Область техники, к которой относится изобретение
Настоящее изобретение относится к производным и способам получения 13-деоксиантрациклинов, конкретно к применению 13-бензенидсульфонил гидразоновых антрациклиновых интермедиатов для синтеза и выделения 13-деоксиантрациклинов, а также к способам получения 13-бензенидсульфонилгидразоновых антрациклинов. Кроме того, настоящее изобретение относится к новым 13-бензенидсульфонилгидразоновым интермедиатам и к способам получения этих интермедиатов.
Уровень техники
Наиболее известными антрациклиновыми противораковыми лекарственными средствами являются доксорубицин и даунорубицин, они оба содержат 13-кетогруппу в составе молекулы. Доксорубицин, описанный в патенте US Pat. No. 3590028, характеризуется широким спектром противораковой активности и используется для лечения лейкемий, лимфом и твердых опухолей. Даунорубицин, описанный в патенте US Pat. No. 3616242, применяется для лечения острых лейкемий. Однако применимость указанных лекарственных средств ограничивается серьезным побочным эффектом - кардиотоксичностью, - поэтому общее количество вводимого пациенту лекарства не может превышать 550 мг/М2 (E.A.Lefrak et al., Cancer, 32:302, 1973). Даже при достижении или приближении к рекомендованной максимальной общей кумулятивной дозе (430-650 мг/М2) у 60% пациентов возникает существенная и устойчивая дисфункция в работе сердца, а у 14% развивается застойная сердечная недостаточность (A.Dresdale et al., Cancer, 52:51, 1983). Таким образом, несмотря на то, что указанные лекарства подходят для ингибирования роста раковых опухолей, пациент может умереть от застойной сердечной недостаточности, вызванной серьезным кардиотоксическим эффектом лекарства.
Обнаружено также, что кардиотоксичность указанных антрациклинов обусловлена восстановлением 13-кетогруппы в 13-дигидроспиртовой метаболит (P.S.Mushlin et al., Fed. Proc., 45:809, 1986). В тестовых системах, в которых не происходит существенного превращения доксорубицина в 3-дигидроспиртовой метаболит (доксорубицинол), ощутимых кардиотоксических эффектов не наблюдается (P.S.Mushlin et al., Fed. Proc, 44:1274, 1985; R.D.Olson et al., Fed. Proc., 45:809, 1986). Напротив, 13-дигидрометаболиты, доксорубицинол и даунорубицинол, вызывают кардиотоксические эффекты в тех же самых тестовых системах при относительно низких концентрациях (1-2 мкг/мл, R.D.Olson et al., Proceed. Am. Assoc. Cancer Res., 26:227, 1985; R.D.Olson et al., Proceed Am. Assoc. Cancer Res. 28:441, 1987).
Если в тестовую систему ввести доксорубицин даже ненадолго, то вещество в некоторой степени метаболизируется, и образовавшегося 13-дигидрометаболита хватает, чтобы вызвать кардиотоксические эффекты (L.Rossini et al., Arch. Toxicol. Suppl., 9:474, 1986; M. Del Tocca et al., Pharmacol. Res. Commun., 17:1073, 1985). Таким образом, в соответствии с накопленными данными, кардиотоксические эффекты таких лекарств, как доксорубицин и даунорубицин, обусловлены сильными кардиотоксическими эффектами их 13-дигидрометаболитов (P.Mushlin et al., FASEB Journal, 2:A1133, 1988; R.Bouceket et al., J. Biol. Chem., 262:15851, 1987, и R.Olson et al., Proc. Natl. Acad. Sci., 85:3585, 1988).
Недавно было обнаружено, что 13-деокси-формы доксорубицина, даунорубицина или других похожих антрациклинов не метаболизируются в кардиотоксические 13-дигидро-формы, и что 5-кетогруппу можно модифицировать так, чтобы она с меньшей вероятностью генерировала свободные радикалы, что дополнительно повышает безопасность. В особенности, см. патенты WO 99/08687, US Patents 5984896 и 5942605 и PCT/US 99/04704, содержание которых включено сюда по ссылке.
Первый документально подтвержденный способ получения некоторых 13-деоксиантрациклинов из 13-пара-метилбензенидсульфонилгидразоновых антрациклинов характеризуется относительно низким выходом, порядка 10% (см. Smith et al., J. Med. Chem. 197821, 280-283). Усовершенствованные способы синтеза 13-деоксиантрациклинов из 13-пара-метилбензенидсульфонилгидразоновых антрациклинов с более высоким выходом описаны в патентах WO 99/08687 и US Patent 5984896. Однако в этих способах используется сравнительно большой избыток реагентов, и реакция занимает относительно много времени. Более того, хотя выход продукта увеличивается, его все-таки недостаточно для коммерческого производства. Далее, при использовании в ходе реакции 13-пара-метил-бензенидсульфонилгидразоновых антрациклинов в конечном 13-деоксиантрациклиновом продукте остается в качестве примеси не менее 3% этого исходного вещества. Описано использование 13- пара-F-бензенидсульфонил гидразоновых антрациклинов, но синтез 13- пара-F-, 13-пара-Cl- или 13-пара-нитробензенидсульфонилгидразоновых антрациклинов из родительских 13-кетоантрациклинов протекает с меньшим выходом по сравнению с 13-пара-метилбензенидсульфонилгидразоновыми антрациклинами и приводит к получению 13-деоксиантрациклинов с меньшим выходом.
Раскрытие изобретения
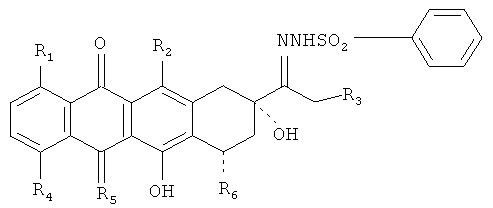
Производные и способы настоящего изобретения позволяют получить 13-деоксиантрациклины из соответствующих 13-кетоантрациклинов с повышенным выходом и чистотой. Один аспект настоящего изобретения относится к производным антрациклинов общей формулы
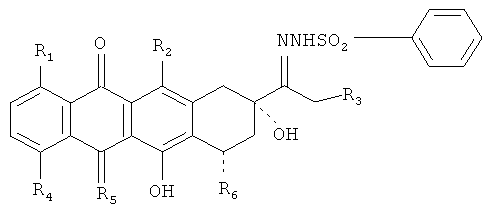
где R1, R2 и R3 представляют собой Н или ОН;
R4 соответствует Н, ОН, алкил, или О-алкил;
R5 означает О или NH и
R6 представляет собой Н, ОН или остаток сахара.
Настоящее изобретение относится также к способу получения 13-бензенидсульфонилгидразоновых антрациклинов, соответствующих приведенной выше формуле, который включает взаимодействие 13-кетоантрациклинов или их кислых солей с бензенидсульфонилгидразидом в спиртовом растворе.
Еще один аспект настоящего изобретения относится к способу получения 13-деоксиантрациклиновых (13-метиленантрациклиновых) производных из 13-бензенидсульфонилгидразоновых антрациклинов, включающий:
1. Образование реакционной смеси путем взаимодействия 13-бензенидсульфонилгидразонового антрациклина с таким восстанавливающим агентом, как, например, цианоборогидрид натрия (NaCNBH), и сильной кислотой, такой как пара-толуолсульфоновая кислота в спиртовой среде, например в метаноле.
2. Нагревание реакционной смеси без перемешивания или взбалтывания.
3. Нейтрализация реакционной смеси водным раствором основания, таким как раствор бикарбоната натрия (NаНСО3) в воде, с образованием 13-деоксиантрациклинового продукта и осаждение солей из реакционной смеси.
4. Фильтрование осажденных солей из реакционной смеси, экстрагирование продукта из осажденных солей с помощью органического растворителя, а также экстракция продукта из фильтрата органическим растворителем.
Еще один аспект настоящего изобретения относится к способу получения 5-имино-13-деоксиантрациклиновых производных из 13-деоксиантрациклинов под действием метанольного раствора аммиака.
Настоящее изобретение позволяет полностью восстановить 13-бензенидсульфонилгидразоновые антрациклины в соответствующие 13-деоксиантрациклины.
Настоящее изобретение позволяет относительно просто выделять 13-деоксиантрациклины.
Настоящее изобретение позволяет получать 5-имино-13-деоксиантрациклины из соответствующих 13-деокси-продуктов.
Настоящее изобретение позволяет синтезировать бензенидсульфонилгидразоновые антрациклины за 16-20 часов.
В соответствии с настоящим изобретением 5-имино-аналоги можно синтезировать из соответствующих неочищенных 13-деоксиантрациклиновых продуктов с использованием метанольного раствора аммиака без необходимости защищать аминогруппу сахарного остатка.
В соответствии с настоящим изобретением было обнаружено, что вместо сильной кислоты для инициирования восстановления исходного вещества можно использовать кислую соль пиридиния, таким образом, реакционную смесь не придется нейтрализовывать или экстрагировать, что облегчает очистку продукта методами препаративной ВЭЖХ.
Осуществление изобретения
Хотя следующее далее описание подробно раскрывает предпочтительные аспекты изобретения, необходимо понимать, что оно не ограничено в данной заявке описанием структур и схем, проиллюстрированных на соответствующих рисунках, поскольку у изобретения могут быть и другие аспекты, и применять на практике их можно по-разному.
Один аспект изобретения относится к производным антрациклинов, представленным следующей общей формулой:
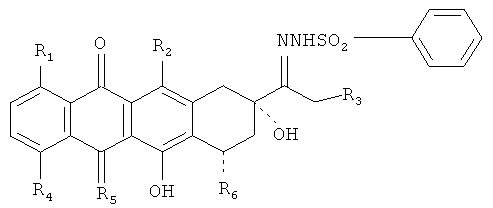
где R1, R2, и R3 в отдельности представляют собой Н или ОН;
R4 выбран из группы, включающей Н, ОН, алкил, и O-алкил;
R5 представляет собой О или NH;
R6 выбран из группы, включающей Н, ОН или остаток сахара.
Как правило, алкильная группа содержит от 1 до 5 атомов углерода, более предпочтительно от 1 до 3 атомов углерода.
O-Алкильная группа, как правило, содержит от 1 до 5 атомов углерода, более предпочтительно, от 1 до 3 атомов углерода, и
R4 обычно представляет собой ОСН3.
Упомянутые соединения представляют собой исходные продукты для получения 13-деоксиантрациклиновых соединений и 5-имино-13-деоксиантрациклиновых производных, применяемых в качестве противораковых лекарств. Примерами антрациклиновых соединений, используемых в способе по настоящему изобретению, являются доксорубицин, даунорубицин, карминомицин, эпирубицин, идарубицин и аннамицин, причем предпочтительны доксорубицин и даунорубицин.
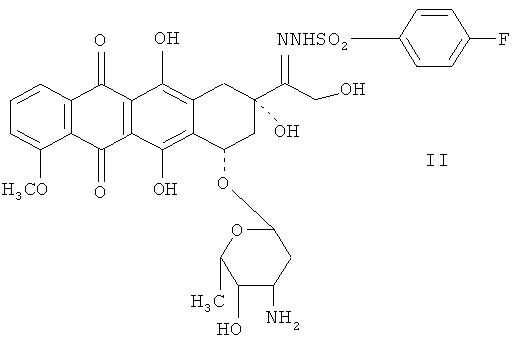
Для превращения 13-кетоантрациклинов в 13-деоксиантрациклины, прежде всего, первые необходимо преобразовать в 13-пара-замещенные бензенидсульфонилгидразоновые антрациклины. 13-пара-Замещенные бензенидсульфонилгидразоновые антрациклины, используемые в качестве исходного материала при синтезе 13-деоксиантрациклинов, - это 13-пара-метилбензенидсульфонилгидразоновый антрациклин и 13-пара-F-бензенидсульфонилгидразоновый антрациклин. Примерами здесь являются 13-пара-метилбензенидсульфонилгидразондоксорубицин (I) и 13-пара-F-бензенидсульфонилгидразондоксорубицин (II):
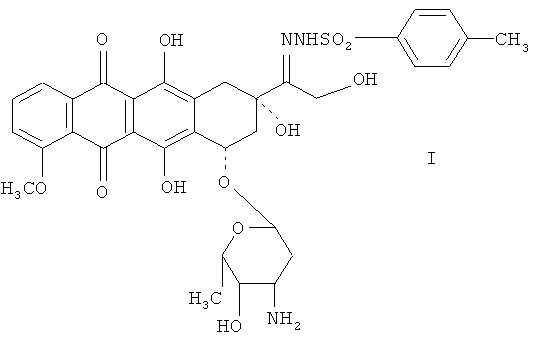
Соединение I содержит электронодонорную группу в бензольном кольце 13-пара-метилбензенидсульфонилгидразоновой группы и не полностью восстанавливается в 13-деоксидоксорубициновый продукт в ходе реакции восстановления. Из полученного 13-деоксидоксорубицинового продукта соединение I выделить трудно, и для его очистки требуется применить хроматографию на кремниевой колонке и препаративную ВЭЖХ.
Соединение II содержит электронно-акцепторную группу в бензольном кольце 13-пара-метилбензенидсульфонилгидразоновой группы и полностью восстанавливается в 13-деоксидоксорубициновый продукт в ходе реакции восстановления настоящего изобретения. Однако синтез соединения II из доксорубицина и пара-F-бензенидсульфонилгидразина приводит к меньшим выходам, по сравнению с соединением I. Кроме того, растворимость соединения II в метаноле относительно мала, и, в зависимости от температуры, может потребоваться несколько часов, чтобы растворить его в требуемой концентрации. Это касается также пара-Cl- и пара-нитро-аналогов. При понижении температуры ниже 20°С раствор соединения II в метаноле становится желатинообразным, что не позволяет работать с реакционными системами при температурах ниже 20°С. Это снижает выход 13-деоксидоксорубицина из соединения II. Другие примеры включают, например, 13-пара-замещенные бензенидсульфонилгидразоновые аналоги даунорубицина, эпирубицина, идарубицина, аннамицина и карминомицина.
Для синтеза 13-пара-замещенных бензенидсульфонилгидразоновых антрациклинов проводят реакцию пара-замещенных бензенидсульфонилгидразинов с 13-кетоантрациклинами в спирте, после чего полученный раствор выдерживают при комнатной температуре в течение 5 дней. Пытаясь найти наиболее подходящий исходный материал для синтеза 13-деоксиантрациклинов, авторы изобретения обнаружили, что использование 13-бензенидсульфонилгидразондоксорубицина (соединение III) без заместителя в пара-положении бензольного кольца, неожиданно привело к решению проблем, связанных с использованием пара-замещенных бензенидсульфонилгидразоновых антрациклинов, таких как соединения I или II.
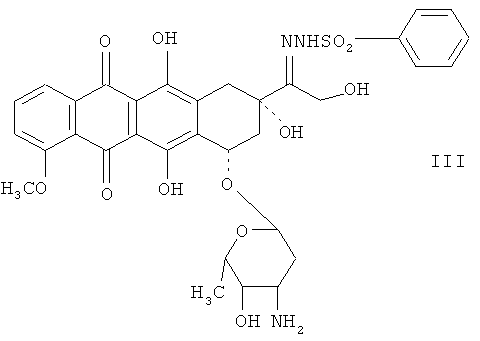
Кроме того, оказалось, что 13-бензенид- или пара-замещенные 13-бензенидсульфонилгидразоновые антрациклины можно синтезировать за 10-24 часа в метаноле при температуре приблизительно 35-60°С, предпочтительно приблизительно 40-45°С, причем выход и чистота продукта соответствуют тем, что получаются при проведении реакции при комнатной температуре в течение 5 дней. Далее показана реакция бензенидсульфонилгидразина с доксорубицином (IV), которая приводит к образованию соединения III и последующему его восстановлению в 13-деоксидоксорубицин (соединение V), как показано далее:
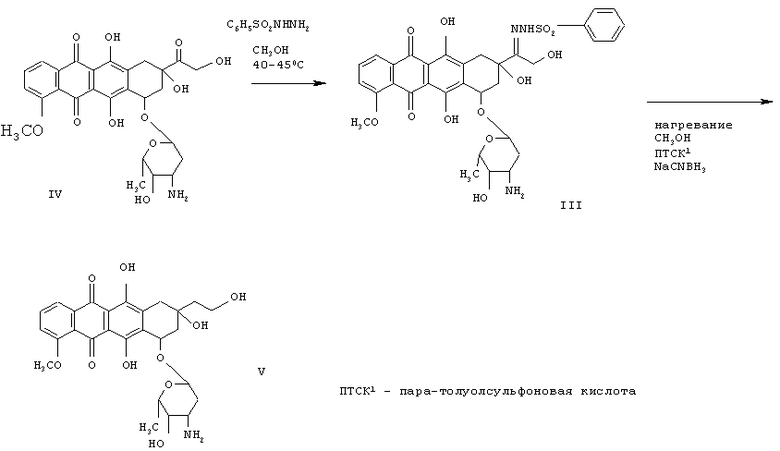
Для превращения 13-пара-замещенных бензенидсульфонилгидразоновых антрациклинов в 13-деоксиантрациклины первые смешивают с восстанавливающим агентом и сильной кислотой, часто это бывает цианоборогидрид и пара-толуолсульфоновая кислота в метаноле. Предположительно восстановление гидразона происходит посредством передачи ему атома водорода от кислоты и еще одного атома от цианоборогидрида. Однако кислота может нейтрализовать цианоборогидрид, поэтому для достижения оптимального выхода 13-деоксиантрациклинов необходимо учитывать концентрации реагентов и температуру реакции. Обычно реакционную смесь в метаноле нагревают до кипения и перемешивают при этой температуре. В конце реакции смесь нейтрализуют добавлением водного раствора основания, которое нейтрализует сильную кислоту и отщепляет от 13-го положения восстановленный гидразин, оставляя в этом положении метиленовую группу.
Введение в реакционную смесь водного раствора основания приводит также к выпадению соли, содержащей связанный с ней 13-деоксиантрациклиновый продукт. Такое связывание продукта с выпавшей в осадок солью приводит к необходимости сложных процедур выделения продукта, таких как экстракция кислотой и множественное разделение на силикагеле, а также хроматография ВЭЖХ. По наблюдениям авторов изобретения взбалтывание реакционной смеси при нагревании приводит к избыточной нейтрализации цианоборогидрида кислотой (пара-толуолсульфоновой) и затем к избыточному отщеплению сахарного остатка от антрациклина, что снижает общий выход продукта. Более того, авторами обнаружено, что смешивание реагентов и нагревание реакционной смеси без перемешивания или взбалтывания приводит к значительно более высоким выходам продукта. Оптимальная температура без перемешивания и взбалтывания составляет от 55 до 64°С. Авторы выяснили также, что 13-деоксиантрациклины можно легко выделить из выпавшей в осадок соли, профильтровав реакционную смесь после добавления водного раствора основания и промыв реакционную смесь такими органическими растворителями, как смесь хлороформа и метанола. Соответствующий процесс описан далее. Исходные вещества, такие как, например, соединение III и цианоборогидрид натрия, растворяют в сухом метаноле, после чего температуру смеси понижают до 0-4°С. пара-Толуолсульфоновую кислоту также растворяют в сухом метаноле, после чего добавляют к холодной реакционной смеси. Затем реакционную смесь нагревают до температуры от 55 до 64°С, предпочтительно от 59 до 60°С, и выдерживают при этой температуре в течение 1-4 часов, предпочтительно 2 часа, без перемешивания или взбалтывания. После этого смесь охлаждают, предпочтительно до 0°С или меньше, после чего к холодной смеси добавляют холодную (0-10°С) насыщенную бикарбонатом воду для нейтрализации кислоты и образования продукта, соединения V. В смеси воды и метанола соль выпадает в осадок, после чего эту смесь профильтровывают на воронке Бюхнера с вакуумной колбой.
На фильтровальной бумаге в воронке Бюхнера соли промывают смесью СНСl3:метанол (соотношение 3,5:1) под вакуумом, тем самым экстрагируя связавшийся с солью продукт, соединение V. Этот экстракт можно собрать в ту же самую вакуумную колбу со смесью метанола и насыщенного раствора бикарбоната в воде или в отдельную вакуумную колбу. В колбу со смесью метанола и насыщенного раствора бикарбоната в воде добавляют достаточное количество СНСl3, чтобы получить смесь хлороформа и метанола в соотношении, приблизительно 3,5:1. Продукт, соединение V, затем экстрагируют из раствора бикарбоната в смесь хлороформ/метанол. Органические экстракты отделяют от воды и упаривают досуха. Остаток, содержащий продукт, соединение V, затем растворяют в метаноле. Это соединение V можно очистить хроматографией (способы хорошо известны в органической химии) или можно осадить добавлением эфира.
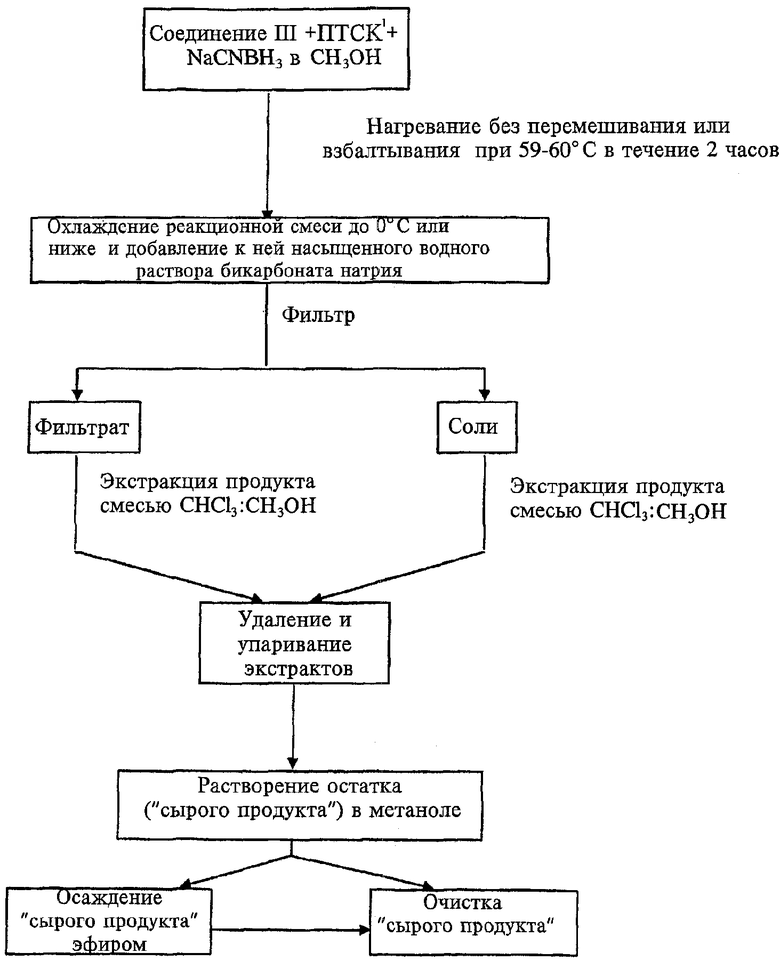
ПТСК1 - пара-толуолсульфоновая кислота
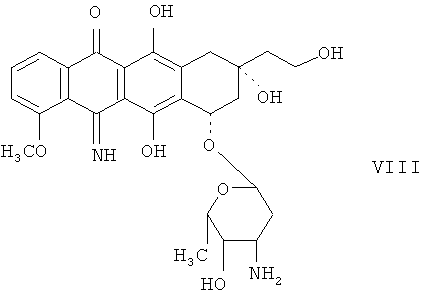
Как известно, 5-иминоантрациклины можно получить из 5-кетоантрациклинов в холодном метанольном растворе аммиака. Известно также, что аминогруппу сахарного остатка 13-кето-14-ОН-антрациклинов необходимо защищать. 5-Имино-аналоги 13-деоксиантрациклинов легко можно получить реакцией с холодным метанольным раствором аммиака, но авторы выяснили, что вводить защиту аминогруппы сахарного остатка не требуется. В соответствии с настоящим изобретением неочищенный 13-деоксиантрациклин можно растворить в метанольном растворе аммиака и выдерживать при температуре менее чем приблизительно 20°С, предпочтительно приблизительно от 0 до 4°С, до завершения реакции, на что обычно требуется от 1 до 5 дней. 5-Имино-аналоги 13-деоксиантрациклинов можно получать до или после образования соли 13-деокси-соединений и соляной кислоты, а также до или после очистки или осаждения. 5-Имино-13-деоксиантрациклины легко можно очистить хорошо известными хроматографическими способами. Ниже показан пример 5-имино-13-деоксиантрациклина, 5-имино-13-деоксидоксорубицина (VIII).
Пример 1. Получение 13-бензенидсульфонилгидразондоксорубицина гидрохлорида (III).
Синтез соединения III сравнивают с синтезом соединений I и II, а также с синтезом 13-пара-метоксибензенидсульфонилгидразондоксорубицина (VI) и 13-пара-нитробензенидсульфонилгидразондоксорубицина (VII).
375 мг соответствующего бензенид- или пара-замещенного бензенидсульфонилгидразина и 500 мг доксорубицина гидрохлорида (IV) растворяют в 15 мл безводного метанола. Растворы нагревают при 40-45°С в течение 16-20 часов, или оставляют при комнатной температуре (23-28°С) примерно на 5 дней, или выдерживают при 0-4°С в течение примерно 10 дней. После завершения реакции к метанольной реакционной смеси добавляют 100 мл диэтилового эфира, чтобы осадить продукты. Выпавшие осадки промывают диэтиловым эфиром, чтобы удалить метанол, и затем высушивают в эксикаторе под вакуумом. По данным ВЭЖХ продукты получают с чистотой 90% или более. Выходы в расчете на доксорубицина гидрохлорид (IV) приведены в таблице 1.

Соединение III было получено с относительно высокими выходами, независимо от температуры реакции, по сравнению с соединением II, которое постоянно характеризовалось относительно низкими выходами. Результаты демонстрируют, что синтез можно выполнять при температуре 40-45°C в течение более короткого времени, по сравнению с низкотемпературным синтезом, и при этом получать такие же высокие выходы.
Соединение III
Масс-спектр:
Спектр был снят на масс-спектрометре Aligent Ion Trap Mass Spectrophotometer (EN 824) (положительная ионизация ESI).
Структура:
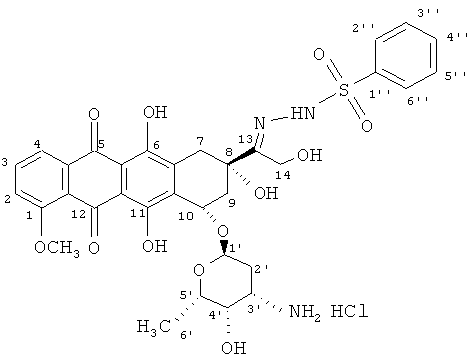
Формула: C33H36N3O12S
Молекулярный вес: 697,7 в виде свободного основания; 734,2 в виде гидрохлорида.
УФ-спектр снят на спектрофотометре Aligent Technologies 8453 UV/Vis (EN-246). Образцы приготовили в метаноле
1H ЯМР (300 MГц, DMSO-d6, δ)
Пример 2. Получение 13-деоксидоксорубицина гидрохлорида (V) из 13-бензенидсульфонилгидразондоксорубицина гидрохлорида (III).
Соединение V синтезируют из соединения III, причем соединение III синтезируют при 23-27°С (комнатная температура), 0-4°С (охлаждение) или 40-45°С (нагревание). Для сравнения соединение V также синтезируют из соединения II при аналогичных условиях. 100 мг соединения III (или соединения II) растворяют в 6 мл сухого метанола, содержащего 100 мг NaCNBH3. Реакционную смесь помещают в баню со льдом. 275 мг пара-толуолсульфоновой кислоты растворяют в 2 мл сухого метанола и затем добавляют к охлажденной реакционной смеси без перемешивания или встряхивания, общее количество метанола в смеси составляет 8 мл. Затем реакционную смесь нагревают при 59-60°С в течение 2 часов без перемешивания или встряхивания. Через 2 часа реакционную смесь помещают в холодильник и охлаждают до температуры 0°С или немного ниже. Затем к 8 мл охлажденной метанольной реакционной смеси добавляют 12 мл насыщенного раствора бикарбонарта натрия, охлажденного до температуры 1-4°С. Водно-метанольную смесь фильтруют через воронку Бюхнера, затем собирают фильтрат в колбу под вакуумом. Соли на фильтровальной бумаге в воронке Бюхнера промывают под вакуумом смесью 20-40 мл хлороформ:метанол в соотношении 3,5:1, чтобы проэкстрагировать продукт из солей в вакуумную колбу, уже содержащую фильтрат водно-метанольной смеси. При необходимости соли можно промывать, собирая фильтрат в отдельную вакуумную колбу. Затем к водно-метанольному фильтрату добавляют 28 мл хлороформа, чтобы создать соотношение хлороформ/метанол 3,5:1. Водно-метанольный фильтрат с добавленным хлороформом помещают в делительную воронку и продукт (соединение V) экстрагируют в смесь хлороформ:метанол. Когда водный и органический слои разделяются, органический слой удаляют и профильтровывают. Органический слой упаривают под вакуумом при 30°С или ниже. Остаток, содержащий продукт, растворяют в 2 мл метанола и помещают в баню со льдом. 0,2 мл 1 М раствора HCl в эфире добавляют к 1 мл сухого метанола и 1 мл диэтилового эфира. Полученную смесь затем добавляют к помещенному в баню со льдом растворенному остатку, получая гидрохлорид соединения V. 30 мл диэтилового эфира добавляют к охлажденному метанолу, чтобы осадить продукт, 13-деоксидоксорубицина гидрохлорид (V). Осадок промывают диэтиловым эфиром, чтобы удалить метанол, и затем высушивают в эксикаторе под вакуумом. Чистоту определяют методом ВЭЖХ. Выходы в расчете на доксорубицина гидрохлорид (IV) приведены в таблице 4.
Выход соединения V был выше, если в качестве исходного вещества взято соединение III, по сравнению с соединением II, независимо от того, при какой температуре синтезированы исходные вещества. Средний выход соединения V, синтезированного из трех разных видов соединения III (получены при разных температурах) составляет 47,3%±2,3 (среднеквадратическая ошибка). Это на 35% выше среднего выхода этого же соединения при его получении из трех разных видов соединения II, 35,0%±1,0 (среднекв. ошибка), р<0,05. Аналогичные эксперименты с соединением VII, синтезированным при высокой температуре, позволяют получить 34% выход соединения V. Эксперименты с соединениями I и VI показывают, что в осадке продукта V присутствует значительное количество этих соединений, снижая чистоту и выход.
Пример 3. Получение 5-имино-13-деоксидоксорубицина гидрохлорида (VIII) из 13-бензенидсульфонилгидразондоксорубицина гидрохлорида (III).
Соединение V синтезировано согласно процедуре, описанной в примере 1, исходя из 200 мг соединения III. "Сырой" продукт V получен с 56,7% выходом и чистотой 67,5%. 100 мг этого продукта растворяют в 2 мл сухого метанола, помещают в баню со льдом и добавляют 6 мл 2 М в метанольного раствора аммиака. Реакционную смесь выдерживают 4 дня при 0-4°С. После этого метанол упаривают в вакууме при температуре 30°С или меньше. Чтобы удалить следы аммиака, остаток растворяют в 15 мл смеси хлороформ:метанол в соотношении 4:1 и упаривают раствор. Процедуру повторяют дважды. Остаток растворяют в 4 мл сухого метанола и продукт, соединение VIII, осаждают добавлением 60 мл диэтилового эфира. Осадок промывают диэтиловым эфиром и высушивают под вакуумом в эксикаторе. Реакция завершается на 81%, чистота продукта составляет 67%, что дает выход 80%.
Хорошо известно, что для реакции восстановления 13-гидразонового антрациклина требуется сильная кислота в присутствии цианоборогидрида. Возможно, это происходит из-за того, что реакция идет при относительно низкой температуре (меньше 100°С), чтобы избежать разложения 13-деоксиантрациклинового продукта. Сильная кислота должна быть нейтрализована по окончании реакции путем добавления основания или отделена от 13-деоксиантрациклинового продукта путем добавления, например, галогенированного углеводорода в качестве растворителя, чтобы предотвратить разложение продукта. Очистка готового продукта сильно облегчается, если реакционную смесь высушить и затем очистить методом препаративной ВЭЖХ. Сильная кислота, видимо, мешает выделению продукта при хроматографии, что дает относительно низкие выходы. Любая попытка высушить реакционную смесь в конце реакции приводит к концентрированию кислоты и разрушению продукта.
В поисках слабой кислоты, которая позволила бы получить продукт со значительным выходом, но не требовала бы нейтрализации или выделения, авторы обнаружили пиридиниевую соль пара-толуолсульфоновой кислоты. Оказалось, что она чрезвычайно эффективна в этом отношении. В конце реакции реакционная смесь стабильна при комнатной температуре, и растворитель можно удалить с получением стабильного сухого остатка. Этот остаток можно сохранить на будущее, а можно растворить в подходящем растворителе с последующей очисткой методами препаративной ВЭЖХ. Реакцию выполняют, как описано выше, за исключением того, что она должна протекать предпочтительно при температуре приблизительно от 65 до 75°С в течение приблизительно 45 минут, а вместо пара-толуолсульфоновой кислоты используют пара-толуолсульфонат пиридиния в количестве приблизительно 66 мг на 100 мг гидразонового исходного вещества. Ранее была неизвестна возможность использования кислых солей пиридиния в реакции восстановления настоящего изобретения, приводящая к описанным выше преимуществам.
Пример 4. Получение 13-деоксидоксорубицина гидрохлорида (V) из 13-бензенидсульфонилгидразондоксорубицина гидрохлорида (III) с использованием пара-толуолсульфоната пиридиния вместо пара-толуолсульфоновой кислоты.
100 мг соединения III растворяют в 6 мл сухого метанола со 100 мг NаСNВН3. Реакционную смесь помещают в баню со льдом. 66 мг пара-толуолсульфоната пиридиния растворяют в 2 мл сухого метанола и добавляют к холодной реакционной смеси, общее количество метанола составляет 8 мл. Реакционную смесь в течение 45 минут выдерживают при температуре приблизительно 72°С. После этого смесь охлаждают до температуры ниже 30°С и добавляют 0,05 мл воды, вызвав гидролиз восстановленного гидразона с получением продукта, 13-деоксидоксорубицина гидрохлорида (V). По данным ВЭЖХ выход 13-деоксидоксорубицина гидрохлорида (V) составляет 55% по сравнению с доксорубицина гидрохлоридом (IV). Реакционную смесь можно очистить непосредственно методами препаративной ВЭЖХ. Метанол удаляют, а остаток растворяют в подходящей для хроматографии среде, реакционную смесь можно нейтрализовать и проэкстрагировать, как показано выше, а также, добавив аммиак, можно получить из нее 5-имино-13-деоксиантрациклиновые производные, как описано выше.
Предшествующее описание ограничено конкретными аспектами изобретения. Однако, очевидно, что компетентный специалист в данной области может внести в описанные аспекты различные изменения, получив некоторые или все описанные преимущества и не отступая от духа и области настоящего изобретения. Например, при экстракции из водно-метанольной смеси или при фильтровании солей можно использовать смеси галогенированного углеводорода и спирта в соотношении от 9:1 до 2:1. Помимо хлороформа, можно использовать и другие растворители - галогенированные углеводороды, например дихлорметан. Помимо метанола, можно использовать и другие спирты, такие как, например этанол. Помимо диэтилового эфира можно использовать и другие простые эфиры, например, третичный метилбутиловый эфир. Помимо пара-толуолсульфоновой кислоты, можно использовать и другие кислоты, например HCl или камфаросульфоновую кислоту. Помимо эфирного раствора HCl, можно использовать метанольный раствор соляной кислоты. Исходя из бензенид- или пара-замещенных бензенидсульфонилгидразоновых агликонов можно получить 13-деоксиагликоны, из которых добавлением сахара можно получить 13-деоксиантрациклины. Соли соляной кислоты могут быть образованы до или после синтеза 5-имино-13-деоксиантрациклинов. 13-Деокси- или 5-имино-13-деоксиантрациклины можно очистить хроматографически до или после получения солянокислой соли. Заместители бензольного кольца 13-бензенидсульфонилгидразоновых антрациклинов могут находиться в орто-, мета- или в пара-положении. Помимо пара-толуолсульфоната пиридиния можно использовать и другие соли пиридиния.
Следует понимать, что компетентный специалист может внести различные изменения в детали, вещества и сочетания частей, описанные и проиллюстрированные выше для разъяснения природы настоящего изобретения, не отступая от принципов и области изобретения, сформулированных в следующей ниже формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТРАЦИКЛИН ГЛИКОЗИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2081878C1 |
| ПРОИЗВОДНЫЕ 5-ИМИНО-13-ДЕЗОКСИ АНТРАЦИКЛИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ РАКА, АУТОИММУННЫХ ЗАБОЛЕВАНИЙ ИЛИ ИММУНОДЕФИЦИТНЫХ НАРУШЕНИЙ | 1999 |
|
RU2239640C2 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНА | 1995 |
|
RU2159619C2 |
| Способ получения хлоргидратов замещенных антрациклинов | 1979 |
|
SU867315A3 |
| ПРОИЗВОДНЫЕ 3'-АЗИРИДИНО-АНТРАЦИКЛИНА, СПОСОБЫ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2149163C1 |
| СПОСОБ АРАЛКИЛИРОВАНИЯ 4'-ГИДРОКСИЛЬНОЙ ГРУППЫ АНТРАЦИКЛИНОВ | 2008 |
|
RU2563453C2 |
| АНТРАЦИКЛИН ГЛИКОЗИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВЫМИ СВОЙСТВАМИ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2043360C1 |
| АНТРАЦИКЛИНОВЫЙ ГЛИКОЗИД И СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 1990 |
|
RU2073681C1 |
| Способ получения гликозидов антрациклина | 1980 |
|
SU993822A3 |
| КОНЪЮГАТЫ АНТРАЦИКЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 1992 |
|
RU2107690C1 |
Изобретение относится к производным антрациклинов общей структурной формулы, приведенной ниже, где R1, R2 и R3 представляют собой Н или ОН; R4 означает Н, ОН, С1-С5алкил, или O-(С1-С5)алкил; R5 соответствует О или NH; и R6 выбран из группы, включающей Н, ОН и остаток сахара, представляющий собой  Изобретение относится также к способу получения данных производных, включающему взаимодействие 13-кетоантрациклина или его кислой соли с бензенидсульфонилгидразидом в спиртовом растворителе при температуре приблизительно от 35 до 50°С в течение приблизительно 10-24 часов. Кроме того, изобретение относится к способу получения 13-деоксиантрациклинов, включающему получение спиртового раствора указанного выше 13-бензенидсульфонилгидразонового антрациклина; добавление к указанному раствору восстановителя и кислоты; нагревание указанного раствора без перемешивания, взбалтывания или дефлегмации с целью восстановления указанного 13-бензенидсульфонилгидразонового антрациклина; нейтрализацию указанного раствора водным раствором основания с получением указанного 13-деоксиантрациклинового производного и выпадением осадка; и, дополнительно, фильтрацию указанного осадка, экстракцию указанного 13-деоксиантрациклина из указанного осадка, а также экстракцию указанного 13-деоксиантрациклина из фильтрата. Изобретение относится также к способу получения 5-имино-13-деоксиантрациклинов, включающему получение 13-деоксиантрациклинов, как указано выше; растворение указанных 13-деоксиантрациклинов в спирте и превращение указанных 13-деоксиантрациклинов в соответствующие 5-имино-13-деоксиантрациклины под действием аммиака при температуре менее чем приблизительно, 20°С. Кроме того, изобретение относится к способу получения 13-деоксиантрациклинов, включающему получение спиртового раствора 13-бензенидсульфонилгидразонового антрациклина структурной формулы, приведенной ниже; добавление к указанному раствору восстановителя и кислой соли пиридиния и нагревание указанного раствора для восстановления указанного 13-бензенидсульфонилгидразонового антрациклина. А также изобретение относится к способу получения 5-имино-13-деоксиантрациклинов, включающему получение указанного 13-деоксиантрациклина и превращение его в соответствующий 5-имино-13-деоксиантрациклин под действием аммиака при температуре менее чем приблизительно 20°С. 6 н. и 15 з.п. ф-лы, 4 табл.
Изобретение относится также к способу получения данных производных, включающему взаимодействие 13-кетоантрациклина или его кислой соли с бензенидсульфонилгидразидом в спиртовом растворителе при температуре приблизительно от 35 до 50°С в течение приблизительно 10-24 часов. Кроме того, изобретение относится к способу получения 13-деоксиантрациклинов, включающему получение спиртового раствора указанного выше 13-бензенидсульфонилгидразонового антрациклина; добавление к указанному раствору восстановителя и кислоты; нагревание указанного раствора без перемешивания, взбалтывания или дефлегмации с целью восстановления указанного 13-бензенидсульфонилгидразонового антрациклина; нейтрализацию указанного раствора водным раствором основания с получением указанного 13-деоксиантрациклинового производного и выпадением осадка; и, дополнительно, фильтрацию указанного осадка, экстракцию указанного 13-деоксиантрациклина из указанного осадка, а также экстракцию указанного 13-деоксиантрациклина из фильтрата. Изобретение относится также к способу получения 5-имино-13-деоксиантрациклинов, включающему получение 13-деоксиантрациклинов, как указано выше; растворение указанных 13-деоксиантрациклинов в спирте и превращение указанных 13-деоксиантрациклинов в соответствующие 5-имино-13-деоксиантрациклины под действием аммиака при температуре менее чем приблизительно, 20°С. Кроме того, изобретение относится к способу получения 13-деоксиантрациклинов, включающему получение спиртового раствора 13-бензенидсульфонилгидразонового антрациклина структурной формулы, приведенной ниже; добавление к указанному раствору восстановителя и кислой соли пиридиния и нагревание указанного раствора для восстановления указанного 13-бензенидсульфонилгидразонового антрациклина. А также изобретение относится к способу получения 5-имино-13-деоксиантрациклинов, включающему получение указанного 13-деоксиантрациклина и превращение его в соответствующий 5-имино-13-деоксиантрациклин под действием аммиака при температуре менее чем приблизительно 20°С. 6 н. и 15 з.п. ф-лы, 4 табл.
1. Производные антрациклинов, охватываемые общей структурной формулой
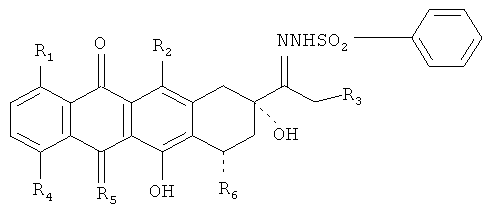
где R1, R2 и R3 представляют собой Н или ОН;
R4 означает Н, ОН, С1-С5алкил, или O-(С1-С5)алкил;
R5 соответствует О или NH и
R6 выбран из группы, включающей Н, ОН и остаток сахара, представляющий собой
2. Производные по п.1, выбранные из группы, включающей производные доксорубицина, даунорубицина, эпирубицина, идарубицина, аннамицина и карминомицина.
3. Способ получения производных по п.1, включающий взаимодействие 13-кетоантрациклина или его кислой соли с бензенидсульфонилгидразидом в спиртовом растворителе при температуре приблизительно от 35 до 50°С в течение приблизительно 10-24 ч.
4. Способ получения 13-деоксиантрациклинов, включающий получение спиртового раствора 13-бензенидсульфонилгидразонового антрациклина по п.1; добавление к указанному раствору восстановителя и кислоты; нагревание указанного раствора без перемешивания, взбалтывания или дефлегмации с целью восстановления указанного 13-бензенидсульфонилгидразонового антрациклина и нейтрализацию указанного раствора водным раствором основания с получением указанного 13-деоксиантрациклинового производного и выпадением осадка.
5. Способ по п.4, который дополнительно включает этап фильтрации указанного осадка, экстракцию указанного 13-деоксиантрациклина из указанного осадка, а также экстракцию указанного 13-деоксиантрациклина из фильтрата.
6. Способ по п.4, в котором указанное нагревание осуществляется при температуре приблизительно от 55 до 64°С, указанный восстановитель представляет собой цианоборогидрид, а указанная кислота паратолуолсульфоновая кислота.
7. Способ по п.6, в котором указанное нагревание осуществляется при температуре приблизительно от 59 до 60°С.
8. Способ по п.4, в котором указанный 13-бензенидсульфонилгидразоновый антрациклин представляет собой производное антрациклина, выбранное из группы, включающей доксорубицин, даунорубицин, эпирубицин, идарубицин, аннамицин и карминомицин.
9. Способ получения 5-имино-13-деоксиантрациклинов, который включает получение 13-деоксиантрациклинов по п.4; растворение указанных 13-деоксиантрациклинов в спирте и превращение указанных 13-деоксиантрациклинов в соответствующие 5-имино-13-деоксиантрациклины под действием аммиака при температуре менее приблизительно 20°С.
10. Способ по п.9, в котором этап превращения указанных 13-деоксиантрациклинов в соответствующие 5-имино-13-деоксиантрациклины под действием аммиака осуществляется при температуре приблизительно от 1 до 4°С в течение 1-4 дней.
11. Способ по п.9, в котором указанный 13-деоксиантрациклин выбран из группы, включающей 13-деоксиформы доксорубицина, даунорубицина, эпирубицина, идарубицина, аннамицина и карминомицина.
12. Способ по п.9, в котором указанный 13-деоксиантрациклин синтезируют из соответствующего 13-бензенидсульфонилгидразонового антрациклина.
13. Способ получения 13-деоксиантрациклинов, включающий получение спиртового раствора 13-бензенидсульфонилгидразонового антрациклина по п.1;
добавление к указанному раствору восстановителя и кислой соли пиридиния и нагревание указанного раствора для восстановления указанного 13-бензенидсульфонилгидразонового антрациклина.
14. Способ по п.13, в котором указанная кислая соль пиридиния представляет собой паратолуолсульфонат пиридиния, а указанный восстановитель представляет собой цианоборогидрид.
15. Способ по п.13, который дополнительно включает этап гидролиза указанного восстановленного 13-бензенидсульфонилгидразонового антрациклина с получением указанного 13-деоксиантрациклина.
16. Способ по п.13, в котором указанное нагревание осуществляется при температуре приблизительно от 65 до 75°С.
17. Способ по п.13, в котором указанный восстановленный 13-бензенидсульфонилгидразоновый антрациклин представляет собой производное антрациклина, выбранное из группы, включающей доксорубицин, даунорубицин, эпирубицин, идарубицин, аннамицин и карминомицин.
18. Способ получения 5-имино-13-деоксиантрациклинов, который включает получение 13-деоксиантрациклина по п.13 и превращение указанного 13-деоксиантрациклина в соответствующий 5-имино-13-деоксиантрациклин под действием аммиака при температуре менее чем приблизительно 20°С.
19. Способ по п.18, в котором этап преобразования указанного 13-деоксиантрациклина в соответствующий 5-имино-13-деоксиантрациклин под действием аммиака протекает при температуре приблизительно от 1 до 4°С в течение 1-4 дней.
20. Способ по п.18, в котором указанный 13-деоксиантрациклин выбран из группы, включающей доксорубицин, даунорубицин, эпирубицин, идарубицин, аннамицин и карминомицин.
21. Способ по п.18, в котором указанный 13-деоксиантрациклин синтезируют из соответствующего 13-бензенидсульфонилгидразонового антрациклина.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| ОПТИЧЕСКИЙ ГАЗОАНАЛИЗАТОР | 2002 |
|
RU2238540C2 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| EP 1600161 A2, 30.11.2005. | |||

