Область техники, к которой относится изобретение
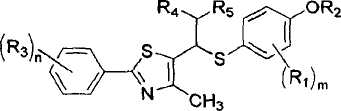
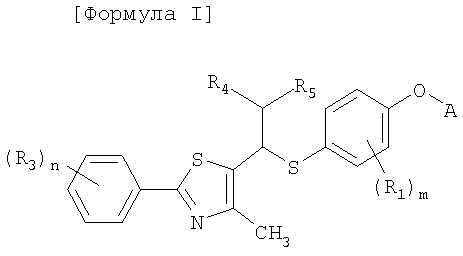
Настоящее изобретение относится к новым тиазольным производным, представленным формулой I, в качестве лигандов, активирующих пролифератор-активируемый рецептор δ пероксисомы (PPARδ), которые могут использоваться для лечения тучности, гиперлипидемии, атеросклероза и диабета, а также к их промежуточным соединениям и к способам их получения:
Формула I
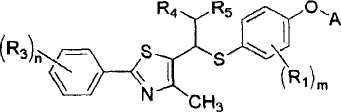
где A представляет собой водород, R2 или
Уровень техники
Среди ядерных рецепторов пролифератор-активируемый рецептор пероксисомы (PPAR) включает три субтипа: PPARα, PPARγ, PPARδ (Nature, 1990, 347, p.645-650, Proc. Natl. Acad. Sci. USA 1994, 91, p.7335-7359). PPARα, PPARγ и PPARδ имеют функции, различающиеся по отношениям к тканям in vivo, и экспрессируются в различных местах. PPARα экспрессируется в основном в сердце, почках, скелетных мышцах и толстой кишке человека (Mol. Pharmacol. 1998, 53, p.14-22, Toxicol. Lett. 1999, 110, p.1l9-127, J. Biol. Chem. 1998, 273, p.16710-16714) и ассоциируется с β-окислением пероксисомы и митохондрии (Biol. Cell. 1993, 77, p.67-76, J. Biol. Chem. 1997, 272, p.27307-27312). PPARγ, как известно, слабоэкспрессируется в скелетных мышцах, но экспрессируется в основном в жировых тканях и, таким образом, является вовлеченным в дифференциацию жировых клеток, хранение энергии в форме жира и регуляцию гомеостаза инсулина и сахара (Moll. Cell. 1999, 4, p.585-594, p.597-609, p.611-617). PPARδ эволюционно сохраняется у позвоночных, таких как млекопитающие, включая людей, грызунов и Ascidiacea. Те, которые обнаружены в настоящее время, известны как PPARβ у Xenopus laevis (Cell 1992, 68, p.879-887) и как NUCI (Mol. Endocrinol. 1992, 6, p.1634-1641), PPARδ (Proc. Natl. Acad. Sci. USA 1994, 91, p.7355-7359), NUCI (Biochem. Biophys. Res. Commun. 1993, 196, p671-677), FAAR (J. Bio. Chem. 1995, 270, p.2367-2371) и тому подобное у людей, но эти наименования недавно стандартизированы как PPARδ. У людей, как известно, PPARδ присутствует в хромосоме 6p21.l-p21.2, в то время как у крыс мРНК PPARδ обнаружена в клетках, в различных местах, но ее количество, как показано, меньше, чем для PPARα и PPARγ (Endocrinology 1996, 137, p.354-366, J. Bio. Chem. 1995, 270, p.2367-2371, Endocrinology 1996, 131, p.354-366). В соответствии с результатами проведенных к настоящему времени исследований PPARδ, как известно, играет важную роль в процессе экспрессирования (Genes Dev. 1999, 13, p.1561-1574) и в осуществлении физиологических функций, включая дифференциацию нервных клеток в центральной нервной системе (CNS) (J. Chem. Neuroanat 2000, 19, p.225-232) и заживление ран посредством противовоспалительного воздействия (Genes Dev. 2001, 15, p.3263-3277, Proc. Natl. Acad. Sci. USA 2003, 200, p.6295-6296). Недавние исследования продемонстрировали, что PPARδ связан с дифференциацией жировых клеток и метаболизмом жиров (Proc. Natl. Acad. Sci. USA 2002, 99, p.303-308, Mol. Cell. Biol. 2000, 20, p.5119-5128), и обнаружено, что PPARδ активирует экспрессию ключевых генов, связанных с β-окислением и расщеплением белков (UCP), связанных с энергетическим метаболизмом, в процессе деградации жирных кислот (Nature 2000, 406, p.415-418, Cell 2003, 113, p.159-170, PLoS Biology 2004, 2, p.1532-1539). Кроме того, активирование PPARδ делает возможным повышение уровней HDL и улучшение состояния при диабете II типа без изменения массы тела (Proc. Natl. Acad. Sci. USA 2001, 98, p.5306-5311, 2003, 100, p.15924-15929), и подавление ассоциированных с атеросклерозом генов, с тем чтобы лечить атеросклероз (Science, 2003, 302, p.453-457). Соответственно регуляция метаболизма жиров посредством PPARδ обеспечивает важное решение, необходимое для лечения тучности, диабета, гиперлипидемии и атеросклероза.
Кристаллическая структура Apo-PPARδ LBD определяется на основе уже известной структуры PPARγ (Nature 1998, 395, p.137-143), и сообщается, что структура LBD демонстрирует представляющее интерес сходство между PPARδ и PPARγ, и в частности, размер карманов связывания лигандов является по существу одинаковым у обеих PPAR (Mol. Cell. 1999, 3, p.397-403). Однако различные лиганды, селективные по отношению к форме кармана, будут связываться с PPAR, и поэтому будет видна разница в функционировании между PPAR. Как результат исследований дополнительных деталей кристаллической структуры PPARδ LBD, он состоит из 13 α-спиралей и 4 маленьких β-нитей, и его карман для связывания лиганда имеет Y-образную форму и имеет размер примерно 1300 Е3. Можно увидеть, что вход кармана для связывания лиганда имеет размер примерно 100 Е2, и его периферия состоит из полярных аминокислот. Анализ связывания природной эйкозопентеновой кислоты (EPA) и синтетического лиганда GW2433 показывает, что аминокислота Y473 на сайте AF-2 кристаллической структуры PPARδ образует водородную связь с карбоновой кислотой лиганда (Proc. Natl. Acad. Sci. USA 2001, 98, p.13919-13924). Это подтверждается тем фактом, что одна сторона структуры большинства PPARδ-активируемых лигандов состоит из функциональной группы, которая может образовывать водородную связь. Соответственно можно предположить, что связывание соактиватора, ассоциированного с PPARδ, хорошо поддерживается посредством стабилизации водородной связи между спиралью AF-2 и лигандом. При исследовании кристаллической структуры кармана для связывания лиганда PPARδ обнаружено также, что активный лиганд требует наличия гидрофобной функциональной группы на другой стороне. Как результат, предполагается, что благодаря размеру кармана для связывания лиганда PPARδ могут связываться различные типы лигандов, и поэтому демонстрируется различие в их активации (Nature 1998, 391, p.79-82).
В случае PPARδ разработка высокоселективных синтетических лигандов является относительно недостаточной по сравнению с PPARα и PPARγ. Селективный лиганд, разработанный на первой стадии, представляет собой L-631033, о котором сообщала группа исследователей Merk Co. (J. Steroid Biochem. Mol. Biol. 1997, 63, p.1-8), при этом лиганд L-631033 был получен посредством введения функциональной группы, способной фиксировать боковую цепь, на основе структуры природных жирных кислот. Кроме того, та же группа исследователей сообщила о более эффективном лиганде L-165041 (J. Med. Chem. 1996, 39, p.2629-2654), который представляет собой соединение, уже известное в качестве агониста лейкотриена, которое действует также как активатор на PPARδ человека. Это вещество демонстрирует в 10 раз более высокую селективность относительно hPPARδ, чем к PPARα и PPARγ, и имеет значение EC50 530 нМ. Однако при исследовании на грызунах он почти не имеет селективности относительно PPARγ. Другие лиганды L-796449 и L-783483 имеют значительно лучшее сродство (EC50=7,9 нМ), но не демонстрируют селективности к другим субтипам hPPAR. Группа исследователей из Glaxo-Smith-Kline Co. сообщала об активаторе PPARα GW2433, лиганде Y-образной формы, имеющем кристаллическую структуру, сходную со структурой кармана лиганда PPARδ (Chem. Biol. 1997, 4, p.909-918). Этот лиганд, как сообщалось, пространственно хорошо связывается с карманом для связывания лиганда, поскольку он имеет Y-образную структуру, содержащую структуру бензола, в отличие от лигандов, разработанных к настоящему времени. Однако этот лиганд представляет собой двойной активирующий лиганд, показывающий активность также по отношению к hPPARα, и показывает пониженную селективность по отношению к PPARδ. PPARδ-селективный лиганд GW501516 ([2-метил-4-[[[4-метил-2-[4-(трифторметил)фенил]-1,3-тиазол-5-ил]метил]сульфанил]фенокси]уксусная кислота), недавно разработанный Glaxo-Smith-Kline Co., показывает превосходную физиологическую активность по сравнению с ранее разработанным лигандом (Proc. Natl. Acad. Sci. USA 2001, 98, p.5306-5311). Лиганд GW501516 имеет очень хорошее сродство (1-10 нМ) к PPARδ и показывает в 1000 раз более высокую селективность по сравнению с PPARα и PPARγ. Соответственно считается, что в будущих экспериментах, связанных с PPARδ, эксперимент на основе GW501516 будет эффективным. Однако активности PPARδ, полученные посредством лигандов, разработанных к настоящему времени, представляют собой результаты, показываемые посредством связывания с 30-40% от общей области кармана для связывания лигандов.
Описание изобретения
Техническая задача
Соответственно для подтверждения конкретных воздействий PPARδ на тучность, гиперлипидемию, атеросклероз и диабет имеется необходимость в разработке новых лигандов, имеющих форму, сходную с карманом для связывания лигандов, и показывающих высокую селективность и активность, а также экономически выгодного способа их получения.
Техническое решение
Настоящее изобретение относится к новым тиазольным производным, представленным формулой I, в качестве лигандов, активирующих пролифератор-активируемый рецептор δ пероксисомы (PPARδ), который может использоваться для лечения тучности, гиперлипидемии, атеросклероза и диабета, а также к их промежуточным соединениям и к способам их получения:
[Формула I]
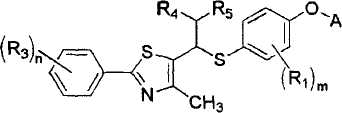
где A представляет собой водород, R2 или
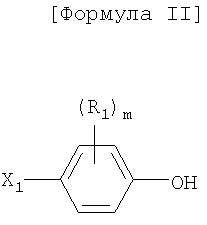
R1 представляет собой атом водорода, C1-4алкильную группу, C1-4алкилоксигруппу, C1-4алкилтиоксигруппу, C1-4алкиламиновую группу, атом фтора или атом хлора;
m представляет собой целое число от 0 до 4;
R2 представляет собой фенол-защитную группу, выбранную из C1-4 низших алкильных групп, аллильных групп, алкилсилильных групп, алкиларилсилильных групп и тетрагидропиранильной группы;
группы R3 являются отличными друг от друга и обозначают атом водорода, атом галогена или C1-4алкильную или алкоксигруппу, замещенную или не замещенную галогеном;
N представляет собой целое число от 0 до 5;
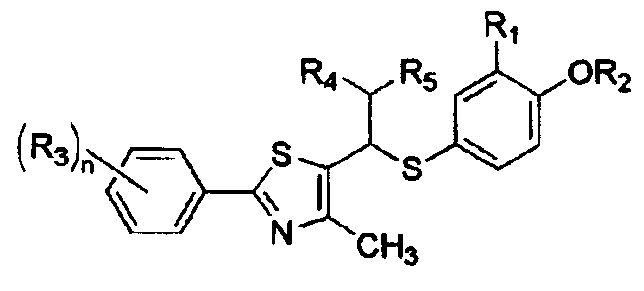
R4 представляет собой
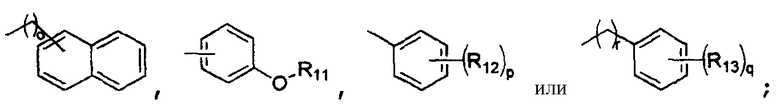
R5 представляет собой атом водорода, гидроксильную группу или C1-4алкильную группу;
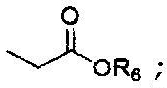
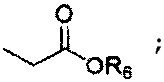
R6 представляет собой защитную группу карбоновой кислоты, имеющую C1-4алкил, аллильную группу, атом водорода или щелочной металл;
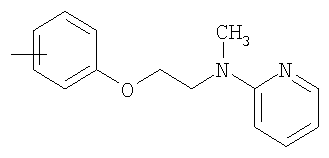
R11 представляет собой ариламиноалкильную группу или алкиламиноалкильную группу;
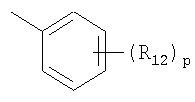
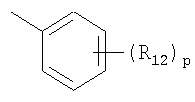
R12 представляет собой атом галогена, цианогруппу или C1-4алкильную или алкоксигруппу, замещенную или не замещенную галогеном;
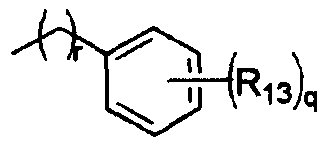
R13 представляет собой атом водорода, атом галогена, цианогруппу, C1-4алкильную или алкоксигруппу, замещенную или не замещенную галогеном;
o, p и q, каждый, независимо представляют собой целое число от 1 до 5; и
r представляет собой целое число от 1 до 9.
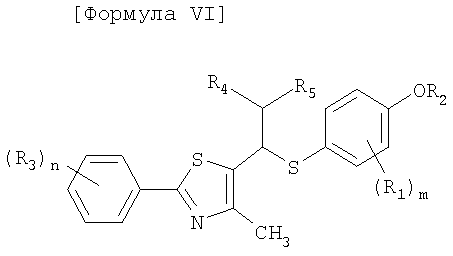
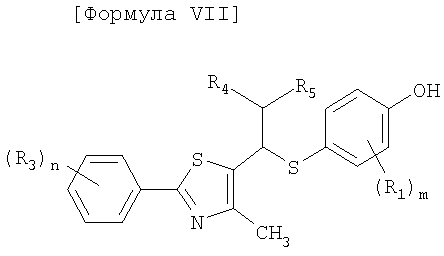
Производные соединения тиазола в соответствии с настоящим изобретения включают рацематы или оптические изомеры, представленные формулой VI, VII и IX, и соединения формулы X, которые могут быть получены из соединений формулы IX:
[Формула VI]
где R1-R5, m и n имеют такие же значения, как описано в формуле I выше;
[Формула VII]
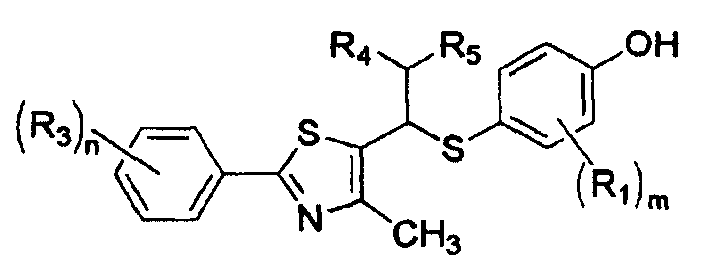
где R1, R3-R5, m и n имеют такие же значения, как описано в формуле I выше;
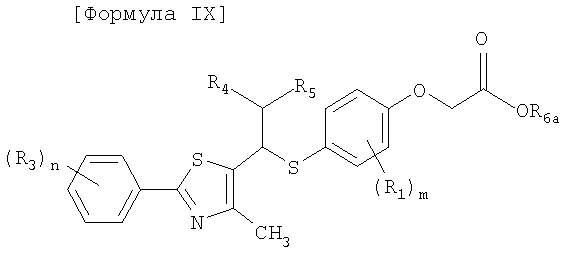
[Формула IX]
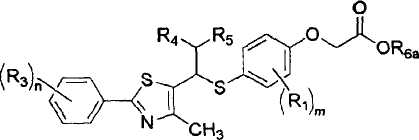
где R1, R3-R5, m и n имеют такие же значения, как описано в формуле I выше, и R6a представляет собой защитную группу карбоновой кислоты, имеющую C1-4алкильную группу или аллильную группу;
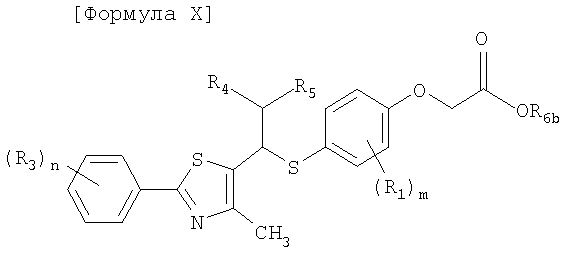
[Формула X]
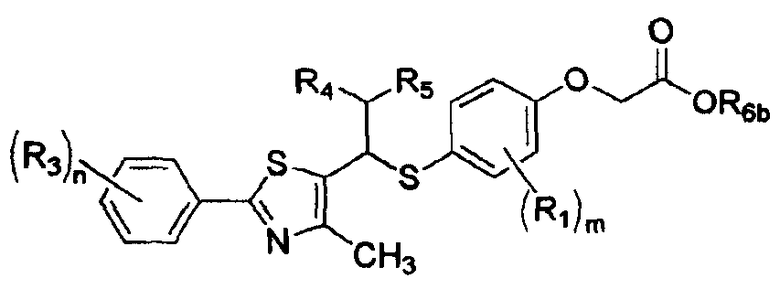
где R1, R3-R5, m, и n имеют такие же значения, как описано в формуле I выше, и R6b представляет собой атом водорода или щелочной металл.
Производные соединения тиазола формулы X в соответствии с настоящим изобретением отличаются тем, что обладают активностью по отношению к пролифератор-активируемому рецептору δ пероксисомы (PPARδ).
Новые соединения в соответствии с настоящим изобретением могут быть получены посредством следующих путей реакций.
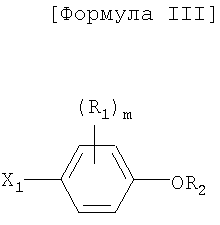
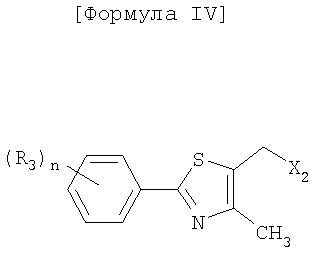
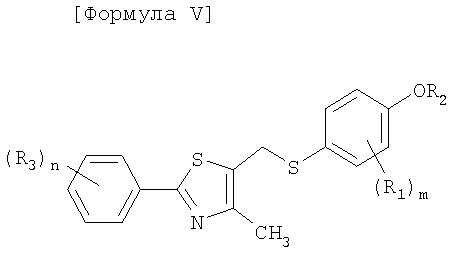
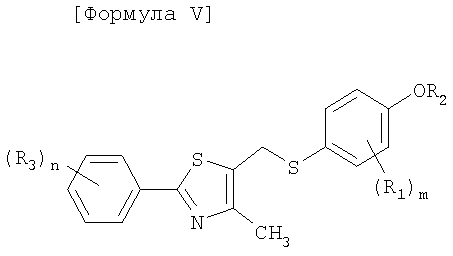
Как показано в следующей далее реакционной схеме, фенольная группа 4-галогенфенольного соединения формулы II, в качестве исходного материала, защищается с помощью алкилсилильной группы, с получением соединения формулы III, которое замещается литием и получает возможность для взаимодействия с серой и соединением формулы IV, с получением соединения формулы V. Соединение формулы V получает возможность для взаимодействия с различными электрофильными соединениями в присутствии сильного основания, с синтезом соединений формулы VI, с последующим удалением силильной защитной группы с фенольной группы, таким образом, получают соединения формулы VII. В другом способе фенольная группа соединения формулы II защищается с помощью реагента Гриньяра, и галоген соединения замещается литием, и полученное соединение получает возможность для взаимодействия с серой и соединением формулы IV с образованием простого тиоэфира. Простой тиоэфир получает возможность для взаимодействия с сильным основанием без разделения, а затем получает возможность для последовательного взаимодействия с различными электрофильными соединениями (O=CR4-R5 или X3-CHR4R5), при этом соединения формулы VII могут быть получены в едином способе. Соединения формулы VII, полученные таким образом, получают возможность для взаимодействия с алкилгалогенацетатом формулы VIII в присутствии неорганической соли, с синтезом соединений формулы IX, с последующим гидролизом сложного эфира, для того чтобы получить соединения формулы X. На основании данных о том, что соединения Формулы X могут быть получены с помощью описанного выше способа, настоящее изобретение завершается.
[Реакционная схема]
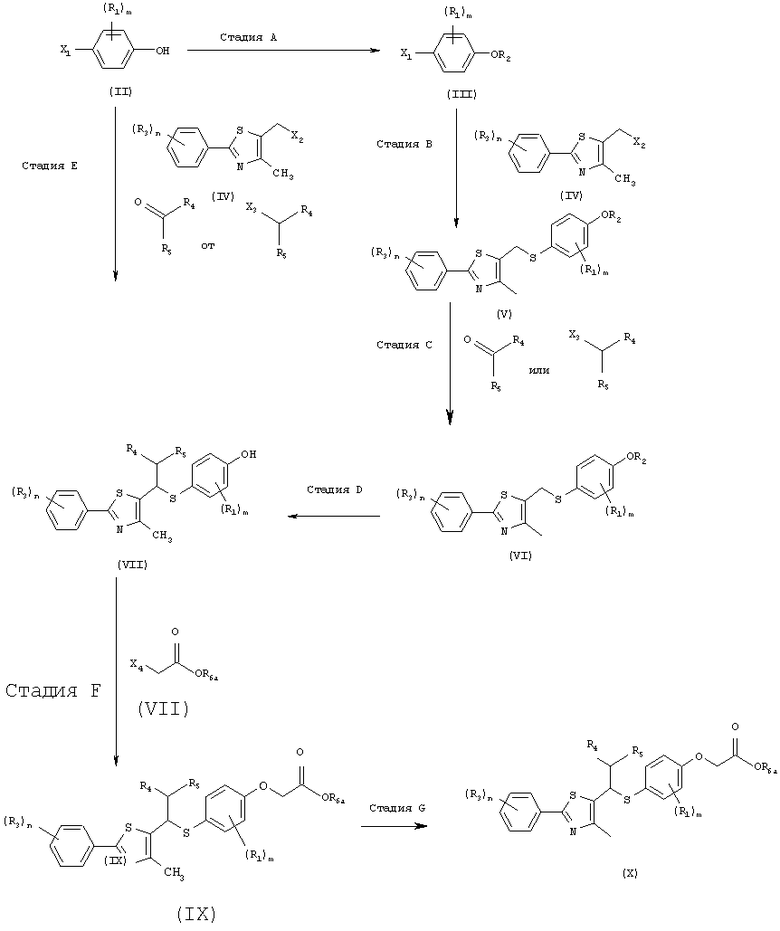
где R1 представляет собой атом водорода, С1-4алкильную группу, С1-4алкилоксигруппу, С1-4алкилтиоксигруппу, C1-4алкиламиновую группу, атом фтора или атом хлора;
m представляет собой целое число от 0 до 4;
R2 представляет собой фенол-защитную группу, выбранную из C1-4 низших алкильных групп, аллильных групп, алкилсилильных групп, алкиларилсилильных групп и тетрагидропиранильной группы;
группы R3 являются отличными друг от друга и обозначают атом водорода, атом галогена или С1-4алкильную или алкоксигруппу, замещенную или не замещенную галогеном;
n представляет собой целое число от 0 до 5;
R4 представляет собой
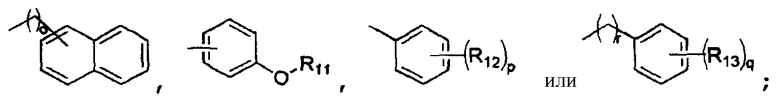
R5 представляет собой атом водорода, гидроксильную группу или C1-4алкильную группу;
R6 представляет собой защитную группу карбоновой кислоты, имеющую C1-4алкил, аллильную группу, атом водорода или щелочной металл;
R11 представляет собой ариламиноалкильную группу или алкиламиноалкильную группу;
R12 представляет собой атом галогена, цианогруппу или C1-4алкильную или алкоксигруппу, замещенную или не замещенную галогеном;
R13 представляет собой атом водорода, атом галогена, цианогруппу или C1-4алкильную или алкоксигруппу, замещенную или не замещенную галогеном;
o, p и q, каждый, независимо представляют собой целое число от 1 до 5; и
r представляет собой целое число от 1 до 9.
Конкретно, целью настоящего изобретения является создание новых PPARδ-активирующих лигандов, представленных формулой X, которые могут использоваться в качестве агентов для лечения тучности, гиперлипидемии, атеросклероза и диабета.
Другой целью настоящего изобретения является создание способа получения соединений формулы VI, который включает взаимодействие соединения формулы II с фенол-защитной группой, с формулы III замещения галогена литием, взаимодействие полученного соединения с серой (S) и соединением формулы IV без разделения и очистки, с получением соединения формулы V, взаимодействие соединения формулы V с сильным основанием, а затем с различными электрофильными соединениями.
Еще одной целью настоящего изобретения является создание способа получения соединения формулы VII посредством удаления фенол-защитной группы из соединений формулы VI.
Еще одной целью настоящего изобретения является создание способа получения соединений формулы VII посредством единого способа удобным образом, способ включает защиту фенольной группы фенольного соединения формулы II реагентом Гриньяра без осуществления специальной реакции для введения защитной группы, воздействие на защищенное соединение замещения галогена литием, взаимодействие полученного соединения с серой (S) , а затем с соединением формулы IV, с получением соединения простого тиоэфира, и взаимодействие соединения простого тиоэфира с сильным основанием и электрофильными соединениями.
Еще одной целью настоящего изобретения является создание способа получения соединения формулы IX, включающего взаимодействие соединений формулы VII с алкилгалогенацетатом и неорганической солью.
Еще одной целью настоящего изобретения является создание способа получения соединения формулы Х посредством гидролиза сложноэфирных соединений формулы IX.
Среди соединений, представленных формулой X, следующие соединения представляют собой новые соединения, и промежуточные соединения формул V, VI, VII и IX для получения этих соединений также являются новыми соединениями: 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-3-фенилпропилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-4-фенилбутилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-5-фенилпентилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-6-фенилгексилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-8-фенилоктилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-11-фенилундецилтио]-2-метилфенокси]уксусная кислота, 2-[4-[2-(2-хлор-6-фторфенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[2-(4-цианофенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(нафталин-3-ил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[2-[4-(трифторметил)фенил]-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(3,5-диметоксифенил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(перфторфенил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[2-(4-бромфенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[2-[2-фтор-6-(трифторметил)фенил]-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(2,6-дифторфенил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[2-(2,6-дихлорфенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(2,4-дифторфенил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(2,3,4-трифторфенил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[2-(2-хлор-5-фторфенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота, 4-[3-(2-хлор-6-фторфенил)-2-гидрокси-1-[4-метил-2-(4-трифторметил-фенил)-тиазол-5-ил]пропилсульфанил]-2-метил-фенокси]уксусная кислота, 4-[2-гидрокси-1-[4-метил-2-(4-трифторметил-фенил)тиазол-5-ил]-11-фенил-ундецилсульфанил]-2-метил-фенокси]уксусная кислота, 4-[2-гидрокси-1-[4-метил-2-(4-трифторметил-фенил)-тиазол-5-ил]-2-фенил-этилсульфанил]-2-метил-фенокси]уксусная кислота, 2-[4-[2-(2-хлор-6-фторфенил)-1-[2-[3-фтор-4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[3-фтор-4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(3,4,5-трифторфенил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[3-фтор-4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(2-фтор-6-(трифторметил)фенил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[3-фтор-4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(2,6-дифторфенил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[2-(2,6-дихлорфенил)-1-[2-[3-фтор-4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[1-[2-[3-фтор-4-(трифторметил)фенил]-4-метилтиазол-5-ил]-2-(2,4-дифторфенил)этилтио]-2-метилфенокси]уксусная кислота, 2-[4-[2-(2-хлор-5-фторфенил)-1-[2-[3-фтор-4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусная кислота и калий 2-[4-[2-(2-хлор-6-фторфенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]ацетат.
Соответственно настоящее изобретение предусматривает новые полезные соединения.
В соединениях по настоящему изобретению R1 обозначает атом водорода, C1-4алкильную группу, C1-4алкилоксигруппу, C1-4алкилтиоксигруппу, C1-4алкиламиногруппу, атом фтора или атом хлора. Каждая из группы R1 находится в орто- или мета-положении по отношению к фенольной группе, и количество (m) групп R1 равно 0-4;
R2 представляет собой фенол-защитную группу, такую как C1-4 низший алкил, аллил, алкилсилил или алкиларилсилил, такой как триметилсилил, трет-бутилдифенилсилил, триизопропилсилил или трет-бутилдиметилсилил, или тетрагидропиранил. Среди этих защитных групп предпочтительными являются трет-бутильная, тетрагидропиранильная или силилированная защитная группа;
группы R3 являются отличными друг от друга и обозначают атом водорода, атом галогена или C1-4алкильную или алкоксигруппу, замещенную или не замещенную галогеном, и количество (n) групп R3 равно 0-5;
R4 обозначает
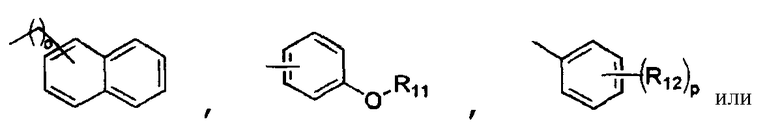
R5 обозначает атом водорода, гидроксильную группу или C1-4алкильную группу;
R6 представляет собой защитную группу карбоновой кислоты, имеющую C1-4алкильную группу (например, метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил или трет-бутил), аллил, атом водорода или щелочной металл (Li+, Na+, K+);
R11 представляет собой ариламиноалкильную группу, такую как метилпиридиниламиноэтил, метилфениламиноэтил или трет-бутилфениламиноэтил, или алкиламиноалкильную группу, такую как метиламиноэтил, трет-бутиламиноэтил или этиламинопропил;
R12 представляет собой атом галогена, цианогруппу или C1-4алкильную или алкоксигруппу, замещенную или не замещенную галогеном;
R13 представляет собой атом водорода, атом галогена, цианогруппу или C1-4алкильную или алкоксигруппу, замещенную или не замещенную галогеном;
o, p и q, каждый, независимо представляют собой целое число от 1 до 5;
r представляет собой целое число от 1 до 9;
X1 представляет собой атом галогена, такой как атом брома (Br) и атом йода (I);
X2 обозначает уходящую группу в нуклеофильной реакции. В качестве уходящей группы могут использоваться обычные уходящие группы, например, атомы галогена, такие как хлор, бром или йод, метансульфонилокси (MsO-) и п-толуолсульфонилокси (TsO-). Среди этих уходящих групп предпочтительными являются хлор и бром;
X3 обозначает уходящую группу. В качестве уходящей группы могут использоваться обычные уходящие группы, например галогены, метансульфонилокси (MsO-) и п-толуолсульфонилокси (TsO-). Галогены включают фтор, хлор, бром и йод. Среди этих уходящих групп предпочтительными являются галогены, а более предпочтительными являются хлор, бром и йод;
X4 обозначает атом галогена, такой как хлор (Cl), бром (Br) или йод (I).
Соединения формул (I) и (II) и электрофильные соединения, используемые в качестве исходных материалов или промежуточных соединений в способе получения в соответствии с настоящим изобретением представляют собой известные соединения, которые могут быть коммерчески легко доступными или легко получаются в соответствии с литературой.
Далее настоящее изобретение будет описываться более подробно.
Стадия A: Получение соединения, представленного формулой III
Для получения соединения, представленного формулой III, соединению, представленному формулой II, дают возможность предпочтительно взаимодействовать с соединением, обычно используемым в качестве фенол-защитной группы, в присутствии основания.
Апротонные полярные растворители, которые могут использоваться на этой стадии, могут включать N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, ацетонитрил, ацетон, этилацетат, четыреххлористый углерод, хлороформ и дихлорметан. Эфирные растворители, которые могут использоваться на этой стадии, могут включать тетрагидрофуран, диоксан, диметоксиэтан, простой диэтиленгликольдиметиловый эфир и простой триэтиленгликольдиметиловый эфир. Ароматические углеводороды могут включать бензол, толуол и ксилол. Среди этих растворителей предпочтительными являются апротонные полярные растворители, а более предпочтительными являются N,N-диметилформамид, хлороформ и дихлорметан.
Основания, которые могут использоваться на этой стадии, включают аминовые основания, такие как пиридин, триэтиламин, имидазол и N,N-диметиламинопиридин, и если алкил или простой аллиловый эфир используется в качестве защитной группы, гидроксид натрия, гидроксид калия, карбонат натрия или калий карбонат будут использоваться в качестве основания. Среди этих оснований предпочтительными основаниями являются имидазол и карбонат калия.
Тетрагидропиранил-защитную группу получают посредством взаимодействия 3,4-дигидро-2H-пирана с алкилом или аллилтрифенилфосфоний бромидом в присутствии катализатора.
Температура реакции может изменяться в зависимости от вида используемого растворителя, но, как правило, составляет от -10 до 80°C, а предпочтительно от 0 до комнатной температуры (25°C). Время реакции может изменяться в зависимости от температуры реакции и вида используемого растворителя, но, как правило, составляет от 1 часа до 1 дня, а предпочтительно 4 часа или меньше.
Стадия B: Получение соединения, представленного формулой V
Соединение, представленное формулой V, получают в едином способе посредством воздействия на соединения формулы III замещения галогена литием, введения серы, а затем взаимодействия с соединением формулы IV.
Безводные растворители, которые могут использоваться на этой стадии, включают простой диэтиловый эфир, тетрагидрофуран, гексан, гептан и смесь двух или более из них. Среди этих растворителей наиболее предпочтительные растворители представляют собой простой диэтиловый эфир, тетрагидрофуран и смешанный растворитель из простого диэтилового эфира и тетрагидрофурана.
Металлические реагенты, которые могут использоваться в реакции замещения галогена металлом, включают металлы, такие как металлический литий и металлический магний, и реагенты на основе органических соединений металлов, такие как н-бутиллитий, втор-бутиллитий и трет-бутиллитий. Среди этих реагентов предпочтительными являются реагенты на основе органических соединений металлов, а более предпочтительными являются н-бутиллитий и трет-бутиллитий.
Температура реакции может изменяться в зависимости от вида используемого растворителя, но, как правило, составляет от -100 до 25°C, а предпочтительно от -75°C до комнатной температуры, для замещения галогена литием и реакции введения серы, и представляет собой комнатную температуру (25°C) для взаимодействия с соединением формулы III. Время реакции может изменяться в зависимости от температуры реакции и вида используемого растворителя, но, как правило, составляет от 30 минут до 4 часов, а предпочтительно 1 час или меньше.
Стадия C: Получение соединения, представленного формулой VI
Соединение, представленное формулой VI, получают посредством обработки α-протона простого тиоэфира соединения формулы V сильным основанием, с получением нуклеофила, которому затем дают возможность для взаимодействия с различными электрофильными соединениями.
Безводные растворители, которые могут использоваться на этой стадии, включают простой диэтиловый эфир, тетрагидрофуран, гексан, гептан и смесь двух или более из них. Среди этих растворителей предпочтительные растворители представляют собой простой диэтиловый эфир, тетрагидрофуран и смешанный растворитель из простого диэтилового эфира и тетрагидрофурана.
Реагенты на основе сильных оснований, которые могут использоваться в реакции извлечения альфа-водорода, включают калий трет-бутоксид (t-BuOK), литий диизопропиламид (LDA), н-бутиллитий, втор-бутиллитий и трет-бутиллитий. Среди этих реагентов, наиболее предпочтительным является трет-бутиллитий.
Электрофильные соединения, которые взаимодействуют с нуклеофильным соединением простого тиоэфира, представляют собой известные соединения, которые могут быть коммерчески легко доступными или легко получаются в соответствии с литературой и содержат галоген, альдегид или кетон. Эти соединения добавляют для взаимодействия после растворения в безводном растворителе или без растворения.
Температура реакции может изменяться в зависимости от вида используемого растворителя, но, как правило, составляет от -78 до 25°C. Предпочтительно извлечение альфа-водорода с помощью сильного основания осуществляют при -75°C, и электрофильные соединения добавляют при -75°C, и они взаимодействуют, в то время как температура медленно поднимается до комнатной температуры (25°C). Время реакции может изменяться в зависимости от стадии реакции, но составляет 10-30 минут для извлечения альфа-водорода с помощью сильного основания и 30-90 минут для взаимодействия с электрофильными соединениями.
Стадия D: Получение соединения, представленного формулой VII
Соединение формулы VII получают посредством удаления фенол-защитной группы из соединения формулы VI.
Полярные растворители, которые могут использоваться на этой стадии, включают N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, ацетонитрил, ацетон, этилацетат, четыреххлористый углерод, хлороформ и дихлорметан. Эфирные растворители могут включать тетрагидрофуран, диоксан, диметоксиэтан и простой диэтиленгликольдиметиловый эфир. Спиртовые растворители могут включать метанол и этанол. Ароматические углеводороды могут включать бензол, толуол и ксилол. Среди этих растворителей предпочтительными являются полярные растворители, а наиболее предпочтительным является тетрагидрофуран.
Для удаления фенол-защитной группы кислоты Льюиса, такие как триметилсилил йодид, натрий этановый тиоспирт, йодид лития, галогенид алюминия, галогенид бора и трифторуксусная кислота, используются для удаления защитных групп, таких как метил, этил, трет-бутил, бензил и группа простого аллилового эфира. Также фториды, такие как тетрабутиламмоний фторид (Bu4N+F-) , галогеновые кислоты (фтористая кислота, хлористоводородная кислота, бромноватая кислота и йодноватая кислота), фторид калия, используются для удаления силилированных защитных групп, таких как триметилсилил, трет-бутилдифенилсилил, триизопропилсилил и трет-бутилдиметилсилил. Среди этих групп для удаления для силилированных защитных групп предпочтительными являются фториды, а более предпочтительным является тетрабутиламмоний фторид.
Температура реакции может изменяться в зависимости от видов используемой группы для снятия защиты и растворителя, но, как правило, составляет от 0 до 120°С, а предпочтительно от 10 до 25°С. Температура реакции может изменяться в зависимости от времени реакции, но, как правило, составляет от 30 минут до 1 дня, а предпочтительно 2 часа или меньше.
Стадия Е: Получение соединения, представленного формулой VII
Для получения соединения, представленного формулой VII, фенольная группа соединения, представленного формулой II, защищается с помощью реагента Гриньяра, и защищенному соединению дают возможность для взаимодействия с реагентом на основе органического соединения металла, серой (S), а затем с соединением формулы IV. Затем полученному соединению дают возможность для взаимодействия с электрофильными соединениями в присутствии сильного основания. Эта стадия Е предлагает очень удобный способ для осуществления реакции в едином способе.
Далее будут описываться подстадии стадии Е.
Защита фенольной группы с помощью реагента Гриньяра (Стадия Е-1)
Безводные растворители, которые могут использоваться на этой стадии, включают простой диэтиловый эфир, тетрагидрофуран, гексан, гептан и смешанный растворитель из двух или более из них. Среди этих растворителей предпочтительными являются простой диэтиловый эфир, тетрагидрофуран или смешанный растворитель простого диэтилового эфира и тетрагидрофурана.
Используемый реагент Гриньяра представляет собой метил, этил, н-пропил, изопропил, н-бутил, втор-бутилмагнийхлорид (R2MgCl) или алкилмагнийбромид, (R2MgBr). Среди этих реагентов наиболее предпочтительным является изопропилмагнийхлорид (СН3)2СНМgСl).
Температура реакции может изменяться в зависимости от вида используемого растворителя, но, как правило, составляет от -20 до 40°С, а предпочтительно от 0°С до комнатной температуры (25°С). Время реакции может изменяться в зависимости от температуры реакции и вида используемого растворителя, но, как правило, составляет 10-60 минут, и предпочтительно 10-30 минут.
Замещение галогена литием и введение серы (S) (Стадии Е-2 и Е-3)
Реагенты на основе органических соединений металла, которые могут использоваться в реакции замещения галогена литием, включают н-бутиллитий, втор-бутиллитий и трет-бутиллитий. Среди этих реагентов на основе органических соединений металла предпочтительным является трет-бутиллитий.
Сера (S) предпочтительно находится в форме порошка из мелкодисперсных частиц и добавляется для взаимодействия после растворения в растворителе из безводного тетрагидрофурана или без растворения.
Температура реакции может изменяться в зависимости от вида используемого растворителя, но, как правило, составляет от -78 до 25°C, а предпочтительно -75°C, для реакции замещения галогена металлом, и доводится до комнатной температуры (25°C), начиная с -75°C, для реакции введения серы. Время реакции составляет 10-30 минут, для реакции замещения галогена металлом и 30-90 минут для реакции введения серы.
Реакция добавления соединения, представленного формулой IV [Стадия E-4]
5-галогенметил-4-метил-2-[4-(трифторметил)фенил]тиазол формулы IV, используемый на этой стадии, синтезируется в соответствии с известным способом (заявка на Международный патент WO 2003/106442). Примеры галогена в формуле IV соединения включают хлор, бром и йод. Среди этих галогенов предпочтительным является хлор.
Температура реакции может изменяться в зависимости от вида используемого растворителя, но, как правило, составляет от -78°C до 25°C, а предпочтительно от 0°C до 10°C. Время реакции, как правило, составляет 10-120 минут, а предпочтительно 10-60 минут.
Реакции с различными электрофильными соединениями (Стадия E-5)
Сильные основания, которые могут использоваться для обработки α-протона простого тиоэфира с получением нуклеофильных соединений, включают трет-бутоксид калия (t-BuO-K+), диизопропиламид лития (LDA), н-бутиллитий, втор-бутиллитий и трет-бутиллитий. Среди этих оснований трет-бутиллитий является наиболее предпочтительным.
Электрофильное соединение, которое взаимодействует с нуклеофильными тиоэфирными соединениями, представляет собой известное соединение, которое является коммерчески легко доступным или легко получается в соответствии с литературой и содержит высокоактивный химически галоген, альдегид или кетон. Это соединение добавляют для взаимодействия после растворения в безводном растворителе или без растворения.
Температура реакции может изменяться в зависимости от вида используемого растворителя, но, как правило, составляет от -78 до 25°C. Предпочтительно извлечение альфа-водорода с помощью сильного основания осуществляют при -75°C, а электрофильные соединения добавляют при -75°C и дают им возможность для взаимодействия, в то же время поднимая температуру до комнатной температуры (25°C). Время реакции изменяется в зависимости от стадии реакции, но составляет 10-30 минут для извлечения альфа-водорода с помощью сильного основания и 30-90 минут для взаимодействия с электрофильными соединениями.
Стадия F: Получение соединения, представленного формулой IX
Для получения соединения, представленного формулой IX, соединению, представленному формулой VII, предпочтительно дают возможность для взаимодействия со сложным алкиловым эфиром галогенуксусной кислоты в присутствии основания.
Сложный алкиловый эфир галогенуксусной кислоты представляет собой известное соединение, которое является коммерчески легко доступным и в котором галоген представляет собой хлор, бром или йод. Наиболее предпочтительный пример сложного алкилового эфира галогенуксусной кислоты представляет собой сложный метиловый эфир бромуксусной кислоты или сложный этиловый эфир бромуксусной кислоты.
Растворители, которые могут использоваться на этой стадии, включают водорастворимые растворители, такие как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, ацетонитрил, ацетон, этанол и метанол или смесь любого из них с 1-10% воды. Среди этих растворителей наиболее предпочтительным является смесь ацетона или диметилсульфоксида с 1-5% воды.
Используемое основание не является конкретно ограниченным, сильное ли это основание или слабое основание, постольку, поскольку оно не влияет отрицательно на реакцию, и его примеры включают гидриды щелочного металла, такие как гидрид натрия и гидрид лития, гидрид щелочноземельного металла, такие как гидрид калия, гидроксиды щелочного металла, такие как гидроксид натрия и гидроксид калия, и карбонаты щелочного металла, такие как карбонат лития, карбонат калия, бикарбонат калия и карбонат цезия. Среди этих оснований предпочтительным является карбонат щелочного металла, а более предпочтительным является карбонат калия.
Температура реакции не является конкретно ограниченной постольку, поскольку она находится ниже температуры кипения используемого растворителя, но реакция при относительно высокой температуре предпочтительно исключается для подавления побочных реакций. Температура реакции, как правило, составляет 0-60°C. Время реакции может изменяться в зависимости от температуры реакции, но, как правило, оно составляет от 30 минут до 1 дня, и предпочтительно 30-90 минут.
Стадия G-1: Получение соединения, представленного формулой X
Соединение, представленное формулой X, получают посредством гидролиза сложного эфира карбоновой кислоты и соединения формулы IX с помощью водорастворимой неорганической соли в спиртовом растворителе.
Растворители, которые могут использоваться на этой стадии, включают водорастворимые спиртовые растворители, такие как метанол и этанол.
Основания, которые могут использоваться на этой стадии, включают примерно 0,1-3 н. водные растворы, полученные с использованием гидроксидов щелочных металлов, таких как гидроксид лития, гидроксид натрия и гидроксид калия, в соответствии с формой щелочных солей карбоновых кислот. В качестве кислоты, используемой для получения соединения формулы X в форме карбоновой кислоты, предпочтительно используется уксусная кислота или 0,1-3 н. водный раствор хлористоводородной кислоты.
Реакцию предпочтительно осуществляют при относительно низкой температуре для ингибирования побочных реакций, и, как правило, она происходит при температуре от 0°C до комнатной температуры. Время реакции может изменяться в зависимости от температуры реакции, но, как правило, составляет от 10 минут до 3 часов, а предпочтительно от 30 минут до 1 часа.
Стадия G-2: Получение соединения, представленного формулой X
Соединение, представленное формулой X, получают посредством замещения сложного аллилового эфира соединения формулы IX металлической солью 2-этилгексаноата в органическом растворителе в присутствии металлического катализатора.
Растворитель, используемый на этой стадии, представляет собой безводные органические растворители, такие как хлороформ, дихлорметан или этилацетат.
В качестве металлического катализатора предпочтительно используется палладий тетракистрифенилфосфин в количестве 0,01-0,1 эквивалента.
Реакцию предпочтительно осуществляют при относительно низкой температуре для ингибирования побочных реакций, и, как правило, осуществляют при температуре от 0°C до комнатной температуры. Время реакции может изменяться в зависимости от температуры реакции, но, как правило, составляет от 10 минут до 3 часов, а предпочтительно от 30 минут до 1 часа.
Это соединение соли выделяют с высокой чистотой посредством центрифугирования. Полученное соединение металла типа соли формулы X легче выделять, чем соединение типа соли, полученное с использованием стадии G-1 (стадия гидролиза).
Y-образные тиазольные соединения формулы X, полученные таким образом, являются важными веществами в качестве лигандов для PPARδ. Также эти соединения имеют хиральный атом углерода, и, таким образом, существуют их стереоизомеры. Среди соединений формулы X, как подтверждается, изомеры R-формы или S-формы являются эффективными по сравнению с рацематами, и рамки настоящего изобретения охватывают соединения формулы X и их стереоизомеры, сольваты и соли.
Преимущества
Как описано выше, новые производные соединения тиазола в соответствии с настоящим изобретением имеют характеристики PPARδ-активирующих лигандов и демонстрируют высокую возможность для использования в качестве агентов для лечения сердечнососудистого заболевания, понижения уровней холестерина и лечения диабета и тучности. Также способ получения по настоящему изобретению является пригодным для получения производных соединений тиазола.
Наилучшая иллюстрация осуществления изобретения
Примеры
Далее способ в соответствии с настоящим изобретением будет описываться более подробно с помощью примеров. Однако специалисту в данной области будет очевидно, что настоящее изобретение не ограничивается этими примерами или с их помощью.
Пример 1: Получение 4-йод-2-метил-фенокси-трет-бутилдиметилсилана (III) [Стадия A]
3,0 г (12,8 ммоль) 4-йод-2-метилфенола и 1,74 г (25,6 ммоль, 2,0 эквивалента) имидазола полностью растворяют в 45 мл диметилформамида. К раствору медленно добавляют 2,12 г (14,1 ммоль, 1,1 эквивалента) трет-бутилдиметилсилил хлорида и смесь перемешивают при комнатной температуре в течение 4 часов. После завершения реакции продукт реакции экстрагируют водным раствором хлорида аммония и этилацетатом и органический слой сушат над сульфатом магния. Остаток очищают на колонке с силикагелем и растворитель удаляют посредством дистилляции при пониженном давлении, получая, таким образом, 4,4 г (выход 98%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDC13) δ 7,47 (д, 1H, J=0,6 Гц), 7,35 (дд, 1H, J=8,4, 2,3 Гц), 6,54 (д, 1H, J=8,4 Гц), 2,18 (с, 3H), 1,03 (с, 9H), 0,22 (с, 6H).
13C ЯМР (75,5 МГц, CDCl3) δ 154,3, 139,9, 135,9, 132,3, 121,1, 83,9, 26,2, 18,7, 17,0, -3,8.
Пример 2: Получение 4-бром-фенокси-трет-бутилдиметилсилана (III) [Стадия A]
500 мг (2,90 ммоль) 4-бромфенола и 409 мг (6,0 ммоль, 2,00 эквивалента) имидазола полностью растворяют в диметилформамиде. К раствору медленно добавляют 436 мг (2,90 ммоль, 1,0 эквивалент) трет-бутилдиметилсилил хлорида и смесь перемешивают при комнатной температуре в течение 4 часов. После завершения реакции продукт реакции экстрагируют водным раствором хлорида аммония и этилацетатом и органический слой сушат над сульфатом магния. Остаток очищают на колонке с силикагелем и растворитель удаляют посредством дистилляции при пониженном давлении, получая, таким образом, 811 мг (выход 97%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDC13) δ 7,32 (д, 2H, J=8,8 Гц), 6,72 (д, 2H, J=10,0 Гц), 0,98 (с, 9H), 0,18 (с, 6H).
13C ЯМР (75,5 МГц, CDC13) δ 155,3, 132,7, 122,3, 114,0, 26,0, 18,6, -4,1.
Пример 3: Получение 5-[4-(трет-бутилдиметилсилилокси)-3-метил-фенилсульфанилметил]-4-метил-2-[(4-трифторметил)фенил]-тиазола (V) [Стадия B]
В атмосфере азота 1,5 г (4,32 ммоль) 4-йод-2-метил-фенокси-трет-бутилдиметилсилана, полученного в Примере 1, растворяют в 120 мл безводного тетрагидрофурана и охлаждают до -78°C. К раствору медленно добавляют 2,54 мл (1,0 эквивалент) трет-бутиллития (1,7 M раствор в гексане). Смесь перемешивают в течение 10 минут, к ней затем добавляют за один раз 138 мг (4,32 ммоль, 1,0 эквивалент) твердофазной серы при такой же температуре. Смеси дают возможность для взаимодействия в течение 40 минут до достижения температуры 15°C, к ней затем медленно добавляют 1,26 г (4,32 ммоль, 1,0 эквивалент) 5-хлорметил-4-метил-2-[(4-трифторметил)фенил]тиазола формулы III, растворенного в 10 мл безводного ТГФ. После взаимодействия в течение дополнительного времени, примерно одного часа, реакцию прекращают с помощью водного раствора хлорида аммония и продукт реакции экстрагируют этилацетатом и водным раствором соли и органический слой сушат над сульфатом магния. После фильтрования растворитель удаляют посредством дистилляции при пониженном давлении и остаток очищают с помощью колоночной хроматографии, получая, таким образом, 1,85 г (выход 84 %) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDC13) δ 7,97 (д, 2H, J=8,0 Гц), 7,65 (д, 2H, J=8,2 Гц), 7,17 (д, 1H, J=1,8 Гц), 7,07 (дд, 1H, J=8,2, 2,3 Гц), 6,67 (д, 1H, J=8,3 Гц), 4,10 (с, 2H), 2,20 (с, 3H), 2,15 (с, 3H), 1,00 (с, 9H), 0,20 (с, 6H).
13C ЯМР (75,5 МГц, CDC13) δ 163,4, 154,9, 151,8, 136,8, 132,6, 130,4, 129,6 (кв, J=32 Гц), 126,8, 126,2 (m), 125,2, 119,6, 33,0, 26,1, 18,7, 17,1, 15,2, -3,9.
Пример 4: Получение 5-[1-[3-метил-4-(трет-бутилдиметил-силилокси)фенилтио]-3-фенилпропил]-2-[4-(трифторметил)фенил]-4-метилтиазола (VI) [Стадия C]
В атмосфере азота 510 мг (1,0 ммоль) 5-[4-(трет-бутилдиметилсиланилокси)-3-метил-фенилсульфанилметил]-4-метил-2-[(4-трифторметил)фенил]тиазола, полученного в Примере 3, растворяют в 20 мл безводного тетрагидрофурана. Реакционный раствор достаточно охлаждают до -78°C, затем к нему медленно добавляют 1,2 мл (2,0 эквивалента) трет-бутиллития (1,7 М раствор гептана). В то время, пока реакционный раствор сохраняет глубокий синий цвет, к нему добавляют 137 мкл (1,0 ммоль) (2-бромэтил)бензола и температуру реакции медленно повышают до комнатной температуры. После восстановления в течение дополнительного времени, примерно 30 минут, реакцию прекращают с помощью водного раствора хлорида аммония, и продукт реакции экстрагируют этилацетатом и водным раствором соли и органический слой сушат над сульфатом магния. После фильтрования растворитель удаляют посредством дистилляции при пониженном давлении и остаток очищают с помощью колоночной хроматографии, получая, таким образом, 388 мг (выход 63%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDC13) δ 7,99 (д, 2H, J=8,5 Гц), 7,67 (д, 2H, J=8,2 Гц), 7,19 (м, 5H), 7,04 (д, 1H, J=2,0 Гц), 6,96 (дд, 1H, J=8,3, 2,4 Гц), 6,59 (д, 1H, J=8,3 Гц), 4,19 (дд, 1H, J=8,9, 6,0 Гц), 2,74 (м, 2H), 2,37 (м, 1H), 2,37 (м, 1H), 2,19 (м, 1H), 2,08 (с, 3H), 1,96 (с, 3H), 0,98 (с, 9H), 0,17 (с, 6H).
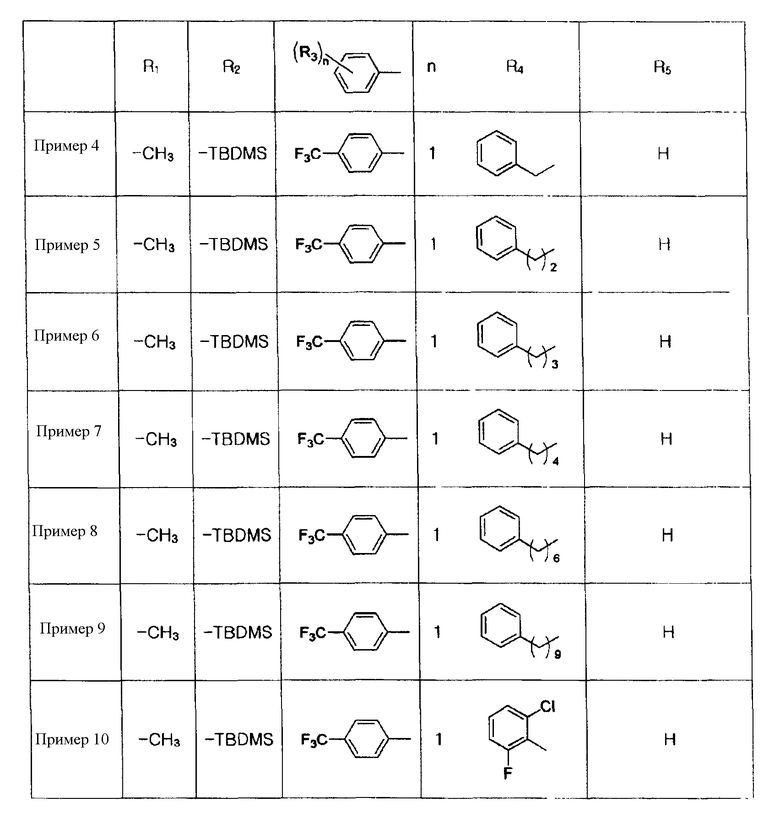
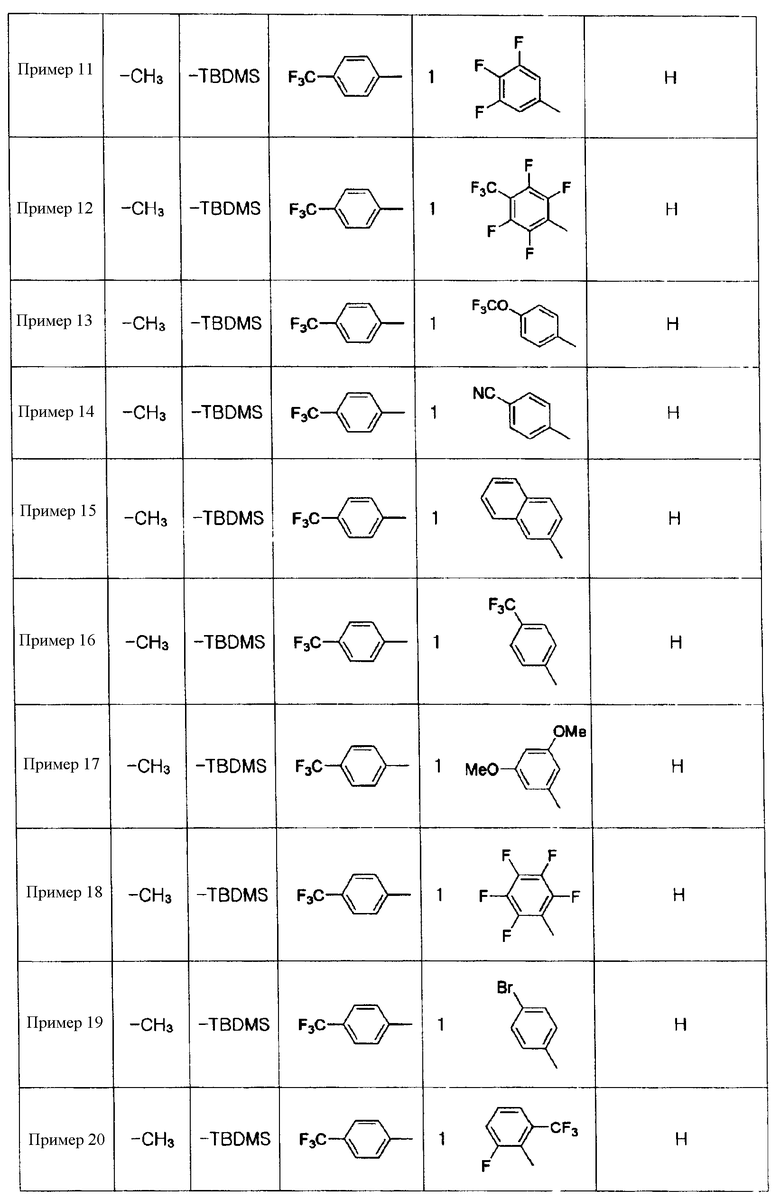
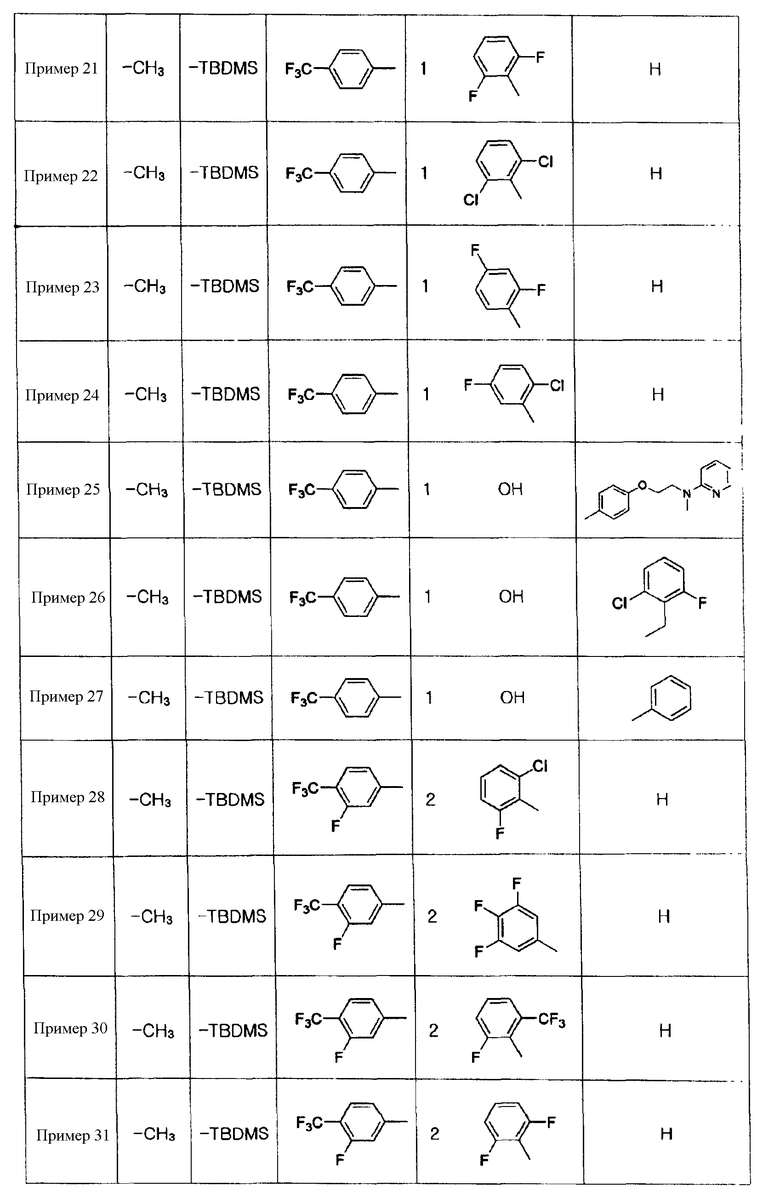
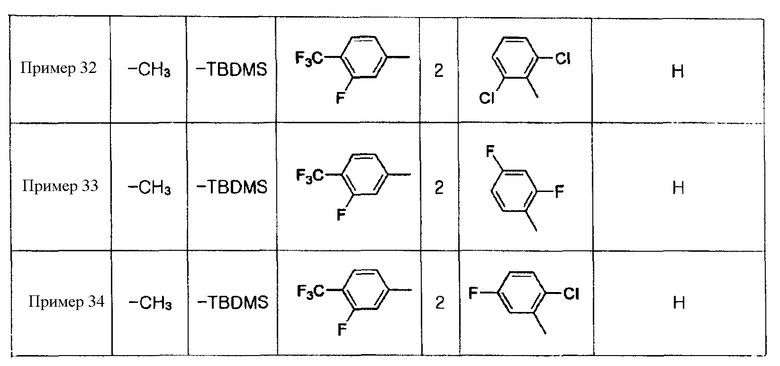
Примеры 5-34
Соединения, показанные в таблице 1 ниже, получают таким же способом, как описано в Примере 4, и данные ЯМР полученных соединений показаны в таблице 2 ниже.
Таблица 1
Пример 35: Получение 4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-6-фенилгексилтио]-2-метилфенола (VII) [Стадия D]
394 мг (0,6 ммоль) 5-[1-[3-метил-4-(трет-бутилдиметил-силилокси)фенилтио]-6-фенилгексил]-2-[4-(трифторметил)фенил]-4-метилтиазола полностью растворяют в 10 мл тетрагидрофурана. К раствору медленно добавляют 1,5 мл (2,5 эквивалента) тетрабутиламмоний фторида (TBAF) (1 M раствор в тетрагидрофуране) при комнатной температуре. После взаимодействия в течение 30 минут продукт реакции экстрагируют водным раствором хлорида аммония и этилацетатом и органический слой сушат над сульфатом магния. После фильтрования растворитель удаляют посредством дистилляции при пониженном давлении и остаток очищают с помощью колоночной хроматографии, получая, таким образом, 306 мг (выход 94%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDC13) δ 7,98 (д, 2H, J=8,0 Гц), 7,66 (д, 2H, J=8,2 Гц), 7,19 (м, 5H), 7,09 (д, 1H, J=1,5 Гц), 6,93 (дд, 1H, J=7,8, 1,9 Гц), 6,57 (д, 1H, J=8,2 Гц), 4,20 (дд, 1H, J=9,1, 5,9 Гц), 2,58 (т, 2H, J=7,5 Гц), 2,18 (с, 3H), 2,04 (м, 1H), 1,97 (с, 3H), 1,85 (м, 1H), 1,60 (м, 2H), 1,39 (м, 4H).
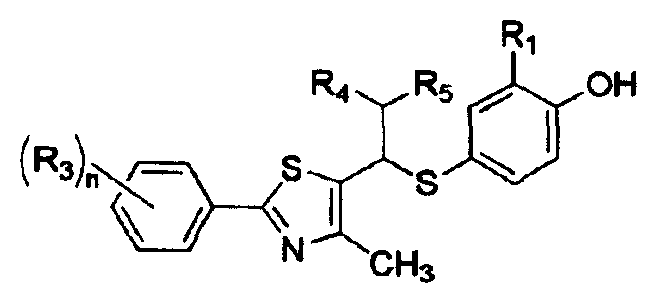
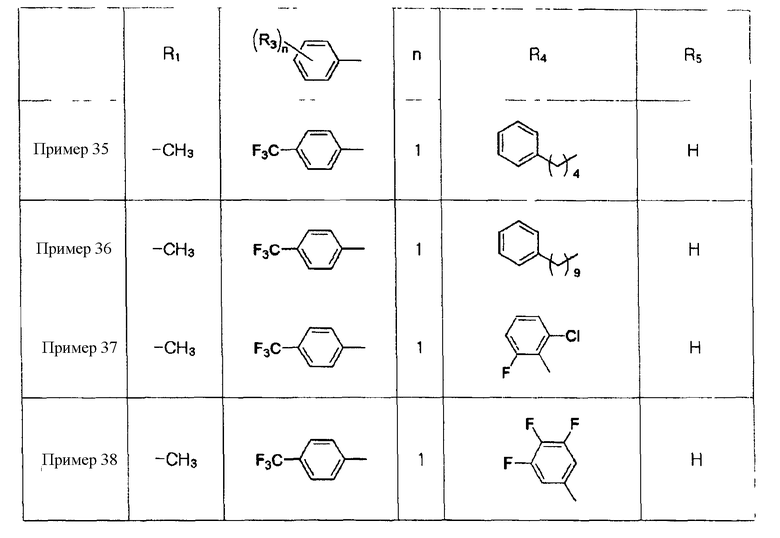
Примеры 36-38
Соединения, показанные в таблице 3 ниже, получают в соответствии со способом Примера 35, и данные ЯМР полученных соединений показаны в таблице 4 ниже.
Таблица 3
Пример 39: Получение 4-[2-(2-хлор-6-фторфенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенола (VII) из соединения формулы II [Стадия E]
В атмосфере азота 585 мг (2,5 ммоль) 4-йод-2-метилфенола растворяют в 35 мл безводного тетрагидрофурана и поддерживают при температуре 0°C. К раствору медленно добавляют 1,3 мл (1,0 эквивалент) изопропилмагнийхлорида (2 M раствор в простом эфире) и смеси дают возможность для взаимодействия в течение 10 минут. После взаимодействия раствор достаточно охлаждают до -78°C, к нему добавляют по каплям 3,0 мл (2,0 эквивалента) трет-бутиллития (1,7 M раствор гептана) и смеси дают возможность для взаимодействия в течение 20 минут. К реакционному продукту добавляют 80 мг (2,5 ммоль, 1,0 эквивалент) твердофазной серы за один раз и реакционной смеси дают возможность для взаимодействия до достижения температуры 15°C. Через 40 минут 730 мг (2,5 ммоль, 1,0 эквивалент) 5-хлорметил-4-метил-2-[(4-трифторметил)-фенил]тиазола формулы IV, растворенного в 3 мл безводного ТГФ добавляют к реакционному продукту при такой же температуре. После взаимодействия в течение дополнительного времени, примерно 20 минут, реакционный материал достаточно охлаждают до -78°C. Затем добавляют по каплям 3,0 мл (2,0 эквивалента) трет-бутиллития (1,7 M раствор гептана) к реакционному раствору, и когда реакционный раствор приобретает синий цвет, добавляют к нему 345 мкл (2,5 ммоль) 2-хлор-6-фторбензил бромида при такой же температуре. Смеси дают возможность для взаимодействия, в то же время медленно повышая температуру до комнатной температуры. Через 20 минут добавляют 30 мл водного раствора хлорида аммония для прекращения реакции. Органический слой отделяют и сушат над сульфатом магния. После фильтрования растворитель удаляют посредством дистилляции при пониженном давлении. Остаток очищают с помощью колоночной хроматографии с использованием гексана/этилацетата (объем/объем =3/1), получая, таким образом, 1,12 г (выход 83%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDC13) δ 7,97 (д, 2H, J=8,1 Гц), 7,64 (д, 2H, J=8,2 Гц), 7,14 (м, 3H), 6,98 (дд, 1H, J=8,2, 2,1 Гц), 6,90 (м, 1H), 6,55 (д, 1H, J=8,3 Гц), 4,74 (дд, 1H, J=8,7, 6,8 Гц), 3,40 (м, 2H), 2,18 (с, 3H), 1,85 (с, 3H).
Примеры 40-41
Соединения, показанные в таблице 5 ниже, получают в соответствии со способом Примера 39, и данные ЯМР полученных соединений показаны в таблице 6 ниже.
Таблица 5
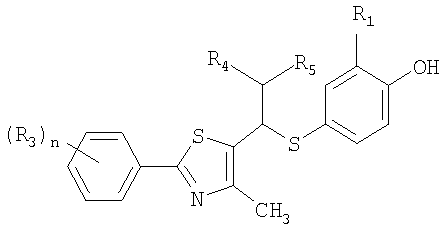
Пример 42: Получение сложного этилового эфира 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-4-фенилбутилтио]-2-метилфенокси]уксусной кислоты формулы IX [Стадия F]
При комнатной температуре 205 мг (0,4 ммоль) 2-метил-4-[1-[4-метил-2-(4-трифторметил-фенил)тиазол-5-ил]-4-фенил-бутилсульфанил]фенола хорошо перемешивают с 10 мл ацетона, содержащего 5% воды и 127 мг (0,92 ммоль, 2,3 эквивалента) карбоната калия. К раствору добавляют 67 мкл (0,6 ммоль, 1,5 эквивалента) сложного этилового эфира бромуксусной кислоты и смесь сильно перемешивают в течение 4 часов. После завершения реакции продукт реакции экстрагируют водным раствором соли и этилацетатом и органический слой сушат над сульфатом магния. После фильтрования растворитель удаляют посредством дистилляции при пониженном давлении и остаток очищают с помощью колоночной хроматографии с использованием гексана/этилацетата (объем/объем =5:1), получая, таким образом, 230 мг (выход 96%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDC13) δ 7,98 (д, 2H, J=8,2 Гц), 7,66 (д, 2H, J=8,5 Гц), 7,19 (м, 6H), 7,01 (дд, 1H, J=8,4, 2,2 Гц), 6,52 (д, 1H, J=8,4), 4,59 (с, 2H), 4,23 (м, 3H), 2,63 (т, 2H, J=8,4 Гц), 2,19 (с, 3H), 2,08 (м, 1H), 2,02 (с, 3H), 1,90 (м, 1H), 1,73 (м, 2H), 1,27 (т, 3H, J=7,2 Гц).
13C ЯМР (75,5 МГц, CDC13) δ 168,9, 163,3, 156,9, 151,2, 141,8, 137,8, 137,1, 137,0, 133,8, 131,6 (кв, J=33 Гц), 128,6, 128,6, 128,3, 126,5, 126,2, 126,0 (кв, J=4 Гц), 124,6, 111,5, 65,7, 61,6, 47,5, 37,3, 35,6, 29,6, 16,3, 15,2, 14,3.
Пример 43: Получение сложного аллилового эфира 2-[4-[2-(2-хлор-6-фторфенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусной кислоты формулы IX [Стадия F]
При комнатной температуре 200 мг (0,37 ммоль) 4-(2-(2-хлор-6-фторфенил)-1-(2-(4-(трифторметил)фенил)-4-метилтиазол-5-ил)этилтио)-2-метилфенола, полученного в Примере 37, хорошо перемешивают с 10 мл ацетона, содержащего 5% воды и 102 мг (0,74 ммоль, 2 эквивалента) карбоната калия. К раствору добавляют 73 мг (0,40 ммоль, 1,1 эквивалента) сложного аллилового эфира бромуксусной кислоты и смесь сильно перемешивают в течение 4 часов. После завершения реакции продукт реакции экстрагируют водным раствором соли и этилацетатом и органический слой сушат над сульфатом магния. После фильтрования растворитель удаляют посредством дистилляции при пониженном давлении и остаток очищают с помощью колоночной хроматографии с использованием смешанного растворителя гексана/этилацетата (объем/объем=5:1), получая, таким образом, 221 мг (выход 94%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDC13) δ 7,60-7,76 (м, 3H), 7,11-7,17 (м, 4H), 6,90 (м, 1H), 6,55 (д, 1H, J=8,4 Гц), 5,89 (м, 1H), 5,34 (м, 1H), 5,24 (м, 1H), 4,79 (дд, 1H, J=8,8, 6,6 Гц), 4,68 (м, 2H), 4,59 (с, 2H), 3,38 (м, 2H), 2,20 (с, 3H), 1,90 (с, 3H).
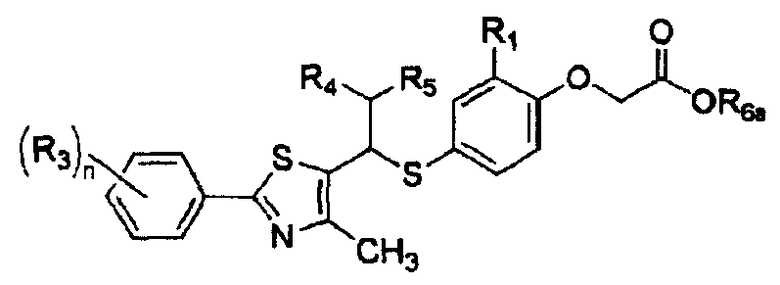
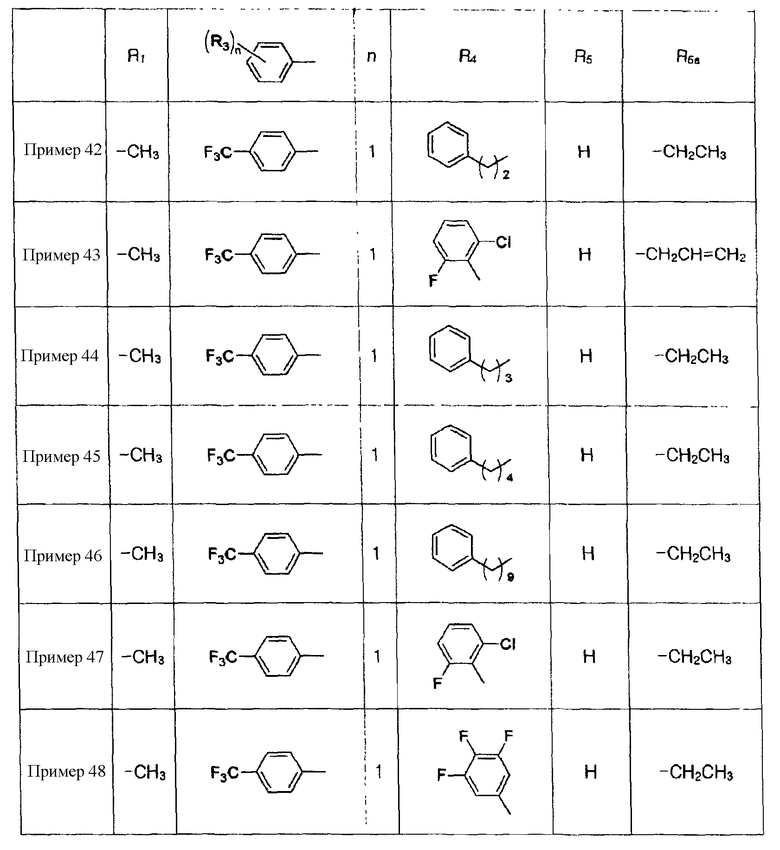
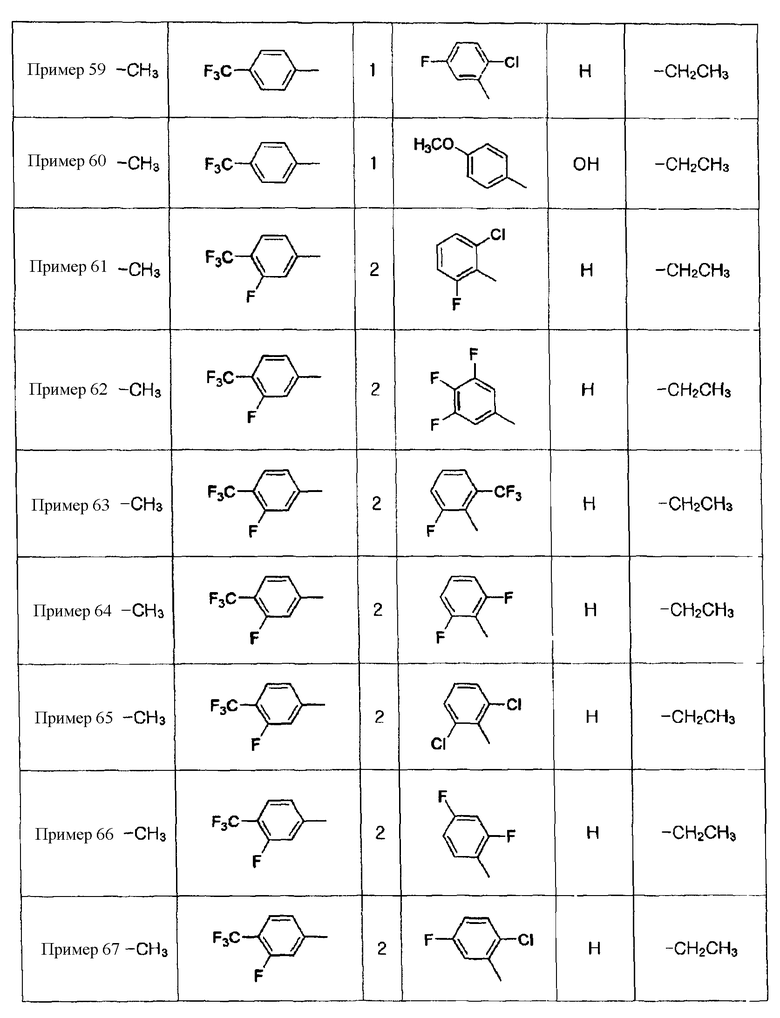
Примеры 44-67
Соединения, показанные в таблице 7 ниже, получают в соответствии со способом Примера 42, и данные ЯМР полученных соединений показаны в таблице 8 ниже.
Таблица 7
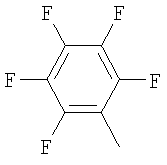
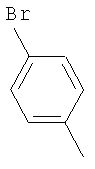
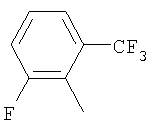
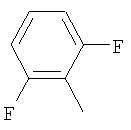
Пример 68: Получение 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-3-фенилпропилтио]-2-метилфенокси]уксусной кислоты формулы X [Стадия G]
178 мг (0,3 ммоль) сложного этилового эфира 2-[4-[1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]-3-фенилпропилтио]-2-метилфенокси]уксусной кислоты хорошо перемешивают с 15 мл этанола. К раствору добавляют 1,0 мл 3 н. водного раствора гидроксида натрия и смесь перемешивают при комнатной температуре в течение 20 минут. После завершения реакции pH продукта реакции доводят до 2,0 с помощью 2 н. HCl, 80% этанолового растворителя удаляют посредством дистилляции при пониженном давлении и оставшийся материал экстрагируют водным раствором соли и этилацетатом. Затем растворитель удаляют посредством дистилляции при пониженном давлении и остаток очищают с помощью колоночной хроматографии на колонке LH-20, получая, таким образом, 166 мг (выход 99%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDC13) δ 7,98 (д, 2H, J=8,1 Гц), 7,68 (д, 2H, J=8,3 Гц), 7,26 (м, 3H), 7,12 (м, 3H), 6,99 (дд, 1H, J=8,4, 2,2 Гц), 6,55 (д, 1H, J=8,5 Гц), 4,64 (с, 1H), 4,20 (дд, 1H, J=8,9, 6,1 Гц), 3,85 (с, 2H), 2,73 (м, 2H), 2,38 (м, 1H), 2,20 (м, 1H), 2,17 (с, 3H), 1,89 (с, 3H).
Пример 69: Получение 2-[4-[2-(2-хлор-6-фторфенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]ацетата калия формулы X [Стадия G]
200 мг (0,31 ммоль) сложного аллилового эфира 2-[4-[2-{2-хлор-6-фторфенил)-1-[2-[4-(трифторметил)фенил]-4-метилтиазол-5-ил]этилтио]-2-метилфенокси]уксусной кислоты и 18 мг (0,015 ммоль, 0,05 эквивалента) палладий тетракистрифенилфосфина растворяют в 10 мл безводного дихлорметана и перемешивают при комнатной температуре. К реакционному раствору медленно добавляют 56 мг (0,31 ммоль, 1,0 эквивалент) 2-этилгексаноата калия, растворенного в 1 мл безводного дихлорметана. Смесь перемешивают при комнатной температуре в течение 1 часа и затем растворитель удаляют посредством центрифугирования. Оставшийся твердый продукт промывают 10 мл дихлорметана и 10 мл н-гексана и сушат, получая, таким образом, 179 мг (выход 91%) указанного в заголовке соединения.
1H ЯМР (300 МГц, D2O) δ 7,96 (д, 2H, J=8,1 Гц), 7,66 (д, 2H, J=8,3 Гц), 7,17 (м, 5H) 6,91 (м, 1H), 6,56 (д, 1H, J=8,5 Гц), 4,79 (дд, 1H, J=8,5, 6,9 Гц), 4,66 (с, 2H), 3,39 (м, 2H), 2,18 (с, 3H), 1,87 (с, 3H).
Примеры 70-97
Соединения, показанные в таблице 9 ниже, получают в соответствии со способом Примера 68, и данные ЯМР полученных соединений показаны в таблице 10 ниже. Здесь спектры ЯМР соединений, где R6b представляет собой щелочной металл (натрий или калий), являются идентичными спектрам соединений, где R6b представляет собой водород.
Таблица 9
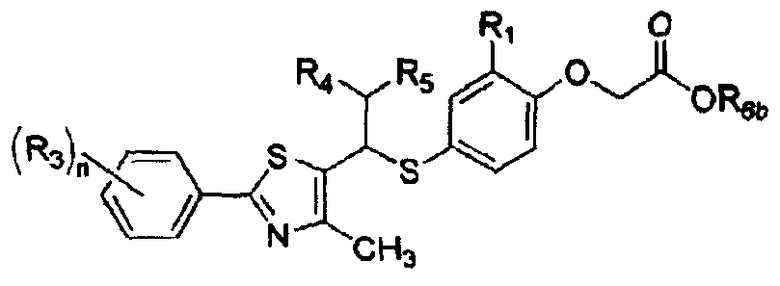
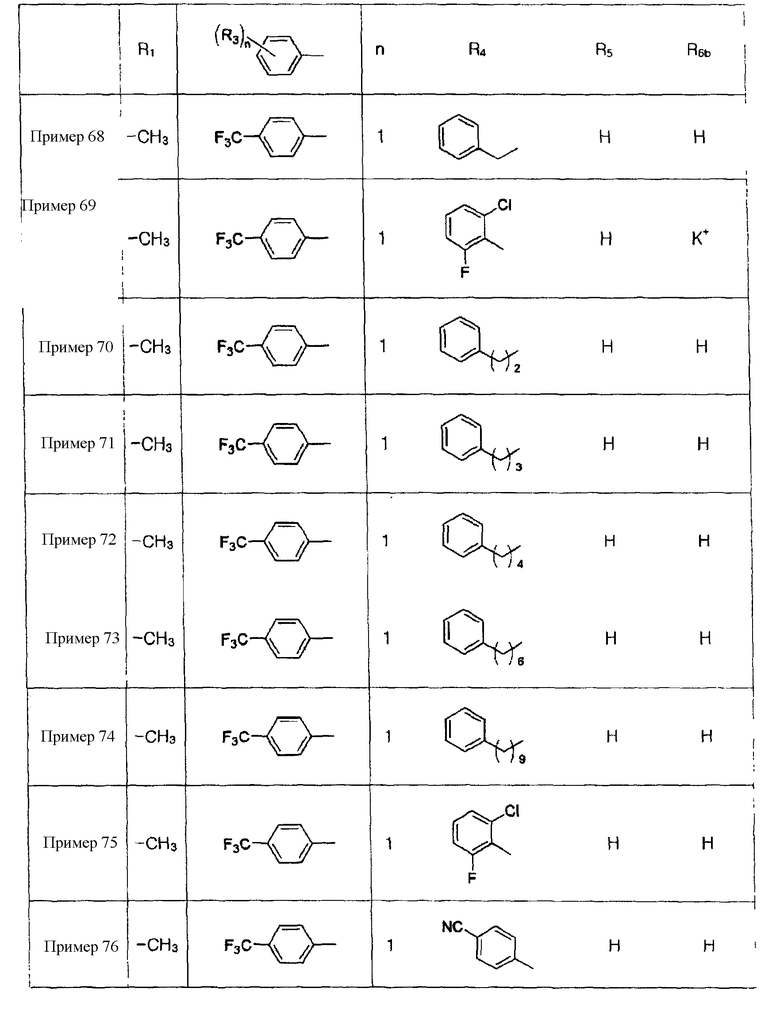
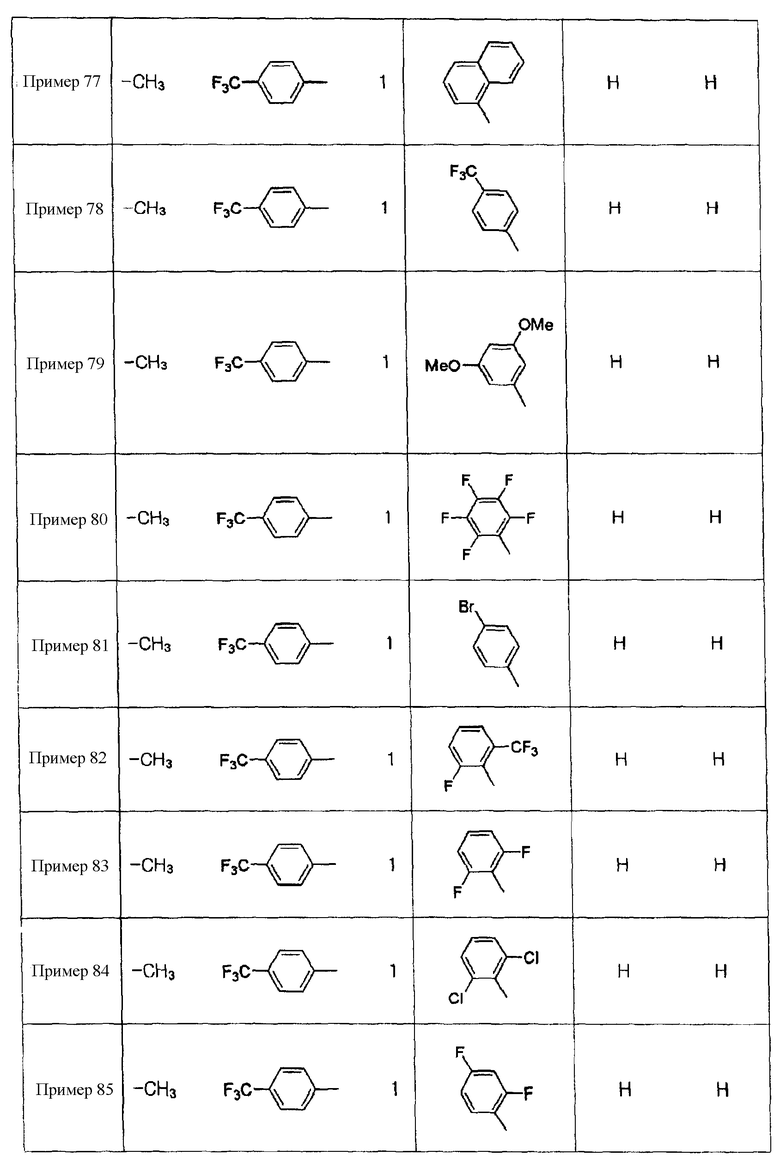
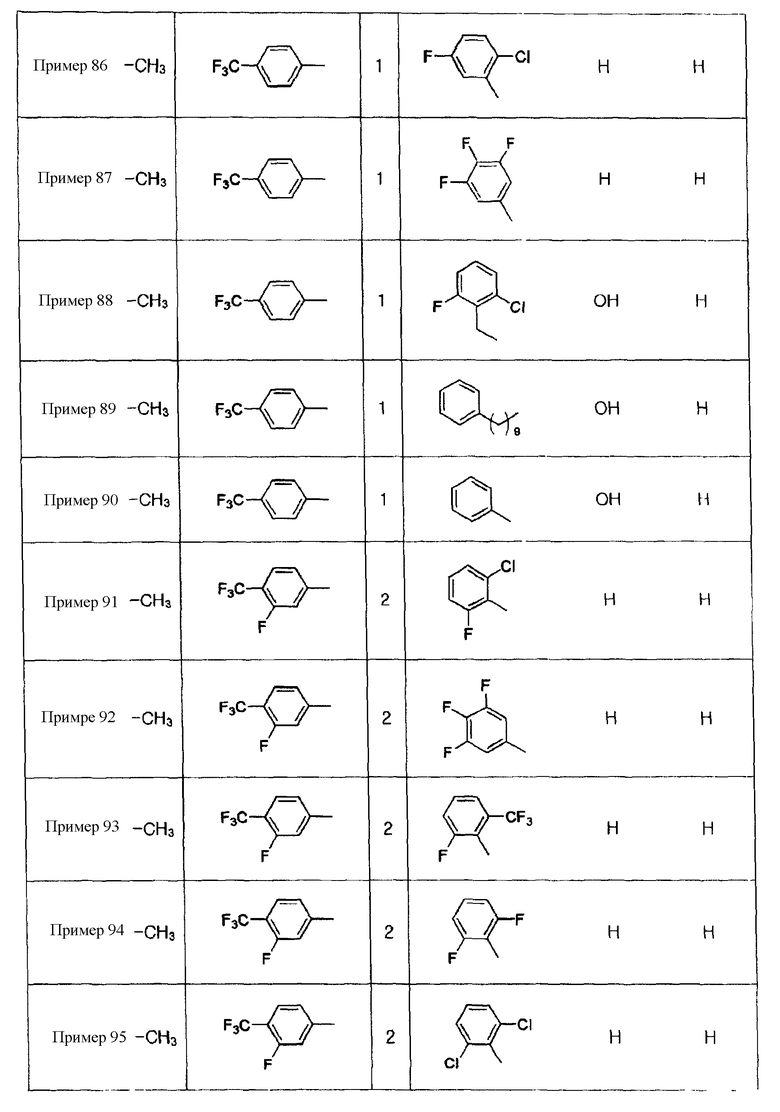
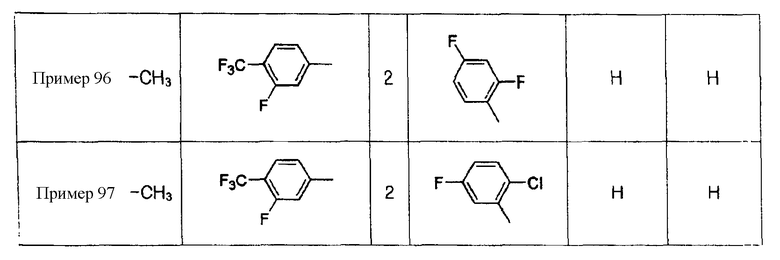
Таблица 10
Пример исследования 1: Исследования активности и цитотоксичности
Соединения, полученные в Примерах, исследуют на активность по отношению к PPARδ посредством трансфекционного анализа. В дополнение к этому соединения исследуют на селективность по отношению к PPARα и PPARγ субтипам PPAR, а также исследуют на токсичность посредством анализа с помощью MTT.
Трансфекционный анализ
Трансфекционный анализ осуществляют с использованием клеток CV-1. Культуру клеток получают с использованием среды DMEM (10% FBS, DBS (делипидированный) и 1% пенициллина/стрептомицина) на 96-луночном планшете, в инкубаторе, содержащем 5% двуокиси углерода, при 37°C. Исследование осуществляют на четырех стадиях, состоящих из инокуляции клеток, трансфекции, обработки соединений и анализа результатов. Конкретно, клетки CV-1 инокулируют на 96-луночном планшете при концентрации 5000 клеток/лунка, и через 24 часа клетки трансфицируют. При трансфицировании клеток полноразмерную плазмиду ДНК PPAR, репортерную ДНК, которая имеет люциферазную активность и, таким образом, делает возможной идентификацию PPAR и ДНК β-галактозидазы, которая обеспечивает информацию об эффективности трансфицирования, используют в качестве реагентов трансфекции. Каждое из соединений, разработанных в настоящем изобретении, растворяют в диметилсульфоксиде (ДМСО) и трансфицируют в клетки при различных концентрациях, используя среды. Клетки культивируют в инкубаторе в течение 24 часов, а затем лизируют с помощью лизисного буфера. Лизированные клетки измеряют на активности люциферазы и β-галактозидазы с использованием люминометра и считывающего устройства для микропланшетов. Измеренные значения для люциферазы нормируют с помощью значений для β-галактозидазы и представляют в виде графика. Из графика определяют значения EC50.
Значения EC50 соединений, полученных в Примерах 47-97 в соответствии с настоящим изобретением, являются, в основном, меньшими, чем 50 нМ, и соединения показывают, по меньшей мере, в 10000 раз большую селективность по сравнению с PPARα и PPARγ.
Анализ MTT
Соединения Примеров 47-97 в соответствии с настоящим изобретением исследуют на цитотоксичность, используя анализ MTT. MTT представляет собой водорастворимое желтое вещество, но, если оно вводится в живые клетки, оно будет дегенерировать в водонерастворимые пурпурные кристаллы благодаря дегидрогеназе, содержащейся в митохондриях. Если это вещество растворяют в диметилсульфоксиде, а затем измеряют на поглощение при 550 нм, может анализироваться цитотоксичность. Способ исследования представляет собой следующее.
Клетки CV-1 сначала инокулируют на 96-луночном планшете при концентрации 5000 клеток/лунку. Инокулированные клетки культивируют в увлажняемом инкубаторе, содержащем 5% двуокиси углерода, при 37°C в течение 24 часов, а затем обрабатывают соединениями по настоящему изобретению при различных концентрациях. После 24 часов культивирования к культивированным клеткам добавляют реагент MTT. После 15 минут культивирования полученный пурпурный кристалл растворяют в диметилсульфоксиде, а затем измеряют на поглощение с использованием считывающего устройства для микропланшетов. По измеренному поглощению анализируют цитотоксичность.
Результаты исследований показывают, что большая часть соединений по настоящему изобретению не имеет цитотоксичности даже при концентрации 90 мкМ.
обозначение «ia» означает «в отсутствие»
обозначение «ND» означает «нет данных»
Промышленное применение
Настоящее изобретение предусматривает новые тиазольные производные в качестве лигандов, активирующих пролифератор-активируемый рецептор δ пероксисомы (PPARδ), которые могут использоваться для лечения тучности, гиперлипидемии, атеросклероза и диабета, а также их промежуточные соединения и способы их получения. Настоящее изобретение является пригодным для получения новых производных соединений тиазола в качестве лигандов, активирующих PPARδ.
Дополнительные примеры
В примерах представлены оздоровительная пищевая добавка, оздоровительный напиток, пищевая добавка и кормовые композиции для животных.
«Активный ингредиент», как указано в нижеприведенных примерах, относится к тиазольному производному формулы (I).
Пример 1
Нагретое до 60°С оливковое масло нагревали и перемешивали с активным ингредиентом. Получали жидкий раствор активного ингредиента. Полученный раствор использовался для изготовления препаратов с жидким содержимым.
Полученную смесь помещали в мягкие капсулы с использование традиционного метода. Одна мягкая капсула содержит:
Активный ингредиент 20 мг
Оливковое масло 350 мг
Пример 2
Активный ингредиент растворяли в диметилсульфоксиде, адсорбированном на кристаллической целлюлозе (мелкодисперсный порошок) и высушивали. Полученный сухой состав использовался для изготовления сухих препаратов, включая кормовые и пищевые композиции, а также в качестве добавки в жидкие суспензионные композиции.
Полученный продукт смешивали с кукурузным крахмалом и преобразовывали в порошок традиционным методом.
Активный ингредиент 10 мг
Кристаллическая целлюлоза 40 мг
Кукурузный крахмал 55 мг
Пример 3
Активный ингредиент растворяли в диметилсульфоксиде, адсорбированном на кристаллической целлюлозе (мелкодисперсный порошок), и высушивали. Полученный продукт смешивали с кукурузным крахмалом, лактозой, карбоксиметилцеллюлозой и стеаратом магния. Добавляли к полученной смеси водный раствор поливинилпирролидона в качестве связующего вещества и полученную смесь преобразовывали в гранулы традиционным методом. Гранулы смешивали с тальком, функционирующим в качестве смазочного вещества. Полученную в результате смесь преобразовывали в таблетки, где каждая таблетка содержала 20 мг активного ингредиента.
Пример 4
Перечисленные ниже ингредиенты смешивали и полученную смесь преобразовывали в гранулы традиционным методом. Полученные гранулы использовались для изготовления сухих препаратов, включая оздоровительные пищевые добавки, оздоровительных напитков, пищевых добавок.
Полученные гранулы паковались в желатиновые твердые капсулы, где каждая капсула содержала 20 мг активного ингредиента.
| название | год | авторы | номер документа |
|---|---|---|---|
| СЕЛЕНОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2371437C2 |
| НОВОЕ ПРОИЗВОДНОЕ АНТРАНИЛОВОЙ КИСЛОТЫ ИЛИ ЕГО СОЛЬ | 2005 |
|
RU2394021C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2013 |
|
RU2640416C2 |
| ДИАРИЛЬНЫЕ СОЕДИНЕНИЯ С МОСТИКОВОЙ СВЯЗЬЮ | 2002 |
|
RU2297409C2 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ БЕНЗАМИДА И ИНГИБИТОР АКТИВНОСТИ ПОДТИПА 1 РЕЦЕПТОРА ВАНИЛЛОИДА (VR1) | 2005 |
|
RU2392278C2 |
| ПРОИЗВОДНОЕ (АЗА)ИНДОЛА И ЕГО ПРИМЕНЕНИЕ В ЛЕЧЕБНЫХ ЦЕЛЯХ | 2008 |
|
RU2477274C2 |
| ИСПОЛЬЗОВАНИЕ ЛИГАНДОВ РЕЦЕПТОРА ЕР4 ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ IL-6 ЗАБОЛЕВАНИЙ | 2003 |
|
RU2285527C2 |
| ПРОИЗВОДНЫЕ 2-ПИРИДОНА | 2010 |
|
RU2551847C2 |
| ПРОИЗВОДНЫЕ 2-ОКСОХИНАЗОЛИНА КАК ИНГИБИТОРЫ МЕТИОНИН АДЕНОЗИЛТРАНСФЕРАЗЫ 2A | 2019 |
|
RU2830169C2 |
| БЕНЗИМИДАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2456276C2 |
Настоящее изобретение относится к тиазольным производным формулы I. Соединения изобретения обладают активностью по отношению к рецептору, активируемому пероксисомальным пролифератором δ (PPARδ). В формуле I
А представляет собой 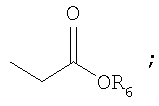 R1 означает С1-4алкильную группу; группы R3 являются отличными друг от друга и обозначают атом галогена или С1-4алкильную группу, замещенную или незамещенную галогеном; R4 означает
R1 означает С1-4алкильную группу; группы R3 являются отличными друг от друга и обозначают атом галогена или С1-4алкильную группу, замещенную или незамещенную галогеном; R4 означает 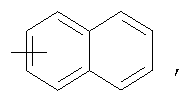
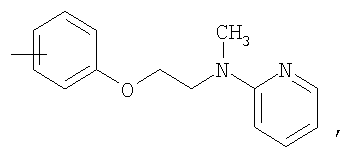
 или
или 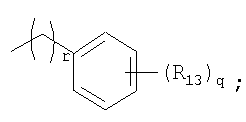 означает R5 представляет собой атом водорода или гидроксильную группу; значения остальных радикалов представлены в формуле изобретения. Изобретение также относится к способам получения соединения I, к промежуточным соединениям VI, VII и их способам получения, к средствам для предотвращения и лечения диабета, тучности, атеросклероза, гиперлипидемии, содержащих в качестве активного ингредиента тиазольное производное формулы I. 19 н. и 2 з.п. ф-лы, 10 табл.
означает R5 представляет собой атом водорода или гидроксильную группу; значения остальных радикалов представлены в формуле изобретения. Изобретение также относится к способам получения соединения I, к промежуточным соединениям VI, VII и их способам получения, к средствам для предотвращения и лечения диабета, тучности, атеросклероза, гиперлипидемии, содержащих в качестве активного ингредиента тиазольное производное формулы I. 19 н. и 2 з.п. ф-лы, 10 табл.
1. Тиазольные производные формулы I в виде рацематов или оптических изомеров:
где А представляет собой 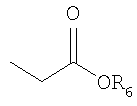
R1 представляет собой С1-4алкильную группу;
m представляет собой целое число 1;
группы R3 являются отличными друг от друга и обозначают атом галогена или С1-4алкильную группу, замещенную или незамещенную галогеном;
n представляет собой целое число от 1 до 5;
R4 представляет собой
 ,
,  ,
,  или
или 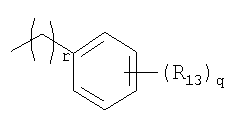 ;
;
R5 представляет собой атом водорода или гидроксильную группу;
R6 представляет собой защитную группу карбоновой кислоты, имеющую С1-4алкил, аллильную группу, атом водорода или щелочной металл;
R12 представляет собой атом галогена, цианогруппу или С1-4алкильную или алкоксигруппу, замещенную или незамещенную галогеном;
R13 представляет собой атом галогена;
р и q, каждый, независимо представляют собой целое число от 0 до 5; и
r представляет собой целое число от 1 до 9.
2. Тиазольные производные формулы VI в виде рацематов или оптических изомеров:
где R1, R3-R5, m и n имеют такие же значения, как определено в формуле I; R2 представляет собой фенол-защитную группу, выбранную из С1-4 низшей алкильной группы, аллильной группы и С1-4 алкилсилильной группы.
3. Тиазольные производные формулы VII в виде рацематов или оптических изомеров:
где R1, R3-R5, m и n имеют такие же значения, как определено в формуле I.
4. Тиазольные производные по п.1, которые представлены формулой IX:
где R1, R3-R5, m и n имеют такие же значения, как определено в формуле I, и R6a представляет собой защитную группу карбоновой кислоты, имеющую С1-4алкильную или аллильную группу.
5. Тиазольные производные по п.1, которые представлены формулой X:
где R1, R3-R5, m и n имеют такие же значения, как определено в формуле I, и R6b представляет собой атом водорода или щелочной металл.
6. Способ получения тиазольного производного формулы VI по п.2, включающий стадии:
a) взаимодействия 4-галогенфенольного соединения формулы II с фенол-защитной алкилсилильной группой в присутствии основания с получением соединения формулы III;
b) замещения атома галогена в соединении формулы III литием, и затем реакции с серой и соединением формулы IV с получением соединения формулы V; и
c) взаимодействия соединения формулы V с O=CR4R5 или Х3-СНR4R5 (электрофильное соединение) в присутствии сильного основания с получением соединения формулы VI:
где X1 обозначает атом брома или атом йода, Х2 обозначает атом хлора, атом брома, атом йода или уходящую группу, имеющую высокую реакционную способность в реакции нуклеофильного замещения, a R1, R3, R4, R5, m и n имеют такие же значения, как определено в формуле I, R2 представляет собой фенол-защитную группу, выбранную из С1-4 низшей алкильной группы, аллильной группы и С1-4 алкилсилильной группы.
7. Способ получения тиазольного производного формулы VII по п.3, включающий стадии:
a) взаимодействия 4-галогенфенольного соединения формулы II с фенол-защитной алкилсилильной группой в присутствии основания с получением соединения формулы III;
b) замещения атома галогена в соединении формулы III литием, и затем реакции с серой и соединением формулы IV с получением соединения формулы V;
c) взаимодействия соединения формулы V с O=CR4R5 или X3-CHR4R5 (электрофильное соединение) в присутствии сильного основания с получением соединения формулы VI; и
а) удаления фенол-защитной силилильной группы соединения формулы VI с получением соединения формулы VII:
где X1 обозначает атом брома или атом йода, Х2 обозначает атом хлора, атом брома, атом йода или уходящую группу, имеющую высокую реакционную способность в реакции нуклеофильного замещения, и R1, R3-R5, m и n имеют такие же значения, как определено в формуле I, R2 представляет собой фенол-защитную группу, выбранную из С1-4 низшей алкильной группы, аллильной группы и C1-4 алкилсилильной группы.
8. Способ получения тиазольного производного формулы VII по п.3, включающий стадии:
взаимодействия 4-галогенфенольного соединения формулы II с реагентом Гриньяра, замещения атома галогена в соединении формулы III литием, и затем взаимодействия продукта реакции с серой и соединением формулы IV с получением соединения простого тиоэфира; и
взаимодействия соединения простого тиоэфира с сильным основанием без выделения и затем с O=CR4R5 или X3-CHR4R5 (электрофильное соединение) с получением соединения формулы VII:
где X1 обозначает атом брома или атом йода, Х2 обозначает атом хлора, атом брома, атом йода или уходящую группу, имеющую высокую реакционную способность в реакции электрофильного замещения, и R1, R3-R5, m и n имеют такие же значения, как определено в формуле I.
9. Способ получения тиазольного производного формулы IX по п.4, включающий стадии:
a) взаимодействия 4-галогенфенольного соединения формулы II с фенол-защитной алкилсилильной группой в присутствии основания с получением соединения формулы III;
b) замещения атома галогена в соединении формулы III литием, и затем реакции с серой и соединением формулы IV с получением соединения формулы V;
c) взаимодействия соединения формулы V с O=CR4R5 или X3-CHR4R5 (электрофильное соединение) в присутствии сильного основания с получением соединения формулы VI;
d) удаления фенол-защитной силилильной группы соединения формулы VI с получением соединения формулы VII; и
e) взаимодействия соединения формулы VII с алкилгалогенацетатом формулы VIII в присутствии неорганической соли с получением соединения формулы IX:
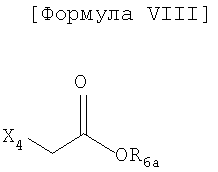
где X1 обозначает атом брома или атом йода, Х2 обозначает атом хлора, атом брома, атом йода или уходящую группу, имеющую высокую реакционную способность в реакции нуклеофильного замещения, и R1, R3-R5, m и n имеют такие же значения, как определено в формуле I, R2 представляет собой фенол-защитную группу, выбранную из С1-4 низшей алкильной группы, аллильной группы и C1-4 алкилсилильной группы, R6а представляет собой защитную группу карбоновой кислоты, имеющую С1-4алкильную или аллильную группу, и Х4 представляет собой атом хлора, атом брома или атом йода.
10. Способ получения тиазольного производного формулы Х по п.5, включающий стадии:
a) взаимодействия 4-галогенфенольного соединения формулы II с фенол-защитной алкилсилильной группой в присутствии основания с получением соединения формулы III;
b) замещения атома галогена в соединении формулы III литием, и затем, реакции с серой и соединением формулы IV с получением соединения формулы V;
c) взаимодействия соединения формулы V с O=CR4R5 или X3-CHR4R5 (электрофильное соединение) в присутствии сильного основания с получением соединения формулы VI;
d) удаления фенол-защитной силилильной группы соединения формулы VI с получением соединения формулы VII;
e) взаимодействия соединения формулы VII с алкилгалогенацетатом формулы VIII в присутствии неорганической соли с получением соединения формулы IX; и
f) гидролиза соединения формулы IX со сложным эфиром карбоновой кислоты в присутствии гидроксидов щелочного металла или кислоты в спиртовом растворе с получением соединения формулы X:
где X1 обозначает атом брома или атом йода, Х2 обозначает атом хлора, атом брома, атом йода или уходящую группу, имеющую высокую реакционную способность в реакции нуклеофильного замещения, и R1, R3-R5, m и n имеют такие же значения, как определено в формуле I, R2 представляет собой фенол-защитную группу, выбранную из C1-4 низшей алкильной группы, аллильной группы и C1-4 алкилсилильной группы, R6a представляет собой защитную группу карбоновой кислоты, имеющую С1-4алкильную или аллильную группу, Х4 представляет собой атом хлора, атом брома или атом йода, и R6b представляет собой атом водорода или щелочной металл.
11. Способ получения тиазольного производного формулы Х по п.5 взаимодействием соединения формулы IX, где R6а представляет собой аллильную группу, с солью щелочного металла в присутствии палладий тетракистрифенилфосфина в органическом растворителе с получением соединения формулы X:
где R1, R3-R5, m и n имеют такие же значения, как определено в формуле I, R6а представляет собой аллильную группу и R6b представляет собой щелочной металл.
12. Средство для лечения диабета, содержащее в качестве активного ингредиента тиазольное производное, представленное формулой I
где А представляет собой , R1, R3-R6, m и n имеют такие же значения, как определено в формуле I.
13. Средство для предотвращения и лечения тучности, содержащее в качестве активного ингредиента тиазольное производное, представленное формулой I
где А представляет собой , R1, R3-R6, m и n имеют такие же значения, как определено в формуле I.
14. Средство для предотвращения и лечения атеросклероза, содержащее в качестве активного ингредиента тиазольное производное, представленное формулой I
где А представляет собой , R1, R3-R6, m и n имеют такие же значения, как определено в формуле I.
15. Средство для предотвращения и лечения гиперлипидемии, содержащее в качестве активного ингредиента тиазольное производное, представленное формулой I
где А представляет собой , R1, R3-R6, m и n имеют такие же значения, как определено в формуле I.
16. Оздоровительная пищевая добавка для предотвращения и лечения тучности, гиперлипидемии, атеросклероза и диабета, содержащая в качестве активного ингредиента тиазольное производное, представленное формулой I
где А представляет собой , R1, R3-R6, m и n имеют такие же значения, как определено в формуле I.
17. Оздоровительный напиток для предотвращения и лечения тучности, гиперлипидемии, атеросклероза и диабета, содержащий в качестве активного ингредиента тиазольное производное, представленное формулой I
где А представляет собой , R1, R3-R6, m и n имеют такие же значения, как определено в формуле I.
18. Пищевая добавка для предотвращения и лечения тучности, гиперлипидемии, атеросклероза и диабета, содержащая в качестве активного ингредиента тиазольное производное, представленное формулой I
где А представляет собой , R1, R3-R6, m и n имеют такие же значения, как
определено в формуле I.
19. Кормовая композиция для животных для предотвращения и лечения тучности, гиперлипидемии, атеросклероза и диабета, содержащая в качестве активного ингредиента тиазольное производное, представленное формулой I
где А представляет собой , R1, R3-R6, m и n имеют такие же значения, как определено в формуле I.
20. Способ получения тиазольного производного формулы IX по п.4, включающий стадии: взаимодействия 4-галогенфенольного соединения формулы II с реагентом Гриньяра, замещения атома галогена в соединении формулы III литием, и затем взаимодействия продукта реакции с серой и соединением формулы IV с получением соединения простого тиоэфира; и
взаимодействия соединения простого тиоэфира с сильным основанием без выделения и затем с O=CR4R5 или Х3-СНR4R5 (электрофильное соединение) с получением соединения формулы VII; и
взаимодействия соединения формулы VII с алкилгалогенацетатом формулы VIII в присутствии неорганической соли с получением соединения формулы IX:
где X1 обозначает атом брома или атом йода, Х2 обозначает атом хлора, атом брома, атом йода или уходящую группу, имеющую высокую реакционную способность в реакции нуклеофильного замещения, R1, R3-R5, m и n имеют такие же значения, как определено в формуле I, R6a представляет собой защитную группу карбоновой кислоты, имеющую С1-4алкильную или аллильную группу, и Х4 представляет собой атом хлора, атом брома или атом йода.
21. Способ получения тиазольного производного формулы Х по п.5, включающий стадии:
взаимодействия 4-галогенфенольного соединения формулы II с реагентом Гриньяра, замещения атома галогена в соединении формулы III литием, и затем взаимодействия продукта реакции с серой и соединением формулы IV с получением соединения простого тиоэфира; и
взаимодействия соединения простого тиоэфира с сильным основанием без выделения и затем с O=CR4R5 или X3-CHR4R5 (электрофильное соединение) с получением соединения формулы VII;
взаимодействия соединения формулы VII с алкилгалогенацетатом формулы VIII в присутствии неорганической соли с получением соединения формулы IX; и
гидролиза соединения формулы IX со сложным эфиром карбоновой кислоты в присутствии гидроксидов щелочного металла или кислоты в спиртовом растворе с получением соединения формулы X:
где X1 обозначает атом брома или атом йода, Х2 обозначает атом хлора, атом брома, атом йода или уходящую группу, имеющую высокую реакционную способность в реакции нуклеофильного замещения, и R1, R3-R5, m и n имеют такие же значения, как определено в формуле I, R6а представляет собой защитную группу карбоновой кислоты, имеющую С1-4алкильную или аллильную группу, Х4 представляет собой атом хлора, атом брома или атом йода, и R6b представляет собой атом водорода или щелочной металл.
| RU 2002135079 А, 10.12.2004 | |||
| ЕА 200400943 A1, 24.02.2005 | |||
| WO 03106442 A1, 24.12.2003 | |||
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОХИНОНА | 0 |
|
SU250047A1 |

