Настоящее изобретение относится к способу приготовления замещенного фенил-карбамата и его фармацевтически приемлемых солей, представляющих в настоящее время фармацевтический интерес. Замещенный фенил-карбамат и его фармацевтически приемлемые соли являются эффективными для повышения холинергической активности в центральной нервной системе и применимыми при лечении таких заболеваний, как болезнь Альцгеймера, синдром Дауна, хорея Гентингтона, атаксия Фридриха и т.д.
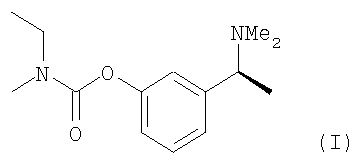
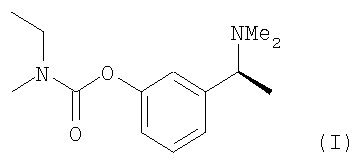
(S)-3-[(1-диметиламино)этил]-фенил-N-этил-N-метилкарбамат (I) является активным ингредиентом фармацевтической композиции, на которую дается ссылка в патенте US 5602176. Указанное соединение также используют для индуцирования селективного ингибирования активности ацетилхолинэстеразы в головном мозгу.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Способ получения соединения по формуле (I), т.е. (S)-3-[(l-диметиламино)этил]-фенил-N-этил-N-метилкарбамата (I) известен в литературе.
Для получения соединения по формуле (I) в соответствии со ссылкой в WO 2004/037771 необходим промежуточный 3-(1-диметиламиноэтил)фенол. Указанный промежуточный продукт получают путем использования Ti(OiPr)4, т.е. тетраизопропоксида титана, борогидрата натрия и диметиламина на первой стадии. На второй стадии используют HBr. Процесс становится дорогостоящим ввиду использования Ti(OiPr)4. Ti(OiPr)4 опасен в обращении, т.к. он представляет собой легковоспламеняющуюся жидкость и, кроме того, является веществом раздражающего действия. Более того, для инициирования реакции используют аммиак и к тому же остающийся по завершении реакции титановый осадок создает проблемы при обработке сточных вод и загрязняет окружающую среду. Борогидрат натрия является одним из дорогостоящих используемых восстанавливающих средств, он опасен в обращении, и для работы с ним требуется исключительно квалифицированный персонал для его добавления в смесь, а также для инициирования реакции. HBr вызывает серьезные ожоги при обращении с ним. HBr также является токсичным. Кроме того, J. Labeled Сотр. and Radiopharm. 1997, 39 (8), 651-668 использует HBr для проведения того же процесса на стадии деметилирования. При использовании HBr для деметилирования метокси-функции недостаток заключается в низком качестве конечного продукта. Для производства в промышленных масштабах требуется исключительно большое количество таких реагентов и, следовательно, необходимо исключить использование таких небезопасных реагентов.
В ЕР 359647 раскрывается способ приготовления аналога морфинана, при котором для метилирования орто-метокси соединения используют сульфоновую кислоту, такую как метаносульфоновую кислоту. Тем не менее, необходимо избегать использования такого дорогостоящего коррозионного и токсичного реагента при производстве в промышленных масштабах.
Способ получения в соответствии с описанием в патентах US 4948807 и US 5602176 соединения по формуле (I) осуществляется путем получения промежуточных соединений изоционатов или карбамоил галогенидов, при котором соединение по формуле (I) имеет заместители на фенилкарбамате в виде низшего алкила. Изоционаты, в частности изоционат с группой низших алкилов в виде заместителей, представляют опасность для здоровья человека ввиду своей токсичности. Применение таких растворителей, как сухой ацетонитрил или бензол, является необходимым для получения соединения по формуле (I) путем получения промежуточных соединений изоционатов или карбамоил галогенидов. Необходимо избегать проведения промышленных процессов с использованием таких дорогостоящих и канцерогенных растворителей, т.к. такие растворители могут привести к самопроизвольным взрывам. Имеется возможность избежать применения основания, например гидрида натрия, при проведении реакций в промышленных масштабах, т.к. при его использовании необходимо принять ряд мер безопасности в плане хранения, транспортировки, а также протекания реакции. Кроме того, гидрид натрия используют в количестве приблизительно 200%, что в процессе инициирования реакции помимо угрозы безопасности вызывает неконтролируемую экзотермичность, приводящую к образованию различных примесей.
В WO 03/101917 раскрывается способ, при котором одним из основных промежуточных продуктов является N-этил-N-метил-4-нитрофенил карбамат, полученный из 4-нитрофенил хлороформата. При описании данного способа не сообщается о получении 3-(1-диметиламиноэтил) фенола. Предпочтительно в способе получения используют [1-(3-метоксифенил)этил]диэтиламин. Реакцию деметилирования проводят с использованием 50% серной кислоты и D1-метионина. Необходимо избегать применения D1-метионина в промышленных масштабах, т.к. он является раздражителем кожи, глаз и дыхательных путей. Кроме того, применение D1-метионина удорожает производство. Помимо этого, продолжительность реакции составляет, как минимум, 28 часов, что увеличивает время до завершения всей последовательности реакции и получения ривастигмина. Следовательно, получение 3-(1-диметиламиноэтил)фенола достигается путем проведения большего количества стадий процесса. Таким образом, существует необходимость в сокращении количества стадий получения 3-(1-диметиламиноэтил)фенола.
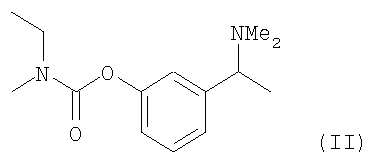
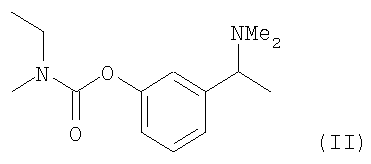
Jiang et. al. (Huadong Shifan Daxue Xuebao, Ziran Kexueban (2001), (1), 61-65. / CAN 136: 183572 AN 2001: 429602) обнаружили, что Ривастигмин может быть получен из кетона путем (i) его преобразования в оксим, (ii) восстановления до амина с последующим (iii) диметилированием амина и, наконец, (iv) путем преобразования амина в карбамат с целью получения рацемического N-этил-3-[(1-диметиламино)этил]-N-метил-фенил-карбамата по формуле (II). Следовательно, существуют практически четыре взаимных превращений функциональных групп исключительно с целью получения рацемического карбамата по формуле (II). Большее количество взаимных превращений функциональных групп приводит, главным образом, к сокращению выхода, а также к высокой стоимости реагентов и растворителей, более длительной производственной загрузке реактора, большему потреблению энергоресурсов, использованию большего количества рабочей силы и т.д. Таким образом, эффективность получения промежуточного продукта и, в свою очередь, конечного продукта ривастигмина является исключительно важным фактором при производстве в промышленных масштабах.
В WO 2005/061446 А1 раскрывается способ получения Ривастигмина, начиная от гидроксифенил кетона. Исходное соединение обрабатывают триэтиламином и этилметил карбамоилхлоридом с целью получения кетона фенилкарбамата, который при обработке диметиламином в присутствии цианоборогидрида натрия позволяет получить аминоалкил-фенилкарбамат, который далее расщепляют с помощью di-O-р-толуилвинной кислоты на водный метанол для получения Ривастигмина. Указанный способ имеет недостаток, заключающийся в том, что в процессе используют токсичное соединение, например цианоборогидрид натрия, который является достаточно опасным при применении в промышленных масштабах. Полученный с применением такого способа Ривастигмин далее подвергается кристаллизации в ряде процессов для достижения требуемой чистоты продукта. Последовательные стадии очистки приводят к значительному сокращению общего выхода продукта.
Следовательно, существует необходимость в создании способа получения с меньшим количеством стадий. Аналогичным образом необходимо исключить применение Ti(OiPr)4, что позволит достичь экономичности способа получения, промышленной и экологической безопасности. Исключение из процесса борогидрата натрия позволило бы упростить процесс и сделать его более безопасным при одновременном достижении высокой экономичности. Кроме того, необходимо исключить из процесса HBr, а также в способе получения не должны быть использованы изоцианат и карбамоилхлорид.
ЦЕЛЬ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является исключение применения i(OiPr)4, борогидрата натрия и HBr.
Другой целью настоящего изобретения является создание экономичного способа для крупномасштабного промышленного производства продукта.
Еще одной целью настоящего изобретения является создание способа, в котором исключено применение изоцианата и карбамоилхлорида.
Дополнительной целью настоящего изобретения является создание Ривастигмина с использованием карбонилдиимидазола.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Способ получения в соответствии с настоящим изобретением осуществляется по СХЕМЕ-1. Соединение по формуле (III) реагирует с аммиаком в присутствии катализатора для получения продукта восстановительного аминирования - 3-(1-аминоэтил)фенола, т.е. соединения по формуле (IV). В качестве катализатора используют никелевый катализатор Ренея. В качестве растворителя в реакции используют спирт, например метанол, этанол, изопропанол, n-пропанол и т.д. Предпочтительным спиртовым растворителем является метанол.
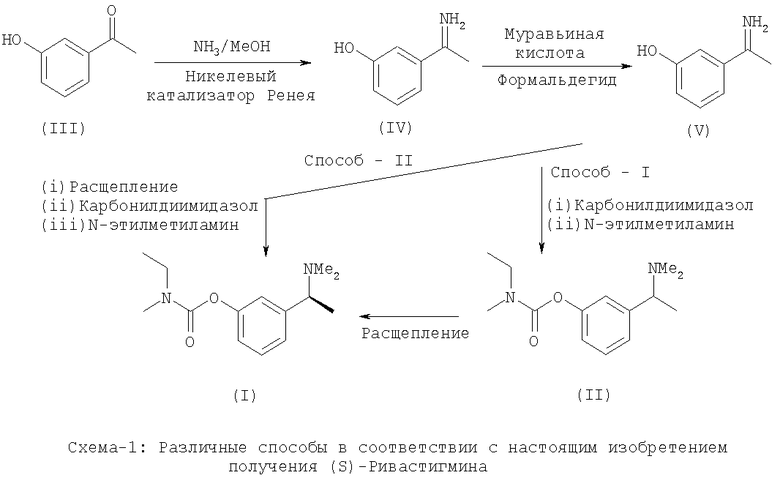
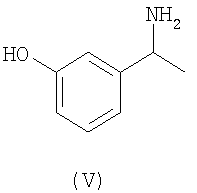
Как показано на CXEME-I, в соответствии с настоящим изобретением рацемический амин по формуле (IV), т.е. 3-(1-аминоэтил)фенол преобразуют в продукт (V) диметиламин, т.е. 3-(1-диметиламиноэтил)фенол. Диметилирование амина проводят с использованием муравьиной кислоты и формальдегида. В результате получают рацемическое соединение (V), т.е. 3-(1-диметиламиноэтил)фенол. При использовании соединения по формуле (V) применяют два способа: СПОСОБ-1: преобразование фенольного соединения (V) в карбамат;
с последующим расщеплением карбамата. СПОСОБ-2: расщепление 3-(1-диметламиноэтил)фенола с последующим образованием карбамата.
СПОСОБ-1:
a) Получение 3-гидроксил-1-фенилэтиламина (III).
Первая стадия в Способе-1 включает восстановительное аминирование соединения (III) с помощью никелевого катализатора Ренея в присутствии аммиака.
В настоящем примере осуществления изобретения способ получения соединения (IV) включает добавление 3-гидроксиацетофенона по формуле (III) к органическому растворителю, выбранному из группы, состоящей из гидроксильных растворителей, например спирта, воды или их смесей.
Используемый спирт выбирают из группы, состоящей из метанола, этанола, изопропанола, n-пропанола, n-бутанола и т.д.
Предпочтительным спиртом является метанол.
Восстановительное аминирование проводят либо только в спирту, либо в водно-спиртовой смеси. Добавляют гидроокись аммония, а затем никелевый катализатор Ренея. Смесь нагревают под давлением в автоклаве в присутствии водорода до завершения реакции. Никелевый катализатор Ренея удаляют путем фильтрации и далее концентрируют фильтрат. Остаток избирательно очищают путем образования солей присоединения кислоты.
b) Получение 3-(1-диметиламиноэтил)фенола (V).
Стадия относится к способу метилирования аминогруппы путем проведения реакции с формальдегидом в присутствии муравьиной кислоты.
3-(1-диметиламиноэтил)фенол по формуле (IV) метилируют в присутствии избыточного количества муравьиной кислоты и формальдегида при температуре флегмы.
Соединение по формуле (V) выделяют путем нейтрализации с помощью водного раствора неорганического основания с последующей экстракцией с помощью органического растворителя и дальнейшей концентрации органического слоя.
с) Получение ((S)-3-[(1-диметиламино)этил]-фенил-N-этил-N-метилкарбамата (II).
Данная стадия относится к получению карбамата путем проведения реакции этилметил карбамоилхлорида с соединением по формуле (V) в органическом растворителе и в присутствии основания.
3-(1-диметиламиноэтил)фенол по формуле (V) добавляют к органическому растворителю, выбранному из группы, включающей алифатические углеводороды, сложные эфиры, хлорированные углеводороды, нитрилы и т.д., предпочтительно нитрил, например ацетонитрил.
Эфиры выбирают из группы, включающей такие эфиры уксусной кислоты как этиловый, метиловый, изобутиловый и т.д.
Этилметил карбамоилхлорид добавляют к реакционной смеси.
Далее реакционную смесь нагревают до завершения реакции. Соединение по формуле (II) выделяют путем охлаждения реакционной смеси до комнатной температуры и экстрагируют с помощью органического растворителя. Органический слой концентрируют и получают соединение по формуле (II).
Полученный в соответствии с вышеописанным способом карбамат по формуле (II) расщепляют для получения (S)-N-этил-3-[(1-диметиламино)этил]-N-метил-фенил-карбамата (I).
СПОСОБ-2:
3-(1-диметиламиноэтил)фенол расщепляют с помощью расщепляющих агентов, например, выбранных из группы, включающей оптически активную винную кислоту, di-O-толилвинную кислоту, дибензилвинную кислоту, миндальную кислоту, камфорсульфоновую кислоту и т.д.
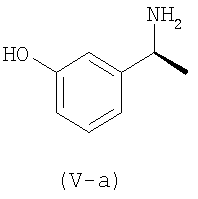
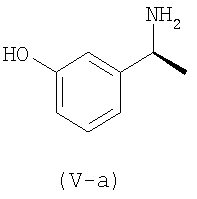
При дальнейшем протекании реакции используют оптически чистое соединение (V-A) для получения требуемого карбамата, т.е. (S)-N-этил-3-[(1-диметиламино)этил]-N-метил-фенил-карбамата (I).
Образование карбамата проводят с помощью различных способов, в которых используются различные вещества для введения карбонильной части между аминовым и фенольным компонентом, например карбонилдиимидазол, трифосген, метилкарбонат и т.д. Реагент для введения карбонила может сначала вступить в реакцию с фенольной ОН группой, а затем в реакцию с аминовым компонентом. По усмотрению реагент для введения карбонила может сначала вступать в реакцию с амином, а затем в реакцию с фенольной ОН группой.
В другом примере осуществления изобретения 4-нитрофенил хлороформат вступает в реакцию с 3-(1-диметиламиноэтил)фенолом, и далее протекает реакция указанного продукта с N-этилметиламином.
Из веществ для введения карбонила выбирают именно карбонилдиимидазол ввиду более высокой скорости протекания реакции и более простого выделения продукта по формуле (II).
Способ-II аналогичен способу-I до процесса получения соединения (V).
i) Расщепление 3-(1-диметиламиноэтил)фенола по формуле (V) для получения (S)-3-(1-диметиламиноэтил)фенола по формуле (V-a).
Первая стадия в способе-II относится к расщеплению соединения (V) путем использования камфорсульфоновой кислоты для получения 3-(1-диметиламиноэтил) фенола по формуле (V) и получения (S)-3-(1-диметиламиноэтил)фенола по формуле (V-a).
ii) Получение Ривастигмина.
а) Получение Ривастигмина путем использования карбонилдиимидазола и этилметиламина.
Получение Ривастигмина включает реакцию (S)-3-(1-(диметиламиноэтил)фенола или его рацемической смеси с этилметиламином в присутствии карбонилдиимидазола (CDI). Данная реакция предусматривает получение производного карбамата соединения (V-a), т.е. ривастигмина по формуле (I) путем введения карбонильной группы между фенольным кислородом и атомом азота этилметиламина.
Следует отметить, что впервые был применен карбонилдиимидазол для получения ривастигмина, при этом исключалось применение таких опасных реагентов, как этилметил карбамоилхлорид, изоцианат, фосген и т.д.
Реакция включает добавление (S)-3-{1-(диметиламиноэтил)фенола по формуле (V-a) или его рацемата по формуле (V) к органическому растворителю.
Органический растворитель выбирают из группы, включающей алифатические углеводороды, сложные эфиры, хлорированные растворители и т.д.
Эфиры выбирают из группы, включающей такие эфиры уксусной кислоты как этиловый, метиловый, изобутиловый и т.д.
Предпочтительным растворителем является хлорированный растворитель.
Хлорированный растворитель выбирают из группы, включающей дихлорметан, этилендихлорид и хлороформ. Предпочтительным хлорированным растворителем является дихлорметан. Количество используемого хлорированного растворителя составляет от 2 объемов до 20 объемов на грамм соединения (V-a) или (V). Предпочтительный объем используемого растворителя составляет от 4 объемов до 10 объемов на грамм соединения (V-a) или (V). К реакционной смеси добавляют карбонилдиимидазол. Количество добавленного карбонилдиимидазола составляет от 1,5 молей до 3,0 молей на моль соединения (V-a) или (V). Предпочтительное количество добавленного карбонилдиимидазола составляет от 1,75 молей до 2,0 молей на моль соединения (V-a) или (V).
Реакционную смесь дефлегмируют в течение 10-15 часов, и процесс реакции контролируют с помощью метода высокоэффективной жидкостной хроматографии. Реакционную смесь охлаждают в диапазоне от 0°С до 10°С и добавляют этилметиламин. Количество добавленного этилметиламина составляет от 1,0 моля до 3,0 молей на моль соединения (V-a) или (V). Предпочтительное количество добавленного этилметиламина составляет от 1,75 молей до 2,25 молей на моль соединения (V-a) или (V).
Реакционную смесь перемешивают при комнатной температуре в течение 4-6 часов. После завершения реакции смесь резко охлаждают водой. Органический слой отделяют и промывают разбавленным водным раствором неорганического основания. Неорганическое основание выбирают из карбоната натрия, карбоната калия, гидроокиси натрия, гидроокиси калия и т.д. Предпочтительным неорганическим основанием является гидроокись натрия. Органический слой промывают разбавленным раствором минеральной кислоты, например соляной кислоты, серной кислоты и т.д. Предпочтительной минеральной кислотой является соляная кислота.
Далее органический слой обрабатывают разбавленным раствором гидроокиси аммония и экстрагируют с помощью органического растворителя. Органический растворитель выбирают из группы, включающей алифатические углеводороды, хлорированные углеводороды, сложные эфиры и т.д. Предпочтительным органическим растворителем является алифатический углеводород, выбранный из группы, включающей гексан, циклогексан, гептан и т.д. После завершения экстрагирования органический слои концентрируют для получения соединения по формуле (I), имеющего 99% оптическую чистоту, либо соединения по формуле (II), т.е. рацемического ривастигмина, который далее расщепляют с использованием стандартных способов и получают Ривастигмин по формуле (I).
Таким образом, за счет исключения процесса расщепления рацемического ривастигмина по формуле (II) на последней стадии и использования значительно менее опасного, безопасного и доступного реагента, например, карбонилдиимидазола и этилметиламина достигается экономичность процесса, экологическая безопасность и его более высокая степень применимости для промышленных целей.
Получение Ривастигмина из (S)-3-{1-(диметиламиноэтил)фенола по формуле (V-a) путем использования этилметил карбамоилхлорида.
Способ включает реакцию (S)-3-{1-(диметиламиноэтил)фенола (V-a) с этилметил карбамоилхлоридом в присутствии слабого неорганического основания или органического основания.
Следует отметить, что в способах известного уровня техники, в которых описывается вышеуказанная реакция, раскрывается использование сильного основания, например гидрида натрия, который является опасным для здоровья человека при его применении в промышленных масштабах.
Кроме того, также важно отметить, что при использовании требуемого изомера промежуточного продукта (S)-3-{1-(диметиламиноэтил)фенола (V-a) для получения Ривастигмина, требуется вдвое меньшее количество этилметил карбамоилхлорида, что позволяет обеспечить экономичный процесс в промышленном масштабе. 3-[1-(диметиламино)этил]фенол по формуле (V) добавляют к органическому растворителю, выбранному из группы, включающей алифатические углеводороды, сложные эфиры, хлорированные углеводороды, нитрилы и т.д.
Предпочтительным растворителем является нитрил. Нитрилы выбирают из группы, включающей ацетонитрил, пропионитрил, бутионитрил и т.д. Предпочтительным растворителем является ацетонитрил. Объем используемого ацетонитрила составляет от 5 до 20 объемов на грамм соединения (V-a). Предпочтительный объем ацетонитрила составляет от 10 до 15 объемов на грамм соединения (V-a).
К смеси добавляют основание, выбранное из группы, включающей неорганическое основание или органическое основание. Предпочтительным основанием является неорганическое основание. Неорганическое основание выбирают из группы, включающей карбонат, бикарбонат, алкоксид щелочных металлов, либо карбонат и бикарбонат щелочно-земельных металлов.
Предпочтительным неорганическим основанием является карбонат щелочных металлов. Неорганическое основание выбирают из группы, включающей карбонат лития, карбонат натрия и карбонат калия. Предпочтительным неорганическим основанием является карбонат калия.
Количество используемого карбоната калия составляет от 1,0 моля до 4,0 молей на моль соединения по формуле (V-a). Предпочтительное количество карбоната калия составляет от 2,75 молей до 3,25 молей на моль соединения по формуле (V-a). К реакционной смеси добавляют этилметил карбамоилхлорид. Количество добавленного этилметил карбамоилхлорида составляет от 1,0 до 1,5 молей на моль соединения по формуле (V-a). Реакцию проводят при температуре в диапазоне от 60°С до 90°С.
Предпочтительная температура составляет 70°С-80°С. После завершения реакции смесь охлаждают до комнатной температуры и концентрируют. Остаток разбавляют водой и экстрагируют органическим растворителем для получения продукта по формуле (I).
Таким образом, преимущество заключается в использовании только половины количества этилметил карбамоилхлорида, что обеспечивает достижение экономической эффективности процесса и его применимости при производстве в промышленных масштабах.
Способ настоящего изобретения приведен ниже со ссылками на примеры, которые имеют чисто иллюстративный характер и никоим образом не должны рассматриваться как ограничивающие объем настоящего изобретения.
ПРИМЕРЫ
Получение 3-окси1-1-фенилэтиламина (IV)
Восстановительное аминирование проводят на 3-оксиацетофеноне (25 г) с аммиачным метанолом (250 мл) в присутствии никелевого катализатора Ренея (5 г) при 80°С и при давлении 10 кг/см2 водородной атмосферы в автоклаве. Спустя 12-14 часов продукт отделяют путем удаления никелевого катализатора Ренея и концентрации фильтрата. Далее продукт очищают путем преобразования его в гидрохлоридную соль, экстракции с помощью этилацетата (100 мл) и нейтрализации каустическим раствором до достижения рН 11-12, в результате чего выход продукта достигает 70%.
Выход: 21,25 грамм.
% выхода: 85%.
Степень чистоты по методу ВЭЖХ: 98-99%.
Получение 3-(1-диметиламиноэтил)фенола (V)
Проводят N-алкилирование на 3-окси-1-фенилэтиламине (10 г) с 2 эквивалентами формальдегида и 4 эквивалентами муравьиной кислоты. Реакцию проводят при 90°С в течение 10-12 часов. Реакционную смесь нейтрализуют каустическим раствором и экстрагируют этилацетатом (3×50 л). Дальнейшую концентрацию обеспечивает получение требуемого продукта при выходе 70%.
Выход: 80-90 грамм.
% выхода: 70%.
Степень чистоты по методу ВЭЖХ: 97%.
Получение 3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамата (II)
Проводят реакцию конденсации 3-(1-диметиламиноэтил)фенола (10 г) и этилметил карбамоилхлорида (10 г) в присутствии карбоната калия (25 г) и ацетонитрила (150 мл) при 70-80°С. После завершения реакции реакционную смесь резко охлаждают в воде (250 мл) при 30-35°С и экстрагируют в этилацетате (100 мл). После выпаривания этилацетата получают требуемое соединение при выходе 70%.
Выход: 80-85 грамм.
% выхода: 66,0%.
Степень чистоты по методу ВЭЖХ: 99%.
Получение (-)-S-3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамата (I)
При расщеплении рацемического соединения (10 г) (+)-O, O′-дитолуилвинной кислотой (16 г) в метаноле (20 мл) и в воде (10 мл) получают 6 г требуемого изомера в холодном состоянии. Чистый изомер получают после кристаллизации (тройной) в смеси воды и метанола (1:2). Соль винной кислоты (6 г) обрабатывают каустическим раствором, экстрагируют дихлорметаном, концентрируют в вакууме и получают 2 г масла бледно-желтого цвета.
Выход: 20-25 грамм.
% выхода: 20% (весовое соотношение).
Степень чистоты по методу ВЭЖХ: 99%.
Получение (S)-3-(1-диметиламиноэтил) фенола (V-a).
3-(1-диметиламиноэтил) фенол (25 г, 0,15 молей) добавляют к этилацетату (125 мл), а затем добавляют D-(+)-10-камфорсульфоновую кислоту (35 г, 0,15 молей). Реакционную массу нагревают до температуры флегмы. Добавляют метанол (17 мл). Реакционную массу дефлегмируют в течение 30 минут, фильтруют при 10°С и получают соединение по формуле (V-a). По выбору соль далее рекристаллизуют из смеси этилацетата и метанола.
Выход: 6,25-7,5 грамм.
% выхода: 25-30%.
Степень чистоты по методу ВЭЖХ: 98%.
Получение Ривастигмина из (S-3-(1-диметиламиноэтил) фенола (V-a) с использованием этилметил карбамоилхлорида.
Проводят реакцию (S)-3-(1-диметиламиноэтил) фенола (V-a; 25 г; 0,15 молей) с этилметил карбамоилхлоридом (20 г, 0,165 молей) в присутствии безводного карбоната калия (31,5 г, 0,228 молей) и ацетонитрила (250 мл) с последующим подогревом. После завершения реакции реакционную смесь фильтруют, фильтрат концентрируют и получают продукт. По выбору продукт очищают путем кислотно-основной обработки.
Выход: 27 грамм.
% выхода: 74%.
Степень чистоты по методу ВЭЖХ: 99%.
Получение 3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамата с использованием карбонилдиимидазола и этилметиламина
3-(1-диметиламиноэтил) фенол (10 г, 0,06 молей) добавляют к дихлорметану (50 мл) с последующим добавлением карбонилдиимидазола (20 г, 0,123 молей). Реакционную массу дефлегмируют в течение 12 часов и затем реакционную массу охлаждают до 5-10°С. К смеси добавляют этилметиламин (7,08 г, 2,0 молей) и смесь перемешивают в течение 4-6 часов до завершения реакции. Добавляют (100 мл) воды для прекращения реакции. Слой дихлорметана отделяют и обрабатывают 10% раствором NaOH (50 мл) при 10-15°С. Далее органический слой обрабатывают разбавленной соляной кислотой, нейтрализуют аммиачным раствором и экстрагируют гексаном. Слой гексана концентрируют и получают соединение (II).
Получение битартрата Ривастигмина
(-)-S-3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамат (I) (100 г, 1,0 моль) и L-винную кислоту (60 г, 1,0 моль) добавляют к ацетону (1000 мл) и смесь нагревают при 60°С в течение 1,0 часа для получения прозрачной смеси, которую далее охлаждают для завершения осаждения соли винной кислоты соединения (I). Соль винной кислоты соединения фильтруют и высушивают.
Выход: 135 грамм.
% выхода: 85%.
Степень чистоты по методу ВЭЖХ: 99%.
Преимущества настоящего способа заключаются в следующем.
1. В способе используется легкодоступное промышленно безопасное сырье, например N,N-карбонилдиимидазол для получения карбамоильной функции. Следует отметить, что N,N-карбонилдиимидазол прежде не использовали для получения ривастигмина.
2. Реагенты, используемые для приготовления карбамата, например N-этил-N-метил карбамоилхлорид, являются исключительно дорогостоящими. Предусматривается сократить вдвое объемы реагентов, используемых для получения карбамата, например N,N-карбонилдиимидазола, в частности ввиду исключения нежелательного изомера (отсутствует) из процесса реакции. Следовательно, способ получения является экономичным в промышленных масштабах.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ N,N'-БИС[2-(1Н-ИМИДАЗОЛ-4-ИЛ)ЭТИЛ]МАЛОНАМИДА | 2018 |
|
RU2679636C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-[2-(ДИМЕТИЛАМИНО)-ЭТИЛ]АКРИДИН-4-КАРБОКСАМИДА И ЕГО ПРОИЗВОДНЫХ | 1997 |
|
RU2178785C2 |
| СПОСОБ СИНТЕЗА N,N-ДИЗАМЕЩЕННЫХ АМИНОМЕТИЛСТИРОЛОВ ИЛИ АЛЬФА-АМИНОМЕТИЛСТИРОЛОВ | 2014 |
|
RU2673231C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-АЛКИЛ-О-АРИЛКАРБАМАТОВ | 2016 |
|
RU2637317C1 |
| Способ получения R-N-[[3-[(диметиламино)карбонил]пиридин-2-ил]сульфонил]карбаматов, в которых заместителем R является метил или этил | 2023 |
|
RU2816572C1 |
| ПРОИЗВОДНОЕ N-ГИДРОКСИФОРМАМИДА И СОДЕРЖАЩЕЕ ЕГО ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2011 |
|
RU2569851C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-АМИНО-N-(2,2,2-ТРИФТОРЭТИЛ)АЦЕТАМИДА | 2011 |
|
RU2581841C2 |
| Способ получения N-метилкарбаматов | 1985 |
|
SU1433410A3 |
| СПОСОБ ПОЛУЧЕНИЯ N-ЗАМЕЩЕННЫХ САЛИЦИЛАМИДОВ | 2005 |
|
RU2411234C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНОГО ЭФИРА N-ЗАМЕЩЕННОЙ КАРБАМИНОВОЙ КИСЛОТЫ И СПОСОБ ПОЛУЧЕНИЯ ИЗОЦИАНАТА С ИСПОЛЬЗОВАНИЕМ СЛОЖНОГО ЭФИРА N-ЗАМЕЩЕННОЙ КАРБАМИНОВОЙ КИСЛОТЫ | 2009 |
|
RU2528423C2 |
Настоящее изобретение относится к способам получения ривастигмина формулы (I),
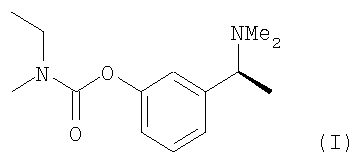
включающим проведение реакции 3-(1-диметиламиноэтил)фенола формулы (V) либо его S-энантиомера формулы (V-a) с карбонилдиимидазолом, последующее взаимодействие продукта реакции с этилметиламином в
органическом растворителе для получения соединения (II) или соединения (I) и произвольное расщепление соединения формулы (II) для получения соединения формулы (I) или проведение реакции S-3-(1-диметиламиноэтил) фенола формулы (V-a) с этилметил карбамоилхлоридом в инертном органическом растворителе при повышенной температуре и в присутствии слабого неорганического или органического основания. Данные способы являются экономичными в промышленных масштабах и позволяют использовать легкодоступное промышленно безопасное сырье. 2 н. и 20 з.п. ф-лы.
1. Способ получения ривастигмина по формуле (I)
включающий:
а) проведение реакции 3-(1-диметиламиноэтил)фенола по формуле (V) либо его S-энантиомера формулы (V-a) с карбонилдиимидазолом,
b) последующее проведение реакции продукта стадии а) с этилметиламином в органическом растворителе для получения соединения (II) или соединения (I) соответственно;
и
с) произвольное расщепление соединения по формуле (II) для получения соединения (I).
2. Способ по п.1, в котором количество используемого карбонилдиимидазола составляет от 1,5 до 3,0 моль на 1 моль соединения по формуле (V) или соединения по формуле (V-a).
3. Способ по пп.1 и 2, в котором количество используемого карбонилдиимидазола составляет предпочтительно от 1,75 до 2,25 моль на 1 моль соединения по формуле (V) или соединения по формуле (V-а).
4. Способ по п.1, в котором количество используемого этилметиламина составляет от 1,0 до 3,0 моль на 1 моль соединения по формуле (V) или соединения по формуле (V-a).
5. Способ по пп.1 и 4, в котором количество используемого этилметиламина составляет предпочтительно от 1,75 до 2,25 моль на 1 моль соединения по формуле (V) или соединения по формуле (V-a).
6. Способ по п.1, в котором используемый растворитель выбирают из группы, включающей алифатические углеводороды, сложные эфиры и хлорированные растворители.
7. Способ по п.6, в котором предпочтительным растворителем является хлорированный углеводород.
8. Способ по п.7, в котором хлорированный растворитель выбирают из группы, включающей дихлорметан, хлороформ и этилендихлорид.
9. Способ по п.8, в котором хлорированный углеводород является предпочтительно дихлорметаном.
10. Способ по п.1, в котором соединение по формуле (II) выделяют из реакционной смеси путем резкого охлаждения смеси водой с последующей экстракцией с помощью органического растворителя и концентрацией органического слоя.
11. Способ получения ривастигмина по формуле (I)
включающий проведение реакции (S)-3-(1-диметиламиноэтил)фенола по формуле (V-a)
с этилметил карбамоилхлоридом в инертном органическом растворителе при повышенной температуре и в присутствии слабого неорганического или органического основания и выделение соединения по формуле (I).
12. Способ по п.11, в котором органический растворитель выбирают из группы, включающей алифатические углеводороды, сложные эфиры и хлорированные углеводороды и нитрилы.
13. Способ по п.12, в котором предпочтительным органическим растворителем является нитрил.
14. Способ по п.13, в котором нитрил выбирают из группы, включающей ацетонитрил, пропионитрил и бутиронитрил.
15. Способ по п.14, в котором предпочтительным нитрилом является ацетонитрил.
16. Способ по п.15, в котором объем используемого ацетонитрила составляет от 10 до 15 объемов на 1 г соединения (V-a).
17. Способ по п.11, в котором слабым неорганическим основанием предпочтительно является карбонат щелочного металла.
18. Способ по п.17, в котором карбонат щелочного металла выбирают из группы, включающей карбонат лития, карбонат натрия и карбонат калия.
19. Способ по п.18, в котором предпочтительным карбонатом щелочного металла является карбонат калия.
20. Способ по п.19, в котором количество используемого карбоната калия составляет от 1,0 до 4,0 моль на 1 моль соединения (V-a).
21. Способ по п.11, в котором количество этилметил карбамоилхлорида, используемого для проведения реакции, составляет от 1,0 до 1,5 моль на 1 моль соединения (V-a).
22. Способ по п.11, в котором соединение по формуле (I) выделяют из реакционной смеси путем резкого охлаждения смеси водой с последующей экстракцией с помощью органического растворителя и концентрацией органического слоя.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| US 4948807 A, 14.08.1990 | |||
| Boezio A.A | |||
| et al | |||
| Asymmetric, catalytic synthesis of alfa-chiral amines using a novel bis(phosphine) monoxide chiral ligand | |||
| Journal of the American chemical society, 2003, 125 (47), 14260-14261 | |||
| KZ 840000588 B1, 24.04.1984 | |||
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННОГО ФЕНИЛОВОГО ЭФИРА N-МЕТИЛКАРБАМИНОВОИ КИСЛОТЫ | 0 |
|
SU303774A1 |

