Изобретение относится к области 2,5-двузамещенных тетрагидротиофенов, тетрагидрофуранов (тетрагидрофурана), пирролидонов и 1,3-двузамещенных циклопентанов. Эти соединения проявляют биологическую активность путем подавления 5-липоксигеназы, действуя в качестве антагонистов рецепторов PAF, или проявляя двойную активность, т.е. действуя как антагонист рецепторов PAF и ингибитор 5-липоксигеназы.
Лейкотриены являются эффективными местными медиаторами, играющими существенную роль в воспалительных и аллергических реакциях, включая артрит, астму, псориаз и тромбоэмболическое заболевание. Лейкотриены являются прямоцепочечными эйкозаноидами, образуемыми окислением арахидоновой кислоты липоксигеназами.
Арахидоновая кислота окисляется 5-липоксигеназой до гидропероксида 5-гидропероксиэйкозатетраеновой кислоты (5-НРЕТЕ), которая превращается в лейкотриен А4, который в свою очередь может превращаться в лейкотриен В4, С4 или D4. Как известно, медленно реагирующее вещество при анафилаксии является смесью лейкотриенов С4, D4 и Е4, все из которых являются сильнодействующими бронхоконстрикторами. Предпринималась попытка разработать в эксперименте специфические антагонисты рецепторов или ингибиторы биосинтеза лейкотриена с целью предотвращения или сведения к минимуму патогенных воспалительных реакций, опосредованных этими соединениями.
В Европейских патентных заявках 90117171.0 и 901170171.0 описаны ингибирующие липоксигеназу соединения индола, бензофурана и бензотиофена.
Недавно появилось сообщение о том, что тетрагидротиофеновое производное L-652,731, транс-2, 5-бис-(3,4,5-триметоксифенил)тетрагидротиофен (L-653,150), является активным антагонистом PAF и умеренным ингибитором 5-липоксигеназы. Было указано, что определенные 2,5-диарилтетрагидротиофены являются антагонистами PAF и ингибиторами синтеза лейкотриена (Biftu et al., Abstr. of 6th Int. Conf. on Prostaglandins and Related Compounds, June 3-6, 1986. Florence, Italy; Патент США 4757084, выданный Biftu); WO 92/15294; WO 94/01430; WO 94/04537 и WO 94/06790.
В заявке WO 92/13848 описан класс ингибирующих рацемическую липоксигеназу гидроксамовую кислоту производных N-гидроксимочевины, имеющих структуру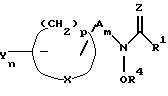
в которой R1 является водородом, алкилом, алкенилом, амино- или замещенной аминогруппой, R4 является водородом, фармацевтически приемлемым катионом, ароилом или алкоилом, А является алкиленом или алкениленом, Х является кислородом или серой, каждая Y является водородом, гало, циано, гидрокси, алкил, алкокси, алкилтио, алкенил, алкоксиалкил, циклоалкил, арил, арилокси, арилалкил, арилалкенил, арилалкокси или замещенным арилом, Z является кислородом или серой, m является 0 или 1, n является от 1 до 5 и р является от 2 до 6,
ингибирует фермент липоксигеназу.
Учитывая значительное количество патологических и воспалительных реакций, которые опосредуются 5-липоксигеназой, сохраняется необходимость идентификации новых соединений и составов, которые ингибируют этот фермент.
Фактор активации тромбоцитов (PAF, 1-о-алкил-2-ацетил-sn-глицерин-3-фосфорилхолин) является мощным фосфолипидным медиатором воспаления с широким разнообразием биологической активности. Первоначально PAF был идентифицирован как водорастворимое соединение, выделяемое кроличьими базофилами, сенсибилизированными иммуноглобулином Е (IgE). В настоящее время известно, что PAF также вырабатывается и выделяется моноцитами, макрофагами, полиморфоядерными лейкоцитами (PMN), эозинофилами, нейтрофилами, лимфоцитами - естественными киллерами, тромбоцитами и эндотелиальными клетками, а также почечными и сердечными тканями под влиянием соответствующей иммунологической и не иммунологической стимуляции (Hwang, "Specific receptors of platelet-activating factor, receptor heterogeneity and signal transduction mechanism", Journal of Lipid Mediators, 1990, Vol.2, p.123. PAF в очень низких концентрациях вызывает агрегацию и дегрануляцию тромбоцитов. Мощность (активность в концентрации от 10-12 до 10-9 М), уровень в тканях (пикомоли) и короткий период полувыведения из плазмы (2-4 минусы) PAF аналогичны таковым других липидных медиаторов, как, например, тромбоксана А2, простагландинов и лейкотриенов.
PAF опосредует биологические реакции путем связывания со специфическими рецепторами PAF, обнаруженными в широком многообразии клеток и тканей. Исследования зависимости активности от структуры PAF и его аналогов, показывают, что способность PAF связываться с этими рецепторами, является структурно-специфичной и стереоспецифичной (Shen et al., "The Chemical and Biological Properties of PAF Agonists, Antagonists and Biosynthetic Inhibitors", Platelet-Activating Factor and Related Lipid Mediators, F. Snyder, Ed Plenum Press, New York, NY 153 (1987)).
Наряду с тем, что PAF опосредует важнейшие биологические реакции, он также, как представляется, играет роль в патологических иммунных и воспалительных реакциях. Во многих опубликованных исследованиях имеются доказательства участия PAF в патогенезе заболеваний человека, включая артрит, острые воспалительные процессы, астму, эндотоксиновый шок, болевой синдром, псориаз, воспалительные поражения глаз, ишемию, изъязвление слизистой желудочно-кишечного тракта, инфаркт миокарда, воспалительные поражения кишечника и острый респираторный дистресс-синдром. На экспериментальных моделях также показано, что выработка PAF происходит или возрастает при определенных патологических состояниях.
Участие PAF в патологических воспалительных и иммунных состояниях стимулировало значительное количество научных исследований с попыткой идентификации антагонистов рецепторов PAF. В 1983 г. появилось сообщение об аналоге фосфолипида, названном CV-3988 (рак-3-(N-n-октадецилкарбамоилокси-ω-метоксипропил-2-тиазолиоэтилфосфат), имеющем свойства антагониста рецепторов PAF (Terashita et al. , Life Sciences, 1983, Vol.32, р.1975). В другой ранней работе в этой области (Shen et al., Proc. Natl. Acad. Sci. (U. S. A. ), 1985, Vol.82, p. 672) сообщалось, что кадсуренон, производное неолигнана, выделенное из перца Piper futokadzura Sieb et Zucc (китайского травяного растения), является активным, специфичным и конкурентным ингибитором активности PAF на рецепторном уровне.
Hwang et al. в 1985 г. показали, что транс-2,5-бис-(3,4,5-триметоксифенил)тетрагидрофуран (L-652,731) ингибирует связывание меченного тритием PAF рецепторными участками PAF (Hwang et al., "Trans-2,5-bis-(3,4,5-trimethoxyphenyl)tetrahydrofuran", Journal of Biological Chemistry, 1985, Vol. 260, p.15639). Как было установлено, L-652,731 был активен при приеме внутрь и ингибировал вызванную PAF проницаемость сосудов кожи в дозе 30 мг/кг массы тела. Как установлено, соединение не влияет на фермент - 5-липоксигеназу. Hwang et al. также сообщили, что транс-L-652,731 (в котором ариловые группы в положениях 2 и 5 расположены на противоположных сторонах плоскости кольца тетрагидрофурана) приблизительно в 100 раз активнее, чем цис-652,731 (в котором заместители арила во 2 и 5 положении находятся на одной стороне плоскости кольца тетрагидрофурана).
В 1988 г. Hwang et al. сообщили, что L-659,989 (транс-2-(3-метокси-4-пропоксифенил-5-метилсульфонил)-5-(3,4,5-триметоксифенил)тетрагидрофуран) является при приеме внутрь активным, сильнодействующим и конкурентным антагонистом рецепторов PAF, при константе равновесного ингибирования, в 10 раз превышающей таковую транс-L-652,731 (Hwang et al., J. Pharmacol. Ther., 1988, Vol.246, р.534).
Из патентов США 4996203, 5001123 и 4539332, выданных Biftu et al., и заявок на Европейский патент 89202593.3, 90306235.4 и 90306234.7 известно, что специфические классы - 2,5-диарилтетрагидрофураны - являются антагонистами рецепторов PAF.
Bowles et al., Synlett, 1993, pp.111, описывают ограниченное количество замещенных тетрагидрофуранов, которые могут обладать антагонизмом по отношению к рецепторам PAF.
Danyoshi et al., Chem. Pharm. Bull., 1989, pp.1969, описывают 2-замещенные N-алкоксикарбонилпирролидины, которые ингибируют вызванную PAF агрегацию кроличьих тромбоцитов.
Поэтому целью настоящего изобретения является обеспечение соединений, которые снижают хемотаксис и респираторный взрыв, ведущие к образованию повреждающих кислородных радикалов во время воспалительной или иммунной реакции.
Еще одной целью настоящего изобретения является обеспечение фармацевтических составов для лечения патологических иммунных или воспалительных расстройств, опосредованных продуктами 5-липоксигеназы.
Еще одной целью настоящего изобретения является обеспечение способа лечения патологических иммунных или воспалительных расстройств, опосредованных продуктами 5-липоксигеназы.
Еще одной целью настоящего изобретения является обеспечение фармацевтических композиций для лечения патологических иммунных или воспалительных расстройств, опосредованных PAF.
Еще одной целью настоящего изобретения является обеспечение способа лечения патологических иммунных или воспалительных расстройств, опосредованных PAF.
Краткое описание изобретения
Предложены соединения формулы I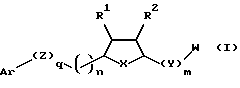
в которой Аr является ариловой или гетероариловой группой, которая необязательно замещена предпочтительно гало (включая, но не ограничивая, фтор), низшим алкокси (включая метокси), низшим арилокси (включая фенокси), W, циано или R3;
m = 0 или 1;
q = 0 или 1;
n = 0-6;
W является независимо -AN(ОМ)С(О)N(R3)R4,
-N(ОM)C(O)N(R3)R4, -AN(R3)С(О)N(ОМ)R4,
-N(R3)С(О)N(ОМ)R4, -AN(ОМ)С(О)R4,
-N(ОМ)С(О)R4-,
АС(О)N(ОМ)R4, -C(О)N(OM)R, -C(О)NHA или
-А-В;
А является группами низшего алкила, низшего алкенила, низшего алкинила, алкиларила или арилалкила, в которых один или более углеродов необязательно могут быть замещены О, N или S (с валентностью, заполняемой, если необходимо, водородом или кислородом), однако -Y-A-, -А- или -AW- не должны включать два примыкающих гетероатома (т.е. -О-ОО-, -S-S-, -О-S- и т. д.). В одном варианте исполнения низший алкил является разветвленной алкильной группой, такой как (СН2)nС(низший алкил)Н-, в которой n является 1-5, и особенно -(СН2)2С(СН3)Н-, или низший алкинил с формулой - (низший алкил)-, включая
(низший алкил)-, включая 
В выбран из группы, состоящей из пиридилимидазола и бензимидазола, каждый из которых необязательно замещен R3 и в которой пиридилимидазол или бензимидазол предпочтительно связан с А через атом азота;
М является водородом, фармацевтически приемлемым катионом или метаболически расщепляемой остаточной группой;
Х является О, S, S(О), S(О)2, NR3 или CHR5;
Y является О, S, S(О), S(О)2, NR3 или CHR5;
Z является О, S, S(О), S(О)2, NR3;
R1 и R2 являются независимо водородом, низшим алкилом, включая метил, циклопропилметил, этил, изопропил, бутил, пентил, гексил и С3-8циклоакил, например циклопентил; гало-низший-алкил, например трифторметил; гало, например фтор, и -СООН;
R3 и R4 являются независимо водородом или алкилом, алкенилом, алкинилом, арилом, арилалкилом, алкиларилом, C1-6алкокси-C1-10алкилом, С1-6алкилтио-C1-10алкилом, гетероарилом или гетероарилалкилом;
R является водородом, низшим алкилом, низшим алкенилом, низшим алкинилом, арилалкилом, алкиларилом, -AN(ОМ)С(О)N(R3)R4, -AN(R3)С(О)N(ОМ)R4, -AN(OM)С(О)R4, -АС(О)N(ОМ)R4, -AS(О)хR3, -AS(О)nCH2C(О)R3, -AS(О)nСН2СН(ОН)R3 или -AC(О)NHR3,
в котором х = 0-2.
В одном варианте исполнения группа Аr выбрана из группы, состоящей из групп фенила, триметоксифенила, диметоксифенила, фторфенила, дифторфенила, пиридила, диметоксипиридила, хинолинила, фурила, имидазолила и тиенила.
В одном варианте исполнения -А-В является
и Аr представляет собой необязательно замещенную арильную или гетероарильную группу, как более подробно описано ниже в разделе I.A.
Не ограничивающими примерами предпочтительных соединений являются
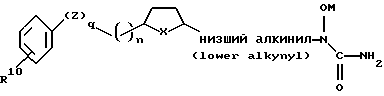
где R является галогеном, -CN, водородом, низшим алкилом, низшим алкенилом, низшим алкинилом, низшим алкокси или низшим арилокси.
Эти соединения в целом снижают хемотаксис и респираторный взрыв, ведущий к образованию повреждающих кислородных радикалов полиморфоядерных лейкоцитов во время воспалительной или иммунной реакции. Соединения проявляют эту биологическую активность путем ингибирования фермента 5-липоксигеназы, действуя как антагонисты рецепторов PAF, или проявляя двойную активность, т. е., действуя как антагонист рецепторов PAF и ингибитор 5-липоксигеназы.
Другим вариантом исполнения настоящего изобретения является фармацевтическая композиция, которая включает эффективное количество соединения формулы I или его фармацевтически приемлемой соли, или производного в комбинации с фармацевтически приемлемым носителем, применяемая по поводу любого из описанных здесь расстройств.
В предпочтительном варианте исполнения соединения используются для лечения расстройств, опосредуемых 5-липоксигеназой, путем введения эффективного количества одного или более из указанных выше соединений или их фармацевтически приемлемых солей или производных, как вариант, в фармацевтически приемлемом носителе.
К удивлению, было установлено, что активность соединения, например активность 5-липоксигеназы, доступность при приеме внутрь и устойчивость in vivo (например, скорость глюкуронизации) могут существенно варьировать среди оптических изомеров описанных соединений. Поэтому в одном варианте исполнения изобретения соединение вводится в энантиоморфно обогащенной форме.
Примеры иммунных, аллергических и сердечно-сосудистых заболеваний включают общее воспаление, сердечно-сосудистые заболевания, включая гипертензию, скелетно-мышечные заболевания, остеоартрит, зоб, астму, отек легких, респираторный дистресс-синдром взрослых, болевой синдром, агрегацию тромбоцитов, шок, ревматоидный артрит, юношеский ревматоидный артрит, псориатический артрит, псориаз, аутоиммунный увеит, аллергический энцефаломиелит, системную красную волчанку, острую некротизирующую геморрагическую энцефалопатию, идиопатическую тромбоцитопению, полиходрит, хронический активный гепатит, идиопатический синдром мальабсорбции, болезнь Крона, офтальмопатию Грэйвса, первичный билиарный цирроз, задний увеит, интерстициальный легочный фиброз, аллергическую астму и неадекватные аллергические реакции на стимулы окружающей среды, такие как яд плюща, пыльца, укусы насекомых и определенные виды пищи, включая атопический дерматит и контактный дерматит.
Соединения, описанные здесь, могут также использоваться в качестве исследовательских инструментов для изучения биологических путей с участием соответственно лейкотриенов или PAF.
Следующие соединения являются не ограничивающими примерами соединений, характеризуемых формулой I. Это просто примеры, которые приведены без намерения ограничить диапазон изобретения:
транс-2-(3,4,5-триметоксифеноксиметил)-5-[4-N'-метил-N'-гидроксиуреидил)бутил]тетрагидрофуран,
транс-2-(3,4,5-триметоксифеноксиметил)-5-[4-N'-метил-N'-гидроксиуреидил)бут-1-инил]тетрагидрофуран,
транс-2-(4-фторфеноксиметил)-5-[4-N'-метил-N'-гидроксиуреидил)бутил] тетрагидрофуран,
транс-2-(4-фторфеноксиметил)-5-[4-N'-метил-N'-гидроксиуреидил)бут-1-инил]тетрагидрофуран,
транс-2-(4-фторфеноксиметил)-5-[4-N'-бутил-N'-гидроксиуреидил)бутил] тетрагидрофуран,
транс-2-(4-фторфеноксиметил)-5-[4-N'-бутил-N'-гидроксиуреидил)бут-1-инил]тетрагидрофуран,
транс-2-(3,4,5-триметоксифеноксиметил)-5-[4-N'-метил-N-гидроксиуреидил)бутил]тетрагидрофуран,
транс-2-(3,4,5-триметоксифеноксиметил)-5-[4-N'-метил-N-гидроксиуреидил)бут-1-инил]тетрагидрофуран,
транс-2-(4-фторфеноксиметил)-5-[4-N'-метил-N-гидроксиуреидил)бутил] тетрагидрофуран,
транс-2-(4-фторфеноксиметил)-5-[4-N'-метил-N-гидроксиуреидил)бут-1-инил] тетрагидрофуран,
транс-2-(4-фторфеноксиметил)-5-[4-N'-бутил-N-гидроксиуреидил)бутил] тетрагидрофуран,
транс-2-(4-фторфеноксиметил)-5-[4-N'-бутил-N-гидроксиуреидил)бут-1-инил] тетрагидрофуран,
транс-2-(3,4,5-триметоксифеноксиметил)-5-[4-N'-метил-N'-гидроксиуреидил)бутил]тетрагидрофуран,
транс-2-(3,4,5-триметоксифеноксиметил)-5-[4-N'-метил-N'-гидроксиуреидил)бут-1-инил]тетрагидротиофен,
транс-2-(4-фторфеноксиметил)-5-[4-N'-метил-N'-гидроксиуреидил)бутил] тетрагидротиофен,
транс-2-(4-фторфеноксиметил)-5-[4-N'-метил-N'-гидроксиуреидил)бут-1-инил]тетрагидротиофен,
транс-2-(4-фторфеноксиметил)-5-[4-N'-бутил-N'-гидроксиуреидил)бутил] тетрагидротиофен,
транс-2-(4-фторфеноксиметил)-5-[4-N'-бутил-N'-гидроксиуреидил)бут-1-инил]тетрагидротиофен,
транс-2-(3,4,5-триметоксифеноксиметил)-5-[4-N'-метил-N-гидроксиуреидил)бутил]тетрагидротиофен,
транс-2-(3,4,5-триметоксифеноксиметил)-5-[4-N'-метил-N-гидроксиуреидил)бут-1-инил]тетрагидротиофен,
транс-2-(4-фторфеноксиметил)-5-[4-N'-бутил-N-гидроксиуреидил)бутил] тетрагидротиофен,
транс-2-(4-фторфеноксиметил)-5-[4-N'-метил-N-гидроксиуреидил)бут-1-инил] тетрагидрофуран,
транс-2-(4-фторфеноксиметил)-5-[4-N'-бутил-N-гидроксиуреидил)бутил] тетрагидротиофен,
транс-2-(4-фторфеноксиметил)-5-[4-N'-бутил-N-гидроксиуреидил)бут-1-инил] тетрагидротиофен,
2-(3,4,5-триметоксифенил)-5-[3-(N'-метил-N'-гидроксиуреидил)пропокси] тетрагидрофуран,
2-(4-фторфенил)-5-[3-(N'-метил-N'-гидроксиуреидил)пропокси] тетрагидрофуран,
2-(3,4,5-триметоксифенил)-5-[3-(N'-н-бутил-N'-гидроксиуреидил)пропокси] тетрагидрофуран,
2-(4-фторфенил)-5-[3-(N'-н-бутил-N'-гидроксиуреидил)пропокси]тетрагидрофуран,
2-(3', 4'-диметоксифенил)-5-[3-(N-бутил-N-гидроксиуреидил)] пропокситетрагидрофуран,
2-(3', 4'-диметоксифенил)-5-[3-(N-метил-N-гидроксиуреидил)] пропокситетрагидрофуран,
2-(2,4,5-триметоксифенил)-5-(3-гидроксиуреидилпропокси)тетрагидрофуран,
2-(4-фторфенил)-5-(3-гидроксиуреидилпропокси)тетрагидрофуран,
2-(4-фторфенил)-5-[3-(N'-метил-N'-гидроксиуреидил)пропокси] тетрагидротиофен и
2-(4-фторфенил)-5-(3-гидроксиуреидилпропокси)тетрагидротиофен.
Дальнейшие не ограничивающие примеры других соединений, которые характеризуются формулой I, представлены ниже в таблицах 1, 2 и 3 и на фигурах 1а и 1б.
Краткое описание чертежей
Фигуры 1а и 1б являются иллюстрациями химических структур с указанной стереохимией отобранных активных соединений.
Фигура 2 иллюстрирует скорость глюкуронидации рацемического соединения 202, а также его энантиомеров, соединений 216, 217, 234 и 236.
Фигура 3 иллюстрирует скорость глюкуронидации следующих проиллюстрированных энантиомеров:



Фигура 4 является иллюстрацией одной технологии синтеза 2S, 5S-транс-2-(4-фторфеноксиметил)-5-(4-N-гидроксиуреидил-1-бутинил)тетрагидрофурана (соединения 401) и 2S, 5S-транс-2-(4-фторфеноксиметил)-5-(4-N-гидроксиуреидилбутил)тетрагидрофурана (соединения 402).
Фигура 5 является графической характеристикой скорости глюкуронидации соединений 401, 403, 404 и 406 (как представлено в таблице 4), выраженной в процентном содержании метаболитов в зависимости от времени (в часах).
I. Описание и синтез соединений.
А. Соединения.
Использованный в настоящем описании термин "энантиоморфно обогащенное", относится к соединению в форме, по крайней мере, 95%, а предпочтительно приблизительно 97%, 98%, 99% или 100% одиночного энантиомера этого соединения.
Термин "алкил", используемый здесь, при отсутствии других уточнений, относится к насыщенному прямому, разветвленному или циклическому углеводороду от C1 до С10, и, в частности, включает метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, циклопентил, изопентил, неопентил, гексил, изогексил, циклогексил, 3-метилпентил, 2,2-диметилбутил и 2,3-диметилбутил. Алкиловая группа может быть необязательно замещена любой соответствующей группой, включая, но не ограничиваясь, R3 или одного или более фрагментов, выбранных из группы, состоящей из гало, гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната либо незащищенных, либо, в случае необходимости, защищенных методами, известными специалистам в этой области, например, как предложено в работе Greene et al., "Protective Groups in Organic Synthesis", John Wiley and Sons, Second Edition, 1991.
Термин "гало", используемый здесь, относится к хлор-, фтор-, йод- или бром-. Термин "низший алкил", используемый здесь, при отсутствии других уточнений, относится к насыщенному прямому, разветвленному или циклическому (в случае С5-6) углеводороду от С1 до C6 и, в частности, включает метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, циклопентил, изопентил, неопентил, гексил, изогексил, циклогексил, 3-метилпентил, 2,2-диметилбутил и 2,3-диметилбутил, как вариант, замещенные, как описано выше, на алкиловые группы.
Термин "алкенил", ссылки на который имеются в данном описании, и при отсутствии других уточнений, относится к прямому, разветвленному или циклическому (в случае С5-6) углеводороду от C1 до С10 с, по крайней мере, одной двойной связью, необязательно замещенный, как описано выше.
Термин "низший алкенил", ссылки на который имеются в данном описании, и при отсутствии других уточнений, относится к алкениловой группе от С2 до С6 и, в частности, включает винил и аллил.
Термин "низший алкиламино" относится к аминогруппе, которая имеет один или два заместителя в виде низших алкилов.
Термин "алкинил", ссылки на который имеются в данном описании, и при отсутствии других уточнений, относится к прямому или разветвленному углеводороду от С2 до С10 с, по крайней мере, одной тройной связью, необязательно замещенный, как описано выше.
Термин "низший алкинил", ссылки на который имеются в данном описании, и при отсутствии других уточнений, относится к алкиниловой группе от C2 до C6 и, в частности, включает ацетиленил, пропинил и 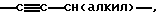 включая
включая 
Термин "арил", ссылки на который имеются в данном описании, и при отсутствии других уточнений, относится к фенилу, бифенилу или нафтилу, предпочтительно к фенилу. Ариловая группа может быть необязательно замещена любой подходящей группой, включая, но не ограничиваясь, один или более фрагментов, выбранных из группы, состоящей из гало, гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната либо незащищенных, либо, в случае необходимости, защищенных методами, известными специалистам в этой области, например, как предложено в работе Greene et al., "Protective Groups in Organic Synthesis", John Wiley and Sons, Second Edition, 1991, и предпочтительно гало (включая, но не ограничиваясь фтором), низшим алкокси (включая метокси), низшим арилокси (включая фенокси), W, циано или R3.
Термин "галоалкил, галоалкенил или галоалкинил" относится к алкиловой, алкениловой или алкиниловой группе, в которой, по крайней мере, один из водородов в группе был замещен атомом галогена.
Термин "гетероарил, гетероцикл или гетероароматический", используемый в настоящем описании, относится к ароматическому фрагменту, который включает, по крайней мере, одну серу, кислород или водород в ароматическое кольцо, которые необязательно замещены, как описано выше, на арильные группы. Не ограничивающими примерами являются пиррил, фурил, пиридил, 1,2,4-тиадиазолил, пиримидил, тиенил, изотиазолил, имидазолил, тетразолил, пиразинил, пиримидил, хинолил, изохинолил, бензотиенил, изобензофурил, пиразолил, индолил, пуринил, карбазолил, бензимидазолил и изоксазолил.
Термин "аралкил" относится к ариловой группе с алкильным заместителем.
Термин "алкарил" относится к алкильной группе с арильным заместителем.
Термин "органический или неорганический анион" относится к органическому или неорганическому фрагменту, который несет отрицательный заряд, и может быть использован как отрицательная часть соли.
Термин "фармацевтически приемлемый катион" относится к органическому или неорганическому фрагменту, который несет положительный заряд, и может вводиться в сочетании с фармацевтическим агентом, например, в качестве противокатиона в соли. Фармацевтически приемлемые катионы известны специалистам в этой области и включают, но ими не ограничиваются, натрий, калий и четвертичный амин.
Термин "метаболически расщепляемая остаточная группа" относится к фрагменту, который может in vivo отщепляться от молекулы, к которой он прикреплен, и включает, но ими не ограничивается, органический или неорганический анион, фармацевтически приемлемый катион, ацил (например, (алкил)С(О), включая ацетил, пропионил и бутирил), алкил, фосфат, сульфат и сульфонат.
Термин "антагонист рецепторов PAF" относится к соединению, которое связывается с рецептором PAF при константе связывания, составляющей 30 мкмоль или ниже.
Термин "ингибитор 5-липоксигеназы" относится к соединению, которое ингибирует фермент при 30 мкмолях или ниже в системе разрушенной клетки.
Термин "фармацевтически активное производное" относится к любому соединению, которое при введении реципиенту способно, прямо или косвенно, обеспечить поступление раскрытых в данном описании соединений.
2,5-Двузамещенные тетрагидротиофены, тетрагидрофураны и пирролидины, а также 1,3-двузамещенные циклопентаны, описанные в настоящей заявке, проявляют активность антагонистов рецепторов PAF или ингибируют фермент 5-липоксигеназу, или имеют двойную активность, и, следовательно, эффективны при лечении людей, которые имеют иммунные, аллергические или сердечно-сосудистые заболевания, которые опосредуются PAF или продуктами 5-липоксигеназы.
Б. Стереохимия.
Было установлено, что активность и свойства активных соединений, включая активность 5-липоксигеназы, возможность приема внутрь и устойчивость in vivo (например, скорость глюкуронидации), могут существенно изменяться среди оптических изомеров описанных соединений. Поэтому в предпочтительном варианте изобретения активное соединение или его предшественник вводится в энантиоморфно обогащенной форме, т.е. преимущественно в форме одного изомера. Предпочтительный энантиомер легко определяется путем оценки различных возможных энантиомеров при выбранных биологических испытаниях, например тех, которые подробно изложены в настоящем описании.
2,5-Двузамещенные тетрагидрофураны и пирролидины отвечают целому ряду стереохимических конфигураций. Атомы углерода 2 и 5 в центральном кольце являются хиральными, и поэтому центральное кольцо существует минимум как диастереометрическая пара. Каждый диастереомер существует как набор энантиомеров. Поэтому, только на основании хиральных атомов C2 и C5, соединение является смесью четырех энантиомеров.
Если на атомах углерода 4 и 5 в центральном кольце расположены не водородные заместители, то атомы С4 и С5 также являются хиральными и также могут существовать как диастереометрическая паpa, которая также является смесью четырех энантиомеров.
Описанные в настоящей заявке 1,3-циклопентаны также проявляют ряд стереохимических конфигураций. Атомы углерода 1 и 3 в центральном кольце являются хиральными, и поэтому центральное кольцо существует минимум как диастереометрическая пара. Каждый диастереомер существует как набор энантиомеров. Поэтому, только на основании хиральных атомов C1 и С3, соединение является смесью четырех энантиомеров.
Если неводородные заместители расположены на атомах углерода 4 и 5 в центральном кольце, то атомы С4 и C5 также являются хиральными и также могут существовать как диастереометрическая пара, которая также является смесью четырех энантиомеров.
Любой специалист в этой области может легко синтезировать и разделить энантиомеры описанных соединений с помощью хиральных реактивов и известных методик и может оценить биологическую активность выделенного энантиомера с помощью описанных здесь методов или других известных методов. Посредством использования хиральных реагентов, вызывающих ЯМР-сдвиг, поляриметрии или хиральной ВЭЖХ можно определить оптическое обогащение соединения.
Классические методы разделения включают множество физических и химических методик. Часто самой простой и наиболее эффективной методикой является повторная перекристаллизация. Перекристаллизацию можно выполнить на любой стадии приготовления соединения или конечного энантиоморфного продукта. В случае успеха этот простой подход представляет метод выбора.
Если перекристаллизация не обеспечивает материал приемлемой оптической чистоты, можно провести оценку других методов. Если соединение является щелочным, можно использовать хиральные кислоты, которые образуют диастереометрические производные, которые могут обладать существенно отличными показателями растворимости. Не ограничивающие примеры хиральных кислот включают яблочную кислоту, миндальную кислоту, дибензоилвинную кислоту, 3-бромкамфор-8-сульфокислоту, 10-камфорсульфокислоту. Аналогичным образом ацилирование свободной гидроксильной группы хиральной кислотой также приводит к образованию диастереометрических производных, физические свойства которых могут отличаться в степени, достаточной для обеспечения возможности разделения.
Энантиоморфно чистые или обогащенные соединения могут быть получены путем пропускания рацемической смеси через хроматографическую колонку, которая была предназначена для хиральных разделений, или путем ферментативного разделения соответствующим образом модифицированных субстратов.
В. Синтез активных соединений.
Описанные здесь 2,5-двузамещенные тетрагидрофураны, тетрагидротиофены и пирролидины, могут быть получены различными путями, известными специалистам в этой области, включая способы, раскрытые Whittaker et al., Synlett, 1993, pp. 111; Biorg. Med. Lett. , 1993, pp. 1499; Achiwa et al., Chem. Pharm. Bull., 1989, pp. 1969.
Например, один способ синтеза энантиоморфно обогащенных материалов описан ниже в схеме 1. При этом способе энантиомерный синтез начинается с хирального восстановления кетона. После замыкания кольца и реакции ОН-группы цис- и транс-изомеры могут быть разделены стандартными методами, известными специалистам в этой области, влияющими на диастереометрическое разделение.
Дополнительные хиральные центры могут быть разделены с помощью методик, известных специалистам в этой области, включая методики, приведенные в схеме А (см. в конце описания).
1,3-Двузамещенные циклопентаны могут быть получены с помощью методики Graham et al. (1,3-Diaryl Cyclopentanes: A New Class of Potent PAF Receptor Antagonists. 197th ACS National Meeting, Dallas, Texas, April 9-14, 1989, Division of Medicinal Chemistry, poster no. 25 (abstract)) или другими известными способами.
Общая методика получения гидроксимочевины показана ниже на схеме 1.
Схема 1. Получение гидроксимочевин.

Общие методики получения реверсивных гидроксимочевин показаны на схеме 2 (см. в конце описания).
Общая методика получения гидроксамовой кислоты показана на схеме 3.
Схема 3. Получение гидроксамовых кислот.

Общая методика получения реверсивной гидроксамовой кислоты показана на схеме 4.
Схема 4. Получение реверсивных гидроксамовых кислот.

На схеме 5 (см. в конце описания) показан синтез 2-(3,4,5-триметоксифенил)-5-[-(N'-замещенного-N'-гидроксиуреидил)пропокси] тетрагидрофурана (91-4) и 2-(4-фторфенил)-5-[N'-замещенного-N'-гидроксиуреидил)пропокси] тетрагидрофурана (9-12).
На схеме 6 (см. в конце описания) показан синтез 2-(2,4,5-триметоксифенил)-5-(3-гидроксиуреидилпропокси)тетрагидрофурана (13) и 2-(4-фторфенил)-5-(3-гидроксиуреидилпропокси)тетрагидрофурана (14,15).
На схеме 7 (см. в конце описания) показан синтез 2-(3,4-диметоксифенил)-5-[3-N'-замещенного-N'-гидроксиуреидилпропокси]тетрагидрофурана (5-8).
Следующие примеры являются просто иллюстративными и не направлены на ограничение диапазона изобретения.
Пример 1. Получение 2-(3,4,5-триметоксифенил)-5-[(N'-замещенного-N'-гидроксиуреидил)пропокси] тетрагидрофурана 91-4) и 2-(4-фторфенил)-5-[N'-замещенного-N'-гидроксиуреидил)пропокси]тетрагидрофурана (9-12).
(а) Получение трет-бутилового эфира 4-(3,4,5-триметоксифенил)-4-кетон-масляной кислоты (соединение 101).
3,4,5-Триметоксибензальдегид (8,0 г, 40,77 ммоля), трет-бутилакрилат (5,29 г, 41,29 ммоля) и катализатор - 3-этил-5-(2-гидроксиэтил)-4-метилтиазолия бромид (3,52 г, 13,95 ммоля) растворяют в 50 мл диметилформамида (ДМФ). К этому раствору добавляют 5,86 мл триэтиламина. Реактивную смесь перемешивают в течение 16 часов при 60oС, охлаждают до комнатной температуры и резко охлаждают для прекращения реакции добавлением 10% НС1 (рН 1-2) и проводят экстракцию дихлорметаном. Органический слой промывают водой и насыщенным раствором NaCl, высушивают над MgSO4, фильтруют и выпаривают в вакууме до масла. Продукт очищают в хроматографической колонке (кварцевой, соотношение гексан/этилацетат 3:1) (4,5 г, 34%).
Н1 ЯМР (СDС13): 1,46 (s, 9Н); 2,70 (t, 2H); 3,24 (t, 2H); 3,92 (s, 9H); 7,25 (s, 2H).
(б) Получение трет-бутилового эфира 4-(4-фторфенил)-4-кетон-масляной кислоты (соединение 119).
Это соединение получают с помощью технологии, аналогичной той, которая описана в примере 1(а), замещая 3,4,5-триметоксибензальдегид 4-фторбензальдегидом.
Н1 ЯМР (СDС13): 1,45 (s, 9H); 2,70 (t, 2H); 3,23 (t, 2H); 7,12 (m, 2H); 8,02 (m, 2H).
(в) Получение трет-бутилового эфира 4-(3,4,5-триметоксифенил)-4-гидроксимасляной кислоты (соединение 102).
Кетоновый эфир 101 (1,09 г, 3,36 ммоля) добавляют к 10 мл тетрагидрофурана и 20 мл метанола. Водный раствор NaBH4 (127,3 мл, 3,36 ммоля в 5 мл воды) по каплям добавляют к этой смеси при 0oC. Реактивную смесь перемешивают при комнатной температуре в течение 4 часов, резко охлаждают водой для прекращения реакции и экстрагируют этилацетатом, органический слой промывают водой, насыщенным раствором NaCl, высушивают над МgSО4, фильтруют и выпаривают в вакууме для получения продукта (1,13 г, 103%).
Н1 ЯМР (СDС13): 1,46 (s, 9H); 2,02 (m, 2H); 2,37 (t, 2H); 3,88 (s, 6Н); 4,70 (m, 1H); 6,58 (s, 2H).
(г) Получение трет-бутилового эфира 4-(4-фторфенил)-4-гидроксимасляной кислоты (соединение 120).
Это соединение получают из соединения 119 с помощью методики, подобной той, которая изложена в примере 1(в), замещая соединение 101 соединением 119.
Н1 ЯМР (СDС13): 1,44 (s, 9H); 2,00 (m, 2H); 2,32 (m, 2H); 4,72 (m, 1H); 7,01 (m, 2H); 7,30 (m, 2H).
(д) Получение 4-(3,4,5-триметоксифенил)-δ-лактона (соединение 104).
Гидроксиэфир 102 (1,13 г, 3,47 ммоля) добавляют к 4 мл метанола, 1,5 мл воды и 5М водного раствора натрия гидроксида (4,5 мл). Реактивную смесь перемешивают при комнатной температуре в течение 30 минут и затем добавляют 12 мл насыщенного водного раствора NаНСО3. Водную фазу смывают эфиром, подкисляют до рН 1-2 добавлением концентрированной НС1 и экстрагируют бензолом (2х30 мл). Бензольный слой контролируют с помощью тонкослойной хроматографии, которая выявляет образование некоторого количества лактона. PPTS (10 мг) добавляют к бензольному экстракту и смесь кипятят в сосуде с обратным холодильником в течение 1 часа для удаления воды. Реактивную смесь промывают насыщенным раствором NаНСО3 и выпаривают в вакууме для получения желаемого лактона в виде твердого вещества (700 мг, 80%).
Н1 ЯМР (СDС13): 2,20 (m, 1H); 2,68 (m, 3Н); 3,85 (s, 3Н); 3,88 (s, 6H); 5,46 (m, 1H); 6,55 (s, 2H).
(е) Получение 4-(4-фторфенил)-δ-лактона (соединение 122).
Это соединение получают из соединения 120 с использованием методики, подобной той, которая представлена в примере 1(д), замещая соединение 102 соединением 120.
Н1 ЯМР (СDС13): 2,20 (m, 1H); 2,68 (m, 3Н); 5,50 (m, 1H); 7,10 (t, 2H); 7,32 (m, 2H).
(ж) Получение 2-(3,4,5-триметоксифенил)-5-гидрокситетрагидрофурана (105).
Лактон 104 (6,86 г, 37,22 ммоля) растворяют в сухом толуоле (100 мл) и раствор охлаждают до -70oС. 1,5 М толуоловый раствор DIBALH (28 мл) по каплям добавляют к раствору. Реактивную смесь перемешивают при -70oС в течение 1 часа. Реакцию прекращают добавлением метанола (11 мл), поддерживая температуру на уровне <-60oС. Смесь нагревают до -20oС с последующим добавлением насыщенного водного раствора калиево-натриевого тартрата (96 мл) при поддержании температуры реакции между -10 и 0oC. Реактивную смесь перемешивают при 0oC в течение 3 часов и затем проводят разделение двух фаз, водный слой экстрагируют этилацетатом. Комбинированные органические слои смывают водой, насыщенным раствором NaCl и затем концентрируют в вакууме для получения продукта (6,51 г, 94%).
Н1 ЯМР (СDС13): 1,82-2,48 (m, 4Н); 3,84 (s, 3Н); 3,88 (s, 6H); 4,97 и 5,20 (m, 1H); 5,65, 5,79 (m, 1H); 6,56, 6,70 (s, 2H).
(з) Получение 2-(4-фторфенил)-5-гидрокситетрагидрофурана (123).
Это соединение получают из соединения 122 с использованием методики, подобной той, которая представлена в примере 1(ж), замещая соединение 104 соединением 122.
Н1 ЯМР (СDС13): 1,79 (m, 1H), 1,95-2,10 (m, 1H); 2,20-2,32 (m, 1H); 2,48 (m, 1H); 5,00 и 5,22 (m, 1H); 5,63 и 5,78 (m, 1H); 7,04 (m, 2H); 7,30 и 7,41 (m, 2H).
(и) Получение транс- и цис-2-(3,4,5-триметоксифенил)-5-(3-фталимидилпропокси)тетрагидрофурана (соединений 107, 108).
Соединение 105 (1,14 г, 4,49 ммоля) растворяют в 4 мл дихлорметана. Триэтиламин (681,4 мг, 6,73 ммоля) добавляют к этому раствору. Реактивную смесь охлаждают на ледяной бане и по каплям добавляют трифторуксусный ангидрид (1,41 г, 6,73 ммоля). Реактивную смесь перемешивают при 0oС в течение 30 минут и затем добавляют 3-фталимидилпропанол (106) (2,4 г, 13,26 ммоля). Реактивную смесь нагревают до комнатной температуры и перемешивают при комнатной температуре в течение 2 часов. Реакцию прекращают добавлением насыщенного водного раствора NаНСО3 и экстрагируют этилацетатом, органический слой промывают водой и насыщенным раствором NaCl, высушивают над МgSО4, фильтруют и выпаривают в вакууме до масла, которое очищают в хроматографической колонке (кварцевой, соотношение гексан/этилацетат 2:1) (107:522 мг (транс); 108:271 мг (цис); смесь 107 и 108 в соотношении 1:1:110 мг; общий выход 46%).
Н1 ЯМР (СDС13):
107: 1,70 (m, 1H); 1,82 (m, 1H); 2,00 (m, 2H); 2,02 (m, 1H); 2,28 (m, 1H); 3,46 (m, 1H); 3,83 (s, 3H); 3,84 (m, 3H); 3,88 (s, 6 H); 4,99 (t, 1H); 5,30 (dd, 1H); 6,56 (s, 2H); 7,72 (m, 2H); 7,85 (m, 2H);
108: 1,95 (m, 3Н); 2,00 (m, 2H); 2,20 (m, 1H); 3,51 (m, 1H); 3,83 (s, 3Н); 3,85 (m, 2H); 3,88 (s, 6Н); 3,92 (m, 1H); 4,90 (m, 1H); 5,16 (dd, 1H); 6,60 (s, 2H); 7,72 (m, 2H); 7,84 (m, 2H).
Для определения стереохимии этой молекулы проводят эксперимент с разницей NOE.
Транс-изомер 107. В этом эксперименте триплет при 4,99 мкг/г облучают расщепляющими импульсами очень низкой радиочастоты и обработку данных проводят так, чтобы измерять только наличие увеличения сигнала. Это должно представлять положительный эффект NOE и указывать на близкое пространственное отношение этих протонов. В этом эксперименте NOE устанавливают для мультиплета при 2,25-2,36 мкг/г, который является протоном фуранового кольца. Другой NOE обнаруживают также для ароматических протонов, что указывает на то, что этот триплет представляет бензильный протон. Для двойного дуплета при 5,30 мкг/г NOE не наблюдают, что указывает на то, что это транс-изомер.
Цис-изомер 108. Для определения стереохимии этой молекулы проводят эксперимент с разницей NOE. В этом эксперименте мультиплет при 4,88-4,93 мкг/г облучают расщепляющими импульсами очень низкой радиочастоты и обработку данных проводят так, чтобы измерять только наличие увеличения сигнала. Это должно представлять положительный эффект NOE и указывать на близкое пространственное отношение этих протонов. В этом эксперименте NOE устанавливают для дуплета при 5,16 мкг/г, который является другим метинфурановым протоном. Другой NOE обнаруживают также для ароматических протонов, что указывает на то, что этот триплет представляет бензильный протон, наблюдают также NOE для мультиплета при 1,93-2,20 мкг/г NOE для других фуранметиленовых протонов.
(к) Получение 2-(4-фторфенил)-5-(3-фталимидилпропокси)тетрагирофурана (соединений 124, 125).
Эти соединения получают из соединения 123 с помощью методики, подобной той, которая изложена в примере 1(и), замещая соединение 105 соединением 123.
Н1 ЯМР (СDС13):
124 (транс): 1,65 (m, 1H); 1,80 (m, 1H); 2,00 (m, 2Н); 2,12 (m, 1H); 2,31 (m, 1H); 3/48 (m, 1H); 3,82 (m, 3H); 5,02 (t, 1H); 5,28 (dd, 1H); 7,00 (t, 2H); 7,29 (m, 2H); 7,71 (m, 2H); 7,85 (m, 2H);
125 (цис): 1,90 (m, 2H); 1,99 (m, 4H); 2,19 (m, 1H); 3,48 (m, 1H); 3,82 (m, 2H); 3,88 (m, 1H); 4,94 (m, 1H); 5,15 (dd, 1H); 7,00 (t, 2H); 7,30 (m, 2H); 7,71 (m, 2H); 7,84 (m, 2H).
(л) Получение 3-фталимидилпропанола (соединение 106).
3-Бромпропанол (4,0 г, 28,78 ммоля), калия фталамид (8,0, 43,17 ммоля) и калия карбонат (4,0 г, 28,78 ммоля) добавляют в 20 мл ДМФ. Реактивную смесь перемешивают при 70oС в течение 4 часов, реакцию резко прекращают водой и экстрагируют этилацетатом. Органический слой смывают водой, насыщенным раствором NaCl и выпаривают в вакууме до твердого вещества, которое кристаллизуют в этилацетате (3,5 г, 67%).
(м) Получение транс- и цис-2-(3,4,5-триметоксифенил)-5-(3-аминопропокси)тетрагидрофурана (соединения 109, 110).
Соединение 107 (455 мг, 1,03 ммоля) и гидразин моногидрат (165,3 мг, 5,16 ммоля) добавляют в 2 мл этанола. Реактивную смесь кипятят в сосуде с обратным холодильником в течение 2 часов, реакцию прекращают водой и экстрагируют дихлорметаном. Органический слой промывают водой и насыщенным раствором NaCl, высушивают над MgSO4, фильтруют и выпаривают в вакууме для получения транс-продукта 109 (225 мг, 70%).
Н1 ЯМР (СDС13): 1,75 (m, 2H); 1,78 (m, 1H); 1,96 (m, 1Н ); 2,20 (m, 1H); 2,40 (m, 1H); 2,82 (t, 2H); 3,55 (m, lH); 3,81 (m, 1H); 3,83 (s, 3Н); 3,87 (s, 6H); 5,00 (t, lH); 5,34 (dd, 1H); 6,56 (s, 2H).
Цис-изомер 110 получают из соединения 108 с помощью методики, аналогичной той, которая описана для соединения 109.
Н1 ЯМР (СDС13): 1,76 (m, 2H); 2,08 (m, 3Н); 2,27 (m, 1H); 2,82 (t, 2H); 3,55 (m, 1H); 3,84 (s, 3Н); 3,88 (s, 6H); 3,92 (m, 1H); 4,95 (m, 1H); 5,20 (m, 1H); 6,64 (s, 2H).
(н) Получение 2-(4-фторфенил)-5-(3-аминопропокси)тетрагидрофурана (соединения 126, 127).
Эти соединения получают из соединений 124 и 125 с помощью методики, аналогичной той, которая описана в примере 1(1), путем замены соединений 107 и 108 соединениями 124 и 125.
Н1 ЯМР (СDС13):
124 (транс): 1,75 (m, 3Н); 1,96 (m, 1H); 2,20 (m, 1H); 2,40 (m, 1H); 2,82 (t, 2H); 3,54 (m, 1H); 3,83 (m, 1H); 5,05 (t, 1H); 5,32 (dd, 1H); 7,01 (t, 2H); 7,30 (m, 2H);
125 (цис): 1,74 (m, 2H); 1,97 (m, 1H); 2,05 (m, 2H); 2,25 (m, 1H); 2,77 (t, 2H); 3,47 (m, 1H); 3,85 (m, 1H); 4,95 (m, 1H); 5,15 (dd, 1H); 7,00 (t, 2H); 7,34 (m, 2H).
(о) Получение транс- и цис-2-(3,4,5-триметоксифенил)-5-[3-(N'-метил-N'-гидроксиуреидил)пропокси]тетрагидрофурана. (соединений 1,3).
Соединение 109 (60 мг, 0,19 ммоля) и трифосген (23 мг, 0,078 ммоля) растворяют в 3 мл дихлорметана. Триэтиламин (29,3 мг, 0,29 ммоля) добавляют к этому раствору. Реактивную смесь кипятят в сосуде с обратным холодильником в течение 2 часов и затем охлаждают на ледяной бане. К этому холодному раствору добавляют триэтиламин (34,0 мг, 0,34 ммоля) и метилгидроксиамингидрохлорид (32,2 мг, 0,39 ммоля). Реактивную смесь перемешивают при комнатной температуре в течение 16 часов, реакцию прекращают водой и экстрагируют дихлорметаном. Органический слой промывают водой и насыщенным раствором NaCl и выпаривают в вакууме до масла, которое очищают с помощью препаративной хроматографии (двуокись кремния, ацетилацетат) для получения транс-продукта 1 (51 мг, 69%).
Н1 ЯМР (СDС13): 1,82 (m, 3Н); 1,95 (m, 1H); 2,22 (m, 1H); 2,40 (m, 1H); 3,15 (s, 3Н); 3,40 (m, 2H); 3,58 (m, 1H); 3,84 (s, 3Н); 3,85 (m, 1H); 3,88 (s, 6H); 5,00 (t, 1H); 5,33 (m, 1H); 6,32 (m, 1H); 6,56 (s, 2H); 7,37 (s, 1H).
Цис-изомер 3 получают из соединения 110 с помощью методики, аналогичной той, которая описана для соединения 1.
Н1 ЯМР (СDС13): 1,83 (m, 2H); 2,07 (m, 3Н); 2,28 (m, 1H); 3,13 (s, 3Н); 3,35 (m, 2H); 3,55 (m, 1H); 3,84 (s, 3Н); 3,87 (s, 6H); 3,88 (m, 1H); 4,97 (m, 1H); 5,20 (m, 1H); 6,22 (m, 1H); 6,63 (s, 2H); 7,37 (s, 1H).
(п) Получение 2-(4-фторфенил)-5-[3-(N'-метил-N'-гидроксиуреидил)пропокси]тетрагидрофурана (соединения 9, 11).
Эти соединения получают из соединений 126 и 127 с помощью методики, аналогичной той, которая описана в примере 1(о), путем замены соединений 109 и 110 соединениями 126 и 127.
Н1 ЯМР (СDС13):
9 (транс): 1,70 (m, 1H); 1,78 (m, 2H); 1,96 (m, 1H); 2,19 (m, 1H); 2,40 (m, 1H); 3,10 (s, 3Н); 3,31 (m, 2H); 3,51 (m, 1H); 3,83 (m, 1H); 5,05 (t, 1H); 5,30 (dd, 1H); 6,38 (t, 1H); 7,01 (t, 2H); 7,28 (m, 2H);
11 (цис): 1,80 (m, 2H); 2,05 (m, 3Н); 2,24 (m, 1H); 3,06 (s, 3Н); 3,30 (m, 2H); 3,48 (m, 1H); 3,86 (m, 1H); 4,98 (m, 1H); 5,16 (dd, 1H); 6,30 (t, 1H); 7,02 (t, 2H); 7,31 (m, 2H); 8,08 (bs, 1H).
(р) Получение транс- и цис-2-(3,4,5-три-метокисфенил)-5-[3-(N'-н-бутил-N'-гидроксиуреидил)пропокси]тетрагидрофурана (соединения 2, 4).
Соединение 109 (60 мг, 0,19 ммоля) и трифосген (23 мг, 0,078 ммоля) растворяют в 3 мл дихлорметана. К этому раствору добавляют триэтиламин (29,3 мг, 0,29 ммоля). Реактивную смесь кипятят в сосуде с обратным холодильником в течение 2 часов и затем охлаждают на ледяной бане. К этому холодному раствору добавляют бутилгидроксиамин (51,4 мг, 0,29 ммоля) и метилгидроксиамина гидрохлорид (32,2 мг, 0,39 ммоля). Реактивную смесь перемешивают при комнатной температуре в течение 16 часов, реакцию прекращают водой и экстрагируют дихлорметаном. Органический слой промывают водой и насыщенным раствором NaCl и выпаривают в вакууме до масла. Транс-продукт отделяют с помощью препаративной хроматографии (двуокись кремния, этилацетат) (46,9 мг, 57%).
Н1 ЯМР (СDС13): 0,92 (t, 3Н); 1,32 (m, 2H); 1,58 (m, 2H); 1,81 (m, 2H); 2,28 (m, 1H); 3,35 (m, 2H); 3,47 (m, 2H); 3,54 (m, 1H); 3,84 (s, 3H); 3,87 (s, 6H); 3,88 (m, 1H); 5,20 (m, 1H); 6,22 (m, 1H); 6,63 (s, 2H).
(с) Получение 2-(4-фторфенил)-5-[3-(N'-н-бутил-N'-гидроксиуреидил)пропокси]тетрагидрофурана (соединения 10, 12).
Эти соединения получают из соединений 126 и 127 с помощью методики, аналогичной той, которая описана в примере 1(о), путем замены соединений 109 и 110 соединениями 126 и 127.
Н1 ЯМР (СDС13):
10 (транс): 0,90 (t, 3Н); 1,30 (m, 2H); 1,55 (m, 2H); 1,70 (m, 1H); 1,78 (m, 2H); 1,96 (m, 1H); 2,19 (m, 1H); 2,40 (m, 1H); 3,31 (m, 2H); 3,44 (m, 2H); 3,52 (m, 1H); 3,82 (m, 1H); 5,05 (t, 1H); 5,30 (dd, 1H); 6,32 (t, 1H); 7,00 (t, 2H); 7,28 (m, 2H); 7,55 (bs, 1H);
12 (цис): 0,90 (t, 3Н); 1,30 (m, 2H); 1,52 (m, 2H); 1,80 (m, 2H); 2,04 (m, 3Н); 2,24 (m, 1H); 3,06 (s, 3Н); 3,30 (m, 2H); 3,40 (m, 2H); 3,48 (m, 1H); 3,85 (m, 1H); 4,98 (t, 1H); 5,16 (dd, 1H); 6,27 (t, 1H); 7,03 (t, 2Н); 7,32 (m, 2H); 7,53 (bs, 1H).
Пример 2. Получение 2-(3,4-диметоксифенил)-5-[3-(N'-замещенный-N'-гидроксиуреидилпропокси]тетрагидрофурана (5-8).
(а) Получение 4-(3',4'-диметоксифенил)-4-оксобутиронитрила (111).
Одну часть чистого акрилонитрила (3,2 мл, 0,048 ммоля) и триэтиламина (5 мл, 0,11 моль) добавляют в перемешанную смесь 3,4-диметоксибензальдегида (7,8 г, 0,047 моль) и 3-бензил-5-(2-гидроксиэтил)-4-метилтиазолия хлорида (5,3 г, 0,02 моль) в сухом диметилформамиде (25 мл) под аргоном. Смесь оставляли на ночь при комнатной температуре. Реактивную смесь разводили водой и экстрагировали этилацетатом (3х100 мл). Органический экстракт промывают водой (3х100 мл), соляным раствором (3х100 мл) и растворитель удаляют под пониженным давлением для получения амбрового масла. Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат : гексаны, 1:1) выявляет смесь трех пятен при Rf 0,80 (начальный альдегид), 0,50 (соединение 1) и 0,30 (неизвестный побочный продукт). Образец очищают с помощью флэш-хроматографии на силикагеле 60 (230-400 меш) для получения желаемого соединения (2,26 г, 22%) в виде желтого твердого вещества.
Н1 ЯМР (СDС13): 2,78 (t, 2H, J=8 Гц), 3,33 (t, 2H, J=8 Гц), 3,96 (s, 3H), 3,98 (s, 3H), 6,90 (d, 1H, J=8,5 Гц), 7,52 (d, J=2 Гц, 2H), 7,58 (dd, J=2 и 8 Гц, 2H).
(б) Получение 4-(3',4'-диметоксифенил)-4-оксимасляной кислоты (112).
Перемешанный раствор 4-(3',4'-диметоксифенил)-4-оксобутиронитрила (111) (2,26 г, 0,01 моль) в уксусной кислоте (15 мл) и хлористоводородной кислоте (12N, 40 мл) нагревают при кипячении в сосуде с обратным холодильником в течение 1,5 часов и охлаждают до комнатной температуры. Растворитель удаляют под пониженным давлением для получения коричневого твердого вещества. Перекристаллизация из воды позволяет получить соединение 112 в виде светло-коричневых кристаллов (1,57 г, 66%).
Н1 ЯМР (СDС13): 2,80 (t, J=7,5 Гц, 2H), 3,30 (t, J=7,5 Гц, 2H), 3,94 (s, 3H), 3,96 (s, 3H), 6,89 (d, 1H, J=9 Гц), 7,55 (d, 1H, J=1 Гц) и 7,64 (dd, 1H, 1 и 9 Гц).
(в) Получение 4-(3',4'-диметоксифенил)бутиролактона (113).
Раствор боргидрида натрия (0,89 г, 0,023 моль) в воде (4 мл) по каплям добавляют (в течение 5 мин) в перемешанный раствор соединения 112 (2,8 г, 0,012 моль) в сухом, свежедистиллированном тетрагидрофуране (40 мл) и метаноле (20 мл) под аргоном. Реактивную смесь оставляют на ночь при комнатной температуре. Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат : метанол : уксусная кислота, 9,5 : 0,5 : несколько капель) показывает присутствие начального материала. Дополнительную загрузку натрия боргидрида (0,5 г, 0,013 моль) в воде (2 мл) добавляют по каплям и реактивную смесь оставляют при комнатной температуре на три часа. Анализ с помощью тонкослойной хроматографии (система аналогична указанной выше) показывает отсутствие начального материала. Реакцию прекращают с помощью хлористоводородной кислоты (6 н. , 25 мл) и оставляют при комнатной температуре на 15 минут. Смесь экстрагируют этилацетатом (3х75 мл). Органический экстракт отмывают водой (3х75 мл), рассолом (3х75 мл) и растворитель удаляют под пониженным давлением для получения твердого вещества рыже-коричневого цвета (2,0 г, 75%).
Н1 ЯМР (CDCl3): 2,18-2,25 (m, 1H), 2,59-2,70 (m, 3H), 5,44-5,49 (m, 1H) и 6,82-6,87 (m, 3H).
(г) Получение 4-(3',4'-диметоксифенил)бутиролактола (114).
Раствор диизобутилалюминийгидрида (1,5 М, 9 мл, 13,5 моль) добавляют по каплям (в течение 30 мин) к соединению 113 (2,0 г, 9 ммоля) в сухом толуоле (40 мл) под аргоном, который охлаждают сухой ледово-ацетоновой баней. Реактивную смесь перемешивают при -78oС в течение одного часа. Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат : гексаны, 1:1) выявляет отсутствие начального материала и присутствие нового пятна у Rf 0,38. Реакцию прекращают метанолом (20 мл) и медленно нагревают до 0oС. Насыщенный раствор натриево-калиевого тартрата (50 мл) добавляют и перемешивают при 0oC в течение 45 минут. Смесь экстрагируют этилацетатом (3х100 мл) и органический экстракт смывают водой (3х75 мл) и рассолом (3х75 мл). Удаление растворителя под пониженным давлением дает темное амбровое масло (1,7 г, 84%).
Н1 ЯМР (СDС13) (смесь цис- и транс-изомеров): 1,71-2,49 (m, 8H), 2,91(br s, 1H), 3,09 (br s, 1H), 3,89 (s, 6Н); 3,90 (s, 6H); 4,97 (m, 1 Н); 5,19 (t, J=7 Гц, 1H); 5,62 (m, 1H); 5,77 (m, 1H) и 6,82-7,28 (m, 6H).
(д) Получение N-(3-гидроксипропил)фталимида (106).
Смесь 3-бромпропанола (4 г, 0,029 моль), калия фталат (8 г, 0,043 моль) и калия карбонат (4 г, 0,029 моль) в сухом ДМФ (50 мл) перемешивают и нагревают при 70oС в течение четырех часов. Смесь разбавляют водой (100 мл) и экстрагируют этилацетатом (3х75 мл). Органический экстракт промывают водой (3х100 мл) и высушивают (Na2SO4). Удаление растворителя под пониженным давлением оставляет белое твердое вещество, которое экстрагируют бензолом. Бензольный экстракт выпаривают до белого твердого вещества и перекристаллизовывают из этилацетат-гексанов для получения белых кристаллов (1,27 г, 27%).
(е) Получение транс- и цис-2-(3',4'-диметоксифенил)-5-[3-(N-фталоил)] пропокси тетрагидрофурана (115 и 116).
Одну часть трифлинового ангидрида (0,68 мл, 4,8 ммоля) добавляют в перемешанный раствор соединения 114 (0,72 г, 3,2 ммоля) в сухом дихлорметане (20 мл) и триэтиламине (0,68 мл, 4,9 ммоля) под аргоном, который охлаждают с помощью ледяной бани. Реактивную смесь перемешивают при 0oС в течение 30 минут. В реактивную смесь добавляют N-(3-гидроксипропил)фталимид (106) (1,27 г, 7 ммолей), раствору дают согреться до комнатной температуры и оставляют при этой температуре на два часа. Реакцию в растворе прекращают водным раствором бикарбоната натрия (насыщенным, 25 мл) и экстрагируют этилацетатом (3х50 мл), рассолом (3х50 мл) и высушивают (натрия сульфат). Удаляют растворитель под пониженным давлением, оставляя амберное масло (2,02 г).
Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат : гексаны, 1: 1) выявляет присутствие четырех пятен при Rf 0,80, 0,60, 0,50 и 0,35. Пятна при Rf 0,60 и 0,50 имеются в соотношении 2:1. Образец очищают с помощью флэш-хроматографии на силикагеле (230-400 меш) и элюируют этилацетатом : гексанами (3:7) для получения сначала вещества при Rf 0,60 в виде прозрачного и бесцветного масла (0,40 г, 30%), идентифицированного как транс-2-(3', 4'-диметоксифенил)-5-[N-фталоил)] пропокситетрагидрофуран (115) (0,40 г, 30%).
Н1 ЯМР (СDС13): 1,34-1,94 (m, 2H); 1,96-2,05 (m, 2H); 2,09-2,20 (m, 1Н); 2,25-2,36 (m, 1H); 3,46-3,53 (m, 1H); 3,84 (t, 9 Гц, 2Н); там также имеется 1 спрятанный протонный мультиплет, 3,88 (s, 3Н); 3,91 (s, 3Н); 5,01 (t, 7,3 Гц, 1H); 5,30 (dd, J=2 и 5 Гц, 1 Гц); 6,82-6,90 (m, 3Н); 7,71-7,74 (m, 2H) и 7,84-7,88 (m, 2Н).
Для определения стереохимии этой молекулы проводят эксперимент с разницей NOE. В этом эксперименте мультиплет при 5,01 мкг/г облучают расщепляющими импульсами очень низкой радиочастоты и обработку данных проводят так, чтобы измерять только наличие увеличения сигнала. Это должно представлять положительный эффект NOE и указывать на близкое пространственное отношение этих протонов. В этом эксперименте NOE устанавливают для дуплета при 2,25-2,36 мкг/г, который является протоном фуранового кольца. Другой NOE обнаруживают также для ароматических протонов, что указывает на то, что этот триплет представляет бензильный протон. Не наблюдают NOE для двойного дуплета при 5,30 мкг/г, что указывает на то, что это транс-изомер.
Продолжающееся элюирование с той же системой растворителя, дает пятно при Rf 0,50 в виде бесцветного масла (0,21 г, 15%), идентифицированного как цис-2-(3',4'-диметоксифенил)-5-[3-(N-фталоил)]пропокситетрагидрофуран (116).
Н1 ЯМР (СDС13): 1,92-2,12 (m, 6Н); 3,44-3,52 (m, 1H); 3,86 (s, 3 Н); 3,88 (s, 3Н); 3,76-3,93 (m, 3Н); 4,89-4,94 (m, 1H); 5,35 (d, J=4 Гц); 6,89 (d, J= 8 Гц); 6,87 (dd, J=2 и 8 Гц); 6,92 (d, J=2 Гц); 7,69-7,72 (m, 2H) и 7,82-7,85 (m, 2H).
Для определения стереохимии этой молекулы проводят эксперимент с разницей NOE. В этом эксперименте мультиплет при 4,89-4,94 мкг/г облучают расщепляющими импульсами очень низкой радиочастоты и обработку данных проводят так, чтобы измерять только наличие увеличения сигнала. Это должно представлять положительный эффект NOE и указывать на близкое пространственное отношение этих протонов. В этом эксперименте NOE устанавливают для дуплета при 5,35 мкг/г, который является другим метинфурановым протоном. Это показывает, что эта молекула является цис-изомером. Другой NOE обнаруживают также для ароматических протонов, что указывает на то, что этот триплет представляет бензильный протон. Присутствует также NOE для мультиплета при 1,92-2,12 мкг/г, который содержит другие фуранметиленовые протоны. Хроматография также дает выход смеси соединений 115 и 116 (0,342 г, 26%).
(ж) Получение транс- и цис-2-(3',4'-диметоксифенил)-5-(3-аминопропокси)тетрагидрофурана (117).
Чистый гидразин гидрат (150 мкл, 3,2 ммоля) добавляют в перемешанный раствор соединения 115 (253 мг, 0,62 ммоля) в абсолютном этаноле (1,5 мл). Раствор нагревают кипячением в сосуде с обратным холодильником в течение 5 минут, после чего из раствора осаждается белое твердое вещество. Смесь нагревают кипячением в сосуде с обратным холодильником в течение двух часов. Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат : гексаны, 1:1) выявляет отсутствие начального материала и присутствие пятна в начале хроматограммы. Реакцию прекращают водой (10 мл) и экстрагируют дихлорметаном (5х10 мл). Органическую фазу промывают водой (2х10 мл), рассолом (2х10 мл) и высушивают (натрия сульфат). Удаление растворителя под пониженным давлением оставляет бесцветное масло (150 мг, 86%).
Н1 ЯМР (СDС13): 1,25 (br s, 2Н); 1,68-l,78 (m, 3Н); 1,81-1,98 (m, 1H); 2,14-2,20 (m, 1H); 2,3-2,36 (m, 1H); 2,80 (t, J=6,5 Гц, 2Н); 3,47-3,55 (m, 1H); 3,78-3,87 (m, частично спрятанный, 1H); 3,86 (s, 3Н), 3,88 (s, 3Н); 4,99 (t, J=7 Гц, 1H); 5,31 (dd, J=2 и 6 Гц, 1H); 6,80-6,88 (m, 3Н).
(з) Получение цис-2-(3',4'-диметоксифенил)-5-(3-аминопропокси)тетрагидрофурана (118).
Чистый гидразин гидрат (125 мкл, 2,57 ммоля) добавляют в перемешанный раствор соединения 116 (210 мг, 0,51 ммоля) в абсолютном этаноле (3,0 мл). Раствор нагревают кипячением в сосуде с обратным холодильником в течение 5 минут, после чего из раствора осаждается белое твердое вещество. Смесь нагревают кипячением в сосуде с обратным холодильником в течение двух часов. Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат : гексаны, 1:1) выявляет отсутствие начального материала и присутствие пятна в начале хроматограммы. Реакцию прекращают водой (10 мл) и экстрагируют дихлорметаном (5х10 мл). Органическую фазу промывают водой (2х10 мл), рассолом (1х10 мл) и высушивают (натрия сульфат). Удаление растворителя под пониженным давлением оставляет густое масло (105 мг, 73%).
Н1 ЯМР (СDС13): 1,45 (br s, 2H); 1,73-1,78 (m, 2H); 2,01-2,12 (m, 3Н); 2,19-2,29 (m, 1H); 2,81 (t, J=7 Гц, 2H); 3,48-3,53 (m, 1H); 3,85-3,93 (m, частично спрятанный, 1H); 3,88 (s, 3Н); 3,90 (s, 3Н); 4,96-5,01 (m, 1H); 5,17 (dd, J=3 и 6 Гц, 1H); 6,83 (d, J=8 Гц, 1H); 6,89 (dd, J=2 и 8 Гц, 1H) и 6,96 (d, J=2 Гц, 1H).
(и) Получение транс-2-(3', 4'-диметоксифенил)-5-[(3-(N-бутил-N-гидроксиуреидил)пропокси]тетрагидрофурана (5).
Триэтиламин (32 мкл, 0,22 ммоля) и затем трифосген (19 мг, 0,06 ммоля) добавляют в перемешанный раствор соединения 117 (53 мг, 0,19 ммоля) в сухом дихлорметане (3 мл) под аргоном. Раствор нагревают кипячением в сосуде с обратным холодильником в течение 30 минут и охлаждают до комнатной температуры. Твердый н-бутилгидроксиламин (34 мг, 0,38 ммоля) добавляют в одной части в раствор, который оставался на ночь при комнатной температуре. Реакцию в растворе прекращают водой (10 мл) и экстрагируют дихлорметаном (3х10 мл). Комбинированную органическую фазу промывают водным раствором бикарбоната натрия (насыщенного, 3х10 мл) и высушивают (натрия сульфат).
Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат) выявляет сложную смесь при Rf 0,90, 0,50, 0,25 и 0,00. Образец очищают с помощью флэш-хроматографии на силикагеле (230-400 меш) и элюируют этилацетатом для получения пятна при Rf 0,50 в виде непрозрачного масла (8 мг, 11%).
Н1 ЯМР (СDС13): 0,92 (t, J=7 Гц, 3Н), 1,27-1,39 (m, 2H); 1,51-1,61 (m, 2H); 1,71-1,86 (m, 3Н); 1,88-2,15 (m, 1H); 2,17-2,29 (m, 1H); 2,32-2,42 (m, 1H), 3,28-3,58 (m, 4H), 3,81-3,94 (m, частично спрятанный, 2H), 3,87 (s, 3Н), 3,90 (s, 3Н); 5,49-5,05 (m, 1H), 5,31-5,38 (m, 1H); 6,28-6,34 (m, 1H) и 6,81-6,86 (m, 3Н).
ИК-спектр (пленка): 3407, 3193, 2933, 1640, 1516, 1263, 1029 см-1.
(к) Получение транс-2-(3',4'-диметоксифенил)-5-[3-(N-метил-N-гидроксиуреидил)пропокси]тетрагидрофурана (6).
Трифосген (12 мг, 0,42 ммоля) и затем сразу триэтиламин (17 мкл, 0,12 ммоля) добавляют в перемешанный раствор соединения 117 (32 мг, 0,011 ммоля) в сухом дихлорметане (3 мл) под аргоном. Раствор нагревают кипячением в сосуде с обратным холодильником в течение 2 часов, охлаждают до комнатной температуры и помещают в ледяную баню. Чистый триэтиламин (32 мкл, 0,23 ммоля), а затем хлористоводородную соль метилгидроксиламина (19 мг, 0,23 ммоля) добавляют в реактивную смесь. Реактивную смесь оставляют на ночь при комнатной температуре. Затем реакцию в растворе прекращают водой (10 мл) и экстрагируют дихлорметаном (3х10 мл). Органический экстракт промывают водой (3х10 мл), рассолом (3х10 мл) и растворитель удаляют под пониженным давлением для получения амберного масла. Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат) выявляет только одно новое пятно при Rf 0,30. Образец очищают с помощью флэш-хроматографии на силикагеле (230-400 меш) и элюируют этилацетатом для получения желаемого соединения в виде амберного масла (12 мг, 30%).
Н1 ЯМР (СDС13): 1,73-1,84 (m, 2H); 1,90-2,01 (m, 1H); 2,03-2,13 (m, 1H); 2,18-2,29 (m, 1H); 2,32-2,43 (m, 1H); 3,13 (s, 3Н); 3,30-3,44 (m, 2H); 3,49-3,59 (m, 1H); 3,82-3,92 (m, частично спрятанный, 3Н); 3,88 (s, 3Н); 3,91 (m, 3H); 4,96-5,04 (m, 1H); 5,34 (dd, J=2 и 5 Гц, 1H); 6,34 (br t, J=5 Гц, 1H) и 6,82-6,68 (m, 3H).
ИК-спектр (пленка): 3407, 3229, 2935, 1636, 1516, 1263, 1029 см-1.
(л) Получение цис-2-(3', 4'-диметоксифенил)-5-[3-(N-бутил-N-гидроксиуреидил)пропокси]тетрагидрофурана (7).
Трифосген (18 мг, 0,06 ммоля) и затем сразу триэтиламин (80 мг, 0,57 ммоля) добавляют в перемешанный раствор соединения 118 (50 мг, 0,18 ммоля) в сухом дихлорметане (3 мл) под аргоном. Раствор нагревают кипячением в сосуде с обратным холодильником в течение 2 часов, охлаждают до комнатной температуры и помещают в ледяную баню. Добавляют чистый триэтиламин (50 мг, 0,35 ммоля), а затем твердый н-бутилгидроксиламин (32 мг, 0,36 ммоля). Реактивную смесь оставляют на ночь при комнатной температуре. Затем реакцию в растворе прекращают водой (10 мл) и экстрагируют дихлорметаном (3х10 мл). Органический экстракт промывают водой (3х10 мл), рассолом (3х10 мл) и растворитель удаляют под пониженным давлением для получения амберного масла. Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат) выявляет два новых пятна в приблизительно равных количествах при Rf 0,85 и 0,45. Образец очищают с помощью флэш-хроматографии на силикагеле 60 (230-400 меш) и элюируют этилацетатом для получения первого пятна при Rf 0,85 в виде амберного масла (26 мг). Продолжающееся элюирование той же системой растворителя затем дает указанное в заголовке соединение в виде амберного масла (25 мг, 35%).
Н1 ЯМР (CDCl3): 1,1 (t, J=7 Гц, 3Н); 1,25-1,37 (m, 2H); 1,49-1,59 (m, 2H); 1,76-1,84 (m, 2H); 1,99-2,1 (m, 3Н); 2,19-2,26 (m, 1H); 3,26-3,54 (m, 5H); 3,84-3,92 (m, частично спрятанный, 1H); 3,87 (s, 3Н); 3,88 (s, 3Н); 4,94-5,02 (m, 1H); 5,17 (d, J=4 Гц, 1H); 6,2 (t, J=4 Гц, 1H); 6,52 (br s, 1H); 6,83 (d, J=8 Гц, 1H) и 6,89-6,95 (m, 2H).
ИК-спектр (пленка): 2913, 1640, 1570, 1463, 1262, 1139 и 1031 см-1.
(м) Получение цис-2-(3', 4'-диметоксифенил)-5-[3-(N-метил-N-гидроксиуреидил)пропокси]тетрагидрофурана (8).
Трифосген (20 мг, 0,07 ммоля) и затем сразу триэтиламин (80 мкл, 0,57 ммоля) добавляют в перемешанный раствор соединения 118 (56 мг, 0,2 ммоля) в сухом дихлорметане (3 мл) под аргоном. Раствор нагревают кипячением в сосуде с обратным холодильником в течение 2 часов, охлаждают до комнатной температуры и помещают в ледяную баню. Добавляют чистый триэтиламин (80 мкл, 0,57 ммоля), а затем твердую хлористоводородную соль метилгидроксиламина (32 мг, 0,39 ммоля). Реактивную смесь оставляют на ночь при комнатной температуре. Затем реакцию в растворе прекращают водой (10 мл) и экстрагируют дихлорметаном (3х10 мл). Органический экстракт промывают водой (3х10 мл), рассолом (3х10 мл) и растворитель удаляют под пониженным давлением для получения амберного масла. Анализ с помощью тонкослойной хроматографии (силикагель; этилацетат) выявляет одно пятно при Rf 0,30 и некоторое количество материала у начала хроматограммы. Образец очищают с помощью флэш-хроматографии на силикагеле 60 (30-400 меш) и элюируют этилацетатом для получения указанного в заголовке соединения в виде амберного масла (30 мг, 42%).
Н1 ЯМР (СDС13): 1,76 (m, 2H); 1,98-2,10 (m, 3Н); 2,18-2,26 (m, 1H); 3,07 (s, 3Н); 3,25-3,37 (m, 2H); 3,46-3,54 (m, 1H); 3,85-3,90 (m, частично спрятанный, 1H); 3,87 (s, 3Н), 3,88 (s, 3Н); 4,93-5,00 (m, 1H); 5,16 (d, J=4 Гц, 1H); 6,27 (t, J=5 Гц, 1H); 6,83 (d, J=8 Гц, 1H) и 6,88-6,93 (m, 2H).
ИК-спектр (чистый): 2933, 1643, 1518, 1261 и 1029 см-1.
Пример 3. Получение 2-(2,4,5-триметоксифенил)-5-(3-гидроксиуреидилпропокси)тетрагидрофурана (13) и 2-(4-фторфенил)-5-(3-гидроксиуреидилпропокси)тетрагидрофурана (14, 15).
(a) Получение 2-(3,4,5-триметоксифенил)-5-(3-бромпропокси)тетрагидрофурана (соединение 128).
Соединение 105 (1,0 г, 3,94 ммоля) растворяют в 4 мл дихлорметана. К этому раствору добавляют триэтиламин (597 мг, 5,90 ммоля). Реактивную смесь охлаждают на ледяной бане и по каплям добавляют трифторуксусный ангидрид (1,24 г, 5,90 ммоля). Реактивную смесь перемешивают при 0oС в течение 30 минут и затем добавляют 3-бромпропанол (1,84 г, 13,27 ммоля). Реактивную смесь согревают до комнатной температуры и перемешивают при комнатной температуре в течение 2 часов. Реакцию прекращают насыщенным водным раствором NаНСО3 и экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором NaCl, высушивают над MgSO4, фильтруют и выпаривают в вакууме до масла, которое очищают на хроматографической колонке (окись кремния, гексан/этилацетат 4:1) (соединение 128: 430 мг и его цис-изомер 250 мг; общий выход 46%).
Н1 ЯМР (СDС13): 128 (транс): 1,77 (m, 1H); 1,98 (m, 1H); 2,15 (m, 2H); 2,20 (m, 1H); 2,40 (m, 1H); 3,53 (t, 2H), 3,60 (m, 1H); 3,83 (s, 3H); 3,87 (m, 1H); 3,89 (s, 6Н); 5,01 (t, 1H); 5,35 (dd, 1H); 6,57 (s, 2H).
(б) Получение 2-(4-фторфенил)-5-(3-бромпропокси)тетрагидрофурана (соединения 129, 130).
Эти соединения получают из соединения 123 с помощью методики, аналогичной той, которая представлена в примере 3(а), замещая соединение 105 соединением 123.
Н1 ЯМР (СDС13):
129 (транс): 1,72 (m, 1H); 1,98 (m, 1H); 2,14 (m, 2H); 2,20 (m, 1H); 2,40 (m, 1H); 3,53 (t, 2H), 3,60 (m, 1H); 3,89 (m, 1H); 5,06 (t, 1H); 5,34 (m, 1H); 7,02 (t, 2H); 7,30 (m, 2H);
130 (цис): 1,98 (m, 1H); 2,07 (m, 2H); 2,14 (m, 2H); 2,26 (m, 1H); 3,52 (t, 2H), 3,58 (m, 1H); 3,93 (m, 1H); 5,20 (dd, 1H); 7,03 (t, 2H); 7,35 (m, 2H).
(в) Получение 2-(3,4,5-триметоксифенил)-5-(3-о-бензилгидроксиламинопропокси)тетрагидрофурана (соединение 131).
Соединение 128 (260 мг, 0,69 ммоля) растворяют в 2 мл 1,3-диметил-3,4,5,6-тетрагидро-2-(1H)-пиримидинона (DMPU). В этот раствор добавляют натрия карбонат (220,4 мг, 2,08 ммоля) и бензилгидроксиламина гидрохлорид (166 мг, 1,04 ммоля). Реактивную смесь перемешивают при 80oС в течение 16 часов, реакцию прекращают водой и экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором натрия хлорида, высушивают над MgSО4, фильтруют и выпаривают до масла, которое очищают с помощью флэш-хроматографии, используя в качестве растворителя этилацетат (114 мг, 40%).
Н1 ЯМР (СDС13): 1,72 (m, 1H); 1,82 (m, 2H); 1,92 (m, 1H); 2,18 (m, 1H); 2,36 (m, 1H); 3,06 (t, 2H), 3,52 (m, 1H); 3,81 (m, 1H); 3,83 (s, 3Н); 3,87 (s, 6H); 4,71 (s, 2H); 4,98 (t, 1H); 5,30 (dd, 1H); 6,55 (s, 2H); 7,35 (m, 5H).
(г) Получение 2-(4-фторфенил)-5-(3-о-бензилгидроксиламинопропокси)тетрагидрофурана (соединения 132, 133).
Эти соединения получают из соединений 129 и 130 с помощью методики, аналогичной той, которая представлена в примере 3(в), замещая соединение 128 соединениями 129 и 130.
Н1 ЯМР (СDС13):
132(транс): 1,70 (m, 1H); 1,83 (m, 2H); 1,94 (m, 1H); 2,17 (m, 1H); 2,38 (m, 1H); 3,07 (t, 2H); 3,52 (m, 1H); 3,82 (m, 2H); 4,71 (s, 2H); 5,02 (t, 1H); 5,30 (ss, 1H); 7,02 (t, 2H); 7,30 (m, 2H); 7,36 (m, 5H);
133 (цис): 1,85 (m, 2H); 1,96 (m, 1H); 2,05 (m, 2H); 2,26 (m, 1H); 3,05 (t, 2H); 3,50 ((m, 1H); 3,88 (m, 2H); 4,70 (s, 2H); 4,99 (m, 1H); 5,17 (dd, 1H); 5,50 (bs, 1H); 7,00 (t, 2H); 7,35 (m, 7H).
(д) Получение 2-(3,4,5-триметоксифенил)-5-(3-о-бензилгидроксиуреидилпропокси)тетрагидрофурана (соединение 134).
Соединение 131 (114 мг, 0,27 ммоля) растворяют в 3 мл дихлорметана. В этот раствор добавляют триметилсилилизоцианат (47,6 мг, 0,41 ммоля). Реактивную смесь перемешивают при комнатной температуре в течение 16 часов и затем кипятят в сосуде с обратным холодильником в течение 4 часов. Реакцию прекращают насыщенным раствором аммония хлорида, экстрагируют этилацетатом и выпаривают до масла. Продукт отделяют с помощью препаративной тонкослойной хроматографии, используя в качестве растворителя этилацетат.
Н1 ЯМР (СDС13): 1,72 (m, 1H); 1,92 (m, 3H); 2,16 (m, 1H); 2,38 (m, 1H); 3,50 (m, 1H), 3,62 (m, 2H); 3,80 (m, 1H); 3,82 (s, 3H); 3,84 (s, 6Н); 4,81 (s, 2H); 4,99 (t, 1H); 5,30 (m, 3H); 6,54 (s, 2H); 7,37 (s, 5H).
(е) Получение 2-(4-фторфенил)-5-(3-о-бензилгидроксиуреидилпропокси)тетрагидрофурана (соединения 135, 136).
Эти соединения получают из соединений 132 и 133 с помощью методики, аналогичной той, которая представлена в примере 3(д), замещая соединение 131 соединениями 132 и 133.
Н1 ЯМР (СDС13):
135 (транс): 1,70 (m, 1H); 1,93 (m, 3Н); 2,16 (m, 1H); 2,39 (m, 1H); 3,50 (m, 1H); 3,62 (m, 2H); 3,80 (m, 1H); 4,82 (s, 2H); 5,04 (t, 1H); 5,30 (ss, 1H); 5,35 (bs, 2H); 7,00 (t, 2H); 7,29 (m, 2H); 7,38 (s, 5H);
136 (цис): 1,98 (m, 4H); 2,08 (m, 1H); 2,25 (m, 1H); 3,48 (m, 1H); 3,62 (m, 2H); 3,83 (m, 1H); 4,81 (s, 2H); 4,98 (m, 1H); 5,17 (dd, 1H); 5,42 (bs, 1H); 7,00 (t, 2H); 7,33 (m, 2H); 7,38 (s, 5H).
(ж) Получение 2-(3,4,5-триметоксифенил)-5-(3-гидроксиуреидилпропокси)тетрагидрофурана (соединения 13).
Соединение 134 (90 мг, 0,19 ммоля) растворяют в 2 мл этилацетата и затем добавляют Pd/C (10%) (18 мг). Реактивную смесь гидрогенируют при давлении в баллоне в течение 16 часов. Продукт отделяют с помощью препаративной тонкослойной хроматографии, используя в качестве растворителя этилацетат (68 мг).
Н1 ЯМР (CDCl3): 1,75 (m, 1H); 1,91 (m, 2H); 1,95 (m, 1H); 2,20 (m, 1H); 2,37 (m, 1H); 3,58 (m, 1H), 3,66 (m, 2H); 3,81 (s, 3Н); 3,85 (m, 1H); 3,87 (s, 6H); 5,00 (t, 1H); 5,35 (dd, 1H); 5,41 (bs, 2H); 6,53 (s, 2H); 8, 39 (s, 1H).
(з) Получение 2-(4-фторфенил)-5-(3-гидроксиуреидилпропокси)тетрагидрофурана (соединения 14, 15).
Соединения 14 и 15 получают из соединений 135 и 136 с помощью методики, аналогичной той, которая представлена в примере 3(ж), замещая соединение 134 соединениями 135 и 136.
Н1 ЯМР (СDС13):
14 (транс): 1,72 (m, 1H); 1,93 (m, 3Н); 2,38 (m, 1H); 3,58 (m, 1H); 3,67 (m, 2H); 3,85 (m, 1H); 5,05 (t, 1H); 5,33 (dd, 1H); 5,48 (bs, 2H); 7,00 (t, 2H); 7,28 (m, 2H); 8,48 (bs, 1H).
15 (цис): 1,92 (m, 2H); 2,01 (m, 1H); 2,10 (m, 2H); 2,26 (m, 1H); 3,53 (m, 1H); 3,64 (m, 2H); 3,87 (m, 1H); 4,98 (m, 1H); 5,20 (dd, 1H); 5,43 (bs, 2H); 7,01 (m, 2H); 7,31 (m, 2H); 8,43 (bs, 1H).
Пример 4. Получение транс-2-(3-(N-гидроксиуреидил)-бут-1-инил)-5-(4-фторфенил) тетрагидрофурана (207).
Схема синтеза соединения 207 проиллюстрирована на схеме 8 (см. в конце описания).
(а) Получение 2-(трет-бутилдиметилсилилокси)-5-(4-фторфенил)тетрагидрофурана (соединение 202).
2-Гидрокси-5-(4-фторфенил)тетрагидрофуран (550 мг, 3,0 ммоля), трет-бутилдиметилсилилхлорид (498 мг, 3,3 ммоля) и имидазол (450 мг, 6,6 ммоля) растворяют в 2 мл сухого ДМФ. Этот раствор в течение ночи перемешивают под сухим аргоном, выливают в 200 мл воды и экстрагируют смесью этилацетата и гексана в соотношении 2:1 (3х100 мл). Комбинированные органические экстракты промывают водой (4х200 мл) и рассолом (100 мл), высушивают над сульфатом натрия и выпаривают для получения 830 мг (93%) 2-(трет-бутилдиметилсилилокси)-5-(4-фторфенил)тетрагидрофурана (202, смесь цис- и транс-изомеров) в виде бесцветного масла, которое не нуждается в какой-либо очистке.
Н1 ЯМР (СDС13): δ 7,40-7,50 (2Н, m, меньший по количеству изомер); 7,25-7,35 (2Н, m, меньший по количеству изомер); 7,00-7,10 (2Н, m, как основной, так и меньший по количеству изомер); 5,71-5,75 (1Н, m, основной изомер); 5,59-5,62 (1Н, m, основной изомер); 5,12-5,20 (1Н, m, основной изомер); 4,90-4,98 (1Н, m, основной изомер); 2,40-2,55 (1Н, m, как основной, так и меньший по количеству изомер); 2,05-2,17 (1Н, m, как основной, так и меньший по количеству изомер); 1,87-2,00 (1Н, m, как основной, так и меньший по количеству изомер); 1,67-1,70 (1Н, m, как основной, так и меньший по количеству изомер); 0,92 (s, 9Н, как основной, так и меньший по количеству изомер); 0,16 (s, 6H, как основной, так и меньший по количеству изомер).
(б) Получение транс-2-(3-тетрагидропиранилокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофурана (соединение 204).
2-(трет-Бутилдиметилсилилокси)-5-(4-фторфенил)тетрагидрофуран (202, 593 мг, 2,0 ммоля) смешивают в 10 мл сухого метиленхлорида (дегазированном перед использованием пробулькиванием аргона). Этот раствор охлаждают до -70oС. При перемешивании при той же температуре под сухим аргоном по каплям добавляют триметилсилилбромид (290 мкл, 2,2 ммоля). Перемешивание дополнительно продолжают 1,5 ч для получения 2-бром-5-(4-фторфенил)тетрагидрофурана (203), который не выделяют, а используют в последующем химизме процесса без дальнейшей очистки (см. ниже).
В отдельной колбе растворяют 3-тетрагидропиранилокси-бут-1-ин (370 мг, 2,4 ммоля) в сухом тетрагидрофуране (5 мл). Раствор охлаждают до -60oС и, продолжая перемешивание при той же температуре под сухим аргоном, по каплям добавляют н-бутиллитий (1,0 мл, 2,4 ммоля). Перемешивание дополнительно продолжают в течение 0,5 часа, полученный раствор набирают в шприц и по каплям добавляют в перемешанный раствор 2-бромтетрагидрофурана (полученный выше) при -70oС. Перемешивание продолжают при -78oС дополнительно в течение 1,5 часов. Реактивную колбу в течение ночи держат в морозильнике (-78oС) (хотя тонкослойная хроматография не выявляет никаких изменений). Реактивную смесь выливают в 2 М раствор аммония хлорида (50 мл) и экстрагируют метиленхлоридом (3х50 мл). Раствор высушивают над сульфатом натрия и растворитель удаляют в условиях вакуума. Остаток очищают с помощью флэш-хроматографии (элюент, 10% этилацетат в гексане) для получения двух компонентов. По данным протонного ЯМР-анализа менее полярный компонент идентифицируют как транс-2-(3-тетрагидропиранилокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофуран (204, 280 мг, 45%), а более полярный компонент (230 мг) является смесью более чем одного соединения. Эту смесь удаляют.
Н1 ЯМР (СDС13): δ 7,27-7,30 (2Н, m,); 7,01 (2Н, t, J=8,7 Гц); 5,09 (1Н, t, J= 7,1 Гц); 4,91-4,95 (2Н, m); 4,57-4,64 (1H, m); 3,78-3,90 (1H, m); 3,50-3,60 (1H, m); 2,30-2,50 (2Н, m); 2,05-2,17 (1H, m); 1,70-1,90 (3Н, m); 1,50-1,65 (4Н, m); 1,48 (3Н, d, J=6,6 Гц).
(в) Получение транс-2-(3-гидрокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофурана (соединение 205).
транс-2-(3-Тетрагидропиранилокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофуран (208, 280 мг, 0,9 ммоля) растворяют в метаноле (15 мл). К этому раствору добавляют п-толуолсульфоновую кислоту (50 мг) и полученный раствор перемешивают в течение 45 минут. Добавляют насыщенный раствор бикарбоната натрия (10 мл). После 5 минут перемешивания раствор добавляют к 10 мл воды, разводят 15 мл солевого раствора и экстрагируют метиленхлоридом (3х30 мл). Комбинированные органические вещества высушивают над сульфатом натрия и растворитель удаляют через роторный испаритель для получения 212 мг (100%) транс-2-(3-гидрокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофурана (205).
Н1 ЯМР (СDС13): δ 7,29 (2Н, dd, J=8,7, 5,2 Гц); 7,01 (2Н, t, J=8,7 Гц); 5,09 (1Н, t, J= 7,4 Гц); 4,92 (1H, t, J=7,4 Гц); 4,59 (1H, q, J=6,6 Гц); 2,30-2,50 (2Н, m); 2,05-2,15 (1H, m); 2,00 (1H, br s); 1,75-1,88 (1H, m); 1,47 (3Н, d, J=6,6 Гц).
(г) Получение транс-2-(3-N-феноксикарбонилокси-N-феноксикарбониламино)-бут-1-инил)-5-(4-фторфенил)тетрагидрофурана (соединение 206).
транс-2-(3-Гидрокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофуран (205, 210 мг, 0,89 ммоля), трифенилфосфин (288 мг, 1,1 ммоля) и N,О-бис(феноксикарбонил)гидроксиламин (283 мг, 1,1 ммоля) растворяют в сухом тетрагидрофуране (5 мл). Раствор охлаждают до 0oС под сухим аргоном и по каплям добавляют диизопропилазодикарбоксилат (216 мл, 1,1 мкмоля). Перемешивание продолжают в течение 1 часа при той же температуре. Растворитель выпаривают и остаток очищают с помощью флэш-хроматографии (элюент: 10% этилацетат в гексане) для получения 250 мг (57%) транс-2-(3-N-феноксикарбонилокси-N-феноксикарбониламино)-бут-1-инил)-5-(4-фторфенил)тетрагидрофурана (206).
Н1 ЯМР (СDС13): δ 7,15-7,45 (12Н, m); 7,02 (2Н, t, J=8,6 Гц); 5,32 (1Н, q, J=7,0 Гц); 5,07 (1H, t, J=6,8 Гц); 4,96 (1H, t, J=5,7 Гц), 2,25-2,50 (2Н, m); 2,05-2,20 (1H, m); 1,70-1,85 (1H, m); 1,66 (3Н, d, J=7,0 Гц).
(д) Получение транс-2-(3-N-гидроксиуреидил)-бут-1-инил)-5-(4-фторфенил)тетрагидрофурана (соединение 207).
транс-2-(3-N-Феноксикарбонилокси-N-феноксикарбониламино)-бут-1-инил)-5-(4-фторфенил)тетрагидрофуран (206, 200 мг, 0,41 ммоля) растворяют в высоконапорной трубе в виде раствора в метиленхлориде. Растворитель выпаривают струей аргона и осадок охлаждают до -78oС. Аммиак (8 мл) конденсируют в этой трубе и добавляют 4 мл трет-бутанола. Трубу герметизируют, медленно согревают до комнатной температуры и перемешивают при комнатной температуре в течение 18 часов. Давление очень медленно снижают до нормального и трубу оставляют открытой на 1 час. Остаток переносят в колбу и дважды упаривают в роторном испарителе с добавленным толуолом. Остаток очищают с помощью флэш-хроматографии (элюент: 3% метанол в этилацетате) и еще раз очищают с помощью препаративной тонкослойной хроматографии (растворитель: 5% метанол в метиленхлориде) для получения 93 мг (78%) транс-2-(3-N-гидроксиуреидил)-бут-1-инил)-5-(4-фторфенил) тетрагидрофурана (207).
ИК-спектр (пленка): 3481, 3269, 2985, 2877, 2249, 1662, 1670, 1510, 1444, 1224, 1172, 1037 см-1.
Н1 ЯМР (СDС13): δ 8,10 (1Н, br s); 7,26 (2Н, dd, J=8,6, 5,4 Гц); 7,00 (2Н, t, J= 8,6 Гц), 5,80 (1H, br s); 5,00-5,20 (2H, m); 4,80-5,00 (1H, m); 2,20-2,50 (2H, m); 2,00-2,20 (1H, m); 1,70-1,90 (1H, m); 1,37 (3Н, dd, J= 6,9, 1,9 Гц).
Пример 5. Получение S,S,S- и S,S,R-изомеров транс-2-{3-(N-гидроксиуреидил)-бут-1-инил}-5-(4-фторфенил)тетрагидрофурана (соединения 216 и 217).
Один способ получения S,S,S- и S,S,R-изомеров транс-2-{3-(N-гидроксиуреидил)-бут-1-инил} -5-(4-фторфенил) тетрагидрофурана проиллюстрирован ниже на схеме 9 (см. в конце описания).
(а) Получение метил-3-(4-фторбензоил)пропионата (соединение 209).
К раствору 3-(4-фторбензоил)пропионовой кислоты (1,98 г, 10 ммолей) в метаноле (25 мл) добавляют 0,5 мл концентрированной серной кислоты. Полученный раствор перемешивают при комнатной температуре под аргоном в течение 2 часов. Реактивную смесь нейтрализуют насыщенным бикарбонатом натрия, метанол удаляют через роторный испаритель и остаток растворяют в 50 мл этилацетата. Полученный раствор промывают насыщенным бикарбонатом натрия (3х50 мл) и рассолом (50 мл), высушивают над сульфатом натрия и растворитель удаляют в вакууме для получения метил 3-(4-фторбензоил)пропионата (2 г, 94%).
ИК-спектр (пленка): 3448, 3111, 3076, 3003, 3958, 1734, 1678, 1601, 1423, 1300, 1240, 1155, 1099 см-1.
Н1 ЯМР (СDС13): δ 7,97 (2Н, dd, J=9,0, 5,5 Гц); 7,10 (2Н, t, J=8,9 Гц); 3,67 (3Н, s); 3,25 (2Н, t, J=6,6 Гц); 2,73 (2Н, t, J=6,6 Гц).
С13 ЯМР (СDС13): δ 196,50; 173,34; 167,54; 164,17; 132,98; 130,77; 115,91; 115,62; 51,91; 33,31; 28,00.
(б) Получение (S)-5-(4-фторфенил)-γ-бутиролактона (соединение 210).
Раствор метил-3-(4-фторбензоил)пропионата (209, 708 мг, 3,67 ммоля) в сухом тетрагидрофуране (2 мл) по каплям добавляют в предварительно охлажденный (0oС) раствор (-)-диизопропилхлорида (2,02 г, 6,25 ммоля) в тетрагидрофуране (2 мл) при перемешивании под сухим аргоном. Полученный раствор перемешивают при той же температуре в течение 2 часов и оставляют на ночь при температуре 0-5oС. Поддерживая перемешиванием температуру на уровне 0oС, по каплям добавляют воду (2 мл), а затем метанол (5 мл) и 5 М раствор NaOH (5 мл). Реактивную смесь перемешивают при комнатной температуре в течение 1,5 часов, охлаждают и добавляют 15 мл насыщенного раствора бикарбоната. Полученную смесь промывают эфиром (3х50 мл) и подкисляют 6 н. НС1. Кислую смесь экстрагируют толуолом (3х50 мл). Комбинированные толуоловые экстракты промывают рассолом (50 мл), высушивают над сульфатом натрия и растворитель удаляют в вакууме. Остаток ресуспендируют в 50 мл толуола и в него добавляют PPTS (10 мг). Полученный раствор кипятят в сосуде с обратным холодильником под ловушкой Дина-Старка (сначала забирают 15 мл дистиллята) в течение 2 часов. Раствор охлаждают, промывают насыщенным раствором бикарбоната (2х50 мл), высушивают над сульфатом натрия и растворитель удаляют в вакууме для получения 620 мг (94%) (S)-5-(4-фторфенил)-γ-бутиролактона.
Н1 ЯМР (СDС13): δ 7,33 (2Н, dd, J=8,8, 5,3 Гц); 7,09 (2H, t, J=8,7 Гц), 5,50 (1H, dd, J=8,4, 5,9 Гц); 2,64-2,71 (3Н, m); 2,17-2,22 (1H, m).
(в) Получение (5S)-2-гидрокси-5-(4-фторфенил)тетрагидрофурана (соединение 211).
(S)-5-(4-Фторфенил)-γ-бутиролактон (210, 620 мг, 3,44 ммоля) азеотропно высушивают (гексаном) и растворяют в сухом метиленхлориде (25 мл). Раствор охлаждают до -78oС и при перемешивании под аргоном по каплям добавляют DIBALH (3,5 мл 1,5 M раствора в толуоле, 5,16 ммоля). Перемешивание продолжают при -78oС в течение 2 часов и затем добавляют насыщенный раствор Na-K-тартрата (25 мл). Охлаждающую баню убирают и перемешивание продолжают еще 2 часа. Реактивную смесь разводят метиленхлоридом (25 мл). Органический слой отделяют, промывают водой (2х50 мл) и солевым раствором (50 мл), высушивают над сульфатом натрия и растворитель удаляют в вакууме для получения 2-гидрокси-5-(4-фторфенил)тетрагидрофурана (620 мг, 100%).
Н1 ЯМР (СDС13): δ 7,30-7,41 (m, 2H); 7,04 (m, 2H); 5,63-5,78 (m, 1H); 5,00-5,22 (m, 1H); 2,48 (m, 1H); 2,20-2,32 (m, 1H); 1,95-2,10 (m, 1H); 1,79 (m, 1H).
(г) Получение (5S)-2-(трет-бутилдиметилсилилокси)-5-(4-фторфенил)тетрагидрофурана (соединение 212).
(5S)-2-Гидрокси-5-(4-фторфенил)тетрагидрофуран (211, 620 мг, 3,5 ммоля), трет-бутилдиметилсилилхлорид (700 мг, 5,25 ммоля) и имидазол (595 мг, 8,75 ммоля) растворяют в 2 мл сухого ДМФ. Полученный раствор перемешивают под сухим аргоном в течение ночи, выливают в 200 мл воды и экстрагируют смесью этилацетата и гексана в соотношении 2:1 (3х100 мл). Комбинированные органические экстракты промывают водой (4х200 мл) и рассолом (100 мл), высушивают над сульфатом натрия и растворитель удаляют в вакууме для получения 1 г (96%) (5S)-2-(трет-бутилдиметилсилилокси)-5-(4-фторфенил)тетрагидрофурана (212, смесь цис- и транс-изомеров) в виде бесцветного масла, которое не нуждается в дальнейшей очистке.
Н1 ЯМР (СDС13): δ 7,40-7,50 (2Н, m, меньший по количеству изомер); 7,25-7,35 (2Н, m, основной изомер); 7,00-7,10 (2Н, m, как основной, так и меньший по количеству изомеры); 5,71-5,75 (1Н, m, основной изомер); 5,59-5,62 (1Н, m, основной изомер); 5,12-5,20 (1Н, m, основной изомер); 4,90-4,98 (1Н, m, меньший по количеству изомер); 2,40-2,55 (1Н, m, как основной, так и меньший по количеству изомеры); 2,05-2,17 (1Н, m, как основной, так и меньший по количеству изомеры); 1,87-2,00 (1Н, m, как основной, так и меньший по количеству изомеры); 1,67-1,70 (1Н, m, как основной, так и меньший по количеству изомеры); 0,92 (s, 9H, как основной, так и меньший по количеству изомеры); 0,16 (s, 6Н, как основной, так и меньший по количеству изомеры).
(д) Получение (2S, 5S)-транс-2-(3-трет-бутилдиметилсилилокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофурана (соединение 213).
(5S)-2-(трет-бутилдиметилсилилокси)-5-(4-фторфенил)тетрагидрофуран (212, 1 г, 3,4 ммоля) растворяют в 10 мл сухого метиленхлорида (дегазированном перед использованием пробулькиванием аргона). Этот раствор охлаждают до -70oС и при перемешивании при той же температуре под сухим аргоном по каплям добавляют триметилсилилбромид (550 мкл, 4,1 ммоля). Перемешивание дополнительно продолжают 1,5 ч для получения (5S)-2-бром-5-(4-фторфенил)тетрагидрофурана, который используют без отделения. В отдельной колбе растворяют 3-трет-бутилдиметилсилилокси-бут-1-ин (840 мг, 4,5 ммоля) в сухом тетрагидрофуране (10 мл). Раствор охлаждают до -60oС и, продолжая перемешивание при той же температуре под сухим аргоном, по каплям добавляют н-бутиллитий (1,8 мл, 2,5 М раствор в гексане, 4,5 ммоля). Перемешивание дополнительно продолжают в течение 0,5 часа. Полученный раствор через канюлю по каплям добавляет в перемешанный раствор 2-бромтетрагидрофурана (полученный выше) при -70oС. Перемешивание продолжают при -78oС дополнительно в течение 1,5 часов. Реактивную колбу в течение ночи держат в морозильнике (-78oС) (хотя тонкослойная хроматография не выявляет никаких изменений). Реактивную смесь выливают в 2 М раствор аммония хлорида (100 мл) и экстрагируют метиленхлоридом (3х75 мл). Раствор высушивают над сульфатом натрия и растворитель удаляют в вакууме. Остаток очищают с помощью флэш-хроматографии (элюент: 10% этилацетат в гексане) для получения двух компонентов. По данным протонного ЯМР-анализа менее полярный компонент идентифицируют как (2S,5S)-транс-2-(3-трет-бутилдиметилсилилокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофуран (213, 765 мг, 65%).
Н1 ЯМР (СDС13): δ 7,29 (2Н, m,); 7,01 (2Н, t, J=8,7 Гц); 5,09 (1Н, t, J= 7,1 Гц); 4,91-4,97 (2Н, m); 4,55-4,62 (1Н, m); 2,26-2,50 (2Н, m); 2,05-2,17 (1Н, m); 1,75-1,88 (1Н, m); 1,38 (3Н, d, J=6,6 Гц); 0,90 (9Н, s); 0,12 (6Н, s).
Более полярный компонент идентифицируют как (2R,5S)-цис-2-(3-трет-бутилдиметилсилилокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофуран (214, 190 мг, 16%).
(е) Получение (2S, 5S)-транс-2-(3-гидрокси-бут-1-инид)-5-(4-фторфенил)тетрагидрофурана (соединение 215).
(2S, 5S)-транс-2-(3-трет-бутилдиметилсилилокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофуран (213, 765 мг, 2,2 ммоля) растворяют в 20 мл тетрагидрофурана. Раствор охлаждают до 0oС и к нему добавляют тетрабутиламмонийфторид (6,6 мл 1 М раствора в тетрагидрофуране). Полученный раствор перемешивают при 0oС в течение 2 ч и растворитель удаляют в вакууме. Остаток растворяют в этилацетате (100 мл), промывают водой (3х100 мл, добавляя по 5 мл солевого раствора каждый раз в отдельные слои), а затем рассолом (50 мл), высушивают над сульфатом натрия и растворитель удаляют в вакууме для получения 500 мг (97%) (2S, 5S)-транс-2-(3-гидрокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофурана (215).
Н1 ЯМР (СDС13): δ 7,29 (2Н, dd, J=8,7, 5,2 Гц); 7,01 (2Н, t, J=8,7 Гц); 5,09 (1H, t, J= 7,4 Гц); 4,92 (1H, t, J=7,4 Гц); 4,59 (1H, q, J=6,6 Гц); 2,30-2,50 (2Н, m); 2,05-2,15 (1H, m); 1,75-1,88 (1H, m); 1,72 (1H, br s); 1,47 (3Н, d, J=6,6 Гц).
(2S, 5S)-транс-2-(3-Гидрокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофуран (215, 500 мг, 2,13 ммоля), (R)-α-метоксифенилуксусную кислоту (1,06 г, 6,4 ммоля) и DMAP (86 мг, 0,7 ммоля) растворяют в сухом метиленхлориде (3 мл). Добавляют DCC (1,5 г, 7,24 ммоля) и полученный раствор перемешивают при комнатной температуре под сухим аргоном в течение 4 ч (в пределах нескольких минут выпадает в осадок большое количество белого твердого вещества). Твердое вещество отфильтровывают и фильтрат концентрируют в вакууме. Остаток очищают с помощью флэш-хроматографии (элюент: 10% этилацетат в гексане) для получения двух диастереомерных эфиров. Менее полярный эфир относят к (2S, 5S)-транс-2-(3-(S)-гидрокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофурану (216, 250 мг, 30%, >95% по данным Н1 ЯМР).
Н1 ЯМР (CDCl3): δ 7,25-7,50 (7Н, m); 7,02 (2Н, t, J=8,5 Гц); 5,52-5,60 (1H, m); 5,06 (1H, t, J=6,8 Гц); 4,88-4,94 (1H, m); 4,78 (1H, s); 3,43 (3Н, s); 2,25-2,47 (2Н, m); 2,00-2,13 (1H, m); 1,75-1,88 (1H, m); 1,37 (3Н, d, J= 6,7 Гц).
Более полярный эфир относят к (2S,5S)-транс-2-(3-(R)-гидрокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофурану (217, 230 мг, 29%, 72% по данным Н1 ЯМР).
Н1 ЯМР (СDС13): δ 7,22-7,50 (7Н, m); 7,01 (2Н, t, J=8,7 Гц); 5,50-5,60 (1H, m); 4,98 (1H, t, J=7,2 Гц); 4,79-4,85 (1H, m); 4,79 (1H, s); 3,44 (3Н, s); 2,20-2,40 (2Н, m); 1,88-1,98 (1H, m); 1,72-1,80 (1H, m); 1,51 (3Н, d, J= 6,7 Гц).
Щелочной гидролиз (перемешивание в 10 мл 1 М этанолового КОН при 50oС в течение 30 мин с последующей обычной обработкой) этих двух эфиров дает их соответствующие спирты: (2S,5S)-транс-2-{3-(S)-гидрокси-бут-1-инил}-5-(4-фторфенил)тетрагидрофуран (218, 150 мг, 98%) и его диастереомер (2S,5S)-транс-2-{ 3-(R)-гидрокси-бут-1-инил)-5-(4-фторфенил)тетрагидрофуран (221, 50 мг, 100%).
Н1 ЯМР спектры обоих этих спиртов были идентичны таковому соединения 218.
(ж) Получение (2S,5S)-транс-2-{3-(R)-(N-феноксикарбонилокси-N-феноксикарбониламино)-бут-1-инил}-5-(4-фторфенил)тетрагидрофурана (соединение 219).
(2S,5S)-транс-2-{3-(S)-гидрокси-бут-1-инил}-5-(4-фторфенил)тетрагидрофуран (218, 150 мг, 0,64 ммоля), трифенилфосфин (200 мг, 0,77 моля) и N,О-бис(феноксикарбонил)гидроксиламин (200 мг, 0,77 моля) растворяют в сухом тетрагидрофуране (3 мл). Раствор охлаждают до 0oС и при перемешивании под сухим аргоном по каплям добавляют диизопропилазодикарбоксилат (142 мкл, 0,77 ммоля). Перемешивание продолжают в течение 1 часа при той же температуре. Растворитель выпаривают в роторном испарителе и остаток очищают с помощью флэш-хроматографии (элюент: 10% этилацетат в гексане) для получения 225 мг (72%) (2S,5S)-транс-2-{3-(R)-(N-феноксикарбонилокси-N-феноксикарбониламино)-бут-1-инил}-5-(4-фторфенил)тетрагидрофурана (219).
Н1 ЯМР (СDС13): δ 7,15-7,45 (12Н, m); 7,02 (2Н, t, J=8,6 Гц); 5,32 (1H, q, J=7,0 Гц); 5,07 (1H, t, J=6,8 Гц); 4,96 (1H, t, J=5,7 Гц); 2,25-2,50 (2Н, m); 2,05-2,20 (1H, m); 1,70-1,85 (1H, m); 1,66 (3Н, d, J=7,0 Гц).
(з) Получение (2S, 5S)-транс-2-{3-(S)-(N-феноксикарбонилокси-N-феноксикарбониламино)-бут-1-инил} -5-(4-фторфенил)тетрагидрофурана (соединение 222).
Начиная с (2S, 5S)-транс-2-{ 3-(R)-гидрокси-бут-1-инил} -5-(4-фторфенил)тетрагидрофурана (221, 150 мг, 0,64 ммоля), следуя той же методике, что и для получения соединения 218, получают 220 мг (70%) (2S,5S)-транс-2-{3-(S)-(N-феноксикарбонилокси-N-феноксикарбониламино)-бут-1-инил} -5-(4-фторфенил)тетрагидрофурана (соединение 222).
Н1 ЯМР-спектр был аналогичен таковому соединения 219.
(и) Получение (2S,5S)-транс-2-{3-(R)-(N-гидроксиуреидил)-бут-1-инил}-5-(4-фторфенил)тетрагидрофурана (CMI-947) (соединение 220).
(2S, 5S)-транс-2-{3-(R)-(N-феноксикарбонилокси-N-феноксикарбониламино)-бут-1-инил}-5-(4-фторфенил)тетрагидрофуран растворяют в высоконапорной трубе в виде раствора в метиленхлориде. Растворитель выпаривают струей аргона и осадок охлаждают до -78oС. 10 мл аммиака конденсируют в этой трубе и добавляют 2 мл трет-бутанола. Трубу герметизируют, медленно согревают до комнатной температуры. Раствор перемешивают при комнатной температуре в течение 18 часов. Давление очень медленно снижают до нормального и трубу оставляют открытой на 1 час. Остаток переносят в колбу и дважды концентрируют с добавленным толуолом. Остаток очищают с помощью препаративной тонкослойной хроматографии (элюент: 5% метанол в метиленхлориде) для получения 120 мг (90%) (2S,5S)-транс-2-{3-(R)-(N-гидроксиуреидил)-бут-1-инил}-5-(4-фторфенил)тетрагидрофурана (CMI-947, 220).
ИК-спектр (пленка): 3209, 2985, 2874, 1653, 1510, 1449, 1336, 1224, 1157, 1037 см-1.
Н1 ЯМР (CDCl3): δ 7,34 (2Н, dd, J=8,7, 5,4 Гц); 7,04 (2Н, t, J=8,8 Гц); 5,00-5,10 (2Н, m); 4,85-4,95 (1Н, m); 2,25-2,50 (2Н, m); 2,00-2,15 (1Н, m), 1,78-1,85 (1H, m); 1,38 (3Н, d, J=7,0 Гц).
(к) Получение (2S,5S)-транс-2-{3-(S)-(N-гидроксиуреидил)-бут-1-инил}-5-(4-фторфенил)тетрагидрофурана (CMI-948) (соединение 220).
Начиная с (2S, 5S)-транс-2-{3-(S)-(N-феноксикарбонилокси-N-феноксикарбониламино)-бут-1-инил} -5-(4-фторфенил)тетрагидрофурана (222, 225 мг), следуя той же методике, что и для получения соединения 219, получают 110 мг (83%) (2S,5S)-транс-2-{3-(S)-(N-гидроксиуреидил)-бут-1-инил}-5-(4-фторфенил)тетрагидрофурана (CMI-948, 223).
ИК-спектр (пленка): 3200, 2985, 2881, 1643, 1510, 1442, 1222, 1035 см-1.
Н1 ЯМР (СDС13): δ 7,34 (2Н, dd, J=8,7, 5,5 Гц); 7,04 (2Н, t, J=8,9 Гц); 5,00-5,10 (2Н, m); 4,85-4,95 (1Н, m); 2,25-2,50 (2Н, m); 2,00-2,15 (1Н, m); 1,70-1,85 (1H, m); 1,38 (3H, d, J=7,0 Гц).
Пример 6. Получение R,R,S- и R,R,R-изомеров транс-2-{3-(N-гидроксиуреидил)-бут-1-инил}-5-(4-фторфенил)тетрагидрофурана (соединения 234 и 236).
Один способ получения R,R,S- и R,R,R-изомеров транс-2-{3-(N-гидроксиуреидил)-бут-1-инил} -5-(4-фторфенил)тетрагидрофурана проиллюстрирован на схеме 10 (см. в конце описания).
(а) Получение 4-(4-фторфенил)-4-оксометилбутаноат (соединение 209).
К перемешанному раствору 3-(4-бензоил)пропионовой кислоты (208) (5,0 г) в метаноле (20 мл) добавляют несколько капель серной кислоты. После перемешивания в течение ночи (19 час) реакцию нейтрализуют насыщенным водным раствором бикарбоната натрия и метанол удаляют под пониженным давлением. Остаток растворяют в этилацетате (50 мл) и промывают насыщенным водным раствором бикарбоната натрия (3х15 мл), водой (2х15 мл) и рассолом (2х15 мл), высушивают (Na2SO4), фильтруют и концентрируют для получения бледного кристаллического твердого вещества (5,3 г, 98%).
Н1 ЯМР: 2,79 (t, 2Н); 3,30 (t, 2Н); 3,7 (s, 3Н); 7,14 (t, 2Н); 8,02 (m, 2H).
(б) Получение R-4-(4-фторфенил)-гамма-бутиролактона (соединение 224).
В охлажденный (0oС), перемешанный раствор (+)-диметиламиноизопропилхлорида (25 г, 77,9 ммоля) в сухом тетрагидрофуране (20 мл) под аргоном медленно добавляют раствор кетоэфира 209 (10,07 г, 48,0 ммолей) в сухом тетрагидрофуране (20 мл). Реактивную смесь помещают в холодильник (4oС) на 30 часов, а затем возвращают в ледяную баню и перемешивают, добавляя воду (10 мл), затем метанол (30 мл), затем 10% водный раствор NaOH (60 мл). Ледяную баню удаляют. Когда происходит гидролиз всего эфира, добавляют насыщенный водный раствор бикарбоната натрия (80 мл). Водную соль экстрагируют эфиром (2х100 мл), затем подкисляют до рН 2 и экстрагируют бензолом (2х180 мл). Пиридиний-п-толуолсульфонат (60 мг) добавляют в комбинированные бензольные слои, которые затем нагревают до возврата флегмы с помощью ловушки Дина-Старка. По окончании реакции раствор бензола промывают насыщенным водным раствором бикарбоната натрия (150 мл) и рассолом (2х50 мл), высушивают, фильтруют и концентрируют для получения белого кристаллического твердого вещества, которое на основании литературных данных относят к конфигурации R (7,92 г, 91%).
Н1 ЯМР: 2,10-2,25 (m, 1H); 2,68 (m, 3H); 5,50 (m, 1H); 7,08 (t, 2H); 7,30 (m, 2H).
(в) Получение цис- и транс-5R-5-(4-фторфенид)-2-гидрокситетрагидрофурана (соединение 225).
В перемешанный раствор лактона 224 (7,25 г, 40,3 ммоля) в сухом толуоле (50 мл), охлажденный в сухой ледовой/ацетоновой бане добавляют диизобутилалюминийгидрид (1,5 М в толуоле) (1,5 экв., 40 мл). По окончании реакции медленно добавляют метанол (10 мл), а затем насыщенный водный раствор натриево-калиевый-L-тартрат (60 мл) и ледяную баню удаляют. Этот раствор выдерживают в течение ночи (16 часов), слои отделяют и водную фракцию экстрагируют этилацетатом (2х50 мл). Комбинированные органические слои промывают водой (3х30 мл) и рассолом (3х30 мл), высушивают (Na2SO4), фильтруют и концентрируют. Получают бесцветное масло, которое является смесью двух диастереомеров (примерно 50/50) (6,32 г, 86%).
Н1 ЯМР: 1,7 (m, 1H); 1,9-2,3 (m, 2H); 2,42 (m, 1H); 3,60 (bs, 0,5H); 3,72 (bs, 0,5Н); 4,98 (t, 0,5H); 5,20 (t, 0,5H); 5,60 (bs, 0,5H); 5,72 (m, 0,5H); 7,00 (t, 2H); 7,25 (m, 1Н); 7,40 (m, 1H).
(г) Получение цис- и транс-5R-5-(4-фторфенил)-2-трет- бутилдиметилсилокситетрагидрофурана (соединение 226).
В перемешанный раствор лактола 225 (6,32 г, 34,7 ммоля) в метиленхлориде (140 мл) добавляют имидазол (1. leq, 38,2 ммоля, 2,60 г) и трет-бутилдиметилсилилхлорид (5,77 г). После перемешивания в течение ночи реактивную смесь фильтруют и концентрируют. Неочищенный продукт фильтруют через пробку окиси кремния для получения бесцветного масла, которое является смесью двух диастереомеров (примерно 2:1) (9,61 г, 93%).
Н1 ЯМР: 0,14 (s, 6H); 0,92 (s, 9H); 1,7 (m, 1H); 1,9-2,2 (m, 2H); 2,4-2,5 (m, 1H); 4,9 (m, 0,33Н); 5,16 (t, 0,66H); 5,59 (m, 0,33H); 5,71 (dd, 0,66H); 7,00 (m, 2H); 7,25 (m, 1,33H); 7,40 (m, 0,66H).
Для образца этого соединения с рацемической смесью и положением 5 выявляют присутствие каждой конфигурации у этого центра с помощью хирального сольватирующего агента [2,2,2-трифтор-1-(9-антрил)этанола, 2,2 мг субстрата, 40 мг камфорсульфоновой кислоты]. Эти условия показывают, что соединение 226 не имеет детектируемого количества 5S-изомера.
Для контроля диастереометрическую смесь соединения 226 (2,2 мг), 2:1, в котором 5-положение является рацемической смесью, обрабатывают камфорсульфоновой кислотой (40 мг). Мультиплет при 4,86-4,92 мкг/г (0,33Н) становится двумя мультиплетами при 4,64-4,72 и 4,78-4,84 мкг/г. Для другого диастереомера (тот же спектр) дуплет дуплетов при 5,66-5,70 мкг/г становится двумя наборами дуплетов при 5,64-5,68 и при 5,70-5,74 мкг/г. Для хирально редуцированного соединения меньший мультиплет (w/CSA) появляется при 4,62-4,70 и дуплет дуплетов появляется при 5,68-5,70 мкг/г. Доказательств присутствия других изомеров не отмечают.
(д) Получение 2R,5R-транс-5-(4-фторфенил)-2-(3-трет-бутилдиметилсилокси-1-бутинил)тетрагидрофурана (соединение 227).
К раствору 226 (500 мг, 1,69 ммоля) в сухом дегазированном метиленхлориде (10 мл), охлажденному до -78oС, добавляют тетраметилсиланбромид (0,25 мл, 1,86 ммоля). Его перемешивают в течение четырех часов. В отдельную колбу, содержащую 3-трет-бутилдиметилсилокси-1-бутин (0,31 г, 1,68 ммоля) и тетрагидрофуран (5 мл) добавляют н-бутиллитий (1,6 М в гексанах, 1,26 мл, 2,02 ммоля). Через 30 минут раствор переносят через канюлю в раствор из указанной колбы. Через два часа реактивную смесь выливают в 2 М водный раствор аммония хлорида (25 мл) и экстрагируют метиленхлоридом (3х25 мл), высушивают (Na2SO4), фильтруют и концентрируют. Флэш-хроматография (5% этилацетат в гексанах) дает их транс-продукт в виде прозрачного масла (280 мг, 48%).
Н1 ЯМР: 0,17 (d, 6H); 0,91 (s, 9H); 1,42 (d, 3H); 1,8 (m, 1H); 2,25-2,50 (m, 2H); 4,58 (m, 1H); 4,91 (m, 1H); 5-09 (m, 1H); 7,0 (t, 2H); 7,30 (m, 2H).
(е) Получение 2R, 5R-транс-5-(4-фторфенил)-2-(3-гидрокси-1-бутинил)тетрагидрофурана (соединение 228).
К перемешанному раствору 227 (0,38 г, 1,1 ммоля) в тетрагидрофуране (5 мл), охлажденному в ледяной бане, добавляют тетрабутиламмонийфторид (0,86 г, 3,3 ммоля). Ледяную баню удаляют. Через 30 минут растворитель удаляют и продукты отделяют с помощью флэш-хроматографии (25% этилацетат в гексанах). Получают бесцветное масло (170 мг, 67%).
Н1 ЯМР: 1,48 (d, 3H); 1,8 (m, 1H); 2,1 (m, 1H); 2,3-2,5 (m, 2H); 4,58 (m, 1H); 4,91 (t, 1H); 5,1 (t, 1H); 7,0 (t, 2H); 7,29 (m, 2H).
Гидроксифункцию соединения 228 этерифицируют R-альфа-метоксифенилуксусной кислотой (1,3-дициклогексилкарбодиимид, 4-диметиламинопиридин, CH2Cl2, 55% после хроматографии) и отделяют полученные диастереомеры (229+230) (флэш-хроматография), отделяя таким образом R- и S-изомеры у углерода карбинола, с помощью щелочного гидролиза (КОН, 78%) удаляют эфир для получения карбинолов 231 и 232. Абсолютные конфигурации определяют на основании модели Мошера.
(ж) Получение 2R,5R-транс-5-(4-фторфенил)-2-(3R-3-N,O-бисфеноксикарбонидгидроксиламино-1-бутинил)тетрагидрофурана (соединение 233).
В охлажденный (ледяная баня) раствор 2R,5R-транс-5-(4-фторфенил)-2-(3S-3-гидрокси-1-бутинил)тетрагидрофурана (231) (29 мг, 0,12 ммоля), трифенилфосфина (39 мг, 0,15 ммоля) и N,О-бисфеноксикарбонилгидроксиламина (37 мг, 0,14 ммоля) в ТГФ (3 мл) медленно добавляют диизопропилазодикарбоксилат (0,029 мл, 0,15 ммоля). Ледяную баню удаляют и когда реакция завершается (через несколько минут), растворитель удаляют. Продукт получают с помощью флэш-хроматографии (15% этилацетат в гексанах) в виде бесцветного масла (32 мг, 53%).
Н1 ЯМР: 1,65 (d, 3Н); 1,8 (m, 1H); 2,1 (m, 1H); 2,4 (m, 2H); 4,94 (m, 1H); 5,08 (m, 1H); 5,30 (m, 1H); 7,0 (t, 2H); 7,15-7,40 (m, 2H).
(з) Получение 2R, 5R-транс-5-(фторфенил)-2-(3R-3-N-гидроксиуреидил-1-бутинил)тетрагидрофурана (соединение 234).
Соединение 233 (32 мг) комбинируют в сосуде с завинчивающейся крышкой при -78oС с помощью палочки для перемешивания концентрированным аммиаком (примерно 3 мл) и трет-бутанолом (примерно 2 мл). Сосуд герметизируют и удаляют холодную баню. После перемешивания в течение ночи при комнатной температуре давление снижают до нормального и растворитель удаляют. Продукт растирают в порошок (25% этилацетат в гексанах) для получения белого твердого вещества (14 мг, 74%).
Н1 ЯМР: 1,41 (d, 3Н); 1,8 (m, 1H); 2,1 (m, 1H); 2,3-2,5 (m, 2H); 4,93 (t, 1H); 5,08 (t, 1H); 5,20 (m, 1H); 5,38 (bs, 1H); 7,0 (t, 2H); 7,29 (m, 2H).
Электрораспылитель MS: M+1=293.
Синтез изомера RRS (CMI-957) продолжают тем же образом из эфира 230, как получают изомер RRR (CMI-954) из эфира 29.
Пример. 7 2R,5R-транс-2-(4-фторфеноксиметил)-5-(4-N-гидроксиуреидил-1-бутинил)тетрагидрофурана (соединение 1, фигура 4) и 2,5R-транс-2-(4-фторфеноксиметил)-5-(4-N-гидроксиуреидилбутил)тетрагидрофурана (соединение 404, фигура 4).
Получение 4S-(4-фторфеноксиметил)-гамма-бутиролактона (соединение 301, фигура 4).
К перемешанному раствору (S)-гамма-бутиролактона (1,0 г, 8,61 ммоля), 4-фторфенола (1,16 г, 10,35 ммоля) и трифенилфосфина (2,49 г, 9,49 ммоля) в ТГФ (10 мл) по каплям добавляют диизопропоксилазодикарбоксилат (1,87 мкл, 9,46 ммоля). После добавления реактивную смесь перемешивают при 80oС в течение 16 часов. Растворитель удаляют и продукт отделяют с помощью флэш-хроматографии (окись кремния, 2:1 гексан/этилацетат) (1,38 г, 76,3%).
Н1 ЯМР (CDCl3): 2,27 (m, 1H); 2,42 (m, 1H); 2,60 (m, 1H); 4,04 (m, 1H); 4,15 (m, 1H); 4,85 (m, lH); 5,20 (m, 1H); 6,84 (m, 2H); 6,98 (m, 2H).
Получение 2S-(4-фторфеноксиметил)-5-гидроокситетрагидрофурана (соединение 302, фигура 4).
В перемешанный раствор лактона 301 (1,38 г, 27,22 ммоля) в сухом толуоле (24 мл) при -78oС по каплям добавляют 1,5 М раствор диизобутилалюминийгидрида (6,76 мл, 10,13 ммоля). Реактивную смесь перемешивают при -78oС в течение 2 часов. Реакцию прекращают путем добавления метанола (1,7 мл), поддерживая температуру на уровне < -60oС. Смесь нагревают до -20oС с последующим добавлением насыщенного водного раствора калиево-натриевого тартрата (10 мл), поддерживая температуру реакции между -10 и 0oС. Реактивную смесь перемешивают при комнатной температуре в течение ночи и затем две фазы отделяют. Водный слой экстрагируют этилацетатом. Комбинированные органические слои промывают водой, насыщенным раствором NaCl, а затем концентрируют в вакууме для оставления масла, которое очищают с помощью флэш-хроматографии (окись кремния, 2:1 гексан/этилацетат) (1,41 г, 101%).
Н1 ЯМР (CDCl3): 1,80 (m, 1H); 2,05 (m, 2H); 2,26 (m, 1H); 3,93 (m, 2H); 4,04 (m, 2H); 4,47 (m, 0,5Н); 4,61 (m, 0,5H); 5,57 (m, 0,5H); 5,66 (m, 0,5H); 6,88 (m, 2H); 6,98 (m, 2H).
Получение 2S-(4-фторфеноксиметил)-5-трет-бутилдиметилсилокси)тетрагидрофурана (соединение 303, фигура 4).
В перемешанный раствор лактола 302 (1,41 г, 6,65 ммоля) в метиленхлориде (25 мл) добавляют имидазол (498,1 мг, 7,32 ммоля) и трет-бутилдиметилсилилхлорид (1,10 г, 7,32 ммоля). Реактивную смесь перемешивают при комнатной температуре в течение ночи, затем реактивную смесь фильтруют и фильтрат концентрируют. Неочищенный продукт очищают с помощью флэш-хроматографии, используя в качестве растворителя гексан/этилацетат в соотношении 9:1, для получения бесцветного масла, которое является смесью двух диастереомеров (примерно 2:1) (1,22 г, 56,2%).
Н1 ЯМР (СDС13): 0,11 (s, 6H); 0,90 (s, 9Н); 1,80-2,10 (m, 3Н); 2,22 (m, 1H), 3,91 (m, 2H); 4,38 (m, 0,33H); 4,50 (m, 0,67H); 5,52 (m, 0,33Н); 5,59 (m, 0,67H); 6,86 (m, 2H); 6,96 (m, 2H).
Получение 2S, 5S-транс-2-(4-фторфеноксиметил)-5-(4-трет-бутилдиметилсилокси)тетрагидрофурана (соединение 304, фигура 4) и 2S,5R-цис-2-(4-фторфеноксиметил-5-(4-трет-бутилдиметилсилокси-1-бутинил)тетрагидрофурана (соединение 305, фигура 4).
В перемешанный раствор 303 (720 мг, 2,21 ммоля) в сухом метиленхлориде (10 мл), охлажденном до -78oС, добавляют тетраметилсиланбромид (349,8 мкл, 2,65 ммоля). Реактивную смесь перемешивают при -78oС в течение 4 часов. В отдельную колбу, содержащую 4-трет-бутилдиметилсилокси-1-бутин (812,8 мг, 4,42 ммоля) и ТГФ (10 мл), добавляют н-бутиллитий (2,5 М в гексане, 2,65 мл, 6,63 ммоля). Через 30 минут содержимое переносят через канюлю в указанную выше смесь. Через два часа реакцию прекращают путем добавления насыщенного водного раствора аммония хлорида и экстрагируют метиленхлоридом, высушивают над MgSO4, фильтруют и концентрируют. После очистки с помощью флэш-хроматографии (окись кремния, гексан/этилацетат в соотношении 95:5) получают два продукта: транс-соединение 304 и цис-соединение 303 (160 мг), и смесь этих двух соединений (50 мг). Общий выход составляет 48,5%.
Н1 ЯМР (СDС13):
304: 0,10 (s, 6H); 0,91 (s, 9H); 1,87 (m, 1H); 2,01 (m, 1H), 2,22 (m, 2H); 2,43 (t, 2H); 3,72 (t, 2H); 3,92 (d, 2H); 4,47 (m, 1H); 4,73 (m, 1H); 6,86 (m, 2H), 6,95 (t, 2H).
305: 0,09 (s, 6H); 0,90 (s, 9H); 1,92-2,20 (m, 4H); 2,42 (m, 2H); 3,70 (t, 2H); 3,92 (m, 1H); 4,07 (m, 1H); 4,29 (m, 1H); 4,62 (m, 1H); 6,86 (m, 2H); 6,96 (t, 2H).
При получении соединений 304 и 305 при желании можно использовать защищающие кислород группы, кроме 4-трет-бутилдиметилсилила, известные специалистам в этой области.
Для определения стереохимии этой молекулы проводят эксперимент с разницей NOE для обоих соединений, 304 и 305. В эксперименте с разницей NOE для соединения 304 мультиплет у 4,73 мкг/г облучают расщепляющими импульсами очень низкой радиочастоты и обработку данных проводят так, чтобы измерить только наличие увеличения сигнала. Это должно представлять положительный эффект NOE и указывать на близкое пространственное отношение этих протонов. В этом эксперименте NOE устанавливают для мультиплета при 2,22 мкг/г, который является протоном фуранового кольца. Когда мультиплет при 4,47 мкг/г облучают расщепляющими импульсами очень низкой радиочастоты и обработку данных проводят так, чтобы измерять только наличие увеличения сигнала, NOE устанавливают для мультиплета при 2,22 мкг/г, которые являются протонами фуранового кольца. Другой NOE также определяют для протонов при 3,92 мкг/г, которые являются протонами на метилене, следующем после этого мультиплета, указывая на то, что этот мультиплет представляет протон, следующий после метилена.
В эксперименте с разницей NOE для соединения 305 триплет у 4,62 мкг/г облучают расщепляющими импульсами очень низкой радиочастоты и обработку данных проводят так, чтобы измерять только наличие увеличения сигнала, это должно представлять положительный эффект NOE и указывать на близкое пространственное отношение этих протонов. В этом эксперименте NOE устанавливают для мультиплета при 4,29 мкг/г, который является другим протоном метинфурана. Другой NOE также обнаруживают для мультиплета при 2,17 мкг/г, которые являются протонами. Когда мультиплет при 4,29 мкг/г облучают расщепляющими импульсами очень низкой радиочастоты и обработку данных проводят так, чтобы измерять только наличие увеличения сигнала, NOE устанавливают для триплета при 4,62 мкг/г, который является другим протоном метинфурана. Другой NOE также определяют для протонов при 3,92 и 4,07 мкг/г, которые являются протонами на метилене, следующем после этого мультиплета, указывая на то, что этот мультиплет представляет протон, следующий после метилена. Другой NOE также обнаруживают для мультиплета при 2,11 мкг/г, которые являются протонами.
Получение 2S, 5S-транс-2-(4-фторфеноксиметил)-5-(4-трет-гидрокси-1-бутил)тетрагидрофурана (соединение 306, фигура 4).
В перемешанный раствор 304 (210 мг, 0,54 ммоля) в ТГФ (1,4 мл), охлажденном в ледяной бане, добавляют тетрабутиламмонийфторид (420,3 мг, 1,61 ммоля). Ледяную баню удаляют и реактивную смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют и продукт отделяют с помощью флэш-хроматографии (окись кремния, гексан/этилацетат 1:1) (124 мг, 83,2%).
Н1 ЯМР (CDCl3): 1,88 (m, 1H); 2,02 (m, 1H); 2,25 (m, 2H); 2,50 (m, 2H); 3,72 (t, 2H); 3,93 (d, 2H); 4,48 (m, 1H); 4,76 (m, 1H); 6,84 (m, 2H); 6,96 (t, 2H).
Получение 2S, 5S-транс-2-(4-фторфеноксиметил)-5-(4-N,О-бисфеноксикарбонилгидроксиламино-1-бутинил)тетрагидрофурана (соединение 307, фигура 4).
В охлажденный (ледяная баня) раствор соединения 306 (124 мг, 0,45 ммоля), трифенилфосфина (128,9 мг, 0,49 ммоля) и N,О-бисфеноксикарбонилгидроксиламина (147,3 мг, 0,54 ммоля) в ТГФ (5 мл) добавляют диизопропоксилазодикарбоксилат (94,1 мкл, 0,48 ммоля). Ледяную баню удаляют и реактивную смесь перемешивают при комнатной температуре в течение 30 минут. Растворитель удаляют и продукт отделяют с помощью флэш-хроматографии (окись кремния, гексан/этилацетат, 4:1) (195 мг, 82,0%).
Н1 ЯМР (СDС13): 1,85 (m, 1H); 2,03 (m, 1H); 2,22 (m, 2H), 2,75 (m, 2Н); 3,92 (d, 2H); 4,05 (m, 2H), 4,47 (m, 1H); 4,76 (m, 1H), 6,84 (m, 2H); 6,95 (m, 2H); 7,26 (m, 5H); 7,41 (m, 5H).
Получение 2S,5S-транс-2-(4-фторфеноксиметил)-5-(4-N-гидроксиуреидил-1-бутинил)тетрагидрофурана (соединение 401, фигура 4).
В сосуд с завинчивающейся крышкой помещают NН3 при -78oС (приблизительно 12 мл). В этот холодный жидкий азот добавляют соединение 307 (195,0 мг, 0,37 ммоля), предварительно растворенное в 20 мл метанола. Сосуд герметизируют и сухую ледяную баню удаляют. Реактивную смесь перемешивают при комнатной температуре в течение 16 часов. Реактивную смесь снова охлаждают сухой ледяной баней, и давление нормализуют. Сосуд открывают и растворитель удаляют. Продукт отделяют с помощью флэш-хроматографии с использованием этилацетата в качестве растворителя для получения твердого вещества (108 мг, 93,7%).
Н1 ЯМР (CDCl3): 1,84 (m, 1H); 2,01 (m, 1H); 2,22 (m, 2H); 2,55 (t, 2H); 3,75 (t, 2H); 3,94 (m, 2Н); 4,48 (m, 1H); 4,74 (t, 1H); 5,25 (bs, 2H); 6,86 (m, 2H); 6,98 (m, 2H).
Получение 2S, 5S-транс-2-(4-фторфеноксиметил)-5-(4-N- гидроксиуреидил-1-бутинил)тетрагидрофурана (402).
Соединение 1 (75 мг, 0,23 ммоля) растворяют в 2 мл этилацетата и затем добавляют Pd/C (10%) (15 мг) и гидрогенируют при баллонном давлении в течение 16 часов. Реактивную смесь фильтруют и фильтрат концентрируют. Продукт отделяют с помощью флэш-хроматографии с использованием этилацетата в качестве растворителя (70 мг, 92,2%).
Н1 ЯМР (CDCl3): 1,50-1,70 (m, 8H); 2,10 (m, 2H); 3,58 (m, 2H); 3,91 (m, 2H); 4,08 (m, 1H); 4,40 (m, 1H); 5,15 (bs, 2H); 6,87 (m, 2H); 6,97 (t, 2H); 7,40 (bs, 1H).
II. ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Людей, лошадей, собак, крупный рогатый скот и других животных, в особенности млекопитающих, страдающих воспалительными заболеваниями, в частности расстройствами, опосредованными PAF или продуктами 5-липоксигеназы, можно лечить введением пациенту эффективного количества одного и более из идентифицированных выше соединений или их фармацевтически приемлемого производного, или соли в фармацевтически приемлемом носителе или растворителе для уменьшения образования кислородных радикалов. Активные материалы могут вводиться любым соответствующим путем внутрь, например парентерально, внутривенно, внутрикожно, подкожно или местно, в виде жидкости, крема, геля или твердого вещества или в аэрозольной форме.
Термин фармацевтически приемлемые соли или комплексы в том смысле, в котором он используется в настоящем описании, относится к солям или комплексам, которые сохраняют желаемую биологическую активность идентифицированных выше соединений и оказывают минимальные нежелательные токсикологические эффекты.
Не ограничивающими примерами таких солей являются:
(а) соли, образуемые в результате присоединения неорганических кислот (например, хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты и тому подобных), и соли, образуемые с органическими кислотами, как, например, уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памоевая кислота, альгиновая кислота, полиглютаминовая кислота, нафталинсульфоновая кислота, нафталиндисульфоновая кислота и полигалактуроновая кислота;
(б) соли, образуемые в результате реакции щелочей с металлическими катионами, как, например, цинком, кальцием, висмутом, барием, магнием, алюминием, медью, кобальтом, никелем, кадмием, натрием, калием и им подобными, или с катионом, образуемым из аммиака, N,N-дибензилэтилендиамина, D-глюкозамина, тетраэтиламмония или этилендиамина, или
(в) комбинаций (а) и (б), например цинковая соль дубильной кислоты или подобные соединения.
Соединения могут также вводиться в виде фармацевтически приемлемых четвертичных солей, известных специалистам в этой области, которые, в частности, включают четвертичную аммониевую соль формулы -NR3 +Z-, в которой R является алкилом или бензилом, a Z является противоионом, включая хлорид, бромид, иодид, -О-алкил, толуолсульфонат, метилсульфонат, сульфонат, фосфат или карбоксилат (например, бензоат, сукцинат, ацетат, глюколат, малеат, малат, нитрат, тартрат, аскорбат, бензоат, циннамоат, манделоат, бензилоат и дифенилацетат).
Активное соединение включается в фармацевтически приемлемый носитель или растворитель в количестве, достаточном для доставки пациенту терапевтически эффективного количества, не оказывая токсического действия на пациента, получающего лечение.
Предпочтительная доза активного соединения для всех упомянутых выше соединений находится в диапазоне от приблизительно 10 нг/кг до 300 мг/кг, предпочтительно от 0,1 до 100 мг/кг в сутки, более обобщенно от 0,5 до приблизительно 25 мг на 1 кг массы тела реципиента в сутки. Предпочтительная доза по сердечно-сосудистым показаниям находится в диапазоне от 10 нг/кг до 20 мг/кг. Обычная доза для местного применения будет находиться в диапазоне от 0,01 до 3 мас.% в подходящем носителе. Диапазон эффективной дозы фармацевтически приемлемых производных можно рассчитать на основании массы родительского соединения, которое предполагается вводить. Если производное само по себе проявляет активность, эффективную дозу можно определить, как указано ниже, используя массу производного или с помощью других методов, известных специалистам в этой области.
Удобно вводить соединение в любой подходящей единичной дозируемой форме, которая включает, но не ограничивается единичной дозой, содержащей от 1 до 3000 мг, предпочтительно от 5 до 500 мг активного ингредиента на единичную дозу. Для приема внутрь обычно удобна доза от 25 до 250 мг.
Активный ингредиент предпочтительно вводится для достижения пиковой концентрации активного соединения в плазме, составляющего от 0,00001 до 30 ммолей, предпочтительно от 0,1 до 30 мкмолей. Это может быть достигнуто, например, путем внутривенного введения раствора или состава активного ингредиента, как вариант, в солевой или водной среде, или болюсного введения активного ингредиента.
Концентрация активного соединения в лекарственной композиции будет зависеть от скорости всасывания, распределения, инактивации и выделения препарата, а также от других факторов, известных специалистам в этой области. Следует отметить, что величины дозы будут также варьировать в зависимости от тяжести состояния, подлежащего лечению. Кроме того, следует понять, что для каждого конкретного субъекта следует подбирать индивидуальный режим и частоту введения доз препарата в соответствии с индивидуальной потребностью и профессиональной оценкой лица, проводящего введение или наблюдающего за введением лекарственных составов, и что диапазоны концентрации, указанные далее в настоящем описании, являются только примером и не предназначены для ограничения сферы или практики применения заявленного состава. Активный ингредиент можно вводить сразу или разделить на ряд более низких доз для введения через различные интервалы времени.
Композиции орального введения обычно включают инертный растворитель или съедобный носитель. Они могут помещаться в желатиновые капсулы или спрессованы в таблетки. Для орального терапевтического применения активное соединение может быть включено в наполнители и использоваться в форме таблеток, драже или капсул. Фармацевтически совместимые связывающие агенты и/или адъювантные материалы могут быть включены в состав в качестве их компонентной части.
Таблетки, пилюли, капсулы, драже и им подобные могут содержать любой из следующих ингредиентов или соединений подобной природы: связующее вещество, как, например, микрокристаллическую целлюлозу, трагант или желатин; наполнитель, как, например, крахмал или лактоза, диспергирующий агент, как, например, альгиновая кислота, Примогель или кукурузный крахмал; смазочный материал, как, например, магния стеарат или Стеротес; глянцеватель, как, например, коллоидная двуокись кремния; подслащивающий агент, как, например, сахароза или сахарин; ароматизирующий агент, как, например, мята перечная, метилсалицилат или апельсиновый ароматизатор. Если формой единицы дозы является капсула, она может содержать, в дополнение к материалу указанного выше типа, жидкий носитель, как, например, жирное масло. Кроме того, формы единиц дозы могут содержать различные другие материалы, которые изменяют физическую форму единицы дозы, например покрытия из сахара, шеллака или защитные покрытия, предохраняющие от действия желудочной среды.
Активное соединение или его фармацевтически приемлемая соль, или производное могут вводиться в качестве компонента эликсира, суспензии, сиропа, вафель, жевательной резинки или им подобных. Кроме активных соединений, сироп может содержать сахарозу в качестве подслащивающего вещества и определенные консерванты, красители, подкрашивающие вещества и ароматизаторы.
Активное соединение или его фармацевтически приемлемые производные, или соли могут также смешиваться с другими активными материалами, которые не нарушают желаемого действия, или с материалами, которые дополняют желаемое действие, как, например, антибиотики, противогрибковые препараты, другие противовоспалительные средства или антивирусные соединения.
Растворы или суспензии, используемые для парентерального, внутрикожного, подкожного или местного применения, могут включать следующие компоненты: стерильный растворитель, как, например, вода для инъекций, солевой раствор, фиксированные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, как, например, бензиловый спирт или метилпарабензоат; антиоксиданты, как, например, аскорбиновая кислота или натрия бисульфит; хелаторы, как, например, этилендиаминтетрауксусная кислота; буферы, как, например, ацетаты, цитраты или фосфаты, и агенты для регулировки тоничности, как, например, натрия хлорид или декстроза. Родительский препарат может быть включен в ампулы, одноразовые шприцы или содержащие множество доз флакончики, изготовленные из стекла или пластика.
При внутривенном введении предпочтительными носителями являются физиологический раствор или физиологический раствор с фосфатным буфером.
В одном варианте реализации активные соединения изготавливают с носителями, которые защитят соединение от быстрого выведения из организма, такими как составы пролонгированного действия, включая имплантаты и микроинкапсулированные системы доставки. Могут использоваться биодеградируемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Способы получения таких составов будут очевидны для специалистов в этой области. Материалы могут также закупаться в компаниях "Alza Corporation" (CA) и "Scios Nova" (Baltimore, MD).
Липосомные суспензии могут также быть фармацевтически приемлемыми носителями. Они могут быть получены с использованием методов, известных специалистам в этой области, например, как описано в патенте США 4522811 (который полностью включен в настоящее описание в виде ссылки). Например, липосомные составы могут быть получены путем растворения соответствующего липида(ов) (таких, как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин, арахадоилфосфатидилхолин и холестерин) в органическом растворителе, который затем испаряется, оставляя тонкую пленку высушенного липида на поверхности резервуара. Затем в резервуар вводятся водный раствор активного соединения или его монофосфатные, дифосфатные и/или трифосфатные производные. Затем резервуар крутят в руке для стряхивания липидного материала со стенок резервуара и для диспергирования липидных агрегатов, образуя таким образом липосомную суспензию.
III. БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
Для оценки способности соединения действовать в качестве антагониста рецепторов PAF, включая способность соединения связывать рецепторы PAF и влияние соединения на различные опосредованные PAF пути, использовалось широкое разнообразие биологических проб. Любую из этих проб можно использовать для оценки способности описанных в настоящем изобретении соединений действовать в качестве антагонистов рецепторов PAF.
Например, как известно, PAF вызывает гемоконцентрацию и возросшую проницаемость микроциркуляторной системы, что ведет к уменьшению объема плазмы. Опосредованный PAF острый циркуляторный коллапс может быть использован в качестве основы теста для оценки способности соединения действовать в качестве антагониста PAF, путем анализа влияния соединения на вызванное PAF снижение объема плазмы на экспериментальной модели, как, например, у мышей.
Эндотоксемия вызывает освобождение химических медиаторов, включающих эйкозаноиды, PAF и фактор опухолевого некроза (TNF), которые стимулируют разнообразные физиологические реакции, включая лихорадку, гипотонию, лейкоцитоз и нарушения метаболизма глюкозы и липидов. Эндотоксемия может привести к тяжелому шоку и смерти. Вызванная эндотоксином смертность мышей является экспериментальной моделью для оценки фармакологического эффекта соединений при эндотоксическом шоке.
Двумя другими обычными методами анализа, используемыми для оценки способности соединения действовать в качестве антагониста рецепторов PAF, являются агрегация тромбоцитов in vitro и гипотония у крыс (Shen, et al., "The Chemical and Biological Properties of PAF Agonists, Antagonists and Biosynthetic Inhibitors", Platelet-Activating Factor and Related Lipid Mediators, F. Snyder, Ed. Plenum Press, New York, NY, 153 (1987)).
Большое разнообразие биологических тестов также использовалось для оценки способности соединения ингибировать фермент 5-липоксигеназу. Например, 5-липоксигеназа цитозоля клеток базофильного лейкоза крыс (RBL) широко использовалась в исследованиях биосинтеза лейкотриена. Соединения, которые ингибируют 5-липоксигеназу, снижают уровни лейкотриенов.
Другой биологический тест, используемый для оценки способности соединения ингибировать фермент 5-липоксигеназу, основан на классической фармакологической модели воспаления, вызванной подавлением LTB4 из цельной крови человека, стимулированной ионофором.
Пример 5. Способность соединения связывать рецепторы PAF.
(а) Выделение мембран тромбоцитов человека.
Мембраны тромбоцитов человека выделяют из концентратов тромбоцитов, полученных в Службе Крови Американского Красного Креста (Dadham, МА). После нескольких промываний раствором для промывания тромбоцитов (150 мкмолей NaCI, 10 мкмолей Трис и 2 мкмоля ЭДТА, рН 7,5), осадок тромбоцитов после центрифугирования ресуспендируют в растворе, содержащем 5 ммолей МgС12, 10 ммолей Трис и 2 ммоля ЭДТД, при рН 7,0. Клетки затем быстро замораживают жидким азотом и медленно оттаивают при комнатной температуре.
Процедуру замораживания и оттаивания повторяют минимум три раза. Для дальнейшего фракционирования фрагментов мембран суспензию лизированных мембран наслаивают на верхушку прерывистого сахарозного градиента плотности 0,25, 1,03 и 1,5 моля сахарозы, полученного в растворе, содержащем 10 ммолей МgСl2, 10 ммолей Трис и 2 ммоля ЭДТА при рН 7,0, и центрифугируют при 63500 х g в течение 2 час. Отдельно собирают фракции мембран, образующие полосы между 0,25 и 1,03 моля (мембрана А) и между 1,03 и 1,5 моля (мембрана Б). Концентрацию белка в препаратах мембран определяют методом Лоури с помощью альбумина бычьей сыворотки (BSA) в качестве стандарта. Мембраны затем отделяют в более мелкие фракции (по 4 мл каждая), хранят при -80oС и перед использованием оттаивают.
(б) Подавление связывания [Н3] PAF.
Способность [Н3] PAF связываться со специфическими рецепторами на мембранах тромбоцитов человека оценивают в оптимальных условиях при рН 7,0 в присутствии 10 ммолей МgСl2. Мембранный белок (100 мкг) добавляют к конечному раствору объемом 0,5 мл, содержащему 0,15 пкмоля (концентрация 0,3 нмоля) [Н3] PAF, и известное количество немеченого PAF или антагониста рецепторов PAF в 10 ммолях MgCl2, 10 ммолях Трис и 0,25% BSA при рН 7,0. После инкубации в течение четырех часов при 0oС отделяют связанный и несвязанный [Н3] PAF через стекловолоконный фильтр Whatman GF/C в вакууме. При этих условиях анализа не должно выявляться разрушение связанного с фильтром [Н3] PAF. Неспецифическое связывание определяют как общее связывание в присутствии избытка немеченого PAF (1 ммоль), когда при более высоких концентрациях как немеченого PAF или аналогов PAF или антагонистов рецепторов PAF, не наблюдается дальнейшее смещение. Специфическое связывание определяют как разницу между общим связыванием и неспецифическим связыванием.
Для определения относительной активности испытуемых соединений связывание [Н3] PAF в присутствии ингибиторов нормализуют в переводе на процент подавления путем определения общего связывания в отсутствие ингибиторов как 0% подавления, а общего связывания в присутствии 1 ммоля немеченого PAF как 100%. Процент подавления соединения можно рассчитать по формуле
% подавления = [(Общее связывание - общее связывание в присутствии соединения)/неспецифическое связывание]•100%.
IС50 рассчитывают как концентрацию ингибитора, необходимую для получения 50% подавления специфического связывания [Н3] PAF, и рассчитывают с помощью компьютерной программы нелинейной регрессии "GraphPad Inplot, версия 3.0" (GraphPad software, San Diego, CA).
Пример 6. Влияние соединения на вызванную PAF гемоконцентрацию.
(а) Животные.
Самок мышей CD-1 с массой тела 16-20 граммов приобретают в "Charles River laboratory" (Wilmington, MA). Водопроводную воду и корм для лабораторных грызунов (5001, Purina Mills, St. Louis, МО) дают без ограничений. Перед использованием мышей держат в среднем в течение четырех дней.
(б) Измерение гематокрита.
PAF (1-О-алкил-2-ацетил-sn-глицерил-3-фосфорилхолин, "Sigma Chemical Co. ") растворяют в 0,25% альбумине бычьей сыворотки (BSA) в 0,9% растворе NaCl. За исключением исследований зависимости реакции от дозы, 10 мкг (10 мл/кг) раствора PAF вводят в хвостовую вену. Все испытуемые соединения растворяют в 0,5% солевом растворе ДМСО и вводят внутривенно в расчете 3 мг/кг массы тела за 15 минут до введения разрешающей дозы PAF. Через 15 минут после введения PAF в гепаринизированную микрогематокритную трубку (наружным диаметром 1,50 мм) собирают от 30 до 50 мкл крови путем отсечения кончика хвоста. Все испытуемые соединения вводят мышам внутривенно в дозе 3 мг/кг за 15 минут до введения PAF (10 мкг/кг, внутривенно) или АА (0,5 мг/ухо).
Пример 7. Влияние соединений на цитозольную 5-липоксигенаву клеток базофильного лейкоза крыс.
(а) Выделение фермента.
Отмытые клетки базофильного лейкоза крыс (4х108) суспензируют в 20 мл 50 М калиевого фосфатного буфера при рН 7,4, содержащего 10% этиленгликоль /1 ммоль ЭДТА (Буфер А). Клеточную суспензию обрабатывают ультразвуком при 20 кГц в течение 30 секунд и обработанную ультразвуком суспензию центрифугируют при 10000 х g в течение 10 минут с последующим дальнейшим центрифугированием при 105000 х g в течение 1 часа. Надосадочный раствор (цитозольная фракция), содержащий 5-липоксигеназу, хранят при -70oС. Концентрацию белка определяют в соответствии с методикой Bradford (красящий реактив Брэдфорда) с альбумином бычьей сыворотки в качестве стандарта.
(б) Количественное определение фермента.
Для рутинного количественного определения 5-липоксигеназы смесь содержит 50 ммолей калиевого фосфатного буфера при рН 7,4, 2 ммоля CaCl2, 2 ммоля АТФ, 25 молей арахидоновой кислоты (0,1 кюри) и фермент (50-100 мг белка) в конечном объеме 200 л. Реакцию приводят при 24oС в течение 3 минут. Смесь экстрагируют с помощью 0,2 мл ледяной смеси этиловый эфир: 0,2 моля лимонной кислоты (30:4:1). Экстракт подвергают тонкослойной хроматографии при -10oС в системе растворителя петролейный эфир : этиловый эфир : уксусная кислота (15: 85: 0,1). Зоны силикагеля, соответствующие аутентичной арахидоновой кислоте и ее метаболитам, соскабливают в сцинтилляционные камеры для подсчета. Активность фермента выражают в переводе на количество арахидоновой кислоты, оксигенированной в течение 3 минут. Репрезентативные соединения 9, 11, 14 и 15, идентифицированные выше, проявляют активность в этом количественном определении.
Пример 8. Подавление растворимой 5-липоксигеназы в экстракте клеток RBL-1.
Клетки RBL-1 выращивают до слияния (2•l06/мл) в центрифужных флаконах в соответствии со спецификациями из АТСС. Клетки собирают и дважды промывают в лишенном кальция и лишенном магния физиологическом растворе с фосфатным буфером. Клетки суспензируют при концентрации 2•l07/мл в 50 ммолях К2НРO4 при рН 7,4, 10% ПЭГ-8000, 1 ммоле ЭДТА, 1 ммоле α-толуолсульфонилфторида, а затем обрабатывают ультразвуком. Лизат центрифугируют при 100000 х g в течение 1 часа при 4oС и надосадочную жидкость (цитозоль) удаляют и хранят в аликвотных количествах при 70oС.
Активность 5-липоксигеназы в цитозоле RBL-1 определяют следующим образом: 0,2 мл реактивных смесей, состоящих из 5 ммолей CaCl2, 2 ммолей АТФ, 50 ммолей К2НР04 при рН 7,4, различных концентраций испытуемого соединения (из 10 мл соединения, растворенного в ДМСО), и концентрации цитозоля RBL-1, который превратит 50% субстрата меченой [С14] арахидоновой кислоты в оксигенированные продукты (определяемые экспериментально для каждого препарата цитозоля), инкубируют в течение 10 минут при комнатной температуре. Реакцию инициируют добавлением 5 мл [С14] арахидоновой кислоты из исходного этанолового материала (конечная концентрация = 9,5 ммолей) и дают ей продолжаться и течение 3 минут. Реакцию прекращают добавлением 0,2 мл холодной смеси этилового эфира : метанола : 0,2 моля лимонной кислоты (30:4:1) с последующим центрифугированием при 1000 х g в течение 1 минуты. 50 мл органической фазы набирают в стеклянную капиллярную пипетку и наносят на пластинки Silica Gel 60A для тонкослойной хроматографии (Whatman #LK6D). Пластинки проявляют в смеси петролейный эфир : этиловый эфир : уксусная кислота (15:85:0,1) в течение 30 минут при комнатной температуре. Пластинки экспонируют на пленку Kodak XAR-5 в течение 24 часов. Пленку проявляют, сканируют с помощью денситометра и подсчитывают площади пиков арахидоновой кислоты и ее продуктов. % подавления определяют по количеству [С14] арахидоновой кислоты, превращенной в оксигенированные продукты, в образцах, содержащих испытуемое соединение, по отношению к таковому в контрольных образцах (без испытуемого соединения).
В таблице 5 представлены данные подавления растворимой 5-липоксигеназы в экстракте клеток RBL-1 рацемическим соединением 202, а также его энантиомерами, соединениями 216, 217, 234 и 236.
Пример 9. Подавление выработки лейкотриена B4 в цельной крови человека, стимулированной Ионофором.
Кровь человека собирают в гепаринизированные трубки для сбора крови, и набирают в аликвотных количествах по 1 мл в микроцентрифужные трубки емкостью 1,5 мл. Пять миллилитров испытуемого соединения в различных концентрациях, растворенного в ДМСО, добавляют в образец крови и инкубируют в течение 15 минут при 37oС. Кальциевый ионофор (5 мл) (А23187) в ДМСО добавляют до конечной концентрации 50 ммолей и образцы инкубируют в течение 30 минут при 37oС. Затем образцы центрифугируют при 1100xg (2500 об/мин, ротор Н1000В, в центрифуге "Sorvall") в течение 10 минут при 4oС. 100 мл надосадочной жидкости переносят в микроцентрифужные трубки емкостью 1,5 мл, добавляют 400 мл холодного метанола и белки преципитируют на лед в течение 30 минут. Образцы центрифугируют при 110xg в течение 10 минут при 4oС и проводят количественный анализ на LTB4 с помощью имеющегося в продаже набора для иммуноферментной пробы (Cayman Chemical) в соответствии со спецификациями производителя.
В таблице 5 представлены данные подавления выработки лейкотриена в стимулированной ионофором цельной крови человека, рацемическим соединением 202, а также его энантиомерами, соединениями 216, 217, 234 и 236.
Пример 10. Оценка активности 5-липоксигеназы цельной крови мышей и человека ex vivo.
Самок мышей линии CD-1 с массой тела 18-25 граммов и самок крыс линии CD с массой тела 150-230 граммов получают из "Charles River Labs". Соединения растворяют в 0,5% ДМСО в 0,9% NaCl для введения мышам и в спиртовом носителе (2% бензиловый спирт, 1% этанол, 40% ПЭГ 300, 10% пропиленгликоль, 47% 5% декстрозы плюс 3,5% плюроновый F-68 в DiH2O) для введения крысам (5 мг/мл). Животным вводят соединение (5 мг/кг) или соответствующий носитель (0,5% ДМСО в физрастворе, 10 мл/кг для мышей; спиртовой носитель, 1 мл/кг для крыс) за 15 минут до того, как их забивают путем декапитации. Гепаринизированную цельную кровь (0,3 мл) добавляют в центрифужную пробирку Эппендорфа емкостью 1,5 мл, содержащую 0,3 мл 2 ммолярного кальциевого ионофора Д23187 (конечная концентрация А23187 20 ммолей). Образец инкубируют в течение 30 минут в водяной бане при 37oС и затем центрифугируют в течение 2 минут. Плазму разводят (x 120) и проводят анализ на LTB4 с помощью иммуноферментной пробы.
В таблице 5 представлены данные по определению ех vivo содержания 5-липоксигеназы в цельной крови мышей и крыс после введения рацемического соединения 202, а также его энантиомеров, соединений 216, 217, 234 и 236.
Пример 11. Скорость глюкуронидации.
Скорость глюкуронидации является мерой метаболической устойчивости in vivo раскрытых в настоящей заявке соединений.
Реакции глюкуронидации in vitro проводят с реактивной смесью, содержащей 2 мг/мл белка микросом человека, 5 ммолей магния хлорида, 100 ммолей Трис-НС1 (рН= 7,4), 0,1-1,0 ммоль субстрата и 3 ммоля Уридин 5'-дифосфатглюкуроновой кислоты. После инкубации при 37oС в течение 0 (контроль), 15, 30, 45, 60, 90, 120, 180, 240 минут аликвотные объемы по 40 мкл реактивной смеси смешивают с 80 мкл ацетонитрила и центрифугируют для удаления преципитированного белка. Аликвоты надосадочной жидкости анализируют с помощью обратнофазной ВЭЖХ для определения исчезновения родительских соединений и образования метаболитов.
В таблице 5 представлены данные, а на фигуре 2 проиллюстрирована скорость глюкуронидации рацемического соединения 202, а также его энантиомеров, соединений 216, 217, 234 и 236.
На фигуре 3 показана скорость глюкуронидации для проиллюстрированных энантиомеров.
Пример 12. Влияние соединений на цитозольную 5-липоксигеназу клеток базофильного лейкоза крыс.
Клетки RBL-2H3 выращивают до слияния в колбе для тканевой культуры по методу Carter et al. (J. Pharm. Exp. Ther, 1991, Vol. 256(3), pp.929-937). Клетки собирают и промывают пять раз в лишенном кальция и лишенном магния физиологическом растворе с фосфатным буфером D. Клетки суспендируют при концентрации 2•l07/мл в 10 ммолях BES, 10 ммолях [1,4-пиперазинбис (этансульфоновой кислоты (PIPES) при рН 6,8, 1 ммоле ЭДТА, а затем обрабатывают ультразвуком. Обработанную ультразвуком суспензию центрифугируют при 20000 х g в течение 20 минут при 4oС. Надосадочную жидкость удаляют и хранят в аликвотных количествах при -70oС.
Активность 5-липоксигеназы в препарате RBL-2H3 определяют следующим образом. 0,1 мл реактивных смесей, состоящих из 0,7 ммоля CaCl2, 100 ммолей NaCl, 1 ммоля ЭДТД, 10 ммолей BES, 10 ммолей PIPES при рН 7,4, различных концентраций испытуемого соединения, растворенного в ДМСО (при анализе конечная концентраци ДМСО 7,5%), и количество препарата RBL-2H3, который превратит 15% субстрата арахидоновой кислоты в оксигенированные продукты (определяемые экспериментально для каждого препарата RBL-2H3), инкубируют в течение 20 минут при комнатной температуре. Реакцию инициируют добавлением 5 мкл смеси субстрата (0,944 нмоля [14С] арахидоновой кислоты и 6,06 нмоля арахидоновой кислоты на анализ в 0,028% NН4OН) и дают ей продолжаться в течение 5 минут при 37oС. Реакцию прекращают добавлением 0,12 мл смеси (I): 1,66 мг/мл трифенилфосфина в этиловом эфире; (II): метанола и (III): 0,2 моля лимонной кислоты (30:4:1) с последующим центрифугированием при 1000 х g в течение 1 минуты. 50 мкл органической фазы набирают в стеклянную капиллярную пипетку и наносят на пластинки Silica Gel 60A для тонкослойной хроматографии (Whatman #LK6D). Пластинки проявляют в смеси этиловый эфир : уксусная кислота (100: 0,1) в течение 25 минут при комнатной температуре. Пластинки экспонируют на пленку Kodak X-OMAT AR в течение 40 часов. Пленку проявляют, сканируют с помощью денситометра и подсчитывают площади пиков арахидоновой кислоты и ее продуктов. % подавления определяют по количеству [С14] арахидоновой кислоты, превращенной в оксигенированные продукты, в образцах, содержащих испытуемое соединение, по отношению к таковому в контрольных образцах (без испытуемого соединения). Результаты представлены в таблице 4.
Пример 13. Подавление выработки лейкотриена В в стимулированной ионофором крови человека.
Кровь человека собирают в гепаринизированные трубки для сбора крови и набирают в аликвотных количествах по 1 мл в микроцентрифужные трубки емкостью 1,5 мл. 5 мл испытуемого соединения в различных концентрациях, растворенного в ДМСО, добавляют в образец крови и инкубируют в течение 15 минут при 37oС. Кальциевый ионофор (5 мл, А23187) и ДМСО добавляют до конечной концентрации 50 ммолей и образцы инкубируют в течение 30 минут при 37oС. Затем образцы центрифугируют при 1100 х g (2500 об/мин, ротор Н1000В, в центрифуге "Sorvall") в течение 10 минут при 4oС. 100 мл надосадочной жидкости переносят в микроцентрифужные трубки емкостью 1,5 мл, добавляют 400 мл холодного метанола и белки преципитируют на лед в течение 30 минут. Образцы центрифугируют при 110 х g в течение 10 минут при 40oС и проводят количественный анализ на LTB4 с помощью имеющегося в продаже набора для иммуноферментной пробы (Cayman Chemical) в соответствии со спецификациями производителя.
Результаты представлены в таблице 5.
Пример 14. Оценка активности 5-липоксигеназы цельной крови мышей ex vivo.
Самок мышей линии CD-1 с массой тела 18-25 граммов и самок крыс линии CD с массой тела 150-230 граммов получают из "Charles River Labs". Соединения растворяют в 0,5% ДМСО в 0,9% NaCl для введения мышам и в спиртовом носителе (2% бензиловый спирт, 1% этанол, 40% ПЭГ 300, 10% пропиленгликоль, 47% 5% декстрозы плюс 3,5% плюроновый F-68 в DiH2O) для введения крысам (5 мг/мл). Животным вводят соединение (5 мг/кг) или соответствующий носитель (0,5% ДМСО в физрастворе, 10 мл/кг для мышей; спиртовой носитель, 1 мл/кг для крыс) за 15 минут до того, как их забивают путем декапитации. Гепаринизированную цельную кровь (0,3 мл) добавляют в центрифужную пробирку Эппендорфа емкостью 1,5 мл, содержащую 0,3 мл 2 ммолярного кальциевого ионофора А23187 (конечная концентрация А23187 20 ммолей). Образец инкубируют в течение 30 минут в водяной бане при 37oС и затем центрифугируют в течение 2 минут. Плазму разводят (х120) и проводят анализ на LTB4 с помощью иммуноферментной пробы.
Пример 15. Исследования глюкуронидации.
Скорость глюкуронидации является мерой метаболической устойчивости in vivo раскрытых в настоящей заявке соединений.
Реакции глюкуронидации in vitro проводят с реактивной смесью, содержащей 2 мг/мл белка микросом человека, 5 ммолей магния хлорида, 100 ммолей Трис НС1 (рН= 7,4), 0,1-1,0 ммоль субстрата и 3 ммоля Уридин-5'-дифосфатглюкуроновой кислоты. После инкубации при 37oС в течение 0 (контроль), 15, 30, 45, 60, 90, 120, 180, 240 минут аликвотные объемы по 40 мкл реактивной смеси смешивают с 80 мкл ацетонитрила и центрифугируют для удаления преципитированного белка. Аликвоты надосадочной жидкости анализируют с помощью обратнофазной ВЭЖХ для определения исчезновения родительских соединений и образования метаболитов. Результаты представлены на фигуре 5.
Пример 16. Анализ эозинофильной инфильтрации.
Накопление воспалительных клеток в легких является одним из патоморфологических признаков астмы. Подъем уровня лейкоцитов, особенно эозинофилов, в крови и в жидкости легочного лаважа наблюдают после вдыхания аллергена пациентами. Эозинофилы представляются важными эффекторными клетками при аллергическом воспалении с цитотоксическими свойствами белков их гранул и способностью освобождать воспалительные медиаторы. Предотвращение вызванного аллергеном притока эозинофилов в легкие расценивается как важная цель при разработке новых противоастматических лекарственных препаратов.
Лейкотриены являются продуктами пути метаболизма 5-липоксигеназы (5-LO) арахидоновой кислоты. Продукты обмена липоксигеназы (LTB4, 5-оксо-15-гидроксиэйкозатетраеноевая кислота), как было установлено, обладают высокой активностью в плане вовлечения в процесс эозинофилов. Ингибитор 5-LO, который способен блокировать немедленное сужение бронхов, а также уменьшить более позднее накопление эозинофилов в ткани легких после стимуляции аллергеном, может быть эффективным в профилактике и лечении астмы.
Эозинофильную инфильтрацию легких можно измерить путем подсчета количества клеток в жидкости бронхоальвеолярного лаважа (BALF) у морских свинок или мышей после стимуляции аллергеном.
Экспериментальная модель ни морских свинках. Самок морских свинок линии Hartley с массой тела 400-500 г активно сенсибилизируют яичным белком (OVA) путем внутрибрюшинной инъекции 20 мкг OVA и 100 мг А1(ОН)3 в 0,5 мл 0,9% NaCl в 1-й и 2-ой день. Провокационную пробу у животных проводят аэрозолем 0,5% OVA (в 0,9% NaCl) в течение 30 сек на 15-й и 16-й день. Соединения готовят в 10% Полиэтиленгликоле 200 или 0,5% карбоксиметилцеллюлозе и вводят внутрь 3 раза (за 1 ч до каждой провокационной пробы и в промежутке между двумя провокационными пробами). Для профилактики вызванной освобождением гистамина смерти за 15 минут до каждой провокационной пробы вводят пириламин (3 мг/кг). Через 24 часа после первой провокационной пробы (или через 4 часа после последней провокационной пробы), под анестезией у животных проводят кровопускание из сонной артерии. Бронхоальвеолярный лаваж проводят с использованием 2х10 мл 0,5 ммоля ЭДТА в ДФБС (водно-масляная эмульсия Са2+, Мg2+) при 37oС через канюлю в трахее. Общее количество клеток в жидкости бронхоальвеолярного лаважа измеряют микросчетчиком клеток "Sysmex" (F-800) и раздельно подсчитывают различные клетки в цитопсиновом препарате.
Процент подавления общего накопления клеток или эозинофилов = [(носитель - ложная провокация) - (леченные - ложная провокация]/(носитель - ложная провокация) х 100.
Экспериментальная модель на мышах. Самцов мышей линии С57 BL/6 с массой тела 21-23 г активно сенсибилизируют OVA путем введения 10 мкг OVA и 1 мг А1(ОН)3 в 0,2 мл 0,9% NaCl в 1-й день. Гиперчувствительность развивается на 14-й и 21-й день после ежедневной ингаляции аэрозоля 1% OVA или физраствора в течение 30 минут. Соединения готовят в 10% Полиэтиленгликоле 200 или 0,5% карбоксиметилцеллюлозе и вводят внутрь в дозе 20 мг/кг b.i.d. на 18-22-й день. Через четыре часа после последней ингаляции OVA у животных в условиях анестезии проводят кровопускание из сонной артерии. BAL проводят с использованием 2х1 мл ДФБС (водно-масляная эмульсия 4 Са2+, Мg2+), содержащая 0,5 ммоля ЭДТА при 37oС через канюлю в трахее. Общее количество клеток в жидкости BAL измеряют микросчетчиком клеток "Sysmex" (F-800). Дифференциальный подсчет клеток в жидкости BAL проводят микросчетчиком клеток "Sysmex" (F-800). Дифференциальный подсчет клеток проводят на препарате цитопсина с окрашиванием белой гимзой.
Процент подавления общего накопления клеток или эозинофилов = [(носитель - ложная провокация) - (леченные - ложная провокация]/(носитель - ложная провокация) х 100
(см. Yeadon M. et al., Agents Actions, 1993, Vol. 39, pp.8-17; Brussells G. G. et al., ALA'94, A754; Schwenk U. еt al., J. Biol. Biochem., 1992, Vol. 267, pp.12482-12488, и Clinic M. et al., Cur. Opin. Immunol., 1994, Vol. 6, pp.860-864).
Модификации и варианты настоящего изобретения, относящиеся к соединениям, которые уменьшают образование кислородных радикалов во время воспалительной или иммунной реакции, будут очевидны специалистам в этой области на основании предшествующего подробного описания изобретения. Имеется в виду, что подобные модификации и варианты входят в объем прилагаемой формулы изобретения.






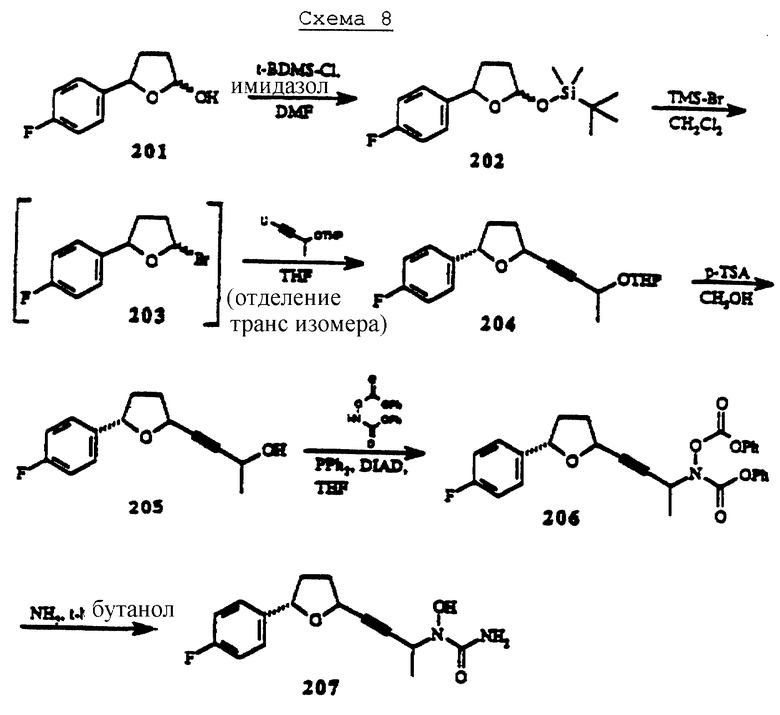


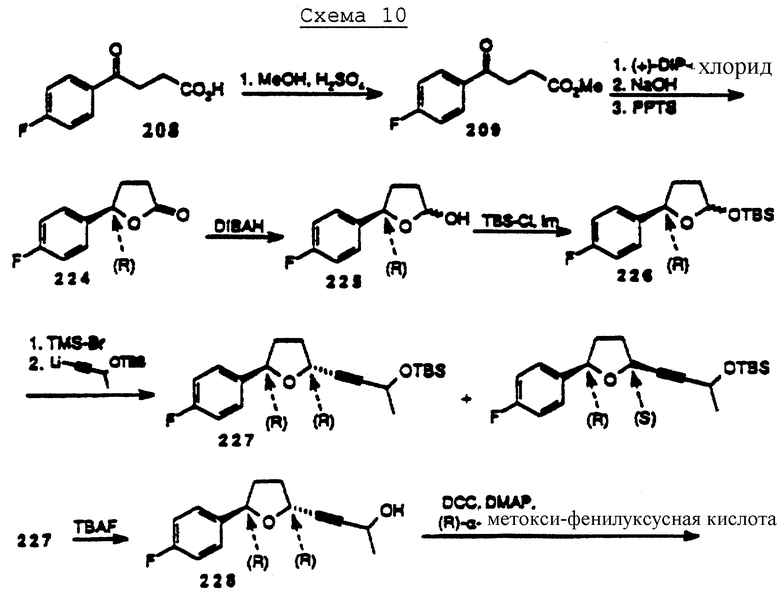











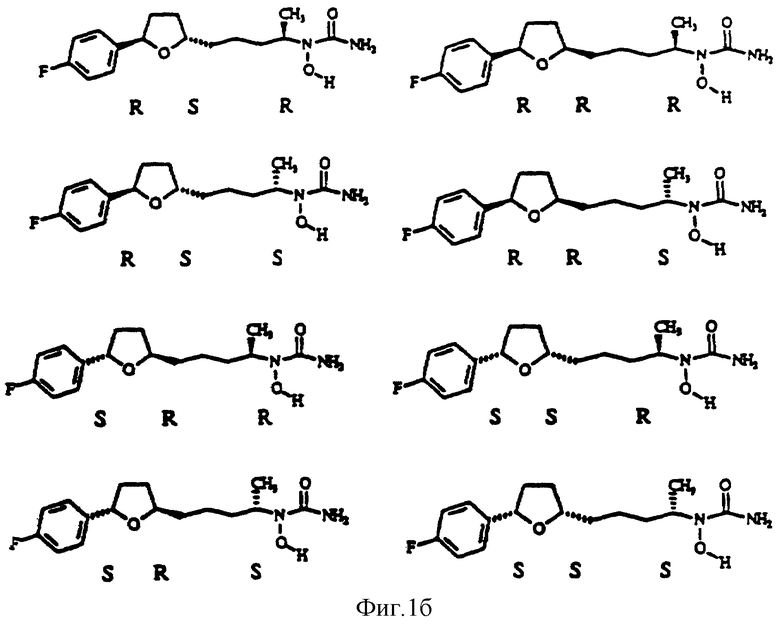


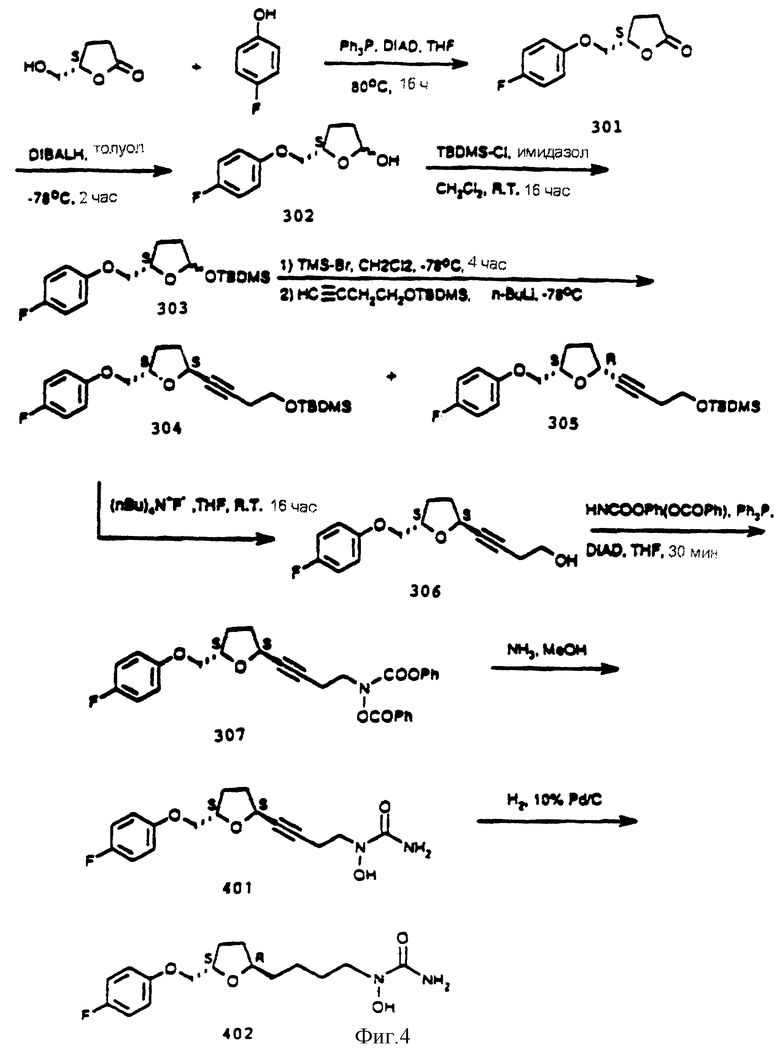

Изобретение относится к новым 2,5-дизамещенным тетрагидрофуранам или тетрагидротиофенам формулы I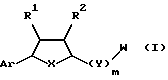
где Ar - фенил, который необязательно замещен, по крайней мере, одной группой, выбираемой из гало (включая, но не ограничиваясь фтором), низшего алкокси (включая метокси), низшего арилокси (включая фенокси), циано или R3;
m = 1;
W независимо - -AN(OM)C(O)N(R3)R4, -AN(R3)C(O)N(OM)R4, -AN(OM)C(O)R4, AC(O)N(OM)R4, -C(O)N(OM)R4 или -C(O)NHA;
А - низший алкил, низший алкенил или низший алкинил, в котором один или более углеродов необязательно могут быть замещены O, N или S;
M водород, фармацевтически приемлемый катион;
X - O,S;
Y - О, S, водород, низший алкил, низший алкенил, низший алкинил, алкарил;
R1 и R2 независимо - водород, низший алкил, гало или -СООН;
R3 и R4 независимо - водород, алкил, алкенил, алкинил, С1-6алкокси-С1-С10 алкил или С1-6 алкилтио-С1-10 алкил,
которые обладают противовоспалительной активностью благодаря подавлению 5-липоксигеназы как антагонисты рецепторов PAF и проявляют двойную активность, т. е. действуя как антогонист рецептора PAF и ингибитор 5-липоксигеназы. Изобретение также относится к фармацевтической композиции на основе этих соединений и способам лечения воспалительных заболеваний. Технический результат - получение новых биологически активных соединений. 10 с. и 28 з. п.ф-лы, 5 табл. 5 ил.
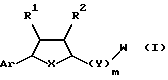
где Ar - фенил, который необязательно замещен, по крайней мере, одной группой, выбранной из гало (включая, но не ограничиваясь фтором), низшего алкокси (включая метокси), низшего арилокси (включая фенокси), циано, или R3;
m = 0;
W независимо - -AN(OM)C(O)N(R3)R4,
-AN(R3)C(O)N(OM)R4,
-AN(OM)C(O)R4, AC(O)N(OM)R4, -C(O)N(OM)R4 или
-C(O)NHA;
А - низший алкинил, в котором один или более атомов углерода, необязательно могут быть замещены O, N или S;
М - водород, фармацевтически приемлемый катион;
Х - O, S;
Y - O, S, водород, низший алкил, низший алкенил, низший алкинил, алкарил;
R1 и R2 независимо - водород, низший алкил, гало или -СООН;
R3 и R4 независимо - водород, алкил, алкенил, алкинил, С1-6алкокси-С1-10алкил или С1-6алкилтио-С1-10алкил.
где Ar - фенил, необязательно замещенный, по крайней мере, одной группой, выбранной из гало (включая, но не ограничиваясь фтором), низшего алкокси (включая метокси), низшего арилокси (включая фенокси), циано, или R3;
m = 0;
W независимо -
-AN(R3 )C(O)N(OM)R4, -AN(OM)C(O)R4,
AC(O)N(OM)R4, -C(O)N(OM)R4 или -C(O)NHA;
А - низший алкил, низший алкенил или низший алкинил, в которых один или более атомов углерода необязательно могут быть замещены O, N или S;
М - водород, фармацевтически приемлемый катион;
Х - O, S;
Y - O, S, водород, низший алкил, низший алкенил, низший алкинил или алкарил;
R1 и R2 независимо - водород, низший алкил, гало или -СООН;
R3 и R4 независимо - водород, алкил, алкенил, алкинил, С1-6алкокси-С1-10алкил или С1-6алкилтио-С1-10алкил.
где Ar - фенил, необязательно замещенный, по крайней мере, одной группой, выбираемой из гало (включая, но не ограничиваясь фтором), низшего алкокси (включая метокси), низшего арилокси (включая фенокси), циано, или R3;
m = 1;
W независимо -
-AN(OM)C(O)N(R3)R4, -AN(R3)C(O)N(OM)R4,
-AN(OM)C(O)R4, AC(O)N(OM)R4, -C(O)N(OM)R4 или
-C(O)NHA;
А - низший алкил, низший алкенил или низший алкинил, в которых один или более атомов углерода необязательно могут быть замещены O, N или S;
М - водород, фармацевтически приемлемый катион;
Х - O, S;
Y - O, S, водород, низший алкил, низший алкенил, низший алкинил или алкарил;
R1 и R2 независимо - водород, низший алкил, гало или -СООН;
R3 и R4 независимо - водород, алкил, алкенил, алкинил, С1-6алкокси-С1-10алкил или С1-6алкилтио-С1-10алкил.
где Ar - фенил, необязательно замещенный по крайней мере одной группой, выбираемой из гало (включая, но не ограничиваясь фтором), низшего алкокси (включая метокси), низшего арилокси (включая фенокси), циано, или R3;
m = 0 или 1;
W независимо -
-AN(OM)C(O)N(R3)R4, -AN(R3)C(O)N(OM)R4,
-AN(OM)C(O)R4, AC(O)N(OM)R4, -C(O)N(OM)R4 или
-C(O)NHA;
А - низший алкил, низший алкенил или низший алкинил, в которых один или более атомов углерода необязательно могут быть замещены O, N или S;
М - водород, фармацевтически приемлемый катион;
Х - O, S;
Y - O, S, водород, низший алкил, низший алкенил, низший алкинил или алкарил;
R1 и R2 независимо - водород, низший алкил, гало или -СООН;
R3 и R4 независимо - водород, алкил, алкенил, алкинил, С1-6алкокси-С1-10алкил или С1-6алкилтио-С1-10алкил.
где Ar - фенил, необязательно замещенный, по крайней мере, одной группой, выбираемой из гало, низшего алкокси, низшего арилокси, циано, или R3;
m = 0 или 1;
W независимо -
-AN(OM)C(O)N(R3)R4, -AN(OM)C(O)N(R3)R4,
-AN(R3)C(O)N(OM)R4, -N(R3)C(O)N(OM)R4,
-AN(OM)C(O)R4, -N(OM)C(O)R4,
-AC(O)N(OM)R4, -C(O)N(OM)R4 или -C(O)NHA;
А - низший алкил, низший алкенил или низший алкинил, в которых один или более атомов углерода необязательно замещены O, N или S;
М - водород, фармацевтически приемлемый катион;
Х - O, S;
Y - O, S, водород, низший алкил, низший алкенил, низший алкинил или алкарил;
R1 и R2 - водород, низший алкил, гало или -СООН;
R3 и R4 независимо - водород, алкил, алкенил, алкинил или арил.

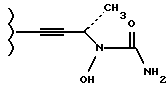
31. Соединение по п.27, где -(Y)mW выбирают из группы, состоящей из

32. Соединение по пп.27-30 или 31 в форме, энантиометрически обогащенной, по крайней мере, на 97%.
Приоритет по пунктам и признакам:
27.06.1994 по пп.1-38;
17.02.1995 по пп.1-38 (разновидности радикалов);
31.05.1995 по пп.1-38 (разновидности радикалов).
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ НИТРОФУРАНА ИЛИ НИТРОТИОФЕНА | 1971 |
|
SU421190A3 |
| Способ получения производных тиено-(3,2-с)пиридина или их солей | 1977 |
|
SU683624A3 |
| US 5037853 А, 06.08.1991 | |||
| US 5110831 А, 05.05.1992 | |||
| US 5175183 А, 29.12.1992 | |||
| US 5183818 А, 02.02.1993 | |||
| US 5187192 А, 16.02.1993 | |||
| US 5288751 А, 22.02.1994 | |||
| US 5169854 А, 08.12.1992. | |||

