ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения 2-амино-N-(2,2,2-трифторэтил)ацетамида и его солей. Настоящее изобретение также относится к промежуточным соединениям вышеуказанного способа и использованию соединения, составляющего предмет настоящего изобретения, в качестве исходного вещества в других способах получения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложен способ получения соединения формулы 1
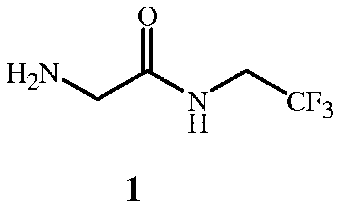 ,
,
включающий (A) взаимодействие соединения формулы 2
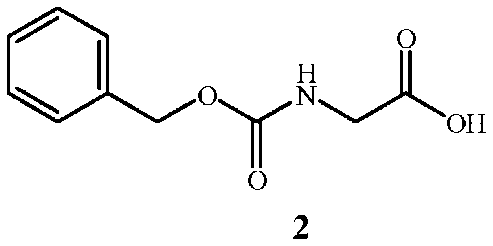
с соединением формулы 3
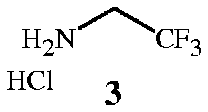
и связующим реагентом с образованием промежуточного соединения формулы 4 в присутствии основания
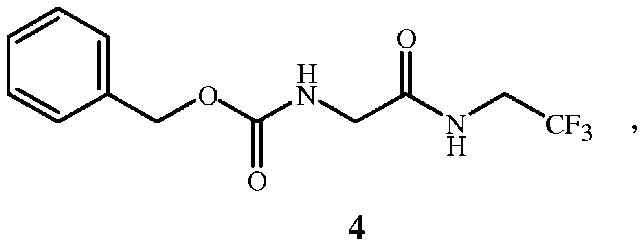
(B) взаимодействие промежуточного соединения формулы 4 с водородом в присутствии катализатора гидрогенолиза с получением соединения формулы 1
и (C) необязательно взаимодействие соединения формулы 1 с кислотой формулы 5
HX,
5
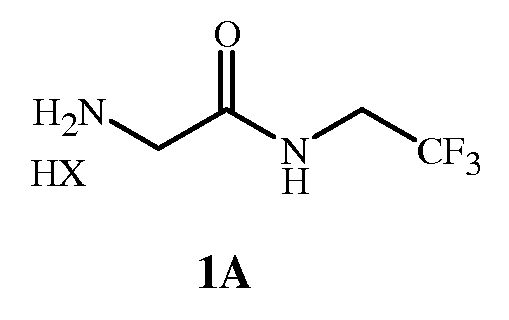
где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3, для получения соединения формулы 1 в форме соли HX (то есть формулы 1A).
Настоящее изобретение также относится к новому соединению - фенилметил N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]карбамату (соединение формулы 4), которое можно использовать как промежуточное соединение в вышеуказанном способе.
В настоящем изобретении также предложен способ получения соединения формулы 1A
,
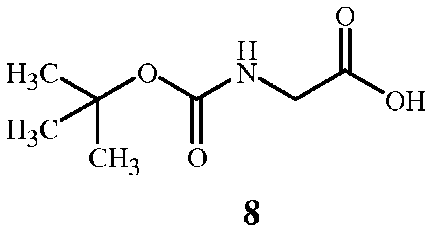
где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3, включающий (A1) взаимодействие соединения формулы 8
с соединением формулы 3
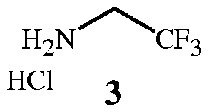
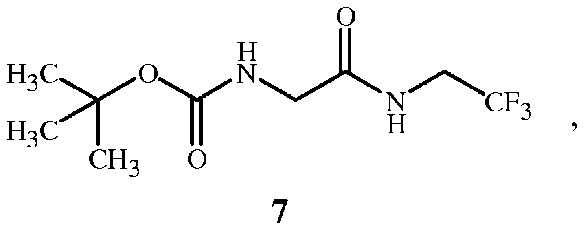
и связующим реагентом с образованием промежуточного соединения формулы 7 в присутствии основания
и (B1) взаимодействие промежуточного соединения формулы 7 с кислотой формулы 5
HX.
5
В настоящем изобретении также предложен способ получения соединения формулы 14
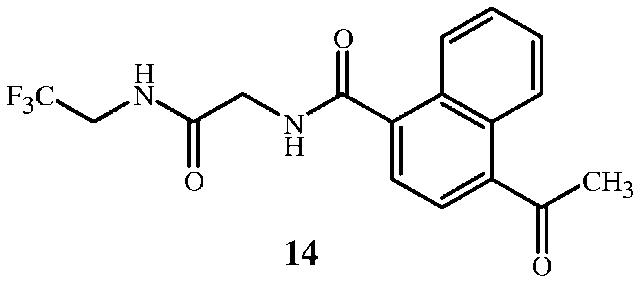 ,
,
включающий взаимодействие соединения формулы 15
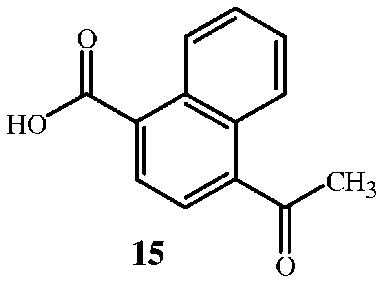
с соединением формулы 1 или 1A
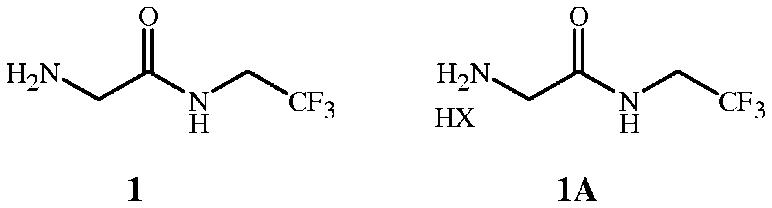 ,
,
где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3, и связующим реагентом в присутствии основания.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Используемые в настоящем документе термины «содержит», «содержащий», «включает», «включающий», «имеет», «имеющий» или любые другие варианты этих терминов означают включение без ограничений. Например, композиция, смесь, процесс, способ, изделие или устройство, содержащее список элементов, необязательно ограничено только ими, но также может включать в себя и другие элементы, не перечисленные в явной форме или присущие такой композиции, смеси, процессу, способу, изделию или устройству. Кроме того, если в явной форме не указано иное, союз «или» относится к включающему «или», а не к исключающему «или». Например, условие «A или B» выполняется в любой из следующих ситуаций: A истинно (или присутствует) и B ложно (или не присутствует), A ложно (или не присутствует) и B истинно (или присутствует), и оба условия A и B истинны (или присутствуют).
Также употребление единственного числа при описании элемента или компонента, составляющего предмет настоящего изобретения, не ограничивает им количество экземпляров элемента или компонента. Следовательно, элемент или компонент в единственном числе должен восприниматься как «один или по меньшей мере один», и употребление элемента или компонента в единственном числе также включает в себя множественное число, за исключением случаев, когда число очевидным образом равно единице.
Термин «связующий реагент» относится к реагенту, который используется для активации функциональной группы карбоновой кислоты для упрощения ее конденсации с функциональной аминогруппой для образования амидной связи.
Соединение формулы 1 в форме соли HX представляет собой соединение формулы 1A
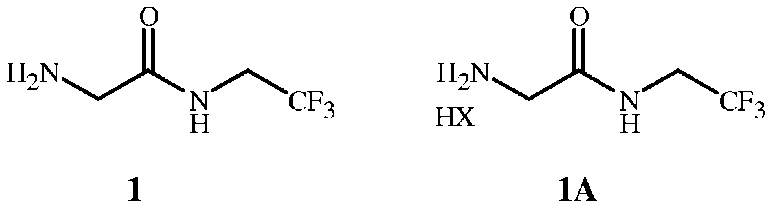 ,
,
где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3.
Под соединением формулы 1A подразумевают соль соединения формулы 1. В альтернативном варианте его можно изобразить в виде формулы 1AA ниже:
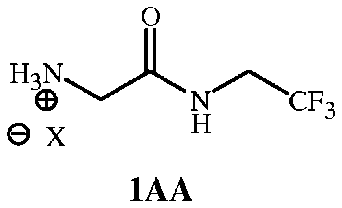 ,
,
где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3.
Если X представляет собой (SO4)1/2, то серная кислота образует сульфат с соединением формулы 1, как показано ниже, где две структуры соответствуют формуле 1AA и формуле 1A.
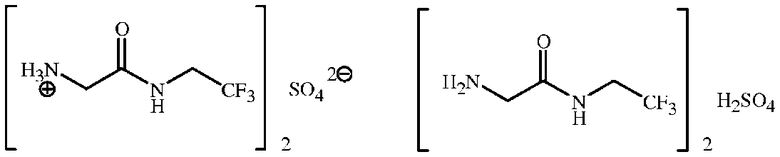
Соединение формулы 1 представляет собой 2-амино-N-(2,2,2-трифторэтил)ацетамид. Соединение формулы 1A представляет собой гидрохлорид 2-амино-N-(2,2,2-трифторэтил)ацетамида. Соединение формулы 4 представляет собой фенилметил N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]карбамат. Соединение формулы 14 представляет собой 4-ацетил-N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]-1-нафталинкарбоксамид.
Варианты осуществления настоящего изобретения включают:
Вариант осуществления 1.0. Способ, описанный на стадии (A) в изложении сущности изобретения, в котором соединения формул 2 и 3 взаимодействуют со связующим реагентом в присутствии основания и не смешивающегося с водой растворителя.
Вариант осуществления 1.1. Способ варианта осуществления 1.0, в котором не смешивающийся с водой растворитель содержит этилацетат или изопропилацетат.
Вариант осуществления 1.2. Способ варианта осуществления 1.1, в котором не смешивающийся с водой растворитель содержит этилацетат.
Вариант осуществления 1.3. Способ, описанный на стадии (A) в изложении сущности изобретения или в любом из вариантов осуществления 1.0-1.3, в котором связующий реагент содержит изобутилхлорформиат или N,N'-карбонилдиимидазол.
Вариант осуществления 1.4. Способ варианта осуществления 1.3, в котором связующий реагент содержит N,N'-карбонилдиимидазол.
Вариант осуществления 1.5. Способ, описанный на стадии (A) в изложении сущности изобретения или в любом из вариантов осуществления 1.0-1.4, в котором основание содержит основной реагент, отличный от соединения, которое является производным связующего реагента.
Вариант осуществления 1.6. Способ варианта осуществления 1.5, в котором основной реагент содержит триэтиламин или N,N-диизопропилэтиламин.
Вариант осуществления 1.7. Способ варианта осуществления 1.6, в котором основной реагент содержит триэтиламин.
Вариант осуществления 1.8. Способ, описанный на стадии (A) в изложении сущности изобретения или в любом из вариантов осуществления 1.0-1.7, в котором основание является производным связующего реагента, а связующий реагент представляет собой N,N'-карбонилдиимидазол.
Вариант осуществления 1.9. Способ, описанный на стадии (A) в изложении сущности изобретения или в любом из вариантов осуществления 1.0-1.8, в котором соединение формулы 2 сначала взаимодействует со связующим реагентом с образованием смеси (например, содержащей ацилимидазол формулы 6), а затем к смеси в присутствии основания добавляют соединение формулы 3.
Вариант осуществления 1.10. Способ, описанный на стадии (A) в изложении сущности изобретения или в любом из вариантов осуществления 1.0-1.9, в котором смесь находится при температуре по меньшей мере приблизительно 15°C.
Вариант осуществления 1.11. Способ, описанный на стадии (A) в изложении сущности изобретения или в любом из вариантов осуществления 1.0-1.10, в котором смесь находится при температуре не более приблизительно 40°C.
Вариант осуществления 1.12. Способ, описанный на стадии (A) в изложении сущности изобретения или в любом из вариантов осуществления 1.0-1.11, в котором молярное соотношение связующего реагента и соединения формулы 2 составляет от приблизительно 1,0 до приблизительно 1,1.
Вариант осуществления 1.13. Способ, описанный на стадии (A) в изложении сущности изобретения или в любом из вариантов осуществления 1.0-1.12, в котором молярное соотношение соединения формулы 3 и соединения формулы 2 составляет приблизительно 1,0.
Вариант осуществления 1.14. Способ, описанный на стадии (B) в изложении сущности изобретения, в котором соединение формулы 4 и водород взаимодействуют в присутствии катализатора гидрогенолиза и не смешивающегося с водой растворителя.
Вариант осуществления 1.15. Способ варианта осуществления 1.14, в котором не смешивающийся с водой растворитель содержит этилацетат или изопропилацетат.
Вариант осуществления 1.16. Способ варианта осуществления 1.15, в котором не смешивающийся с водой растворитель содержит этилацетат.
Вариант осуществления 1.17. Способ, описанный на стадии (B) в изложении сущности изобретения или в любом из вариантов осуществления 1.14-1.16, в котором катализатор гидрогенолиза представляет собой катализатор из благородного металла или катализатор из благородного металла, нанесенный на подложку.
Вариант осуществления 1.18. Способ варианта осуществления 1.17, в котором катализатором гидрогенолиза является палладиевый катализатор на углеродной подложке.
Вариант осуществления 1.19. Способ варианта осуществления 1.18, в котором катализатор гидрогенолиза представляет собой 5% или 10% палладий на углеродной подложке.
Вариант осуществления 1.20. Способ, описанный на стадии (B) в изложении сущности изобретения или в любом из вариантов осуществления 1.14-1.19, в котором гидрогенолиз проводят при температуре окружающей среды.
Вариант осуществления 1.21. Способ, описанный на стадии (B) в изложении сущности изобретения или в любом из вариантов осуществления 1.14-1.20, в котором гидрогенолиз проводят при значении атмосферного давления до приблизительно 0,34 МПа (50 фунтов на кв. дюйм).
Вариант осуществления 1.22. Способ варианта осуществления 1.21, в котором гидрогенолиз проводят при атмосферном давлении.
Вариант осуществления 1.23. Способ, описанный на стадии (C) в изложении сущности изобретения, в котором соединение формулы 1 взаимодействует с кислотой формулы 5 в присутствии не смешивающегося с водой растворителя.
Вариант осуществления 1.24. Способ варианта осуществления 1.23, в котором не смешивающийся с водой растворитель содержит этилацетат или изопропилацетат.
Вариант осуществления 1.25. Способ варианта осуществления 1.24, в котором не смешивающийся с водой растворитель содержит этилацетат.
Вариант осуществления 1.26. Способ, описанный на стадии (C) в изложении сущности изобретения или в любом из вариантов осуществления 1.23-1.25, в котором кислота формулы 5 содержит хлорид водорода, бромид водорода, трифторуксусную кислоту, серную кислоту, метансульфоновую кислоту или фосфорную кислоту.
Вариант осуществления 1.27. Способ варианта осуществления 1.26, в котором кислота формулы 5 содержит хлорид водорода, бромид водорода и серную кислоту.
Вариант осуществления 1.28. Способ варианта осуществления 1.27, в котором кислота формулы 5 содержит хлорид водорода.
Вариант осуществления 1.29. Способ варианта осуществления 1.28, в котором хлорид водорода представляет собой водный раствор (то есть соляную кислоту).
Вариант осуществления 1.30. Способ варианта осуществления 1.28, в котором хлорид водорода является безводным (то есть газообразным хлороводородом).
Вариант осуществления 1.31. Способ, описанный на стадии (C) в изложении сущности изобретения или в любом из вариантов осуществления 1.23-1.30, в котором смесь находится при температуре по меньшей мере приблизительно 20°C.
Вариант осуществления 1.32. Способ, описанный на стадии (C) в изложении сущности изобретения или в любом из вариантов осуществления 1.23-1.31, в котором смесь находится при температуре не более приблизительно 45°C.
Вариант осуществления 1.33. Способ, описанный на стадии (C) в изложении сущности изобретения или в любом из вариантов осуществления 1.23-1.32, в котором молярное соотношение соединения формулы 1 и кислоты формулы 5 составляет по меньшей мере приблизительно 1,0.
Вариант осуществления 1.34. Способ, описанный на стадии (C) в изложении сущности изобретения или в любом из вариантов осуществления 1.23-1.33, в котором молярное соотношение соединения формулы 1 и кислоты формулы 5 составляет не более приблизительно 5,0.
Вариант осуществления 2.0. Способ, описанный на стадии (A1) в изложении сущности изобретения, в котором соединения формул 8 и 3 взаимодействуют со связующим реагентом в присутствии основания и не смешивающегося с водой растворителя.
Вариант осуществления 2.1. Способ варианта осуществления 2.0, в котором не смешивающийся с водой растворитель содержит этилацетат или изопропилацетат.
Вариант осуществления 2.2. Способ варианта осуществления 2.1, в котором не смешивающийся с водой растворитель содержит этилацетат.
Вариант осуществления 2.3. Способ, описанный на стадии (A1) в изложении сущности изобретения или в любом из вариантов осуществления 2.0-2.2, в котором соединение формулы 8 сначала взаимодействует со связующим реагентом с образованием смеси (то есть содержащей ацилимидазол формулы 9), а затем к смеси добавляют соединение формулы 3.
Вариант осуществления 2.4. Способ, описанный на стадии (A1) в изложении сущности изобретения или в любом из вариантов осуществления 2.0-2.3, в котором связующий реагент содержит изобутилхлорформиат или N,N'-карбонилдиимидазол.
Вариант осуществления 2.5. Способ варианта осуществления 2.4, в котором связующий реагент содержит N,N'-карбонилдиимидазол.
Вариант осуществления 2.6. Способ, описанный на стадии (A1) в изложении сущности изобретения или в любом из вариантов осуществления 2.0-2.5, в котором основание содержит основной реагент, отличный от соединения, являющегося производным связующего реагента.
Вариант осуществления 2.7. Способ варианта осуществления 2.6, в котором основной реагент содержит триэтиламин или N,N-диизопропилэтиламин.
Вариант осуществления 2.8. Способ варианта осуществления 2.7, в котором основной реагент содержит триэтиламин.
Вариант осуществления 2.9. Способ, описанный на стадии (A1) в изложении сущности изобретения или в любом из вариантов осуществления 2.0-2.8, в котором основание является производным связующего реагента, а связующий реагент представляет собой N,N'-карбонилдиимидазол.
Вариант осуществления 2.10. Способ, описанный на стадии (A1) в изложении сущности изобретения или в любом из вариантов осуществления 2.0-2.9, в котором соединение формулы 8 сначала взаимодействует со связующим реагентом с образованием смеси, а затем к смеси в присутствии основания добавляют соединение формулы 3.
Вариант осуществления 2.11. Способ, описанный на стадии (A1) в изложении сущности изобретения или в любом из вариантов осуществления 2.0-2.10, в котором смесь находится при температуре по меньшей мере приблизительно 15°C.
Вариант осуществления 2.12. Способ, описанный на стадии (A1) в изложении сущности изобретения или в любом из вариантов осуществления 2.0-2.11, в котором смесь находится при температуре не более приблизительно 40°C.
Вариант осуществления 2.13. Способ, описанный на стадии (A1) в изложении сущности изобретения или в любом из вариантов осуществления 2.0-2.12, в котором молярное соотношение связующего реагента и соединения формулы 8 составляет приблизительно 1,0.
Вариант осуществления 2.14. Способ, описанный на стадии (A1) в изложении сущности изобретения или в любом из вариантов осуществления 2.0-2.13, в котором молярное соотношение соединения формулы 3 и соединения формулы 8 составляет приблизительно 1,0.
Вариант осуществления 2.15. Способ, описанный на стадии (B1) в изложении сущности изобретения, в котором соединения формул 7 и 5 взаимодействуют в присутствии не смешивающегося с водой растворителя.
Вариант осуществления 2.16. Способ варианта осуществления 2.15, в котором не смешивающийся с водой растворитель содержит этилацетат или изопропилацетат.
Вариант осуществления 2.17. Способ варианта осуществления 2.16, в котором не смешивающийся с водой растворитель содержит этилацетат.
Вариант осуществления 2.18. Способ, описанный на стадии (B1) в изложении сущности изобретения или в любом из вариантов осуществления 2.15-2.17, в котором кислота формулы 5 содержит хлорид водорода, бромид водорода, трифторуксусную кислоту, серную кислоту, метансульфоновую кислоту или фосфорную кислоту.
Вариант осуществления 2.19. Способ варианта осуществления 2.18, в котором кислота формулы 5 содержит хлорид водорода, бромид водорода и серную кислоту.
Вариант осуществления 2.20. Способ варианта осуществления 2.19, в котором кислота формулы 5 содержит хлорид водорода.
Вариант осуществления 2.21. Способ варианта осуществления 2.20, в котором хлорид водорода представляет собой водный раствор (то есть соляную кислоту).
Вариант осуществления 2.22. Способ варианта осуществления 2.20, в котором хлорид водорода является безводным (то есть газообразным хлороводородом).
Вариант осуществления 2.23. Способ, описанный на стадии (B1) в изложении сущности изобретения или в любом из вариантов осуществления 2.15-2.22, в котором смесь находится при температуре по меньшей мере приблизительно 20°C.
Вариант осуществления 2.24. Способ, описанный на стадии (B1) в изложении сущности изобретения или в любом из вариантов осуществления 2.15-2.23, в котором смесь находится при температуре не более приблизительно 45°C.
Вариант осуществления 2.25. Способ, описанный на стадии (B1) в изложении сущности изобретения или в любом из вариантов осуществления 2.15-2.24, в котором молярное соотношение соединения формулы 7 и кислоты формулы 5 составляет по меньшей мере приблизительно 1,0.
Вариант осуществления 2.26. Способ, описанный на стадии (B1) в изложении сущности изобретения или в любом из вариантов осуществления 2.15-2.25, в котором молярное соотношение соединения формулы 7 и кислоты формулы 5 составляет не более приблизительно 5,0.
Вариант осуществления 3.0. Способ получения соединения формулы 14, описанный в изложении сущности изобретения, в котором соединения формул 1 или 1A и формулы 15 взаимодействуют со связующим реагентом в присутствии основания и полярного апротонного смешивающегося с водой растворителя.
Вариант осуществления 3.1. Способ варианта осуществления 3.0, в котором полярный апротонный смешивающийся с водой растворитель содержит ацетонитрил, тетрагидрофуран или диоксан.
Вариант осуществления 3.2. Способ варианта осуществления 3.1, в котором полярный апротонный смешивающийся с водой растворитель содержит ацетонитрил.
Вариант осуществления 3.3. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.2, в котором связующий реагент содержит изобутилхлорформиат или N,N'-карбонилдиимидазол.
Вариант осуществления 3.4. Способ варианта осуществления 3.3, в котором связующий реагент содержит N,N'-карбонилдиимидазол.
Вариант осуществления 3.5. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.4, в котором основание содержит основной реагент, отличный от соединения, которое является производным связующего реагента.
Вариант осуществления 3.6. Способ варианта осуществления 3.5, в котором основной реагент содержит триэтиламин или N,N-диизопропилэтиламин.
Вариант осуществления 3.7. Способ варианта осуществления 3.6, в котором основной реагент содержит триэтиламин.
Вариант осуществления 3.8. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.7, в котором основание является производным связующего реагента, а связующий реагент представляет собой N,N'-карбонилдиимидазол.
Вариант осуществления 3.9. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.8, в котором соединение формулы 15 сначала взаимодействует со связующим реагентом с образованием смеси (т.е. содержащей ацилимидазол формулы 16), а затем к смеси в присутствии основания добавляют соединение формулы 1 или 1A.
Вариант осуществления 3.10. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.9, в котором соединение формулы 1 или 1A добавляют к смеси в форме твердого вещества или в виде раствора в полярном апротонном смешивающемся с водой растворителе.
Вариант осуществления 3.11. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.10, в котором соединение формулы 1 или 1A добавляют к смеси в форме раствора или водной суспензии.
Вариант осуществления 3.12. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.12, в котором смесь находится при температуре по меньшей мере приблизительно 20°C.
Вариант осуществления 3.13. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.12, в котором смесь находится при температуре не более приблизительно 45°C.
Вариант осуществления 3.14. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.13, в котором молярное соотношение связующего реагента и соединения формулы 15 составляет от приблизительно 1,0 до приблизительно 1,1.
Вариант осуществления 3.15. Способ получения соединения формулы 14, описанный в изложении сущности изобретения или в любом из вариантов осуществления 3.0-3.14, в котором молярное соотношение соединения формулы 1 или 1A и соединения формулы 15 составляет приблизительно 1,0.
Варианты осуществления настоящего изобретения, в том числе описанные выше варианты осуществления 1.0-3.15, а также любые другие варианты осуществления, описанные в настоящем документе, могут быть скомбинированы любым образом. Описания переменных в вариантах осуществления относятся не только к вышеописанным способам получения соединений формул 1, 1A и 14, но также к исходным соединениям и промежуточным соединениям, подходящим для получения этими способами соединений формул 1, 1A и 14.
В схемах 1-9 ниже X определяется в соединениях формул 1-16 так, как описано выше в изложении сущности изобретения и описаниях вариантов осуществления, если не указано иное.
В способе настоящего изобретения защитная аминогруппа бензилкарбамата (CBZ) используется при получении соединения формулы 1, как показано на схемах 1 и 2. Соединение формулы 1 может дополнительно взаимодействовать с кислотой с образованием кислой соли формулы 1A, как показано на схеме 3 (см. примеры синтеза 1 и 2).
Стадия B способа настоящего изобретения включает в себя удаление в процессе гидрогенолиза защитной группы бензилкарбамата из промежуточного соединения формулы 4 с получением свободного аминосоединения формулы 1, как показано на схеме 1.
Схема 1
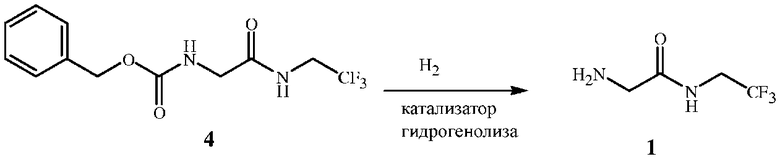
Реакция удаления защитных групп бензилкарбамата может протекать в различных условиях. См., например, Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 2-е изд.; Wiley: г. Нью-Йорк, 1991 г. Одним из наиболее подходящих способов удаления бензильной защитной группы является гидрогенолиз с водородом, который, как правило, проводят при атмосферном давлении. Как правило, используются катализаторы из благородных металлов или катализаторы из благородных металлов, нанесенные на подложку. Гидрогенолиз может быть проведен путем переноса водорода в присутствии катализатора из благородного металла, нанесенного на подложку, и донора водорода (например, формиата аммония или циклогексадиена). Эти способы описаны в работе Rylander, P. N.; Hydrogenation Methods, Academic Press: г. Сан-Диего, 1985 г. Одним из наиболее подходящих катализаторов гидрогенолиза является палладий на углеродной подложке (как правило, 5-10%). Этот способ подробно описан в Harada et al., Bioorganic and Medicinal Chemistry, 2001 г., 9, 2709-2726 и Janda et al., Synthetic Communications, 1990 г., 20, 1073-1082. Защитную группу бензилкарбамата также можно удалить с помощью кислоты, как описано в Lesk et al., Synthetic Communications, 1999 г., 28, 1405-1408.
Способ по схеме 1 можно реализовать в диапазоне температур. Как правило, реакцию проводят при температуре по меньшей мере приблизительно 20°C или при температуре окружающей среды. Гидрогенизация может быть проведена в определенном диапазоне давлений. Как правило, гидрогенизацию проводят при атмосферном давлении, используя водородный баллон. Время протекания реакции составляет от 2 до 24 часов в зависимости от объема реакции.
В настоящем способе реакционная смесь содержит не смешивающийся с водой растворитель. Особенно подходящими растворителями являются этилацетат и изопропилацетат. Особенно подходящими являются полярные апротонные растворители, которые не смешиваются с водой, так как они могут растворять исходное вещество формулы 4. Количество используемого растворителя определяется объемом, необходимым для растворения исходного вещества, и, как правило, находится в диапазоне молярных концентраций от 0,5 до 1,0. Смесь исходного вещества и растворителя можно нагреть до приблизительно 30°C, чтобы обеспечить растворение соединения формулы 4 и добиться концентрации реакционной смеси более 0,5 моль.
Протекание реакции можно контролировать обычными способами, например, с помощью анализа аликвот тонкослойной хроматографией, ГХ, ВЭЖХ и 1H ЯМР. После завершения реакции продукт отделяют от катализатора путем фильтрации. Полученный раствор содержит свободное аминосоединение формулы 1. Для выделения соединения формулы 1 раствор может быть концентрирован. В альтернативном варианте осуществления раствор может дополнительно взаимодействовать с кислотой по схеме 3 с получением соединения формулы 1A. Другим вариантом является добавление к отфильтрованному раствору воды, при этом соединение формулы 1 будет распределяться в воде с образованием водного раствора, который можно разделить и использовать в последующих реакциях.
Стадия A способа настоящего изобретения заключается в реакции исходного вещества формулы 2, защищенного от взаимодействия с бензилкарбаматом, с соединением формулы 3 с получением промежуточного соединения формулы 4, показанного на схеме 2. Стадия A начинается с активации функциональной группы карбоновой кислоты соединения формулы 2 связующим реагентом с получением соединения ацилимидазола формулы 6. Может быть выделено промежуточное соединение ацилимидазола формулы 6, однако в большинстве случаев его не выделяют, а сразу обрабатывают амином формулы 3 с образованием амидной связи и получением соединения формулы 4.
Схема 2
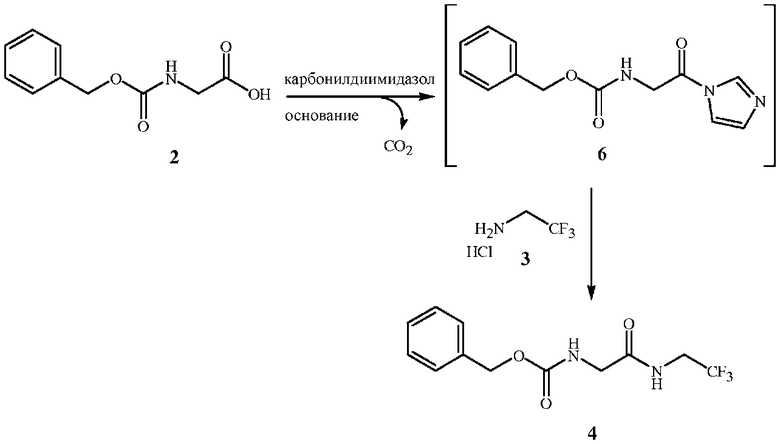
Для получения соединения формулы 4 можно использовать различные связующие реагенты. Особую эффективность продемонстрировали несколько алкилхлорформиатов и карбонилдигетероарилов, которые обеспечивают высокий выход соединений формулы 6. Такие связующие реагенты включают метилхлорформиат, этилхлорформиат, изобутилхлорформиат, N,N'-карбонилдиимидазол и 1,1'-карбонилбис(3-метилимидазолий) трифлат. Предпочтительным является N,N'-карбонилдиимидазол (который также называют карбонилдиимидазолом). N,N'-карбонилдиимидазол (показан на схеме 2) является наиболее эффективным связующим реагентом, так как он дает один эквивалент основания для нейтрализации соли амина формулы 3. Связующие реагенты на основе эфира хлорформиата требуют дополнительного основного реагента для нейтрализации кислоты, образующейся в реакции с соединением формулы 2, а также для высвобождения свободного основания соединения формулы 3. Особенно подходящим основанием для данной реакции является триэтиламин.
Стехиометрический состав данной реакции предполагает эквимолярные количества соединения формулы 2, связующего реагента и основания. В случае если связующим реагентом является N,N'-карбонилдиимидазол, при образовании промежуточного соединения ацилимидазола (соединение формулы 6) выделяется один эквивалент диоксида углерода. При образовании ацилимидазола также высвобождается эквивалент имидазола, который реагирует с одним эквивалентом хлорида водорода при добавлении к реакционной смеси соли амина формулы 3. Таким образом, основание может быть производным связующего реагента, если связующим реагентом является N,N'-карбонилдиимидазол. Эквивалент дополнительного основания (основной реагент, не являющийся производным связующего реагента), такой как триэтиламин, необязателен, если связующим реагентом является N,N'-карбонилдиимидазол. Дополнительное основание (например, триэтиламин или диизопропилэтиламин) ускорит реакцию, так как оно обладает большими основными свойствами по сравнению с имидазолом и быстрее взаимодействует с хлоридом водорода соединения формулы 3 с высвобождением его в форме свободного основания, реагирующего с ацилимидазолом. Молярное соотношение связующего реагента и соединения формулы 2 может находиться в диапазоне от приблизительно 0,95 до приблизительно 1,15, однако предпочтительным является соотношение по меньшей мере 1,0, чтобы обеспечить образование промежуточного соединения ацилимидазола и соединения формулы 6 в полном объеме.
Стехиометрический состав данной реакции дополнительно предполагает эквимолярные количества соединения формулы 3 и соединения формулы 2. Молярное соотношение соединения формулы 3 и соединения формулы 2 может находиться в диапазоне от приблизительно 1,0 до приблизительно 1,15, однако предпочтительным является соотношение по меньшей мере 1,05, которое обеспечивает полное завершение реакции между промежуточным соединением ацилимидазола и соединением формулы 3.
Порядок введения реагентов на стадии A способа настоящего изобретения очень важен. Соединение формулы 2 можно растворить в растворителе с последующим добавлением связующего реагента, или связующий реагент можно растворить в растворителе с последующим добавлением к нему соединения формулы 2. Однако важно предусмотреть достаточное количество времени на образование ацилимидазола до добавления соединения формулы 3. Образование ацилимидазола, как правило, можно контролировать по выделению углекислого газа в течение 1-2 часов в зависимости от объема реакции.
Соединения формулы 2 и формулы 3 доступны в продаже. Соединение формулы 3 является особенно предпочтительным ввиду простоты его использования. Трифторэтиламин можно использовать в его нейтральном состоянии, однако он является летучим веществом (температура кипения 36-37°C), вследствие чего менее удобным в работе.
В настоящем способе реакционная смесь содержит не смешивающийся с водой растворитель. Особенно подходящими растворителями являются этилацетат и изопропилацетат. Особенно подходящими являются полярные апротонные растворители, которые не смешиваются с водой, так как они могут растворять исходное вещество формулы 2 и могут быть отделены от воды при обработке водой. Количество используемого растворителя определяется объемом, необходимым для растворения исходного вещества, и, как правило, находится в диапазоне молярных концентраций от 0,75 до 1,5, особенно подходящей является молярная концентрация 1,0.
Реакция по способу на схеме 2 может протекать в широком диапазоне температур. Как правило, температура реакции составляет по меньшей мере приблизительно 15°C, наиболее часто по меньшей мере приблизительно 20°C. Как правило, температура реакции составляет не более приблизительно 40°C, наиболее часто не более приблизительно 35°C.
Протекание реакции можно контролировать обычными способами, например, с помощью анализа аликвот тонкослойной хроматографией, ГХ, ВЭЖХ и 1H ЯМР. После завершения реакции смесь, как правило, обрабатывают путем добавления водного раствора минеральной кислоты, такой как соляная кислота. Отделение органической фазы, дополнительное промывание соляной кислотой (1,0 Н) для удаления имидазола (и необязательно добавленного триэтиламина), высушивание над осушителями, такими как сульфат магния или молекулярные сита, или азеотропная сушка и затем выпаривание растворителя приводят к получению продукта формулы 4 в форме бесцветного твердого вещества. Выпаривание растворителя необязательно. При азеотропной сушке растворитель не удаляют и далее используют раствор соединения формулы 4.
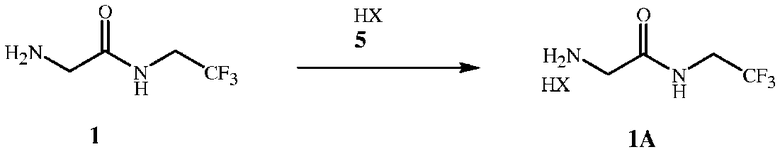
Стадия C способа настоящего изобретения является необязательным и включает в себя взаимодействие свободного амина формулы 1 с кислотой формулы 5 с получением кислой соли формулы 1A, показанной на схеме 3.
Схема 3
 ,
,
где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3.
Свободный амин формулы 1 чувствителен к воздуху. Полученный раствор (на стадии B) соединения формулы 1 можно обработать кислотой для получения более стабильной кислой соли формулы 1A. Затем соединение формулы 1А отделяют фильтрованием и сушат в вакуумной печи (50-60°С) или на воздухе. Соль формулы 1A можно хранить в условиях окружающей среды без последствий в виде увеличения веса в результате воздействия влаги и воздуха и сложностей в использовании вследствие гигроскопичности и липкой консистенции. Для сравнения соединений формулы 1 и 1A и других солей см. пример 12.
Безводные кислоты формулы 5 продемонстрировали особую эффективность, обеспечивая высокий выход соединений формулы 1A. Такие кислоты включают хлорид водорода, бромид водорода, трифторуксусную кислоту, метансульфоновую кислоту, серную кислоту или фосфорную кислоту. Предпочтительным является хлорид водорода вследствие его низкой стоимости. Как правило, кислоту барботируют через реакционную смесь, не содержащую катализатора, или, при использовании жидких кислот, добавляют по каплям. Безводные кислоты формулы 5 добавляют к раствору не смешивающегося с водой растворителя на стадии B с получением твердой соли формулы 1A, которую легко отделить фильтрацией. В альтернативном варианте осуществления водные растворы кислот формулы 5 (например, концентрированной соляной кислоты) можно добавить по каплям к раствору формулы 1 на стадии B с получением водной фазы, содержащей соединение формулы 1A. Такую водную фазу можно отделить от не смешивающегося с водой растворителя и использовать в последующих реакциях.
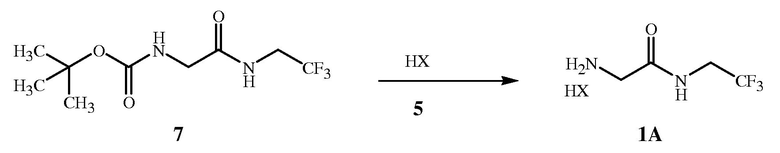
Альтернативой защитной аминогруппе бензилкарбамата (CBZ), которая используется в способе настоящего изобретения на схемах 1 и 2, является защитная аминогруппа третбутилкарбамата (BOC), показанного на схемах 4 и 5 (см. примеры синтеза 3 и 4).
На стадии B способа настоящего изобретения, показанного на схеме 4, соединение формулы 1A получают непосредственно при взаимодействии соединения формулы 7 с кислотой формулы 5. Во время реакции производится удаление защитной группы третбутилкарбамата с одновременным образованием соли функциональной аминогруппы.
Схема 4
 ,
,
где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3.
Стехиометрический состав данной реакции предполагает эквимолярные количества кислоты формулы 5 и соединения формулы 7. Однако для полного удаления защитной группы третбутилкарбамата из соединения формулы 7 и завершения образования кислой соли формулы 1A желателен молярный избыток кислоты формулы 5 от приблизительно 2,0 до приблизительно 5,0.
Безводные кислоты формулы 5 продемонстрировали особую эффективность, обеспечивая высокий выход соединений формулы 1A. Такие кислоты включают хлорид водорода, бромид водорода, трифторуксусную кислоту, метансульфоновую кислоту, серную кислоту или фосфорную кислоту. Предпочтительным является хлорид водорода вследствие его низкой стоимости. Безводные кислоты в форме газа, например, хлороводород (см. стадия B примера синтеза 4), как правило, барботируют через реакционную смесь. При использовании жидких кислот, например трифторуксусной кислоты (см. пример синтеза 7), их добавляют по каплям. Безводные кислоты формулы 5 используют в не смешивающемся с водой растворителе, получая твердую соль формулы 1A, которую легко отделить путем фильтрации реакционной смеси. Образование и отделение полученной соли по описанной выше процедуре не требует выполнения стадии обработки водой. Выделенную твердую соль формулы 1A можно использовать в последующих реакциях.
Водные кислоты формулы 5 также продемонстрировали эффективность, обеспечивая высокий выход соединений формулы 1A. Такие кислоты включают соляную кислоту и бромистоводородную кислоту. Соляная кислота является предпочтительной вследствие ее низкой стоимости (см. стадия B1 примера синтеза 4). Водные кислоты, как правило, добавляют к реакционной смеси по каплям. При использовании водных кислот формулы 5 в не смешивающемся с водой растворителе соль формулы 1A после получения растворяют в водной фазе, которую отделяют от органической фазы. Концентрированный водный раствор соединения формулы 1A можно легко отделить путем выведения более плотной водной фазы через нижнюю часть реакционного сосуда. Концентрированный водный раствор соединения формулы 1A можно использовать в последующих реакциях.
В настоящем способе реакционная смесь содержит не смешивающийся с водой растворитель. Особенно подходящими растворителями являются этилацетат и изопропилацетат. Особенно подходящими являются полярные апротонные растворители, которые не смешиваются с водой, так как они могут растворять исходное вещество формулы 7 и вызывать осаждение продукта формулы 1A. Количество используемого растворителя определяется объемом, необходимым для растворения исходного вещества, и, как правило, находится в диапазоне молярных концентраций от 0,5 до 1,0. Смесь исходного вещества и растворителя можно нагреть до приблизительно 30°C, чтобы обеспечить растворение соединения формулы 7 и добиться концентрации реакционной смеси более 0,5 моль. После растворения исходного вещества источник тепла удаляют, и к реакционной смеси при температуре окружающей среды добавляют кислоту.
Способ, изображенный на схеме 4, можно реализовать в широком диапазоне температур. Как правило, реакцию проводят при температуре по меньшей мере приблизительно 20°C или при температуре окружающей среды. Как правило, реакционную смесь нагревают во время реакции, однако экзотермический эффект, как правило, не требует внешнего охлаждения, и температура реакции, как правило, остается ниже температуры кипения растворителя. Как правило, температура реакции составляет не более приблизительно 45°C, наиболее часто не более приблизительно 40°C.
Протекание реакции можно контролировать обычными способами, например, с помощью анализа аликвот тонкослойной хроматографией, ГХ, ВЭЖХ и 1H ЯМР. После завершения реакции смесь, как правило, охлаждают до комнатной температуры, и продукт отделяют обычными способами, например, фильтрацией. Полученное твердое вещество отделяют фильтрованием и сушат в вакуумной печи (50-60°С) или на воздухе.
На стадии A способа настоящего изобретения, показанного на схеме 5, соединение формулы 7 получают при взаимодействии соединения формулы 8 с соединением формулы 3 и связующим реагентом. Способ получения соединения формулы 7 начинается с активации функциональной группы карбоновой кислоты соединения формулы 8 связующим реагентом с получением соединения ацилимидазола формулы 9. Соединение ацилимидазола формулы 9 можно выделить, однако, как правило, его не выделяют. Оно образует амидную связь с функциональной аминогруппой в соединении формулы 3 с получением соединения формулы 7.
Схема 5
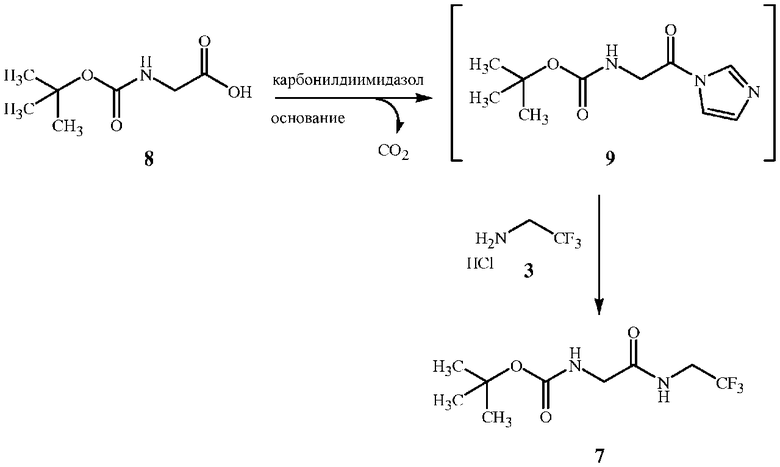
Стехиометрический состав данной реакции предполагает эквимолярные количества соединения формулы 8, связующего реагента и основания. В случае если связующим реагентом является N,N'-карбонилдиимидазол, при образовании промежуточного соединения ацилимидазола (соединение формулы 9) выделяется один эквивалент диоксида углерода. При образовании ацилимидазола также высвобождается эквивалент имидазола, который реагирует с одним эквивалентом хлорида водорода при добавлении к реакционной смеси соли амина формулы 3. Таким образом, основание может быть производным связующего реагента, если связующим реагентом является N,N'-карбонилдиимидазол. Эквивалент дополнительного основания (основной реагент, не являющийся производным связующего реагента), такой как триэтиламин, необязателен, если связующим реагентом является N,N'-карбонилдиимидазол. Дополнительное основание (например, триэтиламин или диизопропилэтиламин) ускорит реакцию, так как оно обладает большими основными свойствами по сравнению с имидазолом и быстрее взаимодействует с хлоридом водорода соединения формулы 3 с высвобождением его в форме свободного основания, реагирующего с ацилимидазолом. Молярное соотношение связующего реагента и соединения формулы 2 может находиться в диапазоне от приблизительно 0,95 до приблизительно 1,15, однако предпочтительным является соотношение по меньшей мере 1,0, чтобы обеспечить образование промежуточного соединения ацилимидазола и соединения формулы 9 в полном объеме. Стехиометрический состав данной реакции предполагает эквимолярные количества соединения формулы 3 и соединения формулы 8. Молярное соотношение соединения формулы 3 и соединения формулы 8 может находиться в диапазоне от приблизительно 1,0 до приблизительно 1,15, однако предпочтительным является соотношение по меньшей мере 1,05, которое обеспечивает полное завершение реакции между промежуточным соединением ацилимидазола и соединением формулы 3.
На стадии A можно использовать различные связующие реагенты. Особую эффективность продемонстрировали несколько алкилхлорформиатов и карбонилдигетероарилов, которые обеспечивают высокий выход соединений формулы 7. Такие связующие реагенты включают метилхлорформиат, этилхлорформиат, изобутилхлорформиат, N,N'-карбонилдиимидазол и 1,1'-карбонилбис(3-метилимидазолий) трифлат. Предпочтительным является N,N'-карбонилдиимидазол (который также называют карбонилдиимидазолом). N,N'-карбонилдиимидазол является наиболее эффективным связующим реагентом, так как он дает один эквивалент основания для нейтрализации соли амина формулы 3. Связующие реагенты на основе эфира хлорформиата требуют дополнительного основного реагента для нейтрализации кислоты, образующейся в реакции с соединением формулы 8, а также для высвобождения свободного основания соединения формулы 3 (см. пример синтеза 6). Особенно подходящим основанием для данной реакции является триэтиламин.
Порядок введения реагентов на стадии A способа настоящего изобретения очень важен. Соединение формулы 8 можно растворить в растворителе с последующим добавлением связующего реагента, или связующий реагент можно растворить в растворителе с последующим добавлением к нему соединения формулы 8. Однако важно предусмотреть достаточное количество времени на образование промежуточного ацилимидазола до добавления соединения формулы 3. Как правило, образование промежуточного соединения ацилимидазола можно контролировать по выделению углекислого газа в течение 1-2 часов в зависимости от объема реакции.
Соединения формулы 8 и формулы 3 доступны в продаже. Соединение формулы 3 является особенно предпочтительным ввиду простоты его использования. Трифторэтиламин можно использовать в его нейтральном состоянии, однако он является летучим веществом (температура кипения 36-37°C), вследствие чего менее удобным в работе. Соединение формулы 7 также можно получить из доступного в продаже N-BOC-глицин-N-карбоксиангидрида (см. пример синтеза 5).
В настоящем способе реакционная смесь содержит не смешивающийся с водой растворитель. Особенно подходящими растворителями являются этилацетат и изопропилацетат. Особенно подходящими являются полярные апротонные растворители, которые не смешиваются с водой, так как они могут растворять исходное вещество формулы 8 и могут быть отделены от воды при обработке водой. Количество используемого растворителя определяется объемом, необходимым для растворения исходного вещества, и, как правило, находится в диапазоне молярных концентраций от 0,75 до 1,5, особенно подходящей является молярная концентрация 1,0.
Реакция по способу на схеме 5 может протекать в широком диапазоне температур. Как правило, температура реакции составляет по меньшей мере приблизительно 15°C, наиболее часто по меньшей мере приблизительно 20°C. Как правило, реакционную смесь нагревают во время реакции, однако экзотермический эффект, как правило, не требует внешнего охлаждения, и температура реакции, как правило, остается ниже температуры кипения растворителя. Как правило, температура реакции составляет не более приблизительно 40°C, наиболее часто не более приблизительно 35°C.
Протекание реакции можно контролировать обычными способами, например, с помощью анализа аликвот тонкослойной хроматографией, ГХ, ВЭЖХ и 1H ЯМР. После завершения реакции смесь, как правило, обрабатывают путем добавления разбавленного водного раствора минеральной кислоты, такой как соляная кислота. Отделение органической фазы, дополнительное промывание соляной кислотой (1,0 Н) для удаления имидазола или другого добавленного основания, высушивание над осушителями, такими как сульфат магния или молекулярные сита, или азеотропная сушка и затем выпаривание растворителя приводят к получению соединения формулы 7 в форме бесцветного твердого вещества. Выпаривание растворителя необязательно. При азеотропной сушке растворитель не удаляют и далее используют раствор соединения формулы 7.
Другой альтернативой защитной аминогруппе бензилкарбамата (CBZ), которая используется в способе настоящего изобретения на схемах 1 и 2, является защитная группа дибензиламина, показанная на схемах 6, 7 и 8 (см. пример синтеза 8).
Альтернативный способ с дибензиламином включает удаление дибензильной защитной группы в промежуточном соединении формулы 10 путем гидрогенолиза с получением свободного аминосоединения формулы 1, как показано на схеме 6.
Схема 6
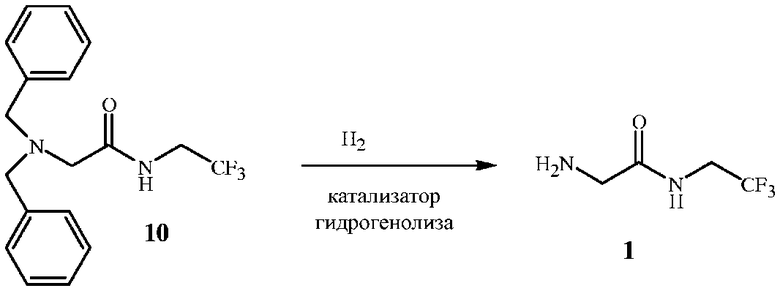
Реакция удаления бензильных защитных групп может протекать в различных условиях. См., например, Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 2-е изд.; Wiley: г. Нью-Йорк, 1991 г. Одним из наиболее подходящих способов удаления бензильной защитной группы с азота является гидрогенолиз с водородом с использованием катализаторов из благородного металла, как правило, под давлением. Этот способ описан в работе Rylander, P. N.; Hydrogenation Methods, Academic Press: г. Сан-Диего, 1985 г. Одним из наиболее подходящих катализаторов гидрогенолиза является палладий на углеродной подложке (5-10%).
Удаление бензильных защитных групп с азота требует более жестких условий по сравнению с удалением бензильной защитной группы с кислорода (как в процедуре с BOC). Как правило, реакцию гидрогенолиза проводят под давлением и при повышенной температуре. Стандартное давление водорода составляет 0,34-0,69 МПа (50-100 фунтов на кв. дюйм). Стандартная температура реакции составляет 50-80°C. Предпочтительны температуры в диапазоне приблизительно 70°C. Реакция не является экзотермической и требует внешнего нагрева для поддержания необходимой температуры.
В способе по схеме 6 реакционная смесь содержит органический растворитель. Особенно подходящими растворителями являются метанол и этанол. Также можно использовать другие растворители, которые, как правило, применяются при гидрогенизации. Количество используемого органического растворителя определяется объемом, необходимым для растворения исходного вещества, и, как правило, находится в диапазоне молярных концентраций от 0,3 до 1,0. Смесь исходного вещества формулы 10 в растворителе нагревают до нужной температуры в водороде при повышенном давлении. Реакционную смесь нагревают до завершения реакции, на которое указывает прекращение поглощения водорода.
Протекание реакции можно контролировать обычными способами, например, с помощью анализа аликвот тонкослойной хроматографией, ГХ, ВЭЖХ и 1H ЯМР, либо по скорости поглощения водорода. После завершения реакции смесь, как правило, охлаждают до температуры окружающей среды и фильтруют, чтобы удалить катализатор на подложке. Полученный продукт формулы 1 выделяют путем концентрации и восстанавливают в форме маслянистой жидкости.
Соединение формулы 10 можно получить при взаимодействии соединения формулы 11 с соединением формулы 12 в присутствии основания. Алкилирование амина формулы 12 алкилхлоридом формулы 11 показано на схеме 7.
Схема 7
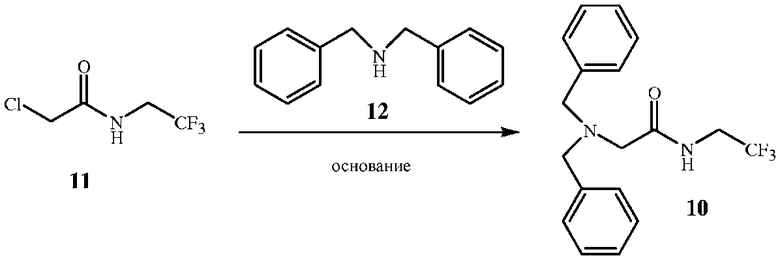
Стехиометрический состав данной реакции предполагает эквимолярные количества хлорацетиламида формулы 11 и амина формулы 12. Однако для полного завершения реакции с хлорацетамидом формулы 11 и полного образования дибензиламина формулы 10 желателен молярный избыток амина формулы 12 от приблизительно 1,1 до приблизительно 1,2. В данной реакции необходимо также эквимолярное количество основания. В зависимости от того, какое основание используется, может потребоваться молярный избыток до 2,0 эквивалентов. Предпочтительным основанием является третичный амин, такой как триэтиламин или основание Хунига (диизопропилэтиламин). Однако можно использовать карбонаты щелочных металлов.
В способе, показанном на схеме 7, реакционная смесь содержит органический растворитель. Особенно подходящим растворителем является метанол, но также можно использовать ароматические растворители, такие как толуол, или полярные апротонные растворители, такие как ацетонитрил. Количество используемого органического растворителя определяется объемом, необходимым для растворения исходных веществ, и, как правило, находится в диапазоне молярных концентраций от 0,5 до 1,0, особенно предпочтительной является молярная концентрация 0,7. Смесь исходных реагентов - хлорацетиламида, дибензиламина и основания в растворителе - нагревают с обратным холодильником или до более высоких температур при повышенном давлении. Предпочтительны температуры в диапазоне приблизительно 80-100°C. Реакционную смесь нагревают до завершения реакции, как правило, 12-24 часа.
Протекание реакции можно контролировать обычными способами, например, с помощью анализа аликвот тонкослойной хроматографией, ГХ, ВЭЖХ и 1H ЯМР. После завершения реакции смесь, как правило, охлаждают до температуры окружающей среды и концентрируют, чтобы удалить растворитель. Маслянистый остаток растворяют в метиленхлориде или аналогичном растворителе и промывают водой по меньшей мере дважды. Продукт выделяют обычными способами, например, концентрацией. Маслянистый продукт, выделенный при концентрации, кристаллизуется при охлаждении.
Исходное вещество дибензиламин (соединение формулы 12) доступно в продаже.
Соединение формулы 11 можно получить при взаимодействии соединения формулы 13 с соединением формулы 3A в присутствии основания. Реакция амина формулы 3A с хлорангидридом формулы 13 показана на схеме 8.
Схема 8
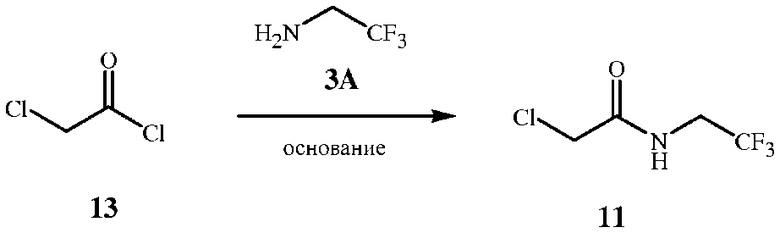
Стехиометрический состав данной реакции предполагает эквимолярные количества хлорангидрида формулы 13 и амина формулы 3A. Однако для полного завершения реакции с амином формулы 3A и полного образования продукта формулы 11 желателен молярный избыток хлорангидрида формулы 13 от приблизительно 1,05 до приблизительно 1,1. В данной реакции необходимо также эквимолярное количество основания. Благоприятным является молярный избыток, аналогичный молярному избытку хлорангидрида. Предпочтительным основным реагентом является карбонат калия. Также можно использовать различные карбонаты или бикарбонаты щелочных металлов.
В способе, показанном на схеме 8, реакционная смесь является двухфазной системой, состоящей из воды и не смешивающегося с водой растворителя. Особенно подходящими растворителями являются этилацетат и диэтиловый эфир. Количество используемого органического растворителя определяется объемом, необходимым для растворения исходных веществ, и, как правило, находится в диапазоне молярных концентраций от 1,0 до 1,5 для амина и от 4,0 до 5,0 для хлорангидрида. Количество используемой воды определяется объемом, необходимым для растворения основного вещества карбоната щелочного метала и варьируется в зависимости от растворимости использованного основания. Для карбоната калия стандартной является молярная концентрация в диапазоне от 1,0 до 3,0. Смесь исходных компонентов - трифторэтиламина (соединение формулы 3A) в растворителе и карбоната в воде - взбалтывают и охлаждают до температуры приблизительно от -5 до 0°C. К охлажденной реакционной смеси добавляют раствор хлорацетилхлорида (соединение формулы 13) в растворителе в течение 0,5-2 часов при поддержании температуры от -5 до 0°C, затем реакционную смесь перемешивают при этой температуре в течение 1 часа.
Реакция по способу на схеме 8 может протекать в узком диапазоне температур. Как правило, температура реакции составляет менее 10°C, в большинстве случаев менее 0°C. Реакция является экзотермической и требует внешнего охлаждения для поддержания необходимой температуры.
Протекание реакции можно контролировать обычными способами, например, с помощью анализа аликвот тонкослойной хроматографией, ГХ и 1H ЯМР. После завершения реакции смесь, как правило, разделяют на фазы, фазу с растворителем промывают водой и выделяют продукт концентрацией растворителя. Маслянистый продукт, выделенный при концентрации, кристаллизуется при отстаивании.
Исходные вещества - хлорацетилхлорид (соединение формулы 13) и трифторэтиламин (соединение формулы 3A) - доступны в продаже.
В другом аспекте настоящего изобретения соединения формулы 14 получают из соединений формулы 1 или формулы 1A. В способе, показанном на схеме 9, соединение формулы 15 взаимодействует со связующим реагентом с образованием промежуточного соединения формулы 16. Можно выделить промежуточное соединение ацилимидазола формулы 16 (см. пример синтеза 9). В большинстве случаев ацилимидазол не выделяют, а сразу обрабатывают соединением формулы 1 или 1A с получением соединения формулы 14.
Схема 9
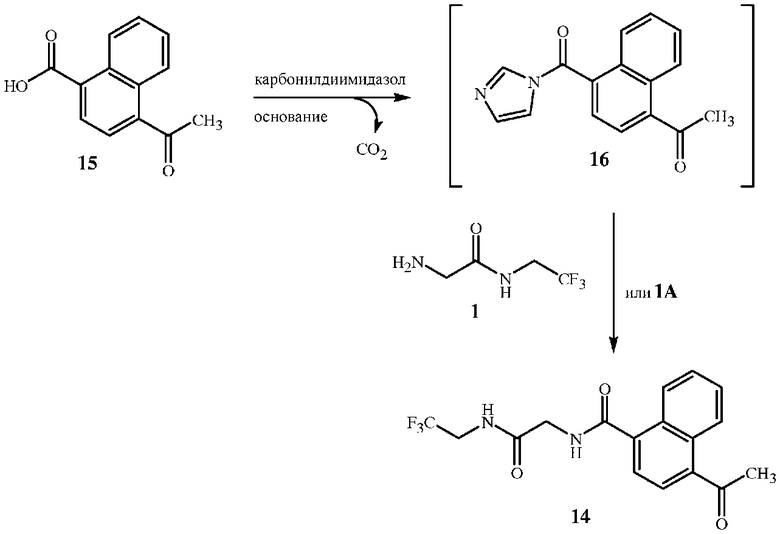
В случае если связующим реагентом является N,N'-карбонилдиимидазол, при образовании промежуточного соединения ацилимидазола (соединение формулы 16) выделяется один эквивалент диоксида углерода. При образовании ацилимидазола также высвобождается эквивалент имидазола, который взаимодействует с одним эквивалентом кислоты (то есть хлоридом водорода, бромидом водорода, трифторуксусной кислотой, метансульфоновой кислотой, серной кислотой или фосфорной кислотой) при добавлении к реакционной смеси соли амина формулы 1A. Таким образом, основание может быть производным связующего реагента, если связующим реагентом является N,N'-карбонилдиимидазол. Эквивалент дополнительного основания (основной реагент, не являющийся производным связующего реагента), такой как триэтиламин, необязателен, если связующим реагентом является N,N'-карбонилдиимидазол. Дополнительное основание (например, триэтиламин или диизопропилэтиламин) ускорит реакцию, так как оно обладает большими основными свойствами по сравнению с имидазолом и быстрее взаимодействует с хлоридом водорода соединения формулы 1A с высвобождением его в форме свободного основания, реагирующего с ацилимидазолом. В альтернативном варианте осуществления ацилимидазол формулы 16 может взаимодействовать со свободным амином формулы 1 вместо его кислой соли формулы 1A. В случае если для получения соединения формулы 14 используют свободный амин формулы 1, дополнительного основания не требуется. Пример синтеза 10 относится к реакции с использованием соединения формулы 1, а пример синтеза 11 относится к реакции с использованием соединения формулы 1A.
Стехиометрический состав реакции со схемы 9 предполагает эквимолярные количества соединения формулы 15, связующего реагента и основания. Молярное соотношение связующего реагента и соединения формулы 15 может находиться в диапазоне от приблизительно 0,95 до приблизительно 1,15, однако предпочтительным является соотношение по меньшей мере 0,97, чтобы обеспечить максимальное количество образованного промежуточного соединения ацилимидазола формулы 16 в отсутствии любого избытка N,N'-карбонилдиимидазола. Стехиометрический состав данной реакции предполагает эквимолярные количества соединения формулы 1 или 1A и соединения формулы 15. Молярное соотношение соединения формулы 1 или 1A и соединения формулы 15 может находиться в диапазоне от приблизительно 1,0 до приблизительно 1,15, однако предпочтительным является соотношение по меньшей мере 1,05, которое обеспечивает завершение реакции между промежуточным соединением ацилимидазола (соединение формулы 16) и соединением формулы 1 или 1A.
В схеме 9 могут быть использованы различные связующие реагенты. Особую эффективность продемонстрировали несколько алкилхлорформиатов и карбонилдигетероарилов, которые обеспечивают высокий выход соединений формулы 14. Такие связующие реагенты включают метилхлорформиат, этилхлорформиат, изобутилхлорформиат, N,N'-карбонилдиимидазол и 1,1'-карбонилбис(3-метилимидазолий) трифлат. Предпочтительным является N,N'-карбонилдиимидазол (который также называют карбонилдиимидазолом). N,N'-карбонилдиимидазол является наиболее эффективным связующим реагентом, так как он дает один эквивалент основания для нейтрализации соли амина формулы 1A. Связующие реагенты на основе эфира хлорформиата требуют дополнительного основного реагента для нейтрализации кислоты, образующейся в реакции с соединением формулы 15, а также для высвобождения свободного основания соединения формулы 3. Особенно подходящим основанием для данной реакции является триэтиламин.
Порядок внесения реагентов является принципиальным. Как правило, связующий реагент растворяют в растворителе, затем к нему добавляют соединение формулы 15. Важно предусмотреть достаточное количество времени на образование ацилимидазола до добавления соединения формулы 1 или 1A. Образование промежуточного соединения ацилимидазола (соединение формулы 16), как правило, можно контролировать по выделению углекислого газа в течение 0,5-2 часов в зависимости от объема реакции.
Соединение формулы 1 или 1A доступно в продаже, либо его можно получить по способу настоящего изобретения, показанному на схемах выше. Соединение формулы 1 или 1A добавляют к смеси в форме твердого вещества или суспензии в полярном апротонном смешивающемся с водой растворителе. Соединение формулы 15 получили по процедуре, указанной в работе F. Feist в Justus Liebigs Annalen der Chemie, 1932 г., 496, 99-122. Соединение формулы 1A особенно предпочтительно ввиду простоты его использования, так как оно не гигроскопично (см. пример 16). Использование нейтрального свободного аминосоединения формулы 1 менее удобно, так как оно является гигроскопичным, и необходимо максимально уменьшить его контакт с воздухом.
В настоящем способе реакционная смесь содержит полярный апротонный смешивающийся с водой растворитель. Подходящими растворителями являются ацетонитрил, тетрагидрофуран и диоксан. Особенно подходящим является ацетонитрил. Количество используемого растворителя определяется объемом, необходимым для растворения исходного вещества, и, как правило, находится в диапазоне молярных концентраций от 0,75 до 1,5, особенно подходящей является молярная концентрация 1,0.
Способ по схеме 9 можно реализовать в широком диапазоне температур. Как правило, температура реакции составляет по меньшей мере приблизительно 20°C, наиболее часто по меньшей мере приблизительно 30°C. Как правило, реакционную смесь нагревают во время реакции, однако экзотермический эффект, как правило, не требует внешнего охлаждения, и температура реакции, как правило, остается ниже температуры кипения растворителя. Как правило, температура реакции составляет не более приблизительно 45°C, наиболее часто не более приблизительно 35°C.
Протекание реакции можно контролировать обычными способами, например, с помощью анализа аликвот тонкослойной хроматографией, ГХ, ВЭЖХ и 1H ЯМР. После завершения реакции смесь, как правило, обрабатывают путем добавления водного раствора минеральной кислоты, такой как соляная кислота (1,1 моль 1 Н). Для гидролиза любого имина, который может быть образован ацетильной группой продукта (соединение формулы 14) и избытком амина соединения формулы 1, проводят быструю обработку кислотой. Затем pH корректируют до 9-10 с помощью основания (гидроксид натрия или карбонат натрия) и получают суспензию. Суспензию охлаждают до температуры 20°C и фильтруют. Полученное твердое вещество промывают водой и высушивают в вакуумной печи (50-60°C).
В альтернативной процедуре получения соединения формулы 14 используют водный раствор соединения формулы 1 или 1A. В реакционной смеси с промежуточным соединением ацилимидазола формулы 16 допустимо содержание значительного количества воды. Промежуточное соединение ацилимидазола формулы 16 взаимодействует быстрее с более нуклеофильным амином формулы 1 (либо при его непосредственном добавлении, либо при его образовании путем нейтрализации соли гидрохлорида формулы 1A), чем с менее нуклеофильной водой, которую вводят вместе с водным раствором формулы 1 или 1A.
Такую реакцию получения соединения формулы 14 с использованием водного раствора соединения формулы 1 или 1A проводят аналогично процедуре с использованием соединения формулы 1 или 1A в твердой форме. Порядок введения реагентов аналогичен вышеописанному. После завершения образования промежуточного соединения ацилимидазола необязательно добавляют небольшое количество воды, чтобы гидролизовать любой избыток N,N'-карбонилдиимидазола (0,26 моль-эквивалента) и предотвратить побочные реакции. После гашения водой избытка N,N'-карбонилдиимидазола при 20°C в течение 1 часа по каплям добавляли концентрированный водный раствор соединения формулы 1 или 1A (приблизительно 50 M) или суспензию соединения формулы 1 или 1A. Реакция между соединением формулы 1 или 1A и промежуточным соединением формулы 16 в водном растворе ацетонитрила, как правило, занимает от 12 до 24 часов. См. примеры синтеза 12, 13, 14 и 15.
Водный раствор соединения формулы 1 или 1A получают путем добавления воды к сухому твердому веществу или непосредственно в соответствии с процедурой, описанной ниже для схемы 1. Соединение формулы 15 получили по процедуре, указанной в работе F. Feist в Justus Liebigs Annalen der Chemie, 1932 г., 496, 99-122.
Другая альтернативная процедура получения соединения формулы 14 с помощью хлорангидрида соединения формулы 15 и соединения формулы 1 описана в примере 7 заявки на патент WO 2009/025983.
Не углубляясь дополнительно в детали, считается, что специалист в данной области может, опираясь на предшествующее описание, в полной мере реализовать весь потенциал настоящего изобретения. Следовательно, следующие примеры следует воспринимать исключительно как иллюстрирующие и не ограничивающие описание настоящего изобретения. Стадии, описанные в следующих примерах, иллюстрируют процедуру для каждой стадии всех синтетических трансформаций. Исходное вещество для каждой стадии не обязательно было получено с помощью определенных процедур подготовки, описанных в других примерах или стадиях. Предполагается, что используются проценты по весу, за исключением смесей растворителей для хроматографии или ситуаций, для которых указано иное. Части и проценты в смесях растворителей для хроматографии указаны по объему, если не указано иное. Спектры 1H ЯМР указаны в м.д. в сторону слабого поля от тетраметилсилана; «с» означает синглет, «д» означает дублет, «т» означает триплет, «кв.» означает квартет, «м» означает мультиплет, «дд» означает дублет дублетов, «дт» означает дублет триплетов, и «уш.» означает «уширенный».
ПРИМЕР 1
Получение гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида
N,N-карбонилдиимидазол (8,2 г, 50,5 ммоль) добавляли к суспензии N-[(фенилметокси)карбонил]глицина (10 г, 47,8 ммоль) в изопропилацетате (100 мл) в течение 14 минут. Полученный раствор перемешивали в течение приблизительно 1 часа, затем добавляли триэтиламин (4,84 г, 47,8 ммоль), после чего по частям добавляли гидрохлорид трифторэтиламина (6,8 г, 50,2 ммоль) в течение 25 минут при температуре ниже 30°C. Суспензию обрабатывали водой (50 мл) и изопропилацетатом (25 мл). Полученной двухфазной смеси позволяли отстояться, после чего фазы разделяли. Водный слой экстрагировали изопропилацетатом (2 x 25 мл). Объединенные органические фазы промывали смесью 1 Н соляной кислоты (50 мл), воды (50 мл), насыщенного водного раствора бикарбоната натрия (50 мл), солевого раствора (50 мл), затем высушивали над сульфатом натрия (25 г) в течение ночи. Суспензию отфильтровывали и остаток промывали изопропилацетатом (30 мл).
К фильтрату с промывочной жидкостью добавляли 10% палладий на углеродной подложке (1,00 г) и помещали смесь в водородную атмосферу (баллон). Приблизительно через 2 часа реакционную суспензию нагревали до 50°C и гидрогенизировали в течение приблизительно 4 часов. Реакционную смесь помещали в атмосферу азота, охлаждали до комнатной температуры и затем фильтровали через фильтр Celite® (15 г), смоченный изопропилацетатом. Остаток промывали изопропилацетатом (30 мл). Фильтрат, объединенный с промывочной жидкостью, обрабатывали газообразным хлороводородом до тех пор, пока pH смеси по индикаторной бумаге не стал равен 1-2. Затем через суспензию при 30-35°C пропускали азот до тех пор, пока pH смеси по индикаторной бумаге не стал равен 4-6. Суспензию охлаждали до температуры <5°C и фильтровали. Остаток промывали изопропилацетатом (20 мл) и сушили в вакуумной печи при 60°C с получением заявляемого соединения в форме твердого вещества серого цвета (7,75 г, выход 84%).
ПРИМЕР 2
Получение фенилметил N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]карбамата
N,N-карбонилдиимидазол (38,72 г, 0,2328 моль) добавляли к суспензии N-[(фенилметокси)карбонил]глицина (50 г, 0,239 моль) в этилацетате (350 мл) в течение 5 минут. Полученный раствор перемешивали в течение 65 минут, затем добавляли по частям гидрохлорид трифторэтиламина (32,9 г, 0,24 моль) при температуре 22°C. Реакционную смесь перемешивали в течение 17 часов, затем гасили водой (250 мл) и экстрагировали этилацетатом (150 мл). Полученной двухфазной смеси позволяли отстояться, после чего фазы разделяли. Органическую фазу дважды промывали 1 Н соляной кислоты (каждый раз по 100 мл) и сушили над сульфатом магния (20 г) в течение ночи. Суспензию фильтровали и остаток промывали 4 порциями этилацетата (50 мл, 100 мл, 100 мл, 50 мл). Фильтрат объединяли с частями промывочной жидкости и концентрировали до получения твердого вещества. Твердое вещество сушили в вакуумной печи при 40°C с получением заявляемого соединения в форме твердого вещества белого цвета (54,1 г, выход 78%).
1H ЯМР (ДМСО-d6): 8,55 (вр. п., J=6,4 Гц, 1H), 7,53 (вр. п., J=6,1 Гц, 1H), 7,43-7,22 (м, 5H), 5,04 (с, 2H), 4,01-3,79 (м, 2H), 3,68 м.д. (д, J=6,1 Гц, 2H); 19F-ЯМР (ДМСО-d6): -70,76 м.д. (вр. п., J=10,1 Гц).
ПРИМЕР 3
Второй способ получения гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида
Раствор трет-бутоксикарбонилглицина (285,7 г, 1,63 моль) в этилацетате (1140 мл) добавляли в течение приблизительно 1 часа к суспензии N,N-карбонилдиимидазола (264,5 г, 1,63 моль) в этилацетате (570 мл) при температуре окружающей среды. Реакционную смесь перемешивали в течение 1 часа, затем гидрохлорид 2,2,2-трифторэтиламина (239,5 г, 1,77 моль) добавляли по частям в течение приблизительно 15 минут. Суспензию перемешивали в течение 5 часов при температуре окружающей среды, затем добавляли 1 Н соляной кислоты (860 мл). Двухфазной смеси позволяли отстояться, после чего фазы разделяли. Органическую фазу последовательно промывали 1 Н соляной кислоты (860 мл) и 5% водным раствором карбоната натрия (860 мл), затем сушили над сульфатом магния и фильтровали. Осадок на фильтре промывали этилацетатом (200 мл). Через объединенные фильтраты в течение 2 часов барботировали газообразный хлороводород (217 г, 5,95 моль) при 20-37°C. Через полученную суспензию пропускали азот и фильтровали. Осадок дважды промывали этилацетатом (каждый раз по 500 мл) и затем сушили в вакуумной печи при температуре 60°C с получением заявляемого соединения в форме твердого вещества белого цвета (235,5 г, выход 75%).
ПРИМЕР 4
Третий способ получения гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида
Стадия А. Получение 1,1-диметилэтилового эфира N-[2-оксо-2-[2,2,2-трифторэтил)амино]этил]карбаминовой кислоты
N,N-карбонилдиимидазол (8,87 г, 54,7 ммоль) добавляли к раствору N-трет-бутоксикарбонилглицина (19 г, 57,1 ммоль) в безводном этилацетате (50 мл) в течение 2 минут. Реакционную смесь перемешивали в течение 33 минут, затем в течение 12 минут добавляли 2,2,2-трифторэтиламин (5,1 мл, 63,5 ммоль). Полученный раствор перемешивали в течение ночи при температуре окружающей среды, затем гасили 1 Н соляной кислоты (25 мл). Реакционной смеси позволяли отстояться, после чего фазы разделяли. Органическую фазу промывали три раза водой (каждый раз по 25 мл), разбавляли этилацетатом (10 мл) и сушили над сульфатом магния (5 г) в течение нескольких часов. Суспензию фильтровали и осадок три раза промывали этилацетатом (10 мл). Фильтрат объединяли с частями промывочной жидкости и концентрировали в вакууме до получения твердого вещества белого цвета (12,7 г).
1H ЯМР (ДМСО-d6): 8,44 (вр. п., J=6,5 Гц, 1H), 7,01 (вр. п., J=6,2 Гц, 1H), 3,87-3,84 (м, 2H), 3,63-3,51 (д, J=6,4 Гц, 2H), 1,21-1,50 м.д. (с, 9H); 19F-ЯМР (ДМСО-d6): -70,75 м.д. (вр. п., J=10 Гц).
Стадия B. Получение гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида
Часть продукта из примера 2 стадии A (11,7 г) разбавляли этилацетатом (50 мл) и обрабатывали газообразным хлороводородом при 18-35,5°C до полного расходования исходного материала. Полученную суспензию охлаждали до 0-5°C, перемешивали в течение приблизительно 1 часа при этой температуре, затем фильтровали. Остаток дважды промывали этилацетатом (каждый раз по 20 мл) и сушили в вакуумной печи при 60°C с получением заявляемого соединения в форме твердого вещества белого цвета (7,22 г, выход 66%).
1H ЯМР (ДМСО-d6): 9,24 (вр. п., J=6,2 Гц, 1H), 8,3 (с, 3H), 4,11-3,89 (м, 2H), 3,64 (с, 2H), 1,21-1,50 м.д. (с, 9H); 19F-ЯМР (ДМСО-d6): -70,69 м.д. (вр. п., J=10,1 Гц).
Стадия B1. Получение гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида
Соляную кислоту (37% вес., 2,1 мл, 25,6 ммоль) добавляли в два приема к смеси 1,1-диметилэтилового эфира N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]карбаминовой кислоты (2,03 г, 7,9 ммоль) в дихлорметане (10 мл) и воде (0,7 мл). Полученную смесь перемешивали при температуре окружающей среды в течение приблизительно 2 часов, затем добавляли раствор карбоната натрия (1,82 г) в воде (6 г). Мутную смесь подкисляли 1 Н соляной кислоты (21 мл) и разбавляли 20 мл дихлорметана. Фазы разделяли, водную фазу концентрировали досуха на роторном испарителе с получением 3,16 г заявляемого соединения в форме твердого вещества белого цвета.
ПРИМЕР 5
Получение 1,1-диметилэтилового эфира N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]карбаминовой кислоты
2,2,2-Трифторэтиламин (2,1 мл, 26,1 ммоль) добавляли по каплям к суспензии 1,1-диметилэтилового эфира 2,5-диоксо-3-оксазолиденкарбоновой кислоты (5,01 г, 24,8 ммоль) в этилацетате (25 мл) при 3-6°C. Реакционную смесь оставляли до достижения температуры окружающей среды и перемешивали в течение ночи. Полученную суспензию разбавляли этилацетатом (35 мл) и последовательно промывали 5% вес. карбонатом натрия (10 мл) и дважды водой (каждый раз по 10 мл). Органическую фазу сушили над сульфатом магния (5 г) и фильтровали через воронку Бюхнера. Остаток на воронке дважды промывали этилацетатом (каждый раз по 10 мл), и промывочную жидкость добавляли к исходному фильтрату. Объединенные органические фазы концентрировали в вакууме и сушили в вакуумной печи при 35°C, немного продувая азотом, с получением заявляемого соединения в форме твердого вещества белого цвета (5,76 г, выход 90,8%).
ПРИМЕР 6
Четвертый способ получения гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида
Триэтиламин (11,67 г, 115 ммоль) добавляли в один прием к раствору трет-бутоксикарбонилглицина (20 г, 114 моль) в дихлорметане (110 мл) при <10°C, затем добавляли изобутилхлорформиат (15,75 г, 115 ммоль) в течение 8 минут. Реакционную смесь оставляли для перемешивания в течение приблизительно 3,3 часа при 10°C, затем по каплям в течение 7 минут добавляли раствор трифторэтиламина (17 г, 171,6 ммоль) и триэтиламина (12,7 г, 122,5 ммоль) в дихлорметане (72 мл). Реакционную смесь перемешивали в течение приблизительно 2 часов, затем гасили 1 Н соляной кислоты (60 мл). Двухфазной смеси позволяли отстояться, после чего фазы разделяли. Органическую фазу последовательно промывали 1 Н соляной кислоты (60 мл) и 5% водным раствором карбоната натрия (60 мл), затем сушили над сульфатом натрия и фильтровали. Осадок на фильтре промывали этилацетатом (30 мл), фильтрат концентрировали в вакууме. К остатку добавляли этилацетат (50 мл), раствор концентрировали до получения маслянистой жидкости (23,81 г). Остаток повторно растворяли в этилацетате (150 мл) и обрабатывали газообразным хлороводородом при 35-41°C до тех пор, пока анализ с помощью ГХ не показал полное удаление защитных групп. Через полученную суспензию пропускали азот и фильтровали. Остаток дважды промывали этилацетатом (каждый раз по 20 мл) и сушили в вакуумной печи при 60°C с получением заявляемого соединения в форме твердого вещества белого цвета (8,9 г, выход 41%).
ПРИМЕР 7
Получение трифторацетата 2-амино-N-(2,2,2-трифторэтил)ацетамида
Раствор трифторуксусной кислоты (4,8 мл, 61,7 ммоль) в дихлорметане (22 мл) добавляли к суспензии 1,1-диметилэтилового эфира N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]карбаминовой кислоты (11,97 г, 46,7 ммоль) в дихлорметане (50 мл) в течение 23 минут при комнатной температуре. Раствор нагревали до 39°C и выдерживали при этой температуре в течение приблизительно 2 часов. Полученный раствор оставляли охлаждаться до температуры окружающей среды, затем добавляли трифторуксусную кислоту (4,8 мл, 61,7 ммоль), и помутневшую реакционную смесь оставляли на ночь для перемешивания. Реакционную смесь охлаждали до 0-5°C, выдерживали при этой температуре 70 минут, затем фильтровали через воронку Бюхнера и получали бесцветный студенистый остаток. Остаток промывали дихлорметаном (1×40 мл, 1×15 мл), затем сушили в вакуумной печи при 35°C, немного продувая азотом, с получением заявляемого соединения в форме липкого твердого вещества белого цвета (6,08 г, 39,4%).
1H ЯМР (ДМСО-d6): 9,13 (вр. п., J=6,3 Гц, 1H), 8,19 (с, 3H), 4,14-3,85 (м, 2H), 3,68 (с, 2H), 19F-ЯМР (ДМСО-d6): -70,83 м.д. (вр. п., J=10 Гц), -74,93 (с).
ПРИМЕР 8
Пятый способ получения гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида
Стадия А. Получение 2-хлор-N-(2,2,2-трифторэтил)ацетамида
Раствор хлорацетилхлорида (60,8 г, 0,52 моль) в этилацетате (120 мл) добавляли в течение 35 минут к предварительно охлажденной (от -5 до 0°C) двухфазной смеси трифторэтиламина (47,6 г, 0,48 моль) в безводном этилацетате (360 мл) и карбоната калия (33,2 г, 0,24 моль) в воде (120 мл). Реакционную смесь перемешивали при этой температуре в течение 60 минут. Реакционной смеси позволяли отстояться, после чего фазы разделяли. Органическую фазу промывали водой и концентрировали в вакууме с получением маслянистой жидкости. Для растворения маслянистой жидкости добавляли метанол. Раствор концентрировали в вакууме до получения бесцветной маслянистой жидкости, которая кристаллизовалась при охлаждении до твердого вещества белого цвета (89,6 г).
1H ЯМР (ДМСО-d6): 8,89 (уш. с., 1H), 4,17 (с, 2H), 3,91-3,99 (м, 2H).
Стадия B. Получение 2-[бис(фенилметил)амино]-N-(2,2,2-трифторэтил)ацетамида
Часть продукта из примера 7 стадии A (40,0 г, 0,23 моль) растворяли в метаноле (300 мл) и вводили в реактор под давлением (реактор Парра, модель 4540, 600 мл, Hasteloy C) вместе с дибензиламином (39,5 г, 0,19 моль) и триэтиламином (22,4 г, 0,22 моль). Через реактор прокачивали азот, и реактор герметично закрывали, затем температуру поднимали до 85°C и удерживали ее на этом уровне в течение 23 часов. Реактор охлаждали до температуры окружающей среды и неочищенный продукт реакции концентрировали в вакууме до получения вязкой маслянистой жидкости красного цвета, которую растворяли в метиленхлориде (400 мл). Раствор дважды промывали водой (общее количество 450 мл) и концентрировали в вакууме до получения маслянистой жидкости янтарного цвета, которая кристаллизовалась при охлаждении (63,5 г).
1H ЯМР (ДМСО-d6): 8,38 (вр. п., 1H), 7,30-7,43 (м, 10H), 3,85-4,0 (м, 2H), 3,63 (с, 4H), 3,07 (с, 2H).
Стадия C. Получение гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида
Часть продукта из примера 7 стадии B (12,0 г) растворяли в метаноле (300 мл) и вводили в реактор под давлением (реактор Парра, модель 4540, 600 мл, Hasteloy C) вместе с катализатором - 5% палладием на углеродной подложке (0,6 г). Через реактор пропускали азот, затем водород, и его нагревали до 70°C под давлением водорода 0,69 МПа (100 фунтов на кв. дюйм) до прекращения поглощения водорода (3 часа). Реактор охлаждали и продували азотом, затем неочищенный продукт реакции фильтровали с помощью фильтра со слоем Celite®, способствующим удалению катализатора, и осадок промывали метанолом. Растворитель и побочный продукт толуол удаляли перегонкой. В результате этого получали маслянистую жидкость янтарного цвета (5,45 г, выход продукта 89% по ГХ).
Неочищенный маслянистый продукт, полученный за два цикла описанной выше процедуры гидрогенолиза (общий выход 10,9 г) разбавляли этилацетатом (50 мл) и обрабатывали газообразным хлороводородом при температуре окружающей среды до полного расходования исходного вещества. Полученную суспензию фильтровали, твердый остаток промывали этилацетатом (20 мл) и сушили на фильтре под слоем азота с получением заявляемого соединения в форме твердого вещества белого цвета (10,0 г).
1H ЯМР (ДМСО-d6): 9,24 (вр. п., J=6,2 Гц, 1H), 8,3 (с, 3H), 4,11-3,89 (м, 2H), 3,64 (с, 2H); 19F-ЯМР (ДМСО-d6): -70,69 м.д. (вр. п., J=10,1 Гц).
ПРИМЕР 9
Получение 1-[4-(1H-имидазол-1-илкарбонил)-1-нафталинил]этанона
1H-Имидазол (1,17 г, 17,2 ммоль) добавляли к раствору 4-ацетил-1-нафталинкарбонилхлорида (2,01 г, 8,6 ммоль) в дихлорметане (35 мл). Полученную суспензию перемешивали при температуре окружающей среды в течение 11,5 часа, затем охлаждали до 0°C в водяной бане со льдом. Добавляли холодную воду (35 мл), после чего реакционную смесь переносили в делительную воронку. Фазы разделяли, органическую фазу промывали водой (35 мл) и сушили над сульфатом магния. Суспензию фильтровали и фильтрат концентрировали в вакууме с получением заявляемого соединения в форме маслянистой жидкости оранжевого цвета.
1H ЯМР (CDCl3): 8,63-8,60 (м, 1H), 7,97-7,91 (м, 3H), 7,72-7,60 (м, 3H), 7,51 (вр. п., 1H, J=1,4 Гц), 7,18-7,17 (м, 1H), 2,80 (с, 3H).
ПРИМЕР 10
Получение 4-ацетил-N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]-1-нафталинкарбоксамид
4-Ацетил-1-нафталинкарбоновую кислоту (680 г, 3,14 моль) добавляли в пять приемов в течение 1 часа при температуре окружающей среды к суспензии N,N-карбонилдиимидазола (505 г, 3,11 моль) в безводном ацетонитриле (2720 мл). Раствор перемешивали в течение 2,5 часов, затем нагревали до 35°C. Добавляли 2-амино-N-(2,2,2-трифторэтил)ацетамид (530 г, 3,73 моль) в пять приемов в течение 30 минут. Реакционную смесь оставляли для перемешивания на 2 часа при 35-40°C, затем охлаждали и оставляли на ночь для перемешивания при температуре окружающей среды. Полученную суспензию в течение 40 минут обрабатывали водой (5540 мл), после чего в течение 30 минут добавляли 1 Н раствор соляной кислоты (5440 мл). Реакционную смесь охлаждали до 5°C, выдерживали при этой температуре 1 час, затем фильтровали. Остаток 3 раза промывали водой (каждый раз по 1360 мл) и сушили в вакуумной печи при 60°C при продувании азотом с получением заявляемого продукта в форме твердого вещества белого цвета (1042,6 г, выход 88,8%).
1H ЯМР (CD3S(=O)CD3): 8,95 (т, J=5,8 Гц, 1H), 8,72 (т, J=6,5 Гц, 1H), 8,55 (дд, J=6,5, 2 Гц, 1H), 8,37-8,33 (м, 1H), 8,13 (д, J=7,3 Гц, 1H), 7,70-7,60 (м, 3H), 4,07-3,95 (м, 4H), 2,75 (с, 3H).
ПРИМЕР 11
Второй способ получения 4-ацетил-N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]-1-нафталинкарбоксамида
4-Ацетил-1-нафталинкарбоновую кислоту (675 г, 3,15 моль) добавляли в пять приемов в течение 32 минут к суспензии N,N-карбонилдиимидазола (486 г, 3,00 моль) в безводном ацетонитриле (2578 мл) при 36°C. Раствор перемешивали в течение приблизительно 2 часов при этой температуре, затем в пять приемов в течение 36 минут добавляли гидрохлорид 2-амино-N-(2,2,2-трифторэтил)ацетамида (629 г, 3,27 моль). Реакционную смесь оставляли для перемешивания на ночь при 35°C, затем охлаждали до приблизительно 18°C, чтобы инициировать кристаллизацию. Полученную суспензию нагревали до 35°C, затем добавляли в течение 90 минут 1 Н соляной кислоты (3064 мл), после чего добавляли в течение 81 минуты 50% раствор гидроксида натрия (514,2 г) в воде (7356 мл). Реакционную смесь охлаждали до приблизительно 18°C, выдерживали при этой температуре в течение 30 минут, затем фильтровали. Остаток 3 раза промывали водой (каждый раз по 700 мл) и сушили в вакуумной печи при 60°C при продувании азотом с получением заявляемого продукта в форме твердого вещества белого цвета (988,6 г, выход 87,7%).
ПРИМЕР 12
Третий способ получения 4-ацетил-N-[2-оксо-2-[2,2,2-трифторэтил)амино]этил]-1-нафталинкарбоксамида
4-Ацетил-1-нафталинкарбоновую кислоту (50 г, 0,2273 моль) добавляли по частям к суспензии N,N-карбонилдиимидазола (39,76 г, 0,2388 моль) в безводном ацетонитриле (200 мл) при 30°C. Раствор перемешивали в течение 2 часов при 30°C и затем охлаждали до 20°C. К смеси добавляли воду (1,06 г, 58,8 ммоль) и перемешивали ее в течение 1 часа. Затем в течение 1 часа при 19-20°C добавляли раствор гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида (45,98 г, 0,2388 моль) в воде (21,5 г). Реакционную смесь оставляли для перемешивания на 17 часов. К полученной суспензии добавляли воду (100 мл), после чего добавляли раствор карбоната натрия (24,1 г, 0,2274 моль) в воде (350 мл) в течение 25 минут и воду (350 мл) в течение 22 минут. Реакционную смесь перемешивали при температуре 20-25°C в течение 6,5 часа и фильтровали. Остаток 3 раза промывали водой (каждый раз по 100 мл) и сушили в вакуумной печи при 50-60°C при продувании азотом с получением заявляемого продукта в форме твердого вещества белого цвета (72,3 г, выход 86,1%).
ПРИМЕР 13
Четвертый способ получения 4-ацетил-N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]-1-нафталинкарбоксамида
Безводный ацетонитрил (40 мл) добавляли к 4-ацетил-1-нафталинкарбоновой кислоте (10,0 г, 46,5 ммоль) и N,N-карбонилдиимидазолу (7,62 г, 46,5 ммоль). Раствор перемешивали в течение 5,75 часа при 25°C и затем нагревали до 30°C. В течение 6 минут добавляли раствор гидрохлорида 2-амино-N-(2,2,2-трифторэтил)ацетамида (9,84 г, 50,8 ммоль) в воде (4,32 г). Реакционную смесь оставляли для перемешивания на 16,3 часа при 30°C, затем охлаждали до 20°C. К полученной суспензии добавляли воду (20 мл), затем добавляли раствор карбоната натрия (9,86 г, 93 ммоль) в воде (140 мл) в течение приблизительно 1 часа. Реакционную смесь перемешивали в течение ночи при 20-25°C, затем выдерживали при 0-8°C в течение 2,25 часа и фильтровали. Остаток 3 раза промывали водой (каждый раз по 20 мл) и сушили в вакуумной печи при 50°C при продувании азотом с получением заявляемого продукта в форме твердого вещества грязно-белого цвета (14,83 г, выход 87,8%).
ПРИМЕР 14
Пятый способ получения 4-ацетил-N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]-1-нафталинкарбоксамида
4-Ацетил-1-нафталинкарбоновую кислоту (10,0 г, 46 5 ммоль) добавляли к суспензии N,N-карбонилдиимидазола (8,21 г, 50,1 ммоль) в безводном ацетонитриле (40 мл). Раствор перемешивали при температуре окружающей среды в течение 1,3 часа. Добавляли воду (0,2 мл, 11,1 ммоль) и перемешивали раствор в течение 30 минут. Готовили раствор сульфата 2-амино-N-(2,2,2-трифторэтил)ацетамида (11,25 г, 54,8 ммоль) в воде (22,6 г) и фильтровали, чтобы удалить нерастворимый остаток, затем добавляли в течение 3 минут к реакционной смеси. Реакционную смесь оставляли для перемешивания на 21,3 часа при 21-23°C. К полученной суспензии добавляли воду (20 мл), затем в течение 15 минут добавляли раствор карбоната натрия (9,82 г, 92,7 ммоль) в воде (140 мл). Реакционную смесь охлаждали и перемешивали при температуре 0-5°C в течение 2,3 часа и затем фильтровали. Остаток 3 раза промывали водой (каждый раз по 20 мл) и сушили в вакуумной печи при 45°C, немного продувая азотом, с получением заявляемого соединения в форме твердого вещества грязно-белого цвета (12,6 г, выход 76,9%).
ПРИМЕР 15
Шестой способ получения 4-ацетил-N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]-1-нафталинкарбоксамида
4-Ацетил-1-нафталинкарбоновую кислоту (10,0 г, 46,5 ммоль) добавляли к суспензии N,N-карбонилдиимидазола (8,22 г, 50,2 ммоль) в безводном ацетонитриле (40 мл). Раствор перемешивали в течение 2 часов 10 минут при 19-21°C. Добавляли воду (0,2 мл, 11,1 ммоль) и перемешивали раствор в течение 1 часа. Готовили раствор 2-амино-N-(2,2,2-трифторэтил)ацетамида (9,03 г, 55,1 ммоль) в воде (16,5 г) и фильтровали, чтобы удалить нерастворимый остаток. Твердый остаток на фильтре промывали водой (1,58 г), части промывочной жидкости объединяли с профильтрованным водным раствором 2-амино-N-(2,2,2-трифторэтил)ацетамида. Водный раствор 2-амино-N-(2,2,2-трифторэтил)ацетамида добавляли в течение 12 минут к реакционной смеси, содержащей промежуточное соединение ацилимидазола. Реакционную смесь оставляли для перемешивания на 20,6 часа при температуре окружающей среды. К полученной суспензии добавляли воду (20 мл), затем по каплям добавляли раствор карбоната натрия (4,91 г, 46,3 ммоль) в воде (70 мл) в течение 16 минут и воду (70 мл) в течение 10 минут. Реакционную смесь охлаждали и перемешивали при температуре 2-7°C в течение 2,5 часов, а затем фильтровали. Остаток 3 раза промывали водой (каждый раз по 20 мл) и сушили в вакуумной печи при 45°C, немного продувая азотом, с получением заявляемого соединения в форме твердого вещества грязно-белого цвета (13,83 г, выход 84,4%).
ПРИМЕР 16
Сравнение стабильности свободного основания и солей 2-амино-N-(2,2,2-трифторэтил)ацетамида
Свободное основание 2-амино-N-(2,2,2-трифторэтил)ацетамида неожиданно продемонстрировало увеличение веса при нахождении в атмосфере окружающей среды, в то время как соответствующий гидрохлорид такого эффекта не продемонстрировал. Такой результат оказался неожиданным, так как гидрохлориды аминов довольно часто являются гигроскопичными. Для дополнительного изучения стабильности свободного амина и солей 2-амино-N-(2,2,2-трифторэтил)ацетамида были проведены следующие эксперименты. Образцы свободного амина и солей оставляли в лаборатории на воздухе в течение определенного периода времени. Было зафиксировано увеличение веса или его уменьшение по сравнению с исходным образцом.
окружающей среды
| название | год | авторы | номер документа |
|---|---|---|---|
| 2,2,2-ТРИФТОРЭТИЛ-ТИАДИАЗИНЫ | 2015 |
|
RU2692102C2 |
| ПРОИЗВОДНЫЕ МОРФОЛИНОПУРИНА, ОБЛАДАЮЩИЕ PI3K И/ИЛИ mTOR ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 2009 |
|
RU2490269C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНЫХ ТИРОЗИНКИНАЗ | 2009 |
|
RU2518089C2 |
| СИНТЕЗ ЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ ПИРАЗОЛИН КАРБОКСАМИДИНА | 2011 |
|
RU2550694C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ (3-ОКСО-2,3-ДИГИДРО-1Н-ИЗОИНДОЛ-1-ИЛ)-АЦЕТИЛГУАНИДИНА | 2004 |
|
RU2397159C2 |
| ПРОИЗВОДНЫЕ 1,7-НАФТИРИДИН-3-КАРБОКСАМИДА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ НЕЙРОГЕННЫХ АГЕНТОВ | 2014 |
|
RU2669943C2 |
| ЦИКЛОАЛКАН-1,3-ДИАМИНОВОЕ ПРОИЗВОДНОЕ | 2019 |
|
RU2793247C2 |
| ИНГИБИТОРЫ КАТЕПСИН-ЦИСТЕИНПРОТЕАЗЫ | 2003 |
|
RU2312861C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ХЛОР-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2013 |
|
RU2653855C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2012 |
|
RU2614976C2 |
Изобретение относится к способам получения соединений формулы 1 и 1А. Способ получения соединений формулы 1 включает (А) взаимодействие соединения формулы 2 с N,N′-карбонилдиимидазолом (связующим реагентом) в полярном апротонном не смешивающемся с водой растворителе, затем добавляют соль формулы 3 в присутствии основания, полученного из связующего реагента, с получением соединения формулы 4; на стадии (В) проводят взаимодействие промежуточного соединения формулы 4 с водородом в присутствии катализатора гидрогенолиза с получением соединения формулы 1. Способ получения солей формулы 1А, где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3, включает (А1) взаимодействие трет-бутоксикарбонилглицина с N,N′-карбонилдиимидазолом (связующим реагентом) в полярном апротонном не смешивающемся с водой растворителе, затем добавляют соль формулы 3 в присутствии основания, полученного из связующего реагента, с получением соединения формулы 7; на стадии (В1) проводят взаимодействие промежуточного соединения формулы 7 с кислотой НХ с получением соединения формулы 1А. Технический результат - усовершенствованные способы получения N-(2,2,2-трифторэтил)ацетамида и его солей, которые используют в синтезе 4-ацетил-N-[2-оксо-2-[(2,2,2-трифторэтил)амино]этил]-1-нафталинкарбоксамида. 2 н. и 12 з.п. ф-лы, 16 пр.
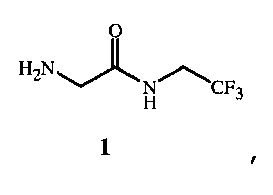
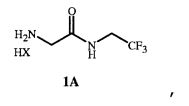
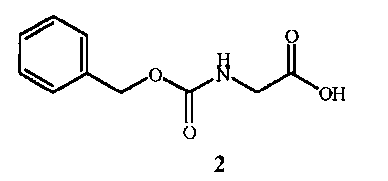 ,
, 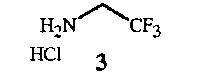 ,
, 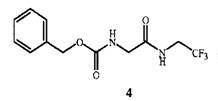 ,
, 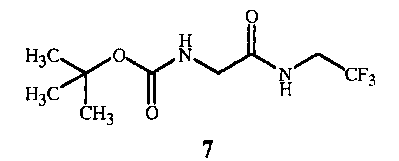
1. Способ получения соединения формулы 1
включающий (А) взаимодействие соединения формулы 2
со связующим реагентом, где связующим реагентом является N,N′-карбонилдиимидазол, в полярном апротонном не смешивающемся с водой растворителе, и затем в присутствии основания, полученного из связующего реагента, добавляют соль формулы 3
с образованием промежуточного соединения формулы 4
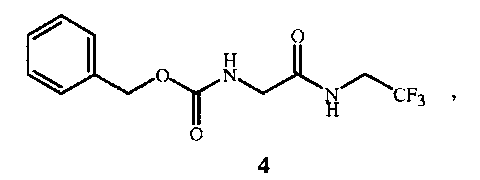
(В) взаимодействие промежуточного соединения формулы 4 с водородом в присутствии катализатора гидрогенолиза с получением соединения формулы 1
и (С) необязательно взаимодействие соединения формулы 1 с кислотой формулы 5
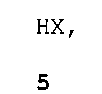
где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3, для получения соединения формулы 1 в форме соли HX.
2. Способ по п. 1, в котором на стадии (А) присутствует дополнительное основание, когда добавляют соль формулы 3.
3. Способ по п. 2, в котором дополнительным основанием является триэтиламин.
4. Способ по п. 1, в котором на стадии (В), соединение формулы 4 и водород взаимодействуют в присутствии катализатора гидрогенолиза и не смешивающегося с водой растворителя.
5. Способ по п. 4, в котором катализатор гидрогенолиза представляет собой палладий на углеродной подложке.
6. Способ по п. 1, в котором на стадии (C), соединение формулы 1 взаимодействует с кислотой формулы 5 в присутствии не смешивающегося с водой растворителя.
7. Способ по п. 6, в котором кислота формулы 5 содержит хлорид водорода.
8. Способ по любому из пп. 2, 4 или 6, в котором не смешивающийся с водой растворитель содержит этилацетат или изопропилацетат.
9. Способ получения соли формулы 1A
где X представляет собой Cl, Br, CF3CO2, CH3SO3, (SO4)1/2 или (PO4)1/3, включающий (А1) взаимодействие соединения формулы 8
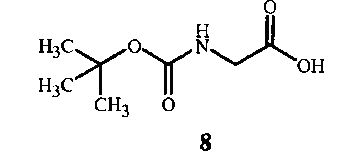
со связующим реагентом, где связующим реагентом является N,N'-карбонилдиимидазол, в полярном апротонном не смешивающемся с водой растворителе, и затем в присутствии основания, полученного из связующего реагента, добавляют соль формулы 3
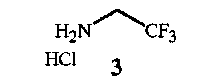
с образованием промежуточного соединения формулы 7
и (В1) взаимодействие промежуточного соединения формулы 7 с кислотой формулы 5
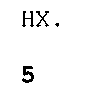
10. Способ по п. 9, в котором на стадии (А1) соединение формулы 8, связующий реагент и соль формулы 3 взаимодействуют в присутствии дополнительного основания.
11. Способ по п. 10, в котором дополнительным основанием является триэтиламин.
12. Способ по п. 9, в котором на стадии (В1) соединения формул 7 и 5 взаимодействуют в присутствии полярного апротонного не смешивающегося с водой растворителя.
13. Способ по п. 12, в котором кислота формулы 5 содержит хлорид водорода.
14. Способ по любому из пп. 9 или 12, в котором полярный апротонный не смешивающийся с водой растворитель содержит этилацетат или изопропилацетат.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| JP 2009173621 А, 06.08.2009 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Марсианский многоканальный диодно-лазерный спектрометр "М-ДЛС" | 2019 |
|
RU2730405C1 |
| Местное применение антиандрогена от выпадения волос и других гиперандрогенных состояний | 2000 |
|
RU2225390C2 |

