ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к новым кристаллическим формам полукальциевой соли [R-(R*,R*)]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1H-пиррол-1-гептановой кислоты в качестве производного пирролгептановой кислоты, их гидратам, способу получения кристаллических форм и фармацевтическим композициям, содержащим кристаллические формы.
УРОВЕНЬ ТЕХНИКИ
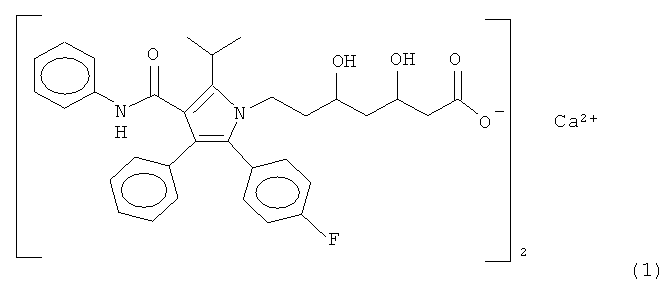
Синтезировано множество производных пирролгептановой кислоты. Из них [R-(R*,R*)]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1H-пиррол-1-гептановая кислота является статиновым лекарственным средством, механизм действия которого выяснен детально и которое, как известно, является наиболее терапевтически эффективным лекарственным средством, пригодным для снижения концентрации липопротеинов низкой плотности (ЛНП), как фактора риска тромбоза. Производное пирролгептановой кислоты в настоящее время коммерчески доступно в виде тригидрата полукальциевой соли. Полукальциевая соль пирролгептановой кислоты представлена формулой 1:
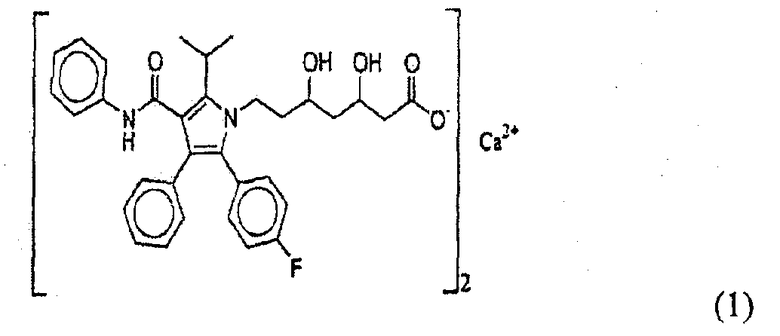
[R-(R*,R*)]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1H-пиррол-1-гептановая кислота была впервые раскрыта в патенте США № 4681893. В патенте США № 5273995 указано, что соединение формулы 1 получают кристаллизацией из концентрированного раствора хлорида натрия, в результате обмена натриевой соли на CaCl2 и дополнительно очищают перекристаллизацией из смеси 5:3 этилацетата и гексана.
В литературе описаны разнообразные способы и ключевые промежуточные соединения для получения аморфных форм соединения формулы 1, например, в патентах США №№ 5003080, 5097045, 5103024, 5124482, 5149837, 5155251, 5216174, 5245047, 5248793, 5280126, 5397792, 5342952, 5298627, 5446054, 5470981, 5489690, 5489691, 5510488, 5998633 и 6087511. Однако аморфные формы обладают не подходящими для крупномасштабного производства параметрами фильтрации и сушки и нуждаются в защите от воздействия тепла, света, кислорода и влаги.
С другой стороны, полиморфы рассматривают как отдельные вещества, потому что они обладают другими физическими свойствами, несмотря на ту же самую молекулярную формулу. Кристаллические формы соединения формулы 1 раскрыты в патентах США №№ 5969156 и 6121461. Кроме того, международная публикация № WO 01/36384 раскрывает полиморфную форму соединения формулы 1. В данной области существует потребность в кристаллических формах соединения формулы 1, которые являются более стабильными и легкими в обращении, чем известные кристаллические формы.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
Таким образом, авторы изобретения намерены раскрыть новые кристаллические формы соединения формулы 1, которые являются более чистыми, удобными для получения и очень стабильными.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ
Настоящее изобретение обеспечивает новые кристаллические формы D1 и D2 соединения формулы 1 и их гидраты.
Новые кристаллические формы по настоящему изобретению теперь будут описаны подробнее.
Было обнаружено, что кристаллические формы D1 и D2 соединения формулы 1 по настоящему изобретению отличаются от других известных кристаллических форм соединения формулы 1, как определено методами порошковой дифракции рентгеновских лучей и твердофазного 13C ядерного магнитного резонанса (ЯМР).
Порошковые рентгенограммы регистрировали на M18HF-SRA дифрактометре (Mac Science) с CuKα излучением, а 13C ЯМР спектры регистрировали на AVANCE-500 спектрометре (Bruker).
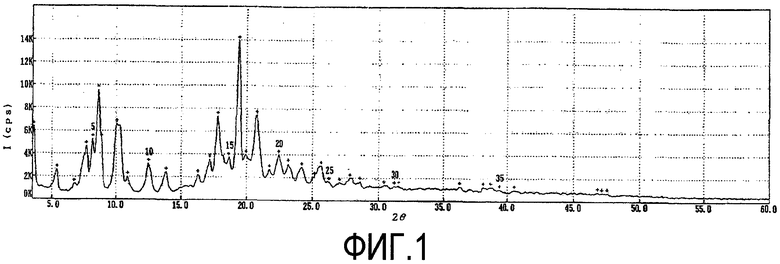
Фиг. 1 представляет собой порошковую рентгенограмму кристаллической формы D1, а таблица 1 дает перечень 2θ углов, d-расстояний и относительных интенсивностей с относительной интенсивностью >10% в порошковой рентгенограмме.
В дополнение к пикам порошковой рентгенограммы, перечисленным в таблице 1, кристаллическая форма D1 может иметь дополнительные пики на порошковой рентгенограмме со слабыми интенсивностями при 2θ=6,8, 10,8, 14,8, 16,2, 21,8, 25,1, 26,1, 27,2, 27,8, 28,5 и 30,4°.
Пики со слабыми интенсивностями определены как пики, имеющие относительную интенсивность <10%. Теоретические сведения о порошковых рентгенограммах можно обнаружить в многочисленных источниках, например, в книге «X-ray diffraction procedures», H.P. Klug и L. E. Alexander, J. Wiley, New York (1974), широко известной в области, к которой относится данная технология.
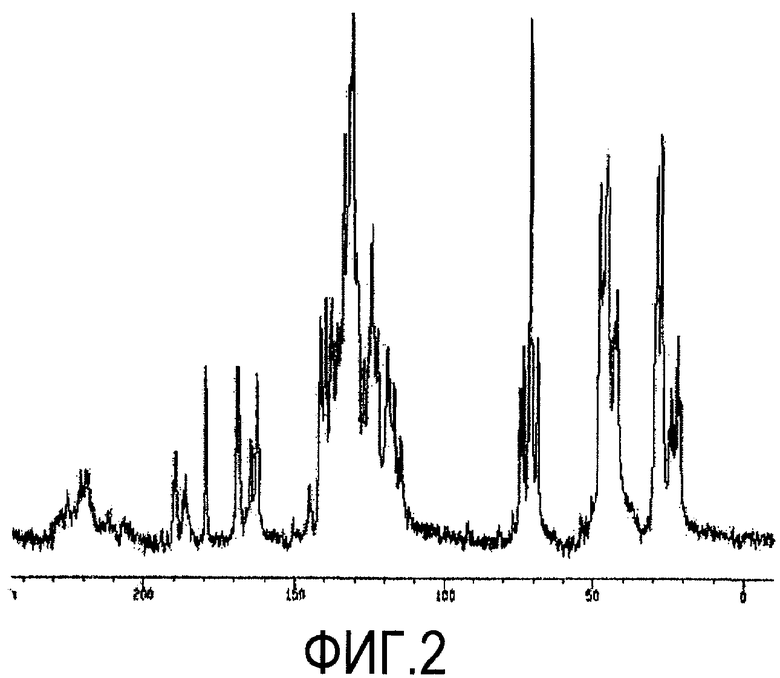
Фиг. 2 представляет собой 13C ЯМР спектр кристаллической формы D1, а таблица 2 дает перечень химических сдвигов атомов углерода кристаллической формы D1 в спектре ЯМР.
С2-С5, С13-С18, С19-С24, С27-С32
С6, С7, С9, С11
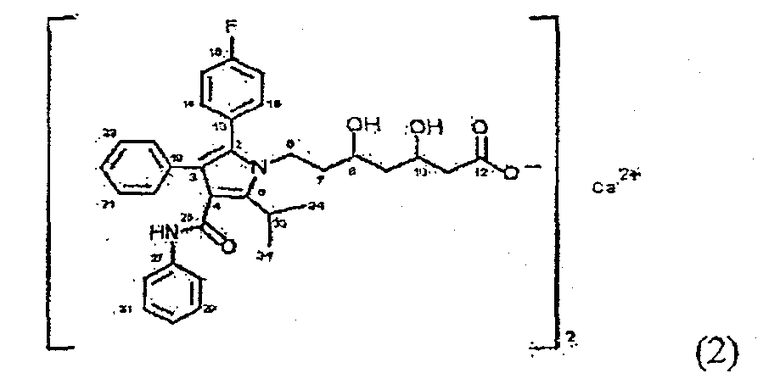
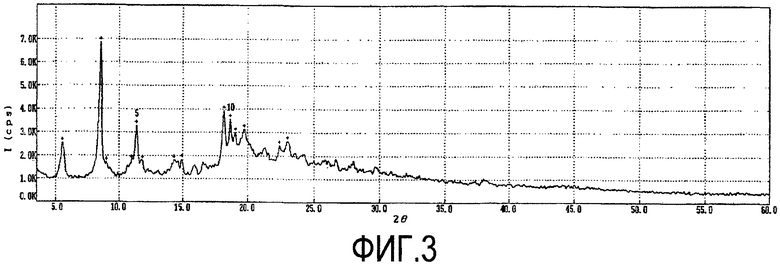
Фиг. 3 представляет собой порошковую рентгенограмму кристаллической формы D2, а таблица 3 дает перечень 2θ углов, d-расстояний и относительных интенсивностей с относительной интенсивностью >10% в порошковой рентгенограмме.
В дополнение к пикам порошковой рентгенограммы, перечисленным в таблице 3, кристаллическая форма D2 может иметь дополнительные пики на порошковой рентгенограмме со слабыми интенсивностями при 2θ=9,0 и 11,8° (w).
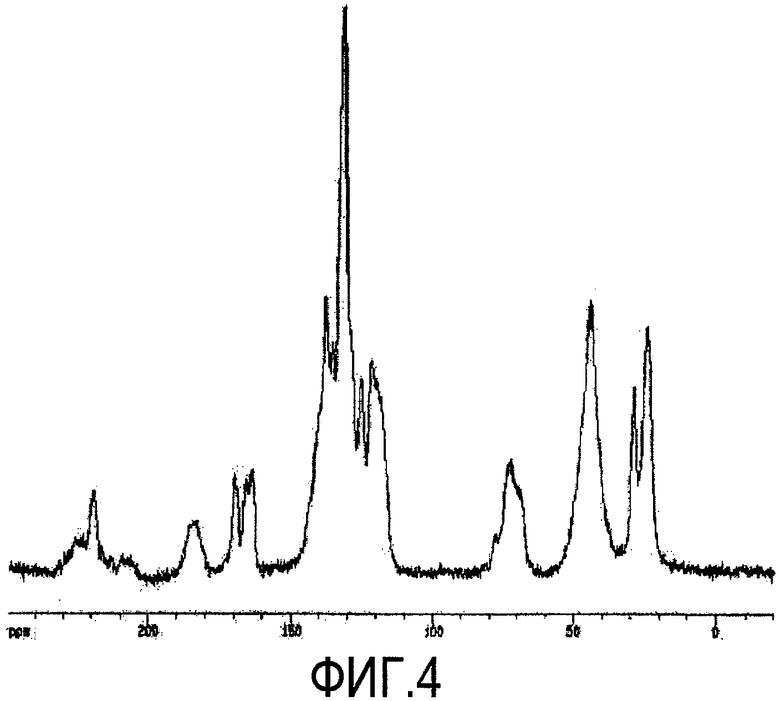
Фиг.4 представляет собой 13C ЯМР спектр кристаллической формы D2, а таблица 4 дает перечень химических сдвигов атомов углерода кристаллической формы D2 в спектре ЯМР.
С2-С5, С13-С18, С19-С24, С27-С32
С6, С7, С9, С11
Настоящее изобретение также обеспечивает гидраты кристаллических форм D1 и D2. Гидраты кристаллической формы D1 и D2 могут содержать 0,5-2,5% и 3,5-5,5% воды, соответственно. Содержание воды может изменяться в процессе хранения в зависимости от различных факторов, таких как температура и относительная влажность.
Кристаллическая форма соединения формулы обладает следующими преимуществами по сравнению с коммерчески доступной в настоящее время кристаллической формой I, описанной в патенте США № 5969156.
Кристаллическую форму I получают по реакции природной формы соединения формулы 1 (см., Пример 10 в патенте США № 5273995) со смесью метанол/вода при 40°C. Однако кристаллическую форму II получают, когда в ходе реакции со смесью метанол/вода температуру реакции понижают до комнатной температуры (см., патент США № 5969156). То есть, температура реакции является фактором, определяющим кристаллическую форму соединения формулы 1. Соответственно, для того чтобы получить требуемую чистую кристаллическую форму, следует с предельной тщательностью контролировать температуру реакции. В противоположность этому, как кристаллическую форму D1, так и кристаллическую форму D2 по настоящему изобретению получают в чистом виде в широком температурном интервале, включая комнатную температуру, исключая, таким образом, необходимость тщательного контроля температуры реакции. Например, кристаллическую форму I соединения формулы 1 получают при повышенной температуре 40°C, в то время как кристаллическую форму D2 соединения формулы 1 можно получить при комнатной температуре. Таким образом, кристаллическая форма D2 соединения формулы 1 является удобной для получения и обладает преимуществами в плане безопасности и стоимости.
Природную форму соединения формулы 1' получают согласно методике, описанной в примере 10 патента США № 5273995, и относят к состоянию, в котором сосуществуют аморфная и кристаллическая формы соединения формулы 1.
Другим преимуществом кристаллической формы D1 является более высокая температура плавления. В частности, коммерчески доступная кристаллическая форма I обладает температурой плавления примерно 176-178°C, в то время как кристаллическая форма D1 обладает температурой плавления 232°C. Эта высокая температура плавления делает возможной дополнительную высокотемпературную обработку кристаллической формы D1.
Кристаллическая форма D1 существует в виде моногидрата и содержание воды в ней меньше, чем в кристаллической форме I в виде тригидрата. То есть, число молекул, присутствующих в кристаллической форме D1 на единицу массы, больше, чем число молекул, присутствующих в кристаллической форме I в виде тригидрата, что обеспечивает большую эффективность кристаллической формы D1 по сравнению с лучшей кристаллической формой I.
Настоящее изобретение также обеспечивает способы получения новых кристаллических форм D1 и D2 соединения формулы 1.
В частности, кристаллическую форму D1 соединения формулы 1 можно получить путем растворения природной формы соединения формулы 1 (см. пример 10 патента США № 5273995) в смеси спирт/тетрагидрофуран и добавления воды в качестве осадителя к раствору, для того, чтобы выкристаллизовать кристаллическую форму D1. Предпочтительно перемешивать суспензию при 0-60°C, в особенности при 30-50°C, в течение длительного периода времени, в частности 10-72 часа. В ходе кристаллизации при необходимости можно ввести затравочный кристалл кристаллической формы D1. Предпочтительно использовать спирт, тетрагидрофуран и воду в объемном соотношении 1:1:8.
Кристаллическую форму D2 соединения формулы 1 можно получить путем растворения природной формы соединения формулы 1 в смеси спирт/метиленхлорид и добавления воды в качестве осадителя к раствору, для того, чтобы выкристаллизовать кристаллическую форму D2. Предпочтительно перемешивать суспензию при 0-40°C, в особенности при 20-30°C, в течение длительного периода времени, в частности 4-48 часов. В ходе кристаллизации при необходимости можно ввести затравочный кристалл кристаллической формы D2. Предпочтительно использовать спирт, метиленхлорид и воду в объемном соотношении 10:3:30.
Спирт, используемый в способах, является предпочтительно C1-C6 низшим спиртом. Наиболее предпочтительным является метанол.
Настоящее изобретение также обеспечивает фармацевтическую композицию для снижения уровня липопротеинов низкой плотности (ЛНП), содержащую кристаллическую форму D1 или D2 или их гидраты в качестве активного ингредиента.
Композиция по настоящему изобретению может дополнительно содержать один или несколько фармацевтически приемлемых носителей. Композиция по настоящему изобретению может дополнительно содержать, по меньшей мере, одно фармацевтически приемлемое вспомогательное средство или добавку, выбираемую из наполнителей, разрыхлителей, подсластителей, связующих, капсулирующих агентов, агентов, вызывающих набухание, смазок, солюбилизаторов и прочее.
Настоящее изобретение также обеспечивает применение композиции для снижения уровня липопротеинов низкой плотности (ЛНП) и способ снижения уровня липопротеинов низкой плотности (ЛНП) с использованием композиции. В частности, способ по настоящему изобретению включает введение композиции нуждающемуся в этом пациенту.
Липопротеины низкой плотности (ЛНП) могут быть теми, которые присутствуют в кровотоке.
Композиция по настоящему изобретению полезна в качестве ингибитора фермента 3-гидрокси-3-метилглутарил-кофермент A редуктазы (HMG-CoA редуктазы) и может использоваться в качестве фармацевтической композиции для лечения и/или предотвращения заболевания, такого как остеопороз, гиперлипопротеинемия, гиперхолестеринемия или болезнь Альцгеймера.
Настоящее изобретение также обеспечивает применение композиции для ингибирования фермента 3-гидрокси-3-метилглутарил-кофермент A редуктазы (HMG-CoA редуктазы). И настоящее изобретение также обеспечивает применение композиции для лечения и/или предотвращения заболевания, такого как остеопороз, гиперлипопротеинемия, гиперхолестеринемия или болезнь Альцгеймера.
Настоящее изобретение также обеспечивает способ ингибирования фермента 3-гидрокси-3-метилглутарил-кофермент A редуктазы (HMG-CoA редуктазы), включающий введение композиции нуждающемуся в этом пациенту. В частности, настоящее изобретение также обеспечивает способ лечения и/или предотвращения заболевания, такого как остеопороз, гиперлипопротеинемия, гиперхолестеринемия или болезнь Альцгеймера, включающий введение композиции нуждающемуся в этом пациенту.
Композицию по настоящему изобретению можно также ввести в состав различных лекарственных форм хорошо известными в данной области способами, например, в порошки, в гранулы, в таблетки, в капсулы, в суспензии, в эмульсии, в сиропы и аэрозоли для перорального применения, препараты для наружного применения, суппозитории и стерильные растворы для инъекций. Предпочтительно можно получить различные составы композиции по настоящему изобретению, в зависимости от типа подлежащего лечению заболевания или ингредиентов, известных в области фармацевтических рецептур, согласно любой из известных методик (см. Remington's Pharmaceutical Science, Mack Publishing Company, Easton PA). Фармацевтическую композицию по настоящему изобретению можно вести различными путями мышам, крысам, домашнему скоту и другим млекопитающим, включая человека. Можно предположить любой способ введения. Композицию по настоящему изобретению можно ввести перорально, ректально или путем инъекции, например, внутривенно, внутримышечно, подкожно, внутриматочно, интратекально или интрацеребровентрикулярно.
Диапазон ежедневной дозы композиции по настоящему изобретению для снижения уровня липопротеинов низкой плотности (ЛНП) или лечения и/или предотвращения заболеваний, таких как гиперлипопротеинемия, гиперхолестеринемия, остеопороз или болезнь Альцгеймера, может обычно включать от 0,5 до 100 мг и предпочтительно от 2,5 до 80 мг кристаллической формы D1 или D2, их гидратов или их смеси. Ежедневная доза может варьироваться в зависимости от типа и тяжести заболевания, возраста, пола и других значимых факторов пациента, но обычно является такой же, как известная ежедневная доза соединения формулы 1.
Преимущества
Кристаллические формы по настоящему изобретению являются чистыми, более удобными для получения и более стабильными, чем аморфные формы соединения формулы 1.
Описание чертежей
Фиг. 1 представляет собой порошковую рентгенограмму кристаллической формы D1 соединения формулы 1 по настоящему изобретению.
Фиг. 2 представляет собой 13C ЯМР спектр кристаллической формы D1 соединения формулы 1 по настоящему изобретению.
Фиг. 3 представляет собой порошковую рентгенограмму кристаллической формы D2 соединения формулы 1 по настоящему изобретению.
Фиг. 4 представляет собой 13C ЯМР спектр кристаллической формы D2 соединения формулы 1 по настоящему изобретению.
СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Следующие примеры и экспериментальные примеры предоставлены для более подробного объяснения настоящего изобретения. Однако эти примеры даны с целью иллюстрации и не предназначены для ограничения настоящего изобретения.
ПРИМЕРЫ
Пример 1 (Получение кристаллической формы D1 полукальциевой соли [R-(R * ,R * )]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1H-пиррол-1-гептановой кислоты)
Один грамм природной формы соединения формулы 1, полученной в примере 10 патента США № 5273995, суспендировали в смеси метанола (5 мл), тетрагидрофурана (5 мл) и воды (40 мл). Суспензии давали прореагировать при 30-50°C в течение, по меньшей мере, десяти часов и фильтровали для получения кристаллов. Высушивание кристаллов под вакуумом при 40-60°C в течение 10-24 часов давало указанную кристаллическую форму D1, моногидрат (выход 80%).
Порошковую рентгенограмму продукта получали на M18HF-SRA дифрактометре (Mac Science) с CuKα излучением: см., Фиг. 1 и таблицу 1.
13C ЯМР (AVANCE-500 спектрометр (Bruker)): см., Фиг. 2 и таблицу 2.
1H ЯМР (Varian 400 МГц, ДМСО-d6): δ 9,7 (с, 1H), 6,8-7,5 (м, 14H), 4,2 (м, 1H), 3,9 (м, 1H), 3,7 (т, J= 6,8 Гц, 2H), 3,4 (м, 1H), 3,3 (с, 3H), 2,5 (д, J= 8,0 Гц, 2H), 1,8 (м, 2H), 1,4 (д, J= 6,8 Гц, 6H), 1,5 (м, 1H).
Определение воды по методу Карла Фишера (Metrohm 831 KF кулометр): 1,5% (1 молекула воды).
Термогравиметрический анализ (sinco TGA N-1000): 1,6% (1 молекула воды).
Пример 2 (Получение кристаллической формы D2 полукальциевой соли [R-(R*,R*)]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1H-пиррол-1-гептановой кислоты)
Один грамм природной формы соединения формулы 1, полученной в примере 10 патента США № 5273995, суспендировали в смеси метанола (10 мл), метиленхлорида (3 мл) и воды (30 мл). Суспензии давали прореагировать при 20-30°C в течение, по меньшей мере, двух часов и фильтровали для получения кристаллов. Высушивание кристаллов под вакуумом при 40-60°C в течение 10-24 часов давало указанную кристаллическую форму D2, тригидрат (выход 85%).
Порошковую рентгенограмму продукта получали на M18HF-SRA дифрактометре (Mac Science) с CuKα излучением: см., Фиг. 3 и таблицу 3.
13C ЯМР (AVANCE-500 спектрометр (Bruker)): см., Фиг. 4 и таблицу 4.
1H ЯМР (Varian 400 МГц, ДМСО-d6): δ 9,7 (с, 1H), 6,8-7,5 (м, 14H), 4,2 (м, 1H), 3,9 (м, 1H), 3,7 (т, J = 6,8 Гц, 2H), 3,4 (м, 1H), 3,3 (с,3H), 2,5 (д, J = 8,0 Гц, 2H), 1,8 (м, 2H), 1,4 (д, J= 6,8 Гц, 6H), 1,5 (м, 1H).
Определение воды по методу Карла Фишера (Metrohm 831 KF кулометр): 4,6% (3 молекулы воды).
Термогравиметрический анализ (sinco TGA N-1000): 4,9% (3 молекулы воды).
Экспериментальный пример 1 (Измерение температур плавления)
Температуры плавления соединений, полученных в примерах 1 и 2, измеряли, используя Mettler Toledo FP90, и результаты представлены в таблице 5.
Высокие температуры плавления кристаллических форм D1 и D2 позволяют проводить дополнительную высокотемпературную обработку кристаллических форм.
Экспериментальный пример 2 (Установление стабильности-измерение содержания)
80 мг новой кристаллической формы соединения, полученной в примере 2, и 80 мг стандартного продукта с чистотой 99,9%, полученного по способу, описанному в примере 10 патента США №5273995, аккуратно взвешивали, помещали в 100 мл мерные колбы и растворяли в диметилформамиде. Объемы растворов доводили до метки на колбах для получения исследуемых растворов и стандартных растворов, соответственно. Каждый из растворов анализировали при помощи колоночной хроматографии при следующих условиях:
- Колонка: Zorbax Rx C8 (4,6 мм × 250 мм, 5 мкм) или ее эквивалент
Подвижная фаза (0,05 М ацетат аммонийный буфер (рН 5,0)/ацетонитрил/тетрагидрофуран)
Подвижная фаза А:
0,05 М ацетат аммонийный буфер
(рН 5,0)/ацетонитрил/тетрагидрофуран (67/21/12)
Подвижная фаза В:
0,05 М ацетат аммонийный буфер (рН 5,0)/ацетонитрил/тетрагидрофуран (54/34/12)
- Детектор: UV адсорбционный спектрофотометр (длина волны: 244 нм)
- Скорость протока: 1,0 мл/мин.
- Температура колонки: 35±2°С
- Хранение: в бутыли из полиэтилена высокой плотности
Содержания соединения, полученного в примере 2, определяли количественно по площади под каждым пиком. Изменения содержания соединения оценивали при хранении при комнатной температуре, в холодильнике, в условиях ускоренного хранения и в условиях экстремального хранения в течение периодов, указанных в таблице 6, и результаты представлены в таблице 6.
(ускоренное хранение (месяцы))
(экстремальное хранение (месяцы))
Из результатов таблицы 6 можно видеть, что содержание исходного вещества сохраняется после хранения в течение шести месяцев при комнатной температуре и в холодильнике и колебания содержания исходного продукта устойчивы в пределах 98-100,2% даже в условиях ускоренного и экстремального хранения. В заключение, кристаллические формы по настоящему изобретению демонстрируют отличную стабильность.
ПРИМЕНЯЕМОСТЬ В ПРОИЗВОДСТВЕННЫХ УСЛОВИЯХ
Кристаллические формы по настоящему изобретению являются чистыми, более удобными для получения и более стабильными, чем аморфные формы соединения формулы 1.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФОРМА VI АТОРВАСТАТИНА КАЛЬЦИЯ ИЛИ ЕЕ ГИДРАТЫ | 2002 |
|
RU2294924C2 |
| КРИСТАЛЛИЧЕСКИЙ КЛОПИДОГРЕЛЬ НАФТАЛИНСУЛЬФОНАТ ИЛИ ЕГО ГИДРАТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2328501C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ [R-(R*,R*)]-2-(4-ФТОРФЕНИЛ)-БЕТА, ДЕЛЬТА-ДИГИДРОКСИ-5-(1-МЕТИЛЭТИЛ)-3-ФЕНИЛ-4-[(ФЕНИЛАМИНО)КАРБОНИЛ]-1Н-ПИРРОЛ-1-ГЕПТАНОВОЙ КИСЛОТЫ | 2004 |
|
RU2315755C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ СТРУКТУРЫ (ПОЛИМОРФНЫЕ МОДИФИКАЦИИ) ПОЛУКАЛЬЦИЕВОЙ СОЛИ АТОРВАСТАТИНА И СПОСОБЫ ПОЛУЧЕНИЯ ЭТИХ И ДРУГИХ ПОЛИМОРФНЫХ МОДИФИКАЦИЙ СОЛИ АТОРВАСТАТИНА | 2001 |
|
RU2344127C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО 1,2-ДИГИДРОПИРИДИНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2323930C1 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА МЕТАНСУЛЬФОНАТ 5-ХЛОР-N-({ (5S)-2-ОКСО-3-[4-(5,6-ДИГИДРО-4H-[1,2,4]ТРИАЗИН-1-ИЛ)ФЕНИЛ]-1,3-ОКСАЗОЛИДИН-5-ИЛ} МЕТИЛ)ТИОФЕН-2-КАРБОКСАМИДА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2014 |
|
RU2663617C1 |
| ТВЕРДЫЕ ФОРМЫ, СОДЕРЖАЩИЕ (-)-О-ДЕСМЕТИЛВЕНЛАФАКСИН, И ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2477269C2 |
| СТРОНЦИЕВАЯ СОЛЬ S-ОМЕПРАЗОЛА ИЛИ ЕЕ ГИДРАТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2006 |
|
RU2372345C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ 6-[2-(МЕТИЛКАРБАМОИЛ)ФЕНИЛСУЛЬФАНИЛ]-3-Е-[2-(ПИРИДИН-2-ИЛ)ЭТЕНИЛ]ИНДАЗОЛА, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ АНОМАЛЬНОГО РОСТА КЛЕТОК У МЛЕКОПИТАЮШИХ | 2008 |
|
RU2518898C2 |
| СОЛИ И ПОЛИМОРФНЫЕ МОДИФИКАЦИИ ИНГИБИТОРА VEGF-R | 2006 |
|
RU2369607C1 |
Изобретение относится к новым кристаллическим формам D1 и D2 [R-(R*,R*)]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты, полукальциевой соли, их гидратам, которые обладают повышенной стабильностью. Описаны способы получения новых кристаллических форм. 7 н. и 8 з.п. ф-лы, 4 ил., 6 табл.
1. Кристаллическая форма (D1) соединения формулы 1:
имеющая пики на порошковой рентгенограмме при 2Θ=5,4, 7,4, 7,7, 8,1, 8,6, 8,9, 10,2, 12,5, 13,8, 17,1, 17,8, 18,2, 18,5, 19,4, 19,9, 20,8, 22,4, 23,2, 24,2 и 25,6°, или ее гидрат.
2. Кристаллическая форма D1 или ее гидрат по п.1, где кристаллическая форма D1 имеет дополнительные пики на порошковой рентгенограмме со слабыми интенсивностями при 2Θ=6,8, 10,8, 14,8, 16,2, 21,8, 25,1, 26,1, 27,2, 27,8, 28,5 и 30,4°.
3. Кристаллическая форма D1 или ее гидрат по п.1, где кристаллическая форма D1 характеризуется сигналами твердофазного 13С ЯМР приблизительно при 179,6, 169,1, 162,5, 140,0, 138,0, 133,8, 131,9, 129,6, 126,0, 119,2, 114,7, 73,6, 71,2, 69,9, 48,0, 45,5, 43,2, 29,0, 28,6, 24,2 и 22,2 м.д.
4. Кристаллическая форма (D2) соединения формулы 1, имеющая пики на порошковой рентгенограмме при 2Θ=5,6, 8,5, 10,9, 11,4, 14,3, 14,9, 18,2, 18,7, 19,0, 19,7, 22,4 и 22,9°, или ее гидрат:
5. Кристаллическая форма D2 или ее гидрат по п.4, где кристаллическая форма D2 имеет дополнительные пики на порошковой рентгенограмме со слабыми интенсивностями при 9,0 и 11,8° (w).
6. Кристаллическая форма D2 или ее гидрат по п.4, где кристаллическая форма D2 характеризуется сигналами твердофазного 13С ЯМР приблизительно при 183,3, 170,3, 164,0 (широкий), 138,1 (широкий), 136,0, 131,7, 125,7, 121,8, 120,2, 73,0 (широкий), 44,3 (широкий), 29,1 и 24,6 м.д.
7. Способ получения новой кристаллической формы D1 по п.1, включающий суспендирование природной формы соединения формулы 1 в смеси спирт/тетрагидрофуран/вода и перемешивание суспензии при температуре от 0 до 60°С:
8. Способ по п.7, в котором спирт, тетрагидрофуран и воду смешивают в объемном соотношении 1:1:8.
9. Способ получения новой кристаллической формы D2 по п.4, включающий суспендирование природной формы соединения формулы 1 в смеси спирт/метиленхлорид/вода и перемешивание суспензии при температуре от 0 до 40°С:
10. Способ по п.9, в котором спирт, метиленхлорид и воду смешивают в объемном соотношении 10:3:30.
11. Способ по любому из пп.7-10, в котором спирт представляет собой C1-С6 низший спирт.
12. Способ по п.11, в котором спирт представляет собой метанол.
13. Фармацевтическая композиция для снижения уровня липопротеинов низкой плотности (ЛНП), содержащая кристаллическую форму D1 или D2 или ее гидрат по любому из пп.1-6 в качестве активного ингредиента и один или несколько фармацевтически приемлемых носителей.
14. Применение композиции по п.13 для снижения уровня липопротеинов низкой плотности (ЛНП).
15. Способ снижения уровня липопротеинов низкой плотности (ЛНП), включающий введение композиции по п.13 нуждающемуся в этом пациенту.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| СПОСОБ ПОЛУЧЕНИЯ АМОРФНОГО АТОРВАСТАТИНА | 2000 |
|
RU2247113C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ VI И VII КАЛЬЦИЕВОЙ СОЛИ АТОРВАСТАТИНА | 2002 |
|
RU2304139C2 |

