Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения монтелукаста, а также к ряду новых промежуточных соединений, используемых в данном способе получения.
Уровень техники
Монтелукаст является международным непатентованным названием (INN) 1-[[[(1R)-1-[3-[(1Е)-2-(7-хлор-2-хинолинил)этенил]фенил]-3-[2-(1-гидрокси-1-метилэтил)фенил]пропил]сульфанил]метил]циклопропан - уксусной кислоты и CAS (реферативная служба по химии) No. 158966-92-8. Монтелукаст натриевую соль (CAS No 151767-02-1) в настоящее время используют при лечении астмы, воспаления, стенокардии, спазмов головного мозга, гломерулярного нефрита, гепатита, эндотоксемии, увеита и отторжения аллотрансплантата.
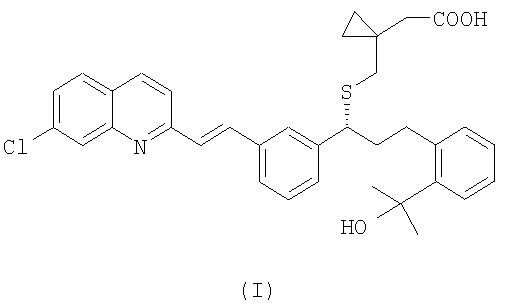
Структура монтелукаста натриевой соли соответствует формуле (I):
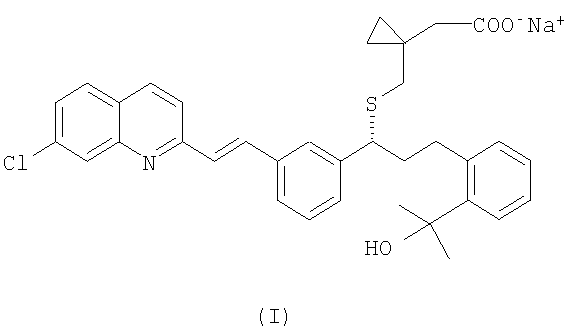
Известны различные стратегии синтеза, предназначенные для получения монтелукаста и его солей. Заявка ЕР 480717 раскрывает ряд замещенных хинолоновых соединений, включая монтелукаст натриевую соль, способы их получения и фармацевтические композиции с использованием данных соединений. В данном документе представлен ряд способов получения монтелукаста натрия. Пример 161 относится к получению монтелукаста натриевой соли. Согласно данному примеру получение монтелукаста натриевой соли осуществляют через соответствующий сложный метиловый эфир, получение которого включает связывание с помощью гидрида натрия или карбоната цезия метил-1-(меркаптометил)-циклопропанацетата с защищенным мезилат(2-(2-(2-(3(S)-(3-(2-(7-хлор-2-хинолинил)-этенил)фенил)-3-(метансульфонилокси)пропил)фенил)-2-пропокси)тетрагидропираном, образующимся in situ. Полученный таким образом сложный метиловый эфир гидролизуют до монтелукаста свободной кислоты, которую затем превращают непосредственно в натриевую соль. Данный способ не очень подходит для крупномасштабного получения, поскольку он требует трудоемкой хроматографической очистки промежуточного сложного метилового эфира и/или конечного продукта, и выходы промежуточных продуктов и конечного продукта являются низкими. Другой способ получения монтелукаста натриевой соли включает реакцию тиоацетата следующей формулы:
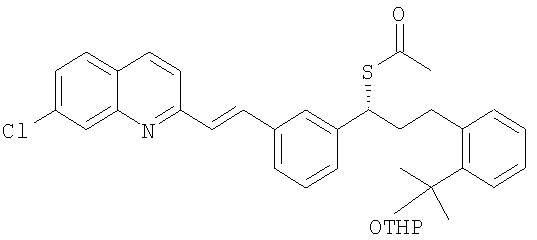
с гидразином или алкоксидом с последующей реакцией с подходящим 1-замещенным циклопропилацетатом, а затем снятием защиты с третичного спирта для получения монтелукаста.
Заявка ЕР 737186 относится к способу получения монтелукаста или его солей, который заключается в реакции дианиона дилития 1-(меркаптометил)циклопропануксусной кислоты с соответствующим мезилатовым спиртом ((2-(2-(2-(3(S)-(3-(2-(7-хлор-2-хинолинил)-этенил)фенил)-3-(метансульфонилокси)пропил)фенил)-2-пропанолом), чтобы получить монтелукаст, который затем превращают в соответствующую натриевую соль через соль дициклогексиламина.
Заявка CN 1420113 А относится к способу получения монтелукаста или его натриевой соли, который заключается в реакции сложного эфира тиоуксусной кислоты с соединением следующей формулы:
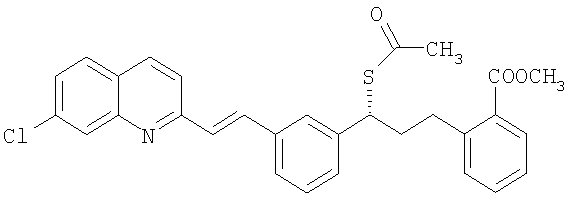
и метилйодидом магния для получения тиолового эфира следующей формулы:
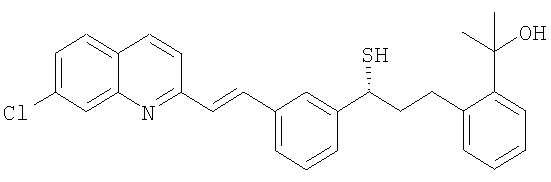
Монтелукаст синтезируют путем реакции между вышеупомянутым промежуточным тиоловым спиртом и соответствующим 1-галогензамещенным циклопропилацетатом с последующим гидролизом сложноэфирной группы. Заявка US 2005107612 описывает способ получения монтелукаста или его соли, который заключается в реакции последнего промежуточного соединения, которое представляет собой 2-[1-[1-R-3-[2-(7-хлорхинолин-2-ил)винил[фенил]-3-[2-метоксикарбонилфенил]пропил-сульфонилметил]циклопропил]уксусную кислоту или его соль, с хлоридом метилмагния или бромидом метилмагния.
Наконец, заявка US 2005/0234241 описывает способ получения монтелукаста, основанный на реакции 2-(2-(3(S)-3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-сульфонилоксипропил)фенил)-2-пропанола с солью 1-(меркаптометил)циклопропанацетонитрила или 1-(меркаптометил) циклопропанацетамида с последующей реакцией гидролиза с помощью основания.
Хотя известен ряд способов получения монтелукаста, остается потребность в новых способах получения монтелукаста и его солей.
Раскрытие изобретения
Авторы обнаружили новый эффективный способ получения монтелукаста из новых промежуточных соединений, которые получают с высокими выходами и без необходимости хроматографической очистки.
Таким образом, согласно одному аспекту настоящего изобретения, в нем представлен способ получения монтелукаста (I):
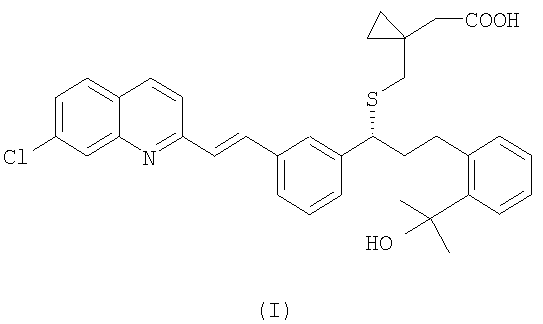
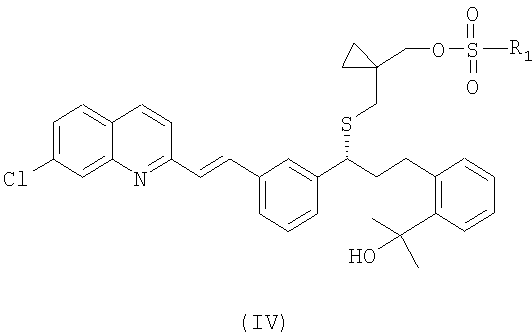
или его фармацевтически приемлемой соли, ее сольвата, включая гидрат, который включает следующие стадии: (а) взаимодействие соединения формулы (IV):
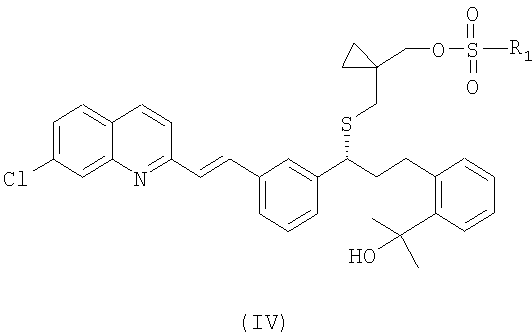
где
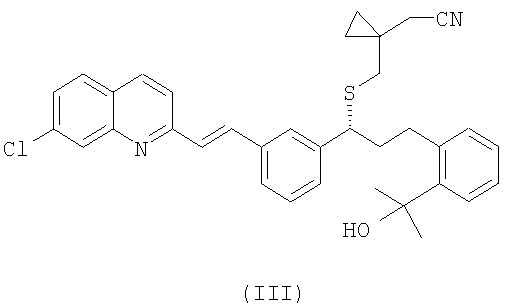
R1 выбран из группы, включающей (С1-С4)-алкил, фенил и фенил, одно- или двузамещенный (С1-С4)-алкильным радикалом, с цианидом щелочного металла с получением соединения формулы (III):
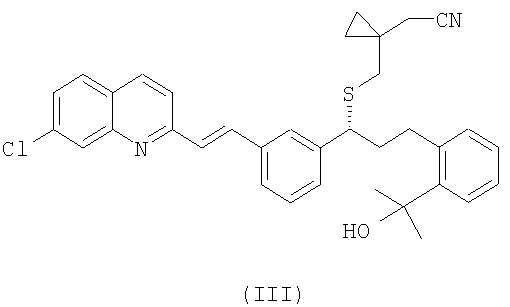
и выделение указанного соединения формулы (III) в виде свободного основания или его соли;
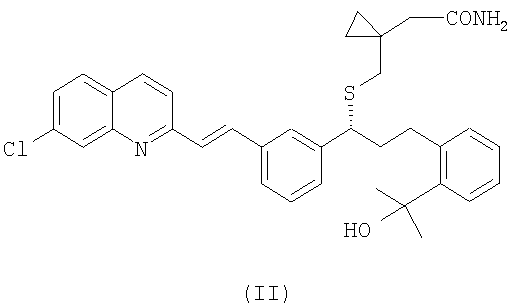
(b) необязательно, гидролиз соединения формулы (III), полученного на стадии (а), с образованием соединения формулы (II):
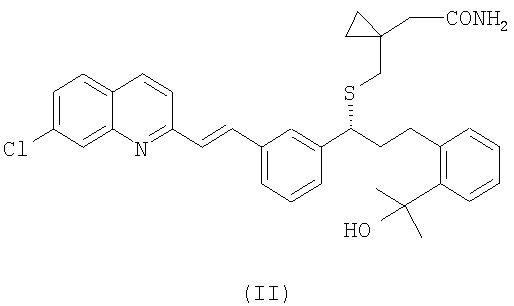
и выделение указанного соединения формулы (II) из реакционной среды;
(c) гидролиз соединения, полученного на стадии (а) или на стадии (b), с образованием монтелукаста (I); и
(d) необязательно, обработка монтелукаста (I) фармацевтически приемлемым основанием с получением соответствующей соли.
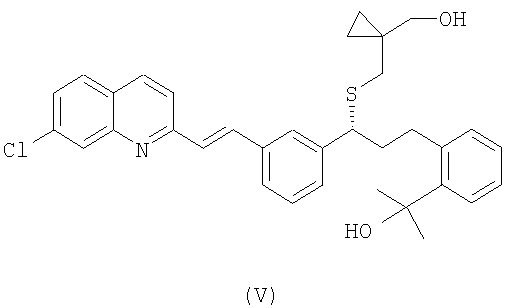
Предварительно, чтобы получить соединение формулы (IV), осуществляют взаимодействие спирта формулы (V):
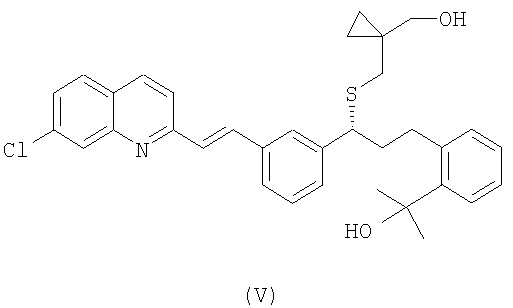
с сульфонилхлоридом формулы Cl-SO2-R1, где R1 имеет значение, как упомянуто выше.
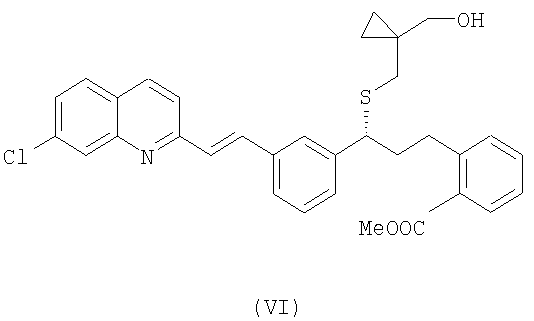
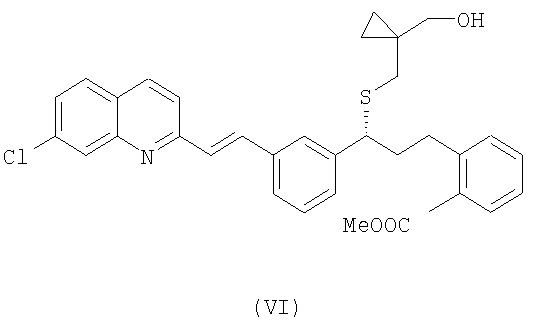
В свою очередь, для того, чтобы получить соединение формулы (V), осущствляют взаимодействие соединения формулы (VI):
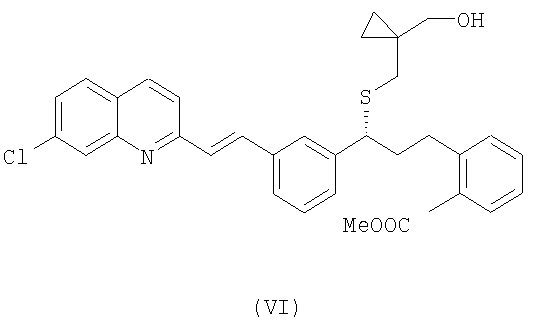
с реагентом Гриньяра, выбранным из группы, включающей галогенид метиллития и галогенид метилмагния, необязательно в присутствии хлорида церия, с возможностью последующего выделения указанного соединения формулы (V) в виде свободного основания или в виде его соли.
Наконец, чтобы получить соединение формулы (VI), проводят реакцию соединения формулы (VII):
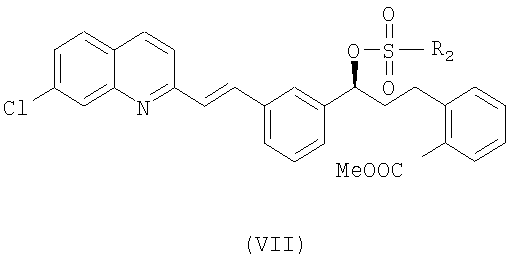
где R2 выбран из группы, включающей (С1-С4)-алкил, фенил и фенил, одно- или двузамещенный (С1-С4)-алкильным радикалом, с соединением формулы (VIII):
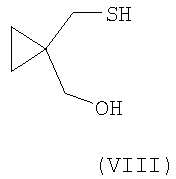
в присутствии основания, выбранного из группы, включающей карбонат цезия, гидроксид натрия и литий бис(триметилсилил)амид, с возможностью последующего выделения соединения формулы (VI) в виде свободного основания или в виде его соли.
Вышеуказанные соединения формул (IV), (V) или их соли и соединение формулы (VI) или его соли являются новыми и также представляют собой часть изобретения. Таким образом, другим аспектом настоящего изобретения является получение новых промежуточных соединений для получения монтелукаста, в частности соединений формулы (IV), (V) и (VI). Часть настоящего изобретения составляют также соли соединения формулы (V), в частности соль лимонной кислоты, соли соединения формулы (VI), в частности соль щавелевой кислоты, и соли, образованные соединением формулы (III) с щавелевой кислотой, L-(-)-яблочной кислотой, L-(+)-винной кислотой, малеиновой кислотой, фумаровой кислотой, янтарной кислотой, бензойной кислотой, 4-гидроксиминдальной кислотой, лимонной кислотой, бензолсульфоновой кислотой и миндальной кислотой.
Способ, соответствующий настоящему изобретению, особенно эффективен в практической промышленной реализации, поскольку он является рентабельным и пригоден для масштабирования. В отличие от других известных способов получения монтелукаста данный способ позволяет избежать внутримолекулярных циклизаций промежуточных соединений. Таким образом, все промежуточные продукты образуются в чистом виде и, вследствие этого, для них не требуется хроматографическое разделение. Более того, конечный продукт получают с высокой химической и оптической чистотой и с высокими выходами.
Осуществление изобретения
Как описано выше, цианосоединение формулы (III) в виде его свободного основания или в виде соли получают из сульфоната формулы (IV). Предпочтительно, когда соединение формулы (III) получают в виде свободного основания из соединения формулы (IV). Превращение осуществляют путем реакции соединения (IV) с цианидом щелочного металла, таким как цианид натрия или калия, в соответствующем растворителе, таком как диметилформамид, диметилсульфоксид, этилацетат или ацетонитрил, и при температуре в интервале от 0°С до температуры кипения с обратным холодильником. Предпочтительно, когда реакцию проводят при приблизительно 60°С. Реакцию можно провести в присутствии катализатора фазового перехода. Примерами подходящих катализаторов фазового перехода являются хлориды три-С8-10-алкилметиламмония, бромид тетрабутиламмония, хлорид гексадецилтриметиламмония, хлорид метилтриоктиламмония, хлорид тетрабутиламмония или гидросульфат тетрабутиламмония. В предпочтительном варианте осуществления в качестве катализатора фазового перехода используют гидросульфат тетрабутиламмония.
В настоящем изобретении предпочтительными сульфонатными соединениями формулы (IV) являются соединения, в которых сульфонат формулы (IV) представляет собой мезилат (R1=метил), безилат (R1=фенил) или тозилат (R1=4-метилфенил). Наиболее предпочтительным сульфонатом формулы (IV) является мезилат.
Соединение формулы (III) можно превратить в его соли, и его соли можно превратить в свободное соединение способами, известными в области техники.
Амидное соединение формулы (II) можно получить из цианосоединения формулы (III) в виде свободного основания или в виде соли путем гидролиза. Предпочтительно, когда соединение формулы (II) получают из соединения формулы (III) в виде свободного основания. Например, гидролиз может достигаться при использовании мягких условий и/или путем сокращения времени реакции. Компетентный специалист в области техники может легко определить условия осуществления гидролиза с помощью рутинных методов. Например, гидролиз можно осуществить, используя такие же условия реакции, как для проведения гидролиза цианосоединения до монтелукаста, но сокращая время реакции, как проиллюстрировано в примере 9, с последующим выделением амидного соединения формулы (II).
Последней стадией способа является реакция гидролиза. Соединение формулы (II) можно гидролизовать с получением монтелукаста. Реакцию гидролиза можно непосредственно осуществить из цианосоединения формулы (III) в виде свободного основания или в виде соли с получением монтелукаста. Предпочтительно, когда гидролиз соединения формулы (II) или соединения формулы (III) в виде свободного основания или в виде соли проводят с помощью основания. Предпочтительно, когда основание представляет собой гидроксид щелочного металла или гидроксид щелочноземельного металла. Более предпочтительно, когда основание представляет собой гидроксид натрия, гироксид калия или гидроксид лития. Наиболее предпочтительным является гидроксид натрия. Реакцию можно осуществить в различных системах растворителей. Предпочтительно, когда система растворителей представляет собой смесь растворителей, включающую С1-С4спирт и воду. Предпочтительно, когда С1-C4спирт представляет собой этанол или изопропанол. Наиболее предпочтительным является этанол. Как правило, гидролиз цианосоединения формулы (III) до монтелукаста формулы (I) проводят при температуре кипения с обратным холодильником, и она в основном завершается в течение 24 часов.
Другая подходящая система растворителей представляет собой двухфазную смесь растворителей, включающую воду и подходящий органический растворитель, не смешивающийся с водой или плохо смешивающийся с водой, необязательно в присутствии катализатора фазового перехода. Предпочтительно, когда органический растворитель представляет собой (С6-С8)-ароматический углеводород. Наиболее предпочтительным является толуол. Подходящий катализатор фазового переноса включает, например, четвертичную соль аммония, такую как бромид тетрабутиламмония, хлориды три-С8-С10-алкилметиламмоний, хлорид метилтриоктиламмония или хлорид гексадецилтриметиламмония. Гидролиз можно осуществить при температуре, находящейся в интервале от комнатной температуры до температуры кипения с обратным холодильником. Как правило, в данных реакционных условиях гидролиз цианосоединения формулы (III) до монтелукаста формулы (I) проводят при 120°С, и он в основном завершается в течение приблизительно семи дней.
Выделение неочищенного монтелукаста можно осуществить посредством разбавления неочищенного продукта толуолом и промыванием раствора водной уксусной кислотой и затем водой при комнатной температуре с последующим упариванием растворителя. Полученный монтелукаст можно очистить различными способами, например экстракциями на основе водной кислоты или перекристаллизацией.
Монтелукаст свободную кислоту, полученный способом, соответствующим настоящему изобретению, можно превратить в фармацевтически приемлемые соли, и соли можно превратить в соединения свободной кислоты известными методами, описанными в области техники. Возможно также выделить соль монтелукаста реакцией гидролиза, т.е. без выделения монтелукаста свободной кислоты.
Сульфонатные соединения формулы (IV) получают из спирта формулы (V) путем реакции с соответствующим сульфонилхлоридом. Данную реакцию проводят в соответствующем растворителе и в присутствии третичного амина при температуре, находящейся в интервале от -20°С до комнатной температуры. Предпочтительно, когда реакцию проводят при низких температурах. Обычные растворители для реакции включают хлорсодержащие растворители, такие как метиленхлорид или 1,2-дихлорэтилен, ароматические углеводороды, такие как толуол или ксилен, и диметилформамид. Примерами подходящих третичных аминов являются диизопропилэтиламин и триэтиламин.
Получение спирта формулы (V) в виде свободного основания или в виде соли из соединения формулы (VI) осуществляют посредством реакции с галогенидом метилмагния, таким как хлорид метилмагния или бромид метилмагния, необязательно в присутствии хлорида церия, или путем реакции с метиллитием в присутствии подходящего растворителя. Примеры подходящих растворителей включают простой эфир, такой как тетрагидрофуран, или ароматический углеводород, такой как толуол или ксилен, или их смеси. Предпочтительно, когда реакцию проводят при температуре в интервале от -78°С до 20°С, более предпочтительно - при -20°С.
Спирт формулы (V) можно превратить в его соли, и его соли можно превратить в свободное соединение методами, известными в области техники. Например, соль лимонной кислоты получают реакцией соединения формулы (V) в виде свободного основания с лимонной кислотой в присутствии подходящего растворителя.
Соединение формулы (VI) в виде свободного основания или в виде соли можно получить из известного соединения формулы (VII), которое реагирует с соединением формулы (VIII) в подходящем растворителе, таком как диметилформамид, диметилсульфоксид, дихлорметан, толуол или тетрагидрофуран, и в присутствии основания, такого как карбонат цезия, гидроксид натрия и бис(триметилсилил)амид лития. Предпочтительно, когда реакцию проводят при температуре в интервале от -10°С до 60°С, более предпочтительно при 0-5°С.
Соединение формулы (VI) можно превратить в его соли, и его соли можно превратить в свободное соединение методами, известными в области техники. Например, соль щавелевой кислоты получают посредством реакции соединения формулы (VI) в виде свободного основания с щавелевой кислотой в присутствии подходящего растворителя.
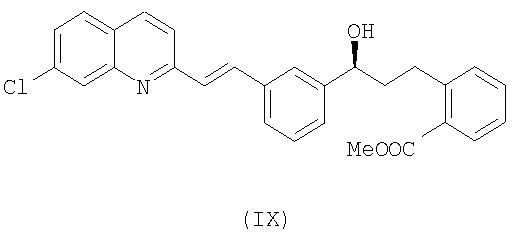
Соединение формулы (VII) можно получить согласно известному методу, описанному в US 2005107612. Метод, описанный в данном документе, заключается в реакции метил-2-((S)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-гидроксипропил)бензоата формулы (IX) с метансульфонилхлоридом или толуолсульфонилхлоридом для образования метил-2-((S)-3-(3-(Е)-(2-(7-хлорхинолин-2-ил)винил)фенил)-3-[(метил-сульфонил)окси]пропил)бензоата или соответствующего тозилата. Аналогичным образом можно получить безилат формулы (VII) из соответствующего бензолсульфонилхлорида.
Соединение формулы (VIII) можно получить из 5,7-диокса-6-тиазо-спиро[2.5]октан 6-оксида согласно способу, кратко представленному на Схеме 1.
Схема 1:
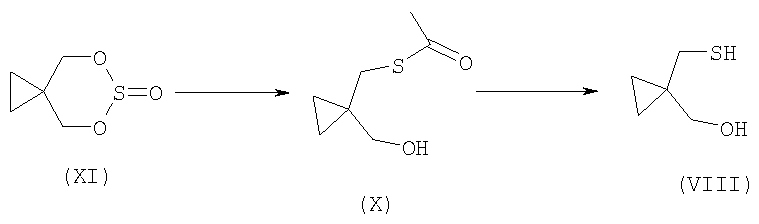
Способ получения соединения формулы (VIII) заключается в реакции соединения формулы (XI) с этантиоатом калия, что дает соединение формулы (X). Как правило, данную реакцию проводят с избытком этантиоата калия в соответствующем растворителе. Примеры подходящих растворителей включают диметилсульфоксид, диметилформамид, этилацетат, ацетонитрил или их смеси. Предпочтительно, когда реакцию осуществляют при температуре в интервале от комнатной температуры до 70°С. После выделения соединения формулы (X) его можно гидролизовать, используя кислотный или основной катализатор, что дает соединение формулы (VIII). Примерами подходящих кислот являются хлористо-водородная кислота, серная кислота, уксусная кислота и муравьиная кислота. Примерами подходящих оснований являются гидроксиды, карбонаты и алкоксид щелочного или щелочноземельного металла. Примерами подходящих растворителей являются (C1-C6)-спирты, (С6-С8)-ароматические углеводороды, диметилформамид, диметилсульфоксид или их смеси.
Оптимальные условия осуществления способа, представленного в настоящем изобретении, варьируют в соответствии с параметрами, предусматриваемыми компетентным специалистом в области техники, такими как исходные материалы, молярное соотношение, температура и т.п. Данные условия реакции может легко определить компетентный специалист в области техники посредством рутинных анализов и с помощью описания примеров, включенных в данный документ.
В тексте описания и формулы изобретения слово "включает" и варианты данного слова не предусматривают исключение других технических признаков, дополнений, компонентов или стадий. Реферат данной заявки включен в данном контексте в виде ссылки. Дополнительные объекты, преимущества и признаки изобретения будут очевидны компетентным специалистам в области техники на основании изучения описания или могут быть изучены при практической реализации изобретения. Следующие примеры предложены в качестве иллюстрации, но они не предназначены для ограничения объема настоящего изобретения.
Примеры
Пример 1
Получение S-(1-(гидроксиметил)циклопропил)метилэтантиоата (X)
200 г 5,7-диокса-6-тиазоспиро[2.5]октан 6-оксида растворяют в 1,2 л диметилсульфоксида и выливают в раствор 308 г этантиоата калия. Затем суспензию нагревают при 40°С в течение 5 часов. После завершения реакции добавляют смесь 3,6 л этилацетата и 3,6 л воды. Органическую фазу отделяют, промывают водой и концентрируют досуха. Выделяют 200 г S-(1-(гидроксиметил)циклопропил)метилэтантиоата. Выход: 78% с поправкой по GC (газовой хроматографии). Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): 3,45 (2Н, d, J: 6,4 Гц); 3,01 (2Н, s); 2,53 (ОН, широкий триплет, J: 6,4 Гц); 2,39 (3Н, s); 0,54 (4Н, m).
Пример 2. Получение (1-(меркаптометил)циклопропил)метанола (VIII)
200 г S-(1-(гидроксиметил)циклопропил)метилэтантиоата растворяют в 2 л метанола. Затем добавляют 111 мл концентрированной HCl в атмосфере азота и перемешивают раствор при комнатной температуре в течение 10 часов. Растворитель отгоняют и остаток перерастворяют в 1,5 л диметилформамида для применения в получении метил 2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил)пропил)бензоата. Выход: 100%. Данные 1H ЯМР-спектроскопии (400 МГц, CDCl3): 3,57 (2Н, s); 2,63 (2Н, d, J: 8,0 Гц); 2,45 (ОН, широкий сигнал); 1,43 (SH, t, J: 8,0 Гц); 0,52 (4Н, m).
Пример 3
Получение метил2-((S)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-[(метилсульфонил)окси]пропил)бензоата (VII с R2: СН3)
12,1 мл диизопропилэтиламина выливают в перемешиваемый раствор 24,5 г метил 2-((S)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-гидроксипропил)бензоата в 120 мл дихлорметана. Смесь охлаждают до -20°С и медленно добавляют 5 мл мезилхлорида. После завершения реакции неочищенный раствор последовательно промывают 120 мл водного 10% раствора NaHCO3 и 120 мл воды. Наконец, растворитель отгоняют, получая 29 г целевого соединения. Продукт перерастворяют в 290 мл диметилформамида для применения в виде раствора на следующей стадии. Выход: 100%.
Пример 4
Получение метил2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил)пропил) бензоата (VI)
Раствор 19 г (1-(меркаптометил)циклопропил)метанола в 190 мл диметилформамида охлаждают до 0°С. Затем добавляют 52 г Cs2CO3 в одной порции. Через 10 минут добавляют конечный раствор, полученный в предшествующем примере, и поддерживают суспензию при той же температуре в течение 18 часов. Наконец, смесь 380 мл этилацетата и 380 мл воды выливают в раствор неочищенного продукта. Органическую фазу промывают 380 мл воды и концентрируют досуха. Выделяют 30 г масла коричневого цвета. Выход: 86% с поправкой после анализа ВЭЖХ (высокоэффективной жидкостной хроматографией). Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 8,38 (1Н, d, J: 8,4 Гц); 8,00-7,26 (14Н, m); 4,50 (ОН, t, J: 5,4 Гц); 3,92 (1H, t, 7,2 Гц); 3,76 (3Н, s); 3,33 (1Н, dd, J: 11,0 Гц, 5,4 Гц); 3,25 (1H, dd, J: 11,0 Гц, 5,4 Гц); 2,98-2,88 (1H, m); 2,83-2,74 (1Н, m); 2,52 (1Н, d, J: 12,7 Гц); 2,39 (1Н, d, J: 12,7 Гц); 2,10 (2Н, m); 0,46-0,18 (4Н, m).
Пример 5
Получение 2-(2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил) фемил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил)пропил) фенил)пропан-2-ола (V)
Продукт, полученный в предшествующем примере, растворяют в 300 мл тетрагидрофурана. Смесь охлаждают до 0°С, добавляют 307 мл 1,4 М раствора бромида метилмагния под азотом. Реакцию поддерживают при 0°С в течение 18 часов. Затем медленно добавляют 400 мл 2 М водного раствора уксусной кислоты, а потом 400 мл этилацетата. Органическую фазу последовательно промывают 400 мл водного 10% раствора NaHCO3 и 400 мл воды. Наконец, растворитель отгоняют, получая 30 г неочищенного продукта. Выход: 81% с поправкой после анализа ВЭЖХ. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 8,39 (1Н, d, J: 8,6 Гц); 8,00-7,06 (14Н, m); 4,91 (ОН, s);4,51 (ОН, t, J: 5,5 Гц); 3,99(1 H, t, 7.3 Гц); 3,34 (1Н, dd, J: 11,0 Гц, 5,5 Гц); 3,26 (1H, dd, J: 11,0 Гц, 5,5 Гц);3,05(1Н, m); 2,74 (1Н, m); 2,54(1H, d, J: 12,7 Гц);2,41 (1Н, d, J: 12,7 Гц); 2,15 (2Н, m); 1,44 (6Н, s); 0,45-0,36 (2Н, m); 0,33-0,22 (2Н, m).
Пример 6
Получение 2-(2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил)пропил) фенил) пропан-2-ола (V)
1,3 г безводного хлорида церия перемешивают в 10 мл сухого тетрагидрофурана в течение 18 часов при комнатной температуре. Затем суспензию охлаждают до -20°С и добавляют 1,5 мл 3 М раствора хлорида метилмагния под азотом. Через некоторый период времени добавляют 2,5 мл раствора в тетрагидрофуране 0,5 г продукта, полученного в примере 4. Смесь перемешивают в течение 18 часов при той же температуре и медленно добавляют 10 мл 2 М водного раствора уксусной кислоты, а затем 10 мл этилацетата. Органическую фазу последовательно промывают 10 мл водного 10% раствора NaHCO3 и 10 мл воды. Наконец, растворитель отгоняют, получая 0,5 г неочищенного продукта. Выход: 80% с поправкой после анализа ВЭЖХ.
Пример 7
Получение (R,E)-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил) циклопропил)метилметансульфоната ((IV) с R1=СН3)
6,5 мл диизопропилэтиламина выливают в перемешиваемый раствор 16 г 2-(2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил)пропил)фенил)пропан-2-ола в 160 мл дихлорметана. Смесь охлаждают до -20°С и медленно добавляют 2,7 мл мезилхлорида. Смесь перемешивают при той же температуре в течение одного часа и затем ее дважды промывают 300 мл воды. Растворитель отгоняют и выделяют 18 г неочищенного продукта. Выход: 100%. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 8,41 (1Н, d, J: 9,6 Гц); 8,02-7,03 (14Н, m); 4,91 (ОН, s); 4,12 (2H, m); 4,02 (1Н, t, J: 7,1 Гц); 3,12 (3Н, s); 3,09-3,02 (1Н, m); 2,78-2,71 (1Н, m); 2,54 (1Н, d, J: 13,2 Гц); 2,46 (1Н, d, J: 13,2 Гц); 2,15 (2Н, m); 1,43 (3Н, s); 1,42 (3Н, s); 0,68-0,45 (4H, m).
Пример 8
Получение (R,E)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил) циклопропил)ацетонитрила (III)
Неочищенный продукт, полученный в предшествующем примере, растворяют в 180 мл диметилформамида и добавляют к раствору 2,1 г цианида натрия в одной порции. Через 18 часов перемешивания при 60°С в смесь, предварительно нагретую до комнатной температуры, выливают 270 мл этилацетата. Затем проводят два промывания 270 мл воды и отгоняют растворитель, получая 14 г неочищенного продукта. Выход: 75% с поправкой после анализа ВЭЖХ. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6):8,39 (1Н, d, J: 8,6 Гц); 8,00-7,05 (14Н, m); 4,90 (ОН, s); 4,04 (1Н, t, J: 7,2 Гц);3,06 (1Н, m); 2,76 (1Н, m); 2,67 (1Н, m); 2,33 (1Н, m); 2,30 (2Н, s); 2,13 (2Н, m); 1,43 (6H, s); 0,59-0,43 (4H, m).
Пример 9
Получение (R,E)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил) сульфанил)метил)циклопропил)ацетамида (II)
1,8 г неочищенного продукта, полученного в примере 8, растворяют в 3,6 мл толуола. Затем добавляют 3,6 мл воды, а потом 14,7 г гидроксида натрия и 0,44 г бромида тетрабутиламония. Смесь перемешивают при 120°С в течение 30 часов. Через данный период времени 10 мл толуола и 10 мл воды добавляют к реакционному раствору, предварительно охлажденному до комнатной температуры. Органическую фазу отделяют и промывают 10 мл воды. Наконец, растворитель отгоняют и неочищенный продукт очищают стандартными методами, чтобы выделить 77 мг целевого соединения. Выход: 6%. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 8,41 (1Н, d, J: 8,6 Гц); 8,03-7,07 (15Н, m); 6,74 (1Н, s); 4,91 (ОН, s); 4,02 (1Н, t, J: 7,3 Гц); 3,04 (1Н, m); 2,75 (1Н, m); 2,57 (1Н, d, J: 13,3 Гц); 2,45 (1Н, d, J: 13,3 Гц); 2,16 (4Н, m); 1,43 (6H, s); 0,50-0,27 (4Н, m).
Пример 10
Получение 2-(1-((((R)-1-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил) циклопропил)уксусной кислоты (Монтелукаст свободная кислота)
5,2 г неочищенного продукта, полученного в примере 8, растворяют в 10 мл толуола. Затем добавляют 10 мл воды с последующим добавлением 14,7 г гидроксида натрия и 0,44 г бромида тетрабутиламмония. Смесь перемешивают при 120°С в течение 7 дней. Через данный период времени 10 мл толуола и 10 мл воды добавляют к реакционному раствору, предварительно охлажденному до комнатной температуры. Органическую фазу отделяют и последовательно промывают 10 мл уксусной кислоты и 10 мл воды. Наконец, растворитель отгоняют, чтобы выделить 4,6 г продукта, который при необходимости можно очистить стандартными методами. Выход: 83%. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 12,02 (1Н, s); 8,39 (1Н, d, J: 8,6 Гц); 8,01-7,04 (14Н, m); 4,90 (ОН, s); 4,00 (1Н, t, J: 7,3 Гц); 3,05 (1Н, m); 2,76 (1H, m); 2,56 (1H, d, J: 12,9 Гц); 2,47 (1Н, d, J: 12,9 Гц); 2,32 (2Н, s); 2,16 (2Н, m); 1,44 (6H, s); 0,52-0,33 (4H, m). ИК-спектр (KBr)=3573,1, 3436,9, 2919,2, 1716,9, 1608,9, 1499.9, 1408,8, 1315,7, 1223,9, 1201,6, 1173,0, 1148,0, 1134,9, 1074,8, 965,9, 950,8, 933,2, 863,6, 842,7, 766,0 см-1.
Пример 11
Получение 2-(1-((((R)-1-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил) циклопропил)уксусной кислоты (Монтелукаст свободная кислота)
1,05 г гидроксида натрия выливают в раствор 1 г (R,Е)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил)циклопропил)ацетонитрила в 5 мл смеси этанол: вода (96:4, об./об.). Полученную в результате суспензию перемешивают при температуре кипения с обратным холодильником в течение 15 часов. Через данный период времени реакционную смесь охлаждают до комнатной температуры и разводят 10 мл толуола. Затем медленно добавляют 20 мл 2 М водного раствора уксусной кислоты и органический слой отделяют и дважды промывают 10 мл воды. Наконец, растворитель отгоняют, чтобы выделить 0,9 г продукта, который при необходимости можно очистить стандартными методами. Выход: 91%.
Пример 12
Получение 2-(1-((((R)-1-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил)циклопропил)ацетата натрия (Монтелукаст натрий)
2,6 г монтелукаста свободной кислоты растворяют в 26 мл толуола и медленно добавляют 8,9 мл 0,5 М раствора NaOH в метаноле при комнатной температуре. Смесь перемешивают в течение 1 часа, и растворитель удаляют под вакуумом, получая остаток. Затем к хорошо перемешанному раствору остатка в 4 мл этилацетата при комнатной температуре в течение 30 минут добавляют гептан (24 мл). Через два часа после добавления твердое вещество не совсем белого цвета отфильтровывают в атмосфере азота и промывают 5 мл гептана. Влажный продукт сушат под вакуумом при 70-80°С в течение 2 дней, получая 2,7 г аморфной твердой формы монтелукаста натрия. Выход: 100%. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 8,38 (1Н, d, J: 8,6 Гц); 8,02-7,04 (14Н, m); 5,19 (ОН, s); 4,01 (1H, t, J: 7,2 Гц);3,08 (1H, m); 2,72 (1H, m); 2,69 (1H, d, J: 12,4 Гц); 2,54 (1H, d, J: 12,4 Гц); 2,22 (1H, m);2,10(1H, d, J: 14,2 Гц); 2,10 (1H, m); 1,96(1H, d, J: 14,2 Гц); 1,45 (3Н, s); 1.44 (3Н, s); 0,46-0,32 (2H, m); 0,28-0,15 (2H, m).
Пример 13 Получение соли шавелевой кислоты метил2-((R)-3-(3-((E)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(((1-(гидроксиметил)циклопропил)метил)-сульфанил)пропил)бензоата (соль щавелевой кислоты соединения (VI))
К раствору 338 г (0,61 моль) метил 2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил) пропил)бензоата в 2,0 л раствора толуола медленно добавляют 75 г (0,60 моль) дигидрата щавелевой кислоты в 1,0 л ацетонитрила при комнатной температуре. Полученную в результате суспензию перемешивают при комнатной температуре в течение десяти часов. Твердое вещество отфильтровывают и несколько раз промывают смесью толуола/ацетонитрила 2/1 (об./об.). Влажное твердое вещество сушат под вакуумом при 25°С. Получают 304 г твердого вещества белого цвета. Выход 78%. DSC: 121,6°С (пик). Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 8,42 (1Н, d, J: 8,6 Гц); 8,00-7,28 (14Н, m); 3,92 (1H, t, 7,3 Гц); 3,76 (3Н, s); 3,31 (1Н, d, J: 11,0 Гц); 3,25 (1Н, d, J: 11,0 Гц); 2,98-2,90 (1Н, m); 2,82-2,75 (1Н, m); 2,52 (1Н, d, J: 12,7 Гц); 2,39 (1Н, d, J: 12,7 Гц); 2,14-2,08 (2Н, m); 0,43-0,19 (4Н, m). Измерение DSC проводят в перфорированном поддоне при скорости сканирования 5°С/мин. В интервале от 25,0°С до 150,0°С с помощью прибора DSC Mettler Toledo DSC822.
Пример 14
Получение метил2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил) фенил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил)пропил) бензоата (VI)
283 г соли щавелевой кислоты, полученной в Примере 13, суспендируют в 1,7 л толуола и 1,7 л воды. 35% водный раствор хлористо-водородной кислоты медленно добавляют к предшествующей смеси до получения рН 6,5. Органическую фазу отделяют и промывают водой. Наконец, растворитель отгоняют под вакуумом. Получают 243 г масла. Выход 100%.
Пример 15
Получение цитрата 2-(2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил) фенил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил)пропил) фенил)пропан-2-ола (соль лимонной кислоты соединения (V))
Раствор 92,5 г 2-(2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил)пропил) фенил)пропан-2-ола (ВЭЖХ: 78% площади) в 910 мл толуола нагревают до 55°С. Затем в одной порции добавляют 34,8 г лимонной кислоты моногидрата. Через 1 час перемешивания при той же температуре смесь постепенно охлаждают до 20°С в течение 2 часов и перемешивают при данной температуре в течение дополнительных 2 часов. Наконец, суспензию фильтруют под вакуумом и остаток дважды промывают 100 мл толуола. Продукт используют в виде влажного твердого вещества на следующей стадии. (ВЭЖХ: 97% площади). Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 12,38 (2Н, s); 8,39 (1Н, d, J: 8,6 Гц); 8,02-7,11 (14Н, m); 4,90 (ОН, s); 4,51 (ОН, s); 3,99 (1H, t, J: 7,3 Гц); 3,34 (1Н, d, J: 11,0 Гц); 3,27 (1Н, d, J:11,0 Гц); 3,06 (1Н, td, J: 12,3 Гц; J: 4,9 Гц); 2,77 (2Н, d, J: 15,4 Гц); 2,73 (1Н, m); 2,66 (2Н, d, J: 15,4 Гц); 2,54 (1Н, d, J: 12,7 Гц); 2,41 (1Н, d, J: 12,7 Гц); 2,17 (2Н, m); 1,44 (6H, s); 0,44-0,36 (2Н, m); 0,32-0,22 (2Н, m). ИК - спектр (KBr)=3411, 3052, 2970, 2922, 2593, 1727, 1632, 1599, 1488, 1437, 1409, 1376, 1318, 1223, 1208, 1153, 1080, 1026, 961, 927, 861, 840, 760, 729, 694, 598,471 см-1.
Пример 16
Получение 2-(2-((R)-3-(3-((Е)-2-(7-хлорхинолин-2-ил)винил)фенил)-3-(((1-(гидроксиметил)циклопропил)метил)сульфанил)пропил)фенил)пропан-2-ола (V)
Влажное твердое вещество, полученное в Примере 15, разделяют с помощью 800 мл толуола и 800 мл 5% водного раствора NaHCO3. Органический слой отделяют, промывают 800 мл воды и концентрируют досуха упариванием на роторном испарителе под вакуумом при 30°С. Выделяют 29,7 г твердого вещества бледно-желтого цвета. Общий выход по 2 стадиям составляет 39% (ВЭЖХ: 96% площади).
Пример 17
Получение (R,E)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил) фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил) циклопропил)ацетонитрила (III)
5 мл воды добавляют к 96 г раствора толуола, содержащего 10 г (R,Е)-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропилтио)метил)циклопропил)метилметансульфоната с последующим добавлением 0,5 г гидросульфата тетрабутиламмония и 0,85 г цианида натрия. Смесь перемешивают при 60°С в течение 20 часов. Затем ее охлаждают до комнатной температуры и органический слой трижды промывают 100 мл воды. Наконец, полученный толуоловый раствор концентрируют досуха упариванием на роторном испарителе под вакуумом при 30°С. Получают 9,5 г продукта.
Общий пример
Получение солей (R,Е)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил) фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил) циклопропил)ацетонитрила (соли соединения (III))
К раствору 0,5 г (8,82 ммоль) (R,Е)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил) винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил) циклопропил)ацетонитрила в 3,5 мл толуола медленно добавляют раствор 8,82 ммоль кислоты в ацетонитриле или тетрагидрофуране при комнатной температуре. Растворитель отгоняют и теоретическое количество соли получают в виде твердого вещества желтого цвета.
Общий пример повторяют, используя соответствующие кислоты, указанные в следующих примерах.
Пример 18
Получение соли щавелевой кислоты соединения (III) с использованием щавелевой кислоты.
Выход: 100%; ИК-спектр (KBr) (см-1): 3420, 2970, 2925, 1719, 1626, 1600, 1486, 1408, 1205, 1157, 1080, 762, 695. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 8,41 (1Н, d, J: 8,7 Гц); 8,0-7,1 (14Н, m); 4,04 (1H, t, 7,3 Гц); 3,1 (1Н, m); 2,8 (1H, m); 2,63 (2H, s); 2,51 (2H, в полудейтерированном ДМСО); 2,2 (2Н, m); 1,44 (3Н, s); 1,43 (3Н, s); 0,60-0,40 (4Н, m).
Пример 19
Получение соли яблочной кислоты соединения (III) с использованием L-(-)-яблочной кислоты
Выход: 100%; ИК-спектр (KBr) (см-1): 3436, 2976, 2930, 1721, 1628, 1607, 1498, 1409, 1161, 1078, 761, 696. Данные 1Н ЯМР-спектроскопии (400 МГц, ДМСО-d6): 12,41 (1Н, широкий); 8,40 (1Н, d, J 8,69 Гц); 8,0-7,1 (14Н, m); 4,91 (1Н, s); 4,26 (1H, dd, J 7,83 Гц, J 4,78 Гц); 4,04 (1Н, t, 7,35 Гц); 3,11-3,01 (1Н, m); 2,80-2.70 (1Н, m); 2,61 (1Н, dd, J 15,66 Гц, J 4,80 Гц); 2,51 (2H, в полудейтерированном ДМСО), 2,44 (1Н, dd, J 15,66 ГЦ, J 7,84 Гц); 2,26-2,10 (2H, m); 1,44 (3Н, s); 1,43 (3Н, s); 0,60-0,40 (4Н, m).
Пример 20
Получение соли винной кислоты соединения (III) с использованием L-(+)-винной кислоты.
Выход: 100%; ИК-спектр (KBr) (см-1): 3401, 2970, 2927, 1726, 1626, 1605, 1487, 1408, 1205, 1130, 1080, 762, 696. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 12,68 (1Н, широкий); 8,41 (1Н, d, J: 8,64 Гц); 8,04-7,04 (14Н, m); 4,91 (1Н, s); 4,31 (2H, s); 4,04 (1Н, t, J 7,34 Гц); 3,12-3,00 (1Н, m); 2,81-2,70 (1Н, m); 2,64 (2H, s); 2,51 (2H, в полудейтерированном ДМСО); 2,26-2,07 (2H, m); 1,44 (3Н, s); 1,43 (3Н, s); 0,60-0,40 (4Н, m).
Используя способ, соответствующий общему примеру, из соответствующих кислот получают следующие соли соединения (III): малеат, фумарат, сукцинат, бензоат, 4-гидроксимандалат, цитрат и манделат.
Пример 21
Получение соли щавелевой кислоты (R,E)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил) пропил)сульфанил)метил)циклопропил)ацетонитрила (соль щавелевой кислоты соединения (III))
К раствору 3,14 г (5,53 ммоль) (R,Е)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил) метил)-циклопропил)ацетонитрила в 21,5 мл толуола добавляют раствор 0,68 г (5,42 ммоль) дигидрата щавелевой кислоты в 10 мл ацетонитрила при комнатной температуре. Раствор концентрируют под вакуумом до половины исходного объема и перемешивают при комнатной температуре в течение ночи. Полученное твердое вещество отфильтровывают и сушат под вакуумом. Получают 2,71 г (выход 71%) твердого вещества желтого цвета. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 8,41 (1Н, d, J: 8,7 Гц); 8,0-7,1 (14Н, m); 4,04 (1H, t, 7,3 Гц); 3,1 (1Н, m); 2,8 (1Н, m); 2,63 (2H, s); 2,51 (2H, в полудейтерированном ДМСО); 2,2 (2H, m); 1,44 (3Н, s); 1,43 (3Н, s); 0,60-0,40 (4Н, m), IR (KBr)=3420, 2970, 2925, 1719, 1626, 1600, 1486, 1408, 1205, 1157, 1080, 762, 695 см-1.
Пример 22
Получение соли бензолсульфоновой кислоты (R,E)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил)винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил) пропил)сульфанил)метил)циклопропил)ацетонитрила (соли бензолсульфоновой кислоты соединения (III))
К раствору 1,0 г (1,76 ммоль) (R,Е)-2-(1-((1-(3-(2-(7-хлорхинолин-2-ил) винил)фенил)-3-(2-(2-гидроксипропан-2-ил)фенил)пропил)сульфанил)метил)-циклопропил)ацетонитрила в 6 мл толуола добавляют раствор 0,303 г (1,73 ммоль) 90% бензолсульфоновой кислоты в 0,6 мл ацетонитрила при комнатной температуре. Раствор перемешивают при комнатной температуре в течение ночи. Полученное твердое вещество отфильтровывают и сушат под вакуумом. Получают 0,96 г (выход 75%) твердого вещества желтого цвета. Данные 1H ЯМР-спектроскопии (400 МГц, ДМСО-d6): 8,74 (1Н, d, J: 8,7 Гц);8,27-7,04 (19Н, m); 4,07 (1H, t, 7.3 Гц); 3,1 (1Н, m); 2,8 (1Н, m); 2,63 (2H, s); 2,51 (2H, в полудейтерированном ДМСО); 2,2 (2H, m); 1,44 (3Н, s); 1,43 (3Н, s); 0,61-0,41 (4H, m). IR (KBr)=3430, 3050, 2910, 1625, 1600, 1480, 1230, 1150, 1125, 1020, 990, 690, 610 см-1.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛИ МОНТЕЛУКАСТА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2020 |
|
RU2840778C2 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2794333C1 |
| СПОСОБ ПОЛУЧЕНИЯ пан-ЦЗК-ИНГИБИТОРОВ ФОРМУЛЫ (I), А ТАКЖЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ | 2011 |
|
RU2585621C2 |
| АРИЛ-N-АРИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2020 |
|
RU2821414C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| ПРОИЗВОДНЫЕ АЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1995 |
|
RU2161612C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВОЙ СОЛИ 1-[[[(R)-M-[(Е)-2-(7-ХЛОР-2-ХИНОЛИЛ)ВИНИЛ]-АЛЬФА-[О-(1-ГИДРОКСИ-1-МЕТИЛЭТИЛ)ФЕНЕТИЛ]БЕНЗИЛ]ТИО]МЕТИЛ]ЦИКЛОПРОПАНУКСУСНОЙ КИСЛОТЫ | 2008 |
|
RU2436773C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2795512C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2739356C2 |
Настоящее изобретение относится к способу получения монтелукаста формулы I или его фармацевтически приемлемой соли или сольвата, включая гидрат, в котором вначале осуществляют взаимодействие соединения формулы (IV), где R1 выбран из группы, включающей (С1-С4)-алкил, фенил и фенил, одно- или двузамещенный (С1-С4)-алкильным радикалом, с цианидом щелочного металла с образованием и последующим выделением соединения формулы (III) в виде свободного основания или его соли, затем необязательно проводят гидролиз полученного соединения формулы (III) с образованием и последующим выделением из реакционной среды соединения формулы (II), далее соединение II или III, полученное на любой из предшествующих стадий, подвергают гидролизу с образованием монтелукаста формулы (I), после этого образовавшийся монтелукаст формулы (I) необязательно обрабатывают фармацевтически приемлемым основанием для получения соответствующей соли. Также изобретение относится к соединениям формул IV, V, VI и к солям соединения формулы III. Технический результат - разработан новый эффективный способ получения монтелукаста, используемого в настоящее время для лечения астмы, воспаления и др. заболеваний, из новых промежуточных соединений, которые получают с высокими выходами. 5 н. и 15 з.п. формулы.
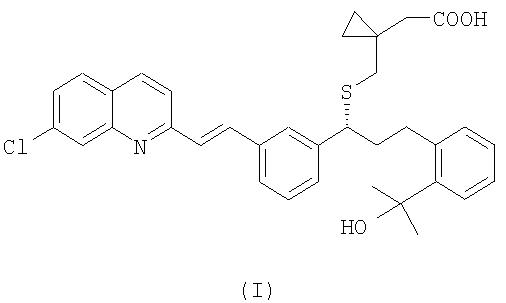
1. Способ получения монтелукаста формулы (I):
или его фармацевтически приемлемой соли или сольвата, включая гидрат, в котором вначале осуществляют взаимодействие соединения формулы (IV):
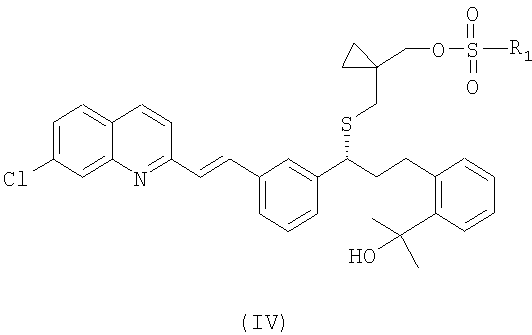
где R1 выбран из группы, включающей (С1-С4)-алкил, фенил и фенил, одно- или двузамещенный (С1-С4)-алкильным радикалом, с цианидом щелочного металла, с образованием и последующим выделением соединения формулы (III), в виде свободного основания или его соли:
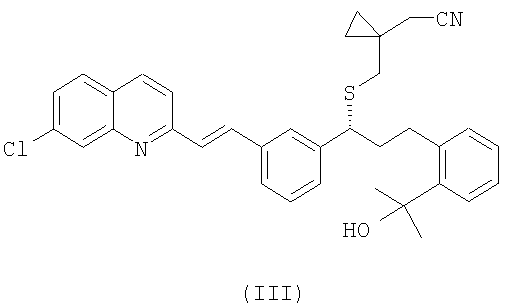
затем необязательно проводят гидролиз полученного на предшествующей стадии соединения формулы (III) с образованием и последующим выделением из реакционной среды соединения формулы (II):
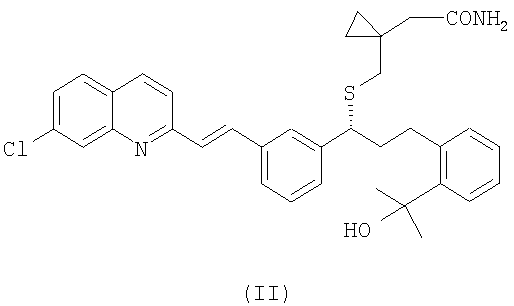
далее соединение, полученное на любой из предшествующих стадий, подвергают гидролизу с образованием монтелукаста формулы (I), и после этого образовавшийся монтелукаст формулы (I) необязательно обрабатывают фармацевтически приемлемым основанием для получения соответствующей соли.
2. Способ по п.1, в котором соединение формулы (III) выделяют в виде свободного основания.
3. Способ по п.1 или 2, в котором R1 выбран из группы, включающей метил, фенил и 4-метилфенил.
4. Способ по п.1, в котором взаимодействие соединения формулы (IV) с цианидом щелочного металла осуществляют в присутствии катализатора фазового перехода.
5. Способ по п.1, в котором реакцию гидролиза осуществляют в присутствии основания.
6. Способ по п.5, в котором реакцию гидролиза осуществляют в смеси, включающей воду и органический растворитель, необязательно в присутствии катализатора фазового перехода.
7. Способ по п.1, в котором для получения соединения формулы (IV) осуществляют взаимодействие соединения формулы (V):
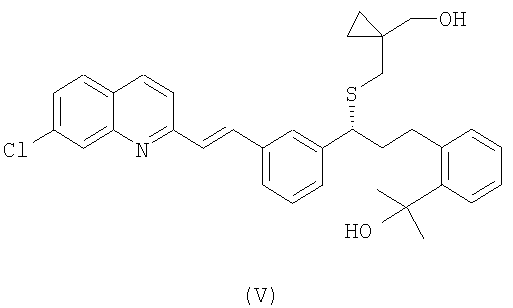
с сульфонилхлоридом формулы Cl-SO2-R1, где R1 имеет значение, как определено в п.1.
8. Способ по п.7, в котором для получения соединения формулы (V) осуществляют взаимодействие соединения формулы (VI):
с реагентом Гриньяра, выбранным из группы, включающей метиллитий и галогенид метилмагния, необязательно, в присутствии хлорида церия, с последующим выделением указанного соединения формулы (V) в виде свободного основания или в виде его соли.
9. Способ по п.8, в котором соединение формулы (V) выделяют в виде свободного основания.
10. Способ по п.8, в котором для получения соединения формулы (VI) осуществляют взаимодействие соединения формулы (VII):
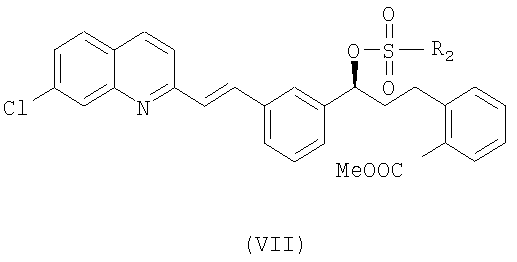
где R2 выбран из группы, включающей (С1-С4)-алкил, фенил и фенил, одно или двузамещенный (С1-С4)-алкильным радикалом с соединением формулы (VIII):
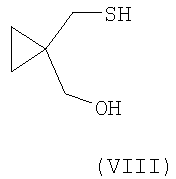
в присутствии основания, с последующим выделением указанного соединения формулы (VI) в виде свободного основания или в виде его соли.
11. Способ по п.10, в котором соединение формулы (VI) выделяют в виде свободного основания.
12. Способ по п.10, в котором соединение формулы (VI) выделяют в виде соли щавелевой кислоты.
13. Способ по п.10, в котором R2 выбран из группы, включающей метил, фенил и 4-метилфенил.
14. Соединение, представленное формулой (IV):
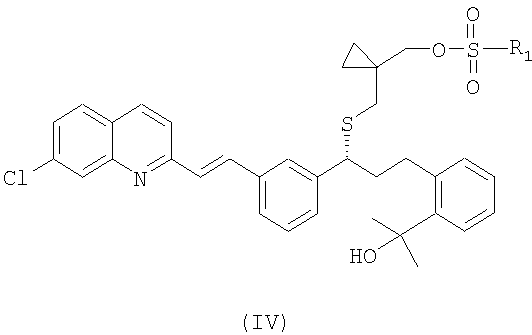
где R1 выбран из группы, включающей (С1-С4)-алкил, фенил и фенил, одно- или двузамещенный (С1-С4)-алкильным радикалом.
15. Соединение по п.14, в котором R1 выбран из группы, включающей метил, фенил и 4-метилфенил.
16. Соединение, представленное формулой (V):
или его соль.
17. Соединение по п.16, в котором указанная соль представляет собой цитрат.
18. Соединение, представленное формулой (VI):
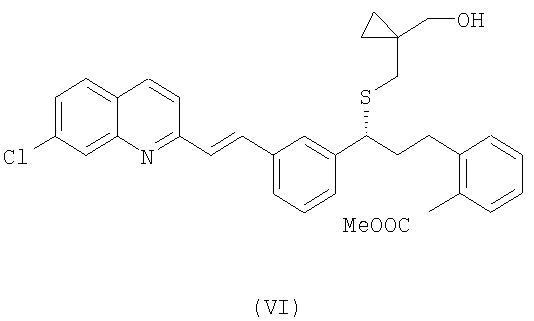
или его соль.
19. Соединение по п.18, в котором указанная соль представляет собой оксалат.
20. Соль соединения, представленного формулой (III):
которая выбрана из группы, включающей оксалат, L-(-)-малат, L-(-)-тартрат, малеат, фумарат, сукцинат, бензоат, 4-гидроксиманделат, цитрат, бензолсульфонат и манделат.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТОВ ЛЕЙКОТРИЕНА, ДИЦИКЛОГЕКСИЛАМИНОВАЯ СОЛЬ, СПОСОБЫ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ НАТРИЕВОЙ СОЛИ И 1-/МЕРКАПТОМЕТИЛ/- ЦИКЛОПРОПАНУКСУСНОЙ КИСЛОТЫ, КРИСТАЛЛИЧЕСКОЕ СОЕДИНЕНИЕ | 1994 |
|
RU2140909C1 |

