Область техники, к которой относится изобретение
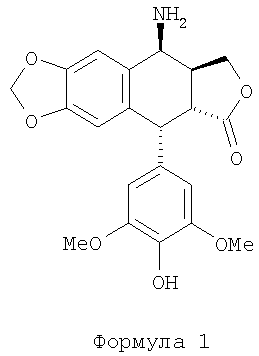
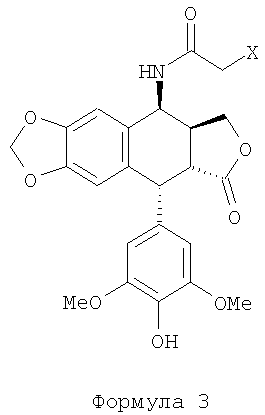
Настоящее изобретение относится к способу получения 4β-амино-4′-деметил-4-дезоксиподофиллотоксина формулы 1 из 4β-галогеноацетамидо-4′деметил-4-дезоксиподофиллотоксина формулы 3 (X=Cl, Br или I) расщеплением в присутствии тиомочевины и кислоты. В частности, изобретение касается способа получения 4β-амино-4′-деметил-4-дезоксиподофиллотоксина формулы 1 из 4′-деметилэпиподофиллотоксина формулы 2 через 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксина формулы 3 (X=Cl, Br или I).
Уровень техники
4β-амино-4′-деметил-4-дезоксиподофиллотоксин представляет собой синтетическое промежуточное соединение, применяемое в производстве противораковых соединений (заявка на патент Франции №0404053).
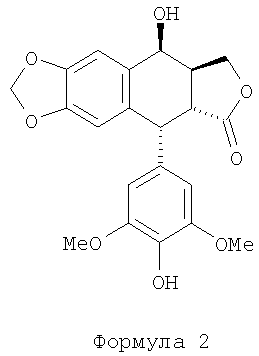
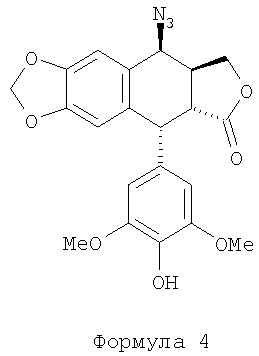
Технология производства этого промежуточного соединения основывается на преобразовании 4′-деметилэпиподофиллотоксина (формулы 2) в 4β-азидо-4′-деметил-4-дезоксиподофиллотоксин формулы 4 с последующим каталитическим окислением этого производного азида в производное амина формулы 1. Проблема при таком преобразовании заключается в отсутствии стереоселективности преобразования активированного производного в позиции 4 (позиция бензила), обеспечивающей получение смеси азидов α и β формулы 4. Эта проблема частично решена за счет применения азида натрия и трифторуксусной кислоты, см. J. Med. Chem. 1991 г., №34, стр.3346. Однако при этом требуется производить очистку промежуточного соединения «азид» формулы 4 с помощью хроматографии, а также и продукта каталитического восстановления, т.е. амина формулы 1. Другой способ раскрыт в Chinese Chemical Letters, 1993 г., №4 (4), стр.289. Эти авторы пользуются методом с применением азида, проводя реакцию между азотисто-водородной кислотой HN3 (приготовленной in situ) в присутствии эфирата BF3 при -10~-15°С. Полученные этими авторами результаты свидетельствуют о высокой стереоселективности преобразования при коэффициенте выхода не менее 80%. Также способ преобразования азида формулы 4 в амин формулы 1 описан в Tet. Let., 1999 г., №40, стр.1967 и Tet. Let., 2000 г., стр.7743. Эти авторы применили йодид самария в t-BuOH и THF (тетрагидрофуране) или же в двойном соединении FeSO4·7H2O/NH3. Недавно появилось сообщение с подтверждением необходимости очистки хроматографией, Bioorg. Med. Chem., 2003 г., №11, стр.5135. Авторами был получен амин формулы 1 при коэффициенте выхода 70%.
Тем не менее эти способы создают две проблемы: 1) применение опасного производного азида, являющегося потенциально взрывчатым, особенно при использовании в крупном масштабе при промышленном производстве медикамента; 2) необходимость проведения одного или даже двух этапов хроматографии для получения соединения «амин» формулы 1 высокого качества для последующего получения целевого продукта, а именно противоракового препарата, которые представляют собой дорогостоящие этапы при промышленном масштабе производства.
Раскрытие изобретения
Задачей настоящего изобретения является решение этих обеих проблем, то есть исключение применения опасных или взрывчатых соединений и этапов очистки хроматографией.
4β-гелогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксин, являющийся промежуточным соединением формулы 3, представляет собой известное соединение (заявка на патент Франции №0404053, WO 2004/073375). Также известен переход соединения формулы 2 в соединение формулы 3 (заявка на патент Франции 0404053). Следовательно, объектом изобретения является способ синтеза соединения формулы 1 из соединения формулы 3.
При традиционном органическом синтезе расщепление хлорацетамидов для получения аминов производится обработкой производного хлорацетамида третичного амида тиомочевиной в этаноле в присутствии уксусной кислоты при оптимальном соотношении 5:1 (A.Jirgensons и др. Synthesis, 2000 г., стр.1709). При этой реакции применяемые этанол и уксусная кислота не содержат воды. Этот метод никогда не применялся для ряда подофиллотоксина и для этого он не пригоден. Действительно, будучи примененным в случае с соединением формулы 3, этот метод приводит через 10 часов при дефлегмации к преобразованию менее 10% исходного материала (соединение формулы 3), и реакционное промежуточное соединение (X=S - изотиоуроний в виде хлорида) более не вступает в реакцию (см. сравнительный пример). Более длительное время реакции является неблагоприятным с точки зрения степени чистоты, так как образуются вторичные продукты.
Потребовалась адаптация и усовершенствование методики для достижения требуемого преобразования. Неожиданно авторами изобретения был создан способ синтеза соединения формулы 1 из соединения формулы 3, позволяющий получать соединение формулы 1 с высокой степенью чистоты без дополнительного этапа очистки (в частности, хроматографией).
Объектом изобретения является способ синтеза 4β-амино-4′-диметил-4-дезоксиподофиллотоксина формулы 1:

отличающийся тем, что он включает в себя следующие последовательные этапы:
а) проведение реакции в среде слабой чистой кислоты без другого растворителя при температуре, превышающей температуру окружающей среды, между тиомочевиной и 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксином формулы 3:

где: X означает атом галогена, выбранного из группы, состоящей из хлора, брома и йода, предпочтительно хлора;
б) извлечение 4β-амино-4′-деметил-4-дезоксиподофиллотоксина.
В отношении способа расщепления хлорацетамидов, описанного в уровне техники (A.Jirgensons и др., Synthesis, 2000 г., стр.1709), было установлено, что эта операция может проводиться в чистой слабой кислоте, т.е. без присутствия воды или органического растворителя. В смысле настоящего изобретения применение термина «кислота» восходит к ее определению по Бронстеду (Bronsted), а именно она представляет собой химическое вещество, способное терять протон Н+. Слабая кислота - это кислота, которая в противоположность сильной кислоте не диссоциируется полностью в воде.
Слабая кислота характеризуется преимущественно показателем кислотности от 4 до 6 при 25°С. В частности, слабой кислотой является преимущественно карбоновая кислота формулы 5 R-COOH, где R означает атом водорода или алкильный радикал с 1 или 2 атомами углерода. Более тяжелые кислоты теперь не применяются в качестве растворителя, они не обладают приемлемой пахучестью (в частности, масляная кислота). В частности, слабая кислота выбирается из группы, состоящей из муравьиной, уксусной или пропионовой кислот, предпочтительно уксусной кислоты.
Приведенные ниже соотношения между разными соединениями соответствуют соотношениям количеств, используемых для этих соединений, если не указано иначе.
В рамках настоящего изобретения выражение «чистая слабая кислота» означает, что кислота является ледяной, т.е. не содержащей воды. Выражение «без другого растворителя» означает, что реакционная среда на этапе а) содержит только чистую слабую кислоту, соединение формулы 3 и тиомочевину, следовательно, она не содержит воду или другой растворитель, такой как спирт или органический растворитель.
На этапе а) реакционную среду предпочтительно нагревают до температуры свыше 60°С, более предпочтительно до температуры от 60 до 100°С. Другим признаком изобретения является то, что применяемая чистая слабая кислота служит растворителем во время реакции. Молярное соотношение между 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксином и слабой кислотой составляет не менее 0,5.
Молярное соотношение между 4β-галогеноацетамидо-4'-деметил-4-дезоксиподофиллотоксином и тиомочевиной составляет предпочтительно от 0,5 до 1. Согласно оптимальному варианту выполнения изобретения на этапе а) 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксин приводят в контакт с чистой слабой кислотой перед добавкой тиомочевины. Согласно другому, более оптимальному варианту 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксин и чистая слабая кислота приводятся в контакт между собой, причем 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксин находится предпочтительно во взвеси в чистой слабой кислоте, а реакционную среду нагревают до требуемой температуры перед добавкой тиомочевины при этой температуре.
Продолжительность реакции на этапе а) составляет предпочтительно от 1 до 3 часов. В случае применения чистой уксусной кислоты продолжительность реакции на этапе а) составляет около 2 часов.
По окончании этапа а) целевой продукт формулы 1 выпадает в осадок в реакционной среде. Его извлекают на этапе b) любым, известным среднему специалисту способом, в частности, простая фильтрация и сушка обычными методами являются достаточными.
После фильтрации и сушки обычными способами соединение формулы 1 в виде хлоргидрата, бромгидрата или йодгидрата получают при среднем молярном выходе свыше 85%, предпочтительно свыше 90% от молярного количества соединения формулы 3. В случае применения чистой уксусной кислоты соединение формулы 1 в виде хлоргидрата, бромгидрата или йодгидрата получают при среднем молярном выходе 93% от молярного количества использованного соединения формулы 3.
Соединение формулы 1 предпочтительно получают при степени чистоты свыше 90%, более предпочтительно 95% или более.
Соединение формулы 1, полученное в виде хлоргидрата, бромгидрата или йодгидрата, является чистым и для него не требуется дополнительный этап очистки хроматографией. Оно может непосредственно применяться на последующих этапах синтеза, чем достигается существенное преимущество с точки зрения рентабельного производства в промышленном масштабе.
Объектом настоящего изобретения является также способ синтеза 4β-амино-4′-деметил-4-дезоксиподофиллотоксина формулы 1:
отличающийся тем, что он включает в себя следующие последовательные этапы:
i) проведение реакции в смеси из кислоты, воды и органического растворителя при температуре свыше температуры окружающей среды между тиомочевиной и 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксином формулы 3:
где Х означает атом галогена, выбранного из группы, состоящей из хлора, брома и йода, предпочтительно хлора;
ii) извлечение 4β-амино-4′-деметил-4-дезоксиподофиллотоксина.
В отношении способа расщепления хлорацетамидов, раскрытого в уровне техники (A.Jirgensons и др., Synthesis, 2000 г., стр.1709), было найдено, что добавление воды в реакционную среду способствует протеканию реакции, сопровождающемуся полным расходом исходного материал без образования продуктов распада.
Предпочтительно, чтобы реакционная среда не содержала другого растворителя или реактива. На этапе i) реакционная среда предпочтительно подогревается до температуры свыше 60°С, более предпочтительно до температуры от 60 до 100°С. Молярное соотношение между 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксином и тиомочевинной составляет предпочтительно от 0,5 до 1. На этапе i) 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксин предпочтительно приводится в контакт со смесью из кислоты, воды и органического растворителя перед добавкой тиомочевины. Более оптимально, когда 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксин и смесь из кислоты, воды и органического растворителя приведет в контакт между собой, при этом предпочтительно, чтобы 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксин находился во взвешенном состоянии в указанной смеси, причем реакционную среду нагревают до требуемой температуры перед добавкой тиомочевины при этой температуре.
Предпочтительно, чтобы органический растворитель, применяемый во втором способе согласно изобретению, представлял собой водорастворимый органический растворитель, более предпочтительно, чтобы он был выбран из группы, состоящей из циклических простых эфиров, в частности диоксана, спиртов, в частности метанола, этанола, пропанола и изопропанола, и N,N-диметилацетамида, диметилформамида и N-метилпирролидона.
Таким образом, по отношению к способу расщепления, описанному в уровне техники (A.Jirgensons и др., Synthesis, 2000 г., стр.1709), было также установлено, что операция может проводиться в присутствии органического растворителя, такого как диоксан или N,N-диметилацетамид, вместо этанола в присутствии воды.
В первом предпочтительном варианте второго способа по изобретению органическим растворителем служит спирт, предпочтительно этанол.
В рамках этого первого варианта кислота представляет собой предпочтительно сильную кислоту, в частности, выбранную из группы, состоящей из соляной, серной и фосфорной кислот. Объемное соотношение спирт/(вода + сильная кислота) составляет предпочтительно 2-5:0,5-2, более предпочтительно 2,5:1, при этом сильная кислота обладает нормальностью от 1 до 2. Соединение формулы 1 предпочтительно получать при молярном выходе свыше 80%, более предпочтительно свыше 85%, в частности 90%. Продолжительность реакции составляет предпочтительно свыше 8 часов, но менее 10 часов, более предпочтительно около 9 часов.
Соединение формулы 1 также предпочтительно получать при степени чистоты свыше 90%, в частности 95%.
В качестве альтернативы в рамках первого варианта предпочтительно применяется слабая кислота, в частности карбоновая кислота формулы 5 R-COOH, где R означает атом водорода или алкильный радикал с 1-2 атомами углерода. Более тяжелые кислоты теперь не применяются в качестве растворителей и не обладают приемлемыми свойствами пахучести (в частности, масляная кислота). В частности, слабая кислота выбирается из группы, состоящей их муравьиной, уксусной или пропионовой кислот, предпочтительно уксусной кислоты. Объемное соотношение спирт/вода/слабая кислота составляет предпочтительно 2-10:0,5-2:0,5-2, более предпочтительно 5:1:1. В частности, объемное соотношение этанол/вода/уксусная кислота составляет предпочтительно 5:1:1.
Соединение формулы 1 получают предпочтительно при молярном выходе свыше 55%, предпочтительно 60%. Продолжительность реакции составляет предпочтительно более 8 часов, но менее 11 часов, более предпочтительно около 10 часов.
Соединение формулы 1 получают предпочтительно при степени чистоты более 90%, в частности 95%.
Во втором предпочтительном варианте второго способа по изобретению органическим растворителем служит циклический простой эфир, в частности диоксан, N,N-диметилацетамид, диметилформамид и N-метилпирролидон.
Объемное соотношение циклический простой эфир (диоксан) или N,N-диметилацетамид, диметилформамид и N-метилпирролидон/вода/слабая кислота (уксусная кислота) составляет предпочтительно 2-10:0,5-2:0,5-2, более предпочтительно 5:1:1. Объемное соотношение диоксан или N,N-диметилацетамид, диметилформамид и N-метилпирролидон/вода/уксусная кислота составляет предпочтительно 5:1:1. Соединение формулы 1 предпочтительно получать при молекулярном выходе свыше 60%, более предпочтительно свыше 65%, в частности 70%. Продолжительность реакции составила предпочтительно более 4 часов, но менее 10 часов, более предпочтительно около 5-6 часов.
Соединение формулы 1 предпочтительно получать при степени чистоты свыше 90%, предпочтительно 95%.
В исследованных случаях целевой продукт выпадал в осадок в реакционной среде. Его извлекали на этапе b) любым, известным среднему специалисту способом, в частности, простая фильтрация и сушка известными способами являются достаточными.
Полученное соединение формулы 1 в виде хлоргидрата, бромгидрата или йодгидрата являлось чистым, и для него не требовался этап дополнительной очистки хроматографией. Он мог использоваться непосредственно на последующих этапах синтеза, что обеспечивает существенное преимущество с точки зрения рентабельного производства в промышленном масштабе.
В рамках первого или второго способа по изобретению 4β-галогеноацетамидо-4′-деметил-4-дезоксиподофиллотоксин формулы 3 получают предпочтительно реакцией между 4′-деметилэпиподофиллотоксином формулы 2:

и галогеноацетонитрилом формулы 6 Х-СН2 - С≡N, где Х означает атом галогена, выбранного из группы, состоящей из хлора, брома и йода, в кислой среде. Затем следует реакция Риттера для прямого получения 4β-хлороацетамидо-4′-деметил-4-дезоксиподофиллотоксина кристаллизацией в конце реакции при коэффициенте выхода предпочтительно свыше 80%, более предпочтительно свыше 90%.
Это промежуточное соединение обладает исключительно стереохимией β на атоме углерода в позиции 4. На этой стадии решается проблема стереохимии. Чистота указанного промежуточного соединения такова, что это соединение может применяться без дополнительной очистки на этапе расщепления для получения 4β-амино-4′-деметил-дезоксиподофиллотоксина формулы 1.
4′-деметилэпиподофиллотоксин формулы 2 (получаемый способом, описанным в патенте Франции №2742439) предпочтительно обрабатывают хлорацетонитрилом, обычным и дешевым реактивом, вместе с серной кислотой. Следовательно, 4β-хлорацетамидо-4′-деметил-4-дезоксидофиллотоксин предпочтительно получают при коэффициенте выхода 93%.
После фильтрации и сушки обычными способами соединение формулы 1 в виде хлоргидрата, бромгидрата или йодгидрата получают в результате применения ледяной чистой уксусной кислоты (без воды и без другого органического растворителя, первый способ согласно изобретению) при среднем молярном выходе 86% от молярного количества использованного 4β-деметилэпиподофиллотоксина (формулы 2), т.е. на двух этапах (от соединения формулы 2 к соединению формулы 3, затем от соединения формулы 3 к соединению формулы 1).
Осуществление изобретения
Приводимые ниже примеры показывают применявшиеся методики.
Пример 1. Получение 4β-хлорацетамидо-4′-деметил-4-дезоксиподофиллотоксина (формулы 3)
В суспензию 30 г (0,075 моля) 4′-деметилэпиподофиллотоксина в 47,5 мл (0,75 моля) хлорацетонитрила добавляют каплями 0,5 мл концентрированной серной кислоты при температуре окружающей среды. Встряхивают в течение одного часа при указанной температуре, при этом наблюдают растворение и повторное выпадение в осадок. Добавляют 300 мл 2-пропанола. Осадок фильтруют, промывают с помощью 200 мл 2-пропанола и воды до достижения показателя рН 7. Полученное твердое вещество белого цвета сушат в вакууме при 40°С и получили 32,9 г соединения хлорацетамид формулы 3. Молярный выход составил 93%.
Точка плавления F составила 240°С.
Анализ ЯМР протона: 1Н ЯРМ (DMSO) δ 8,65 (d, 1H, J=7 Гц, NH), 8,26 (s, 1H, 4′-OH), 6,78 (s, 1H, Н5), 6,54 (s, 1H, H8), 6,24 (s, 2H, Н2′, Н6′), 5,99 (d, 2Н, J=11,3 Гц, OCH2O), 5,17 (dd, 1H, J=4,56 и 7 Гц, Н4), 4,51 (d, 1H, J=5,2 Гц, H1), 4,29 (t, 1H, J=8 Гц, Н11а), 4,10 (s, 2H, CH2Cl), 3,97 (m, 1H, Н3), 3,78 (dd, 1H, J=8 Гц и 10 Гц, H11b), 3,63 (s, 6H, 2×ОСН3), 3,15 (dd, 1H, J=5,2 и 14 Гц, H2).
Другие галогеноацетамиды (X=Br, I) могут быть получены аналогичным образом с помощью бромацетонитрила или йодацетонитрила.
Сравнительный пример. Получение 4-амино-4′-деметил-4-дезоксиподофиллотоксина (формулы 1). Метод с применением этанола/уксусной кислоты 5:1 (Synthesis, 2000 г., стр.1709).
Таблица 1: эксперимент 1.
Суспензию 0,5 г (1,05 ммоля) 4β-хлорацетамидо-4′-деметил-4-дезоксиподофиллотоксина, полученного в примере 1, в смеси из 2,5 мл этанола и 0,5 мл ледяной уксусной кислоты доводят до 80°С при встряхивании. Однократно вводят 0,12 г (1,57 ммоля) тиомочевины. Встряхивают при этой температуре в течение 10 часов. Анализ реакционной среды тонкослойной хроматографией показал наличие лишь менее 10% требуемого продукта 4β-амино-4′-деметилэпиподофиллотоксина (формулы 1) от присутствующего промежуточного соединения изотиоуроний (X=S-изотиоуроний), который более не вступал в реакцию, и продукты разложения.
Пример 2. Получение 4β-амино-4′-деметил-4-дезоксиподофиллотоксина (формулы 1). Метод с применением чистой ледяной уксусной кислоты, первый способ согласно изобретению.
Таблица 1: эксперимент 2.
Суспензию 17 г (0,0358 моля) 4β-хлорацетамидо-4′-деметил-4-дезоксиподофиллотоксина, полученного в примере 1, в 75 мл ледяной уксусной кислоты нагревают до 80°С при встряхивании. Добавляют однократно 4,2 г (0,0537 моля) тиомочевины. При этой температуре встряхивают в течение 1,5 часа, при этом наблюдают растворение и повторное выпадение в осадок. Реакционную среду фильтруют в горячем состоянии, промывают 75 мл ледяной уксусной кислоты и диизопропилового эфира. Полученное твердое вещество белого цвета сушат в вакууме при 40°С. Получено 14,6 г соединения формулы 1 в виде хлоргидрата при молярном выходе 93%.
Точка плавления F составила более 260°С.
Анализ ЯМР протона: 1Н ЯРМ (DMSO) δ 8,63 (m, 2Н), 8,32 (m, 1H), 7,23 (s, 1H, H5), 6,60 (s, 1H, H8), 6,18 (s, 2H, H2′, Н6′), 6,05 (d, 2H, J=2,1 Гц, ОСН2О), 4,73 (d, 1H, J=4,5 Гц, Н4), 4,56 (d, 1H, J=5,2 Гц, H1), 4,34 (m, 2H, H11a и H11b), 3,65 (dd, 1H, J=5,2 Гц, Н2), 3,62 (s, 6H, 2×ОСН3), 3,06 (m, 1H, Н3).
Пример 3. Получение 4β-амино-4′-деметил-4-дезоксиподофиллотоксина (формулы 1). Метод с применением этанола и соляной кислоты 1N, второй способ согласно изобретению, первый вариант, первая альтернатива.
Таблица 1: эксперимент 3.
Суспензию 0,5 г (1,05 ммоля) 4β-хлорацетамидо-4′-деметил-4-дезоксиподофиллотоксина, полученного в примере 1, в смеси из 2,5 мл этанола и 1 мл соляной кислоты 1N нагревают до 80°С при встряхивании. Однократно добавляют 0,12 г (1,57 ммоля) тиомочевины. При указанной температуре встряхивают в течение 9 часов, при этом наблюдают растворение и повторное выпадение в осадок. Охлажденную реакционную среду фильтруют, промывают этанолом и диизопропиловым эфиром. Полученное твердое вещество белого цвета сушат в вакууме при 40°С. Получено 0,4 г соединения формулы 1 в виде хлоргидрата при молярном выходе 90%.
Точка плавления F составила свыше 260°С.
Пример 4. Получение 4β-амино-4′-деметил-4-дезоксиподофиллотоксина (формулы 1). Метод с применением этанола/воды/уксусной кислоты (5:1:1), второй способ согласно изобретению, первый вариант, вторая альтернатива.
Таблица 1: эксперимент 4.
Суспензию 0,5 г (1,05 ммоля) 4β-хлорацетамидо-4′-деметил-4-дезоксиподофиллотоксина, полученного в примере 1, в смеси из 2,5 мл этанола, 0,5 воды и 0,5 ледяной уксусной кислоты нагревают до 80°С при встряхивании. Однократно добавляют 0,12 г (1,57 ммоля) тиомочевины. При указанной температуре встряхивали в течение 10 часов, при этом наблюдают растворение и повторное выпадение в осадок. Охлажденную реакционную среду фильтруют, промывают этанолом и диизопропиловым эфиром. Полученное твердое вещество белого цвета сушат в вакууме при 40°С. Получено 0,27 г соединения формулы 1 в виде хлоргидрата при молярном выходе 60%.
Точка плавления F составила свыше 260°С.
Пример 5. Получение 4β-амино-4′-деметил-4-дезоксиподофиллотоксина (формулы 1). Метод с применением растворителя (N,N-диметилацетамид, диоксан)/воды/уксусной кислоты, второй способ согласно изобретению, второй вариант.
Таблица 1: эксперимент 5.
Суспензию 0,5 г (1,05 ммоля) 4β-хлорацетамидо-4′-деметил-4-дезоксиподофиллотоксина, полученного в примере 1, в смеси из 2,5 мл диоксана или N,N-диметилацетамида, 0,5 мл воды и 0,5 мл ледяной уксусной кислоты нагревают до 80°С при встряхивании. Однократно добавляют 0,12 г (1,57 ммоля) тиомочевины. При указанной температуре встряхивают в течение 5-6 часов, при этом наблюдают растворение и повторное выпадение в осадок. Охлажденную реакционную среду фильтруют, промывают 2-пропанолом и диизопропиловым эфиром. Полученное твердое вещество белого цвета сушат в вакууме при 40°С. Получено 0,31 г соединения формулы 1 в виде хлоргидрата при молярном выходе 70%.
Точка плавления F составила свыше 260°С.
Результаты опытов, полученные в сравнительном примере и в примерах 2-5, представлены в нижеследующей таблице 1.
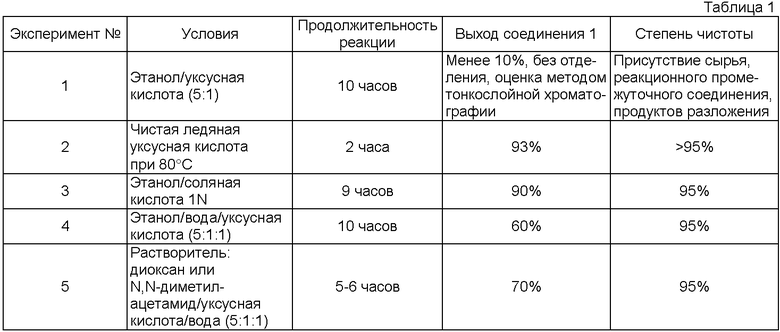
Таблица 1 свидетельствует о значительном преимуществе, достигаемом в случае применения чистой ледяной уксусной кислоты при 80°С при короткой продолжительности реакции, 2 ч, для получения требуемого продукта с превосходным коэффициентом выхода с обеспечением весьма удовлетворительной степени чистоты для последующего применения в синтезе противораковых соединений.
Настоящее изобретение относится к способу синтеза 4β-амино-4'деметил-4-дезоксиподофиллотоксина формулы (1), включающему следующие последующие этапы: а) проведение реакции в среде чистой слабой кислоты или смеси из кислоты, воды и органического растворителя без применения другого растворителя при температуре свыше температуры окружающей среды между тиомочевинной и 4β-галогеноацетамидо-4'-деметил-4-дезоксиподофиллотоксином; б) извлечение 4β-амино-4'-деметил-4-дезоксиподофиллотоксина. Это соединение является промежуточным продуктом в производстве противораковых соединений. 2 н. и 20 з.п. ф-лы, 1 табл.
1. Способ синтеза 4β-амино-4'-деметил-4-дезоксиподофиллотоксина формулы 1
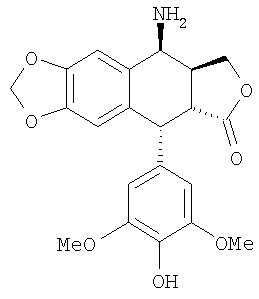
включающий следующие последовательные этапы:
а) проведение реакции в среде чистой слабой кислоты без другого растворителя при температуре выше температуры окружающей среды между тиомочевиной и 4β-галогеноацетамидо-4'-деметил-4-дезоксиподофиллотоксином формулы 3

где Х означает атом галогена, выбранного из группы, состоящей из хлора, брома или йода, предпочтительно хлора;
б) извлечение 4β-амино-4'-деметил-4-дезоксиподофиллотоксина.
2. Способ по п.1, в котором чистой слабой кислотой является карбоновая кислота формулы 5 R-COOOH, где R - атом галогена или алкильный радикал с 1-2 атомами углерода, в частности, уксусная кислота.
3. Способ по п.1 или 2, в котором на этапе а) реакционную среду нагревают до температуры от 60 до 100°С.
4. Способ по п.1 или 2, в котором на этапе а) 4β-галогеноацетамидо-4'-деметил-4-дезоксиподофиллотоксин приводят в контакт с чистой слабой кислотой перед добавкой тиомочевины.
5. Способ по п.3, в котором на этапе а) 4β-галогеноацетамидо-4'-деметил-4-дезоксиподофиллотоксин приводят в контакт с чистой слабой кислотой перед добавкой тиомочевины.
6. Способ по пп.1, 2 или 5, в котором продолжительность реакции на этапе а) составляет от 1 до 3 ч.
7. Способ по п.3, в котором продолжительность реакции на этапе а) составляет от 1 до 3 ч.
8. Способ по п.4, в котором продолжительность реакции на этапе а) составляет от 1 до 3 ч.
9. Способ синтеза 4β-амино-4'-деметил-4-дезоксиподофиллотоксина формулы 1
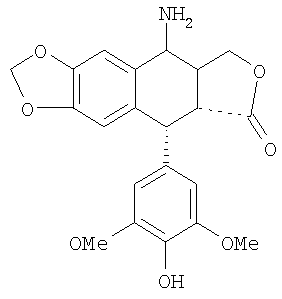
включающий следующие последовательные этапы:
i) проведение реакции в смеси из кислоты, воды и органического растворителя при температуре выше температуры окружающей среды между тиомочевиной и 4β-галогеноацетамидо-4'-деметил-дезоксиподофиллотоксином формулы 3
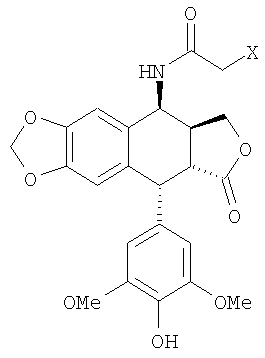
где X означает атом галогена, выбранного из группы, состоящей из хлора, брома и йода, предпочтительно хлора;
ii) извлечение 4β-амино-4'-деметилэпиподофиллотоксина.
10. Способ по п.9, в котором на этапе i) реакционную среду нагревают до температуры 60-100°С.
11. Способ по п.9 или 10, в котором на этапе i) 4β-галогеноацетамидо-4'-деметил-дезоксиподофиллотоксин приводят в контакт со смесью из кислоты, воды и органического растворителя перед добавкой тиомочевины.
12. Способ по п.9 или 10, в котором органическим растворителем является водорастворимый органический растворитель, выбираемый предпочтительно из группы, состоящей из циклических простых эфиров, в частности диоксана, спиртов, в частности этанола, и N,N-диметилацетамида, диметилформамида, N-метилпирролидона.
13. Способ по п.11, в котором органическим растворителем является водорастворимый органический растворитель, выбираемый предпочтительно из группы, состоящей из циклических простых эфиров, в частности диоксана, спиртов, в частности этанола, и N,N-диметилацетамида, диметилформамида, N-метилпирролидона.
14. Способ по любому из пп.9, 10 или 13, в котором растворителем является этанол.
15. Способ по п.11, в котором растворителем является этанол.
16. Способ по п.12, в котором растворителем является этанол.
17. Способ по п.14, в котором кислотой является сильная кислота, выбираемая, в частности, из группы, состоящей из соляной, серной и фосфорной кислот.
18. Способ по п.15 или 16 в котором кислотой является сильная кислота, выбираемая, в частности, из группы, состоящей из соляной, серной и фосфорной кислот.
19. Способ по п.9, в котором кислотой является слабая кислота, в частности, карбоновая кислота формулы 5 R-COOH, где R означает атом водорода, алкильный радикал с 1-2 атомами углерода, предпочтительно уксусная кислота.
20. Способ по п.20, в котором объемное соотношение этанол или диоксин или N,N-диметилацетамид, диметилформамид, N-метилпирролидон : вода : уксусная кислота составляет 5:1:1.
21. Способ по п.1 или 9, в котором молярное соотношение между 4β-галогеноацетамидо-4'-деметил-4-дезоксиподофиллотоксином и тиомочевиной составляет 0,5-1.
22. Способ по п.1 или 9, в котором 4β-галогеноацетамидо-4'-деметил-4-дезоксиподофиллотоксин формулы 3 получают реакцией между 4'-деметилэпиподофиллотоксином формулы 2
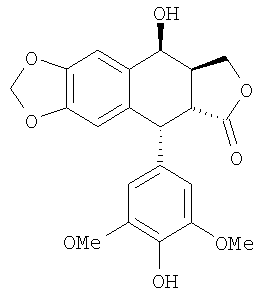
и галогеноацетонитрилом формулы 6Х-СН2-С=М, где Х означает атом галогена, выбранного из группы, состоящей из хлора, брома и йода, в кислой среде.
| KAMAL A | |||
| et al | |||
| Bioorganic & Medicinal Chemistry, 2003, v.11, no.23, 2003 | |||
| JIRGENSONS A | |||
| et al | |||
| Synthesis, 2000, no.12, p.1709-1712 | |||
| ГЕКСАГИДРОФУРО [2,3-B] ФУРАН-3-ИЛ-N-{3-[(1,3-БЕНЗОДИОКСОЛ-5- ИЛСУЛЬФОНИЛ)(ИЗОБУТИЛ)АМИНО]-1-БЕНЗИЛ -2-ГИДРОКСИПРОПИЛ}КАРБАМАТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ИНГИБИРОВАНИЯ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2247123C2 |

