ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области медицинской органической химии, конкретно к улучшенному способу получения гемцитабина гидрохлорида, обладающего антивирусной и противоопухолевой активностью.
УРОВЕНЬ ТЕХНИКИ
Гемцитабин по своей химической природе представляет собой 4-амино-1-[(2R,4R,5R)-3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]-1Н-пиримидин-2-она гидрохлорид и является фторированным аналогом природного нуклеозида цитидина, одного из строительных блоков синтеза нуклеиновых кислот клетки. Предполагаемый механизм действия гемцитабина гидрохлорида состоит во внутриклеточном 5'-фосфорилировании гемцитабина гидрохлорида при участии нуклеозидкиназы до активных дифосфатных и трифосфатных нуклеозидов. Цитотоксический эффект обусловлен встраиванием модифицированных нуклеозидов-метаболитов в цепочки ДНК, и, как следствие, нарушением синтеза ДНК, приводящим к смерти клетки (Справочник Видаль. Лекарственные препараты в России: справочник. М.: АстраФармСервис, 2001, раздел 3, стр.155).
Способы получения гемцитабина гидрохлорида описаны в уровне техники и представляют собой сложные многостадийные синтезы (вариант по европейскому патенту ЕР 0184365 А2 заявителя Элли Лилли энд Компани приведен на фиг.1), при этом полупродукты требуют дополнительной очистки перед введением в следующую стадию.
Впервые гемцитабина гидрохлорид (химическое название 4-амино-1-[(2R,4R,5R)-3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]-1Н-пиримидин-2-она гидрохлорид) был получен компанией Эли Лилли (патент США 4526988 и США 4808614), выпускающей на основе данной субстанции препарат «Гемзар».
Один из вариантов способа заключается в том, что этилбромдифторацетат подвергают взаимодействию с D-глицеральдегидкетонидом, а именно с 2,2-диметил-[1,3]-диоксолан-4-карбальдегидом среде ТГФ и диэтилового эфира в присутствии цинка (реакция Реформатского). При этом для активации цинка можно использовать ультразвуковую энергию. Полученный этил 3-гидрокси-2,2-дифтор-3-[2,2-диметил-[1,3]-диоксолан-4-ил]пропионат подвергают циклизации с помощью ионообменной смолы Dowex 50W-X12 в среде растворителя с получением 2-дезокси-2,2-дифтор-1-оксорибозы ((4R,5R)-4-гидрокси-5-гидроксиметил-3,3-дифтордигидрофуран-2(3Н)-она). После введения защитных трет-бутилдиметилсилильных групп получают 3,5-бис(трет-бутилдиметилсилилокси)-2-дезокси-2,2-дифтор-1-оксорибозу ((4R,5R)-4-трет-бутилдиметилсилилокси-5-((трет-бутилтриметилсилилокси)метил)-3,3-дифтордигидрофуран-2(3Н)-он). Полученный продукт подвергают восстановлению диизобутилалюмогидридом. В полученном продукте модифицируют гидроксигруппу в положении 2 тетрагидрофуранового кольца взаимодействием с метансульфонилхлоридом и получают 3,5-бис-(трет-бутилдиметилсилилокси)-1-метансульфонилокси-2-дезокси-2,2-дифторрибозу ((4R,5R)-2-метансульфонилокси-4-(трет-бутилдиметилсилил)-5-((трет-бутилдиметилсилилокси)метил)-3,3-дифтортетрагидрафуран). Полученный продукт подвергают взаимодействию с 5-метил-2,4-бис-(триметилсилилокси)пиримидином и трифторметансульфонилокситриметилсиланом в качестве инициатора с получением 1-(5-метил-2,4-диоксо-1Н,3Н-пиримидин-1-ил)-1,2-дезокси-2,2-дифторрибозы (4-амино-1-[(2R,4R,5R)-3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]-1Н-пиримидин-2-она, или гемцитабина).
Недостатком этого способа является получение на стадии реакции Реформатского смеси 3S- и 3R-гидрокси-изомеров в соотношениии около 3:1. Для дальнейшего синтеза необходимо преимущественное получение 3-R-гидрокси-изомера. Кроме того, продукт согласно указанному патенту не выделяют в виде фармацевтически приемлемой соли, обладающей достаточной растворимостью в воде для внутривенного введения.
В уровне техники имеются описания улучшений отдельных стадий указанного синтетического метода с заменой защитных групп (патент США 5480992, международная заявка WO 2006095359), улучшениям в соотношении энантиомеров в промежуточных стадиях (патенты США 5401861, 5256798, 5420266).
Улучшения для осуществления ключевой стадии гликозилирования пуринового основания с образованием N-гликозидной связи описаны в патенте РФ 2131880.
В авторском свидетельстве СССР 1442076 описан способ получения гемцитабина из 3,5-бис(трет-бутилдиметилсилилокси)-1-метансульфонилокси-2-дезокси-2,2-дифторрибозы. Неочищенный кристалличесткий продукт, 1-(4-амино-2-оксо-1Н-пиримидин-1-ил)-2-дезокси-2,2-дифторрибозу (гемцитабин), подвергают перекристаллизации из этилацетата.
В уровне техники имеются примеры применения ультразвука для повышения выходов и улучшения соотношения энантиомеров в реакции Реформатского, которая является первой стадией в синтезе гемцитабина гидрохлорида (Фиг.1). Несколько алифатических и ароматических карбонильных соединений были подвергнуты взаимодействию с этилбромацетатом, реакционную смесь подвергали обработке ультразвуком (Organic Sonochemistry. Sonic Acceleration of the Reformatsky Reaction. Byung-Hee Han and Philip Boudjouk, J. Org. Chem. 1982,47, 5030-5032), при этом в большинстве случаев уменьшалась время реакции и увеличивался энантиомерный избыток по отношению к избытку в продукте, полученном при обычном нагревании реакционной смеси.
Однако использование ультразвука в синтезе гемцитабина гидрохлорида в литературе не описано.
Известен способ получения гемцитабина гидрохлорида по патенту РФ 2154648, включающий удаление защитной группы у β-1-(2'-деокси-2',2'-дифтор-3',5'-ди-о-бензоил-D-рибофуранозил)-4-аминопиримидин-2-она (сохранена номенклатура, приведенная в патенте) в присутствии основания и спирта с последующей обработкой полученного раствора соляной кислотой и антирастворителем, и выделение полученного твердого продукта, при этом удаление защитной группы проводят в присутствии каталитического количества алкиламина, в присутствии метанола или этанола в, по существу, безводной среде.
Способ по патенту РФ 2154648 предусматривает выделение гемцитабина гидрохлорида путем перекристаллизации его из метанола и изопропанола, которые в качестве остаточных растворителей могут присутствовать в конечном продукте, что нежелательно для дальнейшего медицинского использования гемцитабина гидрохлорида.
Ввиду многостадийности синтеза и сложности выделения промежуточных продуктов актуальна задача сокращения временных промежутков проведения стадий, а также упрощения стадии выделения чистого кристаллического продукта. Для создания лекарственных композиций, предназначенных для внутривенного введения, важна однородность получаемого продукта по размерам частиц.
Целью настоящего изобретения является разработка экономичного способа получения гемцитабина гидрохлорида, лишенного указанных недостатков, а именно преимущественное получение 3R-изомера в реакции Реформатского, что в конечном счете увеличивает выход целевого гемцитабина гидрохлорида, и получение однородных кристаллических частиц гемцитабина гидрохлорида, не содержащих остатков органических растворителей.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с изобретением описывается способ получения гемцитабина гидрохлорида, состоящий из следующих стадий.
Реакцией бромдифторацетата с 2,2-диметил-[1,3]-диоксолан-4-карбальдегидом в присутствии цинка в условиях реакции Реформатского получают продукт, представляющий собой смесь 3R- и 3S-гидрокси-изомеров этил 3-гидрокси-2,2-дифтор-3-[2,2-диметил-[1,3]диоксолан-4-ил)-пропионата (1) в соотношении не менее 3:1, более предпочтительно в соотношении не менее 5:1. Полученную смесь без разделения используют на следующей стадии.
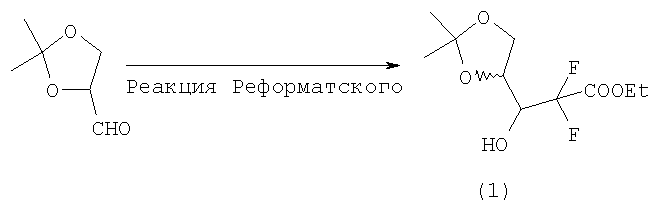
Обнаружено, что продолжительная обработка ультразвуком реакционной смеси на первой стадии синтеза гемцитабина гидрохлорида (Фиг.1) позволяет получить улучшенное соотношение энантиомеров этил 3-гидрокси-2,2-дифтор-3-[2,2-диметил-[1,3]диоксолан-4-ил]пропионата (соотношение 3R-гидрокси-энантиомера к 3S-гидрокси-энантиомеру до 5:1). В уровне техники описано получение соотношения энантиомеров до 3:1 (европейский патент 0122707, стр 14, строки 1-9).
Энергия, передаваемая реакционной системе при облучении ее ультразвуком, отличается от способов передачи энергии, таких как нагревание, тем, что при прохождении через среду ультразвуковых волн в жидкости за счет процесса кавитации накапливается энергия и возникают микроскопические участки с высокой температурой.
Время прохождения многих химических процессов значительно сокращается при таком способе их активации по сравнению, например, с обычным нагреванием, часто ограниченным по температуре за счет температуры кипения растворителя.
В европейском патенте 0184365 упомянута возможность активации металла-реактанта ультразвуковой обработкой, однако в патенте не раскрывается влияние длительности обработки на выход реакции и соотношение 3R- и 3S-гидрокси-изомеров, получаемых в результате реакции. Так, обработка ультразвуком реакционной смеси от 1 до 5 минут практически не влияет на выход продукта и соотношение 3R-гидрокси-энантиомера к 3S-гидрокси-энантиомеру, а обработка ультразвуком от 5 до 15 минут оказывает влияние на выход и соотношение продуктов:
Установки, применяемые для генерации ультразвука, описаны ниже при обсуждении кристаллизации гемцитабина гидрохлорида.
Последующие за реакцией Реформатского стадии синтеза гемцитабина гидрохлорида, как следует из изложенного выше описания уровня техники, подробно изучены ранее. Нами для осуществления дальнейших стадий синтеза была взята за основу схема, изложенная в европейском патенте 0184365 (в описании на стр 8-18 и в примере 1 на стр.19-20), и методики, раскрытые в патентах США 522608 А1 и 4808614, а также авторском свидетельстве СССР 1442076.
Согласно изобретению, следующая стадия синтеза гемцитабина гидрохлорида подразумевает гидролиз продукта реакции Реформатского, этил 3-гидрокси-2,2-дифтор-3-[2,2-диметил-[1,3]-диоксолан-4-ил]пропионат, под действием ионообменной смолы Dowex с последующей циклизацией и получением ((4R,5R)-4-гидрокси-5-гидроксиметил-3,3-дифтордигидрофуран-2(3Н)-она). После формирования рибозного цикла гидроксигруппы защищают путем введения триметилсилильных групп и получением (4R,5R)-4-триметилсилилокси-5-((триметилсилилокси)метил)-3,3-дифтордигидрофуран-2(3Н)-она

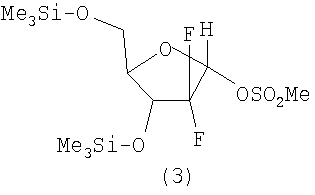
Далее, производят восстановление кетогруппы в полученном соединении (2) литийизопропилалюмогидридом в толуоле. Для осуществления последующих стадий важно получить ((2R,4R,5R)-2-гидрокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофуран). Дробная перекристаллизация из смеси хлористый метилен/гексан позволяет получить ((2R,4R,5R)-2-гидрокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофуран) с энантиомерным избытком более 90%. Взаимодействием с метансульфонилхлоридом получают соединение (3) с уходящей группой в положении 2:
Далее путем SN2-замещения осуществляется замена уходящей группы с обращением конфигурации и получением гемцитабина (4) в виде свободного основания:

Финальная стадия получения гемцитабина гидрохлорида основана на прибавлении к спиртовому раствору основания гемцитабина соляной кислоты (в виде газа или в виде концентрированного водного раствора), что приводит к выпадению в осадок гемцитабина гидрохлорида.
Настоящее изобретение подразумевает использование водной среды для перекристаллизации гемцитабина гидрохлорида, при этом не требуется добавления антирастворителя. Ультразвуковое излучение при кристаллизации позволяет получать высокий выход продукта при кристаллизации из водного раствора.
Процесс кристаллизации подразумевает достижение раствором концентрации, при которой начинается отделение растворенного вещества от растворителя. Достижение такой концентрации осуществляют либо охлаждением раствора, либо добавлением антирастворителя (растворителя, в котором подвергаемое кристаллизации вещество слабо растворимо или нерастворимо), либо внесением затравок - кристаллов чистого вещества. В зависимости от природы кристаллизуемого вещества возможно применение любой комбинации вышеперечисленных методов.
Кристаллизация при помощи обработки ультразвуком имеет ряд преимуществ перед традиционным методами внесения затравки или добавления антирастворителя, потому что не требует столь внимательного контроля момента добавления затравки/разбавления. Например, в случае внесения затравки слишком раннее ее введение часто приводит к растворению затравки до начала выпадения кристаллов. Слишком позднее введение затравки приводит к слишком быстрому выпадению кристаллов и ухудшению качества получаемого кристаллического продукта. Мельчайшие зародыши кристаллов, образующиеся непрерывно во время ультразвуковой обработки, позволяют решить эту проблему.
Применение ультразвука для кристаллизации фармацевтической субстанции пароксетина гидрохлорида в виде сольвата с изопропанолом описано в международной заявке WO 0032597. Способ непрерывной кристаллизации в промышленных масштабах фенотерола гидробромида с применением ультразвука описан в заявке WO 200403943. Применение ультразвука для кристаллизации гемцитабина гидрохлорида позволяет сократить время получения кристаллического продукта высокого качества, регулировать распределение частиц по размерам, а также повысить воспроизводимость кристаллизации. Такие улучшения могут оказаться полезными для получения промышленных количеств высококачественного кристаллического продукта.
Типичная ультразвуковая установка состоит из ультразвукового инструмента, преобразователя энергии электрических колебаний в механические колебания и электронного генератора, вырабатывающего электрические колебания необходимой частоты и мощности. В предпочтительном варианте осуществления изобретения рабочая частота данной установки ультразвуковых колебаний варьируется от примерно 20 до 60 кГц.
В результате обработки ультразвуком концентрированного раствора гемцитабина гидрохлорида в воде, приготовленного при повышенной температуре и быстро охлажденного, образуется густая суспензия, которая отделяется от маточного раствора фильтрованием.
Фильтрование осуществляется известными из уровня техники методами, например, с использованием пористых фильтров.
В результате перекристаллизации гемцитабина гидрохлорида из воды с использованием ультразвукового излучения образуется кристаллический продукт с распределением частиц по размерам, пример которого приведен на Фиг.2. Видно, что при использовании обычных методов кристаллизации (нагревание/охлаждение, использование антирастворителя) максимум пика распределения частиц находится в области 10 мкм, однако его ширина существенно больше ширины пика распределения, для кристаллов, полученных методом ультразвуковой обработки.
Таким образом, техническим результатом изобретения является создание экономичного способа синтеза гемцитабина гидрохлорида, подразумевающее долговременное использование ультразвука на стадии реакции Реформатского, а также использование ультразвуковой обработки водного раствора на стадии перекристаллизации продукта.
Применение ультразвука позволяет сократить время осуществления реакции Реформатского и улучшить соотношение энантиомеров в получаемом продукте до значений 5:1. Применение ультразвука на стадии перекристаллизации гемцтитабина гидрохлорида позволяет исключить органические и спиртовые растворители и получить продукт, характеризующийся распределением частиц, улучшенным по сравнению с традиционными способами кристаллизации, подразумевающими нагревание и охлаждение с необязательным добавлением антирастворителя.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ.
Фиг.1. Способ получения гемцитабина гидрохлорида по Европейскому патенту 0184365 A1. R1 и R2, независимо друг от друга, обозначают C1-С3-алкил, Protect обозначает гидроксильную защитную группу и Leave обозначает уходящую группу.
Фиг.2. Сравнение распределения частиц по размерам в субстанциях гемцитабина гидрохлорида, полученных в примере 2 и в примере 3.
ПРИМЕРЫ ОСУЩЕСТВЛЕНИЯ
Изобретение далее проиллюстрировано примерами его осуществления, которые приведены для раскрытия характерных особенностей и не ограничивают его объем. Растворители и реагенты, указанные в примерах, получены от фирмы Сигма-Олдрич и подвергнуты осушке и перегонке. Для перекристаллизации гемцитабина гидрохлорида использовалась деионизированная вода. Спектры ЯМР 1Н и 13С регистрировали на приборе Broker DRX500 (500 мГц, ДМСО). Для генерации ультразвука использовалась лабораторная установка IKA ULTRA - TURRAX Т 18 basic с рабочей частотой 60 кГц. Данные о размерах частиц кристаллического продукта были получены при помощи анализатора PAMAS DPFS, оснащенного датчиком HCB-LD-100 для определения частиц в диапазоне от 10 до 800 мкм.
ПРИМЕР 1
Получение гемцитабина гидрохлорида
А. Этил 3-гидрокси-2,2-дифтор-3-[2,2-диметил-[1,3]-диоксолан-4-ил]пропионат
В заполненную аргоном колбу помещали 150 мл диоксана, 30 г этил бромдифторацетата и 22 г 2,2-диметил-[1,3]-диоксолан-4-карбальдегида и 17 г цинковой пыли, погружают в раствор источник ультразвука и проводят обработку ультразвуком в течение 5-15 минут, энергично перемешивая реакционную смесь, не давая температуре сильно повышаться.
Ход реакции контролируют по исчезновению сигнала альдегидного протона в спектре ЯМР 1Н.
Реакционную смесь выливают на лед, добавляют небольшими порциями разбавленную соляную кислоту для удаления избытка цинка, затем экстрагируют диэтиловым эфиром.
После высушивания над сульфатом натрия упаривают растворитель. После высушивания выделяют 40 г этил 3-гидрокси-2,2-дифтор-3-[2,2-диметил-[1,3]-диоксолан-4-ил]пропионат в виде смеси 3R- и 3S-гидрокси-изомера в соотношении примерно 4:1 (данные ВЭЖХ).
В. ((4R,5R)-4-гидрокси-5-гидроксиметил-3,3-дифтордигидрофуран-2(3Н)-он
40 г этил 3-гидрокси-2,2-дифтор-3-[2,2-диметил-[1,3]-диоксолан-4-ил]пропионата, полученного в примере 1А, растворяют в 500 мл метанола, 500 мл воды и добавляют 150 г ионообменной смолы Dowex. Смесь перемешивают в течение 5 дней, фильтруют и сушат в вакууме с получением 30 г ((4R,5R)-4-гидрокси-5-гидроксиметил-3,3-дифтордигидрофуран-2(3Н)-она
С. (4R,5R)-4-триметилсилилокси-5-((триметилсилилокси)метил)-3,3-дифтордигидрофуран-2(3Н)-он
15 г продукта, полученного в примере 1В, растворяют в 40 мл дихлорметана и добавляют 12 г триметилхлорсилана в 20 мл дихлорметана, не давая реакционной смеси разогреваться. После окончания прибавления реакционную смесь перемешивают еще 5-6 часов, разбавляют вдвое дихлорметаном, промывают 1 М раствором гидрокарбоната натрия и сушат над сульфатом натрия. Получают 15 г чистого продукта. В ЯМР-спектре (CDCl3) характерные уширенные полосы сигналов гидроксигрупп исчезают и наблюдаются синглеты триметилсилильных групп.
D. (4R,5R)-2-гидрокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофуран
15 г продукта, полученного в примере 1C, растворяют в 200 мл безводного толуола и охлаждают смесью сухой лед/толуол до -70°С. К охлажденному раствору маленькими порциями добавляют 23 г литийдиизопропилалюмогидрида, перемешивают 1 час при охлаждении и затем маленькими порциями добавляют метанол, не убирая охлаждения. Реакционную смесь размораживают, экстрагируют 3 раза по 40 мл этилацетатом, промывают 3 раза насыщенным раствором NaCl и сушат в вакууме, получая 13 г сырого продукта.
Е. (4R,5R)-2-метилсульфонилокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофуран
12 г продукта, полученного в примере 1D, растворяют в пиридине (15 мл), добавляют 1 мл метансульфонилхлорида и убирают в холодильник на 5 часов. Реакционную смесь размораживают, разбавляют водой и экстрагируют 5 раз по 20 мл этилацетатом. Объединенные этилацетатные фракции промывают 4 раза по 20 мл 1 М раствором лимонной кислоты, упаривают и сушат над безводным сульфатом натрия. После упаривания получают 13 г (4R,5R)-2-метилсульфонилокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофурана. В результате дробной кристаллизации из смеси хлористый метилен/гексан получают после высушивания 5 г (2R,4R,5R)-2-метилсульфонилокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофурана
F. (4-амино-1-[(2R,4R,5R)-3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]-1Н-пиримидин-2-он (гемцитабин) и (4-амино-1-[(2R,4R,5R)-3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]-1Н-пиримидин-2-она гидрохлорид (гемцитабина гидрохлорид)
К 5 г (2R,4R,5R)-2-метилсульфонилокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофурана, полученного в примере 1Е, в 70 мл безводного дихлорэтана прибавляют 3 г 5-метил-2,4-(триметилсилилокси)пиримидина. Полученную после добавления 3 г трифторметансульфонилоксиметилсилана реакционную смесь кипятят с обратным холодильником в течение трех часов.
Реакционную смесь остужают до комнатной температуры и разбавляют метанолом, после перемешивания в течение часа осадок фильтруют, фильтрат промывают насыщенным раствором NaCl, 1 M раствором гидрокарбоната натрия, повторяют промывание насыщенным раствором NaCl. Раствор упаривают досуха в вакууме водоструйного насоса. Полученный продукт растворяют в изопропаноле и добавляют концентрированную соляную кислоту по каплям (примерно 2,5 мл). Смесь охлаждают до комнатной температуры и получают после выдерживания в холодильнике 6-8 часов, получают 2,5 г сырого (4-амино-1-[(2R,4R,5R)-3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]-1Н-пиримидин-2-она гидрохлорида (гемцитабина гидрохлорида).
ПРИМЕР 2
Очистка гемцитабина гидрохлорида
Навеску 2,5 г гемцитабина гидрохлорида растворяют при температуре 60°С в 50 мл воды и добавляют активированный уголь. После фильтрации горячий раствор остужают до комнатной температуры и медленно при перемешивании добавляют концентрированный раствор гидроксида натрия до рН~7-8. Затем раствор экстрагируют хлористым метиленом и высушивают экстракт над сульфатом натрия. Дихлорметан упаривают досуха, после чего добавляют воду и концентрированную соляную кислоту в количестве не менее 3 эквивалентов на молекулу гемцитабина. Раствор охлаждают до 5°С. При стоянии в холодильнике при указанной температуре из раствора выпадают мелкие кристаллы гемцитабина гидрохлорида, вес высушенного продукта составляет 4,6 г. Данный продукт содержит более 99,0% гемцитабина гидрохлорида (ВЭЖХ). Спектры ЯМР и масс-спектры продукта совпадают с соответствующими спектрами стандарта, соответствующего требованиям фармакопеи США (USP30, 2007).
ПРИМЕР 3
Кристаллизация гемцитабина гидрохлорида с ультразвуковой обработкой
Навеску 2,5 г гемцитабина гидрохлорида в 20 мл воды нагревали при перемешивании в течение часа до полного растворения суспензии при температуре 60°С. Охлаждали раствор с обработкой ультразвуком в течение 3 минут при температуре раствора около 40°С. Наблюдали образование помутнения.
Выделили после фильтрации и сушки в вакууме 2 г с чистотой субстанции 99,6% (ВЭЖХ).
Сравнительное распределение по размерам частиц в кристаллах, полученных в Примере 2 и в Примере 3, приведено на Фиг.2.
ПРИМЕР 4
Кристаллизация гемцитабина гидрохлорида с ультразвуковой обработкой и добавлением изопропанола
Навеску 2,5 г гемцитабина гидрохлорида в 20 мл воды нагревали при перемешивании в течение часа до полного растворения суспензии при температуре 60°С. Осторожно добавляли подогретый изопропанол при одновременной обработке ультразвуком в течение 3 минут при температуре раствора около 40°С. Прекращали добавлять изопропанол после появления устойчивого помутнения. Смесь охлаждали до комнатной температуры, после чего выдерживали сутки в холодильнике при температуре 5°С. Выделили после фильтрации и сушки в вакууме около 4,8 г с чистотой субстанции 99,6% (ВЭЖХ).
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ (2R,3R,5R)-3-ГИДРОКСИ-(5-ПИРИМИДИН-1-ИЛ)ТЕТРАГИДРОФУРАН-2-ИЛМЕТИЛ АРИЛ ФОСФОРАМИДАТЫ | 2013 |
|
RU2553996C1 |
| Макрогетероциклические нуклеозидные производные и их аналоги, получение и применение | 2017 |
|
RU2731385C1 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| ПРОИЗВОДНЫЕ С-АРИЛГЛЮКОЗИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2606501C2 |
| СПОСОБ ПОЛУЧЕНИЯ РИБОФУРАНОЗИЛ-ПИРИМИДИНОВЫХ НУКЛЕОЗИДОВ | 2007 |
|
RU2421461C2 |
| N-ДИГИДРОКСИАЛКИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 2-ОКСОИМИДАЗОЛА | 2006 |
|
RU2414456C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ФТОРЛАКТОНА | 2014 |
|
RU2656600C2 |
| СОЕДИНЕНИЯ - ПРОИЗВОДНЫЕ СУЛЬФАМАТА ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ ИЛИ СМЯГЧЕНИИ БОЛИ | 2014 |
|
RU2699025C1 |
| ГИПОГЛИКЕМИЧЕСКОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ПОЛИГИДРОКСИБУТИЛПИРАЗИНЫ, НОВЫЕ ПОЛИГИДРОКСИБУТИЛПИРАЗИНЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2186773C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ ПРОТИВООПУХОЛЕВЫХ ЦЕЛЕВЫХ ФЕРМЕНТОВ | 2002 |
|
RU2319482C2 |
Изобретение относится к области медицинской органической химии и касается способа получения гемцитабина гидрохлорида, характеризующегося тем, что 2,2-диметил-[1,3]-диоксолан-4-карбальдегид подвергают взаимодействию с этил бромдифторацетатом в присутствии цинка в среде органического растворителя при обработке реакционной смеси ультразвуком в течение 5-60 минут, полученный этил 3-гидрокси-2,2-дифтор-3-[2,2-диметил-[1,3]диоксолан-4-ил]пропионат подвергают гидролизу и циклизации с помощью ионообменной смолы в водно-спиртовой среде с получением (4R,5R)-4-гидрокси-5-гидроксиметил-3,3-дифтордигидрофуран-2(3Н)-она, который обрабатывают раствором триметилхлорсилана в дихлорметане с получением (4R,5R)-4-триметилсилилокси-5-((триметилсилилокси)метил)-3,3-дифтордигидрофуран-2(3Н)-она, который подвергают восстановлению с помощью литийдиизопропилалюмогидрида в среде органического растворителя при охлаждении до -70°С с получением (4R,5R)-2-гидрокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофурана, который превращают в (4R,5R)-2-метилсульфонилокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофуран обработкой метансульфонилхлоридом в среде растворителя на холоду, полученный (4R,5R)-2-метилсульфонилокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофуран после разделения оптических изомеров обрабатывают бис-триметилсилилацетилцитозином в безводном дихлорэтане и кипятят с трифторметансульфонилоксиметилсиланом с последующим охлаждением и выделением полученного гемцитабина в виде основания или гидрохлорида, а также способа очистки гемцитабина гидрохлорида путем перекристаллизации его из водного раствора при обработке ультразвуком. Изобретение приводит к увеличению соотношения 3-(R)-гидрокси-изомера к 3(S)-гидрокси-изомеру. 2 н. и 4 з.п. ф-лы, 2 ил.
1. Способ получения гемцитабина гидрохлорида, характеризующийся тем, что 2,2-диметил-[1,3]-диоксолан-4-карбальдегид подвергают взаимодействию с этил бромдифторацетатом в присутствии цинка в среде органического растворителя при обработке реакционной смеси ультразвуком в течение 5-60 мин, полученный этил 3-гидрокси-2,2-дифтор-3-[2,2-диметил-[1,3]диоксолан-4-ил]пропионат подвергают гидролизу и циклизации с помощью ионообменной смолы в водно-спиртовой среде с получением (4R,5R)-4-гидрокси-5-гидроксиметил-3,3-дифтордигидрофуран-2(3Н)-она, который обрабатывают раствором триметилхлорсилана в дихлорметане с получением (4R,5R)-4-триметилсилилокси-5-((триметилсилилокси)метил)-3,3-дифтордигидрофуран-2(3Н)-она, который подвергают восстановлению с помощью литийдиизопропилалюмогидрида в среде органического растворителя при охлаждении до -70°С с получением (4R,5R)-2-гидрокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофурана, который превращают в (4R,5R)-2-метилсульфонилокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофуран обработкой метансульфонилхлоридом в среде растворителя на холоду, полученный (4R,5R)-2-метилсульфонилокси-4-(триметилсилилокси)-5-((триметилсилилокси)метил)-3,3-дифтортетрагидрофуран после разделения оптических изомеров обрабатывают бис-триметилсилилацетилцитозином в безводном дихлорэтане и кипятят с трифторметансульфонилоксиметилсиланом с последующим охлаждением и выделением полученного гемцитабина в виде основания или гидрохлорида.
2. Способ очистки гемцитабина гидрохлорида, характеризующийся тем, что гемцитабина гидрохлорид подвергают перекристаллизации из водной среды путем обработки водного раствора гемцитабина гидрохлорида ультразвуком с необязательным добавлением к водному раствору смешивающегося с водой растворителя в количестве до половины объема исходного водного раствора.
3. Способ по п.2, в котором под смешивающимся с водой растворителем подразумевают изопропанол.
4. Способ по п.2, в котором время обработки ультразвуком находится в интервале от примерно 1 до 60 мин.
5. Способ по любому из п.3-4, в котором обработку ультразвуком осуществляют в процессе охлаждения маточного раствора.
6. Способ по любому из пп.3-4, в котором обработку ультразвуком осуществляют в интервале температур маточного раствора от +5 до приблизительно 60°С.
| СПОСОБ ПОЛУЧЕНИЯ ОБОГАЩЕННЫХ БЕТА-АНОМЕРОМ НУКЛЕОЗИДОВ | 1993 |
|
RU2131880C1 |
| Установка для нанесения покрытий | 1978 |
|
SU719788A1 |
| RU 2006129129, A, 20.02.2008. | |||

