Кросс-ссылки на соответствующие патентные заявки
Этот патентный документ заявляет приоритет патентной заявки США номер 60/612,472, поданной 23 сентября 2004 года, которая представлена здесь по этой ссылке.
Предпосылки
Полипренилированные 1,4-бензохиноны и гидрохиноны, такие как убихиноны, пластохиноны и токоферолы, широко распространены в растениях и животных, в которых они играют важную роль в транспорте электронов, фотосинтезе и как антиоксиданты (см. Thomson, R.H. Naturally Occurring Quinones, Academic Press, London, 1971, стр. 1-197; и Pennock, J.F. В: Terpenoids in plants, J.B. Pridham, Ed. Academic Press, London, 1967, стр.129-146).
Природные пренилированные бензохиноны и гидрохиноны, имеющие терпеноидную часть длиной от одной до девяти изопреновых единиц, были выделены ранее из морских организмов, таких как коричневые водоросли рода Fucales, губки и асцидии. Многие водоросли содержат тетрапренил-, трипренил-, и дипренилхиноны и гидрохиноны (см. Ochi, M.; Kotsuki, H.; Inooue, M.; Taniguchi, M.; Tokoroyama, T. Chem. Lett., 1979, 831-832; и Capon, R.J.; Ghisalberti, E.L.; Jeffereis, P.R. Phytochemistry, 1981, 20, 2598-2600). Кроме того, губки содержат линейные незамещенные полипренилированные гидрохиноны и бензохиноны с более длинными боковыми цепями и умеренной антимикробной активностью, а также ингибиторы АТФ-аз, сульфатированные полипренилхиноны (см. Cimino, G.; De Stefano, S.; Minale, L. Tetrahedron, 1972, 28, 1315; Cimino, G.; De Stefano, S.; Minale, L. Experientia, 1972, 28, 1401; Pouches Y.F.; Verbist, J.F.; Biard, J.F.; Boukef, K.J. Nat. Prod., 1988, 51, 188; Lumsdon, D.; Capon, R.J.; Thomas, S.G.; Beveridge, A.A. Aust. J. Chem. 1992, 45, 1321-1325; De Rosa, S.; De Giulio, Iodice, C. J. Nat. Prod. 1994, 57, 1711-1716; Fusetani, N.; Sugano, M.; Matsunaga, S.; Hashimoto, K.; Shikama, H.; Ohta, A., Nagano, H. Experientia, 1987, 43, 1233; Stonik, V.A.; Makarieva, T.N.; Dmitrenok, A.S. J. Nat. Prod., 1992, 55, 1256-1260; и Bifulco, G.; Bruno; I.; Minale; L., Riccio, R.; Debitus, S.; Bourdy, G.; Vassa, A.; Lavayre, J. J. Nat. Prod. 1995, 58, 1444-1449). Из асцидий рода Aplidium были ранее выделены около дюжины пренилированных хинонов, включая наиболее простые из них, монопренилбензохиноны (см. Guella, G; Mancini, I; Pietra, F. Helv. Chim. Acta. 1987, 70, 621-626; Howard, B.M.; Clarkson, K.; Bernstein, R.L. Tetrahedron. Lett. 1979, 4449 - argett, N.M.; Keeran, W.S. J. Nat. Prod., 1984, 47, 556-557; и Faulkner, D.J. Nat. Prod. Rep., 1993, 93, 1771-1791).
Раковые заболевания занимают одну из ведущих позиций по летальным исходам в США. Однако все еще по-прежнему ощущается необходимость в соединениях, обладающих противоопухолевыми свойствами. Также ощущается необходимость в фармакологических инструментах для дальнейшего исследования физиологических процессов, связанных с онкологическими заболеваниями.
Краткое изложение сущности изобретения
В настоящем изобретении заявляются полипренилированные 1,4-бензохиноны (формулы 1 или формулы 3) или полипренилированные 1,4-гидрохиноны (формулы 2 или формулы 4):
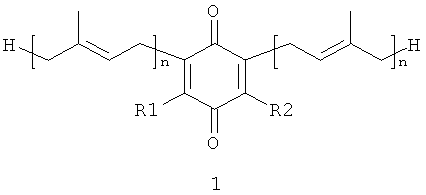
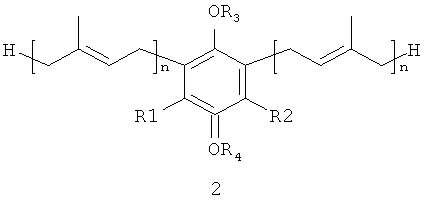
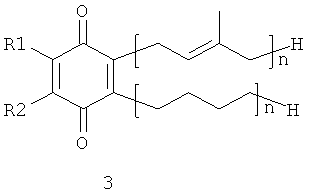

где
каждый из R1 и R2, независимо от других, может являться водородным
радикалом или (С1-С6)-алкокси-группой;
каждый из R3 и R4 независимо от других, может являться водородным радикалом или (С1-С6)-алкильной группой;
n может быть равно 2 или 3;
или их фармацевтически приемлемые соли.
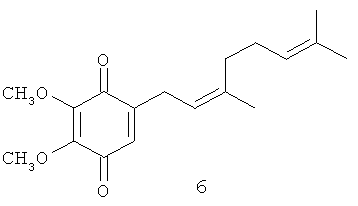
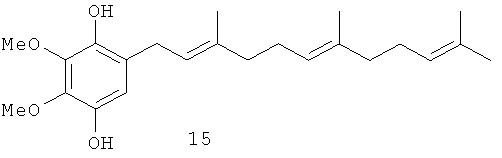
В настоящем изобретении заявляется также применение соединения формулы 1, или формулы 2, или формулы 3, или формулы 4, а также известных полипренилированных 1,4-бензохинонов формулы 5, или формулы 6, или формулы 7, или формулы 8, или формулы 9, или формулы 10, или формулы 11, или формулы 12, или формулы 13, или формулы 14 или полипренилированных 1,4-гидрохинонов формулы 15 или их фармацевтически приемлемых солей для приготовления лекарственного средства, которое может быть использовано для лечения онкологических заболеваний у млекопитающих.





В изобретении также заявляется применение вышеперечисленных новых и известных веществ или их фармацевтически приемлемых солей для приготовления лекарственного средства, используемого для лечения раковых опухолей у млекопитающих, т.е. для уменьшения размера и/или ингибирования роста раковой опухоли у млекопитающих.
В изобретении также заявляется применение вещества изобретения для приготовления лекарственного средства, используемого для индуцирования АР-1-зависимой транскрипционной активности у млекопитающих.
В изобретении также заявляется применение вещества изобретения для приготовления лекарственного средства, используемого для индуцирования NF-κВ-зависимой транскрипционной активности у млекопитающих.
В изобретении также заявляется применение вещества изобретения для приготовления лекарственного средства, используемого для индуцирования одновременно АР-1- и NF-κВ-зависимой транскрипционной активности у млекопитающих.
В изобретении также заявляется применение вещества изобретения для приготовления лекарственного средства, используемого для уменьшения трансформации нормальных клеток в опухолевые у млекопитающих.
В изобретении также заявляется применение вещества изобретения для приготовления лекарственного средства, используемого для уменьшения пролиферации опухолевых клеток у млекопитающих.
В изобретении также заявляется применение вещества изобретения для приготовления лекарственного средства, используемого для индуцирования апоптоза у млекопитающих.
В изобретении также заявляется фармацевтическая композиция, обладающая антираковой активностью, включающая эффективное количество соединения формулы 1, или формулы 2, или формулы 3, или формулы 4, или формулы 5, или формулы 6, или формулы 7, или формулы 8, или формулы 9, или формулы 10, или формулы 11, или формулы 12, или формулы 13, или формулы 15 или фармацевтически приемлемой соли этого соединения в комбинации с фармацевтически приемлемым растворителем или носителем.
В изобретении также заявляется метод лечения онкологических заболеваний, включающий введение млекопитающему, в случае необходимости в таком лечении, эффективного количества соединения формулы 1, или формулы 2, или формулы 3, или формулы 4, или формулы 5, или формулы 6, или формулы 7, или формулы 8, или формулы 9, или формулы 10, или формулы 11, или формулы 12, или формулы 13, или формулы 14, или формулы 15, или эффективного количества фармацевтически приемлемой соли этого соединения или эффективного количества фармацевтической композиции, включающей эти соединения.
В изобретении также заявляется метод лечения онкологических заболеваний, где введение соединения или фармацевтической композиции уменьшает размеры или ингибирует рост раковой опухоли у млекопитающего, предотвращает трансформацию нормальных клеток в опухолевые, уменьшает пролиферацию опухолевых клеток, вызывает апоптоз опухолевых клеток у млекопитающего.
В изобретении также заявляется метод индуцирования в клетке АР-1-зависимой транскрипционной активности в клетках, включающий обработку клеток эффективным количеством вещества изобретения.
В изобретении также заявляется метод индуцирования в клетке NF-κВ-зависимой транскрипционной активности в клетках, включающий обработку клеток эффективным количеством вещества изобретения.
В изобретении также заявляется метод одновременного индуцирования АР-1 и NF-κВ-зависимой транскрипционной активности в клетке, включающий обработку клеток эффективным количеством вещества изобретения.
В изобретении также заявляется метод индуцирования апоптоза или клеточной смерти в клетках, включающий обработку клеток эффективным количеством вещества изобретения.
В изобретении также заявляется метод индуцирования апоптоза или клеточной смерти у млекопитающих, включающий, в случае необходимости такого лечения, введение эффективного количества вещества изобретения.
В изобретении также заявляется метод выделения глабрухинона А (формула 5) и глабрухинона В (формула 6), включающий экстракцию асцидий асцидий Aplidium glabrum этанолом, упаривание этанола, экстракцию водного остатка хлороформом, упаривание хлороформенного слоя до получения коричневого маслообразного остатка, хроматографирование остатка на колонке с силикагелем, в системе гексан - этилацетат, 10:1 и разделение смеси полученных глабрухинонов А и В при помощи ВЭЖХ на колонке с силикагелем в системе гексан - этилацетат, 7:1 с получением индивидуальных глабрухинонов А и В.
Краткое описание фигур.
Фиг.1. Структурные формулы веществ изобретения, заявляемых как новые.
Фиг.2. Структурные формулы веществ 5-15.
Фиг.3. Генеральная схема синтеза глабрухинона А (5) и его аналогов.
Фиг.4. Синтез целевых хинонов и гидрохинонов из производного фенола 35.
Фиг.5. Синтез целевых хинонов и гидрохинонов из производного фенола 36.
Фиг.6. Синтез целевых хинонов и гидрохинонов из производного фенола 37.
Фиг.7. Структуры соединений 5 и 6 и установление структуры 5 с помощью НМВС и 1H-1H-COSY корреляций.
Фиг.8А. Цитотоксическое действие глабрухинона А (5) на JB6 P+ Cl 41 клетки, установленное с помощью MTS-метода. Клетки культивировали в 96-луночных планшетах в 5% FBS/MEM. Затем среду сменили на 0.1% FBS/MEM и клетки обработали указанными концентрациями глабрухинона А (1). Клетки инкубировали с веществом в течение 22 часов и затем добавили по 20 µл MTS-реагента в каждую лунку и инкубировали еще в течение 2 часов. Данные представлены в виде процентного содержания живых клеток в сравнении с контрольными, необработанными глабрухиноном клетками. Каждая точка на графике отражает полученное значение ±SD (стандартное отклонение от среднего значения) и является результатом двух независимых экспериментов, по пять образцов на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.8В. Вычисление IC50 для глабрухинона А (5) при действии на JB6 P+ Cl 41 клетки, произведенное методом линейной регрессии.
Фиг.9А. Цитотоксическое действие глабрухинона А (5) на НТ-460 клетки, установленное с помощью MTS-метода. Клетки культивировали в 96-луночных планшетах в 10% FBS/RPMI. Затем среду сменили на 0.5% FBS/RPMI и далее клетки обрабатывали, как указано для Фиг.8А. Данные представлены в виде процентного содержания живых клеток в сравнении с контрольными, необработанными глабрухиноном А клетками. Каждая точка на графике отражает полученное значение ±SD (стандартное отклонение от среднего значения) и является результатом двух независимых экспериментов, по три образца на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.9В. Вычисление IC50 для глабрухинона А (5) при действии на НТ-460 клетки, произведенное методом линейной регрессии.
Фиг.10А. Ингибирование глабрухиноном А (5) колонеобразования НСТ-116 клеток в мягком агаре. НСТ-116 клетки были обработаны указанными концентрациями глабрухинона А (5) в мягком агаре. Колонии клеток были подсчитаны после инкубации с веществом 5 в течение недели. Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по три образца на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.10В. Вычисление INCC50 для глабрухинона А (5) при действии на НСТ-116 клетки в мягком агаре, произведенное методом линейной регрессии.
Фиг.11А. Ингибирование глабрухиноном А (5) EGF-индуцированной JB6 Р+ Cl 41 клеточной трансформации в мягком агаре. JB6 Р+ Cl 41 клетки были обработаны EGF (10 нг/мл) и указанными концентрациями глабрухинона А (5) в мягком агаре. Колонии клеток были подсчитаны после инкубации с веществом 5 в течение недели. Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по три образца на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.11В. Вычисление INCC50 для глабрухинона А (5) при действии на трансформированные с помощью EGF JB6 Р+ Cl 41 клетки в мягком агаре, произведенное методом линейной регрессии.
Фиг.12А. Ингибирование глабрухиноном А (5) ТРА-индуцированной JB6 P+ Cl 41 клеточной трансформации в мягком агаре. JB6 P+ Cl 41 клетки были обработаны ТРА (10 нг/мл) и указанными концентрациями глабрухинона А (5) в мягком агаре. Колонии клеток были подсчитаны после инкубации с веществом 5 в течение двух недель. Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по три образца на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.12В. Вычисление INCC50 для глабрухинона А (5) при действии на трансформированные с помощью ТРА JB6 P+ Cl 41 клетки в мягком агаре, произведенное методом линейной регрессии.
Фиг.13А. Индукция глабрухиноном А апоптоза в JY клетках (В лимфобласты человека). JY клетки были выращены и обработаны указанными концентрациями глабрухинона А (5). Процент апоптоза был вычислен после 3 часов инкубации с веществом (5). Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по три образца на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.13В. Индукция глабрухиноном А апоптоза в JB6 Р+ Cl 41 клетках мыши. JB6 P+ Cl 41 клетки были выращены и обработаны указанными концентрациями глабрухинона А (5). Процент апоптоза был вычислен после 3 часов инкубации с веществом (5). Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по два образца на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.13С. Индукция глабрухиноном А апоптоза в SK-MEL-28 клетках. Клетки меланомы человека SK-MEL-28 были выращены и обработаны указанными концентрациями глабрухинона А (5). Процент апоптоза был вычислен после 3 часов инкубации с веществом (5). Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по три образца на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.13D. Индукция глабрухиноном А апоптоза в НТ-460 клетках. Клетки рака легких человека НТ-460 были выращены и обработаны указанными концентрациями глабрухинона А (5). Процент апоптоза был вычислен после 3 часов инкубации с веществом (5). Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по три образца на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.13Е. Индукция глабрухиноном А апоптоза в НСТ-116 клетках. Клетки рака кишечника человека НСТ-116 были выращены и обработаны указанными концентрациями глабрухинона А (5). Процент апоптоза был вычислен после 20 часов инкубации с веществом (5). Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по три образца на каждую концентрацию в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.14. Индукция апоптоза глабрухиноном А (5) в COX2+/+или COX2-/- МЭФ, определенная с помощью метода ДНК-лестницы. Клетки COX2+/+ или СОХ2-/- МЭФ (мышиные эмбриональные фибробласты) были выращены, обработаны указанными концентрациями глабрухинона А (5) и инкубированы в течение 24 часов. Затем клетки собрали и поставили тест на апоптоз методом ДНК-лестницы.
Фиг.15А. Глабрухинон А (5) индуцирует NF-κВ-зависимую транскрипционную активность в JB6 Cl 41 клетках. JB6 Cl 41 клетки со стабильно экспрессированным люциферазным репортерным геном, контролируемым NF-κВ ДНК-связанным сиквенсом, были обработаны указанными концентрациями глабрухинона А (5) и инкубированы в течение 24 часов. Затем клетки были экстрагированы лизисным буфером и люциферазная активность была измерена.
Фиг.15В. Глабрухинон А (5) индуцирует АР-1-зависимую транскрипционную активность в JB6 Cl 41 клетках. JB6 Cl 41 клетки со стабильно экспрессированным люциферазным репортерным геном, контролируемым АР-1 ДНК-связанным сиквенсом, были обработаны указанными концентрациями глабрухинона А (5) и инкубированы в течение 24 часов. Затем клетки были экстрагированы лизисным буфером и люциферазная активность была измерена.
Фиг.16. Ингибирование глабрухиноном А (он же деметилубихинон Q2, 5) роста карциномы Эрлиха у белых нелинейных мышей. Мыши были обработаны однократно путем внутрибрюшинного введения, глабрухиноном А, растворенным в 50% ДМСО, за сутки до инокуляции опухоли. Размеры опухоли измеряли на 6-й, 9-й, 12-й, 15-й и 18-й день. Данные представлены в виде процента ингибирования роста опухоли в мышах, обработанных глабрухиноном А, по сравнению с размерами опухоли в необработанных мышах. Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по четыре мыши в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.17. Ингибирование глабрухиноном А (он же деметилубихинон Q2, 5) роста карциномы Эрлиха у белых нелинейных мышей. Мыши были обработаны однократно путем внутрибрюшинного введения, глабрухиноном А, растворенным в 50% этиловом спирте, за сутки до инокуляции опухоли. Размеры опухоли измеряли на 6-й, 9-й, 12-й, 15-й и 18-й день. Данные представлены в виде процента ингибирования роста опухоли в мышах, обработанных глабрухиноном А, по сравнению с размерами опухоли в необработанных мышах. Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по четыре мыши в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Фиг.18. Ингибирование глабрухиноном А (он же деметилубихинон Q2, 5) роста карциномы Эрлиха у белых нелинейных мышей. Мыши были обработаны однократно путем внутрибрюшинного введения, глабрухиноном А, растворенным в 50% ДМСО, через сутки после инокуляции опухоли. Размеры опухоли измеряли на 9-й, 12-й, 15-й и 18-й день. Данные представлены в виде процента ингибирования роста опухоли в мышах, обработанных глабрухиноном А, по сравнению с размерами опухоли в необработанных мышах. Каждый столбец на графике отражает полученное значение ±SD и является результатом двух независимых экспериментов, по четыре мыши в каждом из экспериментов (*р<0,05; Манн-Уитни U-тест).
Детальное описание изобретения
Вещества изобретения показали противораковую превентивную и терапевтическую активность, что было показано с помощью методов определения опухолевой трансформации клеток в мягком агаре, определения апоптоза методами проточной цитометрии и ДНК-лестницы, определения цитотоксической активности MTS-методом, а также в опытах in vivo на белых нелинейных мышах с привитой карциномой Эрлиха. Для экспериментов использовались мышиные эпидермальные JB6 P+ Cl 41 клетки, COX2+/+ и COX2-/- мышиные эмбриональные фибробласты (МЭФ), JY клетки (В лимфобласты человека), опухолевые клетки человека НТ-460 (рак легких), НСТ-116 (рак кишечника), SK-MEL-28 (рак кожи, меланома). Исследуемые полипренилхиноны и гидрохиноны индуцировали в JB6 Cl 41 клетках АР-1- и NF-κВ-зависимую транскрипционную активность. Вещества изобретения также показали раково-превентивную активность в опытах в мягком агаре, в нецитотоксических концентрациях, что свидетельствует о том, что противоопухолевое действие веществ изобретения опосредовано через нетоксический механизм.
Соотношения структура-активность на примере репрезентативной выборки веществ изобретения
Зависимость структура - активность представленных сединений изучалась с использованием статистического анализа (компьютерная программа Statistica 6, Stat Soft Inc., USA). Исследуемые соединения подразделялись на три группы в зависимости от количества изопреновых единиц в терпеноидной части (Табл.2). Группа 1 содержала хиноны с двумя изопреновыми звеньями (10 атомов углерода) в боковой цепи. Группа 2 - хиноны с треми изопреновыми звеньями (15 атомов углерода) в терпеноидной части и группа 3 - хиноны, содержащие от 4 до 6 изопреновых единиц (20-30 атомов углерода) в боковой цепи. Существенные отличия и корреляции между данными о биологической активности, полученные для различных структурных групп хинонов, определялись при помощи метода непараметрической корреляции Спирмана и Манн-Уитни U-тестом при использовании данных из Табл. 2.
Результаты показали, что биологическая активность представленных соединений зависит от длины боковой цепи терпеноидной части молекулы. Статистический анализ данных из Табл.2 показал, что цитотоксичность хинонов из группы 1 имеет в среднем значение IC50 20.0±15.2 Мµ для JB6 Cl 41 клеток. Хиноны из группы 2 имеют среднее значение IC50 9.7±9.0 Mµ. И наконец, хиноны из группы 3 показали среднее значение IC50 84.9±63.8 Mµ. Эти результаты означают, что хиноны с тремя изопреновыми звеньями в терпеноидной части более токсичны для JB6 Cl 41 клеток чем хиноны из группы 1, имеющие 2 изопреновых звена. Противоположный вывод можно сделать при сравнении значений IC50 для хинонов из групп 1 и 3 или 2 и 3. Хиноны из групп 1 или 2 существенно более токсичны для JB6 Cl 41 клеток, чем соединения из группы 3. Следовательно, в соединениях групп 1, 2 с увеличением длины терпеноидной части увеличивается активность соединений, но активность существенно снижается при дальнейшем увеличении длины терпеноидной части от группы 2 к группе 3.
Данные статистического анализа по влиянию представленных соединений на EGF-индуцируемую трансформацию JB6 Cl 41 клеток показали, что хиноны группы 1 имеют среднее значение INCC50 9.4±5.3 Mµ, а хиноны группы 2 - INCC50 24.0±16.7 Мµ. Группа 3 снова показала минимальную активность среди трех групп соединений с INCC50 59.7±33.3 Мµ (Табл.2). Таким образом, учитывая эти данные и данные Манн-Уитни U-теста можно заключить, что хиноны группы 1 обладают наиболее сильным эффектом ингибирования клеточной трансформации (р=0.0283 при сравнении с группой 2; р=0.0253 при сравнении с группой 3; р=0.0084 при сравнении с объединенными данными для групп 2 и 3), а группа 3 наименее эффективна. Существенные корреляции наблюдались между длиной терпеноидной части и INCC50 (р=0.0002, R=0.8556). Это означает, что с увеличением длины терпеноидной части значения INCC50 для клеточной трансформации также увеличиваются.
Сравнивались токсичность и клеточная трансформация JB6 Cl 41 клеток, и было найдено, что хиноны из группы 1 имеют среднее значение INCC50 в 1.4-4 раза меньше, чем IC50 для соответствующих клеток (табл. 2). С другой стороны, большинство хинонов из группы 2 показали значения INCC50 в 4-10 раз выше, чем IC50. На основании этих данных можно сказать, что хиноны группы 1 существенно более токсичны для трансформированных JB6 клеток, чем для нормальных. И наоборот, хиноны группы 2 более токсичны в отношении нормальных JB6 клеток, чем трансформированных. Таким образом, хиноны группы 1 имеют более сильную канцерпревентивную активность чем хиноны группы 2.
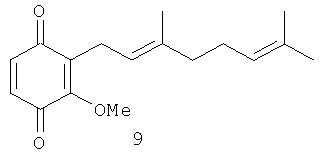
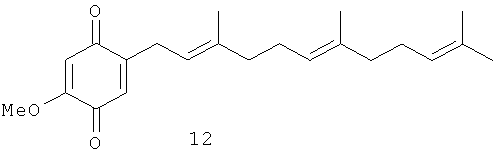
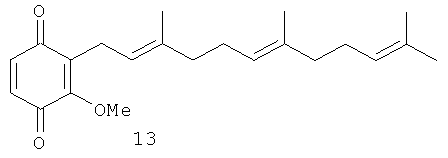
Исследовали также влияние репрезентативной выборки соединений 5, 7-18 на транскрипционную активность, зависимую от некоторых ядерных факторов, в частности, р53, АР-1 и NF-κВ. Было показано, что активность патентуемых соединений зависит в некоторой степени от положения метоксильной группы по отношению к терпеноидной части. Было выбрано несколько пар структурных аналогов с метоксильными группами в одинаковом положении: 1) орто-аналоги, хиноны 9 и 13; 2) мета-аналоги, хиноны 10 и 8; 3) пара-аналоги, хиноны 11 и 12. Из данных таблиц 2 и 3 следует, что канцерпревентивная активность и влияние хинонов на АР-1 зависимую транскрипционную активность возрастает в ряду орто - мета - пара. INCC50 имеет следующие значения: для хинонов 9, 13: 15.1 и 24.6 Мµ соответственно; хинонов 10, 8: 6.6 и 16.7 Мµ, соответственно и для хинонов 11, 12: 3.1 и 7.4 Мµ соответственно.
Индукция АР-1 зависимой транскрипционной активности орто-соединениями 9, 13 составляет 133,8% по отношению к контролю, а мета-соединениями 10, 8 - 188,7%. Пара-соединения 11 и 12 показали самое высокое значение АР-1 зависимой транскрипционной активности - 486,9% по сравнению с контролем. Орто-дизамещенные хиноны 9 и 13 являются наименее активными соединениями по сравнению с мета- и пара-аналогами не только в отношении индукции АР-1 зависимой транскрипционной активности, но и при ингибировании клеточной трансформации. Среди пара-дизамещенных производных хинон 11, имеющий две изопреновые единицы в боковой цепи, показал лучшую активность по сравнению с хиноном 12, имеющим три изопреновые единицы. Кроме того, пара-дизамещенные хиноны 11 и 12 имеют INCC50 3.1 и 7.4 Мµ соответственно против EGF-индуцируемой клеточной трансформации JB6 Р+ Cl 41 клеток. Хинон 9 также показал более высокую индукцию АР-1 зависимой транскрипционной активности (721.7%) по сравнению с хиноном 13 (252.2%).
Канцерпревентивные свойства патентуемых соединений были изучены с использованием мышиных эпителиальных клеток JB6 Cl 41. Хиноны с двумя изопреновыми звеньями в боковой цепи показали специфический эффект против трансформированных в опухолевые JB6 Cl 41 клеток по сравнению с нормальными клетками. Действующие концентрации могли отличаться в 4 раза.
Зависимость структура - активность патентуемых соединений изучалась в отношении цитотоксичности или канцерпревентивной активности. Данное изучение показало, что цитотоксичность хинонов возрастает с увеличением числа атомов углерода, от хинонов с двумя пренильными звеньями в боковой цепи к хинонам с тремя звеньями и затем уменьшается для соединений, имеющих от 4 до 6 изопреновых единиц. Канцерпревентивная активность уменьшалась с увеличением длины полипренильной цепи. Наиболее активные канцерпревентивные полипренилхиноны имеют боковую цепь из двух изопреновых единиц.
Как было показано с помощью методов проточной цитометрии и ДНК-лестницы, репрезентативная выборка патентуемых соединений индуцировала апоптоз в JB6 Р+ Cl 41 клетках и МЭФ. Белок-суппрессор опухолей р53, который является частью "системы безопасности" клетки и функционирует по принципу негативной регуляции клеточного роста в клетках с поврежденной ДНК, часто вовлекается в апоптоз, индуцируемый некоторыми внешними стимулами, включая хемопревентивные агенты и противоопухолевые препараты. Однако патентуемые соединения не только не активируют р53, но даже наоборот, многие из изученных патентуемых соединений демонстрируют значительное ингибирование р53-зависимой транскрипционной активности. В дополнение к этому, патентуемые соединения индуцируют значительную активацию АР-1- или NF-κВ-зависимой транскрипционной активности. АР-1 транскрипционный фактор регулирует множество клеточных процессов, таких как пролиферация, дифференциация, апоптоз, и прежде рассматривался преимущественно как онкоген. Недавно было показано, что некоторые из составляющих АР-1 белков, такие как Jun-B и c-Fos имеют раково-превентивную активность, как in vitro, так и in vivo. Активация другого АР-1 белка, c-Jun требуется для индукции Fas L-опосредованного апоптоза в PC 12 клетках и клетках лейкемии человека HL-60. Активация как АР-1, так и NF-κВ ядерных факторов важна для действия ДНК-повреждающих агентов и апоптоза, индуцируемого церамидами в Т-лимфоцитах и Jurkat Т-клетках. Баланс между белками-членами АР-1 семейства, c-Jun и ATF-2 регулирует выбор между направлениями на дифференциацию или на апоптоз в PC 12 клетках. Противораковые препараты, такие как винбластин, которые ингибируют микротьюбулы, активируют АР-1 фактор транскрипции в клетках карциномы человека КВ-3. Эта активация необходима для эффективного апоптоза, индуцируемого этим лекарством.
NF-κВ, член высокорегулируемого семейства димерных ядерных факторов транскрипции, вовлекается в активацию большого числа генов, которые отвечают на инфекцию, воспаление и другие стрессовые ситуации. Сообщается, что NF-κВ вовлекается как в индукцию, так и в ингибирование апоптоза.
Результаты, представленные в настоящем изобретении, предполагают, что апоптоз, индуцируемый репрезентативной выборкой патентуемых соединений происходит независимо от р53-ядерного фактора транскрипции, но вместо этого может быть опосредован через АР-1 и NF-κВ-ядерные факторы.
Таким образом, полученные результаты показывают, что метоксилированные полипренилхиноны и их синтетические аналоги являются новой перспективной группой противораковых препаратов морского происхождения. Патентуемые соединения показывают цитотоксическую активность и индуцируют апоптоз в JB6 Р+ Cl 41 и МЭФ клетках. Наиболее активные из этих соединений являются мощными индукторами АР-1 и NF-κB-зависимой транскрипционной активности и в то же время, ингибиторами р53-зависимой транскрипционной активности. Канцерпревентивное действие этих соединений может быть объяснено индукцией р53-независимого апоптоза.
Было обнаружено, что хиноны, имеющие боковую цепь, состоящую из 10 атомов углерода, оказывают специфический эффект ингибирования трансформированных JB6 Р+ Cl 41 клеток в отличие от хинонов, имеющих боковую цепь с 15 или от 20 до 30 атомов углерода. Поскольку представленные соединения активны против трансформации эпидермальных JB6 клеток, они могут быть использованы как средства против рака кожи, т.е. средства, предотвращающие рак кожи. Хинон 11, имеющий боковую цепь в пара-положении относительно метокси-группы, обладает наиболее сильным канцерпревентивным эффектом из изученных соединений.
Противораковые и терапевтические свойства исследуемых соединений проявлялись в относительно нетоксичных концентрациях. Это подтверждалось тестом на цитотоксичность с использованием метода MTS, тестом на опухолевую трансформацию клеток в мягком агаре и определением апоптоза методами проточной цитометрии и ДНК-лестницы. Изучение проводилось на мышиных эпидермальных JB6 Cl 41 клетках, JY лимфобластах человека, СОХ2-дефицитных и нормальных мышиных эмбриональных фибробластах (МЭФ), а также некоторых человеческих опухолевых клеточных линиях включая рак легких (НТ460), рак кишечника (НСТ) и меланому (SK-MEL-28). Канцерпромоторный эффект эпидермального фактора роста (EGF) или 12-O-тетрадеканоил-форбол-13-ацетата (ТРА) значительно уменьшался при применении нетоксических доз глабрухинона А или других патентуемых соединений.
Помимо уже упомянутых использовались следующие определения, если другие не описаны: алкил, алкокси и т.д., обозначающие как разветвленные, так и неразветвленные группы, но ссылка на такой радикал как «пропил» означает только неразветвленный радикал, а «изопропил» относится только к разветвленному радикалу.
В основном, термин «выделенное и очищенное» означает, что соединение в значительной степени очищено от биологического материала (например, крови, тканей, клеток, растительного материала и т.д.). В рамках патента в одном случае это означает, что вещество очищено на 75%, в другом - по меньшей мере на 90%, в третьем - на 98 или, наконец, на 99% от биологического материала. В других случаях этот термин относится к синтезированным соединениям.
Термин «лечение» относится как к терапевтическому, так и профилактическому лечению, когда целью является предотвращение или уменьшение нежелательных физиологических изменений или нарушений, таких как развитие и распространение рака. Желательные клинические результаты включают, но не ограничиваются, смягчение симптомов, ограничение распространения болезни, стабилизацию состояния болезни, ограничение или замедление прогресса болезни, улучшение или временное облегчение болезненного состояния, а также ремиссию (частичную или полную), определяемую или неопределяемую. «Лечение» может также означать пролонгирование выживания при сравнении с ожидаемым выживанием при отсутствии лечения. Клинические результаты при необходимости лечения включают результаты уже имеющегося состояния или нарушения, а также склонности к данному состоянию или нарушению или результаты, при которых данное состояние или нарушение предотвращается.
Термины «рак» или «раковый» относятся физиологическому состоянию млекопитающего, при котором характерен неконтролируемый рост клеток. «Рак» может включать одну или больше опухолевых линий. Список видов рака, таких как, к примеру, рак кожи, включен в патент США №6,833,373.
Специфические и предпочтительные значения, перечисленные ниже для радикалов, заместителей и рангов, используются только для иллюстрации, они не исключают других определенных значений или других значений в пределах определенных рангов для радикалов и заместителей.
Специфически (С1-С6)алкил может быть метилом, этилом, пропилом, изопропилом, бутилом, изо-бутилом, втор-бутилом, пентилом, 3-пентилом, или гексилом; (С1-С6)алкокси может быть метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, вторбутокси, пентокси, 3-пентокси или гексилокси.
Процесс получения патентуемых соединений дается в следующих примерах изобретения и иллюстрирован процедурами, в которых радикалы обозначаются так, как указано выше, если не квалифицированы по-другому.
В тех случаях, когда патентуемые соединения являются достаточно сильными основаниями или кислотами для формирования стабильных нетоксичных солей, введение веществ в виде солей может быть предпочтительным. Примерами фармацевтически приемлемых солей могут служить соли органических кислот, которые формируются с кислотами, образующими физиологически приемлемый анион, к примеру тозилат, метансульфонат, ацетат, цитрат, малонат, тартрат, сукцинат, бензоат, аскорбат, α-кетоглутарат, и α-глицерофосфат.Подходящие неорганические соли могут также быть получены, включая гидрохлорид, сульфат, нитрат, бикарбонат и карбонат.
Фармацевтически приемлемые соли могут быть получены с использованием стандартных, широко известных процедур, к примеру, при реакции достаточно основного соединения, такого как амин, с подходящей кислотой, дающей физиологически приемлемый анион. Соли щелочных металлов (к примеру, натрия, калия или лития) или щелочноземельных металлов (к примеру, кальция) и органических кислот, также могут быть получены.
Патентуемые соединения также могут быть сформулированы как фармацевтические композиции и введены млекопитающему, например человеку, в виде различных форм, адаптированных к выбранному пути введения, т.е. орально или парентерально, внутривенно, внутримышечно, местно или подкожно.
Так, представленные соединения могут систематически вводиться, например орально, в комбинации с фармацевтически приемлемым наполнителем, таким, как инертный растворитель или съедобный носитель. Они могут быть оформлены в виде жестких или мягких желатиновых капсул, могут быть сформированы в таблетки или могут быть включены прямо в пищу пациента в виде пищевой добавки. Для орального терапевтического применения активное вещество может быть скомбинировано с одним или более инертным наполнителем и использоваться в форме проглатываемых таблеток, защечных таблеток, капсул, эликсиров, суспензий, сиропов, облаток и т.п. Такие композиции и препараты должны содержать не менее 0,1% активного вещества. Процентное содержание активного вещества в составе композиций и препаратов может, конечно, варьироваться и обычно может составлять от 2 до 60% веса единицы лекарственной формы. Количество активного вещества должно быть таково, чтобы достигался уровень эффективной дозы.
Таблетки, пилюли, капсулы и т.п. могут также содержать следующее: связующие вещества, такие как камедь, акация, желатин; наполнители, такие как фосфат кальция; дезинтегрирующие вещества, например крахмал, альгиновая кислота; смазки, такие как стеарат магния; подсластители, например сахароза, фруктоза, лактоза или аспартам; или отдушки, например мята, винтергрин, вишня. Когда лекарственная форма имеет вид капсулы, она может содержать, кроме вышеупомянутого, жидкий носитель, такой как растительное масло или полиэтиленгликоль. Различные материалы могут быть использованы в качестве оболочек или другой модификации твердой лекарственной формы. Например, таблетки, пилюли или капсулы могут быть покрыты желатином, воском, шеллаком или сахаром и т.п. Сироп или эликсир могут содержать активное вещество, сахарозу или фруктозу в качестве подсластителя, метил- или пропилпарабены в качестве антиоксидантов, красители и отдушки, такие как вишневый или апельсиновый аромат. Конечно, любой материал, используемый для приготовления лекарственных форм, должен быть фармацевтически приемлемым и нетоксичным в используемых количествах. Кроме того, активное вещество может быть помещено в препарат или устройство, откуда будет попадать в организм млекопитающего путем постоянного долговременного высвобождения.
Активное вещество может быть также введено внутривенно или интраперитонеально путем впрыскивания или укола. Растворы активного вещества или его соли могут быть приготовлены в воде, куда может быть добавлено поверхностно-активное вещество. Дисперсии могут быть также приготовлены в глицерине, жидких полиэтиленгликолях, триацетине, их смесях, а также в маслах. При обычных условиях хранения и использования, эти препараты могут содержать антиоксиданты для предотвращения роста микроорганизмов. Фармацевтические формы, пригодные для впрыскивания или укола, могут включать стерильные водные растворы или дисперсии или стерильные пудры, включая активный ингредиент, адаптированный для стерильных инъекций или впрыскиваний, и возможно, инкапсулированный в липосомы. В любом случае, окончательная лекарственная форма должна быть стерильной, жидкой, и стабильной в условиях приготовления и хранения. Жидкий носитель может быть растворителем, или жидкой дисперсионной средой, содержащей, к примеру, воду, этанол, полиол (к примеру, глицерин, пропилен гликоль, жидкие полиэтиленгликоли, и т.п.), растительные масла, нетоксичные эфиры глицерина, и их подходящие смеси. Подходящая вязкость жидкости может быть достигнута, к примеру, путем образования липосом или путем получения частиц нужных размеров в случае дисперсий или путем использования поверхностно-активных веществ. Предотвращение воздействия микроорганизмов может быть достигнуто применением различных антибактериальных и антифунгальных агентов, к примеру парабенов, хлорбутанола, фенола, сорбиновой кислоты, тимерозола и т.п. Во многих случаях предпочтительно включать изотонические агенты, к примеру сахара, буферные растворы или хлорид натрия. Пролонгированная абсорбция впрыскиваемых соединений может быть достигнута применением композиций агентов, замедляющих абсорбцию, к примеру моностеарата алюминия или желатина.
Стерильные растворы для инъекций готовятся путем включения активного вещества в требуемое количество подходящего растворителя с различными другими ингредиентами, перечисленными выше, если требуется, с последующей стерилизацией фильтрованием. В случае стерильных порошков для приготовления стерильных растворов для инъекций, предпочтительными методами приготовления являются вакуумная и лиофильная сушки, которые дают порошок активного ингредиента плюс любой желательный дополнительный ингредиент, присутствующий в предварительно стерилизованных растворах.
Для местного применения соединения могут применяться в чистой форме, в том случае, если это жидкость. Однако может быть желательным наносить их на кожу в виде композиций, в сочетании с дерматологически приемлемым носителем, который может быть твердым или жидким.
Используемые твердые носители включают мелко размолотые твердые вещества, например тальк, глину, микрокристаллическую целлюлозу, силикагель, алюминий и др. Используемые жидкие носители включают воду, спирты, гликоли или смеси воды с гликолями, в которых активные вещества могут быть растворены или диспергированы, возможно с применением нетоксичных ПАВ. Вспомогательные средства, такие как отдушки и дополнительные антимикробные агенты могут быть добавлены, чтобы оптимизировать свойства лекарственной формы. Полученные жидкие композиции могут быть применены с абсорбирующих вспомогательных средств, таких как бандажи и другие повязки или в виде спреев на область воздействия. Загустители, например синтетические полимеры, жирные кислоты, соли и эфиры жирных кислот, алифатические спирты, модифицированные целлюлозы или модифицированные минеральные материалы также могут быть применены вместе с жидкими носителями для формирования паст, гелей, мазей, мыл и пр., для применения непосредственно на кожу пациента.
Примеры используемых дерматологических композиций, применяемых для нанесения патентуемых соединений непосредственно на кожу хорошо известны, к примеру, см. Jacquet et al. (пат. США №4,608,392), Geria (пат. США №4,992,478), Smith et al. (пат. США №4,559,157) и Wortzman (пат. США №4,820,508).
Используемые дозы патентуемых соединений могут быть определены сравнением их in vitro и in vivo активностей на животных моделях. Методы экстраполяции эффективных доз, полученных для мышей и других животных, на человека, также хорошо известны; см., например, пат. США №4,938,949.
Количество соединения или активной соли, или его производного, требуемого для применения при лечении, будет варьировать в зависимости не только от выбранного вида соли, но также в зависимости от выбранного способа введения, от условий введения, от возраста и состояния пациента и будет в конечном счете зависеть от усмотрения врача.
Желательная доза может быть представлена в виде однократной дозы, или может быть введена в виде нескольких доз с определенными интервалами, например две, три, четыре или более субдоз в день. Субдозы сами по себе тоже могут делиться на некоторое количество дискретных введений, со свободными промежутками между ними; это могут быть, например, многочисленные ингаляции или глазные капли.
Таким образом, задачей настоящего изобретения является обеспечить индустриально применимое и эффективное использование моно- ди- или неметоксилированных ди- или три-пренилбензохинонов (вещества формул 1, 3, 5-14) или соответствующих гидрохинонов (вещества формул 2, 4, 15) в качестве противораковых превентивных и терапевтических компонентов медицинских или косметических препаратов для лечения любых млекопитающих, и как научных инструментов для изучения АР-1 или NF-κВ белков.
Патентуемые вещества могут быть получены путем их выделения из природных источников (морские беспозвоночные) или путем химического синтеза. Многие из патентуемых соединений уникальны, так как в их структуре присутствуют метоксильные группы, а полипренильные боковые цепи короче, если сравнивать патентуемые соединения с убихинонами, также имеющими полипренильные боковые цепи. К примеру, глабрухинон А (формула 5) (или, по-другому, деметилубихинон Q2) отличается от убихинонов отсутствием метильной группы в ароматическом ядре и наличием более короткой боковой цепи.
Далее изобретение иллюстрируется следующими примерами.
Основные процедуры
1Н и 13С ЯМР спектры снимали на спектрометрах Bruker WM-250 при 250 и 62,9 Мгц соответственно и Broker DPX 300 при 300 и 75 Мгц, соответственно. Масс-спектры высокого разрешения (HREIMS) получали на масс-спектрометре AMD-604S. Очистку веществ методом высокоэффективной жидкостной хроматографии (ВЭЖХ) проводили на хроматографе DuPont 8800, снабженном детектором-рефрактометром и с использованием силикагельной колонки Ultrasphere Si. ИК-спектры снимали на спектрометре Bruker FT-IR "Vector 22". УФ-спектры снимали в CCLl4 на спектрофотометре Cecil СЕ 7200. Процесс наступления апоптоза анализировали методом проточной цитометрии с использованием проточного цитометра Becton Dickinson FACSCalibur (BD Biosciences, San Jose, CA). MTS-тест на цитотоксичность проводился с использованием планшетного ридера-спектрофотометра Multiscan MS (Labsystems, Finland). Колонии клеток в методе мягкого агара считали с использованием инвертированного микроскопа Leica DM IRB (Leica Mikroskopie und Systeme GmbH, Germany) и компьютерной программы Image-Pro Plus, версия 3.0 для Windows (Media Cybernetics, Silver Spring, MD). Хемилюминесценция в методе определения р53-, АР-1- или NF-кВ-зависимой транскрипционной активности измерялась с использованием планшетного ридера Luminoscan Ascent Type 392 (Labsystems, Finland).
Реагенты
Питательные среды MEM и DMEM были от Gibco Invitrogen Corporation (Carlsbad, CA, США). Сыворотка бычьих эмбрионов (FBS) была от Gemini Bio-Products (Calabasas, США). Пенициллин/стрептомицин и гентамицин были от Bio-Whittaker (Walkersville, MD, США), L-глутамин был от Mediatech, Inc. (Herndon, Virginia, США). Эпидермальный фактор роста (EGF) был от Collaborative Research (Bedford, MA, США). Субстрат для люциферазного метода и MTS-реагент для метода определения цитотоксичности был от Promega (Madison, WA, США). Набор для определения апоптоза методом проточной цитометрии Annexin V-FITC Apoptosis Detection Kit был от Medical & Biological Laboratories (Watertown, MA). Силикагель L (40/100 мкм) для колоночной хроматографии низкого давления был от Chemapol (Praha, Czech Republic). Силикагельные пластинки для тонкослойной хроматографии (4,5×6,0 см, 5-17 мкм) были от Sorbfil (Российская Федерация).
Клеточные культуры
Линии мышиных эпидермальных клеток JB6 Р+ Cl 41 и ее стабильных трансфектантов JB6 Cl 41-NF-κB, JB6 Cl 41-AP-1, JB6 Cl 41-p53 (PG-13) культивировали в монослое при 37°С и 5% СО2 в MEM, содержащей 5% FBS, 2 мМ L-глутамина, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина. Мышиные эмбриональные фибробласты (МЭФ) выращивали при 37°С и 5% CO2 в DMEM, содержащей 10% FBS, 2 мМ L-глутамина, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина. Лимфобластоидные JY клетки человека выращивали при 37°С и 5% CO2 в среде RPMI, содержащей 10% FBS, 2 мМ L-глутамина, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина. Опухолевые клетки человека, НСТ-116, НТ-460 и SK-MEL-28 культивировали при 37°С и 5% СО2 в среде RPMI (для НТ-460), McCoy's (для НСТ-116) или MEM (для SK-MEL-28), содержащей 10% FBS, 2 мМ L-глутамина, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина.
Пример 1. Подготовка и выделение патентуемых соединений.
Выделение глабрухинонов А (5) и В (6) из асцидии Aplidium glabrum.
Замороженные асцидии экстрагировали этанолом (1:1 по весу) этанол упаривали и водный остаток экстрагировали хлороформом (1:1 по объему). Хлороформенный слой упаривали под вакуумом до получения коричневого маслообразного осадка, который хроматографировали на колонке с силикагелем, используя систему гексан - этилацетат, 10:1 (система А). Выделение контролировалось тонкослойной хроматографией (ТСХ) в системе A. Rf смеси глабрухинонов А (5) и В (6) в этих условиях был равен приблизительно 0.35. После упаривания растворителей из соответствующих фракций была получена смесь глабрухинонов А (5) и В (6), которая разделялась при помощи ВЭЖХ на колонке с силикагелем Altex Ultrasphere Si (4,6 мм × 25 см), в системе гексан - этилацетат, 7:1 (система В).
Процедура синтеза
Основное описание.
Синтез полипренилбензохинонов 5-14, 16-21 и гидрохинонов 15, 22-34 осуществлялся по схеме, изображенной на Фиг.3. Она включает алкилирование коммерчески доступных фенолов гераниолом или фарнезолом в условиях кислотного катализа с последующим окислительным деметилированием полученных пренилированных фенолов и восстановлением целевых бензохинонов до соответствующих гидрохинонов. Были получены глабрухиноны А (5), В (6) и ряд аналогов (7-34). Аналоги 7-34 отличаются от глабрухинонов А и В количеством и/или положением метоксильных групп и/или строением и количеством полипренильных боковых цепей или наличием гидрохинонового фрагмента вместо бензохинонового.
Синтез бензохинонов 5-14, 16-21.
Лучшие результаты были получены в результате алкилирования фенолов 35-37 транс-гераниолом (38) или транс-фарнезолом (39) в присутствии эфирата трехфтористого бора в качестве кислотного катализатора (Фиг.4-6). В этих условиях суммарный выход продуктов алкилирования составил около 60%. Окислительное деметилирование полученных фракций церий аммоний нитратом (ЦАН) дало смесь соответствующих бензохинонов (5-13, 16-21), после деления которых были получены индивидуальные соединения. Синтез пренилбензохинона 14 был выполнен из незамещенного монометилового эфира гидрохинона (60) через промежуточные 2-геранил- или 2-фарнезил-4-метоксифенолы (61, 62, на схеме не показаны).
Стадия 1
Эфират трехфтористого бора (0.5 мл) добавляли при перемешивании к смеси 1 ммол соответствующего фенола (35, 36 или 37) и 4 ммол гераниола (38) или фарнезола (39) в 10 мл абсолютного эфира. Смесь выдерживали 12 часов при комнатной температуре, после чего добавляли 30 мл воды и экстрагировали эфиром (3×15 мл). Экстракт промывали 10% NaCl и сушили над Na2SO4. Растворитель удаляли и остаток делили на колонке с силикагелем. Смесь пренилированных хинонов 40, 42, 44 или 41, 43, 45 (Фиг.4), 46, 48, 50 или 47, 49, 51 (Фиг.5), или 52, 54, 56, 58 или 53, 55, 57, 59 (Фиг.6), 61 или 62 (на схеме не показаны) элюировались в градиентной системе гексан:ацетон, 50:1→20:1. Эти смеси были использованы на стадии 2 реакции. Средний выход составил около 60%. В некоторых случаях при помощи ВЭЖХ на колонке Altex Ultrasphere Si (4.6 мм × 25 см) в системе В были выделены индивидуальные пренилированные фенолы. Таким путем были получены индивидуальные пренилированные фенолы 40, 41, 46, 51 и 61, которые использовали для 1Н ЯМР анализа.
Стадия 2
Раствор ЦАН (0.9 ммол) в 3 мл смеси СН3СН:H2O (1:1) добавляли при перемешивании к охлажденному (0°С) раствору фенольных фракций 40, 42, 44 или 41, 43, 45 (Фиг.4), 46, 48, 50 или 47, 49, 51 (Фиг.5), 52, 54, 56, 58 или 53, 55, 57, 59 (Фиг.6), 61 или 62 (0.3 ммол) в 7 мл CH3CN. После перемешивания при 0°С в течение 1-2 часов смесь приливали к 25 мл 10% NaCl и экстрагировали эфиром (3×15 мл). Экстракт сушили над Na2SO4 и упаривали. Соответствующие бензохиноны 5, 17 и 7, 20 (Фиг.4), 11, 9, 21 или 12, 13, 18 (Фиг.5), 9, 10, 11, 19 или 8, 12, 13, 16 (Фиг.6), или 14 (на схеме не показаны) были получены тонкослойной хроматографией на силикагеле в системе гексан:ацетон (8:1). Каждый бензохинон (5, 7-14, 16-21) содержал в качестве примеси (менее 6%) соответствующий 2'-3'- или 2''-3''-цис-изомер. Образование этих изомеров можно объяснить изомеризацией гераниола и/или фарнезола под действием эфирата трехфтористого бора на стадии пренилирования фенолов. Как правило, примесь цис-изомера не отделяли от основного продукта (трамс-изомера), за исключением смеси 5 и 6, которые разделили при помощи ВЭЖХ. Сравнение действия 5 и 6 показало, что они имеют одинаковый уровень активности. Это означает, что для практического использования разделять эти продукты нет необходимости. Выход целевых продуктов на этой стадии составил около 70%. Суммарный выход после двух стадий составил приблизительно 45%.
Синтез гидрохинонов 15, 22-34 (Основной метод)
Раствор Na2S2O4 (3 ммол) в 3 мл воды добавляли к раствору 1 ммол соответствующего бензохинона (5, 7-14, 16-21) в 7 мл ацетона. Смесь перемешивали в течение часа, разбавляли водой и экстрагировали эфиром (3×15 мл). Экстракт сушили над Na2SO4 и упаривали. В результате были получены гидрохиноны 15, 22-34, содержащие небольшое количество примесей (менее 6%) 2'-3' цис- или 2''-3'' цис-изомеров.
Структуры глабрухинонов А (5) и В (6).
Масс-спектры электронного удара (EIMS) глабрухинонов А (5) и В (6) содержали помимо пика молекулярного иона при m/z 304, характерный для бензохинонов пик при m/z 306 (М+2). УФ-спектры показали максимум поглощения при 264 нм (ε=15000), а ИК-спектры - поглощение при 1675, 1657 и 1603 см-1, что подтверждает наличие p-бензохиноновой части молекулы.
1Н ЯМР спектр 5 очень напоминал спектр вераплихинона А из Aplidium sp., но отличался наличием дополнительного синглета при 4.02, характерного для ОМе-группы. Квартеты при 61.2 и 61.3 м.д. и синглеты при 145.2 и 144.9 м.д. в 13С ЯМР-спектре свидетельствовали о наличии двух метоксильных групп в 5. Это означало 5,6- или 3,5-диметоксизамещение в 1,4-бензохиноновой части молекулы. Присоединение терпеноидной части по С-2 бензохиноновой части и 5,6-положение метоксильных групп было подтверждено анализом данных ЯМР-спектров, включая 1H-1Н COSY, NOESY и НМВС (Фиг.7). Мультиплетность Н-3 (узкий триплет при 6.34 м.д.) и кросс-пики Н-3/Н-1' в 1H-1H COSY и Н-3/Н-1' взаимодействие в NOESY-спектре были очень показательны. Сигналы двух тризамещенных двойных связей и трех метильных групп, присоединенных к этим двойным связям в 13С ЯМР-спектре подтвердили наличие дипренильной боковой цепи.
Сравнение ЯМР-спектров 5 и 6 показало, что глабрухиноны А и В содержат геранильный и нерильный типы боковых цепей соответственно (Табл. 1; также см. Shubina et.al., Tetrahedron Letters, 46, 559-562 (2005)). Действительно, эти соединения отличаются друг от друга конфигурацией С2', С3'-двойной связи, что следовало из значений химсдвигов С10'. В более стерически затрудненном. Е-изомере этот сигнал наблюдается в более сильном поле по сравнению с Z-изомером. Значения химсдвигов С-10' в спектрах соединений 5 и 6 существенно отличаются и равны 16.2 и 22.8 м.д. соответственно. Структуры глабрухинонов А и В отличаются от структур вераплихинонов А и В наличием дополнительной метоксильной группы в положении 5. Структуры 5 и 6 были подтверждены синтезом, как описано выше.
Триметоксифенол 35 был получен из коммерчески доступного 2,3,4-триметоксибензальдегида описанным ранее методом (Matsumoto, et. al., 1984.) Кислотно-катализируемое алкилирование фенола 35 trans-геранил бромидом дало trans-геранил фенол 40 с примесью цис-изомера (суммарный выход 18%, соотношение транс:цис=95:5). Пренилированный фенол 40 был очищен при помощи ВЭЖХ и его структура была установлена на основании анализа 1Н ЯМР-спектров и сравнения их со спектрами глабрухинонов 5 и 6. Окислительное деметилирование 40 привело к смеси бензохинонов 5 и 6 (14%). Смесь синтетических 5 и 6 была разделена с помощью ВЭЖХ, и синтетические 5 и 6 были идентифицированы с глабрухинонами А и В сравнением их ЯМР-спектров.
Структуры синтетических полипернилированных соединений.
Структуры целевых пренил-бензохинонов и -гидрохинонов, также как и промежуточных пренилфенолов были установлены с помощью 1Н-ЯМР спектров в сравнении с соответствующими данными для структур глабрухинонов А (5) и В (6). Спектральные характеристики некоторых синтетических аналогов глабрухинонов 5 и 6, а также некоторых промежуточных пренилфенолов даны ниже.
Пример 2
3-Деметилубихинон Q2 или 2,3-диметокси-5-(3',7'-диметил-окта-2'(Е),6'-диенил)-[1,4] бензохинон (5): желтое масло, HREIMS m/z 304.1655 [M]+, вычислено для C18H24O4 304.1675, IR (CHCl3): 1675, 1657, 1603. 1Н-ЯМР (CDCl3, 250 MHz) δ: 6.34 (t, J=1.7, 1H, H-6); 5.13 (m, 1H, H-2'); 5.08 (m, 1H, H-6'); 4.02 (s, 3H, OMe); 4.00 (s, 3H, ОМе); 3.10 (dd, J=7.3, 1.7, 2H, Н-1'); 2.09 (m, 2H, H-5'); 2.08 (m, 2H, H-4'); 1.70 (d, J=1.2, 3H, H-8'); 1.62 (d, J=1.2, 3H, H-10'); 1.60 (br. s, 3H, Н-9'). 13С-ЯМР (CDCl3, 62.9 MHz) δ: 16.20 (q, С-10'), 17.79 (q, C-9'), 25.77 (q, C-8'), 26.52 (t, C-5'), 27.17 (t, С-1'), 39.72 (t, C-4'), 61.20 (q, OMe), 61.30 (q, OMe), 117.78 (d, C-2'). 123.98 (d, C-6'), 130.45 (d, C-6), 131.95 (s, C-7'), 140.17 (s, C-3'), 144.91 (s, C-2 или C-3), 145.16 (s, C-3 или C-2), 146.92 (s, C-5), 184.38 (s, C-4 или C-l), 184.54 (s, C-1 или C-4).
Пример 3
2,3-диметокси-5-(3',7'-диметил-окта-2'(Z),6'-диенил)-[1,4] бензохинон (6): желтое масло, HREIMS m/z 304.1662 [M]+, вычислено для C18H24O4 304.1675, 1Н-ЯМР (CDCl3, 250 MHz) δ: 6.37 (t, J=1.7, 1H, H-6); 5.13 (m, 1H, H-2'); 5.07 (m, 1H, H-6'); 4.02 (s, 3H, ОМе); 4.00 (s, 3H, ОМе); 3.11 (br. d, J=7.1, 1.2, 2H, Н-1'); 2.04 (m, 2H, H-5'); 2.04 (m, 2H, H-4'); 1.75 (q, J=1.2, 3H, H-10'); 1.66 (br. d, J=1.2, 3H, H-8'); 1.59 (d, J=1.2, 3H, H-9'). 13С-ЯМР (CDCl3, 62.9 MHz) δ: 17.79 (q, C-9'), 22.76 (q, C-10'), 25.77 (q, C-8'), 26.52 (t, C-5'), 27.27 (t, С-1'), 32.01 (t, C-4'), 61.20 (q, ОМе), 61.30 (q, ОМе), 117.78 (d, C-2'), 123.98 (d, C-6'), 130.45 (d, C-6), 131.95 (s, C-7'), 140.17 (s, C-3'), 144.91 (s, C-2 или С-3), 145.16 (s, C-3 или C-2), 146.92 (s, C-5), 184.38 (s, C-4 или С-1), 184.54 (s, C-1 или С-4).
Пример 4
2-Метокси-3-(3',7'-диметил-окта-2',6'-диенил)-[1,4] бензохинон (9): желтое масло, HREIMS m/z 274.1558 [M]+, вычислено для C17H22O3 274.1569, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.68 (d, J=10.0, 1H, H-5); 6.59 (d, J=10.0, 1H, H-6); 5.05 (m, 2H, H-2', H-6'); 4.02 (s, 3H, ОМе); 3.15 (br. d, J=7.3, 2H, Н-1'); 2.01 (m, 4H, H-4', H-5'); 1.73 (br. s, 3H, Me); 1.65 (br. s, 3H, Me); 1.58 (br. s, 3H, Me).
Пример 5
2-Метокси-6-(3',7'-диметил-окта-2',6'-диенил)-[1,4] бензохинон (10): желтое масло, HREIMS m/z 274.1576 [M]+, вычислено для C17H22O3 274.1569, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.45 (q, J=2.1, 1H, H-5); 5.87 (d, J=2.4, 1H, H-6); 5.15 (m, 1H, H-2'); 5.08 (m, 1H, H-6'); 3.82 (s, 3H, OMe); 3.14 (br. d, J=7.3, 2H, Н-1'); 2.07 (m, 4H, H-4', H-5'); 1.70 (br. s, 3H, Me); 1.63 (br. s, 3H, Me); 1.60 (br. s, 3H, Me).
Пример 6
2-Метокси-5-(3',7'-диметил-окта-2',6'-диенил)-[1,4] бензохинон (11): желтые кристаллы, HREIMS m/z 274.1582 [M]+, вычислено для C17H22O3 274.1569, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.46 (t, J=1.7, 1H, Н-6), 5.92 (s, 1H, H-3), 5.15 (m, 1H, H-2'), 5.08 (m, 1H, H-6'), 3.82 (s, 3H, OMe), 3.14 (br. d, J=7.3, 2H, Н-1'), 2.08 (m, 4H, H-4', H-5'), 1.70 (br. s, 3H, Me), 1.62 (br. s, 3H, Me), 1.60 (br. s, 3H, Me).
Пример 7
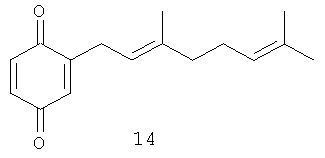
2-(3',7'-Диметил-окта-2',6'-диенил)-[1,4] бензохинон (14): желтое масло, HREIMS m/z 244.1454 [М]+, вычислено для C16H20O2 244.1463, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.77 (d, J=10.2, 1H, H-6); 6.69 (dd, J=10.2, 2.3, 1H, H-5); 6.53 (q, J=1.9, 1H, H-3); 5.15 (m, 1H, H-2'); 5.07 (m, 1H, H-6'); 3.13 (br. d, J=7.5, 2H, Н-1'); 2.08 (m, 4H, H-4', H-5'); 1.69 (br. s, 3H, Me); 1.62 (br. s, 3H, Me); 1.60 (br. s, 3H, Me).
Пример 8
2,3-Диметокси-5-(3',7',11'-триметил-додека-2',6',10'-триенил)-[1,4] бензохинон (7): желтое масло, HREIMS m/z 372.2316 [M]+, вычислено для C23H32O4 372.2300, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.34 (t, J=1.8, 1H, H-6); 5.10 (m, 3H, H-2', H-6', H-10'); 4.02 (s, 3H, ОМе); 4.00 (s, 3H, OMe); 3.11 (br. d, J=7.3, 2H, Н-1'); 2.05 (m, 8H, H-4', Н-5', Н-8', Н-9'); 1.68 (br. s, 3H, Me); 1.62 (br. s, 3H, Me); 1.60 (br. s, 6H, 2Me).
Пример 9
2-Метокси-6-(3',7',11'-триметил-додека-2',6',10'-триенил)-[1,4] бензохинон (8): желтое масло, HREIMS m/z 342.2182 [M]+, вычислено для С22Н30О3 342.2195, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.45 (q, J=2.0, 1H, Н-5); 5.87 (d, J=2.4, 1H, H-3); 5.15 (m, 1H, H-2'); 5.10 (m, 2H, H-6', H-10'); 3.81 (s, 3H, OMe); 3.14 (br. d, J=7.3, 2H, Н-1'); 2.06 (m, 8H, H-4', Н-5', Н-8', Н-9'); 1.67 (br. s, 3H, Me); 1.63 (br. s, 3H, Me); 1.60 (br. s, 6H, 2Me).
Пример 10
2-Метокси-5-(3',7',11'-триметил-додека-2',6',10'-триенил)-[1,4] бензохинон (12): желтые кристаллы, HREIMS m/z 342.2172 [М]+, вычислено для С22Н30О3 342.2195, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.47 (t, J=1.7, 1H, H-6); 5.93 (s. 1H, H-3); 5.16 (m, 1H, H-2'); 5.10 (m, 2H, H-6', H-10'); 3.82 (s, 3H, OMe); 3.14 (br. d, J=7.3, 2H, Н-1'); 2.05 (m, 8H, H-4', Н-5', Н-8', Н-9'); 1.68 (br. s, 3H, Me); 1.62 (br. s, 3H, Me); 1.60 (br. s, 6H, 2Me).
Пример 11
2-Метокси-3-(3',7',11'-триметил-додека-2',6',10'-триенил)-[1,4] бензохинон (13): желтое масло, HREIMS m/z 342.2212 [M]+, вычислено для С22Н30О3 342.2195, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.68 (d, J=10.0, 1H, H-6); 6.57 (d, J = 10.0, 1H, Н-5); 5.07 (m, 3H, H-2', H-6', H-10'); 4.02 (s, 3H, OMe); 3.16 (br. d, J=7.3, 2H, Н-1'); 2.01 (m, 8H, H-4', Н-5', Н-8', Н-9'); 1.73 (br. s, 3H, Me); 1.67 (br. s, 3H, Me); 1.60 (br. s, 3H, Me); 1.57 (br. s, 3H, Me).
Пример 12

2,3-Диметокси-5-(3',7',11'-триметил-додека-2',6',10'-триенил)-бензен-1,4-диол (15): желтое масло, HREIMS m/z 374.2472 [M]+, вычислено для C23H34O4 374.2457, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.49 (s, 1H, H-6); 5.31 (s, 1H, ОН); 5.30 (m, 1H, H-2'); 5.17 (s, 1H, OH); 5.12 (m, 2H, H-6', H-10'); 3.91 (s, 3H, OMe); 3.88 (s, 3H, OMe); 3.28 (br. d, J=7.6, 2H, Н-1'); 2.05 (m, 8H, H-4', H-5', H-8', H-9'); 1.70 (br. s, 3H, Me); 1.68 (br. s, 3H, Me); 1.60 (br. s, 6H, 2Me).
Пример 13
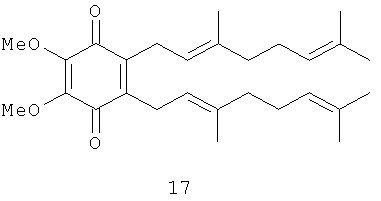
2,3-Диметокси-5,6-бис-(3',7'-диметил-окта-2',6'-диенил)-[1,4] бензохинон (17): желтое масло, HREIMS m/z 440.2944 [М]+, вычислено для C28H40O4, 440.2927, 1Н-ЯМР (250 MHz, CDCl3) δ: 5.04 (m, 2H, H-2', H-2''); 4.94 (m, 2H, H-6', H-6''); 3.99 (s, 6H, 20Me); 3.19 (br. d, J=6.8, 2H, Н-1', H-1''); 2.00 (m, 8H, H-4', H-5', H-4'', H-5''); 1.73 (br. s, 6H, 2Me); 1.66 (br. s, 6H, 2Me); 1.58 (br. s, 6H, 2Me).
Пример 14
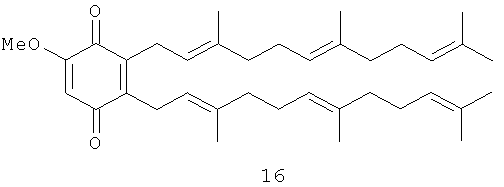
2-Метокси-5,6-бис-(3',7',11'-триметил-додека-2',6',10'-триенил)-[1,4]бензохинон (16): желтое масло, HREIMS m/z 546.4048 [М]+, вычислено для C37H54O3, 546.4073, 1Н-ЯМР (250 MHz, CDCl3) δ: 5.87 (s, 1H, H-3); 5.00 (m, 6H, H-2'. H-6', H-10', H-2'', H-6'', H-10''); 3.79 (s, 3H, OMe); 3.22 (br. d, J=6.8, 4H, Н-1', H-1''); 2.01 (m, 16H, H-4', H-5', H-8', H-9', H-4'', H-5'', H-8'', H-9''); 1.73 (m, 3H, Me); 1.67 (m, 9H, 3Ме); 1.60 (m, 12H, 4Ме).
Пример 15

2-Метокси-3,5-бис-(3',7',11'-триметил-додека-2',6',10'-триенил)-[1,4]бензохинон (18): желтое масло, HREIMS m/z 546.4052 [M]+, вычислено для C37H54O3, 546.4073, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.33 (t, J=1.2, 1H, H-6); 5.09 (m, 6H, H-2', Н-6', Н-10', Н-2'', Н-6'', Н-10''); 4.00 (s, 3Н, ОМе); 3.14 (m, 4Н, Н-1', Н-1''); 2.04 (m, 16H, Н-4', Н-5', Н-8', Н-9', Н-4'', Н-5'', Н-8'', Н-9''); 1.74 (m, 3Н, Me); 1.68 (m, 9H, 3Ме); 1.60 (m, 12H, 4Ме).
Пример 16
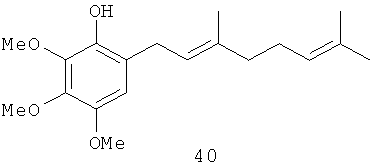
2,3,4-Триметокси-6-(3',7'-диметил-окта-2',6'-диенил)-фенол (40): желтое опалесцирующее масло, HREIMS m/z 320.1974 [M]+, вычислено для C19H28O4 320.1987, ИК (CCl4): 3541, 2935, 1498, 1464 см-1. 1Н-ЯМР (250 MHz, CDCl3) δ: 6.44 (s, 1H, Н-5), 5.45 (s, 1H, ОН), 5.31 (m, 1H, H-2'), 5.11 (m, 1H, Н-6'), 3.95 (s, 3Н, ОМе), 3.86 (s, 6H, ОМе), 3.79 (s, 3Н, ОМе), 3.31 (br. d, J=7.1, 2H, Н-1'), 2.07 (m, 4H, Н-4', Н-5'), 1.72 (d, J=1.2, 3Н, Me), 1.67 (d, J=1.2, 3Н, Me), 1.60 (d, J=0.7. 3Н, Ме). 13С-ЯМР (CDCl3, 62.9 MHz) δ: 16.12 (q, C-10'), 17.66 (q, С-9'), 25.66 (q, C-8'), 26.73 (t, C-5'), 27.90 (t, С-1'), 39.75 (t, C-4'), 56.62 (q, ОМе), 60.89 (q, ОМе), 61.16 (q, ОМе), 108.30 (d, С-5), 121.61 (s, C-6), 121.98 (d, C-2' or C-6'), 124.20 (d C-6' или C-2'), 128.89 (s, C-1), 131.41 (s, C-7'), 136.59 (s, C-4), 140.04 (s, C-3'), 140.81 (s, C-3 или C-2), 146.14 (s, C-2 или С-3).
Пример 17
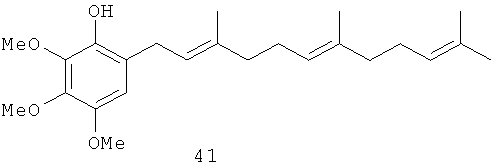
2,3,4-Триметокси-6-(3',7',11'-триметил-додека-2',6',10'-триенил)-фенол (41): желтое опалесцирующее масло, HREIMS m/z 388.2648 [M]+, вычислено для C24H36O4 388.2635, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.44 (s, 1H, Н-5); 5.45 (s, 1H, ОН); 5.32 (br. t, J=7.3, 1H, H-2'); 5.12 (m, 2H, H-6', Н-1''); 3.95 (s, 3Н, ОМе); 3.87 (s, 3Н, ОМе); 3.79 (s, 3Н, ОМе); 3.31 (br.d, J=7.3, 2H, Н-1'); 2.06 (m, 8H, Н-4', Н-5', Н-8', Н-9'); 1.72 (br. s, 3H, Me); 1.67 (br. s, 3H, Me); 1.60 (br. s, 6H, 2Me).
Пример 18
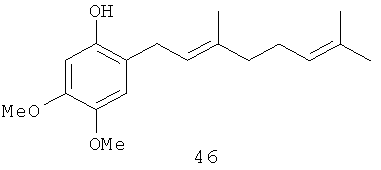
3,4-Диметокси-6-(3',7'-диметил-окта-2',6'-диенил)-фенол (46): желтое опалесцирующее масло, HREIMS m/z 290.1876 [M]+, вычислено для С18Н26О3 290.1882, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.73 (s, 1H, H-2); 6.49 (s, 1H, Н-5); 5.27 (br t, J=7.3, 1H, H-2'); 5.15 (s, 1H, ОН); 5.10 (m, 1H, Н-6'); 3.87 (s, 3H, OMe); 3.79 (s, 3H, OMe); 3.24 (br. d, J=7.3, 2H, Н-1'); 2.05 (m, 4Н, Н-4', Н-5'); 1.68 (br. s, 6H, 2Me); 1.60 (br. s, 3H, Me).
Пример 19
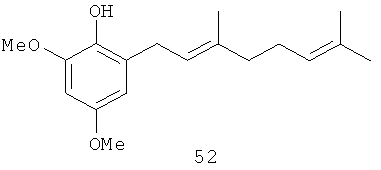
2,4-Диметокси-6-(3',7'-диметил-окта-2',6'-диенил)-фенол (52): желтое опалесцирующее масло, HREIMS m/z 290.1898 [M]+, вычислено для C18H26O3 290.1882, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.36 (d, J=2.7, 1H, Н-3 или Н-5); 6.31 (d, J=2.7, 1H, Н-3 или Н-5); 5.33 (br. t, J=7.1, 1H, H-2'); 5.26 (s, 1H, ОН); 5.11 (br. t, J=6.6, 1H, Н-6'); 3.86 (s, 3H, OMe); 3.75 (s, 3H, OMe); 3.35 (br. d, J=7.3, 2H, Н-1'); 2.08 (m, 4H, Н-4', Н-5'); 1.72 (br. s, 3H, Me); 1.67 (br. s, 3H, Me); 1.60 (br. s, 3H, Me).
Пример 20
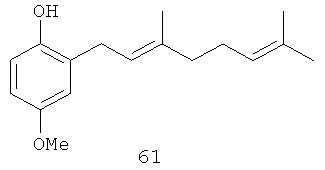
4-Метокси-6-(3',7'-диметил-окта-2',6'-диенил)-фенол (61): желтое опалесцирующее масло, 1Н-ЯМР (250 MHz, CDCl3) δ: 6.69 (m, 3H, Н-3, Н-5, Н-6); 5.31 (br. t, J=7.32, 1H, H-2'); 5.07 (m, 1H, Н-6'); 3.75 (s, 3H, OMe); 3.33 (br. d, J=7.08, 2H, Н-1'); 2.09 (m, 4H, Н-4', Н-5'); 1.76 (br. s, 3H, Me); 1.68 (br. s, 3H, Me); 1.60 (br. s, 3H, Me).
Пример 21. Цитотоксическая активность
Для оценки цитотоксического эффекта репрезентативной выборки патентуемых соединений был использован MTS-метод. Был исследован эффект репрезентативной выборки патентуемых соединений на клеточную выживаемость JB6 Р+ Cl 41 клеток, трансфектных JB6 клеток с NF-κВ люциферазным промотором, трансфектных JB6 клеток с АР-1 люциферазным промотором и клеток рака легких человека НТ-460. Хиноны 5, 9-11, 14, имеющие терпеноидную часть, состоящую из двух изопреновых единиц (или 10 атомов углерода), показали IC50 для JB6 Cl 41 клеток в пределах концентраций от 8,3 до 45,1 µМ, а для НТ-460 клеток - от 25,8 до 81,8 µМ. Хиноны 7, 8, 12, 13, 15, имеющие терпеноидную часть, содержащую 15 атомов углерода (или три изопреновые единицы), показали IC50 для JB6 Cl 41 клеток в пределах концентраций от 3,6 до 29,0 µМ, а для НТ-460 клеток - от 12,1 до 72,0 µМ. Среднее значение IC50 хинонов 5, 9-11, 14 для JB6 Cl 41 клеток было 18,7 µМ, а для НТ-460 клеток - 44,2 µM. Среднее значение IC50 хинонов 7, 8, 12, 13, 15 для JB6 Cl 41 клеток было 10,4 µМ, а для НТ-460 клеток - 30,5 µМ. Следовательно, хиноны 7, 8, 12, 13, 15, характеризующиеся более длинной терпеноидной частью почти в два раза более токсичны для JB6 Cl 41 клеток и в полтора раза более токсичны для НТ-460 клеток, чем хиноны 5, 9-11, 14, имеющие короткую терпеноидную часть. Обе эти группы соединений приблизительно в два раза более токсичны для JB6 Cl 41 клеток (15,9 µМ в среднем), чем для НТ-460 клеток (37,8 µМ в среднем). Третья группа соединений, хиноны 16-18, имеющие две терпеноидных боковых цепи в молекуле, показали наименьшую цитотоксическую активность и для JB6 Cl 41, и для НТ-460 клеток, среди всех трех групп соединений. Цитотоксический эффект хинонов 5, 7-18 по отношению к клеткам рака легких человека (НТ-460) и мышиным эпидермальным JB6 Cl 41 клеткам ранее никем не был опубликован.
Эффект глабрухинона А (5) на клеточную выживаемость оценивался с использованием восстановления MTS-реагента в формазан (CellTiter 96R AQueous One Solution Cell Proliferation Assay, Promega, Madison, WI, США). JB6 P+ Cl 41 или НТ-460 клетки культивировали в течение 12 часов в 96-луночных планшетах (4000 клеток на лунку) с использованием 5% FBS/MEM (для JB6 Р+ Cl 41 клеток) или 10% FBS/RPMI (для НТ-460 клеток). Затем среда была заменена на 0,1% FBS/MEM (для JB6 Р+ Cl 41 клеток) или 0,1% FBS/RPMI (для НТ-460 клеток), содержащую глабрухинон А (5) в различных концентрациях, в объеме 0,1 мл среды на лунку, и клетки инкубировали с веществом в течение 22 часов. Затем 20 µл MTS-реагента добавили в каждую лунку и плотность поглощения продукта восстановления MTS-реагента, формазана, измеряли двумя часами позже, спектрофотометрически, при 492 нм, а также при 690 нм (в качестве фона), с использованием планшетного ридера-спектрофотометра Multiscan MS (Labsystems, Финляндия). Данные представлены на Фиг.8А и 8В в виде процентного содержания живых JB6 P+ Cl 41 или НТ-460 клеток соответственно по сравнению с контрольными, необработанными клетками. На Фиг.9А и 9В показаны IC50, вычисленные для JB6 Р+ Cl 41 или НТ-460 клеток соответственно с использованием линейных регрессий, построенных на основании данных, представленных на Фиг.8А и 8В.
Пример 22. Тест на опухолевую трансформацию нормальных клеток или на фенотипическое проявление раковых клеток в мягком агаре.
Репрезентативная выборка патентуемых соединений ингибирует фенотипическую экспрессию (колонеобразование) в мягком агаре клеток рака легких человека (НТ-460), рака кишечника (НСТ-116), меланомы (SK-MEL-28). Они также ингибируют EGF- или ТРА-индуцированную опухолевую трансформацию JB6 Р+ Cl 41 клеток в мягком агаре. Результаты соответствующих экспериментов представлены в таблице 2 в виде концентраций соответствующих хинонов (в мкМ), при которой на 50% ингибируется опухолевая трансформация JB6 P+ Cl 41 клеток или колонеобразование опухолевых клеток человека по сравнению с необработанными, контрольными клетками. Причем большинство из протестированных веществ 5, 7-18 в значительной степени ингибируют опухолевую трансформацию JB6 P+ Cl 41 клеток в концентрациях, меньших, чем цитотоксические (см. таблицу 2).
Эффект хинонов 5, 7-18 на колонеобразование раковых клеток человка НТ-460, НСТ-116, SK-MEL-28, а также на трансформацию мышиных JB6 P+ Cl 41 клеток в мягком агаре оценивали методом мягкого агара. Ингибирование клеточной трансформации обычно является хорошим показателем того, что вещество будет обладать раково-превентивной активностью. Полученные результаты подтвердили, что репрезентативная выборка патентуемых соединений обладает антираковой профилактической и терапевтической активностями (см. табл. 2). В опытах в мягком агаре хиноны с боковыми цепями, имеющими длину в 10 атомов углерода, продемонстрировали антираковую активность против EGF-индуцированной трансформации JB6 P+ Cl 41 клеток с INCC50 (концентрация, при которой ингибирование числа колоний равно 50% от числа контрольных, необработанных веществами клеток) в 1,4-4 раза меньшей, чем IC50, полученная для этих же веществ на этих же клетках в опытах на цитотоксическую активность. С другой стороны, хиноны с более длинной боковой цепью в 15 атомов углерода показали антираковую превентивную активность по большей части в дозах, которые были равны или даже превосходили d 4-10 раз значения IC50, полученные в опытах на цитотоксическую активность. Следовательно, хиноны, имеющие боковые цепи длиной в 10 атомов углерода, являются более мощными агентами по предупреждению трансформации нормальных клеток в раковые, чем хиноны с боковыми цепями длиной 15 атомов углерода (см. табл. 2). Этот же вывод справедлив и для антираковой терапевтической активности хинонов, которые были исследованы на их действие против колонеобразования НТ-460 клеток в мягком агаре и на цитотоксическую активность против этих же клеток. Данные по IC50 и INCC50 для хинонов 5, 7-18 в этих экспериментах были получены на основании регрессий, построенных с помощью статистической компьютерной программы STATISTICA 6.0 (StatSoft, Inc., США). IC50 означает концентрацию хинона, при которой погибают 50% обработанных клеток по сравнению с необработанными, контрольными клетками. INCC50 означает концентрацию хинона, при которой на 50% ингибируется колонеобразование обработанных клеток по сравнению с необработанными, контрольными клетками.
Антираковые свойства репрезентативной выборки патентуемых соединений по ингибированию колонеобразования человеческих раковых клеток НТ-460 (рак легких), НСТ-116 (рак кишечника), SK-MEL-28 (меланома, рак кожи), а также по ингибированию EGF- или ТРА- индуцированной прораковой трансформации мышиных JB6 P+ Cl 41 клеток ранее никем не был опубликован.
Антираковый профилактический и терапевтический эффект глабрухинона А (5) оценивали с помощью метода мягкого агара. Эксперименты проводили в 6-луночных планшетах. Для оценки антиракового терапевтического эффекта были использованы клетки рака кишечника человека НСТ-116 линии. Для оценки антиракового профилактического эффекта были использованы EGF (10 нг/мл)- или ТРА (20 нг/мл)-активированные JB6 P+ Cl 41 клетки. Метод мягкого агара является широко распространенным инструментом, который применяется для того, чтобы определить -является ли данное вещество потенциально эффективным антираковым агентом для применения на животных или человеке. НСТ-116 или JB6 P+ Cl 41 клетки (8×103 на мл) были обработаны указанными концентрациями глабрухинона А в 1 мл ВМЕ (среда Игла), содержащей 0,33% агара и 10% FBS поверх 3,5 мл ВМЕ, содержащей 0,5% агара и 10% FBS, а также указанные концентрации глабрухинона А. Экспериментальные планшеты инкубировали при 37°С и 5% СО2 в инкубаторе в течение 1 недели (для НСТ-116 или JB6 Р+ Cl 41 клеток, активированных EGF) или 2 недель (JB6 Р+ Cl 41 клеток, активированных ТРА). Колонии клеток подсчитывали затем с помощью LEICA DM IRB инвертированного микроскопа (Leica Mikroskopie und Systeme GmbH, Germany) и компьютерной программы Image Pro Plus 3.0 для Windows (Media Cybernetics, Silver Spring, MD, США). Эффекты глабрухинона А на колонеобразование НСТ-116 клеток (Фиг.10А) или на EGF-индуцированную трансформацию JB6 Р+ Cl 41 клеток (Фиг.11А) или на ТРА-индуцированную трансформацию JB6 Р+ Cl 41 клеток (Фиг.12А) представлены в виде процента колонеобразования по сравнению с необработанными глабрухиноном, контрольными клетками. На Фиг.10В, 11В или 12В представлены вычиления INCC50 глабрухинона А для колонеобразования НСТ-116 клеток, EGF-индуцированной трансформации JB6 P+ Cl 41 клеток или ТРА-индуцированной трансформации JB6 P+ Cl 41 клеток соответственно с использованием регрессий, основанных на данных Фиг.10А, 11А, 12А.
Методы
Канцерпривентивное действие представленных соединений оценивали в шестилуночных планшетах с использованием EGF (10 нг/мл)- или ТРА (20 нг/мл)-активированных JB6 P+ Cl 41 клеток. Метод мягкого агара является широко распространенным инструментом, который применяется для того, чтобы определить -является ли данное вещество потенциально эффективным антираковым агентом для применения на животных или человеке. JB6 P+ Cl 41 клетки (8×103 на мл) были обработаны указанными концентрациями хинонов в 1 мл ВМЕ (среда Игла), содержащей 0,33% агара и 10% FBS поверх 3,5 мл ВМЕ, содержащей 0,5% агара и 10% FBS, а также указанные концентрации хинонов. Экспериментальные планшеты инкубировали при 37°С и 5% СО2 в инкубаторе в течение 1 недели (для JB6 Р+ Cl 41 клеток, активированных EGF) или 2 недель (для JB6 Р+ Cl 41 клеток, активированных ТРА). Колонии клеток подсчитывали затем с помощью LEICA DM IRB инвертированного микроскопа (Leica Mikroskopie und Systeme GmbH, Germany) и компьютерной программы Image Pro Plus 3.0 для Windows (Media Cybernetics, Silver Spring, MD, США). Для каждого соединения были сделаны два независимых эксперимента, в трипликате для каждой концентрации.
Пример 23. Тесты на апоптоз с использованием методов проточной цитометрии и ДНК-лестницы.
Репрезентативная выборка патентуемых соединений индуцирует апоптоз в JB6 Р+ Cl 41 клетках, лимфобластах человека JY-клетках, COX2-/- и СОХ2+/+ МЭФ, а также человеческих раковых клетках НТ-460 (рак легких), НСТ-116 (рак кишечника) и SK-MEL-28 (меланома, рак кожи). К примеру, индукция апоптоза хинонами 5, 7-18 была оценена методом проточной цитометрии в мышиных JB6 Р+ Cl 41 клетках. Индукция апоптоза глабрухиноном А также была исследована методом проточной цитометрии в JY-клетках, а также человеческих раковых клетках НТ-460 (рак легких), НСТ-116 (рак кишечника) и SK-MEL-28 (меланома, рак кожи). Индукция апоптоза глабрухиноном А также была исследована методом ДНК-лестницы в COX2-/- и COX2+/+ МЭФ (Фиг.14).
Способность репрезентативной выборки патентуемых соединений индуцировать апоптоз в JB6 Р+ Cl 41 клетках, лимфобластах человека JY-клетках, СОХ2-/- и COX2+/+ МЭФ, а также человеческих раковых клетках НТ-460 (рак легких), НСТ-116 (рак кишечника) и SK-MEL-28 (меланома, рак кожи) ранее никем не была опубликована.
Индукция раннего и позднего апоптоза глабрухиноном А анализировали на проточном цитометре Becton Dickinson FACs Calibur Flow Cytometer (BD Biosciences, San Jose, CA, США). JB6 P+ Cl 41, JY, COX2-/- или COX2+/+. НТ-460, НСТ-116 или SK-MEL-28 клетки (3×105 клеток на чашку), культивировали в 6-см чашках в течение 24 часов в 10% FBS/MEM для JY и SK-MEL-28 клеток, 5% FBS/MEM для JB6 P+ Cl 41 клеток, 10% FBS/RPMI для НТ-460 или 10% FBS/McCoy's для НСТ-116 клеток. Клетки затем обрабатывали глабрухиноном А в 0,1% FBS/среде в течение 3-х часов для JB6 P+ Cl 41, JY, HT-460, и SK-MEL-28 клеток, или в течение 24-х часов для НСТ-116 клеток. Затем клетки (кроме неприкрепленных JY) трипсинизировали 0,025% трипсином в 0,1% растворе EDTA в PBS-буферном растворе. Трипсинизация была остановлена добавлением 2 мл 5% FBS в PBS-буферном растворе. Клетки были промыты PBS-буферным раствором методом центрифугирования при 1000 об/мин (170 rcf) в течение 5 мин и обработаны для обнаружения раннего и позднего апоптоза с использованием флуоресцентных красителей Annexin V-FITC и PI (пропидиум-иодид) в соответствии с протоколом от производителя. Коротко: 1-5×105 клеток собрали после центрифугирования и ресуспендировали в 500 µл связывающего буферного раствора (Annexin V-FITC Apoptosis Detection Kit, Medical & Biological Laboratories (Waterton, MA, США)). Затем 5 µл Annexin V-FITC и 5 µл PI были добавлены и клетки инкубировали в течение 5 мин в темноте, при комнатной температуре, а затем анализировали в проточном цитометре. Апоптоз, индуцированный глабрухиноном А в вышеупомянутых клетках, показан на Фиг.13А, В, С, D, Е.
СОХ2-/- и СОХ2+/+ МЭФ выращивали в 10 см чашках Петри и обработали глабрухиноном А по достижении 80% конфлюентности в течение 24-х часов. Затем как прикрепленные, так и неприкрепленные клетки были собраны путем соскабливания с последующим центрифугированием. Затем клетки лизировали лизисным буферным раствором (5 мМ Tris-HCl, рН 8.0, 20 мМ EDTA и 0.5% Triton X-100) и оставили на льду в течение 45 мин. После центрифугирования при 14000 об/мин (45 мин, 4°С), супернатант, содержащий фрагментированную ДНК, экстрагировали дважды смесью фенол-хлороформ-изопропиловый спирт (25:24:1, по объему) и один раз хлороформом. Затем фрагментированную ДНК переосадили в течение ночи при -20°С, после добавления двух объемов 100% этанола и 1/10 объема 5 М NaCl. Высажденную ДНК центрифугировали при 14000 об/мин в течение 45 мин, отмыли 70% этанолом, высушили и ресуспендировали в ТЕ-буферном растворе (10 мМ Tris-HCl, 1 мМ EDTA, рН 8.0). После добавления 100 µг/мл РНК-зы A (Sigma), смесь инкубировали при 37°С в течение 2 часов. Фрагменты ДНК разделяли электрофорезом на 1,8% агарозном геле. ДНК-лестницу в геле прокрашивали этидиум-бромидом и фотографировали в УФ-свете. Результат показан на Фиг.14.
Пример 24. Тест на АР-1- и NF-κВ-зависимую транскрипционную активность
Несколько ключевых транскрипционных ядерных факторов, включая р53, АР-1 или NF-κВ обычно участвуют в индукции или ингибировании апоптоза в клетке, в ответ на различные внешние стимулы, включая воздействие хемопревентивных веществ или антиопухолевых препаратов. Поэтому был исследован эффект репрезентативной выборки патентуемых соединений 5, 7-18 на эти три ключевых ядерных фактора транскрипции. Для этого использовали JB6 Cl 41 клеточные линии со стабильно экспрессированным репортерным геном люциферазы, контролируемым р53, АР-1 и/или NF-κВ-связанным сиквенсом. Репрезентативная выборка патентуемых соединений показала значительную (до 8 раз) индукцию АР-1 и/или NF-κВ-зависимой транскрипционной активности и также значительное (до 4-х раз) ингибирование р53-зависимой транскрипционной активности (см. таблицу 3).
Способность репрезентативной выборки патентуемых соединений индуцировать АР-1 и/или NF-κВ-зависимую транскрипционную активность в мышиных JB6 Cl 41 клетках ранее никем не была опубликована. Результаты для глабрухинона А (5) показаны на Фиг.15А, В, где изображены как АР-1 и/или NF-κВ-зависимая транскрипционная активность, выраженная в процентах по отношению к необработанным контрольным клеткам.
Методы. Способность представленных соединений влиять на АР-1-, NF-κВ-, и р53-зависимую транскрипционную активность в мышиных JB6 Cl 41 клетках оценивалась люцефиразным методом. JB6-LucPG-13, JB6-LucAP-l или JB6-LucNF-кB клетки (6×103), суспендированные в 100 мкл 5% FBS-MEM добавили в каждую лунку 96-луночного планшета. Планшеты инкубировали в течение 24 час и затем обработали различными концентрациями хинонов в 100 µл 0,1% FBS/MEM. После инкубирования с хинонами в течение 24 часов, клетки экстрагировали в течение 1 часа при комнатной температуре 100 µл на лунку лизисного буферного раствора (0.1 М Na-фосфатный буферный раствор, рН 7.8, 1% Triton Х-100, 1 мМ DTT, 2 мМ EDTA). Затем 30 µл лизата из каждой лунки были перенесены в планшет для люминесцентного анализа и люциферазную активность измеряли с использованием 100 µл на лунку буферного раствора для люциферазного теста (1 мМ D-люциферина, рН 6.1-6.5; 40 мМ трицин; 2.14 мМ карбоната магния (MgCO3)4×Mg(OH)2×5H2O; 5.34 мМ MgSO4×7H2O, 66.6 мМ DTT, 1.06 мМ АТФ; 0.54 мМ коэнзима А, 0.2 мМ EDTA, рН 7.8) и планшетного ридера Luminoscan Ascent Type 392 (Labsystems, Финляндия). Для каждого соединения были проделаны два независимых эксперимента, по пять образцов для каждой концентрации.
Пример 25. Антираковая превентивная и терапевтическая активность
Изучение антираковой превентивной и терапевтической активности 3-деметилубихинона Q2 (5) проводили с помощью современного метода магнитно-резонансной томографии, на белых нелинейных мышах с привитой карциномой Эрлиха. После всего лишь одной превентивной или терапевтической инъекции 3-деметилубихинона Q2 (5), размеры опухоли значительно (на 25-50%) уменьшались по сравнению с аналогичной опухолью у контрольных, необработанных мышей. Антираковая превентивная и терапевтическая активность 3-деметилубихинона Q2 (5) против карциномы Эрлиха проявлялась при относительно нетоксичных дозах. Этот вывод был подтвержден изучением токсичности 3-деметилубихинона Q2 для белых нелинейных мышей по методу Карбера (G. Karber, Arch. Exp. Pathol. Pharm., 1931, v. 162, p.480).
Токсичность 3-деметилубихинона Q2 (5) для мышей.
С помощью метода Карбера было показано, что LD100 глабрухинона А для мышей составляет 60 мг/кг, в то время как LD50 была равна 35 мг/кг. Медицинская доза глабрухинона А для мышей была выбрана равной 30 мг/кг, раствор в 50% ДМСО или 50% этаноле в воде.
Магнитно-резонансная томография (МРТ).
МРТ является мощным и надежным методом для исследований на животных моделях. Мониторинг роста или ингибирования опухолей в ответ на химиотерапию in vivo - одно из наиболее часто используемых применений МРТ. Преимуществами этого метода являются возможность получения холистических изображений опухоли в любой проекции на животных моделях и в отсутствие радиации.
Изучение ингибирования солидного варианта карциномы Эрлиха после обработки 3-деметилубихиноном Q2 проводили in vivo на белых нелинейных мышах. 3-деметилубихинон Q2 инжектировали в мышей в виде раствора в смеси ДМСО с водой (1:1) или этанола с водой (1:1). После обработки 3-деметилубихиноном Q2 в ДМСО или этаноле с водой мыши теряли около 0,5-1% веса тела. Размер солидной карциномы Эрлиха в мышах, обработанных 3-деметилубихиноном Q2 превентивно (за сутки до прививки опухоли) был значительно (на ≈ 50%) меньше по сравнению с контрольными, необработанными мышами (Фиг.16 и 17). Когда 3-деметилубихинон Q2 использовали как терапевтический препарат и инжектировали в мышей через сутки после прививки опухоли, ингибирование размеров опухоли составляло около 25% (Фиг.18). Таким образом, хинон (5) и другие патентуемые соединения могут быть использованы как канцерпревентивные или терапевтические агенты для лечения карцином.
Кроме измерения размеров опухоли, у экспериментальных мышей также проводили мониторинг прижизненного состояния печени, селезенки и тимуса. Не было обнаружено никаких значительных изменений в размерах, виде или общем состоянии этих иммунокомпетентных органов у мышей, которым вводили 3-деметилубихинон Q2, по сравнению с контрольными, не обработанными мышами. Такие же результаты были получены после усыпления, вскрытия мышей и измерения этих же самых органов.
Методы
Часть 1. Изучение острой токсичности на мышах. Токсичность 3-деметилуби-хинона Q2 для мышей определяли по методу Карбера (G. Karber, Arch. Exp. Pathol. Pharm., 1931, v. 162, p.480). Коротко: для экспериментов использовали белых нелинейных мышей, весом 20 г каждая. 3-Деметилубихинон Q2 растворяли в смеси ДМСО и воды (1:1) или этанола и воды (1:1) и инжектировали в мышь интраперитонеально в указанных концентрациях и в объеме 0.1 мл. Токсичность оценивали по формуле Карбера:
LD50=LD100-Σ(z×d)/m. LD50 или LD100 означает гибель 50% или 100% экспериментальных мышей соответственно; z равно половине числа животных, погибших от двух последних смежных доз; d равно интервалу между каждыми двумя смежными дозами; m равно числу животных, использованных для исследования каждой концентрации вещества.
Часть 2. Магнитно-резонансная томография. Изучение противораковой или раково-превентивной активности 3-деметилубихинона Q2 проводили с использованием современного, неагрессивного технического инструмента, магнитно-резонансного томографа "PharmaScan US 70/16 (Brucker, Germany), который снабжен сверхпроводящим магнитом мощностью 7 Тесла и частотой 300 МГц. Для экспериментов использовали белых нелинейных мышей, весом 20 г каждая. Асцитная опухоль карциномы Эрлиха вводили животным под правую лопатку в концентрации 5 млн/мл. Обработку 3-деметилубихиноном Q2 в одном варианте начинали за сутки до инокуляции опухоли (в случае экспериментов на раково-превентивную активность), а в другом - через сутки после инокуляции опухоли (если исследовали противораковую терапевтическую активность). 3-Деметилубихинон Q2 растворяли в смеси ДМСО и воды (1:1) или этанола и воды (1:1) и инжектировали в мышь интраперитонеально, однократно или двукратно, в объеме 0.1 мл, в обозначенное время и при финальной концентрации 30 мг/кг. Противоопухолевый превентивный и терапевтический эффекты 3-деметилубихинона Q2 оценивали после измерения размеров солидной карциномы Эрлиха на 6-й, 9-й, 12-й, 15-й и 18-й день после трансплантации опухоли.
Мышей анестезировали с помощью раствора ксилазина, 1 мг/мл (SPORA, Praha), при финальной концентрации 0,3 мг/кг и помещали в прибор в положении навзничь так, чтобы опухоль была сосредоточена в центре поверхности катушки. Радиочастота была адаптирована для экспериментов с мышами. Т2-взвешенный спино-эхосиквенс был использован при следующих параметрах: время повтора/ко времени эха, 2579,8/44,5 мсек; FOV 3,2×3,2 см; время аквизиции 3,46 мин; размер матрицы 128×128; толщина слоев 1 мм; расстояние между слоями 1,5 мм. Усиление и отношение сигнал/шум были определялись отдельно для каждой сессии сканирования. Т2-взвешенные изображения RARE_8 давали наилучший контраст между опухолью и окружающими нормальными тканями. Животных, включая необработанных контрольных особей, изучали методом МРТ каждый третий день в течение экспериментов. Объем опухоли определяли исходя из двух ортогональных наборов RARE_8 изображений, включающих всю опухоль. Изображения анализировали с использованием ROI (Region of Interest Tool), компьютерная программа для томографии Para Vision, версия 3.0.1. Статистический анализ данных проводили с использованием непараметрического метода Манн-Уитни U-тест. Раково-превентивный или терапевтический эффекты 3-деметилубихинона Q2 для мышей представлены на Фиг.16-18, как процент ингибирования роста опухоли у экспериментальных мышей по сравнению с контрольными, необработанными веществом мышами.
Пример 26
Следующие иллюстрации представляют состав фармацевтических форм, содержащих патентуемые вещества ("Вещество X"), для терапевтического или профилактического использования человеком. Эти формулировки могут быть получены с помощью обычных процедур, широко известных в кругах фармацевтов.
Все публикации, патенты и патентные документы, поименованные здесь, включены индивидуально, по ссылкам. Изобретение было описано со ссылками на различные специальные и предпочтительные описания и методы. Однако необходимо понимать, что могут быть созданы другие многочисленные вариации и модификации, которые будут сделаны в духе и букве настоящего патента.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ 1,2-ДИТИОЛ-3-ТИОНА ИЛИ 1,2-ДИТИОЛ-3-ОНА И ЛЕКАРСТВЕННЫЕ СРЕДСТВА НА ИХ ОСНОВЕ | 2013 |
|
RU2534525C9 |
| СРЕДСТВО, ВОЗДЕЙСТВУЮЩЕЕ НА АКТИВНОСТЬ НЕКОТОРЫХ ЯДЕРНЫХ ФАКТОРОВ ТРАНСКРИПЦИИ | 2011 |
|
RU2457840C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ КАННАБИГЕРОЛА | 2015 |
|
RU2684913C2 |
| СРЕДСТВО, ПРЕДОТВРАЩАЮЩЕЕ ТРАНСФОРМАЦИЮ НОРМАЛЬНЫХ КЛЕТОК МЛЕКОПИТАЮЩИХ В ОПУХОЛЕВЫЕ | 2010 |
|
RU2429840C1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2009 |
|
RU2414920C1 |
| 6-ГИДРОКСИНАФТОХИНОНЫ ЛАБДАНОВОГО ТИПА, ОБЛАДАЮЩИЕ ЦИТОТОКСИЧЕСКОЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К ОПУХОЛЕВЫМ КЛЕТКАМ ЧЕЛОВЕКА | 2012 |
|
RU2479582C1 |
| СПИРОПРОИЗВОДНЫЕ ПАРТЕНИНА В КАЧЕСТВЕ НОВЫХ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2009 |
|
RU2499798C2 |
| СРЕДСТВО, ПРЕДОТВРАЩАЮЩЕЕ ТРАНСФОРМАЦИЮ НОРМАЛЬНЫХ КЛЕТОК МЛЕКОПИТАЮЩИХ В ОПУХОЛЕВЫЕ | 2010 |
|
RU2429839C1 |
| 16α,17α-ЦИКЛОГЕКСА-17β-(2'-ГИДРОКСИЭТИЛ)-13β-МЕТИЛГОНА-1,3,5(10)-ТРИЕН-3-ОЛ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2015 |
|
RU2601423C1 |
| ПРОИЗВОДНОЕ ТРИАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2022 |
|
RU2832307C2 |
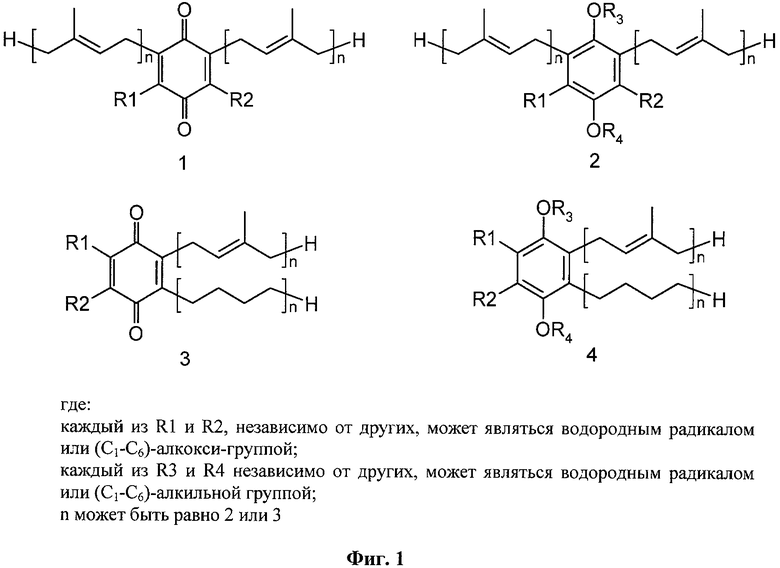
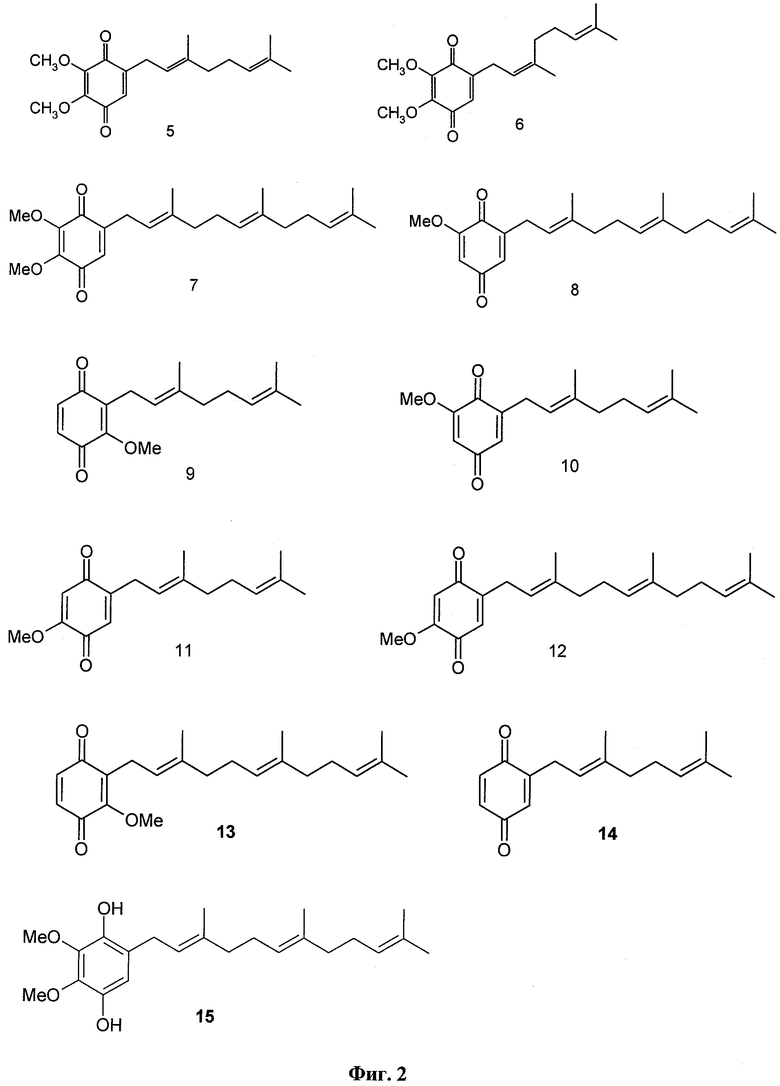
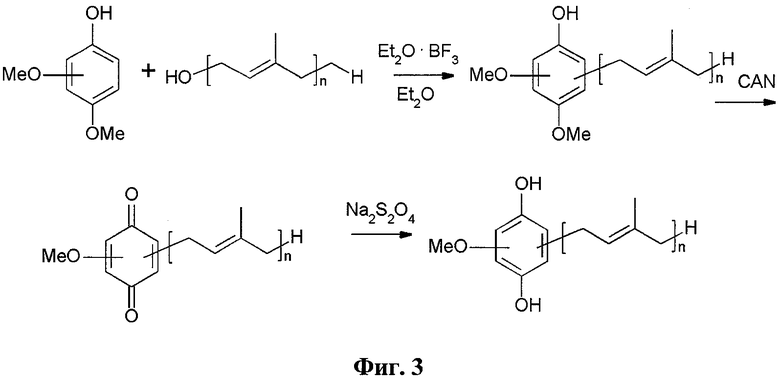

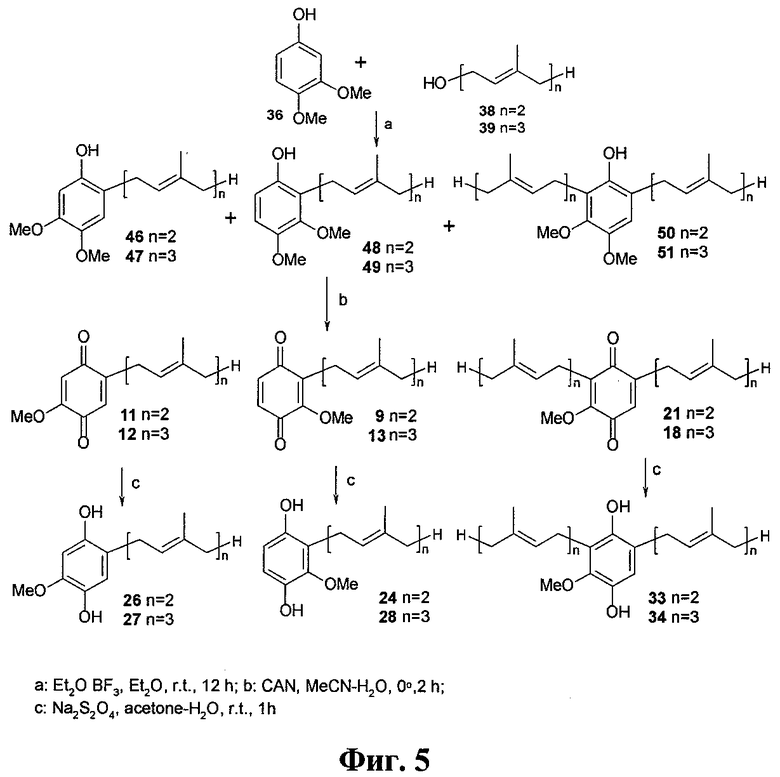
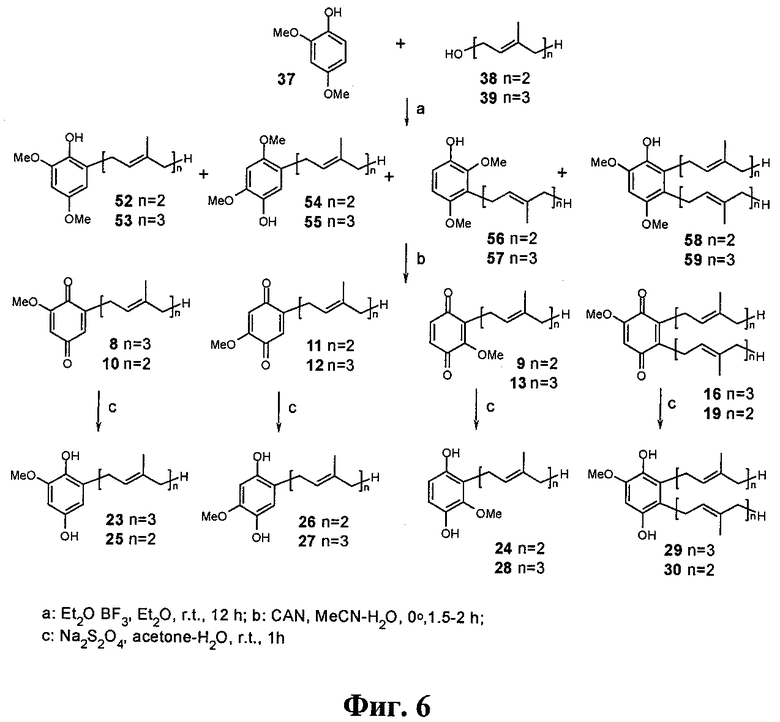
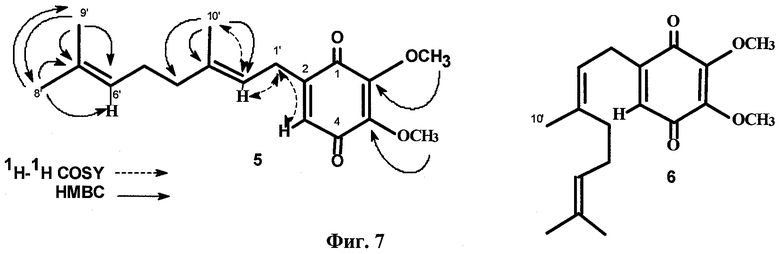

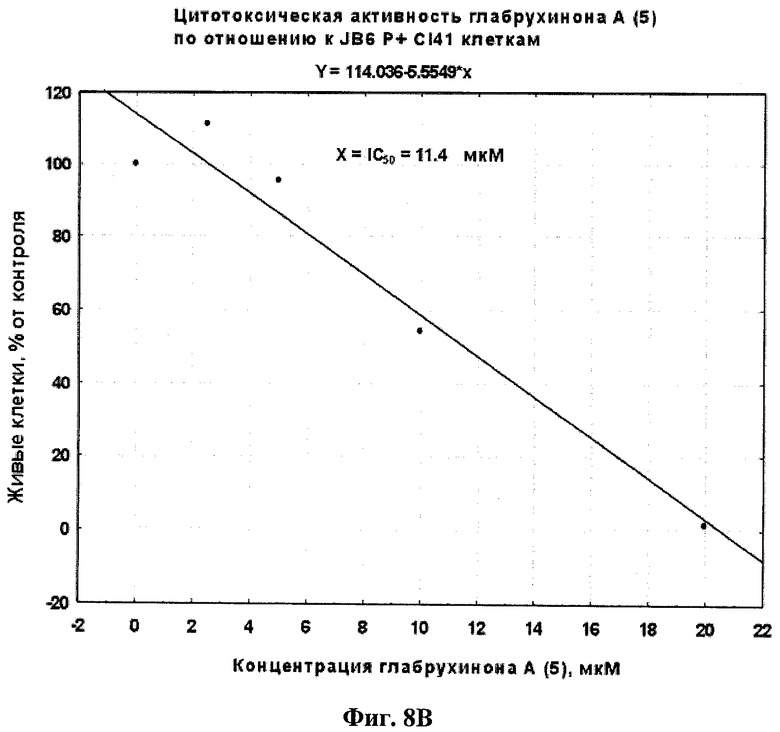
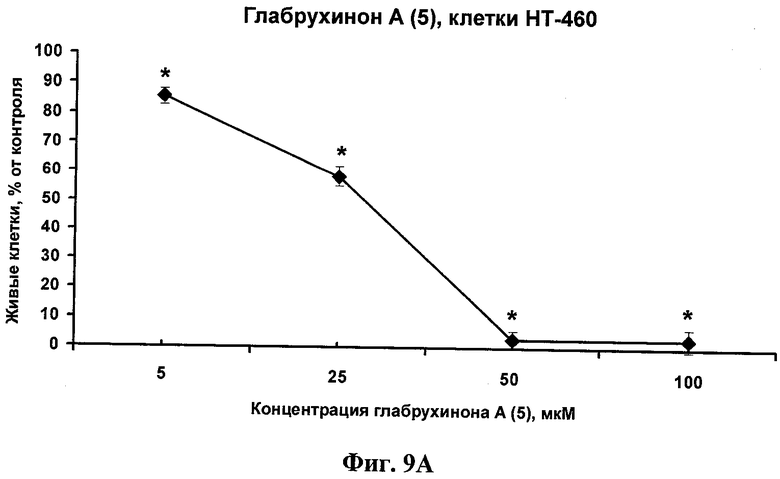
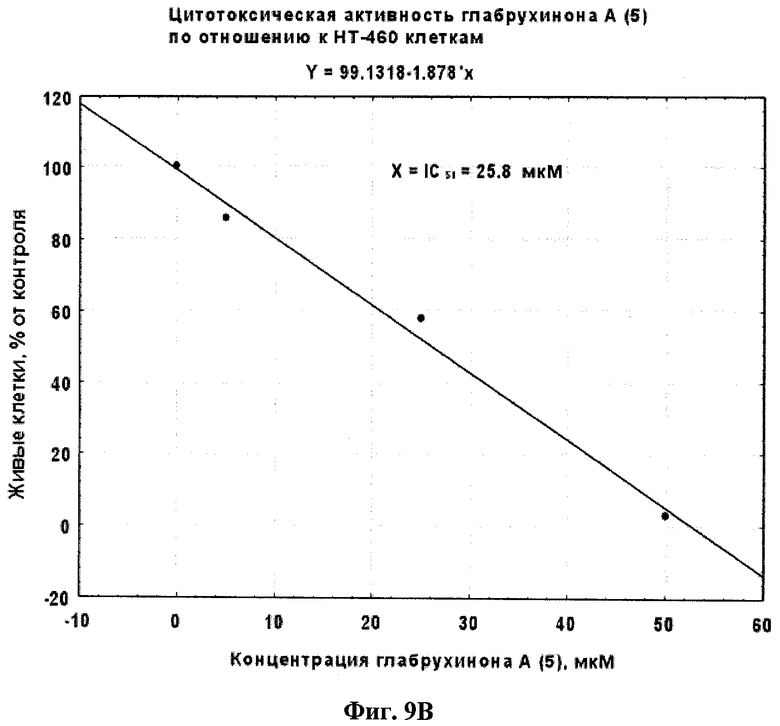

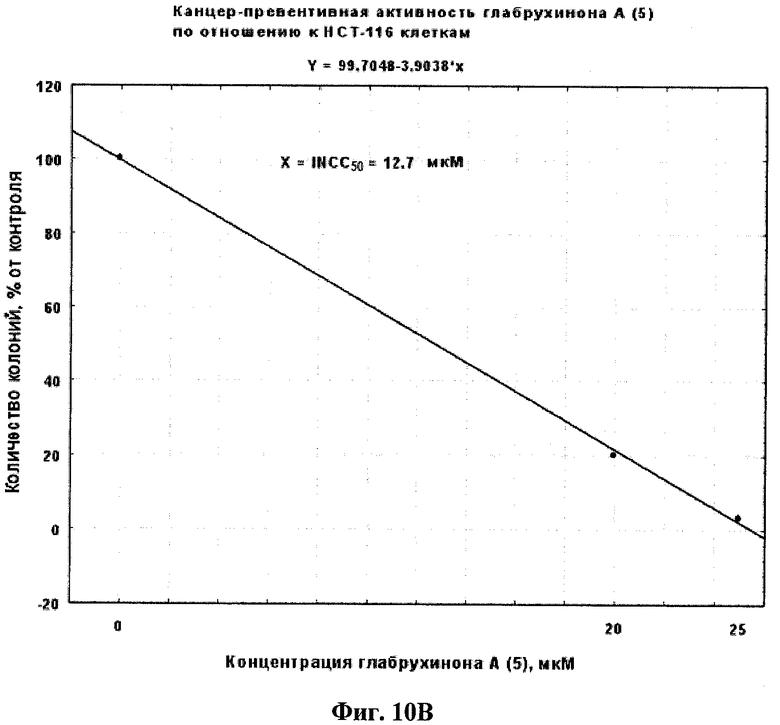
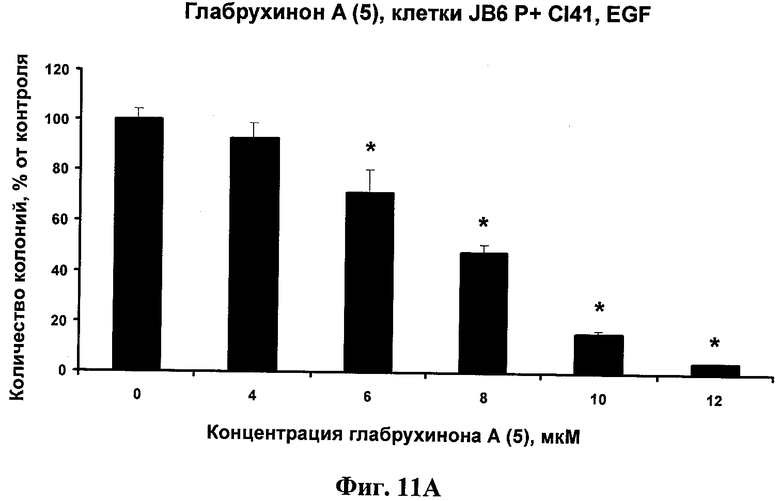
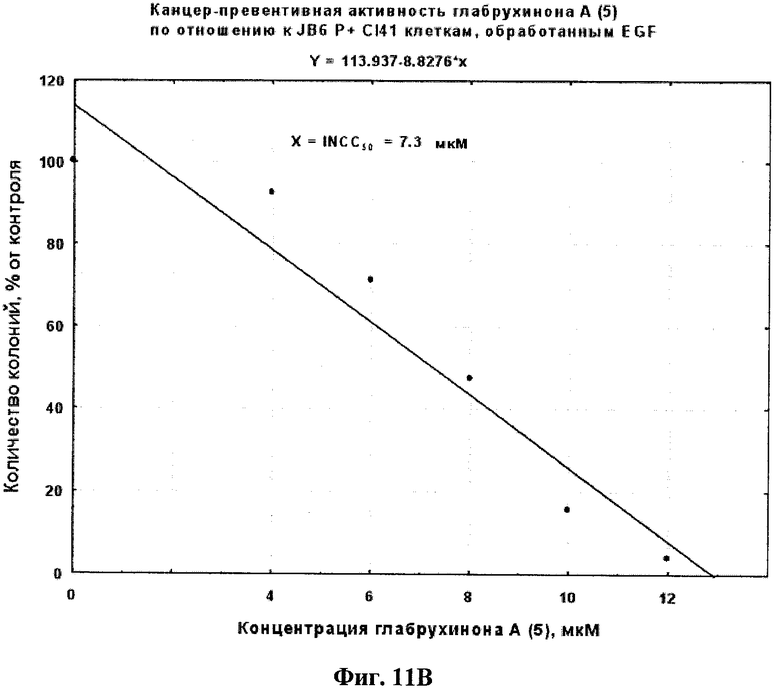
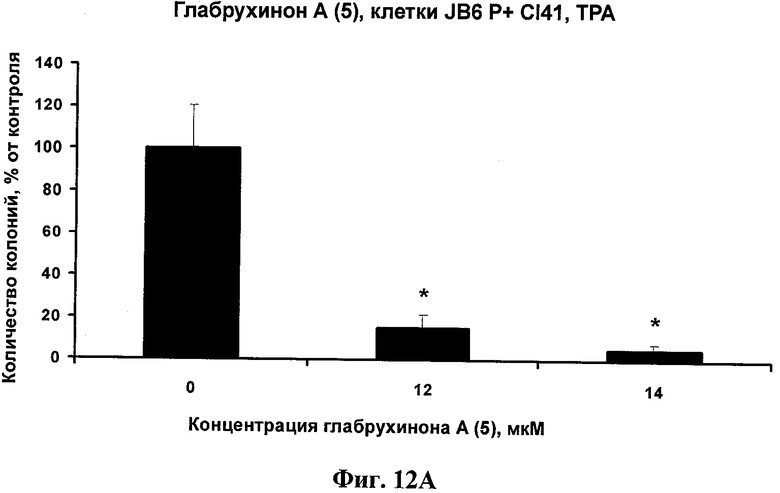

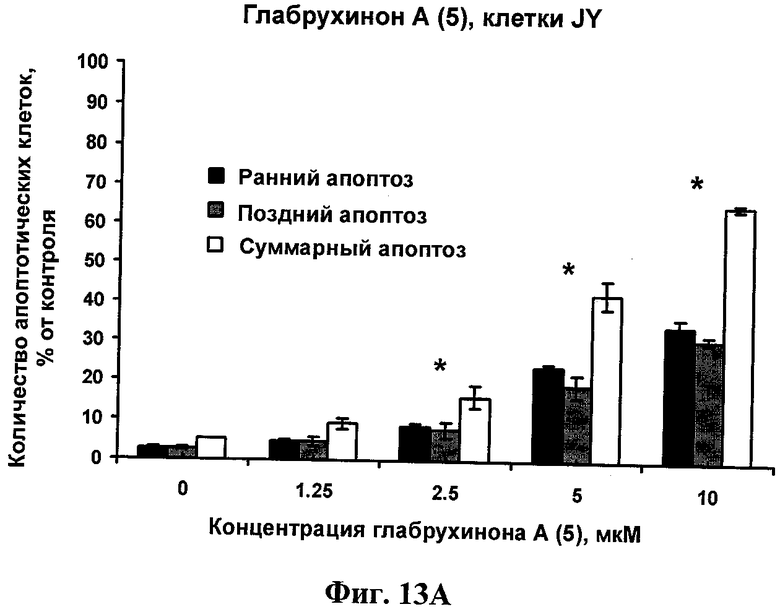
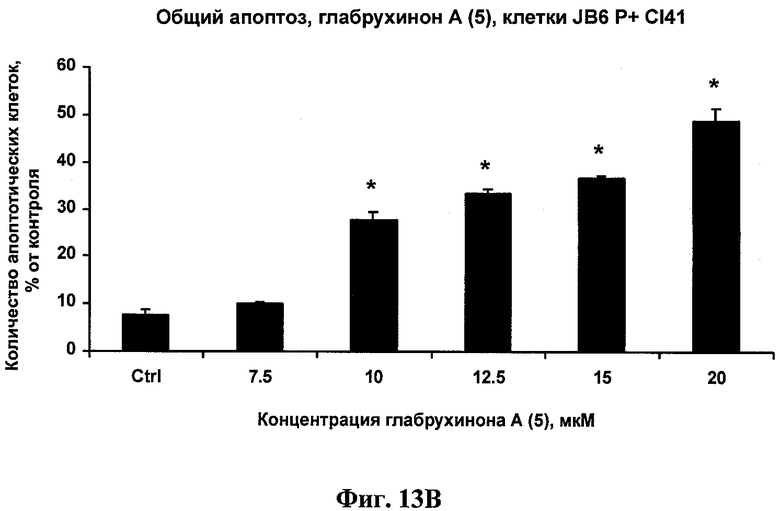
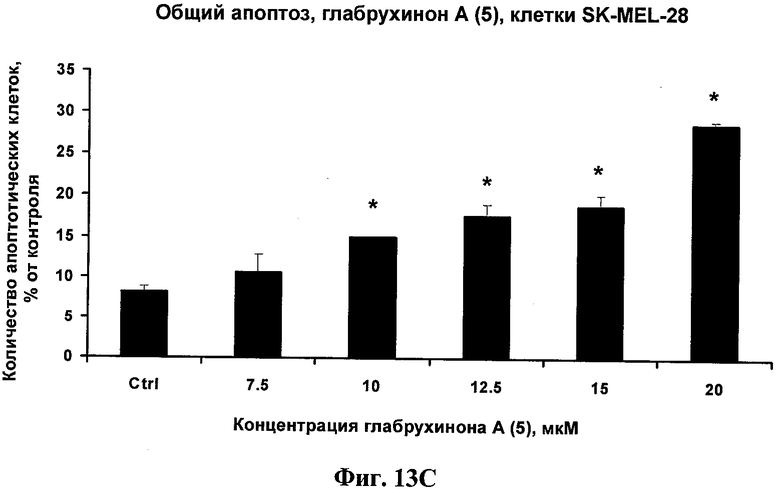
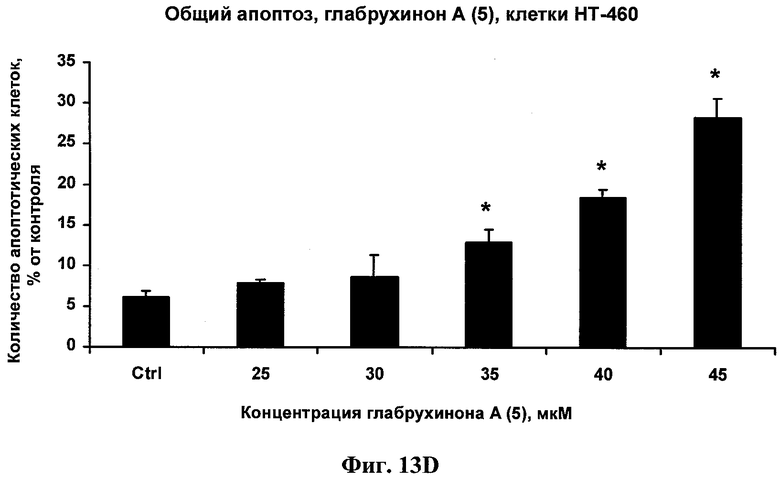
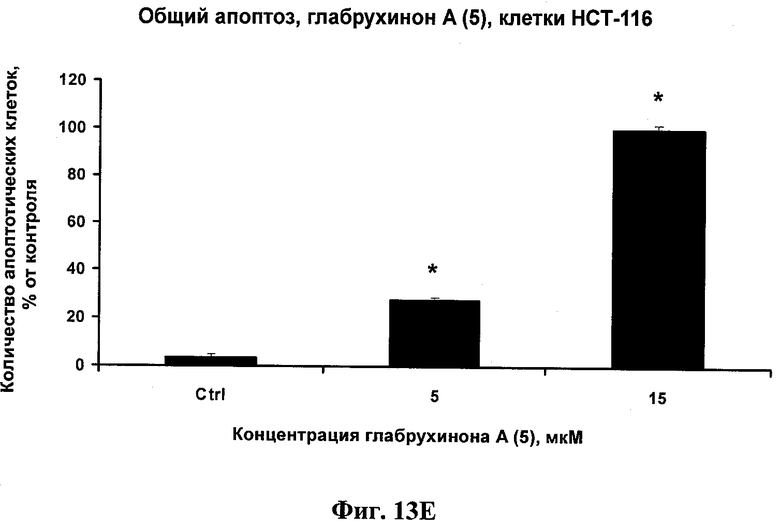
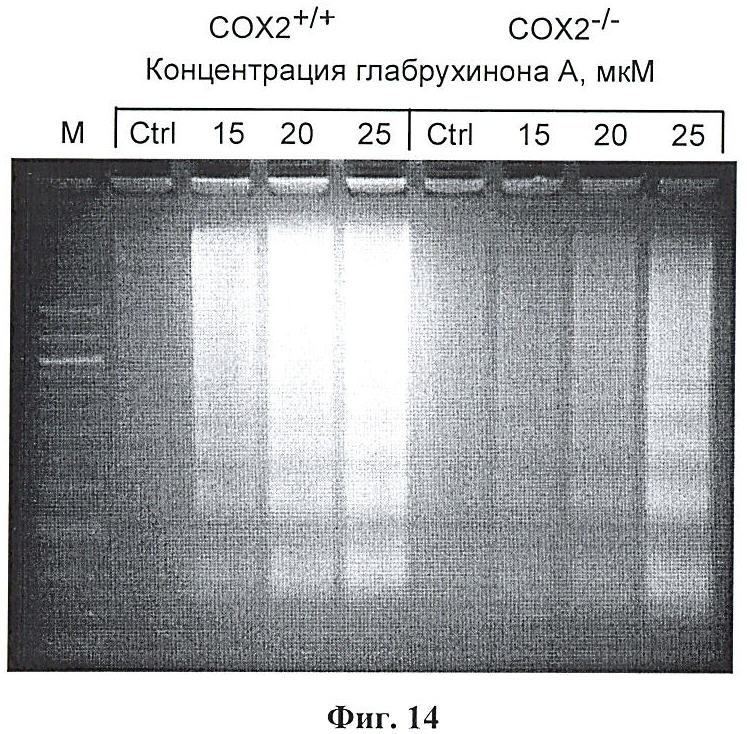

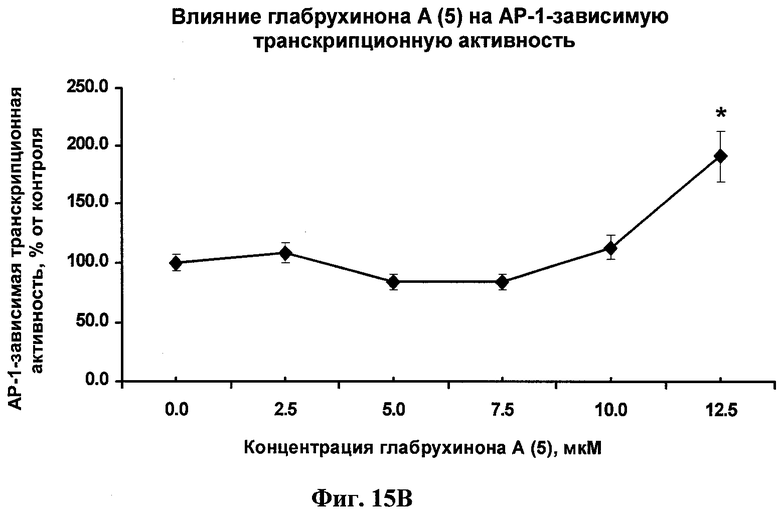
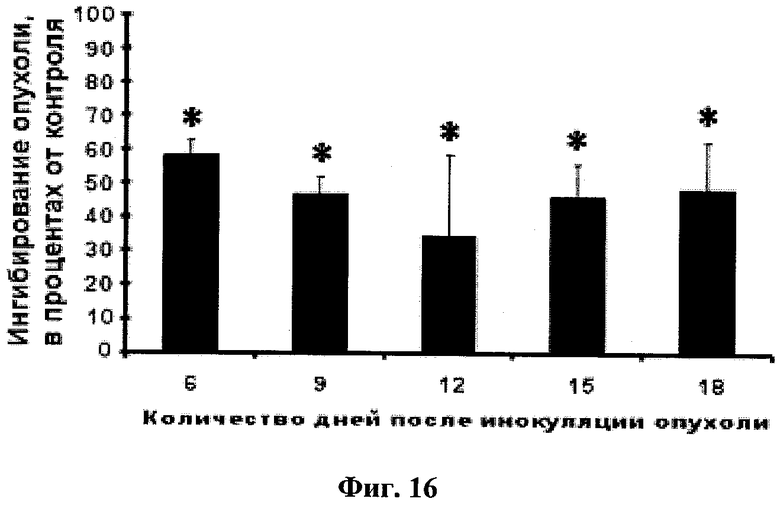
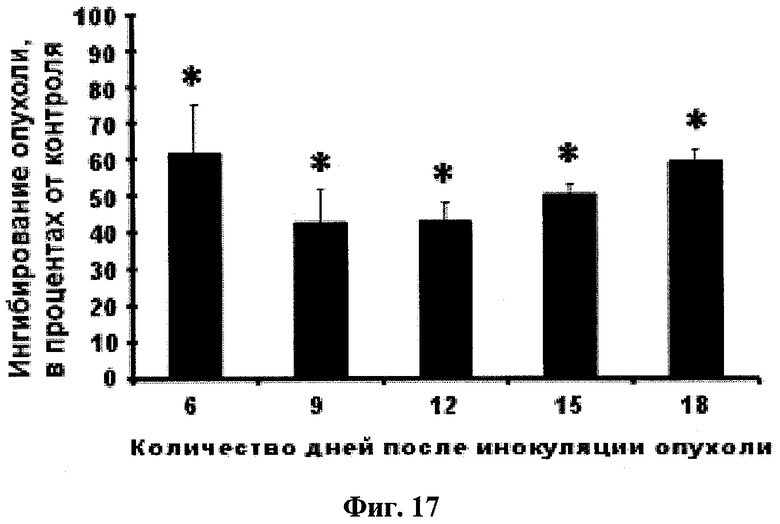
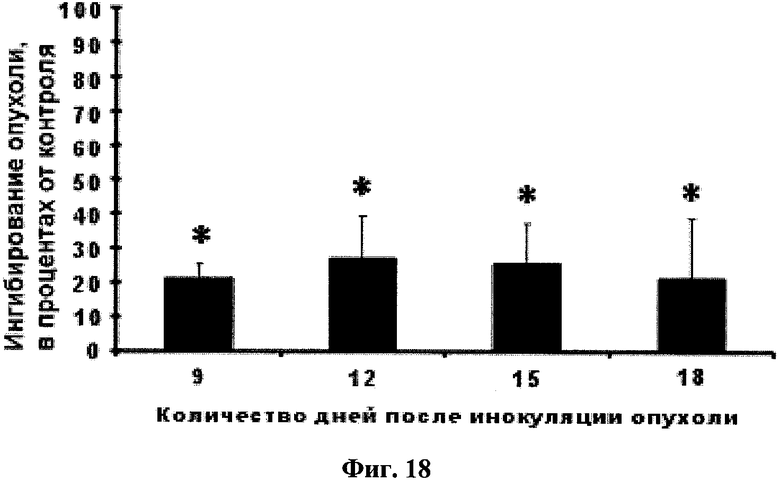
Изобретение относится к новым полипренилированным 1,4-бензохинонам формулы 1 или формулы 3 или полипренилированнам 1,4-гидрохинонам формулы 2 или формулы 4, или к их фармацевтически приемлемым солям, обладающим противораковой активностью
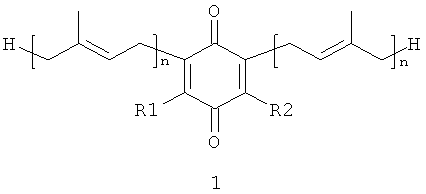
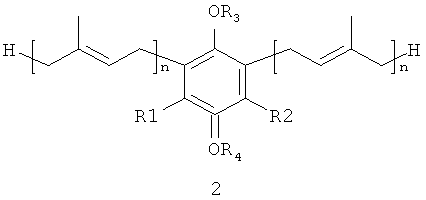

 .
.
В формулах 1-4 каждый из R1 и R2, независимо от других, может являться водородным радикалом или (С1-С6)-алкокси-группой; каждый из R3 и R4 независимо от других может являться водородным радикалом или (C1-С6)-алкильной группой; n может быть равно 2 или 3. Также предлагаемое изобретение относится к применению новых соединений и некоторых известных 1,4-бензохинонов и 1,4-гидрохинонов для приготовления лекарственного средства, к фармацевтической композиции на их основе, к методу лечения онкологических заболеваний, методу индуцирования в клетке АР-1-зависимой или NF-κВ-зависимой транскрипционной активности или обеих активностей одновременно и к методу индуцирования апоптоза в клетке, а также к методу выделения глабрухинонов. 7 н. и 14 з.п. ф-лы, 3 табл., 28 ил.
1. Полипренилированные 1,4-бензохиноны формулы 1 или формулы 3 или полипренилированные 1,4-гидрохиноны формулы 2 или формулы 4
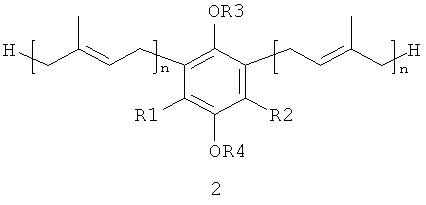

где каждый из R1 и R2, независимо от других, может являться водородным радикалом или (С1-С6)-алкоксигруппой;
каждый из R3 и R4, независимо от других, может являться водородным радикалом или (С1-С6)-алкильной группой;
n может быть равно 2 или 3;
или их фармацевтически приемлемые соли.
2. Применение полипренилированных 1,4-бензохинонов формулы 1 или формулы 3 или полипренилированных 1,4-гидрохинонов формулы 2 или формулы 4


где каждый из R1 и R2, независимо от других, может являться водородным радикалом или (С1-С6)-алкоксигруппой;
каждый из R3 и R4, независимо от других, может являться водородным радикалом или (С1-С6)-алкильной группой;
n может быть равно 2 или 3;
или полипренилированных 1,4-бензохинонов формулы 5, или формулы 6, или формулы 7, или формулы 8, или формулы 9, или формулы 10, или формулы 11, или формулы 12, или формулы 13, или формулы 14 или полипренилированных 1,4-гидрохинонов формулы 15 или их фармацевтически приемлемых солей для приготовления лекарственного средства, которое может быть использовано для лечения онкологических заболеваний у млекопитающих
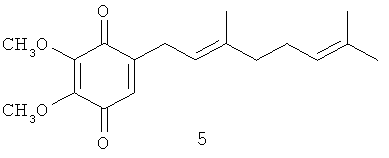

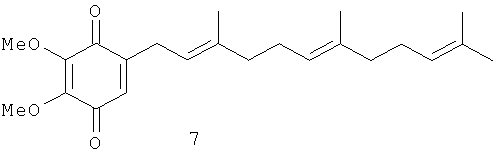
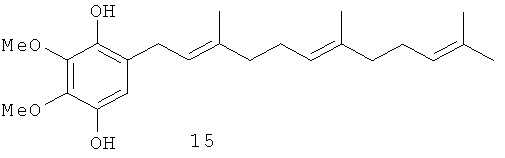 .
.
3. Применение по п.2, где лекарственное средство используется для уменьшения размера раковой опухоли у млекопитающих.
4. Применение по п.2, где лекарственное средство используется для ингибирования роста раковой опухоли у млекопитающих.
5. Применение по п.2 для приготовления лекарственного средства, используемого для индуцирования АР-1-зависимой транскрипционной активности у млекопитающих.
6. Применение по п.2 для приготовления лекарственного средства, используемого для индуцирования NF-κВ-зависимой транскрипционной активности у млекопитающих.
7. Применение по п.2 для приготовления лекарственного средства, используемого для одновременного индуцирования АР-1- и NF-κВ-зависимой транскрипционной активности у млекопитающих.
8. Применение по п.2 для приготовления лекарственного средства, используемого для ингибирования трансформации нормальных клеток в раковые у млекопитающих.
9. Применение по п.2 для приготовления лекарственного средства, используемого для уменьшения пролиферации раковых клеток у млекопитающих.
10. Применение по п.2 для приготовления лекарственного средства, используемого для индуцирования апоптоза у млекопитающих.
11. Фармацевтическая композиция, обладающая антираковой активностью, включающая эффективное количество соединения формулы 1, или формулы 2, или формулы 3, или формулы 4, или формулы 5, или формулы 6, или формулы 7, или формулы 8, или формулы 9, или формулы 10, или формулы 11, или формулы 12, или формулы 13, или формулы 15, как указано в п.2, или фармацевтически приемлемой соли этого соединения в комбинации с фармацевтически приемлемым растворителем или носителем.
12. Метод лечения онкологических заболеваний, включающий введение млекопитающему, в случае необходимости в таком лечении, эффективного количества соединения формулы 1, или формулы 2, или формулы 3, или формулы 4, или формулы 5, или формулы 6, или формулы 7, или формулы 8, или формулы 9, или формулы 10, или формулы 11, или формулы 12, или формулы 13, или формулы 14, или формулы 15, как указано в п.2, или эффективного количества фармацевтически приемлемой соли этого соединения или эффективного количества фармацевтической композиции по п.11, включающей эти соединения.
13. Метод по п.12, где введение соединения или фармацевтической композиции уменьшает размеры или ингибирует рост раковой опухоли у млекопитающего.
14. Метод по п.12, где введение соединения или фармацевтической композиции предотвращает трансформацию нормальных клеток в опухолевые у млекопитающего.
15. Метод по п.12, где введение соединения или фармацевтической композиции уменьшает пролиферацию опухолевых клеток у млекопитающего.
16. Метод по п.12, где введение соединения или фармацевтической композиции вызывает апоптоз опухолевых клеток у млекопитающего.
17. Метод индуцирования в клетке АР-1-зависимой или NF-κВ-зависимой транскрипционной активности или обеих активностей одновременно, включающий обработку клетки эффективным количеством соединения формулы 1, или формулы 2, или формулы 3, или формулы 4, или формулы 5, или формулы 6, или формулы 7, или формулы 8, или формулы 9, или формулы 10, или формулы 11, или формулы 12, или формулы 13, или формулы 14, или формулы 15, как указано в п.2, или эффективным количеством фармацевтически приемлемой соли этого соединения или эффективным количеством фармацевтической композиции по п.11, включающей эти соединения.
18. Метод индуцирования апоптоза в клетке, включающий обработку клетки эффективным количеством соединения формулы 1, или формулы 2, или формулы 3, или формулы 4, или формулы 5, или формулы 6, или формулы 7, или формулы 8, или формулы 9, или формулы 10, или формулы 11, или формулы 12, или формулы 13, или формулы 14, или формулы 15, как указано в п.2, или эффективным количеством фармацевтически приемлемой соли этого соединения или эффективным количеством фармацевтической композиции по п.11, включающей эти соединения.
19. Метод по любому из пп.17 и 18, где клетка является клеткой млекопитающего.
20. Метод по любому из пп.17 и 18, где обработка проводится in vitro или in vivo.
21. Метод выделения глабрухинонов А (формула 5) и В (формула 6), включающий экстракцию асцидий Aplidium glabrum этанолом, упаривание этанола, экстракцию водного остатка хлороформом, упаривание хлороформенного слоя до получения коричневого маслообразного остатка, хроматографирование остатка на колонке с силикагелем, в системе гексан-этилацетат, 10:1 и разделение смеси полученных глабрухинонов А и В при помощи ВЭЖХ на колонке с силикагелем в системе гексан-этилацетат, 7:1 с получением индивидуальных глабрухинонов А и В

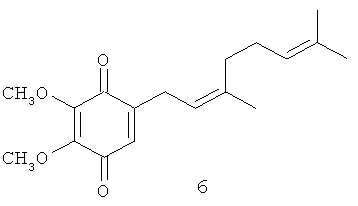
| Сепаратор-центрофуга с периодическим выпуском продуктов | 1922 |
|
SU128A1 |
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
| Food-Drugs Sea, Proc. | |||

