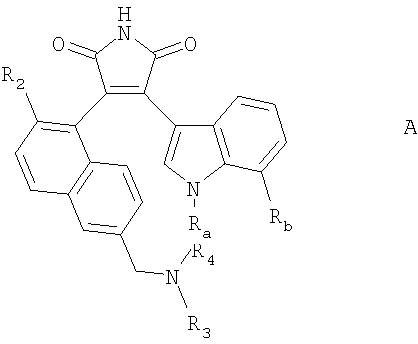
Настоящее изобретение относится к индолилмалеимидным производным, способу их получения и содержащим их фармацевтическим композициям.
В частности настоящее изобретение относится к соединению формулы I:

где
Ra представляет собой Н; С1-4алкил или С1-4алкил, замещенный ОН, NH2, NHC1-4алкилом или N(ди-С1-4алкил)2;
один из Rb, Rc, Rd и Re представляет собой галоген; С1-4алкокси или С1-4алкил, и
другие три заместителя каждый представляет собой Н; или Rb, Rc, Rd и Re все представляют собой H; и
R1 представляет собой радикал формулы (а):

где
R1 представляет собой -(СН2)n-NR3R4, где
каждый R3 и R4 независимо представляет собой Н или С1-4алкил; или R3 и R4 вместе с атомом азота, к которому они присоединены, образуют гетероциклический остаток;
n обозначает 0, 1 или 2; и
R2 представляет собой Н; галоген; С1-4алкил; CF3; ОН; SH; NH2; NO2; C1-4алкокси;
С1-4алкилтио; NHC1-4алкил; N(ди-С1-4алкил)2 или CN.
Соединение формулы I может находиться в свободной форме или в форме соли.
Алкил или алкокси может быть линейным или разветвленным.
Галоген может обозначать F, Cl, Br или I, предпочтительно F, Cl или Br.
Под гетероциклическим остатком подразумевают от трех до восьми, предпочтительно от пяти до восьми членное насыщенное, ненасыщенное или ароматическое гетероциклическое кольцо, содержащее 1 или 2 гетероатома, предпочтительно выбранных из N, О и S, и необязательно замещенное.
Подходящие примеры R1 включают, например, пиридил, например 3- или 4-пиридил, пиперидил, например пиперидин-1-ил, 3- или 4-пиперидил, гомопиперидил, пиперазинил, гомопиперазинил, имидазолил, имидазолидинил, пирролил, пирролидинил или морфолин-4-ил, необязательно замещенный, например моно- или полизамещенный. Когда гетероциклический остаток является замещенным, заместитель может быть у одного или нескольких атомов углерода в цикле и/или у атома азота в цикле, если он присутствует. Примеры заместителя у атома углерода в кольце включают, например, С1-4алкил, например СН3; С3-6циклоалкил, например циклопропил, необязательно также замещенный С1-4алкилом; 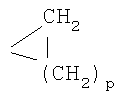 , где p обозначает 1, 2 или 3, предпочтительно 1; CF3; галоген; NH2; -CH2-NR7R8, где каждый из R7 и R8, независимо, представляет собой Н, или С1-4алкил или R7 и R8 вместе с атомом азота, с которым они связаны, образуют гетероциклический остаток или гетероарил; -CH2-OH; -СН2-O-С1-4алкил; -СН2-галоген или -СН2-СН2-галоген. Примерами заместителя у атома азота в цикле являются, например С1-6балкил; ацил, например R'x-CO, где R'x представляет собой Н, С1-6алкил или фенил, необязательно замещенный С1-4алкилом, С1-4алкокси или амино, например формил; С3-6циклоалкил; С3-6циклоалкил-С1-4алкил; фенил; фенил-С1-4алкил, например бензил; гетероциклический остаток, например как описано выше, например ароматический гетероциклический остаток, содержащий 1 или 2 атома азота; или остаток формулы β
, где p обозначает 1, 2 или 3, предпочтительно 1; CF3; галоген; NH2; -CH2-NR7R8, где каждый из R7 и R8, независимо, представляет собой Н, или С1-4алкил или R7 и R8 вместе с атомом азота, с которым они связаны, образуют гетероциклический остаток или гетероарил; -CH2-OH; -СН2-O-С1-4алкил; -СН2-галоген или -СН2-СН2-галоген. Примерами заместителя у атома азота в цикле являются, например С1-6балкил; ацил, например R'x-CO, где R'x представляет собой Н, С1-6алкил или фенил, необязательно замещенный С1-4алкилом, С1-4алкокси или амино, например формил; С3-6циклоалкил; С3-6циклоалкил-С1-4алкил; фенил; фенил-С1-4алкил, например бензил; гетероциклический остаток, например как описано выше, например ароматический гетероциклический остаток, содержащий 1 или 2 атома азота; или остаток формулы β

где R5 представляет собой С1-4алкилен или С2-4алкилен, содержащий О, и Y' представляет собой ОН, NH2, NH(C1-4алкил) или N(C1-4алкил)2. С2-4алкилен, содержащий О, может представлять собой, например, -СН2-СН2-О-СН2-СН2-.
Соединения формулы I могут существовать в свободной форме или в форме соли, например солей с органическими или неорганическими кислотами, например хлористоводородной кислотой, уксусной кислотой, трифторуксусной кислотой.
Следует понимать, что соединения формулы I могут существовать в форме оптических изомеров, рацематов или диастереоизомеров. Например, атом углерода в цикле, содержащем заместитель в положении 3 а пиперазинильного остатка, является асимметричным и может иметь R- или S-конфигурацию. Следует понимать, что настоящее изобретение включает все энантиомеры и их смеси. Энантиомеры являются предпочтительными по сравнению с рацематами. Аналогичные варианты применимы в отношении исходных материалов, имеющих указанные асимметричные атомы углерода.
В соединениях формулы I следующие значения являются предпочтительными отдельно или в любой субкомбинации:
1. Ra представляет собой Н или метил;
2. один из Rb, Rc, Rd и Re представляет собой метил или этил и другие три заместителя каждый представляет собой Н; или Rb, Rc, Rd и Re все представляют собой Н;
3. R2 представляет собой Н, Cl, NO2, CF3, F или метил;
4. n обозначает 1; и
5. каждый из R3 и R4 независимо представляет собой H, метил, этил или изопропил; или R3 и R4 вместе с атомом азота, с которым они связаны, образуют гетероциклический остаток, например необязательно замещенный пиперазинил или пирролидинил.
Настоящее изобретение также включает способ получения соединения формулы I, который включает реакцию соединения формулы II:
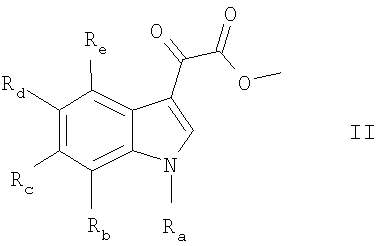
где Ra, Rb, Rc, Rd и Re имеют указанные выше значения, с соединением формулы III:
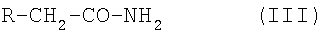
где R является таким, как указано выше,
и, при необходимости, превращение полученного соединения формулы I в свободной форме в форму соли или наоборот, если это возможно.
Способ обычно может осуществляться в присутствии сильного основания, например t-BuOK, например как описано в WO 02/38561 или WO 03/08259, содержание которых приведено здесь в качестве ссылки, и как проиллюстрировано в примерах.
Соединения формулы II и III могут быть получены в соответствии с известными способами, например как описано в WO 02/38561 или WO 03/08259, содержание которых приведено здесь в качестве ссылки, и как проиллюстрировано в примерах.
Несмотря на то, что получение исходных реагентов конкретно не описано, соединения являются известными или могут быть получены аналогично способам, известным из уровня техники или как описано далее.
Следующие примеры иллюстрируют изобретение без его ограничения.
Пример 1
3-(2-Хлор-6-диметиламинометилнафтален-1-ил)-4-(1-метил-lH-индол-3-ил)пиррол-2,5-дион
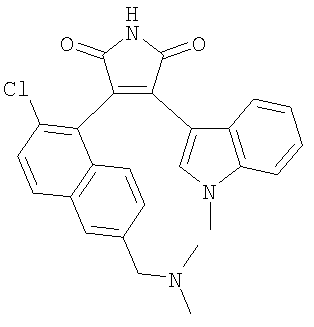
Активированные молекулярные сита 3 Ǻ (50 мг) добавляли к раствору 2-(2-хлор-6-диметиламинометилнафтален-1-ил)ацетамида (54,6 ммоль, 0,20 ммоль) и метилового эфира (1-метил-1Н-индол-3-ил)оксоуксусной кислоты (55,7 мг, 0,26 ммоль) в сухом ТГФ (2,5 мл) в атмосфере аргона. Затем добавляли раствор 1,0 М KOtBu в ТГФ (0,59 мл, 0,59 ммоль) одной порцией при КТ. Через 30 минут при КТ анализ ТСХ показал полное превращение исходных реагентов. Реакционную смесь разбавляли EtOAc и выливали в насыщенный водный раствор NH4Cl. Органический слой отделяли, промывали насыщенным водным раствором хлорида натрия, высушивали Na2SO4 и органический растворитель упаривали. Остаток очищали FCC (EtOAc/АсОН/Н2О 700:110:90) с получением указанного в заголовке соединения. 1Н ЯМР (d6-ДМСО, 400 МГц): δ 2,12 (s, 6H), 3,46 (s, 2H), 3,82 (s, 3Н), 6,16 (d, J=8,8 Гц, 1H), 6,45-6,51 (m, 1H), 6,96-7,02 (m, 1H), 7,32-7,40 (m, 2H), 7,60 - 7,68 (m, 2H), 7,88 (s, 1H), 8,06 (d, J=10 Гц, 1Н), 8,15 (s, 1H). ES+-MS: 445,5, 446,6 [M+H]+.
Получение 2-(2-хлор-6-диметиламинометилнафтален-1-ил)ацетамида
(2-Хлор-6-диметиламинометилнафтален-1-ил)уксусную кислоту (276 мг, 0,99 ммоль) растворяли в атмосфере аргона в ДМФА (3 мл). Добавляли 1,1-карбонилдиимидазол (177 мг, 1,09 ммоль) и прозрачный раствор перемешивали при КТ в течение 3 ч. Добавляли концентрированный водный раствор аммиака (25%, 6 мл) и перемешивание продолжали в течение 10 минут при КТ. Анализ ТСХ показал полное исчезновение исходного материала. Реакционную смесь выливали в воду. Водный слой экстрагировали EtOAc, который затем промывали насыщенным водным раствором хлорида натрия и высушивали Na2SO4. После удаления растворителя остаток представлял собой чистое указанное в заголовке соединение, не нуждающееся в очистке. 1Н ЯМР (d6-ДМСО, 400 МГц): δ 2,18 (s, 6Н), 3,53 (s, 2H), 4,08 (s, 2H), 6,96-7,08 (br, 2H), 7,48-7,68 (m, 2H), 7,78-7,86 (m, 2H), 7,96-8,00 (d, J=10 Гц, 1H). ES+-MS: 277,3, 279,2 [M+H]+.
Получение (2-хлор-6-диметиламинометилнафтален-1-ил)уксусной кислоты
Этиловый эфир (2-хлор-6-диметиламинометилнафтален-1-ил)уксусной кислоты (223 мг, 0,73 ммоль) растворяли в диоксане (2,6 мл). Затем добавляли воду (0,96 мл) и гидроксид лития (21 мг, 0,88 ммоль) и реакционную смесь нагревали при 60°С в течение 4 ч. Анализ ВЭЖХ показал полное исчезновение исходного материала. Реакционную смесь разбавляли водой, значение рН доводили до 6-7 добавлением 1 M водного раствора NaHSO4 и экстрагировали EtOAc. Водный слой затем концентрировали и твердый остаток повторно экстрагировали МеОН с получением чистого указанного в заголовке соединения. ES+ - MS: 278,3, 280,1 [M+H]+.
Получение этилового эфира (2-хлор-6-диметиламинометилнафтален-1-ил)уксусной кислоты
Диметиламин (5,6 М раствор в EtOH, 0,28 мл, 1,53 ммоль) добавляли в атмосфере аргона к раствору этилового эфира (2-хлор-6-формилнафтален-1-ил)уксусной кислоты (284 мг, 1,02 ммоль) в ТГФ (10 мл). Смесь перемешивали при КТ в течение 18 ч перед добавлением раствора цианоборгидрида натрия (78 мг, 1,23 ммоль) в МеОН (2 мл) и ледяной уксусной кислоты (0,29 мл, 5,13 ммоль). После перемешивания при КТ в течение 1 ч анализ ТСХ показал полное исчезновение исходного материала. Реакционную смесь разбавляли водой и значение рН доводили до 8-9 добавлением концентрированного водного раствора NaHCO3. Экстракция EtOAc, промывание насыщенным водным раствором хлорида натрия, сушка Na2SO4 и удаление растворителя приводили к получению сырого реакционного продукта. Очистка FCC (CH2Cl2/EtOH/NH3 190:9:1) приводила к получению указанного в заголовке соединения. 1Н ЯМР (CDCl3, 400 МГц): δ 1,26 (t, J=9 Гц, 3Н), 2,30 (s, 6H), 3,59 (s, 2H), 4,18 (q, J=9 Гц, 2H), 4,30 (s, 2H), 7,49 (d, J=10 Гц, 1Н), 7,54-7,58 (m, 1H), 7,69-7,76 (m, 2H), 7,91 (d, J=10 Гц, 1H). ES+-MS: 306,4, 308,3 [M+Н]+.
Получение этилового эфира (2-хлор-6-формилнафтален-1-ил)уксусной кислоты
Этиловый эфир (2-хлор-6-цианонафтален-1-ил)уксусной кислоты (1,39 г, 5,07 ммоль) растворяли в смеси воды (17 мл), пиридина (33 мл) и ледяной уксусной кислоты (17 мл). Затем добавляли гипофосфит натрия (4,30 г, 40,62 ммоль) и никель Ренея (3,2 г) при КТ. Реакционную смесь нагревали при 100°С в течение 1 ч. Анализ ТСХ показал полное исчезновение исходного материала. Реакционную смесь охлаждали до КТ и отфильтровывали через целит. После добавления силикагеля растворители удаляли на роторном испарителе. Очистка FCC (гексан/EtOAc 5:1) приводила к получению указанного в заголовке соединения. 1Н ЯМР (CDCl3, 400 МГц): δ 1,17 (t, J=8 Гц, 3Н), 4,10 (q, J=8 Гц, 2H), 4,24 (s, 2H), 7,52 (d, J=10 Гц, 1H), 7,82 (d, J=10 Гц, 1H), 7,94-7,98 (m, 2H); 8,26 (s, 1H), 10,09 (s, 1H). ES--MS: 275,2, 277,3 [M-Н]-.
Получение этилового эфира (2-хлор-6-цианонафтален-1-ил)уксусной кислоты
Этиловый эфир (2-хлор-6-трифторметансульфонилоксинафтален-1-ил)уксусной кислоты (3,59 г, 9,04 ммоль) растворяли в ДМФА (30 мл) в атмосфере аргона. После добавления тетракис(трифенилфосфан)палладия (0) (418 мг, 0,36 ммоль) и цианида цинка (II) (2,12 г, 18,09 ммоль) реакционную смесь нагревали до 125°С. Через 1 ч анализ ТСХ показал полное исчезновение исходного материала. Суспензию охлаждали до КТ и выливали в воду. Экстракцию EtOAc осуществляли после промывки органического слоя 1 М водным раствором HCl, насыщенным водным раствором NaHCO3 и насыщенным водным раствором хлорида натрия. После сушки Na2SO4 и удаления растворителя очистка FCC (гексан/EtOAc 3:1) приводила к получению указанного в заголовке соединения. 1Н ЯМР (d6-ДМСО, 400 МГц): δ 1,06 (t, J=8 Гц, 3Н), 3,98 (q, J=8 Гц, 2H), 4,24 (s, 2H), 7,66 (d, J=10 Гц, 1Н), 7,79 (d, J=10 Гц, 1Н), 7,96 (d, J=10 Гц, 1Н), 8,13 (d, J=10 Гц, 1Н), 8,54 (s, 1Н).
Получение этилового эфира (2-хлор-6-трифторметансульфонилоксинафтален-1-ил)уксусной кислоты
Этиловый эфир (2-хлор-6-гидроксинафтален-1-ил)уксусной кислоты (3,39 г, 12,80 ммоль) растворяли в атмосфере аргона в пиридине (35 мл). После охлаждения до 0°С добавляли ангидрид трифторметансульфоновой кислоты (2,32 мл, 14,08 ммоль) по каплям в течение 15 минут. После перемешивания при 0°С в течение 15 минут и при КТ в течение 1 ч анализ ТСХ показал полное исчезновение исходного материала. Реакционную смесь выливали в 1 М водный раствор NaHCO3. После экстракции EtOAc, промывки насыщенным водным раствором хлорида натрия и сушки органического слоя Na2SO4 концентрирование приводило к получению сырого реакционного продукта. Очистка FCC (гексан/EtOAc 4:1) приводила к получению указанного в заголовке соединения. 1Н ЯМР (CDCl4, 400 МГц): δ 1,48 (t, J=9 Гц, 3Н), 4,41 (q, J=9 Гц, 2H), 4,52 (s, 2H), 7,68 (d, J=10 Гц, 1Н), 7,82 (d, J=10 Гц, 1Н), 7,98-8,00 (m, 2H), 8,27 (d, J=10 Гц, 1Н).
Получение этилового эфира (2-хлор-6-гидроксинафтален-1-ил)уксусной кислоты
Этиловый эфир (2-хлор-6-метоксинафтален-1-ил)уксусной кислоты (5,43 г, 19,48 ммоль) и йодид тетрабутиламмония (9,35 г, 25,32 ммоль) растворяли в атмосфере аргона в CH2Cl2 (110 мл). Реакционную смесь охлаждали до -78°С и добавляли 1 М раствор BBr3 в CH2Cl2 (48,7 мл, 48,7 ммоль) в течение 15 минут. После перемешивания при -78°С в течение 10 минут и при КТ в течение 10 минут анализ ТСХ показал полное исчезновение исходного материала. Реакционную смесь выливали в концентрированный водный раствор NaHCO3 и смесь тщательно перемешивали в течение 20 минут при КТ. После экстракции CH2Cl2 органический слой промывали насыщенным водным раствором хлорида натрия и высушивали Na2SO4. Очистка FCC (гексан/EtOAc 2:1) приводила к получению указанного в заголовке соединения. 1Н ЯМР (CDCl3, 400 МГц): δ 1,19 (t, J=9 Гц, 3Н), 4,12 (q, J=9 Гц, 2Н), 4,18 (s, 2H), 5,35-5,60 (br, 1H), 6,93 (s, 1H), 6,99 (d, J=10 Гц, 1H), 7,33 (d, J=10 Гц, 1H), 7,42 (d, J=10 Гц, 1H), 7,70 (d, J=10 Гц, 1H). ES+-MS: 265,2, 266,8 [M+Н]+.
Получение этилового эфира (2-хлор-6-метоксинафтален-1-ил)уксусной кислоты
Смесь этилового эфира (2-хлор-6-метоксинафтален-1-ил)уксусной кислоты и этилового эфира (2-хлор-6-метокси-3,4-дигидронафтален-1-ил)уксусной кислоты (4,07 г, около 14,6 ммоль) растворяли в атмосфере аргона в диоксане (40 мл). Добавляли 2,3-дихлор-5,6-дициано-п-бензохинон (DDQ, 7,30 г, 32 ммоль), и реакционную смесь кипятили с обратным холодильником в течение 4 ч. После охлаждения до КТ добавление МеОН делало реакционную смесь гомогенной. Добавляли силикагель и растворитель удаляли на роторном испарителе. Очистка FCC (гексан/EtOAc от 980:20 до 960:40) приводила к получению указанного в заголовке соединения. 1Н ЯМР (CDCl3, 400 МГц): δ 1,32 (t, J=9 Гц, 3Н), 4,00 (s, 3Н), 4,26 (q, J=9 Гц, 3Н), 4,34 (s, 2H), 7,21 (s, 1H), 7,30 (d, J=10 Гц, 1H), 7,52 (d, J=10 Гц, 1H), 7,71 (d, J=10 Гц, 1H), 7,92 (d, J=10 Гц, 1H). ES+-MS: 279,1, 280,9 [M+H]+.
Получение этилового эфира (2-хлор-6-метоксинафтален-1-ил)уксусной кислоты и этилового эфира (2-хлор-6-метокси-3.4-дигидронафтален-1-ил)уксусной кислоты
Смесь этилового эфира (2-хлор-1-гидрокси-6-метокси-1,2,3,4-тетрагидронафтален-1-ил)уксусной кислоты (5,0 г, 16,64 ммоль), 1,1-дифенилэтена (3,2 мл), 1-метилнафталена (3 мл) и палладия на угле (10%, 500 мг) нагревали в атмосфере аргона при 180°С. Через 3 ч анализ ТСХ показал полное исчезновение исходного материала. Реакционную смесь охлаждали до КТ, разбавляли EtOAc и отфильтровывали. Удаление EtOAc и очистка FCC (от гексана 100 до смеси гексан/EtOAc от 980:20 до 960:40) приводили к получению указанной в заголовке смеси соединений.
Получение этилового эфира (2-хлор-1-гидрокси-6-метокси-1,2,3,4-тетрагидронафтален-1-ил)уксусной кислоты
Раствор EtOAc (7,2 мл, 73,96 ммоль) в ТГФ (20 мл) медленно добавляли в атмосфере аргона при -78°С к раствору литийдиизопропиламина (полученного из 10,5 мл диизопропиламина (73,96 ммоль) и 46,2 мл 1,6 М н-BuLi в гексане (73,96 ммоль)) в ТГФ (20 мл). После перемешивания при -78°С в течение 30 минут медленно добавляли раствор 2-хлор-6-метокси-3,4-дигидро-2Н-нафтален-1-она (7,79 г, 36,98 ммоль) в ТГФ (20 мл) в течение 30 минут. Реакционную смесь перемешивали при -78°С в течение 24 ч. Анализ ТСХ показал полное исчезновение исходного материала. Реакционную смесь разбавляли EtOAc и выливали в насыщенный водный раствор NH4Cl. Органический слой отделяли и промывали насыщенным водным раствором хлорида натрия. После сушки Na2SO4 растворитель удаляли. Очистка FCC (гексан/EtOAc от 920:80 до 880:120) приводила к получению указанного в заголовке соединения. 1Н ЯМР (CDCl3, 400 МГц): δ 1,22 (t, J=9 Гц, 3Н), 2,33-2,41 (m, 2H), 2,80-3,12 (m, 4H); 3,12 (s, 1H), 3,78 (s, 3Н), 4,12 (q, J=9 Гц, 2H), 5,01-5,04 (m, 1H), 6,60-6,62 (m, 1H), 6,78-6,82 (m, 1H), 7,52 (d, J=10 Гц, 1H).
Получение 2-хлор-6-метокси-3,4-дигидро-2Н-нафтален-1-она
Раствор 6-метокси-3,4-дигидро-2Н-нафтален-1-она (5,0 г, 28,37 ммоль) в ТГФ (25 мл) медленно добавляли в атмосфере аргона при -78°С к раствору литийдиизопропиламина в ТГФ (25 мл; полученного из 4,0 мл диизопропиламина (28,37 ммоль) и 17,7 мл 1,6 М н-BuLi в гексане (28,37 ммоль)). Через 30 минут при -78°С добавляли раствор паратолилсульфонилхлорида (5,41 г, 28,37 ммоль) в ТГФ (25 мл) в течение 20 минут. Охлаждающую баню с сухим льдом удаляли, и реакционную смесь нагревали до КТ. Через 1 ч анализ ТСХ показал полное исчезновение исходного материала. Добавляли насыщенный водный раствор NH4Cl (100 мл) и смесь перемешивали при КТ в течение 15 минут. Органический слой отделяли, промывали насыщенным водным раствором хлорида натрия, высушивали Na2SO4 и концентрировали. Очистка FCC (гексан/EtOAc от 920:80 до 880:120) приводила к получению указанного в заголовке соединения. 1Н ЯМР (CDCl3, 400 МГц): δ 2,54-2,63 (m, 1H), 2,68-2,75 (m, 1H), 3,04-3,12 (m, 1H), 3,38-3,46 (m, 1H), 4,02 (s, 3Н); 4,72-4,76 (m, 1H), 6,87 (s, 1H), 7,00-7,04 (m, 1H), 8,22 (d, J=10 Гц, 1H). ES+-MS: 279,1, 280,9 [M+H]+.
Следуя методике, описанной в примере 1, но используя соответствующие исходные реагенты, могут быть получены соединения формулы А, где Ra, Rb, R2, R3 и R4 являются такими, как показано ниже в таблице 1.
Соединения формулы I в свободной форме или в форме фармацевтически приемлемой соли проявляют ценные фармакологические свойства, например, ингибируя протеинкиназу С (РКС), например, РКС изоформы, такие как α, β, δ, ε, η или θ, ингибируя активацию и пролиферацию Т-клеток, например ингибируя выработку Т-клетками или цитокинами, например, IL-2, ингибированием пролиферативного отклика Т-клеток на цитокины, например IL-2, например, как показано в тестах in vitro и in vivo, и следовательно, показаны для лечения.
A. In vitro
1. Анализ с протеинкиназой С
Соединения по изобретению тестировали на их активность на различных изоформах РКС в соответствии со следующим способом. Анализ осуществляли в белых с прозрачным дном 384-луночных микротитровальных плашках с несвязывающей поверхностью. Реакционная смесь (25 мкл) содержала 1,5 мкМ субстрата акцептора тридекапептида, который является миметиком псевдосубстрата последовательности РКСα с замещением Ala→Ser, 10 мкМ 33Р-АТР, 10 мМ Mg(NO3)2, 0,2 мМ CaCl2, PKC с концентрацией белка от 25 до 400 нг/мл (в зависимости от используемого изотипа), липидные связующие (содержащие 30 мол. % фосфатидилсерина, 5 мол.% DAG и 65 мол. % фосфатидилхолина) с конечной концентрацией липида 0,5 мМ в 20 мМ буфера Tris-HCl с рН 7,4+0,1% БСА. Инкубирование осуществляли в течение 60 мин при комнатной температуре. Реакцию останавливали добавлением 50 мкл останавливающей смеси (100 мМ EDTA, 200 мкМ АТФ, 0,1% Triton X-100, 0,375 мг/ячейку покрытых стрептавидином гранул SPA в фосфатном буферном соляном растворе w/o Ca, Mg). Через 10 мин инкубирования при комнатной температуре суспензию раскручивали в течение 10 мин при 300 g. Объединенную радиоактивность измеряли с помощью счетчика Trilux в течение 1 мин. Измерения значений IC50 осуществляли обычным образом, инкубируя серийные разбавления ингибитора в концентрациях в области от 1 до 1000 мкМ. Значения IC50 рассчитывали из графика по кривой с помощью программного обеспечения XL fit®.
2. Анализ с протеинкиназой Cα
Рекомбинантную РКСα человека получали от Oxford Biomedical Research и использовали в условиях анализа, как описано выше в разделе А, 1. В этом анализе соединения формулы I ингибируют РКСα с IC50≤1 мкМ. Например, соединение примера 6 ингибирует РКСβ с IC50 1,1 нМ и соединение примера 5 с IC50 0,9 нМ.
3. Анализ с протеинкиназой Сβ1
Рекомбинантную PKCβ1 человека получали от Oxford Biomedical Research и использовали в условиях анализа, как описано выше в разделе А, 1. В этом анализе соединения формулы I ингибируют PKCβ1 с IC50≤1 мкМ. Например, соединение примера 5 ингибирует PKCβ1 с IC50 2,3 нМ и соединение примера 7 с IC50 2,8 нМ.
4. Анализ с протеинкиназой Сδ
Рекомбинантную РКСδ человека получали от Oxford Biomedical Research и использовали в условиях анализа, как описано выше в разделе А, 1. В этом анализе соединения формулы I ингибируют PKCδ с IC50≤1 мкМ. Например, соединение примера 4 ингибирует РКСδ с IC50 9,4 нМ и соединение примера 5 с IC50 4,5 нМ.
5. Анализ с протеинкиназой Сε
Рекомбинантную PKCε человека получали от Oxford Biomedical Research и использовали в условиях анализа, как описано выше в разделе А, 1. В этом анализе соединения формулы I ингибируют РКСε с IC50≤1 мкМ. Например, соединение примера 1 ингибирует РКСε с IC50 17,6 нМ и соединение примера 6 с IC50 2,3 нМ.
6. Анализ с протеинкиназой Сη
Рекомбинантную РКСη человека получали от Oxford Biomedical Research и использовали в условиях анализа, как описано выше в разделе А, 1. В этом анализе соединения формулы I ингибируют PKCη с IC50≤1 мкМ. Например, соединение примера 3 ингибирует РКСη с IC50 53,9 нМ и соединение примера 4 η с IC50 7,2 нМ.
7. Анализ с протеинкиназой Сθ
Рекомбинантную РКСθ человека получали от Oxford Biomedical Research и использовали в условиях анализа, как описано выше. В этом анализе соединения формулы I ингибируют РКСδθ с IC50≤1 мкМ. Например, соединение примера 1 ингибирует РКС θ с IC50 19,2 нМ и соединение примера 7 с IC50 6,4 нМ.
8. Анализ на совместное стимулирование CD28
Анализ осуществляли с клетками Jurkat, трансфецированными конструкцией гена промотор/репортер интерлейкина-2 человека, как описано Baumann G и др. в Transplant. Proc. 1992; 24: 43-8, причем репортерный ген β-галактозидазы замещен геном люциферазы (de Wet J., и др., Mol. Cell Biol. 1987, 7 (2), 725-737). Клетки стимулировали прикрепленными на твердую фазу антителами или форболмиристатацетатом (РМА) и затем ионофором Са++ иономицином. Для опосредованной антителами стимуляции микротитровальные планшеты Microlite TM1 (Dynatech) покрывали 3 мкг/мл козьими антимышиными антителами IgG Fc (Jackson) в 55 мкл забуференного фосфатом соляного раствора (PBS) на лунку в течение трех часов при КТ. Планшеты блокировали после удаления антител инкубированием с 2% бычьим сывороточным альбумином (БСА) в PBS (300 мкл на лунку) в течение 2 часов при КТ. После трехкратного промывания 300 мкл PBS на лунку добавляли 10 нг/мл антител анти-Т-клеточного рецептора (WT31, Becton & Dickinson) и 300 нг/мл антител анти-CD28 (15Е8) в 50 мкл 2% БСА/PBS в качестве стимулирующих антител и инкубировали в течение ночи при 4°С. Наконец планшеты промывали трижды 300 мкл PBS на лунку. Семь трехкратных серийных разбавлений тестируемых соединений в дубликатах в среде для анализа (RPMI 1640/10% сыворотки зародыша теленка (FCS), содержащей 50 мкМ 2-меркаптоэтанола, 100 единиц/мл пенициллина и 100 мкг/мл стрептомицина), получали в отдельных планшетах, смешивали с трансфецированными клетками Jurkat (клон К22 290_Н23) и инкубировали в течение 30 минут при 37°С в 5% СО2. 100 мкл этой смеси, содержащей 1×105 клеток, затем переносили в аналитические планшеты, покрытые антителами. Параллельно 100 мкл инкубировали 40 нг/мл РМА и 2 мкМ иономицина. После инкубирования в течение 5,5 часов при 37°С в 5% СО2 уровень люциферазы определяли путем измерения биолюминесценции. Планшеты центрифугировали в течение 10 мин при 500 g и супернатант удаляли вытряхиванием. Добавляли лизисный буфер, содержащий 25 мМ Tris-фосфат, рН 7,8, 2 мМ DTT, 2 мМ 1,2-диаминоциклогексан-N,N,N',N-тетраауксусной кислоты, 10% (об.) глицерина и 1% (об.) Triton Х-100 (20 мкл на лунку). Планшеты инкубировали при КТ в течение 10 минут при постоянном встряхивании. Активность люциферазы оценивали при помощи биолюминесцентного счетчика (Labsystem, Helsinki, Finland) после автоматического добавления 50 мкл/лунку люциферазного реакционного буфера, содержащего 20 мМ трицина, 1,07 мМ (MgCO3)4Mg(OH)2×5H2O, 2,67 мМ MgSO4, 0,1 мМ EDTA, 33,3 мМ DTT, 270 мкМ коэнзима А, 470 мкМ люциферина (Chemie Brunschwig AG), 530 мкМ АТФ, рН 7,8. Время удержания составляло 0,5 секунд, общее время измерения составляло 1 или 2 секунды. Низкими контрольными значениями являлись световые единицы от клеток рецептора, стимулируемого анти-Т-клетками или РМА, высокими контрольными значениями являлись уровни от клеток рецептора, стимулируемого анти-Т-клетками/анти-CD28- или РМА/иономицином в отсутствии тестируемого образца. Низкие контрольные значения вычтены из всех величин. Ингибирование, наблюдаемое в присутствии тестируемого соединения, рассчитывалось из процентного ингибирования высокого контрольного значения. Концентрация тестируемых соединений, приводящая к 50% ингибированию (IC50), определялась из кривых зависимости доза-отклик. В этом анализе соединения формулы I ингибируют клетки Jurkat, стимулируемые рецептором анти-Т-клеток/анти-CD28 и РМА/иономицином со значениями IC50≤1 мкМ.
Например, соединение примера 5 ингибирует клетки Jurkat, стимулируемые рецептором анти-Т-клеток/анти-СВ28 и РМА/иономицином со значением IC50 11,5 нМ, и соединение примера 7 со значением IC50 27,5 нМ.
9. Аллогенная реакция смешанных лимфоцитов (MLR)
Двухстадийный MLR осуществляли в соответствии со стандартными методиками (статья в J. Immunol. Methods, 1973, 2, с.279 и Мео Т. и др., Immunological Methods, New York, Academic Press, 1979, cc.227-39). Клетки селезенки от СВА и BALB/c мышей (1,6×105 клеток от каждой линии на лунку в плоскодонных микротитровальных планшетах для культур тканей, всего 3,2×105) инкубировали в среде RPMI, содержащей 10% FCS, 100 U/мл пенициллина, 100 мкг/мл стрептомицина (Gibco BRL, Basel, Switzerland), 50 мкМ 2-меркаптоэтанола (Fluka, Buchs, Switzerland), и серийно разбавленными соединениями. Осуществляли семь трехкратных разбавлений в дубликатах для тестируемого соединения. Через четыре для инкубирования добавляли 3Н-тимидин с активностью 1 мкCi. Клетки высевали после дополнительного пятичасового периода инкубирования, и связавшийся 3Н-тимидин определяли в соответствии со стандартными методиками. Базовыми значениями (низкий контроль) MLR являлась пролиферация BALB/c клеток отдельно. Низкие контрольные значения вычитали из всех значений. Высокие контроли без образца принимали за 100% пролиферацию. Рассчитывали процентное ингибирование образцами и определяли концентрации, необходимые для 50% ингибирования (значения IC50).
Например, соединение примера 5 ингибирует со значением IC50 183 нМ и соединение примера 7 со значением IC50 528 нМ.
Б. In vivo
Трансплантация сердца крысы
Использовали комбинацию линий: Male Lewis (RT1 гаплотип) и BN (RT1 гаплотип). Животных анестезировали с помощью ингаляции изофлураном. Следом за гепаринизацией донорной крысы через брюшную нижнюю вену при одновременном обескровливании через аорту вскрывали грудную клетку и немедленно охлаждали сердце. Аорту лигировали и делили дистально до первого ответвления и плечеголовной ствол разрезали в первом раздвоении. Левую легочную артерию лигировали и разделяли и правую часть отделяли, но оставляли открытой. Все другие сосуды свободно рассекали, лигировали и разделяли и донорское сердце перемещали в охлажденный льдом соляной раствор.
Реципиента подготавливали анатомированием и пережатием инфрапочечной брюшной аорты и вены. Трансплантат инплантировали анастомозом «конец в бок», используя шов хирургической мононитью 10/0, между донорским плечеголовным стволом и аортой реципиента и правой легочной артерией донора к вене реципиента. Зажимы удаляли, трансплантат прикрепляли ретробрюшинно, содержание брюшины промывали теплым соляным раствором и животное зашивали и позволяли восстанавливаться под греющей лампой. Приживаемость трансплантата наблюдали с помощью ежедневной пальпации пульса донорского сердца через брюшинную стенку. Полагали, что отторжение завершается, когда сердце останавливается. Повышение приживаемости трансплантата наблюдали у животных, обработанных соединением формулы I, вводимым перорально в суточной дозе от 1 до 100 мг/кг веса тела, предпочтительно от 1 до 30 мг/кг веса тела.
Модель «трансплантат против хозяина»
Клетки селезенки (2×107) от Wistar/F крыс инъецировали подкожно в правую заднюю ступню гибридных крыс (Wistar/F × Fischer 344)F1. Левую ступню оставляли необработанной. Животным вводили тестируемые соединения через каждые 4 дня (0-3). Подколенные лимфатические узлы удаляли на 7 день и определяли разницу в весе между двумя соответствующими лимфатическими узлами. Результаты выражали в виде ингибирования увеличения лимфатического узла (выраженное в процентах), сравнивая разницы в весе лимфатического узла в экспериментальных группах с разницей в весе между соответствующими лимфатическими узлами от группы животных, не обработанной тестируемым соединением. Воздействие на увеличение лимфатического узла оценивали на животных, обработанных соединением формулы I, вводимым перорально в суточной дозе от 1 до 100 мг/кг веса тела.
Соединения формулы I, следовательно, полезны для лечения и/или профилактики заболеваний или состояний, опосредованных Т-лимфоцитами и/или РКС, например острого или хронического отторжения органа или ткани алло- или ксенотрансплантата, реакции трансплантата против хозяина, атеросклероза, окклюзии сосуда, обусловленной травмой сосуда, такой как ангиопластика, реатеноза, ожирения, синдрома X, ослабленной толерантности к глюкозе, полициститного синдрома яичников, гипертензии, сердечной недостаточности, хронического обструктивного легочного заболевания, заболеваний ЦНС, таких как болезнь Альцгеймера или амиотрофный латеральный склероз, рака, инфекционных заболеваний, таких как СПИД, септический шок или респираторный дистрессовый синдром у взрослых, травмы при ишемии/реперфузии, например инфаркт миокарды, инсульта, кишечной ишемии, почечной недостаточности или геморрагического шока, или травматического шока, например травматической травмы мозга. Соединения формулы I также полезны для лечения и/или профилактики опосредованных Т-клетками острых или хронических воспалительных заболеваний или состояний или аутоиммунных заболеваний, например ревматоидного артрита, остеоартрита, системной красной волчанки, тироидита Хашимото, рассеянного склероза, бульбоспинального паралича, диабетов типа I или II и связанных с ними заболеваний, респираторных заболеваний, таких как астма или воспалительная легочная травма, воспалительной травмы печени, воспалительной гломерулярной травмы, кожных проявлений иммунологически-опосредованных заболеваний или болезней, воспалительных и гиперпролиферативных кожных заболеваний (таких как псориаз, атопический дерматит, аллергический контактный дерматит, раздражающий контактный дерматит и другие экзематозные дерматиты, себорейный дерматит), воспалительных заболеваний глаз, например синдрома Съергена, кератоконьюктивита или ювеита, воспалительного заболевания кишечника, болезни Крона или язвенного колита. Для указанных выше применений необходимая дозировка будет конечно зависеть от способа введения, конкретного излечиваемого состояния и желаемого эффекта. Обычно, удовлетворительные результаты получают при систематическом введении суточных дозировок от около 0,1 до около 100 мг/кг веса тела. Показанная суточная дозировка для более крупных млекопитающих, например людей, находится в области от около 0,5 мг до около 2000 мг, обычно вводимая, например, отдельными дозами до четырех раз в день или в форме замедленного высвобождения.
Соединения по изобретению могут вводиться любым обычным способом, в частности энтерально, например перорально, например в форме таблеток или капсул, или парентерально, например в форме инъекционных растворов или суспензий, местно, например в форме лосьонов, гелей, мазей или кремов, или в форме назального или суппозиторного введения. Фармацевтические композиции, включающие соединение по изобретению в свободной форме или в форме фармацевтически приемлемой соли, вместе с по крайней мере одним фармацевтически приемлемым носителем или разбавителем могут быть получены обычным способом смешения с фармацевтически приемлемым носителем или разбавителем. Единичные дозированные формы для перорального введения включают, например, от около 0,1 мг до около 500 мг активного вещества.
Местное введение обозначает, например, нанесение на кожу. Другой формой местного введения является применение на глаза.
Соединения формулы I могут вводиться в свободной форме или в форме фармацевтически приемлемой соли, например, как показано выше. Такие соли могут быть получены обычным способом и проявляют ту же активность, как и свободные соединения.
В соответствии с вышеизложенным, настоящее изобретение также относится к:
1.1 способу профилактики или лечения заболеваний или болезней, опосредованных Т-лимфоцитами и/или РКС, например таких, которые описаны выше, у субъекта, нуждающегося в таком лечении, который включает введение указанному субъекту эффективного количества соединения по изобретению, например соединения формулы I, или его фармацевтически приемлемой соли;
1.2 способу профилактики или лечения острого или хронического отторжения трансплантата или опосредованных Т-клетками воспалительных или аутоиммунных заболеваний, например указанных выше, у субъекта, нуждающегося в таком лечении, который включает введение указанному субъекту эффективного количества соединения по изобретению, например соединения формулы I, или его фармацевтически приемлемой соли;
2. соединению формулы I, в свободной форме или в форме фармацевтически приемлемой соли, для применения в качестве лекарственного средства, например для любого из способов, описанных выше в 1.1 или 1.2;
3. фармацевтической композиции, например для применения в любом из способов 1.1 или 1.2, включающей соединение формулы I, в свободной форме или в форме фармацевтически приемлемой соли, вместе с фармацевтически приемлемым разбавителем или носителем;
4. соединению формулы I или его фармацевтически приемлемой соли для применения в изготовлении фармацевтической композиций для применения в любом из описанных выше способов 1.1 и 1.2.
Соединения по изобретению, например соединение формулы I, может вводиться в качестве единственного активного ингредиента или вместе с другими лекарственными средствами в иммуномодулирующих режимах, или с другими противовоспалительными агентами, например для лечения или профилактики острого или хронического отторжения алло- или ксенотрансплантата или воспалительных, или аутоиммунных заболеваний. Например, они могут использоваться в комбинации с циклоспоринами или аскомицинами или их иммуносупрессорными аналогами, или производными, например циклоспорином A, ISA Tx247, FK-506, ABT-281, ASM 981; ингибитором mTOR, например рапамицином, 40-O-(2-гидроксиэтил)-рапамицином, CCI779, АВТ578, или рапалогом, например АР23573, АР23464, АР23675, АР23841, TAFA-93, биолимусом 7 или биолимусом 9 и т.д.; кортикостероидами; циклофосфамидом; азатиопреном; метотрексатом; агонистом рецептора EDG со свойствами ускоренного наведения лимфоцитов, например FTY 720 или его аналогом; лефлуномидом или его аналогами; мизорибином; микофенольной кислотой или ее солью, например натриевой солью; микофенолятом мофетила; 15-дезоксиспергуалином или его аналогами; иммуносупрессорными моноклональными антителами, например моноклональными антителами к лейкоцитным рецепторам, например МHС, CD2, CD3, CD4, CD 11a/CD18, CD7, CD25, CD27, B7, CD40, CD45, CD58, CD137, ICOS, CD150 (SLAM), OX40, 4-1BB, или их лигандами, например CD154; или другими иммуномодуляторными соединениями, например рекомбинантной связывающей молекулой, имеющей по крайней мере часть внеклеточного домена CTLA4 или его мутанта, например по меньшей мере внеклеточную часть CTLA4 или его мутанта, связанного с не-CTLA4 белковой последовательностью, например CTLA4Ig (например, известная как АТСС 68629) или его мутанта, например LEA29Y; ингибиторами молекулы адгезии, например mAbs или низкомолекулярные ингибиторы, включая антагонисты LFA-1, антагонистами селектина и антагонистами VLA-4. Соединения формулы I также могут вводиться вместе с антипролиферативным лекарственным средством, например химиотерапевтическим лекарственным средством, например которое используется для лечения рака, включая, но не ограничиваясь ими, ингибиторы ароматазы, антиэстрогены, ингибиторы топоизомеразы I, ингибиторы топоизомеразы II, микротубулярные активные агенты, алкилирующие агенты, ингибиторы гистондезацетилазы, ингибиторы фарнезилтрансферазы, ингибиторы СОХ-2, ингибиторы ММР, ингибиторы mTOR, антинеопластические антиметаболиты, платиновые соединения, соединения, снижающие активность протеинкиназы, и другие антиангиогенные соединения, агонисты гонадорелина, антиадрогены, бенгамиды, бисфосфонаты, антипролиферативные антитела и темозоломид, или с антидиабетическим лекарственным средством, веществом, повышающим секрецию инсулина, или усилителем секреции инсулина, например сульфонилмочевиной, например толбутамидом, хлорпропамидом, толазамидом, ацетогексамидом, 4-хлор-N-[(1-пирролидиниламино)карбонил]бензолсульфонамидом (глюкопирамидом), глибенкамидом (глибуридом), гликлазидом, 1-бутил-3-метанилилмочевиной, карбутамидом, глибонуридом, глипизидом, глихидоном, глисоксепидом, глибутиазолом, глибузолом, глигексамидом, глимидином, глипинамидом, фенбутамидом или толилцикламидом, производным перорального инсулинотропного агента, например усилителем инсулина короткого действия, например меглитинидом, репаглинидом, производным фенилуксусной кислоты, например натеглинидом, ингибитором DPP IV, например 1-{2-[(5-цианопиридин-2-ил)амино]этиламино}ацетил-(2S)-цианопирролидина дигидрохлоридом, LAF237, аналогом агониста GLP-1 или GLP-1, или активатором инсулина, например γ-агонистом рецептора, активируемого пероксисомным пролифератором (PPARγ), например глитазоном, отличным от глитазона соединением, таким как аналог Н-(2-бензоилфенил)-α-тирозина, например GI-262570, или оксолидиндионом, например JTT501, двойным агонистом PPARγ/PPARα, например DRF-554158, NC-2100 или NN-622, агонистом ретиноидного рецептора Х или рексиноидом, например 2-[1-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидро-2-нафтил)-циклопропил]пиридин-5-карбоновой кислотой, 4-[(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидро-2-нафтил)-2-карбонил]бензойной кислотой, 9-цисретиноевой кислотой или ее аналогами, их производным или фармацевтически приемлемой солью, при лечении диабета.
В соответствии с вышеизложенным, настоящее изобретение относится к другому варианту осуществления:
5. способу, как описано выше, включающему совместное введение, например совместно или последовательно, терапевтически эффективного нетоксичного количества ингибитора РКС или Т-клеточной активации и пролиферации, например соединения формулы I в свободной форме или в форме фармацевтически приемлемой соли, и второго лекарственного вещества, где указанное второе лекарственное вещество является иммунодепрессантом, иммуномодулятором, противовоспалительным, антипролиферативным или антидиабетическим лекарственным средством, например как описано выше;
6. терапевтической комбинации, например набору, включающему а) ингибитор РКС или Т-клеточной активации и пролиферации, например соединение формулы I в свободной форме или в форме фармацевтически приемлемой соли, и б) по крайней мере один второй агент, выбранный из иммунодепрессанта, иммуномодулятора, противовоспалительного, антипролиферативного и антидиабетического лекарственного средства. Компонент а) и компонент б) может использоваться одновременно или последовательно. Набор может включать инструкции по его применению.
Если ингибитор РКС или Т-клеточной активации и пролиферации, например соединение формулы I, вводят в сочетании с другим иммунодепресантом/иммуномодулятором, противовоспалительным, антипролиферативным или антидиабетическим лекарственным средством, например для профилактики или лечения острого или хронического отторжения трансплантата или воспалительных или аутоиммунных заболеваний, как описано выше, дозировки совместно вводимого иммунодепрессанта, иммуномодулятора, противовоспалительного, антипролиферативного или антидиабетического соединения будут, конечно, зависеть от типа совместно применяемого лекарственного вещества, например является ли оно стероидом или циклоспорином, от специфичности применяемого лекарственного средства, от состояния болезни и так далее.
Соединения формулы I имеют отличный фармакокинетический профиль и отличные активности in vitro и in vivo.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНДОЛИЛМАЛЕИМИДНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ НАРУШЕНИЙ ИЛИ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ Т-ЛИМФОЦИТАМИ И/ИЛИ ПКС | 2003 |
|
RU2340610C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| МАКРОГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2001 |
|
RU2275373C2 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2794333C1 |
| 3-АРИЛ-5-ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ИЗОХИНОЛИН-1-ОНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2696572C2 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| Имидазопиридиновые соединения в качестве ингибиторов PAD | 2018 |
|
RU2782743C2 |
| СОЕДИНЕНИЯ БЕНЗОТИАЗЕПИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ЖЕЛЧНЫХ КИСЛОТ | 2020 |
|
RU2838460C1 |
| МАКРОЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ГЕПАТИТА C | 2000 |
|
RU2247126C2 |
Изобретение относится к новым производным индолилмалеимида формулы I:
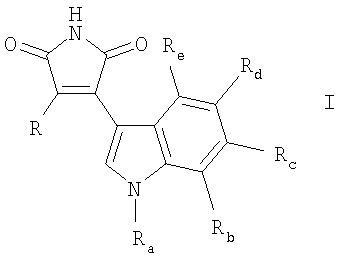
где Ra означает Н, С1-4алкил; один из Rb, Rc, Rd и Re означает С1-4алкил; и другие три заместителя означают H; или Rb, Rd, Re все означают Н; и
R означает радикал формулы (а):
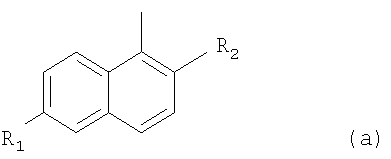
где R1 означает-(СН2)n-NR3R4, где каждый из R3 и R4 независимо означает Н, С1-4алкил; n обозначает 0, 1, 2; R2 означает Н; галоген;
или его фармацевтически приемлемая соль.
4 н. и 4 з.п. ф-лы, 1 табл.
1. Соединение формулы (I)

где Ra представляет собой Н или С1-4алкил;
один из Rb, Rc, Rd и Re представляет собой С1-4алкил; и
другие три заместителя представляют собой Н;
или Rb, Rd и Rg все представляют собой Н; и
R представляет собой радикал формулы (а)
где R1 представляет собой -(СН2)n-NR3R4, где каждый из R3 и R4 независимо представляет собой Н или С1-4алкил;
n обозначает 0, 1 или 2; и
R2 представляет собой Н;
галоген;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где Ra представляет собой Н или метил; один из Rb, Rc, Rd и Re представляет собой метил и другие три заместителя представляют собой Н; или Rb, Rc, Rd и Re все представляют собой Н; R2 представляет собой Н или Cl; n обозначает 1; и каждый из R3 и R4 независимо представляет собой Н, метил, или его фармацевтически приемлемая соль.
3. Соединение по п.1 или 2, выбранное из следующих:
3-(2-хлор-6-диметиламинометилнафтален-1-ил)-4-(1-метил-1Н-индол-3-ил)пиррол-2,5-дион;
3-(2-хлор-6-метиламинометилнафтален-1-ил)-4-(1Н-индол-3-ил)пиррол-2,5-дион;
3-(6-аминометилнафтален-1-ил)-4-(1-метил-1Н-индол-3-ил)пиррол-2,5-дион;
3-(2-хлор-6-диметиламинометилнафтален-1-ил)-4-(1Н-индол-3-ил)пиррол-2,5-дион;
3-(2-хлор-6-диметиламинометилнафтален-1-ил)-4-(7-метил-1Н-индол-3-ил)пиррол-2,5-дион;
3-(2-хлор-6-метиламинометилнафтален-1-ил)-4-(7-метил-1Н-индол-3-ил)пиррол-2,5-дион;
3-(6-аминометилнафтален-1-ил)-4-(1Н-индол-3-ил)пиррол-2,5-дион;
3-(6-аминометилнафтален-1-ил)-4-(7-метил-1Н-индол-3-ил)пиррол-2,5-дион или его фармацевтически приемлемая соль.
4. Соединение по любому одному из пп.1-3 в свободной форме или в форме фармацевтически приемлемой соли для применения в качестве лекарственного средства, ингибирующего протеинкиназу С (РКС) и/или ингибирующего активацию Т-клеток.
5. Фармацевтическая композиция, ингибирующая протеинкиназу С (РКС) и/или ингибирующая активацию Т-клеток, включающая соединение по любому одному из пп.1-3 в свободной форме или в форме фармацевтически приемлемой соли, вместе с его фармацевтически приемлемым разбавителем или носителем.
6. Применение соединения по любому одному из пп.1-3 в свободной форме или в форме фармацевтически приемлемой соли или фармацевтической композиции по п.5 для изготовления лекарственного средства, ингибирующего протеинкиназу С (РКС) и/или ингибирующего активацию Т-клеток.
7. Применение по п.6 упомянутого там соединения или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения и/или профилактики опосредованных Т-клетками острых или хронических воспалительных заболеваний или отторжения трансплантатов.
8. Способ получения соединения формулы I по п.1 или 2, который включает реакцию соединения формулы (II)
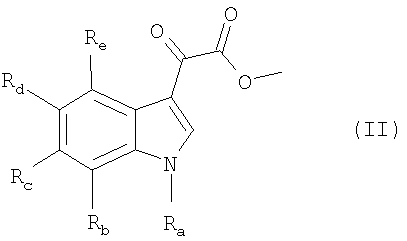
где Ra, Rb, Rc, Rd и Re имеют значения, указанные в пп.1 и 2, с соединением формулы III:
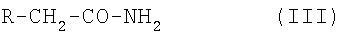
где R имеет значения, определенные в пп.1 и 2,
и, при необходимости, превращение полученного соединения формулы I в свободной форме в форму его фармацевтически приемлемой соли соли или наоборот, как подходит.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| БИС-ИНДОЛМАЛЕИМИДНЫЕ МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2147304C1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

