Изобретение относится к основному органическому синтезу и касается способа получения этиленгликоля, сопряженного с синтезом карбамида.
Известен и широко используется способ получения этиленгликоля (ЭГ) гидратацией этиленоксида (ЭО) (О.Н.Дымент, С.К.Казанский, А.М.Мирошников. Гликоли и другие производные окисей этилена и пропилена, М., Химия, 1976; К.Weissermel, H-J.Arpe, Ind. Organic Chemistry, VCH, 1997). Некаталитический процесс проводят при 140-230°С и при давлении 2-4 МПа с большим (8-15-кратным) избытком воды. Селективность по ЭГ не превышает 90%. Известно много вариантов каталитического процесса с использованием различных классов гомогенных катализаторов в условиях меньшего избытка воды Н2О:ЭО=4:1 при 100°С и давлении 2,0 МПа (J.S.Ledford, et al, US Patent 7196233, 2007, V.F.Shvets et al. Chem. Eng. J., 2005, 107, p.199). Недостатки процесса - низкая стабильность гомогенных катализаторов в присутствии воды, невозможность полного исключения образования высших этиленгликолей (олигомеров этиленоксида) и сложность выделения ЭГ из водных 25%-ных растворов и отделения гомогенных катализаторов.
Использование гетерогенных катализаторов - активированных углей (US Pat. 5798412), глиноземов и гидротальцитов (US Pat. 4967018), цеолитов (US Pat. 4620044) и ионообменных смол (US Pat. 6147265) не решает проблем выделения ЭГ из водных растворов. К тому же низкая стабильность оксидных катализаторов и ионообменных смол в водных растворах при 140-150°С может являться причиной снижения производительности процесса.
В патенте РФ (RU №2122995, 1998) показана возможность использования анионообменных смол как катализаторов процесса гидратации ЭО при отношении вода:этиленоксид 6,7:1 (1 МПа, 140°С). При этом достигается конверсия ЭО 92% при селективности по ЭГ 98%. При более полной конверсии алкиленоксидов в каскаде реакторов селективность не превышает 93%.
С целью избежать образования олигомеров этиленоксида (ди- и триэтиленгликолей) фирма Halcon SD Group Inc. предложила двухстадийный метод синтеза ЭГ через образование и гидролиз этиленкарбоната (ЭК)
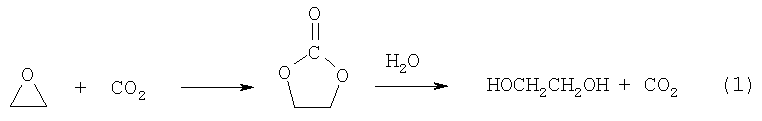
Процесс гидролиза проводят при 120-140°С и давлении 2,5-3 МПа (US Pat. 4519875, 1985) и избытке воды на гетерогенных катализаторах (гидротальциты и др.). Способ не решает проблем выделения ЭГ из разбавленных водных растворов и требует рецикла СО2.
Чтобы избежать участия воды в синтезе ЭГ, фирмы Chi Mei и Asahi Kasei предложили двухстадийный синтез ЭГ, сопряженный с синтезом диметилкарбоната (М. Arai et al, Chapter 3 in "Progress in Catalysis Research", ed. L.B.Bevy):
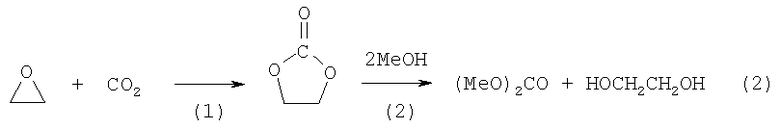
Стадию (2) метанолиза этиленкарбоната (ЭК) проводят с участием гомогенных и гетерогенных (предпочтительно) катализаторов (H.Abimanyu et. al., Catal. Lett, 2007, 118 (1-2), p.30; M.Arai et al, Appl. Catal., 2001, 219 (1-2), p.259). На цеолитах (US Pat. 5436362), ионообменных смолах (US Pat. 4691041) процесс осуществляется при температурах 60-175°С и давлениях 0,3-3,5 МПа. С учетом обратимости второй стадии отгоняют азеотроп диметилкарбонат (ДМС) - метанол при конверсии ЭК ~ 90%. ДМС выделяют экстрактивной ректификацией с водой, а затем используют комплекс колонн для отделения метанола, ЭК и ЭГ.
Усовершенствование катализаторов и технологии метанолиза запатентовано компаниями Shell и Mitsubishi (процесс OMEGA, Shell Magazine, Spring 2006, hppt://www.ChellChemicals.com/magazine, US Pat. 7385070), а также Texaco Dev. Corp., US Pat., 4667088, US Pat. 4691041, EP 0298167, 1992). Отмечается, что одни и те же катализаторы (MgO) можно использовать на обеих стадиях сопряженного процесса. Согласно патенту US Pat. 4519875 для очистки ЭГ от непрореагировавшего ЭК используют процесс каталитического гидролиза ЭК в отдельном аппарате.
Предложен также способ синтеза ЭГ, сопряженный с синтезом СО по реакции (3)

Процесс проводят при 140-180°С в растворе комплекса RuCl2(PPh3)2 в N-метилпирролидоне при давлении 80 атм (K.-I.Tominaga et al., Chem. Commun., 1995, p.1489; Energy, 1997, 22 (2-3), 169). Показано, что реакция осуществляется через образование ЭК и его гидрирование. Выход ЭГ не превышает 76% при образовании большого числа побочных продуктов.
Все рассмотренные способы синтеза ЭГ являются весьма энергоемкими из-за особенностей процесса выделения ЭГ. Так, даже в наиболее перспективном процессе по реакции (2) на второй стадии получается смесь четырех соединений - ЭК, ЭГ, ДМС и метанол с азеотропом ДМС-метанол. Наличие гетерогенных оксидных катализаторов на второй стадии также является недостатком процесса вследствие их быстрого разрушения в жидкой фазе при высоких температурах.
Карбамид получают в промышленности из СО2 и NH3 по реакции Базарова (4)
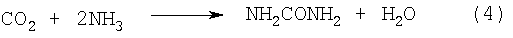
Процесс протекает в очень жестких условиях при 185-200°С и давлении 19,0-20,0 МПа с максимальным равновесным выходом 53% и рециклами СО2 и NH3 (Горловский Д.М., Альтшулер Л.Н., Кучерявый В.И. Технология карбамида, М. Химия, 1981, стр.320).
Известен способ совместного получения ЭГ и диалкилкарбамидов из этиленкарбоната, сопряженный с синтезом этиленкарбоната из диоксида углерода и оксида этилена (5)
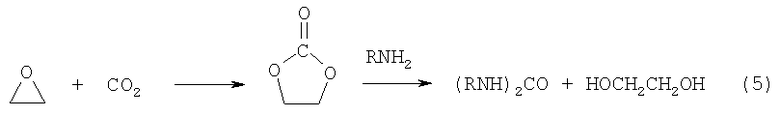
Вторую стадию проводят с гетерогенным катализатором СаО при 100°С (Fujita S.-I. et al, J. Mol. Catal., A: Chemical, 2005, 230 (1-2), p.43) с целью получения диалкилмочевин - малотоннажных продуктов органического синтеза (Т.П.Вишнякова и др. Успехи хим., 1985, 54, 429). В отсутствие катализатора реакция останавливается на присоединении одной молекулы амина с образованием карбамата (уретана), превращение которого в диалкилкарбамид является лимитирующей стадией и в присутствии катализатора. Поэтому карбамат RNHCOOCH2CH2OH и продукты его циклизации (оксазолидинон) всегда присутствуют в продуктах реакции (US Pat. 2627524, 1953; US Pat. 4883854, 1989).
Наиболее близким к предлагаемому способу совместного получения этиленгликоля и карбамида (прототипом) является двухстадийный способ (GB 1414820, опубл. 19.11.1975), заключающийся в том, что на первой стадии проводят реакцию диоксида углерода с оксидом этилена при температурах 0-300°С и давлениях 1-300 атм в присутствии катализаторов с последующим выделением этиленкарбоната, а на второй стадии проводят аммонолиз этиленкарбоната в два этапа при температурах 0-300°С и давлениях 0-200 атм в присутствии оксидных и других типов катализаторов с получением этиленгликоля и карбамида. На первом этапе второй стадии образуется уретан, а на втором этапе - карбамид. Выделение этиленгликоля из смеси ЭГ-карбамид проводят экстракцией ЭГ кетонами (ацетон, метилэтилкетон) (6)
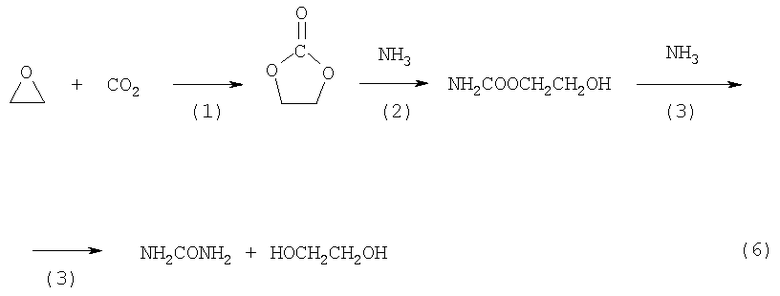
Использование в числе прочих гетерогенных катализаторов на этапах этого процесса и кетонов, относительно хорошо растворяющих карбамид в присутствии ЭГ, существенно усложняет технологию стадий выделения ЭГ и карбамида высокой степени чистоты.
Задача, решаемая изобретением, состоит в создании малоотходного, экологически безопасного и малоэнергоемкого метода синтеза этиленгликоля совместно с карбамидом, обеспечивающего значительное упрощение технологии процесса синтеза обоих продуктов и высокие показатели по конверсии сырья и селективности.
Технический результат от заявляемого способа состоит в улучшении технологичности процесса, практически полном превращении сырья в целевые продукты, а также в значительном снижении энергоемкости вследствие усовершенствования процесса разделения ЭГ и карбамида.
Технический результат достигается использованием двухстадийного метода синтеза ЭГ (реакция 7), сопряженного с синтезом карбамида на стадии аммонолиза этиленкарбоната, без промежуточного этапа синтеза уретана и без использования катализатора, затрудняющего процессы выделения продуктов, и с применением эфирных растворителей, ограниченно растворяющих карбамид
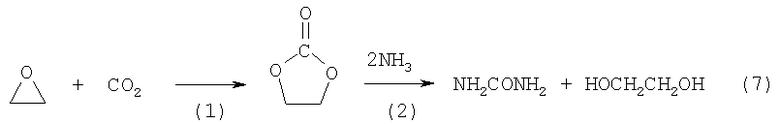
Метод включает синтез ЭК (стадия 1) при давлении 2,1-6 МПа и температуре 80-150°С в присутствии катализатора - комплекса бромида цинка с третичными органофосфинами с практически количественным выходом и аммонолиз ЭК (стадия 2) аммиаком, проводимый в одну технологическую стадию при давлении 2,8-6 МПа и температуре 120-170°С с практически количественным выходом ЭГ и карбамида в растворителях эфирного типа, ограниченно растворяющих карбамид. Синтез этиленкарбоната осуществляют в избытке диоксида углерода в присутствии в качестве гомогенного катализатора комплексов бромида цинка с третичными органофосфинами, с выделением этиленкарбоната дистилляцией или возгонкой. Катализатор вводят в реакционную смесь в виде готовых комплексов или их составляющих. При давлении ниже 2,1 МПа существует риск самопроизвольного взрывного распада оксида этилена. Использование давления более 6 МПа технически нецелесообразно, поскольку это уже превышает давление насыщенных паров жидкого СО2 при комнатной температуре, что требует дополнительного оборудования для подачи этого реагента в реактор. Скорость реакции заметно снижается при температуре ниже 80°С, а при температуре выше 150°С выход продукта уменьшается и становится заметным распад фосфиновой компоненты катализатора. Выбор в качестве катализатора комплексов бромида цинка с органофосфинами объясняется необходимостью обеспечивать отсутствие примесей в промежуточном этиленкарбонате после его выделения дистилляцией или возгонкой, т.е. катализатор должен быть нелетучим и не давать летучие продукты разложения. С другой стороны, катализатор должен быть достаточно активным, чтобы при умеренном расходе обеспечивать количественную конверсию окиси этилена в этиленкарбонат за технологически приемлемое время реакции. Всем этим условиям отвечают именно комплексы бромида цинка с третичными алкилароматическими органофосфинами, взятыми по отношению к бромиду цинка в мольном количестве 2:1. При осуществлении изобретения в качестве катализатора могут быть использованы как готовые комплексы, так и смесь безводного бромида цинка с органофосфинами. В последнем случае истинные катализаторы процесса формируются непосредственно в реакционной смеси. Мольное отношение оксида этилена к катализатору составляет 500-5000:1. Стадию 2 проводят в растворителе, ограниченно растворяющем карбамид. При температуре ниже 120°С аммонолиз этиленкарбоната протекает очень медленно, а при температуре выше 170°С выход этиленгликоля мал из-за побочных реакций. При давлении ниже 2,8 МПа процесс аммонолиза (стадия 2) не происходит, а повышение давления выше 6 МПа не влияет положительно на показатели процесса. При осуществлении стадии 2 технический результат в отношении увеличения выхода, смягчения реакционных условий, повышения чистоты конечного продукта и улучшения технологичности метода достигается использованием такого растворителя, в котором исходные аммиак, этиленкарбонат и образующийся в процессе реакции этиленгликоль хорошо растворимы, а карбамид трудно растворим, что позволяет смещать реакционное равновесие в сторону конечных продуктов и разделять карбамид и ЭГ простым фильтрованием. Указанным условиям удовлетворяет большинство растворителей эфирного типа, в частности тетрагидрофуран и 1,4-диоксан. Фильтрат (ЭГ в эфирном растворителе со следами карбамида) направляется на ректификацию, а карбамид направляют на очистку от следов ЭГ экстракцией диэтиловым эфиром, практически не растворяющим карбамид. Использование эфирных растворителей на стадии синтеза или на стадии выделения ЭГ при проведении процесса без растворителей (см. примеры 12, 13) заметно улучшает процесс по сравнению с методами дистилляции ЭГ или экстракции кетонами (см. прототип).
Осуществление настоящего изобретения иллюстрируют приведенные ниже примеры.
Пример 1
Синтез этиленкарбоната (первая стадия) проводят на установке, состоящей из автоклава производства "Parr Instrument" (объем 300 мл), дозатора жидкой окиси этилена и баллона с диоксидом углерода. Установка снабжена также насосом для перемещения непрореагировавшего диоксида углерода из автоклава обратно в питающий баллон.
В автоклав помещают 0,045 г (0,2 ммоль) безводного бромида цинка и 0,105 г (0,4 ммоль) трифенилфосфина. Установку вакуумируют и с помощью дозатора вводят 17,64 г (0,4 моль, около 20 мл) жидкого оксида этилена. Затем включают перемешивание и подают в автоклав СО2 до общего давления 1,6 МПа, после чего нагревают до 120°С и проводят реакцию. В процессе реакции давление поддерживают в интервале 2,5-3,0 МПа посредством порционной подачи СО2. По завершении реакции автоклав охлаждают и практически количественно перекачивают насосом неизрасходованный диоксид углерода обратно в исходный баллон. Выгруженный сырой продукт отделяют от катализатора перегонкой в вакууме. После шестикратного повторения опыта получают 207,107 г (2,352 моль) этиленкарбоната; т. пл. 35-37°С; посторонние примеси по ГЖХ не обнаружены. По изменению массы баллона найдено, что на химическую реакцию и технологические потери расходуется 106,6 г (2,422 моль) СО2. Выход по израсходованному диоксиду углерода 97,1%, выход по загруженному оксиду этилена 99,1%.
Пример 2
Синтез этиленкарбоната проводят аналогично примеру 1, за исключением того, в качестве катализатора используют 0,15 г (0,2 ммоль) заранее приготовленного комплекса ZnBr2(Ph3P)2, процесс проводят в интервале давлений 4,6-4,9 МПа, саму процедуру выполняют однократно без возвращения непрореагировавшего СО2, а выделение продукта осуществляют возгонкой при остаточном давлении 0,05 мм рт.ст. (40-50°С). Выход этиленкарбоната по загруженному оксиду этилена 98,5%. Посторонние примеси в пределах чувствительности ГЖХ-анализа не обнаружены.
Пример 3
Синтез этиленкарбоната ведут аналогично примеру 2, за исключением того, что в качестве катализатора используют 0,09 г (0,4 ммоль) безводного бромида цинка и 0,244 г (0,8 ммоль) трис(п-толил)фосфина, а сам процесс ведут при 100°С в интервале давлений 2,8-3,2 МПа. Выход продукта по загруженному оксиду этилена 97,8%.
Пример 4
В автоклав емкостью 50 мл загружают 1,1 г (12,5 ммоль) этиленкарбоната, растворенного в 5 мл свежеперегнанного над натрием тетрагидрофурана. Для удаления воздуха автоклав продувают азотом (3×1 МПа) и при перемешивании насыщают растворитель аммиаком под давлением 0,8 МПа в течение 30 мин. Затем общее давление в автоклаве поднимают азотом до 2 МПа и нагревают до 140° (при нагревании давление возрастает до 4,0 МПа). После выдержки в течение 8 часов нагрев и перемешивание выключают, дают автоклаву самопроизвольно охладиться до комнатной температуры, переносят его содержимое на стеклянный пористый фильтр и отфильтровывают нерастворимый продукт. Для сокращения механических потерь автоклав ополаскивают 15 мл тетрагидрофурана (3×5 мл) и промывают этим количеством продукт на фильтре. После этого продукт дополнительно промывают диэтиловым эфиром (3×5 мл) и сушат на фильтре в токе воздуха до постоянной массы. Анализ продукта и маточного раствора на содержание мочевины проводят по стандартной фотоколориметрической методике (Crocker C.L., Amer. J. Med. Technol. 1967, v.33, p.361). Получают 0,711 г карбамида. Содержание основного вещества 96,5 мас.% (11,44 ммоль), выход 91,5% в расчете на загруженный этиленкарбонат. Анализ ЭГ проводят методом ГЖХ. Суммарный выход ЭГ (на загруженный этиленкарбонат) составляет 95%.
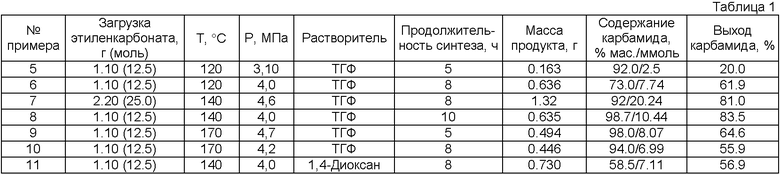
Примеры 5-11
Синтез этиленгликоля и карбамида проводят аналогично примеру 4 при варьировании реакционной температуры, длительности процедуры, загрузки этиленкарбоната и природы растворителя (табл.1).
Примеры 12, 13
Синтез этиленгликоля и карбамида проводят аналогично примеру 4 за исключением того, что эфирный растворитель не используется в синтезе, но применяется на стадии выделения карбамида (табл.2)
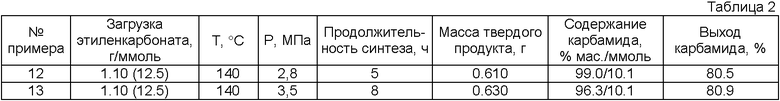
В опытах 5-13 баланс по этиленгликолю не сводили из-за больших потерь этиленгликоля при промывках карбамида вследствие малых загрузок этиленкарбоната.
Пример 14
В автоклав объемом 300 мл помещают 30 г (0,341 моль) этиленкарбоната и 100 мл сухого ТГФ. Автоклав продувают аргоном (3×1 МПа) и при перемешивании с помощью дозатора при комнатной температуре вводят 80 мл (около 49 г, 2,87 моль) жидкого аммиака. Автоклав нагревают до 150°С и выдерживают в течение 3 часов (устанавливается давление 5-6 МПа). После этого нагрев и перемешивание выключают, дают автоклаву самопроизвольно охладиться до комнатной температуры и переносят реакционную массу на стеклянный пористый фильтр. Отфильтрованный карбамид очищают от примесей этиленгликоля обработкой 1,4-диоксаном и диэтиловым эфиром в экстракторе Сокслета, выход 17,119 г (0,285 моль, 83,6%), т. пл. 134-135°С (лит. 132-135°С). Фильтрат и эфирный экстракт объединяют, упаривают легкокипящие растворители, выход этиленгликоля по ГЖХ (внутренний стандарт метанол) близок к 100%. Этиленгликоль выделяют перегонкой в вакууме 1 мм рт.ст., nd 20=1,4315 (лит. 1,431). Из кубового остатка дополнительно выделяют еще 2,257 г (0,038 моль, 11,0%) карбамида. Суммарный выход карбамида в расчете на выделенный продукт 94,6%. Таким образом, предложенный способ получения этиленгликоля совместно с карбамидом обеспечивает получение продуктов с хорошим выходом и с качеством, удовлетворяющим требованиям, предъявляемым к этиленгликолю и карбамиду.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КАРБАМИДА, МЕЧЕННОГО СТАБИЛЬНЫМ ИЗОТОПОМ C | 2009 |
|
RU2415837C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБАМИДА, МЕЧЕННОГО СТАБИЛЬНЫМ ИЗОТОПОМ C | 2010 |
|
RU2440826C1 |
| Способ получения 13 С -мочевины | 2016 |
|
RU2638837C1 |
| Способ получения этиленкарбоната | 2023 |
|
RU2813127C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛИГОМЕРОВ ПЕНТ-1-ЕНА В ПРИСУТСТВИИ ИЕРАРХИЧЕСКОГО ЦЕОЛИТА Н-Y | 2019 |
|
RU2709818C1 |
| ГИДРИД-КАРБОНИЛЬНЫЙ ПОЛИФОСФИТНЫЙ КОМПЛЕКС РОДИЯ СО СМЕШАННЫМИ ФОСФОРОРГАНИЧЕСКИМИ ЛИГАНДАМИ ДЛЯ КАТАЛИЗА ПРОЦЕССА ГИДРОФОРМИЛИРОВАНИЯ ОЛЕФИНОВ | 2015 |
|
RU2584952C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛИГОМЕРОВ ПЕНТ-1-ЕНА В ПРИСУТСТВИИ АМОРФНОГО МЕЗОПОРИСТОГО АЛЮМОСИЛИКАТА ASM | 2019 |
|
RU2697885C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНГЛИКОЛЯ КАТАЛИТИЧЕСКОЙ РЕАКЦИЕЙ ОКСАЛАТА В ПСЕВДООЖИЖЕННОМ СЛОЕ | 2012 |
|
RU2565074C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЕНГЛИКОЛЯ | 2008 |
|
RU2477718C2 |
| Способ получения циклопентенкарбоната каталитическим карбоксилированием 1,2-эпоксициклопентана | 2017 |
|
RU2636940C1 |
Изобретение относится к основному органическому синтезу и касается способа получения этиленгликоля совместно с карбамидом из диоксида углерода, оксида этилена и аммиака. Способ заключается в том, что процесс осуществляют в две стадии: сначала проводят взаимодействие диоксида углерода и оксида этилена при температуре 80-150°С, давлении 2,1-6 МПа в присутствии гомогенного катализатора - комплекса бромида цинка с третичными органофосфинами с последующим выделением этиленкарбоната, а затем проводят аммонолиз этиленкарбоната в одну технологическую стадию в растворителях эфирного типа, ограниченно растворяющих карбамид, при температуре 120-170°С и давлении 2,8-6 МПа с получением этиленгликоля и карбамида. Способ позволяет практически полностью превратить исходное сырье в целевые продукты, а также в значительной степени снизить энергоемкость вследствие усовершенствования процесса разделения этиленгликоля и карбамида. 2 з.п. ф-лы, 2 табл.
1. Способ получения этиленгликоля совместно с карбамидом из диоксида углерода, оксида этилена и аммиака, заключающийся в том, что процесс осуществляют в две стадии: сначала проводят взаимодействие диоксида углерода и оксида этилена при температуре 80-150°С, давлении 2,1-6 МПа в присутствии гомогенного катализатора - комплекса бромида цинка с третичными органофосфинами с последующим выделением этиленкарбоната, а затем проводят аммонолиз этиленкарбоната в одну технологическую стадию в растворителях эфирного типа, ограниченно растворяющих карбамид, при температуре 120-170°С и давлении 2,8-6 МПа с получением этиленгликоля и карбамида.
2. Способ по п.1, отличающийся тем, что компоненты катализатора - бромид цинка и органофосфин вводят в реакционную смесь в виде индивидуальных соединений.
3. Способ по п.1, отличающийся тем, что мольное отношение оксида этилена к катализатору составляет 500-5000:1.
| Разрушающий материал | 1987 |
|
SU1414820A1 |
| Способ одновременного получения 1,3-дизамещенных мочевин и 1,2-диолов | 1978 |
|
SU856379A3 |
| S.Fujita et al, Synthesis of 1,3-dialkylurea from ethylene carbonate and amine using calcium oxide | |||
| Journal of Molecular Catalysis A: Chemical, 2005, т.230, с.43-48. | |||

