Настоящее изобретение относится к органической химии и к продуктам окислительной деструкции кальций аторвастатина, а также к способам их получения. Настоящее изобретение относится также к кальций аторвастатину, который практически не содержит продукты окислительной деструкции, а также к фармацевтическим композициям, содержащим указанный кальций аторвастатин.
Чистота фармацевтически активных соединений всегда является существенным фактором, обеспечивающим безопасность и качество лекарственного средства. Как известно в данной области техники, в результате сложного многостадийного процесса получения фармацевтически активного соединения получают не только требуемый продукт, но также и примеси, которые являются родственными соединениями с близкой структурой. Кроме того, многие фармацевтически активные соединения чувствительны к действию условий окружающей среды, таких как, например, температура, pH, влажность, свет, газы, кислород, диоксид углерода, реакционная способность окружающей среды в течение обработки или хранения. Указанные условия окружающей среды могут привести к превращению фармацевтически активного соединения в продукты деструкции, которые в большинстве случаев являются менее эффективными по сравнению с активным соединением. Кроме более низкой эффективности продукты деструкции вызывают также неблагоприятные побочные действия, таким образом, отрицательно влияя на безопасность лекарственного средства. Даже очень низкое процентное содержание примесей или продуктов деструкции, присутствующих в активном соединении, может в значительной степени снизить безопасность лекарственного средства. В связи с этим большое значение имеет максимально возможная чистота вводимого фармацевтически активного соединения. Это означает, что процентное содержание продуктов деструкции и примесей, присутствующих в фармацевтически активном соединении, должно составлять минимальную величину.
Кроме того, фармацевтические эксципиенты, используемые в фармацевтической лекарственной форме, также оказывают влияние на количество продуктов деструкции и примесей, присутствующих в фармацевтически активных соединениях. Продукты деструкции фармацевтических эксципиентов сами по себе действуют в качестве реакционных центров, инициирующих реакции деструкции фармацевтически активных соединений в фармацевтической лекарственной форме.
Чувствительность различных фармацевтически активных соединений к окислительной деструкции описана в статье Waterman K.C. и др., Stabilization of Pharmaceuticals to Oxidative Degradation, Pharmaceutical Development and Technology, 7, 1, cc.1-32 (2002), где описаны также возможные подходы к стабилизации фармацевтически активных соединений в условиях окислительной деструкции. В упомянутой выше статье и в других публикациях обсуждается сложный механизм окисления в твердых фармацевтических лекарственных формах, который требует дальнейшего исследования, однако установлено, что активное соединение само себе, и более часто активное соединение в фармацевтической лекарственной форме, может окисляться. В статье Byrn S.R. и др. (Solid-State Chemistry of Drugs, 2nd ed., SSCI, West Lafayette, (1999)) установлено, что атмосферный молекулярный кислород взаимодействует с органическими кристаллами и что указанная реакционная способность зависит от формы и морфологии кристаллов активного соединения, которые определяют проницаемость кислорода и его растворимость в кристаллической решетке, соответственно. В некоторых примерах установлено, что реакционная способность снижается при увеличении температуры плавления, что свидетельствует об ингибировании диффузии кислорода в соединениях с высокой энергией кристаллической решетки.
До настоящего времени для предотвращения или снижения окисления активного соединения в фармацевтическом составе используются различные подходы, такие как, например:
1. увеличение концентрации активного соединения в фармацевтическом составе в случае, если окисление вызвано присутствием пероксидных и металлических примесей в эксципиентах,
2. добавление хелатных агентов (например, лимонная кислота, ЭДТУ, фумаровая кислота и малеиновая кислота) для удаления металлических примесей, присутствующих в эксципиентах,
3. использование фармацевтических эксципиентов высокой степени чистоты,
4. использование альтернативных фармацевтических эксципиентов или снижение количества эксципиентов в фармацевтической композиции, прежде всего, если эксципиенты являются причиной окисления за счет содержания в них примеси пероксидов,
5. использование антиоксидантов, которые способны предотвращать или снижать образование пероксидов в фармацевтической композиции, однако указанные антиоксиданты в то же время не снижают уровень уже присутствующих пероксидов. Некоторые известные антиоксиданты включают:
- инициаторы обрыва цепи (как, например, тиолы и фенолы),
- восстановители с более высокой скоростью окисления по сравнению с активным соединением и которые таким образом поглощают присутствующий кислород (например, сульфиты и аскорбиновая кислота), причем при использовании их комбинации наблюдается синергетический эффект (например, комбинация пальмитата аскорбиновой кислоты и токоферола),
- соединения - «поглотители» пероксидов, которые расщепляют пероксиды (например, Fe2+) по методике Фентона, однако их использование ограничено, поскольку в условиях указанной методики возможно образование свободного гидроксильного радикала, который затем инициирует реакции с участием свободных радикалов и, таким образом, деструкцию активного соединения,
- циклодекстрины, которые экранируют участок активного соединения, чувствительный к окислению (Waterman K.C. и др., Stabilization of Pharmaceuticals to Oxidative Degradation, Pharmaceutical Development and Technology, 7, 1, cc.1-32 (2002)).
Однако при использовании конкретных активных соединений невозможно предусмотреть оптимальные условия, и кроме того в современной литературе существует лишь несколько статей о стабилизации соединений (Waterman K.C. и др., Stabilization of Pharmaceuticals to Oxidative Degradation, Pharmaceutical Development and Technology, 7, 1, сс.1-32 (2002)).
Кальций аторвастатин, полукальциевая соль (R-(R*,R*))-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-((фениламино)карбонил)-1Н-пиррол-1-гептановой кислоты, является известным ингибитором редуктазы 3-гидрокси-3-метилглутарилкофермента A (HMG-CoA). Указанное соединение впервые описано в патенте США №5273995. Способы получения кальций аторвастатина и главных промежуточных соединений описаны в патентах США №5003080, 5097045, 5103024, 5124482, 5149837, 5155251, 5216174, 5245047, 5248793, 5280126, 5342952 и 5397792.
Известно, что ингибиторами редуктазы HMG-CoA являются фармацевтически активные соединения, которые чувствительны к условиям окружающей среды, pH, влажности, свету, температуре, диоксиду углерода и кислороду. Указанные соединения являются эффективными терапевтически активными соединениями, предназначенными для лечения дислипидемических и сердечно-сосудистых заболеваний, выбранных из группы, включающей дислепидемию, гиперлипидемию, гиперхолестеринемию, атеросклероз, артериосклероз, заболевания коронарной артерии, коронарную болезнь сердца и т.п., связанных с метаболизмом липидов и холестерина. Механизм действия статиновых соединений заключается в ингибировании биосинтеза холестерина и других стеролов в печени человека или животных. Указанные соединения являются конкурентными ингибиторами редуктазы HMG-CoA или редуктазы 3-гидрокси-3-метилглутарилкофермента А, фермента, который катализирует превращение HMG-CoA в мевалонат в печени человека или животных, причем указанное превращение является важной стадией биосинтеза холестерина в печени. В настоящее время установлено, что кроме выше упомянутого терапевтического действия статины обладают также терапевтическими действиями другого типа и, соответственно, пригодны для лечения заболеваний, патологических состояний и нарушений, которые выбирают из группы, включающей сосудистые нарушения, воспалительное заболевание, аллергическое заболевание, нейродегенеративное заболевание, злокачественное заболевание, вирусное заболевание (заявка WO 0158443), аномальные состояния костной ткани (заявка WO 0137876), нарушения продуцирования β-амилоидного белка-предшественника, такие как болезнь Альцгеймера или синдром Дауна (заявка WO 0132161).
В современной литературе практически отсутствуют данные о снижении содержания продуктов окисления в препаратах аторвастатина и об идентификации продуктов деструкции кальций аторвастатина. Предотвращение окисления кальций аторвастатина за счет проведения процесса его получения в инертной атмосфере и упаковки в инертной атмосфере описано в заявке на выдачу патента Словении SI Р-200200244. Структура одного из продуктов деструкции кальций аторвастатина, фениламида 3-(4-фторбензоил)-2-изобутирил-3-фенилоксиран-2-карбоновой кислоты и его образование при фотодеструкции описано в статье Hurley Т.R. и др., Tetrahedron, 49, cc.1979-1984 (1993).
Так как получение активного соединения с высокой степенью очистки имеет большое значение, существует необходимость идентификации каждой примеси или продукта деструкции, присутствующих в активном соединении и/или в фармацевтической композиции. Идентификация каждой примеси или продукта деструкции, присутствующих в активном соединении или в фармацевтической композиции, прежде всего, является важной в отношении таких активных соединений, для которых уровень чувствительности указанного аналитического метода (например, ЖХВР) при определении примеси и/или продукта деструкции отличается от уровня чувствительности при определении активного соединения. А именно в таких случаях активное соединение считается как фармацевтически приемлемое согласно существующим требованиям, несмотря на то, что действительный уровень примесей или продуктов деструкции находится за пределами допустимых значений.
В настоящем изобретении согласно существующей в данной области техники необходимости предлагается продукт кальций аторвастатина с высокой степенью очистки, характеризующийся низким содержанием продуктов окислительной деструкции, простым способом получения и высоким выходом за счет идентификации трех продуктов окислительной деструкции, присутствующих в кальций аторвастатине и/или в содержащих его фармацевтических композициях.
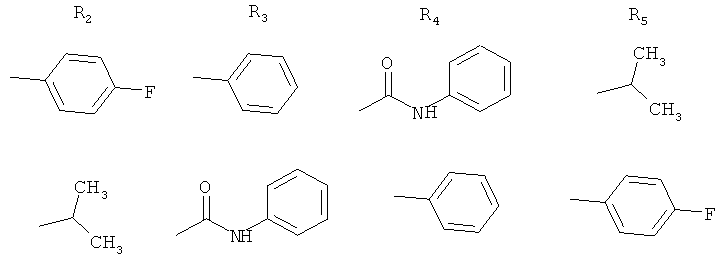
Один объект настоящего изобретения относится к новым соединениям, которые являются продуктами окислительной деструкции кальций аторвастатина следующих формул:
а) соединение формулы I
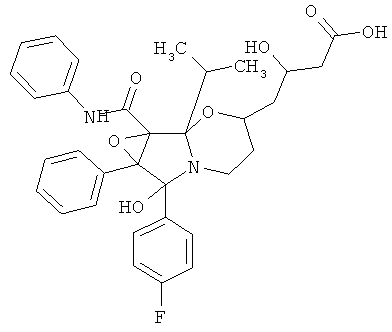
4-[6-(4-фторфенил)-6-гидрокси-1b-изопропил-6а-фенил-1а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляная кислота (сокращенное название ATV-циклоIР),
б) соединение формулы II
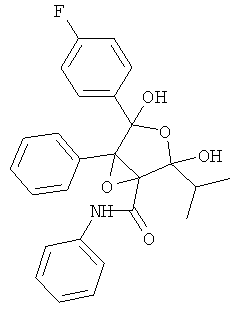
фениламид 4-(4-фторфенил)-2,4-дигидрокси-2-изопропил-5-фенил-3,6-диоксабицикло[3.1.0]гексан-1-карбоновой кислоты (сокращенное название ATV-эпоксифуран),
с) соединение формулы III
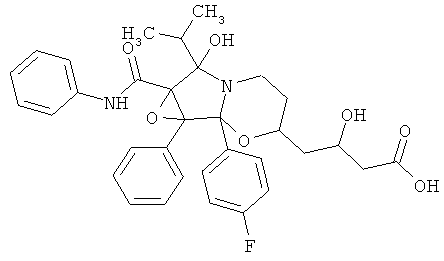
4-[1b-(4-фторфенил)-6-гидрокси-6-изопропил-1а-фенил-6а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляная кислота (сокращенное название ATV-циклоFР).
Продукт окислительной деструкции кальций аторвастатина, описанный в статье Hurley T.R. и др., Tetrahedron, 49, cc.1979-1984 (1993), характеризуется следующей формулой IV:
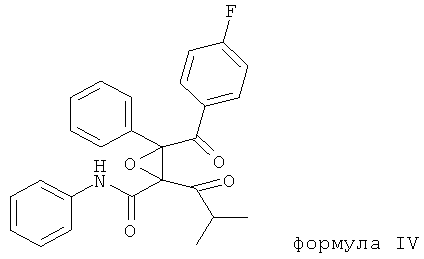 ,
,
фениламид 3-(4-фторбензоил)-2-изобутирил-3-фенилоксиран-2-карбоновой кислоты (сокращенное название ATV-эпоксидион).
Окислительная деструкция кальций аторвастатина представлена на следующей схеме:
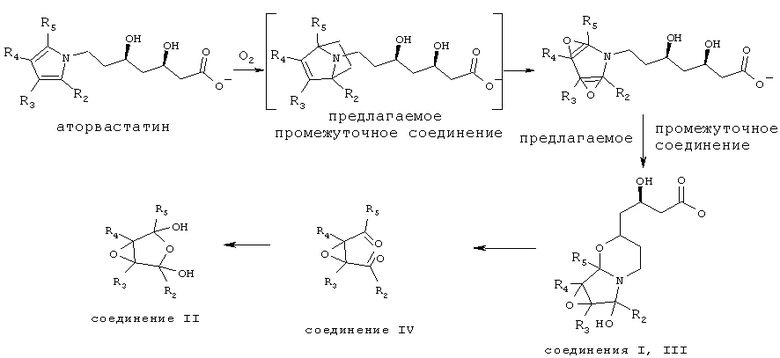
Соединения формул I, II, III и IV по настоящему изобретению присутствуют в препарате кальций аторвастатина в качестве продуктов окислительной деструкции, в связи с этим важно снизить их количество до минимального значения по данным анализа. Такие примеси являются токсичными или в ином отношении оказывают отрицательное воздействие на пациента. В связи с указанными причинами важно снизить содержание таких примесей в соединении до минимального количества. С другой стороны, имеет значение точность детектирования уровня указанных примесей, а их содержание следует определять количественно, например, с использованием стандартов (соединения известной химической структуры и известный метод анализа).
Настоящее изобретение относится также к новым способам получения соединений формул I, II, III и IV по настоящему изобретению.
Новые соединения по настоящему изобретению получают окислением твердого аторвастатина в форме соли (например, в виде соли кальция, натрия, калия, магния или аммония) в атмосфере воздуха или кислорода при повышенной температуре, например от 40 до 90°С. Продолжительность реакции составляет от 1 до нескольких сут. Окисление проводят в растворе соли аторвастатина в воде, и/или в органическом растворителе, и/или в смесях растворителей, таких как, например, ацетонитрил, метанол, этанол, пропанол, дихлорметан или хлористый метилен, при добавлении пероксида водорода или при пропускании воздуха или кислорода через раствор при температуре от приблизительно 40 до 90°С. Твердую соль аторвастатина получают любым известным способом.
Новые соединения по настоящему изобретению получают также фотоокислением аторвастатина в форме соли (например, в виде соли кальция, натрия, калия, магния, аммония), облучая раствор соли аторвастатина солнечным светом или искусственным солнечным светом. Соль аторвастатина получают любым известным способом.
Новые соединения по настоящему изобретению, полученные описанными выше способами, выделяют препаративной нормально-фазовой или обращенно-фазовой хроматографией.
В препаративной нормально-фазовой хроматографии в качестве неподвижной фазы используют силикагель для хроматографии или фазы на основе модифицированного силикагеля, например, содержащего следующие группы: аминопропил, цианопропил, диол или нитрофенил. Мобильная фаза содержит смесь полярного спирта-модификатора, например, метанол, этанол, пропанол или ацетонитрил, и неполярного растворителя, например, гексан, дихлорметан, метилциклогексан, или комбинацию более двух перечисленных выше растворителей.
В препаративной обращенно-фазовой хроматографии используют силикагель, содержащий группы октадецилсилана или октилсилана. Мобильная фаза содержит смесь воды с органическим или неорганическим буферным раствором при концентрации в диапазоне от 5 мМ до 100 мМ и в диапазоне pH от 2 до 8 в смеси с одним или более органических модификаторов, выбранных из спиртов, таких как, например, метанол, этанол и пропанол, или ацетонитрила.
При выделении новых соединений по настоящему изобретению используют одну или более хроматографических стадий. Растворители, использованные на стадиях хроматографии, удаляют упариванием и/или лиофилизацией.
Структуру новых соединений по настоящему изобретению, полученных и выделенных описанными выше способами, определяют методами масс-спектрометрии и ядерного магнитного резонанса. Способы идентификации структуры и их результаты приведены в описанных ниже примерах.
При разработке способа получения устойчивых композиций кальций аторвастатина установлено, что кальций аторвастатин подвергается деструкции при контактировании с воздухом или с кислородом. Неожиданно было установлено, что при использовании различных антиоксидантов, таких как, например, бутилированный гидроксианизол, бутилированный гидрокситолуол, фумаровая кислота, пропилгалат, сульфит натрия, метабисульфит натрия, аскорбат натрия, не наблюдается предотвращения и снижения образования продуктов окислительной деструкции. Неожиданно было также установлено, что при снижении содержания кислорода в окружающей атмосфере можно значительно снизить образование продуктов окислительной деструкции кальций аторвастатина в препарате кальций аторвастатина или в композиции, содержащей кальций аторвастатин. Наблюдаемое снижение количества продуктов окислительной деструкции прямо пропорционально снижению содержания кислорода в окружающей атмосфере. Указанное снижение содержания кислорода достигается за счет замены кислорода на инертный газ, например азот или аргон, или за счет снижения давления атмосферы, окружающей кальций аторвастатин.
Кроме проведения процесса получения кальций аторвастатина в инертной атмосфере и хранения его в инертной атмосфере важно регистрировать количество продуктов окислительной деструкции, присутствующих в препарате кальций аторвастатина и в содержащей его фармацевтической композиции. Для определения количества нежелательных соединений и для проведения точного количественного анализа необходимо получить стандарты указанных соединений (т.е. использовать соединения известной химической структуры и соответствующий метод анализа). Наличие стандартов имеет особое значение в случаях, если уровень чувствительности например, анализа ЖХВР, при идентификации примеси и/или продукта деструкции отличается от уровня чувствительности при определении активного соединения. Для определения примесей в фармацевтически активных соединениях и фармацевтических композициях обычно используют метод ЖХВР.
Неожиданно установлено, что некоторые соединения, присутствующие в препарате кальций аторвастатина, характеризуются уровнем чувствительности, отличающимся от уровня чувствительности самого кальций аторвастатина, если детектирование методом ЖХВР проводят при 250 нм. А именно уровень чувствительности для нового соединения по настоящему изобретению формулы I составляет 0,41, для нового соединения по настоящему изобретению формулы II уровень чувствительности составляет 0,72, для нового соединения по настоящему изобретению формулы III уровень чувствительности составляет 0,48, а для нового соединения по настоящему изобретению формулы IV уровень чувствительности составляет 1,20 в отличие от самого кальций аторвастатина.
Содержание примесей в активном соединении и/или фармацевтической композиции является важным фактором безопасности лекарственного средства, следовательно, следует свести к минимуму содержание примесей. Указанное имеет первостепенное значение для продуктов деструкции, поскольку их содержание в лекарственном средстве увеличивается при хранении лекарственного средства.
В другом объекте настоящего изобретения предлагается кальций аторвастатин, практически не содержащий продукты окислительной деструкции, и фармацевтические композиции, содержащие указанный кальций аторвастатин, а также по крайней мере один фармацевтически приемлемый эксципиент.
В настоящем изобретении предлагается кальций аторвастатин высокой степени очистки, в котором содержание продуктов окислительной деструкции составляет менее приблизительно 0,29 мас.%.
В настоящем изобретении предлагается кальций аторвастатин высокой степени очистки, в котором содержание ATV-циклоIР составляет менее приблизительно 0,09 мас.%.
В настоящем изобретении предлагается кальций аторвастатин высокой степени очистки, в котором содержание ATV-эпоксифурана составляет менее приблизительно 0,05 мас.%.
В настоящем изобретении предлагается кальций аторвастатин высокой степени очистки, в котором содержание ATV-циклоFР составляет менее приблизительно 0,09 мас.%.
В настоящем изобретении предлагается кальций аторвастатин высокой степени очистки, в котором содержание ATV-эпоксидиона составляет менее приблизительно 0,06 мас.%.
В таблице 1 ниже приведено количество каждого продукта окислительной деструкции, присутствующего в препарате кальций аторвастатина, в зависимости от различных атмосферных условий, в которых проводят его получение.
Если кальций аторвастатин получают или хранят на воздухе при комнатной температуре, образуются продукты окислительной деструкции, которые не образуются при хранении кальций аторвастатина в атмосфере азота.
При сравнении данных таблицы 1 (уровень чувствительности для продуктов окислительной деструкции составляет 1,00) и таблицы 2 (различные уровни чувствительности) установлено, что содержание указанных продуктов значительно отличается. Если используется уровень чувствительности 1,00 в отсутствие стандартов примесей, то измеренные значения для продуктов окислительной деструкции в кальций аторвастатине, полученном на воздухе или в атмосфере азота, ниже значений, определенных с использованием более точных уровней чувствительности. Содержание продуктов окислительной деструкции, определенное при уровне чувствительности 1,00, также ниже предельного значения 0,10%, что соответствует стандартным требованиям, принятым в фармацевтике. Более того, при уровне чувствительности выше 1,00 содержание ATV-эпоксидиона выше по сравнению с содержанием, определенным при более точном уровне чувствительности.
Все описанные ниже анализы проводят при следующих уровнях чувствительности: 0,41 для ATV-циклоIР, 0,72 для ATV-эпоксифурана, 0,48 для ATV-циклоFР и 1,20 для ATV-эпоксидиона.
В настоящем изобретении предлагается фармацевтическая композиция, содержащая практически чистый кальций аторвастатин, который содержит менее приблизительно 0,6 мас.% продуктов окислительной деструкции и по крайней мере один фармацевтически приемлемый эксципиент.
В настоящем изобретении предлагается фармацевтическая композиция, содержащая практически чистый кальций аторвастатин, который содержит менее приблизительно 0,2 мас.% ATV-циклоIР и по крайней мере один фармацевтически приемлемый эксципиент.
В настоящем изобретении предлагается фармацевтическая композиция, содержащая практически чистый кальций аторвастатин, который содержит менее приблизительно 0,1 мас.% ATV-эпоксифурана и по крайней мере один фармацевтически приемлемый эксципиент.
В настоящем изобретении предлагается фармацевтическая композиция, содержащая практически чистый кальций аторвастатин, который содержит менее приблизительно 0,2 мас.% ATV-циклоFР и по крайней мере один фармацевтически приемлемый эксципиент.
В настоящем изобретении предлагается фармацевтическая композиция, содержащая практически чистый кальций аторвастатин, который содержит менее приблизительно 0,1 мас.% ATV-эпоксидиона и по крайней мере один фармацевтически приемлемый эксципиент.
Результаты, приведенные в таблицах 3 и 4, свидетельствуют о том, что при хранении кальций аторвастатина или фармацевтического состава, содержащего кальций аторвастатин, в форме таблеток на воздухе при комнатной температуре в течение 24 месяцев содержание продуктов окислительной деструкции значительно возрастает. Указанный эффект можно исключить при хранении кальций аторвастатина в атмосфере азота.
Фармацевтическую композицию по настоящему изобретению вводят млекопитающему в виде лекарственной формы. Лекарственная форма содержит практически чистый кальций аторвастатин по настоящему изобретению и по крайней мере один фармацевтически приемлемый эксципиент, выбранный из группы, включающей разбавители, связующие, дезинтегрирующие вещества, замасливатели, глиданты, ароматизаторы, подсластители, консерванты, красители и другие эксципиенты, используемые при получении фармацевтической композиции. Фармацевтическую композицию по настоящему изобретению перерабатывают в любую лекарственную форму, используемую в фармацевтической промышленности, такую как, например, таблетки, дисперсные составы для перорального применения, капсулы, пеллеты, гранулы и т.д. В качестве инертного газа можно использовать азот или аргон для создания инертной атмосферы. Фармацевтическую композицию можно хранить в инертной атмосфере в блистере алюминий/алюминий, блистере с покрытием из гомополимера Al-полихлор-3-фторэтилен/ПВХ или во флаконах.
Фармацевтическую композицию по настоящему изобретению можно использовать для лечения гиперхолестеринемии и гиперлипидемии.
Настоящее изобретение иллюстрируется следующими примерами, не ограничивающими его объем.
Пример 1
Получение и выделение соединений ATV-циклоIР, ATV-эпоксифуран, ATV-циклоFР и ATV-эпоксидион
Кальций аторвастатин (5 г) хранили в герметично закрытом контейнере объемом 200 мл в атмосфере кислорода при 80°С в течение 30 сут. Полученный таким образом образец растворяли в смеси 50% ацетонитрил/вода (об./об.) и выделяли препаративной хроматографией.
Препаративная хроматография
Выделение продуктов окислительной деструкции проводили обращенно-фазовой хроматографией. Для получения очищенных соединений проводили две стадии хроматографии с различными мобильными фазами.
На первой стадии разделение проводили с использованием хроматографа для препаративной ЖХВР, снабженного колонкой Luna prep С 18(2), 10 мкм (200 мм×50 мм) и УФ-детектором при длине волны 250 нм. В качестве двух мобильных фаз использовали системы А и В: 10 мМ раствор ацетата аммония (pH 4,5) и смесь 95% ацетонитрил/5% тетрагидрофуран (об./об.) соответственно. Скорость потока составляла 140 мл/мин. Элюцию проводили с использованием следующего градиента:
Получали четыре фракции, значение pH первой и второй фракций доводили до 8-9 раствором гидроксида калия (1 М), pH третьей и четвертой фракций доводили до значения 2-3 соляной кислотой (1 М). Фракции упаривали при пониженном давлении. Температуру водяной бани поддерживали ниже 30°С и обратный холодильник охлаждали водой при температуре 0°С.
Для получения очищенных соединений все четыре фракции подвергали дополнительной очистке.
Очистка первой фракции
Первую фракцию очищали в тех же условиях, которые использовали на первой стадии за исключением мобильной фазы А, в качестве которой использовали 10 мМ раствор гидрокарбоната аммония. Элюцию проводили в следующем градиенте:
Получали первую фракцию, pH доводили до 8-9 раствором гидроксида калия (1 М). Фракцию упаривали при пониженном давлении аналогично тому, как описано для первой стадии разделения.
После лиофилизации концентрированной фракции получали очищенное соединение ATV-циклоIР, 4-[6-(4-фторфенил)-6-гидрокси-1b-изопропил-6а-фенил-1а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляную кислоту (170 мг). Хроматографическая чистота составляла 97,2%.
Очистка второй фракции
Вторую фракцию очищали в тех же условиях, которые использовали на первой стадии, за исключением того, что в качестве мобильной фазы А использовали смесь 70% 10 мМ фосфатный буферный раствор (pH 7,0)/25% ацетонитрил/5% тетрагидрофуран (об./об./об.) и следующий градиент:
Получали первую фракцию и упаривали при пониженном давлении. Концентрированную фракцию наносили на колонку для обращенно-фазовой хроматографии, буферные вещества отмывали водой и соединение ATV-циклоFР элюировали из колонки смесью 80% ацетонитрил/20% вода (об./об.).
После лиофилизации концентрированной фракции получали очищенное соединение ATV-циклоFР, 4-[1b-(4-фторфенил)-6-гидрокси-6-изопропил-1а-фенил-6а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляную кислоту (185 мг). Хроматографическая чистота составляла 97,5%.
Очистка третьей фракции
Третью фракцию очищали в тех же условиях, которые использовали на первой стадии, за исключением того, что в качестве мобильной фазы А использовали 5 мМ соляную кислоту и следующий градиент:
Получали первую фракцию и упаривали при пониженном давлении аналогично тому, как описано на первой стадии.
После лиофилизации концентрированной фракции получали очищенное соединение ATV-эпоксифуран, фениламид 4-(4-фторфенил)-2,4-дигидрокси-2-изопропил-5-фенил-3,6-диоксабицикло[3.1.0]гексан-1-карбоновой кислоты (205 мг). Хроматографическая чистота составляла 93,6%.
Очистка четвертой фракции
Четвертую фракцию очищали в тех же условиях, которые использовали для очистки третьей фракции, за исключением того, что использовали следующий градиент:
Получали первую фракцию и упаривали при пониженном давлении аналогично тому, как описано на первой стадии.
После лиофилизации концентрированной фракции получали очищенное соединение ATV-эпоксидион, фениламид 3-(4-фторбензоил)-2-изобутурил-3-фенилоксиран-2-карбоновой кислоты (50 мг). Хроматографическая чистота составляла 96,2%.
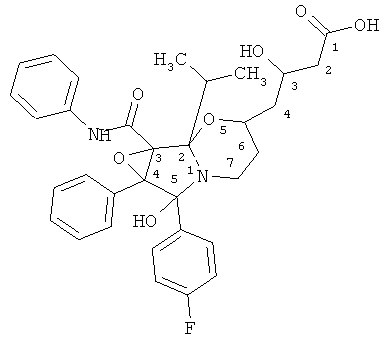
Определение структуры соединения ATV-циклоIР
Масс-спектрометрия
Условия
Масс-спектры высокого разрешения получали с использованием квадрупольного время-пролетного масс-спектрометра Micromass Q TOF Ultima Global. Использовали ионизацию электроспреем. Температуру источника устанавливали равной 100°С, температуру десольватации равной 200°С, скорость газа в распылителе составляла 0 л/ч, а осушающего газа - 200 л/ч. Использовали TOF-анализатор W-образной конфигурации. Прибор калибровали с использованием кластеров формиата Na. Образец растворяли в 50% растворе 5 мМ ацетат аммония/ацетонитрил (об./об.) и подавали в масс-спектрометр с постоянной скоростью потока 10 мкл/мин. Концентрация раствора образца составляла 0,05 мг/мл.
В качестве внутреннего стандарта для высокого разрешения использовали кальций аторвастатин. В раствор с образцом добавляли внутренний стандарт с концентрацией 0,01 мг/мл.
Регистрировали протонированный молекулярный ион 591,2507 m/z. Рассчитанный элементный состав C33H36N2O7F. Отклонение рассчитанного значения от измеренной массы составляло 0,5 мДа. По сравнению с кальций аторвастатином в соединении ATV-циклоIР идентифицированы два дополнительных атома кислорода.
Спектроскопия ядерного магнитного резонанса
Условия
1Н и 13С ЯМР-спектроскопию проводили с использованием прибора Varian INOVA или UNITY 300 при частоте 300 МГц. Прибор INOVA снабжен градиентной ячейкой импульсного поля для инверсного детектирования. 1Н и 13С ЯМР-спектры регистрировали при комнатной температуре.
Образцы растворяли в метаноле, хлороформе или смеси метанол/хлороформ 2:1.
Химические сдвиги в част./млн определяли по отношению к остаточному сигналу растворителя.
Растворитель: CD3OD (1Н и 13С ЯМР-спектры)
Структура:
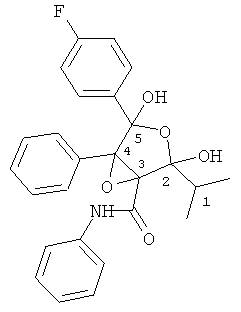
Определение структуры соединения ATV-эпоксифуран
Масс-спектрометрия
Условия
Масс-спектры высокого разрешения регистрировали в аналогичных условиях, как описано для соединения ATV-циклоIР.
В масс-спектре наблюдаются аддукты молекулярного иона с натрием, 472,1536 m/z, и калием, 488,1270, в первом случае рассчитанный элементный состав C26H24NO5FNa (отклонение рассчитанной массы от измеренного значения составляло 0,0 мДа), а во втором случае C26H24NO5FK (отклонение рассчитанной массы от измеренного значения составляло 0,5 мДа).
Протонированный молекулярный ион не наблюдается вследствие быстрого удаления воды из молекулы (М+Н-H2O)+=432,1606 m/z, предполагаемый элементный состав C26H23NO4F. Отклонение рассчитанной массы от измеренного значения составляло 0,5 мДа. Указанный фрагментарный ион также образует аддукты с натрием и калием.
Аддукт двух молекул соединения ATV-эпоксифурана с натрием наблюдается при 921,3131 m/z, предполагаемый элементный состав C52H48N2O10F2Na. Отклонение рассчитанной массы от измеренного значения составляло 4,4 мДа.
Спектроскопия ядерного магнитного резонанса
Условия
1Н и 13С ЯМР-спектры регистрировали, как описано для соединения ATV-циклоIР.
Растворитель: CDCl3 (1H ЯМР)
смесь CD3OD/CDCl3, 2:1 (13С ЯМР)
Структура:
Определение структуры соединения ATV-циклоFР
Масс-спектрометрия
Условия
Масс-спектры высокого разрешения регистрировали, как описано для соединения ATV-циклоIР.
Протонированный молекулярный ион наблюдается при 591,2507 m/z. Интенсивность молекулярного иона значительно ниже по сравнению с молекулярным ионом ATV-циклоIР. Наиболее интенсивный ион в МС-спектре наблюдается при 573,2406 m/z, который образуется при удалении молекулы воды, рассчитанный элементный состав для 591,2507 m/z - C33H36N2O7F. Отклонение рассчитанной массы от измеренного значения составляет 1,4 мДа. В МС-спектре соединения наблюдаются два дополнительных атома кислорода по сравнению с кальций аторвастатином.
Спектроскопия ядерного магнитного резонанса
Условия
1Н и 13С ЯМР-спектры регистрировали, как описано для соединения ATV-циклоIР.
Растворитель: CD3OD (1Н и 13С ЯМР)
Структура:
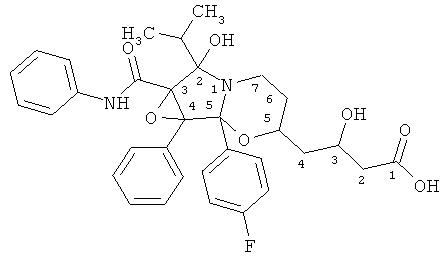
Определение структуры соединения ATV-эпоксидион
Масс-спектрометрия
Условия
Масс-спектры высокого разрешения регистрировали, как описано для соединения ATV-циклоIР.
В спектре наблюдается протонированный молекулярный ион 432,1612 m/z, рассчитанный элементный состав C26H23NO4F. Отклонение рассчитанной массы от измеренного значения 0,1 мДа.
В таблице 5 приведены спектры МС/МС протонированного молекулярного иона.
Спектроскопия ядерного магнитного резонанса
Условия
1Н и 13С ЯМР-спектры регистрировали, как описано для соединения ATV-циклоIР.
Растворитель: смесь CD3OD/CDCl3, 2:1 (1Н и 13С ЯМР)
Структура:
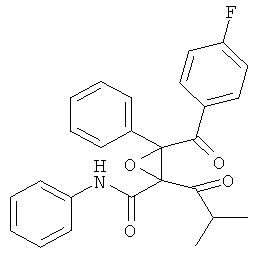
Пример 2
Получение и выделение соединения ATV-циклоIР
Раствор аторвастатина (2 л) получали в смеси 80% ацетонитрил/20% вода (об./об.) при концентрации 1 мг аторвастатина в 1 мл. Раствор помещали в мелкий кристаллизатор и выдерживали на солнце в течение 5 ч, затем раствор немедленно подщелачивали 1 М раствором гидроксида калия до pH 8-9 и упаривали при пониженном давлении до первых признаков помутнения. Температуру водяной бани поддерживали ниже 30°С и обратный холодильник охлаждали водой при температуре 0°С.
Затем раствор осветляли добавлением минимального количества ацетонитрила.
Препаративную хроматографию и определение структуры проводили аналогично тому, как описано в примере 1.
После лиофилизации концентрированной фракции получали очищенное соединение ATV-циклоIР (210 мг). Хроматографическая чистота соединения составляла 96,6%.
Пример 3
Получение и выделение соединений ATV-эпоксифуран и ATV-эпоксидион
Раствор аторвастатина (1 л) получали в смеси 80% ацетонитрил/20% вода (об./об.) при концентрации 1 мг аторвастатина в мл. Раствор помещали в мелкий кристаллизатор и выдерживали на солнце в течение 5 ч, затем раствор немедленно подкисляли 0,5 М фосфорной кислотой до pH 3,0. Смесь выдерживали при комнатной температуре в течение 2 ч и упаривали при пониженном давлении до приблизительно 1/3 первоначального объема.
Препаративную хроматографию и определение структуры проводили аналогично тому, как описано в примере 1.
После лиофилизации концентрированной фракции получали очищенное соединение ATV-эпоксифуран (120 мг). Хроматографическая чистота соединения составляла 92,6%.
После лиофилизации концентрированной фракции получали очищенное соединение ATV-эпоксидион (21 мг). Хроматографическая чистота соединения составляла 95,1%.
Пример 4
Получение и выделение соединения ATV-циклоFР
Раствор аторвастатина (800 мл) получали в ацетонитриле при концентрации 10 мг аторвастатина в мл. Добавляли 12 М раствор гидроксида натрия (4 мл) и 30% раствор пероксида водорода (40 мл). Раствор перемешивали при 55°С в течение 5 ч. Реакционную смесь охлаждали до комнатной температуры и декантировали. Супернатант упаривали при пониженном давлении до приблизительно 50 мл, воду отбрасывали и твердый остаток промывали свежей порцией воды. Затем твердый остаток растворяли в ацетонитриле.
Препаративную хроматографию и определение структуры проводили аналогично тому, как описано в примере 1.
После лиофилизации концентрированной фракции получали очищенное соединение ATV-циклоFР (230 мг). Хроматографическая чистота соединения составляла 98,3%.
Пример 5
Кальций аторвастатин получали любым известным способом. Единственное необходимое условие в течение всего процесса выделения кальций аторвастатина заключается в инертной атмосфере. В полученном таким образом кальций аторвастатине содержание каждого продукта окислительной деструкции (соединений: ATV-циклоIР, ATV-эпоксифуран, ATV-циклоFР и ATV-эпоксидион), определенное по данным ЖХВР при длине волны 250 нм, составляет менее 0,04%.
Пример 6
Кальций аторвастатин, полученный по методике, описанной в примере 1, хранили в атмосфере азота при комнатной температуре в течение 2 лет. В полученном таким образом кальций аторвастатине содержание каждого продукта окислительной деструкции (соединений: ATV-циклоIР, ATV-эпоксифуран, ATV-циклоFР и ATV-эпоксидион), определенное по данным ЖХВР при длине волны 250 нм, составляет менее 0,1%.
Пример 7
Кальций аторвастатин, полученный по методике, описанной в примере 1, хранили на воздухе при комнатной температуре в течение 2 лет. В полученном таким образом кальций аторвастатине содержание продуктов окислительной деструкции (соединений: ATV-циклоIР, ATV-эпоксифуран, ATV-циклоFР и ATV-эпоксидион), определенное по данным ЖХВР при длине волны 250 нм, составляет 0,856%, 0,636%, 0,905% и 0,741% соответственно.
Пример 8
Таблетки получали из кальций аторвастатина, полученного по методике, описанной в примере 1, и по крайней мере одного фармацевтически приемлемого эксципиента.
В полученных таким образом таблетках содержание продуктов окислительной деструкции (соединений: ATV-циклоIР, ATV-эпоксифуран, ATV-циклоFР и ATV-эпоксидион), определенное по данным ЖХВР при длине волны 250 нм, составляет 0,11%, 0,07%, 0,07% и 0,08% соответственно.
Таблетки упаковывали в блистеры алюминий/алюминий в атмосфере азота. Блистеры хранили при комнатной температуре в течение 2 лет. Содержание продуктов окислительной деструкции (соединений: ATV-циклоIР, ATV-эпоксифуран, ATV-циклоFР и ATV-эпоксидион), определенное по данным ЖХВР при длине волны 250 нм, составляет 0,18%, 0,08%, 0,17% и 0,09% соответственно.
Пример 9
Таблетки, полученные, как описано в примере 8, упаковывали в блистеры алюминий/алюминий на воздухе. В полученных таких образом таблетках содержание продуктов окислительной деструкции (соединений: ATV-циклоIР, ATV-эпоксифуран, ATV-циклоFР и ATV-эпоксидион), определенное по данным ЖХВР при длине волны 250 нм, составляет 0,13%, 0,09%, 0,08% и 0,08% соответственно.
Блистеры хранили при комнатной температуре в течение 2 лет. В полученном таким образом кальций аторвастатине содержание продуктов окислительной деструкции (соединений: ATV-циклоIР, ATV-эпоксифуран, ATV-циклоFР и ATV-эпоксидион), определенное по данным ЖХВР при длине волны 250 нм, составляет 1,75%, 0,61%, 1,23% и 0,65% соответственно.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛАМИДА 5-(4-ФТОРФЕНИЛ)-1-[2-(2R, 4R)-4-ГИДРОКСИ-6-ОКСОТЕТРАГИДРОПИРАН-2-ИЛ)ЭТИЛ]-2-ИЗОПРОПИЛ-4- ФЕНИЛ-1-H-ПИ РРОЛ-3- КАРБОНОВОЙ КИСЛОТЫ | 2001 |
|
RU2244714C1 |
| ПИРИМИДИНАМИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2006 |
|
RU2420519C2 |
| 5-ГЕТЕРОЦИКЛО-1,5-БЕНЗОДИАЗЕПИНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1995 |
|
RU2152939C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛАМИДА 5-(4-ФТОРФЕНИЛ)-1-[2-((2R,4R)-4-ГИДРОКСИ-6-ОКСОТЕТРАГИДРОПИРАН-2-ИЛ)ЭТИЛ]-2-ИЗОПРОПИЛ-4-ФЕНИЛ-1H-ПИРРОЛ-3-КАРБОНОВОЙ КИСЛОТЫ | 2004 |
|
RU2337905C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛАМИДА 5-(4-ФТОРФЕНИЛ)-1-[2-(2R,4R)-4-ГИДРОКСИ-6-ОКСОТЕТРАГИДРОПИРАН-2-ИЛ)ЭТИЛ]-2-ИЗОПРОПИЛ-4-ФЕНИЛ-1H-ПИРРОЛ-3-КАРБОНОВОЙ КИСЛОТЫ | 2003 |
|
RU2279430C2 |
| ЗАМЕЩЕННЫЕ ТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИН ТРЕОНИН КИНАЗЫ (ТТК) | 2012 |
|
RU2632464C1 |
| ПРОИЗВОДНЫЕ 1,5-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЙ ИХ ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1994 |
|
RU2135486C1 |
| СПОСОБ СИНТЕЗА АТОРВАСТАТИНА И ПРОМЕЖУТОЧНЫЙ ФЕНИЛБОРОНАТ (ВАРИАНТЫ) | 2001 |
|
RU2269515C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| КРЕМНИЙСОДЕРЖАЩИЕ СОЕДИНЕНИЯ ИЛИ ИХ СОЛИ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2069213C1 |
Настоящее изобретение относится к продуктам окислительной деструкции кальций аторвастатина, а именно к 4-[6-(4-фторфенил)-6-гидрокси-1b-изопропил-6а-фенил-1а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляной кислоте, фениламиду 4-(4-фторфенил)-2,4-дигидрокси-2-изопропил-5-фенил-3,6-диоксабицикло[3.1.0]гексан-1-карбоновой кислоты и 4-[1b-(4-фторфенил)-6-гидрокси-6-изопропил-1а-фенил-6а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляной кислоте. А также изобретение относится к способам их получения, основанных на окислении соли аторвастатина. Технический результат: выявлены продукты окислительной деструкции соли аторвастатина, которые могут быть использованы для идентификации примеси или продукта деструкции соли аторвастатина в соответствии с утвержденными аналитическими процедурами. 6 н. и 9 з.п. ф-лы, 5 табл.
1. 4-[6-(4-Фторфенил)-6-гидрокси-1b-изопропил-6а-фенил-1а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляная кислота.
2. Фениламид 4-(4-фторфенил)-2,4-дигидрокси-2-изопропил-5-фенил-3,6-диоксабицикло[3.1.0]гексан-1-карбоновой кислоты.
3. 4-[1b-(4-Фторфенил)-6-гидрокси-6-изопропил-1а-фенил-6а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляная кислота.
4. Способ получения соединения, выбранного из группы, включающей: 4-[6-(4-фторфенил)-6-гидрокси-1b-изопропил-6а-фенил-1а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляную кислоту, фениламид 4-(4-фторфенил)-2,4-дигидрокси-2-изопропил-5-фенил-3,6-диоксабицикло[3.1.0]гексан-1-карбоновой кислоты, 4-[1b-(4-фторфенил)-6-гидрокси-6-изопропил-1а-фенил-6а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляную кислоту, который характеризуется тем, что твердую соль аторвастатина окисляют в атмосфере воздуха или кислорода при повышенной температуре от 40 до 90°С.
5. Способ по п.4, где твердую соль аторвастатина выбирают из группы, включающей кальций аторвастатин, натрий аторвастатин, калий аторвастатин, магний аторвастатин и аммоний аторвастатин.
6. Способ получения соединения, выбранного из группы, включающей: 4-[6-(4-фторфенил)-6-гидрокси-1b-изопропил-6а-фенил-1а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляную кислоту, фениламид 4-(4-фторфенил)-2,4-дигидрокси-2-изопропил-5-фенил-3,6-диоксабицикло[3.1.0]гексан-1-карбоновой кислоты, 4-[1b-(4-фторфенил)-6-гидрокси-6-изопропил-1а-фенил-6а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляную кислоту, в котором окисление соли аторвастатина проводят в растворе соли аторвастатина в воде и/или в органическом растворителе и/или в смесях растворителей, выбранных из группы, включающей: ацетонитрил, метанол, этанол, пропанол, дихлорметан или хлористый метилен, при добавлении пероксида водорода или при пропускании воздуха или кислорода через раствор при температуре от приблизительно 40 до 90°С.
7. Способ по п.6, где соль аторвастатина выбирают из группы, включающей кальций аторвастатин, натрий аторвастатин, калий аторвастатин, магний аторвастатин и аммоний аторвастатин.
8. Способ получения соединения, выбранного из группы, включающей: 4-[6-(4-фторфенил)-6-гидрокси-1b-изопропил-6а-фенил-1а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляную кислоту, фениламид 4-(4-фторфенил)-2,4-дигидрокси-2-изопропил-5-фенил-3,6-диоксабицикло[3.1.0]гексан-1-карбоновой кислоты, 4-[1b-(4-фторфенил)-6-гидрокси-6-изопропил-1а-фенил-6а-фенилкарбамоилгексагидро-1,2-диокса-5а-азациклопропа[а]инден-3-ил]-3-(R)-гидроксимасляную кислоту, который характеризуется тем, что раствор соли аторвастатина облучают солнечным светом или искусственным солнечным светом.
9. Способ по п.8, где соль аторвастатина выбирают из группы, включающей кальций аторвастатин, натрий аторвастатин, калий аторвастатин, магний аторвастатин и аммоний аторвастатин.
10. Способ по пп.4-9, который, кроме того, включает одну или более стадий выделения.
11. Способ по п.10, где стадию выделения выбирают из группы, включающей препаративную нормально-фазовую и обращенно-фазовую хроматографию.
12. Способ по п.11, где для препаративной нормально-фазовой хроматографии в качестве неподвижной фазы используют силикагель или фазы на основе модифицированного силикагеля, например, содержащего следующие группы: аминопропил, цианопропил, диол и нитрофенил.
13. Способ по п.12, где для препаративной нормально-фазовой хроматографии в качестве мобильной фазы используют смесь полярного спирта-модификатора, выбранного из группы, включающей метанол, этанол, пропанол и ацетонитрил, и неполярного растворителя, выбранного из группы, включающей гексан, дихлорметан, метилциклогексан и любую их комбинацию.
14. Способ по п.13, где для препаративной обращенно-фазовой хроматографии используют силикагель, модифицированный октадецилсиланом или октилсиланом.
15. Способ по п.14, где для препаративной обращенно-фазовой хроматографии в качестве мобильной фазы используют смесь воды с органическим или неорганическим буферным раствором в смеси с одним или более органических модификаторов, выбранных из группы, включающей спирты и ацетонитрил.
| Устройство для избирательного управления с одного конца однопроводной линии несколькими реле | 1918 |
|
SU981A1 |
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2002113564 A, 20.11.2003. | |||

