в течение двадцати четырех часов и затем охлаждают до комнатной температуры. Растворитель удаляют при пониженном давлении и остаток распределяют между этилацетатом (200 мл) и водой (200 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворитель удаляют при пониженном давлении,
получая указанное в заголовке соединение. Неочищенное вещество используют без дополнительной очистки.
2-Нафталин-2-илэтиламин
К раствору 2-(2-азидоэтил)нафталина (3,0 г, 15,2 ммоль) в тетрагидрофуране (100 мл) добавляют трифенилфосфин (6,0 г, 1,5 экв.) и воду (5 мл). Полученный раствор кипятят с обратным холодильником в течение трех часов и затем охлаждают до комнатной температуры. Растворители удаляют при пониженном давлении и неочищенное вещество очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония). Затем очищенное вещество превращают в соль трифторуксусной кислоты, добавляя избыток трифторуксусной кислоты, и последующее удаление избытка кислоты выпариванием при пониженном давлении дает указанное в заголовке соединение.
трет-Бутиловый эфир {1-(S)-[4-нитрогуанидино-1-(2-нафталин-2-илэтилкарбамоил)бутилкарбамоил]-2-(R)-фенилэтил}карбаминовой кислоты
К раствору 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (1,0 экв, 2,14 ммоль) и 2-(2-азидоэтил)нафталина (733 мг, 1,2 экв.) в DMF (50 мл) добавляют гидрохлорид 1-[3-(диметиламино)пропил)-3-этилкарбодиимида (614 мг, 1,5 экв), 1-гидроксибензотриазол (434 мг, 1,5 экв.) и триэтиламин (0,877 мл, 3 экв.). Полученную суспензию перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество распределяют между 10% карбонатом натрия (75 мл) и метиленхлоридом (75 мл).
Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенное вещество очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
(2-Нафталин-2-илэтил)амид 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитроогуанидинопентановой кислоты
К раствору трет-бутилового эфира {1-(S)-[4-нитрогуанидино-1-(2-нафталин-2-илэтилкарбамоил)бутилкарбамоил]-2-(R)-фенилэтил}карбаминовой кислоты (1,5 г, 2,04 ммоль) в метиленхлориде (100 мл) добавляют трифторуксусную кислоту (50 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и растворители удаляют при пониженном давлении. Неочищенное вещество очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
(2-Нафталин-2-илэтил)амид 5-нитрогуанидино-2-(S)-[3-фенил-2-(R)-пропиламино)пропиониламино]пентановой кислоты
К раствору (2-нафталин-2-илэтил)амида 2-(S)-(2-(R)-амино-3-фенилпропиламино)-5-нитрогуанидинопентановой кислоты (600 мг, 1,15 ммоль) в тетрагидрофуране (50 мл) добавляют 3-фенилпропиональдегид (0,121 мл, 0,8 экв.) и молекулярные сита (4 ангстрем, измельченные). Полученную суспензию перемешивают при комнатной температуре в течение двадцати четырех часов и затем доводят рН до 5 уксусной кислотой. К раствору добавляют цианоборгидрид натрия (1,38 мл, 1,2 экв., 1,0M раствор в тетрагидрофуране) при скорости 0,2 мл/час, используя шприц-
насос. Полученную суспензию перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество фильтруют и затем очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
(2-Нафталин-2-илэтил)амид 5-нитрогуанидино-2-(S)-[3-фенил-2-(R)-пропиламино)пропиониламино]пентановой кислоты
К раствору (2-нафталин-2-илэтил)амида 5-нитрогуанидино-2-(S)-[3-фенил-2-(R)-пропиламино)пропиониламино]пентановой кислоты (294 мг, 0,46 ммоль) в 50 мл метанола добавляют уксусную кислоту (5 мл) и 5% палладий на сульфате бария (294 мг). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов, фильтруют через целит и растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 57
Синтез (2-нафталин-1-илэтил)амида 5-гуанидино-2-(S)-[3-фенил-2-(R)-(3-фенилпропиламино)пропиониламино]пентановой кислоты
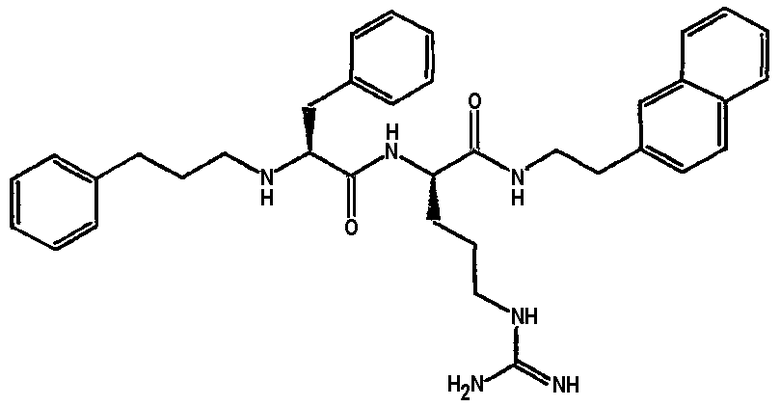
2-Нафталин-1-илэтиловый эфир толуол-4-сульфокислоты
К раствору 2-нафталин-1-илэтанола (3,0 г, 17,4 ммоль) в тетрагидрофуране (50 мл) добавляют ангидрид пара-толуолсульфокислоты (6,8 г, 1,2 экв.) и триэтиламин (7,1 мл, 3
экв.). Полученный раствор перемешивают при комнатной температуре в течение одного часа и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (20% этилацетат/гексаны), получая указанное в заголовке соединение.
1-(2-Азидоэтил)нафталин
К раствору 2-нафталин-1-илэтилового эфира толуол-4-сульфокислоты (5,2 г, 15,9 ммоль) в DMF (100 мл) добавляют азид натрия (1,3 г, 1,3 экв). Полученный раствор нагревают до 80° С в течение двадцати четырех часов, затем охлаждают до комнатной температуры и растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом и водой, органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении, получая указанное в заголовке соединение. Неочищенный продукт используют без дополнительной очистки.
2-Нафталин-1-илэтиламин
К раствору 1-(2-азидоэтил)нафталина (3,0 г, 15,23 ммоль) в тетрагидрофуране (10 мл) добавляют трифенилфосфин (6,0 г, 1,5 экв.) и воду (5 мл). Полученный раствор кипятят с обратным холодильником в течение трех часов и затем охлаждают до комнатной температуры. Растворители удаляют при пониженном давлении и неочищенный продукт очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение. Добавление избыточного количества трифторуксусной кислоты с последующим выпариванием дает соль трифторуксусной кислоты.
трет-Бутиловый эфир {1-(S)-[4-нитрогуанидино-1-(2-нафталин-1-илэтилкарбамоил)бутилкарбамоил]-2-(R)-фенилэтил]карбаминовой кислоты
К раствору метилового эфира 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (1,0 г, 2,1 ммоль) и 2-нафталин-1-илэтиламина (733 мг, 1,2 экв.) в DMF (50 мл) добавляют 1-гидроксибензотриазол (434 мг, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (614 мг, 1,5 экв.) и триэтиламин (0,877 мл, 3 экв.). Полученную суспензию перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между метиленхлоридом и 10% карбонатом натрия, органическую фазу сушат над сульфатом магния, фильтруют и растворители удаляют при пониженном давлении, получая неочищенный продукт. Продукт очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
(2-Нафталин-1-илэтил)амид 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты
К раствору трет-бутилового эфира {1-(S)-[4-нитрогуанидино-1-(2-нафталин-1-илэтилкарбамоил)бутилкарбамоил]-2-(R)-фенилэтил]карбаминовой кислоты (1,3 г, 1,77 ммоль) в метиленхлориде (100 мл) добавляют трифторуксусную кислоту (50 мл). После перемешивания при комнатной температуре в течение двадцати четырех часов, растворители удаляют при пониженном давлении и остаток распределяют между 10% карбонатом натрия (100
мл) и этилацетатом (100 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
(2-Нафталин-1-илэтил)амид 5-нитрогуанидино-2-(S)-[3-фенил-2-(R)-(3-фенилпропиламино)пропиониламино]пентановой кислоты
К раствору (2-нафталин-1-илэтил)амида 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (690 мг, 1,33 ммоль) в тетрагидрофуране (50 мл) добавляют 3-фенилпропиональдегид (0,14 мл, 0,3 экв.) и молекулярные сита (500 мг, 4 Е порошкообразные). Полученную суспензию перемешивают при комнатной температуре в течение двадцати четырех часов и затем рН доводят до 5 уксусной кислотой. Затем медленно (0,2 мл/час) шприцом-насосом добавляют цианоборгидрид натрия (1,6 мл, 1,2 экв., 1,0М раствор в тетрагидрофуране). Спустя двадцать четыре часа, растворители удаляют при пониженном давлении и неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
(2-Нафталин-1-илэтил)амид 5-гуанидино-2-(S)-[3-фенил-2-(R)-(3-фенилпропиламино)пропиониламино]пентановой кислоты
К раствору (2-нафталин-1-илэтил)амида 5-нитрогуанидино-2-(S)-[3-фенил-2-(R)-(3-фенилпропиламино)пропиониламино]пентановой кислоты (280 мг, 0,44 моль) в 50 мл метанола добавляют уксусную кислоту (5 мл) и 5% палладий на сульфате бария (280 мг). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов, фильтруют через целит и
растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 58
Синтез [2-(1Н-индол-3-ил)этил]амида 5-гуанидино-2-(S)-(2-(S)-{[3-(4-гидроксифенил)пропионил]метиламино}-3-фенилпропиониламино)пентановой кислоты
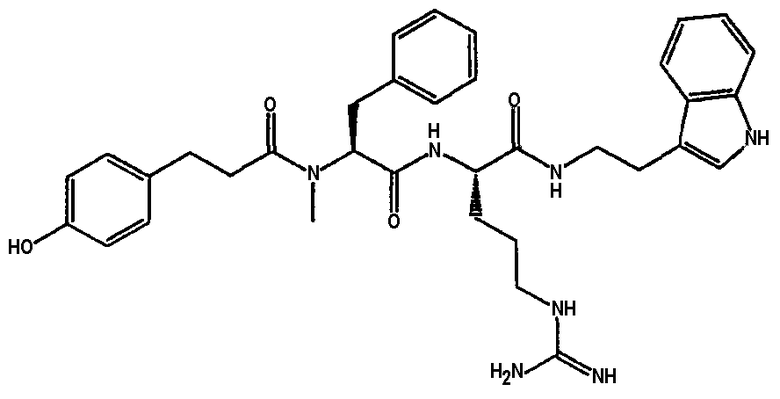
Метиловый эфир 2-(S)-[2-(S)-(трет-бутоксикарбонилметиламино)-3-фенилпропиониламино]-5-нитрогуанидинопентановой кислоты
К раствору 2-(S)-(трет-бутоксикарбонилметиламино)-3-фенилпропионовой кислоты (2,0 г, 7,17 ммоль) и метилового эфира 2-(S)-амино-5-нитрогуанидинопентановой кислоты (1,83 г, 1,1 экв.) добавляют 1-гидроксибензотриазол (1,45 г, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,05 г, 1,5 экв.) и триэтиламин (3,0 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между 10% карбонатом натрия (150 мл) и метиленхлоридом (150 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенное вещество очищают флэш-
хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
Метиловый эфир 5-нитрогуанидино-2-(S)-(2-(S)-метиламино-3-фенилпропиониламино)пентановой кислоты
К раствору метилового эфира 2-(S)-[2-(S)-(трет-бутоксикарбонилметиламино)-3-фенилпропиониламино]-5-нитрогуанидинопентановой кислоты (3,3 г, 6,68 ммоль) в метиленхлориде (200 мл) добавляют трифторуксусную кислоту (100 мл). Полученный раствор перемешивают при комнатной температуре и затем растворители удаляют при пониженном давлении. Остаток распределяют между метиленхлоридом (150 мл) и 10% карбонатом натрия (150 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенное вещество очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
Метиловый эфир 5-нитрогуанидино-2-(S)-(2-(S)-{[3-(4-гидроксифенил)пропионил]метиламино}-3-фенилпропиониламино)пентановой кислоты
К раствору метилового эфира 5-гуанидино-2-(S)-(2-(S)-метиламино-3-фенилпропиониламино)пентановой кислоты (1,0 г, 2,54 ммоль) и 3-(4-гидроксифенил)пропановой кислоты (506 мг, 1,2 экв.) добавляют 1-гидроксибензотриазол (513 мг, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (727 мг, 1,5 экв.) и триэтиламин (1,0 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном
давлении. Остаток распределяют между 10% карбонатом натрия (100 мл) и метиленхлоридом (100 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенное вещество очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
5-Нитрогуанидино-2-(S)-(2-(S)-{[3-(4-гидроксифенил)пропионил]метиламино}-3-фенилпропиониламино)пентановая кислота
К раствору метилового эфира 5-нитрогуанидино-2-(S)-(2-(S)-{[3-(4-гидроксифенил)пропионил]метиламино}-3-фенилпропиониламино)пентановой кислоты (534 мг, 0,98 ммоль) в тетрагидрофуране (50 мл) добавляют моногидрат гидроксида лития (49 мг, 1,1 экв.) и воду (3 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем подкисляют трифторуксусной кислотой (1 экв.). Растворители удаляют при пониженном давлении и неочищенное вещество распределяют между этилацетатом (75 мл) и водой (75 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении, получая указанное в заголовке соединение.
[2-(1Н-Индол-3-ил)этил]амид 5-нитрогуанидино-2-(S)-(2-(S)-{[3-(4-гидроксифенил)пропионил]метиламино}-3-фенилпропиониламино)пентановой кислоты
К раствору 5-нитрогуанидино-2-(S)-(2-(S)-{[3-(4-гидроксифенил)пропионил]метиламино}-3-фенилпропиониламино)пентановой кислоты (460 мг, 0,87 ммоль) и
[2-(1Н-индол-3-ил)этил]амина (168 мг, 1,2 экв.) добавляют 1-гидроксибензотриазол (176 мг, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (250 мг, 1,5 экв.) и триэтиламин (0,36 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом (75 мл) и водой (75 мл), органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
[2-(1Н-Индол-3-ил)этил]амид 5-гуанидино-2-(S)-(2-(S)-{[3-(4-гидроксифенил)пропионил]метиламино}-3-фенилпропиониламино)пентановой кислоты
К раствору [2-(1Н-индол-3-ил)этил]амида 5-нитрогуанидино-2-(S)-(2-(S)-{[3-(4-гидроксифенил)пропионил]метиламино}-3-фенилпропиониламино)пентановой кислоты (250 мг, 0,37 моль) в 50 мл метанола добавляют уксусную кислоту (5 мл) и 5% палладий на сульфате бария (250 мг). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов, фильтруют через целит и растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 59
Синтез бензиламида 5-гуанидино-2-(S)-[3-фенил-2-(R)-(3-фенилпропиламино)пропиониламино]пентановой кислоты
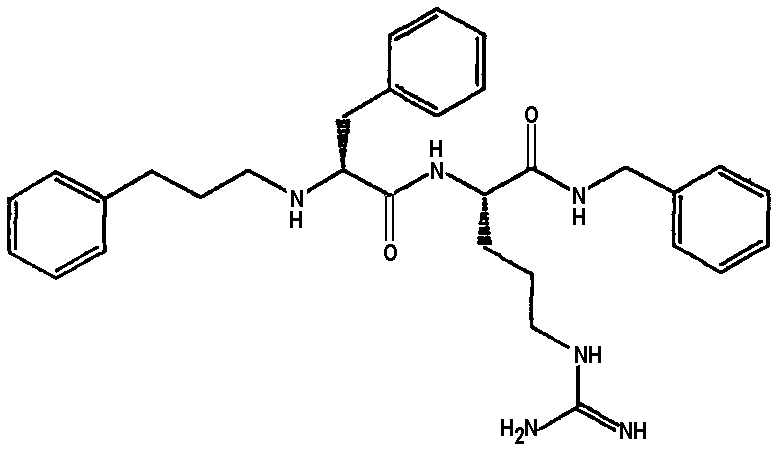
трет-Бутиловый эфир [1-(S)-(1-бензилкарбамоил-4-нитрогуанидинобутилкарбамоил)-2-(R)-фенилэтил]карбаминовой кислоты
К раствору 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (500 мг, 1,29 ммоль), бензиламина (0,155 мл, 1,1 экв.) в DMF (50 мл) добавляют 1-гидроксибензотриазол (261 мг, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (371 мг, 1,5 экв.) и триэтиламин (0,53 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая 526 мг указанного в заголовке соединения.
Бензиламид 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты
К раствору трет-бутилового эфира [1-(S)-(1-бензилкарбамоил-4-нитрогуанидинобутилкарбамоил)-2-(R)-фенилэтил]карбаминовой кислоты (512 мг, 1,12 ммоль) в метиленхлориде (50 мл) добавляют трифторуксусную кислоту (25 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в
заголовке соединение.
Бензиламид 5-нитрогуанидино-2-(S)-[3-фенил-2-(R)-(3-фенилпропиламино)пропиониламино]пентановой кислоты
К суспензии бензиламида 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (370 мг, 0,65 ммоль), 3-фенилпропиональдегида (0,077 мл, 0,9 экв.) и молекулярных сит (370 мг, 4 Е порошкообразные) добавляют триэтиламин (0,177 мл, 2 экв.). Полученную суспензию перемешивают при комнатной температуре в течение двадцати четырех часов и затем рН доводят до 5 уксусной кислотой. Затем медленно (0,2 мл/час) добавляют цианоборгидрид натрия (0,70 мл, 1,0М раствор в тетрагидрофуране, 1 экв.) с использованием шприца-насоса. Через двадцать четыре часа, суспензию фильтруют через целит и растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Бензиламид 5-гуанидино-2-(S)-[3-фенил-2-(R)-(3-фенилпропиламино)пропиониламино]пентановой кислоты
К раствору бензиламида 5-нитрогуанидино-2-(S)-[3-фенил-2-(R)-(3-фенилпропиламино)пропиониламино]пентановой кислоты (80 мг, 0,14 ммоль) в метаноле (50 мл) добавляют уксусную кислоту (5 мл) и 5% палладий на сульфате бария (80 мг). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов, фильтруют через целит и растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 60
Синтез [1-карбамоил-2-(1Н-индол-3-ил)этил]амида 2-(S)-{2-(R)-[2-(S)-ацетиламино-3-(4-гидроксифенил)пропиониламино]-3-фенилпропиониламино}-5-нитрогуанидинопентановой кислоты
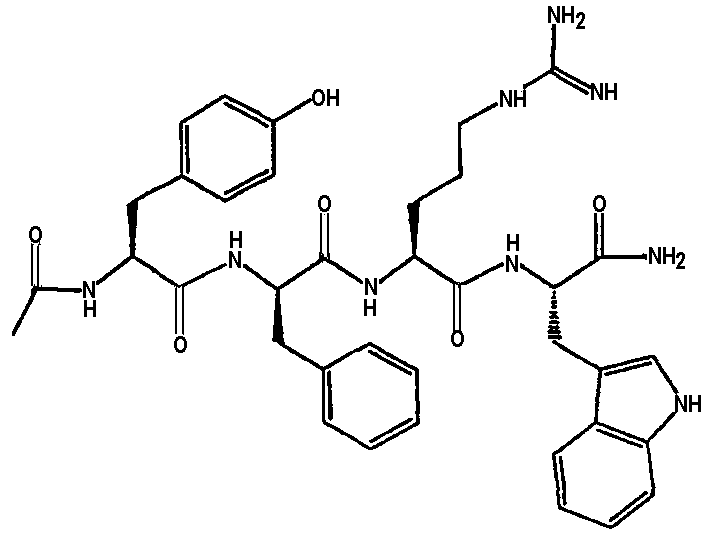
трет-Бутиловый эфир (1-(S)-{1-[1-карбамоил-2-(1Н-индол-3-ил)этилкарбамоил]-4-нитрогуанидинобутилкарбамоил}-2-(R)-фенилэтил)карбаминовой кислоты
К раствору 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (800 мг, 1,7 ммоль) и 2-(S)-амино-3-(1Н-индол-3-ил)пропионамида (500 мг, 1,2 экв.) в DMF (50 мл) добавляют 1-гидроксибензотриазол (350 мг, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (491 мг, 1,5 экв.) и триэтиламин (1,17 мл, 5 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом (75 мл) и 10% карбонатом натрия (75 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
[1-Карбамоил-2-(1Н-индол-3-ил)этил]амид 2-(S)-(2-(R)-амино-
3-фенилпропиониламино)-5-нитрогуаидинопентановой кислоты
К раствору трет-бутилового эфира (1-(S)-{1-[1-карбамоил-2-(1H-индол-3-ил)этилкарбамоил]-4-нитрогуанидинобутилкарбамоил}-2-(R)-фенилэтил)карбаминовой кислоты (1,16 г, 1,78 ммоль) в метиленхлориде (100 мл) добавляют трифторуксусную кислоту (50 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
[1-Карбамоил-2-(1Н-индол-3-ил)этил]амид 2-(S)-{2-(R)-[2-(S)-ацетиламино-3-(4-гидроксифенил)пропиониламино]-3-фенилпропиониламино}-5-нитрогуанидинопентановой кислоты
К раствору [1-карбамоил-2-(1Н-индол-3-ил)этил]амида 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (250 мг, 0,378 ммоль) и 2-(S)-ацетиламино-3-(4-гидроксифенил)пропионовой кислоты (100 мг, 1,2 экв.) в DMF (50 мл) добавляют 1-гидроксибензотриазол (76 мг, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (107 мг, 1,5 экв.) и триэтиламин (0,20 мл, 4 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
[1-Карбамоил-2-(1Н-индол-3-ил)этил]амид 2-(S)-{2-(R)-[2-(S)-ацетиламино-3-(4-гидроксифенил)пропиониламино]-3-фенилпропиониламино}-5-нитрогуанидинопентановой кислоты
К раствору [1-карбамоил-2-(1Н-индол-3-ил)этил]амида 2-(S)-
{2-(R)-[2-(S)-ацетиламино-3-(4-гидроксифенил)пропиониламино]-3-фенилпропиониламино}-5-нитрогуанидинопентановой кислоты (100 мг, 0,13 ммоль) в метаноле (50 мл) добавляют уксусную кислоту (5 мл) и 5% палладий на сульфате бария (70 мг). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов, фильтруют через целит и растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 61
Синтез [2-(S)-(1Н-индол-3-ил)-1-метилкарбамоилэтил]амида 2-(2-(R)-амино-3-фенилпропиониламино)-5-(1-тритил-1Н-имидазол-4-ил)пентановой кислоты

Метиловый эфир 2-амино-5-(3H-имидазол-4-ил)пентановой кислоты
К суспензии 2-трет-бутоксикарбониламино-5-(3Н-имидазол-4-ил)пентановой кислоты (2,0 г, 11,8 ммоль) в метаноле (60 мл) добавляют безводный хлорид водорода до насыщения раствора. Затем раствор кипятят с обратным холодильником в течение двадцати четырех часов и затем охлаждают до комнатной температуры. Растворители удаляют при пониженном давлении, получая указанное в заголовке соединение.
Метиловый эфир 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-
фенилпропиониламино)-5-(3H-имидазол-4-ил)пентановой кислоты
К раствору 2-(R)-2-трет-бутоксикарбониламино-3-фенилпропионовой кислоты (500 мг, 1,88 ммоль) и метилового эфира 2-амино-5-(3Н-имидазол-4-ил)пентановой кислоты (500 мг, 1,1 экв.) добавляют гидроксибензотриазол (381 мг, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (540 мг, 1,5 экв.) и триэтиламин (1,28 мл, 5 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом (75 мл) и 10% карбонатом натрия (75 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
Метиловый эфир 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-(1-тритил-1H-имидазол-4-ил)пентановой кислоты
К раствору метилового эфира 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-(3H-имидазол-4-ил)пентановой кислоты (500 мг, 0,72 ммоль) в тетрагидрофуране (50 мл) добавляют трифенилметилхлорид (220 мг, 1,1 экв.) и триэтиламин (0,2 мл, 2 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (5% метанол/хлороформ), получая указанное в заголовке соединение.
2-(S)-2-(R)-трет-Бутоксикарбониламино-3-фенилпропиониламино)-5-(1-тритил-1Н-имидазол-4-ил)пентановая кислота
К раствору метилового эфира 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-(1-тритил-1Н-имидазол-4-ил)пентановой кислоты (300 мг, 0,45 ммоль) в тетрагидрофуране (30 мл) добавляют моногидрат гидроксида лития (32 мг, 1,2 экв.) и воду (3 мл), полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество используют непосредственно на следующей стадии.
трет-Бутиловый эфир {1-(R)-[1-(2-(S)-(1H-индол-3-ил)-1-метилкарбамоилэтилкарбамоил]-4-(1-тритил-1H-имидазол-4-ил)бутилкарбамоил]-2-фенилэтил}карбаминовой кислоты
К раствору 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-(1-тритил-1Н-имидазол-4-ил)пентановой кислоты (293 мг, 0,45 ммоль) и 2-(S)-амино-3-(1Н-индол-3-ил)пропионамида (117 мг, 1,3 экв.) добавляют PyBOP (301 мг, 1,3 экв.) и триэтиламин (0,18 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом (75 мл) и 19% карбонатом натрия (75 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (5% метанол/хлороформ), получая указанное в заголовке соединение.
[2-(S)-(1Н-Индол-3-ил)-1-метилкарбамоилэтил]амид 2-(2-(R)-амино-3-фенилпропиониламино)-5-(1-тритил-1Н-имидазол-4-ил)пентановой кислоты
К раствору трет-бутилового эфира {1-(R)-[1-[2-(S)-(1Н-индол-3-ил)-1-метилкарбамоилэтилкарбамоил]-4-(1-тритил-1H-имидазол-4-ил)бутилкарбамоил]-2-фенилэтил}карбаминовой кислоты (468 мг, 0,55 ммоль) в метиленхлориде (32 мл) добавляют трифторуксусную кислоту (16 мл). Затем по каплям добавляют триэтилсилан до тех пор, пока не исчезнет ярко-желтая окраска. Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 62
Синтез метилового эфира 2-(R)-[2-(S)-(2-бензил-6-фенилгексаноиламино)-5-гуанидинопентаноиламино]-3-нафталин-2-илпропионовой кислоты
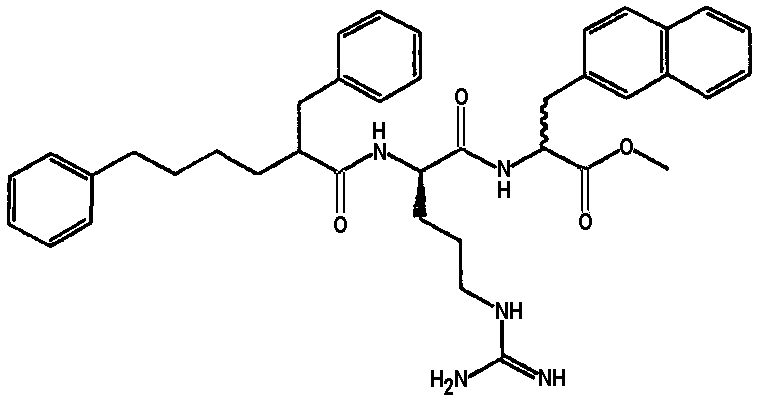
Метиловый эфир 6-фенилгексановой кислоты
Раствор 6-фенилгексановой кислоты (1,9 г, 9,89 ммоль) в метаноле (50 мл) насыщают безводным хлоридом водорода и затем кипятят с обратным холодильником в течение двадцати четырех часов. После охлаждения до комнатной температуры, растворители удаляют при пониженном давлении и остаток распределяют между
хлороформом и 10% карбонатом натрия. Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворитель удаляют при пониженном давлении, получая указанное в заголовке соединение.
Метиловый эфир 2-бензил-6-фенилгексановой кислоты
К охлажденному (-78° С) раствору метилового эфира 6-фенилгексановой кислоты (2,8 г, 13,5 ммоль) в безводном тетрагидрофуране (50 мл) медленно добавляют 2,0M раствор диизопропиламида лития в смеси гексан/тетрагидрофуран (7,5 мл, 1,1 экв.). Полученный раствор перемешивают при –78° С в течение пятидесяти минут и затем медленно добавляют бензилбромид (1,92 мл, 1,2 экв.). Полученный раствор нагревают до комнатной температуры на протяжении ночи и затем растворители удаляют при пониженном давлении. Затем остаток распределяют между этилацетатом и водой. Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
2-Бензил-6-фенилгексановая кислота
К раствору метилового эфира 2-бензил-6-фенилгексановой кислоты (2,17 г, 7,33 ммоль) в тетрагидрофуране (100 мл) добавляют моногидрат гидроксида лития (880 мг, 2 экв.) и воду (15 мл). Полученный раствор кипятят с обратным холодильником в течение сорока восьми часов и затем охлаждают до комнатной температуры. Растворители удаляют при пониженном давлении и остаток распределяют между этилацетатом и 1M лимонной кислотой. Органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении.
Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Метиловый эфир 2-(S)-(2-бензил-6-фенилгексаноиламино)-5-нитрогуанидинопентановой кислоты
К раствору метилового эфира 2-бензил-6-фенилгексановой кислоты (513 мг, 1,82 ммоль) и метилового эфира 2-(S)-амино-5-нитрогуанидинопентановой кислоты (480 мг, 1,1 экв.) в DMF (50 мл) добавляют 1-гидроксибензотриазол (319 мг, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (451 мг, 1,3 экв.) и триэтиламин (0,74 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
2-(S)-(2-Бензил-6-фенилгексаноиламино)-5-нитргуанидинопентановая кислота
К раствору метилового эфира 2-(S)-(2-бензил-6-фенилгексаноиламино)-5-нитрогуанидинопентановой кислоты (700 мг, 1,5 ммоль) в тетрагидрофуране (40 мл) добавляют моногидрат гидроксида лития (180 мг, 2 экв.) и воду (3 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Метиловый эфир 2-амино-3-нафталин-2-илпропионовой кислоты
К суспензии 2-амино-3-нафталин-2-илпропионовой кислоты (300 мг, 1,39 ммоль) в метаноле (40 мл) добавляют безводный хлорид
водорода до насыщения раствора. Полученный раствор кипятят с обратным холодильником в течение двух часов и затем охлаждают до комнатной температуры и растворители удаляют при пониженном давлении, получая указанное в заголовке соединение.
Метиловый эфир 2-(R)-[2-(S)-(2-бензил-6-фенилгексаноиламино)-5-нитрогуанидинопентаноиламино]-3-нафталин-2-илпропионовой кислоты
К раствору 2-(S)-(2-бензил-6-фенилгексаноиламино)-5-нитрогуанидинопентановой кислоты (100 мг, 0,22 ммоль) и метилового эфира 2-амино-3-нафталин-2-илпропионовой кислоты (65 мг, 1,1 экв.) в DMF (30 мл) добавляют 1-гидроксибензотриазол (44 мг, 1 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (63 мг, 1,5 экв.) и триэтиламин (0,09 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Метиловый эфир 2-(R)-[2-(S)-(2-бензил-6-фенилгексаноиламино)-5-гуанидинопентаноиламино]-3-нафталин-2-илпропионовой кислоты
К раствору метилового эфира 2-(R)-[2-(S)-(2-бензил-6-фенилгексаноиламино)-5-нитрогуанидинопентаноиламино]-3-нафталин-2-илпропионовой кислоты (80 мг, 0,11 ммоль) в метаноле (30 мл) добавляют уксусную кислоту (3 мл) и 5% палладий на сульфате бария (75 мг). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов, фильтруют через целит и растворители удаляют при пониженном давлении. Неочищенный
продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение в виде смеси четырех диастереомеров.
Пример 63
Синтез [1-(S)-метилкарбамоил-2-нафталин-2-илэтил]амида 5-(1Н-имидазол-4-ил)-2-[3-фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]пентановой кислоты

Метиловый эфир 3-фенил-2-(R)-(3-фенилпропиониламино)пропионовой кислоты
К раствору 3-фенилпропионовой кислоты (1,0 г, 6,66 ммоль) и метилового эфира 2-(R)-амино-3-фенилпропионовой кислоты (1,19 г, 1 экв.) в DMF (60 мл) добавляют 1-гидроксибензотриазол (1,35 г, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,90 г, 1,5 экв.) и триэтиламин (2,7 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между водой и этилацетатом, сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении, получая указанное в заголовке соединение.
3-Фенил-2-(R)-(3-фенилпропиониламино)пропионовая кислота
К раствору метилового эфира 3-фенил-2-(R)-(3-фенилпропиониламино)пропионовой кислоты (1,5 г, 4,82 ммоль) в тетрагидрофуране (70 мл) добавляют моногидрат гидроксида лития
(434 мг, 1,5 экв.) и воду (5 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Метиловый эфир 5-(3H-имидазол-4-ил)-2-[3-фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]пентановой кислоты
К раствору 3-фенил-2-(R)-(3-фенилпропиониламино)пропионовой кислоты (376 мг, 1,26 ммоль) и метилового эфира 2-амино-5-(3H-имидазол-4-ил)пентановой кислоты (355 мг, 1,1 экв.) в DMF (50 мл) добавляют 1-гидроксибензотриазол (256 мг, 1,5 экв.), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (362 мг, 1,5 экв.) и триэтиламин (0,863 мл, 5 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Метиловый эфир 2-[3-фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]-5-(1-тритил-1Н-имидазол-4-ил)пентановой кислоты
К раствору метилового эфира 5-(3Н-имидазол-4-ил)-2-[3-фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]пентановой кислоты (473 мг, 1,02 ммоль) в тетрагидрофуране (50 мл) добавляют трифенилметилхлорид (341 мг, 1,2 экв.) и триэтиламин (0,42 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный
продукт очищают флэш-хроматографией на силикагеле (5% метанол/хлороформ), получая указанное в заголовке соединение.
2-[3-Фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]-5-(1-тритил-1Н-имидазол-4-ил)пентановая кислота
К раствору метилового эфира 2-[3-фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]-5-(1-тритил-1Н-имидазол-4-ил)пентановой кислоты (380 мг, 0,54 ммоль) в тетрагидрофуране (80 мл) добавляют моногидрат гидроксида лития (48 мг, 1,5 экв.) и воду (10 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество используют непосредственно на следующей стадии.
трет-Бутиловый эфир (1-(S)-метилкарбамоил-2-нафталин-2-илэтил)карбаминовой кислоты
К раствору 2-(S)-трет-бутоксикарбониламино-3-нафталин-2-илпропионовой кислоты (1,5 г, 4,76 ммоль) в DMF (50 мл) добавляют метиламин (3,0 мл 2,0M раствора в тетрагидрофуране), PyBOP (3,7 г, 1,5 экв.) и триэтиламин (1,95 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом и 10% карбонатом натрия, органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (5% метанол/хлороформ), получая указанное в заголовке соединение.
2-(S)-Амино-N-метил-3-нафталин-2-илпропионамид
К раствору трет-бутилового эфира (1-(S)-метилкарбамоил-2-нафталин-2-илэтил)карбаминовой кислоты (1,3 г, 3,96 ммоль) в метиленхлориде (100 мл) добавляют трифторуксусную кислоту (50 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
(1-(S)-Метилкарбамоил-2-нафталин-2-илэтил]амид 2-(S)-[3-фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]-5-(1-тритил-1Н-имидазол-4-ил)пентановой кислоты
К раствору 2-[3-фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]-5-(1-тритил-1Н-имидазол-4-ил)пентановой кислоты (383 мг, 0,53 ммоль) в DMF (50 мл) добавляют 2-(S)-амино-N-метил-3-нафталин-2-илпропионамид (147 мг, 0,8 экв.), PyBOP (420 мг, 1,5 экв.) и триэтиламин (0,220 мл, 3 экв.). Полученный раствор перемешивают при комнатной температуре в течение сорока восьми часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом и 10% карбонатом натрия, органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (5% метанол/хлороформ), получая указанное в заголовке соединение.
(1-(S)-Метилкарбамоил-2-нафталин-2-илэтил]амид 5-(1Н-имидазол-4-ил)-2-[3-фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]пентановой кислоты
К раствору трифторуксусной кислоты (20 мл) в метиленхлориде (40 мл) добавляют (1-метилкарбамоил-2-нафталин-2-илэтил]амид 2-(S)-[3-фенил-2-(R)-(3-фенилпропиониламино)пропиониламино]-5-(1-тритил-1H-имидазол-4-ил)пентановой кислоты (441 мг, 0,489 ммоль). Затем добавляют по каплям триэтилсилан до исчезновения ярко-желтой окраски. Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение в виде смеси диастереомеров. Диастереомеры разделяют ВЭЖХ с обращенной фазой, получая диастереомер, элюирующий раньше, и диастереомер, элюирующий позднее.
Пример 64
Синтез (1-(S)-метилкарбамоил-2-нафталин-2-илэтил]амида 2-(S)-{2-(R)-[2-(S)-ацетиламино-3-(4-гидроксифенил)пропиониламино]-3-фенилпропиониламино}-5-гуанидинопентановой кислоты
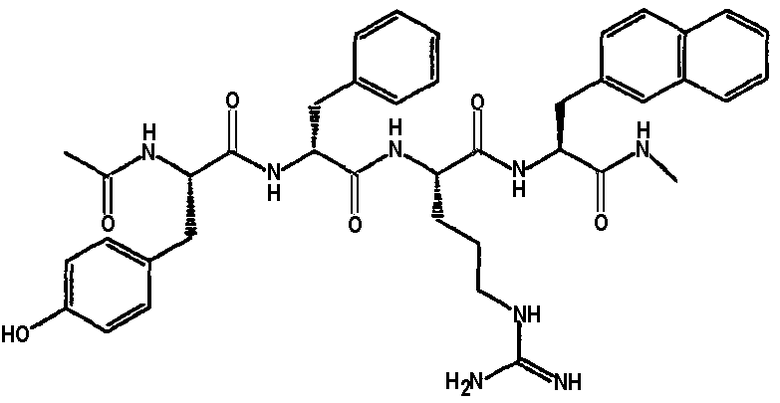
трет-Бутиловый эфир {1-(R)-[4-нитрогуанидино-1-(S)-(1-(S)-метилкарбамоил-2-нафталин-2-илэтилкарбамоил)бутилкарбамоил]-2-фенилэтил}карбаминовой кислоты
К раствору 2-(S)-(2-(R)-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (500 мг, 1,07 ммоль) и 2-(S)-амино-N-метил-3-нафталин-2-илпропионамида
(440 мг, 1,2 экв.) добавляют PyBOP (836 мг, 1,5 экв.) и триэтиламин (0,58 мл, 4 экв.). Полученный раствор перемешивают при комнатной температуре на протяжении ночи и затем растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом и 10% карбонатом натрия, сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
(1-(S)-Метилкарбамоил-2-нафталин-2-илэтил]амид 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты
К раствору трет-бутилового эфира {1-(R)-[4-нитрогуанидино-1-(S)-(1-(S)-метилкарбамоил-2-нафталин-2-илэтилкарбамоил)бутилкарбамоил]-2-фенилэтил}карбаминовой кислоты (500 мг, 0,74 ммоль) в метиленхлориде (60 мл) добавляют трифторуксусную кислоту (30 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
(1-(S)-Метилкарбамоил-2-нафталин-2-илэтил]амид 2-(S)-{2-(R)-[2-(S)-ацетиламино-3-(4-гидроксифенил)пропиониламино-3-фенилпропиониламино}-5-нитрогуанидинопентановой кислоты
К раствору (1-(S)-метилкарбамоил-2-нафталин-2-илэтил]амида 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (400 мг, 0,58 ммоль) в DMF (60
мл) добавляют 2-(S)-ацетиламино-3-(4-гидроксифенил)пропионовую кислоту (155 мг, 1,2 экв.), PyBOP (410 мг, 1,2 экв.) и триэтиламин (0,32 мл, 4 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом и 10% карбонатом натрия, органическую фазу сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле (90:9:1 хлороформ:метанол:гидроксид аммония), получая указанное в заголовке соединение.
(1-(S)-Метилкарбамоил-2-нафталин-2-илэтил]амид 2-(S)-{2-(R)-[2-(S)-ацетиламино-3-(4-гидроксифенил)пропиониламино]-3-фенилпропиониламино}-5-гуанидинопентановой кислоты
К раствору (1-(S)-метилкарбамоил-2-нафталин-2-илэтил]амида 2-(S)-{2-(R)-[2-(S)-ацетиламино-3-(4-гидроксифенил)пропиониламино]-3-фенилпропиониламино}-5-нитрогуанидинопентановой кислоты (330 мг, 0,42 ммоль) в метаноле (50 мл) добавляют уксусную кислоту (5 мл) и 5% палладий на сульфате бария (325 мг). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов и затем фильтруют через целит. Растворители удаляют при пониженном давлении и неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 65
Синтез (1-(S)-метилкарбамоил-2-нафталин-2-илэтил]амида 5-гуанидино-2-(S)-{2-(R)-[3-(4-гидроксифенил)пропиониламино]-3-
фенилпропиониламино}пентановой кислоты
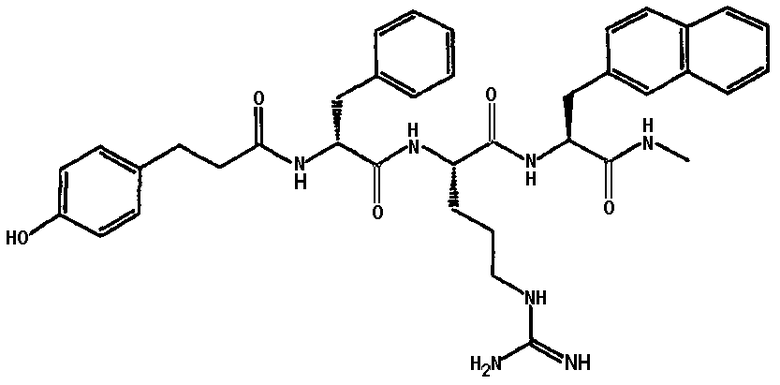
К раствору (1-(S)-метилкарбамоил-2-нафталин-2-илэтил]амида 2-(S)-(2-(R)-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты (268 мг, 3,88 ммоль) и 3-(4-гидроксифенил)пропионовой кислоты (77 мг, 1,2 экв.) добавляют гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (111 мг, 1,5 экв.), 1-гидроксибензотриазол (78 мг, 1,5 экв.) и триэтиламин (0,21 мл, 4 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенное вещество очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
(1-(S)-Метилкарбамоил-2-нафталин-2-илэтил]амид 5-гуанидино-2-(S)-{2-(R)-[3-(4-гидроксифенил)пропиониламино]-3-фенилпропиониламино}пентановой кислоты
К суспензии (1-(S)-метилкарбамоил-2-нафталин-2-илэтил)амида 5-нитрогуанидино-2-(S)-{2-(R)-[3-(4-гидроксифенил)пропиониламино]-3-фенилпропиониламино}пентановой кислоты (141 мг, 0,19 ммоль) и 5% палладия на сульфате бария (100 мг) в метаноле (45 мл) добавляют уксусную кислоту (5 мл). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов, фильтруют через целит и
растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 66
Синтез (1-(S)-метилкарбамоил-2-нафталин-2-илэтил]амида 5-гуанидино-2-(S)-[3-фенил-2-(R)-(2-фенилэтансульфониламино)пропиониламино]пентановой кислоты
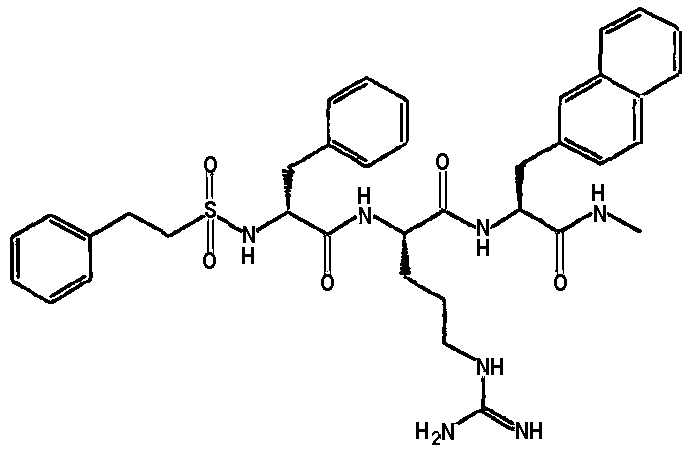
2-Фенилэтансульфонилхлорид
К двухфазному раствору 2-фенилэтантиола (10,0 г, 72,5 ммоль) в 100 мл смеси воды со льдом добавляют уксусную кислоту (20 мл). Затем раствор насыщают газообразным хлором в течение пяти минут. Затем водный раствор экстрагируют этиловым эфиром, сушат над безводным сульфатом магния, фильтруют и растворители удаляют при пониженном давлении, получая указанное в заголовке соединение.
Метиловый эфир 3-фенил-2-(R)-(2-фенилэтансульфониламино)пропионовой кислоты
К раствору метилового эфира 2-амино-3-фенилпропионовой кислоты (700 мг, 3,9 ммоль) в тетрагидрофуране (30 мл) добавляют по каплям 2-фенилэтансульфонилхлорид (1,2 г, 5,88 ммоль). К раствору добавляют триэтиламин (1,55 мл, 3 экв.). Полученный
раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
3-Фенил-2-(R)-(2-фенилэтансульфониламино)пропионовая кислота
К раствору метилового эфира 3-фенил-2-(R)-(2-фенилэтансульфониламино)пропионовой кислоты (890 мг, 2,56 ммоль) в тетрагидрофуране (50 мл) добавляют моногидрат гидроксида лития (260 мг, 1,5 экв.) и воду (5 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
трет-Бутиловый эфир [4-нитрогуанидино-1-(R)-(1-(S)-метилкарбамоил-2-нафталин-2-илэтилкарбамоил)бутил]карбаминовой кислоты
К раствору 2-амино-N-метил-3-нафталин-2-илпропионамида (1,0 г, 2,92 ммоль) и 2-(R)-трет-бутоксикарбониламино-5-нитрогуанидинопентановой кислоты (1,12 г, 1,2 экв.) добавляют гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (837 мг, 1,5 экв.), 1-гидроксибензотриазол (592 мг, 1,5 экв.) и триэтиламин (1,2 мл, 3 экв.). Полученную суспензию перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Остаток распределяют между этилацетатом и 10% карбонатом натрия, органическую фазу сушат над безводным сульфатом магния,
фильтруют и растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
(1-(S)-Метилкарбамоил-2-нафталин-2-илэтил)амид 2-(R)-амино-5-гуанидинопентановой кислоты
К раствору трет-бутилового эфира [4-нитрогуанидино-1-(R)-(1-(S)-метилкарбамоил-2-нафталин-2-илэтилкарбамоил)бутил]карбаминовой кислоты (800 мг, 1,52 ммоль) в метиленхлориде (40 мл) добавляют трифторуксусную кислоту (20 мл). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение в виде соли трифторуксусной кислоты.
(1-(S)-Метилкарбамоил-2-нафталин-2-илэтил)амид 5-нитрогуанидино-2-(S)-[3-фенил-2-(R)-(2-фенилэтансульфониламино)пропиониламино]пентановой кислоты
К раствору 3-фенил-2-(R)-(2-фенилэтансульфониламино)пропионовой кислоты (150 мг, 0,45 ммоль) и (1-(S)-метилкарбамоил-2-нафталин-2-илэтил)амида 2-(R)-амино-5-гуанидинопентановой кислоты (270 мг, 1,1 экв.) добавляют гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (130 мг, 1,5 экв.), 1-гидроксибензотриазол (91 мг, 1,5 экв.) и триэтиламин (0,25 мл, 4 экв.). Полученный раствор перемешивают при комнатной температуре в течение двадцати четырех часов и затем растворители удаляют при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в
заголовке соединение.
(1-(S)-Метилкарбамоил-2-нафталин-2-илэтил)амид 5-гуанидино-2-(S)-[3-фенил-2-(R)-(2-фенилэтансульфониламино)пропиониламино]пентановой кислоты
К раствору (1-(S)-метилкарбамоил-2-нафталин-2-илэтил)амида 5-нитрогуанидино-2-(S)-[3-фенил-2-(R)-(2-фенилэтансульфониламино)пропиониламино]пентановой кислоты (230 мг, 0,31 ммоль) в метаноле (45 мл) добавляют уксусную кислоту (5 мл) и 5% палладий на сульфате бария (200 мг). Полученную суспензию гидрируют при атмосферном давлении в течение двадцати четырех часов и затем фильтруют через целит. Растворители удаляют при пониженном давлении и неочищенный продукт очищают ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение в виде соли трифторуксусной кислоты.
Пример 67
Синтез Ac-(карба-Ff)-RW-NH2 (11)
Указанное соединение получают в соответствии с представленными схемами IA и B:
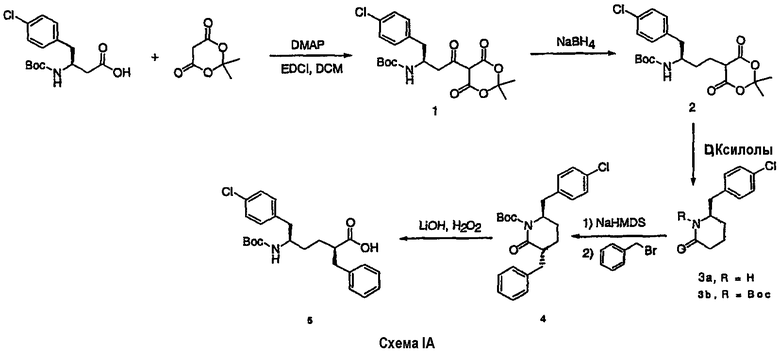
трет-Бутиловый эфир [1-(4-хлорбензил)-3-(2,2-диметил-4,6-диоксо[1,3]диоксан-5-ил)-3-оксопропил]карбаминовой кислоты (1)
К тщательно перемешиваемой смеси Boc-(S)-3-амино-4-(4-хлорфенил)масляной кислоты (5,0 г, 16 ммоль), 2,2-диметил-1,3-диоксан-4,6-диона (2,54 г, 17,6 ммоль) и DMAP (24 ммоль) в DCM (160 мл) при 0° С одной порцией добавляют EDCI (24 ммоль). Полученную смесь перемешивают при 0° С в течение 1 ч, затем при комнатной температуре в течение 18 ч. Добавляют DCM (100 мл) и смесь промывают водой (2× 50 мл), 5% водным гидросульфатом калия (3× 50 мл), 5% водным бикарбонатом натрия (1× 50 мл) и насыщенным раствором соли (1× 50 мл). Органический слой сушат над безводным сульфатом магния и концентрируют, получая 1.
трет-Бутиловый эфир [1-(4-хлорбензил)-3-(2,2-диметил-4,6-диоксо[1,3]диоксан-5-ил)пропил]карбаминовой кислоты (2)
Боргидрид натрия (63,8 ммоль, 4,0 экв.) добавляют порциями на протяжении 1 ч. к тщательно перемешиваемому раствору 1 (15,96 ммоль) в смеси DCM (180 мл) и уксусной кислоты (10 мл, 175 ммоль, 11,0 экв.) при 0° С. Реакционную смесь перемешивают при 0° С в течение 1 ч. и затем при комнатной температуре в течение 64 ч. Реакционную смесь разбавляют DCM (150 мл) и промывают водой (1× 50 мл) и насыщенным раствором соли (2× 50 мл). Органический слой сушат над безводным сульфатом магния и концентрируют на роторном испарителе, получая 2.
6-(4-Хлорбензил)пиперидин-2-он (3a) и трет-бутиловый эфир 2-(4-хлорбензил)-6-оксопиперидин-1-карбоновой кислоты (3b)
Перемешиваемую смесь 2 (15,32 ммоль) и ксилола (140 мл)
кипятят с обратным холодильником в течение 6 ч. Растворитель удаляют выпариванием при 37° С в вакууме, получая неочищенный 3a. Данное вещество смешивают с ди-трет-бутилдикарбонатом (5,0 экв.) и DMAP (0,3 экв.) в DCM (100 мл) и смесь перемешивают при комнатной температуре в течение 40 ч. Растворитель удаляют на роторном испарителе и остаток очищают флэш-хроматографией на силикагеле, используя смесь EtOAc-гексан (1:19, 500 мл и 1:9, 1300 мл) в качестве элюента, получая 3b.
трет-Бутиловый эфир 3-бензил-6-(4-хлорбензил)-2-оксопиперидин-1-карбоновой кислоты (4)
Бис(триметилсилил)амид натрия (5,16 мл 1,0M раствора в THF, 1,0 экв.) добавляют по каплям к тщательно перемешиваемому раствору 3b (1,67 г, 5,16 ммоль) в смеси THF-DME (1:1, 100 мл) при –78° С в атмосфере аргона и полученную смесь перемешивают в течение 0,5 ч при –78° С. К перемешиваемой охлажденной смеси добавляют раствор бензилбромида (0,882 г, 5,16 ммоль, 1,0 экв.) в THF (5 мл) и продолжают перемешивание при –78° С в атмосфере аргона в течение 2 ч. Реакционную смесь гасят насыщенным водным раствором хлорида аммония (20 мл) и продолжают перемешивание в течение 10 мин. Затем смесь распределяют между DCM (80 мл) и водой (40 мл) и водную фазу экстрагируют DCM (2× 40 мл). Объединенную органическую фазу промывают водой (1× 40 мл), сушат над безводным сульфатом магния и концентрируют, получая неочищенное соединение 4, которое очищают флэш-хроматографией на колонке с силикагелем, используя смесь EtOAc-гексан (1:39, 1000 мл и 1:19, 1300 мл) в качестве элюента, получая 4 (единственный
диастереомер).
2-(R)-Бензил-5-(R)-трет-бутоксикарбониламино-6-(4-хлорфенил)гексановая кислота (’Boc-(карба-4-Cl-Ff)-OH’, 5)
К тщательно перемешиваемому раствору 4 (1,152 г, 2,783 ммоль) в смеси THF-вода (4:1) добавляют моногидрат гидроксида лития (0,467 г, 11,132 ммоль, 4 экв.) одной порцией и по каплям добавляют 35% пероксид водорода (1,95 мл, 8,0 экв.). Полученную смесь перемешивают при 0° С в течение 1 ч, а затем при комнатной температуре в течение 16 ч. Реакционную смесь подкисляют водной хлористоводородной кислотой (11,5 мл, 1N) при 0° С и экстрагируют DCM (4× 40 мл). Органический слой сушат над безводным сульфатом магния, концентрируют и остаток растирают, получая 5 (единственный диастереомер).
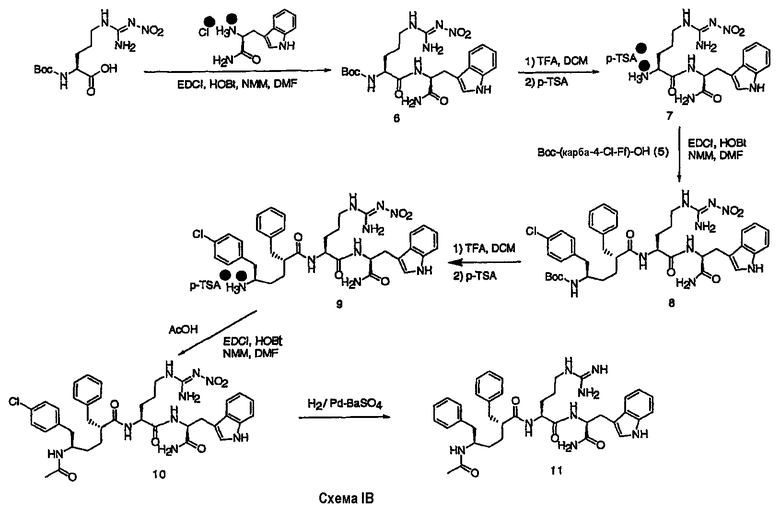
Boc-R(NO2)-W-NH2 (6)
HOBt (1,0 экв.) добавляют к тщательно перемешиваемому
раствору Boc-R(NO2)-OH (0,639 г, 2,0 ммоль) и H-W-NH2Чl· (0,479 г, 2,0 ммоль) в DMF (10 мл) при 0° С с последующим добавлением EDCI (1,1 экв.) и NMM (0,484 мл, 2,2 экв.). Полученную смесь перемешивают при 0° С в течение 1 ч. и затем при комнатной температуре в течение 4-18 ч. Реакционную смесь разбавляют EtOAc (100 мл) и промывают водой (1× 20 мл), 1N водной хлористоводородной кислотой (2× 10 мл), насыщенным водным бикарбонатом натрия (2× 10 мл) и насыщенным раствором соли (1 × 10 мл). Органический слой сушат над безводным сульфатом магния и концентрируют, получая 6.
p-TSA• H-R(NO2)-W-NH2 (7)
Раствор 6 (0,697 г, 1,381 ммоль) в смеси TFA-DCM-вода (10:40:0,5; 15 мл) перемешивают при комнатной температуре в течение 2-4 ч. К реакционной смеси добавляют моногидрат п-толуолсульфокислоты (1,0 экв.). После перемешивания в течение 10 мин смесь концентрируют выпариванием в вакууме и остаток растирают со смесью эфир-гексан (1:1), получая 2.
Boc-(карба-4-Cl-Ff)-R(NO2)-W-NH2 (8)
Следуют методике получения соединения 6 и подвергают 7 (0,5 ммоль) взаимодействию с Boc-(карба-4-Cl-Ff)-OH (5, 0,5 ммоль, 1,0 экв.), получая 8.
p-TSA• H-(карба-4-Cl-Ff)-R(NO2)-W-NH2 (9)
Следуют методике получения соединения 7 и получают 9 из 8.
Ac-(карба-4-Cl-Ff)-R(NO2)-W-NH2 (10)
Следуют методике получения соединения 6 и подвергают 9 (0,472 ммоль) взаимодействию с уксусной кислотой с получением неочищенного соединения 10, которое очищают препаративной ВЭЖХ
(С18), получая 10.
Ac-(карба-Ff)-R-W-NH2 (11)
Смесь 10 (200 мг, 0,263 ммоль) и 300 мг 5% Pd-BaSO4 (невосстановленный) в смеси MeOH-HOAc (10:1, 22 мл) гидрируют при давлении водорода 42 фунт/дюйм2 (250 кПа) при комнатной температуре в течение 17 ч. Катализатор удаляют фильтрованием через подушку целита, которую промывают MeOH. Фильтрат концентрируют, получая неочищенное соединение 11, которое очищают препаративной ВЭЖХ (С18), и затем продукт растирают с эфиром, получая 11 в виде соли TFA.
Примеры 68 и 69
Синтез 3-(4-гидроксифенил)пропаноил-Atc-R-триптамида (17) и 3-фенилпропаноил-Atc-R-триптамида (19)
Соединения указанных примеров получают в соответствии с представленной схемой II:
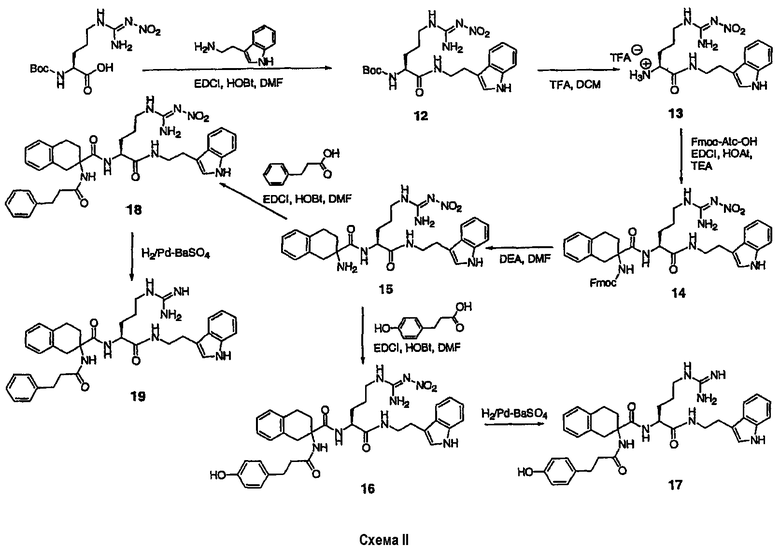
Boc-R(NO2)-триптамид (12)
HOBt (1,0 экв.) и EDCI (0,949 г, 4,95 ммоль, 1,1 экв.) добавляют последовательно к тщательно перемешиваемой смеси Boc-R(NO2)-OH (1,437 г, 4,5 ммоль) и триптамина (0,721 г, 4,5 ммоль) в DMF (20 мл) при 0° С. Полученную смесь перемешивают в течение 30 мин при 0° С и затем при комнатной температуре в течение ~20ч. Смесь разбавляют EtOAc (100 мл) и затем промывают последовательно водой (2× 20 мл), 5% водной лимонной кислотой (3× 10 мл), 5% водным бикарбонатом натрия (2× 10 мл) и насыщенным раствором соли (2× 20 мл). Органический слой сушат над безводным сульфатом натрия и концентрируют на роторном испарителе, получая соединение 12, которое используют непосредственно на следующей стадии.
H-R(NO2)-триптамид (13)
Раствор 12 (1,985 г, 4,3 ммоль) в смеси трифторуксусная кислота-дихлорметан-вода (10:40:0,5; 50 мл) перемешивают при 0° С в течение 15 минут, а затем при комнатной температуре в течение 16 ч. Растворитель удаляют на роторном испарителе и остаток совместно выпаривают с метанолом (3× 40 мл). Затем неочищенный продукт очищают препаративной ВЭЖХ, получая 13.
Fmoc-Atc-R(NO2)-триптамид (14)
HOAt (0,409 г, 3,005 ммоль), EDCI (0,632 г, 3,306 ммоль, 1,1 экв.) и TEA (1,1 экв.) добавляют последовательно к тщательно перемешиваемому раствору 13 (1,086 г, 3,005 ммоль) и Fmoc-Atc-OH (1,242 г, 3,005 ммоль) в DMF (15 мл) при 0° С. Полученную смесь перемешивают в течение 30 мин при 0° С и затем при комнатной
температуре в течение 21 ч. Обработка, как описано выше для 12, дает неочищенный 14. Полученное вещество используют на следующей стадии без дальнейшей очистки.
H-Atc-R(NO2)-триптамид (15)
Раствор 14 (2,27 г, 3,0 ммоль) в смеси DEA-DMF (1:9, 30 мл) перемешивают при комнатной температуре в течение 3 ч. Растворитель удаляют выпариванием при пониженном давлении и остаток растирают со смесью эфир-гексан (1:1, 100 мл), получая 15.
3-(4-Гидроксифенил)пропаноил-Atc-R(NO2)-триптамид (16)
Следуют методике получения 12 и получают 0,603 г неочищенного 16 из 15 (0,535 г, 1,0 ммоль) и 3-(4-гидроксифенил)пропановой кислоты (0,166 г, 1,0 ммоль). Полученный неочищенный продукт очищают препаративной ВЭЖХ, получая 16.
3-(4-Гидроксифенил)пропаноил-Atc-R-триптамид (17)
Смесь 16 (0,270 г, 0,405 ммоль) и 5% Pd-BaSO4 (невосстановленный, 0,270 г) в смеси MeOH-HOAc (10:1, 22 мл) гидрируют при комнатной температуре при атмосферном давлении в течение 17 ч. Катализатор удаляют фильтрованием через подушку целита и фильтрат концентрируют на роторном испарителе. Остаток очищают препаративной ВЭЖХ и растирают с эфиром, получая 17.
3-Фенилпропаноил-Atc-R(NO2)-триптамид (18)
Следуют методике получения 16 и получают 18 из 0,535 г (1,0 ммоль) 15 и 3-фенилпропановой кислоты (0,150 г, 1,0 ммоль).
3-Фенилпропаноил-Atc-R-триптамид (19)
Следуют методике получения 17 и получают 19 в виде
гидратированной соли TFA из 0,270 мг 18.
Пример 70
Синтез 3-фенилпропаноил-f-N(Me)R-триптамида (25)
Соединение данного примера получают в соответствии с представленной схемой III:
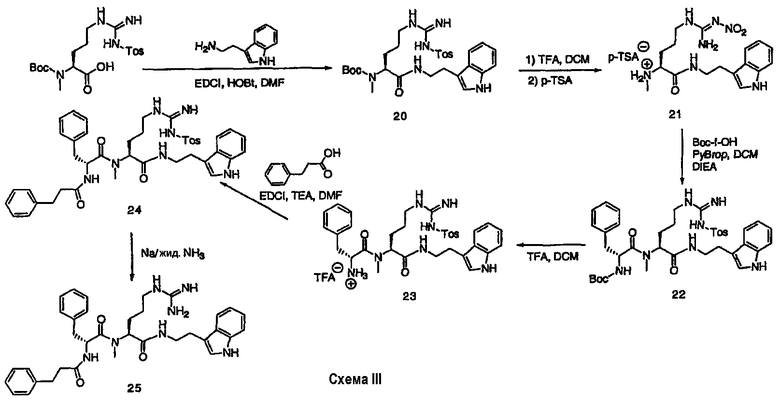
Boc-N(Me)-R(Tos)-триптамид (20)
Следуют методике получения 12 (Схема I) и получают неочищенный 20 из 1,991 г (4,5 ммоль) Boc-N(Me)-R(Tos)-OH и 0,721 г (4,5 ммоль) триптамида. Полученное неочищенное вещество используют на следующей стадии без дополнительной очистки.
p-TSA•H-N(Me)-R(Tos)-триптамид (21)
Следуют методике получения 7 (Схема 0) и получают 21 в виде соли p-TSA из 2,631 г (4,5 ммоль) 20.
Boc-f-N(Me)-R(Tos)-триптамид (22)
DIEA (0,310 мл, 1,772 ммоль, 2,0 экв.) добавляют к тщательно перемешиваемой смеси 21 (0,4293 г, 0,886 ммоль), Boc-f-OH (0,2351 г, 0,886 ммоль) и PyBrop (0,3795 г, 0,886 ммоль) в
DCM (10 мл) при 0° С. Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 ч и затем разбавляют EtOAc (50 мл). Органический слой промывают последовательно водой (1 × 10 мл), 5% водным бикарбонатом натрия (2 × 10 мл) и насыщенным раствором соли (2 × 10 мл) и сушат над безводным сульфатом натрия. Удаление осушителя влаги и выпаривание летучих веществ при пониженном давлении дает неочищенный 22.
H-f-N(Me)-R(Tos)-триптамид (23)
Раствор 22 (0,609 г, 0,833 ммоль), растворенного в смеси TFA-DCM-вода (10:40:0,5; 10 мл), перемешивают при 0° С в течение 1 ч и затем выдерживают в холодильнике при 4° С в течение 72 ч. Растворитель удаляют выпариванием в вакууме и затем остаток дважды совместно выпаривают с DCM (каждый раз 10 мл). Полученное таким образом неочищенное вещество очищают препаративной ВЭЖХ и растирают с эфиром, получая 23.
3-Фенилпропаноил-f-N(Me)-R(Tos)-триптамид (24)
TEA (0,209 ммоль, 1,1 экв.) добавляют к тщательно перемешиваемой смеси 23 (0,120 г, 0,19 ммоль), 3-фенилпропановой кислоты (0,19 ммоль) и EDCI (0,209 ммоль, 1,1. экв) в DMF (3 мл) при 0° С. Полученную смесь перемешивают при 0° С в течение 0,5 ч и затем при комнатной температуре в течение 16 ч. Реакционную смесь разбавляют EtOAc (30 мл) и промывают последовательно водой (2× 5 мл), 5% водной лимонной кислотой (3× 3 мл), 5% водным бикарбонатом натрия (2× 3 мл) и насыщенным раствором соли (2× 5 мл). Органический слой сушат над безводным сульфатом натрия, концентрируют и затем растирают с эфиром, получая 24.
3-Фенилпропаноил-f-N(Me)-R-триптамид (25)
Соединение 24 (100 мг, 0,13 ммоль) растворяют в жидком аммиаке (25 мл) при –78° С и к раствору при энергичном перемешивании магнитной мешалкой добавляют небольшие кусочки металлического натрия до тех пор, пока не исчезнет синяя окраска, в течение 2 ч. Избыточный реагент уничтожают ацетатом аммония и аммиак удаляют выпариванием при комнатной температуре. Остаток растворяют в метаноле и раствор фильтруют через подушку силикагеля, которую затем промывают добавочным метанолом. Объединенные метанольные фильтраты концентрируют на роторном испарителе, получая неочищенное вещество, которое очищают препаративной ВЭЖХ, получая 25 в виде гидратированной соли TFA.
Примеры 71-75
Синтез H-YfRW-NH2 (32), Bc-YfRW-NH2 (34), CH3(CH2)8CO-YfRW-NH2 (36), Bc-YfRW-триптамида (40) и CH3(CH2)8CO-YfRW-триптамида (42)
Соединения указанных примеров получают в соответствии с представленной схемой IV:
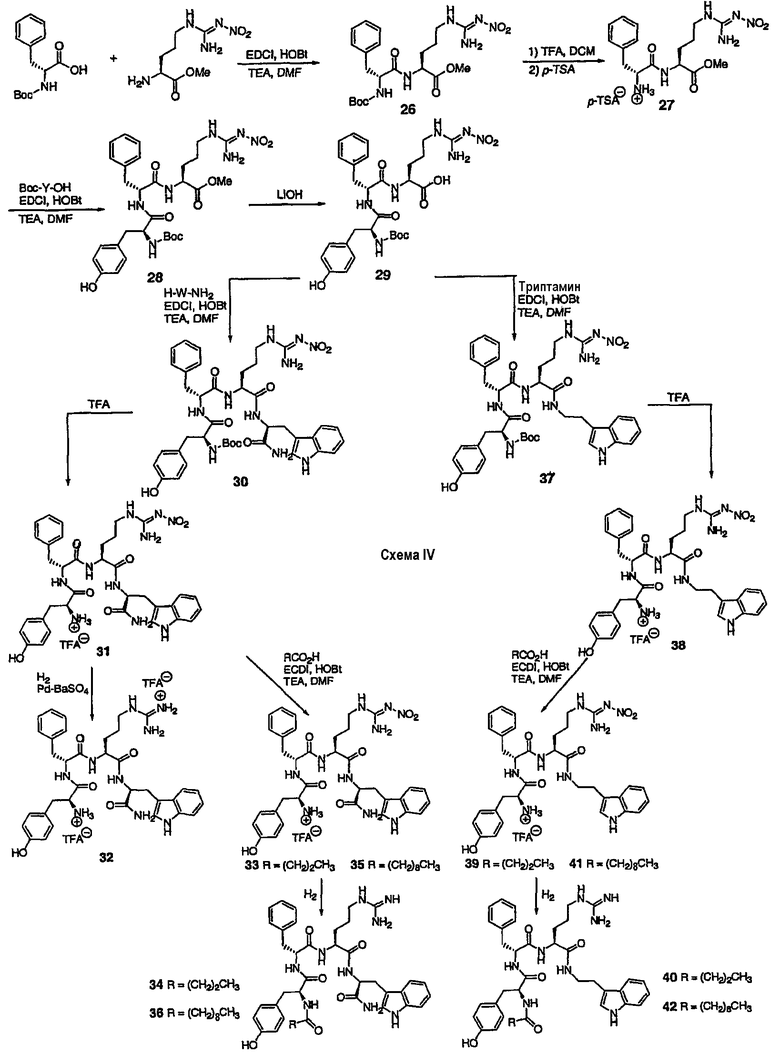
Boc-fR(NO2)-OMe (26)
Boc-f-OH (7,64 г, 28,8 ммоль), H-W(NO2)-OМе· l (6,72 г, 28,8 ммоль), HOBt (3,94 г, 29,2 ммоль), TEA (8 мл, 57,6 ммоль) и
DMF (110 мл, безводный) смешивают, смесь охлаждают при 0° С и в нее при перемешивании добавляют EDCI (5,89 г, 30,8 ммоль). После перемешивания при комнатной температуре в течение 18 ч смесь концентрируют на роторном испарителе, разбавляют водой (350 мл) и экстрагируют EtOAc (4× 80 мл). Объединенный органический экстракт промывают 1N водной HCl (3× 60 мл), насыщенным водным NaHCO3 (2× 50 мл), насыщенным раствором соли (45 мл) и затем сушат безводным Na2SO4. После фильтрования фильтрат концентрируют на роторном испарителе, совместно выпаривают с эфиром (50 мл) и сушат в вакууме, получая 3.
p-TSA• H-fR(NO2)-OMe (27)
TFA (6 мл) добавляют к раствору Boc-fR(NO2)-OMe (26) (1,49 г, 3,1 ммоль) в DCM (20 мл) при 0° С. После перемешивания при 0° С в течение 30 мин реакционную смесь перемешивают при комнатной температуре в течение 5 ч. Растворитель удаляют в вакууме, остаток совместно выпаривают с эфиром (25 мл) и полученный остаток растворяют в метаноле (25 мл). Добавляют моногидрат п-толуолсульфокислоты (0,6 г, 3,1 ммоль), смесь перемешивают в течение 5 мин. при комнатной температуре, летучие вещества удаляют в вакууме и остаток совместно выпаривают с эфиром (2× 25 мл), получая 27.
Boc-YfR(NO2)-OMe (28)
p-TSA• H-fR(NO2)-OMe (27, 3,46 г, 6 ммоль), Boc-Y-OH (1,72 г, 6,12 ммоль), HOBt (0,83 г, 6,18 ммоль), TEA (1,6 мл, 12 ммоль) и DMF (25 мл, безводный) смешивают и охлаждают при 0° С и при перемешивании добавляют EDCI (1,1 г, 6,3 ммоль). После
перемешивания при комнатной температуре в течение 16 ч смесь концентрируют на роторном испарителе, разбавляют водой (110 мл) и экстрагируют EtOAc (4× 23 мл). Объединенный органический экстракт промывают 1N водной HCl (3× 20 мл), насыщенным водным NaHCO3 (2× 25 мл), насыщенным раствором соли (25 мл) и затем сушат безводным Na2SО4. После фильтрования фильтрат концентрируют на роторном испарителе, получая 28.
Boc-YfR(NO2)-OH (29)
Смесь Boc-YfR(NO2)-OCH3 (28, 2,64 г, 4,1 ммоль), LiOH (0,113 г, 4,72 ммоль), воды (0,4 мл) и MeOH (10 мл) перемешивают при комнатной температуре в течение 6 ч. После удаления растворителя остаток растворяют в минимальном количестве воды. Добавляют 1N HCl (приблизительно 4,7 мл), чтобы нейтрализовать смесь до рН 5-6. Отфильтровывают твердое вещество и сушат в вакууме, получая 29.
Boc-YfR(NО2)W-NH2 (30)
Boc-YfR(NO2)-OH (29, 0,99 г, 1,56 ммоль), H-W-NН2·HCl (0,43 г, 1,8 ммоль), HOBt (0,25 г, 1,84 ммоль), TEA (0,55 мл, 3,9 ммоль) и DMF (20 мл, безводный) смешивают и охлаждают при 0° С и при перемешивании добавляют EDCI (0,36 г, 1,87 ммоль). После перемешивания при комнатной температуре в течение 16 ч смесь концентрируют на роторном испарителе, разбавляют водой (120 мл) и экстрагируют EtOAc (4× 25 мл). Объединенный органический экстракт промывают 1N водной HCl (3× 20 мл), насыщенным водным NaHCO3 (2× 25 мл), насыщенным раствором соли (20 мл) и затем сушат безводным Na2SO4. После фильтрования фильтрат концентрируют на роторном испарителе, совместно выпаривают с эфиром (20 мл) и
сушат в вакууме, получая 30.
H-YfR(NO2)-Trp-NH2 (31)
TFA (2 мл) добавляют к раствору пептида 30 (0,95 г, 1,16 ммоль) в DCM (6 мл) при 0° С. После перемешивания при 0° С в течение 30 мин реакционную смесь перемешивают при комнатной температуре в течение 5 ч. Растворитель удаляют на роторном испарителе и полученный остаток очищают препаративной ВЭЖХ, получая 31.
H-YfRW-NH2 (32)
Пептид 31 (0,25 г, 0,35 ммоль), 5% Pd/BaSO4 (0,25 г, невосстановленный) и MeOH (15 мл) смешивают и гидрируют при давлении водорода 40 фунт/дюйм2 (276 кПа) при комнатной температуре в течение 48 ч. После фильтрования фильтрат концентрируют на роторном испарителе и остаток очищают препаративной ВЭЖХ, получая H-YfRW-NH2 (32).
Bc-YfR(NO2)W-NH2 (33)
Используя методику получения 28 с последующей очисткой препаративной ВЭЖХ, получают 33 из H-YfR(NO2)W-NH2 (31, 0,26 г, 0,37 ммоль), масляной кислоты (0,037 г, 0,42 ммоль), HOBt (0,059 г, 0,43 ммоль), TEA (0,1 мл, 0,74 ммоль), EDCI (0,084 г, 0,44 ммоль) и DMF (12 мл).
Bc-YfRW-NH2 (34)
Следуя методике получения 32, получают 34 из 33 (0,16 г, 0,2 ммоль), 5% Pd/BaSO4 (0,15 г, невосстановленный), MeOH (12 мл) и TFA (0,1 мл).
CH3(CH2)8CO-YfR(NO2)W-NH2 (35)
Используя методику получения 28 с последующей очисткой
препаративной ВЭЖХ, получают 35 из H-YfR(NO2)W-NH2 (31, 0,26 г, 0,36 ммоль), декановой кислоты (0,071 г, 0,41 ммоль), HOBt (0,057 г, 0,42 ммоль), TEA (0,1 мл, 0,73 ммоль), EDCI (0,083 г, 0,43 ммоль) и DMF (12 мл).
CH3(CH2)8CO-YfRW-NH2 (36)
Следуют методике получения 31 и получают 36 из 35 (0,2 г, 0,23 ммоль), 5% Pd/BaSO4 (0,18 г, невосстановленный), MeOH (12 мл) и TFA (0,1 мл).
Boc-YfR(NO2)-триптамид (37)
Используют методику получения 30 и получают 37 из Boc-YfR(NO2)-OH (29, 0,95 г, 1,5 ммоль), триптамида (0,276 г, 1,73 ммоль), HOBt (0,239 г, 1,77 ммоль), TEA (0,5 мл, 3,6 ммоль), EDCI (0,34 г, 1,8 ммоль) и DMF (18 мл).
H-YfR(NO2)-триптамид (38)
Пептид 30 (0,98 г, 1,27 ммоль) в DCM (6 мл) и раствор анизол-TFA-DCM (1:8:9, 6 мл) смешивают при 0° С и перемешивают при охлаждении льдом в течение 20 мин. Затем смесь перемешивают при комнатной температуре на протяжении ночи. Растворитель удаляют и полученный остаток совместно выпаривают с эфиром (2 × 25 мл), получая неочищенный 38.
Bc-YfR(NO2)-триптамид (39)
Используя методику получения 28 с последующей очисткой препаративной ВЭЖХ, получают 39 из 38 (0,28 г, 0,41 ммоль), масляной кислоты (0,042 г, 0,47 ммоль), HOBt (0,066 г, 0,49 ммоль), TEA (0,12 мл, 0,83 ммоль), EDCI (0,094 г, 0,49 ммоль) и DMF (10 мл).
Bc-YfR(NO2)-триптамид (40)
Следуют методике получения 32 и получают 40 из 39 (0,19 г, 0,25 ммоль), 5% Pd/BaSO4 (0,18 г, невосстановленный), MeOH (12 мл) и TFA (0,1 мл).
CH3(CH2)8CO-YfR(NO2)-триптамид (41)
Используя методику получения 28 с последующей очисткой препаративной ВЭЖХ, получают 41 из 38 (0,27 г, 0,4 ммоль), декановой кислоты (0,081 г, 0,47 ммоль), HOBt (0,065 г, 0,48 ммоль), TEA (0,11 мл, 0,81 ммоль), EDCI (0,093 г, 0,49 ммоль) и DMF (12 мл).
CH3(CH2)8CO-YfRW-триптамид (42)
Следуют методике получения 32 и получают 42 из 41 (0,21 г, 0,25 ммоль), 5% Pd/BaSO4 (0,18 г, невосстановленный), MeOH (12 мл) и TFA (0,1 мл).
Примеры 76-81
Синтез Ac-YfRW-OMe (47), Ac-YfRW-NHCH3 (49), AcYfRWSar-NH2 (50), 3-(4-OH-Ph)пропаноил-fRW-NHCH3 (53), 3-(4-OH-Ph)пропаноил-fRW-NH(CH2)2OH (54) и 3-(4-OH-Ph)пропаноил-fRW-NH(CH2)2OH (55)
Соединения указанных примеров получают в соответствии с представленной схемой V:
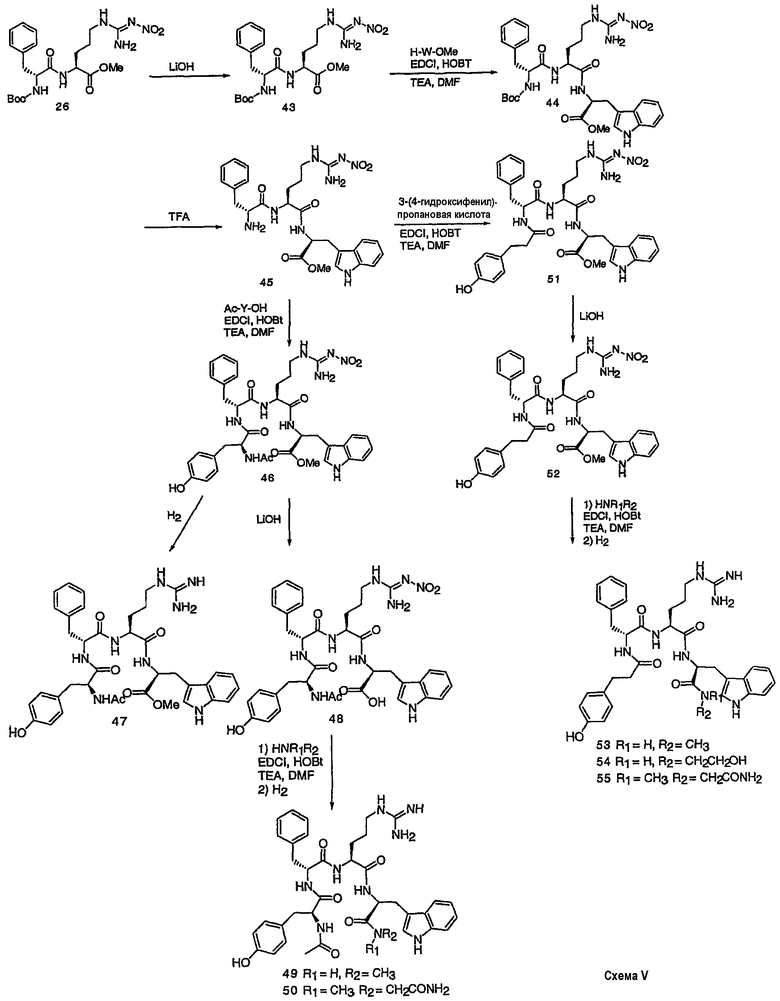
Boc-fR(NO2)-OH (43)
Следуют методике получения 29 и получают 43 из Boc-fR(NO2)-OMe (26, 2,8 г, 5,8 ммоль), LiOH (0,27 г, 11 ммоль) и MeOH (15 мл).
Boc-fR(NO2)W-OMe (44)
Следуют методике получения 26 и получают 44 из Boc-fR(NO2)-OH (43, 4,22 г, 9,06 ммоль), H-W-OMe· l (2,65 г, 10,4 ммоль), HOBt (1,44 г, 10,7 ммоль), TEA (3,15 мл, 22,6 ммоль), EDCI (2,08 г, 10,9 ммоль) и DMF (55 мл).
H-fR(NO2)W-OMe (45)
TFA (12 мл) добавляют к раствору 44 (5 г, 7,5 ммоль) в DCM (30 мл) при 0° С. После перемешивания при 0° С в течение 1 ч реакционную смесь перемешивают при комнатной температуре в течение 16 ч. Растворитель удаляют в вакууме, остаток совместно выпаривают с эфиром (2 × 35 мл) и продукт сушат в вакууме, получая неочищенный 45.
Ac-YfR(NO2)W-OMe (46)
Следуют методике получения 28 и получают неочищенный 46 из H-fR(NO2)W-OMe (45, 3,3 г, 5,8 ммоль), Ac-Y-OH (1,5 г, 6,7 ммоль), HOBt (0,93 г, 6,9 ммоль), TEA (1,7 мл, 12,2 ммоль), EDCI (1,34 г, 7 ммоль) и DMF (25 мл).
Ac-YfRW-OMe (47)
Следуют методике получения 32 и получают 47 из 46 (0,2 г, 0,26 ммоль), 5% Pd/BaSO4 (0,19 г, невосстановленный), MeOH (15 мл) и TFA (0,1 мл).
Ac-YfR(NO2)W-OH (48)
Используют методику получения 29 с последующей очисткой препаративной ВЭЖХ и получают 48 из Ac-YfR(NO2)W-OMe (46, 0,96 г, 1,25 ммоль), LiOH (0,063 г, 2,6 ммоль), воды (0,4 мл) и MeOH (6 мл).
Ac-YfRW-NHCH3 (49)
Следуют методике получения 30, за исключением стадии совместного выпаривания, и получают продукт взаимодействия Ac-YfR(NO2)W-OH (48, 0,3 г, 0,39 ммоль), метиламина (0,014 г, 0,45 ммоль), HOBt (0,058 г, 0,43 ммоль), TEA (0,092 мл, 0,91 ммоль), EDCI (0,089 г, 0,47 ммоль) и DMF (12 мл). Указанный продукт очищают препаративной ВЭЖХ и затем гидрируют, используя методику получения 32 в присутствии 5% Pd/BaSO4 (0,17 г, невосстановленный) в MeOH (12 мл), с получением 49.
Ac-YfRWSar-NH2 (50)
Следуют методике получения 30, за исключением стадии совместного выпаривания, и получают продукт взаимодействия Ac-YfR(NO2)W-OH (19, 0,3 г, 0,39 ммоль), саркозинамида·HCl (0,05 г, 0,4 ммоль), HOBt (0,058 г, 0,43 ммоль), TEA (0,092 мл, 0,91 ммоль), EDCI (0,089 г, 0,47 ммоль) и DMF (12 мл). Продукт очищают препаративной ВЭЖХ и затем гидрируют, используя методику получения 32 в присутствии 5% Pd/BaSO4 (0,18 г, невосстановленный) в MeOH (12 мл), с получением 50.
3-(4-OH-Ph)пропаноил-fRW-OCH3 (51)
Используя методику получения 26 с последующей очисткой препаративной ВЭЖХ, получают 51 из H-fR(NO2)W-OCH3 45 (2,1 г, 3,7 ммоль), 3-(4-гидроксифенил)пропановой кислоты (0,71 г, 4,3 ммоль), HOBt (0,59 г, 4,4 ммоль), TEA (1,1 мл, 0,91 ммоль), EDCI (0,85 г, 4,5 ммоль) и DMF (25 мл).
3-(4-OH-Ph)пропаноил-fRW-ОH (52)
Следуя методике получения соединения 29, получают 52 из 51 (1,15 г, 1,61 ммоль), LiOH (0,081 г, 3,38 ммоль), воды (0,4 мл)
и MeOH (8 мл).
3-(4-OH-Ph)пропаноил-fRW-NHCH3 (53)
Следуя методике получения соединения 30, получают продукт взаимодействия 52 (0,3 г, 0,42 ммоль), метиламина (0,015 г, 0,49 ммоль), HOBt (0,064 г, 0,47 ммоль), TEA (0,14 мл, 0,98 ммоль), EDCI (0,098 г, 0,51 ммоль) и DMF (12 мл). Указанный продукт очищают препаративной ВЭЖХ и гидрируют по методике, используемой для получения 32 в присутствии 5% Pd/BaSO4 (0,16 г) и MeOH (12 мл), получая 53.
3-(4-OH-Ph)пропаноил-fRW-NHCH2CH2OH (54)
Следуя методике получения соединения 30, получают продукт взаимодействия 52 (0,3 г, 0,42 ммоль), этаноламина (0,031 г, 0,51 ммоль), HOBt (0,066 г, 0,49 ммоль), TEA (0,14 мл, 1 ммоль), EDCI (0,1 г, 0,52 ммоль) и DMF (12 мл). Указанный продукт очищают препаративной ВЭЖХ и гидрируют по методике, используемой для получения соединения 32 в присутствии 5% Pd/BaSO4 (0,18 г) и MeOH (12 мл), получая 54.
3-(4-OH-Ph)пропаноил-fRWSar-NH2 (55)
Следуя методике получения соединения 30, получают продукт взаимодействия 52 (0,3 г, 0,42 ммоль), саркозинамида· l (0,061 г, 0,49 ммоль), HOBt (0,068 г, 0,5 ммоль), TEA (0,15 мл, 1,1 ммоль), EDCI (0,098 г, 0,51 ммоль) и DMF (12 мл). Указанный продукт очищают препаративной ВЭЖХ и гидрируют по методике, используемой для получения соединения 32 в присутствии 5% Pd/BaSO4 (0,18 г) и MeOH (12 мл), получая 55.
Примеры 82-84
Синтез Bc-YfRW-NHCH3 (63), Bc-YfRW-NH(CH2)2OH (64) и BcYfRW-N(CH3)(CH2)2OH (65)
Соединения указанных примеров получают в соответствии с представленной схемой VI:
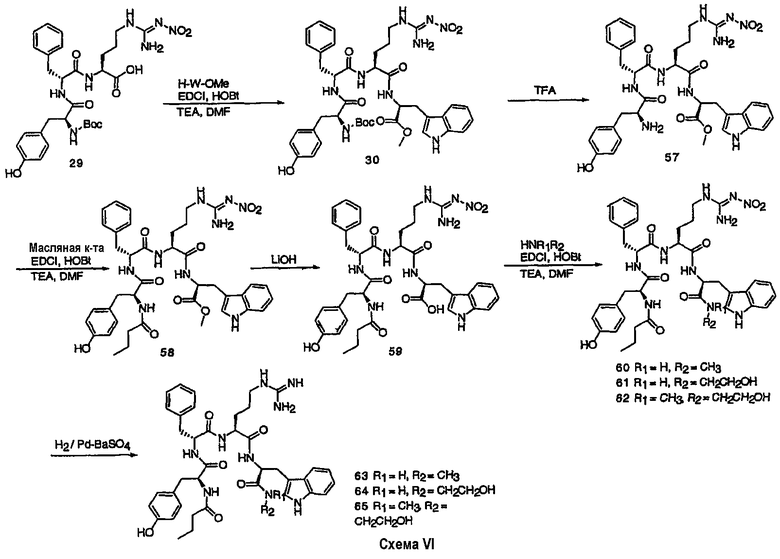
Boc-YfRW-OCH3 (56)
Следуют методике получения соединения 28 и получают 2,1 г (95%) 56 из Boc-YfR(NO2)-OH (29, 1,7 г, 2,6 ммоль), H-W-OCH3·l (0,68 г, 2,7 ммоль), HOBt (0,42 г, 3,1 ммоль), TEA (0,8 мл, 5,8 ммоль), EDCI (0,63 г, 3,3 ммоль) и DMF (25 мл).
H-YfRW-OCH3 (57)
Следуют методике получения соединения 38 и получают 57 из 56 (2,1 г, 2,5 ммоль) и раствора анизол-TFA-DCM (1:8:9, 18 мл).
Bc-YfRW-OСН3 (58)
Следуя методике получения соединения 28, получают неочищенный продукт взаимодействия H-YfR(NO2)W-OCH3 (57, 1,21 г, 1,65 ммоль), масляной кислоты (0,18 г, 2 ммоль), HOBt (0,26 г, 1,95 ммоль), TEA (0,57 мл, 4,1 ммоль), EDCI (0,394 г, 2,06 ммоль) и DMF (25 мл). Указанный продукт очищают препаративной ВЭЖХ, получая 58.
Bc-YfRW-OH (59)
Следуя методике получения соединения 29, получают 59 из 58 (0,1 г, 0,13 ммоль), LiOH (0,004 г, 0,15 ммоль), воды (0,2 мл) и MeOH (2 мл).
Bc-YfRW-NHCH3 (63)
Следуя методике получения соединения 28, получают продукт взаимодействия 60 из 59 (0,1 г, 0,125 ммоль), метиламина (0,006 мл, 0,15 ммоль), HOBt (0,02 г, 0,15 ммоль), TEA (0,04 мл, 0,3 ммоль), EDCI (0,03 г, 0,16 ммоль) и DMF (3 мл). Указанный продукт гидрируют по методике, используемой для получения соединения 32, в присутствии 5% Pd/BaSO4 (0,1 г, невосстановленный) в MeOH (6 мл), получая 63.
Bc-YfR(NO2)W- NH(CH2)2OH (61)
Используя методику получения соединения 28 с последующей очисткой ВЭЖХ, получают 61 из 59 (1,3 г, 1,65 ммоль), этаноламина (0,12 мл, 1,9 ммоль), HOBt (0,263 г, 1,95 ммоль), TEA (0,55 мл, 4 ммоль), EDCI (0,38 г, 2 ммоль) и DMF (20 мл).
Bc-YfRW- NH(CH2)2OH (64)
Следуют методике получения соединения 32 и получают 64 в виде твердого вещества из 61 (0,22 г, 0,26 ммоль), 5% Pd/BaSO4
(0,2 г, невосстановленный), MeOH (12 мл) и TFA (0,1 мл).
Bc-YfR(NO2)W-N(CH3)(CH2)2OH (62)
Используя методику получения соединения 28 с последующей очисткой ВЭЖХ, получают 62 из 59 (0,26 г, 0,33 ммоль), N-метилэтаноламина (0,032 мл, 0,4 ммоль), HOBt (0,053 г, 0,39 ммоль), TEA (0,11 мл, 0,83 ммоль), EDCI (0,076 г, 0,4 ммоль) и DMF (10 мл).
Bc-YfRW-N(CH3)(CH2)2OH (65)
Следуют методике получения соединения 32 и получают 65 из 62 (0,16 г, 0,19 ммоль), 5% Pd/BaSO4 (0,19 г, невосстановленный), MeOH (10 мл) и TFA (0,1 мл).
Пример 85
Синтез 3-(4-OHPh)пропаноил-YfRW-NH2 (69)
Соединение данного примера получают в соответствии с представленной схемой VII:
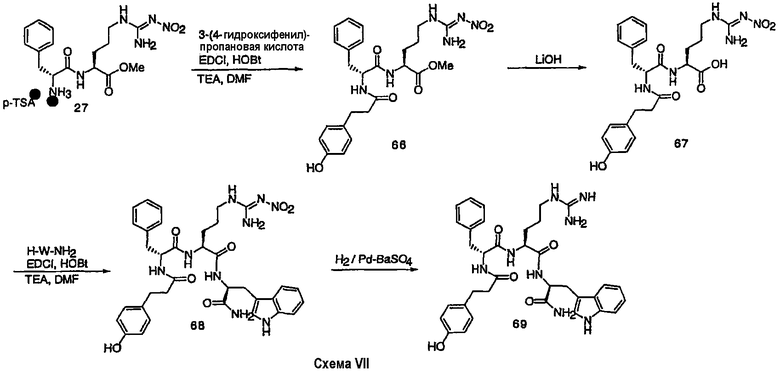
3-(4-OHPh)пропаноил-fR(NO2)-OCH3 (66)
Используя методику получения соединения 28 с последующей очисткой препаративной ВЭЖХ, получают 66 из 27 (1,5 г, 2,6
ммоль), 3-(4-гидроксифенил)пропановой кислоты (0,48 г, 2,9 ммоль), HOBt (0,43 г, 3,2 ммоль), TEA (0,91 мл, 6,6 ммоль), EDCI (0,63 г, 3,3 ммоль) и DMF (28 мл).
3-(4-OHPh)пропаноил-fR(NO2)-OH (67)
Следуя методике получения соединения 29, получают 67 из 66 (0,33 г, 0,63 ммоль), LiOH (0,029 г, 1,2 ммоль), воды (0,4 мл) и MeOH (5 мл).
3-(4-OHPh)пропаноил-fR(NO2)W-NH2 (68)
Используя методику получения соединения 28 с последующей очисткой препаративной ВЭЖХ, получают 68 из 67 (0,32 г, 0,62 ммоль), H-W-NH2·l (0,16 г, 0,68 ммоль), HOBt (0,1 г, 0,73 ммоль), TEA (0,2 мл, 1,6 ммоль), EDCI (0,14 г, 0,75 ммоль) и DMF (13 мл).
3-(4-OHPh)пропаноил-fRW-NH2 (69)
Следуют методике получения соединения 32 и получают 69 из 68 (0,14 г, 0,2 ммоль), 5% Pd/BaSO4 (0,12 г, невосстановленный), MeOH (9 мл) и TFA (0,1 мл).
Пример 86
Синтез Ac-YfR-триптамида (73)
Соединение данного примера получают в соответствии с представленной схемой VIII:
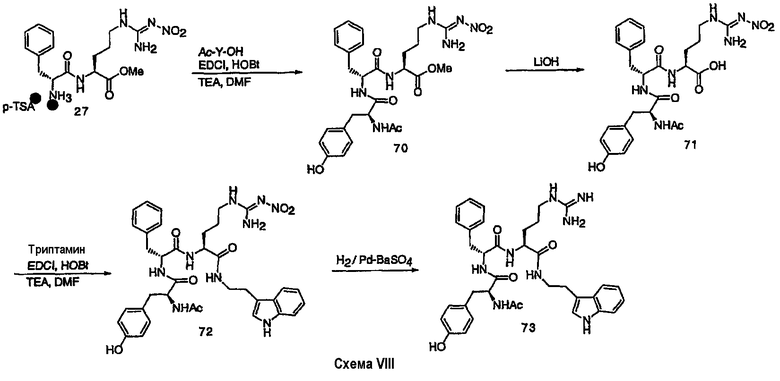
Ac-YfR(NO2)-OCH3 (70)
Используя методику получения соединения 28 с последующей очисткой препаративной ВЭЖХ, получают 70 из 27 (0,36 г, 0,63 ммоль), Ac-Y-OH (0,14 г, 0,63 ммоль), HOBt (0,1 г, 0,74 ммоль), TEA (0,17 мл, 1,2 ммоль), EDCI (0,14 г, 0,76 ммоль) и DMF (12 мл).
Ac-YfR(NO2)-OH (71)
Следуя методике получения соединения 29 с дополнительной стадией совместного выпаривания из THF (5 мл), получают смесь 71 и двух эквивалентов LiCl из 70 (0,08 г, 0,14 ммоль), LiOH (0,007 г, 0,28 ммоль), воды (0,2 мл) и MeOH (4 мл).
Ac-YfR-триптамид (73)
Следуя методике получения соединения 28, получают неочищенный продукт взаимодействия 72 из смеси Ac-YfR(NО2)-OH/2LiCl (71, 0,091 г, 0,136 ммоль), триптамина (0,025 мл, 0,15 ммоль), HOBt (0,021 г, 0,15 ммоль), TEA (0,1 мл, 0,7 ммоль), EDCI (0,03 г, 0,16 ммоль) и DMF (6 мл). Указанный продукт гидрируют по методике, используемой для получения соединения 32,
в присутствии 5% Pd/BaSO4 (0,2 г, невосстановленный) и MeOH (8 мл), с получением 73 в виде твердого вещества.
Пример 87
Синтез гидроциннамоил-fRW-NH2 (77)
Соединение данного примера получают в соответствии с представленной схемой IX:
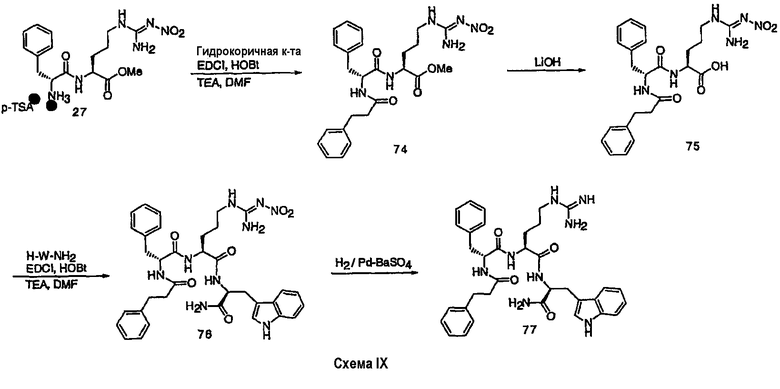
Гидроциннамоил-fR(NO2)-OCH3 (74)
Следуя методике получения соединения 28, получают 74 из 27 (0,25 г, 0,43 ммоль), гидрокоричной кислоты (0,068 г, 0,45 ммоль), HOBt (0,068 г, 0,5 ммоль), TEA (0,16 мл, 1,1 ммоль), EDCI (0,097 г, 0,51 ммоль) и DMF (20 мл).
Гидроциннамоил-fW(NO2)-OH (75)
Следуя методике получения соединения 29, получают 75 из 74 (0,22 г, 0,43 ммоль), LiOH (0,014 г, 0,56 ммоль), воды (0,3 мл) и MeOH (8 мл).
Гидроциннамоил-fRW-NH2 (77)
Следуя методике получения соединения 28, получают неочищенный продукт взаимодействия 76 из 75 (0,2 г, 0,4 ммоль),
H-W-NH2·l (0,1 г, 0,42 ммоль), HOBt (0,068 г, 0,5 ммоль), TEA (0,14 мл, 1 ммоль), EDCI (0,1 г, 0,52 ммоль) и DMF (12 мл). Указанный продукт гидрируют по методике, используемой для получения соединения 32, в присутствии 5% Pd/BaSO4 (0,2 г, невосстановленный) и MeOH (10 мл), с получением 77.
Пример 88
Синтез 3-(4-OHPh)пропаноил-(4-F-f)RW-NH2 (82)
Соединение данного примера получают в соответствии с представленной схемой X:

Boc-(4-F-f)R(NO2)-OMe (78)
Следуя методике получения соединения 26, получают 78 из Boc-(4-F-f)-OH (1,5 г, 5,3 ммоль), H-R(NO2)-OМe·HCl (1,46 г, 5,4 ммоль), HOBt (0,87 г, 6,4 ммоль), TEA (1,86 мл, 13,3 ммоль), EDCI (1,33 г, 6,9 ммоль) и DMF (35 мл).
H-(4-F-f)R(NO2)-OMe (79)
TFA (4 мл) добавляют к раствору 78 (2,48 г, 5 ммоль) в DCM (20 мл) при 0° С. Смесь перемешивают при охлаждении льдом в течение 30 мин. и затем перемешивают при комнатной температуре
на протяжении ночи. Растворитель удаляют, остаток выпаривают совместно с эфиром (2× 30 мл) и полученный остаток сушат в вакууме, получая 79 в виде твердого вещества.
3-(4-OHPh)пропаноил-(4-F-f)R(NO2)-OMe (80)
Следуя методике получения соединения 26, получают 80 из 79 (2 г, 5 ммоль), 3-(4-гидроксифенил)пропановой кислоты (0,92 г, 5,5 ммоль), HOBt (0,8 г, 5,9 ммоль), TEA (1,7 мл, 12,6 ммоль), EDCI (1,15 г, 6 ммоль) и DMF (25 мл).
3-(4-OHPh)пропаноил-(4-F-f)R(NO2)-OH (81)
Следуя методике получения соединения 29, получают 81 из 80 (0,75 г, 1,4 ммоль), LiOH (0,065 г, 2,7 ммоль), воды (0,4 мл) и MeOH (8 мл).
3-(4-OHPh)пропаноил-(4-F-f)R(NO2)W-NH2 (82)
Используя методику получения соединения 30 с последующей очисткой препаративной ВЭЖХ, получают продукт взаимодействия из 81 (0,4 г, 0,75 ммоль), H-W-NH2·HCl (0,2 г, 0,82 ммоль), HOBt (0,12 г, 0,9 ммоль), TEA (0,26 мл, 1,9 ммоль), EDCI (0,17 г, 0,9 ммоль) и DMF (20 мл). Указанный продукт гидрируют по методике, используемой для получения соединения 32, в присутствии 5% Pd/BaSO4 (0,25 г, невосстановленный) и MeOH (15 мл), с получением 82.
Примеры 89-91
Синтез 3-(4-OHPh)пропаноил-fR-триптамида (88), 3-(2-OHPh)пропаноил-fR-триптамида (89) и гидроциннамоил-fR-триптамида (90)
Соединения указанных примеров получают в соответствии с представленной схемой XI:
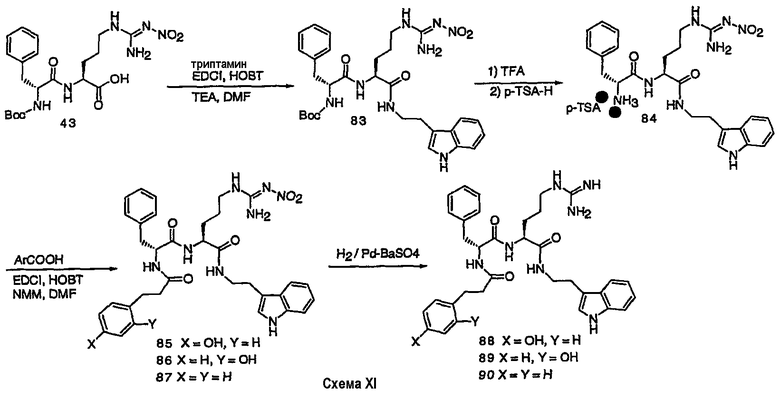
Boc-fR(NO2)-триптамид (83)
Boc-fR(NO2)-OH (43, 507,9 мг, 1,09 ммоль), триптамин (172,9 мг, 1,08 ммоль), HOBt (163,3 г, 1,08 ммоль), TEA (0,16 мл, 0,12 ммоль) и DMF (6 мл, безводный) смешивают, охлаждают при 0° С и при перемешивании добавляют EDCI (226,9 мг, 1,18 ммоль). После перемешивания при 0° С в течение 45 мин баню со льдом удаляют, смесь нагревают и перемешивают при комнатной температуре в течение 14,5 ч. Смесь разбавляют EtOAc (20 мл) и промывают 2N водной HCl (3× 5 мл), водный кислый слой вновь экстрагируют EtOAc (1× 10 мл) и объединенные EtOAc слои промывают 1M NaHCO3 (3× 5 мл) и насыщенным раствором соли (10 мл). Затем органический экстракт сушат безводным Na2SO4, поглотитель влаги удаляют фильтрованием и фильтрат концентрируют на роторном испарителе. Полученное твердое вещество сушат в вакууме, получая 83.
p-TSA• H-fR(NO2)-триптамид (84)
TFA (3 мл) добавляют к раствору Boc-fR(NO2)-триптамида (83)
(0,58 г, 0,95 ммоль) в DCM (6 мл) при 0° С. После перемешивания при 0° С в течение 30 мин. реакционную смесь перемешивают при комнатной температуре в течение 1,25 ч. Добавляют моногидрат п-толуолсульфокислоты (176,2 мг, 0,93 ммоль) и летучие вещества удаляют на роторном испарителе, получая коричневое масло. Масло растирают с эфиром (10 мл) и остаток сушат в вакууме, получая 84.
3-(4-OHPh)пропаноил-fR(NO2)-триптамид (85)
p-TSA• H-fR(NO2)-триптамид (84, 149,9 мг, 220 мкмоль), 3-(4-гидроксифенил)пропановую кислоту (39,6 мг, 238 мкмоль), HOBt (33,7 мг, 233 мкмоль), NMM (0,37 мл, 337 мкмоль) и DMF (1,5 мл, безводный) смешивают, охлаждают при 0° С и при перемешивании добавляют EDCI (44,6 мг, 233 мкмоль). После перемешивания при 0° С в течение 1,3 ч баню со льдом удаляют, смесь нагревают и перемешивают при комнатной температуре в течение 100 ч. Смесь разбавляют EtOAc (20 мл) и промывают 2N водной HCl (3× 5 мл), водный кислый слой вновь экстрагируют EtOAc (1 × 10 мл) и объединенные EtOAc слои промывают 1M NaHCO3 (3× 5 мл) и насыщенным раствором соли (10 мл). Затем органический экстракт сушат безводным Na2SO4, поглотитель влаги удаляют фильтрованием и фильтрат концентрируют на роторном испарителе, получая 85.
3-(2-OHPh)пропаноил-fR(NO2)-триптамид (86)
Следуя методике, используемой для получения соединения 85, получают неочищенный продукт взаимодействия 84 (151,7 мг, 223 мкмоль), 3-(2-гидроксифенил)пропановой кислоты (39,2 мг, 236 мкмоль), HOBt (35,3 мг, 231 мкмоль), NMM (0,40 мл, 364 мкмоль),
EDCI (45,0 мг, 235 мкмоль) и DMF (1,5 мл, безводный). Затем полученное вещество очищают препаративной ВЭЖХ (C4), получая 86.
Гидроциннамоил-fR(NO2)-триптамид (87)
Следуя методике, используемой для получения соединения 85, получают 87 из 84 (150,6 мг, 221 мкммоль), гидрокоричной кислоты (35,6 мг, 237 мкмоль), HOBt (34,4 мг, 225 мкмоль), NMM (0,37 мл, 337 мкмоль), EDCI (45,1 мг, 235 мкмоль) и DMF (1,5 мл, безводный).
3-(4-OHPh)пропаноил-fR-триптамид (88)
Следуя методике, используемой для получения соединения 32, получают 88 из 95 мг (145 мкмоль) 85 и 5% Pd-BaSO4 (68 мг, невосстановленный).
3-(2-OHPh)пропаноил-fR-триптамид (89)
Следуя методике, используемой для получения соединения 32, за исключением того, что полученный продукт лиофилизуют из смеси ацетонитрил-вода, а не очищают ВЭЖХ, получают 89 из 33,7 мг (51 мкмоль) 86 и 5% Pd-BaSO4 (31 мг, невосстановленный).
Гидроциннамоил-fR-триптамид (90)
Следуя методике, используемой для получения соединения 32, получают 90 из 106 мг (165 мкмоль) 87 и Pd-BaSO4 (63 мг, невосстановленный).
Пример 92
Синтез 3-(4-OHPh)пропаноил-Me-fR-триптамида (95)
Соединение данного примера получают в соответствии с представленной схемой XII:
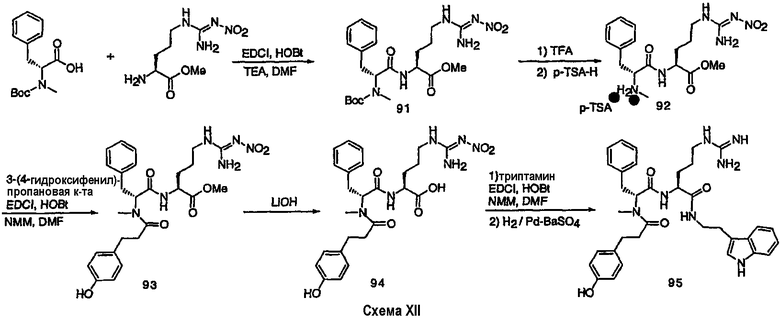
Boc-Me-fR(NO2)-OMe (91)
Boc-Me-f-OH (1,0008 г, 3,6 ммоль), H-R(NO2)-OMe•HCl (0,9659 г, 3,6 ммоль), HOBt (0,5515 г, 3,6 ммоль), TEA (0,525 мл, 3,8 ммоль) и DMF (20 мл, безводный) смешивают, охлаждают при 0° С и при перемешивании добавляют EDCI (0,7211 г, 3,8 ммоль). После перемешивания при 0° С в течение 1 ч. баню со льдом удаляют, смесь нагревают и перемешивают при комнатной температуре в течение 23 ч. Смесь разбавляют EtOAc (20 мл) и промывают 2N водной HCl (3× 8 мл), водный кислый слой вновь экстрагируют EtOAc (1× 10 мл) и объединенные EtOAc слои промывают 1M NaHCO3 (3× 8 мл) и насыщенным раствором соли (10 мл). Затем органический экстракт сушат безводным Na2SO4, поглотитель влаги удаляют фильтрованием и фильтрат концентрируют на роторном испарителе, получая 91.
p-TSA• H-Me-fR(NO2)-OMe (92)
Следуя методике получения соединения 84, получают 92 в виде гидратированной p-TSA соли из 91 (0,4989 г, 1,0 ммоль).
3-(4-OHPh)пропаноил-Me-fR(NO2)-OMe (93)
p-TSA•H-Me-fR(NO2)-OMe (92, 282,8 мг, 500 мкммоль), 3-(4-гидроксифенил)пропановую кислоту (85,7 мг, 516 мкмоль), HOBt
(77,4 мг, 505 мкмоль), NMM (0,60 мл, 546 мкмоль) и DMF (3 мл, безводный) смешивают, охлаждают при 0° С и при перемешивании добавляют EDCI (101,9 мг, 532 мкмоль). После перемешивания при 0° С в течение 1,3 ч баню со льдом удаляют, смесь нагревают и перемешивают при комнатной температуре в течение 24 ч. Добавляют еще EDCI (41,2 мг, 215 мкмоль), смесь перемешивают при комнатной температуре в течение 64 ч, снова добавляют EDCI (29,9 мг, 156 мкмоль) вместе с небольшим количеством NMM (0,60 мл, 546 мкмоль) и смесь нагревают при 50° С в течение 8 ч. Затем смесь оставляют при комнатной температуре в течение 21 ч и обрабатывают по методике получения соединения 91, получая 93.
3-(4-ОHPh)пропаноил-Me-fR(NO2)-OH (94)
Смесь 3-(4-OHPh)пропаноил-Me-fR(NO2)-OMe (93, 96,3 мг, 177 мкмоль), моногидрат LiOH (21,3 мг, 508 мкмоль) в смеси THF-MeOH-вода (6 мл, 4:1:1) перемешивают при комнатной температуре в течение 1 ч. Добавляют 6% водный KHSO4 (1,3 мл) и летучие вещества удаляют на роторном испарителе. К влажному остатку добавляют воду (1,4 мл), доводят рН до ~2 несколькими дополнительными каплями 6% водного KHSO4 и водную смесь экстрагируют EtOAc (3× 2 мл). Объединенные органические слои промывают водой и насыщенным раствором соли и сушат над безводным Na2SО4. Использованный поглотитель влаги удаляют фильтрованием и фильтрат концентрируют на роторном испарителе, получая 94.
3-(4-OHPh)пропаноил-Me-fR-триптамид (95)
3-(4-OHPh)пропаноил-Me-fR(NO2)-OH (94, 77,0 мг, 146 мкмоль),
триптамин (23,9 мг, 149 мкмоль), HOBt (25,1 мг, 164 мкмоль), NMM (0,17 мл, 155 мкмоль) и DMF (5 мл, безводный) смешивают, охлаждают при 0° С и при перемешивании добавляют EDCI (32,4 мг, 169 мкмоль). После перемешивания при 0° С в течение 1,5 ч баню со льдом удаляют, смесь нагревают и перемешивают при комнатной температуре в течение 4 ч. Смесь подвергают обработке по методике получения соединения 85, получая неочищенный продукт взаимодействия, который очищают препаративной ВЭЖХ (С4). Затем очищенный продукт растворяют в смеси 10% уксусная кислота-MeOH (20 мл) и гидрируют при давлении водорода 40 фунт/дюйм2 (276 кПа) при комнатной температуре в течение 15 ч, используя 5% Pd-BaSО4 (31,3 мг, невосстановленный) в качестве катализатора. Катализатор удаляют фильтрованием через целит, летучие вещества удаляют в вакууме, остаток повторно растворяют в смеси 10% ацетонитрил-вода (15 мл) и смесь замораживают и лиофилизуют, получая 95.
Пример 93
Синтез [1-карбамоил-2-(1H-индол-3-ил)этил]амида 5-[2-ацетиламино-3-(4-хлорфенил)пропиониламино]-4-оксо-6-фенил-2-(2-пиридин-2-илэтил)гексановой кислоты (103)
Соединение данного примера получают в соответствии с представленными схемами XIII и XIV:
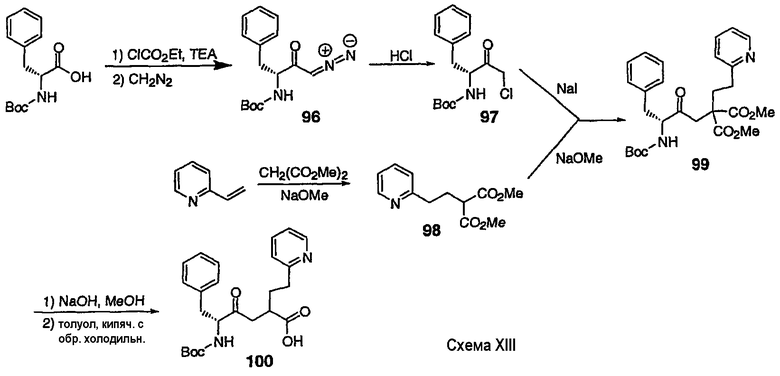
(R)-3-(трет-Бутоксикарбониламино)-1-диазо-4-фенилбутан-2-он (96)
К раствору Boc-f-OH (5,30 г, 20 ммоль) в безводном THF (100 мл) добавляют TEA (2,02 г, 20 ммоль) и этилхлорформиат (2,16 г, 20 ммоль) при –15° С в атмосфере аргона. Смесь перемешивают при -15° С в течение 30 мин, а затем нагревают до 0° С. Добавляют раствор диазометана в эфире [100 мл, полученный из Me(NO)NCONH2 (4,0 г, 40 ммоль) и 50% водного KOH (20 мл)]. Смесь нагревают и перемешивают при комнатной температуре в течение 3 ч. Смесь промывают насыщенным водным NaHCO3 (30 мл), насыщенным водным хлоридом аммония (30 мл) и насыщенным раствором соли (2× 30 мл). Органический слой сушат над Na2SО4 и концентрируют на роторном испарителе. Остаток кристаллизуют из смеси гексаны-EtOAc при 5° С, получая требуемый продукт. Маточный раствор концентрируют и очищают хроматографией (гексаны-EtOAc, 80:20), получая дополнительное количество требуемого продукта.
(R)-3-(трет-Бутоксикарбониламино)-1-хлор-4-фенилбутан-2-он (97)
К раствору диазокетона (96, 115 мг, 0,4 ммоль) в эфире (3 мл) добавляют смесь 4N HCl/1,4-диоксан (0,11 мл, 0,44 ммоль, полученный разбавлением конц. HCl 1,4-диоксаном) по каплям при 0° С. Полученную смесь перемешивают при 0° С в течение 30 мин. Добавляют несколько капель TEA, чтобы нейтрализовать раствор, и смесь разбавляют EtOAc (20 мл), промывают насыщенным водным NaHCO3 (10 мл) и насыщенным раствором соли (3 × 10 мл). Органический слой сушат над Na2SO4 и растворители выпаривают. Остаток очищают хроматографией (гексаны-EtOAc, 90:10), получая 108 мг (92%) бесцветного твердого вещества: т.пл.: 101-102° С; 1H ЯМР (CDCl3, 400 МГц) δ 1,43 (с, 9H), 3,03, 3,10 (АВX, JАВ = 13,8 Гц, JАХ = 7,1 Гц, JBX = 6,8 Гц, 2H), 4,00, 4,19 (AB, JАВ = 16,2 Гц, 2H), 4,70 (м, 1H), 5,04 (шир.д, J = 6,8 Гц 1H), 7,18-7,37 (м, 5H).
Метил 2-метоксикарбонил-4-(2’-пиридинил)бутаноат (98)
К свежеприготовленному NaOMe [из 2,3 г (0,1 моль) натрия] в MeOH (25 мл) добавляют диметилмалонат (32 г, 0,24 ммоль). Раствор 2-винилпиридина (10,5 г, 0,1 моль) в MeOH (15 мл) добавляют по каплям на протяжении 40 мин к раствору NaOMe при кипячении с обратным холодильником. Полученную смесь кипятят с обратным холодильником в течение 2,5 ч. MeOH удаляют при пониженном давлении, остаток обрабатывают 2N HCl (150 мл), затем экстрагируют эфиром (2× 60 мл), чтобы удалить избыточный диметилмалонат. Водную фазу подщелачивают 2N NaOH и экстрагируют эфиром (3× 100 мл). Объединенные органические фазы промывают насыщенным раствором соли (3× 60 мл) и сушат над Na2SO4. Фильтрат концентрируют при пониженном давлении, затем большую
часть избытка 2-винилпиридина удаляют в вакууме. Остаток очищают хроматографией (гексаны-EtOAc, 60:40 до 50:50 до 30:70), получая требуемый продукт.
Метил (R)-5-(трет-бутоксикарбониламино)-2-метоксикарбонил-6-фенил-2-[2’-(2"-пиридинил)этил]-4-оксогексаноат (99)
К раствору α -хлоркетона (97, 150 мг, 0,5 ммоль) в безводном 1,2-диметоксиэтане (3,0 мл) добавляют NaI (75 мг, 0,5 ммоль) и смесь перемешивают в атмосфере аргона в течение 15 мин (Смесь A). К раствору диэфира (98, 142 мг, 0,6 ммоль) в безводном 1,2-диметоксиэтане (3,0 мл) добавляют свежеприготовленный NaOMe (32 мг, 0,6 ммоль) и смесь перемешивают в атмосфере аргона в течение 15 мин (Смесь B). Смесь B добавляют к смеси A и полученную смесь перемешивают при комнатной температуре в течение 1 ч. Смесь разбавляют EtOAc (30 мл) и промывают насыщенным раствором соли (2× 10 мл). Органический слой сушат над Na2SO4, растворители удаляют на роторном испарителе и остаток очищают хроматографией (гексаны-i-PrOH, 80:20), получая 99.
(2RS,5R)-5-(трет-Бутоксикарбониламино)-6-фенил-2-[2’-(2"-пиридинил)этил]-4-оксогексановая кислота (100)
К раствору диэфира (99, 165 мг) в MeOH (10 мл) добавляют 5N NaOH (1,0 мл) и полученную смесь перемешивают при комнатной температуре в течение 2 ч. Смесь концентрируют на роторном испарителе и остаток растворяют в воде (10 мл) и подкисляют 3N HCl до pH=3. Смесь экстрагируют DCM (3× 10 мл) и объединенные органические фазы промывают насыщенным раствором соли (10 мл) и сушат над Na2SO4. Растворители удаляют на роторном испарителе, получая дикислоту. Неочищенную дикислоту суспендируют в толуоле
(10 мл) и смесь кипятят с обратным холодильником в атмосфере аргона в течение 3 ч. Растворитель выпаривают и остаток очищают хроматографией (DCM-MeOH, 98:2 до 95:5), получая смесь двух диастереизомеров, которые могут быть разделены препаративной ВЭЖХ.
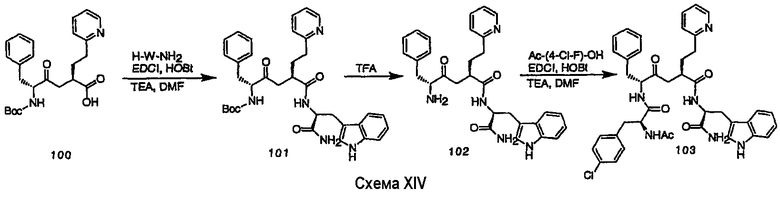
трет-Бутиловый эфир {1-бензил-4-[1-карбамоил-2-(1Н-индол-3-ил)этилкарбамоил]-2-оксо-6-пиридин-2-илгексил}карбаминовой кислоты (101)
Смешивают кислоту 100 (44 мг, 0,1 ммоль), H-W-NH2·l (26 мг, 0,11 ммоль), HOBt (16 мг, 0,12 ммоль), TEA (24 мг, 0,24 ммоль), EDCI (24 мг, 0,12 ммоль) и DMF (1 мл, безводный). Смесь перемешивают при комнатной температуре на протяжении ночи, затем выливают в воду (10 мл) и экстрагируют EtOAc (4× 9 мл). Объединенный экстракт промывают 1N HCl (2 × 8 мл), насыщенным NaHCO3 (1× 8 мл), насыщенным раствором соли (8 мл), затем сушат безводным Na2SO4. После фильтрования фильтрат концентрируют на роторном испарителе, получая 101.
[1-Карбамоил-2-(1Н-индол-3-ил)этил]амид 5-амино-4-оксо-6-фенил-2-(2-пиридин-2-илэтил)гексановой кислоты (102)
Пептид 101 (97 мг, 0,16 ммоль) смешивают с раствором (1 мл) анизол-TFA-DCM (1:8:9). Смесь перемешивают при комнатной температуре на протяжении ночи. После удаления растворителя
полученный остаток совместно выпаривают с эфиром (2 x 8 мл), получая 0,81 г (99%) 102.
Ac-(4-Cl-F)-OH
Смесь Boc-(4-Cl-F)-OH (54 мг, 0,18 ммоль), DCM (0,5 мл) и TFA (0,5 мл) перемешивают при комнатной температуре в течение 2 ч. После удаления растворителя полученный остаток смешивают с DCM (1 мл), уксусным ангидридом (17 мг, 0,2 ммоль) и TEA (40 мг, 0,4 ммоль). Смесь перемешивают при комнатной температуре на протяжении ночи, затем концентрируют на роторном испарителе, получая Ac-(4-Cl-F)-OH.
[1-Карбамоил-2-(1Н-индол-3-ил)этил]амид 5-[2-ацетиламино-3-(4-хлорфенил)пропиониламино]-4-оксо-6-фенил-2-(2-пиридин-2-илэтил)гексановой кислоты (103)
EDCI (36 мг, 0,19 ммоль) добавляют к смеси пептида 102 (81 мг, 16 ммоль), Ac-(4-Cl-F)-OH (41 мг, 0,17 ммоль), HOBt (24 мг, 0,18 ммоль), TEA (35 мг, 0,35 ммоль) и DMF (2 мл, безводный) при 0° С. Реакционную смесь перемешивают при комнатной температуре на протяжении ночи и затем обрабатывают по методике получения соединения 101. Неочищенный продукт очищают препаративной ВЭЖХ, получая 103.
Пример 94
Синтез N-{3-[9-бензил-12-(4-хлорбензил)-3-(1Н-индол-3-илметил-2,5,8,14-тетраоксо-1,4,7,13-тетраазациклотетракос-6-ил]пропил}гуанидина (110)
Указанный пример получают в соответствии с представленной схемой XV:
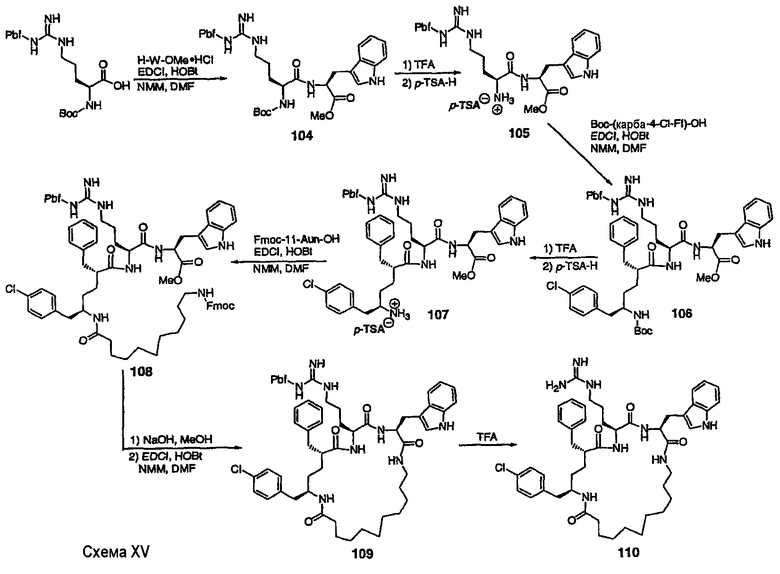
Boc-R(Pbf)W-OMe (104)
NMM (0,48 мл, 4,4 ммоль) добавляют к смеси Boc-R(Pbf)-OH (1,053 г, 2,0 ммоль), H-W-OMe• HCl (0,509 г, 2,0 ммоль), HOBt (0,270 г, 2,0 ммоль), EDCI (0,422 г, 2,2 ммоль) в DMF (10 мл) при 0° С и полученную смесь перемешивают при 0° С в течение 1 ч, а затем при комнатной температуре в течение 4 ч. Реакционную смесь разбавляют EtOAc (100 мл) и промывают последовательно водой (2× 10 мл), 1N HCl (2× 10 мл), насыщенным NaHCO3 (2× 10 мл) и насыщенным раствором соли (2× 10 мл). Органический слой сушат над MgSO4, осушитель влаги удаляют фильтрованием и фильтрат концентрируют на роторном испарителе, получая 1,454 г (100%) неочищенного продукта 104.
p-TSA• H-R(Pbf)W-OMe (105)
Раствор 104 (1,454 г, 2,0 ммоль) в смеси TFA-DCM-вода
(10:40:0,5, 20 мл) и DCM (20 мл) перемешивают при комнатной температуре в течение 6 ч. Добавляют моногидрат п-толуолсульфокислоты (0,380 г, 2,0 ммоль) и смесь перемешивают в течение 10 мин при комнатной температуре. Растворитель удаляют на роторном испарителе и остаток растирают со смесью эфир-гексан (1:1, 100 мл), получая 1,598 г (99,9%) неочищенного 105.
Boc-(карба-4-Cl-Ff)-R(Pbf)W-OMe(106)
EDCI (105,5 мг, 0,55 ммоль) и NMM (0,12 мл, 1,1 ммоль) добавляют к тщательно перемешиваемой смеси 5 (216 мг, 0,5 ммоль), 105 (399,5 мг, 0,5 ммоль) и HOBt (67,6 мг, 0,5 ммоль) в DMF (9 мл) при 0° С. Полученную смесь перемешивают при 0° С в течение 1 ч, а затем при комнатной температуре в течение 5 ч. Смесь обрабатывают по методике получения соединения 104, получая 508 мг (98%) 106.
p-TSA• H-(карба-4-Cl-Ff)-R(Pbf)W-OMe (107)
Раствор 106 (508 мг, 488 мкмоль) в смеси TFA-DCM-вода (10:90:0,5, 15 мл) перемешивают при комнатной температуре в течение 17 ч. Анализ ВЭЖХ указывает на то, что реакция еще полностью не завершена, поэтому добавляют еще смесь TFA-DCM-вода (10:90:0,5, 10 мл) и воду (1 мл) и полученную смесь перемешивают при комнатной температуре в течение 7 д. Добавляют моногидрат п-толуолсульфокислоты (92,8 мг, 488 мкмоль) и смесь перемешивают в течение 10 мин при комнатной температуре. Растворитель удаляют на роторном испарителе и остаток растирают с эфиром (25 мл), получая неочищенный 107.
Fmoc-11-Aun-(карба-4-Cl-Ff)-R(Pbf)W-OMe (108)
Следуют методике получения соединения 104 и получают
неочищенный 108 из Fmoc-11-Aun-OH (207 мг, 488 мкмоль), 107 (543 мг, 488 мкмоль), HOBt (66,0 мг, 488 мкмоль), EDCI (103,0 мг, 537 мкмоль) и NMM (0,118 мл, 1,07 ммоль).
[11-Aun-(карба-4-Cl-Ff)-R(Pbf)W] (109)
1N NaOH (2,2 мл, 2,2 ммоль) добавляют к раствору 108 (520 мг, 387 мкмоль) в смеси THF-MeOH (1:1) при комнатной температуре и полученную смесь перемешивают при комнатной температуре в течение 2 ч. Смесь подкисляют 1N HCl до pH ~3 и распределяют между EtOAc (100 мл) и водой (20 мл). Затем водную фазу экстрагируют EtOAc (2 × 20 мл), объединенную органическую фазу промывают насыщенным раствором соли (20 мл) и сушат над MgSO4. Осушитель влаги удаляют фильтрованием, фильтрат концентрируют на роторном испарителе и остаток растирают с эфиром (20 мл), получая неочищенную аминокислоту, подлежащую циклизации. Добавляют NMM (47 мкл, 460 мкмоль) к смеси указанной выше неочищенной аминокислоты (430 мг, 387 мкмоль), HOBt (53 мг, 387 мкмоль) и EDCI (82,5 мг, 430 мкмоль) в DMF при 0° С. Полученную смесь перемешивают при 0° С в течение 1 ч, а затем при комнатной температуре в течение 14 ч. Смесь обрабатывают по методике получения соединения 104, и очищают препаративной ВЭЖХ, получая 109.
[11-Aun-(карба-4-Cl-Ff)-RW] (110)
Смесь 109 (42 мг, 39 мкмоль) в смеси TFA-DCM-вода (10:10:0,5; 5 мл) перемешивают при комнатной температуре в течение 20 ч, летучие вещества удаляют на роторном испарителе и остаток очищают препаративной ВЭЖХ. Фракции, содержащие продукт, объединяют, концентрируют, замораживают и лиофилизуют, получая
110 в виде соли TFA.
Примеры 95-99
Способ сочетания (CP) для образования пептидной связи, осуществляемый в растворе:
Аминный компонент (1 эквивалент), кислотный компонент (1 эквивалент) и HOBt (2 эквивалента) растворяют в DMF (2 мл/ммоль субстрата). Раствор обрабатывают N-метилморфолином (3-4 эквивалента), EDCI (1,2 эквивалента) и перемешивают при комнатной температуре до тех пор, пока не завершится образование продукта (обычно 1-5 часов). Продукт осаждается после добавления к реакционной смеси воды (6-10 мл/мл DMF), и его отделяют от жидкости фильтрованием или декантацией.
A. Синтез скелета дипептида Boc-DPhe-Arg(NO2)OH (A-2)
A-1. Boc-DPhe-Arg(NO2)OMe
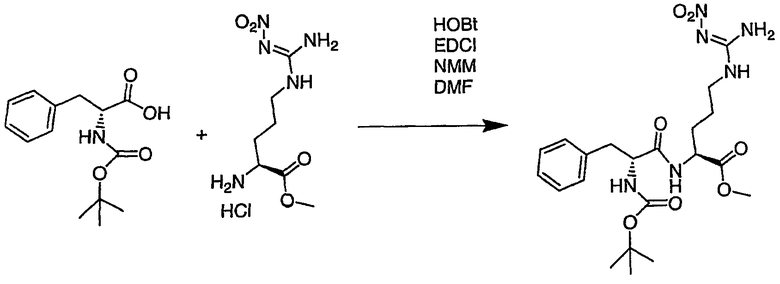
Для образования пептидной связи используют способ сочетания (CP) в растворе.
A-2. Boc-DPhe-Arg(NO2)OH
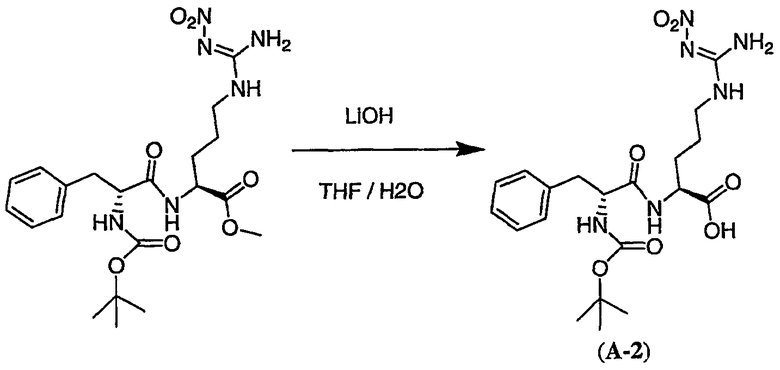
Раствор Boc-DPhe-Arg(NO2)OMe в THF охлаждают на бане со льдом и обрабатывают при 0° С водным раствором LiOH. Реакционную смесь перемешивают на бане со льдом в течение 4 часов. Растворители выпаривают до небольшого остаточного объема, который обрабатывают 1N HCl (приблизительно 25 мл) до pH 2-3. Продукт экстрагируют этилацетатом, промывают смесью вода/насыщенный раствор соли, сушат безводным сульфатом магния; растворитель выпаривают досуха, получая указанное в заголовке соединение.
B. Синтез аминоконцевых групп
B-1. 3-(4-Бензилоксифенил)пропионовая кислота
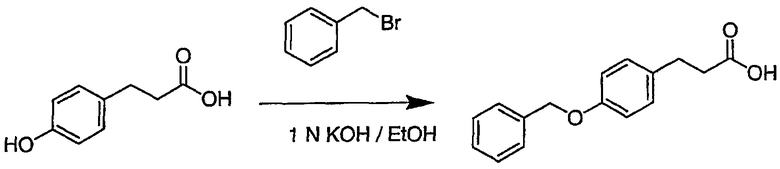
Используют методику, описанную в JACS 1955, 77, p. 4887-4892. Продукт осаждается после того, как реакционную смесь подкисляют до рН 2-3.
B-2. 3-(4-Бензилоксифенил)пропионилхлорид
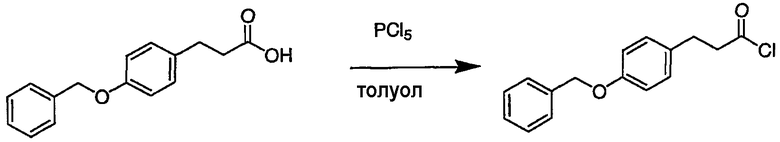
Твердый PCl5 добавляют к раствору 3-(4-бензилоксифенил)пропионовой кислоты в толуоле на протяжении 1 часа. Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и растворитель выпаривают. Остаток перемешивают с гексанами на протяжении ночи, получая кристаллическое вещество, которое отфильтровывают и сушат в вакууме.
B-3. 4-(S)-Бензил-3-[3-(4-бензилоксифенил)пропионил]оксазолидин-2-он
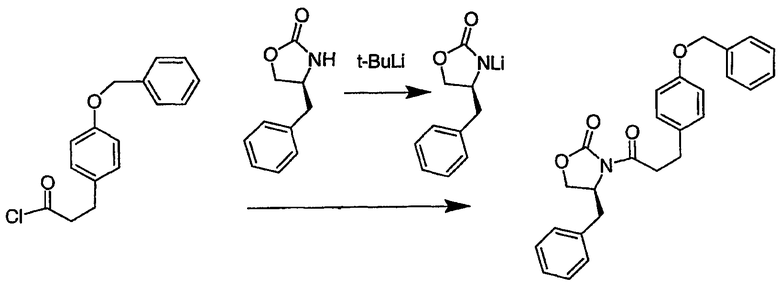
Используют методику, описанную в Tetrahedron 52(43), 1996, p.13733-13738. Соль Li-(S)-(-)-4-бензилоксазолидинона получают при температуре от –65° С до –72° С. Раствор деканоилхлорида в THF охлаждают до –72° С и обрабатывают раствором Li-(S)-(-)-4-бензилоксазолидинона при той же температуре. Реакционную смесь перемешивают при температуре от –70° С до –75° С в течение 1 часа и на протяжении ночи при комнатной температуре, обрабатывают раствором NH4Cl и экстрагируют этилацетатом. Органический слой промывают смесью вода/насыщенный раствор соли, сушат MgSO4 и выпаривают. Остаток очищают на колонке с диоксидом кремния с использованием раствора гексан/этилацетат 7/3 в качестве элюента, получая указанное в заголовке соединение.
B-4. 4-(S)-Бензил-3-[2-(4-бензилоксибензил)пент-4-еноил]оксазолидин-2-он
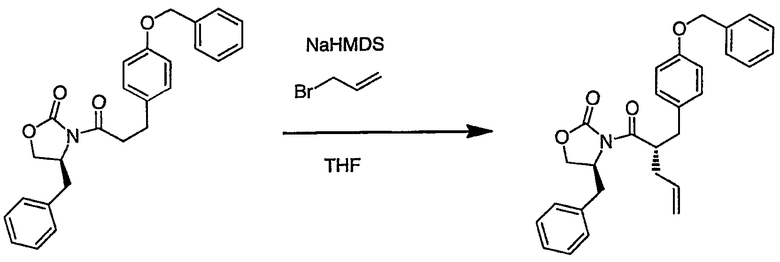
NaHMDS добавляют к раствору 4-(S)-бензил-3-[3-(4-бензилоксифенил)пропионил]оксазолидин-2-она в THF при температуре от –70° С до –75° С на протяжении 15 мин. Смесь перемешивают в течение 1 часа и обрабатывают аллилбромидом при -70° С. Перемешивание продолжают при этой же температуре в течение 1 часа. Реакционной смеси дают нагреться до 0° С в течение 3 часов и гасят 10% NH4Cl. Продукт экстрагируют этилацетатом, промывают смесью вода/насыщенный раствор соли и сушат безводным сульфатом магния. Растворитель выпаривают и неочищенный продукт очищают на колонке с диоксидом кремния, используя смесь гексан/этилацетат 4:1, получая указанное в заголовке соединение.
B-5. 2-(4-Бензилоксибензил)пент-4-еновая кислота

Используют методику, описанную в JOС 1992, 57(10), 2888-2902 (p.2894).
B-6. 4-(4-(S)-Бензил-2-оксооксазолидин-3-ил)-3-(4-бензилоксибензил)-4-оксобутиронитрил
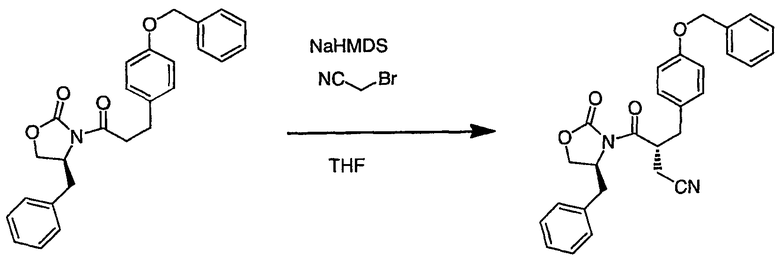
Для получения B-6 используют методику, аналогично используемой для получения B-4.
B-7. Гидрохлорид этилового эфира 4-амино-2-(4-гидроксибензил)масляной кислоты
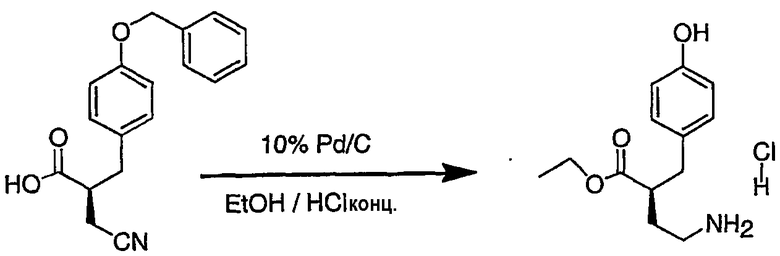
2-Цианометил-3-фенилпропионовую кислоту (8,16 г, 43 ммоль) в растворе этанола (75 мл) и концентрированной HCl (10 мл) гидрируют на протяжении ночи при 40 фунт/дюйм2 (276 кПа) в присутствии 10% Pd/C. Катализатор удаляют фильтрованием; фильтрат концентрируют при пониженном давлении досуха, получая указанное в заголовке соединение.
B-8. Метиловый эфир 3-(4-бензилоксифенил)-2-деканоиламинопропионовой кислоты
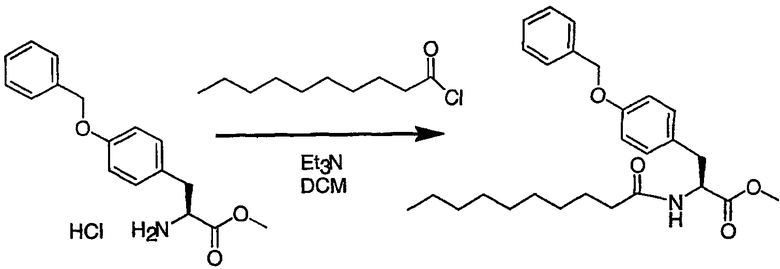
TEA добавляют к раствору остальных реактантов в DCM при температуре от –2° С до +3° С. Реакционную смесь перемешивают при комнатной температуре в течение 4 часов и разбавляют 0,1N HCl. Продукт экстрагируют DCM, промывают водой, сушат MgSO4 и растворитель выпаривают при пониженном давлении. Остаток кристаллизуют из гексанов, получая указанное в заголовке соединение.
B-9. 3-(4-Бензилоксифенил)-2-деканоиламинопропионовая кислота
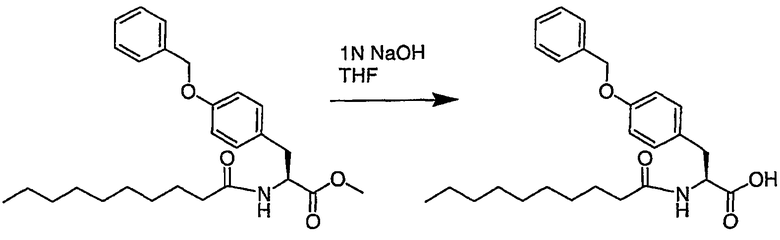
Реактанты перемешивают при комнатной температуре в течение 5 часов. Растворители выпаривают при пониженном давлении; остаток разбавляют водой и подкисляют до рН около 2. Полученный осадок продукта отфильтровывают, промывают водой, пока рН фильтрата не достигнет около 6, и сушат в вакууме на протяжении ночи.
C. Синтез карбоксильных концевых групп
C-1. N1-Бензил-3-(1H-индол-3-ил)-N1-метилпропан-1,2-диамин
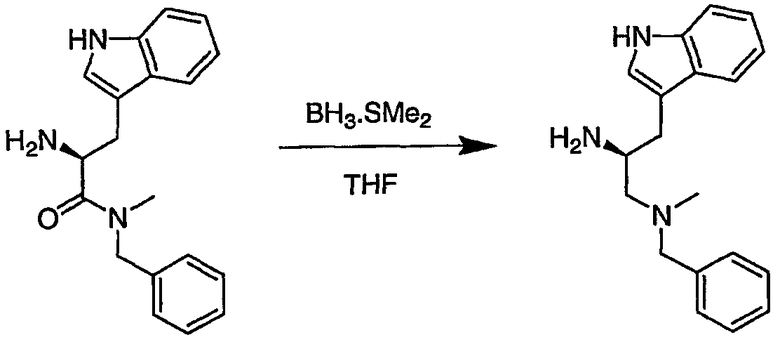
2M раствор комплекса BH3·Me2S в THF (40 мл) добавляют к раствору амидного субстрата (4,5 г, 8,4 ммоль) в безводном THF (50 мл). Реакционную смесь нагревают при 75° С, в то время как медленно отгоняемую жидкость собирают через холодильник. Через 2 часа добавляют новую порцию 2М раствора комплекса BH3·Me2S в THF (10 мл) и продолжают нагревание с одновременной дистилляцией в течение еще 3 часов. Реакционную смесь охлаждают до комнатной температуры и осторожно обрабатывают MeOH, пока не прекратится выделение газа, и 3N NaOH. Неочищенный продукт экстрагируют этилацетатом и очищают на колонке с диоксидом кремния с использованием раствора 1,5% MeOH в AcOEt, а затем раствора EtOAc/DCM/MeOH/Et3N 4/5/0,5/0,3, получая указанное в заголовке соединение.
C-2. N1-Бензил-N1-гексил-3-(1Н-индол-3-ил)пропан-1,2-диамин
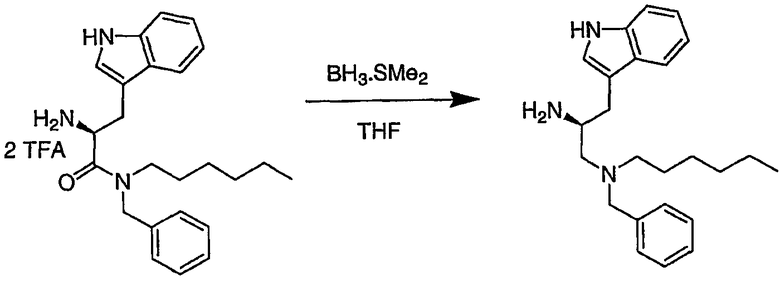
Для получения C-2 используют методику, аналогично используемой для получения C-1.
C-3. N1-Гексил-3-(1Н-индол-3-ил)-N1-метилпропан-1,2-диамин
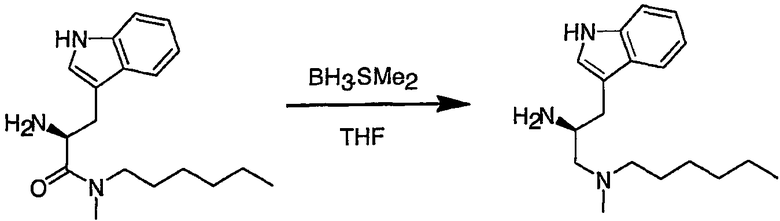
Для получения C-3 используют методику, аналогично используемой для получения C-1.
D. Сборка тетрапептидных миметиков
D-1. трет-Бутиловый эфир (1-{4-нитрогуанидино-1-[2-(гексилметиламино)-1-(1Н-индол-3-илметил)этилкарбамоил]бутилкарбамоил}-2-фенилэтил)карбаминовой кислоты
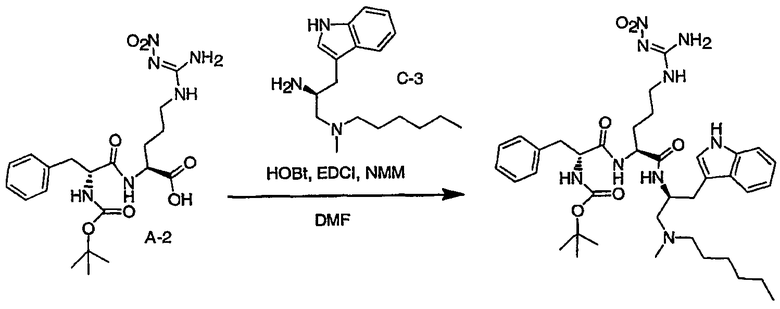
Для образования пептидной связи используют способ сочетания (CP) в растворе. Неочищенный продукт очищают на колонке с диоксидом кремния с использованием смеси гексан/этилацетат 6/1, получая указанное в заголовке соединение.
Для получения следующих соединений D-2, D-3, D-4 и D-5 используют методику, аналогично используемой для получения D-l.
D-2. Метиловый эфир 2-[2-(2-трет-бутоксикарбониламино-3-фенилпропиониламино)-5-нитрогуанидинопентаноиламино]-3-(1Н-индол-3-ил)пропионовой кислоты
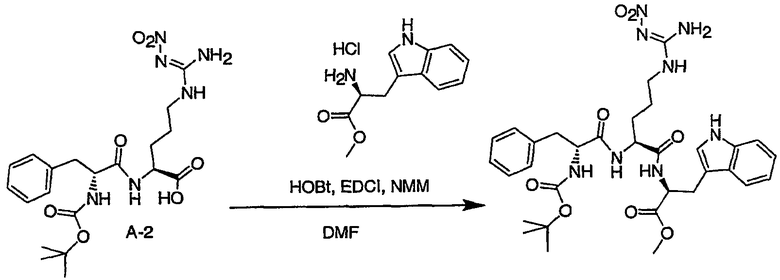
D-3. трет-Бутиловый эфир (1-{1-[2-(бензилгексиламино)-1-(1Н-индол-3-илметил)этилкарбамоил]-4-нитрогуанидинобутилкарбамоил}-2-фенилэтил)карбаминовой кислоты

D-4. трет-Бутиловый эфир (1-{4-нитрогуанидино-1-[2-(1Н-индол-3-ил)-1-(метилпропилкарбамоил)этилкарбамоил]бутилкарбамоил}-2-фенилэтил)карбаминовой кислоты
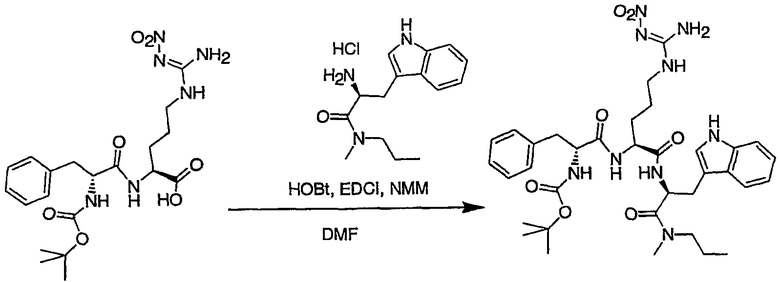
D-5. трет-Бутиловый эфир (1-{1-[1-(карбамоилметилметилкарбамоил)-2-(1Н-индол-3-ил)этилкарбамоил]-4-нитрогуанидинобутилкарбамоил}-2-фенилэтил)карбаминовой кислоты
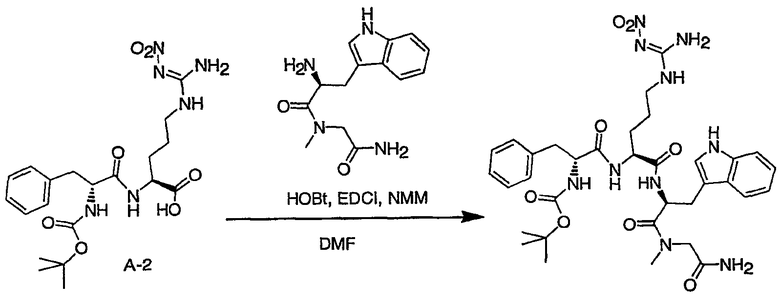
D-6. [2-(гексилметиламино)-1-(1Н-индол-3-илметил)этил]амид 2-(2-амино-3-фенилпропиониламино)-5-нитрогуанидинопентановой кислоты
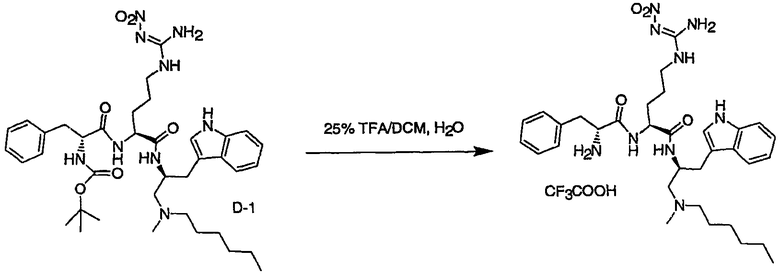
Реакционную смесь перемешивают при комнатной температуре в течение 4 часов и разбавляют 1,2-дихлорэтаном. Растворители выпаривают при пониженном давлении; остаток сушат в вакууме на протяжении ночи.
D-7. (1-{4-нитрогуанидино-1-[2-(гексилметиламино)-1-(1Н-индол-3-илметил)этилкарбамоил]бутилкарбамоил}-2-фенилэтил)амид 2-(4-бензилоксибензил)пент-4-еновой кислоты
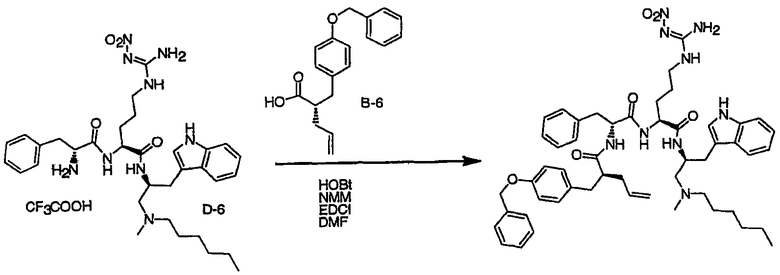
Для образования пептидной связи используют способ сочетания (CP) в растворе. Неочищенный продукт очищают препаративной ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 95
Синтез (1-{4-гуанидино-1-[2-(гексилметиламино)-1-(1H-индол-3-илметил)этилкарбамоил]бутилкарбамоил}-2-фенилэтил)амида 2-(4-гидроксибензил)пентановой кислоты

Реакционную смесь гидрируют при комнатной температуре, при 45 фунт/дюйм2 (310 кПа) на протяжении ночи. Катализатор отделяют фильтрованием через целит. Растворитель выпаривают при пониженном давлении. Неочищенный продукт очищают препаративной ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Пример 96
Синтез [1-(1-{4-гуанидино-1-[2-(гексилметиламино)-1-(1H-индол-3-илметил)этилкарбамоил]бутилкарбамоил}-2-фенилэтилкарбамоил)-2-(4-гидроксифенил)этил]амида декановой кислоты
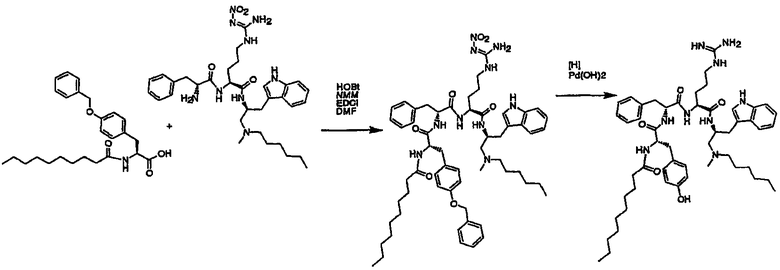
Для образования пептидной связи используют способ сочетания (CP) в растворе. Гидрирование осуществляют в этаноле при 45 фунт/дюйм2 (310 кПа) в течение 48 часов. Катализатор удаляют фильтрованием; неочищенный продукт очищают препаративной ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение.
Для получения соединения Примеров 97-99 используют методику, аналогично используемой для получения соединения Примера 95.
Пример 97
Синтез метилового эфира 2-(5-гуанидино-2-{2-[2-(4-гидроксибензил)пентаноиламино]-3-фенилпропиониламино}пентаноиламино)-3-(1H-индол-3-ил)пропионовой
кислоты
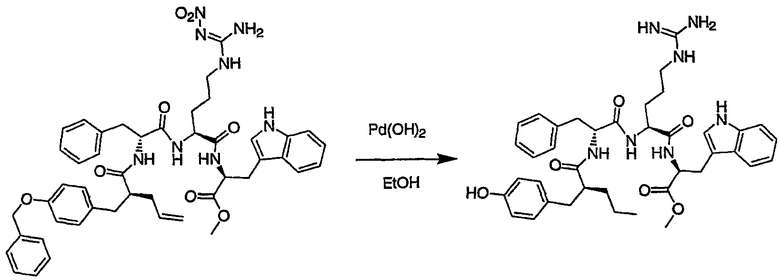
Пример 98
Синтез (1-{1-[1-(бензилметилкарбамоил)-2-(1H-индол-3-ил)этилкарбамоил]-4-гуанидинобутилкарбамоил}-2-фенилэтил)амида 2-(4-гидроксибензил)пентановой кислоты
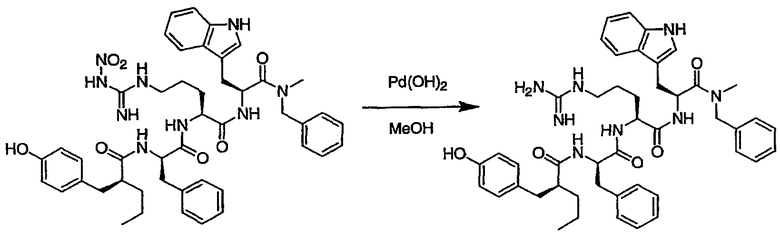
Пример 99
Синтез трет-бутилового эфира [2-(1-{1-[1-(карбамоилметилметилкарбамоил)-2-(1H-индол-3-ил)этилкарбамоил]-4-гуанидинобутилкарбамоил}-2-фенилэтилкарбамоил)-3-(4-гидроксифенил)пропил]карбаминовой кислоты
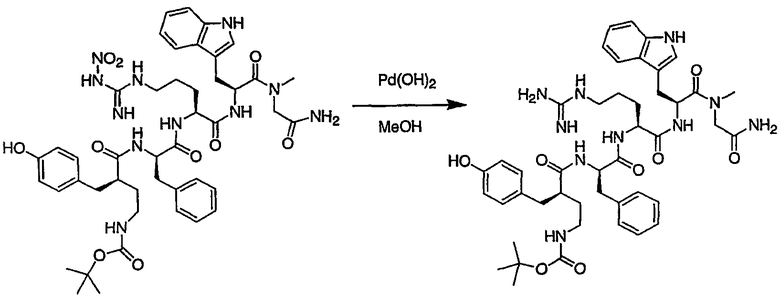
С. Твердофазный синтез пептидов вручную
Следующие пептиды синтезируют вручную, используя химию Fmoc и Rink амидную смолу в качестве твердой подложки. Удаление защитных групп Fmoc достигается 20% пиперидином в DMF в течение 30 мин с последующей промывкой DMF (3× 35 мл), MeOH (3× 35 мл) и DMF (3× 35 мл). Цветную реакцию с нингидрином используют для контроля завершения реакции. Ацетилирование концевой аминогруппы осуществляют смесью 5% Ac2O/0,25% NMM/0,2% HOBt в DMF в течение 30 мин с последующей экстенсивной промывкой DMF и DCM и недолговременной сушкой в вакууме. Неочищенный продукт отщепляют от смолы и защитные группы удаляют, используя 93% TFA и 2,3% этандитиола в воде в течение 3 ч. при комнатной температуре. После удаления смолы фильтрованием и промывания 3% TFA (3× 18 мл) фильтрат экстрагируют эфиром (6× 20 мл), замораживают и лиофилизуют. Затем неочищенный продукт растворяют в 30% водной уксусной кислоте и раствор очищают препаративной ВЭЖХ (размер частиц 10мк, Vydac C18, 10× 250 мм, градиентное элюирование смесью 6-60% ацетонитрил-вода (0,1% TFA) на протяжении 1,5 ч.). Фракции, содержащие продукт, объединяют и лиофилизуют, получая очищенные пептиды.
Пример 100
Синтез Ac-Y-(4-Py)ala-RW-NH2•2TFA
Fmoc-W(Boc)-OH и Fmoc-R(pbf)-OH (каждый в 2-кратном избытке) присоединяют последовательно к Rink амидной смоле (4,28 г, 3 ммоль), используя PyBOP (2-кратный избыток) и NMM (4-кратный избыток). После промывания DMF (3× 35 мл), эфиром (4× 35 мл) и сушки в вакууме достигается увеличение по массе. Смолу
(1,17 г, 0,35 ммоль) суспендируют в DMF (10 мл), удаляют Fmoc группу, последовательно добавляют PyBOP (0,6 г, 1,15 ммоль), NMM (0,26 мл, 2,8 ммоль) и Fmoc-(4-Py)ala-OH (0,447 г, 1,15 ммоль) и смесь встряхивают в течение 1 ч. После удаления защитной группы Fmoc снова повторяют процедуру связывания с Fmoc-Y(t-Bu)-OH (0,528 г, 1,1 ммоль). Затем снимают защиту с пептида, ацетилируют и отщепляют от смолы, получая продукт.
Пример 101
Синтез Ac-Y-(3-Py)ala-RW-NH2•2TFA
Указанное в заголовке соединение получают в соответствии с примером 100 за исключением того, что Fmoc-(3-Py)ala-OH используют вместо Fmoc-(4-Py)ala-OH.
Пример 102
Синтез Ac-Y-(2-Py)ala-RW-NH2•2TFA
Добавление Fmoc-(2-Py)ala-OH к раствору PyBop и NMM в DMF ведет к очень быстрому разложению аминокислоты. Поэтому соединение данного пример получают, используя модификацию методики, используемой для Примера 100, по которой NMM (88 мкл, 0,8 ммоль) добавляют к раствору Fmoc-(2-Py)ala-OH (0,311 г, 0,8 ммоль) и PyBrop (0,373 мг, 0,8 ммоль). Спустя 15 мин добавляют второй эквивалент NMM. По истечении 100 мин любые непрореагировавшие аминоконцы ацетилируют и далее следуют остальным стадиям, описанным в примере 100, получая указанное в заголовке соединение.
VIII. Примеры композиций и способа лечения
Пример А
Страдающую ожирением женщину, массой 130 кг, подвергают
лечению указанным выше способом с целью добиться потери массы тела. В частности, один раз в день на протяжении периода времени 6 месяцев, пациентке вводят, при помощи внутривенной инъекции, 15 мл водного раствора, включающего:
В конце периода лечения пациент демонстрирует измеримую потерю массы тела.
Пример B
Страдающего ожирением мужчину, массой 150 кг, подвергают воздействию программы, направленной на снижение массы тела, которая обеспечивает потерю массы тела вследствие снижения тучности посредством комбинированной терапии, включающей ограниченную диету и повышенную физическую нагрузку. В частности, один раз в день на протяжении периода времени 6 месяцев после потери массы, пациенту вводят, при помощи внутривенной инъекции, 15 мл водного раствора, включающего:
В конце периода лечения пациент демонстрировал устойчивую потерю массы и снижение ожирения.
Пример C
Страдающего ожирением мужчину, массой 165 кг, подвергают воздействию программы, направленной на снижение массы тела, которая обеспечивает потерю массы посредством комбинированной терапии, включающей ограниченную диету, наличие повышенной физической нагрузки и подкожную инъекцию ежедневно 15 мл водного раствора, включающего:
После того как желаемая потеря массы тела была достигнута,
потерю массы у пациента поддерживают при помощи продолжения внутривенной инъекции один раз в день на протяжении дополнительного периода времени 6 месяцев. В конце периода лечения пациент демонстрировал поддерживаемую потерю массы тела и снижение ожирения.
Пример D
Страдающую ожирением женщину, массой 140 кг, подвергают лечению указанным способом с целью добиться потери массы тела. В частности, ее лечат при помощи подкожно имплантируемого насоса, который поставляет 0,1 мг/кг соединения Примера 31 на протяжении 24-часового периода времени. Насос содержит раствор соединения, растворенного в растворе 50% пропиленгликоля и 50% стерильной воды. Насос заменяют ежемесячно, и лечение продолжается в течение шестимесячного периода, и в это время пациентка демонстрирует потерю массы тела и снижение ожирения.
Пример E
Страдающего ожирением мужчину, массой 150 кг, подвергают лечению указанным методом с целью добиться потери массы тела. В частности, его лечат пероральной таблеткой, принимаемой дважды в день, содержащей 300 мг соединения Примера 29. Лечение продолжается в течение 12 месяцев, и в это время пациент демонстрирует потерю массы тела и снижение тучности.
| название | год | авторы | номер документа |
|---|---|---|---|
| Ингибиторы фактора VIIa | 1999 |
|
RU2223967C2 |
| ПРОЛЕКАРСТВА PTH | 2017 |
|
RU2747316C2 |
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2014 |
|
RU2676324C2 |
| РАДИОФАРМАЦЕВТИЧЕСКИЕ СРЕДСТВА, РАДИОВИЗУАЛИЗИРУЮЩИЕ СРЕДСТВА И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2804297C2 |
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2018 |
|
RU2798085C2 |
| Лиганды PSMA для визуализации и эндорадиотерапии | 2018 |
|
RU2807076C2 |
| CNP ПРОЛЕКАРСТВА | 2016 |
|
RU2824988C1 |
| ГОМОДЕТНЫЕ ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ИНТЕГРИН α4β7 | 2018 |
|
RU2773443C2 |
| ПТГ-СОЕДИНЕНИЯ С НИЗКИМИ СООТНОШЕНИЯМИ ПИКА И МИНИМУМА | 2017 |
|
RU2766959C2 |
| РЕЖИМ ДОЗИРОВАНИЯ СОЕДИНЕНИЯ PTH КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ | 2017 |
|
RU2777357C2 |
Представлены лиганды MC-4 и/или MC-3 рецепторов, которые имеют структуру, соответствующую формуле (I):
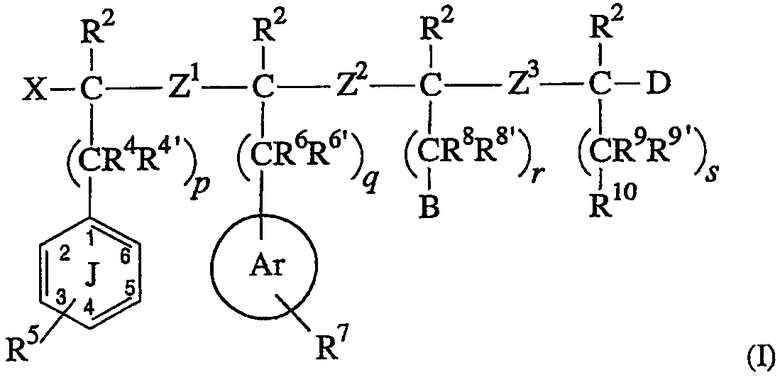
где: X -водород, OR1, -NR1R1’ и –CHR1R1’, где R1 и R1’ выбраны из группы: водород, (C1-C6)алкил и ацил; (1) каждый R2 независимо выбран из группы: водород, (C1-C6)алкил; или (2) (a) R2, связанный с атомом углерода, который связан с X и Z1, и заместитель R5 могут необязательно соединяться, образуя карбоциклическое или гетероциклическое кольцо, которое конденсировано с фенильным кольцом J; или; (b) R2, связанный с атомом углерода, который связан с кольцом Ar, может соединяться с R7, образуя кольцо, конденсированное с кольцом Ar; каждый из Z1, Z2 и Z3 независимо выбран из -N(R3e)C(R3)(R3a)-; -C(R3)(R3a)N(R3e)-; -C(O)N(R3d)-; -N(R3d)C(O)-; -C(R3)(R3a)C(R3b)(R3c)-; -SO2N(R3d)- и -N(R3d)SO2-; где каждый из R3, R3a,R3b и R3с, R3d, R3e когда присутствует, независимо выбран из водорода и (C1-C6)алкила; p целое число от 0 до 5; где, когда p больше, чем 0, R4 и R4’ выбраны из водорода, (C1-C6)алкила и арила; R5 представляет 5 заместителей в фенильном кольце J, где каждый R5 независимо выбран из водорода, гидрокси, галогена, тиола, -OR12, -N(R12)(R12’), (C1-C6)алкила, нитро, арила; где R12 и R12’ выбраны из водорода и (C1-C6)алкила; или два заместителя R5 могут необязательно соединяться с образованием карбоциклического или гетероциклического кольца, которое конденсировано с фенильным кольцом J; q равно 0, 1, 2, 3, 4 или 5; где, когда q больше, чем 0, R6 и R6’ выбраны из водорода и (C1-C6)алкила; Ar выбран из группы, состоящей из фенила, тиофена, фурана, оксазола, тиазола, пиррола и пиридина; R7 - заместители на кольце Ar, где каждый R7 выбран из водорода, галогена, -NR13R13’, (C1-C6)алкила и нитро; где R13 и R13’ выбраны из водорода и (C1-C6)алкила ; r целое число от 0 до 7; где, когда r больше, чем 0, R8 и R8’ выбраны из водорода и (C1-C6)алкила; B выбран из -N(R14)C(=NR15)NR16R17, -NR20R21, гетероарильного кольца, и гетероциклоалкильного кольца, где R14- R17, R20 и R21 независимо выбраны из водорода (C1-C6)алкила; s равно 0, 1, 2, 3, 4 или 5; где, когда s больше, чем 0, R9 и R9’ выбраны из водорода и (C1-C6)алкила; R10 выбран из группы, состоящей из необязательно замещенного бициклического арильного кольца и необязательно замещенного бициклического гетероарильного кольца; D выбран из водорода, амино и -C(O)R11; где R11 выбран из группы: гидрокси; алкокси; амино; алкиламино; -N(R19)CH2C(O)NH2, где R19 представляет (C1-C6) алкил; -NHCH2CH2OH и N(CH3)CH2CH2OH; или его изомеры, соли, гидраты или биогидролизуемый сложный эфир, амид или имид. 3 с. и 15 з.п. ф-лы.
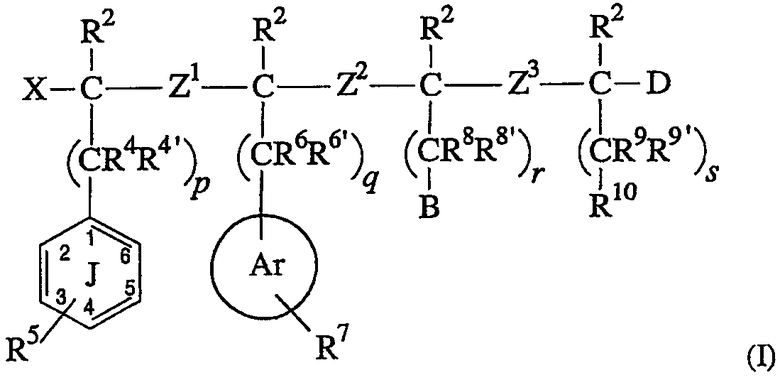
где
(A) X выбран из водорода, OR1, -NR1R1’ и –CHR1R1’, где R1 и R1’ независимо выбраны из группы, состоящей из водорода, (C1-C6) алкила и ацила;
(B) (1) каждый R2 независимо выбран из группы, состоящей из водорода, (C1-C6) алкила; или
(2) (a) R2, связанный с атомом углерода, который связан с X и Z1, и заместитель R5 могут необязательно соединяться, образуя карбоциклическое или гетероциклическое кольцо, которое конденсировано с фенильным кольцом J; или;
(b) R2, связанный с атомом углерода, который связан с кольцом Ar, может соединяться с R7, образуя кольцо, конденсированное с кольцом Ar; или
(C) каждый из Z1, Z2 и Z3 независимо выбран из -N(R3e)C(R3)(R3a)-; -C(R3)(R3a)N(R3e)-; -C(O)N(R3d)-; -N(R3d)C(O)-; -C(R3)(R3a)C(R3b)(R3c)-; -SO2N(R3d)- и -N(R3d)SO2-; где каждый из R3, R3a,R3b и R3с, когда присутствует, независимо выбран из водорода и (C1-C6) алкила;
(2) R3d, когда присутствует, выбран из водорода и (C1-C6) алкила;
(3) R3e, когда присутствует, выбран из водорода и (C1-C6) алкила;
(D) p равно 0, 1, 2, 3, 4 или 5; где, когда p больше, чем 0, каждый R4 и R4’ независимо выбран из водорода, (C1-C6) алкила и арила;
(E) R5 представляет 5 заместителей (т.е. положения 2-6) в фенильном кольце J, где каждый R5 независимо выбран из водорода, гидрокси, галогена, тиола, -OR12, -N(R12)(R12’), (C1-C6) алкила, нитро, арила; где каждый R12 и R12’ независимо выбран из водорода и (C1-C6) алкила; или два заместителя R5 могут необязательно соединяться с образованием карбоциклического или гетероциклического кольца, которое конденсировано с фенильным кольцом J;
(F) q равно 0, 1, 2, 3, 4 или 5; где когда q больше, чем 0, каждый R6 и R6’ независимо выбран из водорода и (C1-C6) алкила;
(G) Ar представляет арильное или гетероарильное кольцо, выбранное из группы, состоящей из фенила, тиофена, фурана, оксазола, тиазола, пиррола и пиридина;
(H) R7 обозначает заместители на кольце Ar, где каждый R7 выбран из водорода, галогена, -NR13R13’, (C1-C6) алкила и нитро; где каждый R13 и R13’ независимо выбран из водорода и (C1-C6) алкила;
(I) r равно 0, 1, 2, 3, 4, 5, 6 или 7; где, когда r больше, чем 0, каждый R8 и R8’ независимо выбран из водорода и (C1-C6) алкила;
(J) B выбран из -N(R14)C(=NR15)NR16R17, -NR20R21, гетероарильного кольца, содержащего, по крайней мере, один атом азота в кольце, и гетероциклоалкильного кольца, содержащего, по крайней мере, один атом азота в кольце, где R14, R15 R16, R17, R20 и R21 независимо выбраны из водорода (C1-C6) алкила;
(K) s равно 0, 1, 2, 3, 4 или 5; где, когда s больше, чем 0, каждый R9 и R9’ независимо выбран из водорода и (C1-C6) алкила;
(L) R10 выбран из группы, состоящей из необязательно замещенного бициклического арильного кольца и необязательно замещенного бициклического гетероарильного кольца;
(M) D независимо выбран из водорода, амино и -C(O)R11; где R11 выбран из группы, состоящей из гидрокси; алкокси; амино; алкиламино; -N(R19)CH2C(O)NH2, где R19 представляет (C1-C6)алкил; -NHCH2CH2OH и -N(CH3)CH2CH2OH;
или его оптический изомер, диастереомер или энантиомер; его фармацевтически приемлемая соль, гидрат или его биогидролизуемый сложный эфир, амид или имид.
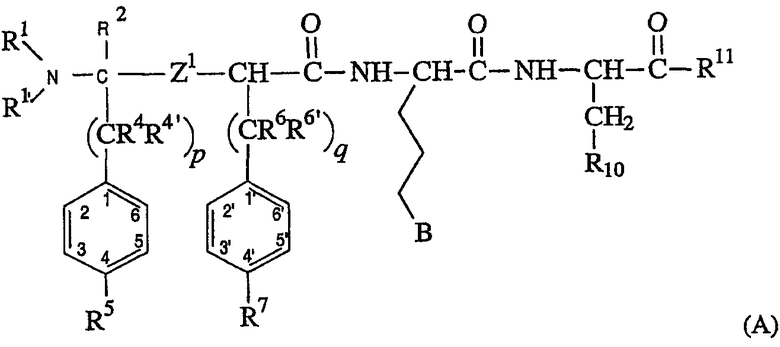
где
(A) R1 и R1’ независимо выбраны из группы, состоящей из водорода, (C1-C6)алкила и ацила;
(B) R2 выбран из группы, состоящей из водорода и (C1-C6) алкила;
(C) Z1 выбран из -N(R3e)C(R3)(R3a)-; -C(R3)(R3a)N(R3e)-; -C(O)N(R3d)-; -N(R3d)C(O)-; -C(R3)(R3a)C(R3b)(R3c)-; -SO2N(R3d)-; -N(R3d)SO2-; где
(1) каждый из R3, R3a, R3b и R3с, если присутствует, независимо выбран из водорода и (C1-C6) алкила;
(2) R3d, если присутствует, выбран из водорода и (C1-C6) алкила;
(3) R3e, если присутствует, выбран из водорода и (C1-C6) алкила;
(D) p равно 1 или 2 и каждый R4 и R4’ независимо выбран из водорода, (C1-C6) алкила и арила;
(E) R5 выбран из водорода, гидрокси, хлора, фтора, -N(R12)(R12’), где R12 и R12’ каждый независимо выбран из водорода и (C1-C6) алкила;
(F) q равно 0, 1 или 2; где, когда q больше, чем 0, каждый R6 и R6’ независимо выбран из водорода и (C1-C6) алкила;
(G) R7 выбран из водорода, галогена, -NR13R13’, (C1-C6) алкила, нитро; где каждый R13 и R13’ независимо выбран из водорода и (C1-C6) алкила;
(H) B выбран из -N(R14)C(=NR15)NR16R17, гетероарильного кольца, содержащего, по крайней мере, один атом азота в кольце, и гетероциклоалкильного кольца, содержащего, по крайней мере, один атом азота в кольце; где R14, R15, R16 и R17 независимо выбраны из водорода и (C1-C6) алкила;
(I) R10 представляет необязательно замещенное бициклическое кольцо, выбранное из 1-нафтила, 2-нафтила, индана, 1Н-индена, бензоциклобутана, бензоциклобутена, индола, индолина, пириндина, дигидропириндина, октагидропириндина, бензотиофена, бензофурана, бензимидазола, бензопирана, хинолина, хинолона и изохинолина;
(J) R11 выбран из группы, состоящей из амино; алкиламино; -N(R19)CH2C(O)NH2, где R19 представляет (C1-C6)алкил; -NHCH2CH2OH; -N(CH3)CH2CH2OH; или его оптический изомер, диастереомер или энантиомер; его фармацевтически приемлемая соль, гидрат или его биогидролизуемый сложный эфир, амид или имид.
| ПРОИЗВОДНЫЕ N-АЦИЛПРОЛИЛДИПЕПТИДОВ | 1993 |
|
RU2119496C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2141483C1 |
| Schioth H.B | |||
| et | |||
| al | |||
| Eur | |||
| J.Pharmacol | |||
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |
| Adan R.A | |||
| et | |||
| al | |||
| Eur | |||
| J.Pharmacol | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |

