Эта заявка является частичным продолжением заявки U.S.S.N. 08/174931, поданной 28 декабря 1993, которая включена сюда полностью.
Предшествующий уровень техники.
Лейкотриены составляют группу локально действующих гормонов, получаемых в живых системах из арахидоновой кислоты. Основными лейкотриенами являются Лейкотриен B4 (сокращенно обозначаемый как LTB4), LTC4, LTD4 и LTF4. Биосинтез этих лейкотриенов начинается с действия фермента 5-липоксигеназы на арахидоновую кислоту с получением эпоксида, известного как Лейкотриен A4 (LTA4), который превращается в другие лейкотриены с помощью последовательных ферментных стадий. Дополнительные детали о биосинтезе, а также о метаболизме лейкотриенов можно найти в книге Leutrienes and Lipoxygenases, под редакцией J. Rokach, Elsevier, Amsterdam (1989). В этой книге под редакцией Rokach также обсуждаются вопросы действия лейкотриенов в живых системах и их вклада в различные состояния заболеваний.
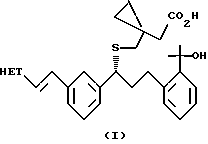
Недавно ряд соединений формулы (I), в которой A представляет собой произвольно (необязательно) замещенный гетероцикл, и их фармацевтически приемлемых солей, были раскрыты как антагонисты лейкотриенов и ингибиторы биосинтеза лейкотриенов.

EP 480717 раскрывает соединения формулы (I), в которой А представляет собой произвольно замещенный хинолин; в частности, раскрывает соединение, в котором A представляет 7-хлор-2-хинолинил. Патент США 5270324 раскрывает два соединения формулы (I), в котором A представляет 6-фтор- или 6,7-дифтор-2-хинолинил. В одновременно рассматриваемой заявке U.S.S.N. 994869, поданной 22 декабря 1992 (опубликованная заявка на ЕП 604114), раскрываются соединения, в которых A представляет галозамещенный тиено-[2,3-b]пиридин, в частности 2,3-дихлортиено[2,3-b]пиридин-5-ил.
Сообщаемые синтезы соединений формулы (I) протекают через получение соответствующих метиловых (сложных) эфиров и включают взаимодействие метил 1-(меркаптометил)циклопропанацетата с мезилатом, примером которого служит соединение формулы (III), генерированного in situ. Метиловые сложные эфиры соединений формулы (I) гидролизуются до свободных кислот, и последние непосредственно превращаются в соответствующие соли натрия. Этот способ, в частности, не приемлем для крупно-масштабного производства, поскольку он требует утомительной хроматографической очистки промежуточных метиловых эфиров и/или конечных продуктов, и выходы продуктов по этому способу низки. Кроме того, конечные продукты, такие как соли натрия, получают в виде аморфных твердых веществ, которые часто не являются идеальными для технологии приготовления лекарственного средства.
Таким образом, существует необходимость в разработке эффективного синтеза соединений формулы (I), что способно увеличить масштаб производства соединения, и обеспечит повышенный суммарный выход продукта, и обеспечит продукт-соли натрия в кристаллической форме.
King et al., J. Org. Chem., 1993, 58:3731-3735, сообщает о синтезе L-699392 путем следующей последовательности превращений:
Сущность изобретения
Данное изобретение относится к улучшенному способу получения соединений формулы (I); к улучшенному способу получения предшественника 1-(меркаптометил)циклопропануксусной кислоты и к промежуточным соединениям.
Соединения формулы (I) являются антагонистами лейкотриенов и полезными средствами для лечения астмы, а также других состояний, опосредованных лейкотриенами, таких как воспаление или аллергии.
Фиг. 1 представляет картину рентгеновской порошковой дифракции соединения Примера 6.
Фиг. 2 представляет картину рентгеновской порошковой дифракции соединения Примера 7.
Фиг. 3 представляет картину рентгеновской порошковой дифракции соединения Примера 8.
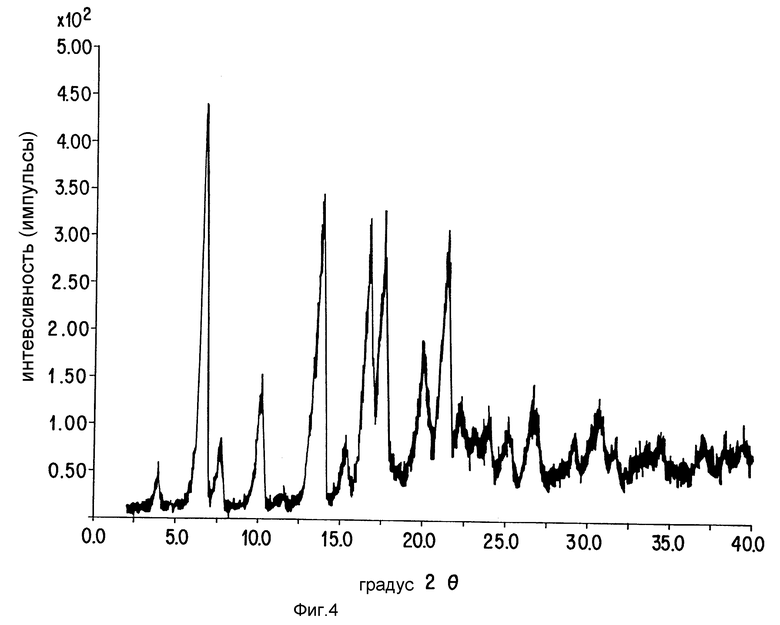
Фиг. 4 представляет картину рентгеновской порошковой дифракции соединения Примера 10.
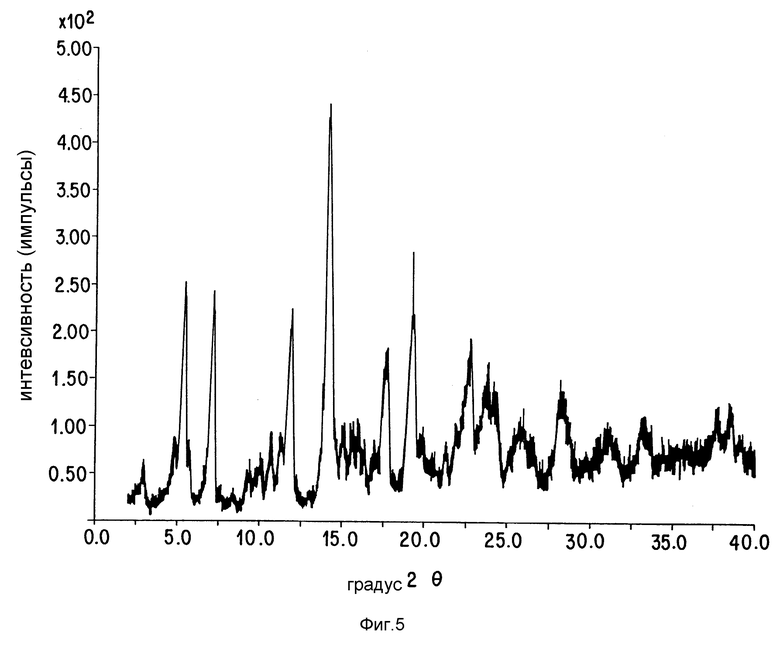
Фиг. 5 представляет картину рентгеновской порошковой дифракции соединения Примера 11.
Фиг. 6 представляет картину рентгеновской порошковой дифракции соединения Примера 13.
Детальное описание изобретения
Целью данного изобретения является разработка способа получения соединения формулы (I) или его натриевой соли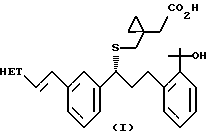
в которой НЕТ представляет 7-хлорхинолин-2-ил или 6,7-дифторхинолин-2-ил, который включает: генерирование динатриевого дианиона 1-(меркаптометил)циклопропануксусной кислоты; взаимодействие указанного аниона с соединением формулы (II)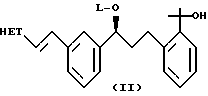
где НЕТ такой, как определено ранее, и L-арилсульфонил или алкилсульфонил. Предпочтительно, НЕТ-7-хлорхинолин-2-ил и L-метансульфонил. В предпочтительном варианте воплощения изобретения способ дополнительно включает: превращение соединения формулы (I) в соль дициклогексиламина; и превращение соли дициклогексиламина соединения формулы (I) в соответствующую соль натрия.
Другая цель данного изобретения - дициклогексиламиновая соль соединения формулы (I). Дициклогексиламиновую соль легко выделяют в кристаллической форме и преимущественно используют как средство для очистки соединения формулы (I), и для получения кристаллической натриевой соли соединения формулы (I). Соответственно другой целью данного изобретения является разработка способа получения кристаллической натриевой соли соединения формулы (I), который включает: обработку дициклогексиламиновой соли соединения формулы (I) кислотой; обработку полученного таким образом продукта источником иона натрия; кристаллизацию натриевой соли соединения формулы (I). В предпочтительном варианте воплощения указанной кислотой является уксусная кислота и указанную кристаллизацию осуществляют из смеси толуол/ацетонитрил.
Данное изобретение также обеспечивает соединение 1-(меркаптометил)циклопропануксусную кислоту, и ее соли, предпочтительно дилитиевую соль. В еще одном аспекте данного изобретения обеспечивается способ получения 1-(меркаптометил)циклопропануксусной кислоты, который включает: получение раствора 1-(ацетилтиометил)циклопропанацетонитрила в органическом растворителе; обработку указанного раствора водным раствором основания с получением двухфазной системы. В предпочтительном варианте воплощения указанным основанием является гидроксид натрия.
Еще одним аспектом данного изобретения является обеспечение кристаллических метансульфонатов формулы (III)
где НЕТ как определен ранее для формулы (I).
Обозначения
AcS (АсТ) = ацетилтио
DСНA (ДЦГА) = дициклогексиламино
DMF (ДМФ) = диметилформамид
DSC (ДСК) = дифференциальная сканирующая калориметрия
HOAc (УК) = уксусная кислота
IPAc (ИПАс) = изопропилацетат
MsCI (McCI) = метансульфонил хлорид = мезил хлорид
RT (КТ) = комнатная температура
THF (ТГФ) = тетрагидрофуран
"Арилсульфонил" означает бензолсульфонильные группы, обычно используемые для превращения гидрокси в отщепляемую группу, и включает замещенный бензолсульфонил, такой как толуолсульфонил.
"Алкилсульфонил" означает низший алкансульфонил, имеющий от одного до четырех углеродных атомов, такой как метансульфонил.
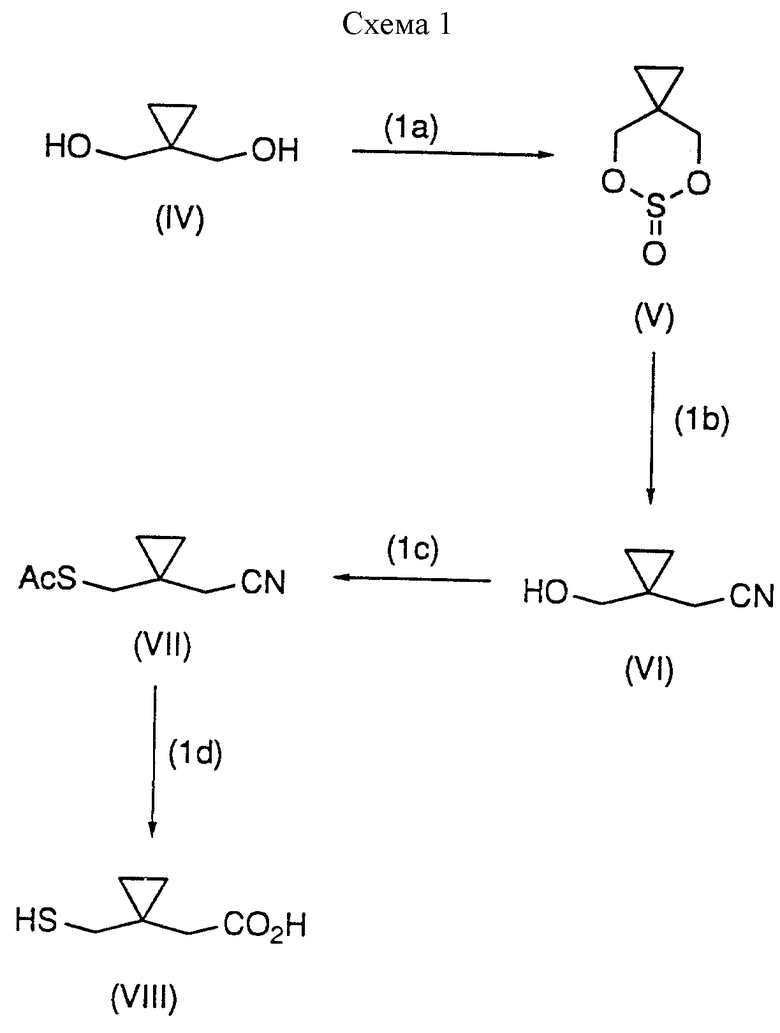
До более детального описания изобретения сначала кратко излагается общая последовательность реакций синтеза соединений формулы (I). Последовательность реакций, исходя из известных веществ, иллюстрируется в Схемах 1-3 (см. в конце описания).
Схема 1 иллюстрирует получение боковой цепи предшественника-тиометилциклопропануксусной кислоты. В стадии (1a), 1,1-циклопропандиметанол (IV) превращают в соответствующий циклический сульфит (V), используя тионил хлорид, и в присутствии основания, такого как диизопропилэтиламин. Реакцию проводят в инертном органическом растворителе, например, галогенированном углеводороде, таком как дихлорметан, или в ароматическом углеводороде, таком как толуол. Реакцию завершают добавлением тионил хлорида.
В стадии (1b) циклический сульфит (V) обрабатывают каталитическим количеством иодида натрия и цианида натрия, получая соответствующий гидрокси-нитрил (VI). Реакцию проводят в смеси диметилформамид/толуол или диметилформамид/изопропил ацетат при температуре, находящейся в диапазоне от около 65 до около 90oC. Предпочтительно, температура реакции составляет около 70oC.
В стадии (1c), сначала гидрокси-нитрил (VI) превращают в его мезилат, используя метансульфонил хлорид, и в присутствии третичного аминового основания, такого как диизопропилэтиламин, триэтиламин, и тому подобное. Затем мезилат обрабатывают тиоацетатом калия, получая 1-(ацетилтиометил)циклопропанацетонитрил (VII). Альтернативно, мезилат обрабатывают тиолуксусной кислотой в присутствии основания, такого как триэтиламин, получая (VII).
В стадии (1d), 1-(ацетилтиометил)циклопропанацетонитрил (VII) превращают в 1-(меркаптометил)циклопропануксусную кислоту (VIII) в бифазной системе растворителя. Эта стадия описана детально позже в описании.
Последовательность реакций Схемы 1 может быть проведена по следующей методике.
Конверсия в циклический сульфит (V) может сопровождаться взаимодействием диола (IV) с диизопропилсульфитом [(CH3)CH]2SO3, который, в свою очередь, получают из тионил хлорида и изопропанола. Реакцию диола и диизопропилсульфита осуществляют в инертном органическом растворителе, таком как диметилформамид/толуол, и в присутствии каталитического количества основания, такого как т-бутоксид натрия. Реакционный раствор предпочтительно сушат (до KF<100 мкг/мл до добавления основания. Получаемый в реакции изопропанол удаляют путем дистилляции, сдвигая реакцию в сторону образования продукта. К раствору циклического сульфита добавляют цианид натрия и каталитическое количество иодида натрия, и реакцию проводят при повышенной температуре, например, при около 70oC, получая гидрокси-нитрил- Соединение (VI). Цианид натрия и иодид натрия предпочтительно сушат до использования.
Гидрокси-нитрил (VI) в смеси толуол/ДМФ превращают в соответствующий мезилат, как описано выше. Отношение толуол/ДМФ для мезирования предпочтительно составляет свыше 1,9:1; обычно используемое отношение составляет около 2,1-2,4: 1. Замещение мезиловой группы тоолуксусной кислотой в присутствии основания, такого как триэтиламин, приводит к получению Соединения (VII), которое затем гидролизуют, как описано позже, получая тиоловую кислоту (VIII).
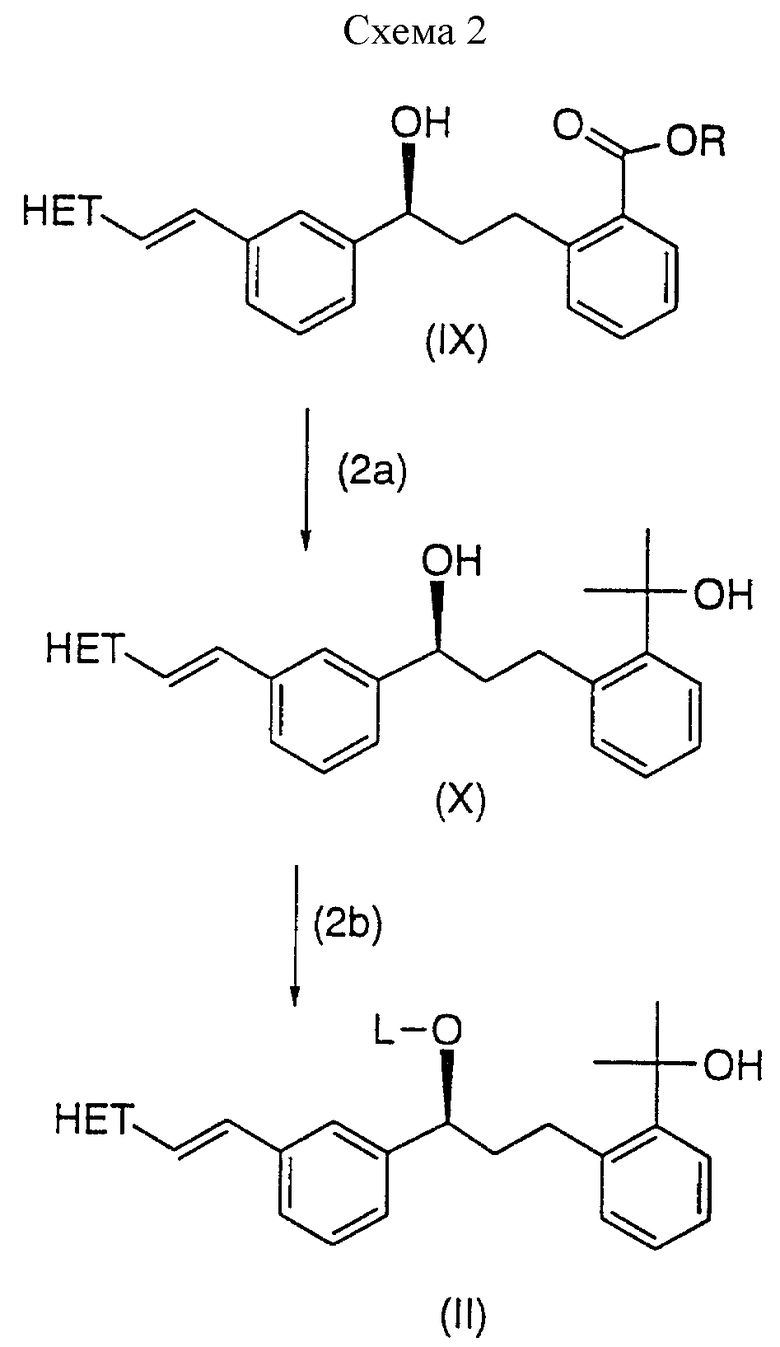
Схема 2 иллюстрирует получение части "главной цепи" соединений формулы (I). В схеме 2, R - низшая алкильная группа, такая как метил или этил; L - арилсульфонил или алкилсульфонил, например, толуолсульфонил или метансульфонил. Предпочтительно, R - метил и L - метансульфонил. В стадии (2a), гидрокси сложный эфир (IX) превращают в диол (X), используя реактив Гриньяра, такой как метилмагний хлорид, и в присутствии хлорида церия. Молярное отношение хлорида церия к метилмагний хлориду (CeCl3:CH3MgCl) может быть от около 1: 1 до около 1: 5, и предпочтительно от около 1:4 до около 1:5; молярное отношение гидрокси эфира (IX) и хлорида церия может варьироваться от около 1: 0,25 до около 1:1, и предпочтительно в диапазоне от около 1:0,5 до около 1: 1. Реакцию проводят в безводных условиях, предпочтительно, используя предварительно высушенные хлорид церия, гидрокси-эфир (IX), и растворители. Реакцию осуществляют в инертном органическом растворителе, таком как ТГФ/толуол при температуре в диапазоне от около -5 до около 5oC. Реакционный раствор, содержащий диол (X), можно сконцентрировать и использовать в следующей стадии, или диол (X) может быть кристаллизован из ароматического растворителя, таком как толуол, и углеводородного растворителя, таком как гексан или гептан, в соотношении от около 1:1 до около 1:3.
В стадии (2b) используют метансульфонил хлорид, чтобы превратить диол (X) в мезилат (II). Эту реакцию детально описывают в описании.
Схема 3 иллюстрирует один аспект данного изобретения, относящийся к улучшенному способу получения соединения формулы (I). В стадии 3(a) дилитиевую соль 1-(меркаптометил)цикло-пропануксусную кислоту (VIIIa) подвергают взаимодействию с сульфонатом формулы (II). Таким образом 1-(меркаптометил)циклопропануксусную кислоту (VIII) сначала превращают в дилитиевый дианион путем контактирования первого с основанием лития, таким как н-бутиллитий, в гексане или гептане, и т.п. Реакцию проводят в инертном органическом растворителе, такой как ТГФ, толуол или их смесь, и при температуре ниже 0oC, обычно при около -5oC или ниже.
Затем к раствору дилитий дианиона добавляют сульфонат (II). Сульфонат может быть добавлен непосредственно в виде твердого вещества или в растворе в инертном органическом растворителе, таком как ТГФ или толуол, предпочтительно ТГФ. Поскольку сульфонат (II) имеет ограниченную стабильность в растворе, раствор сульфоната предпочтительно получают непосредственно перед добавлением к раствору дианиона, и в любом случае его лучше всего использовать в пределах около 30 минут.
Реакционную смесь выдерживают при температуре ниже около 0oC, обычно при около -5oC до тех пор, пока реакция не завершится, обычно реакция завершается в пределах около 10 часов. Реакционный раствор, содержащий требуемый продукт, затем обрабатывают растворимой в воде карбоновой кислотой, например уксусной кислотой, щавелевой кислотой, винной кислотой и т.п., получая свободную кислую форму соединения формулы (I); предпочтительной карбоновой кислотой является винная кислота.
В предпочтительном варианте воплощения вышеупомянутое полученное соединение формулы (I) превращают в соль дициклогексиламина (ДЦГА). Полученный таким образом дициклогексиламин добавляют к раствору соединения формулы (I) в этилацетате, с последующим добавлением гексанов для осуществления кристаллизации соли дициклогексиламина. Предпочтительное отношение этилацетат/гексаны составляет от около 1:1 до около 1:2. Предпочтительно, для ускорения образования кристаллов, к этилацетат/гексановому раствору добавляют затравочный кристалл соли дициклогексиламина. Соли дициклогексиламина кристаллизуются в виде игл.
Вторую кристаллическую форму ДЦГА соли соединения формулы (I) можно получить кристаллизацией из смеси толуол/гептан. Таким образом, свободную кислоту соединения формулы (I) в органическом растворителе, таком как ТГФ, обрабатывают дициклогексиламином; затем добавляют толуол и раствор концентрируют, удаляя ТГФ. После разбавления дополнительным толуолом к раствору толуола добавляют гептан. Отношение толуол/гептан составляет от около 2:1 до около 3: 1. Кристаллизацию можно ускорить добавлением частичек соли ДЦГА, предварительно полученных из смеси толуол/гептан.
Кристаллическая ДЦГА соль соединения формулы (I), в которой НЕТ представляет собой 7-хлорхинолин-2-ил, полученная из смеси толуол-гептан, (Форма B), отличается от ранее упомянутой формы (Форма A), полученной из смеси этилацетат/гексаны. Форма A и Форма B дают различные рисунки рентгеновской порошковой дифракции представленные на фиг. 2 и 6 соответственно. Установлено, что форма B является более термодинамически стабильной полиморфной формой, поскольку она имеет более высокую точку плавления и менее растворима, чем форма A, в различных растворителях при комнатной температуре. Кривая дифференциальной сканирующей калориметрии (ДСК) формы В, при скорости нагревания 10o/мин, показывает единственную эндотерму плавление-разложение с экстраполированной на начало температурой прибл. 139oC, максимальной температурой 143oC, и ассоциированной теплотой (теплотой ассоциации) прибл. 71 Дж/г (J/g). При тех же самых условиях ДСК-кривая формы A показывает единственную эндотерму плавление-разложение с экстраполированной на начало температурой прибл. 117oC, максимальной температурой 124oC и теплотой ассоциации 61 Дж/г. При 25oC растворимость формы A в толуоле составляет 13,5±0,6 мг/мл и 7,5±0,1 мг/мл в этилацетате; растворимость формы B в толуоле составляет 18,7±0,2 мг/мл и 6,6±0,1 мг/мл в этилацетате.
Способность к легкому выделению кристаллической соли дициклогексиламина в любой форме представляет простой и эффективный способ очистки соединения формулы (I), тем самым позволяя обойти потребность в утомительной очистке с помощью хроматографии и добиться более высоких выходов продукта.
Возвратимся теперь к различным другим аспектам данного изобретения. Другой аспект данного изобретения обеспечивает способ получения кристаллической натриевой соли соединения формулы (I) из соответствующей соли дициклогексиламина. Так, соль дициклогексиламина (1a) добавляют к хорошо перемешиваемой смеси органического растворителя и воды. Органическим растворителем может быть, например, ароматический углеводород, предпочтительно толуол; сложный эфир, такой как этилацетат; простой эфир, такой как ТГФ; или их смеси, например, толуол/ТГФ. К этой суспензии при комнатной температуре добавляют растворимую в воде органическую кислоту, например уксусную кислоту, щавелевую кислоту, винную кислоту, и т.п.; предпочтительно используют уксусную кислоту. Органический слой, содержащий свободную кислоту, затем обрабатывают источником иона натрия, например гидроксидом натрия, который используют в приблизительно эквимолярном количестве к свободной кислоте.
Раствор натриевой соли в органическом растворителе сушат с помощью азеотропной перегонки под вакуумом и концентрируют. Ацетонитрил добавляют при повышенной температуре от около 35oC до около 45oC, обычно при около 40oC. Для ускорения кристаллообразования в раствор вводят зародыши заранее полученных кристаллов натриевой соли. Как только образуется хороший слой зародышей кристаллизации (в пределах 2 часов, около 30 - 90 минут при 40oC), добавляют еще ацетонитрил, чтобы получить конечное отношение ацетонитрил/толуол от около 2:1 до около 9:1, предпочтительно около 3:1. Спустя приблизительно 8 - 12 часов при 40oC кристаллическую натриевую соль соединения формулы (I) собирают.
Разработан полунепрерывный способ кристаллизации натриевой соли соединения формулы (I) и он описан в разделе Примеры.
В другом аспекте данного изобретения обеспечивается новое соединение, 1-(меркаптометил)циклопропануксусная кислота (VII), и его соли, предпочтительно дилитиевая соль; и способ его получения. Соединение (VIII) получают из 1-(ацетилтиометил)циклопропанацетонитрила (VII) при помощи катализируемого основанием гидролиза. Установлено, что гидролиз, проводимый в воде, приводит к существенному количеству примесей. Количество примеси в реакционной смеси существенно уменьшается в том случае, когда гидролиз проводят в бифазной системе, содержащей органический растворитель и воду. При бифазном гидролизе требуемый промежуточный тиолат 1-(меркаптометил)циклопропанацетонитрил, содержится в водном слое, в то время как нейтральные примеси остаются в органическом слое, и в соответствии с этим легко удаляются. Кроме того, неочищенный (VII) может быть использован в бифазном гидролизе, избегая тем самым потребность в очистке (VII) с помощью хроматографии.
В соответствии с этим Соединение (VII) растворяют в органическом растворителе; пригодными растворителями являются, например, ароматические углеводороды, такие как толуол, ксилолы, и т.п.; предпочтительный растворитель - толуол. Раствор Соединения (VII) в органическом растворителе обрабатывают водным раствором основания, такого как гидроксид натрия. Реакцию можно проводить при температуре, начиная от комнатной температуры и кончая температурой кипения реакционной смеси. Гидролиз до Соединения (VIII) обычно завершается в пределах нескольких дней при комнатной температуре, обычно около 6 дней; и нескольких часов при кипении флегмы.
Предпочтительно, двухфазную смесь выдерживают при комнатной температуре до тех пор, пока исходное вещество (VII), в основном, не превратится в промежуточный натрий тиолат 1-(меркаптометил)-циклопропанацетонитрил, обычно от около 6 до 18 часов. Водный раствор, содержащий промежуточный продукт, отделяют от органического слоя, содержащего нежелательные примеси. Водный раствор выдерживают при повышенной температуре вплоть до точки кипения, чтобы завершить превращение в (VIII), например, при 80oC в течение от около 12 до 16 часов.
Затем раствор подкисляют, получая (VIII) в виде свободной кислоты, и экстрагируют органическим растворителем, таким как толуол или гептан, затем концентрируют. Концентрированный раствор (VIII) в толуоле стабилен в течение нескольких месяцев, или Соединение (VIII) может быть кристаллизовано из углеводородных растворителей, таких как гексаны, гептан, пентан или т.п., чтобы избавиться от некоторых примесей, присутствующих в исходном веществе (VII). В соответствии с этим смесь Соединения (VIII) в гептане нагревают до 34oC до полного растворения соединения и дают возможность медленно охлаждаться до около 25oC. Введение кристаллов Соединения (VIII) в качестве зародышей кристаллизации может быть использовано для ускорения кристаллообразования. Смесь охлаждают до около -5oC на протяжении около 3 часов для кристаллообразования.
Другой аспект данного изобретения обеспечивает кристаллические мезилаты формулы (III):
где НЕТ такой как определен ранее при формуле (I); предпочтительно НЕТ представляет собой 7-хлор-2-хинолинил.
Мезилат (III) получают из соответствующего диола (X). Реакцию проводят в инертном органическом растворителе, таком как толуол, или толуол и ацетонитрил или ТГФ. Другими приемлемыми растворителями являются, например, ДМФ или ДМФ/ацетонитрил. Реакцию проводят в присутствии третичного аминового основания, такого как диизопропилэтиламин, и при температуре ≤0oC, предпочтительно при между около -25oC до около -15oC. Реакция обычно завершается в пределах около 5 часов. Предпочтительными условиями для селективного мономезилирования во вторичной гидрокси группе являются растворитель толуол: ацетонитрил с предпочтительным соотношением от около 1:2 до около 1:3; диапазон температур реакции от около -25oC до около -15oC; и диизопропилэтиламин в качестве основания.
Мезилат (III) имеет ограниченную стабильность в растворе; поэтому его предпочтительно выделяют в твердой форме, которая при хранении при комнатной температуре от около -15oC и ниже достаточно стабильна в течение шести месяцев или более. Кристаллизацию мезилата (III) предпочтительно осуществляют при температуре 5oC или ниже из толуол/ацетонитрил, предпочтительно при соотношении от около 1:2 до около 1:3. Выделение мезилата (III) облегчает взаимодействие с тиоловой кислотой (VIIIa), осуществляемое в крупномасштабном способе производства соединений формулы (I).
Следующие примеры предназначены для более полной иллюстрации данного изобретения. Следует иметь в виду, что примеры ни коим образом не ограничивают объема изобретения, определенного в формуле изобретения.
Пример 1
1,1-Циклопропандиметанол циклический сульфит
Способ A: В круглодонную колбу, снабженную мешалкой, термопарой, входом для азота и шприцевым насосом, помещают дихлорметан (645 мл) и 1,1-циклопропандиметанол (10,64 г; 97,93 ммол). Смесь перемешивают в течение 10 минут, чтобы обеспечить полное растворение. Добавляют N, N-диизопропилэтиламин (34,21 мл, 195,86 ммол), и раствор охлаждают до 0-5oC. Через тефлоновую иглу при помощи шприцевого насоса добавляют под поверхность раствора тионил хлорид (7,01 мл; 96,04 ммол) на протяжении 60 минут. Реакционный раствор переносят в разделительную воронку, содержащую холодный (0-5oC) фосфатный буфер (pH 7,2; 650 мл). После уравновешивания слои разделяют. Раствор продукта в дихлорметане промывают 2 вес.% раствором хлорида натрия (650 мл), и затем раствор продукта азеотропно сушат и концентрируют при 35 - 40oC при атмосферном давлении до 50 мл. Выход названного соединения = 13,07 г (90%).
Способ B
25 мл градуированный цилиндр, снабженный шлифом, загружают 7,4 мл (97,9 ммол) тионила хлорида и затем разбавляют толуолом до объема 21 мл.
В 1 л круглодонную колбу, снабженную подвесной (проходящей сверху через горловину колбы) мешалкой, термопарой, входом для азота и шприцевым насосом, помещают толуол (636 мл), 1,1-циклопропандиметанол (10,00 г; 97,9 ммол) и N, N-диизопропилэтиламин (32,41 мл; 186,1 ммол). Двухфазную смесь энергично перемешивают при 22oC. Через тефлоновую иглу при помощи шприцевого насоса добавляют под поверхность раствор/раствор тионил хлорид: толуол (21 мл; 97,9 ммол) на протяжении 90 минут, поддерживая температуре реакции ≤40oC. После завершения добавления тионил хлорида, реакционную смесь перемешивают в течение еще 6 - 12 часов для того, чтобы обеспечить максимальное превращение в циклический сульфит. Реакционную смесь переносят в делительную воронку, содержащую холодный (0-5oC) фосфатный буфер (pH 7,2; 650 мл). После уравновешивания слои разделяют и раствор продукта в толуоле промывают 2 вес.% раствором хлорида натрия (650 мл). Раствор продукта затем азеотропно сушат и концентрируют при 40 - 45oC/70 торр, до 70 мл. Выход названного соединения = 12,33 г (85%).
Пример 2
1-(Гидроксиметил)циклопропанацетонитрил
Способ A:
250 мл круглодонную колбу, снабженную подвесной мешалкой, термопарой, дистилляционной головкой и приемником, загружают раствором циклического сульфита Примера 1 в дихлорметане (61 мл; 158,9 мг/мл; 9,69 г). Раствор концентрируют до прибл. 20 мл путем дистилляции при атмосферном давлении. К содержимому добавляют изопропил ацетат (2 x 30 мл) и дистилляцию продолжают до конечного объема 13 мл. Диметилформамид (21 мл) добавляют к раствору при > 55oC и раствор охлаждают до комнатной температуры.
250 мл круглодонную колбу, снабженную подвесной мешалкой, термопарой, обратным холодильником и входом для азота, загружают 40 мл вышеупомянутого раствора циклического сульфита (9,28 г; 62,6 ммол) в ДМФ:ИПАс (4:1). Цианид натрия (4,61 г; 94 ммол) и иодид натрия (3,75 г; 25,0 ммол) добавляют при комнатной температуре. Реакционную смесь нагревают до 70±3oC и выдерживают при этой температуре до тех пор, пока реакция не завершится. Реакционной смеси дают возможность охладиться до комнатной температуры и разбавляют холодным (0 - 5oC) изопропил ацетатом (187 мл). Темно-желтую суспензию (218 мл) переносят в делительную воронку, содержащую холодный (0 - 5oC) 1,0 М гидроксид натрия (107 мл). После уравновешивания слои разделяют. Органический слой промывают соляным раствором (53 мл). Водный слой вновь экстрагируют холодным (0 - 5oC) изопропил ацетатом (107 мл), и органический слой промывают соляным раствором (27 мл). Два органических слоя объединяют, получая раствор названного соединения с концентрацией 17,5 мг/мл. Выход названного соединения = 5,03 г; 72,2%.
Способ B
12 л 3-горлую круглодонную колбу, снабженную подвесной мешалкой, термопарой, дистилляционной головкой и 3 л приемной колбой, загружают раствором циклического сульфита Примера 1 в дихлорметане (2,0 л; 174,0 г/л; 343,6 r). Раствор концентрируют и добавляют вторую порцию циклического сульфита в дихлорметане (2,0 л; 155,9 г/л; 311,8 г) и вновь концентрируют до прибл.2,3 л путем дистилляции при атмосферном давлении. К содержимому добавляют толуол (1,7 л) и продолжают дистилляцию до конечного объема прибл. 1,7 л. Диметилформамид (1,81 л) добавляют к раствору и продолжают концентрирование в вакууме (прибл. 105 торр).
12 л 3-горлую колбу, снабженную подвесной мешалкой, термопарой, дистилляционной головкой и входом для азота, которая содержала 2,2 л вышеупомянутого раствора циклического сульфита (655,3; 4,40 ммол) в ДМФ:толуол (97: 3/об:об), при комнатной температуре загружают цианидом натрия (218,9 г; 4,40 мол) и иодидом натрия (131,9 г; 0,88 мол). Реакционную смесь нагревают до 70 ±3oC на протяжении периода времени 1 час и выдерживают при этой температуре до тех пор, пока не завершится реакция.
Реакционную смесь медленно разбавляют 6,6 л толуола, поддерживая температуру содержимого при прибл. 70oC. В помутневший окрашенный в янтарный цвет раствор загружают 80 мл воды на протяжении периода времени 30 минут. Реакционную смесь охлаждают до 27oC и реакционную колбу снабжают 2 л капельной воронкой, содержащей 2 л толуола. Реакционную смесь концентрируют под вакуумом, в то время как из капельной воронки добавляют толуол. Реакционную смесь охлаждают на протяжении ночи и затем фильтруют через средне-пористую металлокерамическую стеклянную воронку (3 л); затем осадок промывают дополнительными 2,2 л толуола. Выход названного соединения составлял 87,5%.
Пример 3
1-(Ацетилтиометил) циклопропанацетонитрил
Способ A:
500 мл круглодонную колбу, снабженную подвесной мешалкой, термопарой, дистилляционной головкой и приемной колбой, загружают раствором гидрокси-нитрила Примера 2 в изопропил ацетате и ДМФ (118 мл; 91 мг/мл; 10,74 г. Раствор концентрируют до прибл. 50 мл путем дистилляции при атмосферном давлении. К содержимому добавляют изопропил ацетат (200 мл) и продолжают дистилляцию до конечного объема 154 мл.
Установку для дистилляции заменяют дополнительной воронкой. Раствор охлаждают до -3±2oС и добавляют триэтиламин (17,4 мл) на протяжении 1 минуты. Мезил хлорид (8,93 мл) медленно добавляют из дополнительной воронки, поддерживая температуру содержимого ниже 0oC. Добавление занимает 30 минут. Реакционную смесь (прибл. 180 мл) переносят в делительную воронку, содержащую холодную (0 - 5oC) воду (76 мл). После уравновешивания, слои разделяют, и органический слой промывают соляным раствором (76 мл).
Раствор 1-(метансульфонилоксиметил)циклопропанацетонитрила переносят в 500 мл круглодонную колбу, снабженную подвесной мешалкой, термопарой и входом для азота. Твердый тиоацетат калия (14,28 г) добавляют к раствору при 0oC. Гетерогенную смесь нагревают до 20±2oC и выдерживают в течение от 16 часов до 18 часов.
К реакционной смеси добавляют воду (76 мл) и содержимое реакционной колбы переносят в делительную воронку. Слои разделяют и органический слой промывают соляным раствором (76 мл). Раствор названного соединения в изопропил ацетате концентрируют в вакууме (75 торр., 50oC до объема прибл. 50 мл. Добавляют толуол (3 x 75 мл) и продолжают концентрирование под вакуумом (60 торр, 50oC) до тех пор, пока не останется < 1% изопропил ацетата. Выход названного соединения = 13,12 г (81%).
Способ B
Раствор 1-(гидроксиметил)циклопропанацетонитрила (34,2 г, 0,308 мол) в толуол: ДМФ (1,9:1, 210 мл) и триэтиламин (49,4 мл, 0,354 мол) объединяют в 3-горлой, 1-литровой круглодонной колбе, снабженной мешалкой и термопарой, продувают азотом и охлаждают до -15oC. По каплям на протяжении 0,5 часа добавляют мезилхлорид (26 мл), поддерживая температуру ниже 5oC. По возможности быстрее, последовательно добавляют этанол (77 мл), триэтиламин (86 мл, 0,616 мол) и тиолуксусную кислоту (26,4 мл). Смесь удаляют из охлаждающей бани и нагревают до 35oC. Эту температуру поддерживают до тех пор, пока не останется < 1% мезилата, около 7 часов. Добавляют воду (250 мл) и смесь встряхивают. Фазы разделяют водную фазу, повторно экстрагируют толуолом (250 мл), и органические фазы объединяют, получая названное соединение (48,3 г при 103 мг/мл, выход 93%, чистота, около 91%).
Пример 4
1-(Меркаптометил)циклопропануксусная кислота
1 л круглодонная колба, снабженная подвесной мешалкой, термопарой, дистилляционной головкой и приемной колбой, загружают раствором 1-(ацетилтиометил)циклопропан-ацетонитрила в толуоле (248,2 мл; 16,93 г; 100,0 ммол). Раствор концентрируют под вакуумом (75 торр, 50oC) до объема прибл. 100 мл. Установку для дистилляции удаляют, раствор охлаждают в атмосфере азота до 20 - 25oC, и добавляют водный 5N гидроксид натрия (100 мл; 500 ммол). Двухфазную смесь энергично перемешивают при 20 - 25oC в течение 16 - 18 часов.
Водный слой переносят в 250 мл колбу, снабженную подвесной мешалкой, термопарой, входом для азота и обратным холодильником. Раствор кипятят с обратным холодильником в течение прибл. 2 часов, охлаждают до 0 - 5oC и добавляют 8N хлористоводородную кислоту (62,5 мл; 500 ммол) для того, чтобы подвести pH водной среды до 2.0. К водной суспензии при энергичном перемешивании добавляют толуол (190 мл). Двухфазную смесь переносят в делительную воронку, и слои разделяют. К водному слою добавляют толуол (100 мл), и слои разделяют. Два органических слоя объединяют и концентрируют под вакуумом (60 торр, 50oC) до 82 мл, и концентрированный раствор фильтруют. Выход названного соединения = 11,9 г (89%). Раствор названного соединения в толуоле хранят под азотом.
250 мл круглодонную колбу, снабженную подвесной мешалкой, термопарой, дистилляционной головкой и приемной колбой, загружают раствором названного соединения в толуоле (100 мл; 11,50 г; 78,66 ммол). Раствор концентрируют под вакуумом (45 торр, < 40oC) до объема прибл. 23 мл. К раствору добавляют гексан (92 мл) при 20±2oC, и в раствор вводят 10 мг зародышей кристаллизации названного соединения. Смесь выдерживают при 20±2oC в течение приб. 2 часов, чтобы получить хороший слой зародышей кристаллизации. Пробу суспензии анализируют с помощью микроскопии в поляризованном свете с целью подтверждения кристалличности твердого вещества.
Суспензию охлаждают от 0 до - 5oC и выдерживают в течение около 2 часов, затем ей дают возможность нагреться до 20±2oC и выдерживают на протяжении ночи для накопления прекрасных кристаллов. Суспензию охлаждают до -20±5oC на протяжении 3 часов и выдерживают в течение одного часа. Пробу суспензии анализируют с помощью микроскопии в поляризованном свете для подтверждения кристалличности твердого вещества. Суспензию фильтруют и осадок промывают холодными (-20±5oC) гексанами (25 мл), затем сушат при отсасывании в атмосфере азота при 20±2oC.
1H ЯМР (CDCl3 δ 11,8 (шс, 1H), 2,64 (д, 2H), 2,52 (с, 2H), 1,36 (т, 1H), 0,64-0,52 (м, 4H).
ДСК эндотерма плавления с максимумом температуры 49oC и ассоциированной теплотой (теплотой ассоциации) 122 Дж/г.
Рентгеновская порошковая диффракция* - кристалличность.
*Картины порошковой дифракции рентгеновский лучей в этом и последующих примерах получают с помощью установки APD3720 (Philips) при температуре окружающего воздуха и в атмосфере N2.
Пример 5
2-(2-(3(S)-(3-(2-(7-Хлоро-2-хинолинил)-этенил)фенил)-3- гидроксипропил)фенил)-2-пропанол
Стадия 1:
В 5 л колбе, снабженной механической мешалкой и дистилляционной головкой, суспензию гидрата метил 2-(3(S)-(3-(2-(7-хлоро-2-хинолинил) -этенил)фенил)-3-гидроксипропил)бензоата (EP 480717, Пример 146, Стадия 2) (300 г, 0,63 мол) в толуоле (3250 мл) нагревают до температуры кипения флегмы. Твердые вещества растворялись с образованием желто-оранжевого раствора. Азеотроп толуол-Н2О (250 мл) удаляют дистилляцией при атмосферном давлении (темп. 84-110oC, T= 110o после удаления прибл. 200 мл). Прозрачный раствор охлаждают до 20oC KF = 76 мкг/мл.
Стадия 2:
3-горлую 12 л колбу, снабженную механической мешалкой и обратным холодильником, загружают ТГФ (2 л, безволн.) и CeCl3 (160 г, 0,65 мол, безв.) Суспензию серого цвета нагревают при кипячении с обратным холодильником в течение 3 - 5 часов, затем суспензию цвета слоновой кости-белую охлаждают до 0oC. Раствор MeMgCl (3 М в ТГФ, 1100 мл, 3,30 мол) добавляют по каплям на протяжении 30 минут к суспензии CeCl3, поддерживая T = 0±5oC. Раствор выдерживают при 0oC в течение 2 часов. Раствор гидрокси-сложного эфира в толуоле, полученного в Стадии 1, добавляют по каплям на протяжении 1,5 часов, поддерживая -1≤T≤5oC. Раствор выдерживают в течение 0,5 часа, после того как добавление завершается. Затем реакцию гасят путем осторожного добавления к 1: 12 М УК/толуол) 5 л, ea), поддерживая T≤25oC. Бледно-желтый раствор перемешивают при 20 - 25oC в течение 10 мин, затем слои разделяют.
Органический слой промывают 1 x 5 л 10% Na2CO3 последующей промывкой 1 x 5 л H2О. Органический слой (33 мг/мл названного соединения) концентрируют путем дистилляции in vacuo (100 мбар, 40oC), получая желтый раствор названного соединения (прибл. 180-190 мг/мл).
Стадия 3:
Неочищенный раствор названного соединения (диол) в ТГФ/толуол концентрируют с 23,5 мг диола/мл раствора до 253 мг диола/мл раствора путем дистилляции при атмосферном давлении (T = 84 - 110oC). Температуру раствора понижают до и поддерживают при 50oC. Внесение затравки при 50oC приводит к раствору затравки диола. Гексаны (50 мл) добавляют по каплям на протяжении 1 часа и затем в реакционную смесь снова вводят затравку. По-видимому, снова, затравка растворяется. Добавляют дополнительную аликвоту гексанов (25 мл) по каплям на протяжении 15 минут, и в этой точке в сосуде для кристаллизации начинает появляться белое твердое вещество. Кристаллизацию проводят в течение 10 минут с последующим добавлением гексанов (85 мл) на протяжении 30 минут. Кристаллизацию проводят при 50oC в течение 30 минут с последующим добавлением гексанов (160 мл) одной порцией. После 30-минутной выдержки реакционную смесь охлаждают до 25oC на протяжении 60 мин и фильтруют. Названное соединение выделяют с 89% выходом (99,0 аналитический %, чистота 99,6 вес.%).
Пример 6
2-(2-(3(S)-(3-(2-(7-Хлор-2-хинолинил)этенил)фенил)-3- метансульфонилоксипропил)фенил-2-пропанол
100 л круглодонную колбу, снабженную механической мешалкой, термопарой, и воронкой для добавления, продувают N2. Колбу загружают раствором диола в толуоле (продукт Примера 5, 17,7 л, 348,5 г/л), CH3CN (45,4 л), и диизопропилэтиламином (2720 мл). Раствор охлаждают до T=-25±3oC на CO2/метанольной бане. Метансульфонил хлорид (1140 мл) добавляют по каплям на протяжении 2,5 часов, поддерживая T = -25±2oC. После добавления мезил хлорида в реакционную смесь вводят затравку гранулированных затравочных кристаллов названного соединения (5,0 г) и выдерживают при -25oC в течение 2 часов, получая тонкую суспензию (крупные кубические кристаллы согласно микроскопии); анализ супернатанта: 21 г/л мезилат; в последующем эксперимент дает игольчатые кристаллы). Температуру понижают до -35oC на протяжении 1 часа и затем выдерживают в течение 1 часа (анализ супернатанта: 14 г/мл мезилат). Продукт изолируют фильтрацией холодной суспензии в атмосфере N2. Отфильтрованный осадок промывают холодным CH3CN (14 л, -30oC), с последующей промывкой холодным гексаном (16 л, 5oC). После промывок осадок сушат на фильтре путем пропускания N2 через осадок при 5oC в течение 20 часов. Осадок упаковывают при 5oC в двойные полипропиленовые мешки в фибровый барабан и хранят при -18oC, получая продукт в виде бледно-желтого твердого вещества (5844 г, 81% выход, из расчета на вес % чистоты). 1H ЯМР (CDCl3) δ 8,11 (м, 2H), 7,69 (м, 5H), 7,41 (м, 5H), 7,19 (м, 3H), 5,70 (дд, 1H), 3,25 (м, 1H), 3,04 (м, 1H), 2,76 (с, 3H), 2,45 (м, 1H), 1,92 (с, 1H), 1,65 (с, 6H).
Рентгеновская дифракционная картина представлена на фиг. 1. Игольчатая форма.
Пример 7
1-(((1(R)-(3-(2-(7-(хлор-2-хинолинил)этенил)фенил)-3- (2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропануксусная кислота дициклогексиламиновая соль
Стадия 1:
В 100 л реактор, оборудованный механической мешалкой, термопарой, входом для азота и воронкой для добавления помещают тетрагидрофуран (33 л) и 1-(меркаптометил)циклопропануксусную кислоту (1,317 кг, 7,938 мол). Смесь перемешивают в течение 10 минут для того, чтобы обеспечить полное растворение. Получается прозрачный, бледно-желтый раствор. Раствор охлаждают до -15±2oC и добавляют н-бутиллитий (1,56 М в гексанах, 10,5 л, 16,38 мол) на протяжении 75 минут, поддерживая температуру реакционной смеси < -5oC. Суспензию выдерживают при -5oC±2oC в течение 30 минут.
Стадия 2:
В 50 л колбу, снабженную мешалкой, термопарой и входом для азота, помещают тетрагидрофуран (20 л). Растворитель охлаждают до 0 - 5oC. Мезилат Примера 6 (4,501 кг, 7,558 мол) добавляют через воронку порошком и тетрагидрофуран (2,5 л) используют для того, чтобы промыть воронку. Смесь перемешивают в течение 15 минут для того, чтобы обеспечить полное растворение. Получают прозрачный, бледно-желтый раствор.
Стадия 3:
Раствор мезилата из Стадии 2 переносят, используя 0,25 дюймовую н.д. (o. d.) полипропиленовую трубку, под давлением азота в суспензии дианиона Стадии 1 при -5±2oC на протяжении 75 минут. Реакционный раствор выдерживают при 5±2oC в течение 8,5 часов. Реакцию гасят путем выливания прозрачного, желтого реакционного раствора в смесь этилацетата (55 л) и 10% раствора хлорида натрия (55 л). Смесь перемешивают в течение около 30 минут и затем слоям дают возможность разделиться. Получают два прозрачных слоя. Водный бросовый слой сливают. Органический слой продукта промывают 0,5 М винной кислотой (36 л), затем дважды водой (36 л каждый раз). Раствор продукта концентрируют под вакуумом до прибл. 10 л. Продукт растворяют в этилацетате (44 л), и раствору дают возможность уравновеситься до комнатной температуры (20±2oC).
Стадия 4:
В раствор свободной кислоты в этилацетате (54 л) в 2 x 100 л, 3-горлой колбе, снабженной механической мешалкой, термопарой, входом для азота и воронкой для добавления, добавляют дициклогексиламин (1,8 л). Прозрачный раствор затравливают дициклогексиламиновой солью названного соединения (14 г). Полученную смесь выдерживают в течение около часа, и в это время образуется плотная суспензия. Пробу суспензии анализируют с помощью микроскопии в поляризованном свете для подтверждения кристалличности твердого вещества. Гексан (108 л) медленно добавляют на протяжении 2 часов, поддерживая хорошее перемешивание суспензии. Суспензию выдерживают при 20±2oC на протяжении ночи. Пробу суспензии анализируют с помощью микроскопии в поляризованном свете для подтверждения кристалличности твердого вещества. Суспензию фильтруют путем отсасывания, и осадок промывают холодной (0±2oC) смесью 1:2 ацетат: гексаны (32 л). Продукт сушат под вакуумом при 40±2oC с продувкой азота.
Выход = 4,745 кг (99 A%; 96 вес.%; > 99,8% ee; 79% выход).
1H ЯМР (CDCl3) δ 8,25 (д, 1H), 7,95 (д, 1H), 7,86 (д, 1H), 7,83 (д, 1H), 7,77 (д, 1H), 7,70 (шс, 1H), 7,54 (д, 1H), 7,49 (д, 1H), 7,46 - 7,35 (м, 4H), 7,12-7,03 (м, 3H), 4,87 (с, активный H), 4,03 (дд, 1H), 3,11-3,05 (м, 3H), 2,84-2,81 (м, 1H), 2,64 (д, 1H), 2,52 (д, 1H), 2,38 (д, 1H), 2,29 (д, 1H), 2,23 (м, 1H), 2,00 (м, 4H), 1,82 (м, 4H), 1,66 (м, 2H), 1,51 (два с, 6H), 1,37-1,14 (м, 10H), 0,53-0,32 (м, 4H).
Рентгеновская дифракционная картина порошка представлена на фиг. 2.
Пример 8
Натрий 1-(((1(R)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3- (2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропан-ацетат
Толуол (1000 мл) и воду (950 мл) помещают в 12-литровый экстрактор, снабженный подвесной мешалкой, термопарой, входом для азота и воронкой для добавления. При хорошем перемешивании растворителей добавляют твердую дициклогексиламиновую соль Примера 7 (64,3 г, 82,16 ммол) через воронку порошком, и толуол используют для того, чтобы смыть оставшееся твердое вещество. К хорошо перемешиваемой суспензии добавляют уксусную кислоту (2 М, 62 мл, 124 ммол) при комнатной температуре. После приблизительно 10 минут перемешивание останавливают. Две прозрачные фазы (желтый органический слой и бесцветный водный слой) образуются, и водный бросовый слой сливают. Воду (950 мл) загружают в экстрактор, и слои тщательно перемешивают в течение приблизительно 10 минут. Перемешивание останавливают, и водный бросовый слой сливают.
К органическому слою (1270 мл), содержащему свободную кислоту, добавляют титрованный раствор гидроксида натрия в 1% водном этаноле (водный без этанола (0,486 М, 169 мл, 82,13 ммол) непрерывный струей на протяжении 10 минут при комнатной температуре в атмосфере азота. После 10-минутного выдерживания прозрачный раствор требуемой натриевой соли фильтруют через прокладку Solkafloc, используя толуол (100 мл) для переноса и промывки осадка.
Прозрачный фильтрат переносят в атмосфере азота в 3-литровую, 3-горлую колбу, снабженную подвесной мешалкой, термопарой, входом для азота и дистилляционной головкой. Раствор концентрируют под вакуумом до около 400 мл (около 40 мм Hg, ≤40oC). Дистилляционную головку заменяют на обратный холодильник и воронку для добавления. Концентрат поддерживают при 40 ±2 oC и добавляют ацетонитрил (400 мл) на протяжении 20 минут. Прозрачный раствор затравливают 0,5 г кристаллической натриевой соли, и полученную смесь выдерживают при 40oC±2oC в течение 15 минут, и в это время создается хороший слой затравки.
Ацетонитрил (400 мл) медленно добавляют на протяжении 20 минут, поддерживая температуру содержимого при 40±2oC. Белую суспензию перемешивают при 40±2oC в течение 1 часа и медленно на протяжении 20 минут добавляют ацетонитрил (400 мл). Суспензию выдерживают при 40±2oC в течение 12 часов. Пробу суспензии анализируют с помощью микроскопии в поляризованном свете для того, чтобы подтвердить кристалличность твердого вещества. Суспензию охлаждают до комнатной температуры и выдерживают при комнатной температуре в течение 1 часа. Кристаллическую натриевую соль фильтруют путем отсасывания через воронку из спеченного материала под азотом. Осадок промывают ацетонитрилом (400 мл). Осадок кристаллической натриевой соли измельчают в азотной камере с перчаткой и сушат под вакуумом в протоке азота при 40 - 45oC. Продукт (49 г, 80,59 ммол, 98% выход) упаковывают в хорошо герметизируемую бутыль коричневого цвета под азотом. Реакционную смесь и выделенный продукт всегда защищают от света.
ВЭЖХ (HPL С) анализ натриевой соли: > 99,5 A%. Хиральная чистота: 99,8% ee.
1H ЯМР (CD3OD) δ 8,23 (д, 1H), 7,95 (д, 1H), 7,83 (д, 1H), 7,82 (д, 1H), 7,75 (д, H), 7,70 (шс, 1H), 7,54 (дт, 1H), 7,46 (дд, 1H), 7,42-7,35 (м, 3H), 7,37 (д, 1H), 7,14-7,00 (м, 3H), 4,86 (с, активный H), 4,03 (дд, 1H), 3,09 (м, 1H), 2,82 (м, 1H), 2,66 (д, 1H), 2,52 (д, 1H), 2,40 (д, 1H), 2,30 (д, 1H), 2,24-2,14 (м, 2H), 1,51 (два с, 6H), 0,52-0,32 (м, 4H).
ДСК эндотерма плавления с максимумом температуры 133oC и теплотой ассоциации 25 Дж/г.
Рентгеновская дифракционная картина порошка представлена на фиг. 3.
Пример 9.
2-(2-(3(S)-(3-(2-(6,7-дифтор-2-хинолинил)этенил)фенил)-3- метансульфонилоксипропил)фенил)-2-пропанол
Чтобы получить названное соединение, следуют общим методикам, описанным в Примерах 5 и 6, за исключением того, что используют метил 2-(2-(3-(S)-(3-(2-(6,7-дифторо-2-хинолинил)этенил)фенил) -3-гидроксипропил)бензоат, в Примере 5. Стадия 1, от около 750 мл до около 1 л азеотропа толуол-H2О удаляют, и в Примере 6 ДМФ:ацетонитрил (3:1) используют в качестве растворителей.
Пример 10
Дициклогексиламиновая соль 1-(((1(R)-(3-(2-(6,7-дифтор-2-хинолинил) -этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)-метил) циклопропануксусной кислоты
Следуют общей методике Примера 7, за исключением того, что для получения названного соединения используют соединение Примера 9. Рентгеновская дифракционная картина порошка представлена на фиг. 4.
Пример 11
Натрий 1-(((1(R)-(3-(6,7-дифтор-2-хинолинил)этенил)фенил)-3- (2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропанацетат
Чтобы получить названное соединение, следуют общей методике Примера 8, за исключением того, что используют соединение Примера 10. Рентгеновская дифракционная картина порошка: представлена на фиг. 5.
Пример 12
Альтернативный способ получения 1-(меркаптометил)циклопропануксусной кислоты
Стадия 1: Диизопропил сульфит
Толуол (500 мл) и изопропанол (306 мл, 4 мол) комбинируют в атмосфере азота в 2 л колбе, снабженной капельной воронкой и термопарой. Тионил хлорид (73 мл, 1 мол) добавляют из капельной воронки на протяжении 30 минут, поддерживая температуру при 15 - 25oC. После завершения добавления реакционную смесь вакуумируют для того, чтобы удалить HCl. Энергичное выделение HCl отмечается при 150 мм.
Давление медленно понижают. Когда выделение газа прекращается, смесь концентрируют для удаления толуола и избытка изопропанола. Выход = 159 г, 95%. Триэтиламин (1 мл) добавляют для стабилизации продукта, и зарождающийся осадок отфильтровывают. Раствор используют как таковой.
Стадия 2. 1-(Гидроксиметил)циклопропанацетонитрил
Диметилформамид (225 мл) и 1,1-циклопропандиметанол (26,6 г; при 95 вес. %, фактическое количество = 25,5 г, 250 ммол) помещают в 1 л колбу, снабженную устройством для перегонки в вакууме.
ДМФ (25 мл) отгоняют при 75oC/50 торр и к оставшемуся раствору добавляют раствор диизопропил сульфита в толуоле (81,6 мл, 49,9 г, 300 ммол). Толуол (50 мл) отгоняют при 52oC/55 торр и полученный раствор имеет KF 98 мкг/мл.
T-бутилат натрия (2 М в тетрагидрофуране, 2,0 мл) добавляют и вновь начинают дистилляцию при 45oC/50 торр, при этом собирают 30 мл дистиллята. Дистилляцию продолжают, собирая 60 мл при 50 - 70oC/50 торр. T-бутилат (1,0 мл) добавляют и дистилляцию продолжают, собирая 60 мл дистиллята при 60 - 75oC/50 торр. После добавления дополнительного натрий т-бутоксида (0,5 мл) и отгонки 30 мл при 70 - 75oC/50 торр дистилляцию прекращают, и смесь выдерживают при 70oC в течение 1 часа, и затем охлаждают до комнатной температуры. Выход 1,1-циклопропандиметанол циклического сульфита = 33,0 г, 89%.
Натрий цианид (13,5 г, 275 ммол) и натрий иодид (7,5 г, 50 мол) добавляют к полученному выше раствору, и гетерогенную смесь медленно нагревают на протяжении 1 часа до 70oC, и выдерживают в течение около 40 часов при энергичном перемешивании. Толуол (400 мл) добавляют медленно при 70oC, и затем воду (6 мл) добавляют по каплям на протяжении 30 минут. Затем смесь сушат в вакууме, отгоняя 100 мл толуола; когда KF смеси достигнет 200 мкг/мл, ее охлаждают до 10oC и фильтруют. Осадок промывают толуолом (100 мл), и объединенный фильтрат содержит 21,4 г названного соединения (77% от 1,1-циклопропилдиметанола).
Стадия 3:
1-(Ацетилтиометил) циклопропанацетонитрил
Продукт Стадии 2 в толуол/ДМФ (1,9:1) (210 мл для 34,2 г продукта) и триэтиламин (49,4 мл, 0,354 мол) соединяют в 3-горлой 1-литровой круглодонной колбе, снабженной механическим перемешиванием и термопарой, продувают азотом и охлаждают до -15oC. Мезил хлорид (26 мл, 0,339 ммол) добавляют по каплям на протяжении 0,5 часов, поддерживая температуру ниже +5oC.
Триэтиламин (86 мл, 0,616 ммол) и тиолуксусную кислоту (26,4 мл, 0,40 мол) добавляют последовательно по возможности быстрее; смесь удаляют из охлажденной бани и нагревают до 35oC. Эту температуру поддерживают до тех пор, пока не останется < 1% мезилата (около 7 часов). Воду (250 мл) добавляют, смесь встряхивают и две фазы разделяют. Водную фазу экстрагируют толуолом (200 мл), и органические фазы объединяют. Объединенные органические фазы (470 мл) содержат 48,3 г (93%) требуемого продукта.
Стадия 4:
1-(Меркаптометил) циклопропануксусная кислота
Раствор продукта Стадии 3 (447 г, содержащий 48 г продукта) промывают деионизованной водой (2 x 150 мл). В 1 л трехгорлую колбу, снабженную входом для азота и механическим перемешиванием, органический слой дезоксигенируют. Дезоксигенированный 5N NaOH (284 мл) добавляют. Смесь энергично перемешивают при температуре окружающей среды в течение 6 - 10 часов до тех пор, пока не останется 2% исходного вещества. Водный слой отделяют и нагревают при 80oC в течение 12 - 16 часов до тех пор, пока не останется никакого промежуточного 1-(меркаптометил) циклопропанацетамида.
Реакционную смесь охлаждают до 25 - 30oC и 930 мл дезоксигенированного гептана добавляют. Смесь подкисляют до pH 3,5-4,0 с помощью 5 М NaHSO4 раствора на протяжении 1 часа при перемешивании и затем дают возможность нагреться до 30oC. Слои разделяют при 30oC и водный слой вновь экстрагируют 310 мл гептана. Объединенные органические слои концентрируют до 180 мл.
Смесь нагревают до 34oC, чтобы полностью солюбилизировать продукт, и затем дают возможность медленно охлаждаться до 25oC на протяжении 1 часа. Смесь затравливают при 30oC. После перемешивания при 25oC в течение 1 часа для того, чтобы обеспечить хороший затравочный слой, смесь охлаждают до - 5oC на протяжении 3 часов. После перемешивания при -5oC в течение 30 минут смесь фильтруют и промывают 20 мл холодного гептана. Названное соединение получают в виде беловатого кристаллического твердого вещества (34,3 г, 83%).
Пример 13
Дициклогексиламиновая соль 1-(((1(R)-(3-(2-(7-Хлоро-2-хинолинил) -этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио) -метил)циклопропануксусной кислоты, кристаллизованная из смеси толуол/гептана
Стадия 1:
В 2,0 - литровый реактор, снабженный механической мешалкой, термопарой, входом для азота и воронкой для добавления веществ, помещают тетрагидрофуран (132 мл) и 1-(меркаптометил)циклопропануксусную кислоту (9,830 г, 65,98 ммол). Смесь перемешивают в течение 10 минут, чтобы обеспечить полное растворение. Получают прозрачный, бледно-желтый раствор.
Раствор охлаждают до -15 ±2oC. н-Бутиллитий (1,70 М в гексанах, 79,6 мл, 135,26 ммол) добавляют на протяжении 30 минут, поддерживая температуру реакционной смеси ≤ -5oC. Суспензию выдерживают при -5 ±2oC в течение 30 минут.
Стадия 2:
В 250 мл колбу, снабженную мешалкой, термопарой и входом для азота, помещают мезилат Примера 6 (36,52 г, 62,68 ммол) и ТГФ (106 мл). Раствор охлаждают до -5oC. Смесь перемешивают в течение 15 минут, чтобы обеспечить полное растворение. Получают прозрачный, бледно-желтый раствор.
Стадия 3:
Раствор мезилата Стадии 2 переносят через трубку в суспензию дианиона Стадии 1 при -5±2oC на протяжении 5 минут. Реакционный раствор выдерживают при 0±2oC в течение 15 минут. Гетерогенный, желтый реакционный раствор гасят добавлением к раствору 10% соляного раствора (200 мл). Смесь перемешивают в течение около 10 минут, и слоям дают возможность разделиться. Органический слой продукта промывают 0,5 М винной кислотой (280 мл), затем промывают водой (2 x 120 мл).
Раствор продукта переносят в 500 мл 1-горлую колбу. К этому раствору добавляют 250 мл толуола наряду с дициклогексиламином (14,44 мл, 72,60 ммол). Этот прозрачный раствор обрабатывают Darco G-60 (1,8 г), и смесь перемешивают в атмосфере азота в течение часа. Смесь фильтруют через слой solka floc (12 г), используя толуол (20 мл) для промывки и переноса. Фильтрат и промывные воды объединяют и концентрируют под вакуумом до прибл. 200 мл. Затем добавляют еще 200 мл толуола, и снова объем уменьшают до 200 мл.
Вышеупомянутый раствор разбавляют до 640 мл толуолом и переносят в 2-литровую, 3-горлую колбу, снабженную механической мешалкой, термопарой, входом для азота, и воронкой для введения. Прозрачный раствор затравливают солью дициклогексиламина названного соединения (200 мг), ранее кристаллизованной из толуол/гептана. Полученную смесь выдерживают в течение около 3 часов, и в это время получают плотную суспензию. Пробу суспензии анализируют с помощью микроскопии в поляризованном свете, чтобы подтвердить кристалличность твердого вещества. Медленно добавляют гептан (280 мл) на протяжении 2 часов, поддерживая хорошее перемешивание суспензии. Суспензию выдерживают при 20±2oC в течение ночи. Пробу суспензии анализируют с помощью микроскопии в поляризованном свете для подтверждения кристалличности твердого вещества. Суспензию фильтруют путем отсасывания, и осадок промывают 1:1 гептан:толуол (200 мл). Продукт сушат под вакуумом при 40±2oC с продувкой азота. Выход выделенной названной дициклогексиламиновой соли = 40,39 г (чистота: 99,3 A%, > 99,8% ee; 80,6% выход).
В случае чистоты продукта ниже около 90% продукт может быть дополнительно очищен путем покачивания (Swishing со смесью толуол/гептана. Например, ДЦГА соль (98,6 A% чистоты, 10,03 г) was swished толуол/гептаном (1,5:1, 300 мл) при комнатной температуре в течение 5 часов. Суспензию фильтруют и сушат как ранее описано, получая дополнительно очищенную ДЦГА соль (9,63 г, 99,44 A%).
Рентгеновская дифракционная картина порошка: представлена на фиг. 6.
Пример 14
Альтернативный способ получения натрий 1-(((1(R)-(3-(2-(7-хлор-2- хинолинил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил) фенил)пропил)-тио)метил)циклопропанацетата
В 1-литровую, круглодонную, 3-горлую колбу, снабженную подвесной мешалкой и барботером для азота, загружают 285 мл толуола, 85 мл ТГФ и 215 мл деионизованной воды. К этому загружают 25,0 грамм твердой ДЦГА соли 1-(((1(R)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3- (2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропануксусной кислоты (97,3 вес % чистоты). К полученной суспензии добавляют 23,3 мл 2,04 М водной уксусной кислоты.
Колбу продувают 3 раза азотом и вакуумируют, оставляя в атмосфере азота. Двухфазную смесь перемешивают в течение 15 минут. Перемешивание прекращают и смесь переносят в 1000 мл делительную воронку, содержимое отстаивают в течение 15 минут и водный слой удаляют.
Органический слой промывают деионизованной водой (2 x 215 мл), как указано выше, и органический слой возвращают в 1-литровую круглодонную колбу и продувают 3 раза азотом и вакуумируют. 0,500 М раствор NaOH в 1% водном этаноле (63,3 мл) добавляют к органическому слою. По окончании добавления наблюдают образование одной прозрачной фазы. Полученный раствор фильтруют через 0,45 мкм найлоновый мембранный фильтр (предварительно покрытый 2,5 г Solka Floc) во вторую 1-литровую круглодонную колбу. Воронку промывают 50 мл толуола, промывные воды объединяют с начальным фильтратом. Полученный раствор дистиллируют в вакууме (T≤40oC) до объема прибл. 165 мл. Толуол (165 мл, 0,45 мкм фильтрованный) добавляют, и раствор концентрируют до 165 мл. Стадию разбавление толуолом/концентрирование повторяют, получая раствор с конечным объемом 165 мл.
Слой затравочной суспензии получают в 1-литровом полимерном реакторе, снабженном подвесной мешалкой и барботером для азота. В полимерный реактор загружают 32 мл толуола, фильтрованного через 0,45 мкм фильтр, 64 мл ацетонитрила, фильтрованного через 0,45 мкм фильтр, и 3,86 г затравки названной натриевой соли.
Концентрат натриевой соли (165 мл) и высушенный на сите, фильтрованный через 0,45 мкм фильтр, ацетонитрил (330 мл, KF < 100 мкг/мл) загружают одновременно к затравочному слою на протяжении 8 часов при помощи двух шприцевых насосов. Температуру слоя затравки поддерживают при 20oC во время добавления, и скорости потока подбирают так, чтобы поддерживать соотношение растворителей для кристаллизации при прибл. 2:1 ацетонитрил: толуол. Появление (микроскопическое) суспензии и концентрацию супернатанта контролируют посредством одновременного добавления. После завершения добавления полученную суспензию выдерживают на протяжении ночи при 20oC (16 часов).
Кристаллическую суспензию фильтруют под вакуумом при введении азота, оставляя примерно 100 мл суспензии, которую используют в качестве слоя затравки для последующей кристаллизации. Отфильтрованный осадок промывают 238 мл ацетонитрила, высушенного просеиванием путем фильтрации через 0,45 мкм фильтр (KF < 100 мкг/мл). Полученный осадок сушат в вакуумной печи при 40 - 45oC в течение 48 часов. Суммарно выделяют 17,75 г натриевой соли (99,3 вес. %).
Второй цикл образования натриевой соли и кристаллизации проводят так же, как описано выше, используя слой затравки для кристаллизации, получаемый из цикла #1. После завершения цикла #2 кристаллизации, всю суспензию фильтруют, не оставляя после этого слой для затравки. Суммарное количество продукта, выделенного из цикла #2, составляет 20,38 г (99,7 вес.%). Суммарный материальный баланс для двух циклов составляет 95,2%, с выходом 92,1% (скорректированный на механические потери при отборе проб из-за задержки в кристаллизаторе).




Описывается способ получения соединения формулы (I), где НЕТ представляет собой 7-хлорхинолин-2-ил или 6,7-дифторхинолин-2-ил, отличающийся тем, что осуществляют генерирование дилитиевого дианиона 1-(меркаптометил)циклопропануксусной кислоты реакцией 1-(меркаптометил)циклопропануксусной кислоты с литиевым основанием; взаимодействие указанного дианиона с соединением формулы (II), где НЕТ имеет значение, указанное выше, и L означает арилсульфонил или алкилсульфонил, с последующей обработкой кислотой для генерации свободной кислоты соединения формулы (I), и необязательно превращают свободную кислоту в соответствующую соль дициклогексиламина и затем в натриевую соль. Технический результат - создание эффективного способа, обеспечивающего повышенный суммарный выход продукта и возможность получения кристаллической формы. 5 с. и 9 з.п.ф-лы., 6 ил.



где НЕТ представляет собой 7-хлорхинолин-2-ил или 6,7-дифторхинолин-2-ил,
отличающийся тем, что осуществляют генерирование дилитиевого дианиона 1-(меркаптометил) циклопропануксусной кислоты реакцией 1-(меркаптометил)циклопропануксусной кислоты с литиевым основанием, взаимодействие указанного дианиона с соединением формулы (II)
где НЕТ имеет значение, указанное выше;
L означает арилсульфонил или алкилсульфонил,
с последующей обработкой кислотой для генерации свободной кислоты соединения формулы (I), и необязательно превращают свободную кислоту в соответствующую соль дициклогексиламина и затем в натриевую соль.
где НЕТ представляет 7-хлор-2-хинолинил, имеющий рентгеновскую порошковую дифракцию, представленную на фиг.1.
Приоритет по пунктам:
28.12.93 по пп.1-7, 9-14;
09.12.94 по п.8.
| Способ получения производных фторметилхинолина | 1983 |
|
SU1299507A3 |
| Способ получения производных хинолинкарбоновых кислот или их фармацевтически приемлемых солей или эфиров | 1984 |
|
SU1393314A3 |
| Способ получения диалкил - -имидометилдитиофосфатов | 1973 |
|
SU480717A1 |
| Способ получения диалкилфосфатов | 1974 |
|
SU480716A1 |
| Способ автоматического управления процессом сушки сыпучих материалов | 1972 |
|
SU565185A1 |

