Изобретение относится к области синтеза практически важных оптически активных ациклических изопреноидов, а именно к новому энантиоселективному способу получения (3R,7R)-1-бром-3,7,11-триметилдо декана, или (3R,7R)-гексагидрофарнезилбромида 1.

Соединение 1 является ключевым синтоном для витамина Е, витамина K1, (R,R)-фитола, феромонов и других природных соединений [Fujisawa Т., Sato Т., Kawara Т., Ohashi К. // Tetrahedron Lett. - 1981. - V.22. - №48. - P.4823-4826; Takano S., Sugihara Т., Ogasawara K. // Synlett. - 1991. - P.279-282; Schmid R., Antoulas S., Rüttiman A., Schmid M., Vecchi M., Weiser H. // Helv. Chim. Acta. - 1990. - V.73. - №5. - P.1276-1299; Nozawa M., Takahashi K., Kato K., Akita H. // Chem. Pharm. Bull. - 2000. - V.48 - №2. - P.272-277; Leuenberger H.G.W., Boguth W., Barner R., Schmid M., Zell R. // Helv. Chim. acta - 1979. - V.62. - №45. - P.455-472].
(3R,7R)-Гексагидрофарнезилбромид 1 получают бромированием природного сесквитерпенола - (R,R)-гексагидрофарнезола ((3R,7R)-3,7,11-триметил-1-додеканола) 2, синтез которого базируется на сочетании С4-, С5- и С10-хиральных строительных блоков, которые, в свою очередь, получают микробиологическими методами, классическим оптическим расщеплением рацемических смесей, путем биохимических трансформаций и с использованием асимметрического металлокомплексного катализа [Netscher Т.// Chimia. - 1996. - V.50. - P.563-567].
В энантиоселективном синтезе оптически чистого (3R,7R)-гексагидрофарнезилбромида 1, построенном на хемоэнзиматическом подходе, в качестве блок-синтонов использованы С10-изопреноид 3, полученный из (R)-цитронеллола, и хиральный бифункциональный изопреноид 4, который получали кинетическим разделением (±)-син-β-ацетокси-α-метилпропионата 5 (с получением (2S,3S)-5) или (±)-анти-1,3-диола 13 (с получением (2R,3R)-14), катализируемым липазами «Amano А» или «Amano Р» (схема 1) [Chen С.Y., Nagumo S., Akita H. // Chem. Pharm. Bull. - 1996. - V.44. - №11. - P.2153-2156; Nozawa M., Takahashi K., Kato K., Akita H. // Chem. Pharm. Bull. - 2000. - V.48 - №2. - P.272-277].
Катализируемый липазой «Amano А» гидролиз (±)-син-β-ацетокси-α-метилпропионата 5 привел к гладкому кинетическому разделению рацемата на целевой спирт (2S,3S)-6 высокой оптической чистоты [энантиомерный избыток (ее 94%)] и непрореагировавший ацетат (2R,3R)-5. Полученный спирт (2S,3S)-6 подвергли ацетилированию с последующим восстановлением ацетата (2S,3S)-5 в диол (2S,3S)-7, гидрогенолиз которого позволил получить спирт (2S)-8. Его взаимодействие с N-бромсукцинимидом (NBS) привело к бромиду (2S)-9, который обработали фенилсульфонатом натрия (PhSO2Na·2H2O), получив фенилсульфон (2S)-10. Озонолиз 10, протекающий по ароматическому кольцу, и последующая обработка полученной кислоты диазометаном дали эфир (3S)-11, который восстановили LiAlH4 до спирта 12. С целью дифференцирования функциональных групп спиртовую группу защитили, обработав дигидропираном (DHP) в присутствии пиридиний пора-толуолсульфоната (PPTS). Сочетанием сульфона 4 с блок-синтоном, полученным из цитронеллола 3, завершили сборку молекулы (3R,7R)-гексагидрофарнезола 2, который под действием CBr4 трансформировали в целевой бромид 1.
Схема 1
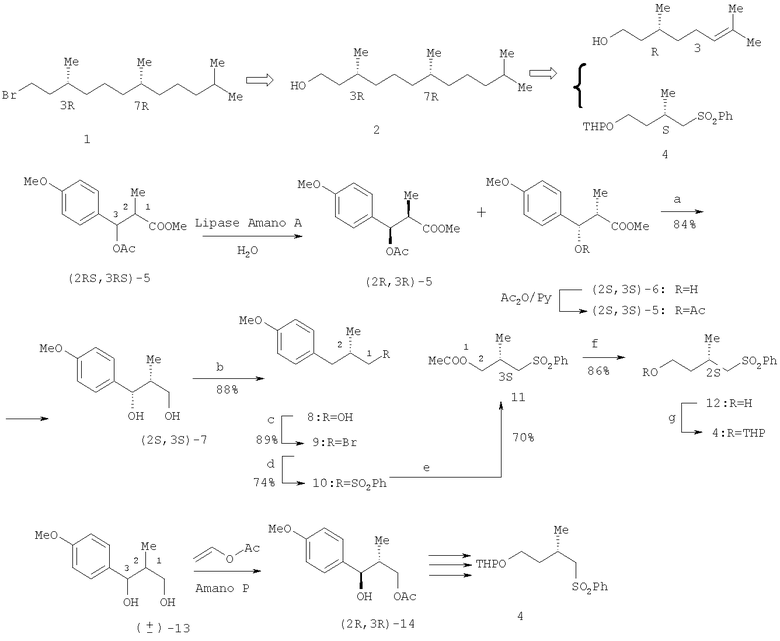
Реагенты и условия: a. LiAlH4/THF; b. Н2/20% Pd(OH)2/H+; с. NBS/Ph3Р; d.
PhSO2Na·2H2O/DMF; е. 1) О3/AcOEt 2) 30% Н2O2, 3) CH2N2/Et2O; f. LiAlH4/THF; g. DHP/PPTS.
Энантиоселективный хемоэнзиматический синтез (3R,7R)-гексагидрофарнезола 2 путем асимметрической индукции прохиральных 1,3-пропандиолов, катализируемой липазами PS и PPL осуществлен в работе [Takabe К., Sawada H., Satani Т., Yamada Т., Katagiri Т., Yoda H. Chemoenzymatic synthesis of optically active α-tocopherol side chain. // Bioorg. Med. Chem. Lett. - 1993. - V.3. - №2 - P.-157-160].
С использованием на ключевой стадии пекарских дрожжей осуществлен энантиоселективный синтез (3R,7R)-гексагидрофарнезола 2 из ненасыщенного альдегида 15. [Fuganti С., Grasselli P. Efficient stereoselective synthesis of natural α-tocopherol (vitamin E). // J. Chem. Soc., Chem. Commun. - 1979. - №19 - P.995-997]. Восстановлением пекарскими дрожжами альдегида 15 получен хиральный (2S,4Е)-2-метил-5-фенилпента-4-ен-1-ол 16, который после превращения в тозилат и сочетания с изопентилмагнийбромидом, катализируемым Li2CuCl4, трансформировали в хиральный углеводород 17. Его озонолиз и восстановительная обработка дали (3R)-3,7-диметилоктан-1-ол 18. Сочетание магнийорганического соединения, полученного из 18 (после его превращения в соответствующий бромид), с тозильным производным спирта 16, дала углеводород 19, который последующими трансформациями, описанными для 17, превратили в целевой спирт (3R,7R)-2 (схема 2).
Схема 2
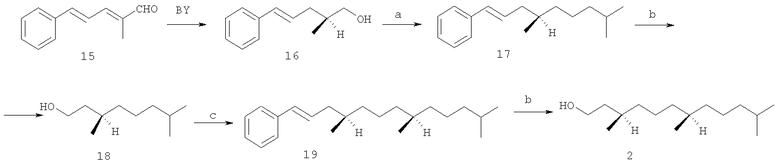
Реагенты и условия: а 1) TsCl/Py; 2) iAmMgBr/Li2CuCl4, Et2O-THF; b 1) O3/гексан, - 30°C, 2) LiAlH4/Et2O, - 30°C; с 1) NBS-Ph3Р, CH2Cl2, 2) Mg/Et2O, 3) тозилат спирта 16.
Элегантный синтез бромида 1 осуществлен с использованием лактона 20, который в свою очередь получали из бифункционального соединения 21 путем восстановления двойной связи и ацетальной функции пекарскими дрожжами [Rüttiman A. Recennt advances in the synthesis of K-vitamins. // Chimia. - 1986. - V.40. - №9. - P.290-306.]. Раскрытие лактона 20 действием HBr дало γ-бромэфир 22. Восстановлением сложноэфирной группы диизобутилалюминийгидридом с последующей обработкой дигидропираном и затем магнием получен реагент Гриньяра 23, сочетание которого с тозилатом 24 привело (после кислотного гидролиза и тозилирования) к соединению 25, превращенному под действием реагента Гриньяра 23 в гексагидрофарнезол 2. Его бромирование N-бромсукцинимидом дало целевой бромид 1 (схема 3).
Схема 3
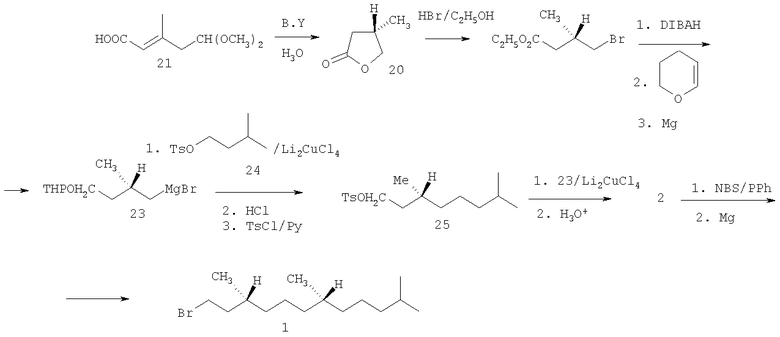
Хемоэнзиматический подход к синтезу гексагидрофарнезилбромида 1 позволяет получать целевой продукт, как правило, с высокой оптической чистотой. Вместе с тем описанные методы имеют ряд серьезных недостатков. Они чрезвычайно сложны и многостадийны, поскольку основаны на сочетании малых хиральных блоков путем многочисленных манипуляций с функциональными группами. Кроме того, в биохимических трансформациях часто используются малодоступные, дорогостоящие биокатализаторы.
В последние десятилетия в синтезе гексагидрофарнезилбромида 1 и родственных ему изопреноидов широко представлены работы, основанные на использовании асимметрического металлокомплексного катализа. Наиболее известная методология синтеза хиральных изопреноидов, в том числе (3R,7R)-гексагидрофарнезола 2 и (3R,7R)-гексагидрофарнезилбромида 1, разработанная в пионерских работах Otsuka и Noyori, базируется на асимметрической изомеризации аллиламинов в енамины под действием катионных комплексов Rh(I) с хиральными фосфиновыми лигандами. Среди последних наиболее эффективным является комплекс [Rh{(+)-binap}]+ClO4 -, содержащий хиральный фосфиновый лиганд-2,2'-бис(дифенилфосфин)-1,1'-бинафтил(binap) [Tani К., Yamagata Т., Tatsuno Y., Yamagata Y., Tomita К., Akutagawa S., Kumobayashi H., Otsuka S. Bis[(R)-(+)-binap]rhodium(I)-perchlorat, ein hochwirksamer katalysator für die asymmetrische isomerisierung von allyiaminen. // Angew. Chem. - 1985. - V.97 - №3. - P.231-234]. С использованием этого комплекса был синтезирован ряд оптически активных енаминов с оптической чистотой ≥99% [Tani К., Yamagata Т, Otsuka S., Akutagawa S., Kumobayashi H., Taketomi Т., Takaya H., Miyashita A., Noyori R. Cationic rhodium (I) complex-catalysed asymmetric isomerisatio of allylamines to optically active enamines. // J. Chem. Soc., Chem. Commun. - 1982. - P.600-601].
Эта методология была применена в асимметрическом синтезе (3R,7R)-3,7,11-триметилдодеканаля 31, необходимого для получения бромида 1 (схема 4). Исходным материалом служил (R)-цитронеллаль 26, который восстановили NaBH4, а затем гидрировали над Pd-катализатором. (R)-3,7-Диметилоктанол 27 обработали HBr, полученный бромид превратили в соответствующий реагент Гриньяра, который сочетали с E-аминосульфоном 28 в присутствии ацетилацетоната меди (II). Асимметрическую изомеризацию полученного аллиламина 29 в енамин 30 осуществили под действием каталитических количеств катионного комплекса Rh. Кислотный гидролиз енамина 30 дал альдегид 31 с оптической чистотой 97.4% ее.
Схема 4
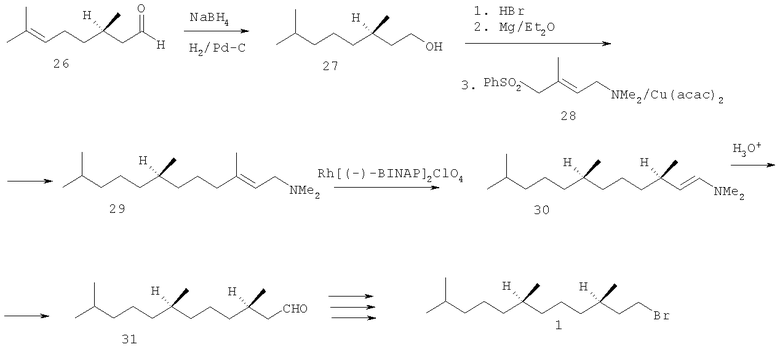
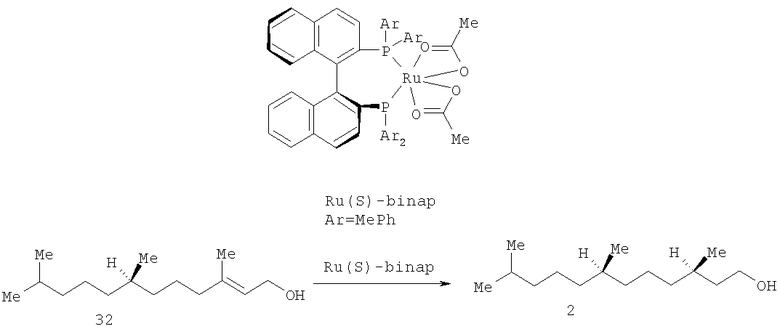
С высокой оптической чистотой спирт 2 был получен при использовании асимметрического гидрирования аллильных спиртов [Takaya H., Ohta Т., Sayo N., Kumobayashi H., Akutagawa S., Kasahara I., Noyori R. Enantioselective hydrogenation of allylic and homoallylic alcohols. // J. Am. Chem. Soc. - 1987. - V.109. - №15. P.1596-1597], эфиров, α,β-ненасыщенных карбоновых кислот и енамидов под действием дикарбоксилатных комплексов Ru(II), содержащих в качестве лигандов BINAP и его аналоги [Kawano H., Ishii Y., Ikaria Т., Saburi M., Yoshikawa S., Uchida Y. Ruthenium (II)-BINAP complex catalyzed asymmetric hydrogenation of unsaturated dicarboxylic acids. // Tetrahedron Lett. - 1987. - V.28 - №17. - P.1905-1908]. Так, селективным гидрированием (R,Е)-аллильного спирта 32 получили (3R,7R)-гексагидрофарнезол 2 с оптической чистотой 99% ее (схема 5).
Схема 5
Энантиконтролируемый многостадийный синтез (3R,7R)-гексагидрофарнезилбромида 1 был осуществлен с использованием на ключевых стадиях реакции асимметрического эпоксидирования по Шарплессу и [3,3]-сигматропной перегруппировки Кляйзена [Takano S., Sugihara Т., Ogasawara K. An enantiocontrolled synthesis of phytol by reiterative application of the chiral 3-hydroxyalkyne formation reaction. // Synlett. - 1991. - P.279-282].
Описанные методы синтеза изопреноидов 1 и 2 с применением асимметрического катализа имеют ряд общих недостатков. Как и в случае хемоэнзиматических трансформаций, они многостадийны и приводят к целевым продуктам с низким выходом. Кроме того, они основаны на использовании агрессивных или чувствительных к действию влаги и воздуха металлоорганических реагентов и катализаторов. В связи с этим данные методы большого препаративного значения не имеют.
Известно несколько примеров синтеза изопреноидов 1 и 2, когда использовались оптически активные природные продукты, например хлорофилл [Scott J.W., Bizzarro F.T., Parrish D., Saucy G. Syntheses of (2R,4'R,8'R)-α-tocopherol and (2R,3'E,7'E)-α-tocotrienol. // Helv. Chim. Acta. - 1976. - V.59. - №1. - P.270-306] (R)-пулегон [Fujisawa Т., Sato T., Kawara Т., Ohashi К. A stereocontrolled total synthesis of optically active (R,R)-phytol. // Tetrahedron Lett. - 1981. - V.22. - №48. - P.4823-4826] или мевалонолактон [Takano S., Shimazaki Y., Iwabuchi Y., Ogasawara K. A convergent enantiocontrolled route to mevalonolactone and vitamin E from (S)-O-benzylglycidol. // Tetrahedron Lett. - 1990. - V.31 - №25. - Р.3619-3622].
В работах [Mayer Н., Schudel P., Rüegg R., Isler O. Uber die chemia des vitamins E. Die total syntheese von (2R,4'R,8'R)- und (2S',4'R,8'R)-α-tocopherol. // Helv. Chim. Acta. - 1963. - V.46. - P.650-671; Scott J.W., Bizzarro F.T., Parrish D., Saucy G. Syntheses of (2R,4'R,8'R)-α-tocopherol and (2R,3'Е,7'Е)-α-tocotrienol. // Helv. Chim. Acta. - 1976. - V.59. - №1. - P.270-306] синтез изопреноидов 1 и 2 осуществлен из (7R,11R)-фитола 33. Стратегия синтеза строилась на дегидратации фитола под действием фталевого ангидрида и TsOH. Полученную смесь фитадиенов 34 и 35 (оба - смесь (E)- и (Z)-изомеров) озонировали и полученные альдегиды 36 и 37 восстановили в спирты 2 и 38 (2:38=1:3), которые разделили ректификацией. Недостатком данного способа является низкая региоселективность реакции дегидратации и образование трудно разделимых гомологов, что снижает практическую значимость метода (схема 6).
Схема 6
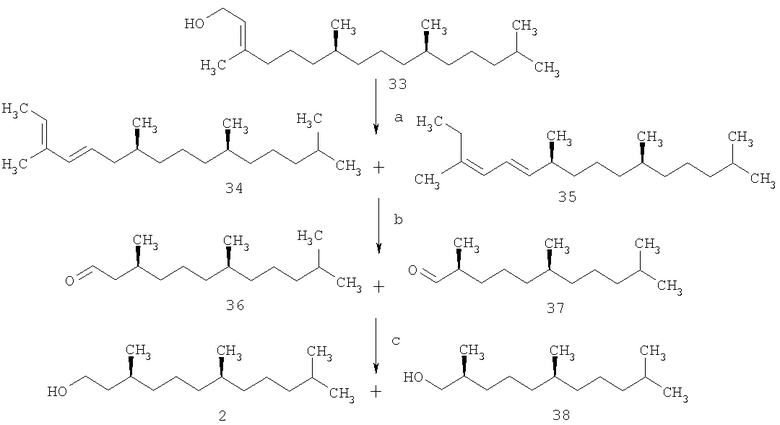
Реагенты и условия: а.фталевый ангидрид/TsOH, С6Н6; b. О3/н-C5Н12, -50°С;
с. NaAlH2(OCH2CH2OCH3)2 (Red-Al).
Как видно, в стереоконтролируемом синтезе гексагидрофарнезилбромида 1 существует большое разнообразие синтетических схем, в которых использованы очень тонкие энантиоселективные трансформации исходных и промежуточных веществ. Наиболее широко в последние годы представлены работы, в которых использованы хемоэнзиматические реакции или асимметрические превращения с применением металлокомплексного катализа. Все эти синтезы многостадийны и пока не приобрели серьезного практического значения.
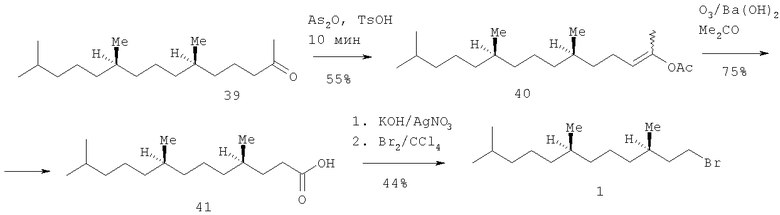
Предлагается новый энантиоселективный короткий способ получения (3R,7R)-гексагидрофарнезилбромида 1, основанный на использовании оптически чистого (6R,10R)-6,10,15-триметилпентадекан-2-она ((R,R)-фитона), полученного из хлорофилла по разработанному нами ранее методу с применением озонолиза [Одиноков В.Н., Маллябаева М.И., Спивак А.Ю., Емельянова Г.А., Джемилев У.М. Новый подход к синтезу (2RS,4'R8'R)-α-токоферола (витамина Е). // Докл. АН. - 2001. - Т.380. - №2. - 201-203]. Сущность изобретения заключается в региоселективном превращении (R,R)-фитона 39 в енолацетат 40 под действием уксусного ангидрида, каталитических количеств кристаллогидрата p-TsOH, при мольном соотношении фитон: Ас2O:TsOH=1.1:4.4:0.08 и при промотировании реакции СВЧ-облучением в микроволновом генераторе домашнего типа при максимальной мощности 750 W в течение 10 мин. Озонолиз енолацетата 40 в ацетоне в присутствии Ba(OH)2 при комнатной температуре привел к С16-кислоте 41. Ее взаимодействие с Br2 по реакции Хунсдикера в ее классическом варианте через серебряную соль кислоты дает оптически чистый бромид 1. Общий выход бромида 1 в расчете на фитон 39 составил 11%.
Преимущества предлагаемого способа:
1. Доступность и высокая оптическая чистота исходного хирального соединения - (R,R)-фитона, который получают из природного возобновляемого сырья, каковым является крапива двудомная с высоким выходом без предварительной очистки хлорофилла от сопутствующих липоидов.
2. Высокая оптическая чистота целевого продукта - (3R,7R)-гексагидрофарнезилбромида, которая обеспечивается полным сохранением конфигурации хиральных центров исходного (R,R)-фитона в трехстадийном маршруте его превращений.
3. Сравнительно короткий и перспективный для практического использования путь синтеза целевого бромида в сравнении с известными в литературе способами получения.
4. Использование реакции енолизации, промотируемой микроволновым облучением при выполнении необходимого сокращения C18-углеродной цепи (R,R)-фитона. Для реакции енолизации (R,R)-фитона, исследованной нами впервые, найдены эффективные условия региоселективной трансформации в целевой термодинамически контролируемый енолацетат. Использование микроволнового нагрева, имеющего чрезвычайно важное значение для развития «зеленой химии» и ресурсосберегающих технологий, в нашем случае позволило получить целевое соединение при незначительных расходах уксусного ангидрида, каталитических количеств TsOH (мольное соотношение (R,R)-фитон: Ас2O:TsOH=1.1:4.4:0.08), малых энергетических расходах и короткого времени (10 мин). Хотя конверсия фитона составляет 57%, образующийся енолацетат легко отделяется от не вступившего в реакцию исходного соединения с помощью колоночной хроматографии на силикагеле, а возвращенный обратно фитон подвергают повторному превращению.
5. Помимо реакции енолизации предлагаемая схема синтеза (3R,7R)-гексагидрофарнезилбромида включает реакцию озонолиза енолацетата в С16-кислоту и ее трансформацию в целевой бромид, которые выполняются по типовым нетрудоемким методикам и протекают селективно с хорошим выходом требуемых продуктов.
Изобретение поясняется следующим примером:
Стадия 1
Получение -(6R,10R)-2-ацетокси-6,10,14-триметилпента-2E/Z-децена (40). Смесь 0.3 г (1.1 ммоля) фитона 39, 0.44 г (4.4 ммоля) Ас2О и 0.002 г (0.08 ммоля 7 мол.%) TsOH·H2O помещают в термостойкий химический стакан и подвергают микроволновому облучению в бытовой микроволновой печи типа «Samsung» при 750 W в течение 10 минут. Затем реакционную смесь экстрагируют 10 мл EtOAc, раствор последовательно промывают насыщенным раствором NaHCO3 и NaCl, сушат MgSO4, упаривают в вакууме, получив 0.28 г коричневого масла, содержащего 57% енолацетатов (E/Z~2:1, ГЖХ) и 43% непрореагировавшего фитона. Хроматографированием на колонке с SiO2 (30 г, элюент СН2Cl2 получают 0,12 г енолацетатов 40, [α]D 20+6.00 (с 4.78, CHCl3). Найдено (%): С, 77.31, Н, 12.21, С20Н38O2. Вычислено (%): С, 77.36; Н 12.33. ИК-спектр, ν/см-1: 1750 (OC=O), 1650 (С=С). Спектр ЯМР 1H (δ, м.д., J/Гц): 0.75-0.90 (м, 12Н, МеС(6), МеС(10), МеС(14) и Н(15)); 1.0-1.60(м, 17Н, Н(5)-Н(14)); 1.90(м, 2Н. Н(4)), 1.89(с, 3Н, Н(1)), 2.10(с, ~1Н, СН3СО Z-изомера); 2.15(с, ~2Н, СН3СО Е-изомера); 5.00(т, ~0.7 Н, Н(3) Е-изомера, J=7.5), и 5.10(т, ~0.3 Н, Н(3) Z-изомера, J=8). Спектр ЯМР 13С (δ, м.д.): 18.99, 19.20 МеС(6), МеС(10); 22.08, 22.17 и 22.44 (МеС(1), МеС(14) и С(15)); 23.85 и 24.26 (С(8) и С(12)); 27.44 С(14); 29.16 С(1); 31.82 и 32.26 (С(6) и С(10)); 35.70, 36.68, 36.76 и 36.91 (С(5), С(7), С(9) и С(11)); 38.85 (С(13)); 117.26 (С(3) Е-изомера), 117.81 (С(3) Z-изомера), 144.59 (С(2) Е-изомера), 145.02 (С(2) Z-изомера); 169.85 (С=O, Е-изомера), 169.60 (СО, Z-изомера).
Стадия 2
Получение (4R,8R)-4,8,12-триметилтридекановой кислоты (41). Через смесь, состоящую из 0.14 г (0.45 ммоля) енолацетата 2, 0.12 г (Ва(ОН)2 и 8 мл ацетона пропускают озонокислородную смесь со скоростью 30 л·ч-1 в течение 2 мин (1 ммоль О3, производительность озонатора 30 ммоль О3·ч-1). Реакционную смесь продувают Ar, фильтруют, затем упаривают на роторном испарителе, получив 0.12 г подвижного желтого масла. Остаток растворяют в 10 мл EtOAc, обрабатывают 1н NaOH (3×20 мл), водный слой отделяют, подкисляют конц. HCl до рН~6 и экстрагируют диэтиловым эфиром (3×20 мл). Экстракт промывают насыщенным раствором NaCl, сушат MgSO4 и упаривают. Получают 0.09 г (75%) кислоты 41 виде вязкого бесцветного масла, [α]D 18+2.80 (c 19.0, CHCl3). Найдено (%): С, 74.75; Н, 12.69. С16Н32O2. Вычислено (%): С, 74.94; Н.12.54. ИК-спектр, ν/см-1: 1700 (С=О), 3100-3400 (ОН). Спектр ЯМР 1H (5, м.д.,J/Гц):0.80-1.0 (м, 12Н, МеС(4), МеС(8), МеС(12) и Н(13)); 1.05-1.90 (м, 17Н, Н(3)-Н(12)); 2.40(т, 2Н, Н(2), J=7.4); 10.7 (уш. с, 1Н, CO2Н). Спектр ЯМР 13С (δ, м.д.): 19 20 и 19.63(МеС(4) и Ме(С8)); 22.62 и 22.72 (МеС(12) и С(13)); 24.32 и 24.81 (С(6) и С(10)); 27.96(С(12)); 31.57 и 31.90 (С(2) и С(3)); 32.33 и 32.75 (С(4) и С(8)); 36.95 и 37.28 (С(5), С(7) и С(9)); 39.36 (С(11)); 180.70 (С(1)).
Стадия 3
Получение (3R,7R)-1-бром-3,7,11-триметилдодекана (42).
К 0.5 г (2 ммоля) кислоты 41 и 11 г КОН в 5 мл Н2O при перемешивании прибавляют раствор 0.34 г AgNO3 в 4 мл Н2O, осадок фильтруют, промывают на фильтре МеОН и выдерживают 16 ч при 90-100°С в вакууме (~ 5 мм рт.ст.). Полученную таким образом сухую серебряную соль кислоты 41 переносят в 5 мл CCl4 и при перемешивании прибавляют по каплям 0.25 г (3.1 ммоль) Br, затем реакционную смесь кипятят 1 ч, осадок (AgBr) фильтруют и промывают CCl4. Объединенный фильтрат промывают 10% NaHCO3, сушат MgSO4 и упаривают. Остаток хроматографируют на колонке с 20 г SiO2 (элюент -гексан-этилацетат, 3:1) получив 0.25 г (55%) бромида 42 в виде желтого масла (Rf 0.8, н-гексан-AcOEt, 3:1), [α]D 24-3.10 (с 2.15, CHCl3). Найдено(%): С, 61.02; Н, 10.35, Br, 27.33. C15H31Br. Вычислено (%): С, 61.85, Н, 10.73, Br, 27.43.. ИК-спектр, ν/см-1: Спектр ЯМР 1Н (δ, м.д., J/Гц): 0.84-0.99 (м, 12Н, МеС(3), МеС(7), 2 МеС(11)); 1.10-1.40 (м, 14Н, Н(4)-Н(11)); 1.50-1.75 (м, 1Н, Н (3) и 2Н, Н(2)); 3.43 (м, 2Н, Н(1)). Спектр ЯМР 13С (δ, м.д.): 19.00 и 19.72 (МеС(3) и МеС(7)); 22.60 и 22.69(МеС(11) и С(12)); 24.23 и 24.78 (С(5) и С(9)); 27.97 (С(11)); 31.72 (С(3)); 31.92 (С(1)); 32.13 (С(4)); 32.77 (С(7)); 36.85 (С(6)) 37.29 (С(8)); 39.37 (С(10)); 40.08 (С(2)).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (S)-(-)-6-БЕНЗИЛОКСИ-3,4-ДИГИДРО-2,5,7,8-ТЕТРАМЕТИЛ-2Н-1-БЕНЗОПИРАН-2-ИЛМЕТАНОЛА | 2010 |
|
RU2443695C1 |
| СПОСОБ ПОЛУЧЕНИЯ (6R,10R)-6,10,14-ТРИМЕТИЛПЕНТАДЕКАН-2-ОНА (ФИТОНА) | 2001 |
|
RU2197465C2 |
| СПОСОБ ПОЛУЧЕНИЯ (S)-3-(АМИНОМЕТИЛ)-5-МЕТИЛГЕКСАНОВОЙ КИСЛОТЫ | 2015 |
|
RU2643373C2 |
| СПОСОБ ЭНАНТИОСЕЛЕКТИВНОГО СИНТЕЗА (R)-ДИЭТИЛ(2-НИТРО-1-ФЕНИЛЭТИЛ) МАЛОНАТА В ПРИСУТСТВИИ КОМПЛЕКСА НИКЕЛЯ | 2011 |
|
RU2488576C2 |
| СПОСОБ ЭНАНТИОСЕЛЕКТИВНОГО СИНТЕЗА (S)-ПРЕГАБАЛИНА | 2012 |
|
RU2529996C2 |
| СПОСОБ ПОЛУЧЕНИЯ НЕРАЦЕМИЧЕСКОГО 1-(АДАМАНТ-1-ИЛ)-2-(2-НИТРО-1-ФЕНИЛЭТИЛ)БУТАН-1,3-ДИОНА | 2015 |
|
RU2612966C1 |
| СПОСОБ ОБУСЛОВЛЕННОЙ ДИФФЕРЕНЦИАЛЬНОЙ РАСТВОРИМОСТЬЮ АСИММЕТРИЧЕСКОЙ ТРАНСФОРМАЦИИ ЗАМЕЩЕННЫХ 2H-ХРОМЕН-3-КАРБОНОВЫХ КИСЛОТ | 2020 |
|
RU2792894C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНОГО 5-МЕТОКСИ-2-((4-МЕТОКСИ-3,5-ДИМЕТИЛПИРИДИН-2-ИЛ)МЕТИЛСУЛЬФИНИЛ)-1Н-БЕНЗО[d]ИМИДАЗОЛА | 2007 |
|
RU2341524C1 |
| СПОСОБ РАЗДЕЛЕНИЯ ХИРАЛЬНЫХ СУЛЬФОКСИДОВ С ПОМОЩЬЮ ЭНАНТИОСЕЛЕКТИВНОЙ ХРОМАТОГРАФИИ | 2006 |
|
RU2310505C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОКСИДОВ | 2010 |
|
RU2448954C1 |
Изобретение относится к способу получения (3R,7R)-гексагидрофарнезилбромида или (3R,7R)-1-бром-3,7,11-триметилдодекана. Процесс включает превращение (6R,10R)-6,10,14-триметилпентадекан-2-он[(R,R)-фитона] в енолацетат путем взаимодействия [(R,R)-фитона] с уксусным ангидридом в присутствии n-толуолсульфокислоты при мольном отношении фитон: Ас20: n-TsOH=1,1:4,4:0,08 под действием СВЧ-облучения в бытовом микроволновом генераторе при мощности 750 W в течение 10 мин, затем окисление енолацетата в соответствующую C16-кислоту в присутствии Ва(ОН)2 в ацетоне при комнатной температуре, последующее превращение С16-кислоты в целевой продукт путем взаимодействия с бромом по реакции Хунсдикера. Технический результат - высокая оптическая чистота продукта, короткий синтез и хороший выход продукта.
Способ получения (3R,7R)-гексагидрофарнезилбромида формулы 1
характеризующийся тем, что (6R,10R)-6,10,14-триметилпентадекан-2-он[(R,R)-фитон] превращают в енолацетат путем взаимодействия [(R,R)-фитона] с уксусным ангидридом в присутствии n-толуолсульфокислоты при мольном отношении фитон: Ас2O:n-TsOH=1,1:4,4:0,08 под действием СВЧ-облучения в бытовом микроволновом генераторе при мощности 750 W в течение 10 мин, затем енолацетат окисляют озоном в соответствующую С16-кислоту в присутствии Ва(ОН)2 в ацетоне при комнатной температуре, с последующим превращением С16-кислоты в 1 путем взаимодействия с бромом по реакции Хунсдикера.
| Scott J.W | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Chim | |||
| Acta., 1976, v.59, №1, p.270-306 | |||
| Takano S | |||
| et al | |||
| A convergent enantiocontrolled route to mevalonolactone and vitamin E from (S)-O-benzylglycidol, Tetrahedron Lett., 1990, v.31, №25, p.3619-3622 | |||
| Fujisawa T | |||
| et al | |||
| A | |||

