ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способу получения кислоты Монтелукаст и ее натриевой соли.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Известно, что кислота Монтелукаст формулы I или ее фармацевтически приемлемая соль останавливает или ингибирует синтез и активность лейкотриенов, и ее натриевая соль на текущий момент продается в компания Merk как Singulair (зарегистрированная торговая марка).

Лейкотриены представляют собой группу локальных гормонов, образованных из арахидоновой кислоты в организме, и характерные примеры лейкотриенов включают лейкотриен В4 (LTB4), лейкотриен С4 (LTC4), лейкотриен D4 (LTD4) и лейкотриен Е4 (LTE4). Есть сообщения о том, что синтез таких лейкотриенов включает в себя метаболизм арахидоновой кислоты посредством 5-липоксигеназы, приводящий к вырабатыванию одного из известных эпоксидов, то есть лейкотриена А4 (LTA4), который сразу же превращается в другие лейкотриены через последовательные ферментативные стадии. О метаболизме и биосинтезе лейкотриенов, а также о их роли в некоторых заболеваниях подробно изложено в работе [Leukotrienes and Lipoxygenases, ed. J. Rokach, Elsevier, Amsterdam (1989)].
EP № 480717 раскрывает способ получения соединения формулы 1 с использованием соответствующего метилового сложного эфира, который показан на реакционной схеме 1 и который включает стадии: проведения реакции сочетания между метил-1-(меркаптометил)циклопропанилацетатом формулы (II) и промежуточным соединением метансульфонила формулы (I), в котором гидроксигруппа является защищенной тетрагидропиранильной группой (THP), с получением метилового сложного эфира формулы (III); и гидролизирования метилового сложного эфира с получением соответствующей свободной кислоты, которая сразу же превращается в соответствующую натриевую соль формулы (IV). Однако вышеупомянутый способ требует выполнения нескольких сложных техник, таких как введение защитной группы; снятие защитной группы и колоночное хроматографическое выделение продукта, что приводит к низкому общему выходу конечного продукта.
Реакционная схема 1

Далее, для того чтобы решить вышеупомянутую задачу, EP № 737186 дает идею способа использования соединения метансульфонила формулы (V) с незащищенной гидроксигруппой и дилитиевой соли 1-(меркаптометил)циклопропанилуксусной кислоты формулы (VI), что показано на реакционной схеме 2, для того чтобы избежать трудоемких стадий введения защитной группы; снятия защитной группы, использованных в реакционной схеме 1. Этот способ, кроме того, включает стадии добавления дициклогексиламина к кислоте Монтелукаст, полученной в реакции сочетания, с получением соли дициклогексиламина и кислоты Монтелукаст формулы (VII) с хорошим выходом; и обработки этой соли посредством NaOH с получением натриевой соли кислоты Монтелукаст формулы (IV).
Реакционная схема 2
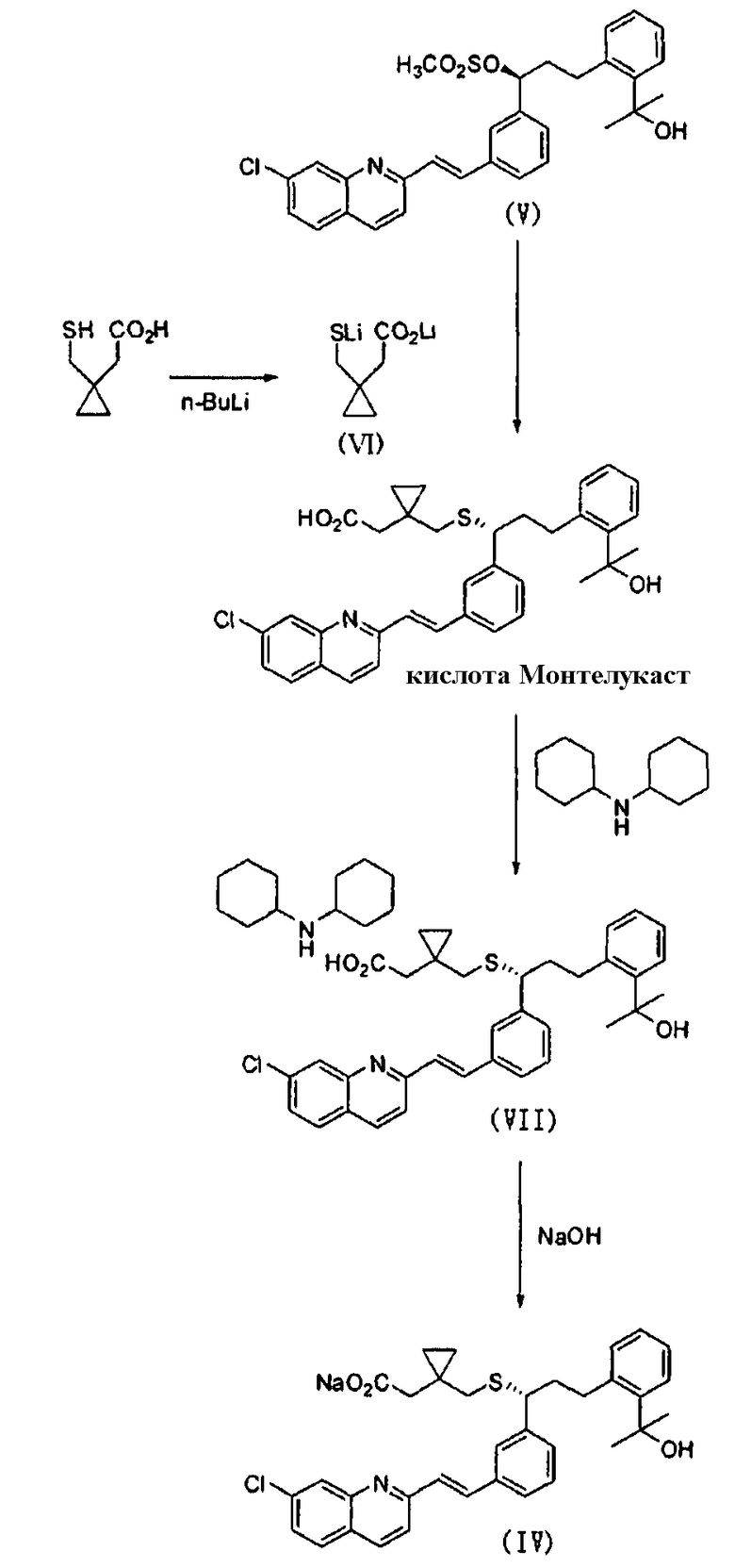
Однако, очень трудно получить соединение формулы (VI), использованное в реакции сочетания вышеупомянутого способа, вследствие использования самовоспламеняющегося н-бутиллития. Кроме того, реакция должна быть проведена без промедления при очень низкой температуре, -30ºС, по причине чувствительности соединения формулы (VI) к влаге и воздуху.
При этом WO 2005/105751 раскрывает способ получения натриевой соли кислоты Монтелукаст формулы (IV), что показано на реакционной схеме 3, который включает стадии: проведения реакции сочетания между соединением метансульфонила формулы (V) и метил-1-(меркаптометил)циклопропилацетатом формулы (II) в присутствии основания, например, LiOH, NaOH, NaH, NaOCH3, BuLi, LiOCH3, LiNPr2 и трет-бутоксида калия (KOt-Bu), с получением метилового сложного эфира формулы (VIII); гидролизирования и подкисления соединения формулы (VIII) с получением кислоты Монтелукаст; и обработки получающегося в результате соединения посредством NaOH, NaOCH3 или трет-бутоксида натрия (NaOt-Bu) с получением натриевой соли кислоты Монтелукаст формулы (IV).
Реакционная схема 3

Однако, вышеупомянутый патент сообщает о том, что чистота и выход кислоты Монтелукаст, полученной вышеупомянутым способом, составляют 94% и 64%, соответственно, а чистота и выход натриевой соли кислоты Монтелукаст составляют 97% и 50%, соответственно, что говорит о том, что чистота натриевой соли кислоты Монтелукаст, полученной этим способом, не может достигнуть необходимой чистоты вещества, равной 99,3%. Следовательно, такой способ требует дополнительной стадии очистки, которая является очень сложной, что дает в результате общий выход лишь 20% или менее.
Во время получения кислоты Монтелукаст могут образовываться различные структурно родственные примеси, содержание которых должно соответствовать нормативным уровням, которые показаны в таблице.






Более того, суммарный уровень всех примесей не должен превышать 0,7% продукта, натриевой соли кислоты Монтелукаст, и следовательно, чистота кислоты Монтелукаст, соединения-предшественника натриевой соли кислоты Монтелукаст, должна быть высокой, в диапазоне от 98% до 99%. Однако, трудно удовлетворить такому требованию при использовании традиционных общепринятых способов.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Таким образом, целью настоящего изобретения является обеспечение эффективного и экономичного способа получения высокочистой кислоты Монтелукаст и ее натриевой соли с высоким выходом.
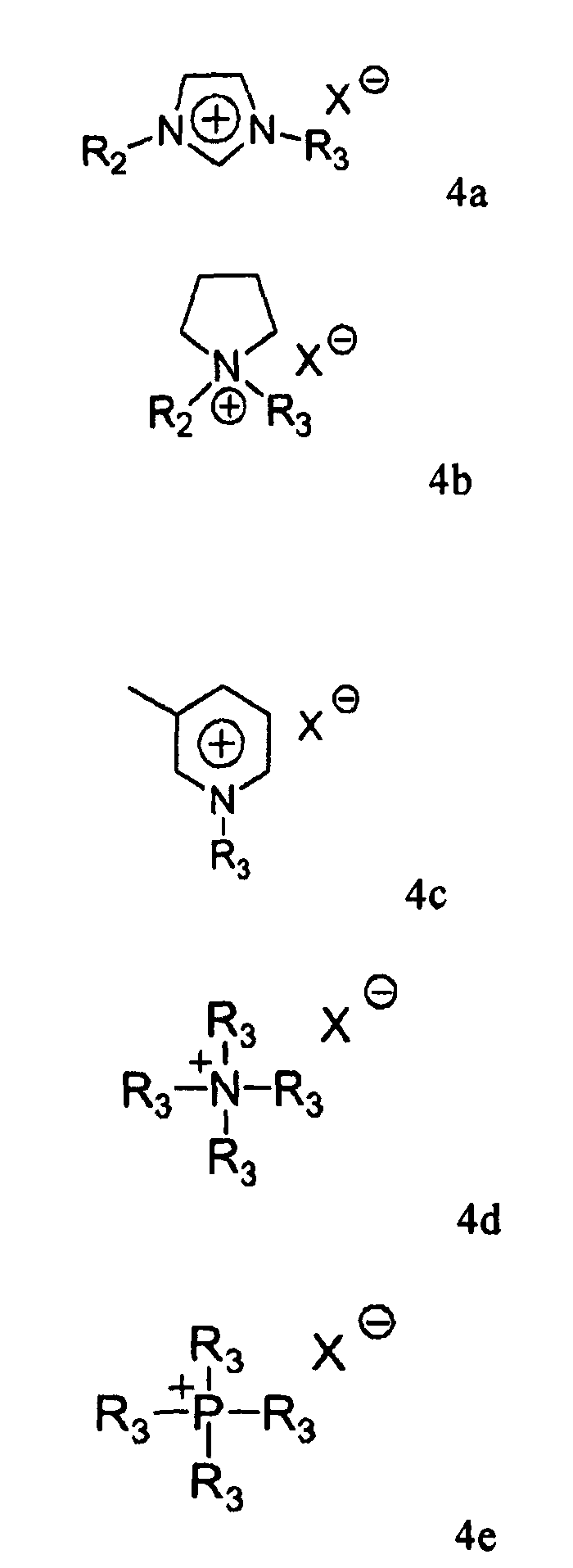
В соответствии с одним аспектом настоящего изобретения обеспечивают способ получения кислоты Монтелукаст формулы 1 или ее натриевой соли, включающий стадию проведения реакции сочетания между тиольным соединением формулы 2 и промежуточным соединением Монтелукаст формулы 3 в присутствии основания в среде, содержащей ионное жидкое соединение, выбранное из группы, состоящей из соединений, имеющих формулы 4а-4е:


в которых R1 означает водород, метил или этил;
L-O представляет собой уходящую группу;
R2 и R3 представляют собой, каждый независимо, водород или С1-12 алкил; и
Х означает Cl, Br, BF4, PF6, SbF6, бис((трифторметан)сульфонил)имид) (NTf2), (трифторметан)сульфонат (OTf), ацетат (OAc), NO3 или метансульфонат.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
R1 тиольного соединения формулы 2 может представлять собой водород, метил или этил, предпочтительно водород, который позволяет осуществлять способ изобретения без использования стадии гидролизирования. В настоящем изобретении тиольное соединение может быть применено в количестве 1 эквивалента или более, предпочтительно в количестве 1-3 эквивалентов, исходя из соединения формулы 3. Тиольное соединение формулы 2 является коммерчески доступным (например, Iffect, и так далее, в компании Changzhou United Chemical), или оно может быть получено общепринятым способом (патенты US №№ 5614632 и 5523477).
Характерные примеры L в соединении формулы 3 включают алкилсульфонил, арилсульфонил, диалкилфосфорил и диарилфосфорил, в которых алкил может представлять собой метил или этил, и арил может представлять собой фенил или п-толил. Сульфонильная группа может представлять собой метансульфонил или п-толуолсульфонил, и метансульфонильное соединение может быть получено традиционным способом, описанным в патенте US № 5614632. Фосфорильная группа может представлять собой диметилфосфорил, диэтилфосфорил или дифенилфосфорил, предпочтительно дифенилфосфорил, который может быть получен традиционным способом, описанным в корейской заявке на патент № 2006-0127942.
Характерные примеры ионных жидких соединений, имеющих формулы 4а-4е, включают бромид 1-этил-3-метилимидазолия, гексафторфосфат 1-этил-3-метилимидазолия, бромид N-бутил-N-метилпирролидия, гексафторфосфат N-бутил-N-метилпирролидия, бромид N-бутил-3-метилпиридия, гексафторфосфат N-бутил-3-метилпиридия, бромид тетра-N-бутиламмония, гексафторфосфат тетра-N-бутиламмония, бромид тетра-N-бутилфосфония и гексафторфосфат тетра-N-бутилфосфония, предпочтительно гексафторфосфат N-бутил-N-метилпирролидия и бромид 1-этил-3-метилимидазолия.
В настоящем изобретении ионное жидкое соединение может быть применено в количестве, находящемся в диапазоне от 0,1 до 100-кратной, предпочтительно от 0,5 до 10-кратной массы соединения формулы 3, и в том случае, когда количество ионного жидкого соединения выходит за рамки вышеупомянутого диапазона, трудно добиться ожидаемых чистоты и выхода продукта.
Основание, использованное для образования тиольного аниона во время реакции сочетания, может представлять собой трет-бутоксид калия (KOt-Bu), трет-бутоксид натрия (NaOt-Bu), NaH, NaOH или KOH, предпочтительно KOt-Bu, который может быть применен в количестве 1 или более эквивалентов, исходя из соединения формулы 2, и в количестве, находящемся в диапазоне от 1,5 до 3,0 эквивалентов, предпочтительно 1,8-2,0 эквивалента, исходя из соединения формулы 2 в том случае, когда R1 представляет собой водород. Дополнительно, в том случае, когда R1 представляет собой метил или этил, основание может быть использовано в количестве, находящемся в диапазоне от 0,6 до 2,0 эквивалентов, предпочтительно 0,9-1,0 эквивалент, исходя из соединения формулы 2. В том случае, когда количество является меньшим, чем количество, определяемое вышеупомянутым диапазоном, реакция замедляется, и в том случае, когда количество является большим, чем количество, определяемое вышеупомянутым диапазоном, чистота продукта ухудшается.
Реакция может быть проведена в растворителе, который может представлять собой диметилсульфоксид, диметилформамид, ацетонитрил или тетрагидрофуран, предпочтительно диметилсульфоксид, при температуре, находящейся в диапазоне от -10ºС до 50ºС. Реакция может быть доведена до конца за 1 час при проведении при комнатной температуре.
Если соединение формулы 2, в которой R1 представляет собой водород, подвергают реакции сочетания с промежуточным соединением формулы 3 без ионного жидкого соединения, образуются различные примеси в избыточных количествах. Также, в том случае, когда продлевается время реакции, тиольное соединение подвергается нежелательным расщеплениям.
Следует отметить, что при проведении реакции сочетания без ионного жидкого соединения реакция замедляется и не протекает нормально за короткое время, тогда как при использовании ионного жидкого соединения настоящего изобретения реакция протекает хорошо за 1 час даже тогда, когда основание и соединение формулы 2 используют в минимально требуемых количествах. Кроме того, продукт, полученный способом изобретения, показывает высокую чистоту, и способ изобретения дает заметно больший выход, чем выходы, получаемые при использовании традиционных способов.
Натриевая соль кислоты Монтелукаст может быть легко получена путем обработки кислоты Монтелукаст, полученной способом изобретения, посредством NaOH, NaOCH3 или трет-бутоксида натрия (NaOt-Bu) в соответствии с общепринятым способом.
Следующие примеры предназначены для дополнительной иллюстрации настоящего изобретения без ограничения его объема.
Пример получения 1: Получение 2-(2-(3(S)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-метансульфонилоксипропил)фенил)-2-пропанола
100 г 2-(2-(3(S)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-гидроксилпропил)фенил)-2-пропанола (Sinochem Ningbo, China) растворяют в смеси 285 мл толуола и 712 мл ацетонитрила, и к этому добавляют по каплям 44 мл диизопропилэтиламина. Затем, после охлаждения получающейся в результате смеси до -25ºС, к этому добавляют медленно по каплям 18,4 мл метансульфонилхлорида и перемешивают при той же температуре в течение 2,5 часов. После визуального обнаружения того, что продукт образовался, смесь дополнительно перемешивают при -25ºС в течение 2 часов, и затем при -35ºС в течение 2 часов до завершения реакции. Получающуюся в результате смесь фильтруют в атмосфере азота при 0ºС-5ºС, и фильтрат концентрируют при пониженном давлении при 0ºС-5ºС в течение 12 часов с получением 91 г соединения, указанного в заголовке, в виде желтого твердого вещества (выход: 78,1%).
1Н ЯМР (300 МГц, CDCl3): δ 8,1 (2Н, м), 7,69 (5Н, м), 7,41 (5Н, м), 7,19 (3Н, м), 5,70 (1Н, дд), 3,25 (1Н, м), 3,04 (1Н, м), 2,76 (3Н, с), 2,45 (1Н, м), 1,92 (1Н, с), 1,65 (6Н, с).
Пример получения 2: Получение 2-(2-(3-(S)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-дифенилфосфатоксипропил)фенил)-2-пропанола
20 г 2-(2-(3-(S)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-гидроксипропил)фенил)-2-пропанола (Sinochem Ningbo, China) растворяют в 240 мл смеси метиленхлорида и толуола (2:1), и к этому добавляют по каплям 7,31 мл триэтиламина. Затем к получающейся в результате смеси добавляют медленно по каплям 13,6 мл дифенилхлорфосфата, с последующим добавлением 1,06 г 4-диметиламинопиридина. После подтверждения завершения реакции с помощью тонкослойной хроматографии (TLC) по истечении 1 часа, получающуюся в результате смесь объединяют со 100 мл метиленхлорида и 200 мл дистиллированной воды, органический слой отделяют, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток перекристаллизовывают с использованием 60 мл смеси этилацетата и н-гексана (1:3), фильтруют, промывают посредством 40 мл дистиллированной воды и сушат теплым воздухом, с получением 29,5 г соединения, указанного в заголовке, в виде желтого твердого вещества (выход: 97,8%).
Температура плавления (М.Р.): 127ºС
1Н ЯМР (300 МГц, CDCl3): δ 8,4 (1Н, д), 7,94 (1Н, д), 7,75 (3Н, м), 6,97-7,35 (20Н, м), 5,70-5,72 (1Н, м), 3,02-3,09 (2Н, м), 2,29-2,34 (2Н, м), 1,65 (3Н, с), 1,59 (3Н, с).
Получение 1-(((1-(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропануксусной кислоты (кислота Монтелукаст)
Пример 1
15 г Бромида 1-этил-3-метилимидазолия растворяют в 60 мл диметилсульфоксида, к этому добавляют 3,15 г 1-(меркаптометил)циклопропанилуксусной кислоты (Changzhou United Chemical, China), и затем к смеси быстро добавляют 2,42 г трет-бутоксида калия при 10ºС с последующим перемешиванием получающейся в результате смеси в течение 5 минут. После быстрого добавления 2,42 г трет-бутоксида калия, получающуюся в результате смесь дополнительно перемешивают при той же температуре в течение 10 минут. Затем, к смеси добавляют 10 г соединения, полученного в примере получения 2, при 15~17ºС, с последующим перемешиванием получающейся в результате смеси в течение 1 часа до завершения реакции.
После добавления 30 мл н-гептана к смеси, полученной ранее, последовательно добавляют 500 мл ледяной воды и 300 мл тетрагидрофурана, органический слой отделяют, и водный слой экстрагируют посредством 500 мл этилацетата. Объединенный органический слой промывают 3 раза посредством 200 мл насыщенного жидкого хлорида аммония, сушат над сульфатом натрия, и концентрируют при пониженном давлении. Остаток, полученный таким образом, перекристаллизовывают с использованием 60 мл изопропанола и 30 мл очищенной воды, и получающееся в результате твердое вещество отфильтровывают и сушат под вакуумом с получением 7,9 г соединения, указанного в заголовке (выход: 93%, чистота: 98,6%).
1Н ЯМР (300 МГц, CD3OD): δ 8,27 (1Н, д), 7,98 (1Н, с), 7,78 (2Н, д), 7,73 (2Н, д), 7,38-7,56 (6Н, м), 7,07-7,14 (3Н, м), 4,84 (1Н, т), 3,30-3,33 (1Н, м), 2,84-2,87 (1Н, м), 2,52 (2Н, с), 2,18-2,23 (2Н, м), 1,55 (6Н, с), 0,37-0,52 (4Н, м).
Пример 2
14 г Бромида N-бутил-N-метилпирролидия растворяют в 60 мл диметилсульфоксида, к этому добавляют 2,7 г 1-(меркаптометил)циклопропанилуксусной кислоты (Changzhou United Chemical, China), и затем к смеси добавляют быстро по каплям 4,0 г трет-бутоксида калия при комнатной температуре, с последующим перемешиванием получающейся в результате смеси в течение 30 минут. К этому добавляют 5 г соединения, полученного в примере получения 2, получающуюся в результате смесь перемешивают в течение 30 минут до завершения реакции, и к этому добавляют 50 мл очищенной воды и 50 мл этилацетата. Объединенный органический слой отделяют, сушат над сульфатом натрия, концентрируют при пониженном давлении и перекристаллизовывают с использованием 30 мл изопропанола и 10 мл очищенной воды. Получающееся в результате твердое вещество отфильтровывают и сушат под вакуумом с получением 2,98 г соединения, указанного в заголовке (выход: 70,2%, чистота: 98,7%).
1Н ЯМР-данные являются такими же, как 1Н ЯМР-данные, описанные в примере 1.
Пример 3
15 г Гексафторфосфата N-бутил-N-метилпирролидия растворяют в 60 мл диметилсульфоксида, к этому добавляют 3,7 г метил 1-(ацетилтиометил)циклопропанилацетата, и затем к смеси добавляют быстро по каплям 2,4 г трет-бутоксида калия при комнатной температуре, с последующим перемешиванием получающейся в результате смеси в течение 30 минут. Затем, после добавления к этому 10 г соединения, полученного в примере получения 2, получающуюся в результате смесь перемешивают в течение 30 минут до завершения реакции, и к этому добавляют 100 мл очищенной воды и 100 мл этилацетата. Объединенный органический слой отделяют, сушат над сульфатом натрия и концентрируют при пониженном давлении. Получающийся в результате остаток растворяют в смеси, состоящей из 29 мл тетрагидрофурана и 29 мл метанола, к этому медленно добавляют 29 мл 10%-ного водного раствора NaOH при 10ºС, и получающуюся в результате смесь перемешивают при комнатной температуре в течение 5 часов до завершения реакции. После добавления 100 воды и 100 мл этилацетата к смеси, полученной ранее, объединенный органический слой отделяют, сушат над сульфатом натрия, концентрируют при пониженном давлении и перекристаллизовывают с использованием 60 мл изопропанола и 20 мл очищенной воды. Получающееся в результате твердое вещество отфильтровывают и сушат под вакуумом с получением 7,7 г соединения, указанного в заголовке (выход: 90,2%, чистота: 99,0%).
1Н ЯМР-данные являются такими же, как 1Н ЯМР-данные, описанные в примере 1.
Пример 4
30 г Бромида 1-этил-3-метилимидазолия растворяют в 1200 мл диметилсульфоксида, к этому добавляют 9,6 г метил 1-(ацетилтиометил)циклопропанил-ацетата, и затем к смеси добавляют быстро по каплям 6,3 г трет-бутоксида калия при комнатной температуре, с последующим перемешиванием получающейся в результате смеси в течение 30 минут. Затем к смеси добавляют 20 г соединения, полученного в примере получения 1, получающуюся в результате смесь перемешивают в течение 30 минут с завершением реакции, и к этому добавляют 200 мл очищенной воды и 200 мл этилацетата. Объединенный органический слой отделяют, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток, полученный таким образом, растворяют в смеси, содержащей 58 мл тетрагидрофурана и 58 мл метанола, к этому медленно добавляют 58 мл 10%-ного водного раствора NaOH при 10ºС, и получающийся в результате раствор перемешивают при комнатной температуре в течение 5 часов с завершением реакции. После добавления к этому 200 мл очищенной воды и 200 мл этилацетата, объединенный органический слой отделяют, сушат над сульфатом натрия, концентрируют при пониженном давлении и перекристаллизовывают с использованием 120 мл изопропанола и 40 мл очищенной воды. Получающееся в результате твердое вещество отфильтровывают и сушат под вакуумом с получением 18,0 г соединения, указанного в заголовке (выход: 82,6%, чистота: 98,1%).
1Н ЯМР-данные являются такими же, как 1Н ЯМР-данные, описанные в примере 1.
Сравнительный пример 1 (традиционный способ, описанный в WO 2005/105751)
Стадия 1
2,04 г 60%-ного раствора гидрида натрия добавляют к 80 мл тетрагидрофурана, к этому быстро добавляют смесь, полученную смешением 11,7 г метил 1-(ацетилтиометил)циклопропанилацетата с 20 мл тетрагидрофурана, при комнатной температуре, с последующим перемешиванием получающейся в результате смеси в течение 1 часа. После добавления к этому 100 мл диметилформамида добавляют 80 мл тетрагидрофурана, содержащего 25% соединения, полученного в примере получения 1, который был охлажден до -5ºС, и получающуюся в результате смесь перемешивают при комнатной температуре в течение 4 часов. После добавления к этому 200 мл этилацетата и 400 мл 5%-ного водного раствора NaCl, объединенный органический слой отделяют, промывают 2 раза посредством 100 мл очищенной воды, сушат над сульфатом натрия и концентрируют при пониженном давлении для удаления растворителя, с получением 29,5 г масла, содержащего 75% метилового сложного эфира 1-(((1(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропанилуксусной кислоты.
Стадия 2
Остаток, полученный на стадии 1, растворяют в 58,9 мл тетрагидрофурана и 29,5 мл метанола, и к этому добавляют 58,9 г 10%-ного раствора NaOH, с последующим перемешиванием смеси в течение ночи при комнатной температуре. После добавления 85,4 мл толуола к получающейся в результате смеси отделяют объединенный органический слой, и корректируют рН с доведением до 4 при использовании 0,5 М раствора винной кислоты. Объединенный органический слой отделяют, концентрируют при пониженном давлении с доведением его общего объема до приблизительно 60 мл, и перемешивают при комнатной температуре для перекристаллизации. Получающееся в результате твердое вещество отфильтровывают и сушат под вакуумом с получением 18,2 г соединения, указанного в заголовке (выход: 83,1%, чистота: 93,3%).
Сравнительный пример 2 (способ, в котором не используется ионное жидкое соединение)
5,5 г 1-(меркаптометил)циклопропанилуксусной кислоты (Changzhou United Chemical, China) растворяют в 60 мл диметилсульфоксида, к этому быстро добавляют 8,2 г трет-бутоксида калия при комнатной температуре, с последующим перемешиванием смеси в течение 30 минут. Затем, после добавления к смеси 10 г соединения, полученного в примере получения 2, получающуюся в результате смесь перемешивают в течение 8,5 часов, и к этому добавляют 100 мл очищенной воды и 100 мл этилацетата. Объединенный органический слой сушат над сульфатом натрия, концентрируют при пониженном давлении с удалением растворителя и перекристаллизовывают с использованием 60 мл изопропанола и 20 мл очищенной воды. Получающееся в результате твердое вещество отфильтровывают и сушат под вакуумом с получением 1,2 г соединения, указанного в заголовке (выход: 14,1%, чистота: 92%).
Сравнительный пример 3 (способ получения дициклогексиламин 1-(((1(R)-(3-(2-(7-хлор-2-хинолидил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропанилацетата, описанный в EP № 737186)
5,9 г 1-(меркаптометил)циклопропануксусной кислоты растворяют в 140 мл тетрагидрофурана и охлаждают до -15ºС, и к этому добавляют медленно по каплям 47 мл гексана, содержащего 1,6 М н-бутиллития, с последующим перемешиванием смеси в течение 30 минут. Затем к этому добавляют по каплям раствор, полученный растворением 20 г 2-(2-(3(S)-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-метансульфонилоксипропил)фенил)-2-пропанола в 80 мл тетрагидрофурана, при -5ºС в течение 30 минут, и получающийся в результате раствор перемешивают при той же температуре в течение 8,5 часов. После добавления к этому 240 мл этилацетата и 240 мл 10%-ного водного раствора NaCl, объединенный органический слой отделяют, промывают посредством 10%-ного раствора винной кислоты, сушат над сульфатом натрия и концентрируют при пониженном давлении с удалением растворителя. Остаток, полученный таким образом, смешивают с 240 мл этилацетата и 8 мл дициклогексиламина, с последующим перемешиванием получающейся в результате смеси в течение 2 часов. Затем, после добавления к получающейся в результате смеси 240 мл н-гексана, полученный раствор перемешивают в течение ночи с получением 17,4 г соединения, указанного в заголовке (выход: 60%, чистота: 74%).
1Н ЯМР (300 МГц, CD3OD): δ 8,30 (1Н, д), 8,01 (1Н, с), 7,85-7,92 (2Н, м), 7,85-7,92 (2Н, м), 7,79 (1Н, с), 7,73 (1Н, с), 7,53-7,54 (2Н, м), 7,40-7,45 (4Н, м), 7,11-7,16 (3Н, м), 4,05 (1Н, т), 3,12-3,13 (3Н, м), 3,10-3,12 (1Н, м), 2,65 (1Н, д), 2,56 (1Н, д), 2,38 (1Н, д), 2,36 (1Н, д), 2,28-2,33 (2Н, м), 2,03-2,06 (4Н, м), 1,87-1,88 (4Н, м), 1,84 (2Н, д), 1,28-1,39 (10Н, м), 1,54 (6Н, с), 0,39-0,52 (4Н, м).
Хотя изобретение было описано относительно вышеупомянутых конкретных вариантов осуществления, следует понимать, что в отношении изобретения специалистами в данной области могут быть сделаны различные модификации и изменения, которые также будут попадать в пределы объема изобретения, которое определено прилагаемой формулой изобретения.
Настоящее изобретение относится к области органической химии, а именно к способу получения кислоты Монтелукаст 1 или ее натриевой соли путем проведения реакции между тиольным соединением формулы 2 и промежуточным соединением Монтелукаст формулы 3 в присутствии основания в среде, содержащей ионное жидкое соединение, выбранное из группы соединений формул 4а-4е. Технический результат: разработан новый способ получения кислоты Монтелукаст или ее натриевой соли, отличающийся экономичностью и высоким выходом целевого продукта. 7 з.п. ф-лы, 1 табл.

1. Способ получения кислоты Монтелукаст формулы 1 или ее натриевой соли, включающий стадию сочетания тиольного соединения формулы 2 с промежуточным соединением Монтелукаст формулы 3 в присутствии основания в среде, содержащей ионное жидкое соединение, выбранное из группы, состоящей из соединений, имеющих формулы 4а-4е:

в которых, R1 означает водород, метил или этил;
L-0 представляет собой уходящую группу;
R2 и R3 представляют собой, каждый независимо, водород или C1-12 алкил; и
Х означает Cl, Br, BF4, PF6, SbF6, бис((трифторметан)сульфонил)имид) (NTf2), (трифторметан)сульфонат (OTf), ацетат (ОАс), NO3 или метансульфонат.
2. Способ по п.1, где L выбирают из группы, состоящей из метансульфонила, п-толуолсульфонила, диметилфосфонила, диэтилфосфонила и дифенилфосфонила.
3. Способ по п.2, где L представляет собой дифенилфосфонил.
4. Способ по п.1, где ионное жидкое соединение выбирают из группы, состоящей из бромида 1-этил-3-метилимидазолия, гексафторфосфата 1-этил-3-метилимидазолия, бромида N-бутил-N-метилпирролидия, гексафторфосфата N-бутил-N-метилпирролидия, бромида N-бутил-3-метилпиридия, гексафторфосфата N-бутил-3-метилпиридия, бромида тетра-N-бутиламмония, гексафторфосфата тетра-N-бутиламмония, бромида тетра-N-бутилфосфония и гексафторфосфата тетра-N-бутилфосфония.
5. Способ по п.4, где ионное жидкое соединение представляет собой бромид 1-этил-3-метилимидазолия или гексафторфосфат N-бутил-N-метилпирролидия.
6. Способ по п.1, где ионное жидкое соединение применяют в количестве, находящемся в диапазоне от 0,1 до 100-кратной массы соединения формулы 3.
7. Способ по п.1, где основание выбирают из группы, состоящей из трет-бутоксида калия, трет-бутоксида натрия, NaH, NaOH и КОН.
8. Способ по п.7, где основание представляет собой трет-бутоксид калия.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| ХИНОЛИН-ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ АНТАГОНИСТА ЛЕЙКОТРИЕНА | 1993 |
|
RU2143426C1 |

