Область техники
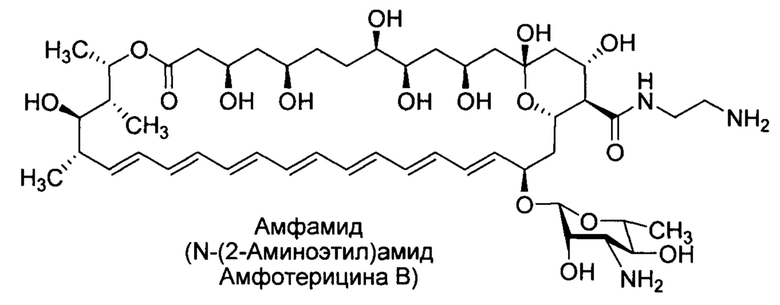
Настоящее изобретение относится к химико-фармацевтической промышленности и касается способа получения производного амфотерицина В - N-(2-аминоэтил)амида амфотерицина В (амфамида), формулы I, обладающего противогрибковыми свойствами и низкой токсичностью в сравнении с амфотерицином В. Способ получения N-(2-аминоэтил)амида амфотерицина В (амфамида) включает реакцию амидирования амфотерицина В этилендиамином в виде свободного основания в присутствии конденсирующего агента и соли органического основания и минеральной кислоты, а также очистку получающегося амфамида-сырца методом обращено-фазовой хроматографии. Технический результат - повышение выхода и снижение себестоимости производства целевого продукта.
Уровень техники
На сегодняшний день грибковые заболевания - одни из самых распространенных, ими страдает каждый четвертый житель нашей планеты [Biotechnology and Bioprocess Engineering 24: 436-444 (2019)]. Заболеваемость инвазивными грибковыми инфекциями значительно ниже, чем поверхностными, однако инвазивные заболевания ассоциируются с недопустимо высокими показателями летальности. Увеличение числа инвазивных микозов имеет многочисленные основополагающие причины, такие как рост числа иммунокомпрометированных больных, в том числе пациентов после иммуносупрессивной терапии при трансплантации, онкологических больных с индуцированной химиотерапией нейтропенией, а также ВИЧ-инфицированных пациентов. Кроме того, за последние несколько десятилетий увеличилось число онкологических больных, склонных к грибковым инфекциям [Open Forum Infectious Diseases, 2018, 5(8), ofyl87]. В настоящее время препаратом выбора для лечения тяжелых системных грибковых инфекций является амфотерицин В, клиническое использование которого значительно ограничено серьезными побочными эффектами лечения (нефро- и гепатотоксичность, поражения центральной нервной системы) и его крайне низкой растворимостью в воде. Таким образом, разработка лекарственного средства для лечения системных грибковых инфекций, подходящего, в частности, иммунокомпрометированным пациентам, обладающего высокой противогрибковой активностью при сниженной токсичности и повышенной растворимостью в воде и высокой стабильностью, является актуальной задачей современной фармацевтики и медицины. Ранее был описан N-(2-аминоэтил)амид амфотерицина В (Амфамид, Формула I), продемонстрировавший преимущества перед исходным амфотерицином В, в том числе, улучшенные фармакологические свойства и повышенную растворимость в воде [ACS Infect. Dis. 2020, V. 6, P. 2029-2044; Химико-фармацевтический журнал, 2019, 53, №10, 50-54; Химико-фармацевтический журнал, 2019, 53, №11, 30-33; Патент на изобретение РФ №2688658]. Амфамид обладает более высокой противогрибковой активностью в сравнении с амфотерицином В в экспериментах in vitro на панели штаммов грибков и дрожжей, играющих ведущую роль в патогенезе системных грибковых инфекций человека, а также демонстрирует высокую эффективность в эксперименте in vivo на модели кандидозного сепсиса мышей [ACS Infect. Dis. 2020, V. 6, P. 2029-2044]. Установлено, что амфамид обладает значительно сниженной токсичностью в сравнении с амфотерицином В: дозы, характеризующие острую токсичность амфотерицина В при внутривенном введении составляют: LD50 1,2 мг/кг (мыши), 1,6 мг/кг (крысы), в то время как для Амфамида - LD50 13,8 мг/кг (мыши). Таким образом, Амфамид превосходит по специфической фармакологической активности Амфотерицин В при действии на клинически важные грибные патогены при улучшенных фармакокинетических характеристиках.
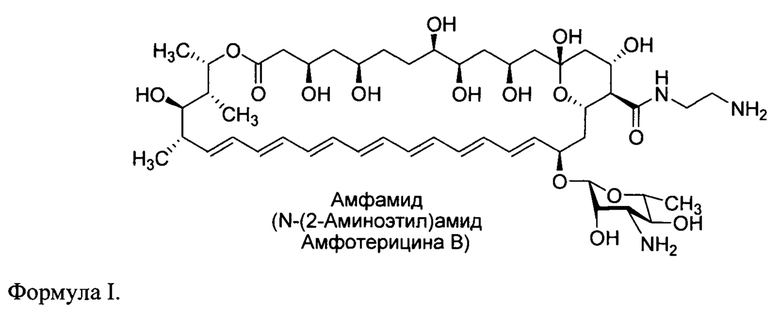
Исходя из уровня техники процесс получения Амфамида из амфотерицина В включает три синтетических стадии (Рис. 1) [ACS Infect. Dis. 2020, V. 6, P. 2029-2044; Патент на изобретение РФ №2688658]. Синтез включает получение N'-Fmoc-амфотерицина В реакцией амфотерицина В с N-(9Н-флуорен-2-ил-метоксикарбонил)сукцинимидом, амидирование N'-Fmoc-амфотерицина В этилендиамином (2 экв.) в ДМСО в присутствии РуВОР (бензотриазол-1-ил-оксотрипирролидинофосфониум гексафторфосфата) (1.5 экв.) в качестве конденсирующего агента и триэтиамина в качестве основания (для поддержание рН реакционной среды около рН 7-8) и удаление Fmoc-защитных групп на заключительной стадии действием пиперидина (3 экв.) [ACS Infect. Dis. 2020, V. 6, P. 2029-2044; Патент на изобретение РФ №2688658].
Для получения конечного амфамида требуемой степени чистоты (около 95% по данным ВЭЖХ) требовалась очистка промежуточных интермедиатов, N'-Fmoc-амфотерицина В (II) и N'-Fmoc-N-(2-аминоэтил)амида амфотерицина В (III), методом колоночной хроматографии на силикагеле. Лабильность интермедиатов и конечного соединения (Амфамида), крайне низкая растворимость в воде и органических растворителях полиеновых антибиотиков, необратимая сорбция на хроматографических сорбентах, а также высокие требования к чистоте конечного препарата (не менее 95%) обуславливают невысокий выход (36%) Амфамида исходя из амфотерцина В при получении его по трехстадийной схеме, что повышает стоимость конечной фармацевтической субстанции Амфамида [ACS Infect. Dis. 2020, V. 6, P. 2029-2044].
Синтез N-(2-аминоэтил)амида амфотерицина В описан также в патенте WO 2015/164289 А1. Синтез включает получение N'-Fmoc-амфотерицина В реакцией амфотерицина В с N-(9H-флуорен-2-ил-метоксикарбонил)сукцинимидом, амидирование N'-Fmoc-амфотерицина В гидрохлоридом моно N-Fmoc-этилендиамина (1.5 экв.) при 0°C в ДМФА в присутствии диизопропилэтиламина (2.5 экв.) в качестве основания и COMU (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолино-карбениум гексафторфосфата) (1 экв.) в качестве конденсирующего агента и удаление Fmoc-защитных групп добавлением пиперидина (3 экв.). Очистку получающегося N-(2-аминоэтил)амида амфотерицина В вели методом ВЭЖХ, выход целевого соединения составил 20%.
Таким образом, разработка более эффективного способа получения Амфамида, позволяющего повысить выход целевого продукта, является актуальной задачей, решение которой позволит организовать рентабельное производство фармацевтической субстанции и лекарственной формы Амфамида для лечения пациентов с системными грибковыми инфекциями.
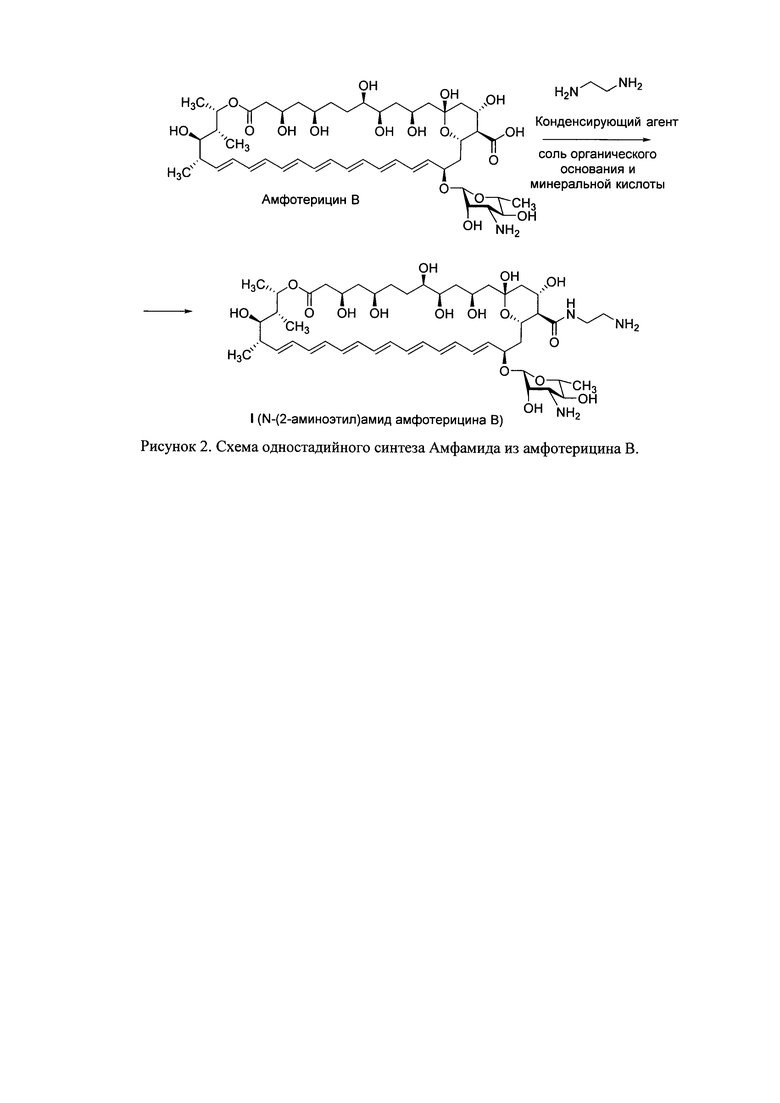
Указанная техническая задача решена путем перехода к одностадийной схеме синтеза Амфамида из амфотерицина В и подбору условий реакции амидирования С-16 карбоксильной группы антибиотика, обеспечивающих высокую селективность протекания реакции и минимальное количество побочных продуктов при отсутствии защитных групп в молекуле антибиотика и диамина.
Необходимо отметить, что в литературе описаны способы получения амидов амфотерицина В напрямую реакцией амидирования аминами различного строения в присутствии конденсирующих агентов [PLoS ONE 11(9): е0162171; The Journal of Antibiotics (2016), 1-12; J. Am. Chem. Soc. 1995,117, 6249-6253], однако, случай получения амидов амфотерицина В с свободными диаминами является особым случаем реакции амидирования, поскольку в этом случае возможно образование побочных продуктов димерных амидов, содержащих два антибиотика, присоединенных амидной связью по каждой из аминогрупп аминосахала или диамина, как это описано, например, в работе Yamaju N. с соавт. [Tetrahedron Letters, 2007, 49(19), 3393-3396].
В литературе имеется два примера осуществления реакции амидирования амфотерицина В напрямую аминами, имеющими две свободные аминогруппы.
Jarzebski А. с соавт. [The Journal of Antibiotics, 1982, 35(2), 220-229] описали синтез N-(3-аминопропил)амида амфотерицина В реакцией конденсации антибиотика с 10-кратным избытком 1,3-диаминопропана в N,N-диметилацетамиде в присутствии 10 эквивалентов дифенилфосфорилазида (DPPA) и 10 эквивалентов триэтиламина. Очистку производного проводили методом противоточного распределения (хлороформ : метанол : вода, 2:2:1). Чистота конечного амида была неудовлетворительной и составила всего 66%, выход конечного продукта не указан.
Тевяшова с соавт. [ACS Infect. Dis. 2020, V. 6, P. 2029-2044] описали синтез 1-(пиперазин-1-ил)амида амфотерицина В, N-(2-((2-гидроксиэтил)амино)этил)амида амфотерицина В и N-(2-(2-аминоэтокси)этил)амида амфотерицина В. Синтез вели добавлением соответствующего амина (0.44 ммоль, 2 экв.) к раствору амфотерицина В AmB (200 мг, 0.22 ммоль) в ДМСО (5 мл), добавлением РуВОР (бензотриазол-1-ил-оксотрипирролидинофосфониум гексафторфосфата) (137 мг, 0.26 ммоль, 1.18 экв.) в качестве конденсирующего агента и триэтиамина в качестве основания (для поддержания рН реакционной среды около рН ~8). Очистку целевых амидов проводили методом колоночной хроматографии на силикагеле, выходы амидов составили 12-25%.
Таким образом, известные из уровня техники способы синтеза амидов амфотерицина В, исходя незащищенного антибиотика и свободных аминов, имеющих две первичных или вторичных аминогруппы, не позволяют получать такие амиды с высоким выходом и приемлемой чистоты, а, следовательно, не могут быть использованы для препаративного синтеза амфамида в одну стадию из амфотерицина В.
Раскрытие сущности изобретения
Техническая задача получения амфамида (I) с высоким выходом и высокой степени чистоты решена путем перехода к одностадийной схеме синтеза амфамида из амфотерицина В, подбора условий реакции амидирования амфотерицина В, в частности нахождении оптимальной буферной системы для проведения реакции и очистке получающегося амфамида-сырца методом обращенно-фазовой хроматографии (Рис. 2).
Серия экспериментов по оптимизации количества используемого аминного компонента и буферной системы при получении амфамида показала, что использование свободного 1,2-этилендиамина в качестве аминного компонента и соли органического основания с минеральной кислотой для поддержания оптимального для проведения реакции рН приводят к неожиданному результату - а именно, повышению выхода и качества (чистоты) конечного продукта, и лучшей воспроизводимости результатов синтеза. Использование такой буферной системы, позволяет создать в реакционной смеси оптимальное значение рН, обеспечивающее минимальное количество побочных реакций, таких как конденсация двух молекул антибиотика или получения димеров амфотерицина В, сшитых этилендиаминовым спейсером, что приводит к повышению чистоты амфамида-сырца, выделяемого осаждением из реакционной смеси, и в дальнейшем облегчает процесс хроматографической очистки, в частности, уменьшает число смешанных фракций, что в конечном итоге позволяет значительно повысить выход целевого амфамида по сравнению с трехстадийной схемой, известной из уровня техники. В качестве органического основания могут быть использованы такие органические основания, как третичные амины, пиридин и алкилпиридины, хинолины и другие. В качестве минеральной кислоты могут быть использованы такие минеральные кислоты как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота и другие. Соль органического основания и минеральной кислоты может быть предварительно получена смешением эквимолярных количеств органического основания и минеральной кислоты. Предпочтительным является использование коммерчески доступных солей органического основания и минеральной кислоты, таких как гидрохлорид триэтиламина, гидробромид триэтиламина, гидрохлорид триметиламина, трипропиламин фосфат, гидрохлорид пиридина, гидробромид пиридина. В предпочтительном варианте осуществления изобретения в качестве соли органического основания и минеральной кислоты используется соль третичного амина или пиридина и минеральной кислоты, в наиболее предпочтительном варианте - гидрохлорид триэтиламина.
Кроме того, повысить выход целевого амфамида позволил переход от хроматографической очистки на силикагеле полупродуктов II и III, получаемых по трехстадийной схеме, к очистки конечного N-(2-аминоэтил)амида амфотерицина В (I) методом обращенно-фазовой хроматографии. Введение в молекулу амфотерицина В дополнительной группы, способной к протонированию, значительно повысило растворимость производного I в воде, что позволило осуществлять его очистку на силанизированном силикагеле или методом препаративной ВЭЖХ на обращенно-фазовых носителях (С9 или С18). Дополнительным преимуществом такого метода является использование в качестве элюентов водно-органических смесей, таких как вода - ацетонитрил, вода - изопропанол, вода - этанол, вода - н-пропанол, являющихся гораздо менее токсичными, чем хлороформ и метанол, используемые при очистке интермедиатов II и III методом хроматографии на силикагеле. Кроме того, более высокая растворимость в воде амфамида обеспечивает его меньшую сорбцию на обращенно-фазовом носителе, что, во-первых, позволяет, повысить выход целевого препарата, а во-вторых, делает возможным повторное использование носителей для обращенно-фазовой хроматографии после их регенерации.
Использованные в совокупности приемы, а именно, переход на одностадийную схему синтеза амфамида, проведение реакции амидирования амфотерицина В этилендиамином в виде свободного основания в присутствии конденсирующего агента и соли третичного основания или придина и минеральной кислоты, а также очистка получающегося амфамида-сырца методом обращено-фазовой хроматографии позволили достичь выхода N-(2-аминоэтил)амида амфотерицина В (I) в 65-70% при соблюдении требования к чистоте не менее 95% по данным ВЭЖХ, что значительно превышает выход амфамида при получении его по трехстадийной схеме, а также выходы амидов амфтерицина В, получаемых прямым амидированием антибиотика аминами, содержащими две свободные аминогруппы.
Одностадийный синтез Амфамида I, описываемый в настоящем изобретении, может быть осуществлен методами органической химии, известными из уровня техники, и включает активацию С16-карбоксильной группы амфотерицина В реагентами Кастро (ВОР или РуВОР) или другими конденсирующими агентами (TBTU, HBTU, COMU), предпочтительно РуВОР, с последующим взаимодействием с этилендиамином в виде свободного основания в присутствии гидрохлорида триэтиламина. Реакцию проводят в полярном апротонном растворителе, таком как диметилсульфоксид (ДМСО), диметилформамид (ДМФА), диметилацетамид (ДМАА) или их смеси, предпочтительно, в ДМСО. Соотношение реагентов может варьироваться в пределах антибиотик : этилендиамин : конденсирующий агент : соль третичного амина или пиридина и минеральной кислоты от 1:1:1:1 до 1:10:10:10, в более предпочтительном варианте 1:5-7:1.5-2:5-8, предпочтительно 1:5:1.5:6 (моль/моль/моль/моль). Ключевым фактором, обеспечивающий неожиданный результат - максимальный выход целевого амфамида - является добавление в реакционную смесь соли органического основания и минеральной кислоты, предпочтительно третичного амина или пиридина и минеральной кислоты, более предпочтительно, гидрохлорида триэтиламина, от в количестве от 1 от до 10 эквивалентов в расчет на исходный амфотерицин В, предпочтительно от 6 до 8 эквивалентов, более предпочтительно 6 эквивалентов.
Выделение сырца полученного антибиотика (в виде основания из реакционной смеси ведут, осаждая антибиотик растворителем, таким как диэтиловый эфир, гексан, петролейный эфир, ацетон, предпочтительно, диэтиловый эфир, и отфильтровывая выпавший осадок. Очистку выделяемого амфамида-сырца проводят методом обращенно-фазовой хроматографии, в том числе на силанизированном силикагеле или с С9 или С18 силанизированном силикагеле, предпочтительно силанизированном силикагеле. Фракции, содержащие целевой амфамид объединяют, удаляют органический растворитель, и выделяют целевой амфамид осаждением из водного раствора добавлением ацетона или оставшийся водный раствор замораживают при температуре -18°C, и высушивают с помощью лиофильной сушки.
Описанная последовательность операция позволяет получать позволяет получить целевой N-(2-аминоэтил)амид амфотерицина В с выходом 65-70% и чистотой 93-95% по данным ВЭЖХ.
Нижеприведенные неограничивающие примеры даны для демонстрации предпочтительных вариантов осуществления настоящего изобретения.
Пример 1. Синтез N-(2-аминоэтил)амида амфотерицина В (Амфамида I)
К раствору амфотерицина В (460 мг, 0.5 ммоль) в ДМСО (10 мл) прибавляют этилендиамин (166 μл, 2.5 ммоль, 5 экв.), гидрохлорид триэтиламина (411 мг, 3 ммоль, 6 экв.) и конденсирующий агент РуВОР (388 мг, 0.75 ммоль. 1.5 экв.). Реакционную смесь перемешивают 1 ч при комнатной температуре (контроль методом ТСХ, система хлороформ-метанол 7:1). К реакционной смеси прибавляют ацетон (10 мл) и диэтиловый эфир (25 мл), после чего фильтруют образовавшийся осадок, промывают его ацетоном и диэтиловым эфиром и сушат в вакууме. Выход ~500 мг (95%) амфамида-сырца.
Пример 1. Синтез N-(2-аминоэтил)амида амфотерицина В (Амфамида I)
К раствору амфотерицина В (460 мг, 0.5 ммоль) в ДМСО (10 мл) прибавляют этилендиамин (166 μл, 2.5 ммоль, 5 экв.), гидрохлорида пиридина (346 мг, 3 ммоль, 6 экв.) и конденсирующий агент РуВОР (388 мг, 0.75 ммоль. 1.5 экв.). Реакционную смесь перемешивают 1 ч при комнатной температуре (контроль методом ТСХ, система хлороформ-метанол 7:1). К реакционной смеси прибавляют ацетон (10 мл) и диэтиловый эфир (25 мл), после чего фильтруют образовавшийся осадок, промывают его ацетоном и диэтиловым эфиром и сушат в вакууме. Выход ~500 мг (95%) амфамида-сырца.
Пример 3. Очистка N-(2-аминоэтил)амида амфотерицина В (Амфамида I)
Исходный образец Амфамида-сырца (~500 мг) растворяли в 10 мл воды, к которой добавляли уксусную кислоту до рН раствора ~3-4. Раствор наносят на колонку с силанизированным силикагелем, продукт элюируют, используя в качестве элюента смесь 0.01% водного раствора уксусной и ацетонитрила, градиаент ацетонитрила от 0% до 95%. Собирают фракции по 9 мл. Фракции, содержащие целевой Амфамид объединяли, отгоняли органический растворитель на роторном вакуумном испарителе при температуре не выше 40°C, оставшийся водный раствор замораживали при температуре -18°C, и лиофилизировали. Чистота полученного Амфамида составила 95% по данным ВЭЖХ, общий выход - 355 мг (65% в расчет на исходный амфотерицин В).
Тпл 115-118°C (разл). Вычислено для C49H79N3O16: С, 60,91; Н, 8,24; N, 4,35; О, 26,49. Найдено: С, 60,88; Н, 8,26; N, 4,33. 1Н NMR (400 MHz, DMSO-d6) δ: 7,01 (1H, m, 3'-NH); 6,43 (1H, m, 24-CH); 6,38 (1H, m, 22-CH); 6,32 (1H, m, 26-CH); 6,29 (1H, m, 29-CH); 6,29 (1H, m, 30-CH); 6,28 (1H, m, 25-CH); 6,25 (1H, m, 23-CH); 6,16 (1H, m, 31-CH); 6,15 (1H, m, 27-CH); 6,08 (1H, m, 21-CH); 6,07 (1H, m, 32-CH); 5,96 (1H, m, 20-CH); 5,43 (1H, m, 33-CH); 5,20 (3H, m, 37-CH3); 4,47 (1H, m, 1'-СН); 4,37 (m, 1H, 19-CH); 4,22 (1H, m, 11-CH); 4,19 (1H, m, 17-CH); 4,05 (1H, m, 3-CH); 3,96 (1H, m, 15-CH); 3,66 (m, 2C-CH); 3,52 (m, 1H, 5-CH); 3,45 (m, 1H, 9-CH); 3,42 (m, 1H, C3'-H); 3,17 (1H, m, 1H, 4'-CH); 3,17 (1H, m, 5'-CH); 3,09 (1H, m, 35-CH); 3,08 (1H, m, 8-CH); 2,54 (4H, m, NH-CH2-CH2-NH); 2,28 (1H, m, 34-CH); 2,16 (2H, m, 2-CH2); 2,06 (1H, m, 18-CH); 1,9 (1H, m, 16-CH); 1,86 (1H, m, 14-CH); 1,72 (1H, m, 36-CH); 1,57 (1H, m, 7-CH); 1,57 (1H, m, 18-CH); 1,55 (1H, m, 10-CH); 1,53 (2H, m, 12-CH2); 1,39 (1H, m, 6-СН); 1,39 (1H, m, 4-CH); 1,31 (1H, m, 4-CH); 1,30 (1H, m, 10-CH); 1,25 (1H, m, 6-CH); 1,24 (1H, m, 7-CH); 1,16 (3H, m, 6'-CH3); 1,11 (3H, m, 37-CH3); 1,09 (1H, m, 14-CH); 1,03 (3H, m, 34-CH3); 0,91 (3H, m, 36-CH3). 13C NMR (100 MHz, DMSO-d6) δ: 136,6 (C20); 136,5 (C33); 133,7 (C24; 133,4 (C26); 133,3 (C22); 132,9 (C30); 132,0 (C29); 131,9 (C27); 131,9 (C25); 131,6 (C23); 131,6 (C31); 131,0 (C32); 128,4 (C21); 96,7 (С1'); 76,9 (C8); 74,5 (19); 73,5 (C9); 73,3 (C35); 73,0 (C5'); 69,4 (C4'); 69,0 (5); 68,8 (C2'); 68,6 (C37); 66,0 (C3); 65,4 (С11); 65,2 (C15); 65,1 (C17); 56,7 (C3'); 56,5 (C16); 50,0 (CNH); 48,0 (CNH); 46,0 (C12); 44,5 (C4); 44,1 (C14); 42,2 (C34); 41,8 (C2); 39,4 (C36); 39,3 (C10); 37,0 (С18); 35,9 (С6); 28,8 (С7); 18,2 (34Ме); 17,9 (С6'); 16,7 (С37Ме); 11,8 (С36Ме). ЭСП (0,01 мг/мл) λмакс., нм: 235, 345, 365, 385, 406. ИК-спектр, λмакс.: 3394, 3008, 2924, 1774, 1720, 1705, 161, 1635, 1558, 1543, 1458, 1442, 1381, 1319, 1265, 1180, 1111, 1072, 1018, 956, 887, 848, 802 см-1. MW (HR ESI-MS) вычислено для [M+H]+1 C49H79N3O16: 966,5539. Найдено: 966,5775 [M+H]+1. Rt 7.24 мин (Колонка 4×250 мм с октадецилсиланом (С-18) с зернением 5 мкм, подвижная фаза: 0,01 М раствор фосфорной кислоты (рН 2,6): ацетонитрил, линейный градиент ацетонитрила от 30 до 60% за 15 мин).
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИДЫ НАТАМИЦИНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ | 2022 |
|
RU2803742C1 |
| Противогрибковый полусинтетический полиеновый антибиотик, его водорастворимые соли и фармацевтические композиции на их основе | 2018 |
|
RU2688658C1 |
| ПРОИЗВОДНЫЕ НИСТАТИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОГРИБКОВЫХ АГЕНТОВ | 2008 |
|
RU2488590C2 |
| ПРОИЗВОДНЫЕ АНТИБИОТИКА ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ ОЛИВОМИЦИНА 1, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2350621C1 |
| СОЕДИНЕНИЕ ПЕПТИДНОЙ ПРИРОДЫ, ОБЛАДАЮЩЕЕ СПОСОБНОСТЬЮ СВЯЗЫВАТЬСЯ С ПСМА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2823164C2 |
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2014 |
|
RU2676324C2 |
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2018 |
|
RU2798085C2 |
| ПОЛУСИНТЕТИЧЕСКИЕ АНАЛОГИ АНТИБИОТИКА ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ ОЛИВОМИЦИНА А, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2010 |
|
RU2453552C2 |
| НОВЫЙ ЭФФЕКТИВНЫЙ АМИНОГЛИКОЗИДНЫЙ АНТИБИОТИК ПРОТИВ БАКТЕРИЙ С МНОЖЕСТВЕННОЙ ЛЕКАРСТВЕННОЙ РЕЗИСТЕНТНОСТЬЮ | 2016 |
|
RU2751634C2 |
| ПРОИЗВОДНОЕ АНТИБИОТИКА А 40926, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2125058C1 |

Изобретение относится к способу получения N-(2-аминоэтил)амида амфотерицина В (Амфамида) формулы I, который заключается в том, что синтез проводят путем конденсации амфотерицина В с этилендиамином в виде свободного основания в присутствии конденсирующего агента и соли органического основания и минеральной кислоты, а очистку образующегося амфамида-сырца проводят методом обращенно-фазовой хроматографии. Технический результат - разработан новый способ получения амфамида с высоким выходом, который может найти свое применение в медицине в качестве средства, обладающего противогрибковыми свойствами. 5 з.п. ф-лы, 2 ил., 3 пр.
1. Способ получения N-(2-аминоэтил)амида амфотерицина В (Амфамида) формулы I
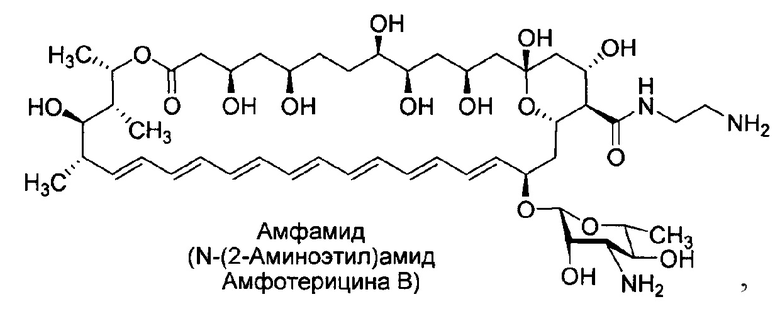
отличающийся тем, что синтез амфамида проводят путем конденсации амфотерицина В с этилендиамином в виде свободного основания в присутствии конденсирующего агента и соли органического основания и минеральной кислоты, а также очистку образующегося амфамида-сырца проводят методом обращенно-фазовой хроматографии.
2. Способ получения соединения I по п. 1, в котором в качестве органического основания используется третичный амин или пиридин.
3. Способ получения соединения I по п. 1, в котором в качестве соли минеральной кислоты и третичного амина используется гидрохлорид триэтиламина.
4. Способ получения соединения I по п. 1, в котором в качестве конденсирующего агента используется РуВОР (бензотриазол-1-ил-оксотрипирролидинофосфониум гексафторфосфат).
5. Способ получения соединения I по п. 1, в котором в качестве растворителя используется ДМСО.
6. Способ получения соединения I по п. 1, в котором мольное соотношение реагентов амфотерицин В:этилендиамин:РуВОР:гидрохлорид триэтиламина выбрано в диапазоне от 1:1:1:1 до 1:10:10:10, предпочтительно около 1:5:1.5:6.
| Противогрибковый полусинтетический полиеновый антибиотик, его водорастворимые соли и фармацевтические композиции на их основе | 2018 |
|
RU2688658C1 |
| TEVYASHOVA A | |||
| N | |||
| ET AL, ACS Infect | |||
| Dis | |||
| vol | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Устройство разъемной опоры для подшипников велосипедных осей | 1924 |
|
SU2029A1 |
| WO 2015164289 A1, 29.10.2015 | |||
| WO 2016112260 A1, 14.07.2016 | |||
| WO 2013186384 A1, 19.12.2013 | |||
| Амиды антибиотиков группы полиеновых макролидов и их производных,обладающие противогрибковой активностью | 1980 |
|
SU1152954A1 |

