ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым амидным производным, способу их получения и содержащим их фармацевтическим композициям, а также их применению в качестве лечебного средства, в частности в качестве ингибитора микросомальной простагландин Е-синтазы-1 (mPGES-1), и их применению для получения лекарственного средства для лечения и/или предупреждения заболеваний или расстройств, таких как воспаление и/или боль, и т.д.
УРОВЕНЬ ТЕХНИКИ
Многие заболевания и/или расстройства по существу являются воспалительными. Основной существующей проблемой, связанной с лечением воспалительных заболеваний, является недостаточная эффективность и/или наличие частых побочных явлений. Воспалительные заболевания, возникающие у людей, включают астму, воспалительное заболевание кишечника, ревматоидный артрит, остеоартрит, ринит, конъюнктивит, дерматит и тому подобные. Воспаление является частой причиной боли, которая может быть обусловлена различными причинами, такими как инфекция, хирургическая операция или другие травмы. При этом некоторые заболевания, к числу которых относятся злокачественные опухоли и сердечно-сосудистые заболевания, также имеют симптомы воспаления.
Простагландин Е2 (простагландин Е2, PGE2) является одним из наиболее распространенных простагландинов (PG), представляющий собой сильный провоспалительный медиатор, способный вызывать повышение температуры тела и боль, участвовать в различных физиологических и патологических процессах в организме. Его химический синтез состоит из трех последовательных ферментативных реакций: (1) арахидоновая кислота (АА) высвобождается из глицерофосфолипида на мембране посредством катализа фосфолипазы A2(PLA2); (2) АА образует PGG2 и PGH2 под воздействием циклооксигеназы (СОХ); (3) PGH2 образует PGE2, PGF2, PGD2, простациклин и тромбоксан А2 посредством катализа PGE2-синтазы (PGES).
Существуют две формы циклооксигеназы (СОХ). Одна конститутивно экспрессируется как СОХ-1 во многих клетках и тканях, а другая представляет собой СОХ-2, индуцируемую провоспалительными стимулянтами, такими как цитокины, во время воспалительной реакции. В настоящее время существуют виды ингибиторов СОХ-1 и/или СОХ-2 для контроля воспаления посредством уменьшения конечного образования PGE2, такие как "НПВП" (нестероидные противовоспалительные препараты) и "коксиб" (селективный ингибитор СОХ-2). Однако ингибирование целевого СОХ уменьшает образование всех метаболитов арахидоновой кислоты (АА), включая некоторые метаболиты, которые являются полезными для организма человека. Таким образом, ингибиторы СОХ могут оказывать неблагоприятные биологические воздействия на организм человека. Следовательно, разработка новых, более безопасных и эффективных лекарственных средств для лечения воспалительных заболеваний обладает высокой клинической значимостью и рыночным интересом.
Во время синтеза PGE2 используют PGES, которая представляет собой терминальный фермент, ограничивающий скорость синтеза PGE2. Как хорошо Известно, существует по меньшей мере три вида PGES, которые называют цитоплазмичной PGES (cPGES) (или называемая PGE-3), мембранно-связанной PGES-1 (mPGES-1) и мембранно-связанной PGES-2 (mPGES-2). cPGES представляет собой GSH-зависимый конститутивный фермент, который относится к ферментам, широко экспрессируемым конститутивными генами во множестве тканей и клеток, и на который не оказывает влияния фактор, стимулирующий воспаление. mPGES-2 представляет собой GSH-независимый конститутивный фермент, экспрессируемый в основном в тканях с относительно низкой экспрессией mPGES-1, таких как мозг, сердце, почки, кишечник, и не индуцируемый воспалением и повреждением ткани. mPGES-1 относится к GSH-зависимым ферментам с индуцируемой экспрессией, который может экспрессироваться в больших количествах при индуцировании фактором воспаления, и играет важную роль в различных заболеваниях, таких как артрит, связанные с воспалением повышение температуры тела и боль, атеросклероз и патологические и физиологические процессы развития рака. Ген mPGES-1 находится на хромосоме 9q34.3 и содержит три экзона и два интрона, имеет длину приблизительно 14,8 т.н. Его кДНК кодирует полипептид, содержащий 152 аминокислоты. Первичная белковая структура mPGES-1 у разных видов обладает гомологичностью более 80%. Исследование показало, что экспрессия СОХ-2 и mPGES-1 существенно увеличивается в различных культивируемых клетках при стимуляции фактора воспаления (LPS, IL-1 и т.д.), что сопровождается усиленным синтезом PGE2. Иммуногистохимические эксперименты также показывают, что и СОХ-2 и mPGES-1 локализованы в микросомальной мембране, что указывает на то, что mPGES-1 связывается в основном с СОХ-2, и опосредует увеличение синтеза PGE2 в замедленной реакции, вызванной воспалительными факторами. Тем не менее, исследование ферментативной кинетики показало, что индукции СОХ-2 и mPGES-1 не полностью одинаковы, и в некоторых случаях СОХ-2 может связываться с mPGES-2, и в то же самое время mPGES-1 также может связываться с СОХ-1. Более того, из PGH2, образуемого катализом СОХ-2, под воздействием mPGES-1 можно синтезировать PGE2 или другие типы простагландинов. Таким образом, регуляторные механизмы экспрессии СОХ-2 и mPGES-1 являются как перекрывающимися, так и разными.
В настоящий момент существует два вида ингибиторов mPGES-1: AAD-2004 от корейской фармацевтической компании GNT (Neurotech) и LY-3023703 от компании Eli Lilly. Показания к применению AAD-2004 не относятся к боли, но он представляет собой молекулу, представляющую эффективную спиновую ловушку и ингибитор микросомальной простагландин Е-синтазы-1, который может быть использован при лечении болезни Альцгеймера, болезни Паркинсона и болезни двигательных нейронов. LY -3023703, вошедший в фазу клинических испытаний в июне 2016, применяют при лечении остеоартритной боли. В настоящее время существует только две связанные с mPGES-1 патентные заявки: WO 2012087771 и WO 2012161965, раскрытые компанией Eli Lilly.
Хотя к настоящему моменту раскрыта серия ингибиторов микросомальной простагландин Е-синтазы-1 (mPGES-1), новые соединения с улучшенной эффективностью все еще не разработаны. В настоящем изобретении в результате продолжительных усилий получили серию соединений общей формулы (I) и обнаружили, что эти соединения обладают прекрасной эффективностью и действием.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы (I), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смесям, или его фармацевтически приемлемым солям,
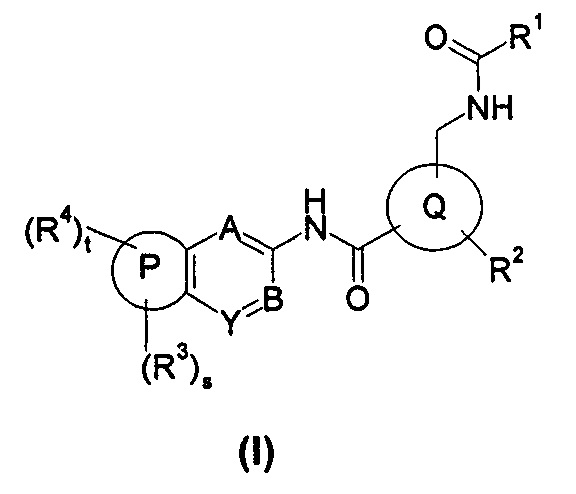
где:
кольцо Р выбрано из пятичленного гетероарила и пятичленного гетероциклила;
кольцо Q выбрано из арила и гетероарила, предпочтительно фенила, пиридила или пиримидинила;
А, В или Y выбран из -СН и N;
R1 выбран из алкила и циклоалкила, где указанный алкил или циклоалкил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена и галогеналкила;
R2 выбран из галогена и галогеналкила;
R3 являются одинаковыми или разными, и каждый из них независимо выбран из водорода, галогена, алкокси, циано, нитро, алкила, циклоалкила, гетероциклила, оксо, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 или -C(O)NR6R7, где каждый из указанных алкила, циклоалкила и гетероциклила необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидроксила, алкокси, циано, нитро, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 и -C(O)NR6R7;
R4 выбран из арила и гетероарила, предпочтительно фенила, где каждый из указанных арила и гетероарила необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, алкокси, гидроксила, циано, нитро, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R6, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 и -C(O)NR6R7, где указанный галогеналкил предпочтительно представляет собой трифторметил;
R5 выбран из алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанных алкила, циклоалкила, арила и гетероарила необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоновой кислоты и карбоксилатной группы;
каждый из R6 и R7 независимо выбран из водорода, алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанных алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоновой кислоты и карбоксильной группы; или
R6 и R7 могут в совокупности с атомом азота, к которому они присоединены, образовывать гетероциклил, где указанный гетероциклил может содержать один или более чем один гетероатом, выбранный из группы, состоящей из N, О и S(O)m, и где указанный гетероциклил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, карбоновой кислоты и карбоксильной группы;
m представляет собой 0, 1 или 2;
s представляет собой целое число от 0 до 3;
t представляет собой 0 или 1.
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, когда указанный R2 представляет собой галоген, t представляет собой 1.
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли, представляет собой соединение формулы (II), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли:
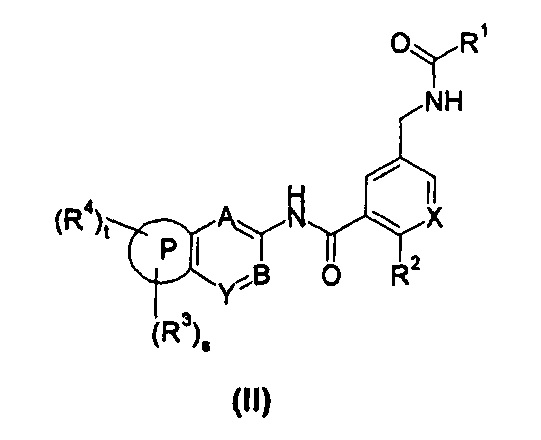
где:
X выбран из -СН- и N;
кольцо Р, А, В, Y, s, t и R1-R4 являются такими, как определено в общей формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли, представляет собой соединение формулы (III), формулы (IV) или формулы (V), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли:

где:
X выбран из -СН- и N;
каждый из Е, G и W независимо выбран из CRa, NRb, N, О и S;
каждый из Ra и Rb независимо выбран из водорода, галогена, алкокси, циано, нитро, алкила, циклоалкила, гетероциклила, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 или -C(O)NR6R7, где каждый из указанных алкила, циклоалкила и гетероциклила необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, алкокси, циано, нитро, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 и -C(O)NR6R7; и
A, B, Y, t, R1-R2 и R4-R7 являются такими, как определено в общей формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли, представляет собой соединение формулы (VI), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли:
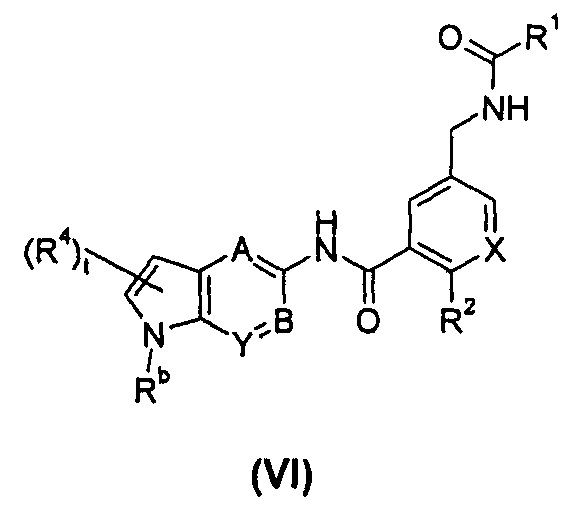
где:
X выбран из -СН- и N;
Rb выбран из водорода, галогена, алкокси, циано, нитро, алкила, циклоалкила, гетероциклила, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)0R5, -NR6R7, -OC(O)NR6R7 и -C(O)NR6R7, где каждый из указанных алкила, циклоалкила и гетероциклила необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, алкокси, циано, нитро, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 и -C(O)NR6R7; предпочтительно С1-С4 алкила, C1-C4 алкокси или тетрагидрофурила;
А, В, Y, t, R1-R2 и R4-R7 являются такими, как определено в общей формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли, представляет собой соединение формулы (VII), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли:
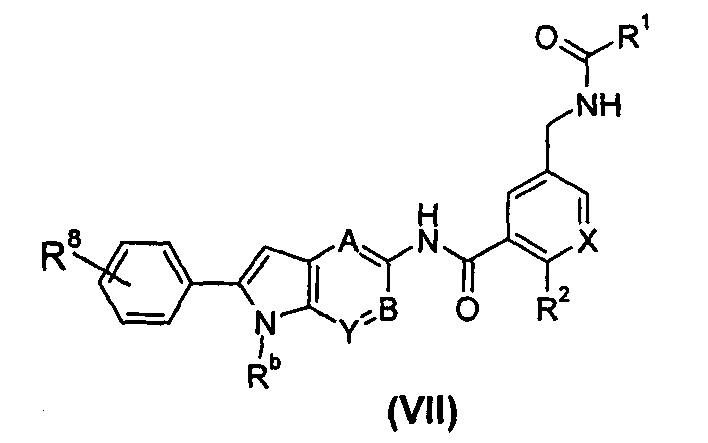
где:
X выбран из -СН- и N;
Rb выбран из водорода, галогена, алкокси, циано, нитро, алкила, циклоалкила, гетероциклила, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 и -C(O)NR6R7, где каждый из указанных алкила, циклоалкила и гетероциклила необязательно дополнительно замещен одной или более чем одной группой, выбранной из галогена, гидрокси, алкокси, циано, нитро, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 или -C(O)NR6R7;
R8 выбран из галогена, алкокси, гидркосила, циано, нитро, алкил, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 или -C(O)NR6R7, где указанный галогеналкил предпочтительно представляет собой трифторметил; и
А, В, Y, R1-R2 и R5-R7 являются такими, как определено в общей формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (VII), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли представляет собой соединение формулы (VII-A) или формулы (VII-B), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли:
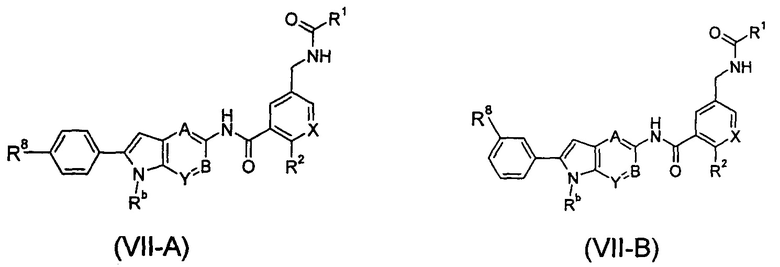
А, В, X, Y, R1-R2, R8 и Rb являются такими, как определено в формуле (VII).
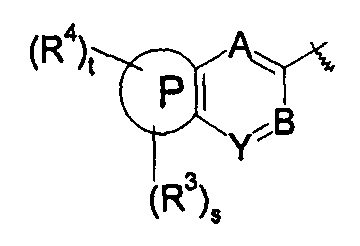
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, указанные А, В и Y выбраны из СН.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, один из А, В и Y выбран из N, а два других выбраны из -СН-.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, указанные группы
включают, но не ограничиваются следующими:
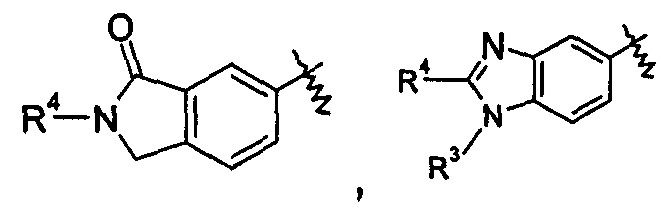 или
или 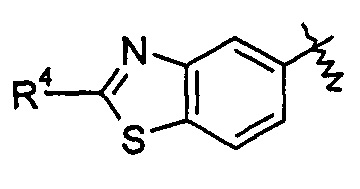 ;
;
R3 и R4 являются такими, как определено в общей формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, указанный R1 выбран из алкила и галогеналкила, предпочтительно третичного бутила, изопропила, 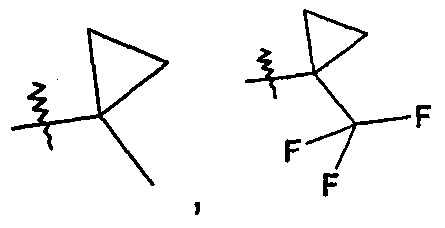 и
и 
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, указанный R1 выбран из циклоалкила, где указанный циклоалкил дополнительно замещен галогеналкилом, R1 предпочтительно представляет собой 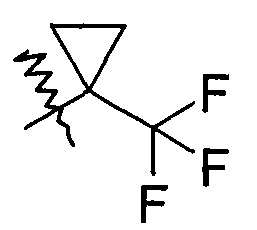 .
.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, указанный R2 выбран из галогеналкила, предпочтительно -CHF2.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, указанный R2 выбран из галогеналкила, и t представляет собой 0.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, указанный R2 выбран из галогена, предпочтительно хлора.
В другом предпочтительном варианте осуществления настоящего изобретения, в соединении формулы (III), или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смесях, или его фармацевтически приемлемых солях, указанное t представляет собой 1.
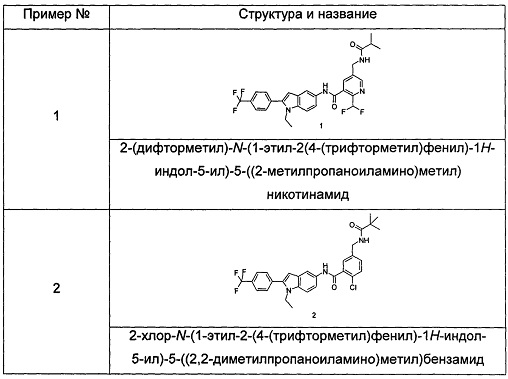
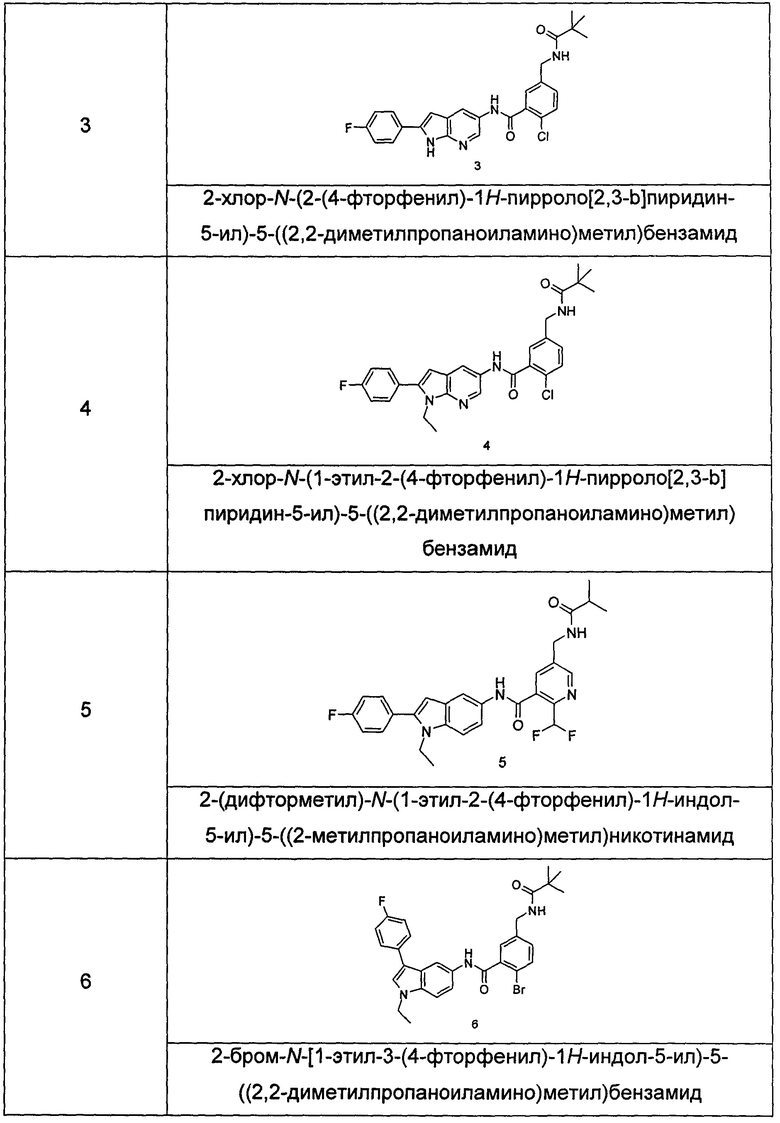
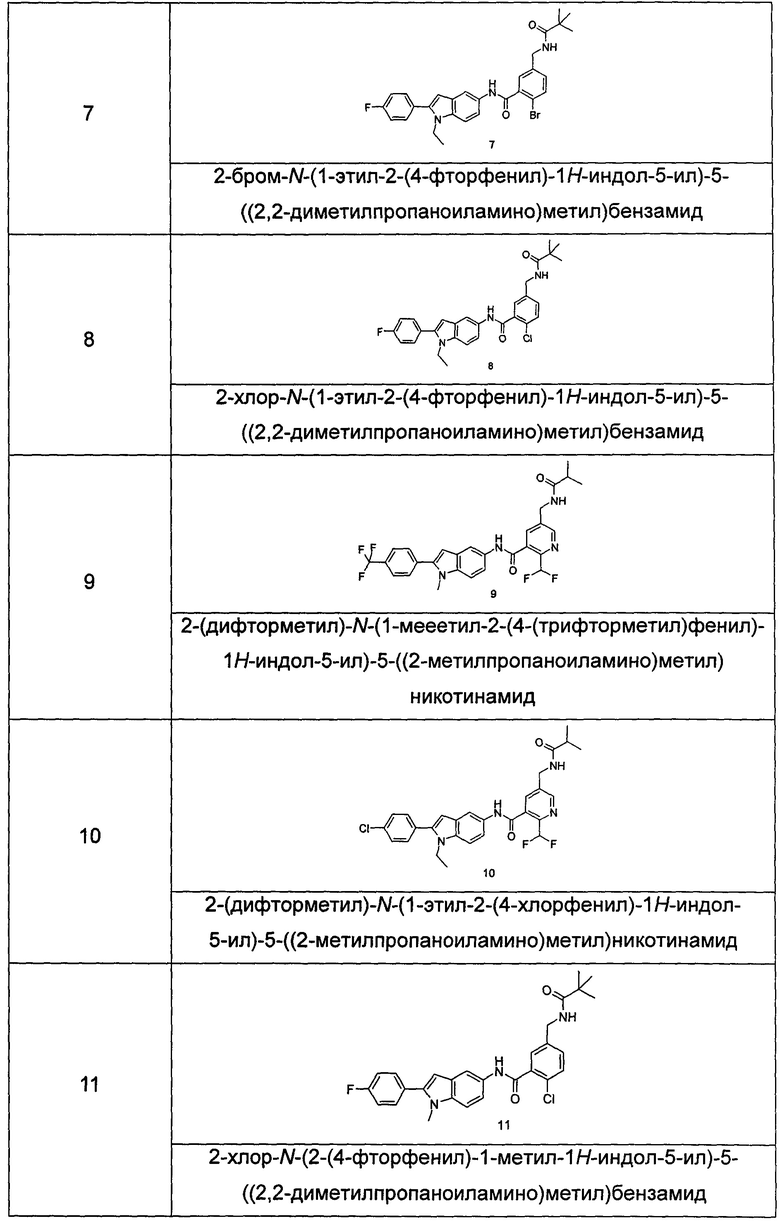
Типичные соединения по настоящему изобретению включают, но не ограничиваются следующими:
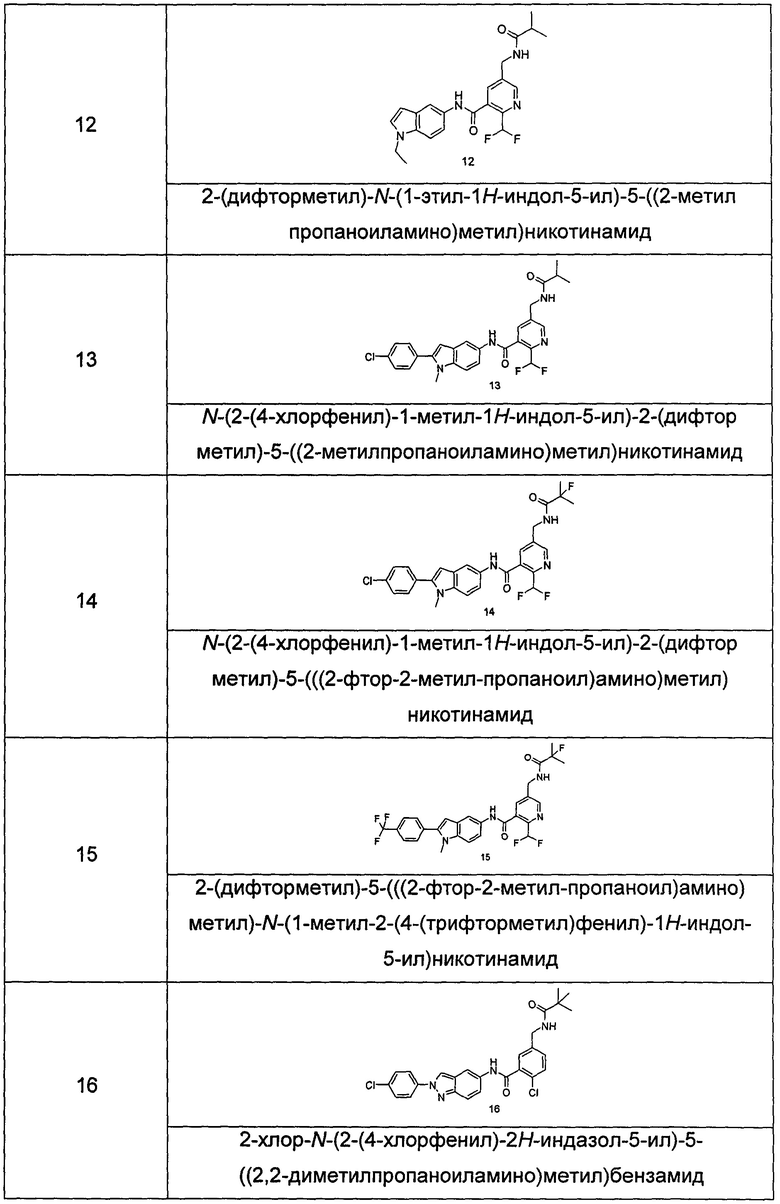
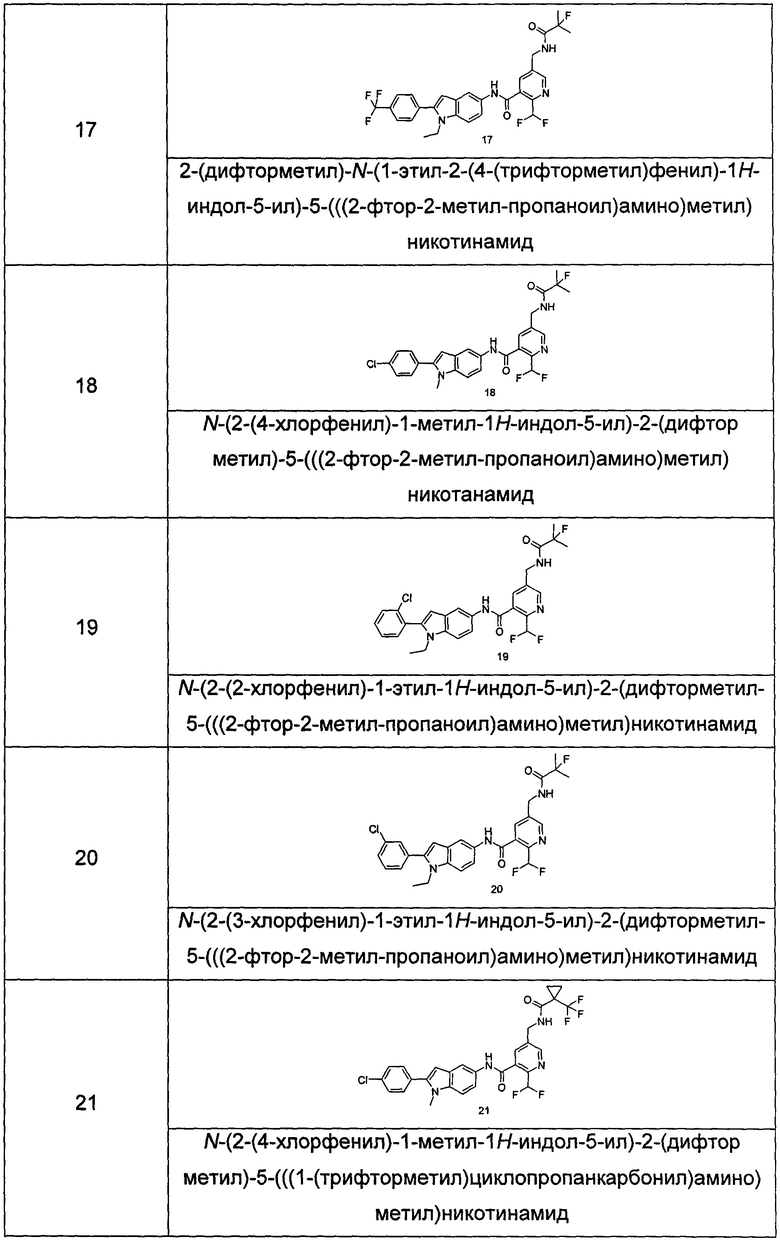
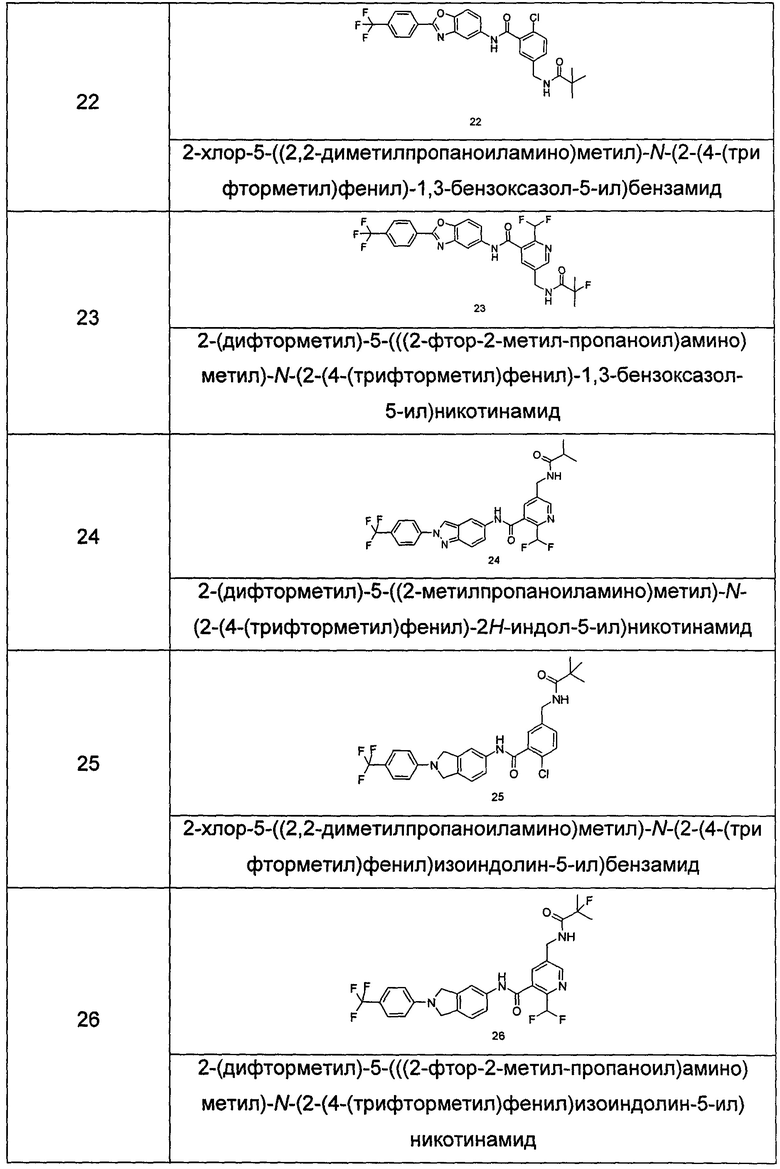
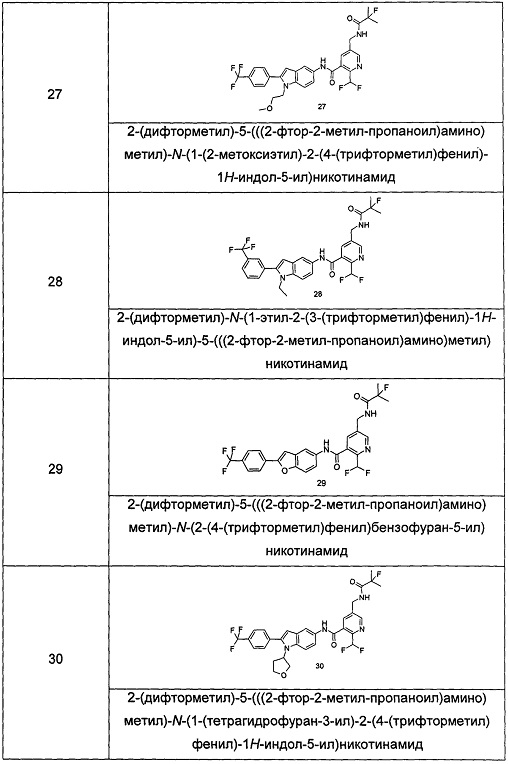
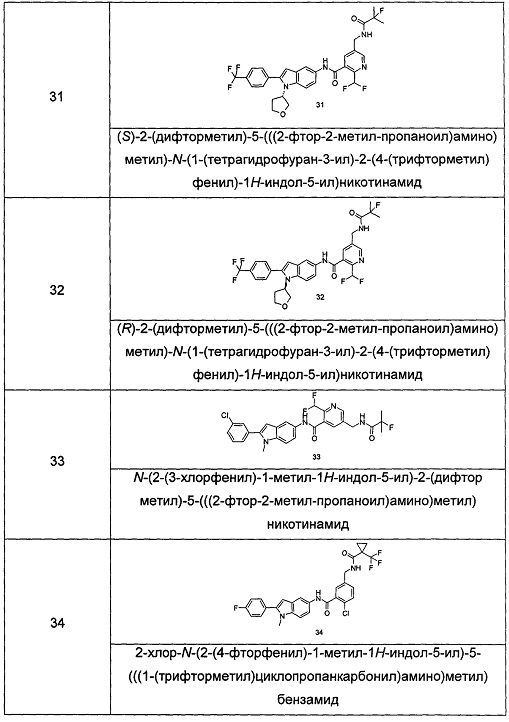
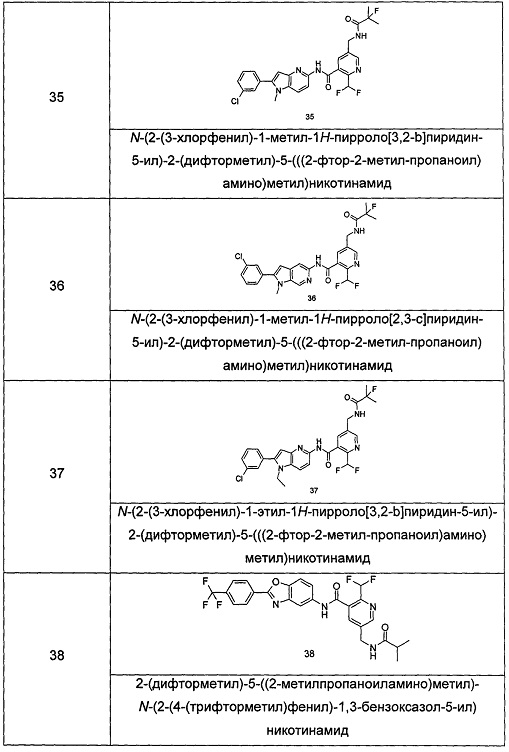
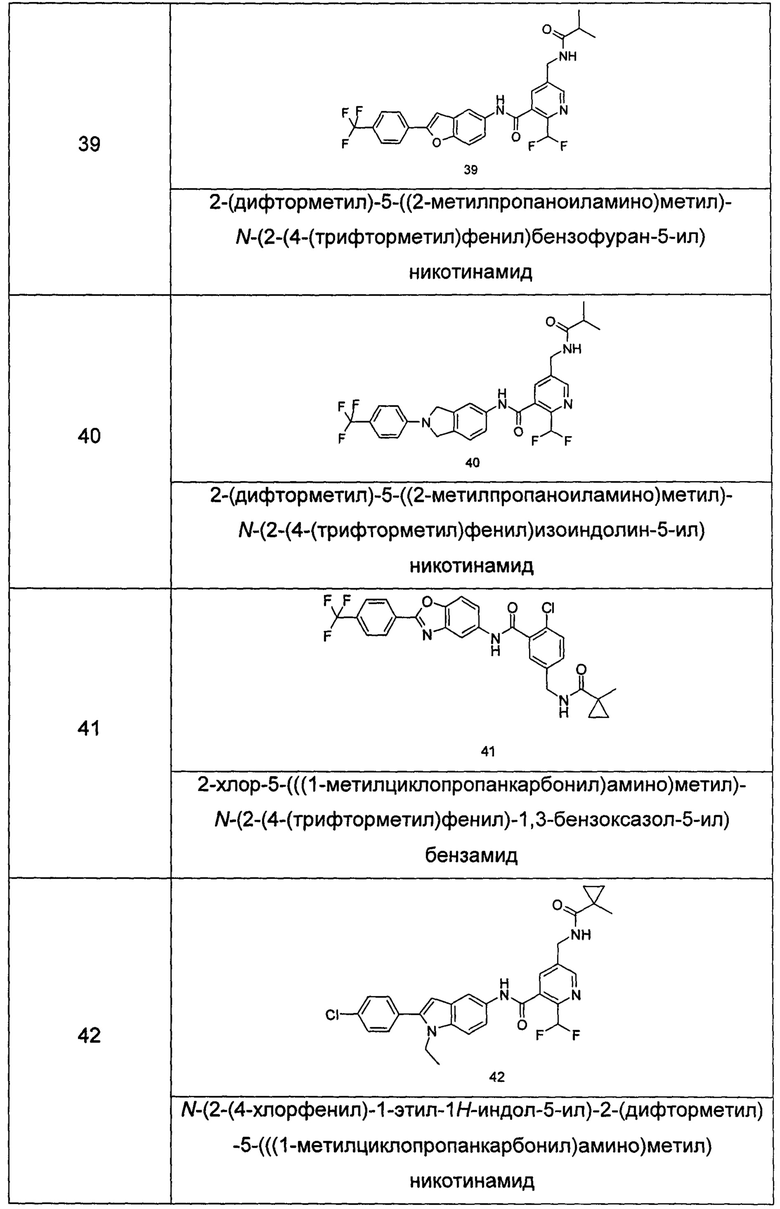
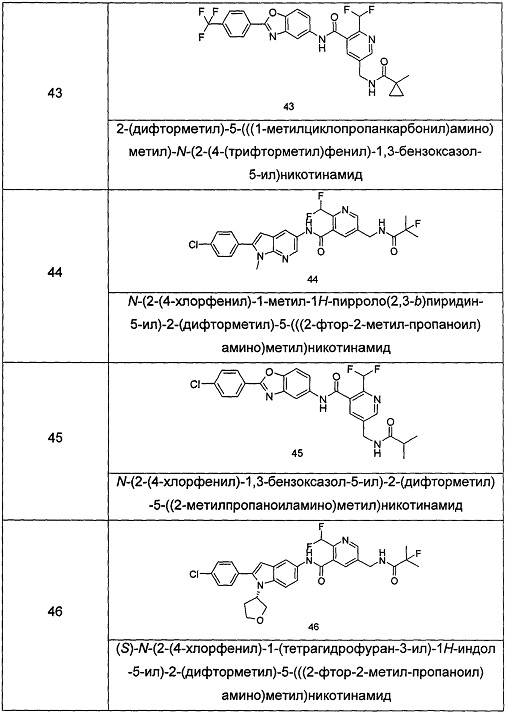
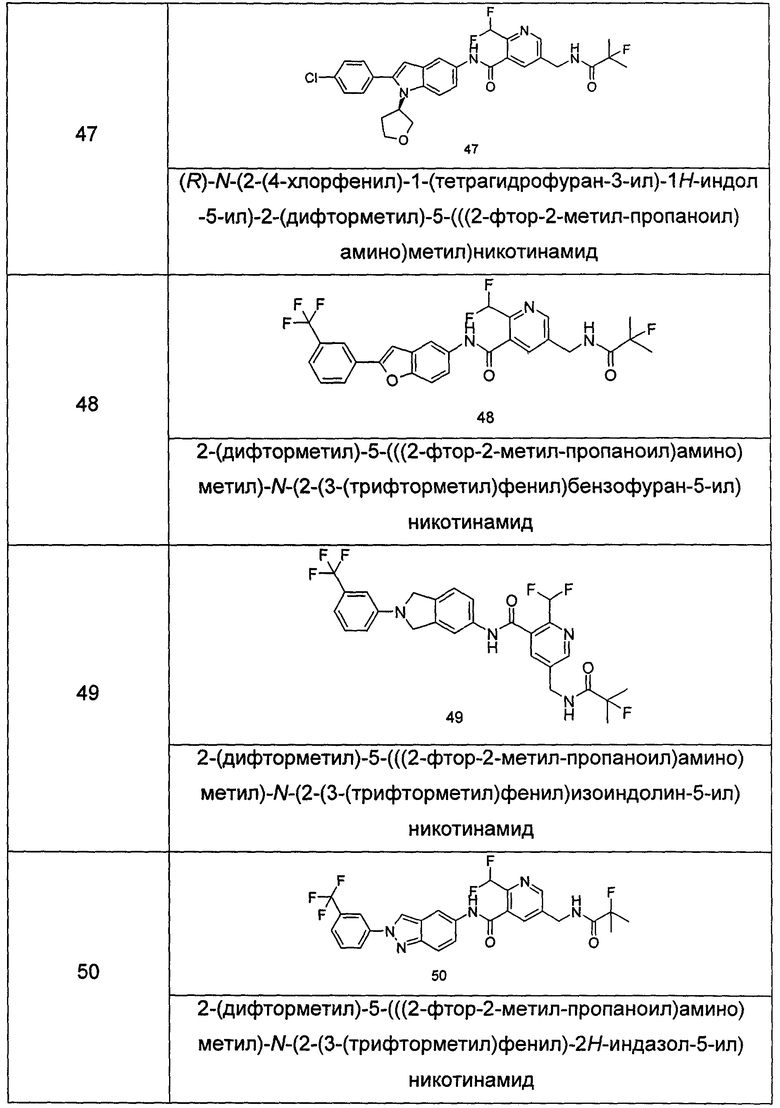
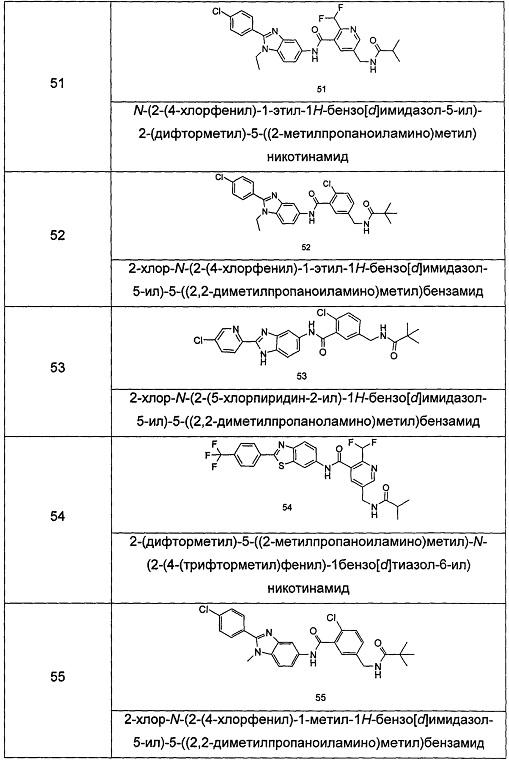
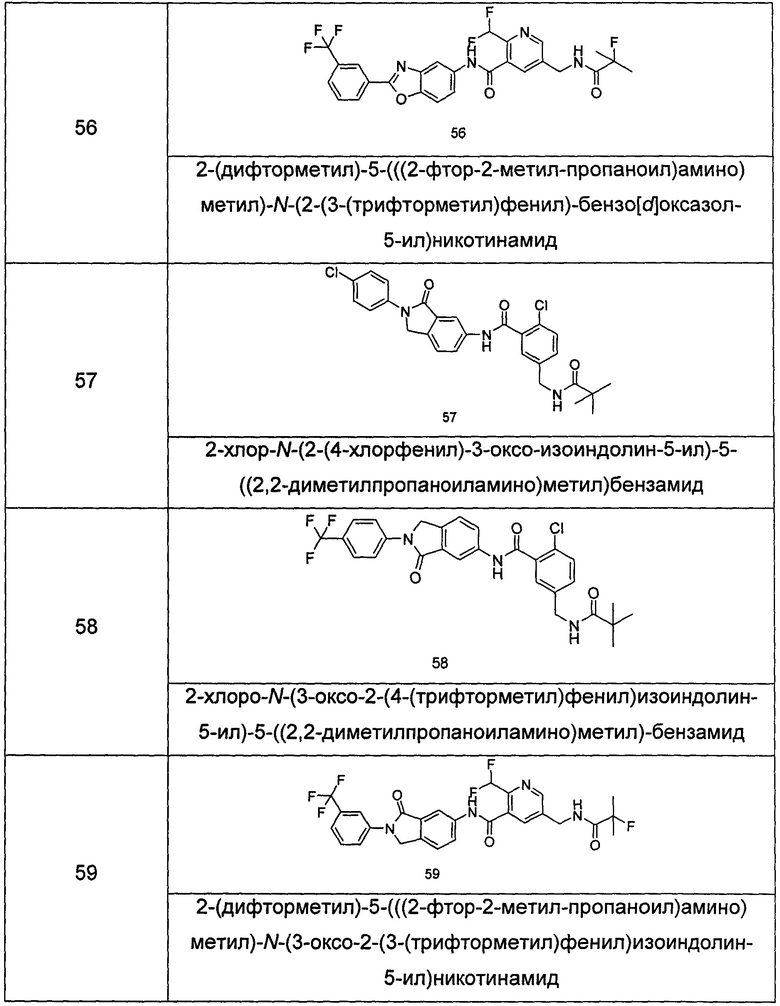
или их таутомеры, мезомеры, рацематы, энантиомеры, диастереомеры, или их смеси, или их фармацевтически приемлемые соли.
В еще одном аспекте настоящего изобретения предложен способ получения соединения формулы (I), или его таутомера мезомера, рацемата, энантиомера, диастереомера, или их смесей, или его фармацевтически приемлемых солей, включающий стадию:
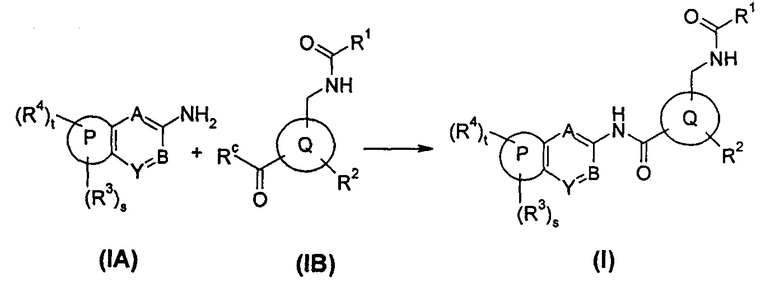
соединение общей формулы (IA) или его соли подвергают реакции конденсации с соединением общей формулы (IB) в щелочной среде с получением соединения формулы (I);
где:
Rc выбран из гидрокси и галогена;
кольцо Р, кольцо Q, А, В, Y, s, t и R1-R4 являются такими, как определено в общей формуле (I).
Щелочные реагенты включают органическое основание и неорганическое основание, где указанное органическое основание включает, но не ограничивается следующими, триэтиламин, N,N-диизопропилэтиламин, пиридин, бис(триметилсилил)амид натрия, н-бутиллитий, трет-бутанолят калия или бромид тетрабутиламмония, где указанное неорганическое основание включает, но не ограничивается следующими, гидроксид лития, гидроксид натрия, гидроксид калия, гидрид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, или карбонат цезия, предпочтительно триэтиламин.
Катализаторы включают, но не ограничиваются следующими, Pd/C, никель Ренея.
Конденсирующие агенты включают, но не ограничиваются следующими, 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат, 2-(7-азо-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат, бензотриазол-1-ил-окси-трис(диметиламино)-фосфония гексафторфосфат, или бензотриазол-1-ил-окси-трипирролидинил-фосфония гексафторфосфат.
В еще одном аспекте настоящего изобретения предложено соединение формулы (IA), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли,
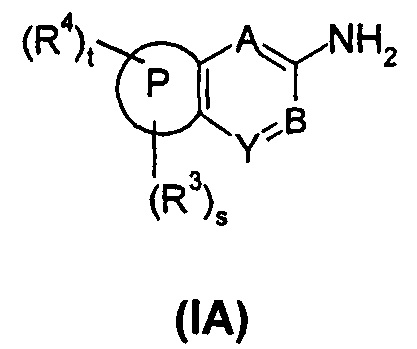
содержащее следующие группы:
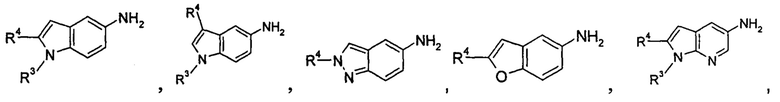
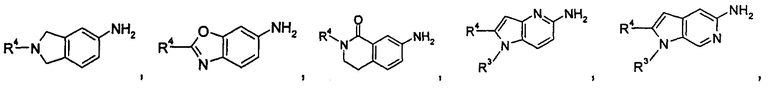
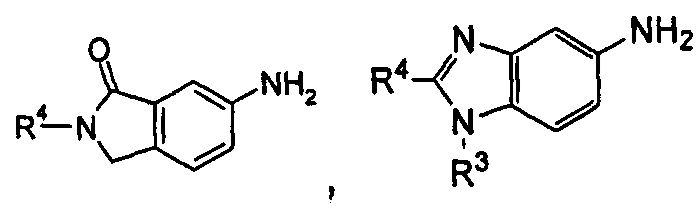 или
или 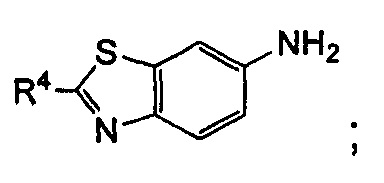
где:
R3 выбран из водорода, алкила и гетероциклила, где указанный алкил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкокси и гетероциклила, предпочтительно С1-С4 алкила, С1-С4 алкокси или тетрагидрофуранила;
R4 выбран из арила и гетероарила, где указанный арил или гетероарил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена и галогеналкила, где указанный галогеналкил предпочтительно представляет собой трифторметил; R4 предпочтительно представляет собой фенил, где указанный фенил дополнительно замещен одним галогеном или одним галогеналкилом;
t представляет собой 1;
при условии, что
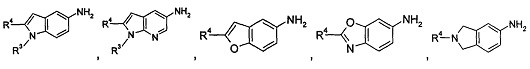
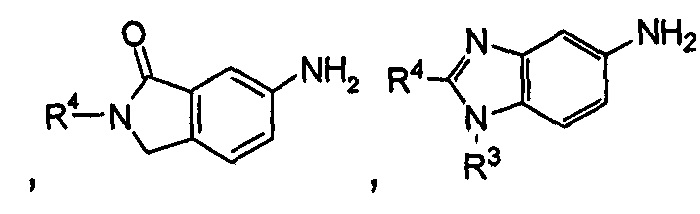
когда общая формула (IA) представляет собой следующие группы:
 или
или 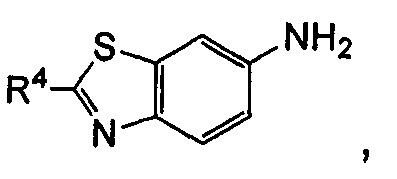
R3 выбран из водорода и алкила, где указанный алкил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкокси и гетероциклила;
R4 представляет собой фенил, где указанный фенил дополнительно замещен одним галогеналкилом, где указанный галогеналкил предпочтительно представляет собой трифторметил; или
R3 представляет собой гетероциклил;
R4 представляет собой фенил, где указанный фенил дополнительно замещен одним галогеном или одним галогеналкилом.
Соединения общей формулы (IA) включают, но не ограничиваются следующими:
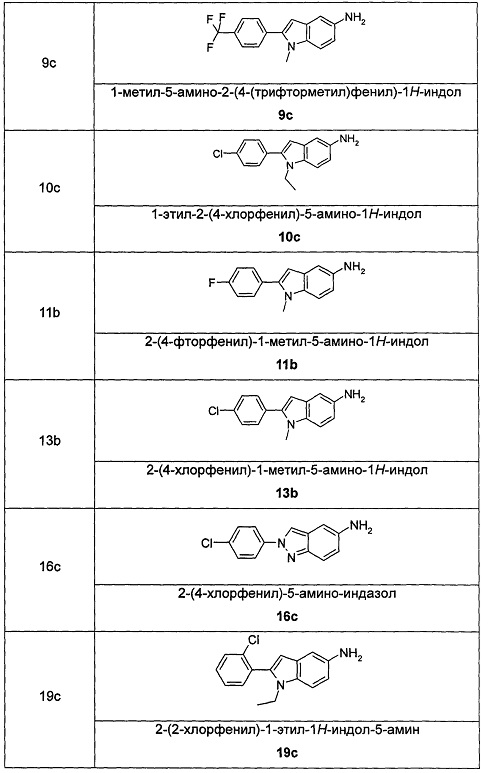
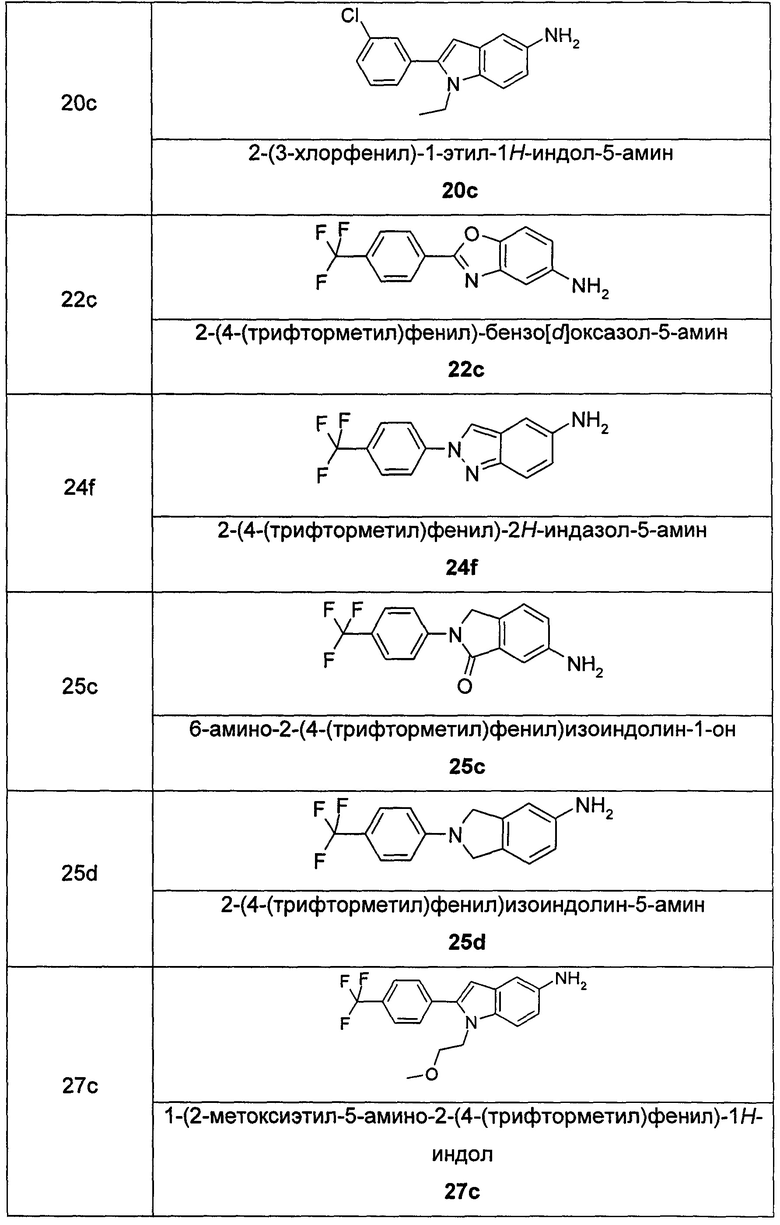
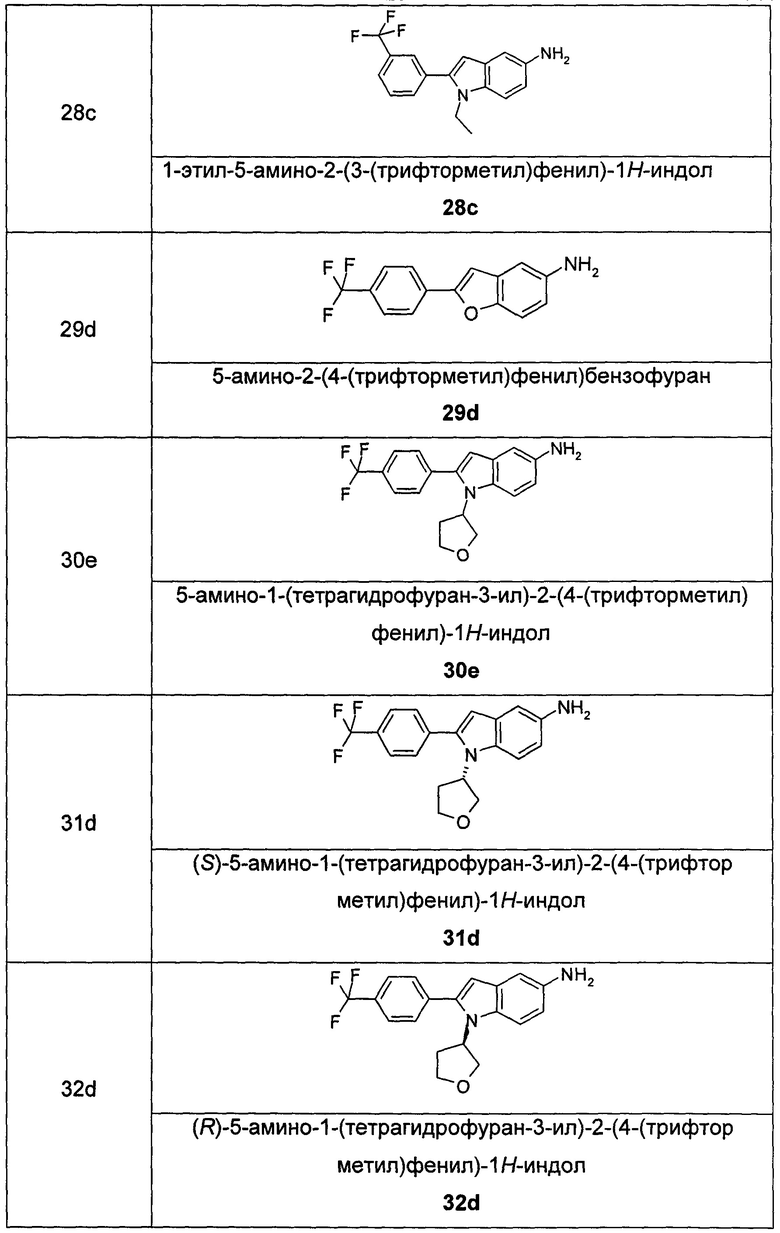
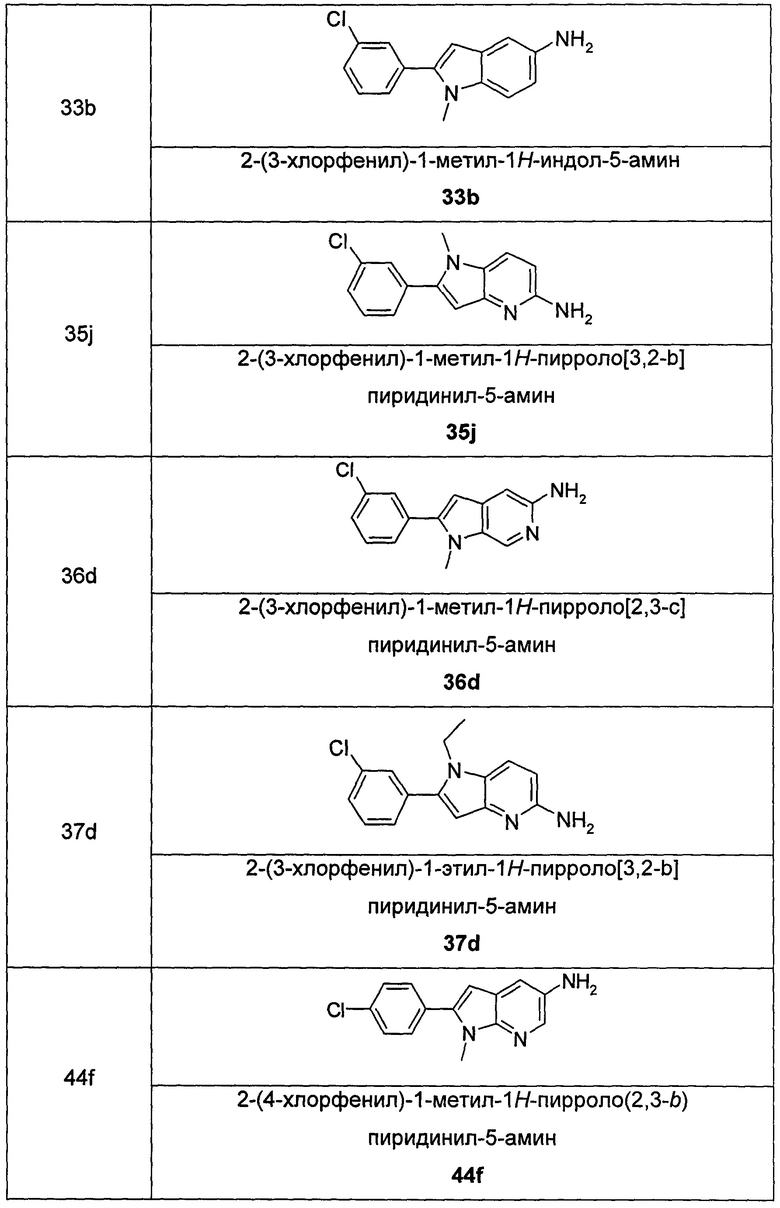
или их таутомеры, мезомеры, рацематы, энантиомеры, диастереомеры, или их смеси, или их фармацевтически приемлемые соли.
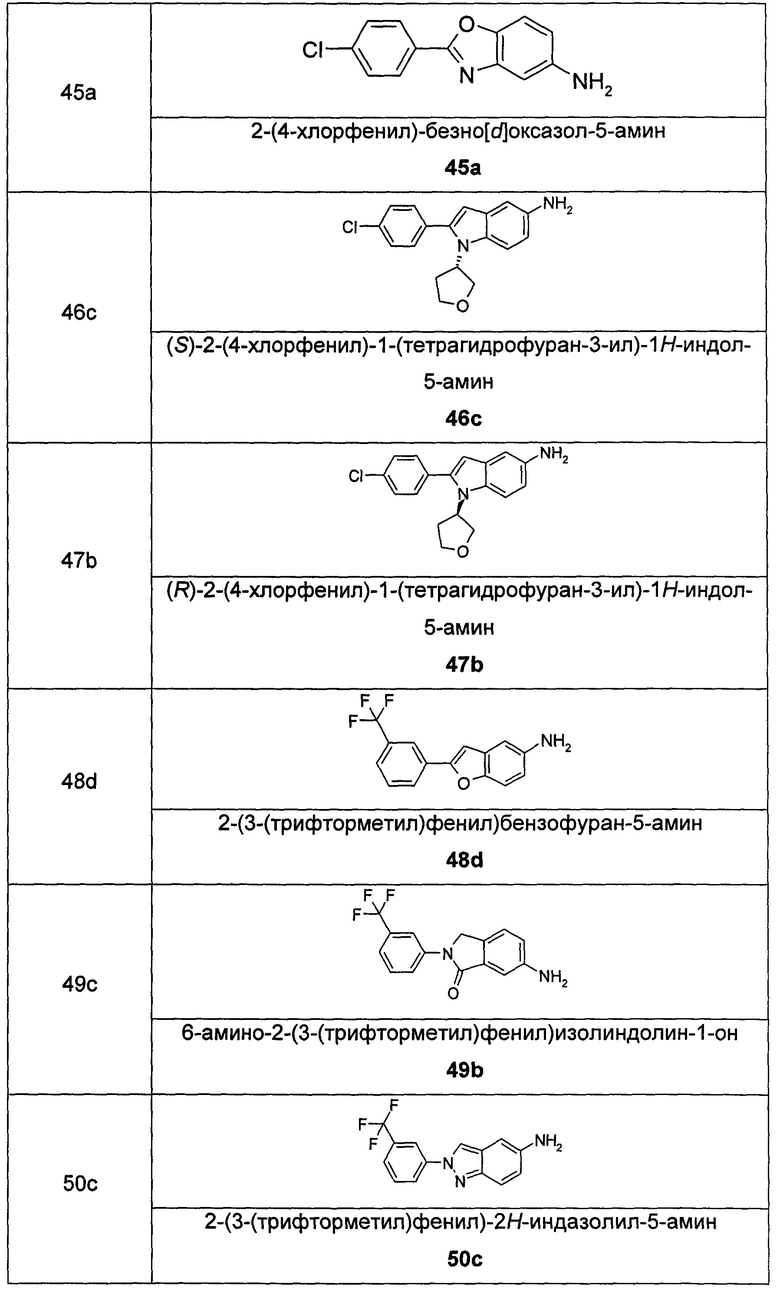
В еще одном аспекте настоящего изобретения предложено соединение формулы (IB), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или его фармацевтически приемлемые соли:
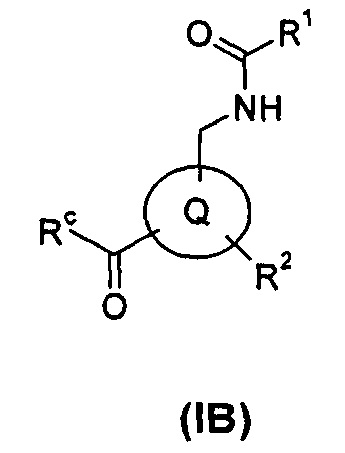
где
Rc выбран из гидрокси и галогена; и
кольцо Q, R1 и R2 являются такими, как определено в общей формуле (I).
Соединения общей формулы (IB) включают, но не ограничиваются следующими:
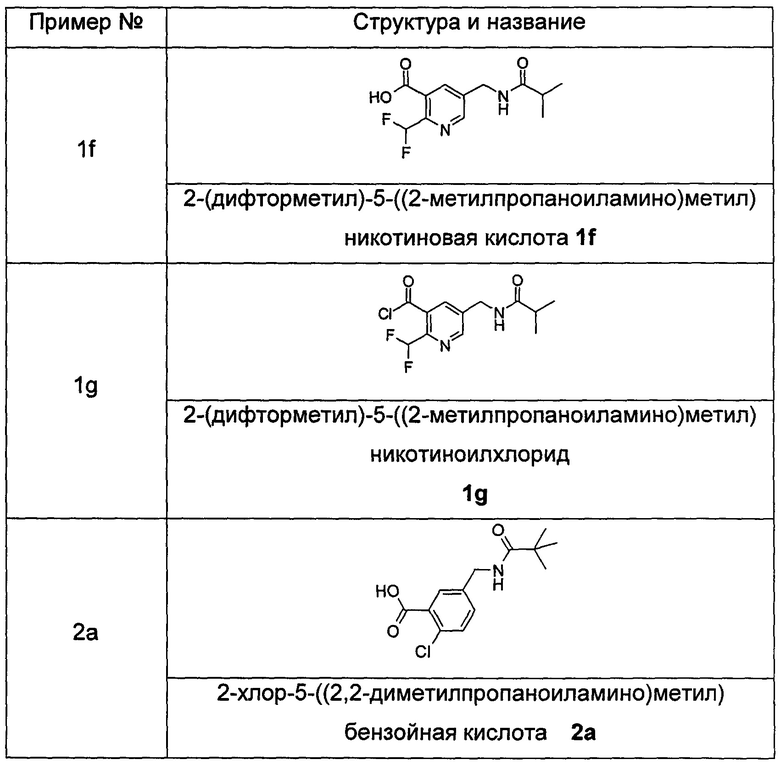
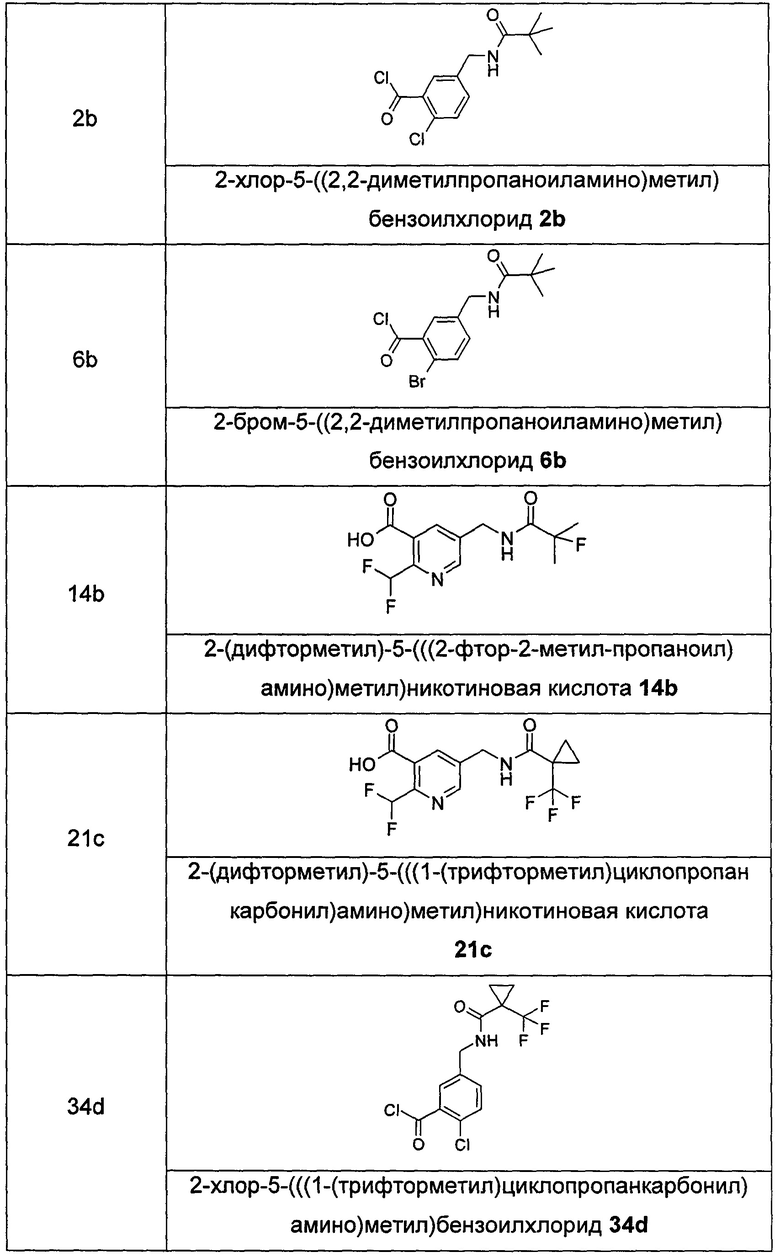
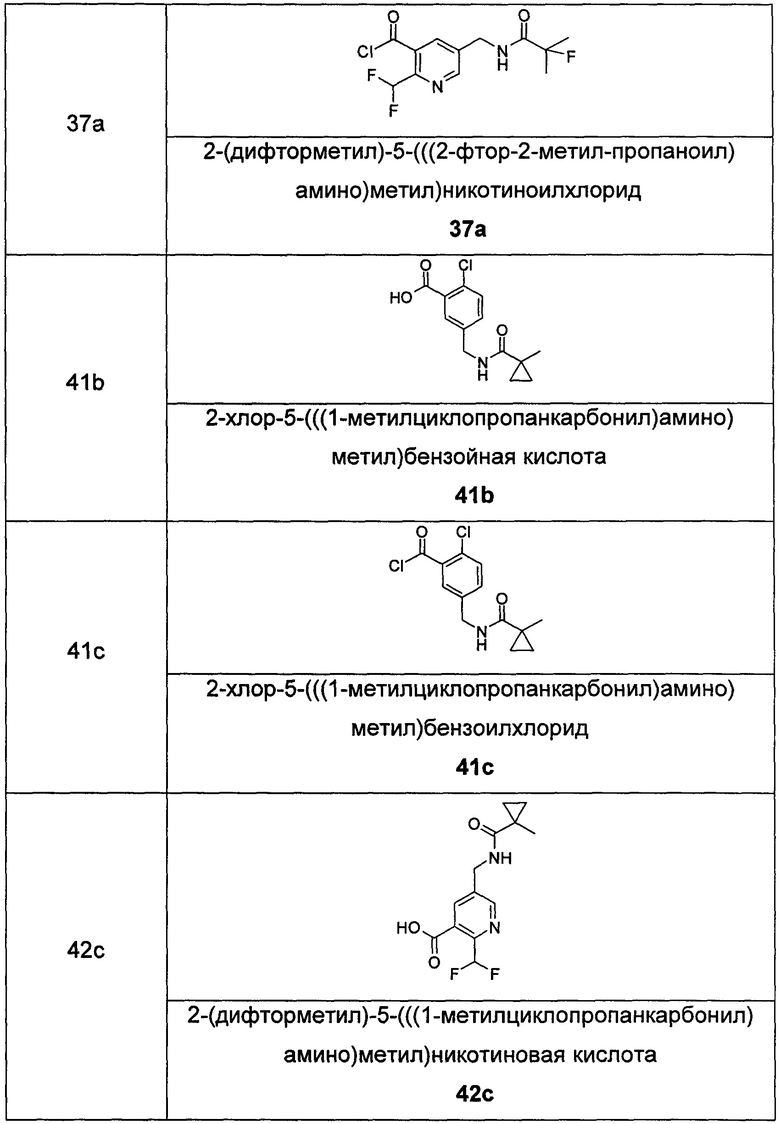
или их таутомеры, мезомеры, рацематы, энантиомеры, диастереомеры, или их смеси, или их фармацевтически приемлемые соли.
Еще один аспект настоящего изобретения направлен на фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или его фармацевтически приемлемых солей, или его фармацевтически приемлемые соли и фармацевтически приемлемые носители, разбавители или вспомогательные вещества.
Еще один аспект настоящего изобретения направлен на применение соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или его фармацевтически приемлемых солей, или содержащей его фармацевтической композиции, для получения лекарственного средства для лечения заболеваний, опосредованных микросомальной простагландин Е-синтазой-1 (mPGES-1).
Еще один аспект настоящего изобретения направлен на применение соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или его фармацевтически приемлемых солей, или содержащей его фармацевтической композиции, для получения лекарственного средства для ингибирования микросомальной простагландин Е-синтазы-1 (mPGES-1).
Еще один аспект настоящего изобретения направлен на применение соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или его фармацевтически приемлемых солей, или содержащей его фармацевтической композиции, для получения лекарственного средства для лечения или предупреждения заболеваний или расстройств, где указанные заболевания или расстройства выбраны из группы, состоящей из воспаления, боли, рака, диабета и осложнений диабета или нейродегенеративных расстройств и тому подобных, где указанное воспаление включает связанное с воспалением аутоиммунное заболевание, кожное заболевание, заболевание легких, заболевание внутренних органов, заболевание уха, носа, рта и горла или сердечнососудистое заболевание и тому подобные; где аутоиммунное заболевание включает артрит, остеоартрит, ювенильный артрит, ревматоидный артрит, анкилозирующий спондилит, подагру, ревматическую атаку, бурсит, системную красную волчанку (СКВ) или множественный склероз и тому подобные; указанное кожное заболевание включает дерматит, экзему, псориаз, ожоги или травмы ткани, и тому подобные; указанное заболевание легких включают астму, хроническую обструктивную болезнь легких (ХОБЛ), или пневмофиброз, и тому подобные; указанное заболевание внутренних органов включает воспалительное заболевание кишечника, болезнь Крона, язвенный колит, синдром раздраженного кишечника (СРК), язвенную болезнь, цистит, простатит, панкреатит или нефрит, и тому подобные; указанное заболевание уха, носа, рта и горла включает грипп, вирусную инфекцию, бактериальную инфекцию, повышение температуры тела, ринит, фарингит, тонзиллит, конъюнктивит, ирит, склерит или увеит, и тому подобные; указанное сердечнососудистое заболевание включает атеросклероз, тромбоз, инсульт или коронарную болезнь сердца, и тому подобные; указанная боль включает нейропатическую боль, боль при воспалении, боль внутренних органов, боль, связанную с раковым заболеванием, боль, связанную с химиотерапией, боль в результате травмы, боль в результате хирургической операции, послеоперационную боль, боль при родах, послеродовую боль, хроническую боль, постоянную боль, боль периферического генеза, боль центрального генеза, хроническую головную боль, мигрень, головную боль в области придаточных пазух носа, головную боль напряжения, фантомную боль в ампутированных конечностях, периферическая нервная травма или их комбинацию, и тому подобные; указанный рак включает рак простаты, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак груди, рак легких, рак головы и шеи, рак щитовидной железы, глиобластому, меланому, лимфому, лейкемию, Т-клеточную лимфому кожи или В-клеточную лимфому кожи, и тому подобные; указанный диабет и осложнения диабета включают диабетическую васкулопатию, диабетическую нейропатию или диабетическую ретинопатию, и тому подобные; указанное нейродегенеративное расстройство включает болезнь Альцгеймера или болезнь Паркинсона; где указанные заболевания или расстройства предпочтительно выбраны из воспаления и боли, более предпочтительно выбраны из остеоартрита, ревматоидного артрита, бурсита, анкилозирующего спондилита, или боли, связанной с любым из заболеваний или расстройств, перечисленных выше.
Еще один аспект настоящего изобретения направлен на способ лечения заболеваний, опосредованных микросомальной простагландин Е-синтазой-1 (mPGES-1), где способ включает введение субъекту, который в этом нуждается, терапевтически эффективной дозы соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или его фармацевтически приемлемых солей, или содержащей его фармацевтической композиции.
Еще один аспект настоящего изобретения направлен на способ ингибирования микросомальной простагландин Е-синтазы-1 (mPGES-1), где способ включает введение субъекту, который в этом нуждается, терапевтически эффективной дозы соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или его фармацевтически приемлемых солей, или содержащей его фармацевтической композиции.
Еще один аспект настоящего изобретения направлен на способ лечения или предупреждения заболеваний или расстройств, где способ включает введение субъекту, который в этом нуждается, терапевтически эффективной дозы соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или его фармацевтически приемлемых солей, или содержащей его фармацевтической композиции; где указанные заболевания или расстройства выбраны из группы, состоящей из воспаления, боли, рака, диабета и осложнений диабета или нейродегенеративных расстройств и тому подобных, где указанное воспаление включает связанное с воспалением аутоиммунное заболевание, кожное заболевание, заболевание легких, заболевание внутренних органов, заболевание уха, носа, рта и горла или сердечнососудистое заболевание и тому подобные; где аутоиммунное заболевание включает артрит, остеоартрит, ювенильный артрит, ревматоидный артрит, анкилозирующий спондилит, подагру, ревматическую атаку, бурсит, системную красную волчанку (СКВ) или множественный склероз и тому подобные; указанное кожное заболевание включает дерматит, экзему, псориаз, ожоги или травмы ткани, и тому подобные; указанное заболевание легких включают астму, хроническую обструктивную болезнь легких (ХОБЛ), или пневмофиброз, и тому подобные; указанное заболевание внутренних органов включает воспалительное заболевание кишечника, болезнь Крона, язвенный колит, синдром раздраженного кишечника (СРК), язвенную болезнь, цистит, простатит, панкреатит или нефрит, и тому подобные; указанное заболевание уха, носа, рта и горла включает грипп, вирусную инфекцию, бактериальную инфекцию, повышение температуры тела, ринит, фарингит, тонзиллит, конъюнктивит, ирит, склерит или увеит, и тому подобные; указанное сердечнососудистое заболевание включает атеросклероз, тромбоз, инсульт или коронарную болезнь сердца, и тому подобные; указанная боль включает нейропатическую боль, боль при воспалении, боль внутренних органов, боль, связанную с раковым заболеванием, боль, связанную с химиотерапией, боль в результате травмы, боль в результате хирургической операции, послеоперационную боль, боль при родах, послеродовую боль, хроническую боль, постоянную боль, боль периферического генеза, боль центрального генеза, хроническую головную боль, мигрень, головную боль в области придаточных пазух носа, головную боль напряжения, фантомную боль в ампутированных конечностях, травму периферического нерва или их комбинацию, и тому подобные; указанный рак включает рак простаты, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак груди, рак легких, рак головы и шеи, рак щитовидной железы, глиобластому, меланому, лимфому, лейкемию, Т-клеточную лимфому кожи или В-клеточную лимфому кожи, и тому подобные; указанный диабет и осложнения диабета включают диабетическую васкулопатию, диабетическую нейропатию или диабетическую ретинопатию, и тому подобные; указанное нейродегенеративное расстройство включает болезнь Альцгеймера или болезнь Паркинсона; где указанные заболевания или расстройства предпочтительно выбраны из воспаления и боли, более предпочтительно выбраны из остеоартрита, ревматоидного артрита, бурсита, анкилозирующего спондилита или боли, связанной с любым из заболеваний или расстройств, перечисленных выше.
Еще один аспект настоящего изобретения направлен на соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или его фармацевтически приемлемые соли, или содержащую его фармацевтическую композицию, для применения в качестве лекарственного средства для лечения заболеваний, опосредованных микросомальной простагландин Е-синтазой-1 (mPGES-1).
Еще один аспект настоящего изобретения направлен на соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или его фармацевтически приемлемые соли, или содержащую его фармацевтическую композицию, для применения в качестве лекарственного средства для ингибирования микросомальной простагландин Е-синтазы-1 (mPGES-1).
Еще один аспект настоящего изобретения направлен на соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или его фармацевтически приемлемые соли, или содержащую его фармацевтическую композицию, для применения в качестве лекарственного средства для лечения или предупреждения заболеваний или расстройств, где указанные заболевания или расстройства выбраны из группы, состоящей из воспаления, боли, рака, диабета и осложнений диабета или нейродегенеративных расстройств и тому подобных, где указанное воспаление включает связанное с воспалением аутоиммунное заболевание, кожное заболевание, заболевание легких, заболевание внутренних органов, заболевание уха, носа, рта и горла или сердечнососудистое заболевание и тому подобные; где аутоиммунное заболевание включает артрит, остеоартрит, ювенильный артрит, ревматоидный артрит, анкилозирующий спондилит, подагру, ревматическую атаку, бурсит, системную красную волчанку (СКВ) или множественный склероз и тому подобные; указанное кожное заболевание включает дерматит, экзему, псориаз, ожоги или травмы ткани, и тому подобные; указанное заболевание легких включают астму, хроническую обструктивную болезнь легких (ХОБЛ), или пневмофиброз, и тому подобные; указанное заболевание внутренних органов включает воспалительное заболевание кишечника, болезнь Крона, язвенный колит, синдром раздраженного кишечника (СРК), язвенную болезнь, цистит, простатит, панкреатит или нефрит, и тому подобные; указанное заболевание уха, носа, рта и горла включает грипп, вирусную инфекцию, бактериальную инфекцию, повышение температуры тела, ринит, фарингит, тонзиллит, конъюнктивит, ирит, склерит или увеит, и тому подобные; указанное сердечнососудистое заболевание включает атеросклероз, тромбоз, инсульт или коронарную болезнь сердца, и тому подобные; указанная боль включает нейропатическую боль, боль при воспалении, боль внутренних органов, раковую боль, связанную с раковым заболеванием, боль, связанную с химиотерапией, боль в результате травмы, боль в результате хирургической операции, послеоперационную боль, боль при родах, послеродовую боль, хроническую боль, постоянную боль, боль периферического генеза, боль центрального генеза, хроническую головную боль, мигрень, головную боль в области придаточных пазух носа, головную боль напряжения, фантомную боль в ампутированных конечностях, травму периферического нерва или их комбинацию, и тому подобные; указанный рак включает рак простаты, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак груди, рак легких, рак головы и шеи, рак щитовидной железы, глиобластому, меланому, лимфому, лейкемию, Т-клеточную лимфому кожи или В-клеточную лимфому кожи, и тому подобные; указанный диабет и осложнения диабета включают диабетическую васкулопатию, диабетическую нейропатию или диабетическую ретинопатию, и тому подобные; указанное нейродегенеративное расстройство включает болезнь Альцгеймера или болезнь Паркинсона; где указанные заболевания или расстройства предпочтительно выбраны из воспаления и боли, более предпочтительно выбраны из остеоартрита, ревматоидного артрита, бурсита, анкилозирующего спондилита или боли, связанной с любым из заболеваний или расстройств, перечисленных выше.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, термины, используемые в настоящем описании, имеют следующие значения.
"Алкил" относится к линейной или разветвленной насыщенной алифатической углеводородной группе, содержащей от 1 до 20 атомов углерода, предпочтительно к алкилу, содержащему от 1 до 10 атомов углерода, более предпочтительно к алкилу, содержащему от 1 до 6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, содержащий от 1 до 6 атомов углерода, и неограниченные примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, и тому подобные. Алкильная группа может быть замещенной или незамещенной. В случае замещения, замещающая(ие) группа(ы) могут быть замещены в любой доступной точке присоединения. Замещающая(ие) группа(ы) предпочтительно представляет(ют) одну или более чем одну группу, выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклической алкокси, циклоалкилтио, гетероциклилтио, оксо, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты, -C(O)OR5, -OC(O)OR5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 или -C(O)NR6R7.
"Алкенил" относится к алкилу, определенному так, как указано выше, имеющему по меньшей мере два атома углерода и по меньшей мере одну двойную связь углерод-углерод, например, этенилу, 1-пропенилу, 2-пропенилу, 1-, 2- или 3-бутенилу и тому подобным, предпочтительно алкенилу, содержащему от 2 до 10 атомов углерода, более предпочтительно алкенилу, содержащему от 2 до 6 атомов углерода, наиболее предпочтительно алкенилу, содержащему от 2 до 4 атомов углерода. Алкенильная группа может быть замещенной или незамещенной. В случае замещения замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилтио, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклилтио, оксо, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты, -C(O)OR5, -OC(O)OR5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 или -C(O)NR6R7.
"Алкинил" относится к алкилу, определенному так, как указано выше, имеющему по меньшей мере два атома углерода и по меньшей мере одну тройную связь углерод-углерод, например, этинилу, 1-пропинилу, 2-пропинилу, 1-, 2- или 3-бутинилу и тому подобным, предпочтительно алкинилу, содержащему от 2 до 10 атомов углерода, более предпочтительно алкинилу, содержащему от 2 до 6 атомов углерода, наиболее предпочтительно алкинилу, содержащему от 2 до 4 атомов углерода. Алкинильная группа может быть замещенной или незамещенной. В случае замещения замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилтио, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклилтио, оксо, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты, -C(O)OR5, -OC(O)OR5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7, или -C(O)NR6R7.
"Циклоалкил" относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, содержащей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода, и наиболее предпочтительно от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил, и тому подобные, предпочтительно циклопропил и циклогексенил. Полициклический циклоалкил включает циклоалкил, имеющий спирановую, конденсированную или мостиковую структуру.
"Спироциклоалкил" относится к пол и циклической группе с числом атомов углерода от 5 до 20, с кольцами, соединенными через один общий атом углерода (называемый спироатомом), где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, предпочтительно спироциклоалкилу, с числом атомов углерода от 6 до 14, и более предпочтительно спироциклоалкилу, с числом атомов углерода от 7 до 10. В соответствии с числом общих спироатомов спироциклоалкил можно разделить на моноспироциклоалкил, диспироциклоалкил, или полиспироциклоалкил, и предпочтительно моноспироциклоалкил или диспироциклоалкил, более предпочтительно моноспироциклоалкил с числом атомов углерода в каждом кольце 4 и 4, 4 и 5, 4 и 6, 5 и 5, или 5 и 6. Неограничивающие примеры спироциклоалкилов включают, но не ограничиваются следующими:
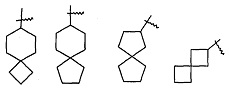 и
и 
"Конденсированный циклоалкил" относится к полностью состоящей из атомов углерода пол и циклической группе, с числом атомов углерода от 5 до 20, где каждое кольцо в системе имеет общую с другим кольцом смежную пару атомов углерода, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, предпочтительно конденсированному циклоалкилу, с числом атомов углерода от 6 до 14, более предпочтительно конденсированному циклоалкилу, с числом атомов углерода от 7 до 10. В соответствии с числом колец конденсированный циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, предпочтительно бициклический или трициклический конденсированный циклоалкил, и более предпочтительно бициклический конденсированный циклоалкил с числом атомов углерода в каждом кольце 5 и 5, или 5 и 6. Неограничивающие примеры конденсированного циклоалкила включают, но не ограничиваются следующими:
 и
и 
"Мостиковый циклоалкил" относится к полностью состоящей из атомов углерода полициклической группе, с числом атомов углерода от 5 до 20, где каждые два кольца в системе имеют два общих не связанных между собой атома, где кольца могут иметь одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, предпочтительно мостиковому циклоалкилу, с числом атомов углерода от 6 до 14, и более предпочтительно мостиковому циклоалкилу, с числом атомов углерода от 7 до 10. В соответствии с числом колец мостиковый циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, и предпочтительно бициклический, трициклический или тетрациклический мостиковый циклоалкил, и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостиковых циклоалкилов включают, но не ограничиваются следующими:
 и
и  .
.
Указанный циклоалкил может быть конденсированным с арилом, гетероарилом или гетероциклилом, где кольцо, связанное с исходной структурой, представляет собой циклоалкил. Неограниченные примеры включают инданил, тетрагидронафтил, бензоциклогептил и тому подобные. Циклоалкил может быть необязательно замещенным или незамещенным. В случае замещения замещающая(ие) группа(ы) представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклической алкокси, циклоалкилтио, гетероциклилтио, оксо, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты, -C(O)OR5, -OC(O)OR5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7, или -C(O)NR6R7.
"Гетероциклил" относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, с числом атомов колец от 3 до 20, имеющей один или более чем один гетероатом, выбранный из группы, состоящей из N, О, и S(O)m (где m представляет собой целое число, выбранное из диапазона от 0 до 2) в качестве атома кольца, но исключая -O-O-, -O-S- или -S-S- в кольце, где оставшиеся атомы кольца представляют собой атомы углерода. Предпочтительно, гетероциклил имеет от 3 до 12 атомов с числом гетероатомов от 1 до 4, более предпочтительно от 3 до 10 атомов, и наиболее предпочтительно от 5 до 6 атомов. Неограничивающие примеры моноциклического гетероциклила включают, но не ограничиваются следующими, пирролидинил, пиперидил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил, тетрогидрофуранил, и тому подобные. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или кольцо с внутренней мостиковой связью.
"Спирогетероциклил" относится к полициклическому гетероциклилу, с числом атомов колец от 5 до 20, с кольцами, соединенными через один общий атом углерода (также называемый спироатомом), где указанные кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей N, О, и S(O)m (где m представляет собой целое число, выбранное из диапазона от 0 до 2) в качестве атома кольца, и оставшиеся атомы кольца представляют собой атомы углерода, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы; предпочтительно спирогетероциклилу, с числом атомов колец от 6 до 14, и более предпочтительно спирогетероциклилу, с числом атомов колец от 7 до 10. В соответствии с числом общих спироатомов спирогетероциклил можно разделить на моноспирогетероциклил, диспирогетероциклил или полиспирогетероциклил, предпочтительно моноспирогетероциклил или диспирогетероциклил, и более предпочтительно моноспирогетероциклил с числом атомов каждого кольца 4 и 4, 4 и 5, 4 и 6, 5 и 5, или 5 и 6. Неограничивающие примеры спирогетероциклилов включают, но не ограничиваются следующими:
 или
или 
"Конденсированный гетероциклил" относится к полициклической гетероциклильной группе, с числом атомов колец от 5 до 20, где каждое из колец в системе имеет общую с другим кольцом смежную пару атомов углерода, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, и где указанные кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О, и S(O)m (где m представляет собой целое число, выбранное из диапазона от 0 до 2) в качестве атома кольца, где оставшиеся атомы кольца представляют собой атомы углерода; предпочтительно конденсированному гетероциклилу, с числом атомов колец от 6 до 14, и более предпочтительно конденсированному гетероциклилу, с числом атомов колец от 7 до 10. В соответствии с числом колец конденсированный гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, предпочтительно бициклический или трициклический конденсированный гетероциклил, и более предпочтительно бициклический конденсированный гетероциклил с числом атомов каждого кольца 5 и 5, или 5 и 6. Неограничивающие примеры конденсированного гетероциклила включают, но не ограничиваются следующими:
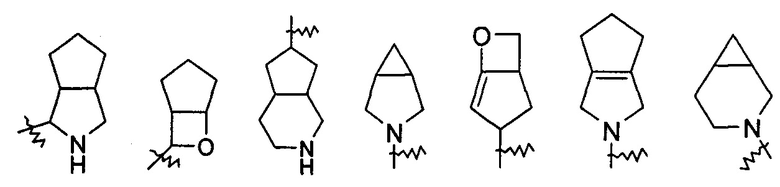
 и
и 
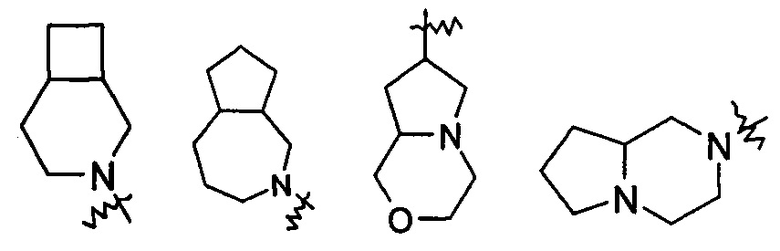
"Мостиковый гетероциклил" относится к полициклической гетероциклильной группе с числом атомов колец от 5 до 14, где каждые два кольца в системе имеют два общих не связанных между собой атома, где кольца могут иметь одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, и кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О, и S (O)m (где m представляет собой целое число, выбранное из диапазона от 0 до 2) в качестве атома кольца, где оставшиеся атомы кольца представляют собой атомы углерода; предпочтительно мостиковому гетероциклилу, с числом атомов колец от 6 до 14, и более предпочтительно мостиковому гетероциклилу, с числом атомов колец от 7 до 10. В соответствии с числом колец мостиковый гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, и предпочтительно бициклический, трициклический или тетрациклический мостиковый гетероциклил, и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостиковых гетероциклилов включают, но не ограничиваются следующими:
 и
и 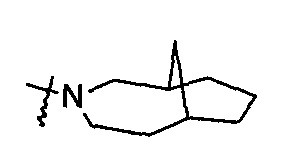 .
.
Указанный гетероциклил может быть конденсированным с арилом, гетероарилом или циклоалкилом, где кольцо, связанное с исходной структурой, представляет собой гетероциклил. Неограничивающие примеры включают, но не ограничиваются следующими:
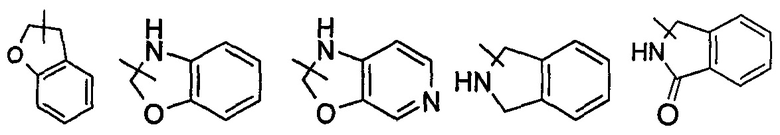 и
и  , и т.д.
, и т.д.
Гетероциклил может быть необязательно замещенным или незамещенным. В случае замещения замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклической алкокси, циклоалкилтио, гетероциклилтио, оксо, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты, -C(O)OR5, -OC(O)OR5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 или -C(O)NR6R7.
"Арил" относится к полностью состоящей из атомов углерода моноциклической кольцевой или полициклической конденсированной кольцевой (т.е. каждое кольцо в системе имеет смежную пару атомов углерода с другим кольцом в системе) группе с числом атомов углерода от 6 до 14, имеющей полностью сопряженную пи-электронную систему; предпочтительно арилу с числом атомов углерода от 6 до 10, более предпочтительно фенилу и нафтилу, и наиболее предпочтительно фенилу. Арил может быть конденсированным с гетероарилом, гетероциклилом или циклоалкилом, где кольцо, связанное с исходной структурой, представляет собой арил. Неограничивающие примеры включают, но не ограничиваются следующими:
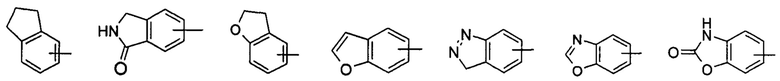
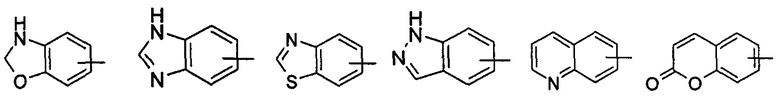 и
и 
Арил может быть необязательно замещенным или незамещенным. В случае замещения замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклической алкокси, циклоалкилтио, гетероциклилтио, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты, -C(O)OR5, -OC(O)OR5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 или -C(O)NR6R7.
"Гетероарил" относится к арилу с числом атомов колец от 5 до 14, имеющему от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N в качестве атомов кольца, где оставшиеся атомы представляют собой атомы углерода; предпочтительно гетероарилу с числом атомов колец от 5 до 10, более предпочтительно гетероарилу с числом атомов колец от 5 до 6, такому как фурил, тиенил, пиридил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил, тетразолил, оксазолил, тиазолил, пиразолил и тому подобные. Гетероарил может быть конденсированным с арилом, гетероциклилом или циклоалкилом, где кольцо, связанное с исходной структурой, представляет собой гетероарил. Неограничивающие примеры включают, но не ограничиваются следующими:
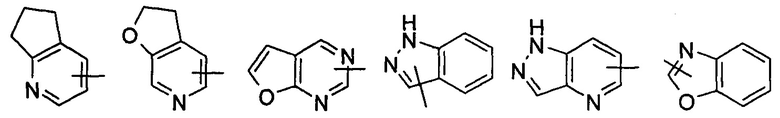
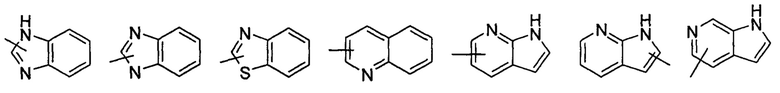
 и
и  .
.
Гетероарил может быть необязательно замещенным или незамещенным. В случае замещения замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклической алкокси, циклоалкилтио, гетероциклилтио, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты, -C(O)OR5, -OC(O)OR5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7 или -C(O)NR6R7.
"Алкокси" относится к -О-(алкил) или -O-(незамещенный циклоалкил) группе, где алкил является таким, как определено выше. Неограничивающие примеры включают, но не ограничиваются следующими, метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси, и тому подобные. Алкокси может быть необязательно замещенной или незамещенной. В случае замещения заместитель предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклической алкокси, циклоалкилтио, гетероциклилтио, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты, -C(O)OR5, -OC(O)OR5, -NHS(O)mR5, -C(O)R5, -NHC(O)R5, -NHC(O)OR5, -NR6R7, -OC(O)NR6R7, или -C(O)NR6R7.
"Галогеналкил" относится к алкилу, замещенному одним или более чем одним галогеном, где алкил является таким, как определено выше.
"Гидрокси" относится к -ОН группе.
"Гидроксиалкил" относится к алкилу, замещенному гидроксигруппой, где алкил является таким, как определено выше.
"Галоген" относится к фтору, хлору, брому или йоду.
"Амино" относится к -NH2 группе.
"Циано" относится к -CN группе.
"Нитро" относится к -NO2 группе.
"Бензил" относится к -СН2-фенил группе.
"Оксо" относится к =O группе.
"Карбоксил" относится к -С(O)ОН группе.
"Эфир карбоновой кислоты" относится к -С(O)O(алкил) или (циклоалкил) группе, где алкил и циклоалкил являются такими, как определено выше.
"Аминозащитная группа" относится к группе, предотвращающей аминогруппу от взаимодействия, когда другие части молекулы подвергают реакции, которая может быть легко удалена. Неограничивающие примеры включают, но не ограничиваются следующими, формил, алкилкарбонил, алкоксикарбонил, бензоил, аралкилкарбонил, аралкоксикарбонил, тритил, фталильную группу, N,N-диметиламинометиленил, замещенный силицил, и тому подобные. Эти труппы могут быть необязательно замещенными 1-3 группами, независимо выбранными из группы, состояще из галогена, алкокси или нитро. Аминозащитная группа предпочтительно представляет собой t-бутилоксикарбонил.
"Необязательный" или "необязательно" означает, что явление или условие, описанное далее, может, но не обязательно должно произойти, и такое описание включает ситуацию, в которой явление или условие может произойти или может не произойти. Например, "гетероциклическая группа, необязательно замещенная алкилом" означает, что алкильная группа может, но не обязательно должна присутствовать, и такое описание включает ситуацию, где гетероциклическая группа замещена алкилом и где гетероциклическая группа не замещена алкилом.
"Замещенный" относится к одному или более чем одному атому водорода, предпочтительно до 5, более предпочтительно атомам водорода от 1 до 3, независимо замещенным соответствующим числом заместителей. Само собой разумеется, что заместители находятся только в возможном для них химическом положении. Специалист способен определить, возможно замещение или нет, путем экспериментирования или теоретически, без приложения чрезмерных усилий. Например, комбинация амино или гидрокси, имеющего свободный атом водорода, с атомами углерода, имеющими ненасыщенные связи (такими как олефиновые) может быть нестабильной.
"Фармацевтическая композиция" относится к смеси одного или более чем одного соединения по настоящему изобретению или его физиологически/фармацевтически приемлемых солей, или его пролекарств, и других химических компонентов, таких как физиологически/фармацевтически приемлемые носители и эксципиенты. Назначение фармацевтический композиции заключается в облегчении введения соединения в организм, что способствует абсорбции активного ингредиента и таким образом проявлению биологической активности.
m и R5-R7 являются такими, как определено в соединении формулы (I).
СПОСОБ СИНТЕЗА ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Для достижения цели настоящего изобретения в настоящем изобретении применяют следующие технические решения для синтеза.
Схема 1
Способ получения соединения формулы (I) по настоящему изобретению, или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или его фармацевтически приемлемых солей, включающий следующие стадии:
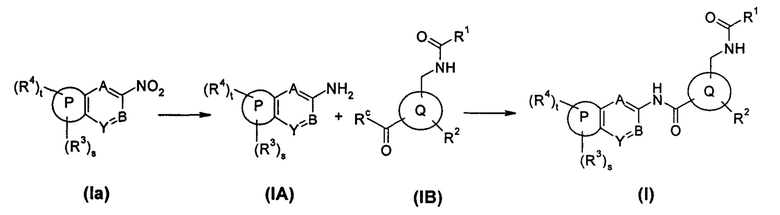
соединение формулы (Ia) подвергают реакции восстановления в присутствии катализатора с получением соединения формулы (IA) или его солей; соединение формулы (IA) или его соли подвергают реакции конденсации с соединением формулы (IB) в щелочной среде с получением соединения формулы (I);
где:
Rc выбран из гидрокси и галогена;
кольцо Р, кольцо Q, А, В, Y, s, t и R1-R4 являются такими, как определено в общей формуле (I).
Схема 2
Способ получения соединения формулы (III), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или его фармацевтически приемлемых солей, включающий следующие стадии:
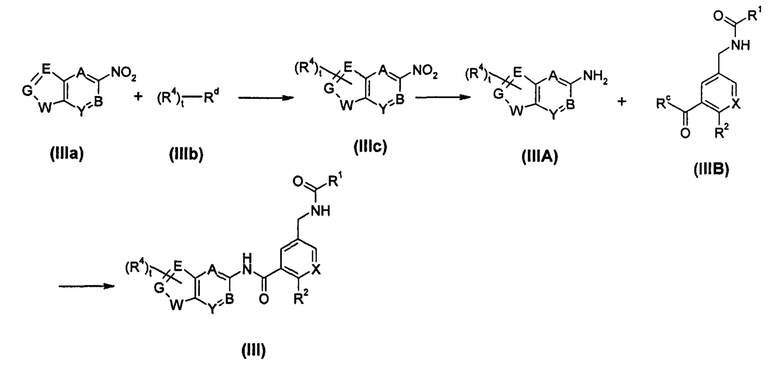
соединение формулы (IIIa) подвергают реакции сочетания с соединением формулы (IIIb) в присутствии катализатора с получением соединения формулы (IIIc); соединение формулы (IIIc) подвергают реакции восстановления в присутствии катализатора с получением соединения формулы (IIIA) или его солей; соединение формулы (IIIA) или его соли подвергают реакции конденсации с соединением формулы (IIIB) в щелочной среде с получением соединения формулы (III);
где:
Rc выбран из гидрокси и галогена;
Rd выбран из галогена, предпочтительно брома и йода;
каждый из Е, G и W независимо выбран из CRa, NRb, N, О и S;
А, В, X, Y, t, R1, R2 и R4 являются такими, как определено в формуле (I).
Способ получения соединения формулы (IV) и формулы (V) является таким же или схожим со способом получения соединения формулы (III).
Схема 3
Способ получения соединения формулы (VI), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или его фармацевтически приемлемых солей, включающий следующие стадии:
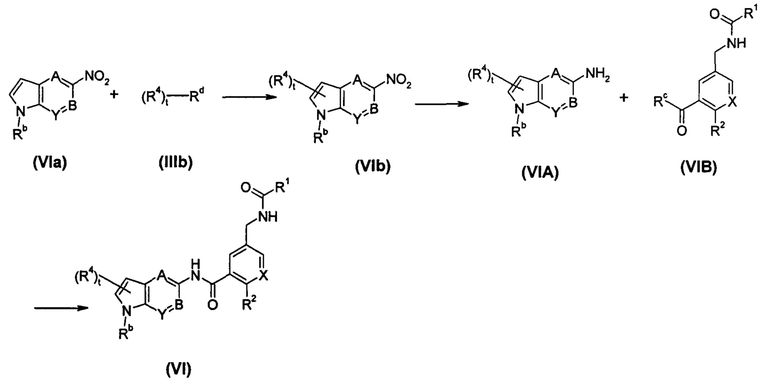
соединение формулы (VIa) подвергают реакции сочетания с соединением формулы (IIIb) в присутствии катализатора с получением соединения формулы (VIb); соединение формулы (VIb) подвергают реакции восстановления в присутствии катализатора с получением соединения формулы (VIA) или его солей; соединение формулы (VIA) или его соли подвергают реакции конденсации с соединением формулы (VIB) в щелочных условиях с получением соединения формулы (VI);
Rc выбран из гидрокси и галогена;
Rd выбран из галогена, предпочтительно брома и йода;
А, В, X, Y, t, R1, R2 и R4 являются такими, как определено в формуле (I);
Rb является таким, как определено в формуле (IV).
Схема 4
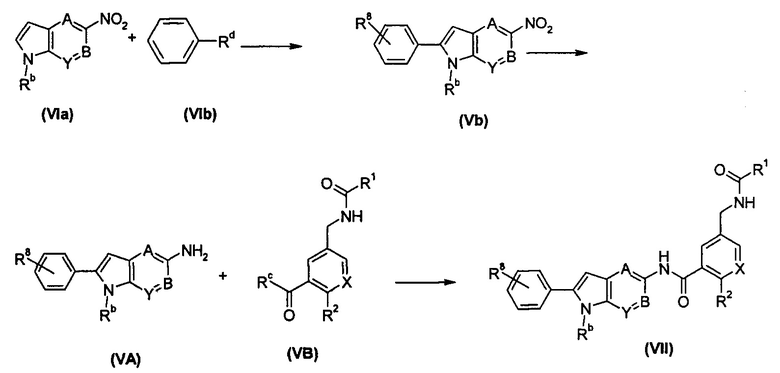
соединение формулы (VIa) подвергают реакции сочетания с соединением формулы (VIb) в присутствии катализатора с получением соединения формулы (Vb); соединение формулы (Vb) подвергают реакции восстановления в присутствии катализатора с получением соединения формулы (VA) или его солей; соединение формулы (VA) или его соли подвергают реакции конденсации с соединением формулы (VB) в щелочных условиях с получением соединения формулы (VII);
Rc выбран из гидрокси и галогена;
Rd выбран из галогена, предпочтительно брома и йода;
А, В, X, Y, R1 и R2 являются такими, как определено в формуле (I);
Rb является таким, как определено в формуле (IV);
R8 является таким, как определено в формуле (VII).
Щелочные реагенты включают органическое основание и неорганическое основание, где указанное органическое основание включает, но не ограничивается следующими, триэтиламин, N,N-диизопропилэтиламин, пиридин, бис(триметилсилил)амид натрия, н-бутиллитий, трет-бутанолят калия, или бромид тетрабутиламмония, где указанное неорганическое основание включает, но не ограничивается следующими, гидроксид лития, гидроксид натрия, гидроксид калия, гидрид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, или карбонат цезия, предпочтительно триэтиламин.
Катализаторы включают, но не ограничиваются следующими, Pd/C, никель Ренея, тетракис(трифенилфосфин)палладий, хлорид палладия, диацетат палладия, (1,1'-бис(дибензилфосфино)ферроцен)дихлорпалладий(II), трис(дибензилиденацетон)дипалладий.
Конденсирующие агенты включают, но не ограничиваются следующими, 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, О-бензотриазол-N,N,N',N'-тетраметилурония гексафторфосфат, 2-(7-азобензотриазол-1-ил)-N,N,N',N-тетраметилурония гексафторфосфат, бензотриазол-1-ил-окси-трис(диметиламино)-фосфония гексафторфосфат, бензотриазол-1-ил-окси-трипирролидинофосфония гексафторфосфат.
Настоящее изобретения будет дополнительно описано следующими примерами, которые не следует рассматривать как ограничивающие объем изобретения.
Условия, не указанные в примерах, являются общеизвестными в уровне техники условиями или рекомендованными изготовителем продукта условиями для исходных веществ. Реагенты, для которых не указан источник происхождения, являются коммерчески доступными общеизвестными реагентами.
Примеры
Структуры соединений были установлены путем ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). ЯМР был определен на Bruker AVANCE-400. Растворители представляли собой дейтерированный диметилсульфоксид (DMSO-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD) с тетраметилсиланом (ТМС) в качестве внутреннего стандарта. ЯМР химические сдвиги (δ) даны в 10-6 (млн-1).
МС определяли на FINNIGAN LCQAd (ESI) масс-спектрометре (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Высокоэффективная жидкостная хроматография (ВЭЖХ) проводилась на спектрометре высокого давления для жидкостной хроматографии Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и спектрометре высокого давления для жидкостной хроматографии Waters 2695-2996 (хроматографическая колонка Gimini С18 150×4,6 мм).
Средняя скорость ингибирования киназы и значения IC50 определяли на NovoStar ELISA (BMG Co., Германия).
Для тонкослойной гель-хроматографии на силикагеле (ТСХ) использовали пластины с силикагелем Yantai Huanghai HSGF254 или Qingdao GF254. Измерения пластины с силикагелем, использованной в ТСХ, составляют от 0,15 мм до 0,2 мм, и измерения пластины с силикагелем, использованной для очищения продукта, составляют от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии использовали силикагель Yantai Huanghai с размером частиц 200-300 меш.
Известные исходные вещества по настоящему изобретению можно получить общеизвестными в уровне техники способами получения, или можно приобрести у производителей ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc., или Dari chemical Company, и т.д.
Если не указано иное, реакции проводили в атмосфере азота или в атмосфере аргона.
Термин "атмосфера азота" или "атмосфера аргона" означает, что реакционный сосуд оборудован баллоном с азотом или аргоном объемом 1 л.
Термин "атмосфера водорода" означает, что реакционный сосуд оборудован баллоном с водородом объемом 1 л.
Реакции гидрирования под давлением проводили с аппаратом для гидрирования Parr 3916EKX и генератором водорода QL-500 или аппаратом для гидрирования HC2-SS.
В реакциях гидрирования в реакционной системе, как правило, создают вакуум и заполняют ее водородом, при этом указанную операцию повторяют три раза.
В реакции, активируемой микроволновым излучением, используют микроволновый реактор типа СЕМ Discover-S 908860.
Если не указано иное, раствор, использованный в реакциях, относится к водному раствору.
Если не указано иное, температура проведения реакции в реакциях относится к комнатной температуре, и диапазон температуры составлял от 20°С до 30°С.
За ходом реакции наблюдали при помощи тонкослойной хроматографии (ТСХ), где элюирующая система включает А - дихлорметан и метанол, В - н-гексан и этилацетат, С - петролейный эфир и этилацетат, D - ацетон. Соотношение объемов растворителей можно регулировать в соответствии с полярностью соединений.
Элюирующая система для очищения соединений посредством колоночной хроматографии и тонкослойной хроматографии включает А - дихлорметан и метанол, В - н-гексан и этилацетат, С - н-гексан и ацетон, D - н-гексан, Е - этилацетат. Соотношение объемов растворителей можно регулировать в соответствии с полярностью соединений, и в некоторых случаях можно добавлять небольшое количество щелочного реагента, такого как триэтиламин или кислотный реагент.
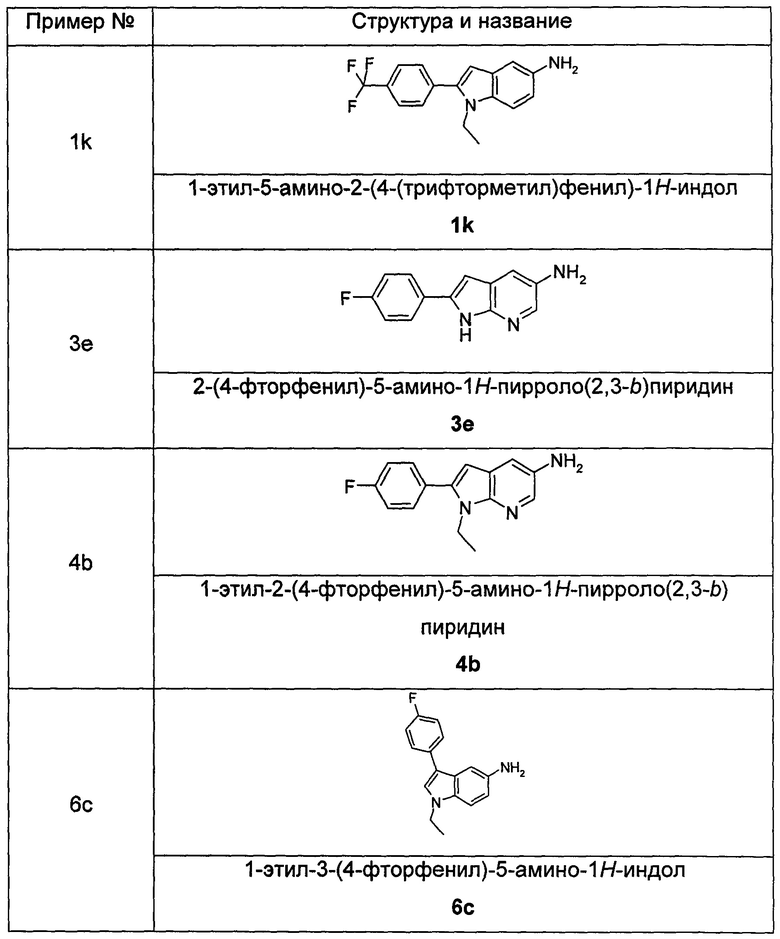
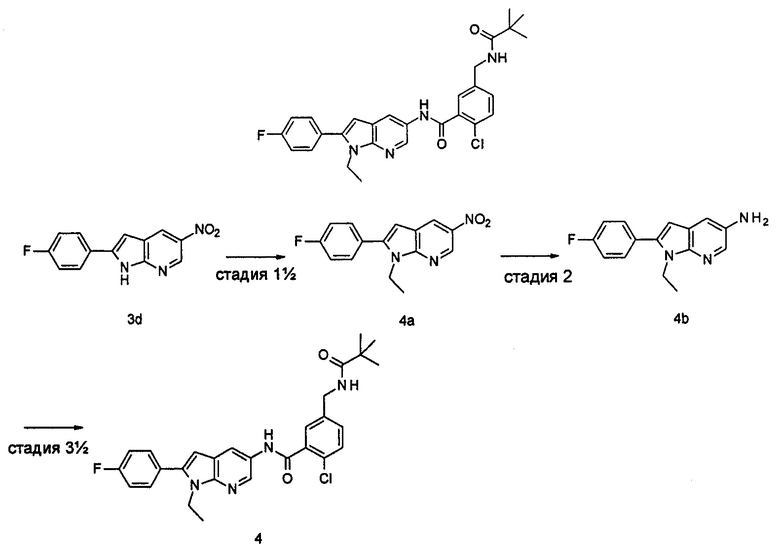
Пример 1
2-(дифторметил)-N-(1-этил-2-(4-(трифторметил)фенил)-1H-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамид
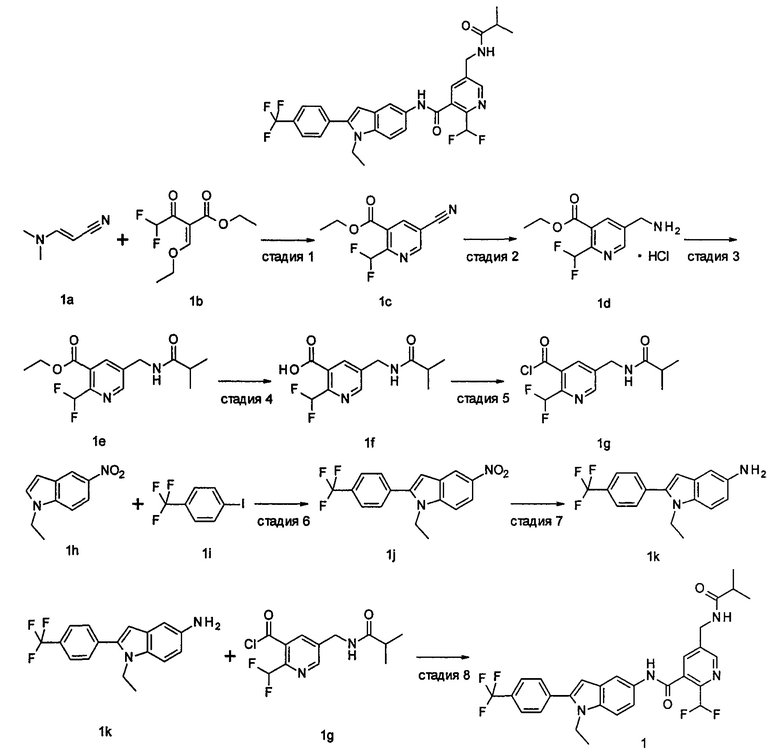
Стадия 1
Этил 5-циано-2-(дифторметил)никотинат
3-Диметиламиноакрилонитрил 1а (865 мг, 9,0 ммоль, полученный в соответствии со способом, раскрытым в "Pharma Chemica, 2010, 2(3), 178-186") растворяли в 20 мл N,N-диметилформамида, и нагревали до 65°С. 5 мл раствора этил 2-(этоксиметилен)-4,4-дифтор-3-оксо-бутаноата 1b (2,0 г, 9,0 ммоль, полученного в соответствии со способом, раскрытым в патентной заявке WO 2012025469) в N,N-диметилформамиде по каплям добавляли в раствор и перемешивали в течение 1 часа. Затем к реакционному раствору добавляли ацетат аммония (1,1 г, 14,0 ммоль) и перемешивали еще в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, к остатку добавляли 100 мл воды и экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали посредством тонкослойной хроматографии (ТСХ) с элюирующей системой С с получением целевого соединения этил 5-циано-2-(дифторметил)никотината 1с (606 мг, 30%) в виде бледно-желтого масла.
МС m/z (ESI (электроспрей ионизация)): 227,1 [М+1]
Стадия 2
Этил 5-(аминометил)-2-(дифторметил)никотината гидрохлорид
Этил 5-циано-2-(дифторметил)никотинат 1с (606 мг, 2,7 ммоль) растворяли в 15 мл этанола и добавляли концентрированную хлороводородную кислоту (1,0 мл, 37%) и Pd/C(180 мг, 10%). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционный раствор фильтровали через Celatom, и концентрировали фильтрат при пониженном давлении с получением указанного целевого соединения этил 5-(аминометил)-2-(дифторметил)никотината гидрохлорида 1d (709 мг, 99%) в виде желтого твердого вещества.
МС m/z (ESI): 231,1 [М+1]
Стадия 3
Этил 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинат
Этил 5-(аминометил)-2-(дифторметил)никотината гидрохлорид 1d (709 мг, 2,7 ммоль) растворяли в 50 мл дихлорметана, и добавляли N,N-диизопропилэтиламин (1,9 мл, 10,6 ммоль). По завершению добавления к реакционной смеси по каплям добавляли раствор изобутирилхлорида в дихлорметане (0,7 М, 5 мл) и затем перемешивали в течение 2 часов. Реакционную смесь последовательно промывали водой (50 мл) и концентрированным раствором бикарбоната натрия (50 мл). Органическую фазу концентрировали при пониженном давлении с получением указанного целевого соединения этил 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотината 1е (790 мг, 99%) в виде желтого твердого вещества.
МС m/z (ESI): 301,1 [М+1]
Стадия 4
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)никотиновая кислота
Этил 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинат 1е (790 мг, 2,6 ммоль) растворяли в 10 мл 1,4-диоксана и добавляли 5 мл воды и гидрат гидроксида лития (291 мг, 6,9 ммоль). Реакционную смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли 5 мл воды и доводили до рН2 5М раствором хлороводородной кислоты. Большое количество твердого вещества осадили и отфильтровали. Фильтрат экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток объединяли с вышеуказанным фильтровальным осадком, промывали водой, и сушили с получением указанного целевого соединения 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновой кислоты 1f (420 мг, 56%) в виде желтого твердого вещества.
МС m/z (ESI): 273,1 [М+1]
Стадия 5
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)никотиноилхлорид
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (150 мг, 0,55 ммоль) растворяли в 5 мл дихлорметана и добавляли одну каплю N,N-диметилформамида и тионилхлорида (197 мг, 1,65 ммоль). По завершении добавления реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении с получением целевого соединения 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиноил-хлорида 1g (160 мг) в виде бледно-желтого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 6
1-Этил-5-нитро-2-(4-(трифторметил)фенил)-7Н-индол
1-Этил-5-нитро-1H-индол 1h (500 мг, 2,63 ммоль, полученный в соответствии со способом, раскрытым в "Bioorganic & Medicinal Chemistry, 2005, 13(10), 3531-3541") растворяли в 5 мл N,N-диметилацетамида, и последовательно добавляли 4-йодтрифтортолуола 1i (790 мг, 2,92 ммоль), трифенилфосфина (140 мг, 0,53 ммоль), ацетата палладия (30 мг, 0,13 ммоль) и ацетата цезия (1,6 г, 5,21 ммоль). По завершении добавления полученную смесь нагревали до 140°С и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли 50 мл этилацетата, промывали водой (20 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения 1-этил-5-нитро-2-(4-(трифторметил)фенил)-1H-индола 1j (130 мг, 14,8%) в виде желтого твердого вещества.
МС m/z (ESI): 335,1 [М+1]
Стадия 7
1-Этил-5-амино-2-(4-(трифторметил)фенил)-7Н-индол
1-Этил-5-нитро-2-(4-(трифторметил)фенил)-7Н-индол 1j (130 мг, 0,39 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и добавляли никель Ренея (30 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и концентрировали фильтрат при пониженном давлении с получением неочищенного целевого соединения 1-этил-5-амино-2-(4-(трифторметил)фенил)-7Н-индола 1k (120 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 305,1 [М+1]
Стадия 8
2-(Дифторметил)-N-(1-этил-2-(4-(трифторметил)фенил)-1H-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамид 1-Этил-5-амино-2-(4-(трифторметил)фенил)-1H-индол 1k (120 мг, 0,39 ммоль) растворяли в 10 мл тетрагидрофурана и добавляли триэтиламин (0,10 мл, 0,78 ммоль), и по каплям 5 мл раствора 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиноилхлорида 1g (160 мг, 0,55 ммоль) в тетрагидрофуране. Реакционную смесь перемешивали в течение 1 часа. Реакционную смесь фильтровали, и концентрировали фильтрат при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-N-(1-этил-2-(4-(трифторметил)фенил)-1H-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамида 1 (25 мг, 11,5%) в виде желтого твердого вещества.
МС m/z (ESI): 559,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,61 (s, 1Н), 8,68 (s, 1Н), 8,46 (t, 1Н), 8,07 (s, 1Н), 8,01 (s, 1Н), 7,93-7,86 (d, 2Н), 7,85-7,78 (d, 2Н), 7,61-7,55 (d, 1Н), 7,47-7,42 (d, 1Н), 7,19 (t, 1Н), 6,70 (s, 1Н), 4,47-4,39 (d, 2Н), 4,31-4,20 (m, 2Н), 2,49-2,41 (m, 1Н), 1,21 (t, 3Н), 1,09-1,03 (d, 6Н).
Пример 2
2-хлор-N-(1-этил-2-(4-(трифторметил)фенил)-7Н-индол-5-ил)5-((2,2-диметилпропаноиламино)метил)-бензамид
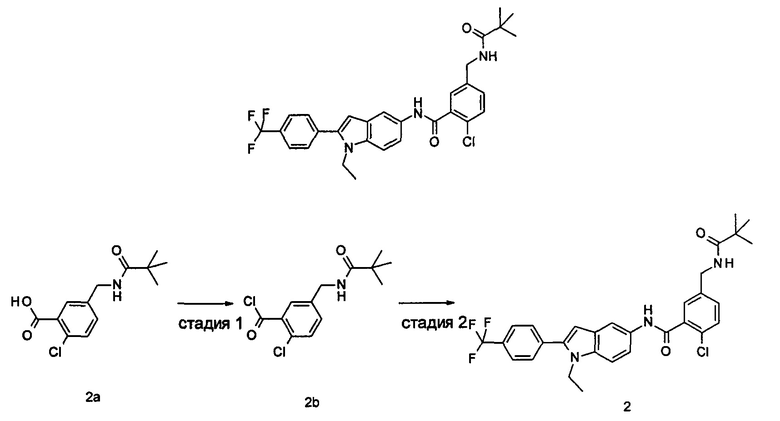
Стадия 1
2-Хлор-5-((2,2-диметилпропаноиламино)метил)бензоилхлорид
2-хлор-5-((2,2-диметилпропаноиламино)метил)бензойную кислоту 2а (500 мг, 1,86 ммоль, полученную в соответствии со способом, раскрытым в патентной заявке WO 2012025469), растворяли в 10 мл дихлорметана, и по каплям добавляли тионилхлорид (0,4 мл, 5,58 ммоль) и одну каплю N,N-диметилформамида. По завершении добавления реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении с получением целевого соединения 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензоилхлорида 2b (550 мг) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 2
2-Хлор-N-(1-этил-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)-5-((2,2-диметилпропаноиламино)метил)-бензамид
1-Этил-5-амино-2-(4-(трифторметил)фенил)-1Н-индол 1k (90 мг, 0,27 ммоль) растворяли в 5 мл тетрагидрофурана и по каплям добавляли триэтиламин (75 мкл, 0,54 ммоль) и 2 мл раствора 2-хлор-5-((2,2-диметилпропаноиламино)метил) бензоилхлорида 2b (77 мг, 0,27 ммоль) в тетрагидрофуране. По завершению добавления реакционную смесь перемешивали в течение 1 часа. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(1-этил-2-(4-(трифторметил)фенил)-7H-индол-5-ил)-5-((2,2-диметилпропаноиламино)метил)-бензамида 2 (45 мг, 30%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 557,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,40 (s, 1Н), 8,18 (t, 1Н), 8,10 (s, 1Н), 7,92-7,87 (d, 2Н), 7,84-7,79 (d, 2Н), 7,58-7,53 (d, 1Н), 7,52-7,48 (d, 1Н), 7,47-7,41 (m, 2Н), 7,36-7,30 (d, 1Н), 6,69 (s, 1Н), 4,34-4,29 (d, 2Н), 4,29-4,21 (m, 2Н), 1,21 (t, 3Н), 1,13 (s, 9Н).
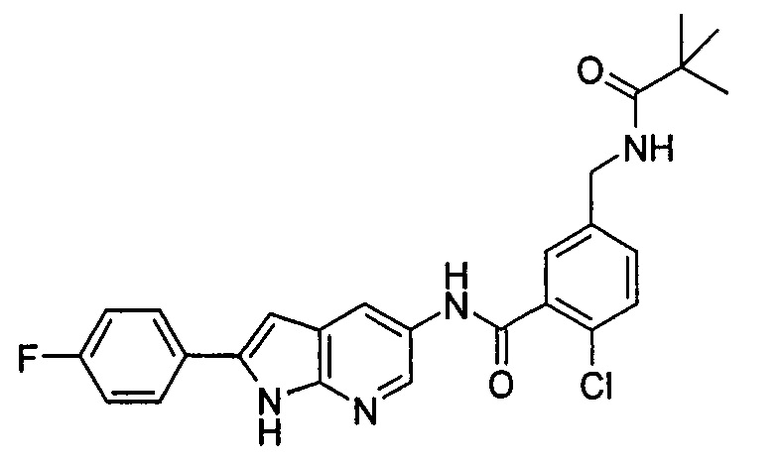
Пример 3
2-хлор-N-(2-(4-фторфенил)-1H-пирролл(2,3-b)пиридин-5-ил)-5-((2,2-диметилпропаноиламино)метил)-бензамид
Стадия 1
3-((4-Фторфенил)этинил)-5-нитро-пиридин-2-амин
2-Амино-3-бром-5-нитро-пиридин 3b (1,0 г, 4,6 ммоль), 1-этинил-4-фтор-бензол 3а (1,24 г, 10,3 ммоль), хлорид бис(трифенилфосфин)палладия(II) (0,25 г, 0,35 ммоль), йодид меди (7 мг, 0,35 ммоль) и триэтиламин (0,7 мл, 4,6 ммоль) добавляли к 20 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 16 часов в атмосфере аргона. Реакционную смесь фильтровали через Celatom, и концентрировали фильтрат при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением неочищенного целевого соединения 3-((4-фторфенил)этинил)-5-нитро-пиридин-2-амина 3с (1,7 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 256,0 [М-1]
Стадия 2
2-(4-Фторфенил)-5-нитро-1H-пирроло[2,3-b]пиридин
3-((4-Фторфенил)этинил)-5-нитро-пиридин-2-амин 3с (1,7 г, 4,6 ммоль) и трет-бутоксид калия (1,0 г, 9,2 ммоль) растворяли в 20 мл N,N-диметилформамида. Реакционную смесь нагревали до 70°С и перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 200 мл воды и затем фильтровали через Celatom. Фильтровальный осадок очищали колоночной хроматогафией на силикагеле с элюирующей системой С, затем добавляли 20 мл дихлорметана и затем фильтровали. Остаток сушили с получением неочищенного целевого соединения 2-(4-фторфенил)-5-нитро-1H-пирроло[2,3-b]пиридина 3d (564 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 3
2-(4-фторфенил)-5-амино-1H-пирроло[2,3-b]пиридин
2-(4-Фторфенил)-5-нитро-1Н-пирроло[2,3-b]пиридин 3d (64 мг, 0,25 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (30 мг). Реакционную смесь перемешивали в течение 1 часа в атмосфере водорода и затем фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(4-фторфенил)-5-амино-1H-пирроло[2,3-b]пиридина 3е (60 мг) в виде коричневого масла, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 226,1 [М-1]
Стадия 4
2-Хлор-N-(2-(4-фторфенил)-1H-пирроло[2,3-b]пиридин-5-ил)
5-((2,2-диметилпропаноиламино)метил)-бензамид
2-(4-Фторфенил)-5-амино-1H-пирроло[2,3-b]пиридин 3е (60 мг, 0,25 ммоль) растворяли в 8 мл тетрагидрофурана и по каплям добавляли триэтиламин (0,43 мл, 0,31 ммоль) и 5 мл раствора 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензоилхлорида 2b (220 мг, 0,76 ммоль) в тетрагидрофуране. Реакционную смесь перемешивали в течение 1 часа и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(2-(4-фторфенил)-1Н-пирроло[2,3-b]пиридин-5-ил)5-((2,2-диметилпропаноиламино)метил)-бензамида 3 (5 мг, 4,2% для двух стадий) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 479,4 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 9,16 (s, 1Н), 8,06 (s, 1Н), 7,94 (s, 1Н), 7,65-7,56 (m, 1Н), 7,55-7,46 (m, 2Н), 7,48-7,36 (m, 2Н), 7,27-7,16 (m, 3Н), 6,41 (s, 1Н), 5,15 (m, 1Н), 4,26-4,21 (m, 2H), 1,13 (s, 9Н).
Пример 4
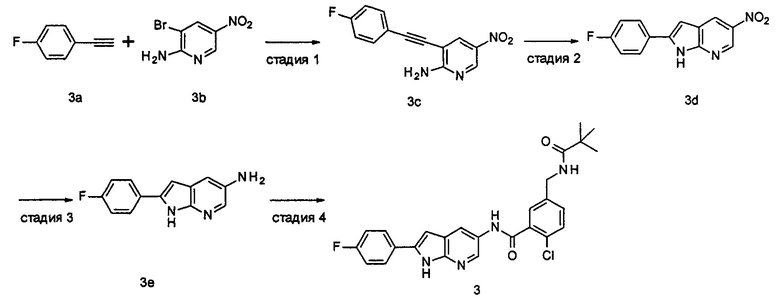
2-Хлор-N-(1-этил-2-(4-фторфенил)-1H-пирроло[2,3-b]пиридин-5-ил)
5-((2,2-диметилпропаноиламино)метил)-бензамид
Стадия 1
1-Этил-2-(4-фторфенил)-5-нитро-1Н-пирроло[2,3-b]пиридин
2-(4-Фторфенил)-5-нитро-1Н-пирроло[2,3-b]пиридин 3d (60 мг, 0,23 ммоль), йодэтан (40 мкл, 0,47 ммоль) и карбонат цезия (150 мг, 0,47 ммоль) добавляли к 5 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 16 часов, затем добавляли 20 мл воды, экстрагировали этилацетатом (20 мл×4). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×2), сушили над безводным сульфатом натрия, фильтровали, и концентрировали фильтрат при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением неочищенного целевого соединения 1-этил-2-(4-фторфенил)-5-нитро-1H-пирроло[2,3-b]пиридина 4а (70 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 286,1 [М+1]
Стадия 2
1-Этил-2-(4-фторфенил)-5-амино-1H-пирроло[2,3-b]пиридин
1-Этил-2-(4-фторфенил)-5-нитро-1Н-пирроло[2,3-b]пиридин 4а (70 мг, 0,23 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (20 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода, и затем фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 1-этил-2-(4-фторфенил)-5-амино-1Н-пирроло[2,3-b]пиридина 4b (60 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 256,2 [М+1]
Стадия 3
2-Хлор-N-(1-этил-2-(4-фторфенил)-1Н-пирроло[2,3-b]пиридин-5-ил)-5-((2,2-диметилпропаноиламино)метил)-бензамид
1-Этил-2-(4-фторфенил)-5-амино-1Н-пирроло[2,3-b]пиридин 4b (60 мг, 0,23 ммоль) растворяли в 5 мл тетрагидрофурана и по каплям добавляли триэтиламин (65 мкл, 0,47 ммоль) и 5 мл раствора 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензоилхлорида 2b (220 мг, 0,76 ммоль) в тетрагидрофуране. Реакционную смесь перемешивали в течение 1 часа и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(1-этил-2-(4-фторфенил)-1H-пирроло[2,3-b]пиридин-5-ил)-5-((2,2-диметилпропаноиламино)метил)-бензамида 4 (5 мг, 4,2% для трех стадий) в виде желтого твердого вещества.
MC m/z (ESI): 507,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 9,15 (s, 1Н), 8,05 (s, 1Н), 7,93 (s, 1Н), 7,66-7,56 (m, 1Н), 7,55-7,48 (m, 2Н), 7,48-7,38 (m, 2Н), 7,27-7,17 (m, 3Н), 6,56 (s, 1Н), 4,34-4,28 (d, 2Н), 4,29-4,20 (m, 2Н), 1,23 (t, 3Н), 1,12 (s, 9Н).
Пример 5
2-(Дифторметил)-N-(1-этил-2-(4-фторфенил)-1Н-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамид
Стадия 1
1-Этил-2-(4-фторфенил)-5-нитро-1H-индол 5b
1-Этил-3-(4-фторфенил)-5-нитро-1Н-индол 5с
1-Этил-5-нитро-1Н-индол 1h (1,64 г, 6,17 ммоль) растворяли в 20 мл N,N-диметилацетамида и затем последовательно добавляли 1-фтор-4-йод-бензол 5а (4,65 г, 21,0 ммоль), трифенилфосфин (360 мг, 1,36 ммоль), ацетат палладия (70 мг, 0,31 ммоль) и ацетат цезия (4,0 г, 12,3 ммоль). Реакционную смесь нагревали до 140°С и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевых соединений 1-этил-2-(4-фторфенил)-5-нитро-1Н-индола 5b (0,51 г, 29,1%) в виде желтого твердого вещества и 1-этил-3-(4-фторфенил)-5-нитро-1H-индола 5с (125 мг, 7,1%) в виде желтого твердого вещества.
5b: МС m/z (ESI): 285,0 [М+1]
5с: МС m/z (ESI): 285,1 [М+1]
Стадия 2
1-Этил-2-(4-фторфенил)-5-амино-1Н-индол
1-Этил-2-(4-фторфенил)-5-нитро-1Н-индол 5b (35 мг, 0,12 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (10 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода и затем фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 1-этил-2-(4-фторфенил)-5-амино-1Н-индола 5d (31 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 255,2 [М+1]
Стадия 3
2-(Дифторметил)-N-(1-этил-2-(4-фторфенил)-1Н-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамид
1-Этил-2-(4-фторфенил)-5-амино-1Н-индол 5d (31 мг, 0,12 ммоль) растворяли в 5 мл ацетонитрила, и затем по каплям добавляли триэтиламин (31 мкл, 0,22 ммоль) и 5 мл раствора 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиноилхлорида 1g (32 мг, 0,11 ммоль) в ацетонитриле. Реакционную смесь перемешивали в течение 1 часа и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-N-(1-этил-2-(4-фторфенил)-1H-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамида 5 (35 мг, 62,5%) в виде желтого твердого вещества.
МС m/z (ESI): 509,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 8,70 (s, 1Н), 8,07 (s, 1Н), 7,94 (s, 1Н), 7,63-7,54 (m, 2Н), 7,46-7,51 (d, 2Н), 7,44-7,39 (d, 2Н), 7,32-7,25 (m, 2Н), 7,12 (t, 1Н), 6,51 (s, 1Н), 5,36 (t, 1Н), 4,53 (s, 2Н), 4,30-4,21 (m, 2Н), 1,27 (t, 3Н), 1,20-1,14 (d, 6Н).
Пример 6
2-Бром-N-(1-этил-3-(4-фторфенил)-1Н-индол-5-ил)5-((2,2-диметилпропаноиламино)метил)-бензамид
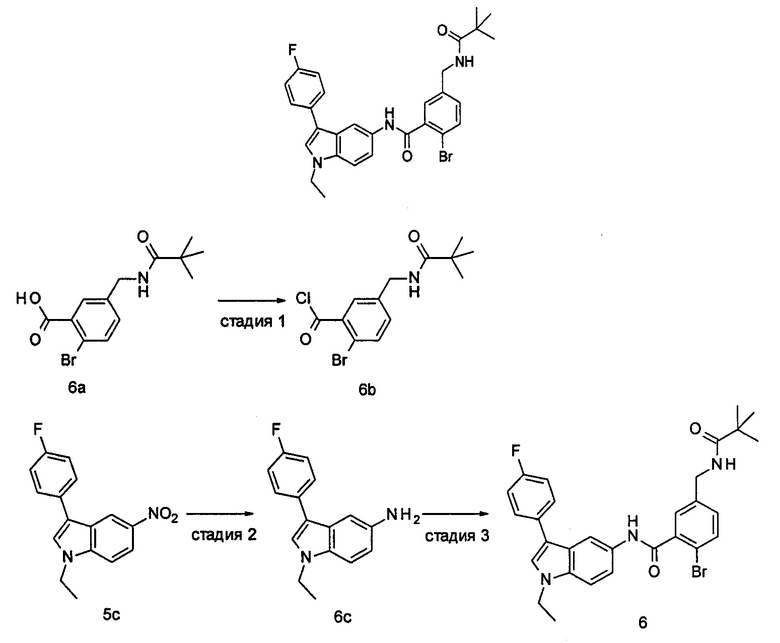
Стадия 1
2-Бром-5-((2,2-диметилпропаноиламино)метил)бензоилхлорид
2-Бром-5-((2,2-диметилпропаноиламино)метил)бензойную кислоту 6а (500 мг, 1,59 ммоль, полученную в соответствии со способом, раскрытым в патентной заявке "US 20120157506") растворяли в 10 мл дихлорметана, и затем добавляли тионилхлорид (0,4 мл, 4,78 ммоль) и одну каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 2 часов и затем концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-бром-5-((2,2-диметилпропаноиламино)метил)бензоилхлорида 6b (530 мг) в виде светло-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 331,1 [М-1]
Стадия 2
1-Этил-3-(4-фторфенил)-5-амино-1Н-индол
1-Этил-3-(4-фторфенил)-5-нитро-1Н-индол 5с (110 мг, 0,39 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и добавляли никель Ренея (20 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода и затем фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 1-этил-3-(4-фторфенил)-5-амино-1Н-индола 6с (100 мг) в виде серого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 255,2 [М+1]
Стадия 3
2-Бром-N-(1-этил-3-(4-фторфенил)-1Н-индол-5-ил)-5-((2,2-диметилпропаноиламино)метил)-бензамид
1-Этил-3-(4-фторфенил)-5-амино-1Н-индол 6с (100 мг, 0,39 ммоль) растворяли в 5 мл тетрагидрофурана, и по каплям добавляли триэтиламин (63 мкл, 0,45 ммоль) и 5 мл раствора 2-бром-5-((2,2-диметилпропаноиламино)метил)бензоилхлорида 6b (50 мг, 0,15 ммоль) в тетрагидрофуране. Реакционную смесь перемешивали в течение 16 часов и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-бром-N-(1-этил-3-(4-фторфенил)-1H-индол-5-ил)-5-((2,2-диметилпропаноиламино)метил)-бензамида 6 (15 мг, 18,2%) в виде желтого твердого вещества.
МС m/z (ESI): 551,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 8,17 (s, 1Н), 7,99 (s, 1Н), 7,66-7,56 (m, 4Н), 7,55-7,50 (m, 3Н), 7,43-7,37 (m, 1Н), 7,25-7,19 (m, 1Н), 7,15 (t, 1Н), 6,28 (s, 1Н), 4,46-4,36 (d, 2Н), 4,30-4,20 (m, 2Н), 1,55 (t, 3Н), 1,25 (s, 9Н).
Пример 7
2-Бром-N-(1-этил-2-(4-фторфенил)-1H-индол-5-ил)5-((2,2-диметилпропаноиламино)метил)-бензамид
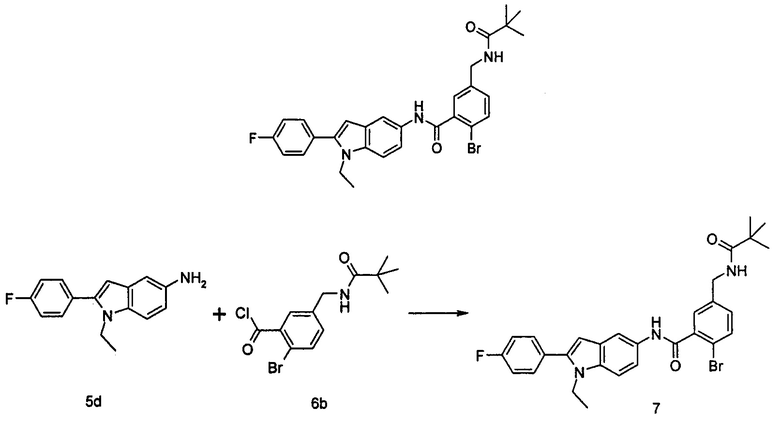
1-Этил-2-(4-фторфенил)-5-амино-1H-индол 5d (30 мг, 0,12 ммоль) растворяли в 5 мл тетрагидрофурана, и затем по каплям добавляли триэтиламин (63 мкл, 0,45 ммоль) и 5 мл раствора 2-бром-5-((2,2-диметилпропаноиламино)метил)бензоилхлорида 6b (50 мг, 0,15 ммоль) в тетрагидрофуране. Реакционную смесь перемешивали в течение 16 часов и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-бром-N-(1-этил-2-(4-фторфенил)-1Н-индол-5-ил) 5-((2,2-диметилпропаноиламино)метил)-бензамида 7 (5 мг, 10,4%) в виде желтого твердого вещества.
МС m/z (ESI): 551,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 8,00 (s, 1Н), 7,90 (s, 1Н), 7,65-7,56 (m, 2Н), 7,54-7,48 (m, 2Н), 7,47-7,38 (m, 2Н), 7,28-7,17 (m, 3Н), 6,52 (s, 1Н), 6,22 (s, 1Н), 4,50-4,40 (d, 2Н), 4,25-4,16 (m, 2Н), 1,33 (t, 3Н), 1,27 (s, 9Н).
Пример 8
2-Хлор-N-(1-этил-2-(4-фторфенил)-1Н-индол-5-ил)5-((2,2-диметилпропаноиламино)метил)-бензамид
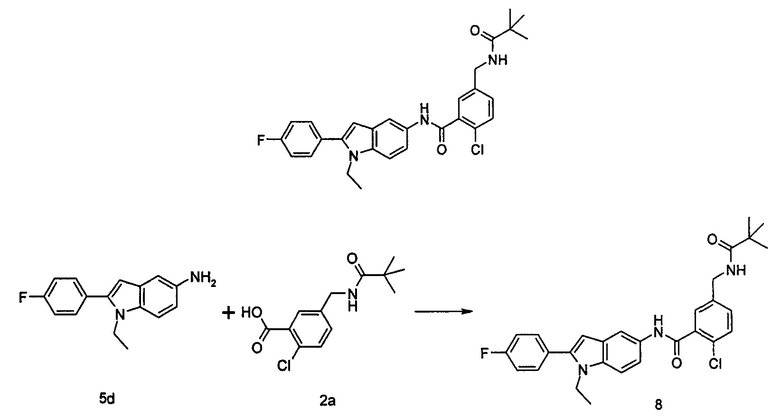
1-Этил-2-(4-фторфенил)-5-амино-1Н-индол 5d (38 мг, 0,15 ммоль), 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензойную кислоту 2а (40 мг, 0,15 ммоль), 1-этил-(3-диметиламинопропил)-карбодиимида гидрохлорид (57 мг, 0,3 ммоль), 1-гидроксибензотриазол (2 мг, 0,015 ммоль) и N,N-диизопропилэтиламин (38 мг, 0,3 ммоль) растворяли в 5 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 16 часов и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(1-этил-2-(4-фторфенил)-1H-индол-5-ил) 5-((2,2-диметилпропаноиламино)метил)-бензамида 8 (5 мг, 6,7%) в виде желтого твердого вещества.
МС m/z (ESI): 506,0 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,64 (s, 1Н), 8,12 (s, 1Н), 8,09 (s, 1Н), 7,95-7,90 (d, 2Н), 7,82-7,77 (d, 2Н), 7,57-7,47 (d, 2Н), 7,48-7,45 (m, 2Н), 7,30-7,22 (m, 1Н), 6,72 (s, 1Н), 4,40-4,36 (d, 2Н), 4,33-4,29 (m, 2Н), 1,21 (t, 3Н), 1,12 (s, 9Н)
Пример 9
2-(Дифторметил)-N-(1-метил-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)
5-((2-метилпропаноиламино)метил)-никотинамид
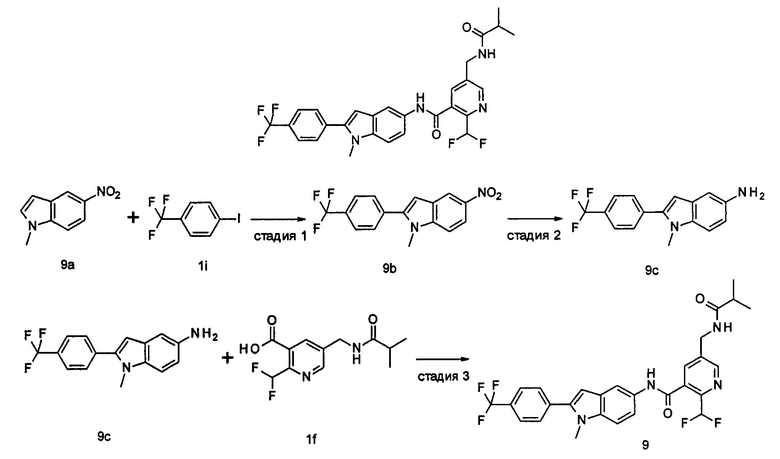
Стадия 1
1-Метил-5-нитро-2-(4-(трифторметил)фенил)-1Н-индол 9b
1-Метил-5-нитро-1Н-индол 9а (1,0 г, 5,7 ммоль) растворяли в 40 мл N,N-диметилацетамида, и последовательно добавляли 4-йодтрифторбензол 1i (1,7 г, 6,2 ммоль), трифенилфосфин (300 мг, 1,4 ммоль), ацетат палладия (130 мг, 0,57 ммоль) и ацетат цезия (2,2 г, 1,4 ммоль). Реакционную смесь нагревали до 140°С, и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли 50 мл этилацетата, промывали водой (20 мл×2), сушили над безводным сульфатом натрия, и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюирующей системой В с получением целевого соединения 1-метил-5-нитро-2-(4-(трифторметил)фенил)-1H-индола 9b (350 мг, 19,4%) в виде желтого твердого вещества.
МС m/z (ESI): 321,1 [М+1]
Стадия 2
1-Метил-5-амино-2-(4-(трифторметил)фенил)-1Н-индол
1-Метил-5-нитро-2-(4-(трифторметил)фенил)-1H-индол 9b (350 мг, 1,1 ммоль) растворяли в 10 мл тетрагидрофурана, и затем добавляли никель Ренея (35 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 1-метил-5-амино-2-(4-(трифторметил)фенил)-1Н-индола 9с (300 мг) в виде коричневого масла, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 291,2 [М+1]
Стадия 3
2-(Дифторметил)-N-(1-метил-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)
5-((2-метилпропаноиламино)метил)-никотинамид
1-Метил-5-амино-2-(4-(трифтрметил)фенил)-1Н-индол 9с (150 мг, 0,52 ммоль), 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (141 мг, 0,52 ммоль), O-бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний (250 мг, 0,78 ммоль) и N,N-диизопропилэтиламин (100 мг, 0,78 ммоль) растворяли в 5 мл N,N-диметилформамида. Реакционную смесь нагревали до 75°С и затем перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-N-(1-метил-2-(4-(трифторметил)фенил)-1H-индол-5-ил) 5-((2-метилпропаноиламино)метил)-никотинамида 9 (10 мг, 2,0%) в виде коричневого твердого вещества.
МС m/z (ESI): 545,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,64 (s, 1Н), 8,68 (s, 1Н), 8,50-8,47 (m, 1Н), 8,09 (s, 1Н), 8,03 (s, 1Н), 7,90-7,85 (m, 4Н), 7,56-7,54 (m, 1Н), 7,48-7,45 (m, 1Н), 7,28-7,20 (m, 1Н), 6,75 (s, 1Н), 4,44-4,42 (d, 2Н), 3,80 (s, 3Н), 2,47-2,46 (m, 1Н), 1,07-1,05 (d, 6Н)
Пример 10
2-(Дифторметил)-N-(1-этил-2-(4-хлорфенил)-1Н-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамид
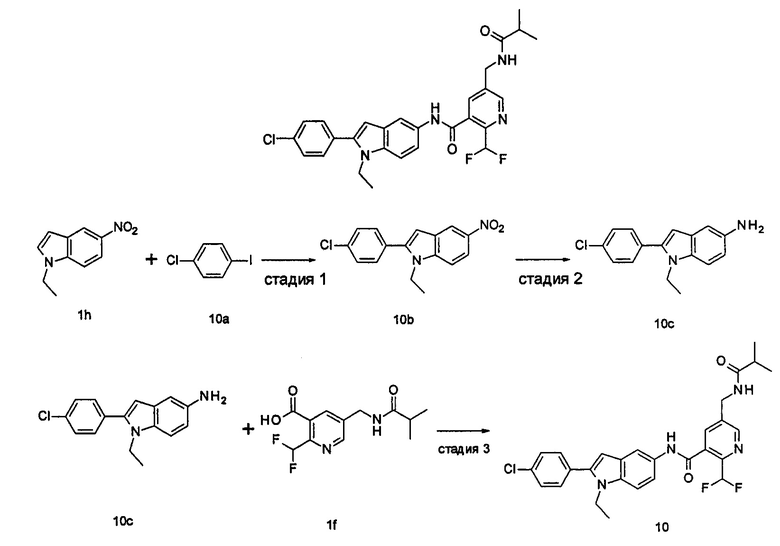
Стадия 1
1-Этил-2-(4-хлорфенил)-5-нитро-1Н-индол
1-Этил-5-нитро-1Н-индол 1h (439 мг, 2,3 ммоль) растворяли в 5 мл N,N-диметилацетамида, и последовательно добавляли 1-хлор-4-йод-бензол 10а (500 мг, 2,1 ммоль), трифенилфосфин (110 мг, 0,42 ммоль), ацетат палладия (24 мг, 0,11 ммоль), и ацетат цезия (806 мг, 4,2 ммоль). Реакционную смесь нагревали до 140°С и затем перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь фильтровали. К фильтрату добавляли 150 мл этилацетата и затем промывали водой (20 мл×1). Органическую фазу сушили над безводным сульфатом натрия, фильтровали, и концентрировали фильтрат при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением целевого соединения 1-этил-2-(4-хлорфенил)-5-нитро-1Н-индола 10b (80 мг, 12,7%) в виде желтого твердого вещества.
Стадия 2
1-Этил-2-(4-хлорфенил)-5-амино-1Н-индол
1-Этил-2-(4-хлорфенил)-5-нитро-1Н-индол 10b (80 мг, 0,27 ммоль) растворяли в 20 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (10 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода и затем фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 1-этил-2-(4-хлорфенил)-5-амино-1Н-индола 10 с (72 мг) в виде серого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 271,1 [М+1]
Стадия 3
2-(Дифторметил)-N-(1-этил-2-(4-хлорфенил)-1Н-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамид
1-Этил-2-(4-хлорфенил)-5-амино-1Н-индол 10с (50 мг, 0,18 ммоль), 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (55 мг, 0,18 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (70 мг, 0,37 ммоль) и 1-гидроксибензотриазол (25 мг, 0,18 ммоль) последовательно растворяли в 3 мл N,N-диметилформамида. Реакционную смесь нагревали до 40°С, и затем перемешивали в течение 5 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-N-(1-этил-2-(4-хлорфенил)-1Н-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамида 10 (20 мг, 27,8%) в виде желтого твердого вещества.
МС m/z (ESI): 525,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,57 (s, 1Н), 8,65 (s, 1Н), 8,44 (t, 1Н), 8,02 (s, 1Н), 7,89 (s, 1Н), 7,59 (s, 2Н), 7,52 (d, 1Н), 7,41 (d, 1Н), 7,39 (s, 1Н), 6,57 (s, 1Н), 4,42 (d, 2Н), 4,21 (d, 2Н), 3,89 (s, 1Н), 1,25 (d, 3Н), 1,22 (t, 2Н), 1,03 (d, 6Н).
Пример 11
2-Хлор-N-(2-(4-фторфенил)-1-метил-1Н-индол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
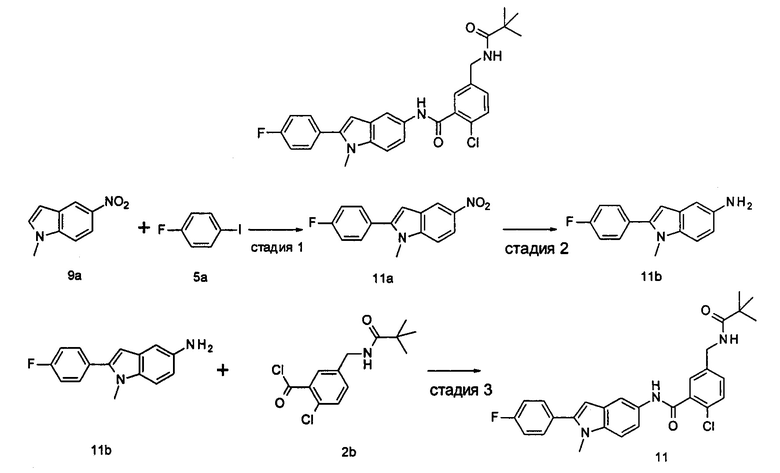
Стадия 1
2-(4-Фторфенил)-1-метил-5-нитро-1Н-индол
1-Метил-5-нитро-1Н-индол 9а (0,8 г, 4.5 ммоль) растворяли в 10 мл N,N-диметилацетамида, и затем последовательно добавляли 1-фтор-4-йод-бензол 5а (1,12 г, 5,0 ммоль), трифенилфосфин (238 мг, 0,9 ммоль), ацетат палладия (51 мг, 0,23 ммоль), и ацетат цезия (1,7 г, 0,91 ммоль). Реакционную смесь нагревали до 140°С, и затем перемешивали в течение 18 часов в атмосфере аргона. Реакционный раствор охлаждали до комнатной температуры и затем добавляли 50 мл воды, и экстрагировали этилацетатом (50 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали, и концентрировали фильтрат при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением целевого соединения 2-(4-фторфенил)-1-метил-5-нитро-1Н-индола 11а (130 мг, 10,7%) в виде желтого твердого вещества.
МС m/z (ESI): 271,1 [М+1]
Стадия 2
2-(4-Фторфенил)-1-метил-5-амино-1Н-индол
2-(4-Фторфенил)-1-метил-5-нитро-1Н-индол 11а (130 мг, 0,48 ммоль) растворяли в 16 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (15 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода и затем фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(4-фторфенил)-1-метил-5-амино-1Н-индола 11b (130 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 3
2-Хлор-N-(2-(4-фторфенил)-1-метил-1Н-индол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
2-(4-Фторфенил)-1-метил-5-амино-1Н-индол 11b (70 мг, 0,29 ммоль) растворяли в 20 мл тетрагидрофурана, и затем по каплям добавляли триэтиламин (85 мкл, 0,60 ммоль) и 5 мл раствора 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензоилхлорида 2b (83 мг, 0,29 ммоль) в тетрагидрофуране. Реакционную смесь перемешивали в течение 2 часов и затем фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(2-(4-фторфенил)-1-метил-1Н-индол-5-ил)-5-((2,2-диметилпропаноилами но)метил)бензамида 11 (40 мг, 28,0%) в виде желтого твердого вещества.
МС m/z (ESI): 492,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 8,01 (s, 1Н), 7,91 (s, 1Н), 7,65-7,56 (m, 2Н), 7,54-7,49 (m, 2Н), 7,48-7,38 (m, 2Н), 7,28-7,17 (m, 3Н), 6,52 (s, 1Н), 6,22 (s, 1Н), 4,25-4,16 (m, 2Н), 3,83 (t, 3Н), 1,27 (s, 9Н).
Пример 12
2-(Дифторметил)-N-(1-этил-1H-индол-5-ил)-5-((2-метилпропаноиламино)метил)никотинамид
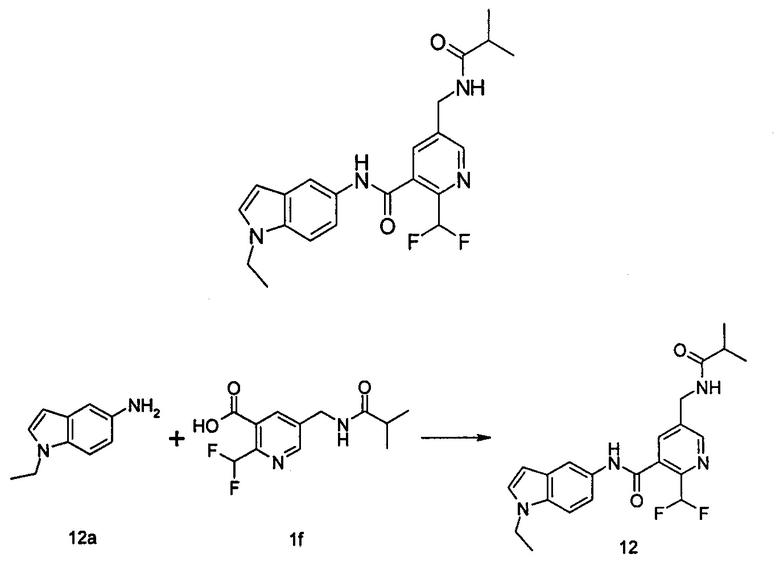
1-Этил-1Н-индол-5-амин 12а (50 мг, 0,31 ммоль, полученный в соответствии с "Bioorganic & Medicinal Chemistry, 2005, 13(10), 3531-3541"), 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (70 мг, 0,26 ммоль), O-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (124 мг, 0,39 ммоль), и N,N-диизопропилэтиламин (100 мг, 0,78 ммоль) последовательно растворяли в 5 мл N,N-диметилформамида. Реакционную смесь нагревали до 75°С и затем перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-N-(1-этил-1H-индол-5-ил)-5-((2-метилпропаноиламино)метил)ник отинамида 12 (20 мг, 18,9%) в виде коричневого твердого вещества.
МС m/z (ESI): 415,1 [(М+1])
1Н ЯМР (400 МГц, DMSO-d6): δ 10,53 (s, 1Н), 8,67 (s, 1Н), 8,46 (t, 1Н), 7,99 (s, 2Н), 7,50-7,44 (d, 1Н), 7,42-7,34 (m, 2Н), 7,18 (t, 1Н), 6,46-6,40 (d, 1Н), 4,47-4,39 (d, 2Н), 4,25-4,15 (m, 2Н), 2,49-2,41 (m, 1Н), 1,36 (t, 3Н), 1,10-1,01 (d, 6Н).
Пример 13
N-(2-(4-Хлорпентил)-1-метил-1H-индол-5-ил)-2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинамид
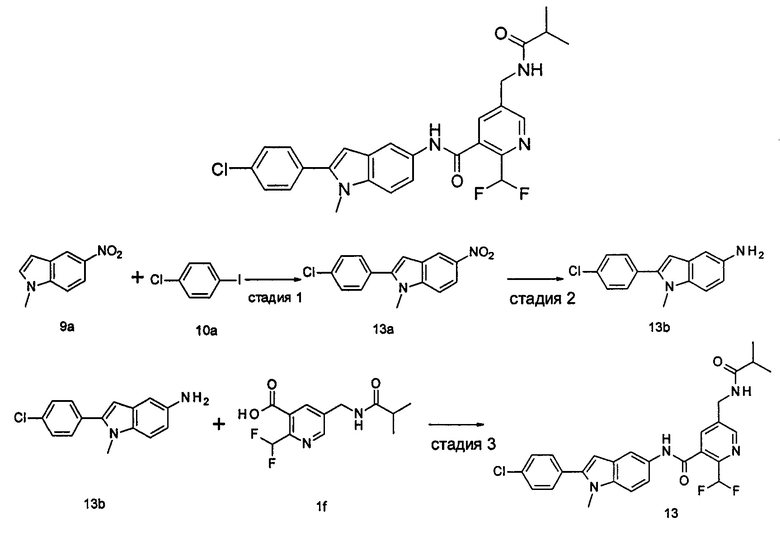
Стадия 1
2-(4-Хлорфенил)-1-метил-5-нитро-1Н-индол
1-Метил-5-нитро-1Н-индол 9а (3,30 г, 18,7 ммоль) растворяли в 20 мл N,N-диметилацетамида и затем последовательно добавляли 1-хлор-4-йод-бензол 10а (4,96 г, 20,8 ммоль), трифенилфосфин (982 мг, 3,75 ммоль), ацетат палладия (210 мг, 0,94 ммоль), и ацетат цезия (7,20 г, 3,75 ммоль). Реакционную смесь нагревали до 140°С, и затем перемешивали в течение 18 часов в атмосфере аргона. Реакционный раствор охлаждали до комнатной температуры и затем добавляли 50 мл воды, экстрагировали этилацетатом (50 мл). Органическую фазу промывали насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением целевого соединения 2-(4-хлорфенил)-1-метил-5-нитро-1Н-индола 13а (270 мг, 5,0%) в виде желтого твердого вещества.
Стадия 2
2-(4-Хлорфенил)-1-метил-5-амино-1Н-индол
2-(4-Хлорфенил)-1-метил-5-нитро-1Н-индол 13а (80 мг, 0,28 ммоль) растворяли в 20 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добаляли никель Ренея (8 мг). Реакционную смесь перемешивали в течение 1 часа в атмосфере водорода. Реакционную смесь фильтровали через Celatom и концентрировали фильтрат при пониженном давлении с получением неочищенного целевого соединения 2-(4-хлорфенил)-1-метил-5-амино-1Н-индола 13b (72 мг) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 3
N-(2-(4-Хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинамид
2-(4-Хлорфенил)-1-метил-5-амино-1Н-индол 13b (72 мг, 0,28 ммоль), 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (77 мг, 0,28 ммоль), 1-этил-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (81 мг, 0,42 ммоль) и 1-гидроксибензотриазол (38 мг, 0,28 ммоль) последовательно растворяли в 5 мл N,N-диметилформамида. Реакционную смесь нагревали до 40°С и затем перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой Ас получением твердого вещества. К твердому веществу добавляли 20 мл дихлорметана и перемешивали в течение 20 минут с получением целевого соединения N-(2-(4-хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинамида 13 (20 мг, 14,0%) в виде желтого твердого вещества.
МС m/z (ESI): 511,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,59 (s, 1Н), 8,68 (s, 1Н), 8,45 (s, 1Н), 8,04 (d, 2Н), 7,62 (dd, 4Н), 7,47 (dd, 2Н), 7,20 (t, 1Н), 6,64 (s, 1Н), 4,43 (d, 2Н), 3,76 (s, 3Н), 2,46 (m, 1Н), 1,06 (d, 6H).
Пример 14
N-(2-(4-Хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
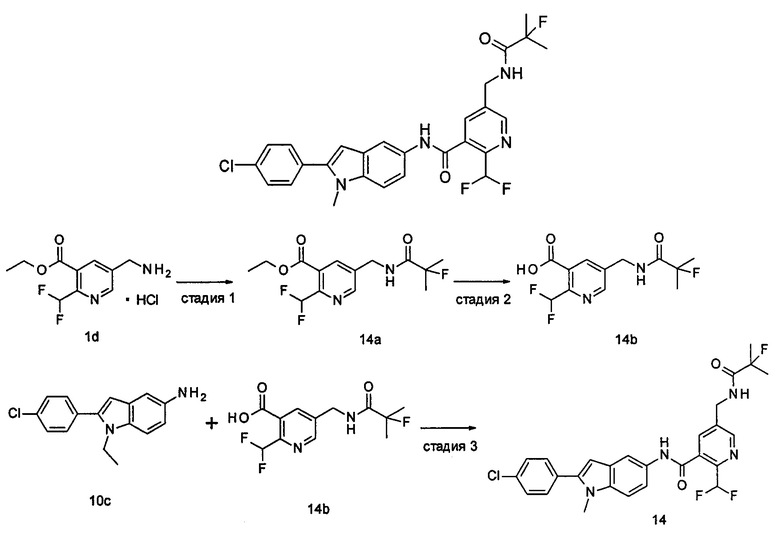
Стадия 1
Этил 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинат Этил 5-(аминометил)-2-(дифторметил)никотината гидрохлорид 1d (2,18 г, 8,20 ммоль) растворяли в 20 мл N,N-диметилформамида, и затем последовательно добавляли 2-фторизомасляную кислоту (1,04 г, 9,83 ммоль), 1-(3-диметиламинопропил)-3-этилакарбодиимида гидрохлорид (2,36 г, 12,30 ммоль), 1-гидроксибензотриазол (1,66 г, 12,30 ммоль) и триэтиламин (7 мл, 49,2 ммоль). Реакционную смесь перемешивали в течение 16 часов и затем добавляли 100 мл этилацетата и 50 мл воды. Органическую фазу концентрировали при пониженном давлении. Остаток очищали посредством ТСХ с элюирующей системой А с получением целевого соединения этил 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотината 14а (1,6 г, 61,3%) в виде желтого твердого вещества.
МС m/z (ESI): 319,1 [М+1]
Стадия 2
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновая кислота
Этил 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил) никотинат 14а (1,6 г, 5,03 ммоль) растворяли в 50 мл 1,4-диоксана, и затем добавляли 25 мл воды и гидрат гидроксида лития (529 мг, 12,6 ммоль). Реакционную смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли 5 мл воды, и затем доводили до рН 3 5М раствором хлороводородной кислоты. Большое количество твердого вещества осадили из реакционного раствора и отфильтровали. Фильтрат экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток объединяли с вышеуказанным фильтровальным осадком, и затем промывали водой с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновой кислоты 14b (500 мг, 34,2%) в виде белого твердого вещества.
МС m/z (ESI): 291,1 [М+1]
Стадия 3
N-(2-(4-Хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
1-Этил-2-(4-хлорфенил)-5-амино-1Н-индол 10с (100 мг, 0,37 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (107 мг, 0,37 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (142 мг, 0,74 ммоль) и 1-гидроксибензотриазол (191 мг, 1,48 ммоль) последовательно растворяли в 5 мл N,N-диметилформамида. Реакционную смесь нагревали до 70°С и затем перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения N-(2-(4-хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 14 (40 мг, 19,9%) в виде белого твердого вещества.
МС m/z (ESI): 543,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,60 (s, 1Н), 8,87 (s, 1Н), 8,69 (m, 1Н), 8,04-8,02 (m, 2Н), 7,60-7,58 (m, 4Н), 7,54-7,53 (m, 1Н), 7,43-7,42 (m, 1Н), 7,19 (t, 1Н), 6,59 (s, 1Н), 4,48-4,46 (m, 2H), 4,24-4,19 (m, 2H), 1,54-1,48 (d, 6H), 1,26-1,15 (m, 3Н).
Пример 15
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-метил-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамид
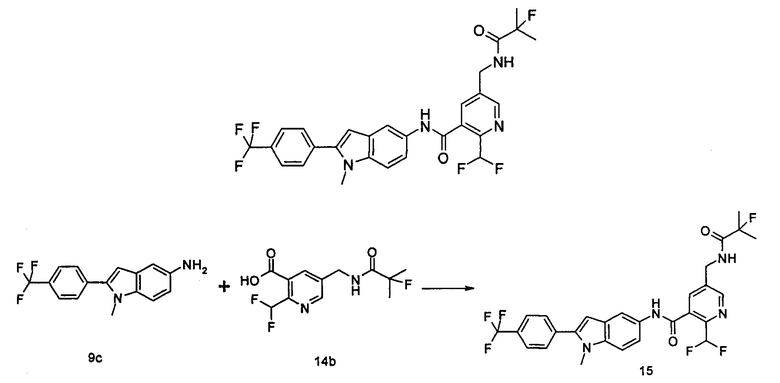
1-Метил-5-амино-2-(4-(трифторметил)фенил)-1Н-индол 9с (50 мг, 0,17 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (50 мг, 0,17 ммоль), 1-этил-(3-диметиламинопропил)-3-карбодиимида гидрохлорид (55 мг, 0,29 ммоль), 1-гидроксибензотриазол (2,3 мг, 0,017 ммоль) и N,N-диизопропилэтиламин (44 мг, 0,34 ммоль) последовательно растворяли в 5 мл N,N-диметилформамида. Реакционную смесь нагревали до 70°С, и затем перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-метил-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамида 15 (4 мг, 4,2%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 563,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,62 (s, 1Н), 8,87 (s, 1Н), 8,69 (s, 1Н), 8,03 (s, 1Н), 7,90-7,85 (m, 4Н), 7,56-7,54 (m, 1Н), 7,47-7,44 (m, 1Н), 7,20 (s, 1Н), 6,75 (s, 1Н), 4,48-4,46 (m, 2Н), 3,80 (s, 3Н), 1,54-1,48 (d, 6Н).
Пример 16
2-Хлор-N-(2-(4-хлорфенил)индазол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
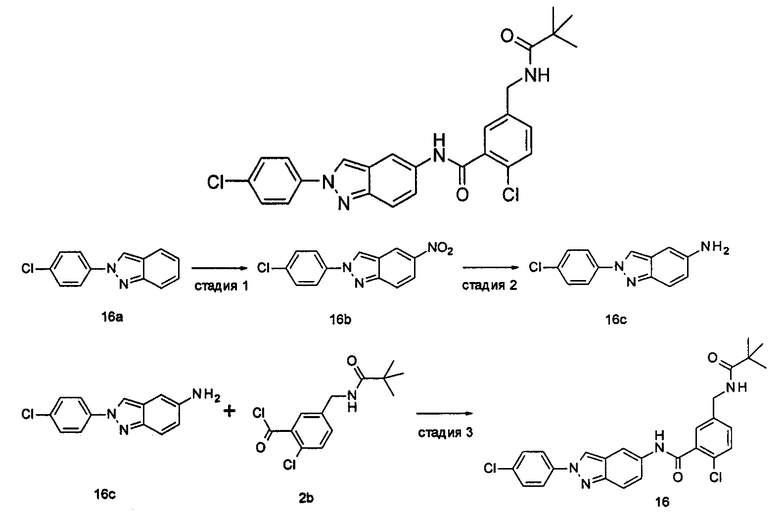
Стадия 1
2-(4-Хлорфенил)-5-нитро-индазол
Нитрат натрия (1,6 г, 19,2 ммоль) добавляли к 10 мл серной кислоты в ледяной бане. Затем частями добавляли 2-(4-хлорфенил)-2Н-индазол 16а (2,2 г, 9,6 ммоль). Затем реакционную смесь нагревали до 70°С и перемешивали в течение 1 часа. Реакционную смесь выливали в 30 мл ледяной воды, и доводили до рН более 7 насыщенным раствором бикарбоната натрия. Смесь экстрагировали этилацетатом (40 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, и затем фильтровали. Фильтрат концентрировали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле с элюирующей системой В с получением целевого соединения 2-(4-хлорфенил)-5-нитро-индазола 16b (500 мг, 19,2%) в виде желтого твердого вещества.
МС m/z (ESI): 274,0 [М+1]
Стадия 2
2-(4-Хлорфенил)-5-амино-индазол
2-(4-Хлорфенил)-5-нитро-индазол 16b (500 мг, 1,8 ммоль) растворяли в 20 мл тетрагидрофурана, и затем добаляли никель Ренея (50 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и промывали фильтровальный осадок этилацетатом (5 мл×3). Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(4-хлорфенил)-5-амино-индазола 16с (200 мг, 45%) в виде коричневого твердого вещества.
МС m/z (ESI): 244,1 [М+1]
Стадия 3
2-Хлор-N-(2-(4-хлорфенил)-2Н-индазол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
2-(4-хлорфенил)-5-амино-индазол 16с (200 мг, 0,37 ммоль), 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензоилхлорид 2b (300 мг, 1,04 ммоль) и N,N-диизопропилэтиламин (300 мг, 2,32 ммоль) последовательно растворяли в 10 мл дихлорметана. Полученную смесь перемешивали в течение 1 часа и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(2-(4-хлорфенил)-2Н-индазол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамида 16 (5 мг, 1,3%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 495,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,56 (s, 1Н), 9,12 (s, 1Н), 8,38 (s, 1Н), 8,19-8,11 (m, 3Н), 7,73-7,66 (m, 3Н), 7,51 (m, 1Н), 7,45-7,44 (m, 2Н), 7,36-7,34 (m, 1Н), 4,31-4,30 (m, 2Н), 1,14 (s, 9Н)
Пример 17
2-(Дифторметил)-N-(1-этил-2-(4-(трифторметил)фенил)-1H-индол-5-ил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
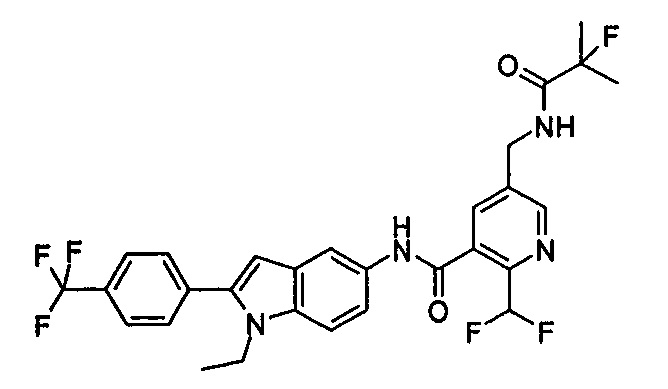
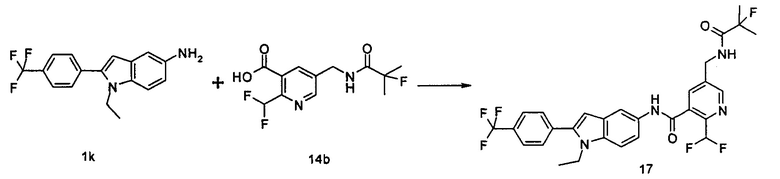
1-Этил-5-амино-2-(4-(трифторметил)фенил)-1Н-индол 1k (105 мг, 0,34 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (100 мг, 0,34 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (97 мг, 0,51 ммоль), 1-гидроксибензотриазол (4,6 мг, 0,034 ммоль) и N,N-диизопропилэтиламин (88 мг, 0,68 ммоль) последовательно растворяли в 5 мл N,N-диметилформамида. Полученную смесь перемешивали в течение 2 часов и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-N-(1-этил-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 17 (20 мг, 25,5%) в виде желтого твердого вещества.
МС m/z (ESI): 577,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,60 (s, 1Н), 8,86 (s, 1Н), 8,68 (s, 1Н), 8,06 (s, 1Н), 8,01 (s, 1Н), 7,90-7,88 (m, 2Н),7,82-7,80 (m, 2Н), 7,58-7,56 (m, 1Н), 7,45-7,43 (m, 1Н), 7,32 (t, 1Н), 6,69 (s, 1Н), 4,47-4,46 (m, 2Н), 4,28-4,26 (m, 2Н), 1,53-1,48 (d, 6Н), 1,22-1,09 (m, 3Н).
Пример 18
N-(2-(4-Хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
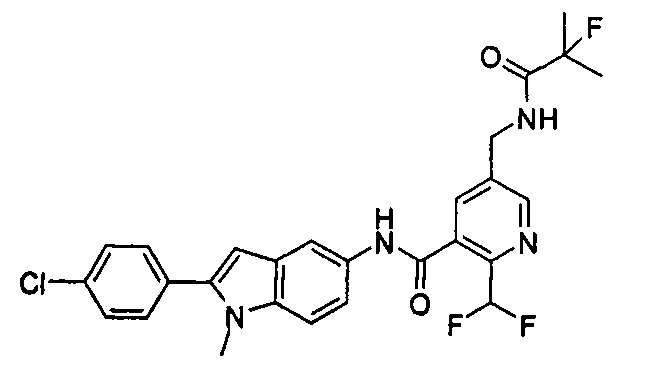
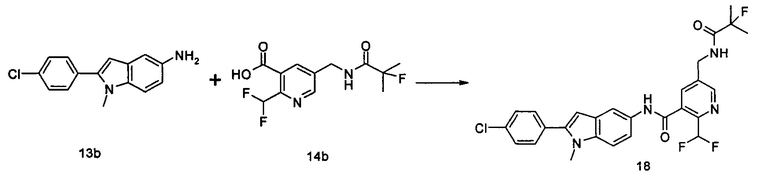
2-(4-Хлорфенил)-1-метил-5-амино-1Н-индол 13b (45 мг, 0,18 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (51 мг, 0,18 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (51 мг, 0,26 ммоль), 1-гидроксибензотриазол (24 мг, 0,18 ммоль) и триэтиламин (71 мг, 0,70 ммоль) последовательно растворяли в 5 мл N,N-диметилформамида. Полученную смесь перемешивали в течение 3 часов и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения N-(2-(4-хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 18 (43 мг, 46,2%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 529,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,60 (s, 1Н), 8,87 (s, 1Н), 8,68 (s, 1Н), 8,04 (d, 2Н), 7,62 (dd, 4Н), 7,47 (dd, 2Н), 7,20 (t, 1Н), 6,64 (s, 1Н), 4,47 (d, 2Н), 3,76 (s, 3Н), 1,54 (s, 3Н), 1,48 (s, 3Н).
Пример 19
N-(2-(2-Хлорфенил)-1-этил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
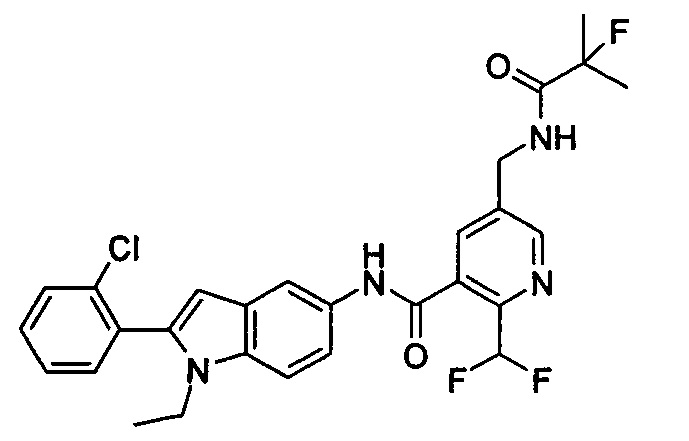
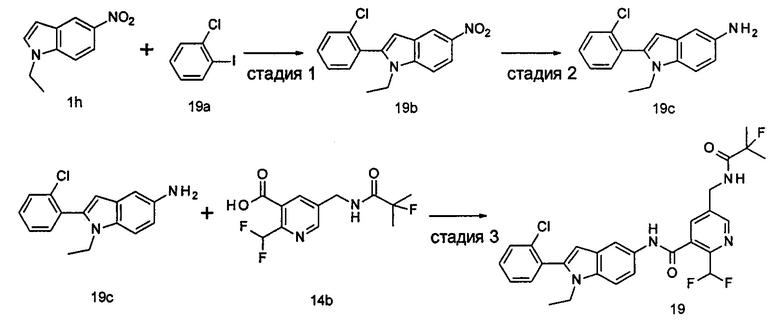
Стадия 1
2-(2-Хлорфенил)-1-этил-5-нитро-1Н-индол
1-Этил-5-нитро-1Н-ндол 1h (1,0 г, 5,3 ммоль) растворяли в 10 мл N,N-диметилацетамида, и затем последовательно добавляли 1-хлор-2-йод-бензол 19а (1,25 г, 5,3 ммоль), трифенилфосфин (300 мг, 1,1 ммоль), ацетат палладия (120 мг, 0,53 ммоль) и ацетат цезия (2,1 г, 11 ммоль). Реакционную смесь нагревали до 140°С и затем перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой В с получением целевого соединения 2-(2-хлорфенил)-1-этил-5-нитро-1Н-индола 19b (300 мг, 18,9%) в виде желтого твердого вещества.
МС m/z (ESI): 301,3 [М+1]
Стадия 2
2-(2-Хлорфен ил)-1-этил-1Н-индол-5-амин
2-(2-Хлорфенил)-1-этил-5-нитро-1Н-индол 19b (300 мг, 1,0 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (50 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(2-хлорфенил)-1-этил-1Н-индол-5-амина 19с (270 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 271,1 [М+1]
Стадия 3
N-(2-(2-Хлорфенил)-1-этил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
2-(2-Хлорфенил)-1-этил-1Н-индол-5-амин 19с (80 мг, 0,30 ммоль), 2-(дифторметил)-5-(((2-фтор-2-етил-пропаноил)амино)метил)никотиновую кислоту 14b (86 мг, 0,30 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (115 мг, 0,60 ммоль), 1-гидроксибензотриазол (40 мг, 0,30 ммоль) и триэтиламин (118 мг, 1,17 ммоль) последовательно растворяли в 5 мл N,N-диметилформамида. Реакционную смесь нагревали до 40°С и перемешивали в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения N-(2-(2-хлорфенил)-1-этил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 19 (10 мг, 14,3%) в виде желтого твердого вещества.
МС m/z (ESI): 543,9 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,59 (s, 1Н), 8,87 (s, 1Н), 8,68 (s, 1Н), 8,02 (d, 2Н), 7,65 (d, 1Н), 7,52-7,49 (m, 3Н), 7,42 (d, 1Н), 6,48 (s, 1Н), 4,46 (d, 2Н), 3,99-3,97 (m, 2Н), 2,88 (s, 1Н), 2,73 (s, 1Н), 1,53 (s, 3Н), 1,46 (s, 3Н), 1,09 (t, 3Н)
Пример 20
N-(2-(3-Хлорфенил)-1-этил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
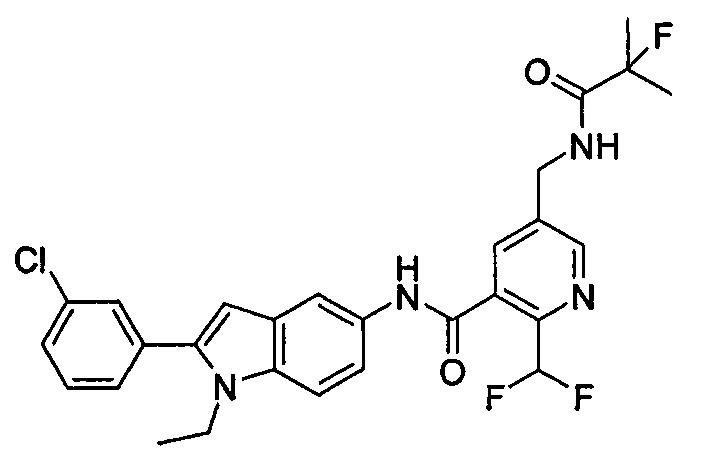
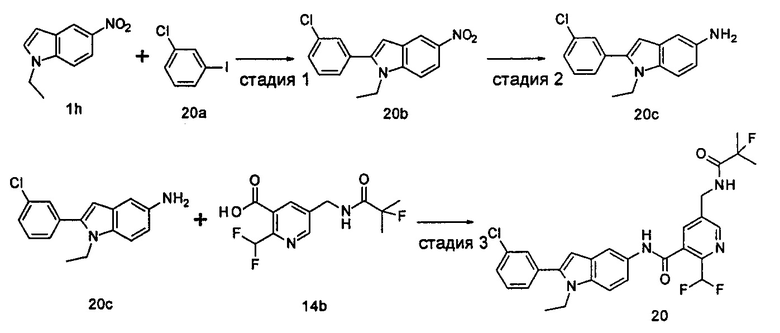
Стадия 1
2-(3-Хлорфенил)-1-этил-5-нитро-1Н-индол
1-Этил-5-нитро-1Н-индол 1h (1,0 г, 5,3 ммоль) растворяли в 10 мл N,N-диметилацетамида, и затем последовательно добавляли 1-хлор-3-йод-бензол 20а (1,4 г, 5,8 ммоль), трифенилфосфин (276 мг, 1,1 ммоль), ацетат палладия (119 мг, 0,53 ммоль) и ацетат цезия (2,0 г, 11 ммоль). Реакционную смесь нагревали до 140°С и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением целевого соединения 2-(3-хлорфенил)-1-этил-5-нитро-1Н-индола 20b (120 мг, 7,6%) в виде желтого твердого вещества.
МС m/z (ESI): 301,1 [М+1]
Стадия 2
2-(3-Хлорфенил)-1-этил-1Н-индол-5-амин
2-(3-Хлорфенил)-1-этил-5-нитро-1Н-индол 20b (50 мг, 0,17 ммоль) растворяли й 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (5 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(3-хлорфенил)-1-этил-1Н-индол-5-амина 20с (45 мг) в виде красного масла, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 271,1 [М+1]
Стадия 3
N-(2-(3-Хлорфенил)-1-этил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
2-(3-Хлорфенил)-1-этил-1H-индол-5-амин 20с (45 мг, 0,17 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (49 мг, 0,17 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (48 мг, 0,25 ммоль), 1-гидроксибензотриазол (23 мг, 0,17 ммоль) и триэтиламин (68 мг, 0,66 ммоль) последовательно растворяли в 5 мл N,N-диметилформамида. Полученную смесь перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения N-(2-(3-хлорфенил)-1-этил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 20 (45 мг, 50% для двух стадий) в виде белого твердого вещества.
МС m/z (ESI): 543,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6): δ 10,62 (s, 1Н), 8,88 (s, 1Н), 8,69 (s, 1Н), 8,03 (d, 2Н), 7,57-7,55 (m, 6Н), 7,43 (t, 1Н), 7,19 (s, 1Н), 6,64 (d, 2Н), 4,49-4,47 (m, 2Н), 1,54 (s, 3Н), 1,49 (s, 3Н), 1,19 (t, 3Н).
Пример 21
N-(2-(4-Хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)никотинамид
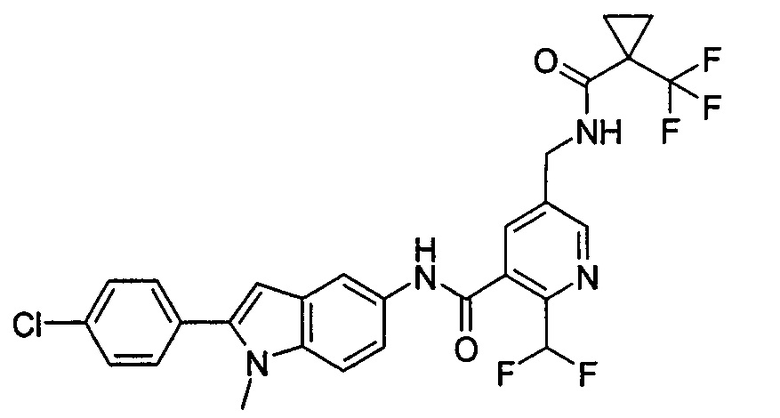
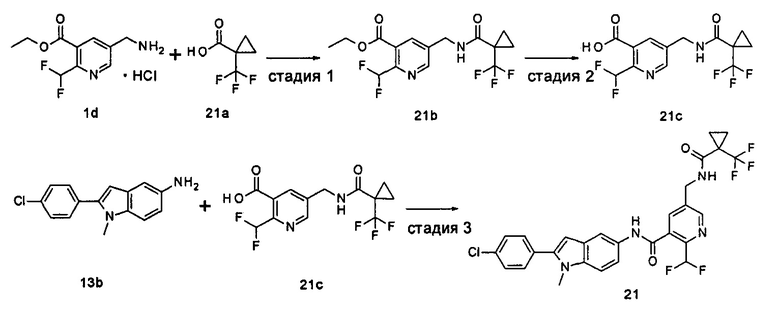
Стадия 1
Этил 2-(дифторметил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)никотинат
Этил 5-(аминометил)-2-(дифторметил)никотината гидрохлорид 1d (4,0 г, 13,2 ммоль) растворяли в 50 мл N,N-диметилформамида, и затем последовательно добавляли 1-(трифторметил)циклопропан-1-карбоновую кислоту 21а (2,03 г, 13,2 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (5,07 г, 26,4 ммоль), 1-гидроксибензотриазол (178 мг, 1,32 ммоль) и триэтиламин (5,3 г, 52,8 ммоль). Полученную смесь перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Остаток промывали этилацетатом (50 мл) и водой (50 мл), сушили с получением неочищенного целевого соединения этил 2-(дифторметил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)никотината 21b (4,83 г) в виде коричневого масла, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 367,1 [М+1]
Стадия 2
2-(Дифторметил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)никотиновая кислота
Этил 2-(дифторметил)-5-(((1-(трифторметил)циклопропанкарбонил) амино)метил) никотинат 21b (4,83 г, 13,2 ммоль) растворяли в 150 мл смеси 1,4-диоксана и воды (соотношение объемов 2:1), и затем добавляли гидрат гидроксида лития (1,38 г, 33,0 ммоль). Реакционную смесь перемешивали в течение 1 часа. Этанол удаляли при пониженном давлении, и остаток доводили до рН 4-5 6М раствором хлороводородной кислоты. Большое количество твердого вещества осаждали, и к нему добавляли 100 мл зтилацетата. Твердое вещество отфильтровывали и сушили с получением целевого соединения 2-(дифторметил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)никотиновой кислоты 21с (2,0 г, 44,8% для двух стадий) в виде белого твердого вещества.
МС m/z (ESI): 339,1 [М+1]
Стадия 3
N-(2-(4-Хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)никотинамид
2-(4-Хлорфенил)-1-метил-5-амино-1Н-индол 13b (45 мг, 0,17 ммоль), 2-(дифторметил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)никоти новую кислоту 21с (59 мг, 0,17 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (51 мг, 0,26 ммоль), 1-гидроксибензотриазол (24 мг, 0,17 ммоль) и триэтиламин (71 мг, 0,70 ммоль) последовательно растворяли в 5 мл N,N-диметилформамида. Полученную смесь перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения N-(2-(4-хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)никотинамида 21 (20 мг, 20%) в виде бледно-желтого твердого вещества.
MC m/z (ESI): 577,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,61 (s, 1Н), 8,67 (s, 1Н), 8,59-8,57 (m, 1Н), 8,03 (d, 2Н), 7,62 (dd, 4Н), 7,47 (dd, 2Н), 7,19 (t, 1Н), 6,64 (s, 1Н), 4,46 (d, 2Н), 3,76 (s, 3Н), 1,28-1,26 (m, 4Н).
Пример 22
2-Хлор-5-((2,2-диметилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)бензамид
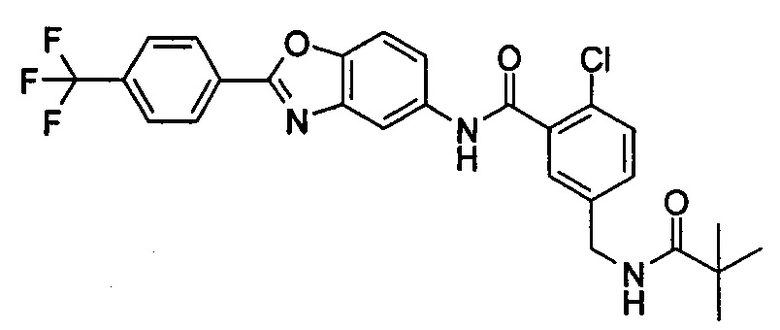
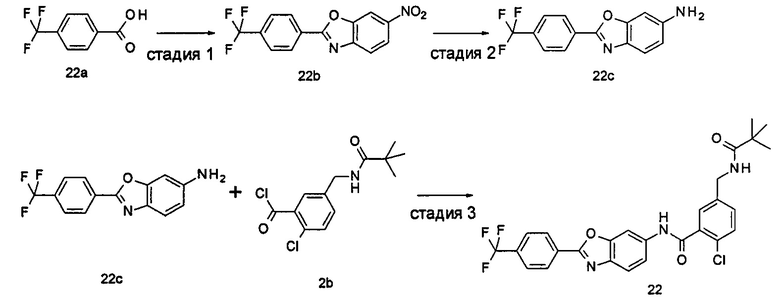
Стадия 1
5-Нитро-2-(4-(трифторметил)фенил)бензо[d]оксазол
4-(Трифторметил)бензойную кислоту 22а (2,0 г, 10,5 ммоль) растворяли в 14 мл полифосфорной кислоты, и затем добавляли 2-амино-4-нитрофенол (1,62 г, 10,5 ммоль). Реакционную смесь нагревали до 120°С и перемешивали в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, и затем выливали в 500 мл воды. Раствор доводили до рН 7, по частями добавляя гидроксид натрия, и затем экстрагировали этилацетатом (500 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением целевого соединения 5-нитро-2-(4-(трифторметил)фенил)бензо[d]оксазола 22b (1,5 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 2
2-(4-(Трифторметил)фенил)бензо[d]оксазол-5-амин
5-Нитро-2-(4-(трифторметил)фенил)бензо[d]оксазол 22b (170 мг, 0,55 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (20 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(4-(трифторметил)фенил)бензо[d]оксазол-5-амина 22с (150 мг) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 279,1 [М+1]
Стадия 3
2-Хлор-5-((2,2-диметилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)бензамид
2-(4-(Трифторметил)фенил)бензо[d]оксазол-5-амин 22с (50 мг, 0,18 ммоль) растворяли в 10 мл тетрагидрофурана, и затем по каплям добавляли трифторуксусную кислоту (54 мкл, 0,39 ммоль) и 2 мл раствора 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензоилхлорида 2b (52 мг, 0,18 ммоль) в тетрагидрофуране. Полученную смесь перемешивали в течение 1 часа. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением твердого вещества, к которому добавляли 10 мл этилацетата и фильтровали. Фильтровальный осадок сушили с получением целевого соединения 2-хлор-5-((2,2-диметилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)бензамида 22 (15 мг, 15,3% для двух стадий) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 530,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,41 (s, 1Н), 8,19 (t, 1Н), 8,11 (s, 1Н), 7,91-7,87 (d, 2Н), 7,83-7,79 (d, 2Н),7,59-7,53 (d, 2Н), 7,53-7,48 (d, 2Н), 7,48-7,41 (m, 1Н), 4,29-4,22 (m, 2Н), 1,13 (s, 9H).
Пример 23
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)никотинамид
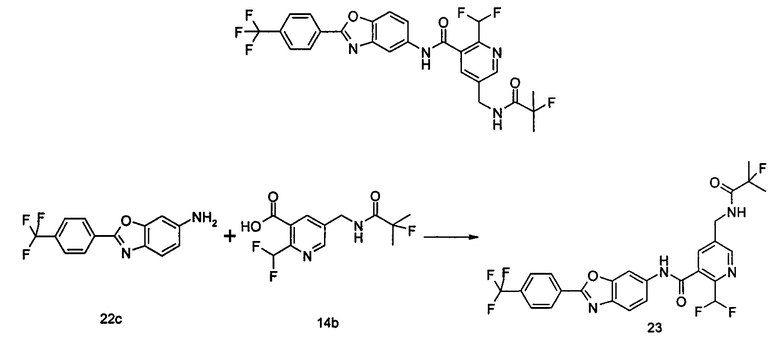
2-(4-(Трифторметил)фенил)бензо[d]оксазол-5-амин 22с (50 мг, 0,18 ммоль) растворяли в 10 мл N,N-диметилформамида, и затем последовательно добавляли 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (52 мг, 0,18 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (75 мг, 0,39 ммоль), 1-гидроксибензотриазол (2,4 мг, 0,02 ммоль) и триэтиламин (73 мг, 0,72 ммоль). Полученную смесь нагревали до 70°С и перемешивали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением твердого вещества. К твердому веществу добавляли 10 мл диэтилового эфира и фильтровали. Фильтровальный осадок сушили с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)никотинамида 23 (10 мг, 10,1%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 551,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,60 (s, 1Н), 8,69 (s, 1Н), 8,45 (t, 1Н), 8,08 (s, 1Н), 8,01 (s, 1Н), 7,92-7,86 (d, 2Н), 7,85-7,79 (d, 2Н), 7,61-7,56 (d, 1Н), 7,46-7,42 (d, 1Н), 7,20 (t, 1Н), 4,31-4,21 (m, 2Н), 2,49-2,40 (m, 1Н), 1,09-1,02 (d, 6Н).
Пример 24
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)-2Н-индазол-5-ил)никотинамид
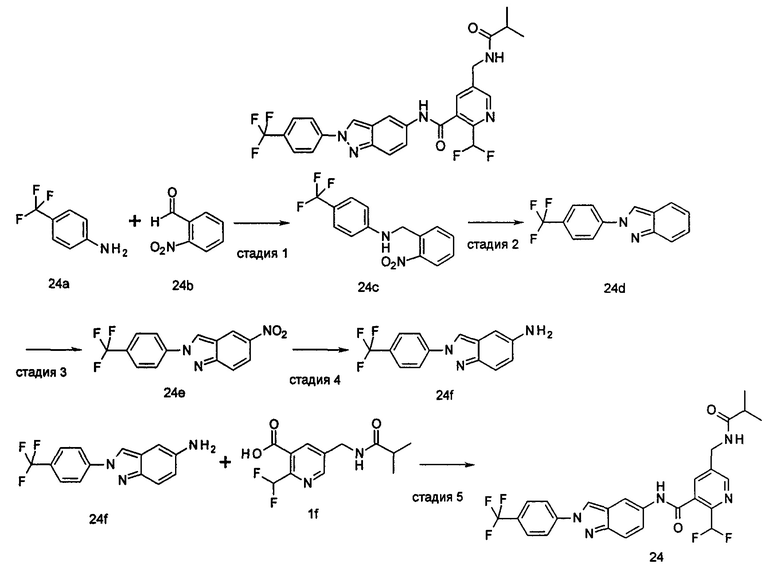
Стадия 1
N-(2-Нитробензил)-4-(трифторметил)анилин
4-Аминобензотрифторид 24а (2,35 г, 14,56 ммоль) растворяли в 50 мл 1,2-дихлорэтана, и затем добавляли 2-нитробензальдегид 24b (2,0 г, 13,23 ммоль). Полученную смесь перемешивали в течение 0,5 часа и затем добавляли триацетоксиборгидрид натрия (5,6 г, 26,46 ммоль), и перемешивали еще в течение 16 часов. К реакционной смеси добавляли 100 мл дихлорметана и 100 мл воды. Органическую фазу концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения N-(2-нитробензил)-4-(трифторметил)анилина 24с (3,5 г, 89,4%) в виде желтого масла.
МС m/z (ESI): 297,1 [М+1]
Стадия 2
2-(4-(Трифторметил)фенил)-2Н-индазол
Порошок цинка (1,73 г, 27,04 ммоль) добавляли к 50 мл тетрагидрофурана, и затем добавляли тетрахлорид титана (2,57 г, 13,55 ммоль). Реакционную смесь нагревали до 70°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры и доводили до рН 8 триэтиламином. N-(2-Нитробензил)-4-(трифторметил)анилин 24с (1,0 г, 3,38 ммоль) растворяли в 20 мл тетрагидрофурана и затем добавляли в полученную выше смесь. Реакционную смесь непрерывно пермешивали еще в течение 30 минут и затем доводили до рН 3 6М раствором хлороводородной кислоты. Раствор экстрагировали дихлорметаном (100 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения 2-(4-(трифторметил)фенил)-2Н-индазола 24d (100 мг, 11,3%) в виде белого твердого вещества.
МС m/z (ESI): 263,1 [М+1]
Стадия 3
5-Нитро-2-(4-(трифторметил)фенил)-2Н-индазол
Нитрат натрия (29 мг, 0,67 ммоль) добавляли к 1 мл серной кислоты в ледяной бане, и затем частями добавляли 2-(4-(трифторметил)фенил)-2Н-индазол 24d (50 мг, 0,19 ммоль). Реакционную смесь нагревали до 70°С и перемешивали в течение 1 часа. Реакционную смесь выливали в 30 мл ледяной воды, и доводили до рН более 7. Раствор экстрагировали этилацетатом (40 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения 5-нитро-2-(4-(трифторметил)фенил)-2Н-индазола 24е (30 мг, 25,6%) в виде желтого твердого вещества.
МС m/z (ESI): 308,1 [М+1]
Стадия 4
2-(4-(Трифторметил)фенил)-2Н-индазол-5-амин
5-Нитро-2-(4-(трифторметил)фенил)-2Н-индазол 24е (30 мг, 0,098 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (3 мг). Реакционную смесь перемешивали в течение 1 часа в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(4-(трифторметил)фенил)-2Н-индазол-5-амина 24f (27 мг) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 278,1 [М+1]
Стадия 5
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)-2H-индазол-5-ил)никотинамид
2-(4-(Трифторметил)фенил)-2Н-индазол-5-амин 24f (27 мг, 0,098 ммоль), 2-(дифтормеил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (27 мг, 0,10 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (29 мг, 0,15 ммоль) и 1-гидроксибензотриазол (14 мг, 0,10 ммоль) последовательно добавляли к 5 мл N,N-диметилформамида. Полученную смесь нагревали до 40°С и перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением твердого вещества. К твердому веществу добавляли 20 мл дихлорметана и перемешивали в течение 20 минут. Смесь фильтровали и фильтровальный осадок промывали дихлорметаном (5 мл×3), и сушили с получением целевого соединения 2-(дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)-2Н-индазол-5-ил)никотинамида 24 (27 мг, 50,9%) в виде белого твердого вещества.
МС m/z (ESI): 532,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,78 (s, 1Н), 9,25 (s, 1Н), 8,70 (s, 1Н), 8,47 (t, 1Н), 8,38-8,33 (m, 3Н), 8,08-7,97 (m, 3Н), 7,76 (d, 1Н), 7,49 (d, 1Н), 7,19 (t, 1Н), 4,44 (d, 2Н), 2,49-2,41 (m, 1Н), 1,06 (d, 6Н).
Пример 25
2-Хлор-5-((2,2-диметилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)изоиндолин-5-ил)бензамид
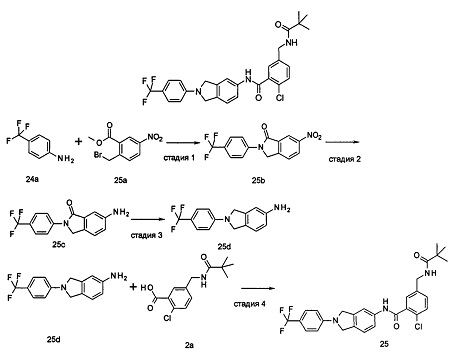
Стадия 1
6-Нитро-2-(4-(трифторметил)фенил)изоиндолин-1-он
4-Аминобензотрифторид 24а (193 мг, 1,2 ммоль) растворяли в 3 мл этанола, и затем добавляли метил 2-(бромметил)-5-нитро-бензоат 25а (274 мг, 1,0 ммоль) и N,N-диизопропилэтиламин (258 мг, 2,0 ммоль). Реакционную смесь нагревали до 110°С и перемешивали в течение 16 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрационный осадок сушили с получением целевого соединения 6-нитро-2-(4-(трифторметил)фенил)изоиндолин-1-она 25b (70 мг, 21,7%) в виде желтого твердого вещества.
МС m/z (ESI): 321,0 [М-1]
Стадия 2
6-Амино-2-(4-(трифторметил)фенил)изоиндол ин-1-он
6-Нитро-2-(4-(трифторметил)фенил)изоиндолин-1-он 25b (70 мг, 0,098 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (7 мг). Реакционную смесь перемешивали в течение 1 часа в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 6-амино-2-(4-(трифторметил)фенил)изоиндолин-1-она 25с (64 мг) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 293,1 (М+1)
Стадия 3
2-(4-(Трифторметил)фенил)изоиндолин-5-амин
6-Амино-2-(4-(трифторметил)фенил)изоиндолин-1-он 25с (64 мг, 0,22 ммоль) растворяли в 10 мл тетрагидрофурана, и затем добавляли алюмогидрид лития (51 мг, 1,32 ммоль). Реакционную смесь нагревали до 65°С, и перемешивали в течение 16 часов. К реакционной смеси добавляли 0,1 мл раствора гидроксида натрия (15%) и 0,4 мл воды, а затем сульфат магния, и перемешивали еще в течение 5 мин. Смесь фильтровали и экстрагировали этилацетатом. Органическую фазу концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(4-(трифторметил)фенил)изоиндолин-5-амина 25d (11 мг, 18,0%) в виде серого твердого вещества.
МС m/z (ESI): 279,1 [М+1]
Стадия 4
2-Хлор-5-((2,2-диметилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)изоиндолин-5-ил)бензамид
2-(4-(Трифторметил)фенил)изоиндолин-5-амин 25d (11 мг, 0,04 ммоль), 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензойную кислоту 2а (11 мг, 0,04 ммоль), 1-этил-(3-диметиламинопропил) карбодиимида гидрохлорид (12 мг, 0,06 ммоль), 1-гидроксибензотриазол (6 мг, 0,04 ммоль) и триэтиламин (17 мг, 0,16 ммоль) последовательно добавляли к 5 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-5-((2,2-диметилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)изоиндолин-5-ил)бензамида 25 (6 мг, 28,6%) в виде белого твердого вещества.
МС m/z (ESI): 530,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,61 (s, 1Н), 8,18 (t, 1Н), 7,89 (s, 1Н), 7,60-7,50 (m, 4Н), 7,42-7,34 (m, 3Н), 6,80 (d, 2Н), 4,68 (d, 4Н), 4,30 (d, 2Н), 1,14 (s, 9Н).
Пример 26
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(4-(трифторметил)фенил)изоиндолин-5-ил)никотинамид
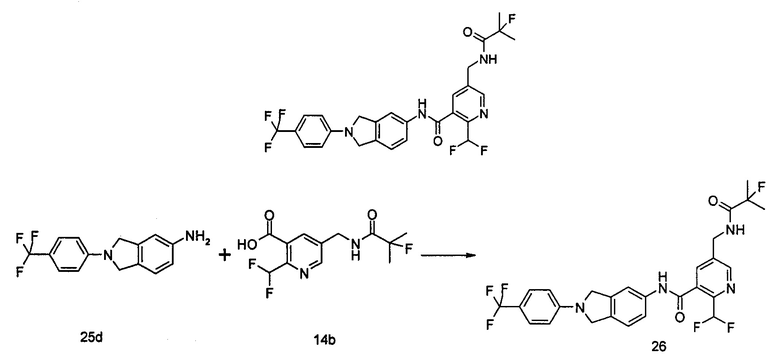
2-(4-(Трифторметил)фенил)изоиднолин-5-амин 25d (63 мг, 0,23 ммоль) растворяли в 5 мл N,N-диметилформамида и последовательно добавляли 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (66 мг, 0,23 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (67 мг, 0,35 ммоль), 1-гидроксибензотриазол (27 мг, 0,23 ммоль) и триэтиламин (93 мг, 0,92 ммоль). Реакционную смесь перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(4-(трифторметил)фенил)изоиндолин-5-ил)никотинамида 26 (50 мг, 39,7%) в виде белого твердого вещества.
МС m/z (ESI): 551,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,79 (s, 1Н), 8,87 (s, 1Н), 8,70 (s, 1Н), 8,00 (s, 1Н), 7,87 (s, 1Н), 7,57 (d, 3Н), 7,42 (d, 1Н), 7,16 (t, 1Н), 6,80 (d, 2Н), 4,69 (d, 4Н), 4,66 (d, 2H), 1,53 (s, 3Н), 1,48 (s, 3Н).
Пример 27
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(2-метоксиэтил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамид
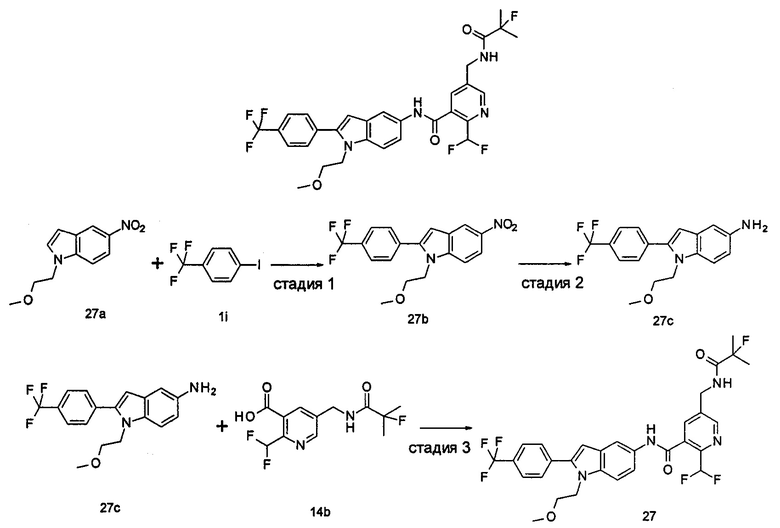
Стадия 1
1-(2-Метоксиэтил-5-нитро-2-(4-(трифторметил)фенил)-1Н-индол
1-(2-Метоксиэтил)-5-нитро-1Н-индол 27а (2,2 г, 10 ммоль, полученный в соответствии со способом, описанным в патентной заявке US 20090076275) растворяли в 10 мл N,N-диметилацетамида и затем последовательно добавляли 1-йод-4-(трифторметил)бензол 1i (2,7 г, 10 ммоль), трифенилфосфин (564 мг, 2,0 ммоль), ацетат палладия (225 мг, 1,0 ммоль) и ацетат цезия (3,8 г, 20 ммоль). Реакционную смесь нагревали до 140°С и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении, и остаток промывали этилацетатом (5 мл×3). Фильтрат концентрировали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением целевого соединения 1-(2-метоксиэтил-5-нитро-2-(4-(трифторметил)фенил)-1H-индола 27b (300 мг, 8,2%) в виде желтого твердого вещества.
МС m/z (ESI): 365,1 (М+1)
Стадия 2
1-(2-Метоксиэтил-5-амино-2-(4-(трифторметил)фенил)-1Н-индол
1-(2-Метоксиэтил-5-нитро-2-(4-(трифторметил)фенил)-1Н-индол 27b (100 мг, 0,27 ммоль) растворяли в 20 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (10 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и фильтровальный осадок промывали этилацетатом (30 мл). Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 1-(2-метоксиэтил-5-амино-2-(4-(трифторметил)фенил)-1Н-индола 27с (90 мг) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 335,1 [М+1]
Стадия 3
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(2-метоксиэтил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамид
1-(2-Метоксиэтил-5-амино-2-(4-(трифторметил)фенил)-1Н-индол 27с (150 мг, 0,44 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил) никотиновую кислоту 14b (128 мг, 0,44 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (169 мг, 0,88 ммоль), 1-гидроксибензотриазол (6 мг, 0,04 ммоль) и триэтиламин (178 мг, 1,76 ммоль) последовательно растворяли в 10 мл N,N-диметилацетамида. Реакционную смесь нагревали до 75°С и перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(2-метоксиэтил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамида 27 (60 мг, 22,4%) в виде желтого твердого вещества.
МС m/z (ESI): 607,3[М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,624 (s, 1Н), 8,879 (s, 1Н), 8,692 (s, 1Н), 8,021-8,062 (m, 2Н), 7,871-7,921 (m, 4Н), 7,592-7,614 (m, 1Н), 7,421-7,438 (m, 1Н), 7,192-7,416 (m, 1Н), 6,689 (s, 1Н), 4,376-4,815 (m, 4Н), 3,548-3,575 (m, 2Н), 3,058-3,575 (s, 3Н), 1,443-1,541 (d, 6Н)
Пример 28
2-(Дифторметил)-N-(1-этил-2-(3-(трифторметил)фенил)-1Н-индол-5-ил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-никотинамид
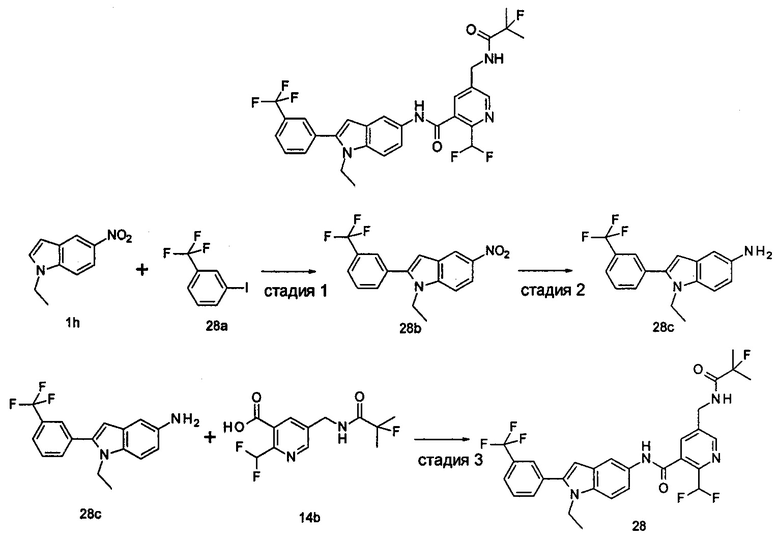
Стадия 1
1-Этил-5-нитро-2-(3-(трифторметил)фенил)-1Н-индол
1-Этил-5-нитро-1Н-индол 1h (500 мг, 2,63 ммоль) растворяли в 5 мл N,N-диметилацетамида, и затем посделовательно добавляли 1-йод-3-(трифторметил)бензол 28а (860 мг, 3,16 ммоль), трифенилфосфин (140 мг, 0,53 ммоль), ацетат палладия (119 мг, 0,13 ммоль), и ацетат цезия (2,0 г, 11 ммоль). Реакционную смесь нагревали до 140°С и перемешивали в течение 16 часов в атмосфере аргона. К реакционному раствору добавляли 40 мл воды и экстрагировали этилацетатом (40 мл×2). Органические фазы объединяли, сушили над безводным сульфтом натрия и филтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением целевого соединения 1-этил-5-нитро-2-(3-(трифторметил)фенил)-1Н-индола 28b (100 мг, 11,4%) в виде желтого твердого вещества.
МС m/z (ESI): 335,1 [М+1]
Стадия 2
1-Этил-5-амино-2-(3-(трифторметил)фенил)-1Н-индол
1-Этил-5-нитро-2-(3-(трифторметил)фенил)-1Н-индол 28b (110 мг, 0,33 ммоль) растворяли в смеси 8 мл тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (30 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 1-этил-5-амино-2-(3-(трифторметил)фенил)-1Н-индола 28 с (100 мг) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 305,1 [М+1]
Стадия 3
2-(Дифторметил)-N-(1-этил-2-(3-(трифторметил)фенил)-1Н-индол-5-ил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
1-Этил-5-амино-2-(3-(трифторметил)фенил)-1Н-индол 28с (50 мг, 0,16 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил) никотиновую кислоту 14b (50 мг, 0,16 ммоль), 1-этил-(3-диметиламинопропил) карбодиимида гидрохлорид (70 мг, 0,33 ммоль) и 1-гидроксибензотриазол (25 мг, 0,16 ммоль) последовательно добавляли к 3 мл N,N-диметилацетамида. Реакционную смесь нагревали до 40°С и перемешивали в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-N-(1-этил-2-(3-(трифторметил)фенил)-1Н-индол-5-ил)-5-(((2-фтор-2-метил-пропаноил)амино) метил)никотинамида 28 (20 мг, 21,1%) в виде желтого твердого вещества.
МС m/z (ESI): 575,5 [М-1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,59 (s, 1Н), 8,85 (s, 1Н), 8,67 (s, 1Н), 8,03 (d, 2Н), 7,85-7,83 (m, 1Н), 7,81-7,79 (m, 2Н), 7,56 (d, 1Н), 7,44 (d, 1Н), 6,68 (s, 1Н), 4,46 (d, 2Н), 4,22-4,18 (m, 2Н), 2,87 (s, 1Н), 2,72 (s, 1Н), 1,52 (s, 3Н), 1,46 (s, 3Н), 1,22 (t, 3Н)
Пример 29
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензофуран-5-ил)никотинамид
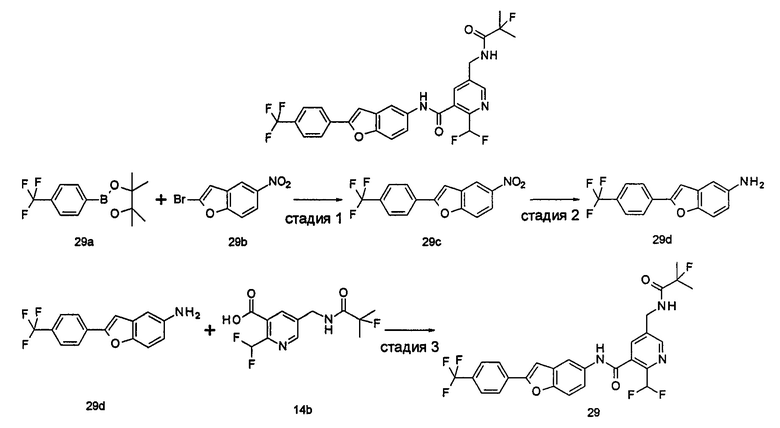
Стадия 1
5-Нитро-2-(4-(трифторметил)фенил)бензофуран
4-(Трифторметил)(пинаколборил)бензол 29а (170 мг, 0,62 ммоль) растворяли в смеси 6 мл 1,4-диоксана и воды (соотношение объемов 5:1), и затем добавляли 2-бром-5-нитро-бензофуран 29b (100 мг, 0,43 ммоль, полученный в соответствии со способом, раскрытым в "European Journal of Organic Chemistry, 2013, 2013(9), 1644-1648"), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (30 мг, 0,04 ммоль) и карбонат натрия (88 мг, 0,83 ммоль). Реакционную смесь нагревали до 100°С и перемешивали в течение 16 часов в атмосфере аргона. К реакционному раствору добавляли 50 мл дихлорметана, и фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения 5-нитро-2-(4-(трифторметил)фенил)бензофурана 29с (80 мг) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 2
5-Амино-2-(4-(трифторметил)фенил)бензофуран
5-Нитро-2-(4-(трифторметил)фенил)бензофуран 29с (40 мг, 0,13 ммоль) растворяли в смеси 10 мл тетрагидрофурана и воды (соотношение объемов 1:1), и затем добавляли никель Ренея (40 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и фильтрат концентрировали при пониженном давлении с получением целевого соединения 5-амино-2-(4-(трифторметил)фенил)бензофурана 29d (36 мг) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 278,2 [М+1]
Стадия 3
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензофуран-5-ил)никотинамид
5-Амино-2-(4-(трифторметил)фенил)бензофуран 29d (33 мг, 0,12 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (35 мг, 0,12 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (45 мг, 0,24 ммоль) и 1-гидроксибензотриазол (16 мг, 0,12 ммоль) последовательно добавляли к 3 мл N,N-диметилацетамида. Реакционную смесь нагревали до 40°С и перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензофуран-5-ил)никотинамида 29 (20 мг, 30,3%) в виде белого твердого вещества.
МС m/z (ESI): 550,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,7 (s, 1Н), 8,71 (s, 1Н), 8,49 (t, 1Н), 8,12 (s, 1Н), 8,03 (s, 1Н), 7,93-7,88 (m, 2Н), 7,86-7,80 (m, 2Н), 7,61-7,56 (m, 1Н), 7,46-7,42 (m, 1Н), 7,31-7,01 (m, 1Н), 7,15 (s, 1H), 4,44-4,38 (m, 2Н), 1,55-1,49 (m, 6Н).
Пример 30
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-тетрагидрофуран-3-ил-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамид
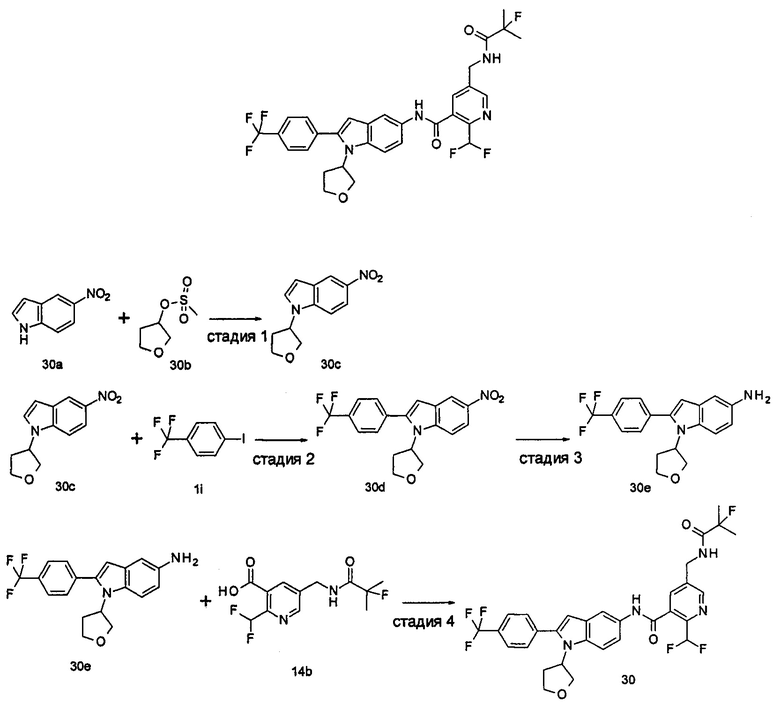
Стадия 1
5-Нитро-(1-тетрагидрофуран-3-ил)-1Н-индол
5-Нитроиндол 30а (810 мг, 5,0 ммоль) растворяли в 10 мл N,N-диметилацетамида в ледяной бане и затем добавляли гидрид натрия (300 мг, 7,5 ммоль). Реакционный раствор подогревали до комнатной температуры и затем перемешивали в течение 10 минут. Добавляли тетрагидрофуран-3-ила метансульфонат 30b (1,66 г, 10,0 ммоль, полученный в соответствии со способом, раскрытым в "Journal of Organic Chemistry, 2008, 73(14), 5397-5409"), нагревали смесь до 50°С и перемешивали в течение 16 часов. К реакционной смеси добавляли 100 мл воды, хорошо перемешивали, и затем экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли и промывали насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением целевого соединения 5-нитро-1-тетрагидрофуран-3-ил-1H-индола 30 с (1,16 г) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 233,1 [М+1]
Стадия 2
5-Нитро-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1H-индол
5-Нитро-1-(тетрагидрофуран-3-ил)-1Н-индол 30с (1,16 г, 5,0 ммоль) растворяли в 10 мл N,N-диметилацетамида, и затем последовательно добавляли 1-йод-4-(трифторметил)бензол 1i (1,36 г, 5,0 ммоль), трифенилфосфин (282 мг, 1,0 ммоль), ацетат палладия (113 мг, 0,5 ммоль) и ацетат цезия (1,9 г, 10 ммоль). Реакционную смесь нагревали до 140°С и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением целевого соединения 5-нитро-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индола 30d (200 мг, 10,6%) в виде желтого твердого вещества.
МС m/z (ESI): 377,1 [М+1]
Стадия 3
5-Амино-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол
5-Нитро-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол 30d (100 мг, 0,27 ммоль) растворяли в 10 мл тетрагидрофурана и затем добавляли никель Ренея (10 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и промывали фильтровальный осадок этилацетатом (30 мл). Фильтрат концентрировали при пониженном давлении с получением целевого соединения 5-амино-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индола 30е (85 мг, 92,4%) в виде желтого твердого вещества.
МС m/z (ESI): 347,1 [М+1]
Стадия 4
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамид
5-Амино-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол 30е (70 мг, 0,20 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (58 мг, 0,20 ммоль), 1-этил-(3-диметиламинопропил) карбодиимида гидрохлорид (58 мг, 0,30 ммоль), 1-гидроксибензотриазол (3 мг, 0,02 ммоль) и N,N-диизопропилэтиламин (52 мг, 0,40 ммоль) последовательно добавляли к 10 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотин амида 30 (20 мг, 16,1%) в виде бледно-желтого твердого вещества.
MC m/z (ESI): 619,2 [M+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,64 (s, 1Н), 8,87 (s, 1Н), 8,69 (t, 1Н), 8,13 (s, 1Н), 8,01 (s, 1Н), 8,01-7,91 (m, 2Н), 7,78-7,76 (m, 2Н), 7,72-7,69 (m, 1Н), 7,40-7,39 (m, 1Н), 7,18 (t, 1Н), 6,65 (s, 1Н), 5,13 (m, 1Н), 4,68-4,67 (m, 2Н), 4,48-4,46 (m, 1Н), 4,32-4,17 (m, 1Н), 3,96-3,91 (m, 1Н), 3,67-3,65 (m, 1Н), 2,40-2,33 (m, 2Н), 1,54-1,42 (d, 6Н).
Пример 31
(S)-2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамид
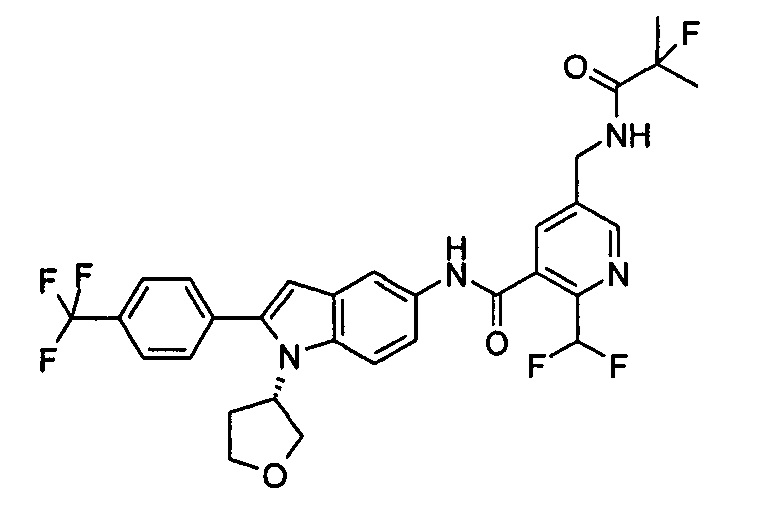
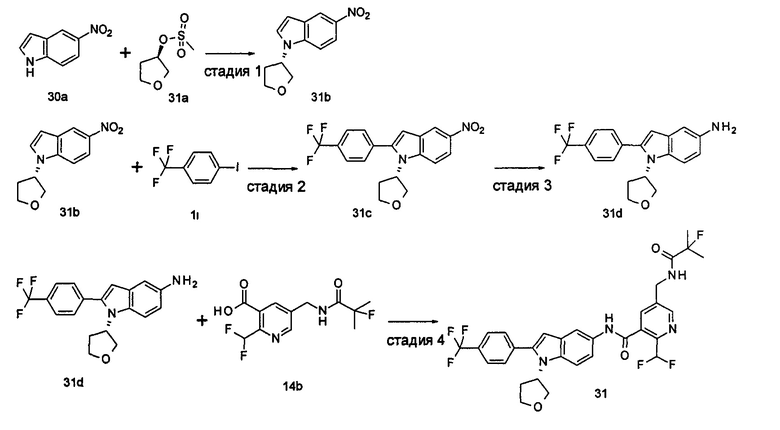
Стадия 1
(S)-5-Нитро-1-(тетрагидрофуран-3-ил)-1Н-индол
5-Нитроиндол 30а (9,0 г, 55,3 ммоль) и карбонат цезия (36,0 г, 110,6 ммоль) растворяли в 100 мл N,N-диметилацетамида, и затем добаляли (R) тетрагидрофуран-3-ил метансульфонат 31а (18,4 г, 110,6 ммоль, полученный в соответствии со способом, раскрытым в "Nature Chemical Biology, 2008, 4(11), 691-699"). Реакционную смесь нагревали до 70°С и перемешивали в течение 16 часов. Реакционный раствор выливали в 400 мл ледяной воды. Большое количество твердого вещества осаждали и затем отфильтровывали. Фильтрационный осадок растворяли в этилацетате и фильтровали. Фильтрат сушили над безводным сульфатом натрия, и затем фильтровали. Фильтрат концентрировали при пониженном давлении с получением целевого соединения (S)-5-нитро-1-(тетрагидрофуран-3-ил)-1Н-индола 31b (12 г, 93,8%) в виде желтого твердого вещества.
МС m/z (ESI): 233,0 [М+1]
Стадия 2
(S)-5-Нитро-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол
(S)-5-Нитро-1-(тетрагидрофуран-3-ил)-1Н-индол 31b (2,0 г, 8,6 ммоль) растворяли в 15 мл N,N-диметилацетамида и затем последовательно добавляли 1-йод-4-(трифторметил)бензол 1i (2,58 г, 9,5 ммоль), трифенилфосфин (450 мг, 1,7 ммоль), ацетат палладия (97 мг, 0,4 ммоль) и ацетат цезия (5,0 г, 25,9 ммоль). Реакционную смесь нагревали до 140°С, и перемешивали в течение 18 часов в атмосфере аргона. К реакционному раствору добавляли 200 мл воды и экстрагировали этилацетатом (200 мл×2). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением целевого соединения (S)-5-нитро-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индола 31с (200 мг, 5,3%) в виде желтого твердого вещества.
МС m/z (ESI): 377,1 [М+1]
Стадия 3
(S)-5-Аамино-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол
(S)-5-Нитро-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол 31с (200 мг, 0,53 ммоль) растворяли в смеси 10 мл тетрагидрофурана и метанола (соотношение объемов 1:1) и затем добавляли никель Ренея (20 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционный раствор фильтровали через Celatom, и концентрировали фильтрат при пониженном давлении с получением целевого соединения (S)-5-амино-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индола 31d (130 мг, 70,7%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 347,2 [М+1]
Стадия 4
(S)-2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамид
(S)-5-Амино-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол 31d (130 мг, 0,38 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (58 мг, 0,20 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (58 мг, 0,30 ммоль), 1-гидроксибензотриазол (4 мг, 0,03 ммоль) и триэтиламин (139 мг, 1,38 ммоль) последовательно добавляли к 5 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением целевого соединения (S)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамида 31 (100 мг, 46,9%) в виде белого твердого вещества.
МС m/z (ESI): 619,3 [M+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,63 (s, 1Н), 8,86 (s, 1Н), 8,68 (s, 1Н), 8,12 (s, 1Н), 8,01-8,12 (d, 1Н), 7,89-7,91 (d, 2Н), 7,75-7,77 (d, 2Н), 7,69-7,71 (d, 1Н), 7,39-7,40 (d, 1Н), 7,31 (t, 1Н), 6,64 (s, 1Н), 4,46-4,48 (d, 2Н), 4,32-4,35 (m, 1Н), 4,13-4,17 (m, 1Н), 3,91-3,95 (m, 1Н), 3,65-3,67 (m, 1Н), 2,42-2,45 (m, 1Н), 2,32-2,36 (m, 1Н), 1,98-2,04 (m, 1Н), 1,54 (s, 3Н), 1,48 (s, 3Н).
Пример 32
(R)-2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1H-индол-5-ил)никотинамид
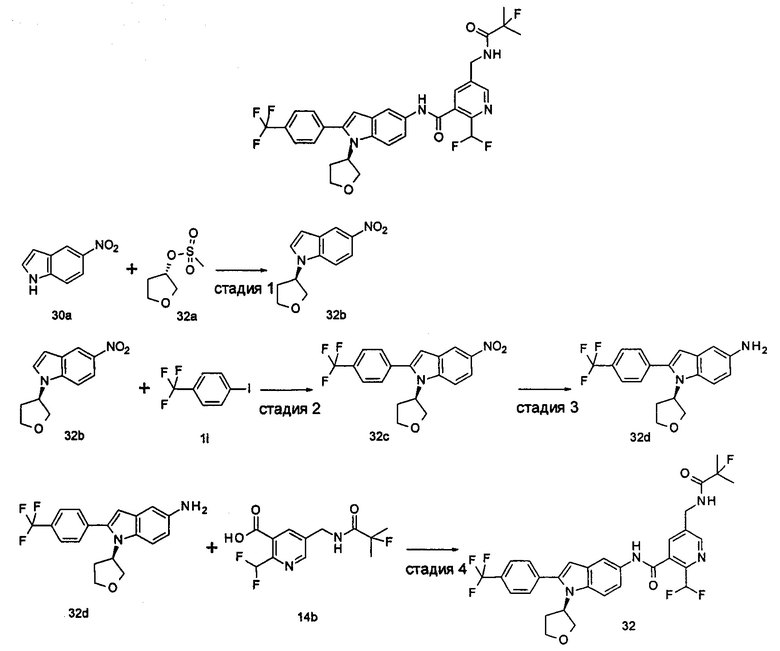
Стадия 1
(R)-5-Нитро-1-(тетрагидрофуран-3-ил)-1Н-индол
5-Нитроиндол 30а (5,0 г, 30,8 ммоль) и карбонат цезия (20,0 г, 61,3 ммоль) растворяли в 70 мл N,N-диметилацетамида, и затем добавляли (S)-тетрагидрофуран-3-ил метансульфонат 32а (10,0 г, 60,2 ммоль, полученный в соответствии со способом, раскрытым в "Nature Chemical Biology, 2008, 4(11), 691-699"). Реакционную смесь нагревали до 70°С, и перемешивали в течение 16 часов. Реакционный раствор выливали в 400 мл ледяной воды. Большое количество твердого вещества осаждали и отфильтровали. Фильтровальный осадок промывали водой (50 мл×3), и сушили с получением целевого соединения (R)-5-нитро-1-(тетрагидрофуран-3-ил)-1Н-индола 32b (7,0 г) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 233,0 [М+1]
Стадия 2
(R)-5-Нитро-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол
(R)-5-Нитро-1-(тетрагидрофуран-3-ил)-1Н-индол 32b (2,0 г, 8,6 ммоль) растворяли в 80 мл N,N-диметилацетамида, и затем последовательно добавляли 1-йод-4-(трифторметил)бензол 1i (2,34 г, 8,6 ммоль), трифенилфосфин (485 мг, 1,7 ммоль), ацетат палладия (200 мг, 0,86 ммоль) и ацетат цезия (2,5 г, 12,9 ммоль). Реакционную смесь нагревали до 140°С и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали при помощи ТСХ с элюирующей системой С с получением целевого соединения (R)-5-нитро-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индола 32с (300 мг, 9,4%) в виде желтого твердого вещества.
МС m/z (ESI): 377,3 [М+1]
Стадия 3
(R)-5-Амино-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол
(R)-5-Нитро-1-(тетргаидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол 32с (200 мг, 0,53 ммоль) растворяли в смеси 10 мл тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (20 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и фильтрат концентрировали при пониженном давлении с получением целевого соединения (R)-5-амино-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индола 32d (184 мг) в виде желтого твердого вещества, который использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 347,0 [М+1]
Стадия 4
(R)-2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамид
(R)-5-Амино-1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол 32d (160 мг, 0,55 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (190 мг, 0,55 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (158 мг, 0,83 ммоль), 1-гидроксибензотриазол (7 мг, 0,05 ммоль) и N,N-диизопропилэтиламин (142 мг, 1,1 ммоль) последовательно добавляли к 10 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением целевого соединения (R)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(1-(тетрагидрофуран-3-ил)-2-(4-(трифторметил)фенил)-1Н-индол-5-ил)никотинамида 32 (100 мг, 29,4%) в виде белого твердого вещества.
МС m/z (ESI):619,2 [M+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,64 (s, 1Н), 8,88 (s, 1Н), 8,69 (t, 1Н), 8,13 (s, 1Н), 8,01 (s, 1Н), 7,91-7,89 (m, 2Н), 7,78-7,76 (m, 2Н), 7,71-7,69 (m, 1Н), 7,40-7,39 (m, 1Н), 7,31 (t, 1Н), 6,65 (s, 1Н), 5,13 (m, 1Н), 4,48-4,46 (m, 2Н), 4,34-4,30 (m, 1Н), 4,17-4,14 (m, 1Н), 3,95-3,93 (m, 1Н), 3,67-3,65 (m, 1Н), 2,45-2,33 (m, 2Н), 1,53-1,48 (d, 6Н).
Пример 33
N-(2-(3-Хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
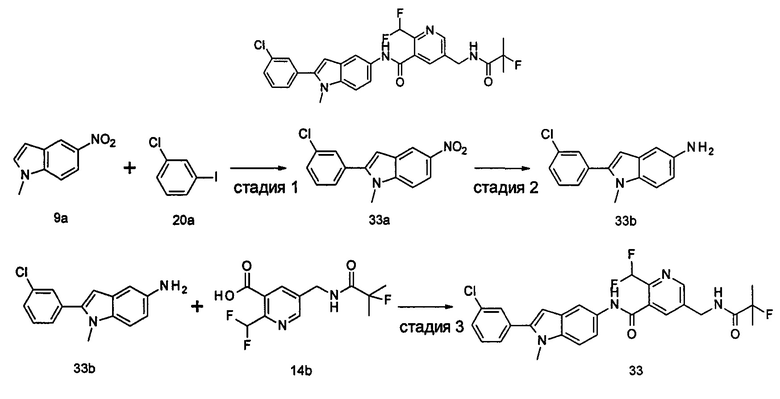
Стадия 1
2-(3-Хлорфенил)-1-метил-5-нитро-1Н-индол
1-Метил-5-нитро-1Н-индол 9а (2,0 г, 11,4 ммоль) растворяли в 20 мл N,N-диметилацетамида, и затем последовательно добавляли 1-хлор-3-йод-бензол 20а (2,7 г, 11,3 ммоль), трифенилфосфин (620 мг, 2,2 ммоль), ацетат палладия (250 мг, 1,1 ммоль) и ацетат цезия (4,2 г, 22,0 ммоль). Реакционную смесь нагревали до 140°С и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали при помощи ТСХ с элюирующей системой В с получением целевого соединения 2-(3-хлорфенил)-1-метил-5-нитро-1Н-индола 33а (300 мг, 9,5%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 287,1 [М+1]
Стадия 2
2-(3-Хлорфенил)-1-метил-1Н-индол-5-амин
2-(3-Хлорфенил)-1-метил-5-нитро-1Н-индол 33а (300 мг, 1,05 ммоль) растворяли в смеси 20 мл тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (30 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(3-хлорфенил)-1-метил-1Н-индол-5-амина 33b (269 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 257,1 [М+1]
Стадия 3
N-(2-(3-Хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
2-(3-Хлорфенил)-1-метил-1Н-индол-5-амин 33b (269 мг, 1,05 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (305 мг, 1,05 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (302 мг, 1,58 ммоль), 1-гидроксибензотриазол (14 мг, 0,10 ммоль) и N,N-диизопропилэтиламин (271 мг, 2,1 ммоль) последовательно добавляли к 10 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой Ас получением целевого соединения N-(2-(3-хлорфенил)-1-метил-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 33 (30 мг, 5,4% для двух стадий) в виде белого твердого вещества.
МС m/z (ESI): 529,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,60 (s, 1Н), 8,87 (s, 1Н), 8,69 (s, 1Н), 8,06 (d, 2Н), 7,69 (s, 1H), 7,56-7,51 (m, 4Н), 7,45-7,44 (m, 1Н), 7,20 (s, 1Н), 6,69 (s, 1Н), 4,48-4,46 (m, 2Н), 3,77 (s, 3Н), 1,54 (s, 3Н), 1,48 (s, 3Н)
Пример 34
2-Хлор-N-(2-(4-фторфенил)-1-метил-1Н-индол-5-ил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)бензамид
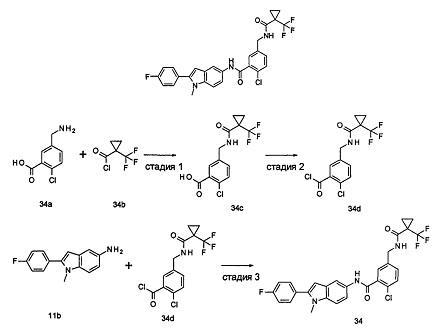
Стадия 1
2-Хлор-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)бензойная кислота
5-(Аминометил)-2-хлор-бензойную кислоту 34а (1,5 г, 6,8 ммоль, полученную в соответствии со способом, раскрытым в патентной заявке WO 2011048004) и N,N-диизопропилэтиламин (2,63 г, 20,3 ммоль) растворяли в 4 мл дихлорметана, и охлаждали до 0°С в ледяной бане. Добавляли 1-(трифторметил) циклопропанкарбонил хлорид 34b (1,28 г, 7,5 ммоль, полученный в соответствии со способом, раскрытым в патентной заявке WO 2005023773), подогревали реакционную смесь до комнатной температуры и перемешивали в течение 5 часов. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-хлор-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)бензойной кислоты 34с (2,4 г) в виде бледно-желтого масла, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 320,1 [М-1]
Стадия 2
2-Хлор-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)бензоилхлорид
2-Хлор-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)бензойную кислоту 34с (200 мг, 0,62 ммоль) и тионилхлорид (222 мг, 1,86 ммоль) растворяли в 20 мл дихлорметана, и затем добавляли одну каплю N,N-диметилформамида. Реакционную смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-хлор-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)бензоилхлорида 34d (210 мг) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 3
2-Хлор-N-(2-(4-фторфенил)-1-метил-1Н-индол-5-ил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)бензамид
2-(4-Фторфенил)-1-метил-5-амино-1Н-индол 11b (85 мг, 0,35 ммоль) растворяли в 10 мл дихлорметана, и затем добавляли 2-хлор-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)бензоилхлорид 34d (120 мг, 0,35 ммоль) и N,N-диизопропилэтиламин (90 мг, 0,70 ммоль). Реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(2-(4-фторфенил)-1-метил-1Н-индол-5-ил)-5-(((1-(трифторметил)циклопропанкарбонил)амино)метил)бензамида 34 (20 мг, 10,5%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 544,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,37 (s, 1Н), 8,47 (s, 1Н), 8,06 (s, 1Н), 7,66-7,64 (m, 2H), 7,53-7,35 (m, 7H), 6,58 (s, 1H), 4,34-4,32 (m, 2H), 3,73 (s, 3H), 1,34-1,24 (m, 4H).
Пример 35
N-(2-(3-Хлорфенил)-1-метил-1Н-пирроло[3,2-b]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
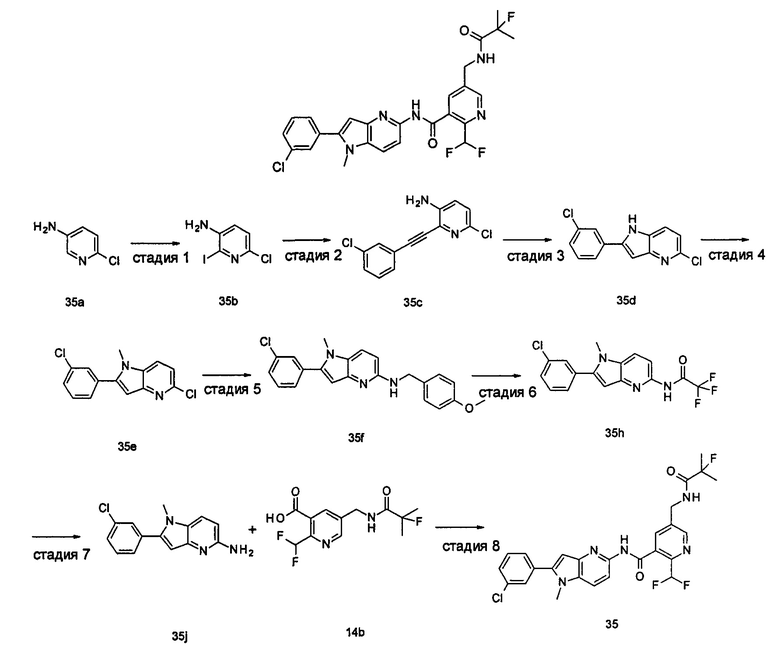
Стадия 1
6-Хлор-2-йод-пиридин-3-амин
6-Хлорпиридин-3-амин 35а (1 г, 7,78 ммоль) растворяли в 20 мл N,N-диметилформамида, и затем добавляли N-йодсукцинимид (2,27 г, 10,11 ммоль). Реакционный раствор перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения 6-хлор-2-йод-пиридин-3-амина 35b (1,37 г, 69,2%) в виде коричневого твердого вещества.
Стадия 2
6-Хлор-2-(2-(3-хлорфенил)этинил)пиридин-3-амин
6-Хлор-2-йод-пиридин-3-амин 35b (1,37 г, 5,38 ммоль), 3-хлорфенилацетилен (882 мг, 6,46 ммоль), бис(трифенилфосфин)палладия(II) хлорид (378 мг, 0,54 ммоль), йодид меди(I) (205 мг, 1,08 ммоль) и N,N-диизопропилэтиламин (1,39 г, 10,76 ммоль) добавляли к 10 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 16 часов при 100°С в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения 6-хлор-2-(2-(3-хлорфенил)этинил)пиридин-3-амина 35с (871 мг, 61,6%) в виде коричневого твердого вещества.
Стадия 3
5-Хлор-2-(3-хлорфенил)-1Н-пирроло[3,2-b]пиридин
6-Хлор-2-(2-(3-хлорфенил)этинил)пиридин-3-амин 35с (871 мг, 3,32 ммоль) и трет-бутанолят калия (746 мг, 6,64 ммоль) растворяли в 20 мл N,N-диметилформамида. Полученную смесь перемешивали в течение 16 часов при 70°С. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли 10 мл этилацетата и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 5-хлор-2-(3-хлорфенил)-1Н-пирроло[3,2-b]пиридина 35d (1,17 г) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 4
5-Хлор-2-(3-хлорфенил)-1-метил-1Н-пирроло[3,2-b]пиридин
5-Хлор-2-(3-хлорфенил)-1Н-пирроло[3,2-b]пиридин 35d (1,17 г, 4,46 ммоль) растворяли в 15 мл N,N-диметилформамида и затем добавляли 60% гидрид натрия (268 мг, 6,69 ммоль). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Добавляли йодметан (761 мг, 5,36 ммоль) и непрерывно перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения 5-хлор-2-(3-хлорфенил)-1-метил-1Н-пирроло[3,2-b]пиридина 35е (650 мг, 52,8%) в виде желтого твердого вещества.
Стадия 5
2-(3-Хлорфенил)-N-(4-метоксибензил)-1-метил-1H-пирроло[3,2-b]пиридин-5-амин
5-Хлор-2-(3-хлорфенил)-1-метил-1H-пирроло[3,2-b]пиридин 35е (276 мг, 1,0 ммоль), 4-метоксибензиламин (172 мг, 1,25 ммоль), трис(дибензилиденацетон)дипалладий (92 мг, 0,1 ммоль), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (125 мг, 0,2 ммоль), фосфат калия (425 мг, 2 ммоль) растворяли в 15 мл 1,4-диоксана. Реакционную смесь перемешивали в течение 16 часов при 100°С в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(3-хлорфенил)-N-(4-метоксибензил)-1-метил-1Н-пирроло[3,2-b]пиридин-5-амина 35f (221 мг, 58,5%) в виде желтого твердого вещества.
Стадия 6
N-(2-(3-Хлорфенил)-1-метил-1Н-пирроло[3,2-b]пиридин-5-ил)-2,2,2-трифтор-ацетамид
2-(3-Хлорфенил)-N-(4-метоксибензил)-1-метил-1Н-пирроло[3,2-b]пиридин-5-амин 35f (221 мг, 0,58 ммоль) растворяли в 5 мл трифторуксусной кислоты. Полученную смесь перемешивали в течение 16 часов при 60°С. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного целевого соединения N-(2-(3-хлорфенил)-1-метил-1Н-пирроло[3,2-b]пиридин-5-ил)-2,2,2-трифтор-ацетамида 35h (207 мг) в виде коричневого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 7
2-(3-Хлорфенил)-1-метил-1H-пирроло[3,2-b]пиридин-5-амин
N-(2-(3-Хлорфенил)-1-метил-1Н-пирроло[3,2-b]пиридин-5-ил)-2,2,2-трифтор-ацетамид 35h (207 мг, 0,58 ммоль) и карбонат калия (162 мг, 1,17 ммоль) растворяли в 10 мл этанола. Реакционную смесь перемешивали в течение 1 часа при 80°С. Реакционную смесь концентрировали при пониженном давлении, остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(3-хлорфенил)-1-метил-1H-пирроло[3,2-b]пиридин-5-амина 35j (30 мг, 20%) в виде бледно-желтого твердого вещества.
Стадия 8
N-(2-(3-Хлорфенил)-1-метил-1Н-пирроло[3,2-b]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (34 мг, 0,12 ммоль) и 2-(3-хлорфенил)-1-метил-1Н-пирроло[3,2-b]пиридин-5-амин 35j (30 мг, 0,12 ммоль) растворяли в 15 мл N,N-диметилформамида, и затем добавляли триэтиламин (47 мг, 0,46 ммоль), 1-гидроксибензотриазол (16 мг, 0,12 ммоль) и 1-этил-(3-диметиламинопропил) карбодиимида гидрохлорид (34 мг, 0,17 ммоль). Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения N-(2-(3-хлорфенил)-1-метил-1H-пирроло[3,2-b]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 35 (3 мг, 4,8%) в виде белого твердого вещества.
МС m/z (ESI): 530,1 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,63 (s, 1Н), 8,34 (s, 1Н), 7,96 (s, 1Н), 7,82 (s, 1Н), 7,54-7,43 (m, 4Н), 7,31 (s, 1Н), 6,93 (s, 1Н), 6,42 (s, 1Н), 4,43 (s, 2Н), 3,83 (s, 3Н), 1,64 (s, 3Н), 1,59 (s, 3Н).
Пример 36
N-(2-(3-Хлорфенил)-1-метил-1Н-пирроло[2,3-с]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
Стадия 1
4-Йод-6-нитро-пиридин-3-амин
5-Амино-2-нитропиридин 36а (1 г, 7,2 ммоль), йодат калия (770 мг, 3,6 ммоль) и йодид калия (1,2 г, 7,2 ммоль) последовательно добавляли к 30 мл серной кислоты (2N). Реакционный раствор перемешивали в течение 16 часов при 80°С и затем охлаждали до комнатной температуры. Реакционную смесь доводили до рН 10 водным раствором гидроксида натрия (2N). Большое количество твердого вещества осаждали и отфильтровывали. Фильтровальный осадок промывали водой, и сушили с получением целевого соединения 4-йод-6-нитро-пиридин-3-амина 36b (1,5 г, 79%) в виде желтого твердого вещества.
Стадия 2
4-((3-Хлорфенил)этинил)-6-нитро-пиридин-3-амин
4-Йод-6-нитро-пиридин-3-амин 36b (1,5 г, 5,7 ммоль), 3-хлорфенилацетилен (850 мг, 6,2 ммоль), йодид меди (1,1 г, 5,7 ммоль), (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладий(II) (834 мг, 1,14 ммоль) и триэтиламин (1,6 мл, 11,4 ммоль) последовательно добавляли к 20 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 16 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением целевого соединения 4-((3-хлорфенил)этинил)-6-нитро-пиридин-3-амина 36с (1,5 г, 97%) в виде желтого твердого вещества.
Стадия 3
2-(3-Хлорфенил)-5-нитро-1H-пирроло[2,3-с]пиридин
4-((3-Хлорфенил)этинил)-6-нитро-пиридин-3-амин 36с (200 мг, 0,73 ммоль), тетрабутанолят калия (123 мг, 1,1 ммоль) и 10 мл N,N-диметилформамида добавляли в колбу объемом 50 мл. Полученную смесь перемешивали в течение 16 часов при 60°С. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(3-хлорфенил)-5-нитро-1H-пирроло[2,3-с]пиридина 36d (300 мг) в виде коричневого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 4
2-(3-Хлорфенил)-1-метил-5-нитро-1H-пирроло[2,3-с]пиридин
2-(3-Хлорфенил)-5-нитро-1Н-пирроло[2,3-с]пиридин 36d (300 мг, 1,1 ммоль), йодметан (233 мг, 1,64 ммоль), карбонат цезия (717 мг, 2,2 ммоль) и 10 мл N,N-диметилформамида добавляли в колбу объемом 100 мл. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(3-хлорфенил)-1-метил-5-нитро-1Н-пирроло[2,3-с]пиридина 36е (50 мг, 15,8%) в виде желтого твердого вещества.
Стадия 5
2-(3-Хлорфенил)-1-метил-1Н-пирроло[2,3-с]пиридин-5-амин
2-(3-Хлорфенил)-1-метил-5-нитро-1Н-пирроло[2,3-с]пиридин 36е (50 мг, 0,17 ммоль), никель Ренея (5 мг) и 10 мл тетрагидрофурана добавляли в колбу объемом 50 мл. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре в атмосфере водорода. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением целевого соединения 2-(3-хлорфенил)-1-метил-1Н-пирроло[2,3-с]пиридин-5-амина 36f (40 мг, 88,9%) в виде бледно-желтого твердого вещества.
Стадия 6
N-(2-(3-Хлорфенил)-1-метил-1H-пирроло[2,3-с]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (45 мг, 0,16 ммоль), 2-(3-хлорфенил)-1-метил-1H-пирроло[2,3-c]пиридин-5-амин 36f (40 мг, 0,16 ммоль), 1-гидроксибензотриазол (2,2 мг, 0,016 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (46 мг, 0,24 ммоль) и N,N-диизопропилэтиламин (31 мг, 0,24 ммоль) растворяли в N,N-диметилформамиде. Реакционную смесь перемешивали в течение 2 часов при 70°С. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой Ас получением целевого соединения N-(2-(3-хлорфенил)-1-метил-1Н-пирроло[2,3-с]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил) никотинамида 36 (20 мг, 23,5%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 530,1 [(М+1])
1Н ЯМР (400 МГц, DMSO-d6): δ 11,15 (s, 1Н), 8,82-8,80 (m, 1Н), 8,67 (s, 1Н), 8,07-8,05 (m, 3Н), 7,75 (s, 1Н), 7,66-7,65 (m, 1Н), 7,57-7,55 (m, 2Н), 7,21 (s, 1Н), 6,72 (s, 1Н), 4,46-4,44 (d, 2Н), 3,83 (s, 3Н), 1,53 (s, 3Н), 1,48 (s, 3Н).
Пример 37
N-(2-(3-Хлорфенил)-1-этил-1Н-пирроло[3,2-b]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
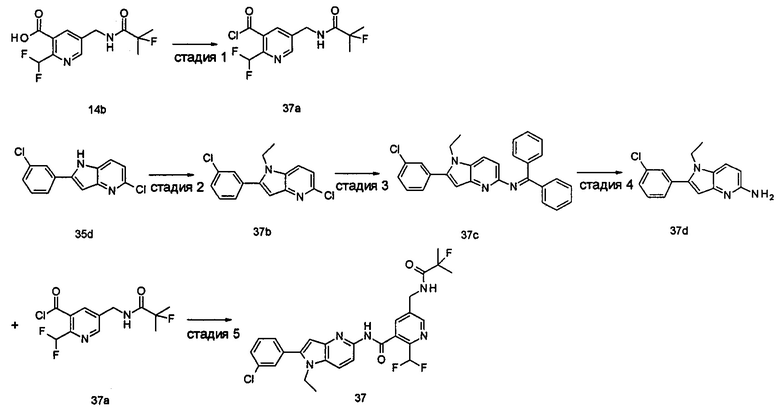
Стадия 1
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиноилхлорид
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (1,5 г, 5,17 ммоль) растворяли в 10 мл дихлорметана, и затем добавляли тионилхлорид (1,2 мл, 15,5 ммоль) и 2 капли N,N-диметилформамида. Реакционный раствор перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиноилхлорида 37а (1,6 г) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 2
5-Хлор-2-(3-хлорфенил)-1-этил-1Н-пирроло[3,2-b]пиридин
5-Хлор-2-(3-хлорфенил)-1Н-пирроло[3,2-b]пиридин 35d (0,87 г, 3,32 ммоль) растворяли в 20 мл N,N-диметилформамида и затем добавляли 60% гидрид натрия (0,2 г, 4,98 ммоль). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре и затем добавляли йодэтан (0,62 г, 3,98 ммоль) и непрерывно перемешивали еще в течение 2 часов при комнатной температуре. К реакционной смеси добавляли 10 мл воды и концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 5-хлор-2-(3-хлорфенил)-1-этил-1Н-пирроло[3,2-b]пиридина 37b (0,35 г, 36,3%) в виде желтого масла.
Стадия 3
(2-(3-Хлорфенил)-N-(дифенилметиленил)-1-этил-1H-пирроло[3,2-b]пиридин-5-амин
5-Хлор-2-(3-хлорфенил)-1-этил-1H-пирроло[3,2-b]пиридин 37b (30 мг, 0,1 ммоль), бензофенон имин (22 мг, 0,12 ммоль), трис(дибензилиденацетон)дипалладий (9,5 мг, 0,01 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (12 мг, 0,021 ммоль) и фосфат калия (44 мг, 0,21 ммоль) растворяли в 2 мл 1,4-диоксана. Полученную смесь перемешивали в течение 16 часов при 100°С в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли небольшое количество тетрагидрофурана и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения (2-(3-хлорфенил)-N-(дифенилметиленил)-1-этил-1Н-пирроло[3,2-b]пиридин-5-амина 37с (50 мг) в виде серого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 4
2-(3-Хлорфенил)-1-этил-1Н-пирроло[3,2-b]пиридин-5-амин
(2-(3-Хлорфенил)-N-(дифенилметиленил)-1-этил-1H-пирроло[3,2-b]пиридин-5-амин 37с (50 мг, 0,12 ммоль) растворяли в 10 мл тетрагидрофурана и затем добавляли 4 мл 2 М раствора хлороводородной кислоты. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, и затем доводили до рН более 7 насыщенным раствором бикарбоната натрия. Смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением неочищенного целевого соединения 2-(3-хлорфенил)-1-этил-1Н-пирроло[3,2-b]пиридин-5-амина 37d (62 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 5
N-(2-(3-Хлорфенил)-1-этил-1Н-пирроло[3,2-b]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиноилхлорид 37а (13,5 мг, 0,044 ммоль) растворяли в 10 мл дихлорметана, и затем добавляли триэтиламин (94 мг, 0,088 ммоль) и 2-(3-хлорфенил)-1-этил-1Н-пирроло[3,2-b]пиридин-5-амин 37d (11,9 мг, 0,044 ммоль). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения N-(2-(3-хлорфенил)-1-этил-1Н-пирроло[3,2-b]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 37 (5 мг, 20,8%) в виде белого твердого вещества.
МС m/z (ESI): 544,1 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 13,16 (s, 1Н), 8,84-8,82 (d, 2Н), 8,72-8,70 (d, 1Н), 8,26-8,24 (d, 1Н), 7,57-7,52 (m, 3Н), 7,43-7,42 (d, 1Н), 7,36-7,31 (m, 2Н), 6,81 (s, 1Н), 4,71-4,69 (d, 2Н), 4,39-4,33 (m, 2Н), 1,64 (s, 3Н), 1,59 (s, 3Н), 1,44-1,42 (m, 3Н).
Пример 38
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)никотинамид
2-(4-(Трифторметил)фенил)бензо[d]оксазол-5-амин 22с (120 мг, 0,44 ммоль) растворяли в 5 мл N,N-диметилформамида, и затем добавляли 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (100 мг, 0,37 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (140 мг, 0,74 ммоль), 1-гидроксибензотриазол (5 мг, 0,037 ммоль) и триэтиламин (200 мкл, 1,47 мкмоль). Полученную смесь нагревали до 70°С и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)никотинамида 38 (60 мг, 30,6%) в виде белого твердого вещества.
МС m/z (ESI): 533,2 [M+1]
1H ЯМР (400 МГц, DMSO-d6): δ 11,00 (s, 1Н), 8,81 (s, 1Н), 8,61 (t, 1Н), 8,21-8,27 (m, 3Н), 8,10 (s, 1Н), 7,83 (d, 1Н), 7,72 (d, 3Н), 7,22 (t, 1Н), 4,44 (d, 2Н), 2,49 (d, 1Н), 1,06 (d, 6H).
Пример 39
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)бензофуран-5-ил)никотинамид
5-Амино-2-(4-(трифторметил)фенил)бензофуран 29d (70 мг, 0,25 ммоль), 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (69 мг, 0,25 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (97 мг, 0,51 ммоль), 1-гидроксибензотриазол (3 мг, 0,025 ммоль) и триэтиламин (140 мкл, 0,80 мкмоль) последовательно добавляли к 5 мл N,N-диметилформамида. Полученную смесь нагревали до 70°С и перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-((2-метилпропаноиламино) метил)-N-(2-(4-(трифторметил)фенил)бензофуран-5-ил)никотинамида 39 (70 мг, 52,2%) в виде белого твердого вещества.
MC m/z (ESI): 532,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,81 (s, 1Н), 8,70 (s, 1Н), 8,48 (s, 1Н), 8,20 (s, 1Н), 8,15 (d, 2Н), 8,04 (s, 1Н), 7,89 (d, 2Н), 7,73 (s, 1Н), 7,69 (s, 1Н), 7,59 (s, 1Н), 7,19 (s, 1Н), 4,44 (d, 2Н), 2,47 (d, 1Н), 1,06 (d, 6Н).
Пример 40
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)изоиндолин-5-ил)никотинамид
2-(4-(Трифторметил)фенил)изоиндолин-5-амин 25d (10 мг, 0,036 ммоль) и 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (10 мг, 0,036 ммоль) растворяли в 5 мл N,N-диметилформамида, и затем добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (11 мг, 0,054 ммоль) и 1-гидроксибензотриазол (5 мг, 0,036 ммоль). Полученную смесь перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)изоиндолин-5-ил)никотинамида 40 (10 мг, 52,6%) в виде белого твердого вещества.
МС m/z (ESI): 533,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,62 (s, 1Н), 8,66 (s, 1Н), 8,44 (d, 1Н), 8,07 (s, 1Н), 8,00 (s, 1Н), 7,88-7,85 (m, 4Н), 7,54 (d, 1Н), 7,47 (d, 1Н), 7,42 (t, 1Н), 4,60 (s, 4Н), 4,41 (d, 2Н), 3,18-3,17 (m, 1Н), 1,05 (s, 6Н)
Пример 41
2-Хлор-5-(((1-метилциклопропанкарбонил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)бензамид
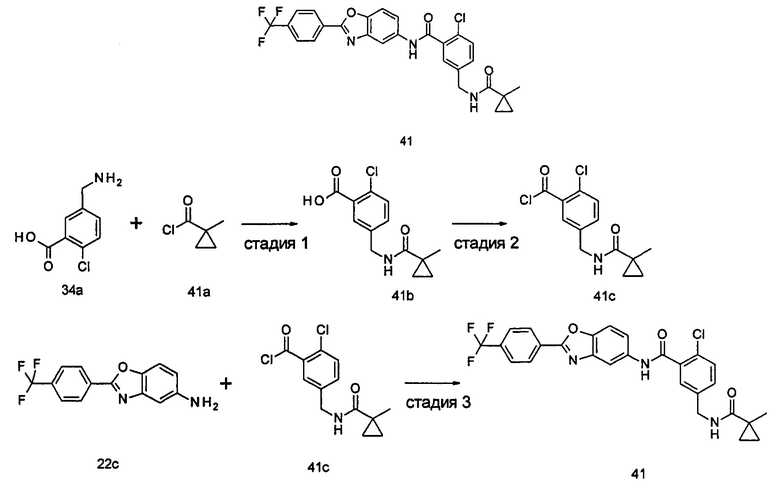
Стадия 1
2-Хлор-5-(((1-метилциклопропанкарбонил)амино)метил)бензойная кислота
5-(Аминометил)-2-хлор-бензойную кислоту 34а (1,5 г, 6,8 ммоль, полученную в соответствии со способом, раскрытым в патентной заявке WO 2011048004) и триэтиламин (3,2 мл, 23,2 ммоль) растворяли в 15 мл тетрагидрофурана и затем охлаждали до 0°С в ледяной бане. Раствор 1-метилциклопропанкарбонилхлорида 41а (890 мг, 7,47 ммоль, полученный в соответствии со способом, раскрытым в "Journal of Medicinal Chemistry, 2014, 57(22), 9323-9342") в 5 мл тетрагидрофурана по каплям добавляли к полученной выше смеси. Реакционную смесь подогревали до комнатной температуры и затем перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-хлор-5-(((1-метил циклопропанкарбонил)амино) метил)бензойной кислоты 41b (4,78 г,) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 266,1 [М-1]
Стадия 2
2-Хлор-5-(((1-метилциклопропанкарбонил)амино)метил)бензоилхлорид
2-Хлор-5-(((1-метилциклопропанкарбонил)амиино)метил)бензойную кислоту 41b (200 мг, 0,75 ммоль) растворяли в 10 мл дихлорметана и затем добавляли тионилхлорид (0,2 мл, 2,75 ммоль). Реакционный раствор перемешивали в течение 15 минут при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-хлор-5-(((1-метилциклопропанкарбонил)амино)метил)бензоилхлорида 41с (220 мг) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 3
2-Хлор-5-(((1-метилциклопропанкарбонил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)бензамид
2-(4-(Трифторметил)фенил)бензо[d]оксазол-6-амин 22с (90 мг, 0,33 ммоль) и триэтиламин (90 мкл, 0,65 мкмоль) растворяли в 5 мл тетрагидрофурана, и затем по каплям добавляли раствор 2-хлор-5-(((1-метилциклопропанкарбонил) амино)метил)бензоилхлорида 41с (220 мг, 0,72 ммоль) в 5 мл тетрагидрофурана. Реакционную смесь перемешивали в течение 30 минут. Реакционную смесь фильтровали и концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-5-(((1-метилциклопропанкарбонил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)бензамида 41 (30 мг, 17,4% для двух стадий) в виде желтого твердого вещества.
МС m/z (ESI): 528,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,78 (s, 1Н), 8,42 (d, 2Н), 8,33 (s, 1Н), 8,23 (t, 1Н), 8,01 (d, 2Н), 7,84 (d, 1Н), 7,72 (d, 1Н), 7,54 (d, 1Н), 7,49 (s, 1Н), 7,39 (d, 1Н), 4,31 (d, 2Н), 1,30 (s, 3Н), 0,97 (d, 2Н), 0,54 (d, 2Н).
Пример 42
N-(2-(4-Хлорфенил)-1-этил-1Н-индол-5-ил)-2-(дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)никотинамид
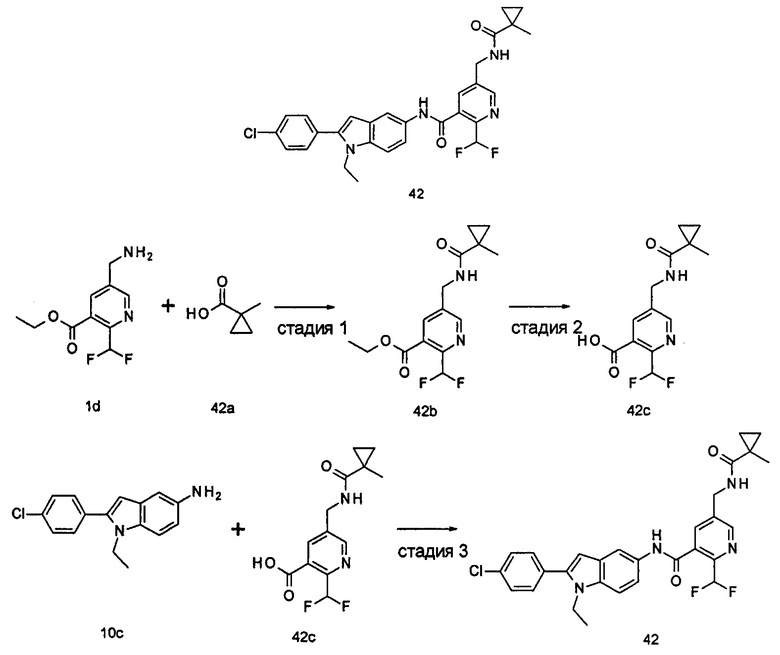
Стадия 1
Этил 2-(дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)никотинат
Этил 5-(аминометил)-2-(дифторметил)никотината гидрохлорид 1d (1,8 г, 5,94 ммоль) растворяли в 25 мл N,N-диметилформамида, и затем последовательно добавляли 1-метилциклопропан-1-карбоновую кислоту 42а (594 мг, 5,94 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (2,28 н, 11,9 ммоль), 1-гидроксибензотриазол (80 мг, 0,59 ммоль) и триэтиламин (3,3 мл, 23,8 ммоль). Полученную смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли 50 мл воды и экстрагировали этилацетатом (25 мл×3). Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с элюирующей системой С с получением целевого соединения этил 2-(дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)никотината 42b (1,2 г, 64,9%) в виде желтого масла.
МС m/z (ESI): 313,2 (M+1)
Стадия 2
2-(Дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)никотиновая кислота
Этил 2-(дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил) никотинат 42b (1,2 г, 3,85 ммоль) растворяли в 30 мл смеси 1,4-диоксана и воды (соотношение объемов 2:1), и затем добавляли гидрат гидроксида лития (400 мг, 9,62 ммоль). Реакционную смесь перемешивали в течение 16 часов. 1,4-Диоксан удаляли при пониженном давлении, и остаток доводили до рН 4-5 6М раствором хлороводородной кислоты. Часть твердого вещества осадили, к ней добавляли 30 мл этилацетата и фильтровали. Фильтровальный осадок собирали и фильтрат наслаивали. Органическую фазу сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток объединяли с фильтровальным осадком, полученным выше, с получением целевого соединения 2-(дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)пиридин-3-карбоновой кислоты 42 с (1,0 г, 91,7%) в виде белого твердого вещества.
МС m/z (ESI): 285,1 [М+1].
Стадия 3
N-(2-(4-Хлорфенил)-1-этил-1H-индол-5-ил)-2-(дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)никотинамид
2-(Дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)никотиновую кислоту 42с (52 мг, 0,19 ммоль), 1-этил-2-(4-хлорфенил)-5-амино-1Н-индол 10с (50 мг, 0,19 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (71 мг, 0,37 ммоль), 1-гидроксибензотриазол (2,5 мг, 18,5 мкмоль) и триэтиламин (100 мкл, 0,74 ммоль) растворяли в 5 мл N,N-диметилформамида. Реакционную смесь нагревали до 70°С и перемешивали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения N-(2-(4-хлорфенил)-1-этил-1Н-индол-5-ил)-2-(дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)никотинамида 42 (40 мг, 40,4%) в виде желтого твердого вещества.
МС m/z (ESI): 538,2 [М+1]
1H ЯМР (400 МГц, DMSO-d6): δ 10,62 (s, 1Н), 8,69 (s, 1Н), 8,31 (s, 1Н), 8,04 (d, 2Н), 7,55-7,69 (m, 3Н), 7,44 (d, 2Н), 7,19 (t, 1Н), 6,60 (s, 1Н), 4,43 (d, 2Н), 4,22 (d, 2Н), 1,32 (s, 3Н), 1,20 (t, 3Н), 1,00 (s, 3Н), 0,56 (s, 2Н).
Пример 43
2-(Дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил)никотинамид
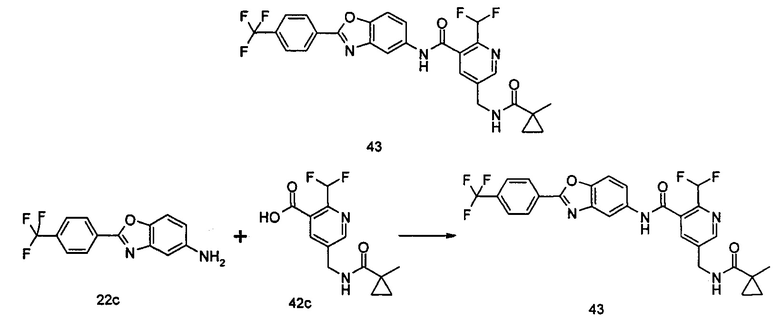
2-(4-(Трифторметил)фенил)бензо[d]оксазол-5-амин 22с (50 мг, 0,18 ммоль) растворяли в 5 мл N,N-диметилформамида и затем добавляли 2-(дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)никотиновую кислоту 42с (51 мг, 0,18 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (75 мг, 0,39 ммоль), 1-гидроксибензотриазол (2,4 мг, 0,018 ммоль) и триэтиламин (0,1 мл, 720 мкмоль). Реакционную смесь нагревали до 70°С, и перемешивали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((1-метилциклопропанкарбонил)амино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]оксазол-5-ил) никотинамида 43 (20 мг, 20,4%) в виде желтого твердого вещества.
МС m/z (ESI): 545,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,95 (s, 1Н), 8,70 (d, 1Н), 8,43 (d, 2Н), 8,30 (t, 2Н), 8,03 (d, 3Н), 7,87 (d, 1Н), 7,72 (d, 1Н), 7,19 (t, 1Н), 4,43 (d, 2Н), 1,31 (s, 3Н), 0,99 (d, 2Н), 0,56 (d, 2Н).
Пример 44
N-(2-(4-Хлорфенил)-1-метил-1H-пирроло(2,3-b)пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
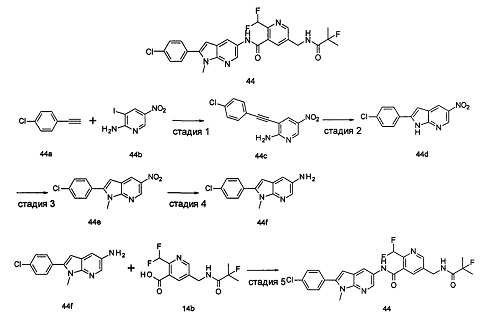
Стадия 1
3-((4-Хлорфенил)этинил)-5-нитро-пиридин-2-амин
2-Амино-3-йод-5-нитро-пиридин 44b (300 мг, 1,13 ммоль), 4-хлорфенилацетилен 44а (325 мг, 2,38 ммоль), бис(трифенилфосфин)палладия(II) хлорид (40 мг, 56,5 мкмоль), йодид меди(I) (11 мг, 56,5 мкмоль) и триэтиламин (1,6 мл, 11,3 ммоль) последовательно добавляли к 20 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 16 часов в атмосфере аргона. К реакционной смеси добавляли 100 мл этилацетата, и промывали водой (50 мл×2). Органические фазы сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 3-((4-хлорфенил)этинил)-5-нитро-пиридин-2-амина 44с (250 мг, 80,9%) в виде желтого твердого вещества.
МС m/z (ESI): 272,0 [М-1]
Стадия 2
2-(4-хлорфенил)-5-нитро-1H-пирроло(2,3-b)пиридин
3-((4-Хлорфенил)этинил)-5-нитро-пиридин-2-амин 44с (160 мг, 0,58 ммоль) и третбутанолят калия (746 мг, 6,64 ммоль) растворяли в 10 мл N,N-диметилформамида. Реакционную смесь нагревали до 70°С и перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток смешивали с 50 мл этилацетата и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(4-хлорфенил)-5-нитро-1H-пирроло(2,3-b)пиридина 44d (160 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 273,0 [М-1]
Стадия 3
2-(4-Хлорфенил)-1-метил-5-нитро-1H-пирроло[2,3-b]пиридин
2-(4-Хлорфенил)-5-нитро-1Н-пирроло[2,3-b]пиридин 44d (160 мг, 0,58 ммоль) растворяли в 5 мл N,N-диметилформамида и затем добавляли карбонат цезия (380 мг, 1,17 ммоль) и йодметан (73 мкл, 1,17 ммоль). Реакционную смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения 2-(4-хлорфенил)-1-метил-5-нитро-1H-пирроло[2,3-b]пиридина 44е (30 мг, 17,9% для двух стадий) в виде желтого твердого вещества.
МС m/z (ESI): 290,0 [М+1]
Стадия 4
2-(4-Хлорфенил)-1-метил-1H-пирроло[2,3-b]пиридин-5-амин
2-(4-Хлорфенил)-1-метил-5-нитро-1H-пирроло[2,3-b]пиридин 44е (50 мг, 0,17 ммоль) растворяли в смеси 10 мл тетрагидрофурана и метанола (соотношение объемов 1:1) и затем добавляли никель Ренея (30 мг). Реакционную смесь перемешивали в течение 1 часа в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и концентрировали фильтрат при пониженном давлении с получением неочищенного целевого соединения 2-(4-хлорфенил)-1-метил-1Н-пирроло[2,3-b]пиридин-5-амина 44f (45 мг) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 259,1 [М+1]
Стадия 5
N-(2-(4-Хлорфенил)-1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
2-(4-Хлорфенил)-1-метил-1H-пирроло[2,3-b]пиридин-5-амин 44f (50 мг, 0,17 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил) никотиновую кислоту 14b (50 мг, 0,17 ммоль), 1-этил-(3-диметиламинопропил)-карбодиимида гидрохлорид (67 мг, 0,35 ммоль) и 1-гидроксибензотриазол (2,3 мг, 17,4 мкмоль) последовательно добавляли к 5 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения N-(2-(4-хлорфенил)-1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 44 (3 мг, 3,3%) в виде желтого твердого вещества.
МС m/z (ESI): 531,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,86 (s, 1Н), 8,71 (s, 1Н), 8,51 (t, 1Н), 8,20-8,27 (m, 2Н), 8,05 (s, 2Н), 7,82 (d, 1Н), 7,71 (d, 2Н), 7,19 (t, 1Н), 6,55 (s, 1Н), 4,47 (d, 2Н), 3,83 (s, 3Н), 1,54 (s, 3Н), 1,48 (s, 3Н).
Пример 45
N-(2-(4-Хлорфенил)бензо[d]оксазол-5-ил)-2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинамид
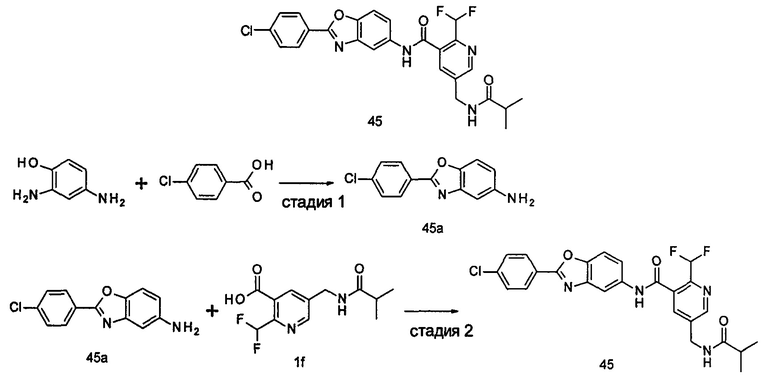
Стадия 1
2-(4-Хлорфенил)бензо[d]оксазол-5-амин
2,4-Диаминофенол (800 мг, 6,45 ммоль) и 4-хлорбензойную кислоту (1,1 г, 7,1 ммоль) растворяли в 10 мл полифосфорной кислоты. Реакционную смесь нагревали до 95°С и перемешивали в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и выливали в 50 мл ледяной воды, и доводили до рН 7, добавляя по каплям раствор гидроксида натрия. Смесь экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(4-хлорфенил)бензо[d]оксазол-5-амина 45а (90 мг, 5,7%) в виде серого твердого вещества.
МС m/z (ESI): 259,1 [М+1]
Стадия 2
N-(2-(4-Хлорфенил)бензо[d]оксазол-5-ил)-2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинамид 2-(4-Хлорфенил)бензо[d]оксазол-5-амин 45а (80 мг, 0,33 ммоль) и 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (81 мг, 0,30 ммоль) растворяли в 5 мл N,N-диметилформамида и затем добавляли 1-(3-диметиламинопропил)-3-этил-карбодиимида гидрохлорид (67 мг, 0,35 ммоль), 1-гидроксибензотриазол 4 мг, 0,030 ммоль) и триэтиламин (0,16 мл, 1,19 мкмоль). Реакционную смесь нагревали до 70°С и перемешивали в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, и затем концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения N-(2-(4-хлорфенил)бензо[d]оксазол-5-ил)-2-(дифторметил)-5-((2-метил пропаноиламино)метил)никотинамида 45 (6 мг, 4,1%) в виде коричневого твердого вещества.
МС m/z (ESI): 499,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,96 (s, 1Н), 8,70 (s, 1Н), 8,51 (t, 1Н), 8,20-8,27 (m, 3Н), 8,05 (s, 1Н), 7,82 (d, 1Н), 7,71 (d, 3Н), 7,20 (t, 1Н), 4,44 (d, 2Н), 2,49 (d, 1Н), 1,06 (d, 6H).
Пример 46
(S)-N-(2-(4-Хлорфенил)-1-(-тетрагидрофуран-3-ил)-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
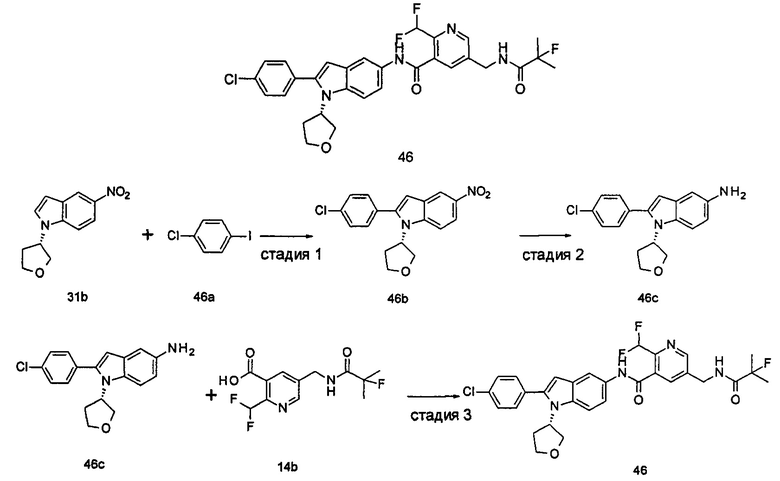
Стадия 1
(S)-2-(4-Хлорфенил)-5-нитро-1-(тетрагидрофуран-3-ил)-1Н-индол
(S)-5-Нитро-1-(тетрагидрофуран-3-ил)-1Н-индол 31b (2,0 г, 8,6 ммоль) растворяли в 15 мл N,N-диметилацетамида и затем последовательно добавляли 4-хлор-йод-бензол 46а (2,3 г, 9,5 ммоль), трифенилфосфин (450 мг, 1,7 ммоль), ацетат палладия (97 мг, 0,4 ммоль) и ацетат цезия (4,1 г, 21,6 ммоль). Реакционную смесь нагревали до 140°С и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь охлаждали до комнатной температуры и добавляли 200 мл воды, затем экстрагировали этилацетатом (200 мл×). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением целевого соединения (S)-2-(4-хлорфенил)-5-нитро-1-(тетрагидрофуран-3-ил)-1Н-индола 46b (100 мг, 3,4%) в виде желтого твердого вещества.
МС m/z (ESI): 343,1 [М+1]
Стадия 2
(S)-2-(4-Хлорфенил)-1-(тетрагидрофуран-3-ил)-1Н-индол-5-амин
(S)-2-(4-Хлорфенил)-5-нитро-1-(тетрагидрофуран-3-ил)-1Н-индол 46b (70 мг, 0,20 ммоль) растворяли в 10 мл смеси тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (20 мг). Реакционную смесь перемешивали в течение 3 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения (S)-2-(4-хлорфенил)-1-(тетрагидрофуран-3-ил)-1Н-индол-5-амина 46с (64 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 313,1 [М+1]
Стадия 3
(S)-N-(2-(4-Хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
(S)-2-(4-Хлорфенил)-1-(тетрагидрофуран-3-ил)-1Н-индол-5-амин 46с (64 мг, 0,21 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (100 мг, 0,35 ммоль), 1-этил-(3-диметиламинопропил) карбодиимида гидрохлорид (63 мг, 0,69 ммоль), 1-гидроксибензотриазол (4,7 мг, 0,03 ммоль) и триэтиламин (0,2 мл, 1,38 ммоль) последовательно добавляли к 5 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения (S)-N-(2-(4-хлорфенил)-1-(тетрагидрофуран-3-ил)-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил) никотинамида 46 (30 мг, 14,9%) в виде желтого твердого вещества.
МС m/z (ESI): 586,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,61 (s, 1Н), 8,86 (br, 1Н), 8,69 (d, 1Н), 8,09 (d, 1Н), 8,01 (s, 1Н), 7,67 (d, 2Н), 7,61 (d, 2Н), 7,55 (d, 1Н), 7,36 (d, 1Н), 7,18 (t, 1Н), 6,55 (s, 1Н), 5,11 (br, 1Н), 4,47 (d, 2Н), 4,32 (t, 1Н), 4,13 (dd, 1Н), 3,91 (t, 1Н), 3,66 (d, 1Н), 2,36-2,45 (m, 1Н), 2,26-2,32 (m, 1Н), 1,54 (s, 3Н), 1,48 (s, 3Н).
Пример 47
(R)-N-(2-(4-Хлорфенил)-1-(тетрагидрофуран-3-ил)-1Н-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
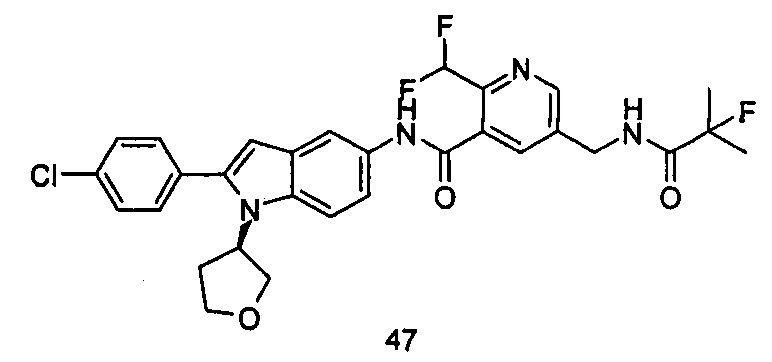
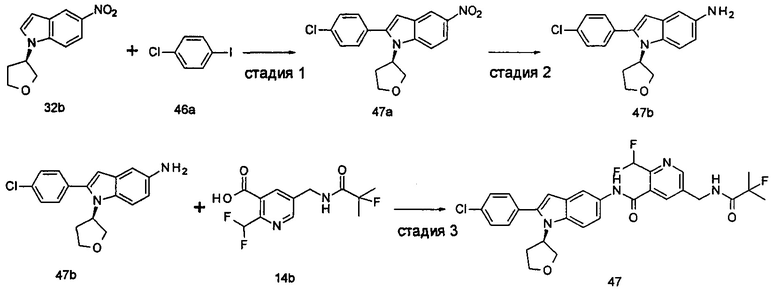
Стадия 1
(R)-2-(4-Хлорфенил)-5-нитро-1-(тетрагидрофуран-3-ил)-1Н-индол
(R)-5-Нитро-1-(тетрагидрофуран-3-ил)-1Н-индол 32b (2,0 г, 8,6 ммоль) растворяли в 30 мл N,N-диметилацетамида и затем последовательно добавляли 4-хлор-йод-бензол 46а (2,1 г, 8,6 ммоль), трифенилфосфин (508 мг, 1,8 ммоль), ацетат палладия (200 мг, 0,86 ммоль) и ацетат цезия (3,5 г, 18 ммоль). Реакционную смесь нагревали до 140°С и перемешивали в течение 18 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой В с получением целевого соединения (R)-2-(4-хлорфенил)-5-нитро-1-(тетрагидрофуран-3-ил)-1Н-индола 47а (280 мг, 9,0%) в виде желтого твердого вещества.
МС m/z (ESI): 343,9 [М+1]
Стадия 2
(R)-2-(4-Хлорфенил)-1-(тетрагидрофуран-3-ил)-1Н-индол-5-амин
(R)-2-(4-Хлорфенил)-5-нитро-1-(тетрагидрофуран-3-ил)-1Н-индол 47а (280 мг, 0,82 ммоль) растворяли в 20 мл и затем добавляли никель Ренея (28 мг). Реакционную смесь перемешивали в течение 3 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением целевого соединения (R)-2-(4-хлорфенил)-1-(тетрагидрофуран-3-ил)-1Н-индол-5-амина 47b (230 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 313,1 [М+1]
Стадия 3
(R)-N-(2-(4-Хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамид
(R)-2-(4-Хлорфенил)-1-(тетрагидрофуран-3-ил)-1Н-индол-5-амин 46с (230 мг, 0,74 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (213 мг, 0,74 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (211 мг, 1,1 ммоль), 1-гидроксибензотриазол (10 мг, 0,074 ммоль) и N,N-диизопропилэтиламин (200 мг, 1,47 ммоль) последовательно добавляли к 5 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения (R)-N-(2-(4-хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-индол-5-ил)-2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотинамида 47 (100 мг, 23,3%) в виде белого твердого вещества.
МС m/z (ESI): 586,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,61 (s, 1Н), 8,86 (t, 1Н), 8,68 (s, 1Н), 8,09 (s, 1Н), 8,01 (s, 1Н), 7,76 (d, 1Н), 7,61 (d, 2Н), 7,55 (d, 2Н), 7,38 (d, 1Н), 7,18 (t, 1Н), 6,55 (s, 1Н), 5,10 (br, 1Н), 4,47 (d, 2Н), 4,31 (t, 1Н), 4,13 (d, 1Н), 3,92 (t, 1Н), 3,65 (dd, 1Н), 2,40 (d, 1Н), 2,32 (d, 1Н), 1,54 (s, 3Н), 1,48 (s, 3Н).
Пример 48
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)бензофуран-5-ил)никотинамид
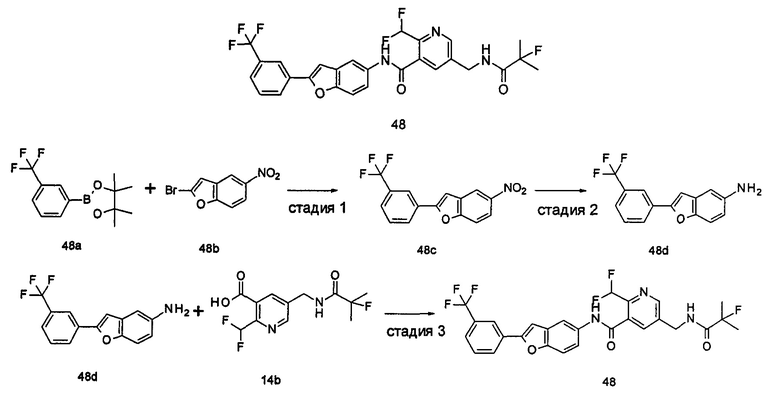
Стадия 1
5-Нитро-2-(3-(трифторметил)фенил)бензофуран
3-(Трифторметил)(пинаколборил)бензол 48а (143 мг, 0,53 ммоль) и 2-бром-5-нитро-бензофуран 48b (85 мг, 0,35 ммоль, полученный в соответствии со способом, раскрытым в "Organic & Biomolecular Chemistry, 2013,11(24), 4095-4101") растворяли в смеси 6 мл 1,4-диоксана и воды (соотношение объемов 5:1), и затем добавляли (1,1'-бис(дифенилфосфино)ферроцен)-дихлорпалладий(II) (26 мг, 0,036 ммоль) и карбонат натрия (75 мг, 0,71 ммоль). Реакционную смесь нагревали до 100°С и перемешивали в течение 16 часов в атмосфере аргона. Реакционную смесь охлаждали до комнатной температуры. Реакционную смесь фильтровали и концентрировали фильтрат при пониженном давлении. Остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой С с получением целевого соединения 5-нитро-2-(3-(трифторметил)фенил)бензофурана 48с (64 мг, 64,0%) в виде желтого твердого вещества.
Стадия 2
2-(3-(Трифторметил)фенил)бензофуран-5-амин
5-Нитро-2-(3-(трифторметил)фенил)бензофуран 48с (64 мг, 0,21 ммоль) растворяли в смеси 10 мл тетрагидрофурана и метанола (соотношение объемов 1:1) и затем добавляли никель Ренея (20 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением целевого соединения 2-(3-(трифторметил)фенил)бензофуран-5-амина 48d (45 мг, 77,6%) в виде желтого масла.
Стадия 3
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)бензофуран-5-ил)никотинамид
2-(3-(Трифторметил)фенил)бензофуран-5-амин 48d (23 мг, 0,083 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (24 мг, 0,083 ммоль), 1-этил-(3-диметиламинопропил)-карбодиимида гидрохлорид (32 мг, 0,17 ммоль) и 1-гидроксибензотриазол (11,2 мг, 0,083 ммоль) последовательно добавляли к 3 мл N,N-диметилацетамида. Реакционную смесь нагревали до 40°С. Реакционную смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)бензофуран-5-ил) никотинамида 48 (10 мг, 21,7%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 550,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 8,68 (s, 1Н), 8,15-8,11 (d, 2Н), 8,04-8,00 (d, 3Н), 7,61-7,52 (m, 3Н), 7,39-7,38 (d, 1Н), 7,15-6,85 (m, 3Н), 4,58-4,57 (d, 2Н), 1,64 (s, 3Н), 1,58 (s, 3Н).
Пример 49
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)изоиндолин-5-ил)никотинамид
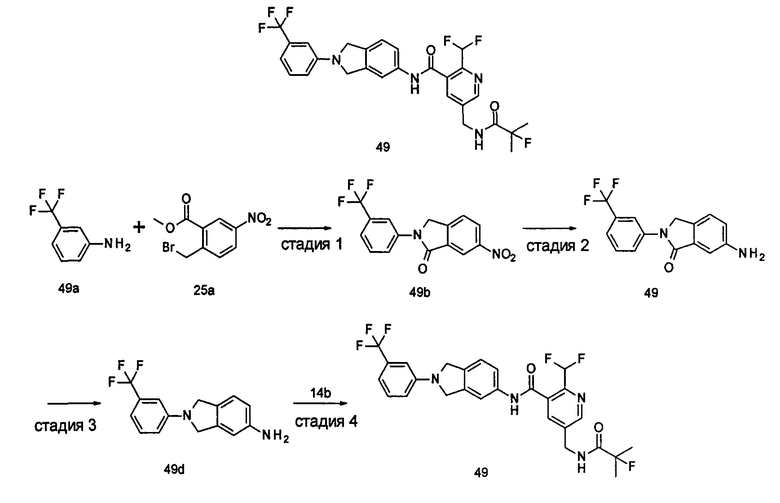
Стадия 1
6-Нитро-2-(3-(трифторметил)фенил)изоиндолин-1-он
3-(Трифторметил)анилин 49а (773 мг, 4,8 ммоль) растворяли в 15 мл уксусной кислоты и затем добавляли метил 2-(бромметил)-5-нитро-бензоат 25а (1,1 г, 4,0 ммоль). Реакционную смесь нагревали до 110°С и перемешивали в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры. Часть твердого вещества осаждали и отфильтровывали. Фильтровальный осадок сушили с получением целевого соединения 6-нитро-2-(3-(трифторметил)фенил) изоиндолин-1-она 49b (300 мг, 23,2%) в виде белого твердого вещества.
МС m/z (ESI): 323,0 [М+1]
Стадия 2
6-Амино-2-(3-(трифторметил)фенил)изоиндолин-1-он
6-Нитро-2-(3-(трифторметил)фенил)изоиндолин-1-он 49b (300 мг, 0,93 ммоль) растворяли в смеси 40 мл тетрагидрофурана и метанола (соотношение объемов 1:1) и затем добавляли никель Ренея (30 мг). Реакционную смесь перемешивали в течение 16 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и фильтровальный осадок промывали 30 мл этилацетата. Фильтрат объединяли и концентрировали при пониженном давлении с получением неочищенного целевого соединения 6-амино-2-(3-(трифторметил)фенил)изоиндолин-1-она 49с (272 мг) в виде черного твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 293,2 [М+1]
Стадия 3
2-(3-(Трифторметил)фенил)изоиндолин-5-амин
К алюмогидриду лития (70 мг, 1,86 ммоль) добавляли 10 мл тетрагидрофурана и затем охлаждали в ледяной бане. К раствору медленно по каплям добавляли 10 мл раствора 6-амино-2-(3-(трифторметил)фенил) изоиндолин-1-она 49с (271 мг, 0,93 ммоль) в тетрагидрофуране. Ледяную баню удаляли и нагревали реакционную смесь до 65°С, и затем перемешивали в течение 1 часа. К реакционной смеси добавляли 2 мл 1М раствора гидроксида натрия и фильтровали. Фильтровальный осадок промывали 20 мл этилацетата. Фильтрат объединяли и концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(3-(трифторметил)фенил)изоиндолин-5-амина 49d (258 мг) в виде черного твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 279,1 [М+1]
Стадия 4
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)изоиндолин-5-ил)никотинамид
2-(3-(Трифторметил)фенил)изоиндолин-5-амин 49d (258 мг, 0,93 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (270 мг, 0,93 ммоль), 1-этил-(3-диметиламинопропил)-карбодиимида гидрохлорид (357 мг, 01,86 ммоль), 1-гидроксибензотриазол (13 мг, 0,093 ммоль) и триэтиламин (375 мг, 3,72 ммоль) последовательно добавляли к 5 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)изоиндолин-5-ил)никотинамида 49 (25 мг, 4,9% для трех стадий) в виде желтого твердого вещества.
МС m/z (ESI): 551,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,79 (s, 1Н), 8,87 (br, 1Н), 8,69 (s, 1Н), 8,00 (s, 1Н), 7,84 (s, 1Н), 7,58 (d, 1Н), 7,47 (t, 1Н), 7,41 (d, 1Н), 7,16 (s, 1Н), 6,97 (t, 2Н), 6,89 (s, 1Н), 4,68 (d, 3Н), 4,47 (d, 2Н), 4,39-4,44 (m, 1Н), 1,54 (s, 3Н), 1,48 (s, 3Н).
Пример 50
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)-2Н-индазол-5-ил)никотинамид
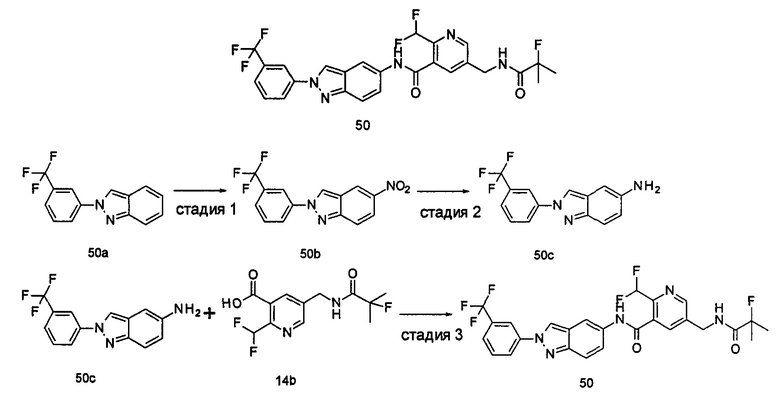
Стадия 1
5-Нитро-2-(3-(трифторметил)фенил)-2H-индазол
2-(3-(Трифторметил)фенил)индазол 50а (28 мг, 0,11 ммоль) и нитрат натрия (14,5 мг, 0,17 ммоль) добавляли к 1 мл концентрированной серной кислоты. Реакционную смесь нагревали до 70°С и перемешивали в течение 0,5 часа. Реакционную смесь охлаждали до комнатной температуры и затем выливали в 20 мл насыщенного раствора карбоната натрия, затем экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением целевого соединения 5-нитро-2-(3-(трифторметил)фенил)-2H-индазола 50b (30 мг, 91,5%) в виде желтого твердого вещества.
МС m/z (ESI): 308,1 [М+1]
Стадия 2
2-(3-(Трифторметил)фенил)-2Н-индазол-5-амин
5-Нитро-2-(3-(трифторметил)фенил)-2Н-индазол 50b (30 мг, 0,098 ммоль) растворяли в смеси 10 мл тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (10 мг). Реакционную смесь перемешивали в течение 1 часа в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(3-(трифторметил)фенил)-2Н-индазол-5-амина 50с (25 мг, 92,6%) в виде желтого твердого вещества.
МС m/z (ESI): 278,1 [М+1]
Стадия 3
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)-2H-индазол-5-ил)никотинамид
2-(3-(Трифторметил)фенил)-2Н-индазол-5-амин 50 с (25 мг, 0,090 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (26 мг, 0,090 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (35 мг, 0,18 ммоль), 1-гидроксибензотриазол (1,2 мг, 9,02 мкмоль) и триэтиламин (50 мкл, 0,36 ммоль) последовательно добавляли к 5 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)-2Н-индазол-5-ил)никотин амида 50 (14 мг, 28,6%) в виде желтого твердого вещества.
МС m/z (ESI): 550,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,80 (s, 1Н), 9,30 (s, 1Н), 8,89 (br, 1Н), 8,70 (s, 1Н), 8,44 (d, 2Н), 8,37 (s, 1Н), 7,85 (s, 1Н), 7,76-7,81 (m, 2Н), 7,77 (d, 1Н), 7,48 (d, 1Н), 7,20 (t, 1Н), 4,47 (d, 2Н), 1,54 (s, 3Н), 1,48 (s, 3Н).
Пример 51
N-(2-(4-Хлорфенил)-1-этил-1Н-бензо[d]имидазол-5-ил)-2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинамид
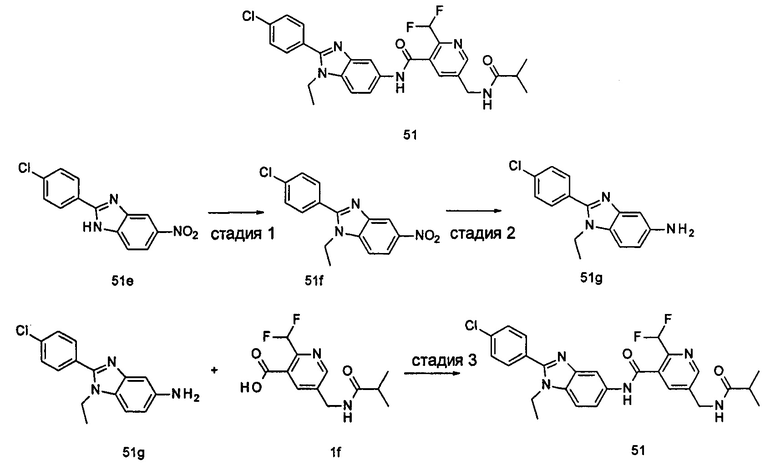
Стадия 1
2-(4-Хлорфенил)-1-этил-5-нитро-бензо[d]имидазол
2-(4-Хлорфенил)-5-нитро-1Н-бензо[d]имидазол 51е (600 мг, 2,29 ммоль, полученный в соответствии со способом, раскрытым в "Monatshefte fuer Chemie, 2009, 140(5), 547-552") растворяли в 40 мл N,N-диметилформамида, и затем добавляли гидрид натрия (87 мг, 2,29 ммоль). Реакционную смесь перемешивали в течение 0,5 часа, и затем добавляли йодэтан (512 мг, 3,28 ммоль) и непрерывно перемешивали еще в течение 3 часов. После завершения реакции в реакционную смесь добавляли 30 мл воды и экстрагировали этилацетатом (30 мл×3), промывали насыщенным раствором хлорида натрия (30 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(4-хлорфенил)-1-этил-5-нитро-бензо[d]имидазола 51f (540 мг, 82,0%) в виде желтой пасты.
МС m/z (ESI): 302,1 [М+1]
Стадия 2
2-(4-Хлорфенил)-1-этил-1Н-бензо[d]имидазол-5-амин
2-(4-Хлорфенил)-1-этил-5-нитро-бензо[d]имидазол 51f (150 мг, 0,49 ммоль) растворяли в 10 мл метанола и затем добавляли никель Ренея (15 мг). Реакционную смесь перемешивали в течение 14 часов при комнатной температуре в атмосфере водорода. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого 2-(4-хлорфенил)-1-этил-1Н-бензо[d]имидазол-5-амина 51g (100 мг) в виде коричневой пасты, которую использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 272,2 [М+1]
Стадия 3
N-(2-(4-Хлорфенил)-1-этил-1Н-бензо[d]имидазол-5-ил)-2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинамид
2-(4-Хлорфенил)-1-этил-1Н-бензо[d]имидазол-5-амин 51g (50 мг, 0,87 ммоль), 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (80 мг, 0,87 ммоль), 1-гидроксибензотриазол (25 мг, 0,87 мкмоль) и 1-этил-3-(3-диметиламинопропил)-карбодиимида гидрохлорид (70 мг, 0,367 ммоль) последовательно добавляли к 3 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 3,5 часов при 40°С. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения N-(2-(4-хлорфенил)-1-этил-1H-бензо[d]имидазол-5-ил)-2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотинамида 51 (40 мг, 40%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 526,7 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,50 (br, 1Н), 8,89 (s, 1Н), 8,77 (s, 1Н), 8,55 (s, 1Н), 8,13 (d, 2Н), 8,10 (s, 1Н), 7,66 (d, 1Н), 7,58 (d, 1Н), 7,56 (s, 2Н), 6,47 (s, 1Н), 4,51 (s, 2Н), 4,15 (m, 2Н), 2,71 (m, 1Н), 1,30 (m, 3Н), 1,15 (m, 6Н).
Пример 52
2-Хлор-N-(2-(4-хлорфенил)-1-этил-1Н-бензо[d]имидазол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
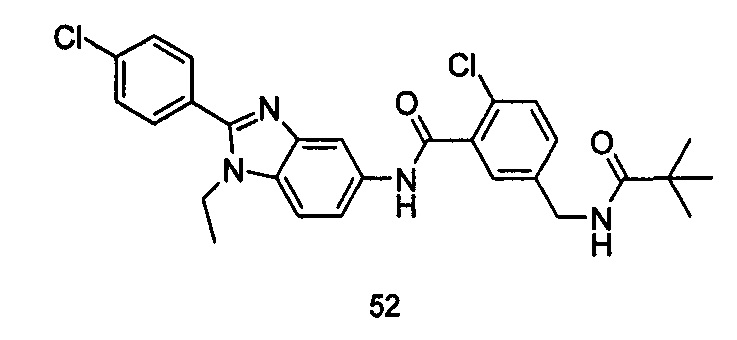
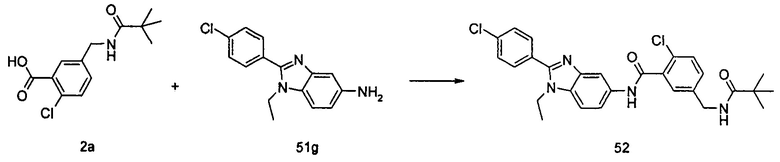
2-(4-Хлорфенил)-1-этил-1Н-бензо[d]имидазол-5-амин 51g (50 мг, 0,87 ммоль), 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензойную кислоту 2а (100 мг, 0,87 ммоль), 1-гидроксибензотриазол (25 мг, 0,87 мкмоль), 1-этил-3-(3-диметиламинопропил)-карбодиимида гидрохлорид (70 мг, 0,367 ммоль) последовательно добавляли к 3 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 3,5 часов при 40°С.Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(2-(4-хлорфенил)-1-этил-1Н-бензо[d]имидазол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамида 52 (10 мг, 10,7%) в виде насыщнно- желтого твердого вещества.
МС m/z (ESI): 523,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,39 (br, 1Н), 8,18 (d, 2Н), 8,10 (s, 1Н), 7,70 (s, 1Н), 7,66 (d, 1Н), 7,58 (m, 2Н), 7,56-7,53 (m, 3Н), 4,26 (s, 2Н), 4,16 (m, 2Н), 1,30 (m, 4Н), 1,21 (s, 9Н).
Пример 53
2-Хлор-N-(2-(5-хлорпиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
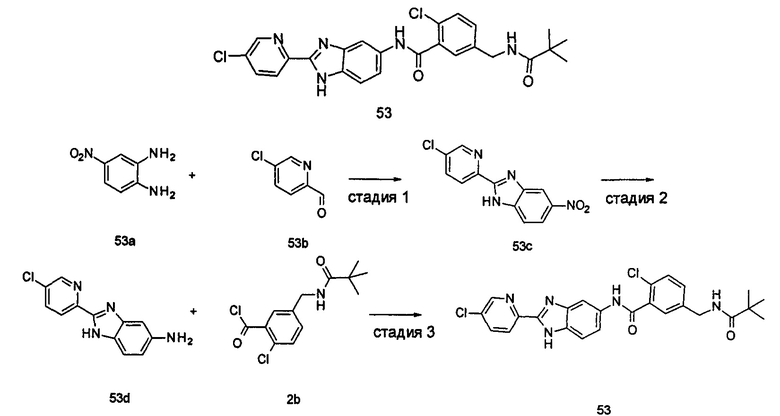
Стадия 1
2-(5-Хлорпиридин-2-ил)-5-нитро-1Н-бензо[d]имидазол
4-Нитробензол-1,2-диамин 53а (400 мг, 2,6 ммоль), 5-хлорпиридин-2-карбальдегид 53b (92 мг, 0,65 ммоль) и 10 мл хлороводородной кислоты (6N) хорошо смешивали. Полученную смесь перемешивали в течение 12 часов при 65°С. Реакционную смесь фильтровали и фильтровальный осадок промывали водой (20 мл) и сушили с получением неочищенного целевого соединения 2-(5-хлорпиридин-2-ил)-5-нитро-1Н-бензо[d]имидазола 53с (350 мг) в виде желтого твердого вещества, которое использовали в следующей стадии без дополнительной очистки.
МС m/z (ESI): 275,1 [М+1]
Стадия 2
2-(5-Хлорпиридин-2-ил)-1Н-бензо[d]имидазол-5-амин
2-(5-Хлор-2-пиридил)-5-нитро-1Н-бензо[d]имидазол 53с (120 мг, 0,44 ммоль) растворяли в 20 мл тетрагидрофурана и 20 мл метанола, и затем добавляли никель Ренея (100 мг). Реакционную смесь перемешивали в течение 12 часов в атмосфере водорода. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого 2-(5-хлорпиридин-2-ил)-1Н-бензо[d]имидазол-5-амина 53d (110 мг) в виде черно-желтого масла, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 245,1 [М+1]
Стадия 3
2-Хлор-N-(2-(5-хлорпиридин-2-ил)-1Н-бензо[d]имидазол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
2-Хлор-5-((2,2-диметилпропаноиламино)метил)бензоилхлорид 2b (58 мг, 0,20 ммоль) растворяли в 10 мл тетрагидрофурана и затем добавляли N,N-диизопропилэтиламин (53 мг, 0,41 ммоль) и 2-(5-хлорпиридин-2-ил)-1Н-бензо[d]имидазол-5-амин 53d (50 мг, 0,20 ммоль). Полученную смесь перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(2-(5-хлорпиридин-2-ил)-1H-бензо[d]имидазол-5-ил)-5-((2,2-диметил пропаноиламино)метил)бензамида 53 (20 мг, 20%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 496,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,82 (s, 1Н), 8,71 (s, 1Н), 8,35 (d, 1Н), 8,18 (s, 1Н), 8,13 (s, 1Н), 7,95 (d, 1Н), 7,57-7,52 (m, 5Н), 5,5 (s, 1Н), 4,30 (d, 2Н), 1,13 (s, 9Н).
Пример 54
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]тиазол-6-ил)никотинамид
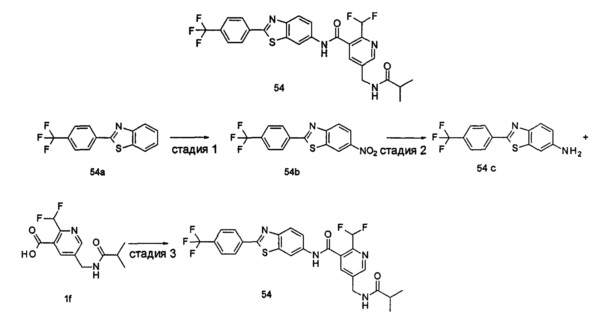
Стадия 1
6-Нитро-2-(4-(трифторметил)фенил)бензо[d]тиазол
Нитрат натрия (27 мг, 0,32 ммоль) и 1 мл концентрированной серной кислоты смешивали, и затем медленно добавляли 2-(4-(трифторметил)фенил)бензо[d]тиазол 54а (50 мг, 0,18 ммоль, полученный в соответствии со способом, раскрытым в "Journal of Molecular Structure, 2012, 1011, 81-93"), при этом поддерживали температуру ниже 75°С. По завершению добавления реакционную смесь перемешивали в течение 1 часа при 70°С. Реакционную смесь доводили до рН 9 раствором гидроксида натрия (50%) и затем добавляли 50 мл этилацетата и 20 мл воды. Органическую фазу концентрировали при пониженном давлении с получением неочищенного целевого соединения 6-нитро-2-(4-(трифторметил)фенил)бензо[d]тиазола 54b (58 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 325,1 [М+1]
Стадия 2
2-(4-(Трифторметил)фенил)бензо[d]тиазол-6-амин
6-Нитро-2-(4-(трифторметил)фенил)бензо[d]тиазол 54b (58 мг, 0,179 ммоль) растворяли в 5 мл тетрагидрофурана и 5 мл метанола, и затем добавляли никель Ренея (5 мг). Реакционную смесь перемешивали в течение 20 минут в атмосфере водорода. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(4-(трифторметил)фенил)бензо[d]тиазол-6-амина 54с (53 мг) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 295,1 [М+1]
Стадия 3
2-(Дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]тиазол-6-ил)никотинамид
2-(4-(Трифторметил)фенил)бензо[d]тиазол-6-амин 54 с (53 мг, 0,18 ммоль), 2-(дифторметил)-5-((2-метилпропаноиламино)метил)никотиновую кислоту 1f (49 мг, 0,18 ммоль), 1-гидроксибензотриазол (25 мг, 0,18 ммоль) и 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (52 мг, 0,27 ммоль) добавляли к 5 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 1 часа при 40°С. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-((2-метилпропаноиламино)метил)-N-(2-(4-(трифторметил)фенил)бензо[d]тиазол-6-ил)никотинамида 54 (20 мг, 20%) в виде желтого твердого вещества.
МС m/z (ESI): 529,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,60 (s, 1Н), 8,67 (s, 1Н), 8,45 (d, 1Н), 8,08 (s, 1Н), 8,00 (s, 1Н), 7,89-7,87 (m, 4Н), 7,54 (d, 1Н), 7,46 (d, 1Н), 7,43 (t, 1Н), 4,42 (d, 2Н), 3,17-3,16 (m, 1Н), 1,05 (s, 6Н).
Пример 55
2-Хлор-N-(2-(4-хлорфенил)-1-метил-1H-бензо[d]имидазол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
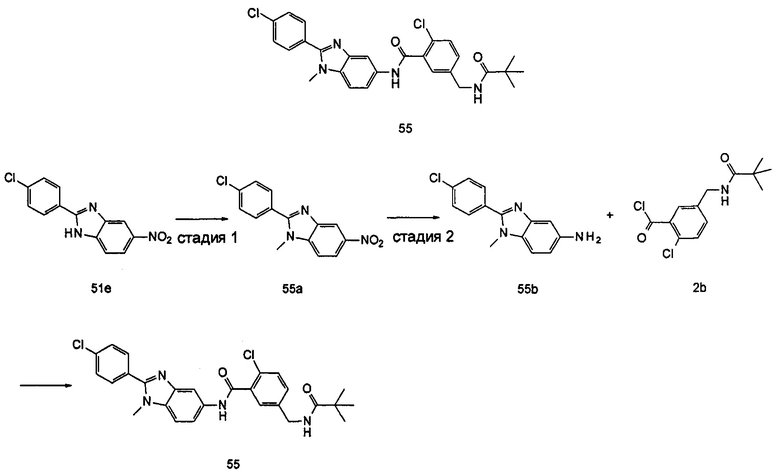
Стадия 1
2-(4-Хлорфенил)-1-метил-5-нитро-1Н-бензо[о]имидазол
Смешивали 2-(4-хлорфенил)-5-нитро-1H-бензо[d]имидазол 51е (600 мг, 2,2 ммоль), йодметан (0,47 мг, 3,3 ммоль) и карбонат цезия (2,15 г, 6,6 ммоль). Реакционную смесь перемешивали в течение 12 часов. После завершения реакции реакционную смесь выливали в 50 мл воды и фильтровали. Фильтровальный осадок промывали водой (20 мл), и сушили с получением неочищенного целевого соединения 2-(4-хлорфенил)-1-метил-5-нитро-1Н-бензо[d]имидазола 55а (0,35 г) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 288,2 [М+1]
Стадия 2
2-(4-Хлорфенил)-1-метил-1Н-бензо[d]имидазол-5-амин
2-(4-Хлорфенил)-1-метил-5-нитро-1H-бензо[d]имидазол 55а (320 мг, 1,11 ммоль) растворяли в 1 мл тетрагидрофурана и 1 мл метанола и затем добавляли никель Ренея (50 мг). Реакционную смесь перемешивали в течение 12 часов в атмосфере водорода. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(4-хлорфенил)-1-метил-1H-бензо[d]имидазол-5-амина 55b (112 мг) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 258,1 [М+1]
Стадия 3
2-Хлор-N-(2-(4-хлорфенил)-1-метил-1Н-бензо[d]имидазол-5-ил)-5-ь((2,2-диметилпропаноиламино)метил)бензамид
2-(4-Хлорфенил)-1-метил-1Н-бензо[d]имидазол-5-амин 55b (50 мг, 0,19 ммоль) растворяли в 10 мл тетрагидрофурана и затем добавляли 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензоилхлорид 2b (56 мг, 0,19 ммоль), N,N-диизопропилэтиламин (52 мг, 0,39 ммоль). Полученную смесь перемешивали в течение 2 часов при 65°С. После завершения реакции реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А, и затем посредством жидкостной хроматографии высокого давления с получением целевого соединения 2-хлор-N-(2-(4-хлорфенил)-1-метил-1Н-бензо[d]имидазол-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамида 55 (10 мг, 10,1%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 509,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,37 (br, 1Н), 8,16 (d, 2Н), 8,12 (s, 1Н), 8,00 (s, 1Н), 7,72 (s, 1Н), 7,68 (d, 1Н), 7,59 (m, 2Н), 7,58-7,55 (m, 3Н), 4,28 (d, 2Н), 3,98 (s, 3Н), 1,23 (s, 9H).
Пример 56
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)бензо[d]оксазол-5-ил)никотинамид
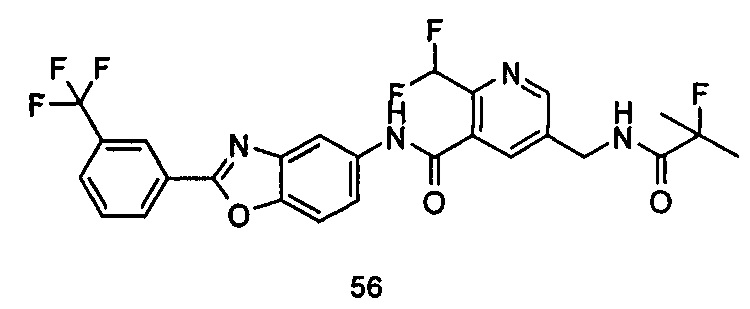
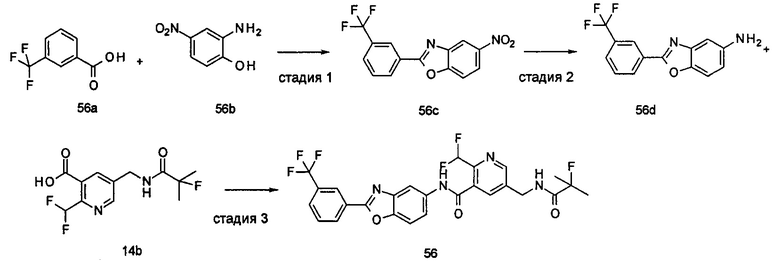
Стадия 1
5-Нитро-2-(3-(трифторметил)фенил)бензо[d]оксазол
В колбу добавляли 10 мл полифосфорной кислоты и нагревали до 120°С. Частями добавляли 3-(трифторметил)бензойную кислоту 56а (1,48 г, 7,79 ммоль) и 2-амино-4-нитро-фенол 56b (1,0 г, 6,49 ммоль). Полученную смесь перемешивали в течение 12 часов при 120°С. Реакционную смесь охлаждали до комнатной температуры и выливали в 50 мл раствора гидроксида натрия (1N). Раствор доводили до рН 8-9 раствором гидроксида натрия (1N) на ледяной бане, и затем экстрагировали этилацетатом (50 мл×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой В с получением целевого соединения 5-нитро-2-(3-(трифторметил)фенил)бензо[d]оксазола 56с (0,26 г, 10%) в виде белого твердого вещества.
МС m/z (ESI): 308,9 [М+1]
Стадия 2
2-(3-(Трифторметил)фенил)бензо[о]оксазол-5-амин
5-Нитро-2-(3-(трифторметил)фенил)бензо[d]оксазол 56с (100 мг, 0,3 ммоль) растворяли в 5 мл тетрагидрофурана и 5 мл метанола и затем добавляли никель Ренея (30 мг). Реакционную смесь перемешивали в течение 1 часа в атмосфере водорода. Реакционную смесь фильтровали через Celatom. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 2-(3-(трифторметил)фенил)бензо[d]оксазол-5-амина 56d (100 мг) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 279,1 [М+1]
Стадия 3
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)бензо[d]оксазол-5-ил)никотинамид
2-(3-(Трифторметил)фенил)бензо[d]оксазол-5-амин 56d (100 мг, 0,325 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил) никотиновую кислоту 14b (94 мг, 0,325 ммоль), 1-гидроксибензотриазол (4,0 мг, 0,03 мкмоль), 1-этил-(3-диметиламинопропил)-карбодиимида гидрохлорид (125 мг, 0,65 ммоль) и триэтиламин (0,18 мл, 1,3 ммоль) добавляли к 10 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(2-(3-(трифторметил)фенил)бензо[d]оксазол-5-ил)никотинамида 56 (190 мг, 56%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 551,3 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,94 (s, 1Н), 8,88 (br, 1Н), 8,71 (d, 1Н), 8,51 (d, 1Н), 8,46 (s, 1Н), 8,27 (d, 1Н), 8,05 (d, 2Н), 7,89-7,97 (m, 1Н), 7,84-7,88 (m, 1Н), 7,72 (dd, 1Н), 7,20 (t, 1Н), 4,48 (d, 2Н), 1,54 (s, 3Н), 1,49 (s, 3Н).
Пример 57
2-Хлор-N-(2-(4-хлорфенил)-3-оксо-изоиндолин-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
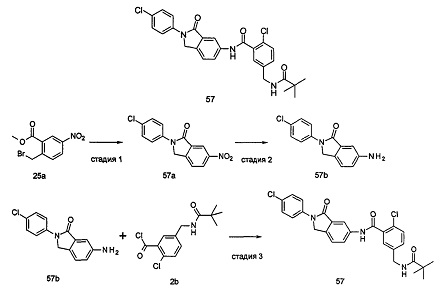
Стадия 1
2-(4-Хлорфенил)-6-нитро-изоиндолин-1-он
Метил 2-(бромметил)-5-нитро-бензоат 25а (200 мг, 0,73 ммоль), 4-хлоранилин (112 мг, 0,88 ммоль) и N,N-диизопропилэтиламин (113 мг, 0,88 ммоль) добавляли к 5 мл этанола. Реакционную смесь нагревали до 110°С и перемешивали в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтровальный осадок промывали этанолом (0,5 мл×2) и сушили с получением целевого соединения 2-(4-хлорфенил)-6-нитро-изоиндолин-1-она 57а (160 мг, 76,2%) в виде желтого твердого вещества.
МС m/z (ESI): 289,0 [М+1]
Стадия 2
6-Амино-2-(4-хлорфенил)изоиндолин-1-он
2-(4-Хлорфенил)-6-нитро-изоиндолин-1-он 57а (160 мг, 0,55 ммоль) растворяли в смеси 20 мл тетрагидрофурана и метанола (соотношение объемов 1:1), и затем добавляли никель Ренея (50 мг). Реакционную смесь перемешивали в течение 4 часов в атмосфере водорода. Реакционную смесь фильтровали через Celatom, и концентрировали фильтрат при пониженном давлении с получением неочищенного целевого соединения 6-амино-2-(4-хлорфенил)изоиндолин-1-она 57b (160 мг) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
МС m/z (ESI): 259,1 [М+1]
Стадия 3
2-Хлор-N-(2-(4-хлорфенил)-3-оксо-изоиндолин-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамид
6-Амино-2-(4-хлорфенил)изоиндолин-1-он 57b (80 мг, 0,31 ммоль) и триэтиламин (0,2 мл, 1,49 ммоль) растворяли в 10 мл ацетонитрила и затем на ледяной бане добавляли 2 мл раствора 2-хлор-5-((2,2-диметилпропаноиламино)метил)бензоил хлорида 2b (74 мг, 0,26 ммоль) в дихлорметане. Реакционную смесь перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(2-(4-хлорфенил)-3-оксо-изоиндолин-5-ил)-5-((2,2-диметилпропаноиламино)метил)бензамида 57 (15 мг, 15,9% для двух стадий) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 511,2 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,82 (s, 1Н), 8,27 (s, 1Н), 8,19 (t, 1Н), 7,97 (d, 2Н), 7,65 (d, 1Н), 7,54 (d, 1Н), 7,50-7,53 (m, 3Н), 7,46 (s, 1Н), 7,37 (d, 1Н), 5,01 (s, 2Н), 4,30 (s, 2Н), 1,13 (s, 9Н).
Пример 58
2-Хлор-N-(3-оксо-2-(4-(трифторметил)фенил)изоиндолин-5-ил)-5-((2,2-диметил-пропаноил)амино)метил)-бензамид
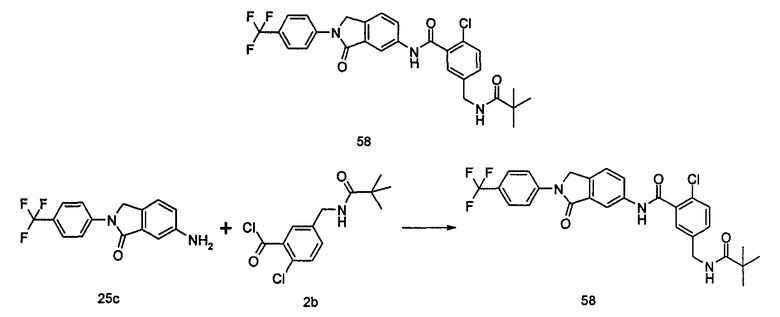
6-Амино-2-(4-(трифторметил)фенил)изоиндолин-1-он 25с (82 мг, 0,28 ммоль) и триэтиламин (0,2 мл, 1,49 ммоль) растворяли 10 мл в дихлорметане и затем на ледяной бане добавляли 10 мл раствора 2-хлор-5-((2,2-диметилпропаноиламино) метил)бензоилхлорида 2b (80 мг, 0,28 ммоль) в дихлорметане. Реакционную смесь перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-хлор-N-(3-оксо-2-(4-(трифторметил)фенил)изоиндолин-5-ил)-5-((2,2-диметил-пропаноил)амино)метил)-бензамида 58 (4 мг, 2,6%) в виде бледно-желтого твердого вещества.
МС m/z (ESI): 544,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 10,84 (s, 1Н), 8,30 (d, 1Н), 8,17 (d, 3Н), 7,87 (d, 1Н), 7,82 (s, 2Н), 7,70 (d, 1Н), 7,55 (d, 1Н), 7,47 (s, 1Н), 7,38 (d, 1Н), 5,08 (s, 2Н), 4,30 (d, 2Н), 1,13 (s, 9Н)
Пример 59
2-(Дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(3-оксо-2-(3-(трифторметил)фенил)изоиндолин-5-ил)никотинамид
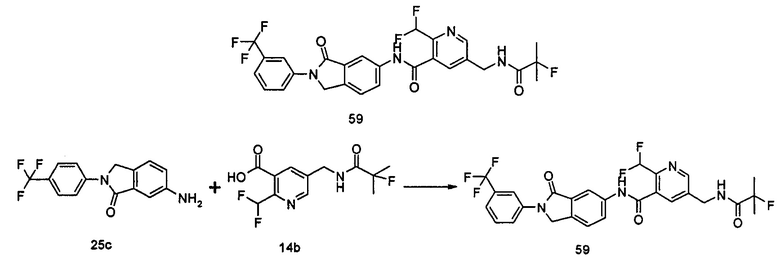
6-Амино-2-(4-(трифторметил)фенил)изоиндолин-1-он 25с (100 мг, 0,34 ммоль), 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)никотиновую кислоту 14b (100 мг, 0,34 ммоль), 1-этил-(3-диметиламинопропил)-карбодиимида гидрохлорид (130 мг, 0,69 ммоль), 1-гидроксибензотриазол (5,0 мг, 37 мкмоль) и триэтиламин (140 мкл, 1,03 ммоль) добавляли к 5 мл N,N-диметилацетамида. Реакционную смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией (ТСХ) с элюирующей системой А с получением целевого соединения 2-(дифторметил)-5-(((2-фтор-2-метил-пропаноил)амино)метил)-N-(3-оксо-2-(3-(трифторметил)фенил)изоиндолин-5-ил)никотинамида 59 (80 мг, 41,5%) в виде бледно-желтого твердого вещества.
МС m/z (ESI); 565,1 [М+1]
1Н ЯМР (400 МГц, DMSO-d6): δ 11,02 (s, 1Н), 8,89 (br, 1Н), 8,71 (d, 1Н), 8,45 (s, 1Н), 8,27 (d, 1Н), 8,13 (d, 1Н), 8,06 (s, 1Н), 7,91 (d, 1Н), 7,70 (d, 2Н), 7,55 (d, 1Н), 7,20 (t, 1Н), 5,11 (s, 2Н), 4,48 (d, 2Н), 1,54 (s, 3Н), 1,48 (s, 3Н).
ТЕСТОВЫЕ ПРИМЕРЫ
БИОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ
Тестовый пример 1. Ингибирующая активность соединений по настоящему изобретению в отношении mPGES-1 человека
Описанный здесь способ используется для определения ингибирующей активности соединений по настоящему изобретению в отношении mPGES-1 человека.
I. Материалы и прибор
1. Набор реактивов для PGE2 (Cisbio, #62Р2АРЕВ)
2. Простагландин Н2 (sigma, #P7867-1MG)
3. Ридер для микропланшетов FlexStation3.
II. Методика эксперимента
1. Получение мембранных белков фермента mPGES-1
Плотность клеток HEK-293 F приводили к 6×105 на миллилитр и на следующий день трансфектировали плазмидой, содержащей ген mPGES-1 человека, при помощи трансфекционного реагента ПЭИ. Клетки непрерывно культивировали в течение 72 часов при 37°С при встряхивании. После центрифугирования при 1100 g в течение 5 минут клетки отбирали для исследования. После ультразвуковой обработки на ледяной бане и центрифугирования при 5000 g в течение 10 минут получали супернатант. Супернатант центрифугировали при 100000 g в течение 1 часа с получением преципитата. Преципитат ресуспендировали в буфере для хранения, содержащем 10% глицерина, упаковывали и быстро замораживали жидким азотом, и хранили при -80°С.
2. Исследование фермента mPGES-1
mPGES-1 разбавляли буфером для исследования и добавляли в планшет в концентрации 49 мкл/лунка с добавкой 1 мкл соединения (конечная концентрация каждого соединения в семи равномерно понижающихся концентрациях составила 10000 нМ, 1000 нМ, 100 нМ, 10 нМ, 1 нМ, 0,1 нМ и 0,01 нМ). В каждую лунку добавляли 3,8 мкл PGH (20 мкг/мл), предварительно охлажденного на льду после встряхивания в течение 30 секунд, и инкубировали в течение 7 минут на льду. Затем для остановки реакции в каждую лунку добавляли 53,8 мкл SnCl2 (6 мг/мл). Образец разбавляли буфером для разбавления в соотношении 1:400. 10 мкл разбавленного образца, 5 мкл PGE2-d2 и 5 мкл анти-PGE2-криптата добавляли в черный 384-луночный планшет и инкубировали при 4°С в течение ночи. Гомогенную флуоресценцию с временным разрешением (HTRF) определяли с использованием ридера flexstation, и значения IC50 соединений получали с использованием программного обеспечения для обработки данных.
Ингибирующую активность соединений по настоящему изобретению в отношении mPGES-1 определяли по методике исследования, описанной выше. Значения IC50 приведены в таблице 1 ниже.
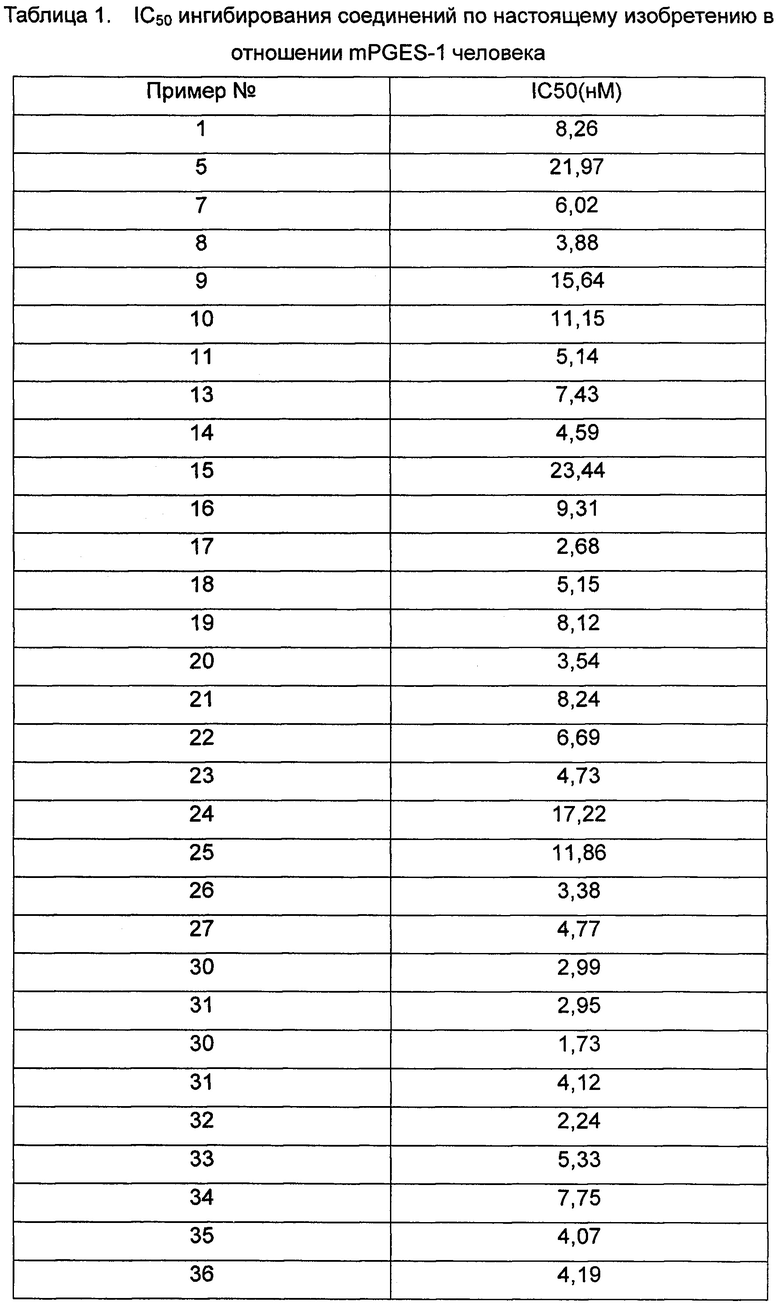
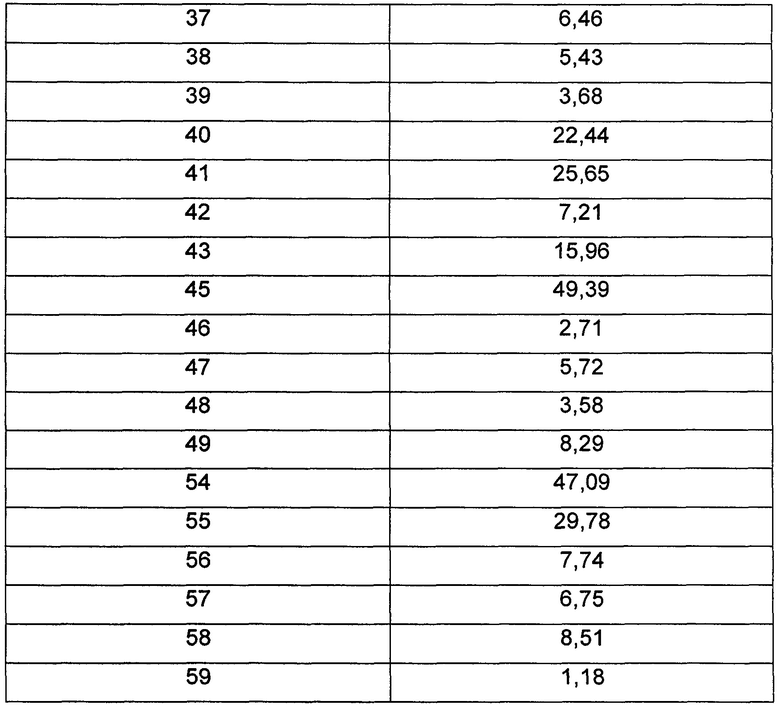
Вывод: соединения по настоящему изобретению обладают существенной ингибирующей активностью в отношении белка mPGES-1 человека.
Тестовый пример 2. Ингибирующая активность соединений по настоящему изобретению в отношении mPGES-1 морской свинки
Описанный здесь способ использовали для определения ингибирующей активности соединений по настоящему изобретению в отношении mPGES-1 морской свинки.
I. Материалы и прибор
1. Набор реактивов PGE2 (Cisbio, #62Р2АРЕВ)
2. Простагландин Н2 (sigma, #P7867-1MG)
3. Ридер для микропланшетов FlexStation3.
II. Методика эксперимента
1. Получение мембранных белков фермента mPGES-1
Плотность клеток HEK-293 F приводили к 6×105 на миллилитр и на следующий день трансфектировали плазмидой, содержащей ген mPGES-1 человека, при помощи трансфекционного реагента ПЭИ. Клетки непрерывно культивировали в течение 72 часов при 37°С при встряхивании. После центрифугирования при 1100 g в течение 5 минут клетки отбирали для исследования. После ультразвуковой обработки на ледяной бане и центрифугирования при 5000 g в течение 10 минут получали супернатант. Супернатант центрифугировали при 100000 g в течение 1 часа с получением преципитата. Преципитат ресуспендировали в буфере для хранения, содержащем 10% глицерина, упаковывали и быстро замораживали жидким азотом, и хранили при -80°С.
2. Исследование фермента mPGES-1
mPGES-1 разбавляли буфером для исследования и добавляли в планшет в концентрации 49 мкл/лунка с добавкой 1 мкл соединения (конечная концентрация каждого соединения в семи равномерно понижающихся концентрациях составила 10000 нМ, 1000 нМ, 100 нМ, 10 нМ, 1 нМ, 0,1 нМ и 0,01 нМ). В каждую лунку добавляли 3,8 мкл PGH (20 мкг/мл), предварительно охлажденной на льду после встряхивания в течение 30 секунд, и инкубировали в течение 7 минут на льду. Затем для остановки реакции в каждую лунку добавляли 53,8 мкл 6 мг/мл SnCl2. Образец разбавляли буфером для разбавления в соотношении 1:400.
10 мкл разбавленного образца, 5 мкл PGE2-d2 и 5 мкл анти-PGE2-криптата добавляли в черный 384-луночный планшет и инкубировали при 4°С в течение ночи. HTRF определяли с использованием ридера flexstation и значения IC50 соединений получали с использованием программного обеспечения для обработки данных, Ингибирующую активность соединений по настоящему изобретению в отношении mPGES-1 определяли по методике исследования, описанной выше. Значения IC50 приведены в таблице 2 ниже.
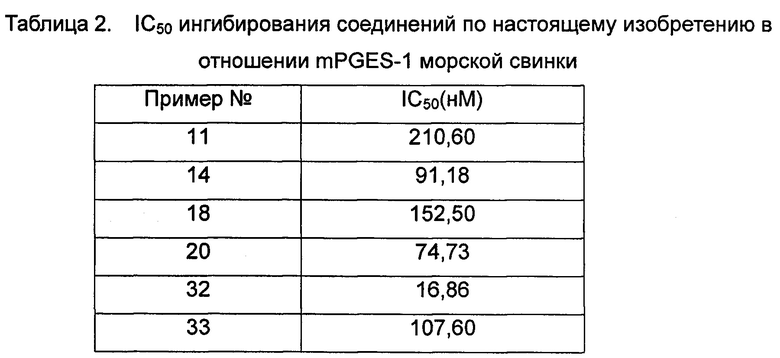
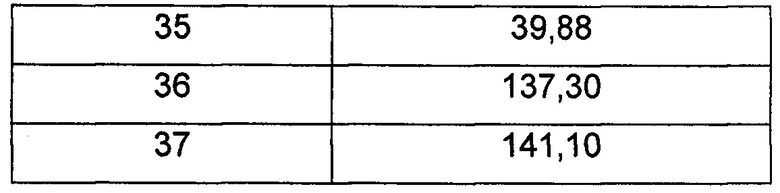
Вывод: соединения по настоящему изобретению обладают существенной активностью в отношении белка mPGES-1 морской свинки.
Тестовый пример 3. Ингибирующая активность соединений по настоящему изобретению в отношении секреции PGE2 клетками А549 при стимуляции IL-1β.
Описанный здесь способ используют для определения ингибирующей активности соединений по настоящему изобретению в отношении секреции PGE2 клетками А549 при стимуляции IL-1β.
I. Материалы и прибор
1. Набор реактивов PGE2 (Cisbio, #62Р2АРЕВ)
2. IL-1β (Peprotech, # AF-200-01B)
3. Клеточная линия: А549 (АТСС: CCL-185)
4. Ридер для микропланшетов FlexStation3.
II. Методика эксперимента
В первый день клетки А549 засеивали в 96-луночный планшет в концентрации 40000/лунка. На второй день питательную среду для культур удаляли из 96-луночного планшета и затем добавляли 90 мкл соединений, разбавленных питательной средой для культур (концентрация каждого соединения в семи равномерно понижающихся концентрациях составила 11111 нМ, 11111 нМ, 111 нМ, 11,11 нМ, 1,11 нМ, 0,11 нМ и 0,011 нМ). Клетки инкубировали в течение 30 минут при 37°С в инкубаторе и затем добавляли IL-1β до конечной концентрации 0,2 нг/мл. Затем клетки инкубировали еще в течение 24 часов при 37°С. На третий день в черный 384-луночный планшет добавляли 10 мкл супернатанта, 5 мкл PGE2-d2 и 5 мкл анти-PGE2-криптата и затем инкубировали при 4°С в течение ночи. HTRF определяли с использованием ридера flexstation и значения IC50 соединений получали с использованием программного обеспечения для обработки данных.
Ингибирующая активность соединений по настоящему изобретению в отношении секреции PGE2 клетками А549 при стимуляции IL-1β определяли по методике исследования, описанной выше. Значения IC50 приведены в таблице 3 ниже.
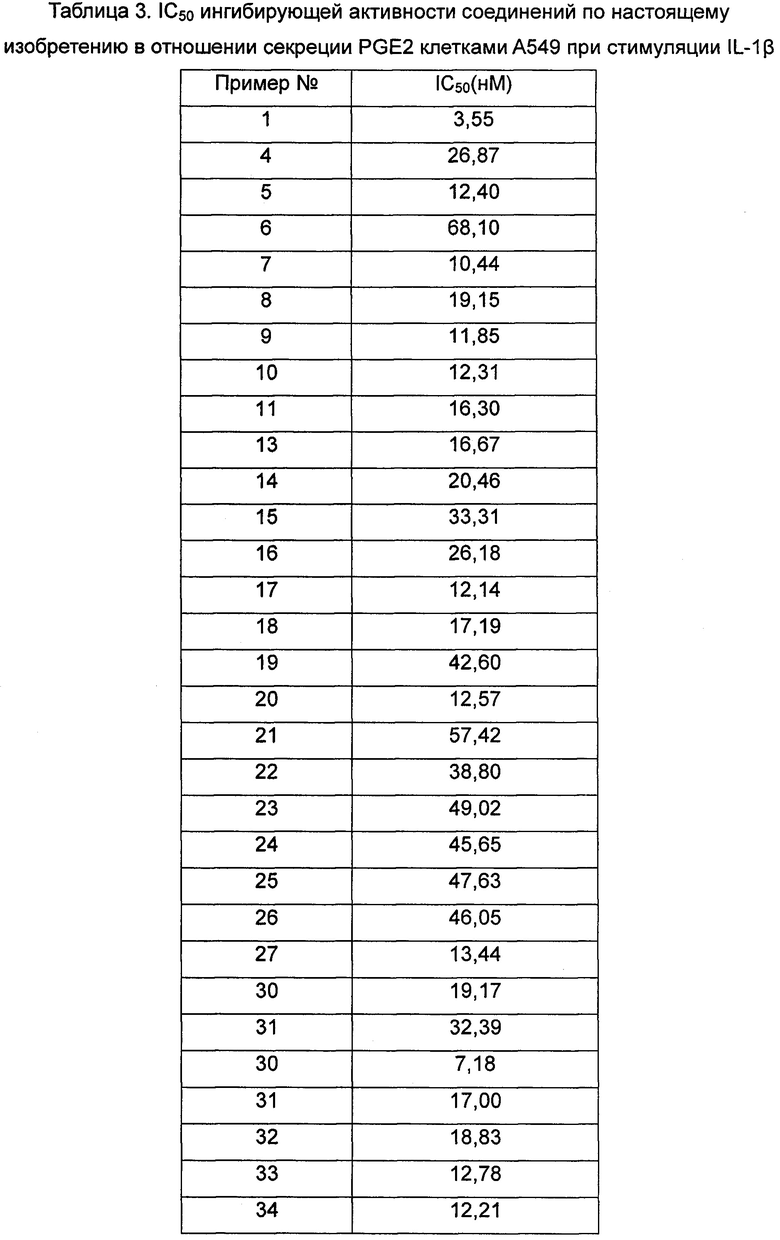
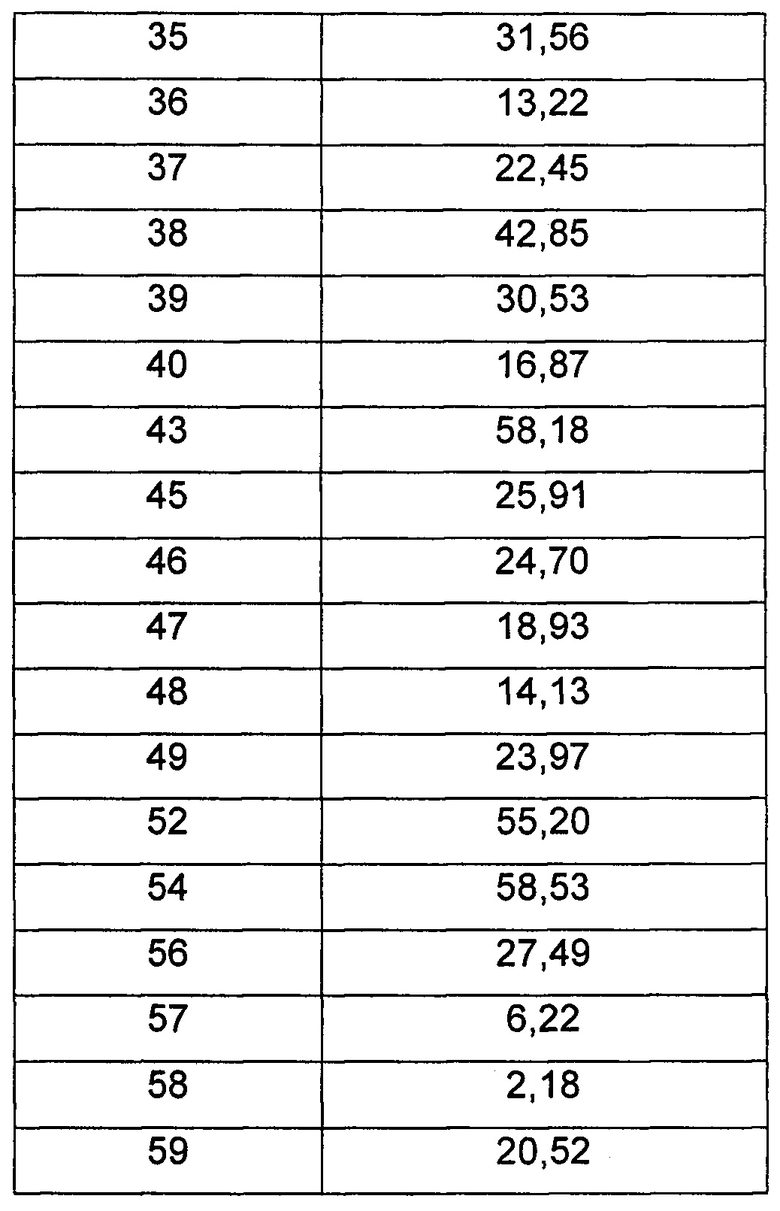
Вывод: Соединения по настоящему изобретению обладают существенной ингибирующей активностью в отношении секреции PGE2 клетками А549 при стимуляции IL-1β.
ИССЛЕДОВАНИЕ ФАРМАКОКИНЕТИКИ
Тестовый пример 4. Исследование фармакокинетики соединений по настоящему изобретению
1. Реферат
В качестве подопытных животных были использованы крысы. Концентрацию лекарственного средства в плазме в различные контрольные моменты времени определяли при помощи ЖХ/МС/МС после введения соединений по Примеру 8, Примеру 11, Примеру 14, Примеру 17, Примеру 20, Примеру 27, Примеру 33, Примеру 35, Примеру 35, Примеру 37 и Примеру 48. Фармакокинетическое поведение соединений по настоящему изобретению изучали и оценивали на крысах.
2. Протокол
2.1 Образцы
Соединения по Примеру 8, Примеру 11, Примеру 14, Примеру 17, Примеру 20, Примеру 27, Примеру 33, Примеру 35, Примеру 36, Примеру 37 и Примеру 48.
2.2 Подопытные животные
44 здоровых взрослых крыс Спрег-Доули (СД), половину из которых составляли самцы и половину самки, приобретенных в SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО, с сертификатом №: SCXK (Шанхай) 2008-0016, разделяли на 11 групп, по 4 крысы в каждой группе.
2.3 Получение исследуемых соединений
Надлежащее количество исследуемых соединений взвешивали и смешивали с 25 мкл Твин-80 и Лабразола (Labrasol) с получением 0,6 мг/мл суспензии путем ультразвуковой обработки.
2.4 Введение
После голодания в течение ночи 44 крысы СД, половину из которых составляли самцы и половину самки, разделяли на 11 групп, и осуществляли внутрижелудочное введение дозы 5,0 мг / кг и с объемом введения 10 мл/кг.
3. Способ
Образцы крови (0,1 мл) отбирали из ретроорбитального сплетения до введения и через 0,5 ч, 1,0 ч, 2,0 ч, 4,0 ч 6,0 ч, 8,0 ч, 11,0 ч, и 24,0 ч после введения. Образцы хранили в пробирках с ЭДТА, препятствующих свертыванию, и центрифугировали в течение 5 минут при 3,500 об/мин для отделения плазмы крови. Образцы плазмы хранили при -20°С. Крыс кормили через 2 часа после введения.
Концентрацию исследуемых соединений в плазме крыс после внутрижелудочного введения определяли при помощи ЖХ/МС/МС. Линейность способа составляла 5,00-2000 нг/мл и 1,00-2000 нг/мл, и минимальная количественная оценка составила 5,00 нг/мл и 1,00 нг/мл. Образцы плазмы анализировали после предварительной обработки осаждением белков.
4. Полученные фармакокинетические параметры Фармакокинетические параметры соединений по настоящему изобретению приведены в следующей таблице.
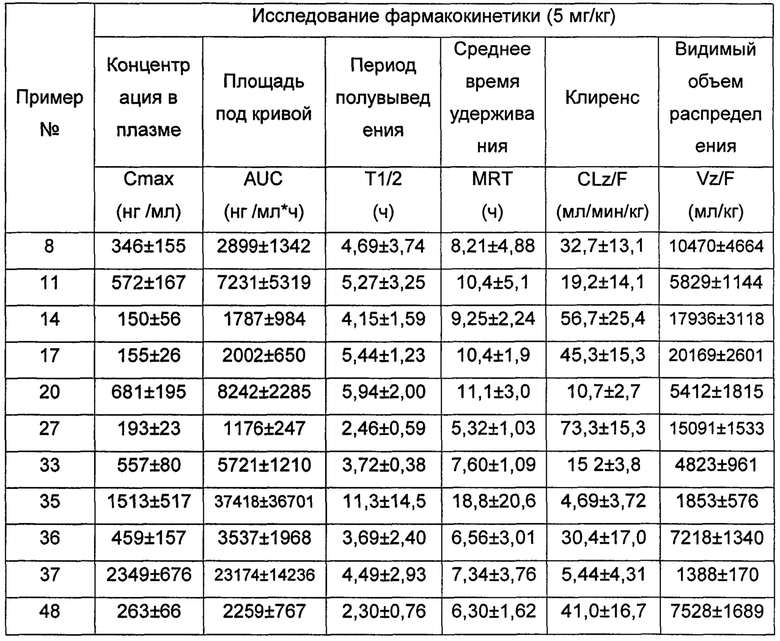
Вывод: Соединения по настоящему изобретению обладают хорошими фармакокинетическими характеристиками и существенным фармакокинетическим абсорбционным эффектом.
Настоящее изобретение относится к cоединению общей формулы (I), или его энантиомеру, или их смеси, или его фармацевтически приемлемым солям:
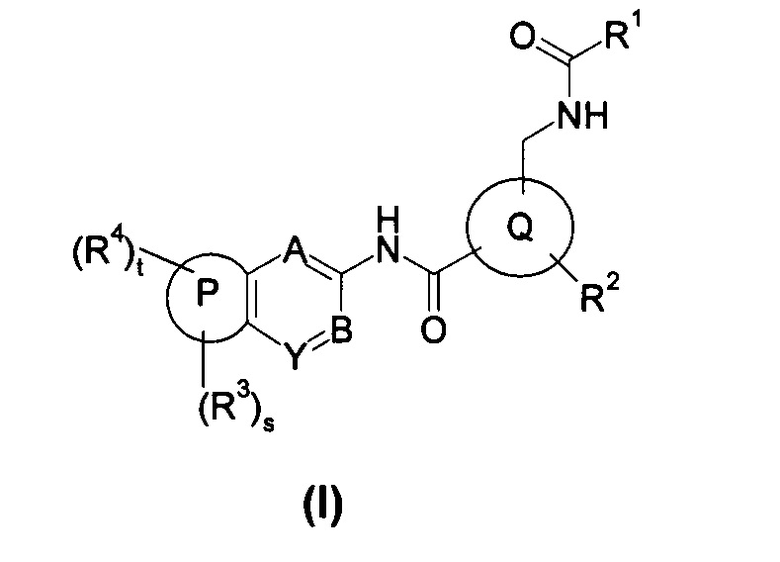
В формуле (I) кольцо Р выбрано из пятичленного гетероарила, имеющего от одного до двух гетероатомов, выбранных из группы, состоящей из N, О и S, в качестве атома кольца, и пятичленного гетероциклила, имеющего один гетероатом N в качестве атома кольца; кольцо Q выбрано из фенила и пиридила; А, В или Y выбран из -СН- и N; R1 выбран из алкила, содержащего от 1 до 6 атомов углерода, и циклоалкила, содержащего от 3 до 6 атомов углерода, где указанный алкил, содержащий от 1 до 6 атомов углерода, или циклоалкил, содержащий от 3 до 6 атомов углерода, необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, содержащего от 1 до 6 атомов углерода, галогена и галогеналкила, содержащего от 1 до 6 атомов углерода; R2 выбран из галогена и галогеналкила, содержащего от 1 до 6 атомов углерода; R3 являются одинаковыми или разными и каждый из них независимо выбран из водорода, алкила, содержащего от 1 до 6 атомов углерода, циклоалкила, содержащего от 3 до 6 атомов углерода, пяти- или шестичленного гетероциклила, имеющего один гетероатом О в качестве атома кольца, и оксо, где указанный алкил, содержащий от 1 до 6 атомов углерода, необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидроксила, алкокси, содержащего от 1 до 6 атомов углерода, и гидроксиалкила, содержащего от 1 до 6 атомов углерода; R4 выбран из фенила и пиридила, где каждый из указанных фенила и пиридила необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена и галогеналкила, содержащего от 1 до 6 атомов углерода, где указанный галогеналкил, содержащий от 1 до 6 атомов углерода, предпочтительно представляет собой трифторметил; s представляет собой целое число от 0 до 3; t представляет собой 0 или 1. Также предложены способ получения соединения формулы (I), промежуточные соединения формул (IA), (IB), фармацевтическая композиция и применение соединения формулы (I). Предложенные соединения формулы (I) обладают способностью ингибировать микросомальную простагландин Е-синтазу-1 и могут быть использованы для получения лекарственного средства. 8 н. и 9 з.п. ф-лы, 4 табл., 63 пр.
1. Соединение общей формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли:
где кольцо Р выбрано из пятичленного гетероарила, имеющего от одного до двух гетероатомов, выбранных из группы, состоящей из N, О и S, в качестве атома кольца, и пятичленного гетероциклила, имеющего один гетероатом N в качестве атома кольца;
кольцо Q выбрано из фенила и пиридила;
А, В или Y выбран из -СН- и N;
R1 выбран из алкила, содержащего от 1 до 6 атомов углерода, и циклоалкила, содержащего от 3 до 6 атомов углерода, где указанный алкил, содержащий от 1 до 6 атомов углерода, или циклоалкил, содержащий от 3 до 6 атомов углерода, необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, содержащего от 1 до 6 атомов углерода, галогена и галогеналкила, содержащего от 1 до 6 атомов углерода;
R2 выбран из галогена и галогеналкила, содержащего от 1 до 6 атомов углерода;
R3 являются одинаковыми или разными и каждый из них независимо выбран из водорода, алкила, содержащего от 1 до 6 атомов углерода, циклоалкила, содержащего от 3 до 6 атомов углерода, пяти- или шестичленного гетероциклила, имеющего один гетероатом О в качестве атома кольца, и оксо, где указанный алкил, содержащий от 1 до 6 атомов углерода, необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидроксила, алкокси, содержащего от 1 до 6 атомов углерода, и гидроксиалкила, содержащего от 1 до 6 атомов углерода;
R4 выбран из фенила и пиридила, где каждый из указанных фенила и пиридила необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена и галогеналкила, содержащего от 1 до 6 атомов углерода, где указанный галогеналкил, содержащий от 1 до 6 атомов углерода, предпочтительно представляет собой трифторметил;
s представляет собой целое число от 0 до 3;
t представляет собой 0 или 1.
2. Соединение формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли по п. 1, представляющее собой соединение формулы (II), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли:
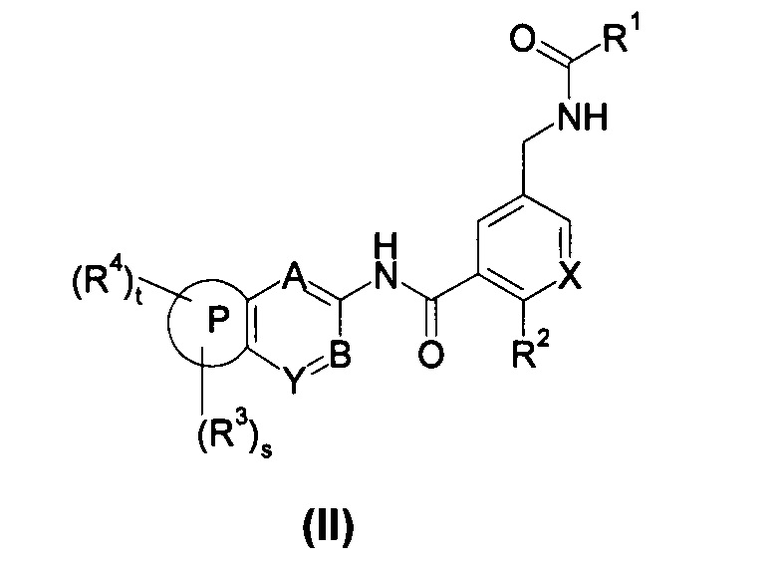
где X выбран из -СН- и N;
кольцо Р, А, В, Y, s, t и R1-R4 являются такими, как определено в п. 1.
3. Соединение формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли по п. 1, представляющее собой соединение формулы (III), формулы (IV) или формулы (V), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли:
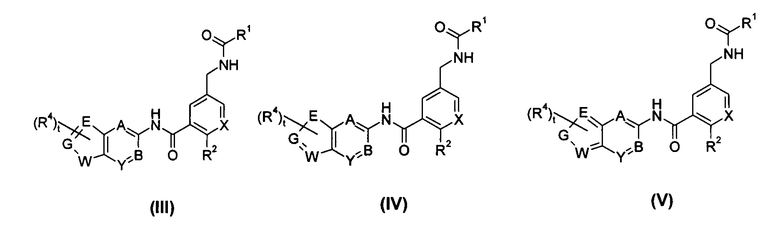
где X выбран из -СН- и N;
каждый из Е, G и W независимо выбран из CRa, NRb, N, О и S;
каждый из Ra и Rb независимо выбран из водорода, алкила, содержащего от 1 до 6 атомов углерода, циклоалкила, содержащего от 3 до 6 атомов углерода, пяти- или шестичленного гетероциклила, имеющего один гетероатом О в качестве атома кольца, где указанный алкил, содержащий от 1 до 6 атомов углерода, необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, алкокси, содержащего от 1 до 6 атомов углерода, и гидроксиалкила, содержащего от 1 до 6 атомов углерода, и
А, В, Y, t, R1-R2 и R4 являются такими, как определено в п. 1.
4. Соединение формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли по п. 1, представляющее собой соединение формулы (VI), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли:
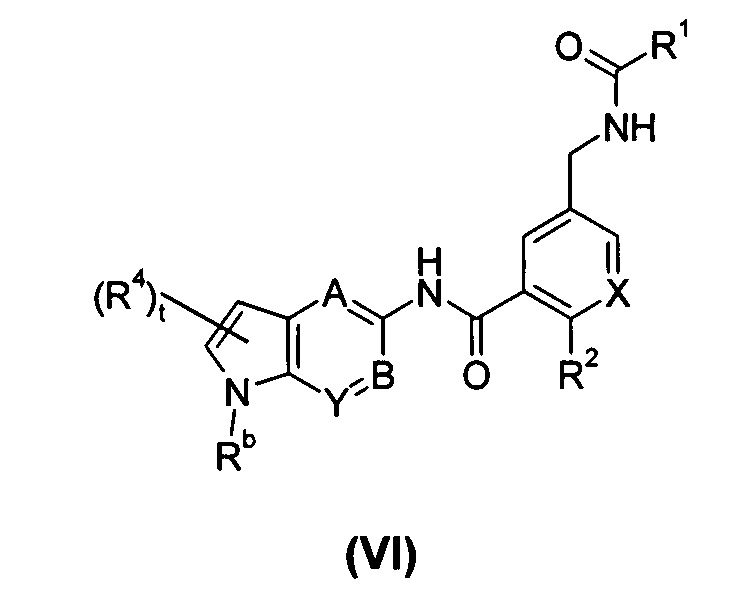
где X выбран из -СН- и N;
Rb выбран из водорода, алкила, содержащего от 1 до 6 атомов углерода, циклоалкила, содержащего от 3 до 6 атомов углерода, пяти- или шестичленного гетероциклила, имеющего один гетероатом О в качестве атома кольца, где указанный алкил, содержащий от 1 до 6 атомов углерода, необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, алкокси, содержащего от 1 до 6 атомов углерода, и гидроксиалкила, содержащего от 1 до 6 атомов углерода, и
А, В, Y, t, R1-R2 и R4 являются такими, как определено в п. 1.
5. Соединение формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли по п. 1, представляющее собой соединение формулы (VII), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли:
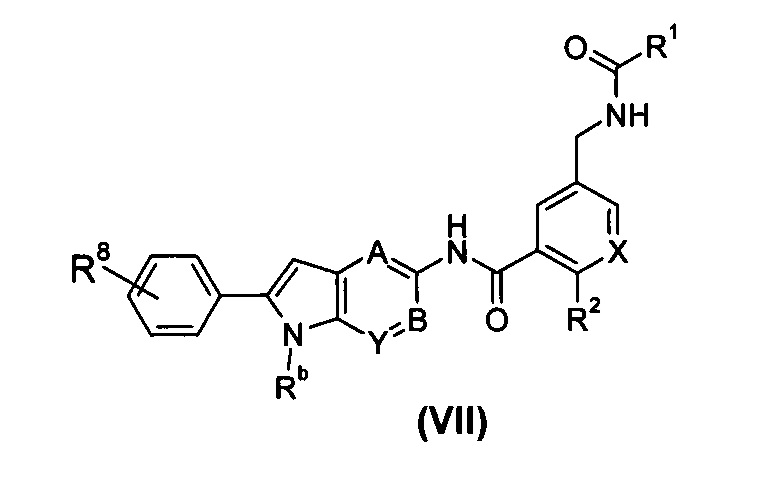
где X выбран из -СН- и N;
Rb выбран из водорода, алкила, содержащего от 1 до 6 атомов углерода, циклоалкила, содержащего от 3 до 6 атомов углерода, пяти- или шестичленного гетероциклила, имеющего один гетероатом О в качестве атома кольца, где указанный алкил, содержащий от 1 до 6 атомов углерода, необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, алкокси, содержащего от 1 до 6 атомов углерода, и гидроксиалкила, содержащего от 1 до 6 атомов углерода;
R8 выбран из галогена или галогеналкила, содержащего от 1 до 6 атомов углерода, где указанный галогеналкил, содержащий от 1 до 6 атомов углерода, предпочтительно представляет собой трифторметил, и
А, В, Y и R1-R2 являются такими, как определено в п. 1.
6. Соединение формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли по любому из пп. 1-5, где А, В и Y представляют собой -СН-.
7. Соединение формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли по любому из пп. 1-5, где один из А, В и Y представляет собой N, а два других представляют собой -СН-.
8. Соединение формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли по п. 1 или 2, где указанные группы
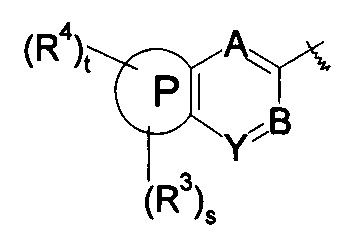
выбраны из:
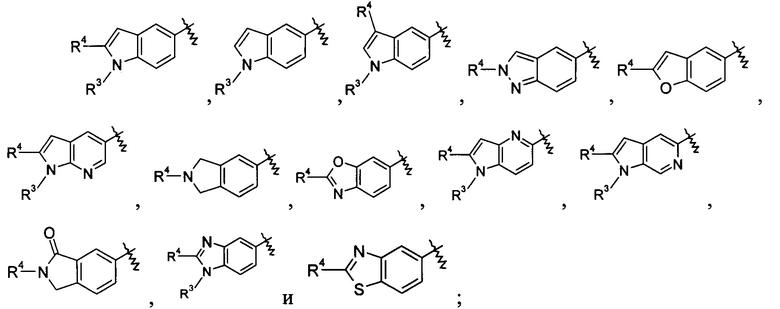
R3 и R4 являются такими, как определено в п. 1.
9. Соединение формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли по п. 1, где указанный R1 выбран из алкила, содержащего от 1 до 6 атомов углерода, и галогеналкила, содержащего от 1 до 6 атомов углерода, предпочтительно третичного бутила, изопропила, 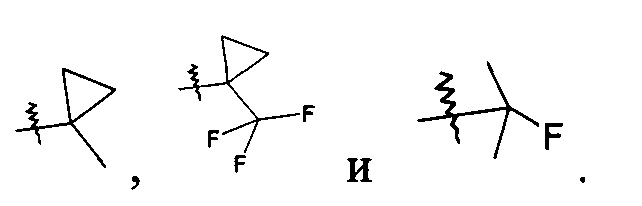
10. Соединение формулы (I), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли по любому из пп. 1-5, где указанные соединения выбраны из:
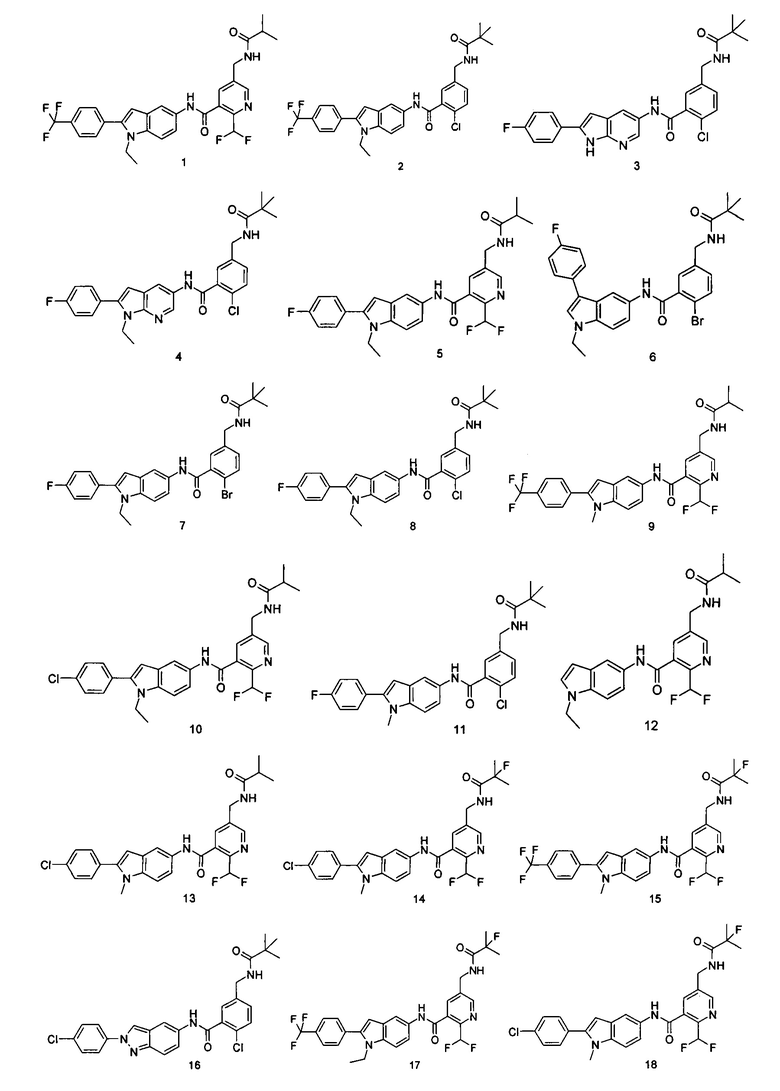
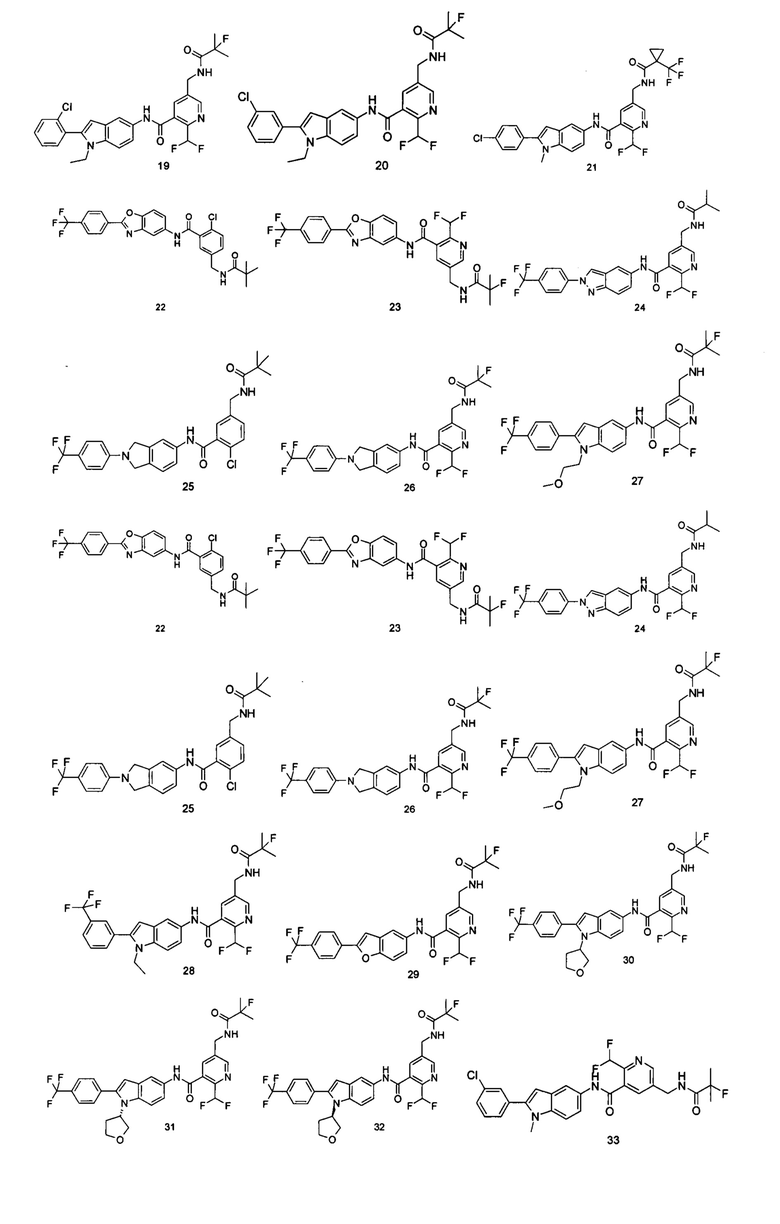
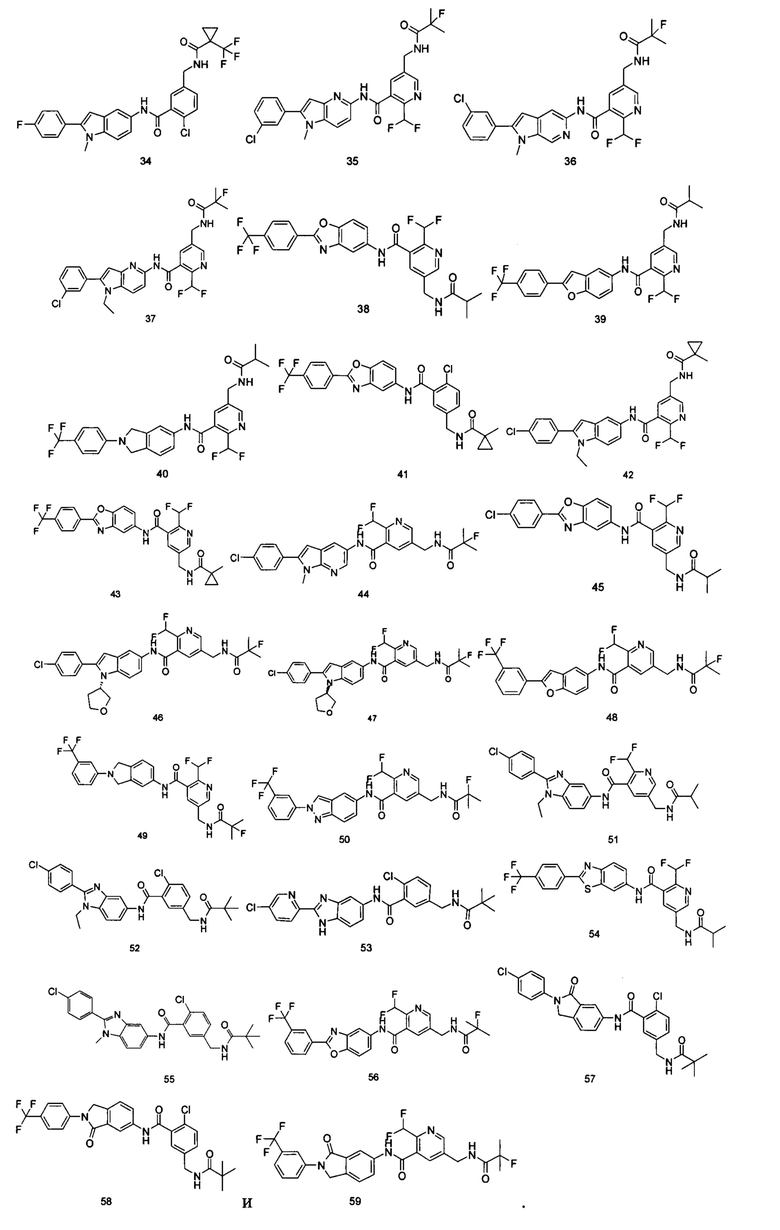
11. Способ получения соединения формулы (I), или его энантиомера, или их смесей, или его фармацевтически приемлемых солей по п. 1, включающий стадию
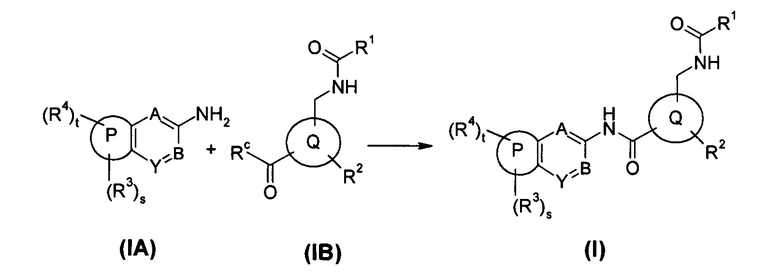
взаимодействия соединения формулы (IA) или его солей с соединением формулы (IB) с получением соединения формулы (I),
где Rc выбран из гидрокси и галогена;
кольцо Р, кольцо Q, А, В, Y, s, t и R1-R4 являются такими, как определено в п. 1.
12. Соединение формулы (IA), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли:
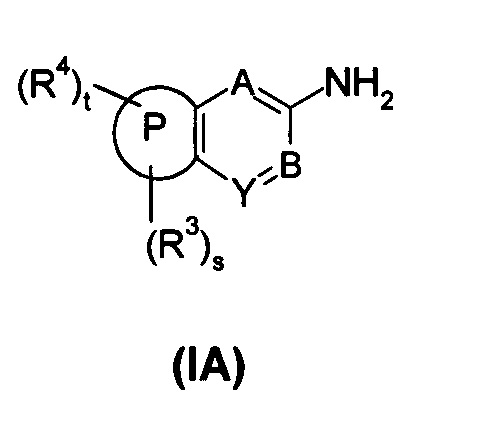 ,
,
где указанные соединения выбраны из:
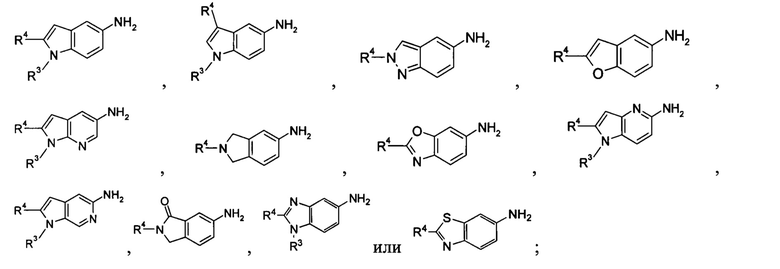
при этом, если соединения выбраны из
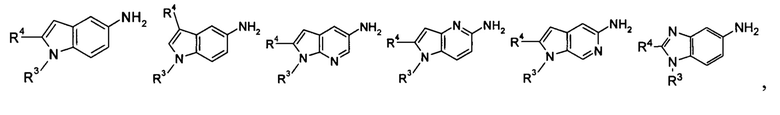
R3 выбран из водорода и алкила, содержащего от 1 до 6 атомов углерода, где указанный алкил, содержащий от 1 до 6 атомов углерода, необязательно дополнительно замещен одной или более чем одной группой, выбранной из алкокси, содержащего от 1 до 6 атомов углерода;
R4 выбран из фенила, где указанный фенил дополнительно замещен одним галогеналкилом, содержащим от 1 до 6 атомов углерода, где указанный галогеналкил, содержащий от 1 до 6 атомов углерода, предпочтительно представляет собой трифторметил; или
R3 представляет собой пяти- или шестичленный гетероциклил, имеющий один гетероатом О в качестве атома кольца;
R4 представляет собой фенил, где указанный фенил дополнительно замещен одним галогеном или галогеналкилом, содержащим от 1 до 6 атомов углерода;
и, если соединения выбраны из:
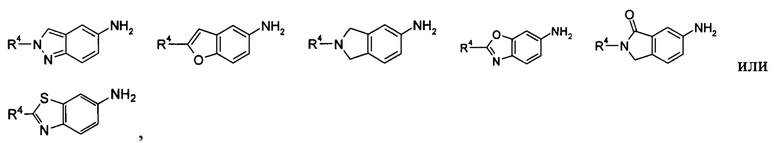
R4 выбран из фенила, где указанный фенил дополнительно замещен одним галогеном или галогеналкилом, содержащим от 1 до 6 атомов углерода, где указанный галогеналкил, содержащий от 1 до 6 атомов углерода, предпочтительно представляет собой трифторметил.
13. Соединение, или его энантиомер, или их смеси, или его фармацевтически приемлемые соли, выбранное из:
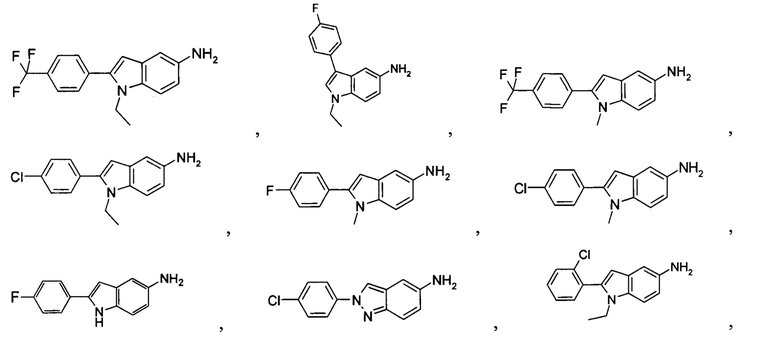
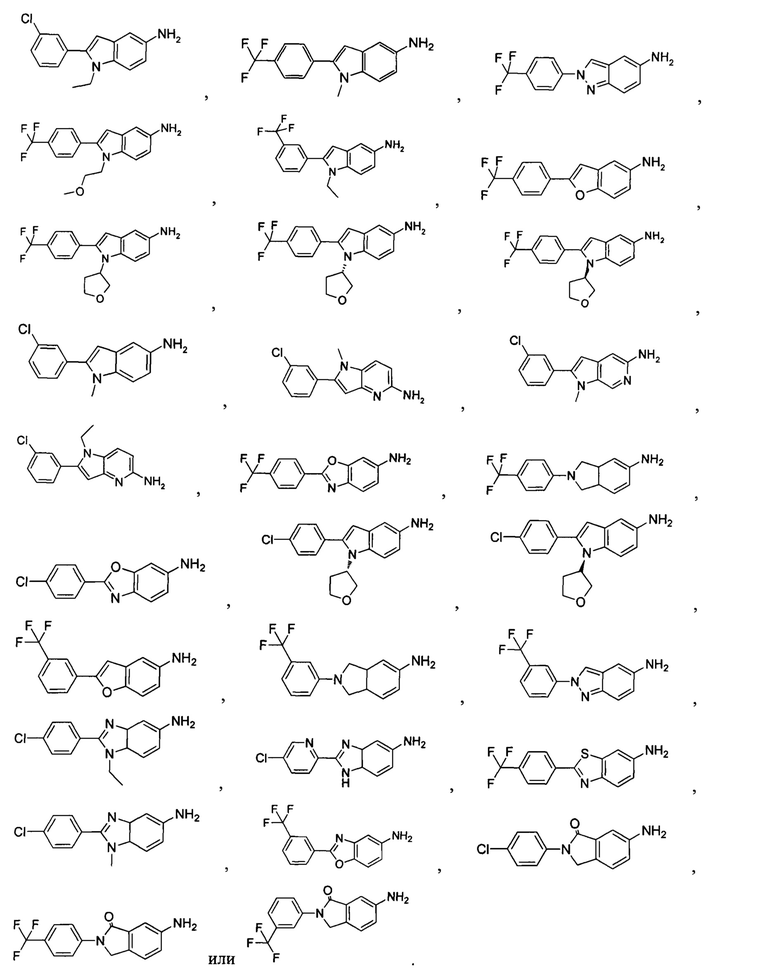
14. Соединение формулы (IB), или его энантиомер, или их смеси, или его фармацевтически приемлемые соли:
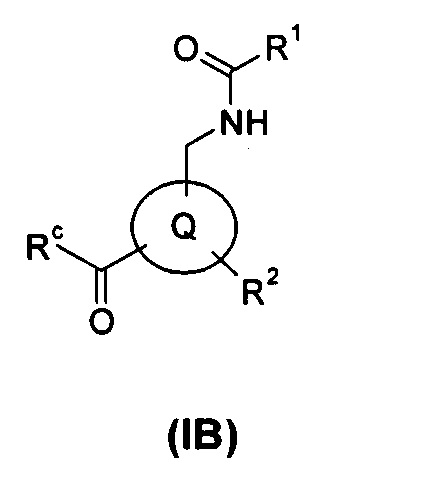 ,
,
где Rc выбран из гидрокси и галогена и
кольцо Q, R1 и R2 являются такими, как определено в п. 1,
где указанные соединения выбраны из:
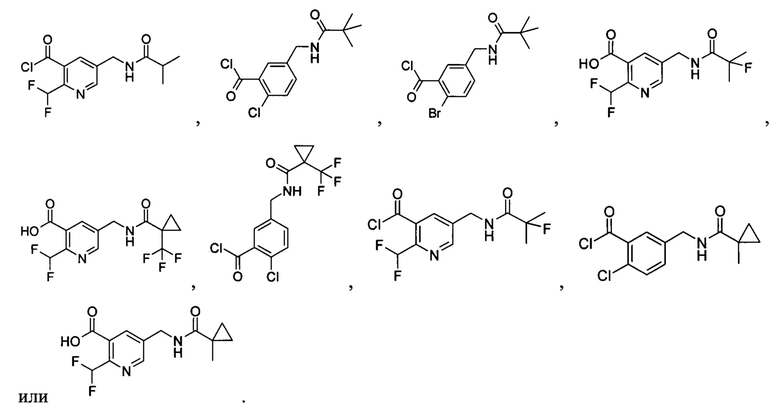
15. Фармацевтическая композиция для ингибирования микросомальной простагландин Е-синтазы-1, содержащая терапевтически эффективное количество соединения формулы (I), или его энантиомера, или их смесей, или его фармацевтически приемлемых солей по п. 1, и фармацевтически приемлемый носитель, разбавители или вспомогательное вещество.
16. Применение соединения формулы (I), или его энантиомера, или их смесей, или его фармацевтически приемлемых солей по любому из пп. 1-10, или фармацевтической композиции по п. 15 для получения лекарственного средства для ингибирования микросомальной простагландин Е-синтазы-1.
17. Применение соединения формулы (I), или его энантиомера, или их смесей, или его фармацевтически приемлемых солей по любому из пп. 1-10, или фармацевтической композиции по п. 15 для получения лекарственного средства для лечения и/или предупреждения заболеваний или расстройств, где указанные заболевания или расстройства выбраны из группы, состоящей из воспаления, боли и остеоартрита.
| Устройство перепада для сопряжения бьефов водотока | 1927 |
|
SU15689A1 |
| WO 2013118071 A1, 15.08.2013 | |||
| WO 2013038308 A1, 21.03.2013 | |||
| WO 2013072825 A1, 23.05.2013. | |||

