ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтики и, в частности, к производному триазола, способу его получения и его применению.
УРОВЕНЬ ТЕХНИКИ
В настоящее время злокачественные новообразования остаются одними из основных заболеваний, угрожающих жизни людей. Несмотря на достижение больших успехов при лечении рака, до сих пор нет возможностей лечить рак радикальным способом. Хотя имеющиеся в настоящее время на рынке противораковые препараты обладают определенной лечебной эффективностью, большинство из них являются цитотоксическими препаратами с серьезными токсическими и побочными эффектами. Поэтому изучение новых нацеленных противораковых препаратов на основе использования эффективных опухолевых мишеней стало первоочередной задачей для фармацевтических исследователей.
Клетки большинства основных солидных и гематологических злокачественных новообразований человека проявляют аномальную клеточную локализацию множества онкогенных белков, белков-супрессоров опухолей и регуляторов клеточного цикла. Например, некоторые мутации белка р53 могут приводить к его локализации в цитоплазме, а не в ядре. Это приводит к потере нормальной регуляции роста, несмотря на неизменную функцию супрессора опухоли. В других опухолях белок р53 дикого типа связывается в цитоплазме или быстро деградирует, снова вызывая потерю его супрессорной функции. Восстановление соответствующей ядерной локализации функционального белка р53 способно нормализовать некоторые свойства опухолевых клеток, восстанавливать чувствительность раковых клеток к агентам, повреждающим ДНК, и приводить к регрессии сформированных опухолей. Аналогичные данные были получены для других белков-супрессоров опухолей, таких как белки forkhead (Sullivan) и c-Abl. Кроме того, аномальная локализация некоторых белков-супрессоров опухолей, регулирующих рост, может быть связана с патогенезом аутоиммунных заболеваний. Ингибирование CRM1 может быть особенно полезным при семейных раковых синдромах (например, синдроме Ли-Фраумени, вызванном делецией одного аллеля p53, и раковом синдроме BRCA1 или BRCA2), при которых специфические белки-супрессоры опухолей (TSP) удалены или дисфункциональны, а увеличение уровней TSP, достигаемых системным (или местным) применением ингибиторов CRM1, способно восстанавливать нормальную функцию супрессора опухолей.
Определенные белки и РНК транспортируются в ядро и из ядра специфическими транспортными молекулами, которые классифицируются как импортины, если они транспортируют молекулы в ядро, и как экспортины, если они транспортируют молекулы из ядра. Белки, которые транспортируются в ядро и из ядра, содержат последовательности ядерного импорта/локализации (NLS) или последовательности ядерного экспорта (NES), которые позволяют им взаимодействовать с соответствующими транспортными факторами. Белок 1 поддержания структуры хромосом (CRM1), также известный как экспортин-1 или XPO1, является основным экспортином. Ингибиторы CRM1 блокируют ядерный экспорт белков-супрессоров и регуляторов роста, таких как p53, c-Ab1, p21, p27, pRB, BRCA1 IkB, ICp27, E2F4, KLF5, YAP1, ZAP, KIF5, HDAC4, HDAC5 или белки forkhead (например, FOXO3a), которые связаны с экспрессией генов, пролиферацией клеток, ангиогенезом и эпигенетикой. Было показано, что ингибиторы CRM1 индуцируют апоптоз в раковых клетках даже в присутствии активирующих сигналов или сигналов, активирующих рост, не затрагивая нормальные (нетрансформированные) клетки. Большинство исследований функции CRM1 было выполнено с использованием природного вещества лептомицина B (LMB). Лептомицин сам по себе очень токсичен для опухолевых клеток, но его сложно использовать в клинической практике из-за его высокой токсичности для желудочно-кишечного тракта. Дериватизация LMB для улучшения его лекарственных свойств может привести к получению соединений, которые сохраняют противоопухолевую активность и лучше переносятся моделями опухолей животных. Таким образом, ингибиторы ядерного экспорта могут оказывать благотворное влияние на неопластические заболевания и другие пролиферативные нарушения. Однако, на сегодняшний день, низкомолекулярные лекарственные ингибиторы CRM1 для применения in vitro и in vivo по-прежнему имеют определенные недостатки.
Помимо белков-супрессоров опухолей, CRM1 также экспортирует несколько ключевых белков, участвующих во многих воспалительных процессах, включая IkB, NF-kB, Cox-2, RXRa, Commal, HIFI, HMGBI, FOXO, FOXP и т. п. Транскрипционные активаторы семейства ядерного фактора xB (NF-kB/rel), названные благодаря обнаружению их способности запускать экспрессию гена иммуноглобулина x, могут регулировать экспрессию мРНК различных генов, связанных с воспалением, пролиферацией, иммунитетом и выживанием клеток. В основных условиях ингибитор белка NF-kB, называемый IkB, связывается с NF-kB в ядре, и комплекс IKB-NF-kB инактивирует транскрипционную функцию NF-kB. В ответ на воспалительные стимулы IkB диссоциирует из комплекса IkBNF-kB, высвобождая NF-kB и восстанавливая его потенциальную транскрипционную активность. Многие сигналы, которые активируют NF-kB, делают это путем нацеливания IkB на протеолиз (фосфорилирование IkB делает его «помеченным» для прохождения убиквитинирования, а затем протеолиза). Ядерный комплекс IkBa-MF-kB может экспортироваться с помощью CRM1 в цитоплазму, где комплекс диссоциирует, приводя к реактивации NF-kB. Убиквитинированный IkB также может диссоциировать из комплекса NF-kB, восстанавливая транскрипционную активность NF-kB. Ингибирование CRM1-индуцированного экспорта в нейтрофилах и макрофагоподобных клетках человека (U937) с помощью IMB не только приводит к накоплению транскрипционно-неактивного ядерного комплекса IkBa-NF-kB, но также предотвращает начальную активацию NF-kB даже при клеточной стимуляции. В другом исследовании лечение с помощью LMB ингибировало IL-1β-индуцированное связывание ДНК NF-kB (первая стадия активации транскрипции NF-kB), экспрессию 1L-8 и экспрессию молекул межклеточной адгезии в эндотелиальных клетках микрососудов легких. COMMD1 является еще одним ядерным ингибитором транскрипционной активности как NF-kB, так и индуцируемого гипоксией фактора 1 (HIF1). Блокирование ядерного экспорта COMMD1 путем ингибирования CRM1 может привести к усилению ингибирования транскрипционной активности NF-kB и HIF1.
CRM1 также опосредует транспорт ретиноидного X-рецептора a (RXRa). RXRa интенсивно экспрессируется в печени и играет центральную роль в регуляции метаболизма желчных кислот, холестерина, жирных кислот, стероидов и ксенобиотиков, а также в гомеостазе. Во время гепатита уровни ядерного RXRa значительно снижены, в основном из-за опосредованного воспалением ядерного экспорта RXRa с помощью CRM1. В клетках печени человека LepB способен предотвращать индуцированное L-1B повышение цитоплазматических уровней RXRa.
Роль CRM1-опосредованного ядерного экспорта в передаче сигналов NF-kB, HIF-1 и RXRa предполагает, что блокирование ядерного экспорта может быть потенциально полезным при многих воспалительных процессах во многих тканях и органах, включая заболевания сосудов (васкулит, артериит, ревматическую полимиалгию и атеросклероз), заболевания кожи и ревматические заболевания (ревматоидный и родственные артриты, псориатический артрит, спондилоартропатию, кристаллическую артропатию, системную красную волчанку, смешанное заболевание соединительной ткани, миозитный синдром, дерматомиозит, миозит с тельцами-включениями, недифференцированное заболевание соединительной ткани, синдром Шегрена, склеродермию, перекрестный синдром и т. п.).
Ингибирование CRM1 может влиять на экспрессию генов путем ингибирования/активации ряда факторов транскрипции, таких как ICp27, E2F4, KL5, YAP1 и ZAP.
Ингибирование CRM1 оказывает потенциальное терапевтическое воздействие на многие дерматологические синдромы, включая воспалительные дерматозы (атопический дерматит, аллергический дерматит, химический дерматит и псориаз), повреждение солнцем (ультрафиолетовое (УФ) повреждение) и инфекции. Ингибирование CRM1, изученное наиболее полно с помощью LMB, показало минимальное влияние на нормальные кератиноциты и оказало противовоспалительное действие на кератиноциты, находившиеся под воздействием УФ, TNFα или других воспалительных стимулов. Ингибирование CRM1 может также повышать активность NRF2 (фактор 2, связанный с ядерным фактором 2 эритроидного происхождения), и NRF2 может защищать кератиноциты от окислительного повреждения. LMB индуцирует апоптоз кератиноцитов, инфицированных онкогенными штаммами вируса папилломы человека (PV), такими как IPV16, но не индуцирует апоптоз неинфицированных кератиноцитов.
CRM1 также опосредует транспорт ключевых нейропротекторных белков, которые могут быть полезны при нейродегенеративных заболеваниях, включая болезнь Паркинсона (БП), болезнь Альцгеймера и боковой амиотрофический склероз. Например, путем (1) принудительного удержания в ядре ключевых регуляторных нейропротекторных факторов, таких как NRF2, привязки к нейронным клеткам и/или (2) ингибирования транскрипционной активности NF-kB путем связывания 1XB с ядром глиальных клеток, ингибирование CRM1 может замедлять или предотвращать гибель нейронов, выявляемую при этих заболеваниях. Имеются также данные, свидетельствующие о том, что аномальная пролиферация глиальных клеток связана с аномалиями уровней CRM1 или функций CRM1.
Интактное созревание многих вирусов также требует интактного ядерного экспорта, опосредованного главным образом CRM1. Вирусы, жизненный цикл которых задействует ядерный экспорт и/или сам CRM1, включают вирус иммунодефицита человека (ВИЧ), аденовирус, ретровирус обезьян I типа, вирус болезни Борна, вирус гриппа (обычные штаммы, а также штаммы HINL и птичьего HN1), вирус гепатита В (HBV) и вирус гепатита C (HCV), вирус папилломы человека (HPV), респираторно-синцитиальный вирус (RSV), вирус Денге, коронавирус тяжелого острого респираторного синдрома, вирус желтой лихорадки, вирус Западного Нила, вирус простого герпеса (HSV), цитомегаловирус (CMV) и полиомавирус клеток Меркеля (MCV).
Белок Rev ВИЧ-1, который проходит через ядрышко и перемещается между ядром и цитоплазмой, способствует экспорту несплайсированных и однократно сплайсированных транскриптов ВИЧ, содержащих РНК Rev-ответного элемента (RRE), через путь экспорта CRM1. Ингибирование Rev-опосредованного транспорта РНК, достигаемое с помощью ингибиторов CRM1, таких как LepB или PKF050-638, может предотвращать процесс транскрипции ВИЧ-1, ингибировать продукцию новых вирионов ВИЧ-1 и, тем самым, снижать уровни ВИЧ-1.
Вирус Денге (DENV) является возбудителем распространенного переносимого членистоногими вирусного заболевания, лихорадки Денге (DF), и ее более тяжелой и потенциально смертельной формы, геморрагической лихорадки Денге (DHF). DHF, по-видимому, является результатом чрезмерно сильной воспалительной реакции на DENV. NS5 является самым крупным и наиболее консервативным белком DENV. CRM1 регулирует транспорт NS5 из ядра в цитоплазму, где опосредуется большинство функций NS5. Ингибирование опосредованного CRM1 экспорта NS5 может приводить к изменению кинетики продукции вируса и снижению индукции воспалительного хемокина - интерлейкина-8 (IL-8), что обеспечивает новый подход к лечению заболеваний, вызванных DENV и другими важными с медицинской точки зрения флавивирусами, включая вирус гепатита С.
Другие кодируемые вирусом РНК-связывающие белки, которые выходят из ядра с помощью CRM1, включают тегументный белок типа HSVI (P13/14 или HUA7), белок pp65 человеческого CMV, белок ORF3b коронавируса SARS и белок матрикса (M) RSV.
Интересно, что многие из этих заболеваний связаны с определенными типами раковых заболеваний человека, включая гепатоцеллюлярную карциному (ГЦК) из-за хронической инфекции HBV или HCV, рак шейки матки из-за HPV и карциному из клеток Меркеля, связанную с MCV. Следовательно, ингибиторы CRM1 оказывают благотворное влияние как на подавление процесса вирусной инфекции, так и процесса неопластической трансформации, вызываемых этими вирусами.
CRM1 контролирует локализацию в ядре и, следовательно, активность различных ферментов, метаболизирующих ДНК, включая гистоновые деацетилазы (HDAC), гистоновые ацетилтрансферазы (HAT) и гистоновые метилтрансферазы (HMT).
CRM1 также связан с другими нарушениями. Болезнь Лебера, наследственное заболевание, характеризующееся дегенерацией ганглиозных клеток сетчатки и потерей зрения, связано с бездействием переключателя CRM1. Имеются также данные, свидетельствующие о том, что нейродегенеративные нарушения связаны с аномальным ядерным транспортом.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на предоставление производного триазола, способа его получения и его применения, при этом производное триазола можно использовать в качестве ингибитора CRM1 для получения лекарственного средства для лечения заболевания, связанного с активностью CRM1.
Для достижения вышеуказанной цели в настоящем изобретении используются следующие технические решения:
Предложено производное триазола или его фармацевтически приемлемая соль, причем производное триазола имеет структуру, показанную в формуле I:
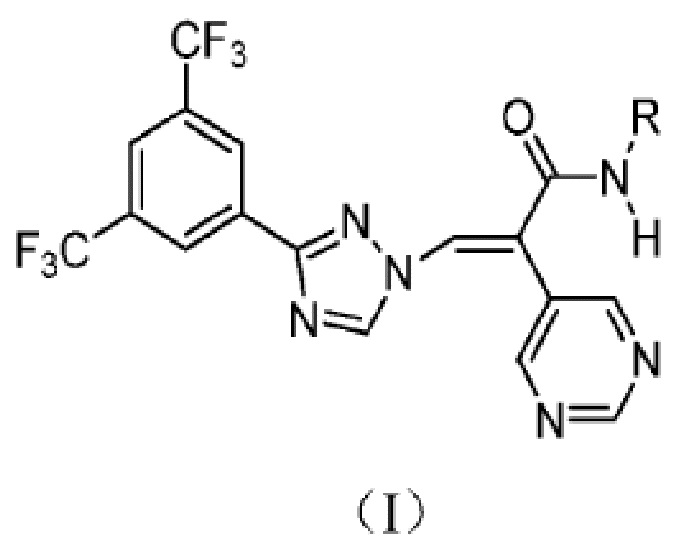
где:
R выбран из гидрокси, C1-6 алкила, C1-6 алкокси и -C(OR1)(=NR2);
R1 и R2 независимо представляют собой водород, C1-6 алкил, C1-6 галогеналкил или C1-6 алкокси.
Кроме того, R выбран из гидрокси, C1-4 алкила, C1-4 алкокси и -C(OR1)(=NR2);
R1 и R2 независимо представляют собой водород или C1-4 алкил.
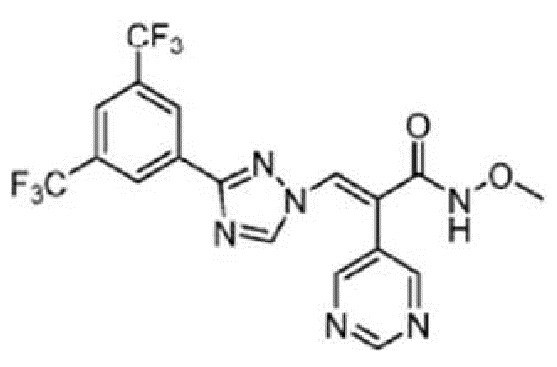
Кроме того, производное триазола выбрано из следующих соединений:
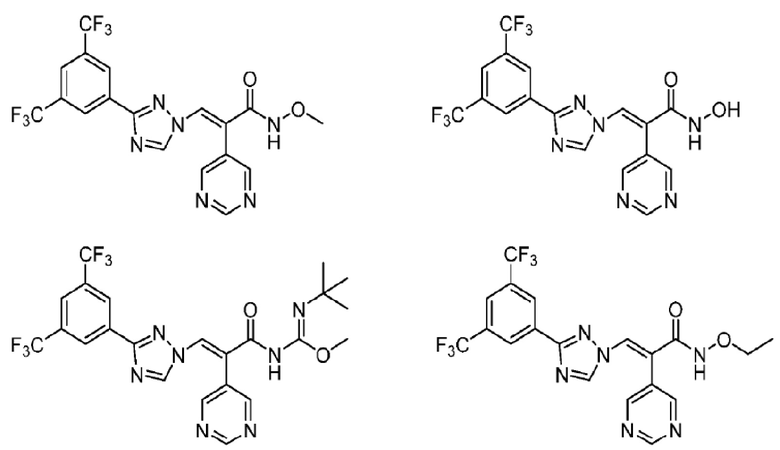
Предложен способ получения производного триазола или его фармацевтически приемлемой соли, описанных выше, который осуществляется в соответствии со следующей
схемой:
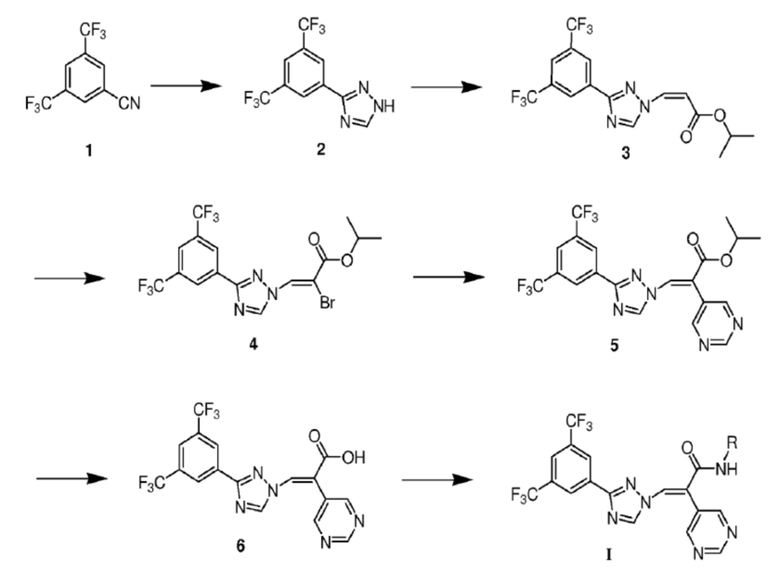
Предложена фармацевтическая композиция для лечения заболевания, нарушения или симптома, связанного с активностью CRM1, содержащая производное триазола или его фармацевтически приемлемую соль, описанные выше, а также фармацевтически приемлемый носитель.
Предложено применение производного триазола или его фармацевтически приемлемой соли, описанных выше, для получения лекарственного средства для лечения нарушения, связанного с активностью CRM1.
Кроме того, нарушение, связанное с активностью CRM1, выбрано из пролиферативного нарушения, рака, воспалительного нарушения, аутоиммунного нарушения, вирусной инфекции, офтальмологического нарушения, нейродегенеративного нарушения, нарушения из-за аномального роста тканей, расстройства, связанного с приемом пищи, аллергии и респираторного заболевания.
Кроме того, нарушение, связанное с активностью CRM1, представляет собой рак.
Кроме того, нарушение, связанное с активностью CRM1, представляет собой множественную миелому.
Положительные эффекты заключаются в следующем: результаты клеточной активности показывают, что соединения I и IV по настоящему изобретению по существу эквивалентны KPT-8602. Периоды полувыведения соединения I при пероральном и внутривенном введении больше, чем у KPT-8602, а именно, период полувыведения при пероральном приеме примерно в 2 раза больше, и AUC соединения I при внутривенном и пероральном введении намного выше, чем у KPT-8602, что демонстрирует лучшие фармакокинетические свойства. Кроме того, соединение I показывает хорошую безопасность.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
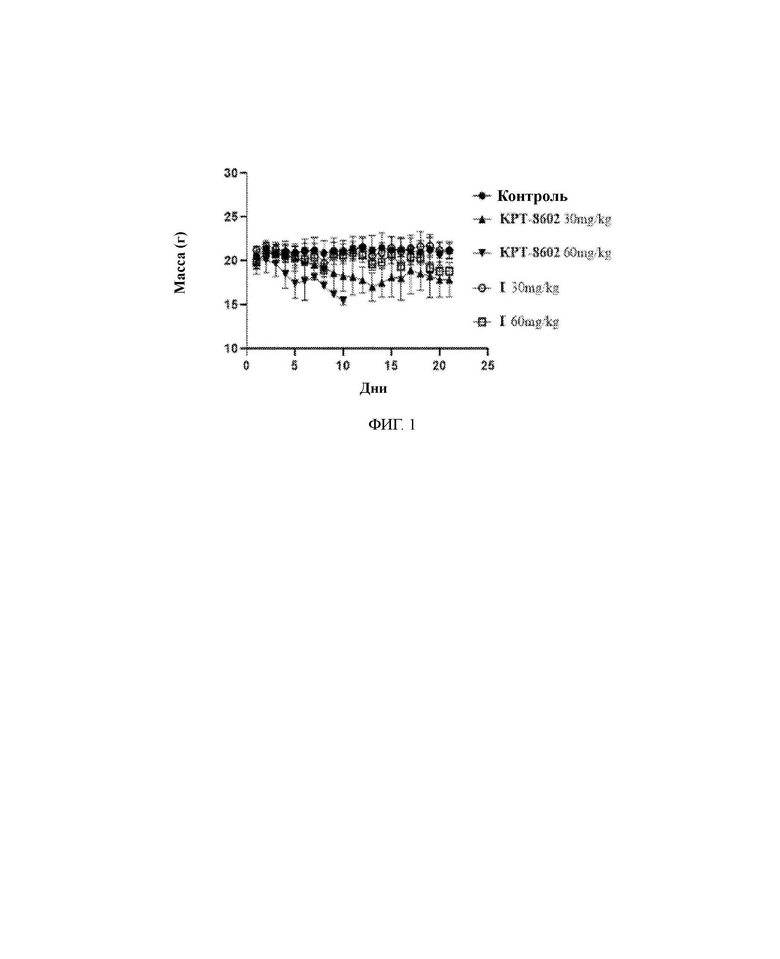
На ФИГ. 1 показаны результаты анализа токсичности соединения I in vivo у мышей BALB/C.
ПОДРОБНОЕ ОПИСАНИЕ
Определение соединений
Соединения по настоящему изобретению включают те, которые, в целом, описаны выше, и дополнительно описаны классами, подклассами и видами, раскрытыми в настоящем документе. В настоящем документе должны использоваться следующие определения, если не указано иное. Для целей настоящего изобретения эти химические элементы идентифицированы в соответствии с Периодической таблицей элементов (версия CAS) и Справочником по химии и физике (75-е издание).
Если в данном описании не указано иное, номенклатура, используемая в этом описании, обычно соответствует примерам и правилам, изложенным в Номенклатуре органической химии, разделы A, B, C, D, E, F и H, которая включена в настоящий документ посредством ссылки, для получения типовых названий химических структур и правил наименования химических структур.
Соединения по настоящему изобретению могут иметь асимметричные центры, хиральные оси и хиральные плоскости, и существовать в виде рацематов, рацемических смесей и индивидуальных диастереомеров или энантиомеров, при этом все возможные изомеры и их смеси (включая оптические изомеры) включены в настоящее изобретение.
Используемый термин «галоген» включает атомы фтора, хлора, брома и иода. В частности, предпочтительным является атом фтора или хлора.
Используемый в настоящем документе термин «алкил» включает в себя линейные или разветвленные углеводородные группы, содержащие 1-15 атомов углерода, предпочтительно 1-10 атомов углерода, более предпочтительно 1-6 атомов углерода и еще более предпочтительно 1-4 атома углерода. Примеры алкила включают: метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил, н-гептил, изогептил, н-октил, изооктил, н-нонил, н-децил и т. п.
Используемый термин «алкокси» означает группу, в которой указанный выше «алкил» связан с атомом кислорода. Примеры алкокси включают: метокси, этокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси, изобутокси, втор-бутокси, пентилокси, изопентилокси, гексилокси и т.п.
Предпочтительные варианты «алкокси» включают: метокси, этокси, н-пропокси, изопропокси и трет-бутокси.
Используемый в настоящем документе термин «галогеналкил» включает группу, в которой атом(-ы) водорода, связанный(-е) с атомом(-ами) углерода описанного выше «алкила», заменены одним или большим количеством «галогенов», описанных выше. Примеры галогеналкила включают: монофторметил, монофторэтил, монофторпропил, 2,2,3,3,3-пентафторпропил, монохлорметил, трифторметил, трихлорметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил, 1,2-дибромэтил, 1,1,1-дифторпропан-2-ил и т. п.
Варианты осуществления «галогеналкила» включают трифторметил и трихлорметил.
Настоящее изобретение предоставляет композицию, включающую соединение по настоящему изобретению или его фармацевтически приемлемое производное, а также фармацевтически приемлемый носитель, адъювант или наполнитель. Количество соединения в композиции по настоящему изобретению таково, что оно эффективно для умеренного ингибирования CRM1 в биологическом образце или у пациента. В некоторых вариантах осуществления композиция по настоящему изобретению составлена для введения пациенту, нуждающемуся в такой композиции. Используемый в настоящем документе термин «пациент» означает животное. В некоторых вариантах осуществления животное представляет собой млекопитающее. В некоторых вариантах осуществления пациент является ветеринарным пациентом (т. е. пациентом, являющимся отличным от человека млекопитающим).
Термин «фармацевтически приемлемый носитель, адъювант или наполнитель» относится к нетоксичному носителю, адъюванту или наполнителю, который не разрушает фармакологическую активность соединения, с которым он образует состав. Фармацевтически приемлемые носители, адъюванты или наполнители, которые можно использовать в композиции по настоящему изобретению, включают, помимо прочего, ионообменники, оксид алюминия, стеарат алюминия, лецитин, белки сыворотки (такие как сывороточный альбумин человека), буферные вещества (такие как фосфаты), глицин, сорбиновую кислоту, сорбат калия, частичные глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты (такие как сульфат протамина, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка), коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, карбоксиметилцеллюлозу натрия, полиакрилаты, воски, блок-полимеры полиэтилена-полиоксипропилена, этиленгликоль и ланолин.
Способ получения производного триазола или его фармацевтически приемлемой соли осуществляют в соответствии со следующей схемой:
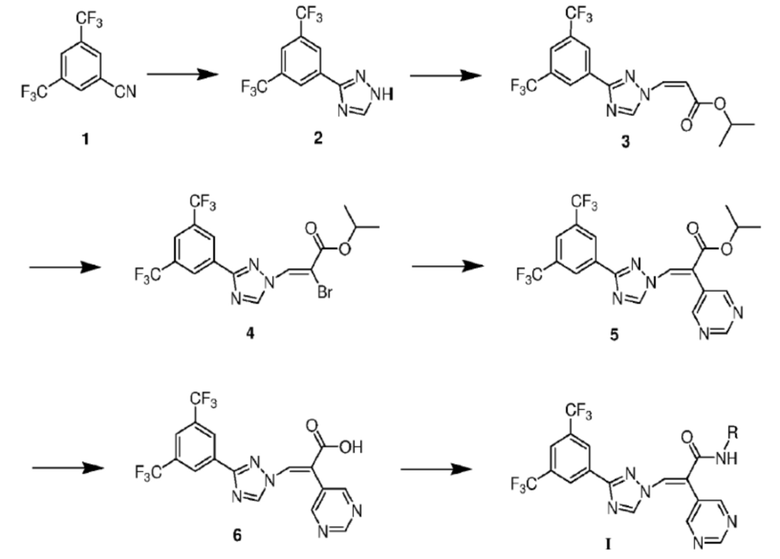
где в соединениях формул реакций каждая группа R определена, как описано ранее; соединение формулы (1) вступает в реакцию под действием гидросульфида натрия с получением формулы (2); соединение формулы (2) вступает в реакцию нуклеофильного замещения с получением соединения формулы (3); затем соединение формулы (3) вступает в реакцию под действием бромной воды и триэтиламина с получением соединения (4); соединение формулы (4) вступает в реакцию сочетания с получением соединения (5); соединение формулы (5) гидролизуется в карбоновую кислоту формулы (6) под действием LiOH; и соединение формулы (6) конденсируется с аминовым субстратом с получением соединения формулы I.
Способ получения соединения по настоящему изобретению подробно описан ниже:
Цианогруппа соединения 1 реагирует под действием гидросульфида натрия и хлорида магния с образованием тиоформамида, а затем в присутствии гидразингидрата и муравьиной кислоты образуется триазол 2. Затем триазол 2 соединяется с (Z)-изопропил 3-иодакрилатом под каталитическим действием триэтилендиамина с получением соединения 3. Затем фрагмент с двойной связью соединения 3 вступает в реакцию присоединения с жидким бромом, и одна молекула брома удаляется под действием триэтиламина с получением ключевого промежуточного соединения 4. Затем промежуточное соединение 4 отдельно подвергается сочетанию Сузуки под каталитическим действием бис(трифенилфосфин)палладия дихлорида с множеством азотсодержащих ароматических групп со структурой борной кислоты с получением соединений 5, связанных с различными азотсодержащими ароматическими группами. Наконец, сложноэфирная связь соединения 5 гидролизуется в соединение карбоновой кислоты 6 под действием LiOH, и соединение карбоновой кислоты 6 далее конденсируется с получением целевого соединения формулы I.
Настоящее изобретение дополнительно подробно проиллюстрировано ниже со ссылкой на прилагаемые графические материалы и конкретные примеры, но настоящее изобретение не следует рассматривать как ограничивающееся ими. Все модификации или замены в отношении способов, процедур или условий настоящего изобретения без отклонения от сущности и объема настоящего изобретения должны входить в объем настоящего изобретения. В примерах экспериментальные процедуры без определенных условий и реагенты без указанных составов соответствуют общепринятым условиям в данной области.
Пример 1
I. Синтез соединений
Получение соединений по настоящему изобретению можно осуществить следующим образом:
Схема превращения и синтез соединения I
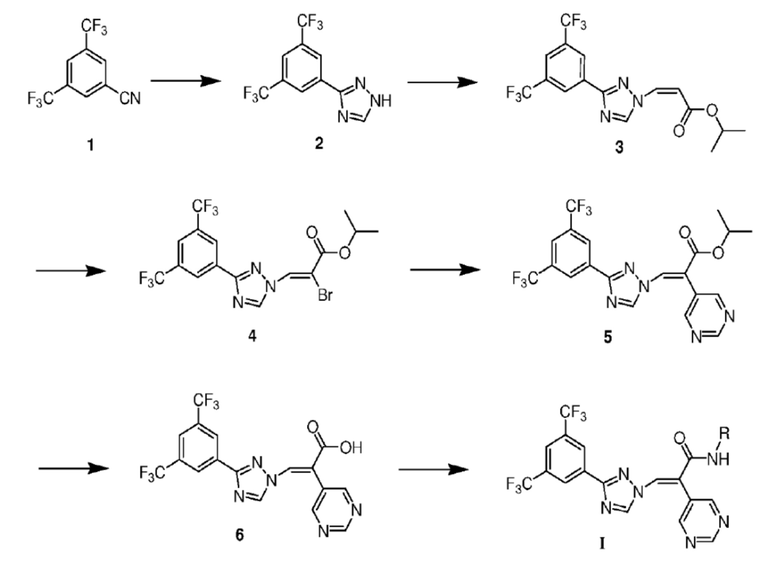
Синтез (Z)-изопропил 3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)акрилата (3)
В колбу в форме баклажана объемом 250 мл добавляли 3,5-бис(трифторметил)бензонитрил 1 (10 г, 41,8 ммоль) и растворяли его в растворе ДМФ (50 мл). Последовательно добавляли NaSH (7,8 г, 83,7 ммоль) и MgCl2 (8,5 г, 41,8 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. После завершения реакции по данным ТСХ реакционный раствор выливали в смесь льда и воды (500 мл). Смесь экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл × 1), сушили над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении для удаления растворителя, что давало неочищенный 3,5-бис(трифторметил)бензотиоамид (10,9 г, выход 95,1%, чистота 84%) в виде желтой маслянистой жидкости (МС (ESI) m/z 274,35 [M+H]+), которую использовали непосредственно на следующей стадии.
В колбу в виде баклажана объемом 250 мл добавляли 3,5-бис(трифторметил)бензотиоамид (10,9 г, 39,8 ммоль), растворяли его в растворе DMF (30 мл) и добавляли в колбу по каплям 80% гидразингидрата (5,1 мл, 83,6 ммоль) при комнатной температуре. Смесь перемешивали в течение 1 ч, а затем к ней по каплям добавляли муравьиную кислоту (30 мл). Затем реакционную смесь перемешивали при 90°С еще 3 ч. После завершения реакции по данным ТСХ реакционный раствор охлаждали до комнатной температуры и затем выливали в очищенную воду (600 мл). Смесь экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли, промывали насыщенным раствором бикарбоната натрия (300 мл × 3) и насыщенным раствором хлорида натрия (100 мл × 1), сушили над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении для удаления растворителя, что привело к получению неочищенного соединения. Неочищенное соединение перемешивали с н-гексаном (200 мл × 3) для промывки, фильтровали и сушили с получением 3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазола 2 (8,5 г, выход 76,0%, чистота 90%) в виде белого твердого вещества. 1 H ЯМР (400 МГц, CDCl3) δ 8,63 (с, 2H, Ph), 8,40 (с, 1H, NCH), 8,02 (д, J=13,8 Гц, 1H, NCHCH), 7,95 (с, 1H, Ph), 6,74 (д, J=13,8 Гц, 1H, NCHCH) МС (ESI) m/z 279,89 [M+H]+.
В колбу в форме баклажана объемом 250 мл добавляли 3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол 2 (8,5 г, 30,2 ммоль) и растворяли его в DMF (40 мл), и в колбу добавляли DABCO (8,5 г, 75,5 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, а затем к реакционному раствору по каплям добавляли (Z)-этил-3-иодакрилат (7,5 г, 33,2 ммоль). Затем реакционную смесь перемешивали при комнатной температуре еще 3 ч. После завершения реакции по данным ТСХ реакционный раствор выливали в смесь льда и воды (400 мл). Смесь экстрагировали этилацетатом (80 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл × 1), сушили над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении для удаления растворителя, получая, таким образом, неочищенное соединение. Полученный неочищенный продукт очищали колоночной хроматографией (PE/EtOAc=25:1) с получением соединения (Z)-этил 3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)акрилата 3 (7,9 г, выход 68,2%, чистота 98%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,63 (с, 2H, Ph), 8,40 (с, 1H, NCH), 8,02 (д, J=13,8 Гц, 1H, NCHCH), 7,95 (с, 1H, Ph), 6,74 (д, J=13,8 Гц, 1H, NCHCH), 4,87 (м, 1H, CH), 1,36 (т, J=7,1 Гц, 6H, Ме). МС (ESI) m/z 394,23 [M+H]+.
Синтез (Z)-изопропил 3-(3-(3,5-бис(трифторметил)фенилтриазол-1-ил)-2-бромакрилата (4)
В колбе в форме баклажана объемом 250 мл растворяли полученный ранее (Z)-этил-3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)акрилат 3 (7,9 г, 20,6 ммоль) в дихлорметане (40 мл), и медленно по каплям в течение 30 мин добавляли жидкий бром (6,6 г, 41,2 ммоль). Затем реакционную смесь перемешивали при комнатной температуре в течение 8 ч. После завершения реакции по данным ТСХ реакционный раствор выливали в смесь льда и воды (100 мл). Смесь экстрагировали дихлорметаном (50 мл × 3). Органические фазы объединяли, промывали насыщенным раствором бисульфита натрия (100 мл × 3) и насыщенным раствором хлорида натрия (50 мл × 1), сушили над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении для удаления растворителя. Проводили разделение и очистку колоночной хроматографией (PE/EtOAc=50:1) с получением изопропил 3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)-2,3-дибромпропионата (10,3 г, выход 92,7%, чистота 95%) в виде белого твердого вещества. МС (ESI) m/z 551,97 [M+H]+.
Промежуточный изопропил 3-(3-(3,5-бис(трифторметил)фенил)-1Н-1,2,4-триазол-1-ил)-2,3-дибромпропионат (103 г, 19,1 ммоль), полученный на предыдущей стадии, взвешивали в колбе в форме баклажана объемом 250 мл и растворяли его в тетрагидрофуране (40 мл). Реакционный раствор перемешивали в течение 10 мин на ледяной бане, а затем к нему по каплям добавляли триэтиламин (3,9 г, 38,2 ммоль). Реакционную смесь перемешивали еще 30 мин, а затем оставляли при комнатной температуре и перемешивании еще 6 ч. После завершения реакции по данным ТСХ к реакционному раствору добавляли смесь льда и воды (100 мл). Смесь экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл × 1), сушили над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении для удаления растворителя. Разделение и очистку колоночной хроматографией (PE/EtOAc=50:1) проводили с получением (Z)-этил-3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)-2-бромакрилата 4 (7,7 г, выход 88,2%, чистота 96%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,75 (с, 1H, NCH), 8,56 (с, 2H, Ph), 7,93 (с, 1H, Ph), 7,65 (с, 1H, CBrCH), 4,38 (м, 1H, CH), 1,37 (т, J=7,1 Гц, 6H, Me). МС (ESI) m/z 473,09 [M+H]+.
Синтез (E)-изопропил 3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)-2-(пиримидин-5-ил)акрилата (5)
Важное промежуточное соединение 4 (200 мг, 0,44 ммоль) и 5-пиримидинбороновую кислоту (81,9 мг, 0,66 ммоль) взвешивали в трехгорлой колбе объемом 25 мл и растворяли его в смешанном растворе диоксана (5 мл) и воды (1 мл). Затем в реакционный раствор вносили навеску ацетата натрия (86,4 мг, 0,88 ммоль). Смесь продували азотом 3 раза и перемешивали при комнатной температуре. Затем к реакционному раствору добавляли Pd(PPh3)Cl2 (30,9 мг, 0,04 ммоль), смесь снова продували азотом 3 раза и перемешивали в течение ночи при 80°C. После завершения реакции по данным ТСХ реакционный раствор охлаждали до комнатной температуры и добавляли к нему очищенную воду (50 мл). Смесь экстрагировали этилацетатом (15 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл × 1), сушили над безводным Na2SO4, фильтровали и перегоняли под вакуумом для удаления растворителя, получая при этом неочищенное соединение. Неочищенное соединение отделяли и очищали колоночной хроматографией (PE/EtOAc=8:1) с получением (E)-этил-3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)-2-(пиримидин-5-ил)акрилата 5 (125,3 мг, выход 62,3%, чистота 93%) в виде белого твердого вещества. ПЯМР (400 МГц, CDCl3) δ 9,28 (с, 1H, пиримидин), 8,89 (с, 2H, пиримидин), 8,72 (с, 1H, NCH), 8,59 (с, 2H, Ph), 7,96 (с, 1H, Ph), 7,40 (с, 1H, COCCH), 4,85 (м, 1H, CH), 1,36 (т, J=7,2 Гц, 6H, Me). МС (ESI) m/z 472,17 [M+H]+.
Синтез (E)-3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)-2-(пиримидин-5-ил)пропенкарбоновой кислоты (6)
В колбе в форме баклажана объемом 25 мл растворяли соединение 5 (125,3 мг, 0,27 ммоль) в тетрагидрофуране (3 мл). Реакционный раствор перемешивали в течение 10 мин на бане со льдом, а затем к нему по каплям добавляли раствор LiOH H2O (45,3 мг, 1,08 ммоль) в воде (1 мл). После перемешивания еще в течение 30 мин реакционную смесь оставляли при комнатной температуре и перемешивании в течение ночи. После завершения реакции по данным ТСХ в реакционный раствор выливали смесь льда и воды (10 мл). Раствор доводили до рН 2-3 с помощью 4 н. соляной кислоты и экстрагировали этилацетатом (5 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл × 1), сушили над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении для удаления растворителя, что давало относительно чистое соединение (Е)-3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)-2-(пиримидин-5-ил) акриловой кислоты (99,4 мг, выход 85,8%), чистота 89%) в виде белого твердого вещества, которое использовали непосредственно на следующей стадии. МС (ESI) m/z 427,92 [M-H]-.
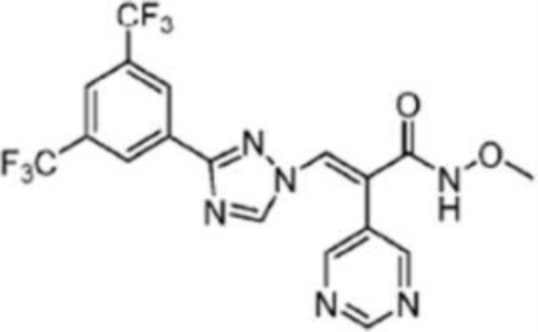
Синтез (E)-3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)-N-метокси-2-(пиримидин-5-ил)акриламида (I)
В колбе в форме баклажана объемом 25 мл растворяли соединение 6 (125,3 мг, 0,27 ммоль) в дихлорэтане (3 мл). Реакционный раствор перемешивали в течение 10 мин на бане со льдом, а затем к нему добавляли EDCI (20,3 мг, 1,08 ммоль) и HOBT (70,5 мг, 1,5 ммоль). После перемешивания в течение еще 30 мин реакционную смесь оставляли при комнатной температуре. Добавляли по каплям DIPEA (54,5 мг, 3,0 ммоль) и гидрохлорид метоксигидроксиламина (23,4 мг, 1,0 ммоль), и смесь перемешивали в течение ночи. После завершения реакции по данным ТСХ в реакционный раствор выливали смесь льда и воды (10 мл). Смесь экстрагировали этилацетатом (5 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл × 1), сушили над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении для удаления растворителя, получая таким образом соединение (Е)-3-(3-(3,5-бис(трифторметил)фенил)-1H-1,2,4-триазол-1-ил)-N-метокси-2-(пиримидин-5-ил)акриламида (35,4 мг, 40% выход, чистота 89%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,78 (д, J=5,9 Гц, 2H, пиридин), 8,40 (с, 2H, Ph), 8,33 (с, 1H, NCH), 7,92 (с, 1H, Ph), 7,87 (с, 1H, COCCH), 7,28 (д, J=1,6 Гц, 2H, пиридин), 4,34 (т, J=7,1 Гц, 3H, Ме). МС (ESI) m/z 459,10 [M+H]+.
Конкретные синтезированные соединения и их названия приведены в таблице ниже.
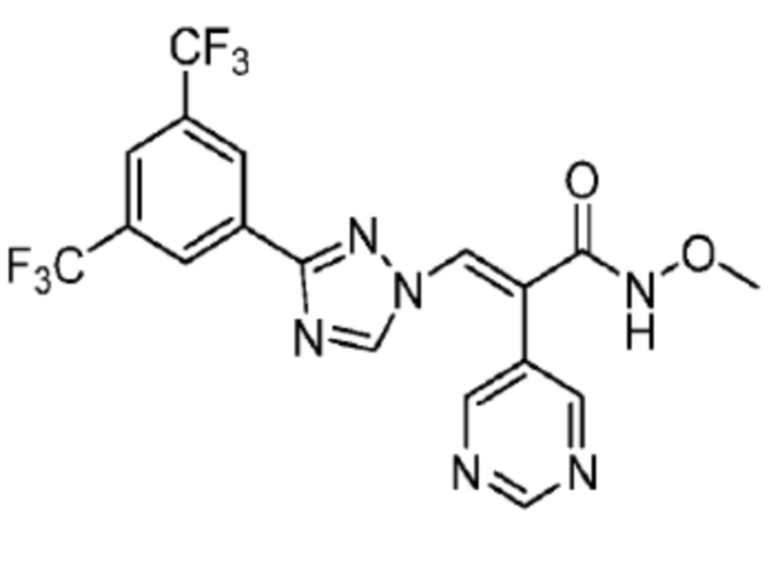
1H ЯМР (400 МГц, CDCl3) δ 8,78 (д, J=5,9 Гц, 2H, пиридин), 8,40 (с, 2H, Ph), 8,33 (с, 1H, NCH), 7,92 (с, 1H, Ph), 7,87 (с, 1H, COCCH), 7,28 (д, J=1,6 Гц, 2H, пиридин), 4,34 (т, J=7,1 Гц, 3H, Me). 13C ЯМР (CDCl3) δ 159,8; 158,1; 154,4; 147,7; 131,8; 131,2; 129,4; 128,6; 124,7; 123,1; 111,6; 130,4; 121,0; 64,4; масс-спектрометрия: HRMS-EI (m/z): рассчитано для C18H12F6N6O2 [M+H]+, 459,0998. Обнаружено 459,0999.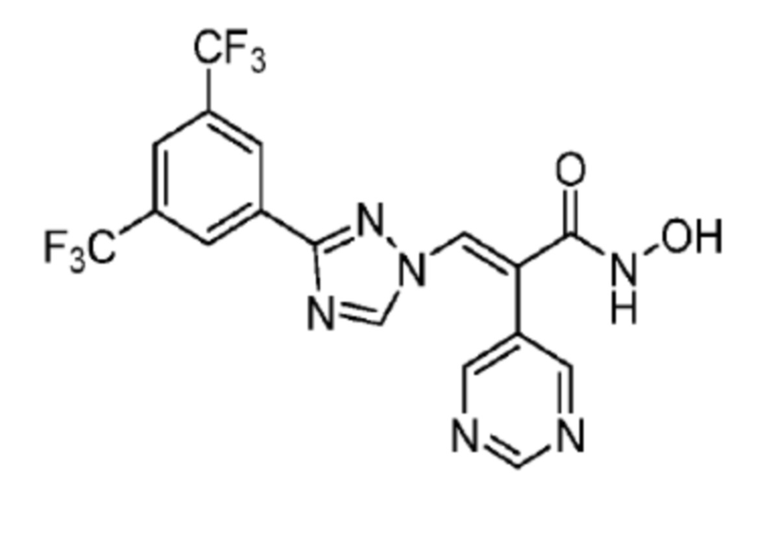
1H ЯМР (400 МГц, CDCl3) δ 8,78 (д, J=5,9 Гц, 2H, пиридин), 8,40 (с, 2H, Ph), 8,33 (с, 1H, NCH), 7,92 (с, 1H, Ph), 7,87 (с, 1Н, СОССН), 7,28 (д, J=1,6 Гц, 2Н, пиридин); 13C ЯМР (CDCl3) δ 161,6; 159,8; 158,1; 154,4; 147,7; 131,8; 131,2; 130,4; 129,4; 128,6; 124,7; 123,1; 121,0; масс-спектрометрия: HRMS-EI (m/z): рассчитано для C17H10F6N6O2 [M+H]+, 445,0842. Обнаружено 445,0849.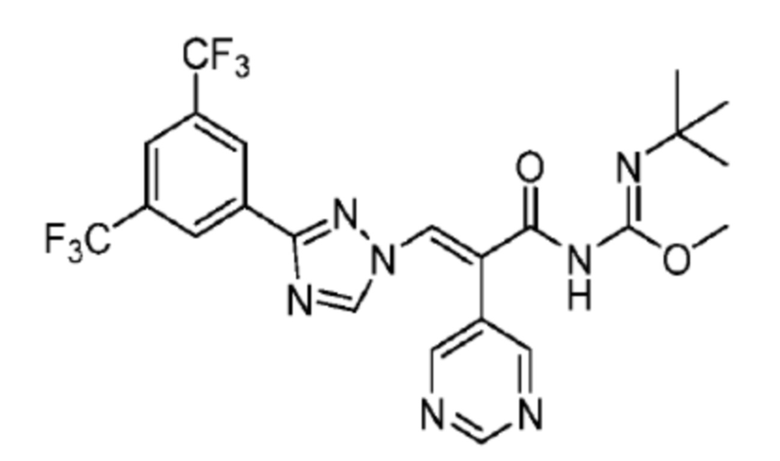
1H ЯМР (400 МГц, CDCl3) δ 8,78 (д, J=5,9 Гц, 2H, пиридин), 8,40 (с, 2H, Ph), 8,33 (с, 1H, NCH), 7,92 (с, 1H, Ph), 7,87 (с, 1H, COCCH), 7,28 (д, J=1,6 Гц, 2H, пиридин), 1,17 (с, 9H, Me); 13С ЯМР (CDCl3) δ 161,6; 159,8; 158,1; 154,4; 147,7; 131,8; 131,2; 130,4; 129,4; 128,6; 124,7; 123,1; 121,0; 84,2; 28,4; масс-спектрометрия: HRMS-EI (m/z): рассчитано для C22H18F6N6O2 [M+H]+, 545,1366. Обнаружено 545,1370.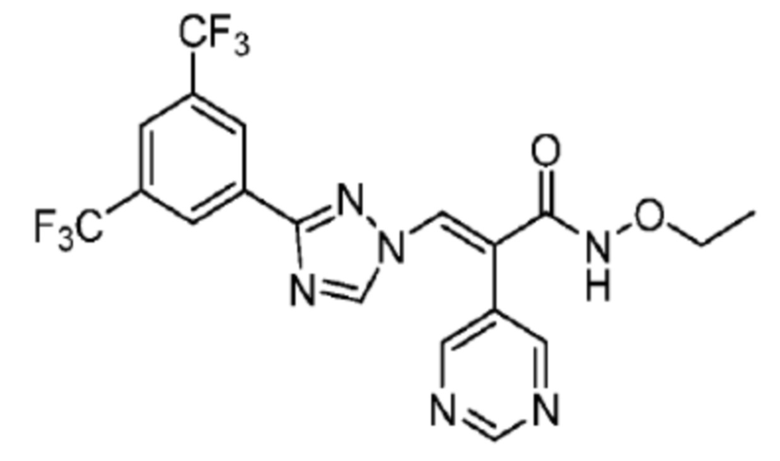
1H ЯМР (400 МГц, CDCl3) δ 8,78 (д, J=5,9 Гц, 2H, пиридин), 8,40 (с, 2H, Ph), 8,33 (с, 1H, NCH), 7,92 (с, 1H, Ph), 7,87 (с, 1H, COCCH), 7,28 (д, J=1,6 Гц, 2H, пиридин), 3,57 (м, 2H, CH2) 1,34 (м, 3H, Me). 13C ЯМР (CDCl3) δ 159,8; 158,1; 154,4; 147,7; 131,8; 131,2; 129,4; 128,6; 124,7; 123,1; 111,6; 130,4; 121,0; 65,4; 12,3; масс-спектрометрия: HRMS-EI (m/z): рассчитано для C19H14F6N6O2 [M+H]+, 473,1155. Обнаружено 473,1159.
II. Ингибирующая активность в линии клеток
1. Криоконсервация клеток
(1) Клетки собирали, центрифугировали при 1000 об/мин в течение 5 мин при комнатной температуре и промывали PBS.
(2) Затем клетки ресуспендировали в среде 1640, содержащей 7% ДМСО и 10% фетальной бычьей сыворотки.
(3) Клеточную суспензию разделяли на аликвоты в пробирки для криоконсервации, помещали в контейнеры для криоконсервации клеток, криоконсервировали при температуре -80°C в течение ночи, а затем хранили в жидком азоте.
2. Восстановление клеток и культивирование
(1) Пробирки для криоконсервации извлекали из резервуара с жидким азотом, помещали в теплую воду при температуре 37°C и осторожно встряхивали, чтобы оттаяла жидкость с клетками.
(2) Клетки переносили в стерильную центрифужную пробирку и добавляли среду 1640, содержащую 10% фетальной бычьей сыворотки. Смесь осторожно пипетировали, получая суспензию.
(3) Клетки центрифугировали при 1000 об/мин в течение 5 мин при комнатной температуре, и отбрасывали надосадочную жидкость. Добавляли среду 1640, содержащую 10% фетальной бычьей сыворотки, и смесь осторожно пипетировали, получая суспензию.
(4) Суспензию переносили в колбу для культивирования и культивировали в инкубаторе (37°C, 5% CO2, насыщенный влагой воздух). Через 1-2 дня культивирования среду заменяли.
3. Клеточный пассаж
Исходную среду утилизировали. Клетки однократно промывали стерильным PBS, инкубировали с 1 мл 0,25% панкреатина в течение примерно 1 мин и затем наблюдали под микроскопом. После того, как большинство клеток округлилось, панкреатин осторожно аспирировали, и добавляли свежую среду, чтобы остановить расщепление. Клетки пипетировали с получением гомогенной клеточной суспензии, и клеточную суспензию переносили в инкубатор клеток для дальнейшего культивирования.
4. Эксперимент по определению цитотоксичности
(1) Клетки RPMI8226 в логарифмической фазе роста добавляли на 384-луночный планшет по 1000 клеток/лунку в объеме 18 мкл/лунку и инкубировали при 37°C, 5% CO2 в течение 24 ч.
(2) Тестируемое соединение в исходной концентрации 10 мМ разбавляли в 10 раз ДМСО, а затем в 100 раз бессывороточной средой 1640, чтобы получить разведение рабочей концентрации 10 мкМ, содержащее 1% ДМСО. Затем путем 2-кратного серийного разведения получали 10 концентраций с использованием бессывороточной среды 1640, содержащей 1% ДМСО, при этом десятая точка концентрации представляла собой группу контроля с растворителем (без лекарственного средства). Разбавленное соединение добавляли в планшет с высеянными клетками в количестве 2 мкл/лунку с получением конечных концентраций соединения от 1000 нМ (2-кратное градиентное разведение, 10 градиентов концентрации), и для каждой концентрации устанавливали 4 повторения. Использовали 1% ДМСО в качестве контроля с растворителем и KPT-8602 в качестве положительного контроля.
(3) Клетки инкубировали при 37°C в течение 72 ч, и в каждую лунку добавляли по 10 мкл реагента для анализа Cell-Titer с последующей инкубацией в инкубаторе клеток в течение 10 мин.
(4) Смесь тщательно перемешивали путем встряхивания, а затем анализировали с помощью программы Cell-Titer, работающей на устройстве для считывания микропланшетов. Значения % ингибирования и IC50 (мкМ) рассчитывали с использованием программы GraphPad Prism 5.
Результаты клеточной активности с изучаемыми соединениями показаны в таблице ниже:
Результаты клеточной активности показывают, что соединения I и IV по существу эквивалентны KPT-8602.
III. Фармакокинетические результаты изучения лекарственных средств у крыс SD
Вывод: периоды полувыведения соединения I при пероральном и внутривенном введении были больше, чем у KPT-8602, а именно, период полувыведения при пероральном приеме был примерно в 2 раза больше, и AUC соединения I при внутривенном и пероральном введении были намного выше, чем у KPT-8602, что демонстрирует лучшие фармакокинетические свойства.
IV. Анализ токсичности соединений-кандидатов in vivo у мышей BALB/C
Случайным образом 25 мышей BALB/C разделяли на следующие 5 групп: группа контроля с растворителем (контроль), группа положительного контроля с KPT-8602 (30 мг/кг, один раз в сутки), группа KPT-8602 (60 мг/кг, один раз в сутки), группа соединения I (30 мг/кг, один раз в сутки) и группа соединения I (60 мг/кг, один раз в сутки), по 5 мышей в каждой группе. Каждой группе внутрижелудочно вводили тестируемое соединение в соответствующей концентрации в дозе 3 мл/кг. KPT-8602 и соединение I вводили ежедневно в течение 21 дня подряд.
Растворы соединений готовили следующим образом:
Получение 10% сульфобутил-β-циклодекстрина:
Взвешивали 5,0 г порошка сульфобутил-β-циклодекстрина в лабораторном стакане и пипеткой добавляли в стакан 50 мл буфера лимонной кислоты. После растворения порошка полученный раствор переносили в емкость.
Подготовка соединения I:
Взвешивали 24 мг соединения I и добавляли 1,6 мл 20%-ного водного раствора полиэтиленгликоля. После полного растворения добавляли 6,4 мл 10%-ного водного раствора сульфобутил-β-циклодекстрина, получая испытуемый раствор соединения I (3 мг/мл).
Подготовка КРТ-8602:
Взвешивали 24 мг соединения КРТ-8602 и добавляли 1,6 мл 20%-ного водного раствора полиэтиленгликоля. После полного растворения добавляли 6,4 мл 10% водного раствора сульфобутил-β-циклодекстрина, получая таким образом испытуемый раствор КРТ-8602 (3 мг/мл).
Результаты изменения массы тела мышей после последовательного введения показаны на ФИГ. 1.
Вывод:
Как показано на фигуре, две мыши в группе, получавшей 60 мг/кг KPT-8602, умерли на 7-й день, и все умерли на 10-й день. Группа, получавшая 30 мг/кг КРТ-8602, показала значительное снижение массы тела. Напротив, группы, получавшие 30 мг/кг и 60 мг/кг соединения I, сохраняли массу тела, сравнимую с контрольной группой, после 21 дня лечения, подтверждая хорошую безопасность.
| название | год | авторы | номер документа |
|---|---|---|---|
| АКРИЛСОДЕРЖАЩИЕ МОДУЛЯТОРЫ ЯДЕРНОГО ТРАНСПОРТА И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2838463C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ БЛОКАДЫ РЕЦЕПТОРОВ НЕЙРОКИНИНА-1 У МЛЕКОПИТАЮЩИХ, СПОСОБ ДЛЯ БЛОКАДЫ РЕЦЕПТОРОВ НЕЙРОКИНИНА-1 У МЛЕКОПИТАЮЩИХ | 1993 |
|
RU2140914C1 |
| ЗАМЕЩЕННЫЕ МОРФОЛИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПРОТИВОДЕЙСТВИЯ ВЕЩЕСТВУ Р ИЛИ БЛОКИРОВАНИЯ РЕЦЕПТОРОВ НЕЙРОКИНИНА-1 | 1995 |
|
RU2170233C2 |
| ДИФЕНИЛЬНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1997 |
|
RU2175319C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ СНИЖЕНИЯ КОЛИЧЕСТВА ТАХИКИНИНОВ ПРИ ЛЕЧЕНИИ ИЛИ ПРОФИЛАКТИКЕ ФИЗИОЛОГИЧЕСКИХ СОСТОЯНИЙ, АССОЦИИРУЕМЫХ С ИЗБЫТКОМ ТАХИКИНИНОВ | 1994 |
|
RU2131426C1 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| СОЕДИНЕНИЯ, ОКАЗЫВАЮЩИЕ ВОЗБУЖДАЮЩЕЕ ДЕЙСТВИЕ НА РЕЦЕПТОР АКТИВАТОРА ПРОЛИФЕРАЦИИ ПЕРОКСИСОМ ПОДТИПА Б, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ УКАЗАННЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2522450C2 |
Изобретение относится к области фармацевтики, а именно к производному триазола формулы I. Также изобретение относится к фармацевтической композиции на основе соединения формулы I и применению соединения формулы I для получения лекарственного средства для лечения нарушения, связанного с активностью CRM1. Технический результат – получение соединения формулы I, обладающего ингибирующей активностью в отношении CRM1 для лечения заболеваний, опосредованных CRM1. 3 н. и 3 з.п. ф-лы, 1 ил., 3 табл., 1 пр.
 I
I
1. Производное триазола формулы:
2. Фармацевтическая композиция для лечения заболевания, нарушения или симптома, связанного с активностью CRM1, содержащая производное триазола по п. 1 и фармацевтически приемлемый носитель.
3. Применение производного триазола по п. 1 для получения лекарственного средства для лечения нарушения, связанного с активностью CRM1.
4. Применение по п. 3, где нарушение, связанное с активностью CRM1, выбрано из пролиферативного нарушения, рака, воспалительного нарушения, аутоиммунного нарушения, вирусной инфекции, офтальмологического нарушения, нейродегенеративного нарушения, нарушения из-за аномального роста тканей, расстройства, связанного с приемом пищи, аллергии и респираторного нарушения.
5. Применение по п. 4, где нарушение, связанное с активностью CRM1, представляет собой рак.
6. Применение по п. 5, где нарушение, связанное с активностью CRM1, представляет собой множественную миелому.
| WO 2014205389 A1, 24.12.2014 | |||
| WO 2012099807 A1, 26.07.2012 | |||
| WO 2013019561 A1, 07.02.2013 | |||
| СПОСОБ РЕГЕНЕРАЦИИ ДУБЛЕНОГО ГАЛАЛИТА | 1931 |
|
SU36639A1 |

