Область техники
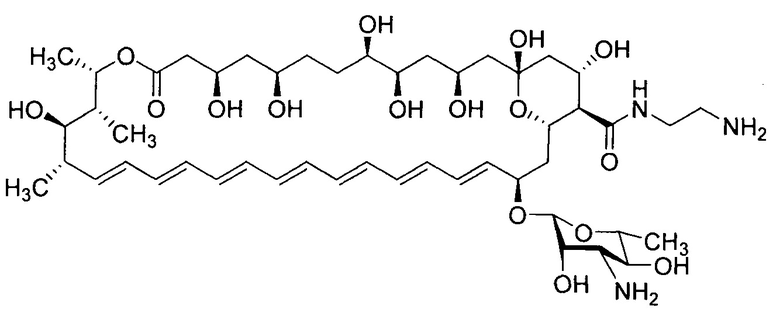
Настоящее изобретение относится к медицине, фармации и химико-фармацевтической промышленности, а более конкретно к новому производному амфотерицина В - N-(2-аминоэтил)амиду амфотерицина В (амфамиду), формулы I, обладающему более выраженными противогрибковыми свойствами и более низкой токсичностью в сравнении с амфотерицином В, его солям, а также фармацевтическим композициям на их основе и их медицинскому применению.
Уровень техники
В течение последних лет отмечено значительное увеличение числа грибковых инфекций. Особенно тяжело протекают грибковые инфекции у пациентов с иммунодепрессивными состояниями, в том числе у недоношенных детей, пациентов после трансплантации органов или прогрессирования ВИЧ, а также после химиотерапии онкологических заболеваний. Так, в крупных онкологических учреждениях тяжелые формы кандидоза составляют приблизительно 10-25% среди всех инфекционных осложнений, при этом инвазивный кандидоз характеризуется тяжестью клинических проявлений и высокой (30-70%) летальностью. В настоящее время препаратом выбора для лечения тяжелых системных грибковых инфекций является амфотерицин В, клиническое использование которого значительно ограничено серьезными побочными эффектами лечения (нефро- и гепатотоксичность, поражения центральной нервной системы) и его крайне низкой растворимостью в воде.
Таким образом, разработка лекарственного средства для лечения системных грибковых инфекций, подходящего, в частности, иммунокомпрометированным пациентам, обладающего высокой противогрибковой активностью при сниженной токсичности и повышенной растворимостью в воде и высокой стабильностью, является актуальной задачей современной фармацевтики и медицины.
Целью настоящего изобретения является повышение растворимости амфотерицина В в воде и улучшение его фармакологических характеристик (снижение токсичности при сохранении высокой активности или ее увеличении относительно амфотерицина В) за счет химической модификации С16-карбоксильной группы антибиотика с получением N-(2-аминоэтил)амида амфотерицина В (амфамида) формулы I) и его солевых форм.
Другой целью изобретения является улучшение стабильности фармацевтической субстанции амфотерицина В за счет его вышеуказанной модификации, а также разработка на основе производного I и его солей фармацевтических композиций с улучшенными свойствами, применимых для лечения грибковых заболеваний.
Раскрытие сущности изобретения
Настоящее изобретение направлено на решение технических проблем, представленных выше. Полиеновый антибиотик амфотерицин В обладает высокой противогрибковой активностью, однако, его применение ограничивается крайне низкой растворимости в воде и относительно высокой токсичностью. Указанные проблемы могут быть решены получением полусинтетического производного амфотерицина В, модифицированного по карбоксильной группе - N-(2-аминоэтил)амида амфотерицина В (амфамид I), а также его солей.
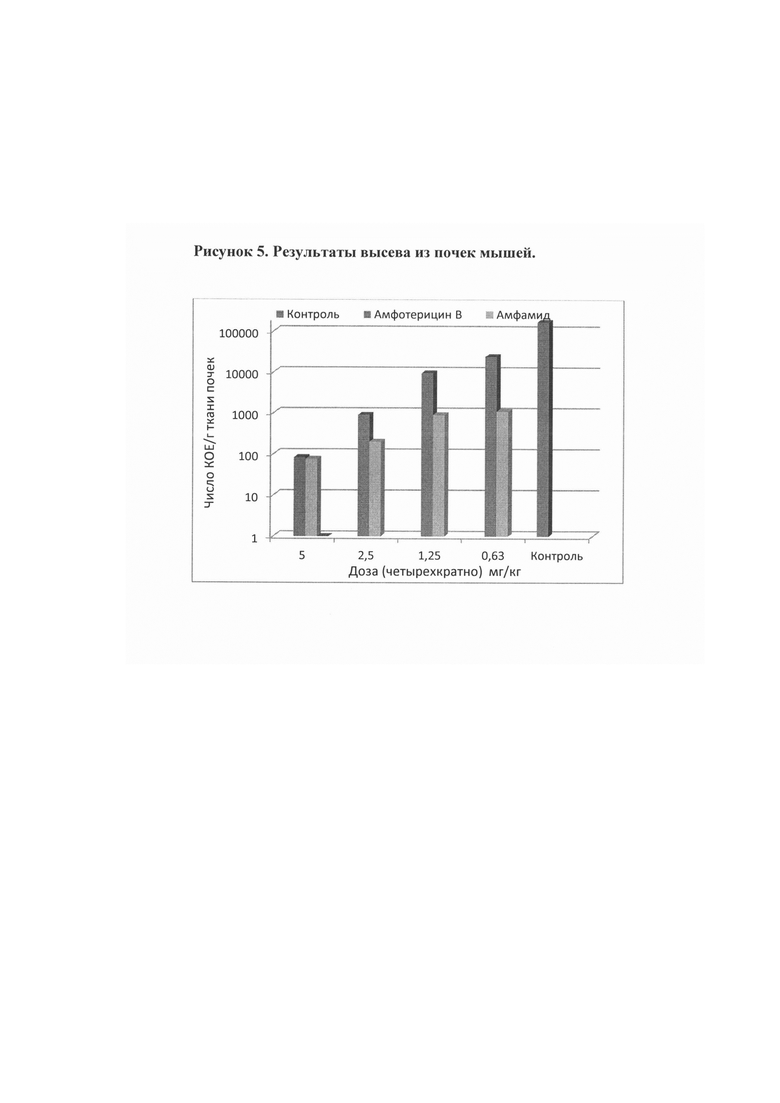
N-(2-Аминоэтил)амид амфотерицина В, раскрываемый в настоящем изобретении обладает более высокой противогрибковой активностью в сравнении с амфотерицином В, в экспериментах in vitro на панели штаммов грибков и дрожжей, играющих ведущую роль в патогенезе системных грибковых инфекций человека, а также высокую эффективность в эксперименте in vivo на модели кандидозного сепсиса мышей. Так, в сравнительных экспериментах in vitro на панели штаммов дрожжей и грибков показано, что амфамид I обладает широким спектром противогрибковой активности, превосходя или не уступая по значениям минимальных подавляющих концентраций амфотерицину В (МПК=0,06-0,5 мкг/мл для I; 0,12-2 мкг/мл для амфотерицина В соответственно). На модели кандидозного сепсиса мышей химиотерапевтическая эффективность амфамид I в два-три выше, чем препарата сравнения амфотерицина В в дозах 2,5, 1,25 и 0,63 мг/кг (по критерию высева Candida albicans из почек), так амфотерицин В продемонстрировал низкую противогрибковую активность в условиях эксперимента (падение количества колониеобразующих единиц на грамм почек (КОЕ/г) на два порядка) даже в максимально высокой дозе (2,5 мг/кг в сутки), в то время как новое производное I продемонстрировало высокую противогрибковую активность в диапазоне доз 0,62-2,5 мг/кг в сутки.
Кроме того, указанная модификация привела к снижению токсичности исходного амфотерицина В. Так, дозы, характеризующие острую токсичность амфотерицина В при внутривенном введении составляют: LD50 1,2 мг/кг (мыши), 1,6 мг/кг (крысы), в то время как для N-(2-аминоэтил)амида амфотерицина В (I) и его глутамата - LD50 13,8 мг/кг (мыши), для фармацевтической композиции LD50 - 10,7 мг/кг (крысы).
Таким образом, созданное авторами изобретения новое производное N-(2-аминоэтил)амид амфотерицина В (I) и его соли обладает большей растворимостью в воде, большей противогрибковой активностью и эффективностью в сравнении с исходным амфотерицином В и меньшей токсичностью.
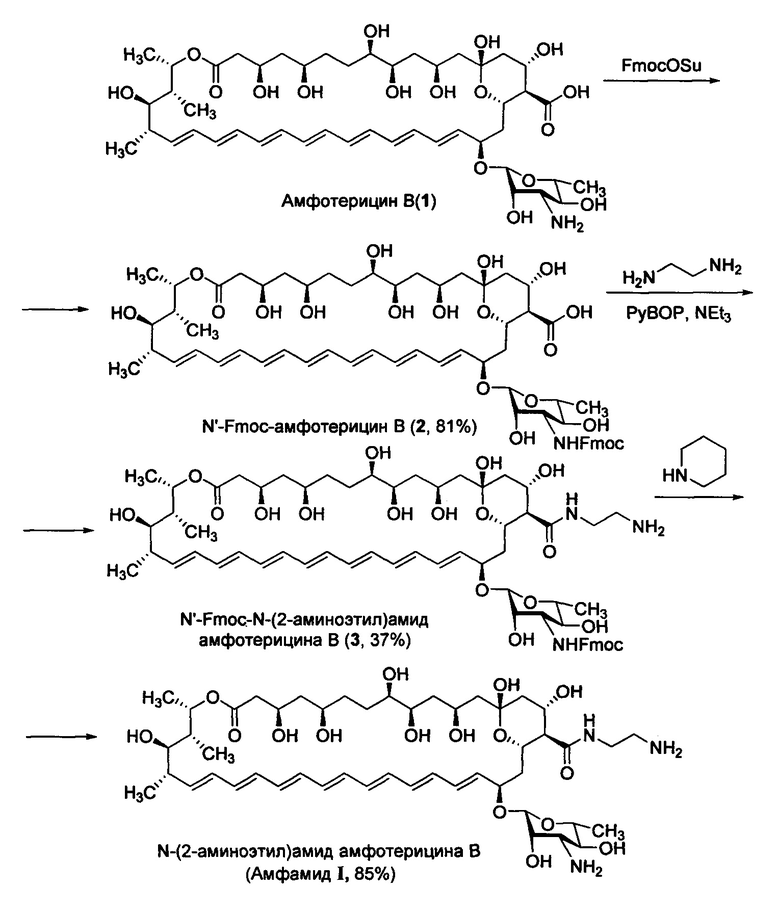
Амфамид I, описываемый в настоящем изобретении можно получить различными способами. Один из типичных подходов включает активацию С16-карбоксильной группы амфотерицина В методами, известными из уровня техники, например, производными карбодиимида (например DCC или EDC), дифенилфосфорилазидом (DPPA), реагентами Кастро (ВОР или РуВОР) или другими конденсирующими агентами (TBTU, HBTU, COMU) с последующим взаимодействием с этилендиамином. Другой типичный подход включает введение защитной группы для 3'-аминогруппы амфотерицина В, в качестве которой можно использовать различные защитные группы, хорошо известные специалистам в области органического синтеза, предпочтительно 9-флуоренилметилоксикарбонильную группу (Fmoc-группу), активацию С16-карбоксильной группы амфотерицина В методами, известными из уровня техники, например, производными карбодиимида (например DCC или EDC), дифенилфосфорилазидом (DPPA), реагентами Кастро (ВОР или РуВОР) или другими конденсирующими агентами (TBTU и HBTU) с последующим взаимодействием с этилендиамином и удаление Fmoc-защитной группы методами, хорошо известными специалистам в области органического синтеза.
Одна из возможных схем синтеза N-(2-аминоэтил)амида амфотерицина В (I) (схема 1), подробно описанная в примерах настоящего изобретения и приведенная на схеме А, включает:
1. Получение 3'-N-Fmoc-амфотерицина В (2) взаимодействием амфотерицина В (1) с
9-флуоренилметилоксикарбонил-сукцинимидом (Fmoc-OSu);
2. Взаимодействие 3'-N-Fmoc-амфотерицина В (2) с этилендиамином в присутствии
конденсирующего реагента с образованием 3'-N-Fmoc-(N-(2-аминоэтил)амида) амфотерицина В (3);
3. Удаление защитной Fmoc-группы 3'-N-Fmoc-(N-(2-аминоэтил)амида) амфотерицина В (3) действием пиперидина, приводящее к образованию целевому амфамиду I.
Недостатки фармацевтических композиций на основе амфотерицина обусловлены его амфотерностью, и проявляются в недостаточной растворимости активного агента в фармацевтически приемлемых водных средах. Это сказывается на его биологической доступности, приводит к образованию резких колебаний концентрации при введении и уменьшает время пребывания в организме, что в свою очередь нередко приводит к недостижению необходимого терапевтического эффекта. Кроме того такие композиции нестабильны при длительном хранении [Wiest D.B., Maish W.A., Garner S.S., el-Chaar G.M. Stability of amphotericin В in four concentrations of dextrose injection. Am J Hosp Pharm., 1991, v. 48(11), pp.2430-2433].
Указанные недостатки также могут быть решены получением N-(2-аминоэтил)амида амфотерицина В, а также его солей, обладающих высокой растворимостью в фармацевтически приемлемых водных средах и созданием их противогрибковых композиций на их основе.
Применение в составе композиции N-(2-аминоэтил)амида амфотерицина В (I) и/или его солевой формы также способствует повышению растворимости терапевтического агента, увеличению биологической доступности, позволяет контролировать профиль его растворения, чтобы нивелировать резкие скачки концентраций препарата в системном кровотоке, а также увеличить время пребывания лекарственного вещества в организме. Соли амфамида I, входящие в состав фармацевтических композиций имеют противогрибковую активность, не меньшую чем активность исходного соединения.
Таким образом, технический результат настоящего изобретения относится к получению N-(2-аминоэтил)амида амфотерицина В (I) и его солей, обладающих хорошей растворимостью в водных средах, превышающей растворимость амфотерицина В, характеризующихся высокой противогрибковой активностью, меньшей токсичностью и гемолитической активностью в сравнении с амфотерицином В, а также высокой стабильностью при хранении. Фармацевтические композиции, содержащие по меньшей мере одно из вышеуказанных соединений, также имеют высокую противогрибковую активность и меньшую гемолитическую активность в сравнении с амфотерицином В, обладают высокой стабильностью при хранении и хорошей растворимостью.
Подходящими солями в контексте решаемой технической проблемы являются соли амфамида I с дикарбоновыми аминокислотами, предпочтительно аспарагиновой или глутаминовой кислотами, карбоновыми кислотами, предпочтительно уксусной и пропионовой кислотами, карбоновыми ненасыщенными органическими кислотами, предпочтительно фумаровой кислотой, трикарбоновыми органическими кислотами, предпочтительно лимонной кислотой.
Фармацевтические композиции для парентерального введения обычно являются стерильными изотоническими растворами для инъекций, которые, помимо терапевтически эффективного количества лекарственного средства, содержат фармакологически приемлемый носитель (растворитель), а также различные эксципиенты (вспомогательные вещества). Фармацевтические композиции могут являться как жидкими готовыми растворами для инъекций, так и суспензиями, стерильными порошками или лиофилизированными композициями, для применения которых необходимо добавление стерильного растворителя (например, воды для инъекций) непосредственно перед использованием. Методики получения парентеральных лекарственных композиций хорошо известны из уровня техники.
В качестве носителя (растворителя) для приготовления жидких фармацевтических композиций в настоящем изобретении предпочтительно используется «раствор глюкозы» («изотоническом растворе глюкозы (5%) для внутривенных инъекций»). Кроме «раствора глюкозы», для приготовления жидких лекарственных форм могут быть применены и другие менее предпочтительные носители, включая водные изотонические солевые растворы.
Помимо лекарственного средства и носителя, фармацевтические композиции могут содержать один или несколько известных из уровня техники фармацевтически приемлемых эксципиентов (вспомогательных компонентов). В частности, в настоящем изобретении эксципиенты могут быть выбраны из солюбилизаторов, соединений, способствующих поддержанию рН и/или изотоничности, наполнителей, эмульгаторов, консервантов, антиоксидантов и других веществ. Эти хорошо известные из уровня техники компоненты фармацевтических композиций [Modern Pharmaceutics (Fourth Edition). G. Banker et al. (eds.), 2002, 864; Remington's Pharmaceutical Sciences (21th Edition) D B. Troy, P. Beringer (ed.), 2006, 2393, A.J. Spiegel et al. J. Pharm. Sci., 1963, 52 (10), 917 - 927] способствуют улучшению их потребительских или полезных свойств за счет облегчения применения, повышения стабильности, регулирования значения рН, изменения времени удерживания лекарственного соединения в месте введения.
Так, в соответствии с еще одним аспектом настоящего изобретения растворимость N-(2-аминоэтил)амида амфотерицина В (I), а также стабильность его фармацевтических композиций и их эффективность дополнительно могут быть повышены за счет одного или нескольких эксципиентов, выбранных независимо из группы солюбилизаторов, наполнителей, эмульгаторов, консервантов, антиоксидантов, буферных солей и веществ для поддержания изотоничности. Количество N-(2-аминоэтил)амида амфотерицина В (I) или его фармацевтически приемлемой соли в фармацевтической композиции не ограничивается, но предпочтительно составляет от 0,1% до 20% (мас./об.), более предпочтительно от 5% до 20% (мас./об.)- Количество растворителя (наполнителя или разбавителя) не ограничивается, но может достигать до 99 масс. % в расчете на общую массу композиции, как хорошо известно в технологии приготовления лекарственных форм. Количество сорастворителя в композиции не ограничивается и может варьироваться, например, от 1% до 60%. Количество солюбилизирующего агента в композиции не ограничивается и может варьироваться, например, от 0,1% до 20%. Количество стабилизатора и антиоксиданта в композиции не ограничивается, но может достигать, например, от 0,1 до 1%. Количество агента для поддержания изотоничности в композиции не ограничивается, но может достигать, например, от 0,5 до 10%. Количество консерванта не ограничивается и может составлять, например от 0,001 до 5%.
В качестве эксципиентов, повышающих растворимость N-(2-аминоэтил)амида амфотерицина В, его фармацевтические композиции могут содержать один или несколько фармакологически приемлемых солюбилизирующих агентов, хорошо знакомых специалистам в области фармацевтики, такие как (без ограничения перечисленным) поливинилпирролидон (ПВП), декстран, полисорбат 80 (твин 80), кремофор ЕН, гидроксиалкилированые β-циклодекстрины и γ-циклодекстрины и т.п. Полиэтиленгликоли (ПЭГ400 или ПЭГ200), а также β- или γ-гидроксипропилциклодекстрины не только стабилизируют раствор действующего вещества, но и способствуют его растворению при комнатной температуре. Поэтому в предпочтительных вариантах осуществления настоящего изобретения для приготовления стабильных фармацевтических композиций для парентерального применения N-(2-аминоэтил)амида амфотерицина В (I), являющихся предметом настоящего изобретения, используются ПВП, твин 80, кремофор ЕН, полиэтиленгликоли и β- или γ-(2-гидроксипропил)циклодекстрины.
В качестве консервантов, предохраняющих композицию от воздействия микроорганизмов, в настоящем изобретении могут быть применены различные противомикробные вещества, хорошо знакомые специалистам в области фармацевтики, такие как (без ограничения перечисленным) сульфиты, бензиновый спирт, хлорбутанол, сорбит, ксилит, сорбиновая кислота, бензойная кислота, тимеросал, парабены и их различные соли.
В качестве вспомогательных буферных компонентов в фармацевтических композициях настоящего изобретения могут быть предпочтительно использованы кислоты (например, лимонная, уксусная, фосфорная и др.) или их соли с щелочными металлами (например, цитрат, ацетат или дигидрофосфат натрия и др.), или их сочетания (например, лимонная кислота и цитрат натрия). Ионная сила буферного раствора, используемого как компонент жидкой композиции в настоящем изобретении, не ограничивается и может быть, например, на уровне 0,01-0,6.
В качестве антиоксидантов могут быть использованы, например, глюкоза, аскорбиновая кислота, метабисульфит натрия, бисульфит натрия, сульфит натрия, фенолы и тиофенолы.
Таким образом, настоящее изобретение описывает готовые фармацевтические композиции для парентерального применения, содержащие N-(2-аминоэтил)амид амфотерицина В (I). В некоторых вариантах осуществления композиция является сухой (как, например, лиофилизированной) композицией, которая может быть восстановлена, ресуспендирована или регидратирована для получения жидкой композиции фармацевтического вещества. Так, авторы настоящего изобретения путем лиофилизации жидких композиций получили ряд лиофилизированных препаратов, обладающих превосходной лекарственной стабильностью в процессе приготовления и хранения и легко восстанавливающихся в жидкую композицию при добавлении водного растворителя. Соответственно, настоящее изобретение также включает лиофилизированные композиции амфамида I. В некоторых вариантах осуществления композиция может быть промежуточной жидкой (как, например, водной) композицией, которую можно высушить (как, например, лиофилизировать) или жидкой (как, например, водной) композицией, полученной посредством восстановления или ресуспендирования сухой композиции.
Для получения лиофилизированных композиций, являющихся предметом настоящего изобретения, жидкую композицию амфамида I и/или его солевой формы, полученную, как описано выше, заливают в жесткий сосуд, такой как стерильная ампула, флакон или пузырек и подвергают лиофилизации традиционным способом. Количество жидкого препарата, которым заполняют сосуд, составляет, например от 5 до 50% (об./об.) от объема сосуда, особенно предпочтительно 10-25% (об./об.). Внешнюю температуру при лиофилизации предпочтительно поддерживают от -70 до 0°С, особенно предпочтительно от -60 до -40°С, и применяемое давление для сублимации растворителя предпочтительно составляет от 0,01 до 0,2 мм рт. ст., более предпочтительно от 0,01 до 0,1 мм рт. ст. Предпочтительно скорость лиофилизации регулируется таким образом, чтобы растворитель (в расчете на раствор) сублимировался со скоростью от 10 мкл до 100 мкл на 1 см2 площади поверхности, с которой сублимируется растворитель в течение 1 часа.
Лиофилизируемые композиции могут содержать дополнительные криозащитные компоненты, предотвращающие осаждение лекарственного соединения, такие как поверхностно-активные вещества, смачивающие или эмульгирующие агенты (как, например, лецитин, полисорбат 80, полиоксиэтиленстеарат). Содержание указанных эксципиентов может составлять 0,01-10% от веса лекарственного препарата.
Лиофилизированные композиции, являющиеся предметом настоящего изобретения, приготовленные, как указано выше, обладают отличными свойствами в отношении стабильности действующего вещества N-(2-аминоэтил)амида амфотерицина В) в процессе приготовления или хранения. Вышеописанные лиофилизированные формы, как и жидкие композиции для парентерального применения амфамида I и/или его солевых форм, могут быть упакованы в стерильные флаконы, ампулы или пакеты, пузырьки для однократного или многократного применения. Фармацевтические композиции в сухой (лиофилизированной) или жидкой концентрированной форме, как известно из уровня техники, перед использованием могут быть восстановлены или разбавлены посредством добавления стерильного фармацевтически приемлемого растворителя с получением необходимой концентрации терапевтического агента. Это дает возможность, при необходимости, легко приготовить стерильную жидкую композицию для парентерального применения N-(2-аминоэтил)амида амфотерицина В, которая может быть непосредственно введена пациенту.
N-(2-аминоэтил)амид амфотерицина В и жидкие фармацевтические композиции, содержащие от 0,1 до 5% соединения формулы I или его соли, могут быть применены парентерально (например, внутривенно, внутриартериально, внутримышечно, подкожно, ректально, интраспинально, интраперитонеально, внутриполостно) для лечения инфекций, вызванных грибковыми патогенами.
Термином "защитная группа" в настоящем изобретении обозначается группа, подходящая для блокирования функциональной группы в условиях проведения реакций, как описано литературе [Green, Т.W.; Wuts, P.G. М. Protective Groups in Organic Synthesis. 1991, 351]. Пример таких групп для блокирования аминогруппы включает, без ограничения перечисленным, трет-бутоксикарбонильную (Boc),
адамантилоксикарбонильную (Adoc), флуоренилметоксикарбонильную (Fmoc), ацетил, карбонибензилокси (Cbz), метоксикарбонильную, этоксикарбонильную.
Термин "фармацевтическая композиция", использованный в настоящем описании, означает, например, смесь, содержащую определенное количество терапевтического агента, например, терапевтически эффективное количество лекарственного соединения, по меньшей мере, с одним фармацевтически приемлемым носителем, предназначенную для введения млекопитающему, например человеку, для лечения инфекций, вызванных грибковыми патогенами.
Термин "фармацевтически приемлемый", использованный в настоящем описании, относится к соединениям, композициям и/или лекарственным формам, которые при контакте с тканями млекопитающих, прежде всего, тканями человека, не вызывают аллергических реакций, раздражения, осложнений или других токсических проявлений, и указанные соединения характеризуются достаточно высоким соотношением польза/риск.
"Фармацевтически приемлемая соль" в настоящем изобретении означает обычные соли, получаемые прибавлением кислоты к основанию, которые сохраняют биологическую эффективность и свойства N-(2-аминоэтил)амида амфотерицина В и образуются из его свободного основания и подходящих органических кислот. Примеры солей, получаемых прибавлением кислоты, включают соли, полученные из соли дикарбоновых аминокислот, например, аспарагиновой или глутаминовой кислот, органических кислот, например, уксусной кислоты, карбоновой ненасыщенной органической кислоты, например, фумаровой кислоты, трикарбоновой органической кислоты, например, лимонной кислоты. Кроме того, термин "фармацевтически приемлемая соль" также включает фармацевтически приемлемые сольваты, предпочтительно гидраты. Химическое превращение N-(2-аминоэтил)амида амфотерицина В в соль осуществляется способом, хорошо известным химикам-фармацевтам обеспечивающим улучшенную физическую и химическую стабильность, гигроскопичность, сыпучесть и растворимость соединений [см., например, Pharmaceutical Dosage Forms and Drug Delivery Systems (6th Edition). R.C. Rowe (Ed.), 1995,1456-1457].
Термин "фармакологически приемлемый носитель" в настоящем изобретении означает один или несколько совместимых жидких или твердых разбавителей или наполнителей, которые подходят для введения млекопитающему, предпочтительно человеку. Предпочтительно в качестве фармацевтически приемлемого носителя в фармацевтических композициях по изобретению используются протон-содержащие среды, более предпочтительно водные среды.
Концентрация терапевтического агента в фармацевтической композиции составляет определенное значение, обеспечивающее введение терапевтически эффективного количества лекарственного средства, которое зависит от скорости абсорбции, инактивации и выведения из организма препарата, а также от тяжести состояния пациента или от других факторов, известных специалистам в данной области техники.
"Эффективное количество" или "терапевтически эффективное количество" соединения в настоящем изобретении означает количество N-(2-аминоэтил)амида амфотерицина В или его фармацевтически приемлемой соли, которое значительно подавляет рост клеток грибков или дрожжей и эффективно для предупреждения, ослабления или смягчения симптомов заболевания или увеличения продолжительности жизни субъекта, подвергающегося лечению. Определение терапевтически эффективного количества относится к компетенции специалиста в данной области техники. Терапевтически эффективное количество или дозировка соединения, предлагаемого в настоящем изобретении, может меняться в широких пределах и может определяться способом, известным в данной области техники.
Для конкретного реципиента соответствующий курс лечения подбирается с учетом индивидуальной потребности и мнения врача, который вводит фармацевтические композиции пациенту или назначает введение фармацевтических композиций. Суточную дозу терапевтического агента можно вводить однократно в виде разовой дозы или многократно в виде разделенных доз, которые вводят через определенные периоды времени, или, при парентеральном введении, ее можно вводить путем непрерывного вливания. Таким образом, необходимое количество терапевтического агента, например необходимое терапевтически эффективное количество, определяет специалист в данной области медицины. Например, доза терапевтического агента может варьироваться в зависимости от возраста, веса тела или условий в интервале от приблизительно 1 мг до приблизительно 100 мг в расчете на N-(2-аминоэтил)амида амфотерицина В (I) или его фармацевтически приемлемую соль на килограмм массы тела реципиента в сутки.
Нижеприведенные неограничивающие примеры даны для демонстрации предпочтительных вариантов осуществления настоящего изобретения. Специалисты в данной области техники легко поймут, что для стабилизации лекарственных композиций на основе N-(2-аминоэтил)амида амфотерицина В (I) могут быть использованы разные эксципиенты и варианты осуществления настоящего изобретения.
Краткое описание чертежей
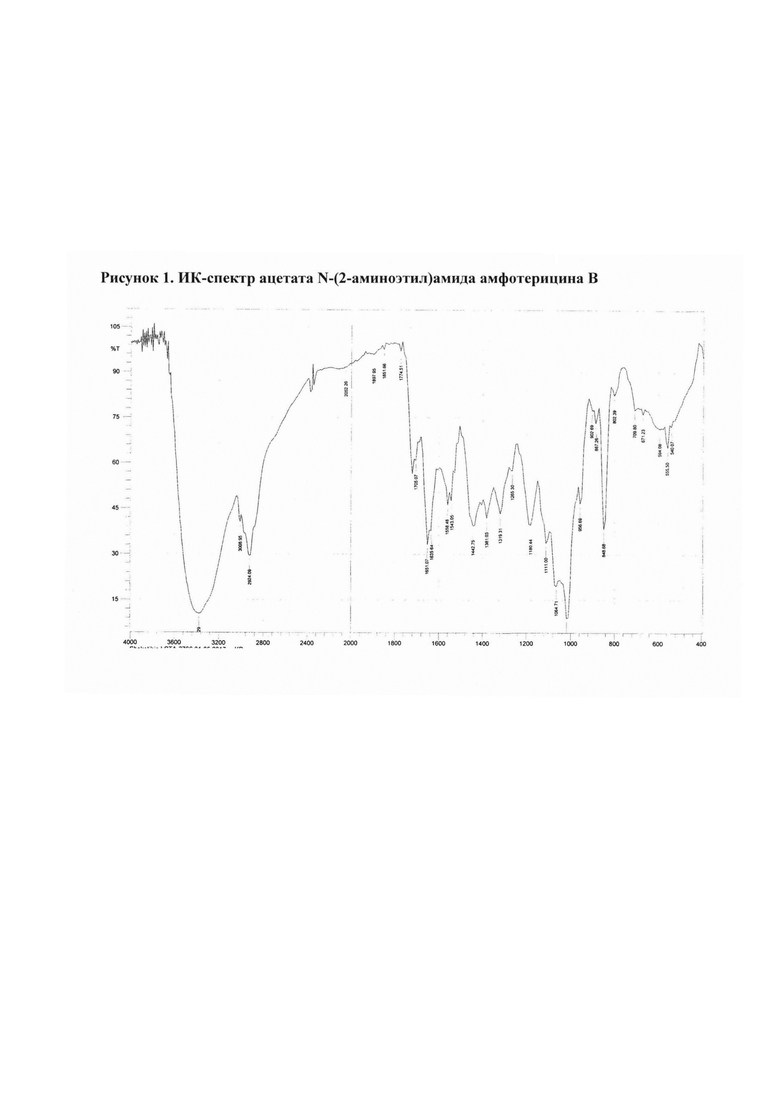
Рис. 1 ИК-спектр ацетата N-(2-аминоэтил)амида амфотерицина В
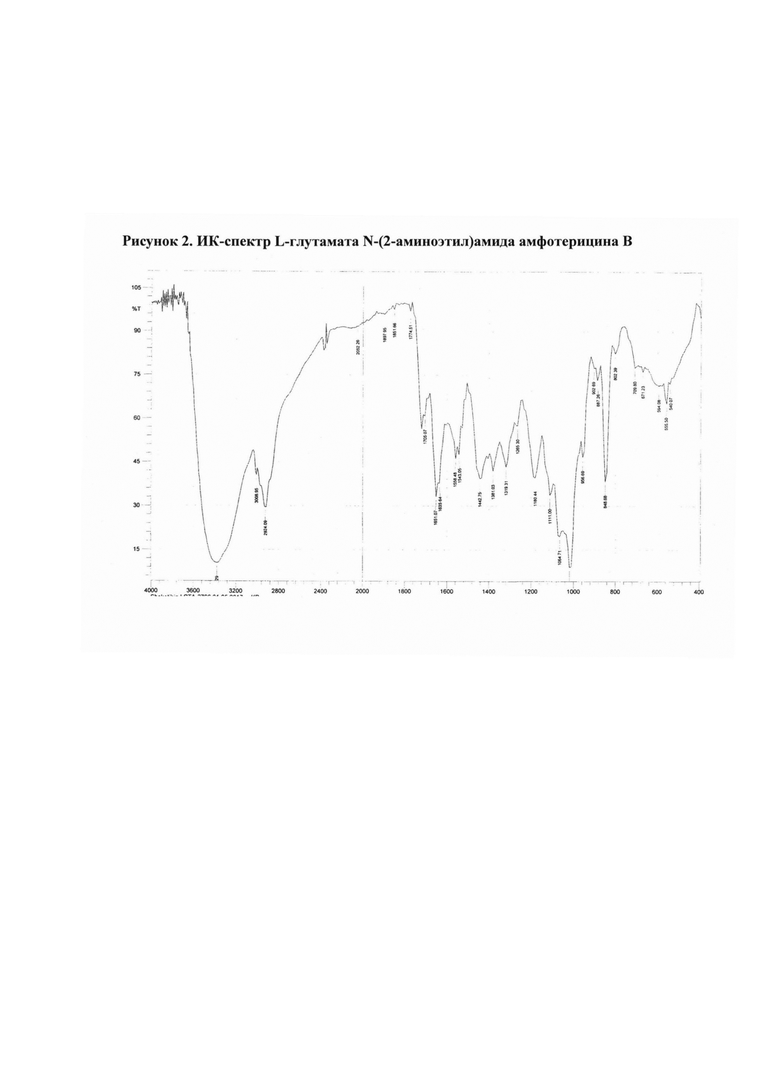
Рис. 2 ИК-спектр L-глутамата N-(2-аминоэтил)амида амфотерицина В
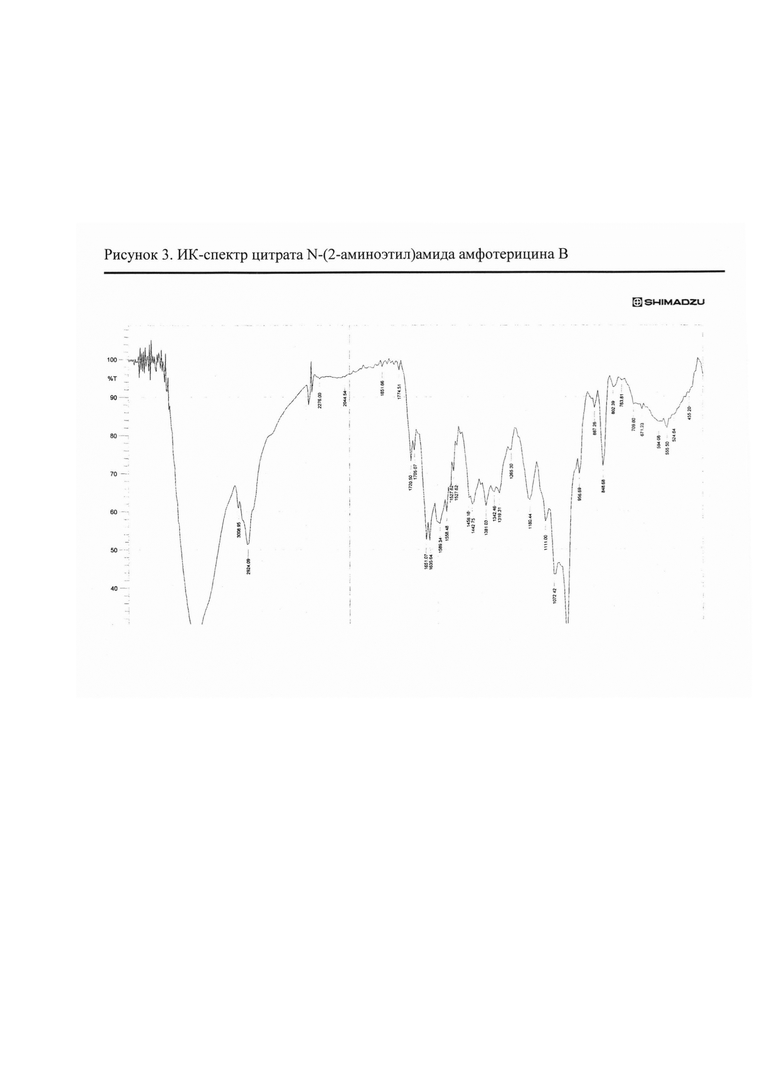
Рис. 3 ИК-спектр цитрата N-(2-аминоэтил)амида амфотерицина В
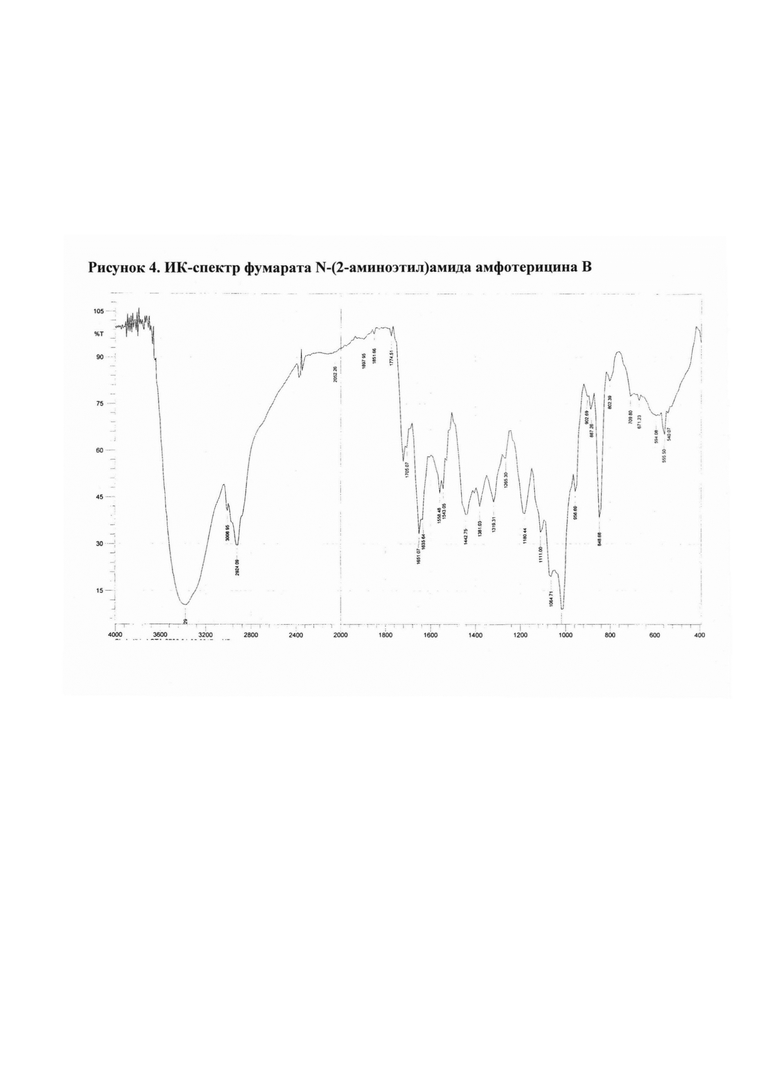
Рис. 4 ИК-спектр фумарата N-(2-аминоэтил)амида амфотерицина В
Рис. 5. Результаты высева из почек мышей.
Пример 1. Синтез N-(2-аминоэтил)амида амфотерицина В (Амфамида I)
Стадия 1. N'-Fmoc-амфотерицин В (2)
В круглодонную колбу объемом 250 мл вносят амфотерицин В (6,00 г) и диметилформамид (60,0 мл), к полученной суспензии прибавляют пиридин (0,62 мл) и N-(9-флуоренилметоксикарбонилокси)сукцинимид (2,32 г). Реакционную массу перемешивают 1,5 ч при комнатной температуре, контролируя протекание реакции методом ТСХ (CHCl3-МеОН-H2O-НСООН, 13:6:1:0,1, система А1). Добавляют метанол (10 мл) и диэтиловый эфир (100 мл). Образовавшийся осадок отфильтровывают, дополнительно промывая его диэтиловым эфиром (20 мл), получая сырец N'-Fmoc-амфотерицин, который далее очищают методом колоночной хроматографии (силикагель, элюент смесь CHCl3-МеОН-H2O-НСООН; 13:6:0,5:0,05; 700 мл). Фракции, содержащие целевое вещество (Rf=0,60, система А1) объединяют, концентрируют до объема 10 мл, добавляют диэтиловый эфир (100 мл). Образовавшийся осадок отфильтровывают, промывая диэтиловым эфиром (20 мл). Полученный желтый осадок высушивают в вакууме, получая целевой N'-Fmoc-амфотерицин В (2). 1Н NMR (400 MHz, DMSO-d6) δ: 8,20-7,33 (8Н, m, Ar-Fmoc); 7,01 (1H, m, 3'-NH); 6,43 (1H, m, 24-CH); 6,38 (1H, m, 22-CH); 6,32 (1H, m, 26-CH); 6,29 (1H, m, 29-CH); 6,29 (1H, m, 30-CH); 6,28 (1H, m, 25-CH); 6,25 (1H, m, 23-CH); 6,16 (1H, m, 31-CH); 6,15 (1H, m, 27-CH); 6,08 (1H, m, 21-CH); 6,07 (1H, m, 32-CH); 5,96 (1H, m, 20-CH); 5,43 (1H, m, 33-CH); 5,20 (3H, m, 37-CH3); 5,0 (2H, m, CH2-Fmoc); 4,66 (1H, m, CH-Fmoc); 4,47 (1H, m, 1'-CH); 4,37 (m, 1H, 19-CH); 4,22 (1H, m, 11-CH); 4,19 (1H, m, 17-CH); 4,05 (1H, m, 3-CH); 3,96 (1H, m, 15-CH); 3,66 (m, 2C'-CH); 3,52 (m, 1H, 5-CH); 3,45 (m, 1H, 9-CH); 3,42 (m, 1H, C3'-H); 3,17 (1H, m, 1H, 4'-CH); 3,17 (1H, m, 5'-CH); 3,09 (1H, m, 35-CH); 3,08 (1H, m, 8-CH); 2,28 (1H, m, 34-CH); 2,16 (2H, m, 2-CH2); 2,06 (1H, m, 18-CH); 1,9 (1H, m, 16-CH); 1,86 (1H, m, 14-CH); 1,72 (1H, m, 36-CH); 1,57 (1H, m, 7-CH); 1,57 (1H, m, 18-CH); 1,55 (1H, m, 10-CH); 1,53 (2H, m, 12-CH2); 1,39 (1H, m, 6-СН);1,39 (1H, m, 4-CH); 1,31 (1H, m, 4-CH); 1,30 (1H, m, 10-CH); 1,25 (1H, m, 6-CH); 1,24 (1H, m, 7-CH); 1,16 (3H, m, 6'-CH3); 1,11 (3H, m, 37-CH3); 1,09 (1H, m, 14-CH); 1,03 (3H, m, 34-CH3); 0,91 (3H, m, 36-CH3). 13C NMR (100 MHz, DMSO-d6) δ: 136,6 (C20); 136,5 (C33); 133,7 (C24; 133,4 (C26); 133,3 (C22); 132,9 (C30); 132,0 (C29); 131,9 (C27); 131,9 (C25); 131,6 (C23); 131,6 (C31); 131,0 (C32); 128,4 (C21); 96,7 (С1'); 76,9 (C8); 74,5 (19); 73,5 (C9); 73,3 (C35); 73,0 (C5'); 69,4 (C4'); 69,0 (5); 68,8 (C2'); 68,6 (C37); 66,0 (C3); 65,4 (С11); 65,2 (C15); 65,1 (C17); 56,7 (C3'); 56,5 (C16); 46,0 (C12); 44,5 (C4); 44,1 (C14); 42,2 (C34); 41,8 (C2); 39,4 (C36); 39,3 (C10); 37,0 (C18); 35,9 (C6); 28,8 (C7); 18,2 (34Me); 17,9 (C6'); 16,7 (C37Me); 11,8 (C36Me).
Стадия 2. N'-Fmoc-N-(2-аминоэтил)амид амфотерицина В (3)
N'-Fmoc-амфотерицин В (2, 1,7 г) растворяют в ДМСО (20 мл), добавляют при перемешивании гидрохлорид этилендиамина (3 экв., 430 мг) и порциями бензотриазол-1-ил-окситрипирролидинофосфониум гексафторфосфат (РуВОР, 1,5 экв., 1,16 г (1,5 эквивалента) РуВОР. Реакционную смесь перемешивают в течение 2 часов, поддерживая рН смеси около 8 добавлением триэтиламина. По окончании реакции в реакционную смесь добавляют ацетон (5 мл) и диэтиловый эфир (30 мл). Выпавший осадок отфилтровывают, промывают диэтиловым эфиром и высушивают. Очистку целевого соединения проводят методом колоночной хроматографии на силикагеле в системе: хлороформ-метанол-вода-муравьиная кислота 1(3:6:0,5:0,05). Фракции, содержащие целевое вещество объединяют, концентрируют до объема около 10 мл, добавляют диэтиловый эфир (40 мл). Выпавший осадок отфильтровывают, промывают диэтиловым эфиром и высушивают в вакууме, получая 650 мг (37%) целевого соединения. 1Н NMR (400 MHz, DMSO-d6) δ: 8,20-7,33 (8Н, m, Ar-Fmoc); 7,01 (1Н, m, 3'-NH); 6,43 (1H, m, 24-CH); 6,38 (1H, m, 22-CH); 6,32 (1H, m, 26-CH); 6,29 (1H, m, 29-CH); 6,29 (1H, m, 30-CH); 6,28 (1H, m, 25-CH); 6,25 (1H, m, 23-CH); 6,16 (1H, m, 31-CH); 6,15 (1H, m, 27-CH); 6,08 (1H, m, 21-CH); 6,07 (1H, m, 32-CH); 5,96 (1H, m, 20-CH); 5,43 (1H, m, 33-CH); 5,20 (3H, m, 37-CH3); 5,0 (2H, m, CH2-Fmoc); 4,66 (1H, m, CH-Fmoc); 4,47 (1H, m, 1'-CH); 4,37 (m, 1H, 19-CH); 4,22 (1H, m, 11-CH); 4,19 (1H, m, 17-CH); 4,05 (1H, m, 3-CH); 3,96 (1H, m, 15-CH); 3,66 (m, 2C-CH); 3,52 (m, 1H, 5-CH); 3,45 (m, 1H, 9-CH); 3,42 (m, 1H, C3'-H); 3,17 (1H, m, 1H, 4'-CH); 3,17 (1H, m, 5'-CH); 3,09 (1H, m, 35-CH); 3,08 (1H, m, 8-CH); 2,54 (4H, m, NH-CH2-CH2-NH); 2,28 (1H, m, 34-CH); 2,16 (2H, m, 2-CH2); 2,06 (1H, m, 18-CH); 1,90 (1H, m, 16-CH); 1,86 (1H, m, 14-CH); 1,72 (1H, m, 36-CH); 1,57 (1H, m, 7-CH); 1,57 (1H, m, 18-CH); 1,55 (1H, m, 10-CH); 1,53 (2H, m, 12-CH2); 1,39 (1H, m, 6-СН);1,39 (1H, m, 4-CH); 1,31 (1H, m, 4-CH); 1,30 (1H, m, 10-CH); 1,25 (1H, m, 6-CH); 1,24 (1H, m, 7-CH); 1,16 (3H, m, 6'-CH3); 1,11 (3H, m, 37-СН3); 1,09 (1H, m, 14-CH); 1,03 (3H, m, 34-CH3); 0,91 (3H, m, 36-CH3). 13C NMR (100 MHz, DMSO-de) δ: 136,6 (C20); 136,5 (C33); 133,7 (C24; 133,4 (C26); 133,3 (C22); 132,9 (C30); 132,0 (C29); 131,9 (C27); 131,9 (C25); 131,6 (C23); 131,6 (C31); 131,0 (C32); 128,4 (C21); 96,7 (С1'); 76,9 (C8); 74,5 (19); 73,5 (C9); 73,3 (C35); 73,0 (C5'); 69,4 (C4'); 69,0 (5); 68,8 (C2'); 68,6 (C37); 66,0 (C3); 65,4 (С11); 65,2 (C15); 65,1 (C17); 56,7 (C3'); 56,5 (C16); 50,0 (CNH); 48,0 (CNH); 46,0 (C12); 44,5 (C4); 44,1 (C14); 42,2 (C34); 41,8 (C2); 39,4 (C36); 39,3 (C10); 37,0 (C18); 35,9 (C6); 28,8 (C7); 18,2 (34Me); 17,9 (C6'); 16,7 (C37Me); 11,8 (C36Me).
Стадия 3. N-(2-аминоэтил)амид амфотерицина В (I)
N'-Fmoc-N-(2-аминоэтил)амид амфотерицина В (3) (650 мг) растворяют смеси ДМСО:МеОН (7 мл, 5:2), добавляют пиперидин (200 мкл). Реакционную смесь перемешивают 1 ч, затем добавляют ацетон (3 мл) и диэтиловый эфир (20 мл). Выпавший осадок отфильтровывают, промывают диэтиловым эфиром и высушивают. Выход 450 мг (85%) целевого соединения. Тпл, 115-118°С (разл). Вычислено для C49H79N3O16: С, 60,91; H, 8,24; N, 4,35; O, 26,49. Найдено: С, 60,88; Н, 8,26; N, 4,33. 1Н NMR (400 MHz, DMSO-d6) δ: 7,01 (1H, m, 3'-NH); 6,43 (1H, m, 24-CH); 6,38 (1H, m, 22-CH); 6,32 (1H, m, 26-CH); 6,29 (1H, m, 29-CH); 6,29 (1H, m, 30-CH); 6,28 (1H, m, 25-CH); 6,25 (1H, m, 23-CH); 6,16 (1H, m, 31-CH); 6,15 (1H, m, 27-CH); 6,08 (1H, m, 21-CH); 6,07 (1H, m, 32-CH); 5,96 (1H, m, 20-CH); 5,43 (1H, m, 33-CH); 5,20 (3H, m, 37-CH3); 4,47 (1H, m, 1'-CH); 4,37 (m, 1H, 19-CH); 4,22 (1H, m, 11-CH); 4,19 (1H, m, 17-CH); 4,05 (1H, m, 3-CH); 3,96 (1H, m, 15-CH); 3,66 (m, 2C-CH); 3,52 (m, 1H, 5-CH); 3,45 (m, 1H, 9-CH); 3,42 (m, 1H, C3'-H); 3,17 (1H, m, 1H, 4'-CH); 3,17 (1H, m, 5'-CH); 3,09 (1H, m, 35-CH); 3,08 (1H, m, 8-CH); 2,54 (4H, m, NH-CH2-CH2-NH); 2,28 (1H, m, 34-CH); 2,16 (2H, m, 2-CH2); 2,06 (1H, m, 18-CH); 1,9 (1H, m, 16-CH); 1,86 (1H, m, 14-CH); 1,72 (1H, m, 36-CH); 1,57 (1H, m, 7-CH); 1,57 (1H, m, 18-CH); 1, 55 (1H, m, 10-CH); 1,53 (2H, m, 12-CH2); 1,39 (1H, m, 6-СН);1,39 (1H, m, 4-CH); 1,31 (1H, m, 4-CH); 1,30 (1H, m, 10-CH); 1,25 (1H, m, 6-CH); 1,24 (1H, m, 7-CH); 1,16 (3H, m, 6'-CH3); 1,11 (3H, m, 37-СН3); 1,09 (1H, m, 14-CH); 1,03 (3H, m, 34-CH3); 0,91 (3H, m, 36-CH3). 13C NMR (100 MHz, DMSO-d6) δ: 136,6 (C20); 136,5 (C33); 133,7 (C24; 133,4 (C26); 133,3 (C22); 132,9 (C30); 132,0 (C29); 131,9 (C27); 131,9 (C25); 131,6 (C23); 131,6 (C31); 131,0 (C32); 128,4 (C21); 96,7 (С1'); 76,9 (C8); 74,5 (19); 73,5 (C9); 73,3 (C35); 73,0 (C5'); 69,4 (C4'); 69,0 (5); 68,8 (C2'); 68,6 (C37); 66,0 (C3); 65,4 (С11); 65,2 (C15); 65,1 (C17); 56,7 (C3'); 56,5 (C16); 50,0 (CNH); 48,0 (CNH);46,0 (C12); 44,5 (C4); 44,1 (CI4); 42,2 (C34); 41,8 (C2); 39,4 (C36); 39,3 (C10); 37,0 (C18); 35,9 (C6); 28,8 (C7); 18,2 (34Me); 17,9 (C6'); 16,7 (C37Me); 11,8 (С36Ме). ЭСП (0,01 мг/мл) 1 λмакс., нм: 235, 345, 365, 385, 406. ИК-спектр, λмакс.: 3394, 3008, 2924, 1774, 1720, 1705, 161, 1635, 1558, 1543, 1458, 1442, 1381, 1319, 1265, 1180, 1111, 1072, 1018, 956, 887, 848, 802 см-1. MW (HR ESI-MS) вычислено для [M+H]+1 C49H79N3O16: 966,5539. Найдено: 966,5775 [M+H]+1. Rt 7.24 мин (Колонка 4×250 мм с октадецилсиланом (С-18) с зернением 5 мкм, подвижная фаза: 0,01 М раствор фосфорной кислоты (рН 2,6): ацетонитрил, линейный градиент ацетонитрила от 30 до 60% за 15 мин).
Пример 2. Синтез солевых форм N-(2-аминоэтил)амида амфотерицина В (I)
Солевые формы получали взаимодействием субстанции N-(2-аминоэтил)амида амфотерицина В и соответствующей кислоты.
2.1 Ацетат N-(2-аминоэтил)амида амфотерицина В
Амфамид (50 мг) добавляли к раствору уксусной кислоты (5,37 мкл) в воде (25 мл). Смесь перемешивали 15 мин при комнатной температуре, затем разливали в стеклянные флаконы по 5 мл раствора. Флаконы выдерживали при -18°С в течение 24 ч, затем высушивали досуха в лиофильной сушке (Рис. 1).
2.2. L-глутамат N-(2-аминоэтил)амида амфотерицина В
N-(2-Аминоэтил)амид амфотерицина В (50 мг) добавляли к раствору L-глутаминовой уксусной кислоты (15,2 мг) в воде (25 мл). Смесь перемешивали 15 мин при комнатной температуре, затем разливали в стеклянные флаконы по 5 мл раствора. Флаконы выдерживали при -18°С в течение 24 ч, затем высушивали досуха в лиофильной сушке (Рис. 2).
2.3. Цитрат N-(2-аминоэтил)амида амфотерицина В
N-(2-Аминоэтил)амид амфотерицина В (50 мг) добавляли к раствору лимонной кислоты (19,9 мг) в воде (25 мл). Смесь перемешивали 15 мин при комнатной температуре, затем разливали в стеклянные флаконы по 5 мл раствора. Флаконы выдерживали при -18°С в течение 24 ч, затем высушивали досуха в лиофильной сушке (рис. 3).
2.4. Фумарат N-(2-аминоэтил)амида амфотерицина В
N-(2-Аминоэтил)амид амфотерицина В (50 мг) добавляли к раствору фумаровой кислоты (12,0 мг) в воде (25 мл). Смесь перемешивали 15 мин при комнатной температуре, затем разливали в стеклянные флаконы по 5 мл раствора. Флаконы выдерживали при -18°С в течение 24 ч, затем высушивали досуха в лиофильной сушке (Рис. 4).
Полученные соли охарактеризованы ИК-спектрами* (см. рис. 1-4).
ИК-спектры регистрировали на приборе ИК-Фурье-спектрометр «Nicolet-iS10». Измерение проводили при разрешении 4 см-1; зона спектра 3000-650 см-1. Спектры обрабатывали с использованием программы OMNIC-7.0.
Пример 3. Определение стабильности N-(2-аминоэтил)амида амфотерицина В (I) и его солевых форм
Стабильность при хранении определяли методом ускоренного старения. Для этого три серии каждой соли (пять образцов по 10 мг в каждой серии) в стеклянном пенициллиновом флаконе, закрытом резиновой пробкой и закатанном алюминиевой крышкой помещали в термостат при температуре 60±1°С. Выдерживали в течение 1 месяца в термостате при указанной температуре, оценивая физико-химические характеристики (внешний вид, чистота методом ВЭЖХ) каждой соли в нулевой точке (день закладки на хранение) и через 1 месяц.
Данные о стабильности солей при хранении в условиях ускоренного старения, полученные методом ВЭЖХ, представлены на рис. 5-10. ВЭЖХ-анализ проводили на хроматографе Shimadzu (Япония) LC-20 AD с УФ детектором на колонке Kromasil (Швеция) С18 размером 4×250 мм, с зернением 5 мкм или аналогичной. Концентрация вводимого раствора - 0,1 мг/мл; объем петли - 20 мкл. Хроматографирование проводили в системе состоящей из 0,01М фосфорной кислоты (рН 2,6) и ацетонитрила. Элюцию проводили в режиме линейного градиента с увеличением концентрации ацетонитрила от 30 до 60% за 15 мин при скорости потока 1 мл/мин. Результаты представлены в таблице 1.

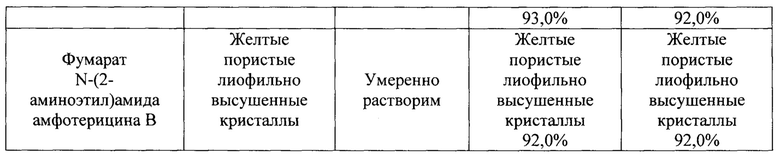
Данный пример демонстрирует, что N-(2-аминоэтил)амид амфотерицина В (I) обладает более высокой растворимостью в воде и более высокой стабильностью в сравнении с амфотерицином В, а данные солей превосходят показатели N-(2-аминоэтил)амида амфотерицина В.
Пример 4. Противогрибковая активность N-(2-аминоэтил)амида амфотерицина В (I) и его солевых форм in vitro
Для сравнительного исследования противогрибковой активности N-(2-аминоэтил)амида амфотерицина В (I), его солевых форм и амфотерицина В проведено определением минимальной подавляющей концентрации (МПК) с помощью микрометода серийных разведений в бульоне [Руководство по проведению доклинических исследований лекарственных средств, часть первая, под ред. А.Н. Миронова, Москва, Гриф и К, 2012] для панели грибков и дрожжей, являющихся типичными возбудителями грибковых инфекций. Тестируемая панель патогенов включала штаммы С. albicans АТСС 24433, C. parapsilosis АТСС 22019, С. krusei 432М, С. tropicalis 3019, C. glabrata 61Л, M.canis В-200, Т. rubrum 2002, A. niger 31а
Результаты оценки чувствительности контрольных штаммов патогенов и клинических изолятов к амфамиду (I) и его солям в сравнении с Амфотерицином В приведены в таблице 2.
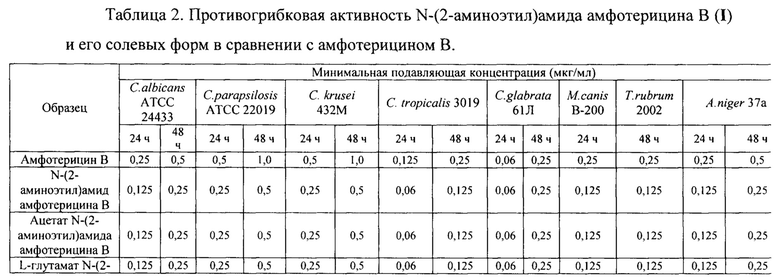

На основании результатов экспериментов согласно примерам 3-4 можно сделать вывод о том, что N-(2-аминоэтил)амид амфотерицина В (I) и его солевые формы, являющиеся предметом настоящего изобретения, обладают высокой растворимостью в воде, высокой противогрибковой активностью и стабильностью. При этом солевые формы амфамида обладают лучшей растворимостью и большей стабильностью, чем свободное основание амфамида.
Пример 5. Получение лиофилизированных фармацевтических композиций солевых форм N-(2-аминоэтил)амида амфотерицина В (I) для парентерального применения
Растворы амфамида I для получения лиофилизированных фармацевтических композиций готовят в воде для инъекций в стерильной посуде. В мерный сосуд наливают 3/4 от требуемого объема воды, растворяют в ней отвешенное количество N-(2-аминоэтил)амида амфотерицина В (I) и вспомогательных компонентов, при перемешивании смеси. Растворы амфамида I стерилизуют фильтрованием раствора через микропористый фильтр в асептических условиях. Полученный раствор переносят в мерный сосуд и доводят стерильной водой до требуемого объема. После проверки содержания N-(2-аминоэтил)амида амфотерицина В (I) методом ВЭЖХ (требуемая концентрация 5±0,2 мг/мл) растворы дозируют по 2 мл в стерильные флаконы из нейтрального стекла. Флаконы с растворами закрывают стерильными марлевыми тампонами и выдерживают при -70°С 12 ч. Флаконы с замороженным раствором вносят в установку для лиофильной сушки и выдерживают 12 ч при давлении 0,01 мм рт.ст. После извлечения из установки флаконы укупоривают стерильными резиновыми пробками и обкатывают алюминиевыми колпачками.
Иллюстративные рецептуры лиофилизированных фармацевтических композиций N-(2-аминоэтил)амида амфотерицина В для приготовления инъекционных растворов приведены ниже (представлен состав композиций N-(2-аминоэтил)амида амфотерицина В из расчета на один флакон объемом 10 мл).
Состав 1: 10 мг глутамата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 20 мг маннита.
,
Состав 2: 10 мг глутамата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 20 мг поливинилпирролидона К-15.
Состав 3: 10 мг глутамата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 10 мг маннита; 10 мг поливинилпирролидона К-15.
Состав 4: 10 мг глутамата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 20 мг поливинилпирролидона К-9.
Состав 5: 10 мг глутамата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 20 мг лактозы.
Состав 6: 10 мг глутамата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 50 мг (2-гидроксипропил)-β-циклодекстрина.
Состав 7: 10 мг ацетата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 20 мг маннита.
Состав 8: 10 мг ацетата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 20 мг поливинилпирролидона К-15.
Состав 9: 10 мг ацетата а N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 10 мг маннита; 10 мг поливинилпирролидона К-15.
Состав 10: 10 мг ацетата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 20 мг поливинилпирролидона К-9.
Состав 11: 10 мг ацетата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 20 мг лактозы.
Состав 12: 10 мг ацетата N-(2-аминоэтил)амида амфотерицина В (в расчете на основание); 50 мг (2-гидроксипропил)-β-циклодекстрина.
Приготовленные составы стабильны при хранении как в естественных условиях (1 год, 25°С, в затемненном месте), так и в условиях ускоренного старения (30 дней при 60°С). Лиофилизированные композиции легко восстанавливаются в исходный гомогенный раствор при комнатной температуре менее чем через 5 минут при добавлении стерильной воды или «изотонического раствора глюкозы (5%) для внутривенных инъекций». Для приготовления 10 мл 0,1% раствора амфамида I к содержимому флакона прибавляют 10 мл «изотонического раствора глюкозы (5%) для внутривенных инъекций», периодически помешивая содержимое флакона до полного растворения композиции.
Пример 6. Противогрибковая активность композиций солевых форм in vivo Целью исследования являлась определение специфической активности препаратов (композиций солевых форм) амфамида I в сравнении с амфотерицином В на модели кандидозного сепсиса мышей (мыши SHK весом 18-20 г). В качестве инфекционного агента использовали Candida albicans (штамм 14053 АТСС) при внутривенном пути заражения [Руководство по проведению доклинических исследований лекарственных средств, часть первая, под ред. А.Н. Миронова, Москва, Гриф и К, 2012]. Мышей заражали инфекционным инокулюм в дозе 1,0 млн. КОЕ на мышь в объеме 0,1 мл. Через 30 мин после заражения мышам проводили первое внутривенное введение испытуемых препаратов в соответствующих дозах в объеме 0,1 мл. Каждую дозу вводили ежедневно в течение четырех дней, начиная со дня заражения (0, 1, 2 и 3 дни). На 5 день опыта определяли высеваемость Candida albicans на 1 г ткани почек.
Полученные данные, показанные на рис. 5 показывают, что композиция на основе амфамида I в отношении Candida albicans проявляет более высокую противогрибковую эффективность в экспериментах in vivo по сравнению с амфотерицином В.
Пример 7. Изучение острой токсичности N-(2-аминоэтил)амида амфотерицина В и его солевых форм на мышах
Изучение острой токсичности глутамата амфамида I проведено на мышах SHK, самках в соответствии с требованиями действующего Руководства по проведению доклинических исследований лекарственных средств (2012) [Руководство по проведению доклинических исследований лекарственных средств, часть первая, под ред. А.Н. Миронова, Москва, Гриф и К, 2012] и согласно Правилам лабораторной практики в Российской Федерации (Национальный стандарт Российской федерации, ГОСТ Р 53434 - 2009). Введение животным глутамата амфамида I проводили однократно внутривенно в диапазоне доз от 8 до 20 мг/кг в 0,05% концентрации. Для всех данных рассчитано среднее значение и стандартное отклонение, различия считаются достоверными при р≤0,05. Дозы, характеризующие токсичность глутамата амфамида I, рассчитаны по методу Литчфилда и Уилкоксона:
ЛД50=13,8 (113÷16,3) мг/кг
МПД (ЛД10)=9,25 мг/кг
ЛД16=10,26 мг/кг
ЛД84=17,4 мг/кг
ЛД100=19,2 мг/кг
Из литературных данных [Proffitt R.T., Satorius A., Chiang S.-M., Sullivan L., Adler-Moore J.-P. Pharmacology and toxicology of a liposomal formulation of amphotericin В (AmBisome) in rodents. J.Antimicrob. Chemoth., 1991, v. 28, s._B, pp. 49-61] доза, характеризующая острую токсичность амфотерицина В на мышах, при внутривенном введении составляет:
ЛД50=2,3 мг/кг.
Таким образом, полученные данные свидетельствуют, что глутамат амфамида I обладает достоверно меньшей острой токсичностью в сравнении в амфотерицином В.
Пример 8. Изучение острой токсичности фармацевтической композиции N-(2-аминоэтил)амида амфотерицина В на крысах
Изучение острой токсичности фармацевтической композиции на основе амфамида I (состав 6) проведено на беспородных крысах, самках, в соответствии с требованиями действующего Руководства по проведению доклинических исследований лекарственных средств (2012) [Руководство по проведению доклинических исследований лекарственных средств, часть первая, под ред. А.Н. Миронова, Москва, Гриф и К, 2012] и согласно Правилам лабораторной практики в Российской Федерации (Национальный стандарт Российской федерации, ГОСТ Р 53434 - 2009). Введение животным фармацевтической композиции на основе амфамида I (состав 6) проводили однократно внутривенно в диапазоне доз от 1 до 15 мг/кг в 0,1% концентрации в пересчете на амфамид I. Для всех данных рассчитано среднее значение и стандартное отклонение, различия считаются достоверными при р≤0,05. Дозы, характеризующие токсичность глутамата амфамида I, рассчитаны по методу Литчфилда и Уилкоксона:
ЛД50=10,7 (8,0÷13,4) мг/кг
МПД (ЛД10)=3,8 мг/кг
ЛД16=5,3 мг/кг
ЛД84=16,1 мг/кг
ЛД100=18,9 мг/кг
Из литературных данных [Proffitt R.T., Satorius A., Chiang S.-M., Sullivan L., Adler-Moore J.-P. Pharmacology and toxicology of a liposomal formulation of amphotericin В (AmBisome) in rodents. J.Antimicrob. Chemoth., 1991, v. 28, s. B, pp. 49-61] доза, характеризующая острую токсичность амфотерицина В на крысах, при внутривенном введении составляет:
ЛД50=1,6 мг/кг.
Таким образом, полученные данные свидетельствуют, что фармацевтическая композиция N-(2-аминоэтил)амид амфотерицина В (состав 6) обладает достоверно меньшей острой токсичностью в сравнении в амфотерицином В.
Противогрибковый полусинтетический полиеновый антибиотик, его водорастворимые соли и фармацевтические композиции на их основе
Формула I
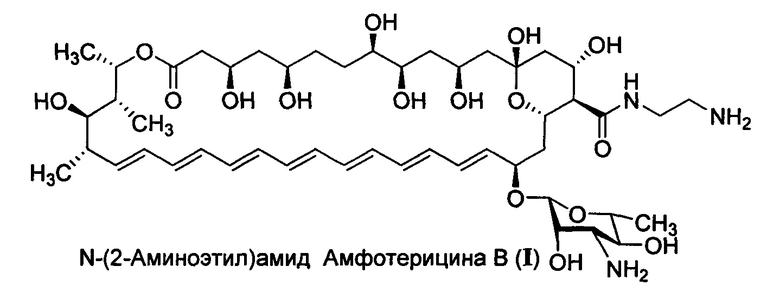
Схема 1
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения противогрибкового полусинтетического полиенового антибиотика | 2020 |
|
RU2751333C1 |
| АМИДЫ НАТАМИЦИНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ | 2022 |
|
RU2803742C1 |
| ПРОТИВООПУХОЛЕВЫЙ АНТРАФУРАНДИОН И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ЕГО ОСНОВЕ | 2014 |
|
RU2554939C1 |
| ПРОИЗВОДНЫЕ НИСТАТИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОГРИБКОВЫХ АГЕНТОВ | 2008 |
|
RU2488590C2 |
| ЗАМЕЩЕННЫЕ В ИНДОЛЬНОМ ЯДРЕ ПРОИЗВОДНЫЕ ТРИИНДОЛИЛМЕТАНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ АНТИБАКТЕРИАЛЬНАЯ И ПРОТИВОГРИБКОВАЯ АКТИВНОСТЬ | 2008 |
|
RU2388749C2 |
| СИСТЕМЫ ДОСТАВКИ ЛЕКАРСТВ НА ОСНОВЕ МАЙТАНЗИНОИДА | 2018 |
|
RU2783076C2 |
| РАДИОФАРМАЦЕВТИЧЕСКИЕ СРЕДСТВА, РАДИОВИЗУАЛИЗИРУЮЩИЕ СРЕДСТВА И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2804297C2 |
| Производные пиримидина - ингибиторы репродукции вирусов, относящихся к роду Orthoflavivirus | 2023 |
|
RU2831118C1 |
| 2-НИТРОГЕТЕРИЛТИОЦИАНАТЫ ДЛЯ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2475481C1 |
| Соль гликопептидного антибиотика и фармацевтические композиции на ее основе | 2023 |
|
RU2827992C1 |
Настоящее изобретение относится к солям ацетату, L-глутамату, цитрату или фумарату N-(2-аминоэтил)амид амфотерицина В формулы I
 ,
,
которые могут быть использованы в медицине. Предложены новые противогрибковые соединения со сниженной токсичностью и улучшенной стабильностью, фармацевтические композиции на их основе для лечения грибковых инфекций, эффективные в отношении заболеваний, вызванных С. albicans, C. parapsilosis, С. krusei, С. tropicalis, C. glabrata, M. canis, Т. Rubrum, Aspergillus. 3 н. и 7 з.п. ф-лы, 5 ил., 2 табл., 8 пр.
1. Ацетат, L-глутамат, цитрат или фумарат N-(2-аминоэтил)амид амфотерицина В структурной формулы I
 .
.
2. Фармацевтическая композиция, обладающая противогрибковой активностью, включающая соль N-(2-аминоэтил)амид амфотерицина В формулы I по п. 1, в терапевтически эффективном количестве.
3. Фармацевтическая композиция по п. 2, дополнительно включающая фармакологически приемлемый носитель и один или более фармацевтических эксципиентов.
4. Фармацевтическая композиция по п. 3, в которой фармакологически приемлемый носитель содержит воду.
5. Фармацевтическая композиция по п. 3, дополнительно включающая один или более эксципиентов, выбранных из сорастворителей, диспергаторов, поверхностно-активных веществ, солюбилизаторов, эмульгаторов, стабилизаторов, консервантов, антиоксидантов, буферных соединений, веществ для поддержания изотоничности.
6. Фармацевтическая композиция по п. 5, в которой эксципиентом, является β-циклодекстрин или его производное, γ-циклодекстрина или его производное, поливинилпирролидона, полисорбата, кремофора, декстрана.
7. Фармацевтическая композиция по п. 2, выполненная в лиофилизированной форме.
8. Фармацевтическая композиция, полученная растворением композиции по п. 7 в водной среде.
9. Фармацевтическая композиция по п. 2 для лечения грибковой инфекции вызванной С. albicans, C. parapsilosis, С. krusei, С. tropicalis, C. glabrata, M. canis, Т. Rubrum, Aspergillus.
10. Способ лечения грибковой инфекции, предусматривающий парентеральное введение нуждающемуся пациенту терапевтически эффективного количества соли N-(2-аминоэтил)амида амфотерицина В формулы I по п. 1 или фармацевтических композиций по пп. 2-8.
| WO 2015164289 A1, 29.10.2015 | |||
| WO 2016112260 A1, 14.07.2016; | |||
| US 6284736 B1, 04.09.2001 | |||
| WO 2013186384 A1, 19.12.2013 | |||
| RU 2007148974 A, 20.07.2009. |

