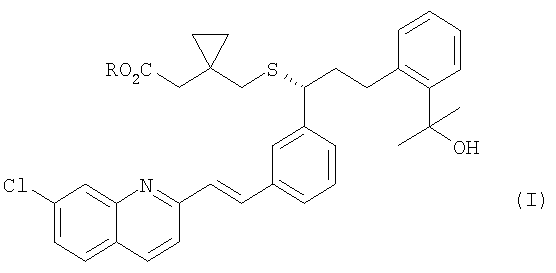
Настоящее изобретение относится к улучшенному способу получения монтелукаста и промежуточному соединению, используемому в данном способе.
УРОВЕНЬ ТЕХНИКИ
Лейкотриены образуют группу локально действующих гормонов, образующихся из арахидоновой кислоты in vivo, основными лейкотриенами являются лейкотриен B4 (LTB4), C4 (LTC4), D4 (LTD4) и E4 (LTE4). Биосинтез лейкотриенов включает образование эпоксида, известного как лейкотриен A4 (LTA4), из арахидоновой кислоты под действием 5-липоксигеназы, а LTA4 затем превращается в различные лейкотриены посредством серии ферментативных реакций (см. Leukotrienes and lipoxygenases, ed. J. Rokach, Elsevier, Amsterdam, 1989).
В настоящее время монтелукаст или его фармацевтически приемлемая соль известны своим действием в качестве антагониста, а также в качестве ингибитора биосинтеза лейкотриенов. Натриевая соль монтелукаста коммерчески доступна от фирмы Merck под торговой маркой Singulair® и используется для лечения астмы.
В EP 480717 раскрывается способ получения указанной натриевой соли монтелукаста, как показано на реакционной Схеме 1, метил-1-(меркаптометил)циклопропилацетат формулы (B) объединяют с соединением формулы (A) для получения соединения формулы (С) в качестве промежуточного соединения, и соединение формулы (С) затем гидролизуют с получением его в форме свободной кислоты, с последующей обработкой указанной свободной кислоты NaOH. Однако этот способ дает низкий выход или требует слишком высокие производственные затраты.
Реакционная Схема 1
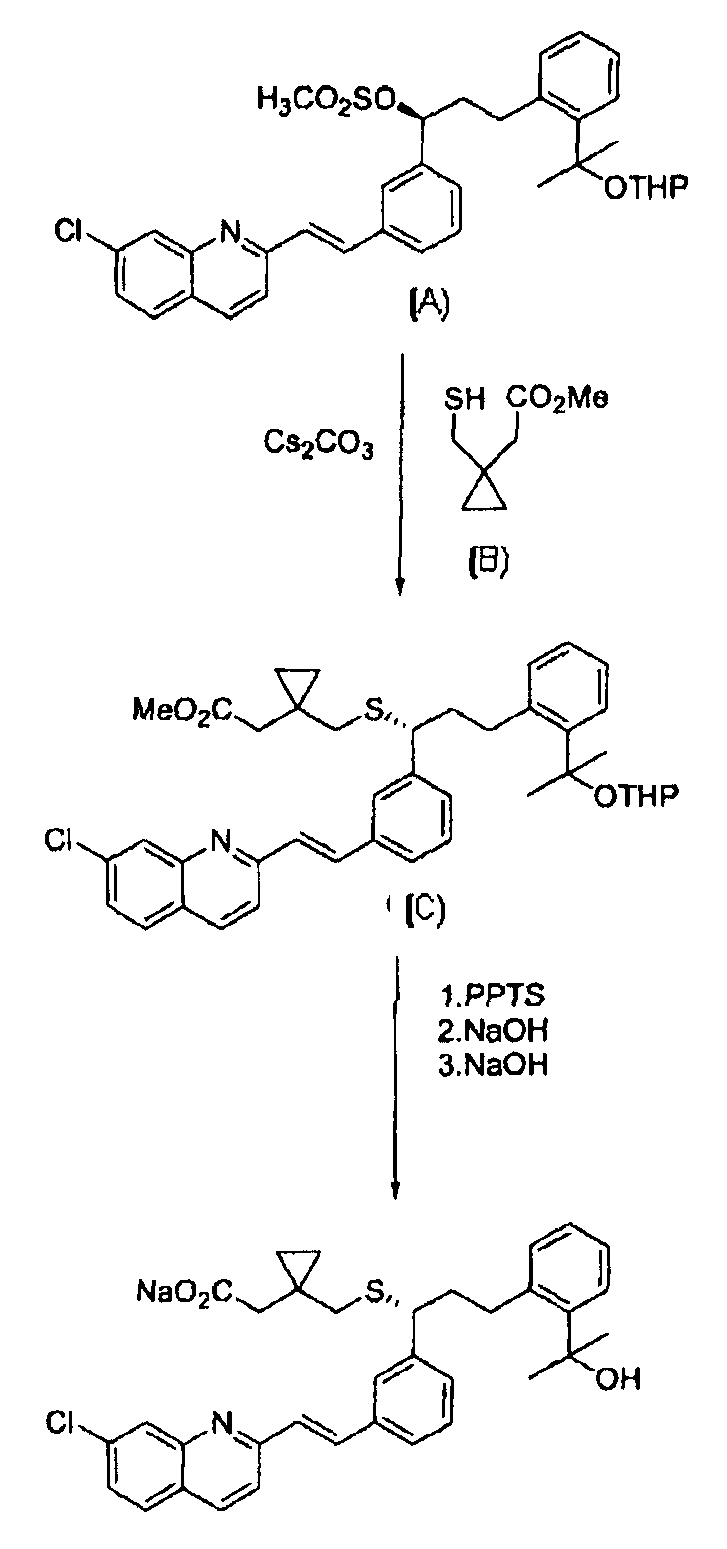
THP: тетрагидропиранил.
PPTS: п-толуолсульфонат пиридиния.
Для решения вышеуказанных проблем в EP 737186 предлагается способ, проиллюстрированный реакционной Схемой 2. В этом способе используется метансульфонильное соединение формулы (A'), имеющее незащищенную гидроксильную группу вместо THP-защищенного соединения формулы (A). Также в этом способе используется дилитиевая соль 1-(меркаптометил)циклопропилацетата формулы (B'), вместо метил-1-(меркаптоэтил)циклопропилацетата формулы (B), вследствие чего последующая стадия снятия защиты не требуется. Затем к соединению формулы (C') добавляют дициклогексиламин для получения соединения формулы (D), которое превращают в желаемую натриевую соль.
Реакционная Схема 2
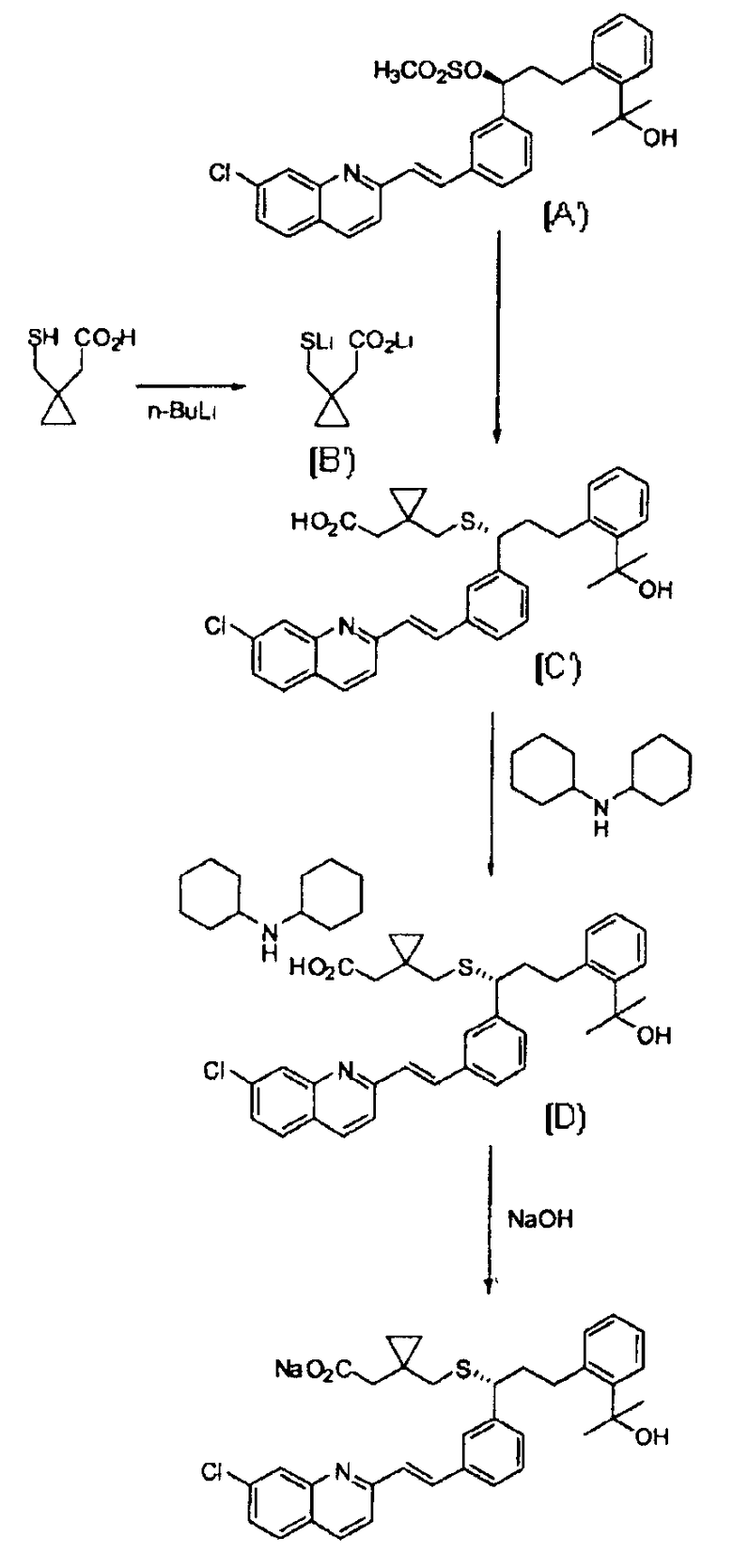
Однако метансульфонильное соединение формулы (A'), используемое выше в качестве исходного материала, является очень нестабильным, что значительно осложняет весь процесс. А именно реакцию с образованием соединения формулы (A') следует проводить только при низкой температуре, при приблизительно -30°С, а полученный продукт необходимо хранить при приблизительно -15°С. Таким образом, полученное соединение формулы (A') является нестабильным по отношению к влаге и воздуху и поэтому его взаимодействие должно проводиться быстро и в строго контролируемых условиях. Кроме того, синтез соединения формулы (B') требует использования н-бутиллития, который является взрывоопасным при контакте с влагой и воздухом. Таким образом, способ, показанный на реакционной Схеме выше, не подходит для крупномасштабного производства.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Соответственно, задачей настоящего изобретения является предоставление эффективного способа получения монтелукаста с высокой степенью чистоты и его натриевой соли с высоким выходом, который может быть использован в крупномасштабном производстве.
Другой задачей настоящего изобретения является предоставление нового фосфатного промежуточного соединения, применяемого для получения монтелукаста.
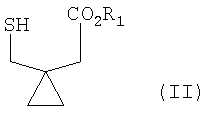
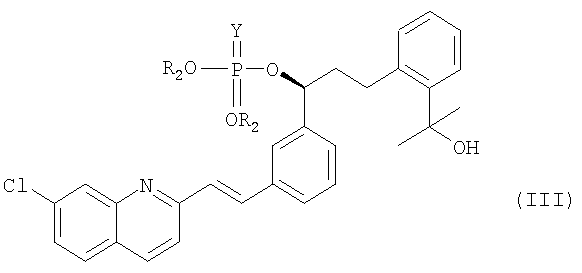
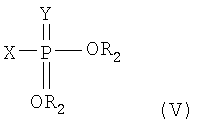
В соответствии с одним аспектом настоящего изобретения предлагается способ получения монтелукаста или его натриевой соли формулы (I), включающий следующие стадии: (а) взаимодействие галогенфосфатного соединения формулы (V) с диольным соединением формулы (IV) в растворителе в присутствии основания, с получением фосфатного соединения формулы (III); и (б) объединение фосфатного соединения формулы (III) с соединением тиокарбоновой кислоты формулы (II) в растворителе в присутствии основания:
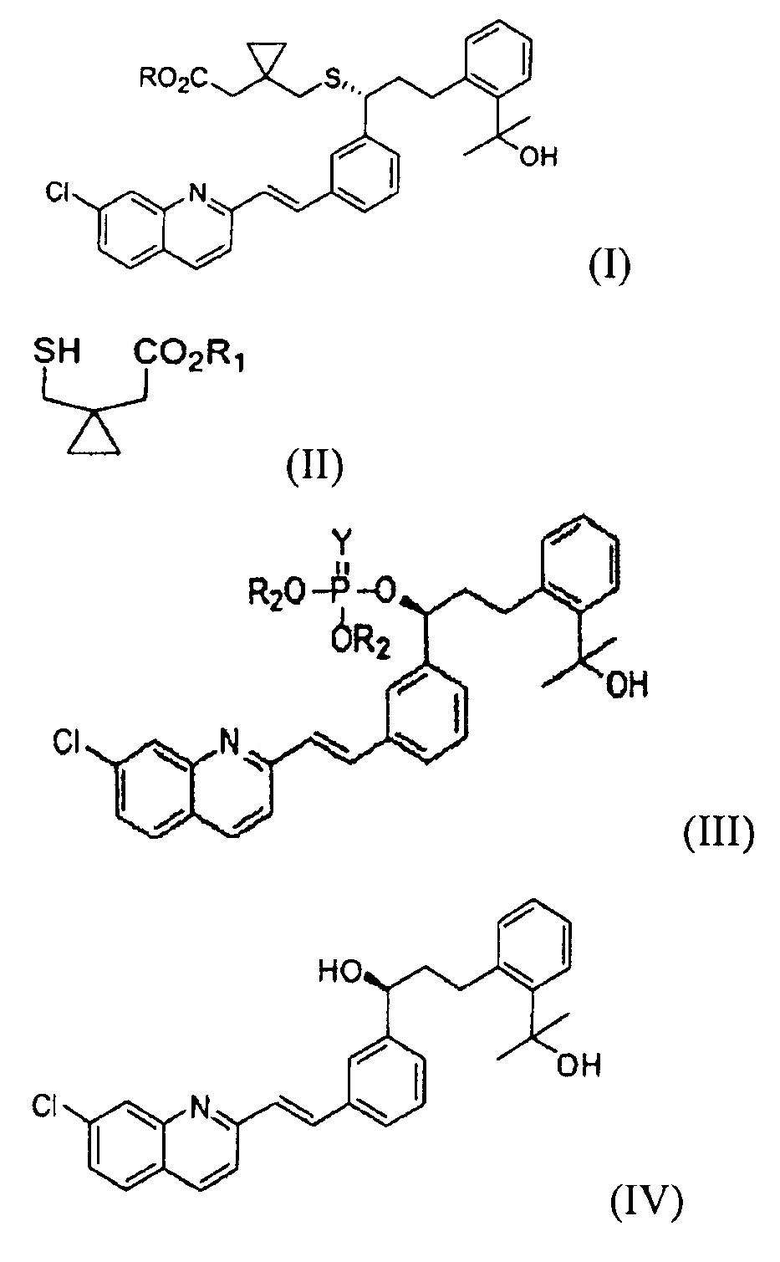

где
R представляет собой H или Na;
R1 представляет собой H, метил или этил;
R2 представляет собой метил, этил или фенил;
X представляет собой галоген;
Y представляет собой серу или кислород.
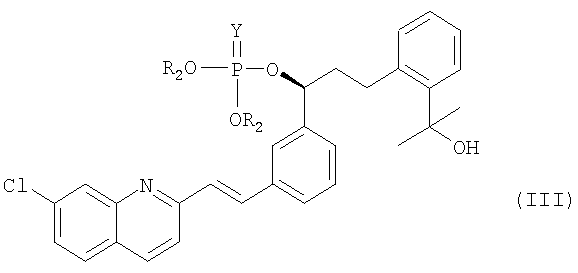
В соответствии с другим аспектом настоящего изобретения, предлагается фосфатное соединение формулы (III), применяемое в качестве промежуточного соединения при получении монтелукаста:
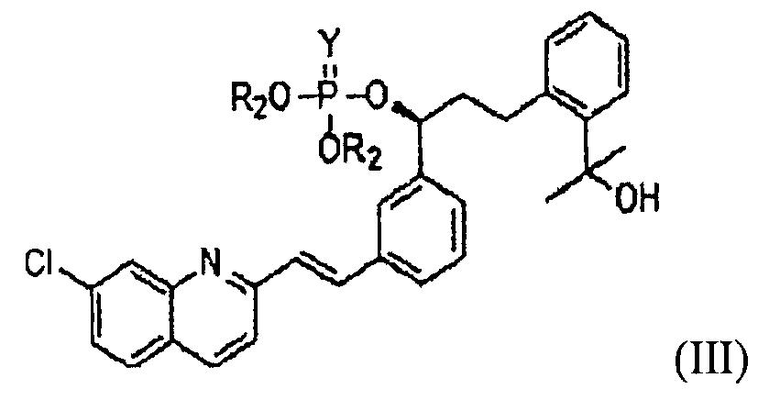
где
R2 представляет собой метил, этил или фенил; и
Y представляет собой серу или кислород.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
В настоящем изобретении соединение формулы (I) может быть получено, как показано в реакционной Схеме 3.
Реакционная Схема 3

где
R представляет собой H или Na;
R1 представляет собой H, метил или этил;
R2 представляет собой метил, этил или фенил;
X представляет собой галоген; а
Y представляет собой серу или кислород.
На реакционной стадии (а) соединение формулы (IV) объединяют с галогенфосфатным соединением формулы (V) для получения фосфатного соединения формулы (III), имеющего уходящую фосфатную группу. Галогенфосфатное соединение формулы (V) является коммерчески доступным или оно может быть легко получено с использованием общепринятых методик синтеза. Предпочтительное галогенфосфатное соединение формулы (V) представляет собой соединение, в котором X означает хлор, Y означает кислород, а R2 представляет собой фенил. Наиболее предпочтительной уходящей фосфатной группой является дифенилфосфатная группа.
Кроме того, галогенфосфатное соединение формулы (V) может быть использовано на стадии (а) в количестве от 1,1 до 1,5 эквивалентов, в расчете на диольное соединение (IV).
Кислота, образующаяся на стадии (а), может быть удалена с помощью основания, выбранного из пиридина, триэтиламина, трибутиламина, диизопропилэтиламина, метилпиперидина и их смеси. Предпочтительное основание представляет собой триэтиламин. Основание может быть использовано в количестве от 1,2 до 2 эквивалентов, в расчете на диольное соединение формулы (IV). Также, если необходимо, в качестве катализатора на стадии (а) может быть использован 4-диметиламинопиридин или 4-пирролидинопиридин.
Реакционную стадию (а) можно проводить в растворителе, выбранном из бензола, толуола, ацетонитрила, тетрагидрофурана, метиленхлорида, хлороформа, этилацетата и их смеси, предпочтительно в смеси толуола и метиленхлорида, при комнатной температуре, варьирующейся в интервале от -25 до 50°С.
Фосфатное соединение (III), полученное на стадии (а), может быть выкристаллизовано из растворителя, выбранного из метилацетата, этилацетата, н-гексана, циклогексана, диэтилового эфира, изопропилового эфира и их смеси, предпочтительно из смеси этилацетата и н-гексана.
Фосфатное соединение (III) обладает высокой стабильностью, в отличие от метансульфонильного соединения, полученного с использованием традиционного способа. В целом для получения метансульфонильного соединения традиционным способом реакцию необходимо проводить при приблизительно -30°С, а продукт необходимо держать при приблизительно -15°С. В отличие от этого, фосфатное соединение (III), полученное согласно настоящему изобретению, можно хранить при комнатной температуре. Более того, оно не подвергается деградации во время сушки при 40°С. Кроме того, фосфатное соединение (III) характеризуется высокой чистотой и содержит лишь незначительное количество примесей.
На реакционной стадии (б) фосфатное соединение формулы (III) объединяют с производным тиокарбоновой кислоты формулы (II). Репрезентативные примеры соединения формулы (II) включают 1-(меркаптометил)циклопропилуксусную кислоту и ее алкиловые эфиры.
Кроме того, основание, применяемое на стадии (б), может быть выбрано из гидроксида натрия, гидрида натрия, трет-бутоксида калия, метоксида натрия, этоксида натрия, триэтиламина, пиридина и их смеси. Предпочтительным основанием является гидрид натрия. Растворитель, применяемый на стадии (б), может быть выбран из диметилформамида, диметилсульфоксида, ацетонитрила, хлороформа, метиленхлорида, тетрагидрофурана, 1,4-диоксана, этилацетата, этанола, метанола, и их смеси, предпочтительно с диметилформамидом.
В случае применения 1-(меркаптометил)циклопропил уксусной кислоты в качестве соединения (II) на реакционной стадии (б) количества 1-(меркаптометил)циклопропилуксусной кислоты и применяемого основания могут находиться в пределах от 2 до 4 эквивалентов и от 3 до 5 эквивалентов, соответственно, в расчете на диольное соединение формулы (III).
С другой стороны, в случае применения метил-1-(меркаптометил)циклопропилацетата в качестве соединения (II), количества и метил-1-(меркаптометил)циклопропилацетата и применяемого основания могут быть от 1 до 3 эквивалентов и от 1 до 2 эквивалентов, соответственно, в расчете на диольное соединение формулы (III).
Таким образом, полученный продукт монтелукаст может быть гидролизован в присутствии основания, такого как гидроксид натрия, для получения натриевой соли монтелукаста. Для получения натриевой соли монтелукаста с высокой чистотой обработку гидроксидом натрия осуществляют, предпочтительно, после отделения монтелукаста в форме свободной кислоты.
Как описано выше, в предложенном способе получения монтелукаста применяют новое фосфатное промежуточное соединение вместо метансульфонатного производного, используемого в традиционном способе, которое обеспечивает преимущества целевого продукта, обладающего значительно улучшенной стабильностью и существенно сниженной степенью загрязнения.
Приведенные ниже Примеры даны только для целей иллюстрации и не предназначены для ограничения границ настоящего изобретения.
Пример 1: Получение 2-(2-(3-(S)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-дифенилфосфат-оксипропил)фенил)-2-пропанола
20 г 2-(2-(3-(S)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-гидроксипропил)фенил)-2-пропанола растворили в 240 мл смеси метиленхлорида и толуола (2:1), и медленно добавили к ней 7,31 мл (1,2 экв.) триэтиламина. Затем к полученной смеси по каплям добавили 13,6 мл дифенилхлорфосфата и 1,06 г 4-диметиламинопиридина. После истечения приблизительно 1 часа окончание реакции было подтверждено с помощью тонкослойной хроматографии (ТСХ). Реакционную смесь обработали 100 мл метиленхлорида и 200 мл дистиллированной воды. При перемешивании органический слой отделили и высушили над сульфатом натрия, затем удалили растворитель при пониженном давлении. Полученный таким образом остаток растворили в 60 мл смеси этилацетата и н-гексана (1:3) и перекристаллизовали из него продукт. Кристаллизовавшийся продукт профильтровали, промыли 40 мл дистиллированной воды и высушили с получением 29,5 г (97,8%) заявленного соединения в виде твердого вещества желтого цвета.
Т.пл.: 127°С.
1H-ЯМР (300 МГц, CDCl3): δ 8,4 (1H, д), 7,94 (1H, д), 7,75 (3H, м), 6,97-7,35 (20H, м), 5,70-5,72 (1H, м), 3,02-3,09 (2H, м), 2,29-2,34 (2H, м), 1,65 (3H, с), 1,59 (3H, с).
Пример 2: Получение 1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропилуксусной кислоты
12,7 г 1-(меркаптометил)циклопропилуксусной кислоты, растворенной в 90 мл диметилформамида медленно по каплям добавили к раствору 6,26 г 60% гидрида натрия, растворенного в 90 мл диметилформамида при 0-5°С. К полученной смеси медленно по каплям добавили 30 г 2-(2-(3-(S)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-дифенилфосфат-оксипропил)фенил)-2-пропанола, полученного в Примере 1 и растворенного в 120 мл диметилформамида. После того как температура медленно повысилась до комнатной, реакция была запущена и проводилась в течение 18-20 часов. Затем реакционную смесь нейтрализовали насыщенным водным раствором хлорида аммония и обработали этилацетатом и дистиллированной водой. При перемешивании органический слой отделили и высушили над сульфатом натрия, затем удалили растворитель при пониженном давлении. Полученный таким образом остаток растворили в 270 мл циклогексана и перекристаллизовали из него продукт. Кристаллизовавшийся продукт профильтровали, промыли 40 мл дистиллированной воды и высушили с получением 22,2 г (87,1%) заявленного соединения в виде твердого вещества желтого цвета.
1H-ЯМР (300 МГц, CD3OD): δ 8,27 (1H, д), 7,98 (1H, с), 7,78 (2H, д), 7,73 (2H, д), 7,38-7,56 (6H, м), 7,07-7,14 (3H, м), 4,84 (1H, т), 3,30-3,33 (1H, м), 2,84-2,87 (1H, м), 2,52 (2H, с), 2,41 (2H, с), 2,18-2,23 (2H, м), 1,55 (6H, с), 0,37-0,52 (4H, м).
Пример 3: Получение 1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)-тио)метил)циклопропилацетата натрия
Стадия 1: Получение метил-1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропилацетата.
2,1 г метил-1-(ацетилтиометил)циклопропилацетата, растворенного в 35 мл диметилформамида, медленно добавили к раствору 0,71 г 60%-ного хлорида натрия, растворенного в 35 мл диметилформамида при температуре, изменяющейся в пределах от 0 до 5°С. К полученной смеси при температуре, изменяющейся в пределах от 0 до 5°С, медленно по каплям добавили 7,73 г 2-(2-(3-(S)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-дифенилфосфат-оксипропил)фенил)-2-пропанола, полученного в Примере 1 и растворенного в 35 мл диметилформамида. После истечения приблизительно 1 часа реакционную смесь обработали этилацетатом и дистиллированной водой. При перемешивании органический слой отделили и высушили над сульфатом натрия, затем удалили растворитель при пониженном давлении, с образованием 5,68 г (84,5%) заявленного соединения в виде жидкости желтого цвета.
1H-ЯМР (300 МГц, CDCl3): δ 8,12 (2H, д), 7,66-7,74 (4H, м), 7,37-7,48(6H, м), 7,12-7,20 (3H, м), 3,96 (1H, т), 3,14-3,16 (1H, м), 2,88 (1H, м), 2,53 (2H, с), 2,43 (2H, с), 1,62 (6H, д), 0,41-0,54 (4H, м).
Стадия 2: Получение 1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропилуксусной кислоты.
12 г метил-1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропилацетата, полученного на стадии 1, растворили в смеси 60 мл тетрагидрофурана и 30 мл метилового спирта. После доведения температуры до 10-15°С к полученной смеси медленно добавили 24 г 10%-ного раствора NaOH. Затем температуру медленно повысили до комнатной температуры (24-27°С) и реакционную смесь перемешивали в течение 20 часов. После завершения реакции органический слой отделили и высушили, затем удалили растворитель при пониженном давлении. Полученный таким образом остаток снова смешали с водным слоем и добавили к нему 120 мл толуола. После этого значение pH продукта реакции довели до 4 посредством добавления 300 мл уксусной кислоты. Органический слой снова отделили и высушили над сульфатом натрия, затем удалили растворитель при пониженном давлении. Полученный таким образом остаток растворили в 96 мл смеси изопропанола и дистиллированной воды (2:1) и перекристаллизовали из нее продукт. Кристаллизовавшийся продукт отфильтровали с получением 9,82 г (83%) заявленного соединения в виде твердого вещества желтого цвета.
1H-ЯМР (300 МГц, CD3OD): δ 8,27 (1H, д), 7,98 (1H, с), 7,78 (2H, д), 7,73 (2H, д), 7,38-7,56 (6H, м), 7,07-7,14 (3H, м), 4,84 (1H, т), 3,30-3,33 (1H, м), 2,84-2,87 (1H, м), 2,52 (2H, с), 2,41 (2H, с), 2,18-2,23 (2H, м), 1,55 (6H, с), 0,37-0,52 (4H, м).
Т. пл.: 154°С, чистота >99%.
Стадия 3: Получение 1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)-метил)циклопропилацетата натрия.
5 г 1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропилуксусной кислоты, полученной на стадии 2, смешали с 10 мл толуола, с последующим удалением растворителя при пониженном давлении. К полученному таким образом остатку затем добавили 14,5 мл толуола и 13 мл 0,5 н раствора NaOH/MeOH. Полученную смесь перемешивали в течение 30 минут с последующим удалением растворителя при пониженном давлении. Остаток растворили в смеси 10 мл толуола и 50 мл н-гексана и перекристаллизовали из нее продукт. Кристаллизовавшийся продукт отфильтровали с получением 5,1 г (98%) заявленного соединения в виде твердого вещества желтого цвета.
1H-ЯМР (300 МГц, CD3OD): δ 8,29 (1H, д), 7,99 (1H, с), 7,83-7,91 (3H, м), 7,72 (1H, с), 7,49-7,52 (2H, м), 7,38-7,44 (4H, м), 7,10-7,15 (3H, м), 4,04 (1H, т), 3,08 (1H, м), 2,82 (1H, м), 2,66 (1H, д), 2,52 (1H, д), 2,43 (1H, д), 2,29 (1H, д), 2,16-2,24 (2H, м), 1,52 (6H, с), 0,33-0,52 (4H, м).
Сравнительный Пример 1: Получение 1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропилацетата дициклогексиламиновой соли
Стадия 1: Получение 2-(2-(3-(S)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-метансульфонилоксипропил)фенил)-2-пропанола.
10 г 2-(2-(3-(S)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-гидроксипропил)фенил)-2-пропанола добавили к смеси 28,5 мл толуола и 71,3 мл ацетонитрила в атмосфере азота. К полученной смеси добавили 4,41 мл диизопропилэтиламина. После понижения температуры до -25°С добавляли по каплям 4,41 мл метансульфонилхлорида в течение 30 минут для получения смеси, с последующим перемешиванием в течение 2,5 часов. После понижения температуры до -35°С реакционную смесь дополнительно перемешивали в течение 2 часов до образования кристаллов. Затем полученные кристаллы быстро отфильтровали и остаток отмыли ацетонитрилом при -30°С и н-гексаном при -5°С. Полученный продукт высушили в атмосфере азота, с образованием 8,97 г (76,7%) заявленного соединения. Полученное таким образом соединение хранили в двойном полипропиленовом пакете при -18°С.
1H-ЯМР (300 МГц, CDCl3): δ 8,11 (м, 2H), 7,69 (м, 5H), 7,4l (м, 5H), 7,19 (м, 3H), 5,70 (дд, 1H), 3,25(м, 1H), 3,04 (м, 1H), 2,76 (с, 3H), 2,45 (м, 1H), 1,92 (с, 1H), 1,65 (с, 6H).
Стадия 2: Получение 1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)-этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)-метил)циклопропилацетата дициклогексиламиновой соли.
1 г 1-(меркаптометил)циклопропилуксусной кислоты добавили к 23 мл тетрагидрофурана. После перемешивания в течение 10 минут полученную смесь охладили до -15±2°С. Затем при температуре -5°С в течение 30 минут добавляли к ней 10,6 мл раствора н-бутиллития гексана с получением суспензии. Полученную суспензию оставили при температуре -5±2°С в течение 30 минут. Затем 3,11 г метансульфоната, полученного на стадии 1, растворили в 14 мл тетрагидрофурана при температуре, изменяющейся в интервале от 0 до 5°С, с получением прозрачного раствора лимонно-желтого цвета. Полученный раствор переносили в суспензию в течение 30 минут и хранили в течение 8,5 часов при температуре -5 ± 2°С. Для завершения реакции к реакционной смеси добавили 42 мл этилацетата и 42 мл 10%-ного раствора NaCl. После перемешивания в течение 30 минут органический слой отделили, затем отмыли 27 мл 0,5 М винной кислотой и два раза 30 мл воды, с последующим концентрированием под вакуумом. 8 мл полученного продукта растворили в 38 мл этилацетата. Температуру довели до комнатной (20±2°С) и добавили к полученному раствору 1,4 мл дициклогексиламина. После внесения затравки дициклогексиламиновой соли полученную смесь оставили на приблизительно 1 час для образования концентрированной суспензии. К полученной суспензии медленно в течение 2 часов добавляли 85 мл н-гексана при перемешивании. Полученную суспензию оставили при 20±2°С на ночь и отфильтровали. Полученный таким образом осадок промыли 25 мл смеси этилацетата и н-гексана (1:2) при 0±2°С и высушили при 40°С под вакуумом, с получением 1,63 г (43%) заявленного соединения.
1H-ЯМР (CD3OD): δ 8,25 (д, 1H), 7,95 (д, 1H), 7,86 (д, 1H), 7,83 (д, 1H), 7,77 (д, 1H), 7,70 (ушир.с, 1H), 7,54 (д, 1H), 7,49 (д, 1H), 7,46-7,35 (м, 4H), 7,12-7,03 (м, 3H), 4,87 (с, актив. H), 4,03 (дд, 1H), 3,1l-3,05 (м,3H), 2,84-2,81 (м, 1H), 2,64 (д, 1H), 2,52 (д, 1H), 2,38 (д, 1H), 2,29 (д, 1H), 2,23 (м, 1H), 2,00 (м, 4H), 1,82 (м, 4H), 1,66 (м, 2H), 1,51 (2 с, 6H), 1,37-1,14 (м, 10H), 0,53-0,32 (м, 4H).
Хотя изобретение было описано в отношении вышеуказанных конкретных вариантов осуществления, следует понимать, что специалистами в данной области могут быть произведены различные модификации и изменения, которые также охвачены границами настоящего изобретения, как определено в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КИСЛОТЫ МОНТЕЛУКАСТ В ИОННОЙ ЖИДКОЙ СРЕДЕ | 2008 |
|
RU2436774C1 |
| СПОСОБ ПОЛУЧЕНИЯ МОНТЕЛУКАСТА И СОЕДИНЕНИЯ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2006 |
|
RU2402532C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВОЙ СОЛИ 1-[[[(R)-M-[(Е)-2-(7-ХЛОР-2-ХИНОЛИЛ)ВИНИЛ]-АЛЬФА-[О-(1-ГИДРОКСИ-1-МЕТИЛЭТИЛ)ФЕНЕТИЛ]БЕНЗИЛ]ТИО]МЕТИЛ]ЦИКЛОПРОПАНУКСУСНОЙ КИСЛОТЫ | 2008 |
|
RU2436773C2 |
| ПРОИЗВОДНЫЕ 1-α-ГАЛОГЕН-2,2-ДИФТОР-2-ДЕЗОКСИ-D-РИБОФУРАНОЗЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2346948C2 |
| АГОНИСТЫ РЕЦЕПТОРА СФИНГОЗИН-1-ФОСФАТА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВНОГО СРЕДСТВА | 2014 |
|
RU2654483C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО D-ЭРИТРО-2,2-ДИФТОРО-2-ДЕЗОКСИ-1-ОКСОРИБОЗЫ | 2005 |
|
RU2337917C1 |
| СОЕДИНЕНИЯ ИНДОЛА И ИНДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА НЕКРОЗА КЛЕТКИ | 2008 |
|
RU2437883C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТОВ ЛЕЙКОТРИЕНА, ДИЦИКЛОГЕКСИЛАМИНОВАЯ СОЛЬ, СПОСОБЫ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ НАТРИЕВОЙ СОЛИ И 1-/МЕРКАПТОМЕТИЛ/- ЦИКЛОПРОПАНУКСУСНОЙ КИСЛОТЫ, КРИСТАЛЛИЧЕСКОЕ СОЕДИНЕНИЕ | 1994 |
|
RU2140909C1 |
| НОВЫЕ СОЕДИНЕНИЯ, ИХ ИЗОМЕР ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ В КАЧЕСТВЕ АНТАГОНИСТА ВАНИЛОИДНОГО РЕЦЕПТОРА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2448108C2 |
| ПРОИЗВОДНЫЕ 5,6-ДИГИДРОПИРОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2140917C1 |
Настоящее изобретение относится к способу получения монтелукаста формулы (I), применяемого в качестве ингибитора биосинтеза лейкотриенов для лечения астмы. Способ включает стадии: (а) взаимодействия галогенфосфатного соединения формулы (V) с диольным соединением формулы (IV) в растворителе в присутствии основания с образованием фосфатного соединения формулы (III); и (б) объединения фосфатного соединения формулы (III) с соединением тиокарбоновой кислоты формулы (II) в растворителе в присутствии основания:

где R представляет собой Н или Na; R1 представляет собой Н, метил или этил; R2 представляет собой метил, этил или фенил; Х представляет собой галоген; Y представляет собой серу или кислород. Технический результат - разработка нового способа получения монтелукаста или его натриевой соли высокой степени чистоты с высоким выходом. 2 н. и 9 з.п. ф-лы.
1. Способ получения монтелукаста или его натриевой соли формулы (I), включающий следующие стадии:
(а) взаимодействие галогенфосфатного соединения формулы (V) с диольным соединением формулы (IV) в растворителе в присутствии основания, с образованием фосфатного соединения формулы (III); и
(б) объединение фосфатного соединения формулы (III) с соединением тиокарбоновой кислоты формулы (II) в растворителе в присутствии основания:
где R представляет собой Н или Na;
R1 представляет собой Н, метил или этил;
R2 представляет собой метил, этил или фенил;
Х представляет собой галоген;
Y представляет собой серу или кислород.
2. Способ по п.1, в котором применяемое на стадии (а) основание выбрано из группы, состоящей из пиридина, триэтиламина, трибутиламина, диизопропилэтиламина, метилпиперидина и их смеси.
3. Способ по п.2, в котором основание представляет собой триэтиламин.
4. Способ по п.1, в котором количество галогенфосфатного соединения формулы (V), применяемого на стадии (а), составляет 1 или более эквивалентов в расчете на диольное соединение формулы (IV).
5. Способ по п.1, в котором дополнительно применяют 4-диметиламинопиридин или 4-пирролидинопиридин в качестве катализатора на стадии (а).
6. Способ по п.1, в котором галогенфосфатное соединение представляет собой соединение формулы (V), где Х означает Cl, Y означает О, a R2 означает фенил.
7. Способ по п.1, в котором применяемый на стадии (а) растворитель выбран из группы, состоящей из бензола, толуола, ацетонитрила, тетрагидрофурана, метиленхлорида, хлороформа, этилацетата и их смеси.
8. Способ по п.1, в котором применяемое на стадии (б) основание выбрано из группы, состоящей из гидроксида натрия, гидрида натрия, трет-бутоксида калия, метоксида натрия, этоксида натрия, триэтиламина, пиридина и их смеси.
9. Способ по п.1, в котором применяемый на стадии (б) растворитель выбран из группы, состоящей из диметилформамида, диметилсульфоксида, ацетонитрила, хлороформа, метиленхлорида, тетрагидрофурана, 1,4-диоксана, этилацетата, этанола, метанола, и их смеси.
10. Способ по п.9, в котором растворитель представляет собой диметилформамид.
11. Фосфатное соединение формулы (III), которое применяется в качестве промежуточного соединения при получении монтелукаста или его натриевой соли по п.1:
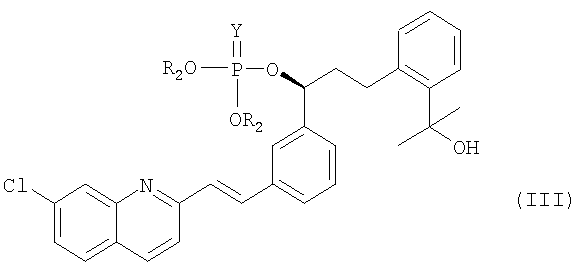
где R2 представляет собой метил, этил или фенил; а
Y представляет собой серу или кислород.
| RU 94045911 A1, 07.10.1994 | |||
| WO 2005105751 А, 10.11.2005 | |||
| US 5952347 А, 14.09.1999 | |||
| WO 2006008751 А2, 26.01.2006. |

