Область изобретения
Данное изобретение относится к энантиоселективному синтезу (+)-транс энантиомера пирролидинов, замещенных флавонами, представленых соединениями формулы 1 или их солями, которые являются ингибиторами циклин-зависимых киназ, и могут применяться для лечения пролиферативных нарушений, таких как рак.
Предпосылки изобретения
Циклин-зависимые киназы (Cdk) являются основными ферментами для контроля развития клеточного цикла. Ожидалось, что ингибиторы циклин-зависимых киназ обладают терапевтической применимостью относительно широкого ряда пролиферативных заболеваний, особенно рака. В результате этого CDK были нацелены на поиск лекарственного средства, и ряд небольших молекулярных ингибиторов CDK идентифицировали и изучали. Ингибиторы комплексов CDK/циклин, представленные следующей общей формулой 1:
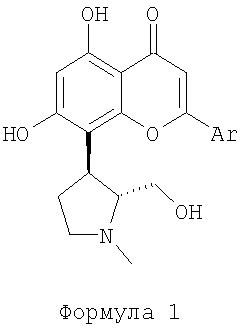
где Ar определено в подробном описании,
были описаны в РСТ заявке на патент № PCT/IB 2006/052002, которая включена в данное описание ссылкой. Эти соединения проявляют хорошую селективность и цитотоксичность к различным линиям пролиферативных клеток. Новые соединения, которые раскрыты в вышеупомянутой заявке на патент, имеют два хиральных центра и, следовательно, могут существовать в виде четырех энантиомеров, а именно (+)-транс, (-)-транс, (+)-цис и (-)-цис. Хиральность приобрела возрастающее значение для фармацевтической промышленности, подтверждаемое тем фактом, что более 80% лекарственных средств, разработанных до этого времени, обладают хиральными свойствами. Различные энантиомеры могут развивать совершенно разные эффекты в организме, так что только один из двух или более энантиомерных введенных форм могут быть эффективными. Наблюдалось, что в случае соединений формулы 1 активны только (+)-транс энантиомеры, тогда как (-)-транс энантиомеры не активны. Обширное исследование данными изобретателями эффективности рацемических соединений формулы 1 и их отдельных энантиомеров привело к РСТ заявке на патент № PCT/IB 2006/052002 данного заявителя. Введение активного (+)-транс энантиомера любого из соединений, представленных формулой 1, главным образом свободных от других изомеров, позволит существенно снизить дозу лекарственного средства. Из-за важности (+)-транс энантиомеров соединений, представленных формулой 1, как ингибиторов циклин-зависимых киназ, существует потребность разработки экономного и эффективного способа синтеза для их получения.
РСТ заявка на патент № PCT/IB 2006/052002 данного заявителя описывает способ получения (+)-транс энантиомера пирролидина, замещенного флавоном, представленного следующей формулой 1:
где Ar определено в детальном описании.
Способ, как описано в РСТ заявке на патент № РСТ/IB 2006/052002, включает расщепление промежуточного соединения и последующее превращение расщепленного промежуточного соединения в соединение, представленное формулой 1. Например, (+)-транс-2-(2-хлорфенил)-5,7-дигидрокси-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-хромен-4-он получили расщеплением промежуточного соединения, а именно (±)-транс-[1-метил-3-(2,4,6-триметокси-фенил)-пирролидин-2-ил]-метанола, и последующим превращением (-)-трансизомера промежуточного соединения в (+)-транс-2-(2-хлорфенил)-5,7-дигидрокси-8-(2-гидроксиметил-1 -метил-пирролидин-3-ил)-хромен-4-он. Получение (-)-транс-изомера промежуточного соединения включает этапы обработки его рацемата хиральным вспомогательным элементом для получения соответствующих (+)- и (-)-транс диастереомерных солей, последующее разделение необходимой диастереомерной соли кристаллизацией и обработку его основанием для получения желательного (-)-транс энантиомера. Этот способ расщепления включает существенную обработку, а также применение расщепляющего агента делает способ дорогостоящим. Частичная рециркуляция расщепляющего агента является выполнимой, но дорогостоящей, поскольку она требует дополнительной обработки, а также связана с образованием отходов. Нежелательный энантиомер не может быть рециркулирован и отбрасывается. Максимальный теоретический выход полученного ключевого промежуточного соединения составляет всего лишь 50% при синтезе в лабораторных масштабах из-за потери половины рацемата. Этот выход может дополнительно снижаться из-за потребности в высокой хиральной чистоте (>95% энантиомерного избытка). Таким образом, существует четкая потребность в развитии альтернативного асимметричного синтеза, который обеспечил бы необходимый (+)-транс энантиомер эффективным и более специфичным способом.
Целью данного изобретения является обеспечение альтернативного способа получения (+)-транс энантиомера соединений, представленных формулой 1, который представляет собой энантиоселективный способ. Способ по данному изобретению позволяет эффективный крупномасштабный синтез путем преодоления недостатков традиционных методик расщепления.
Краткое описание изобретения
Данное изобретение обеспечивает новый способ энантиоселективного синтеза (+)-транс энантиомера соединения, представленного формулой 1:
где Ar определено в подробном описании.
Способ по данному изобретению также включает энантиоселективный синтез соединения следующей формулы А, которое является хиральным предшественником соединения формулы 1:
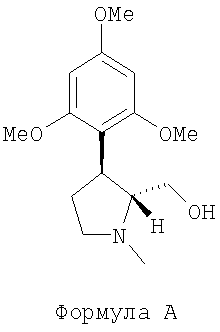 .
.
Способ по данному изобретению обеспечивает энантиоселективный синтез (+)-транс энантиомеров соединений формулы 1, который избегает недостатки вышеупомянутого способа.
Способ по данному изобретению также обладает дополнительным преимуществом с точки зрения стоимости и времени, поскольку все промежуточные соединения в способе кристаллические и не требуют дополнительной очистки.
Детальное описание изобретения
Данное изобретение конкретно направлено на способ для энантиоселективного синтеза (+)-транс энантиомера соединения, представленного формулой 1:
где Ar представляет собой фенил, который не замещен или замещен 1, 2 или 3 идентичными или разными заместителями, выбранными из: галогена, нитро, циано, C1-C4-алкила, фторметила, дифторметила, трифторметила, гидроксила, C1-C4-алкокси, карбокси, C1-C4-алкоксикарбонила, C1-C4-алкиленгидроксила, CONH2, CONR1R2, SO2NR1R2, циклоалкила, NR1R2 и SR3,
где R1 и R2 каждый независимо выбран из: водорода, C1-C4-алкила, C1-C4-алкилкарбонила и арила или R1 и R2, вместе с атомом азота, с которым они связаны, образуют 5- или 6-членное кольцо, которое может необязательно содержать, по меньшей мере, один дополнительный гетероатом; и
R3 выбран из водорода, C1-C4-алкила, арила и SR4, где R4 представляет собой C1-C4-алкил или арил.
С целью раскрытия перечисленное ниже является определениями различных выражений, применяемых для описания соединений по данному изобретению. Эти определения относятся к выражениям, поскольку они применяются по всему описанию (если они в других случаях не ограничены определенными значениями) либо индивидуально, либо как часть большей группы. Они не должны интерпретироваться в буквальном смысле. Они не являются основными определениями и релевантны только для данной заявки.
Выражение "алкил" относится к радикалу насыщенных алифатических групп, включая алкильные группы с прямой цепью или алкильные группы с разветвленной цепью. Более того, если не указано другое, выражение "алкил" включает незамещенные алкильные группы, так же как и алкильные группы, которые замещены одним или более различными заместителями. Примерами алкильных остатков, содержащих от 1 до 20 атомов углерода, являются: метил, этил, пропил, бутил, пентил, гексил, гептил, октил, нонил, децил, ундецил, додецил, тетрадецил, гексадецил, октадецил и эйкозил, n-изомеры всех этих остатков, изопропил, изобутил, 1-метилбутил, изопентил, неопентил, 2,2-диметилбутил, 2-метилпентил, 3-метилпентил, изогексил, 2,3,4-триметилгексил, изодецил, sec-бутил или t-бутил.
Выражение "циклоалкил" относится к неароматической моно- или полициклической кольцевой системе приблизительно с 3-7 атомами углерода, которые могут быть не замещены или замещены одним или более различными заместителями. Примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил и подобное.
Выражение "алкокси", как применяется в данном описании, относится к алкильной группе, как определено выше, имеющей кислородный радикал, присоединенный к ней. Характерные алкоксильные группы включают метокси, этокси, пропокси, t-бутокси и подобное.
Выражение "галоген" относится к хлору, брому, фтору и йоду.
Выражение "гетероатом" относится к азоту, кислороду, сере и фосфору.
Выражение "энантиомерный избыток" относится к разнице между количеством одного энантиомера и количеством другого энантиомера, которые находятся в смеси продукта. Таким образом, например, энантиомерный избыток 96% относится к смеси продукта, имеющей 98% одного энантиомера и 2% другого энантиомера.
Там, где стереохимия изображена в структурах, она представлена относительно, а не в абсолютной конфигурации.
В одном варианте осуществления данного изобретения обеспечили способ энантиоселективного синтеза соединения (-)-транс-(1-метил-3-(2,4,6-триметоксифенил)пирролидин-2-ил)метанола, представленного следующей формулой А:
(далее называется соединение А), или его фармацевтически приемлемой соли, причем способ включает этапы, на которых:
(а) проводят стереоспецифическую реакцию Майкла диметилмалоната с (Е)-метил-2-нитро-3-(2,4,6-триметоксифенил)акрилатом в растворителе в присутствии каталитического комплекса, щелочи и молекулярного сита, где каталитический комплекс включает хиральный лиганд бис(оксазолина) и комплексное соединение металла, для получения (+)-триметил 3-нитро-2-(2,4,6-триметоксифенил) пропан-1,1,3-трикарбоксилата, представленного следующей формулой В:
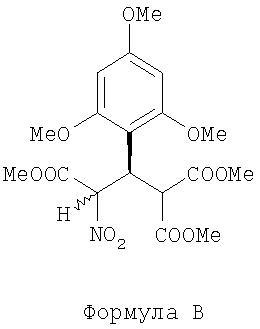
(далее называется соединение В);
(b) обрабатывают соединение В, полученное на этапе (а), восстанавливающим агентом в приемлемом растворителе для получения (+)-диметил 5-оксо-3-(2,4,6-триметоксифенил)-пирролидин-2,4-дикарбоксилата, представленного следующей формулой С:
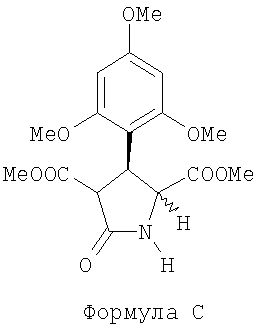 ,
,
(далее называется соединение С);
(с) обрабатывают соединение С хлоридом натрия в растворителе и нагревают полученную реакционную смесь до температуры в диапазоне 120-170°С для получения (+)-метил-5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилата как смеси цис- и транс-изомеров, представленного следующей формулой D:
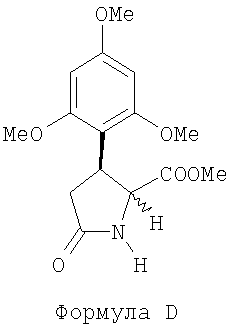 ,
,
(далее называется соединение D);
(d) подвергают реакции соединение D с метилирующим агентом и основанием, выбранным из: гидрида щелочного металла и карбоната щелочного металла, в растворителе, затем подвергают полученную смесь цис- и транс-соединений щелочному гидролизу с гидроксидом щелочного металла в спирте с нагреванием до температуры в диапазоне 50-100°С для получения (-)-транс-1-метил-5-оксо-3-(2,4,6-триметоксифенил)-пирролидин-2-карбоновой кислоты, представленной следующей формулой Е:
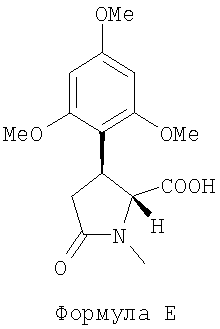
(далее называется соединение Е) как отдельный транс-изомер;
(е) обрабатывают соединение Е восстанавливающим агентом в растворителе для получения желательного (-)-транс-(1-метил-3-(2,4,6-триметоксифенил)пирролидин-2-ил)-метанола, представленного формулой А.
В одном варианте осуществления данное изобретение обеспечивает применение соединения А, как получено новым описанным способом, для получения соединения, представленного формулой 1.
По другому варианту осуществления данного изобретения обеспечен способ получения (+)-транс энантиомера соединения, представленного формулой 1:
где Ar представляет собой фенил, который не замещен или замещен 1, 2 или 3 идентичными или разными заместителями, выбранными из: галогена, нитро, циано, C1-C4-алкила, фторметила, дифторметила, трифторметила, гидроксила, C1-C4-алкокси, карбокси, C1-C4-алкоксикарбонила, C1-C4-алкиленгидроксила, CONH2, CONR1R2, SO2NR1R2, циклоалкила, NR1R2 и SR3,
где R1 и R2 каждый независимо выбран из: водорода, C1-C4-алкила, C1-C4-алкилкарбонила и арила, или R1 и R2, вместе с атомом азота, с которым они связаны, формируют 5- или 6-членное кольцо, которое может необязательно содержать, по меньшей мере, один дополнительный гетероатом; и
R3 выбран из водорода, C1-C4-алкила, арила и SR4, где R4 представляет собой C1-C4-алкил или арил,
или его фармацевтически приемлемой соли,
причем способ включает этапы, на которых:
(i) обрабатывают соединение А (выше) уксусным ангидридом в присутствии катализатора для получения (-)-транс-уксусной кислоты 3-(3-ацетил-2-гидрокси-4,6-диметокси-фенил)-1-метил-пирролидин-2-ил метилового эфира, представленного следующей формулой F:
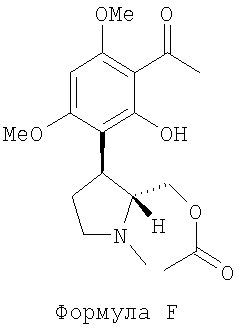
(далее называется соединение F);
(ii) обрабатывают соединение F водным раствором щелочи и повышают температуру реакционной смеси до приблизительно 50°С для получения (-)-транс-1-[2-гидрокси-3-(2-гидроксиметил-1-метил-пирролидин-3-ил)-4,6-диметокси-фенил)-этанона, представленного следующей формулой G:
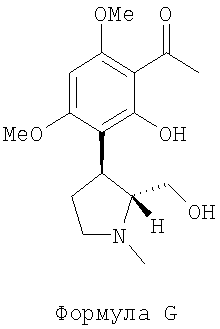
(далее называется соединение G);
(iii) подвергают реакции соединение G со сложным эфиром формулы ArCOOCH3 (где Ar как определено в формуле 1) в присутствии основания и приемлемого растворителя в атмосфере азота, с последующей циклизацией, катализированной кислотой, для получения диметокси соединения, представленного следующей формулой 2:
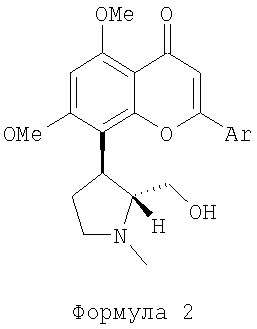
(далее называется соединение 2);
(iv) подвергают соединение 2 деметилированию нагреванием его с деметилирующим агентом при температуре в диапазоне 120-180°С для получения необходимого (+)-транс энантиомера соединения, представленного формулой 1.
В наиболее предпочтительном варианте осуществления данное изобретение обеспечивает способ энантиоселективного синтеза (+)-транс-2-(2-хлор-фенил)-5,7-дигидрокси-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-хромен-4-она, представленного формулой 1А ниже, где в соединениях общей формулы 1 Ar группа представляет собой фенил, замещенный хлором:
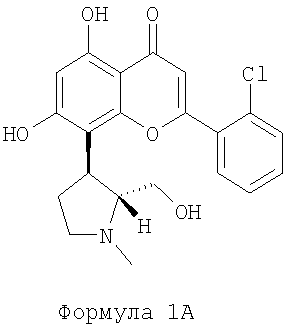
(далее называется соединение 1А), причем способ включает этапы, на которых:
(i) обрабатывают соединение А уксусным ангидридом в присутствии катализатора для получения (-)-транс-уксусной кислоты 3-(3-ацетил-2-гидрокси-4,6-диметокси-фенил)-1-метил-пирролидин-2-ил метилового эфира, представленного следующей формулой F:
(далее называется соединение F);
(ii) обрабатывают соединение F водным раствором щелочи и повышают температуру реакционной смеси до приблизительно 50°С для получения (-)-транс-1-[2-гидрокси-3-(2-гидроксиметил-1 -метил-пирролидин-3-ил)-4,6-диметокси-фенил)-этанона, представленного следующей формулой G:
(далее называется соединение G);
(iii) подвергают реакции соединение G с метил 2-хлорбензоатом в присутствии основания и приемлемого растворителя в атмосфере азота с последующей циклизацией, катализированной кислотой, для получения (+)-транс-2-(2-хлорфенил)-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-5,7-диметокси-хромен-4-она, представленного следующей формулой 2А:

(далее называется соединение 2А);
(iv) подвергают соединение 2А деметилированию нагреванием его с пиридингидрохлоридом при температуре в диапазоне 120-180°С для получения соединения 1А и
(v) необязательно, преобразовывают соединение 1А в его фармацевтически приемлемую соль, например, его гидрохлоридную соль, (+)-транс-2-(2-хлорфенил)-5,7-дигидрокси-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-хромен-4-он гидрохлорид, обычными средствами.
Соединение (Е)-метил-2-нитро-3-(2,4,6-триметоксифенил)акрилат, применяемое на этапе (а), можно получить реакцией между 2,4,6-триметоксибензальдегидом и метилнитроацетатом в присутствии ацетата аммония и сульфата магния. Соединение 2,4,6-триметоксибензальдегид можно получить обычными способами из 2,4,6-триметоксибензола реакцией с фосфорилхлоридом и N,N-диметилформамидом. Соединение метилнитроацетата можно получить из нитрометана традиционными способами, например, нагреванием нитрометана с основанием, например, гидроксида калия, при 160°С с последующей обработкой при 15°С серной кислотой и метанолом.
Каталитический комплекс, применяемый на этапе (а) выше, включает хиральный лиганд бис(оксазолина) и комплексное соединение металла. Применение хиральных лиганд бис(оксазолина) в каталитическом асимметрическом синтезе подробно рассматривалось (Ghosh, А.К.; Mathivanan, P.; Cappiello, J. Tetrahedron: Asymmetry 1998, 9, 1-45). По данному изобретению предпочтительным хиральным лигандом бис(оксазолина) является (3aS,3a'S,8aR,8a'R)-2,2'(циклопропан-1,1-диил)бис(8,8а-дигидро-3аН-индено[1,2d]оксазол), который можно получить по способу, который рассматривался в J. Am. Chem. Soc. 2002, 124(44), 13097-13105, включенному в данное описание ссылкой. Реакцию можно проводить с помощью только 4-6 мол. % хирального лиганда бис(оксазолина).
Комплексные соединения металла, приемлемые для обеспечения каталитического комплекса, включают трифторметансульфонат магния, перхлорат магния, трифторметансульфонат меди, трифторметансульфонат цинка, трифторметансульфонат лантана, трифторметансульфонат никеля, бромид магния, бромид меди, бромид цинка, бромид никеля, йодид магния, йодид меди, йодид цинка, йодид никеля, ацетилацетонат магния, ацетилацетонат меди, ацетилацетонат цинка и ацетилацетонат никеля. По данному изобретению предпочтительным комплексным соединением металла является трифторметансульфонат магния.
Основание, применяемое на этапе (а), может быть выбрано из: триэтиламина, диизопропиламина, 2,6-лутидина, N-метилморфолина, N-этилпиперидина, имидазола и 5,6-диметилбензимидазола. Предпочтительно, N-метилморфолин применяют как основание.
Восстанавливающим агентом, применяемым на этапе (b), может быть хлорид олова или никель Ренея. Когда хлорид олова применяют как восстанавливающий агент, соединение С получают как отдельный изомер. Когда никель Ренея применяют как восстанавливающий агент, соединение С получают как смесь изомеров, как определено с помощью 1Н NMR (ядерно-магнитный резонанс). Если небольшой образец смеси изомеров очистят колоночной хроматографией для отделения изомеров, можно подтвердить, что один из изомеров является идентичным отдельному изомеру, который получен с помощью хлорида олова как восстанавливающего агента. Растворителем, применяемым на этапе (b), предпочтительно является апротонный растворитель, такой как этилацетат, диоксан, N,N-диметилформамид и тетрагидрофуран. Когда восстановление проводят с хлоридом олова, применяемым растворителем, предпочтительно является этилацетат, а когда восстановление проводят с никелем Ренея, применяемый растворитель предпочтительно выбран из: тетрагидрофурана, диоксана и N,N-диметилформамида.
Применяемым растворителем на этапе декарбоксилирования (с) предпочтительно является полярный апротонный растворитель, такой как N-метилпирролидон и диметилсульфоксид.
Метилирующим агентом, применяемым на этапе (d), может быть метилйодид или диметилсульфат. Растворителем, применяемым на этапе (d), предпочтительно является полярный апротонный растворитель, который может быть выбран из: N,N-диметилформамида, тетрагидрофурана и диоксана. Карбонатом щелочного металла может быть карбонат натрия или карбонат калия. Гидридом щелочного металла может быть гидрид натрия. Гидроксидом щелочного металла может быть гидроксид натрия или гидроксид калия. Применяемым спиртом предпочтительно является ациклический спирт. Более предпочтительно, спирт выбран из: этанола, метанола и изопропанола.
Восстанавливающим агентом, применяемым на этапе (е), предпочтительно является гидрид, более предпочтительно гидрид, выбранный из: литийалюминиевого гидрида, диизобутилалюминиевого гидрида и боргидрида натрия. Растворителем, применяемым на этапе восстановления, предпочтительно является эфир. Более предпочтительно растворитель выбран из: тетрагидрофурана, диоксана и диэтилового эфира.
В способе получения соединений формулы 1 из промежуточных соединений формулы А катализатор, применяемый на этапе (i), может быть выбран из кислоты Льюиса и полифосфорной кислоты. Катализатор кислоты Льюиса может быть выбран из хлорида цинка, хлорида алюминия, трифторида бора и трибромида бора. Самым предпочтительным катализатором кислоты Льюиса является трифторид бора.
Щелочами, применяемыми на этапе (ii), могут быть гидроксид натрия или гидроксид калия.
Основание, применяемое на этапе (iii), может быть выбрано из: гидрида натрия, n-бутиллития, гексаметилдисилазиды лития и диизопропиламида лития. Применяемым основанием предпочтительно является гидрид натрия. Растворитель, применяемый на этапе (iii), может быть выбран из: тетрагидрофурана, N,N-диметилформамида и диоксана. Применяемым растворителем предпочтительно является N,N-диметилформамид.
Деметилирующий агент, применяемый на этапе (iv), может быть выбран из пиридингидрохлорида, трибромида бора, эфирата трифторида бора и трихлорида алюминия. Предпочтительным деметилирующим агентом является пиридингидрохлорид.
Таким образом, по способу данного изобретения соединение формулы А получено с хиральной чистотой более 97% ее (энантиомерный избыток) с доведением до соединений формулы 1 с хиральной чистотой более 99% ее.
Соединения формулы 1, полученные новым способом по данному изобретению, могут быть необязательно преобразованы в соответствующие фармацевтически или токсикологически приемлемые их соли, в частности, в фармацевтически применяемые их соли.
Соединения формулы 1, которые содержат одну или более основных групп, например, групп, которые могут быть протонированы, могут применяться по данному изобретению в форме их аддитивных солей с нетоксическими неорганическими или органическими кислотами. Примеры приемлемых неорганических кислот включают: борную кислоту, хлорную кислоту, соляную кислоту, бромистоводородную кислоту, серную кислоту, сульфамовую кислоту, фосфорную кислоту, азотную кислоту и другие неорганические кислоты, известные специалистам данной области. Примеры приемлемых органических кислот включают: уксусную кислоту, глюконовую кислоту, пропионовую кислоту, янтарную кислоту, гликолевую кислоту, стеариновую кислоту, молочную кислоту, яблочную кислоту, винно-каменную кислоту, лимонную кислоту, аскорбиновую кислоту, памовую кислоту, малеиновую кислоту, гидроксималеиновую кислоту, фенилуксусную кислоту, глутаминовую кислоту, бензойную кислоту, салициловую кислоту, сульфаниловую кислоту, 2-ацетоксибензойную кислоту, фумаровую кислоту, толуолсульфоновую кислоту, метансульфоновую кислоту, этандисульфоновую кислоту, щавелевую кислоту, изетионовую кислоту, кетоглутаровую кислоту, бензолсульфоновую кислоту, глицерофосфорную кислоту и другие органические кислоты, известные специалистам данной области. Соединения формулы 1, которые содержат кислотные группы, могут применяться по данному изобретению, например, как соли щелочных металлов, типа соли Li, Na и К. Фармацевтически приемлемые соли по данному изобретению могут быть синтезированы из испытуемого соединения, которое содержит основную и кислотную части, традиционными химическими способами. Как правило, соли получают при контакте свободного основания или кислоты со стехиометрическими количествами или с избытком необходимой солеобразующей неорганической или органической кислоты или основания в приемлемом растворителе или дисперганте, или анионным обменом, или катионным обменом с другими солями. Пригодными растворителями являются, например, этилацетат, эфир, спирты, ацетон, тетрагидрофуран, диоксан или смеси этих растворителей.
Понятно, что модификации в условиях реакции, которые не влияют на хиральность различных вариантов осуществления данного изобретения, включены в раскрытое здесь изобретение. Следовательно, следующие примеры предназначены иллюстрировать, но не ограничивать данное изобретение.
Примеры
Пример 1:
(Е)-Метил-2-нитро-3-(2,4,6-триметоксифенил)акрилат
2,4,6-триметоксибензальдегид (20,75 г, 0,105 моль) растворили в дихлорметане (300 мл) и к этому раствору добавили сульфат магния (15 г, 0,124 моль), ацетат аммония (10 г, 0,129 моль) и метилнитроацетат (12,60 г, 0,105 моль) и перемешивали при комнатной температуре на протяжении 2 часов. Через два часа воду (300 мл) добавили к реакционной массе, органический слой отделили и водный слой экстрагировали дихлорметаном (2×100 мл). Органические слои объединили и концентрировали под пониженным давлением для получения твердого вещества, которое кристаллизовали из метанола (100 мл).
Выход: 22 г (66,82%)
1H NMR (CDCl3): δ 8,37 (s, 1H), 6,08 (s, 2H), 3,86 (s, 3H), 3,84 (s, 3H), 3,82 (s, 6H).
MS (ES+): 298 (M+1)
Пример 2:
(+)-Триметил 3-нитро-2-(2,4,6-триметоксифенил)пропан-1,1,3-трикарбоксилат
В двугорлую круглодонную колбу на 500 мл, которую держивали под азотом, добавили хлороформ (10 мл), трифталат магния (0,161 г, 0,5 ммоль) и воду (0,036 мл, 2,0 ммоль). К этому перемешанному раствору добавили (3aS,3a'S,8aR,8a'R)-2,2'(циклопропан-1,1-диил)бис(8,8а-дигидро-3аН-индено[1,2d]оксазол) (бис(оксазолин)) (0,196 г, 0,55 ммоль) и реакционную смесь перемешивали на протяжении 1 часа. Через 1 час добавили хлороформ (30 мл) и молекулярные сита (2 г) и смесь перемешивали на протяжении еще 90 минут. Добавили (Е)-метил-2-нитро-3-(2,4,6-триметоксифенил)акрилат (3,1 г, 0,01 моль), диметилмалонат (1,92 г, 0,014 моль) и N-метилморфолин (0,06 г, 0,6 ммоль) и реакционную смесь перемешивали на протяжении 12 часов, после чего нагревали при 40°С на протяжении 4 часов. Петролейный эфир (15 мл) добавили в реакционную смесь, перемешивали на протяжении 10 минут и смесь отфильтровали. Молекулярные сита промыли метил-t-бутиловым эфиром и объединенный органический слой промыли 5% фосфорной кислотой (10 мл) и рассолом (15 мл). Органический слой концентрировали под пониженным давлением для получения масла. Масло растворили в метаноле (10 мл), охладили и отфильтровали для получения белого кристаллического твердого вещества.
Выход: 2,9 г (67,82%).
1H NMR (CDCl3): δ (6,05 (br.s, 1H), 6,03 (br.s, 1H), 6,0 (d, 1H, 12,0 Hz), 5,24 (dd, 1H, 9,0 Hz, 12,0 Hz), 4,26 (d, 1H, 9,0 Hz), 3,83 (s, 6H), 3,77 (s, 3H), 3,76 (s, 3H), 3,72 (s, 3H), 3,4 (s, 3H).
MS (ES+): 430 (M+1).
Пример 3:
(+)-Диметил 5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2,4-дикарбоксилат
Способ 1
(+)-Триметил 3-нитро-2-(2,4,6-триметоксифенил)пропан-1,1,3-трикарбоксилат (7,8 г, 0,018 моль) растворили в этилацетате (100 мл). К этому раствору добавляли в течение 10 минут при перемешивании дигидрат хлорида олова (25 г, 0,118 моль) частями. Реакционную смесь нагревали до 55°С на протяжении 2 часов. Смесь охладили до 10°С, подщелочили 10% раствором гидроксида натрия до рН 9, отфильтровали через целитную прокладку, а прокладку промыли этилацетатом (50 мл). Водный слой экстрагировали этилацетатом (2×100 мл). Органические слои объединили, высушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения названного соединения как белого твердого вещества.
Выход: 4,5 г (67,44%).
1H NMR (CDCl3): δ 6,06 (br.s, 2Н), 6,00 (br.s, 1Н), 4,98 (dd, 1H), 4,59 (d, 1H), 3,96 (d, 1H), 3,79 (s, 3H), 3,76 (s, 9H), 3,35 (s, 3H).
MS (ES+): 368 (M+1).
Способ 2
В реактор высокого давления емкостью 1 л добавили тетрагидрофуран (100 мл) и никель Ренея (20 г), после чего добавили раствор (+)-триметил 3-нитро-2-(2,4,6-триметоксифенил)пропан-1,1,3-трикарбоксилата (32 г, 0,074 моль) в тетрагидрофуране (300 мл). При перемешивании реактор продули трижды азотом, после этого водородом. Реакционную смесь перемешивали всю ночь под давлением водорода 80 psi. В конце реакции никель Ренея отфильтровали и промыли тетрагидрофураном (150 мл) в азоте. Органический слой концентрировали под пониженным давлением для получения белого твердого вещества. 1H NMR выявил присутствие смеси изомеров. Смесь цис- и транс-изомеров получили в выходе 25 г (91,32%). Небольшую часть реакционной смеси очистили колоночной хроматографией, применяя 5% метанол в хлороформе как элюирующий агент для отделения изомеров, и обнаружили, что один из отделенных изомеров являлся идентичным изомеру, полученному при восстановлении хлоридом олова, что подтверждено 1Н NMR, масс-спектрами и ВЭЖХ (высокоэффективная жидкостная хроматография).
1H NMR (CDCl3): δ 6,06 (br.s, 2H), 6,00(br.s, 1H), 4,98 (dd, 1H), 4,59 (d, 1H), 3,96 (d, 1H), 3,79 (s, 3H), 3,76 (s, 9H), 3,35 (s, 3H).
MS (ES+): 368 (M+1)
Пример 4:
(+)-Метил 5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилат
(+)-Диметил 5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2,4-дикарбоксилата (4,0 г, 0,0109 моль) растворили в N-метилпирролидоне (15 мл). Добавили хлорид натрия (0,631 г, 0,0109 моль) и воду (0,196 мл, 0,0109 моль) и реакционную смесь нагревали до 170°С на протяжении 5 часов. Реакционную смесь вылили на лед (50 г), а твердое вещество отфильтровали и высушили.
Выход: 1,5 г (44,5%).
Продукт являлся смесью цис- и транс-изомеров, как видно из 1H NMR. Смесь изомеров применяли без разделения для следующей реакции. Небольшое количество смеси очистили колоночной хроматографией (5% метанол в хлороформе) для спектральной характеристики цис- и транс-изомеров.
(+)-цис-Метил 5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилат
1Н NMR (CDCl3): δ 6,08 (s, 2H), 5,89 (br.s, 1H), 4,62 (m, 1H), 4,48 (d, 1H, 9,6Hz), 3,79 (s, 3H), 3,76 (s, 6H), 3,34 (s, 3H), 2,74 (dd, 1H), 2,60 (dd, 1H).
MS (ES+): 310 (M+1).
(+)-транс-Метил 5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилат
1H NMR (CDCl3): δ 6,15 (s, 2H), 5,87 (br.s, 1H), 4,42 (d, 1H, 7,5Hz), 4,26 (m, 1H), 3,82 (s, 3H), 3,81 (s, 6H), 3,68 (s, 3H), 2,76 (dd, 1H), 2,53 (dd, 1H).
MS (ES+): 310 (M+1).
Пример 5:
(+)-Метил-1-метил-5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилат
(+)-Метил-5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилат (1,7 г, 0,0055 моль) растворили в N,N-диметилформамиде (15 мл) и раствор охладили 0°С. Гидрид натрия (0,134 г, 0,0056 ммоль) добавляли частями на протяжении 10 минут и перемешивали на протяжении еще 20 минут при 0°С. Метилйодид (0,514 мл, 0,0082 моль) добавили по каплям, а реакционную смесь нагрели до комнатной температуры за 1 час. Реакционную смесь медленно вылили на смесь дробленого льда (20 г) и 1:1 раствора соляной кислоты (5 мл). Смесь экстрагировали этилацетатом (2×50 мл), промыли рассолом, высушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения масла. Масло перетерли в порошок с петролейным эфиром, а полученное твердое вещество отфильтровали.
Выход: 1,7 г (96,04%).
Продукт являлся смесью цис- и транс-изомеров, как видно из 1H NMR. Смесь изомеров применяли без разделения для следующей реакции. Небольшое количество смеси очистили колоночной хроматографией (5% метанол в хлороформе) для спектральной характеристики цис- и транс-изомеров.
(+)-цис-метил 1-метил-5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилат
1Н NMR (CDCl3): δ 6,07 (s, 2H), 4,44 (dd, 1H), 4,27 (d, 1H, 9,6Hz), 3,79 (s, 3H), 3,74 (s, 6H), 3,38 (s, 3H), 3,20 (dd, 1H), 2,90 (s, 3H), 2,45 (dd, 1H) MS (ES+): 324 (M+1).
(+)-транс-Метил-1-метил-5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилат
1H NMR (CDCl3): δ 6,12 (s, 2H), 4,13 (d, 1H, 6,3Hz), 4,05 (dd, 1H), 3,80 (s, 3H), 3,76 (s, 6H), 3,70 (s, 3H), 2,88 (s, 3H), 2,64 (m, 2H).
MS (ES+): 324 (M+1)
Пример 6:
(-)-транс-1-Метил-5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоновая кислота
Смесь цис- и транс-изомеров метил- 1-метил-5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилата (1,6 г, 0,0049 моль) растворили в метаноле (15 мл). К ней добавили раствор гидроксида калия (0,96 г, 0,017 моль) в воде (4 мл) и реакционную смесь нагревали при 65°С на протяжении 3 часов. Метанол удалили под пониженным давлением, добавили 15 мл воды и смесь подкислили 1:1 раствором соляной кислоты до рН 2. Полученное твердое вещество отфильтровали, промыли водой и высушили.
Выход: 0,94 г (61,44%).
1Н NMR (CDCl3): δ 6,13 (s, 2Н), 4,16 (m, 2H), 3,80 (S, 3Н), 3,77 (S, 6H), 2,93 (S, 3Н), 2,74 (m, 1H), 2,62 (m, 1H).
MS (ES+); 310 (M+1).
[α]D 25: -37,83° (с=0,518, МеОН)
Пример 7:
(-)-транс-(1-Метил-3-(2,4,6-триметоксифенил)пирролидин-2-ил)метанол
Литийалюминий гидрид (0,304 г, 0,008 моль) перемешивали в тетрагидрофуране (40 мл) в атмосфере азота. (-)-транс-1-Метил-5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоновую кислоту (1,0 г, 0,0032 моль) добавили частями и реакционную смесь перемешивали с нагреванием при 50°С на протяжении 90 минут. Реакционную смесь охладили до 10°С и разбавили водой (2,5 мл) и 15% раствором гидроксида натрия (0,6 мл) при перемешивании. Твердое вещество отфильтровали и промыли этилацетатом (10 мл). Органические слои объединили и концентрировали под пониженным давлением для получения белого твердого вещества.
Выход: 0,91 г (100%).
1Н NMR (CDCl3): δ 6,16 (s, 2H), 3,98 (m, 1H), 3,64 (s, 9H), 3,62 (dd, 1H), 3,43 (d, 1H), 3,21 (m, 1H), 2,78 (m, 1H), 2,63 (m, 1H), 2,44 (s, 3H), 2,04 (m, 2H).
MS (ES+): 282 (M+1).
[α]D 25: -20° (с=0,2, МеОН).
Пример 8:
(-)-транс-Уксусной кислоты 3-(3-ацетил-2-гидрокси-4,6-диметокси-фенил)-1-метил-пирролидин-2-ил метиловый эфир
Диэтиловый эфират трифторида бора (25,2 г, 0,178 моль) добавили по каплям при перемешивании при 0°С в атмосфере азота в раствор (-)-транс-(1-метил-3-(2,4,6-триметоксифенил)пирролидин-2-ил)метанола (10 г, 0,0356 моль) в уксусном ангидриде (18 г, 0,178 моль). Реакционную смесь перемешивали при комнатной температуре на протяжении 2 часов. Смесь вылили на дробленый лед (1 кг), подщелочили с помощью насыщенного водного раствора карбоната натрия и экстрагировали этилацетатом (3×200 мл). Органический экстракт промыли рассолом, высушили (безводным сульфатом натрия) и концентрировали для получения названного соединения.
Выход: 10 г (80%).
1Н NMR (CDCl3): δ 14,20 (s, 1H), 5,96 (s, 1H), 4,10 (d, 2H), 3,90 (s, 3H), 3,89 (s, 3H), 3,85 (m, 1H), 3,26 (m, 1H), 2,82 (m, 1H), 2,74 (m, 1H), 2,66 (s, 3H), 2,52 (s, 3H), 2,21 (m, 2H), 2,10 (s, 3H).
Пример 9:
(-)-транс-1-[2-Гидрокси-3-(2-гидроксиметил-1-метил-пирролидин-3-ил)-4,6-диметокси-фенил)-этанон
К раствору (-)-транс-уксусной кислоты-3-(3-ацетил-2-гидрокси-4,6-диметокси-фенил)-1-метил-пирролидин-2-ил метилового эфира) (10 г, 0,0284 моль) в метаноле (25 мл) добавили при перемешивании при комнатной температуре 10% водный раствор гидроксида натрия (25 мл). Температуру реакционной смеси повышали до 50°С на протяжении 45 минут, охладили до комнатной температуры, подкислили с помощью 1:1 раствора соляной кислоты и концентрировали для удаления метанола. Смесь подщелочили насыщенным водным раствором карбоната натрия. Осевшее соединение отфильтровали, промыли водой и высушили.
Выход: 7,14 г (81,1%).
IR (Инфракрсный, ИК) (KBr): 3400, 3121, 3001, 1629, 1590 см-1.
1H NMR (CDCl3): δ 5,96 (s, 1H), 3,93 (m, 1H), 3,90 (s 3H), 3,88 (s, 3H), 3,59 (dd, 1H), 3,37 (d, 1H), 3,13 (m, 1H), 2,75 (m, 1H), 2,61 (s, 3H), 2,59 (m, 1H), 2,37 (s, 3H), 2,00 (m, 2H).
MS(ES+): m/z 310 (M+1).
Пример 10:
(+)-транс-2-(2-Хлорфенил)-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-5,7-диметокси-хромен-4-он
Гидрид натрия (50%, 0,54 г, 0,01125 моль) добавили частями к раствору (-)-транс-уксусной кислоты 3-(3-ацетил-2-гидрокси-4,6-диметокси-фенил)-1-метил-пирролидин-2-ил метилового эфира (0,7 г, 0,0022 моль) в N,N-диметилформамиде (15 мл) при 0°С в атмосфере азота при перемешивании. Через 10 минут добавили метил 2-хлорбензоат (1,15 г, 0,00675 моль). Реакционную смесь перемешивали при 25°С на протяжении 2 часов. Метанол осторожно добавили при ниже 20°С. Реакционную смесь вылили на дробленый лед (300 г), подкислили 1:1 раствором соляной кислоты до рН 2 и экстрагировали этилацетатом (2×100 мл). Водный слой подщелочили насыщенным раствором карбоната натрия до рН 10 и экстрагировали с помощью хлороформа (3×200 мл). Органический слой высушили над безводным сульфатом натрия и концентрировали. К этому остатку добавили концентрированную соляную кислоту (25 мл) и перемешивали при комнатной температуре на протяжении 2 часов. Реакционную смесь вылили на дробленый лед (300 г) и сделали щелочной с помощью насыщенного раствора карбоната натрия. Смесь экстрагировали с помощью хлороформа (3×200 мл). Органический экстракт промыли водой, высушили над безводным сульфатом натрия и концентрировали для получения названного соединения.
Выход: 0,67 г (68,88%).
mp (точка плавления): 95-97°С.
IR (KBr): 3400, 1660 см-1.
[α]D 25=+5,8° (с=0,7, метанол).
1Н NMR (CDCl3) δ 7,7 (dd, 1Н), 7,41 (m, 1H), 7,45 (m, 2H), 6,55 (s, 1H), 6,45 (s, 1H), 4,17 (m, 1H), 4,05 (s, 3H), 3,95 (s, 3H), 3,65 (dd, 1H), 3,37 (dd, 1H), 3,15 (m, 1H), 2,77 (d, 1H), 2,5 (m, 1H), 2,3 (s, 3H), 2,05 (m, 2H).
MS: m/e 430 (M+), 398 (M-31).
Пример 11:
(+)-транс-2-(2-Хлорфенил)-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-5,7-дигидрокси-хромен-4-он
Расплавленный пиридингидрохлорид (4,1 г, 0,0354 моль) добавили к (+)-транс-2-(2-хлорфенил)-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-5,7-диметокси-хромен-4-ону (0,4 г, 0,0009 моль) и нагревали при 180°С на протяжении 1,5 часа. Реакционную смесь охладили до 25°С, разбавили метанолом (10 мл) и подщелочили карбонатом натрия до рН 10. Смесь отфильтровали, а органический слой концентрировали. Остаток суспендировали в воде (5 мл), перемешивали на протяжении 30 минут, отфильтровали и высушили для получения названного соединения.
Выход: 0,25 г (66,86%).
IR (KBr): 3422, 3135, 1664, 1623, 1559 см-1.
1H NMR (CDCl3): δ 7,56 (d, 1H), 7,36 (m, 3Н), 6,36 (s, 1H), 6,20 (s, 1H), 4,02 (m, 1H), 3,70 (m, 2H), 3,15 (m, 2H), 2,88 (m, 1H), 2,58 (s, 3Н), 2,35 (m, 1H), 1,88 (m, 1H).
MS (ES+): m/z 402 (M+1).
Исследование: C21H20ClNO5 C, 62,24 (62,71); H, 5,07 (4,97); N, 3,60 (3,48); Cl, 9,01 (8,83).
Пример 12:
(+)-транс-2-(2-Хлорфенил)-5,7-дигидрокси-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-хромен-4-он гидрохлорид
(+)-транс-2-(2-Хлорфенил)-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-5,7-диметокси-хромен-4-он (0,2 г, 0,48 ммоль) суспендировали в метаноле (2 мл) и добавили эфирный HCl (5 мл). Суспензию перемешивали для получения прозрачного раствора. Раствор концентрировали под пониженным давлением для получения названного соединения.
Выход: 0,21 г (97%).
[α]D 25=+21,2° (с=0,2, метанол).
1Н NMR (CD3OD, 300 MHz): δ 7,80 (d, 1H), 7,60 (m, 3Н), 6,53 (s, 1H), 6,37 (s, 1H), 4,23 (m, 1H), 3,89 (m, 2H), 3,63 (m, 1H), 3,59 (dd, 1H), 3,38 (m, 1H), 2,90 (s, 3H), 2,45 (m, 1H), 2,35 (m, 1H).
MS (ES+): m/z 402 (M+1), свободное основание.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЭНАНТИОМЕРНО ЧИСТЫЕ ПРОИЗВОДНЫЕ ФЛАВОНА ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ НАРУШЕНИЙ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2418786C2 |
| 2,5-ДИЗАМЕЩЕННЫЕ ТЕТРАГИДРОФУРАНЫ ИЛИ ТЕТРАГИДРОТИОФЕНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ | 1995 |
|
RU2190607C2 |
| Новые производные имидазо[4,5-с]хинолина в качестве ингибиторов LRRK2 | 2018 |
|
RU2773516C2 |
| 6-ГЕТЕРОЦИКЛИЛ-4-МОРФОЛИН-4-ИЛПИРИДИН-2-ОНЫ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА И ДИАБЕТА | 2017 |
|
RU2762968C2 |
| АРИЛСУЛЬФОНИЛМЕТИЛЬНЫЕ ИЛИ АРИЛСУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВ, ВОСПРИИМЧИВЫХ К ЛЕЧЕНИЮ ЛИГАНДАМИ ДОФАМИНОВЫХ D РЕЦЕПТОРОВ, С ИХ ПОМОЩЬЮ | 2005 |
|
RU2442781C2 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
| БЕНЗОКОНДЕНСИРОВАННЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО ПРОДУКТА | 1992 |
|
RU2114110C1 |
| ПРОИЗВОДНЫЕ 2-АМИНО-4-ФЕНИЛ-4-ОКСОМАСЛЯНОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2139850C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-АЦИЛ-4-ФЕНИЛСУЛЬФОНИЛПРОЛИНАМИДА И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2615997C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2545080C2 |
Данное изобретение относится к энантиоселективному синтезу промежуточных соединений, а именно (-)-транс-(1-метил-3-(2,4,6-триметоксифенил) пирролидин-2-ил)-метанолу, представленному формулой А:
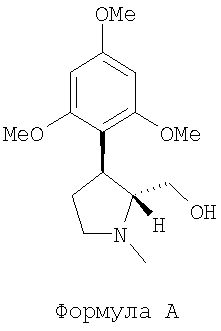 ,
,
включающему этапы, на которых обрабатывают (-)-транс-1-метил-5-оксо-3-(2,4,3-триметоксифенил) пирролидин-2-карбоновую кислоту следующей формулы Е:
восстанавливающим агентом в растворителе. Эти соединения используются для получения (+)-транс энантиомера пирролидинов, замещенных флавонами, представленного формулой 1, или его солей, которые являются ингибиторами циклин-зависимых киназ, и могут применяться для лечения пролиферативных нарушений, таких как рак:
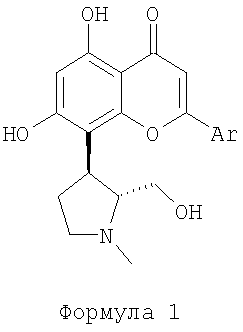
Данный способ позволяет провести более эффективный крупномасштабный синтез соединения формулы 1 путем исключения методик расщепления рацемата. 14 з.п. ф-лы.
1. Способ получения соединения (-)-транс-(1-метил-3-(2,4,6-триметоксифенил) пирролидин-2-ил)-метанола, представленного формулой А
,
включающий этапы, на которых обрабатывают (-)-транс-1-метил-5-оксо-3-(2,4,6-триметоксифенил) пирролидин-2-карбоновую кислоту следующей формулы E
(далее называется соединение Е), восстанавливающим агентом в растворителе.
2. Способ по п.1, где восстанавливающим агентом является гидрид.
3. Способ по п.2, при котором гидрид выбран из литийалюминиевого гидрида, диизобутилалюминиевого гидрида и боргидрида натрия.
4. Способ по п.1, при котором применяемым растворителем является эфир.
5. Способ по п.4, при котором эфир выбран из тетрагидрофурана, диоксана и диэтилового эфира.
6. Способ по любому из пп.1-5, при котором соединение Е получают способом, при котором:
(а) проводят стереоспецифическую реакцию Майкла диметилмалоната с (Е)-метил-2-нитро-3-(2,4,6-триметоксифенил)акрилатом в растворителе в присутствии каталитического комплекса, основания и молекулярных сит, где каталитический комплекс включает хиральный лиганд бис(оксазолина) и комплексное соединение металла, для получения (+)-триметил-3-нитро-2-(2,4,6-триметоксифенил) пропан-1,1,3-трикарбоксилата, представленного следующей формулой В
(далее называется соединение В);
(b) обрабатывают соединение В, что получено на этапе (а), восстанавливающим агентом в растворителе для получения (+)-диметил-5-оксо-3-(2,4,6-триметоксифенил)-пирролидин-2,4-дикарбоксилата, представленного следующей формулой С
(далее называется соединение С);
(с) обрабатывают соединение С хлоридом натрия в растворителе и нагревают полученную реакционную смесь до температуры в диапазоне 120 - 170°С для получения (+)-метил-5-оксо-3-(2,4,6-триметоксифенил)пирролидин-2-карбоксилата как смеси цис- и трансизомеров, представленного следующей формулой D
(далее называется соединение D);
(d) подвергают реакции соединение D с метилирующим агентом в растворителе и с основанием, выбранным из гидрида щелочного металла и карбоната щелочного металла; затем подвергают полученную смесь цис- и транссоединений щелочному гидролизу с гидроксидом щелочного металла в спирте и нагревают полученную реакционную смесь до температуры в диапазоне 50 - 100°С для получения соединения Е как отдельного трансизомера.
7. Способ по п.6, при котором хиральный лиганд бис(оксазолина), применяемый на этапе (а), представляет собой (3аS,3а'S,8аR,8а'R)-2,2'(циклопропан-1,1-диил)бис(8,8а-дигидро-3аН-индено[1,2d]оксазол).
8. Способ по п.6, при котором комплексное соединение металла, применяемое на этапе (а), выбирают из трифторметансульфоната магния, перхлората магния, трифторметансульфоната меди, трифторметансульфоната цинка, трифторметансульфоната лантана, трифторметансульфоната никеля, бромида магния, бромида меди, бромида цинка, бромида никеля, йодида магния, йодида меди, йодида цинка, йодида никеля, ацетилацетоната магния, ацетилацетоната меди, ацетилацетоната цинка и ацетилацетоната никеля.
9. Способ по п.8, при котором комплексное соединение металла представляет собой трифторметансульфонат магния.
10. Способ по п.6, при котором основание, применяемое на этапе (а) п.6, выбирают из триэтиламина, диизопропиламина, 2,6-лутидина, N-метилморфолина, N-этилпиперидина, имидазола и 5,6-диметилбензимидазола.
11. Способ по п.10, при котором основание представляет собой N-метилморфолин.
12. Способ по п.6, при котором на этапе (b) п.6 проводят обработку соединения В восстанавливающим агентом в растворителе, применяя хлорид олова в качестве восстанавливающего агента.
13. Способ по п.12, где растворитель представляет собой этилацетат.
14. Способ по п.6, при котором на этапе (b) п.6 проводят обработку соединения В восстанавливающим агентом в растворителе, применяя никель Ренея в качестве восстанавливающего агента.
15. Способ по п.14, при котором растворитель выбирают из тетрагидрофурана, диоксана и N,N-диметилформамида.
| WO 2004004632 A2, 15.01.2004 | |||
| Naik et al, Tetrahedron, 1988, v.44, no.7, p.2081-2086 | |||
| Оксо-2-пирролидино-1-уксусные кислоты,или их амиды, или их дициклогексиламиновые соли, проявляющие активность по отношению к мнемоническим процессам | 1979 |
|
SU969701A1 |
| Способ получения цис- или транс- 3-(5- @ -2-пирролидил)пропанолов-1 | 1982 |
|
SU1139727A1 |

