Настоящее изобретение относится к новым нафталиновым производным, способам их получения, к их применению в качестве фармацевтических средств и к содержащим их фармацевтическим композициям.
Более конкретно одним из объектов настоящего изобретения является соединение формулы I
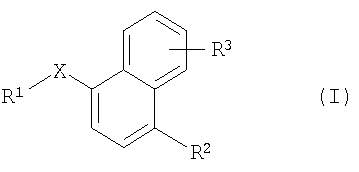
где
Х обозначает -S-, -S(O)-, -S(O)2-, -S(O)2NH-, -Р(O)(ОСН3)-, -Р(O)(ОН)-, -NH-, -N(CH3)-, -NHC(O)NH-, -C(O)-, -C(O)O-, -NHC(O)-, -СН(ОН)-, -CH=N-, -СН=СН-, -CH2NH- или -C(=NH)-;
R1 обозначает арил или гетероарил;
R2 обозначает водород, OR4 или NR5R6;
R4 обозначает С1-С8алкил или С2-С8алкенил;
R5 и R6 независимо друг от друга обозначают водород, С1-С8алкил или C(O)(C1-С8)алкил; и
R3 обозначает водород, цианогруппу, гетероарил, гетероциклоалкил, C(O)R7, OR8 или NR9R10;
R7 обозначает ОН, С1-С4алкокси, NH2, NHCH2C(O)OH или арил;
R8 обозначает водород, С1-С8алкил, С(O)С1-С4алкил или С(O)-арил; и
R9 и R10 независимо друг от друга обозначают водород, С1-С8алкил или С2-С4алкенил; при условии, что если Х обозначает -С(O)- и R2 и R3 обозначают водород или R2 обозначает Н и R3 обозначает 4-метокси, то R1 не обозначает ни 1-нафтил, ни 4-метокси-1-нафтил;
в форме свободного основания или в форме кислотно-аддитивной соли.
В контексте настоящего описания понятия арил или гетероарил относятся к 6-членному кольцу или бициклу, состоящему из двух сконденсированных колец или одного 6-членного и одного 5-членного кольца, где один или несколько атомов углерода независимо друг от друга могут быть замещены на атом, выбранный из ряда, включающего кислород, азот и серу. Примерами являются С6-С10арил, С1-С9гетероарил и С6арил, сконденсированный с 5- или 6-членным алифатическим или гетероалифатическим кольцом, например нафтил, 1,2,3,4-тетрагидронафталинил, фенил, индолил, хинолинил, изохинолинил, 1,2,3,4-тетрагидрохинолинил, бензотиазолил, имидазолил, бензимидазолил, бензоксадиазолил, бензотриазолил, инданил, оксадиазолил, пиразолил, триазолил и тетразолил.
Примерами гетероциклоалкила являются пиперидинил, пиперазинил и морфолинил.
Следует иметь в виду, что вышеуказанные соединения могут нести в своей структуре заместители, например один или несколько заместителей, выбранных из ряда, включающего ОН; нитро; галоген; циано; СООН; C(O)NH2; C(O)NHNHC(O)CH3; C(NH2)=NOH; С1-С4алкил; S-C1-C4алкил; С1-С8алкокси; C5-С10арил, такой как фенил; С1-С4-гетероарил, такой как оксадиазолил; C1-C5-N-гетероциклоалкил, такой как морфолинил или пиперидинил; C(O)O-C1-С4алкил или NR11R12, где R11 и R12 независимо друг от друга обозначают водород, С1-С4алкил, С(O)NHOC1-С4алкил, С(O)С1-С4алкил или SO2-С1-С4алкил; причем заместители в свою очередь могут быть замещены заместителем, выбранным из ряда, включающего ОН; нитрогруппу; NH2; С1-С4алкил; С1-С4алкоксигруппу; С1-С4алкоксигруппу, замещенную ОН; С3-С6циклоалкил; N-(С1-С4алкил)2; фенил или морфолинил.
Например, в радикале R арил или гетероарил могут быть незамещенными или могут быть замещены одним или несколькими заместителями, выбранными из ряда. включающего ОН; СООН; C(O)NH2; нитро; галоген; циано; C(NH2)=NOH; С1-С4-N-гетероарил; C1-C5-N-гетероциклоалкил; С1-С4алкил; S-С1-С4алкил; С1-С8алкокси и NR11R12, где R11 и R12 независимо друг от друга обозначают водород, С1-С4алкил, C(O)NHOC1-C4алкил, С(О)С1-С4алкил или SO2-С1-С4алкил; где С1-С4алкил, C1-C8алкокси и С1-С5-N-гетероциклоалкил в свою очередь могут быть незамещенными или могут быть замещены ОН; C1-С4алкилом; С1-С4алкоксигруппой; С1-С4алкоксигруппой, замещенной ОН; С3-С6циклоалкилом; N-(С1-С4алкилом)2; фенилом; или морфолинилом;
причем в радикале R3 оксадиазолил, пиперазинил или тетразолил могут быть замещены метилом;
в радикале R4 С1-С8алкил может быть незамещенным или может быть замещен ОН, С(O)O-С1-С4алкилом, морфолинилом, пиперидинилом, фенилом или оксадиазолилом; причем фенил и оксадиазолил в свою очередь могут быть незамещенными или могут быть замещены С1-С4алкилом, С1-С4алкокси-, нитрогруппой, NH2 или N(С1-С4алкилом)2;
в радикале R5 или R6 С1-С4алкил может быть незамещенным или может быть замещен морфолинилом;
в радикале R8 С1-С4алкил может быть незамещенным или может быть замещен С(O)ОН, С(O)ОСН3, C(O)NHNHC(O)CH3 или оксадиазолом, замещенным С1-С4алкилом.
Соединения по изобретению могут находиться в свободной форме или в форме кислотно-аддитивной соли. Следует иметь в виду, что под объем изобретения подпадают соединения формулы I как в свободной форме, так и в форме соли, например в форме трифторацетата или гидрохлорида. Пригодные для фармацевтического применения согласно изобретению фармацевтически приемлемые кислотно-аддитивные соли включают прежде всего гидрохлориды.
В формуле I радикалы независимо друг от друга, в сочетании друг с другом или в любой комбинации или субкомбинации предпочтительно имеют следующие значения:
(а) Х обозначает, -S-, -S(O)-, S(O)2-, -S(O)2NH-, -Р(O)(ОСН3)-, -Р(O)(ОН)-, -NH-, -N(СН3)-, -NHC(O)NH-, -NHC(O)-, -C(O)-, -C(O)O-, -СН(ОН)-, -CH-N-, -СН=СН-, -CH2NH- или =NH)-; предпочтительно -NH, -С(O)-, -С(O)O- или -CH2NH-, более предпочтительно -С(O)- или -С(O)O-;
(б) R1 обозначает фенил, 4-метоксифенил, 2-гидрокси-3-метоксифенил, 2,3-диметоксифенил, 3,4-диметоксифенил, 4-[2-(морфолин-4-ил)этокси]фенил, 4-[3-(гидрокси)пропокси]фенил, 4-бутоксифенил, 3-(NHC(O)NHOCH3)-4-пентоксифенил, 4-тиометилфенил, 4-ацетамидофенил, нафтил, 4-карбоксинафтил, 4-аминокарбонилнафтил, 4-гидроксинафтил, 4-(C(NH2)=NOH)-нафтил, 4-фторнафт-1-ил, 4-цианонафтил, 3-нитронафт-1-ил, 4-нитронафт-1-ил, 3-аминонафт-1-ил, 4-аминонафт-1-ил, 4-диметиламинонафт-1-ил, 4-метоксинафт-1-ил, 4-[4-(гидрокси)бутокси]нафтил, 4-пентоксинафтил, 4-[2-(морфолин-4-ил)этокси]нафтил, 3-(диметиламино)нафтил, 3-метилсульфониламидонафтил, 4-метилсульфонамидонафтил, 4-(1,2,4-триазол-1-ил)нафт-1-ил, 4-(1H-тетразол-5-ил)нафтил, 4-(пиразол-1-ил)нафтил, 4-(имидазол-1-ил)нафтил, 1,2,3,4-тетрагидронафталин-5-ил, индан-4-ил, индол-7-ил, хинолин-8-ил, хинолин-4-ил, хинолин-3-ил, хинолин-5-ил, изохинолин-5-ил, изохинолин-1-ил, 1,2,3,4-тетрагидрохинолин-1-ил, 1,2,3,4-тетрагидрохинолин-8-ил, 6-метокси-1,2,3,4-тетрагидрохинолин-1-ил, 5-гидрокси-1,2,3,4-тетрагидрохинолин-1-ил, 7-пентоксибензотриазол-4-ил, 5,7-диметил-2,1,3-бензотиадиазол-4-ил, 5-хлор-2,1,3-бензотиадиазол-4-ил, 2,1,3-бензотиадиазол-4-ил, 2,1,3-бензоксадиазол-4-ил, 7-пентокси-2,1,3-бензоксадиазол-4-ил, 2-оксо-7-пентокси-1,3-дигидробензимидазол-4-ил, 2-(NHCH2фенил)-7-пентоксибензимидазол-4-ил, 2-(NHCH2циклогексил)-7-пентоксибензимидазол-4-ил, 2-(NH(СН2)3N(СН2СН3)2)-7-пентоксибензимидазол-4-ил, 2-(NH(СН2)3СН3)-7-пентоксибензимидазол-4-ил, 2-(4-метилпиперазин-1-ил)-7-пентоксибензимидазол-4-ил, 2-(NH(СН2)2OH)-7-пентоксибензимидазол-4-ил, 2-(NH(CH2)2O(CH2)2OH)-7-пентоксибензимидазол-4-ил, 2-метил-7-пентоксибензимидазол-4-ил, 7-пентоксибензимидазол-4-ил; прежде всего нафтил, 4-гидроксинафтил, 4-фторнафт-1-ил, 4-цианонафт-1-ил, 4-нитронафт-1-ил, 4-диметиламинонафт-1-ил, 4-метоксинафт-1-ил, 4-[4-(гидрокси)бутокси]нафтил, 4-(1,2,4-триазол-1-ил)нафт-1-ил, 4-(пиразол-1-ил)нафтил, 4-(имидазол-1-ил)нафтил, 1,2,3,4-тетрагидронафталин-5-ил, индан-4-ил, хинолинил, хинолин-8-ил, хинолин-4-ил, изохинолин-5-ил, 7-пентоксибензотриазол-4-ил, 5-хлор-2,1,3-бензотиадиазол-4-ил, 2-(NHCH2фенил)-7-пентоксибензимидазол-4-ил, 2-(NH(CH2)3CH3)-7-пентоксибензимидазол-4-ил или 7- пентоксибензимидазол-4-ил, более предпочтительно нафтил, 4-фторнафт-1-ил, 4-цианонафт-1-ил, 4-диметиламинонафт-1-ил, 4-(1,2,4-триазол-1-ил)нафт-1-ил, 4-(имидазол-1-ил)нафтил, 1,2,3,4-тетрагидронафталин-5-ил, индан-4-ил, хинолин-8-ил, изохинолин-5-ил или 5-хлор-2,1,3-бензотиадиазол-4-ил;
(в) R2 обозначает водород, -O-(СН2)2СН3, -O-(СН2)3СН3, -O-(СН2)4СН3, -O-(СН2)5СН3, -O-(СН2)6СН3, -O-(СН2)3СН(СН3)2, 2-(морфолин-4-ил)этокси, 2-(пиперидин-1-ил)этокси, 2-(4-метоксифенил)этокси, 2-(фенил)этокси, 2-(4-нитрофенил)этокси, 2-(4-диметиламинофенил)этокси, 2-(4-аминофенил)этокси, 2-(2-нитрофенил)этокси, 2-(2-аминофенил)этокси, 2-(2-диметиламинофенил)этокси, 3-(морфолин-4-ил)пропилокси, 3-(пиперидин-1-ил)пропилокси, -O-(СН2)3С(O)ОСН2СН3, -O-(СН2)4С(O)ОСН2СН3, -O-(СН2)2OCH2СН3, -O-СН2С(O)ОСН3, -O-СН2-(2-метил)оксадиазол-5-ил, -O-СН2-(2-этил)оксадиазол-5-ил, -O-СН2-(2-пропил)оксадиазол-5-ил, O-СН2СН=СНСН2СН3 (Z) и (Е), -O-(СН2)3ОН, -O-(СН2)4OH, -O-(СН2)5OH, -N-[2-(морфолин-4-ил)этил]-N-(СН2)3СН3, -NH-(СН2)3СН3, -NH-(СН2)4СН3, -NHC(O)(CH2)3CH3, -N(СН3)(СН2)3СН3 или -N(СН3)(СН2)4СН3; предпочтительно водород, -O-(СН2)2СН3, -O-(СН2)3СН3, -O-(СН2)4СН3, -O-(СН2)5СН3, -O-(СН2)3СН(СН3)2, 2-(морфолин-4-ил)этокси, O-СН2СН=СНСН2СН3 (Z) и (Е), -NH-(СН2)3СН3, -NH-(СН2)4СН3, -N(СН3)(СН2)3СН3 или -N(СН3)(СН2)4СН3; более предпочтительно -O-(СН2)3СН3, -O-(СН2)4СН3, -O-(СН2)3СН(СН3)2, -O-СН2СН=СНСН2СН3 (Z) и (Е), -NH-(СН2)3СН3, -NH-(СН2)4СН3, -N(СН3)(СН2)3СН3 или -N(CH3)(CH2)4CH3;
(г) R3 обозначает водород, 7-ОН, 8-ОН, 7-ОСН3, 7-ОСН2С(O)ОН, 7-ОСН2С(O)ОСН3, 7-OCH2C(O)NHNHC(O)CH3, 7-[-O-СН2-(2-метил)-1,3,4-оксадиазол-5-ил], 7-ОС(O)СН3, 7-ОС(O)-нафтил, 3-С(O)ОН, 7-С(O)ОН, 3-С(O)ОСН3, 7-C(O)NH2, 8-ОС(O)-нафтил, 3-C(O)NHCH2C(O)OH, 7-циано, 6-NH2, 7-NH2, 6-N(CH3)2, 7-N(CH3)2, 6-NHCH2CH=CH2, 6-N(CH2CH=CH2)2, 7-(пиперазин-1-ил), 7-(4-метилпиперазин-1-ил), 7-(1H-тетразол-5-ил), 7-(1-метил)тетразол-5-ил), 7-(2-метил)тетразол-5-ил) или 7-(2-метил)-1,3,4-оксадиазол-5-ил; предпочтительно водород, 7-ОН, 8-ОН, 7-ОС(O)СН3 или 6-NHCH2CH=CH2; более предпочтительно водород, 7-ОН или 7-ОС(O)СН3.
Кроме того, в настоящем изобретении предложен способ получения соединения формулы I, который заключается в том, что арильный или гетероарильный фрагмент подвергают взаимодействию с соответствующим замещенным нафталином, после чего при необходимости осуществляют дополнительную дериватизацию с помощью методов, известных специалистам в данной области.
Более конкретно в изобретении предложен способ получения соединения формулы I, заключающийся в том, что осуществляют следующие стадии (а) взаимодействие соединения формулы II

где R1 имеет указанные выше значения и R13 обозначает -ОН, -SH, -I, -Cl, 1,8-бис(диметиламино)нафтил-, -СООН, -NH2, -Н, -карбонитрил, -O-трифторметансульфонил или -С(O)Cl, с соединением формулы III
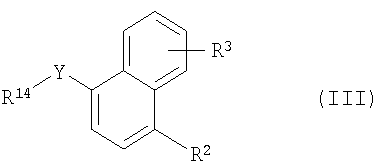
где R2 и R3 имеют указанные выше значения, Y обозначает -O-, -S(O)2O-, -Р(O)(ОСН3)-, простую связь, -С(O)O-, -С(O)- или -В(ОН)2- и R14 обозначает, например, водород, -I, -Cl, получая соединение формулы Iа
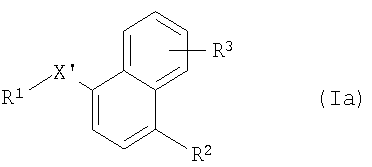
где R1, R2 и R3 имеют указанные выше значения и X' обозначает -СО-, -S-, -Р(O)(ОСН3)-, -NH-, -S(O)2NH-, -C(O)O-, -CH=N-, -СН(ОН)-, -NHC(O)NH-, -C(=NH)-, или (в том случае, если указанный радикал связан с атомом азота R1) -S(O)2-; или
(б) превращение соединения формулы Iа в соединение формулы Iб
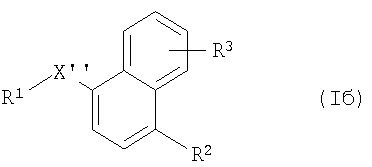
где R1, R2 и R3 имеют указанные выше значения и X'' обозначает -SO-, -S(O)2- (которое можно получать с помощью процесса (б), если в компоненте, с которым происходит связывание, R1 обозначает С), -N(СН3)-, -Р(O)ОН-, -CH2NH-, -СН=СН- или (при связывании через атом углерода радикала R1) -S(O)2-
и выделения полученного таким образом соединения формулы Iа и формулы Iб в свободной форме или в форме соли.
Процесс (а) можно осуществлять обычным образом, например так, как это описано в примерах 1-14.
Для процесса (б),
(I) для получения соединения формулы Iб, где X'' обозначает -SO- или -S(O)2-, можно применять соединение формулы Iа, где X' обозначает -S- и мета-хлорбензойную кислоту, например, как это описано в примере 2;
(II) для получения соединения формулы Iб, где X'' обозначает -Р(O)ОН-, можно применять соединение формулы Iа, где X' обозначает -Р(O)(ОСН3)- и триметилсилилйодид, например, как это описано в примере 3;
(III) для получения соединения формулы Iб, где X'' обозначает -N(СН3)-, можно применять соединение формулы Iа, где X' обозначает -NH- и метилйодид, например, как это описано в примере 4;
(IV) для получения соединения формулы Iб, где X'' обозначает -CH2NH-, можно применять соединение формулы Iа, где X' обозначает -CH=N- и ВН3 - пиридин, например, как это описано в примере 8.
Обработку реакционных смесей и очистку полученных таким образом соединений можно осуществлять с помощью известных методов.
Кислотно-аддитивные соли можно получать из свободных оснований известным методом и наоборот. В качестве пригодных кислотно-аддитивных солей по настоящему изобретению можно применять, например, гидрохлориды.
Исходные соединения формул II и III можно получать, например, согласно методам, описанным в примерах 2, 3, 5, 6, 12, 13 и 14; или можно использовать известные соединения, или их можно получать методами, аналогичными известным методам.
Соединения по изобретению и их фармацевтически приемлемые кислотно-аддитивные соли, называемые в настоящем описании как агенты по изобретению, обладают ценными фармакологическими свойствами, как это установлено в опытах in vitro и на животных, и поэтому их можно применять в качестве фармацевтических средств.
В частности, агенты по изобретению обладают активностью в отношении связывания каннабиноидного (СВ) рецептора. Более конкретно агенты по изобретению обладают активностью в отношении человеческого CB1-рецептора. Взаимодействие агентов по изобретению с каннабиноидным рецептором можно продемонстрировать в опытах по оценке их активности в отношении вытеснения, например, [3H]CP55940 из человеческих каннабиноидных рецепторов, экспрессируемых, например, в клетках линии рЕАК, например, согласно приведенному ниже методу тестирования.
Тест I: анализ связывания с рецептором CB1
Смесь для анализа содержит 75 мкл суспензии мембран [мембраны из клеток линии рЕАК, трансфектированных человеческими CB1-рецепторами, полученными от фирмы Receptor Biology, Белтсвилл, штат Мэриленд; 133 мкг/мл в буфере для анализа (50 мМ Трис-HCl, 2,5 мМ ЭДТК, 5 мМ MgCl2, 5 мг/мл БСА, рН 7,4), приблизительно 10 мкг/лунку)], 25 мкл WGA-YS гранул [гранулы силиката иттрия, сенсибилизированные агглютинином из проростков пшеницы, фирма Amersham (40 мг/мл, 1 мг/лунку)], 50 мкл тестируемого соединения в 4%-ном ДМСО и 50 мкл радиолиганда {[3Н]СР55940 (180 Ки/ммоль), фирма New England Nuclear; конечная концентрация 0,125 нМ, в буфере для анализа}. Все компоненты смешивают, встряхивают при комнатной температуре в течение 2 ч, затем подсчитывают радиоактивность с помощью счетчика Topcount. Ненасыщающее связывание оценивают в присутствии 10 мкМ (R)-(+)-[2,3-дигидро-5-метил-3-[(4-морфолинил)метил]пирроло[1,2,3-de]-1,4-бензоксазин-6-ил](1-нафталинил)метанон (Tocris).
Для агентов по изобретению значения Ki составляют от 1 нМ до 100 мкМ, предпочтительно от 10 нМ до 2 мкМ. Значения IC50 вычисляют с помощью программы ORIGIN с использованием аппроксимации экспериментальных данных логарифмической кривой. Значения Ki рассчитывают на основе величин IC50 с использованием уравнения Ченга-Пруссова (Ki=IC50/(1+([L]/Kd)), где [L] обозначает концентрацию лиганда.
Агенты по изобретению наиболее целесообразно применять для лечения или предупреждения хронической боли, прежде всего воспалительной, например хронической воспалительной боли, воспалительных заболеваний, например воспалительных заболеваний дыхательных путей, например хронического обструктивного заболевания легких (ХОЗЛ), или астмы, ринита, воспалительного заболевания кишечника, цистита, например интерстициального цистита, панкреатита, увеита, воспалительных заболеваний кожи и ревматоидного артрита.
Удельную активность соединений в качестве аналгетиков можно подтверждать с помощью стандартных методов, например с помощью описанного ниже теста.
Тест II: модель невропатической боли
Действие в отношении гипералгезии оценивают с использованием модели невропатической боли, индуцированной наложением частичной лигатуры на седалищный нерв согласно методу, описанному Seltzer и др. (1990). В целом метод состоит в том, что крыс линии Wistar (весом 120-140 г) подвергают анестезии, обнажают левый седалищный нерв на уровне середины бедра с помощью небольшого разреза и пережимают его на 1/3-1/2 толщины нерва путем наложения плотной лигатуры из шовного материала 7.0. Рану закрывают с помощью обычного шовного материала для мышечной ткани и кожных скобок и опыливают порошком антибиотика ауреомицина. Животным дают восстановиться, и через 12-15 дней после хирургического вмешательства на них проводят опыты.
Механическую гипералгезию оценивают на основе измерения предельных величин силы, при которой происходит отдергивание лапы при увеличении давления на дорсальную поверхность лапы, с использованием аналгезиметра (фирма Ugo-Basile, Милан), рассчитанного на предельную нагрузку 250 г. Пороговые значения силы, при которой происходит отдергивание лапы, измеряют как на ипсилатеральной (на которую наложена лигатура), так и на контрлатеральной (на которую не наложена лигатура) лапе до (перед введением дозы) и через 6 ч после введения лекарства или носителя. Данные выражают в виде предельных величин силы (г), при которой происходит отдергивание лапы и процента реверсии гипералгезии, который рассчитывают по следующей формуле:

Эффективность выражают в виде значения D50, т.e. дозы, необходимой для вызывания 50%-ной реверсии гипералгезии.
Для агентов по изобретению величины D50 составляют от 0,1 до 100 мг/кг.
Таким образом, агенты по изобретению можно применять в качестве агонистов каннабиноидного рецептора, например, для лечения боли различного происхождения или этиологии и в качестве противовоспалительных агентов и/или антиэдемических (противоотечных) средств для лечения воспалительных реакций, заболеваний или состояний, а также для лечения аллергических реакций. Благодаря аналгетическому/противовоспалительному профилю их можно применять для лечения связанной с воспалением боли, для лечения гипералгезии и прежде всего для лечения серьезной хронической боли. Их можно применять, например, для лечения боли, воспаления и/или отека, например, связанных с травмой, например, связанных с ожогами, растяжениями, переломами или т.п., последствиями хирургического вмешательства, например, в качестве послеоперационных аналгетиков, а также для лечения связанной с воспалением боли другого происхождения, например, для лечения боли в костях и суставах (остеоартрит), ревматоидного артрита, ревматического заболевания, тендосовинита, подагры, боли при раке, миофасциальной боли (повреждение мышц, фибромиалгия), хронической невропатической боли, например, диабетической невропатии, фантомной боли конечности и периоперативной (возникшей в процессе хирургической операции) боли (общая хирургия, гинекологическая операция). Кроме того, их также можно применять для лечения боли, связанной, например, со стенокардией, менструацией или раком. Кроме того, их можно применять в качестве противовоспалительных/противоотечных агентов, например, для лечения воспалительных заболеваний кожи, например псориаза и экземы.
Агенты по настоящему изобретению можно применять также в качестве миорелаксантов гладких мышц, например, для лечения спазмов желудочно-кишечного тракта или матки, например, для лечения глаукомы/внутриглазного давления, например, для терапии болезни Крона, неспецифического язвенного колита или панкреатита и для лечения мышечной спастичности и тремора, например, при рассеянном склерозе.
Для перечисленных выше показаний дозы агентов по изобретению, конечно, варьируются в зависимости, например, от хозяина, пути введения и природы и серьезности подлежащего лечению состояния, а также от относительной эффективности конкретного применяемого агента по изобретению. Требуемое количество действующего вещества можно оценивать, например, с помощью известных методов анализа in vitro и in vivo, определяя, насколько долго концентрация конкретного действующего вещества в плазме крови остается на приемлемом для оказания терапевтического действия уровне. В целом установлено, что удовлетворительные результаты на животных получают при введении пероральным путем (р.о.) суточных доз от приблизительно 0,01 до приблизительно 20,0 мг/кг. Для человека соответствующая суточная доза составляет от приблизительно 0,7 до приблизительно 1400 мг/день р.о., например, от приблизительно 50 до 200 мг (человек весом 70 кг), которую вводят в виде однократной дозы или в виде разделенных доз до 4-х раз в день или с помощью формы с непрерывным высвобождением. Пригодные для этой цели пероральные лекарственные средства содержат от приблизительно 1,75 или 2,0 до приблизительно 700 или 1400 мг агента по изобретению в сочетании с соответствующим фармацевтически приемлемым разбавителем или носителем.
В альтернативном варианте агенты по изобретению можно применять местно, например, в форме крема, геля или т.п., например, для лечения описанных выше состояний кожи, или путем ингаляции, например, в форме сухого порошка, например, для лечения астмы.
Примерами композиций, содержащих агент по изобретению, являются, например, твердая дисперсия, водный раствор, например, содержащий солюбилизирующий агент, микроэмульсия и суспензия, например, гидрохлорида соединения формулы I в концентрации от 0,1 до 1%, например 0,5%. Композиции можно забуферивать с помощью пригодного буфера до значения рН, составляющего, например, от 3,5 до 9,5, например до рН 4,5.
Агенты по изобретению можно применять также в качестве химических препаратов при проведении научных исследований.
Агенты по изобретению можно вводить in vivo либо индивидуально, либо в сочетании с другими фармацевтическими агентами, эффективными в отношении лечения заболеваний и состояний, обусловливающих или сопровождающих активацию CB1-рецептора, включая ингибиторы циклооксигеназы-2 (СОХ-2), такие как специфические ингибиторы СОХ-2 (например, целекоксиб и рофекоксиб) и нестероидные противовоспалительные лекарственные средства (НСПВС) (например, ацетилсалициловая кислота, производные пропионовой кислоты), антагонисты ваниллоидного рецептора, трициклические антидепрессанты (например, Anafranil®, Asendin®, Aventyl®, Elavil®, Endep®, Norfranil®, Norpramin®, Pamelor®, Sinequan®, Surmontil®, Tipramin®, Tofranil®, Vivactil®, Tofranil-PM®), антиконвульсивные средства (например, габапентин) и агонисты ГАМКВ (гамма-аминомасляная кислота) (например, L-баклофен).
Фармацевтические композиции по изобретению, предназначенные для раздельного введения компонентов, входящих в состав композиции, и для введения в виде фиксированной композиции, т.е. одной галеновой композиции, содержащей по меньшей мере два компонента, можно получать хорошо известным методом, и их можно вводить энтерально, например перорально или ректально, а также парентерально, млекопитающим, включая человека, причем они содержат терапевтически эффективное количество по меньшей мере одного обладающего фармакологической эффективностью компонента индивидуально или в сочетании с одним или несколькими фармацевтически приемлемыми носителями, прежде всего пригодными для энтерального или парентерального введения.
Новая фармацевтическая композиция содержит, например, от приблизительно 0,1 до приблизительно 99,9%, предпочтительно от приблизительно 20 до приблизительно 60% действующих веществ. Фармацевтические композиции для совместной терапии, вводимые энтеральным или парентеральным путем, представляют собой, например, композиции, находящиеся в виде стандартных дозируемых форм, таких как покрытые сахаром таблетки, таблетки, капсулы или суппозитории, а также ампулы. Если не указано иное, их получают хорошо известным методом, например, с использованием общепринятых методов смешения, грануляции, нанесения сахарного покрытия, растворения или лиофилизации. Следует иметь в виду, что количество входящего в состав композиции компонента, содержащегося в одной стандартной дозе каждой дозируемой формы, не обязательно должно представлять собой эффективное количество, поскольку необходимое эффективное количество можно вводить с использованием нескольких стандартных доз.
В частности, терапевтически эффективное количество каждого из компонентов, входящих в композицию, можно вводить одновременно или последовательно и в любом порядке, причем компоненты можно вводить по отдельности или в виде фиксированной композиции. Например, способ по изобретению замедления развития или лечения пролиферативного заболевания может предусматривать (I) введение компонента (а), входящего в состав композиции в свободной форме или в форме фармацевтически приемлемой соли, и (II) введение компонента (б), входящего в состав композиции в свободной форме или в форме фармацевтически приемлемой соли, одновременно или последовательно в произвольном порядке в количествах, которые в совокупности обладают терапевтической эффективностью, предпочтительно в количествах, обладающих синергетическим действием, например, в суточных дозах, соответствующих указанным в настоящем описании количествам. Индивидуальные компоненты, входящие в состав композиции, можно вводить по отдельности в различные моменты времени в процессе лечения или одновременно в виде разделенных или объединенных композиций. Кроме того, понятие введение включает также применение пролекарства компонента, входящего в состав композиции, которое превращается in vivo в компонент, входящий в состав композиции. Таким образом, следует иметь в виду, что под объем настоящего изобретения подпадают все такие режимы одновременного или чередующегося лечения и понятие «введение» следует интерпретировать соответствующим образом.
Эффективная доза каждого из входящих в композицию компонентов может варьироваться в зависимости от конкретного соединения или применяемой фармацевтической композиции, пути введения, состояния, подлежащего лечению, степени серьезности состояния, подлежащего лечению. Таким образом, схему приема лекарственного средства выбирают с учетом различных факторов, включая путь введения и почечную и печеночную функцию пациента. Лечащий врач, клиницист или ветеринар, специализирующиеся в данной области, могут легко определять и предписывать эффективное количество индивидуальных действующих веществ, необходимых для предупреждения, противодействия или прекращения развития состояния. Оптимальную концентрацию действующих веществ, находящуюся в диапазоне, в котором можно достичь эффективного действия, не сопровождающегося токсичностью, получают с использованием схемы приема лекарственного средства, разработанной с учетом кинетики достижения действующими веществами областей-мишеней. В целом установлено, что для животных удовлетворительные результаты получают при введении пероральным путем (р.о.) суточных доз, составляющих от приблизительно 0,01 до приблизительно 20,0 мг/кг. Для человека суточная доза составляет от приблизительно 0,7 до приблизительно 1400 мг/день р.о., например от приблизительно 50 до 200 мг (для человека весом 70 кг), которую удобно вводить в виде однократной дозы или разделенных доз (до 4-х раз в день) или в форме с непрерывным высвобождением. Соответственно пригодные пероральные дозируемые формы содержат от приблизительно 1,75 или 2,0 до приблизительно 700 или 1400 мг.
В соответствии с вышеизложенным объектами настоящего изобретения являются:
(1) агент по изобретению, предназначенный для применения в качестве агониста каннабиноидного рецептора, например для применения при любом из указанных выше показаний;
(2) Фармацевтическая композиция, включающая в качестве действующего вещества агент по изобретению наряду с фармацевтические приемлемым разбавителем или носителем. Такую композицию можно приготавливать общепринятым методом.
(2') Фармацевтическая композиция, предназначенная для лечения или предупреждения заболевания или состояния, в которых играет роль или участвует активация каннабиноидного рецептора, включающая агент по изобретению и носитель.
(3) Способ лечения любого из перечисленных выше заболеваний у пациента, нуждающегося в таком лечении, заключающийся в ведении эффективного количества агента по изобретению;
(3') Способ лечения или предупреждения заболевания или состояния, при котором играет роль или принимает участие активация каннабиноидного рецептора, заключающийся в том, что нуждающемуся в этом млекопитающему вводят терапевтически эффективное количество агента по изобретению.
(4) Применение агента по изобретению для приготовления лекарственного средства, предназначенного для лечения или предупреждения заболевания или состояния, при котором играет роль или принимает участие активация каннабиноидного рецептора;
(5) Способ, как он определен выше, предусматривающий совместное введение, например, одновременно или последовательно терапевтически эффективного количества агониста СВ, например агента по изобретению, и второй лекарственной субстанции, применяемой, например, для указанных в настоящем описании показаний.
(6) Композиция, содержащая терапевтически эффективное количество агониста СВ, например агента по изобретению и второй лекарственной субстанции, например, предназначенная для применения при любом из указанных выше показаний.
Предпочтительным соединением формулы I для применения согласно изобретению является соединение из примера 1. Это соединение является эффективным агонистом СВ, в частности CB1-агонистом in vitro (Ki=0,015±0,004 мкМ). Величина D50 для модели невропатической боли, используемой в тесте II, для соединения из примера 1 составляет 0,18 мг/кг р.о.
Сокращения, используемые в примерах:
Изобретение более подробно проиллюстрировано на приведенных ниже примерах.
Пример 1: Получение нафталин-1-ил-(4-пентилоксинафталин-1-ил)метанона
(а) 20 г 1-нафтола, 21,2 мл NEt3 и 1,7 г диметиламинопиридина растворяют в 300 мл метиленхлорида при комнатной температуре (КТ). Раствор охлаждают до 10°С. В течение 15 мин по каплям добавляют 20,9 мл нафтоилхлорида в 100 мл метиленхлорида. После общепринятой обработки получают нафталин-1-ил-(нафталинокси-1-ил)метанон.
(б) 29,0 г нафталин-1-ил-(нафталинокси-1-ил)метанона добавляют порциями к суспензии, содержащей 14,3 г хлорида алюминия в 100 мл толуола, и перемешивают в течение 2 ч при 140°С. После обычной обработки получают нафталин-1-ил-(4-пентилоксинафталин-1-ил)метанон.
(в) 11,0 г нафталин-1-ил-(нафталинокси-1-ил)метанона и 6,1 г карбоната калия в 130 мл ацетона перемешивают в течение 15 мин при температуре дефлегмации. Затем в течение 2 мин добавляют раствор, содержащий 6,8 мл 1-бромпентана в 20 мл ацетона, и суспензию перемешивают еще в течение 22 ч при температуре дефлегмации. После общепринятой обработки и последующей хроматографии получают нафталин-1-ил-(4-пентилоксинафталин-1-ил)метанон. Температура плавления: 72-75°С (пропан-2-ол); ЖХВР: время удерживания (мин): 8,15 [ЖХВР-метод: колонка типа Kingsorb 3 micron C18 (30×4,6 мм). Градиент элюирования: 10-100% ацетонитрила в 0,1% трифторуксусной кислоты в воде в течение 7 мин, затем 100% ацетонитрила в течение 3 мин].
1H ЯМР (400 MHz, CDCl3): δ 9,02 (d, 1H), 8,43 (d, 1H), 8,25 (d, 1H), 8,01 (d, 1H), 7,95 (d, 1H), 7,70 (t, 1H), 7,62-7,50 (m, 6H), 6,68 (d, 1H), 4,19 (t, 2H), 2.0-1,94 (m, 2H), 1,6-1,54 (m, 2H), 1,49-1,44 (m, 2H), 0,99 (t, 3H).
MC m/z (%): 369,1 (M+H, 100); ИК (ν, см-1): 1633 (C=O)
В следующих примерах получают соединения формулы I согласно изобретению, где R2 обозначает -O-(СН2)4СН3 (Прим. обозначает пример).
В следующих примерах представлены полученные согласно изобретению соединения формулы I, где Х обозначает С(O):
В следующих примерах представлены полученные согласно изобретению соединения формулы I, где Х обозначает С(O) и R обозначает водород:
В следующем примере представлено полученное согласно изобретению соединение формулы I. где Х обозначает C(O):
Соединения получают прежде всего согласно следующим методам:
Метод получения 1: синтез кетонов
Метод получения, описанный в примере 1, применяют для получения соединений из примеров: 29, 81, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 121, 122, 123, 124, 125, 128, 129, 130, 131, 132, 135, 136, 137, 138, 139, 140, 142, 143, 144, 147, 148, 149.
Пример 2: Синтез сульфидов, сульфонов и сульфоксидов
Метод применяют для получения соединений из примеров: 2, 3, 4, 7, 8, 9, 10, 12, 17, 18, 19.
(а) 1-йод-4-пентилоксинафталин: Раствор 1-пентилоксинафталина (6,41 г, 29,9 ммоль) в ацетонитриле (120 мл) обрабатывают N-йодсукцинимидом (10,1 г, 44,9 ммоль) и перемешивают в течение 6 ч при 82°С. После охлаждения до комнатной температуры реакционную смесь распределяют между 1М КНСО3 (185 мл) и толуолом (2×185 мл). Органическую фазу промывают водой, сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (циклогексан) получают 9,0 г (89%) продукта в виде кристаллов слегка розоватого цвета. EI-МС (m/z) 340 (М+).
(б) 1-(1-Нафталинсульфанил)-4-пентилоксинафталин: Смесь, содержащую 1-йод-4-пентилоксинафталин (0,68 г, 2,0 ммоль), трет-BuOK (0,40 г, 3,0 ммоль), 1-нафттиол (0,48 г, 3,0 ммоль), ДФЭфос (120 мг) и Pd2dba3 (80 мг) в толуоле (16 мл), выдерживают в течение 2 ч при 90°С. После охлаждения до комнатной температуры реакционную смесь промывают водой (16 мл) и фильтруют через Hyflo. Органическую фазу сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (циклогексан/ацетон) получают 0,62 г (80%) продукта в виде бесцветных кристаллов.
(в) 1-(1-нафталинсульфинил)-4-пентилоксинафталин: Раствор 1-(1-нафталинсульфанил)-4-пентилоксинафталина (112 мг, 0,3 ммоль) в ДХМ (3 мл) перемешивают с МХПБК (74 мг, 0,3 ммоль) в течение 2 ч при 0°С. Реакционную смесь распределяют между ДХМ (3 мл) и 1М КНСО3 (6 мл). Органическую фазу промывают водой (3 мл), сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (циклогексан/ацетон) получают 94 мг (80%) продукта в виде бесцветных кристаллов.
(г) 1-(1-Нафтилсульфонил)-4-пентилоксинафталин: Раствор 1-(1-нафталинсульфинил)-4-пентилоксинафталина (112 мг, 0,3 ммоль) в ДХМ (3 мл) перемешивают с МХПБК (148 мг, 0,9 ммоль) в течение 2 ч при 0°С, а затем еще в течение 2 ч при комнатной температуре. Реакционную смесь распределяют между ДХМ (3 мл) и 1М КНСО3 (6 мл). Органическую фазу промывают водой (3 мл), сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (циклогексан/ацетон) получают 91 мг (73%) продукта в виде бесцветных кристаллов.
Пример 3: Синтез эфиров фосфиновой кислоты
Метод применим для получения соединений из примеров: 5, 6, 14, 15, 16, 20, 21, 23.
(а) Метиловый эфир (4-пентилоксинафталин-1-ил)фосфиновой кислоты: Раствор безводного кристаллического Н3РО2 (1,46 г, 21,9 ммоль) в смеси толуол/ТГФ (1:1, 11 мл) обрабатывают НС(ОМе)3 (9,6 мл, 87,7 ммоль) и перемешивают в течение 1 ч при 0°С, а затем еще в течение 2 ч при комнатной температуре. Смесь добавляют к раствору 1-йод-4-пентилоксинафталина (3,65 г, 10,7 ммоль) и NEt3 (1,64 мл, 11,8 ммоль) в ацетонитриле (27 мл). После добавления (Ph3Р)2PdCl2 (376 мг, 0,54 ммоль) реакционную смесь выдерживают при 90°С в течение 4 ч. После охлаждения до комнатной температуры реакционную смесь концентрируют. После экспресс-хроматографии (ДХМ/метанол) получают 2,16 г (69%) продукта в виде масла коричневатого цвета. EI-MC (m/z) 292 (М+).
(б) Метиловый эфир нафталин-1-ил-(4-пентилоксинафталин-1-ил)фосфиновой кислоты: Смесь, содержащую метиловый эфир (4-пентилоксинафталин-1-ил)фосфиновой кислоты (339 мг, 1,2 ммоль), NEt3 (0,18 мл, 1,3 ммоль), 1-нафтилйодид (0,17 мл, 1,2 ммоль), ДФЭфос (81 мг) и Pd2dba3 (60 мг) в ацетонитриле (3 мл), выдерживают при 90°С в течение 3 ч. После охлаждения до комнатной температуры реакционную смесь распределяют между водой (6 мл) и толуолом (2×6 мл). Объединенные органические фазы промывают водой (6 мл), сушат над Na2S04 и концентрируют. После экспресс-хроматографии (ДХМ/метанол) получают 246 мг (50%) продукта в виде масла желтоватого цвета.
(в) Нафталин-1-ил-(4-пентилоксинафталин-1-ил)фосфиновой кислота: Раствор метилового эфира нафталин-1-ил-(4-пентилоксинафталин-1-ил)фосфиновой кислоты (156 мг, 0,38 ммоль) в ацетонитриле (1,5 мл) обрабатывают триметилсилилйодидом (0,1 мл, 0,75 ммоль) и перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь распределяют между 1М Na2CO3 (4 мл) и толуолом (4 мл). Водную фазу подкисляют с помощью раствора HCl (1,5 мл) и экстрагируют толуолом (2×4 мл). Объединенные экстракты сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (ДХМ/метанол/NH3) получают 127 мг (83%) продукта в виде бесцветной пены.
Синтез аминов
Метод применим для получения соединений из примеров; 24, 26, 28.
(а) Нафталин-1-ил-(4-пентилоксинафталин-1-ил)амин: Смесь, содержащую 1-йод-4-пентилоксинафталин (1,02 г, 3,0 ммоль), трет-BuONa (0,29 г, 4,2 ммоль), 1-нафтиламин (0,43 г, 3,6 ммоль), 2-(ди-трет-бутилфосфин)бифенил (53,7 мг) и Pd2dba3 (155,3 мг) в толуоле (6 мл), выдерживают в течение 40 мин при 80°С. После охлаждения до комнатной температуры реакционную смесь фильтруют через силикагель и концентрируют. После экспресс-хроматографии (циклогексан/этилацетат) получают 0,85 г (80%) продукта в виде бесцветных кристаллов.
(б) Метилнафталин-1-ил-(4-пентилоксинафталин-1-ил)амин: Раствор нафталин-1-ил-(4-пентилоксинафталин-1-ил)амина (154 мг, 0,40 ммоль) в ДМФ (1,7 мл) обрабатывают NaH (75%, 18 мг, 0,56 ммоль) и метилйодидом (0,13 мл, 2,2 ммоль) и перемешивают при 50°С в течение 18 ч. После охлаждения до комнатной температуры реакционную смесь распределяют между водой (4 мл) и толуолом (2×4 мл). Объединенные органические фазы сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (ДХМ/метанол) получают 70 мг (48%) продукта в виде пены светло-коричневого цвета.
Пример 5: Синтез сульфонамидов
Метод применим для получения соединений из примеров: 11, 13, 22, 25.
(а) Натриевая соль 4-пентилоксинафталин-1-сульфоновой кислоты: Смесь 4-гидроксинафталин-1-сульфоновой кислоты (14,07 г, 40 ммоль), NaOH (3,2 г, 80 ммоль), н-пентилбломида (10 мл, 80 ммоль) и ДМСО (200 мл) перемешивают при 60°С в течение 2 ч. После охлаждения до комнатной температуры реакционную смесь обрабатывают водой (400 мл) и нейтрализуют с помощью 6н. HCl (15 мл). После перемешивания в течение 30 мин при 0°С продукт собирают фильтрацией, промывают водой и сушат в вакууме, получая 12,6 г (100%) бесцветных кристаллов, tпл. 275-285°С.
(б) 1-(4-Пентилоксинафталин-1-сульфонил)-1,2,3,4-тетрагидрохинолин: Смесь натриевой соли 4-пентилоксинафталин-1-сульфоновой кислоты (147 мг, 0,5 ммоль) и ДХМ (3 мл) обрабатывают тионилхлоридом (43 мкл, 0,6 ммоль) и перемешивают при комнатной температуре в течение 30 мин. Образовавшийся прозрачный раствор обрабатывают ДИЭА (86 мкл, 0,5 ммоль) и 1,2,3,4-тетрагидрохинолином (95 мкл, 0,75 ммоль) и перемешивают при комнатной температуре в течение 18 ч. Реакционную смесь распределяют между водой (3 мл) и ДХМ (2×3 мл). Органическую фазу промывают водой (3 мл), сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (толуол) получают 79 мг (39%) продукта в виде бесцветного масла.
Пример 6: Синтез амидов
Метод применим для получения соединений из примеров: 79, 80, 163.
(а) 4-Пентилоксинафталин-1-карбальдегид: Смесь 4-гидроксинафталин-1-карбальдегида (1,72 г, 10 ммоль), NaOH (0,48 г, 12 ммоль), н-пентилбромида (1,5 мл, 12 ммоль) и ДМСО (10 мл) перемешивают при 50°С в течение 4 ч. После охлаждения до комнатной температуры реакционную смесь обрабатывают водой (20 мл) и 2н. HCl (1,5 мл, рН 4). После экстракции толуолом (2×20 мл) объединенные органические фазы промывают водой, сушат над Na2SO4 и концентрируют. После кристаллизации (циклогексан) получают 2,15 г (89%) продукта в виде кристаллов коричневатого цвета, tпл. 67-68°С.
(б) 4-Пентилоксинафталин-1-карбоновая кислота: Раствор, содержащий 4-пентилоксинафталин-1-карбальдегид (1,9 г, 7,8 ммоль) и 2-метил-2-бутен (39 мл) в трет-BuOH (150 мл), обрабатывают раствором NaClO2 (7,05 г, 78 ммоль) и NaH2РО4·Н2О (7,53 г, 55 ммоль) в воде (62 мл). После перемешивания при комнатной температуре в течение 17 ч продукт собирают фильтрацией, промывают водой и сушат в вакууме, получая 1,92 г (95%) кристаллов коричневатого цвета, tпл. 190-202°С.
(в) (3,4-Дигидро-2H-хинолин-1-ил)-(4-пентилоксинафталин-1-ил)метанон: Смесь 4-пентилоксинафталин-1-карбоновой кислоты (103 мг, 0,4 ммоль) и ДХМ (2 мл) обрабатывают тионилхлоридом (34,6 мкл, 0,48 ммоль) и ДМФ (0,2 мл) и перемешивают при 40°С в течение 1 ч. Образовавшийся прозрачный раствор обрабатывают ДИЭА (103 мкл, 0,6 ммоль), 1,2,3,4-тетрагидрохинолином (80 мг, 0,6 ммоль) и ДМАП (4,9 мг, 0,04 ммоль). После выдерживания при температуре дефлегмации 42°С в течение 3 ч реакционную смесь распределяют между 1М КНСО3 (4 мл) и ДХМ (2×4 мл). Объединенные органические фазы промывают водой, сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (циклогексан/ацетон) получают 78 мг (52%) продукта в виде кристаллов зеленоватого цвета.
Пример 7: Синтез сложных эфиров
Метод применим для получения соединений из примеров: 27, 32, 33, 35, 36, 37, 38, 46
Нафталин-1-иловый эфир 4-пентилоксинафталин-1-карбоновой кислоты:
Раствор 4-пентилоксинафталин-1-карбальдегида (121 мг, 0,5 ммоль) в CCl4 (2 мл) обрабатывают трет-BuOCl (8,82М, 170 мкл, 1,5 ммоль) и перемешивают при 50°С в течение 1 ч. После добавления ДИЭА (0,3 мл, 1,7 ммоль) и 1-нафтола (216 мг, 1,5 ммоль) смесь кипятят с обратным холодильником в течение 2 ч и распределяют между 1М КНСО3 (5 мл) и ДХМ (2×5 мл). Объединенные органические фазы сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (циклогексан/ацетон) получают 82 мг (43%) продукта в виде бесцветных кристаллов.
Пример 8: Синтез иминов и аминов
Метод применим для получения соединений из примеров: 30, 34, 42, 43, 44.
(а) Нафталин-1-ил-[1-(4-пентилоксинафталин-1-ил)метилиден]амин: Раствор, содержащий 4-пентилоксинафталин-1-карбальдегид (48,5 мг, 0,2 ммоль), 1-нафтиламин (28,6 мг, 0,2 ммоль) в ДХМ (1 мл), обрабатывают молекулярными ситами с размером частиц 4Å (80 мг) и перемешивают при комнатной температуре в течение 2 ч. Смесь фильтруют через Hyflo, сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (циклогексан/этилацетат) получают 60 мг (82%) продукта в виде кристаллов желтого цвета.
(б) Нафталин-1-ил-(4-пентилоксинафталин-1-илметил)амин: раствор, содержащий нафталин-1-ил-[1-(4-пентилоксинафталин-1-ил)метилиден]амина (24 мг, 0,07 ммоль) и ВН3 пиридин (16,3 мкл, 0,13 ммоль) в ТГФ (0,65 мл), перемешивают при комнатной температуре в течение 16 ч. Реакционную смесь концентрируют и распределяют между водой (2 мл) и ДХМ (2 мл). Органическую фазу сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (циклогексан/ацетон) получают 14 мг (58%) продукта в виде бесцветного масла.
Синтез мочевин
Метод применим для получения соединений из примеров: 39, 40, 41.
1-Нафталин-1-ил-3-(4-пентилоксинафталин-1-ил)мочевина: Раствор, содержащий 4-пентилоксинафталин-1-карбоновую кислоту (103 мг, 0,4 ммоль) и 1,8-бис(диметиламино)нафталин (86 мг, 0,4 ммоль) в ТГФ (0,8 мл), перемешивают при комнатной температуре в течение 30 мин. После добавления ДФФА (86 мкл, 0,4 ммоль) и (1 1-нафтиламина (229 мг, 1,6 ммоль) смесь выдерживают при 100°С в течение 6 ч и распределяют между 2М HCl (8 мл) и ДХМ (2×8 мл). Объединенные органические фазы промывают 1М Na2СО3 и водой, сушат над Na2SO4 и концентрируют. После экспресс-хроматографии (циклогексан/ацетон) получают 78 мг (49%) продукта в виде кристаллов коричневатого цвета.
Пример 10: Синтез бисарилкетонов по методу Фриделя-Крафтса
Метод применим для получения соединений из примеров: 48, 49, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 141, 145, 146.
(4-Фторнафталин-1-ил)-(4-пентилоксинафталин-1-ил)метанон: Раствор 4-фтор-1-нафтойной кислоты (0,5 г, 2,63 ммоль) в безводном ДХМ (10 мл) обрабатывают при перемешивании при комнатной температуре оксалилхлоридом (0,52 г, 4,1 ммоль), после чего добавляют несколько капель безводного ДМФ. После прекращения выделения пузырьков газа прозрачный раствор охлаждают до 4°С в ледяной бане и добавляют в виде одной порции хлорид алюминия (0,7 г, 5,25 ммоль). После перемешивания при 4°С в течение 20 мин добавляют 1-пентилоксинафталин (0,563 г, 2,63 ммоль) и реакционной смеси дают постепенно в течение ночи нагреться до температуры окружающей среды. Реакционную смесь распределяют между этилацетатом (50 мл) и водой (250 мл) и осуществляют экстракцию. Водную фазу дополнительно промывают свежим этилацетатом (2×50 мл). Объединенные органические фазы сушат (безводный MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле с помощью устройства Biotage (фирма Dyax Corp.), применяя в качестве элюента циклогексан:этилацетат (9:1), в результате чего получают требуемый продукт (0,996 г, 98%).
Пример 11: Синтез алкиламинобисарилкетонов
Метод применим для получения соединений из примеров: 60, 61, 64, 120, 126, 127, 133, 134.
(а) 4-(нафталин-1-карбонил)нафталин-1-иловый эфир трифторметансульфоновой кислоты: Трифторметансульфоновый ангидрид (3,1 мл, 18,43 ммоль) медленно добавляют к раствору (4-гидроксинафталин-1-ил)нафталин-1-илметанона (5,0 г, 16,76 ммоль) в пиридине (15 мл) при 0°С в атмосфере инертного газа. Реакционную смесь перемешивают при 0°С в течение 30 мин и затем дают нагреться до температуры окружающей среды в течение 24 ч. После этого реакционную смесь сливают на воду и трижды экстрагируют ДХМ. Объединенные органические экстракты промывают последовательно водой, разбавленным водным раствором HCl, водой и соляным раствором. Органическую фазу сушат над безводным MgSO4 и концентрируют в вакууме. Остаток очищают с помощью экспресс-хроматографии (10% простой эфир/циклогексан), получая требуемый продукт (5,56 г, 77%).
(б) Нафталин-1-ил-(4-бутиламинонафталин-1-ил)метанон: Раствор 4-(нафталин-1-карбонил)нафталин-1-илового эфира трифторметансульфоновой кислоты (308 мг, 0,716 ммоль) и н-бутиламина (62,8 мг, 0,859 ммоль) в безводном толуоле (3 мл) добавляют в атмосфере инертного газа к смеси, содержащей ацетат палладия(II) (3,2 мг, 0,014 ммоль), БИНАФ (10 мг, 0,016 ммоль) и трет-бутоксид натрия (96 мг, 1,002 ммоль). Смесь выдерживают при 80°С в течение 4 ч. После охлаждения смесь разбавляют этилацетатом и фильтруют через фильтр из целита. Фильтрат упаривают в вакууме, получая розовато-коричневый остаток. Его очищают с помощью экспресс-хроматографии (10% простой эфир/циклогексан), получая требуемый продукт (85 мг, 34%) и 30 мг регенерированного исходного продукта.
(в) [4-{Бутил(2-морфолин-4-илэтил)амино}нафталин-1-ил]-нафталин-1-илметанон: Раствор нафталин-1-ил-(4-бутиламинонафталин-1-ил)метанона (65 мг, 0,18 ммоль) в безводном ДМФ (4 мл) в атмосфере инертного газа обрабатывают NaH (60%, 28,8 мг, 0,72 ммоль). Через 20 мин добавляют в виде одной порции гидрохлорид N-(2-хлорэтил)морфолина (37 мг, 0,2 ммоль) и реакционную смесь перемешивают при 80°С в течение 2 ч. После охлаждения до комнатной температуры реакционную смесь распределяют между водой и этилацетатом. Объединенные органические фазы сушат над безводным MgSO4 и концентрируют в вакууме. После экспресс-хроматографии (циклогексан/этилацетат) получают 29 мг (34%) требуемого продукта и 26 мг регенерированного исходного продукта.
(г) 8-(Нафталин-1-карбонил)-5-пентилоксинафталин-2-иловый эфир трифторметансульфоновой кислоты: Раствор (7-гидрокси-4-пентилоксинафталин-1-ил) нафталин-1-илметанона (1,2 г, 3,13 ммоль) в безводном пиридине (12 мл) обрабатывают при перемешивании при комнатной температуре трифторметансульфоновым ангидридом (0,88 г, 3,13 ммоль) и смесь перемешивают в атмосфере азота в течение 48 ч. Растворитель удаляют при пониженном давлении и остаток разбавляют раствором бикарбоната натрия и дважды экстрагируют этилацетатом. Объединенные органические экстракты промывают водой, сушат (MgSO4) и удаляют растворитель при пониженном давлении. Остаток очищают с помощью экспресс-хроматографии на силикагеле (циклогексан:этилацетат 9:1), получая требуемый продукт (1,0 г, 67%).
(е) [7-(4-Метилпиперазин-1-ил)-4-пентилоксинафталин-1-ил]нафталин-1-илметанон: Смесь, содержащую 8-(нафталин-1-карбонил)-5-пентилоксинафталин-2-иловый эфир трифторметансульфоновой кислоты (40 мг, 0,084 ммоль), N-метилпиперазин (20 мг, 0,2 ммоль), карбонат цезия (38 мг, 0,12 ммоль), ацетат палладия(II) (2 мг, 10 мол.%) и БИНАФ (8 мг, 15 мол.%) в безводном диоксане (0,5 мл), при перемешивании выдерживают при 80°С в атмосфере аргона в течение 30 ч. Смесь охлаждают до комнатной температуры, разбавляют водой и трижды экстрагируют этилацетатом. Объединенные органические экстракты промывают водой, сушат (MgSO4) и растворитель удаляют при пониженном давлении. Остаток очищают с помощью ЖХВР. Все фракции, содержащие продукт, подщелачивают с помощью бикарбоната натрия и экстрагируют этилацетатом. Органические экстракты объединяют, сушат (MgSO4) и упаривают, получая продукт в виде свободного основания (12 мг, 31%).
Пример 12: Синтез замешенных бисарилкетонов
Метод применим для получения соединений из примеров: 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 62, 63, 65, 66, 67, 68, 69, 70, 71, 74, 76, 77, 78.
(а) 8-(Нафталин-1-карбонил)-5-пентилоксинафталин-2-карбонитрил: Смесь, содержащую 8-(нафталин-1-карбонил)-5-пентилоксинафталин-2-иловый эфир трифтор метансульфоновой кислоты (1,0 г, 2,09 ммоль), цианид цинка (0,294 г, 2,51 ммоль) и Pd(PPh3)4 (0,121 мг, 0,1 ммоль, 5 мол.%) в безводном ДМФ (10 мл), выдерживают при перемешивании в атмосфере аргона при 90°С в течение 3 ч. Смесь охлаждают до комнатной температуры, разбавляют водой, трижды экстрагируют этилацетатом и отфильтровывают нерастворившийся продукт через фильтр из целита. Объединенные органические экстракты промывают водой, сушат (MgSO4) и растворитель удаляют при пониженном давлении. Остаток очищают с помощью хроматографии на силикагеле (циклогексан:этилацетат 9:1), получая требуемый продукт (0,53 г, 65%).
(б) Диаллил-(4-бром-3-фторфенил)амин: Смесь, содержащую 4-бром-3-фторанилин (17,47 г, 91,9 ммоль), аалилбромид (23,72 г, 251,1 ммоль) и карбонат калия (26,7 г, 193,5 ммоль) в ацетоне (200 мл), при перемешивании кипятят с обратным холодильником в течение 24 ч. Растворитель удаляют при пониженном давлении и остаток разбавляют водой и дважды экстрагируют этилацетатом. Объединенные органические экстракты промывают водой, сушат (MgSO4) и упаривают в вакууме. Остаток очищают с помощью хроматографии на силикагеле (циклогексан), получая требуемый продукт (15,27 г, 62%).
(в) Диаллил-(11-оксатрицикло[6.2.1.0_2,7]ундека-2,4,6,9-тетраен-4-ил)амин: Раствор диаллил-(4-бром-3-фторфенил)амина (15,55 г, 57,6 ммоль) в безводном простом эфире (30 мл) и безводном фуране (30 мл) обрабатывают при перемешивании раствором н-бутиллития в гексане (36 мл, 57,6 ммоль; 1,6М раствор) при -70°С в атмосфере аргона. Через 1 ч смеси дают нагреться до комнатной температуры и перемешивают еще в течение 4 ч. Реакцию прекращают добавлением воды и трижды экстрагируют этилацетатом. Объединенные органические экстракты промывают соляным раствором, сушат (MgSO4) и растворитель удаляют при пониженном давлении. Остаток очищают с помощью хроматографии на силикагеле (исходный элюент циклогексан, конечный элюент циклогексан:этилацетат 19:1), получая требуемый продукт (5,4 г, 39%).
(г) 7-Диаллиламинонафталин-1-ол: Раствор диаллил-(11-оксатрицикло [6.2.1.0_2,7]ундека-2,4,6,9-тетраен-4-ил)амин (4,48 г, 18,74 ммоль) в метаноле (45 мл) и концентрированной соляной кислоте (4,5 мл) при перемешивании кипятят с обратным холодильником в течение 5 ч. Растворитель удаляют при пониженном давлении и остаток разбавляют водой, нейтрализуют твердым бикарбонатом натрия и трижды экстрагируют этилацетатом. Объединенные экстракты промывают соляным раствором, сушат (MgSO4) и растворитель удаляют при пониженном давлении. Остаток очищают с помощью хроматографии на силикагеле (исходный элюент циклогексан, конечный элюент циклогексан:этилацетат 19:1), получая требуемый продукт (3,67 г, 82%).
(д) Диаллил-(8-пентилоксинафталин-2-ил)амин: К раствору, содержащему н-пентанол (0,18 г, 2,1 ммоль) и трифенилфосфин (0,55 г, 2,1 ммоль) в безводном ТТФ (10 мл), добавляют при перемешивании раствор 7-диаллиламинонафталин-1-ола (0,5 г, 2,1 ммоль) и ДИАД (0,45 мл, 2,1 ммоль) в безводном ТГФ (10 мл). После перемешивания в течение ночи смесь разбавляют соляным раствором и трижды экстрагируют этилацетатом. Объединенные органические экстракты промывают соляным раствором, сушат (MgSO4) и упаривают досуха. Остаток очищают с помощью хроматографии на силикагеле (исходный элюент циклогексан, конечный элюент циклогексан:этилацетат 98:2), получая требуемый продукт (0,28 г, 43%).
(е) (6-Диаллиламино-4-пентилоксинафталин-1-ил)нафталин-1-илметанон; К суспензии, содержащей безводный хлорид алюминия (0,24 г, 1,81 ммоль) в безводном ДХМ (30 мл), при перемешивании добавляют нафтоилхлорид (0,205 мл, 1,36 ммоль) при 0°С в атмосфере азота. Через 15 мин по каплям добавляют раствор диаллил-(8-пентилоксинафталин-2-ил)амина (0,28 г, 0,906 ммоль) в безводном ДХМ (5 мл) и смеси дают нагреться до комнатной температуры и перемешивают в атмосфере азота в течение ночи. Смесь промывают насыщенным раствором бикарбоната натрия (рН 8) и водную фазу дополнительно трижды экстрагируют диэтиловым эфиром. Органические фазы объединяют, промывают водой, сушат (MgSO4) и растворитель удаляют при пониженном давлении. Остаток очищают с помощью хроматографии на силикагеле (исходный элюент циклогексан, конечный элюент циклогексан:этилацетат 98:2), получая требуемый продукт (0,32 г, 75%).
(ж) 5-Пентилоксинафталин-2-ол: Смесь, содержащую нафталин-1,6-диол (10,0 г, 62,5 ммоль), 1-бромпентан (7,75 мл, 62,5 ммоль) и гидроксид натрия (2,5 г, 62,5 ммоль) в ДМСО (100 мл), выдерживают при 100°С в течение 6 ч. После охлаждения до комнатной температуры смесь разбавляют водой и трижды экстрагируют этилацетат. Объединенные органические экстракты промывают несколько раз водой, сушат (MgSO4) и растворитель удаляют при пониженном давлении. Остаток очищают с помощью хроматографии на силикагеле (исходный элюент циклогексан:этилацетат 97:3, конечный элюент циклогексан:этилацетат 90:10), получая неразделенную смесь требуемого продукта и изомера 6-пентилоксинафталин-1-ола (6,18 г, 43%), причем сначала элюируется дважды алкилированный продукт, 1,6-бис(пентилокси)нафталин.
(з) 5-пентилоксинафталин-2-иловый эфир уксусной кислоты: Раствор, содержащий смесь 5-пентилоксинафталин-2-ол/6-пентилоксинафталин-1-ол (6,18 г, 26,8 ммоль) в ДХМ (100 мл), при перемешивании в присутствии NEt3 (4,4 мл, 31,6 ммоль) обрабатывают по каплям при 0°С раствором ацетилхлорида (2,24 мл, 31,5 ммоль) в ДХМ (30 мл). После нагрева до комнатной температуры и перемешивания в течение 3 ч реакционную смесь промывают соляным раствором, сушат (MgSO4) и растворитель удаляют при пониженном давлении. Остаток очищают с помощью хроматографии на силикагеле (1,5% этилацетат в циклогексане), получая требуемый продукт (4,56 г, 56%) и изомер 6-пентилоксинафталин-1-илового эфира уксусной кислоты (1,0 г, 13%).
(и) 8-(нафталин-1-карбонил)-5-пентилоксинафталин-2-иловый эфир уксусной кислоты: К суспензии безводного хлорида алюминия (4,42 г, 33,09 ммоль) в безводном ДХМ (290 мл) при перемешивании при 0°С в атмосфере азота добавляют по каплям раствор нафтоилхлорида (3,7 мл, 24,8 ммоль) в безводном ДХМ (35 мл). Через 15 мин добавляют раствор 5-пентилоксинафталин-2-илового эфира уксусной кислоты (4,5 г, 16,54 ммоль) в безводном ДХМ (70 мл) и реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 20 ч. Смесь промывают насыщенным раствором бикарбоната натрия, разделяют фазы и удаляют ДХМ при пониженном давлении. Остаток растворяют в диэтиловом эфире и промывают водой, сушат (MgSO4) и растворитель удаляют при пониженном давлении. После очистки с помощью хроматографии на силикагеле (1-3% этилацетат в циклогексане) получают требуемый продукт (4,8 г, 68%) и 8-(нафталин-1-карбонил)-5-пентилоксинафталин-2-иловый эфир нафталин-1 -карбо новой кислоты (2,1 г), который образуется вследствие замещения ацетильной группы нафтоилом.
(л) (7-Гидрокси-4-пентилоксинафталин-1-ил)нафталин-1-илметанон: Раствор, содержащий 8-(нафталин-1-карбонил)-5-пентилоксинафталин-2-иловый эфир уксусной кислоты (4,8 г, 11,2 ммоль) и 8-(нафталин-1-карбонил)-5-пентилоксинафталин-2-иловый эфир нафталин-1-карбоновой кислоты (2,1 г, 3,9 ммоль) в метаноле (70 мл), кипятят с обратным холодильником при перемешивании в присутствии 5М раствора NaOH (20 мл) в течение 3 ч. После охлаждения до комнатной температуры смесь разбавляют водой, подкисляют с помощью уксусной кислоты и трижды экстрагируют этилацетатом. Объединенные экстракты промывают водой, сушат (MgSO4) и растворитель удаляют при пониженном давлении. Остаток перекристаллизовывают из этилацетата, получая требуемый продукт (3,7 г, 64%) в виде твердого вещества ярко-желтого цвета.
Пример 13: Синтез арил-гетероарильных кетонов
Метод применим для получения соединений из примеров: 45, 92, 93.
(а) Изохинолин-1-ил-(4-пентилоксинафталин-1-ил)метанон: К раствору 1-йод-4-пентилоксинафталина (419 мг, 1,232 ммоль) в ТГФ (8 мл) при -78°С (баня ацетон/сухой лед) добавляют по каплям н-BuLi (0,99 мл, 2,5М в гексане). Через несколько минут образуется осадок желтого цвета. После перемешивания в течение 30 мин добавляют по каплям с помощью шприца раствор изохинолин-1-карбонитрила (210 мг, 1,364 ммоль) в ТГФ (2 мл), получая раствор красного цвета. Реакционную смесь удаляют из охлаждающей бани и дают нагреться до температуры выше 3 г. Образуется раствор ярко-синего цвета. После этого добавляют разбавленную серную кислоту (2,5 мл, 10 об.%) и смесь перемешивают при комнатной температуре в течение 45 мин. Затем реакционную смесь разбавляют этилацетатом и раствор промывают насыщенным водным раствором бикарбоната натрия до щелочного значения рН (индикаторная бумага), водным раствором тиосульфата натрия (×2) и соляным раствором; сушат над безводным Na2SO4 и концентрируют на роторном испарителе. Неочищенный продукт хроматографируют на силикагеле (градиент элюирования: циклогексан/этилацетат 9/1, затем 5/1, затем 2/1), получая указанное в заголовке соединение в виде вязкого масла ярко-желтого цвета (270 мг, 59%).
(б) 4-Пентилокси-1-нафталинбороновой кислоты: К охлажденному (баня: сухой лед/ацетон) раствору 1-йод-4-пентилоксинафталина (0,993 г, 2,92 ммоль) в ТГФ (10 мл) в атмосфере безводного аргона добавляют по каплям с помощью шприца н-BuLi (2,5М в гексане, 2,4 мл, 6,0 ммоль). Реакционная смесь приобретает темно-желтый цвет, и образуется осадок. После выдерживания в течение 0,5 ч при температуре охлаждающей бани по каплям с помощью шприца добавляют триметилборат (0,66 мл, 5,8 ммоль). Реакционную колбу удаляют из охлаждающей бани, и продукт желтого цвета быстро в течение нескольких минут становится бесцветным. Через 1,5 ч добавляют серную кислоту (20 об.%, 3 мл) и образовавшуюся суспензию распределяют между этилацетатом и водой. Органический слой промывают водным раствором тиосульфата натрия (×2) и соляным раствором, сушат (над безводным Na2SO4) и упаривают на роторном испарителе. Остаток растворяют в минимальном объеме ДХМ и вносят в силикагелевую колонку, которую элюируют циклогексаном/этилацетатом (1/1), получая бороновую кислоту (267 мг, 35%).
(в) (4-Пентилоксинафталин-1-ил)хинолин-8-илметанон: В трехгорлую высушенную с помощью пламени колбу, снабженную впускным устройством для газа и мембраной, вносят 8-гидроксихинолинтрифторметансульфонат (122,8 мг, 0,442 ммоль), 4-пентилокси-1-нафталинбороновую кислоту (124,5 мг, 0,482 ммоль), безводный карбонат калия (199,7 мг, 1,447 ммоль), комплекс PdCl2dppf·CH2Cl2 (10,5 мг, 0,0128 ммоль, фирма Avocado) и йодид натрия (150 мг). Реакционную колбу подвергают вакуумированию (неглубокий вакуум) и прочищают монооксидом углерода из баллона (3 цикла). С помощью шприца добавляют 3 мл анизола и после перемешивания реакционную смесь оранжевого цвета помещают в предварительно нагретую до 80°С масляную баню. Через 3 ч добавляют дополнительную порцию анизола (1 мл) и реакционную смесь перемешивают в течение ночи при 80°С. Реакционной смеси, которая приобретает черный цвет, дают нагреться до комнатной температуры и разбавляют этилацетатом и водой. Органический слой промывают соляным раствором (×2), сушат над безводным Na2SO4 и концентрируют на роторном испарителе. Неочищенный продукт хроматографируют на силикагеле (циклогексан/этилацетат, 5/1), получая указанное в заголовке соединение в виде масла зеленого цвета (52 мг, 32%).
Пример 14: Синтез бензимидазолонов. бензимидазолов и бензотриазолов
Метод применим для получения соединений из примеров: 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 161, 162.
(а) N-(2-Пентилокси-фенил)ацетамид: 2-Ацетамидофенол (5 г, 33,09 ммоль) растворяют в безводном ДМФ (35 мл) при комнатной температуре. Добавляют карбонат цезия (17,25 г, 52,53 ммоль), а затем 1-бромпентан (6,15 мл, 49,61 ммоль) и смесь перемешивают при 60°С в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют водой (400 мл) и экстрагируют этилацетатом (3×100 мл). Этилацетатные экстракты объединяют, промывают насыщенным соляным раствором, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая продукт с достаточной степенью чистоты (6,02 г, 82%).
(б) N-[5-(Нафталин-1-карбонил)-2-пентилоксифенил]ацетамид: В сухой колбе, продутой безводным азотом, суспендируют хлорид алюминия (5,45 г, 40,86 ммоль) в безводном 1,2-дихлорэтане (50 мл). Суспензию охлаждают с помощью бани лед/вода, после чего добавляют в виде одной порции раствор 1-наффтоилхлорида (4,51 мл, 29, 96 ммоль) в безводном 1,2-дихлорэтане (10 мл). Через 10 мин добавляют N-(2-пентилоксифенил)ацетамид (6,02 г, 27,24 ммоль) и реакционной смеси дают нагреться до комнатной температуры в течение ночи. Смесь сливают на смесь, содержащую лед-воду и 5М водный раствор гидроксида натрия (в количестве, достаточном для подщелачивания водного слоя), перемешивают в течение 15 мин и экстрагируют этилацетатом (4×100 мл). Органические экстракты объединяют и промывают насыщенным соляным раствором (100 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью хроматографии на силикагеле, используя устройство типа Biotage (колонка 90 г; фирма Dyax Corp.) и циклогексан:этилацетат (2:1) в качестве элюента, получая продукт в виде густого масла (3,68 г, 36%). Получают также еще 5,64 г содержащего небольшое количество примесей продукта, который обладает достаточной чистотой для использования в последующих реакциях.
(в) (3-Амино-4-пентилоксифенил)-нафталин-1-илметанон: N-[5-(нафталин-1-карбонил)-2-пентилоксифенил]ацетамид (1,78 г, 4,75 ммоль) растворяют в метаноле (20 мл) при комнатной температуре. Добавляют соляную кислоту (10М, 20 мл) и смесь выдерживают при температуре дефлегмации в течение 1 ч. Реакционную смесь упаривают досуха при пониженном давлении, распределяют между насыщенным водным раствором бикарбоната натрия и этилацетатом и экстрагируют еще одной порцией этилацетата (3×100 мл). Этилацетатные экстракты объединяют, промывают насыщенным соляным раствором, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт в виде невязкого масла коричневого цвета. Его очищают на силикагеле, используя устройство типа Biotage (90 г колонка) и циклогексан:этилацетат (4:1) в качестве элюента, в результате чего получают чистый продукт (0,97 г, 61%).
(г) 3-[5-(Нафталин-1-карбонил)-2-пентилоксифенил]-1-метоксимочевина: ди-трет-бутилдикарбонат (1,833 г, 8,4 ммоль) растворяют в безводном ДХМ (20 мл) при комнатной температуре и добавляют ДМАП (0,733 г, 6 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 5 мин, после чего добавляют раствор (3-амино-4-пентилоксифенил)нафталин-1-илметанона (2,0 г, 6 ммоль) в безводном ДХМ (10 мл). Смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют ДИЭА (1,045 мл, 6 ммоль) и гидрохлорид метоксиламина (0,501 г, 6 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 4 ч. Реакционную смесь обрабатывают водой (200 мл) и экстрагируют ДХМ (3×75 мл). ДХМ-экстракты объединяют, промывают насыщенным соляным раствором, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт. Этот продукт хроматографируют на силикагеле, используя устройство типа Biotage (с картриджем 40 г) и циклогексан:этилацетат (4:1) в качестве элюента, в результате чего получают 1,25 г требуемого продукта наряду с 0,66 г продукта, содержащего дополнительную трет-бутоксикарбонильную группу. Продукт растворяют в смеси ДХМ-трифторуксусная кислота (1:1, 6 мл) и перемешивают при комнатной температуре в течение 2 ч. Летучие вещества удаляют при пониженном давлении и остаток распределяют между ДХМ (20 мл) и насыщенным водным раствором бикарбоната натрия (50 мл). Эту смесь экстрагируют еще одной порцией ДХМ (3х50 мл) и ДХМ-экстракты объединяют, промывают насыщенным соляным раствором, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая требуемый продукт (1,88 г, 77%).
(д) 1-Метокси-7-(нафталин-1-карбонил)-4-пентилокси-1,3-дигидробензимидазол-2-он: 3-[5-(нафталин-1-карбонил)-2-пентилоксифенил]-1-метоксимочевина (650 мг, 1,6 ммоль) растворяют в безводном ДХМ (60 мл) в атмосфере азота, раствор охлаждают до 0°С и порциями добавляют бис(трифторацетокси)йодбензол (757 мг, 1,76 ммоль). Реакционной смеси дают нагреться до комнатной температуры в течение 1,5 ч, после чего добавляют воду (200 мл). Смесь экстрагируют ДХМ (3×100 мл) и ДХМ-экстракты объединяют, промывают насыщенным соляным раствором (100 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт в виде масла коричневого цвета. Продукт хроматографируют на силикагеле, используя устройство типа Biotage (с картриджем 40 г) и циклогексан:этилацетат (3:1) в качестве элюента, в результате чего получают требуемый продукт (0,34 г, 53%).
(е) 4-(Нафталин-1-карбонил)-7-пентилокси-1,3-дигидробензимидазол-2-онтрифторацетат: 1-Метокси-7-(нафталин-1-карбонил)-4-пентилокси-1,3-дигидробензимидазол-2-он (800 мг, 1,98 ммоль) растворяют в ледяной уксусной кислоте (10 мл) и добавляют порошкообразный цинк (5,18 г, 79,22 ммоль). Смесь выдерживают при 50°С и обучают ультразвуком в течение 2 ч, охлаждают до комнатной температуры и фильтруют через подушку из целита. Подушку из целита промывают этилацетатом и затем удаляют летучие компоненты при пониженном давлении, получая продукт в виде масла оранжевого цвета. После очистки с использованием устройства типа Biotage (с картриджем 40 г) и смеси ДХМ:метанол (20:1) в качестве элюента получают требуемый продукт (0,67 г, 90%).
(ж) (2-Хлор-7-пентилокси-3H-бензимидазол-4-ил)нафталин-1-илметанон: 4-(Нафталин-1-карбонил)-7-пентилокси-1,3-дигидробензимидазол-2-он (500 мг, 1,34 ммоль) растворяют в оксихлориде фосфора (10 мл) и кипятят с обратным холодильником (температура масляной бани 105°С) в течение 30 мин. Реакционную смесь охлаждают до комнатной температуры, сливают на охлажденный на льду 2М водный раствор гидроксида натрия для нейтрализации смеси и экстрагируют ДХМ (3×100 мл). Объединенные ДХМ-экстракты промывают насыщенным водным раствором бикарбоната натрия (3×50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт (500 мг, 95%), который используют непосредственно без дополнительной очистки.
(з) Трифторацетат(2-бензиламино-7-пентилокси-3H-бензимидазол-4-ил)нафталин-1-илметанона: (2-Хлор-7-пентилокси-3H-бензимидазол-4-ил)нафталин-1-илметанон (55 мг, 0,14 ммоль) и бензиламин (1 мл), не содержащие примесей, выдерживают вместе при 135°С в течение 4 ч и затем охлаждают до комнатной температуры. Неочищенную реакционную смесь сливают на воду (10 мл), добавляют 10%-ную водную соляную кислоту (10 мл) и смесь экстрагируют ДХМ (4×20 мл). ДХМ-экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт, который очищают с помощью препаративной хроматографии с обращенной фазой (колонка типа Dynamax 300A С 18; градиент от 20% ацетонитрила в воде (+0,1% трифторуксусной кислоты) до 100% ацетонитрила в течение 30 мин), получая 15,4 мг требуемого продукта.
(и) 2,3-Бисацетиламинофениловый эфир уксусной кислоты: 2,3-Диаминофенол (3,226 г, 25,99 ммоль) растворяют в уксусном ангидриде (50 мл) и смесь выдерживают при 70°С в течение 4 ч. Смесь охлаждают до комнатной температуры и дают выстояться в течение 48 ч. Образовавшийся осадок собирают фильтрацией, промывают этилацетатом и сушат в вакууме, получая твердое вещество белого цвета (3,99 г, 61%).
(л) N-(2-ацетиламино-6-гидроксифенил)ацетамид (CAS регистрационный №116345-46-1): 2,3-Бисацетиламинаминофениловый эфир уксусной кислоты (3,99 г, 15,96 ммоль) растворяют в безводном метаноле (50 мл) в атмосфере азота. Добавляют раствор метоксида натрия (полученного из металлического натрия (0,404 г, 17,56 ммоль) в безводном метаноле (10 мл)) и смесь перемешивают при комнатной температуре в течение 16 ч. Растворитель удаляют при пониженном давлении и к остатку добавляют воду, затем подкисляют до рН 1 с помощью 1М соляной кислоты. Водный слой концентрируют при пониженном давлении для осаждения продукта, который выделяют фильтрацией и сушат в вакууме, получая твердое вещество белого цвета (1,96 г, 59%).
(м) N-(2-ацетиламино-6-пентилоксифенил)ацетамид: N-(2-ацетиламино-6-гидроксифенил)ацетамид (1,46 г, 7,02 ммоль) растворяют в безводном ДМФ (50 мл) при комнатной температуре. Добавляют карбонат цезия (2,97 г, 9,13 ммоль) и 1-бромпентан (1,04 мл, 8,42 ммоль) и смесь выдерживают при 60°С в течение 20 ч и затем перемешивают при комнатной температуре в течение 4 дней. Добавляют воду (800 мл) и раствор экстрагируют ДХМ (4×100 мл). ДХМ-экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая 1,95 г неочищенного продукта. Его перекристаллизовывают из этилацетата/циклогексана, получая чистый продукт (0,9 г, 46%).
(н) N-(2-Ацетиламино-3-йод-6-пентилоксифенил)ацетамид: К раствору N-(2-ацетиламино-6-пентилоксифенил)ацетамида (0,9 г, 3,24 ммоль) и гидрата перйодной кислоты (129 мг, 0,57 ммоль) в смеси уксусная кислота:вода:серная кислота (100:20:3; 10 мл) добавляют йод (332 мг, 1,31 ммоль). Смесь перемешивают при комнатной температуре в течение 16 ч, разбавляют 10%-ным водным раствором тиосульфата натрия (100 мл) и затем экстрагируют ДХМ (1×100 мл), этилацетатом (1×100 мл) и диэтиловым эфиром (1×100 мл). Органические экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт, который перекристаллизовывают из этилацетата, получая чистый продукт (550 мг, 42%).
(о) 7-Йод-2-метил-4-пентилокси-1H-бензимидазол: N-(2-Ацетиламино-3-йод-6-пентилоксифенил)ацетамид (100 мг, 0,248 ммоль) добавляют к раствору гидроксида калия (139 мг, 2,48 ммоль) в этаноле (5 мл) и воде (1 мл). Смесь выдерживают при температуре дефлегмации в течение 3 ч, дают выстояться в течение 2 дней и затем выдерживают при температуре дефлегмации еще в течение 6 ч, после чего дают выстояться при комнатной температуре в течение 8 дней. Летучие компоненты удаляют при пониженном давлении и остаток распределяют между этилацетатом (10 мл) и водой (10 мл) и экстрагируют еще одной аликвотой этилацетата (3×10 мл). Этилацетатные экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт. После очистки с помощью хроматографии на силикагеле (Biotage, картридж 40 г) с использованием циклогексана:этилацетата (3:1) в качестве элюента получают указанное в заголовке соединение (35,5 мг, 42%).
(п) (2-Метил-7-пентилокси-3H-бензимидазол-4-ил)нафталин-1-илметанон: 7-Йод-2-метил-4-пентилокси-1H-бензимидазол (35 мг, 0,102 ммоль), безводный карбонат калия (42 мг, 0,306 ммоль), 1-нафталинбороновую кислоту (19 мг, 0,112 ммоль) и PdCl2dppf·СН1Cl2 (3 мг, 0,003 ммоль) смешивают в безводном анизоле (5 мл) и помещают в атмосферу монооксида углерода. Смесь выдерживают при 80°С в течение 20 ч, охлаждают до комнатной температуры и разбавляют водой (10 мл). Смесь экстрагируют ДХМ (2×10 мл) и этилацетатом (3×10 мл), органические экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт. Его очищают с помощью препаративной ЖХВР с обращенной фазой (колонка типа Dynamax 300Å С 18 от 20% ацетонитрила в воде (+0,1% уксусной кислоты) до 100% ацетонитрила в течение 30 мин), получая 12,7 мг требуемого продукта.
(р) N-[2-(Ацетилокси)-6-нитрофенил]ацетамид (CAS регистрационный №69194-51-0); 2-Амино-3-нитрофенол (3 г, 19,46 ммоль) растворяют в уксусном ангидриде (20 мл) и смесь выдерживают при 50°С в течение 16 ч. После охлаждения до комнатной температуры добавляют воду (400 мл) и смесь экстрагируют ДХМ (3×100 мл). ДХМ-экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая указанное в заголовке соединение (4,14 г, 89%).
(с) N-(2-гидрокси-6-нитрофенил)ацетамид (CAS Reg. No. 59820-29-0): N-[2-(Ацетилокси)-6-нитрофенил]ацетамид (4,13 г, 17,35 ммоль) растворяют в безводном метаноле (20 мл) и добавляют свежий раствор метоксида натрия (полученный из натрия (0,6 г, 26,03 ммоль) в безводном метаноле (15 мл)). Реакционную смесь перемешивают при 50°С в течение 2 ч, охлаждают до комнатной температуры и метанол удаляют при пониженном давлении. Добавляют воду (100 мл), значение рН доводят до рН 1 с помощью 2М водного раствора соляной кислоты и раствор экстрагируют этилацетатом (3×100 мл). Этилацетатные экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и дают выстояться при комнатной температуре в течение 7 дней. Образовавшиеся кристаллы собирают фильтрацией и сушат, получая чистый продукт (1,5 г, 44%). Маточный раствор концентрируют при пониженном давлении, получая дополнительную порцию неочищенного продукта (2,3 г), который обладает достаточной чистотой для использования в последующих реакциях.
(т) N-(2-Нитро-6-пентилоксифенил)ацетамид: N-(2-гидрокси-6-нитрофенил)ацетамид (3,8 г, 19,39 ммоль) растворяют в безводном ДМФ (25 мл). Добавляют карбонат цезия (8,83 г, 27,1 ммоль) и 1-бромпентан (23,26 ммоль) и смесь перемешивают при 80°С в течение 2 ч. После охлаждения до комнатной температуры добавляют воду (400 мл) и смесь экстрагируют этилацетатом (3×100 мл). Этилацетатные экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт. Продукт перекристаллизовывают из этилацетата/н-гексана при 4°С, получая указанное в заголовке соединение (1,74 г, 34%). Маточный раствор концентрируют при пониженном давлении, получая дополнительную порцию неочищенного продукта, который очищают с помощью хроматографии на силикагеле (Biotage, картридж 40 г), используя смесь ДХМ:метанол (50:1) в качестве элюента. В результате получают еще 0,79 г (15%) указанного в заголовке соединения и 0,31 г (7%) деацетилированного продукта, 2-нитро-6-пентилоксифениламина.
(у) 2-Нитро-6-пентилоксифениламин: N-(2-Нитро-6-пентилоксифенил)ацетамид (1,74 г, 6,53 ммоль) растворяют в метаноле (50 мл) и добавляют 10М соляную кислоту (25 мл). Смесь выдерживают при температуре дефлегмации в течение 4 ч, охлаждают до комнатной температуры и удаляют метанол при пониженном давлении. Значение рН остатка доводят до рН 12 с помощью 5М водного гидроксида натрия и экстрагируют этилацетатом (3×100 мл). Этилацетатные экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая чистый продукт (1,46 г, 100%).
(ф) 3-Пентилоксибензол-1,2-диамин: 2-Нитро-6-пентилоксифениламин (1,46 г, 6,52 ммоль) растворяют в этилацетате (20 мл) и колбу продувают азотом. Добавляют 10%-ный палладий на активированном угле (50 мг) и реакционную смесь вакуумируют и трижды продувают водородом. Смесь перемешивают в течение 24 ч в атмосфере водорода с использованием баллона с газообразным водородом. Добавляют метанол (20 мл) для облегчения солюбилизации и реакционную смесь перемешивают еще в течение 2 ч при комнатной температуре. Реакционную смесь продувают азотом, фильтруют через подушку из целита и концентрируют при пониженном давлении, получая твердое вещество белого цвета, из которого можно путем перекристаллизации из этилацетата/метанола получить указанное в заголовке соединение (1,007 г, 80%).
(х) 7-Пентилокси-1H-бензимидазол: 3-Пентилоксибензол-1,2-диамин (200 мг, 1,03 ммоль) и триметилортоформиат (2 мл) смешивают в пробирке из стекла типа пирекс и подвергают микроволновому облучению мощностью 100 Вт в течение 30 с в лабораторной микроволновой установке типа Labwell MW10. После удаления летучих компонентов при пониженном давлении получают чистый продукт в виде твердого вещества кремового цвета (217 мг, 100%).
(ц) 4-Йод-7-пентилокси-1H-бензимидазол: 7-Пентилокси-1H-бензимидазол (100 мг, 0,49 ммоль) растворяют в смеси уксусная кислота:вода: серная кислота (100:20:3; 5 мл) и добавляют гидрат перйодной кислоты (22 мг, 0,098 ммоль), а затем йод (50 мг, 0,196 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 4 ч и при 80°С в течение 16 ч. После охлаждения до комнатной температуры добавляют 10%-ный водный раствор тиосульфата натрия (100 мл) и смесь экстрагируют этилацетатом (3×25 мл). Этилацетатные экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт. Продукт очищают с помощью хроматографии на силикагеле (Biotage, картридж 40 г), получая указанное в заголовке соединение (65 мг, 40%).
(ч) Нафталин-1-ил-(7-пентилокси-3H-бензимидазол-4-ил)метанон: 4-Йод-7-пентилокси-1H-бензимидазол (65 мг, 0,197 ммоль), безводный карбонат калия (82 мг, 0,591 ммоль), 1-нафталинбороновую кислоту (37 мг, 0,217 ммоль) и PdCl2dppf·CH2Cl2 (9 мг, 0,011 ммоль) смешивают в безводном анизоле (5 мл) и помещают в атмосферу монооксида углерода. Смесь выдерживают при 80°С в течение 18 ч, охлаждают до комнатной температуры и разбавляют водой (10 мл). Смесь экстрагируют ДХМ (2×10 мл) и этилацетатом (3×10 мл) и органические экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт. Продукт очищают с помощью хроматографии на силикагеле (Biotage, картридж 40 г), используя в качестве элюента смесь циклогексан:этилацетат (3:1), в результате чего получают указанное в заголовке соединение (15 мг, 21%).
(ш) 7-Пентилокси-1H-бензотриазол: 3-Пентилоксибензол-1,2-диамин (100 мг, 0,516 ммоль) растворяют в ледяной уксусной кислоте (5 мл) и воде (5 мл). Реакционную смесь охлаждают до 0°С и добавляют в виде одной порции холодный раствор нитрита натрия (39 мг, 0,568 ммоль) в воде (5 мл). Реакционной смеси в течение ночи дают медленно нагреться до комнатной температуры, разбавляют водой (20 мл) и экстрагируют ДХМ (3×50 мл). ДХМ-экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая указанное в заголовке соединение (90 мг, 85%), которое можно использовать без дополнительной очистки.
(щ) 4-Йод-7-пентилокси-1H-бензотриазол: 7-Пентилокси-1H-бензотриазол (90 мг, 0,439 ммоль) растворяют в смеси уксусная кислота:вода:серная кислота (100:20:3; 10 мл) и добавляют гидрат перйодной кислоты (20 мг, 0,088 ммоль), а затем йод (45 мг, 0,176 ммоль). Реакционную смесь перемешивают при 80°С в течение 5 ч. После охлаждения до комнатной температуры добавляют 10%-ный водный раствор тиосульфата натрия (10 мл) и смесь экстрагируют этилацетатом (3×25 мл). Этилацетатные экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт. Продукт очищают с помощью хроматографии на силикагеле (Biotage, картридж 40 г), получая указанное в заголовке соединение (67 мг, 46%), а также 124 мг в основном дийодированного продукта (4,6-дийод-7-пентилокси- 1H-бензотриазол), являющегося примесью к указанному в заголовке соединению.
(э) Нафталин-1-ил-(7-пентилокси-3H-бензотриазол-4-ил)метанон: 4-Йод-7-пентилокси-1H-бензотриазол (67 мг, 0,202 ммоль), безводный карбонат калия (84 мг, 0,607 ммоль), 1-нафталинбороновую кислоту (38 мг, 0,223 ммоль) и PdCl2dppf·СН2Cl2 (17 мг, 0,02 ммоль) смешивают в безводном анизоле (5 мл) и помещают в атмосферу моноксида углерода. Смесь выдерживают при 80°С в течение 20 ч, охлаждают до комнатной температуры и разбавляют водой (20 мл). Смесь экстрагируют ДХМ (2×10 мл) и этилацетатом (3×10 мл), органические экстракты объединяют, промывают насыщенным соляным раствором (50 мл), сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт. Продукт очищают на силикагеле (Biotage, картридж 40 г), используя в качестве элюента смесь циклогексан:этилацетат (4:1), в результате чего получают указанное в заголовке соединение (44 мг, 61%).
Характеристики соединений
Установлено, что соединения из представленных выше таблиц имеют указанные ниже температуры плавления, времена удержания (мин) при анализе с помощью ЖХВР и/или ионную массу:
9,4, С
9,2, С
10,1, С
10,8, С
7,8, С
3,4, С
10,0, С
6,8, С
10,0, С
5,9, С
5,8, С
8,0, С
8,0, С
7,3, С
7,1, С
[М+Н+Н2O]+
8,7, С
3,6, С
[М+Н+Н2O+MeCN]+
8,8, С
[М+Н+Н2O+MeCN]+
5,3, С
5,9, С
[М+Н+Н2O]+
5,8, С
[M+H+H2O]+
7,6, С
5,9 С
Условия ЖХВР В: колонка типа Kingsorb 3,5, мкм С18, 50×4,6 мм, градиент элюирования от 10 to 100% ацетонитрила в воде (+0,1% трифторуксусной кислоты) в течение 10 мин.
Условия ЖХВР С: колонка типа Kingsorb 3 мкм С18, 30×4,6 мм, градиент элюирования от 10 to 100% ацетонитрила в воде (+0,1% трифторуксусной кислоты) в течение 12 мин.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 4-АМИНОПИПЕРИДИНА | 2005 |
|
RU2396257C2 |
| БЕНЗОКСАЗИНИЛ-АМИДОЦИКЛОПЕНТИЛ-ГЕТЕРОЦИКЛИЧЕСКИЕ МОДУЛЯТОРЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2301802C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ ПРОИЗВОДНЫЕ ПЕПТИДОВ КАК ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ ЛАМИНИНА/НИДОГЕНА | 2000 |
|
RU2380371C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ ПРОИЗВОДНЫЕ ПЕПТИДОВ КАК ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ ЛАМИНИНА/НИДОГЕНА | 2000 |
|
RU2262509C2 |
| СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2244709C2 |
| ЛИГАНДЫ МЕЛАНОКОРТИНОВЫХ РЕЦЕПТОРОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2277535C2 |
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
| СУЛЬФОНАМИДНЫЕ ПЕРИ-ЗАМЕЩЕННЫЕ БИЦИКЛЫ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ПОРАЖЕНИЯ АРТЕРИЙ | 2005 |
|
RU2403240C2 |
| НОВЫЕ, СОДЕРЖАЩИЕ ГЕТЕРОАТОМ ТЕТРАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ЭСТРОГЕНА | 2003 |
|
RU2331645C2 |
| ИНГИБИТОРЫ IAP | 2008 |
|
RU2491276C2 |
Изобретение относится к новым соединениям формулы (I) и его фармацевтически приемлемым кислотно-аддитивным солям. Соединения настоящего изобретения обладают активностью в отношении связывания каннабиноидного (СВ) рецептора. В общей формуле (I)
Х обозначает -S-, -S(=O)-, -S(=O)2-, -S(=O)2N(H)-, -Р(=O)(ОСН3)-, -Р(=O)(ОН)-, -N(H)-, -N(СН3)-, -N(H)C(=O)N(H)-, -С(=O)-, -C(=O)O-, -N(H)C(=O)-, -C(H)(OH)-, -C(H)=N-, -С(Н)=С(Н)-, -CH2N(H)- или -C(=NH)-; R1 обозначает фенил, нафтил, 1,2,3,4-тетрагидронафтил, индолил, хинолинил, 1,2,3,4-тетрагидрохинолинил, изохинолинил, бензимидазолил, 2-оксо-1,3-дигидробензимидазолил, бензоксадиазолил, бензотиадиазолил, бензотриазолил или инданил, которые необязательно могут быть замещены; R2 обозначает водород, -OR4 или -N(R5)R6; R3 обозначает водород; циано; оксадиазолил, пиперазинил или тетразолил, необязательно замещенные метилом; -C(=O)R7, -OR8 или -N(R9)R10. Изобретение также относится к способу получения соединения формулы I и к фармацевтической композиции, обладающей активностью в отношении связывания каннабиноидного (СВ) рецептора, содержащей в качестве активного компонента соединение формулы I. 3 н. и 2 з.п. ф-лы.
1. Соединение формулы I
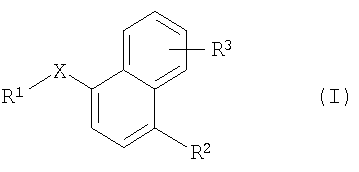
где X обозначает -S-, -S(=O)-, -S(=O)2-, -S(=O)2N(H)-, -Р(=O)(ОСН3)-, -Р(=O)(ОН)-, -N(H)-, -N(СН3)-, -N(H)C(=O)N(H)-, -С(=O)-, -С(=O)O-, -N(H)C(=O)-, -C(H)(OH)-, -C(H)=N-, -C(H)=C(H)-, -СН2N(Н)- или -C(=NH)-;
R1 обозначает фенил, нафтил, 1,2,3,4-тетрагидронафтил, индолил, хинолинил, 1,2,3,4-тетрагидрохинолинил, изохинолинил, бензимидазолил, 2-оксо-1,3-дигидробензимидазолил, бензоксадиазолил, бензотиадиазолил, бензотриазолил или инданил, где указанные фенил, нафтил, 1,2,3,4-тетрагидронафтил, индолил, хинолинил, 1,2,3,4-тетрагидрохинолинил, изохинолинил, бензимидазолил, 2-оксо-1,3-дигидробензимидазолил, бензоксадиазолил, бензотиадиазолил, бензотриазолил или инданил являются незамещенными или могут быть замещены одним или несколькими заместителями, выбранными из группы, включающей гидрокси; карбокси; аминокарбонил; нитро; галоген; циано; -C(NH2)=N-OH; тетразолил; 1,2,4-триазолил; пиразолил; имидазолил; пиперазинил, замещенный С1-С4алкилом; C1-С4алкил; С1-С4алкилтио; C1-C8алкокси, который является незамещенным или может быть замещен гидрокси или морфолинилом; и -N(R11)R12, где R11 и R12 независимо друг от друга выбраны из группы, включающей водород; С1-С4алкил, который является незамещенным или может быть замещен гидрокси, фенилом, С3-С6циклоалкилом, -N-(С1-С4алкилом)2 или гидроксиС1-С4алкокси; -С(=O)N(Н)ОС1-С4алкил; С1-С4алкилкарбонил и -S(=O)2-C1-С4алкил;
R2 обозначает водород; -OR4 или -N(R5)R6;
где R4 обозначает С2-С8алкенил или С1-С8алкил, который является незамещенным или может быть замещен гидрокси, С1-С4алкокси, -С(=O)O-С1-С4алкилом, морфолинилом, пиперидинилом, фенилом или оксадиазолилом, где фенил или оксадиазолил в свою очередь являются незамещенными или могут быть замещены С1-С4алкилом, С1-С4алкокси, нитро, амино или -N(С1-С4алкилом)2;
R5 и R6 независимо друг от друга выбраны из группы, включающей водород; С1-С8алкил, который является незамещенным или может быть замещен морфолинилом; и -C(=O)C1-С8алкил;
R3 обозначает водород; циано; оксадиазолил, пиперазинил или тетразолил, где указанные оксадиазолил, пиперазинил или тетразолил являются незамещенными или могут быть замещены метилом; -C(=O)R7, -OR8 или -N(R9)R10;
где R7 обозначает гидрокси; С1-С4алкокси; амино или -N(H)-CH2-C(=O)OH;
R8 обозначает водород; С1-С8алкил, который является незамещенным или может быть замещен карбокси, метоксикарбонилом, -C(=O)N(H)-N(H)-C(=O)-CH3; или обозначает С1-С4алкил, замещенный оксадиазолилом; -С(=O)-С1-С4алкил или -С(=O)-нафтил; и R9 и R10 независимо друг от друга выбраны из группы, включающей водород; C1-C8алкил или С2-С4алкенил;
при условии, что (а) если Х обозначает -С(=O)- и R2 и R3 обозначают водород или R2 обозначает Н и R3 обозначает 4-метокси, то R1 не обозначает ни 1-нафтил, ни 4-метокси-1-нафтил; (б) если Х обозначает -С(=O)- или -С(Н)(ОН)-, то R1 не обозначает фенил; (в) если Х обозначает -С(=O)- или -C(=NH) и R2 или R3 обозначают -N(R5)R6, то R1 не обозначает ни диметиламинофенил, ни диэтиламинофенил; (г) если Х обозначает -С(Н)=С(Н)- или -C(H)=N-, то R2 не обозначает водород; (д) если Х обозначает -CH2N(H)-, то R1 не обозначает 2,4-диамино-5-метилпиридо[2,3-d]пиримидинил; (е) если Х обозначает -N(H)C(=O)-, то R2 не обозначает аминогруппу; (ж) если Х обозначает -S-, -S(=O)2-, -S(=O)2N(H)-, -N(СН3)-, -Р(=O)(ОСН3)- или -С(=O)O-, то R1 не обозначает фенил; (з) если Х обозначает -N(H)-, то R1 не обозначает ни фенил, ни 4,6-диметилпиримидил; (и-1) если Х обозначает -N(H)-C(=O)-N(H)-, то R2 обозначает метокси и R3 обозначает водород, то R1 не обозначает 4-метоксинафтил-1-ил; (и-2) если Х обозначает -N(H)-C(=O)-N(H)-, R2 обозначает этокси и R3 обозначает водород, то R1 не обозначает 4-этоксинафтил-1-ил; (к) если Х обозначает -C(H)=N-, то R2 не обозначает ни метокси-, ни диметиламино; (л) если Х обозначает -Р(=O)(ОН)- и R2 и R3 обозначают водород, то R1 не обозначает фенил; и (м) если Х обозначает -CH2-N(H)-, и R2 и R3 обозначают водород, или R2 обозначает метокси, а R3 обозначает водород, или R2 обозначает водород, а R3 обозначает 2-метокси, то R1 не обозначает 2,4-диаминопиридо[2,3-d]пиримидин-6-ил;
в форме свободного основания или фармацевтически приемлемой кислотно-аддитивной соли.
2. Соединение формулы I по п.1, где Х обозначает -С(=O)-, R1 обозначает нафтил, R2 обозначает -O-(СН2)4СН3 и R3 обозначает водород.
3. Соединение формулы I по п.2, где Х обозначает -С(=O)-, R1 обозначает 1-нафтил, R2 обозначает -O-(СН2)4СН3 и R3 обозначает водород.
4. Способ получения соединения формулы I по п.1, включающий взаимодействие соединения формулы II

где R1 имеет значения, указанные в п.1 для формулы I, и R13 обозначает -ОН, -SH, -I, -Cl, 1,8-бис(диметиламино)нафтил-, -СООН, -NH2, -Н, -карбонитрил, -O-трифторметансульфонил, -C(=O)Cl, с соединением формулы III
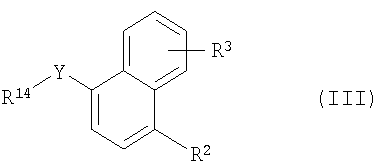
где R2 и R3 имеют значения, указанные в п.1 для формулы I, Y обозначает -O-, -S(=O)2O-, -Р(=O)(ОСН3)-, простую связь, -С(=O)-O-, -С(=O)- или -O-В(ОН)2- и R14 обозначает водород, -I или -Cl,
с получением соединения формулы Iа
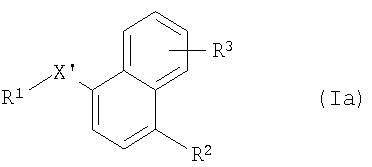
где R1, R2 и R3 имеют значения, указанные в п.1 для формулы I, и X' обозначает -С(=O)-, -S-, -Р(=O)(ОСН3)-, -N(H)-, -S(=O)2- (при связывании через атом N в R1), -S(=O)2-N(H)-, -C(=O)-O-, -C(H)=N-, -C(H)(OH)-, -N(H)-C(=O)-N(H)- или -C(=NH)-, которое в случае необходимости превращают в соединение формулы Iб
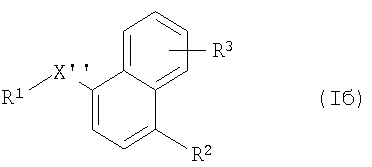
где R1, R2 и R3 имеют значения, указанные в п.1 для формулы I, и X'' обозначает -S(=O)-, -S(=O)2-(при связывании через атом С в R1), -N(СН3)-, -Р(=O)(ОН)-, -CH2-N(H)-, -C(H)=C(H)-,
с последующим выделением полученных таким образом соединений формулы Iа или формулы Iб в свободной форме или в форме фармацевтически приемлемой кислотно-аддитивной соли.
5. Фармацевтическая композиция, обладающая активностью в отношении связывания каннабиноидного (СВ) рецептора, содержащая в качестве активного ингредиента соединение формулы I по п.1 в форме свободного основания или в форме фармацевтически приемлемой кислотно-аддитивной соли и по крайней мере один фармацевтически приемлемый носитель или разбавитель.
| Nakayama Т | |||
| et al | |||
| "Synthesis and Structure-Activity Study of Protease Inhibitors | |||
| II | |||
| Amino - and Guanidino-Substituted Naphthoates and Tetrahydronaphthoates" CHEMICAL & PHARMACEUTICAL BULLETIN, 1984, Vol.32, №10, 3968-3980 | |||
| 5-ЗАМЕЩЕННЫЕ НАФТАЛИН-1-СУЛЬФОНИЛАМИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2051146C1 |
| WO 9832732 A1, 30.07.1998 | |||
| Устройство для ввода информации | 1984 |
|
SU1200273A1 |

