Изобретение относится к полиморфам мотилида и способам получения и применения таких полиморфов.
Желудочно-кишечная ("GI") двигательная функция регулирует правильное продвижение проглоченного вещества через кишечник для обеспечения адекватного всасывания питательных веществ, электролитов и жидкостей. Надлежащее прохождение содержимого желудочно-кишечного тракта через пищевод, желудок, тонкий кишечник и толстую кишку зависит от регионального регулирования внутрипросветного давления и некоторых сфинктеров, которые регулируют продвижение вперед и предотвращают обратное движение. Нормальная картина GI двигательной функции может быть нарушена в различных обстоятельствах, включая заболевание и хирургическую операцию.
Расстройства GI двигательной функции включают гастропарез и гастроэзофагеальный рефлюкс ("GERD"). Гастропарез, симптомы которого включают расстройство желудка, изжогу, тошноту и рвоту, представляет собой задержку опорожнения содержимого желудка. GERD относится к разнообразным клиническим проявлениям обратного тока содержимого желудка и двенадцатиперстной кишки в пищевод. Наиболее общими симптомами являются изжога и дисфазия, причем также известно, что они могут протекать с кровопотерей в результате эрозии пищевода. Другие примеры GI расстройств, в которые вовлечена нарушенная GI двигательная функция, включают анорексию, стаз желчного пузыря, послеоперационную паралитическую непроходимость кишечника, склеродермию, кишечную псевдообструкцию, синдром раздраженного кишечника, гастрит, рвоту и хронический запор (атонию толстой кишки).
Мотилин представляет собой пептидный гормон из 22 аминокислот, секретируемый эндокринными клетками в слизистую оболочку кишечника. Связывание его с рецептором мотилина в GI тракте стимулирует GI двигательную функцию. Введение терапевтических агентов, которые действуют как агонисты мотилина ("прокинетические агенты"), было предложено в качестве лечения GI расстройств.
Эритромицины представляют собой семейство макролидных антибиотиков, получаемых путем ферментации актиномицетов Saccharopolyspora erythraea. Эритромицин А, обычно используемый антибиотик, является наиболее распространенным и важным членом этого семейства.

Побочные эффекты эритромицина А включают тошноту, рвоту и абдоминальный дискомфорт. Эти эффекты связаны с действием эритромицина А (1) как агониста мотилина и еще в большей степени с первоначальным продуктом (5) катализируемой кислотой деградации. (Вторичный продукт деградации, спирокеталь (6), является неактивным.)
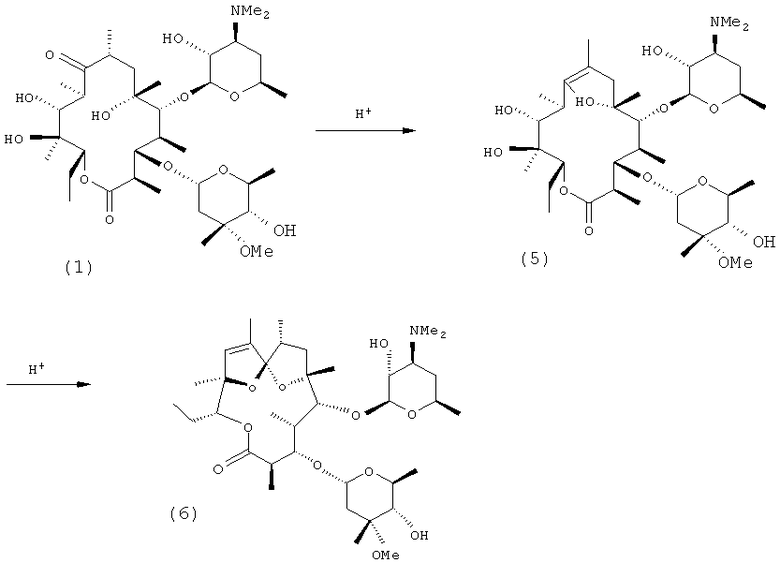
Вдохновленные открытием у эритромицина А и продукта 5 его деградации агонистической активности в отношении мотилина исследователи приложили усилия с целью обнаружения новых мотилидов, как называют макролиды с прокинетической активностью. В значительной степени данное исследование было сконцентрировано на создании новых аналогов эритромицина, или посредством постферментационной химической трансформации продуцируемого в природе эритромицина, или посредством модификации (включая генно-инженерную) процесса ферментации. Иллюстративные описания, относящиеся к мотилидам, включают: Omura et al., US 5008249 (1991) и US 5175150 (1992); Harada et al., US 5470961 (1995); Freiberg et al., US 5523401 (1996); US 5523418 (1996); US 5538961 (1996); и US 5554605 (1996); Lartey et al., US 5578579 (1996); US 5654411 (1997); US 5712253 (1998); и US 5834438 (1998); Кода et al., US 5658888 (1997); Miura et al., US 5959088 (1998); Premchandran et al., US 5922849 (1999); Keyes et al., US 6084079 (2000); Ashley et al., US 2002/0025936 A1 (2002); Ashley et al., US 2002/0094962 A1 (2002); Carreras et al., US 2002/0192709 A1 (2002); Ito et al., JP 60-218321 (1985) (соответствующий реферат №104:82047 в Chemical Abstracts); Santi et al., US 2004/138150 A1 (2004); Carreras et al., US 2005/0113319 A1 (2005); Carreras et al., US 2005/0119195 A1 (2005); Liu et al., US 2005/0256064 A1 (2005); Omura et al., J. Antibiotics 1985, 38, 1631-2; Faghih et al., Biorg. & Med. Chem. Lett., 1998, 8, 805-810; Faghih et al., J. Med. Chem., 1998, 41, 3402-3408; Faghih et al., Synlett., Jul. 1998, 751; и Lartey et at., J. Med. Chem., 1995, 38, 1793-1798. Описания всех вышеупомянутых документов включены в данное описание посредством ссылки.
Также возможно подходят и другие соединения с эритромициновым каркасом, даже в тех случаях, когда они не были предназначены в качестве агонистов мотилина; при этом иллюстративными описаниями являются: Krowicki et al., US 3855200 (1974); Radobolja et al., US 3939144 (1976); Kobrehel et al., US 3983103 (1976); Toscano, US 4588712 (1986); Agouridas et al., US 5444051 (1995); Agouridas et al., US 5561118 (1996); Agouridas et al., US 5770579 (1998); Asaka et al., US 6169168 B1 (2001); Kobrehel et al., DE 2402200 (1974); Pliva Pharmaceuticals, GB 1416281 (1975); Pliva Pharmaceuticals, GB 1461032 (1977); Asaga et al., JP 2002/241391 (2002); Ryden etal., J. Med. Chemistry, 1973, 16 (9), 1059-1060; Naperty et al., Roczniki Chemii, 1977, 51 (6), 1207-10; Kobrehel et al., Eur. J. Med. Chemistry, 1978, 13 (1), 83-7; Egan et al., J. Antibiotics, 1978, 31 (1), 55-62; Matijasevic et al., Croatica Chemica Acta, 1980, 53 (3), 519-24; Radobolja et al., Croatica Chemica Acta, 1985, 58 (2), 219-25; Hunt et al., J. Antibiotics, 1989, 42 (2), 293-298; Myles et al., J. Org. Chem., 1990, 55, 1636-1648. Описания всех вышеупомянутых документов включены в данное описание посредством ссылки.
Специалистам в данной области техники будет очевидно, что при разработке мотилидов ряд параметров является существенным. Во-первых, эволюция эритромицинового каркаса в природных продуцирующих микроорганизмах обусловлена антибактериальной эффективностью, а не прокинетической эффективностью. Таким образом, остаются значительные возможности для оптимизации зависимости структура-активность для агонистов мотилина. Во-вторых, на самом деле нежелательно, чтобы мотилид обладал антибактериальной активностью. GI тракт представляет собой "хозяина" для огромной популяции бактерий, воздействие на которые мотилида, обладающего антибактериальной активностью, может индуцировать развитие у них устойчивости к эритромициновым антибиотикам. Или мотилид, обладающий антибактериальной активностью, может убивать полезные для кишечника бактерии. Таким образом, желательно, чтобы в результате разработки мотилида его прокинетическая активность была повышена, а антибактериальная активность была исключена. В-третьих, недостатком, обычно встречающимся среди мотилидов по оценкам на сегодняшний день, является их склонность десенсибилизировать рецептор мотилида, а это означает, что после первой дозы последующие дозы мотилида вызывают более слабый ответ или не вызывают ответа (тахифилаксия). В-четвертых, важны стабильность и биодоступность - доказательством служит быстрая деградация эритромицина А в желудке и отсутствие активности у его вторичного продукта деградации. В-пятых, сообщали, что некоторые соединения эритромицинового семейства демонстрируют нежелательные проаритмические эффекты, включая пролонгацию интервала QT и индукцию желудочковых аритмий. Желательно ограничить эти эффекты до приемлемого уровня. Таким образом, существует необходимость в разработке новых мотилидов, удовлетворяющих различным требованиям к рабочим параметрам.
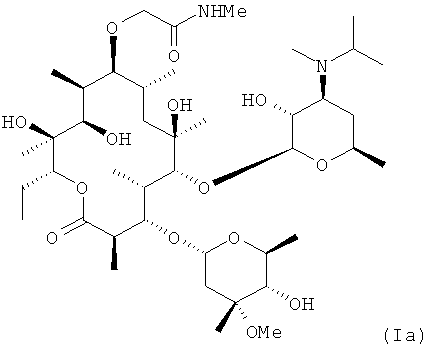
В заявке Liu et at., US 2006/0270616 A1 (2006), включенной в данное описание посредством ссылки (далее "заявка Liu '616''), описано семейство мотилидов, представленных общей формулой I, где RA, RB, RC, RD и RE представляют собой структурные переменные. Конкретным соединением, описанным в указанной заявке, является соединение (Ia), которое обладает привлекательным балансом свойств для мотилида.
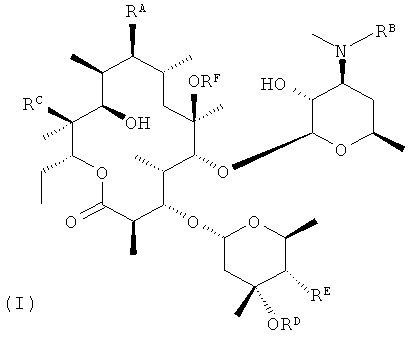
После того как соединение выбрано для разработки в качестве возможного кандидата для клинического применения, должно быть уделено внимание технологии изготовления его в составе соответствующей фармацевтической композиции. В свою очередь это означает, должно быть уделено внимание возможности существования полиморфов. Если полиморфы существуют, то они могут отличаться по своим фармацевтически важным свойствам, включая растворимость, стабильность при хранении, гигроскопичность, плотность и биодоступность. В процессе хранения один полиморф может более или менее спонтанно превращаться в другой полиморф. В результате такого превращения композиция, предназначенная для доставки конкретного полиморфа, может в конечном счете содержать другой полиморф, который несовместим с данной композицией. Гигроскопичный полиморф может захватывать воду в процессе хранения, что вносит ошибки в операции взвешивания и влияет на возможность его обработки. Методика приготовления, предназначенная для применения с конкретным полиморфом, может не подходить для применения с другим полиморфом. Даже если не происходит никаких взаимопревращений, изготовить композицию с одним полиморфом может быть легче, чем с другим, что делает выбор правильного полиморфа критическим. Таким образом, выбор полиморфа является важным фактором в разработке фармацевтической композиции. (Как использовано в данном описании, термин "полиморф" включает аморфные формы и несольватированные и сольватированные кристаллические формы, как подробно изложено в руководстве Q6A(2) ICH (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use)).
Настоящее изобретение относится к полиморфам соединения Iа, которые особенно желательны для использования в фармацевтических композициях.
Соединение Iа, если его получают согласно заявке Liu '616, получают в форме, которая не оптимизирована для создания композиций (эта форма обозначена в данном описании как Полиморф I - см. Пример 3, ниже). Авторы изобретения обнаружили дополнительные полиморфы соединения Ia, включая полиморф (упоминаемый в данном описании как Полиморф IV), который имеет улучшенные свойства для использования в фармацевтической композиции. Другой полиморф, обозначенный как Полиморф II, также имеет подходящие свойства для использования в фармацевтической композиции. Таким образом, в одном из воплощений согласно этому изобретению предложен очищенный полиморф IV соединения Ia. В другом воплощении предложен очищенный полиморф II соединения Ia.
В другом воплощении согласно этому изобретению предложен способ получения очищенного Полиморфа IV Соединения Ia, включающий подвергание полиморфа Соединения Ia, упоминаемого в данном описании как Полиморф II, множественным циклам нагревания и охлаждения в присутствии среды, выбранной из диизопропилового эфира ("DIPE") и С5-С7алкана или -алкена (предпочтительно гептана).
В другом воплощении согласно этому изобретению предложен способ получения очищенного Полиморфа IV Соединения Ia, включающий получение этилацетатного раствора Соединения Ia и добавление к этому раствору C5-С7алкана или -алкена для того, чтобы вызвать кристаллизацию Соединения la в виде очищенного Полиморфа IV.
В другом воплощении согласно этому изобретению предложена фармацевтическая композиция, содержащая очищенный Полиморф IV Соединения Ia и фармацевтически приемлемый эксципиент.
В другом воплощении согласно этому изобретению предложена фармацевтическая композиция, содержащая очищенный Полиморф II Соединения Iа и фармацевтически приемлемый эксципиент.
Согласно изобретению также предложены: способ лечения заболевания нарушенной двигательной функции желудка, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества очищенного Полиморфа IV Соединения Iа; очищенный Полиморф IV Соединения Iа для применения в качестве лекарственного средства; очищенный Полиморф IV Соединения Iа для применения в лечении заболевания нарушенной двигательной функции желудка; применение очищенного Полиморфа IV Соединения Iа для изготовления лекарственного средства для лечения заболевания нарушенной двигательной функции желудка; и фармацевтическая композиция для лечения заболевания нарушенной двигательной функции желудка, содержащая очищенный Полиморф IV Соединения Iа.
Кроме того, согласно изобретению предложены: способ лечения заболевания нарушенной двигательной функции желудка, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества очищенного Полиморфа II Соединения Iа; очищенный Полиморф II Соединения Iа для применения в качестве лекарственного средства; очищенный Полиморф II Соединения Iа для применения в лечении заболевания нарушенной двигательной функции желудка; применение очищенного Полиморфа II Соединения Iа для изготовления лекарственного средства для лечения заболевания нарушенной двигательной функции желудка; и фармацевтическая композиция для лечения заболевания нарушенной двигательной функции желудка, содержащая очищенный Полиморф II Соединения Iа.
Иллюстративные примеры расстройств, которые представляют собой заболевания нарушенной двигательной функции желудка, включают (без ограничения) гастропарез, гастроэзофагеальный рефлюкс ("GERD"), анорексию, стаз желчного пузыря, послеоперационную паралитическую непроходимость кишечника, склеродермию, кишечную псевдообструкцию, синдром раздраженного кишечника, гастрит, рвоту и хронический запор (атонию толстой кишки). Полиморфы по изобретению особенно эффективны в лечении GERD.
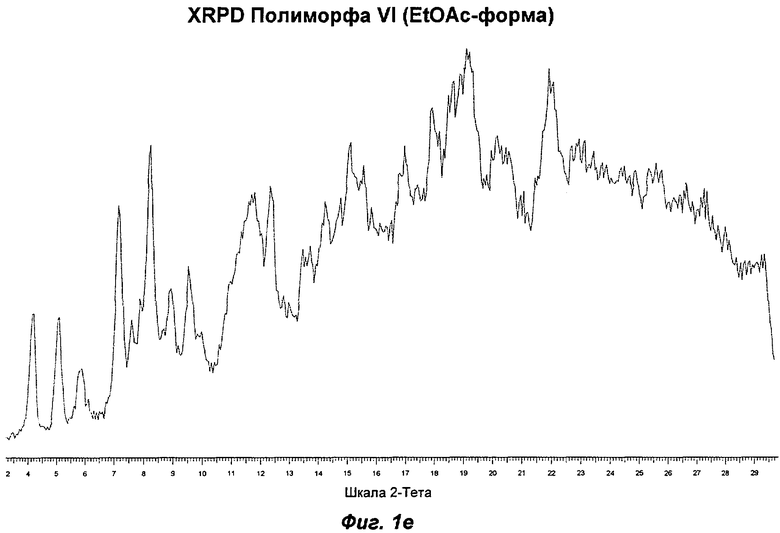
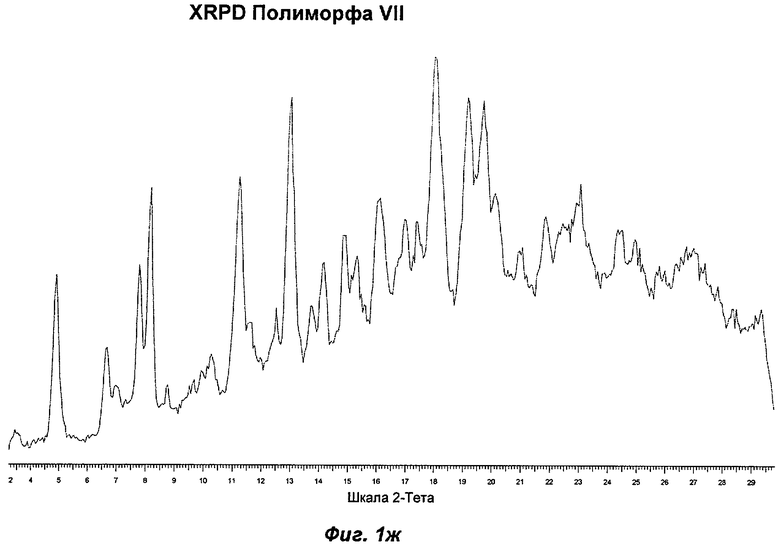
На Фиг.1а, 1б, 1в, 1г, 1д, 1е и 1ж представлены репрезентативные картины дифракции рентгеновских лучей на порошке ("XRPD") для Полиморфов I, II, III, IV, V, VI (этилацетатная форма) и VII соответственно Соединения Ia.
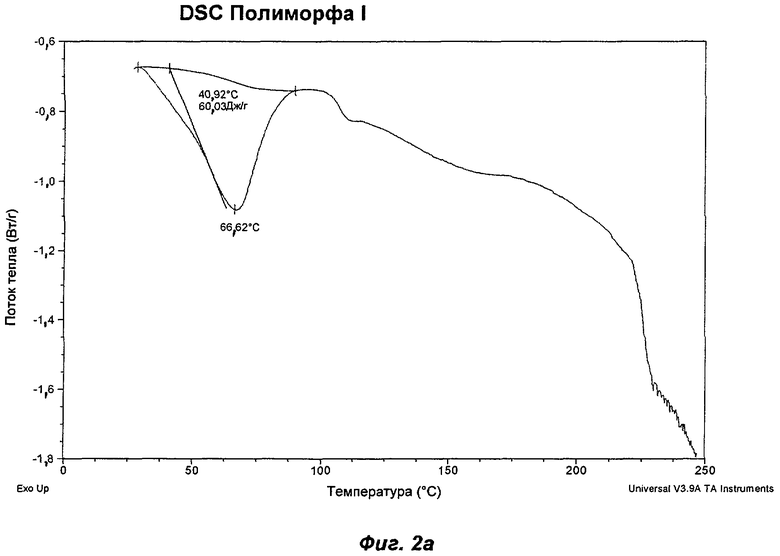
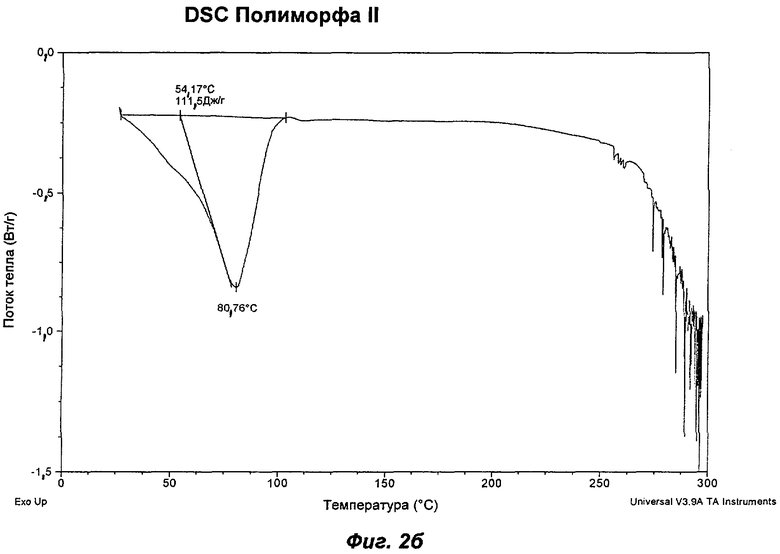
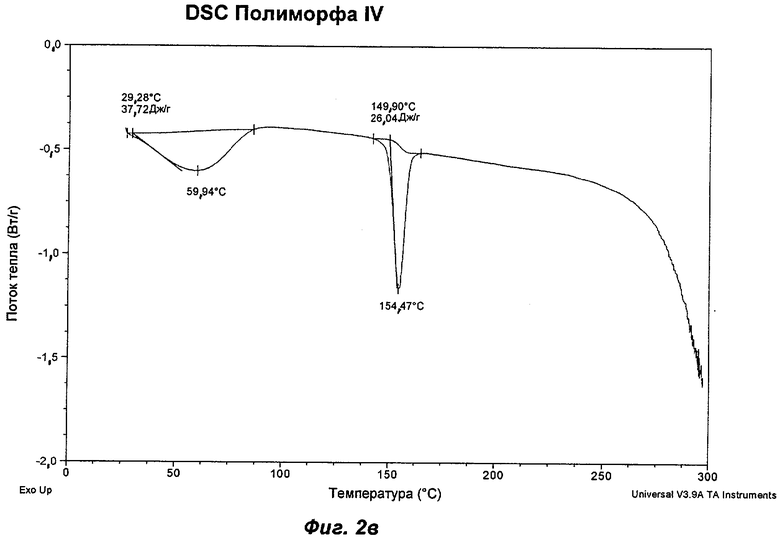
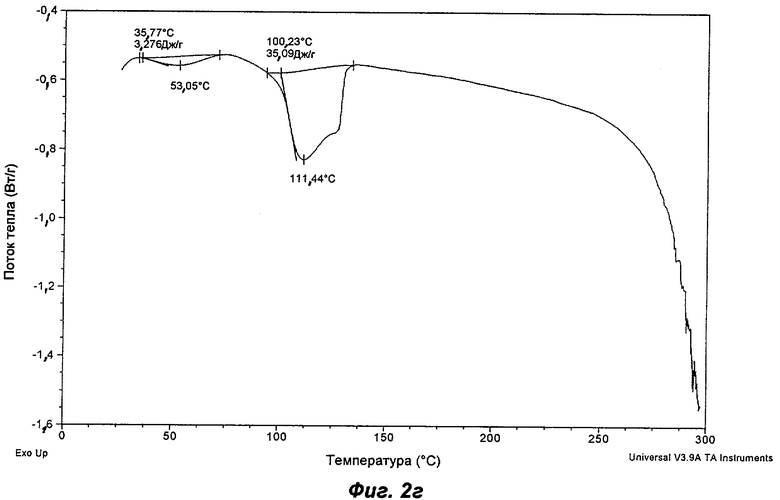
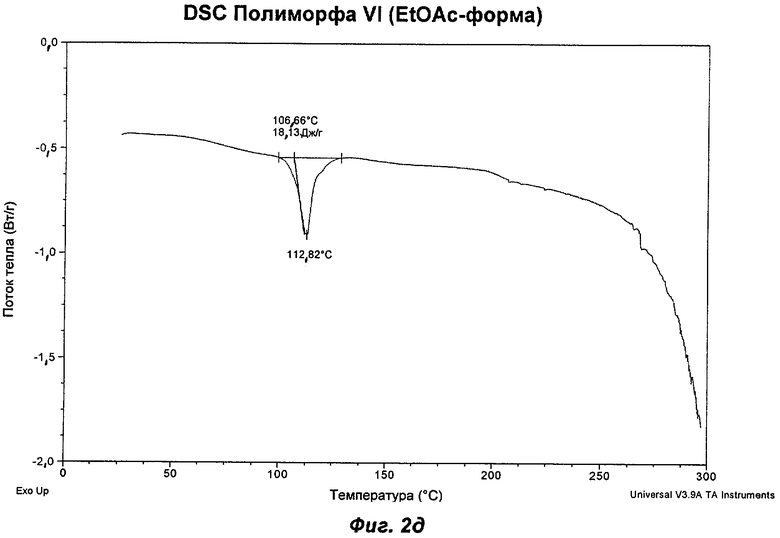
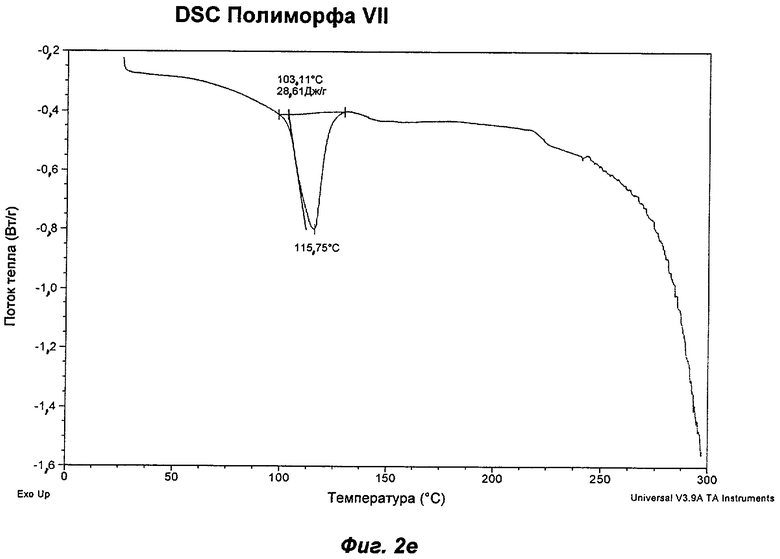
На Фиг.2а, 2б, 2в, 2г, 2д и 2е представлены репрезентативные полученные сканированием результаты дифференциальной сканирующей калориметрии ("DSC") для Полиморфов I, II, IV, V, VI (этилацетатная форма) и VII соответственно Соединения Ia.
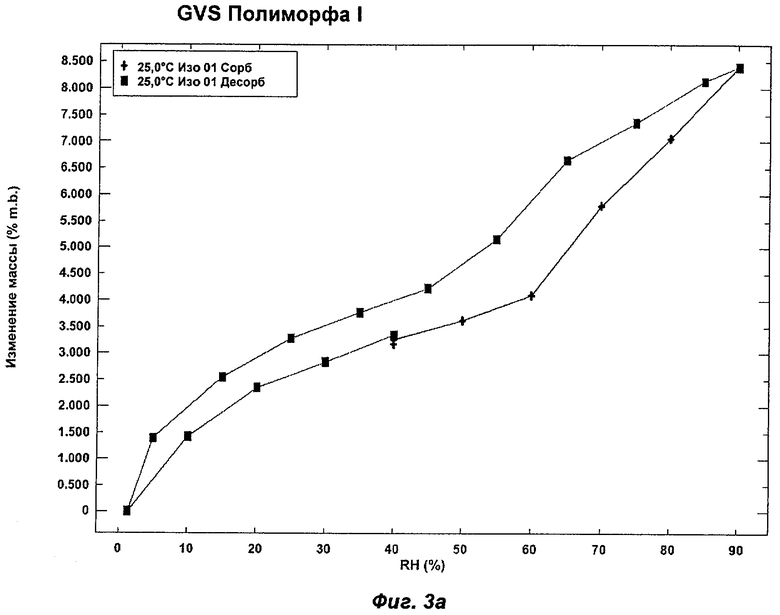
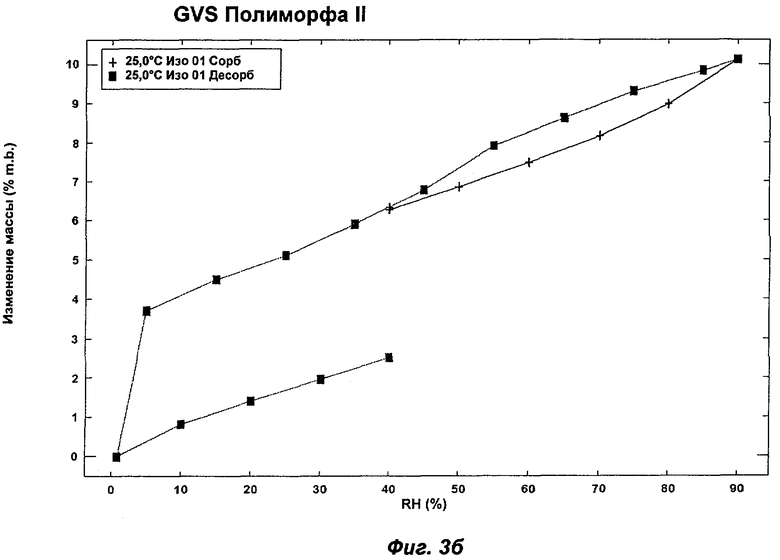
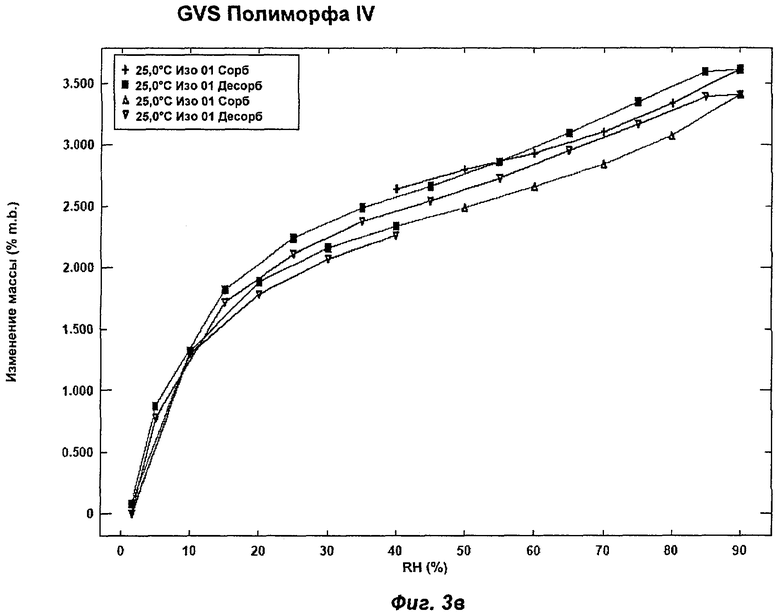
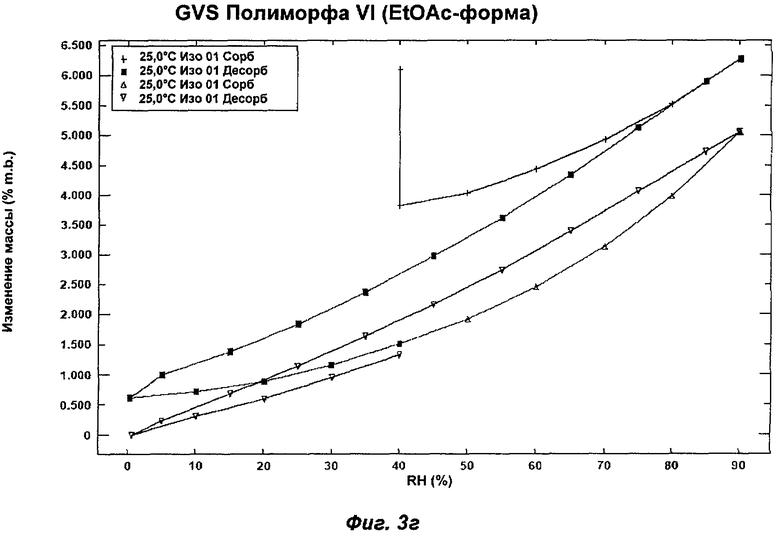
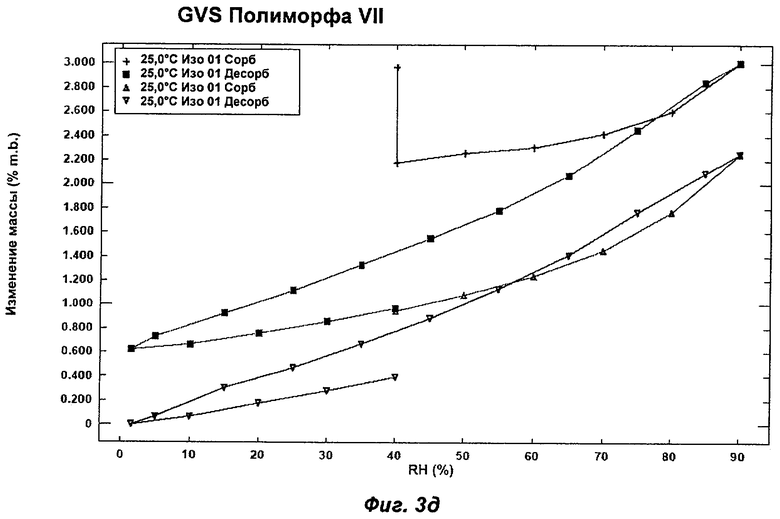
На Фиг.3а, 3б, 3в, 3г и 3д представлены репрезентативные полученные сканированием результаты гравиметрических исследований сорбции пара ("GVS") для Полиморфов I, II, IV, VI (этилацетатная форма) и VII соответственно Соединения Ia.
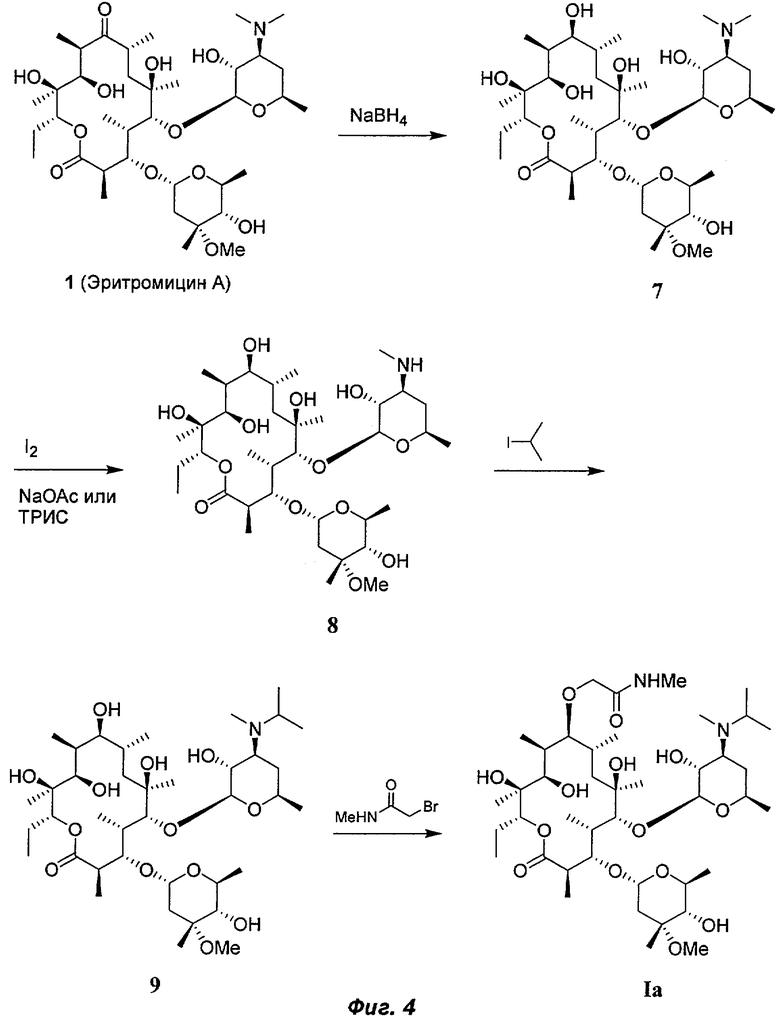
На Фиг.4 показана схема синтеза для получения соединения la.
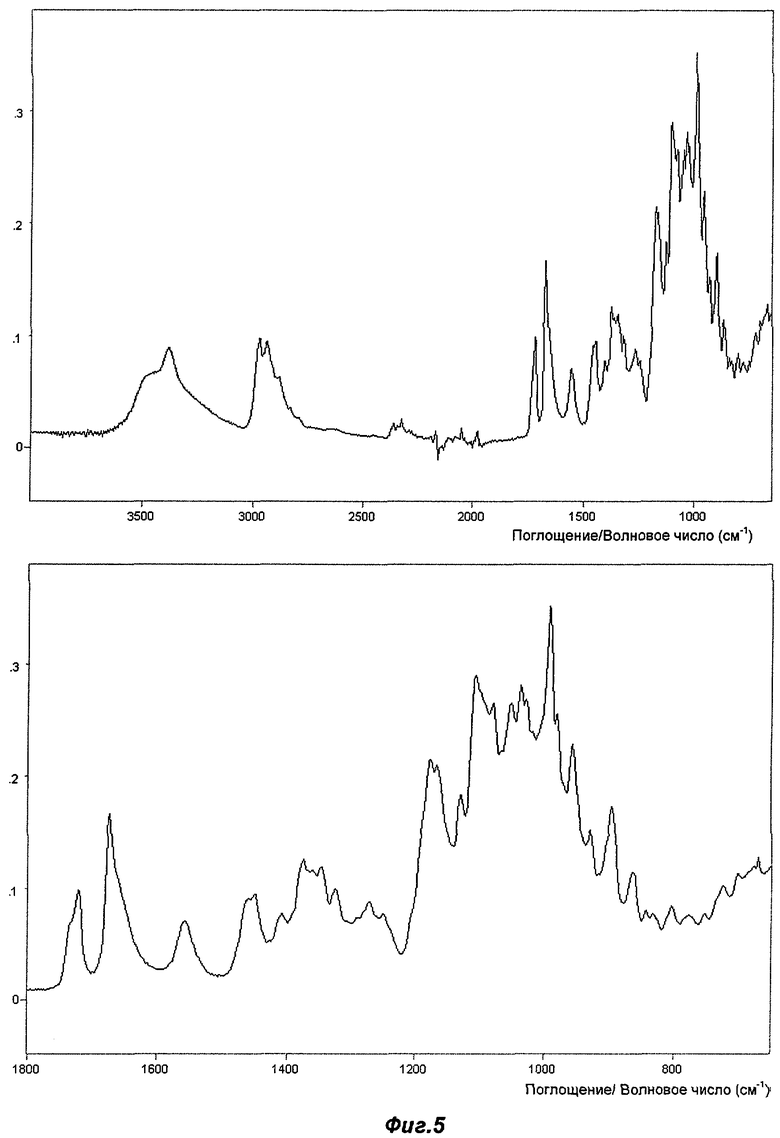
На Фиг.5 представлен репрезентативный FT-IR-спектр (инфракрасный спектр с Фурье-преобразованием) для Полиморфа IV.
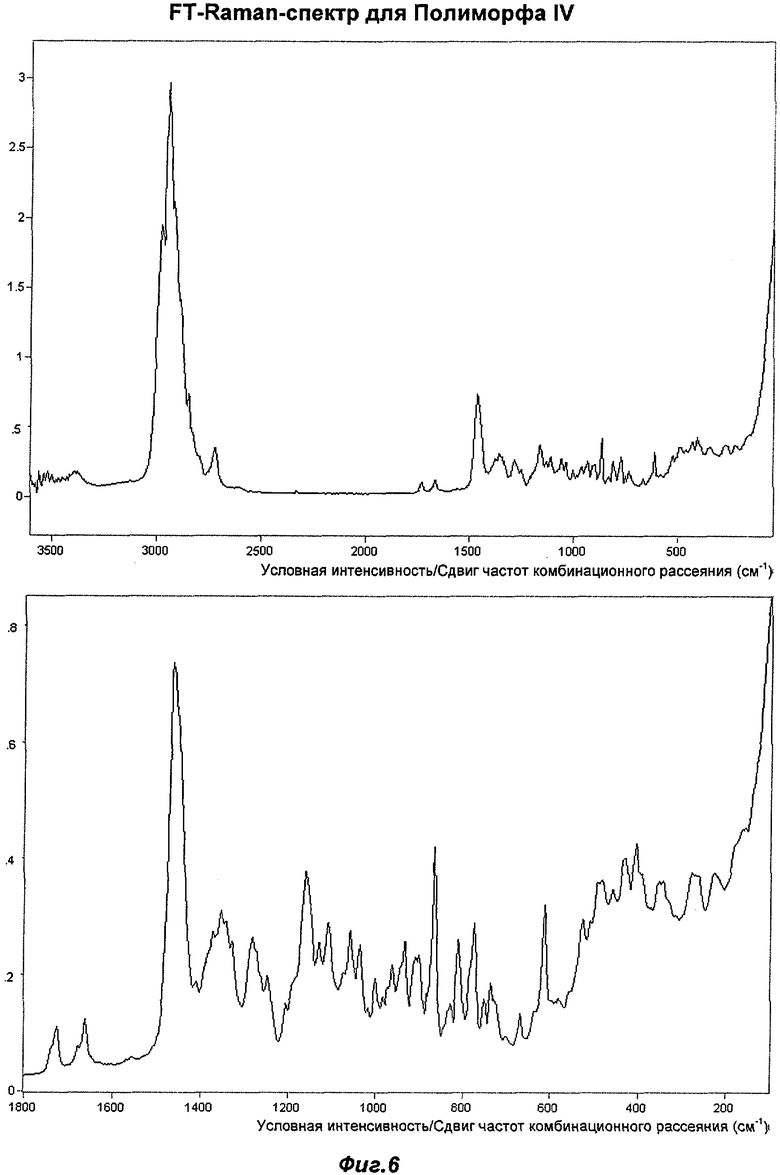
На Фиг.6 представлен репрезентативный FT-Raman-спектр (спектр комбинационного рассеяния с Фурье-преобразованием) для Полиморфа IV.
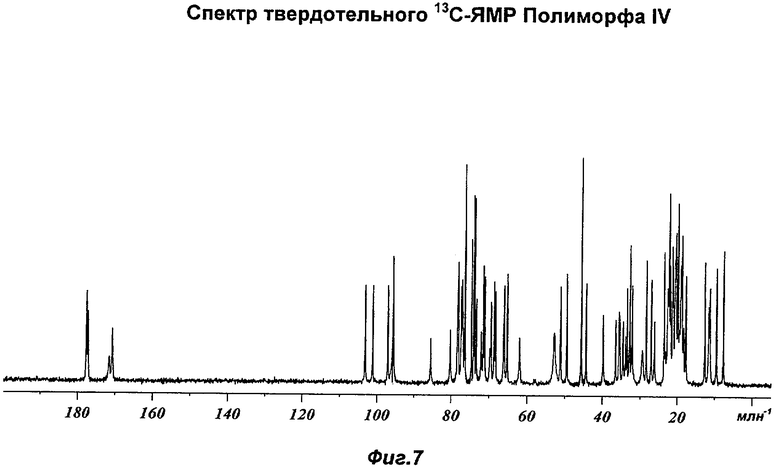
На Фиг.7 представлен репрезентативный спектр твердотельного 13С-ЯМР (ядерного магнитного резонанса) для Полиморфа IV.
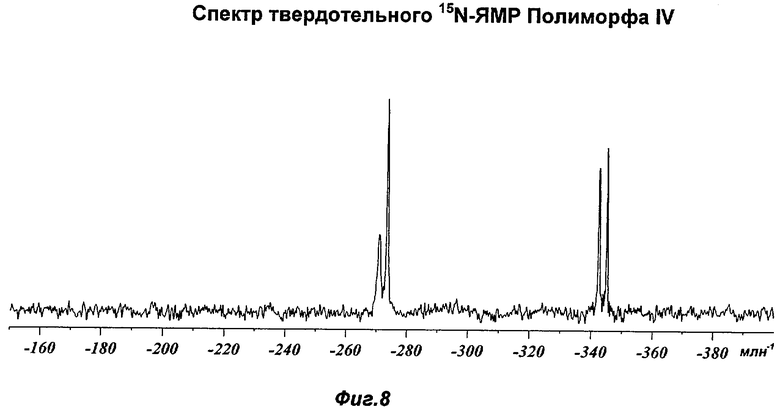
На Фиг.8 представлен репрезентативный спектр твердотельного 15N-ЯМР для Полиморфа IV.
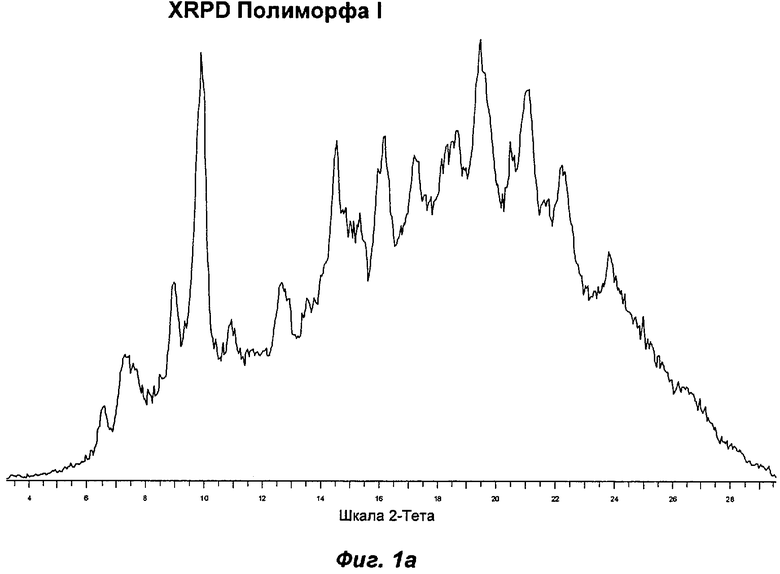
Полиморф I был охарактеризован как белый порошок, который в значительной степени был аморфным, при этом слабо кристаллическим по данным XRPD. Он был относительно гигроскопичным и демонстрировал увеличение массы на 8,5% в диапазоне RH (относительная влажность) от 0 до 90%. Термический анализ показал эндотерм в интервале от температуры окружающей среды до 90°С, обусловленный потерей растворителя. Потеря массы, сопровождающая эндотерм, составляла 3,0%, что соответствует 1,4 моль воды. При нагревании до температуры в диапазоне от 75 до 100°С Полиморф I терял кристалличность. В водных условиях Полиморф I превращался во второй полиморф, упоминаемый как Полиморф II. Эти два последних наблюдения свидетельствуют против выбора Полиморфа I в качестве полиморфа для создания композиций. Репрезентативные данные XRPD, DSC и GVS для Полиморфа I показаны на Фиг.1а, 2а и 3а соответственно.
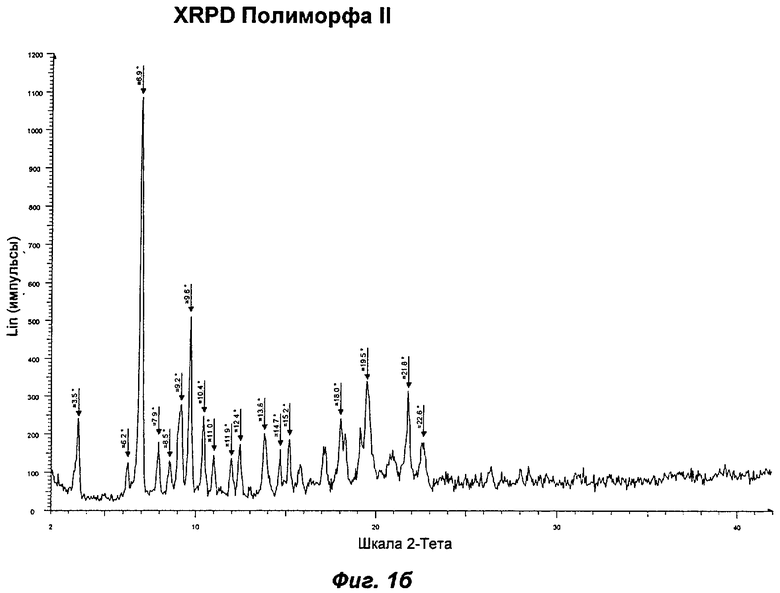
Полиморф II был охарактеризован как белый порошок с частицами небольшого размера (<10 мкм) и без какой-либо различимой морфологии. XRPD показала, что он является кристаллическим с некоторым содержанием аморфного вещества. При выдерживании в интервале значений RH от 5 до 0% Полиморф II показал потерю массы на 4%, эквивалентную 2 моль воды на моль соединения (Ia). Этому соответствовала потеря кристалличности, о чем свидетельствовали результаты повторного анализа с использованием XRPD в условиях окружающей среды, указывая на то, что Полиморф II представляет собой дигидрат. Термический анализ показал широкий эндотерм при температуре в диапазоне от температуры окружающей среды до 100°С, обусловленный потерей растворителя (воды). Эта потеря соответствует 5,0% потере массы, что эквивалентно 2,5 моль воды, дополнительному содержанию воды, свойственному Полиморфу II, являющемуся гигроскопичным. Наблюдали потерю кристалличности в диапазоне 50-75°С. Кроме того, Полиморф II терял кристалличность в процессе вакуумной сушки при 30°С в течение 72 ч. Репрезентативные данные XRPD, DSC и GVS для Полиморфа II показаны на Фиг.1б, 2б и 3б соответственно.
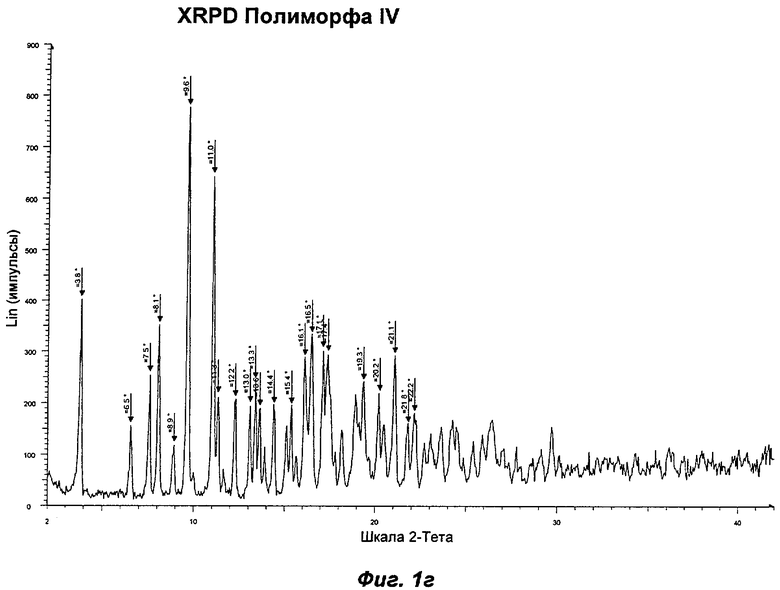
Полиморф IV был охарактеризован как белый порошок с размером частиц вплоть до 50 мкм и игольчатой морфологией. Он был кристаллическим по данным XRPD. Его растворимость в воде составляла 0,77 мг/мл. При степени чистоты 97,9% он не был сильно гигроскопичным, демонстрируя увеличение массы на 3,5% в диапазоне RH 0-90%. Увеличение массы не приводило к изменению картины XRPD при проведении повторного анализа в условиях окружающей среды. Термический анализ показал широкий эндотерм в диапазоне от температуры окружающей среды до 65°С, обусловленный потерей растворителя (воды) (потеря массы 1,5%). Имелся переход при плавлении (melting transition) с началом при 150°С без какого-либо изменения картины XRPD при нагревании вплоть до плавления. Ни при хранении при 40°С и RH 75% в течение 10 недель, ни при манипулировании во время проведения анализа растворимости не происходило каких-либо значительных изменений. Сохранение кристалличности при нагревании и стабильность при хранении делают Полиморф IV хорошим кандидатом для создания на его основе фармацевтических композиций. Репрезентативные данные XRPD, DSC и GVS для Полиморфа IV показаны на Фиг.1г, 2в и 3в соответственно. Репрезентативные данные FT-IR, FT-Raman, твердотельного 13С-ЯМР и твердотельного 15N-ЯMP для Полиморфа IV показаны на Фиг.5, 6, 7 и 8 соответственно.
Полиморф IV может быть получен из Полиморфа II в результате созревания (повторяющиеся циклы нагревания и охлаждения) в диизопропиловом эфире. Также можно использовать С5-С7-алкан или -алкен, такой как (предпочтительно) гептан, - получаемое таким образом вещество первоначально содержало некоторое количество Полиморфа II, который, впрочем, был удален (что определено по данным XRPD) после сушки под вакуумом. Число циклов составляет по меньшей мере два, предпочтительно 3, хотя может быть использовано и большее число циклов (например, вплоть до 12). Температурный диапазон для циклов обычно составляет от 5 до 50°С, предпочтительно от 25 до 50°С в течение периода времени 24 ч.
В дополнение к этому авторы изобретения также открыли несколько других полиморфов Соединения Ia, при этом получение и характеристики таких других полиморфов суммированы ниже. По различным причинам эти полиморфы менее желательны, чем Полиморфы II и IV, для создания композиций.
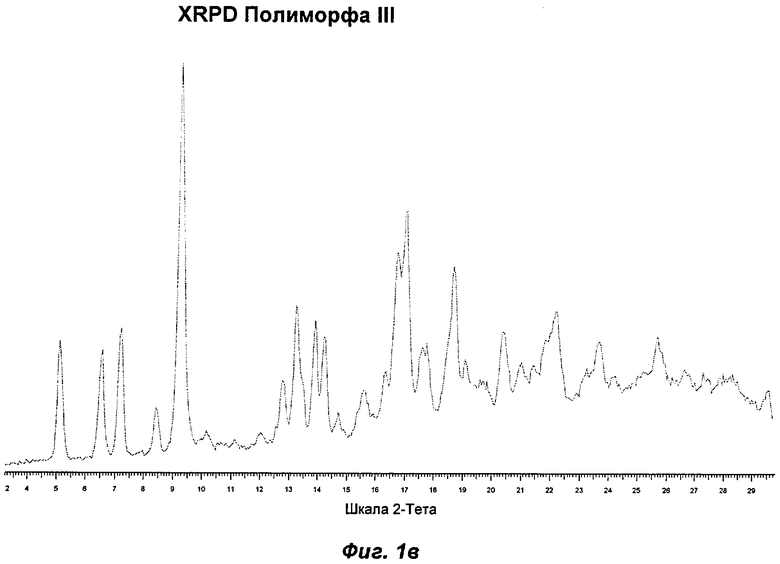
Полиморф III представляет собой полиморф, полученный после созревания (повторяющиеся циклы нагревания и охлаждения) аморфной стеаратной соли Соединения Ia в DIPE. Этот полиморф не смогли выделить в больших количествах и дополнительно не исследовали. На Фиг.1в показаны репрезентативные данные XRPD для Полиморфа III.
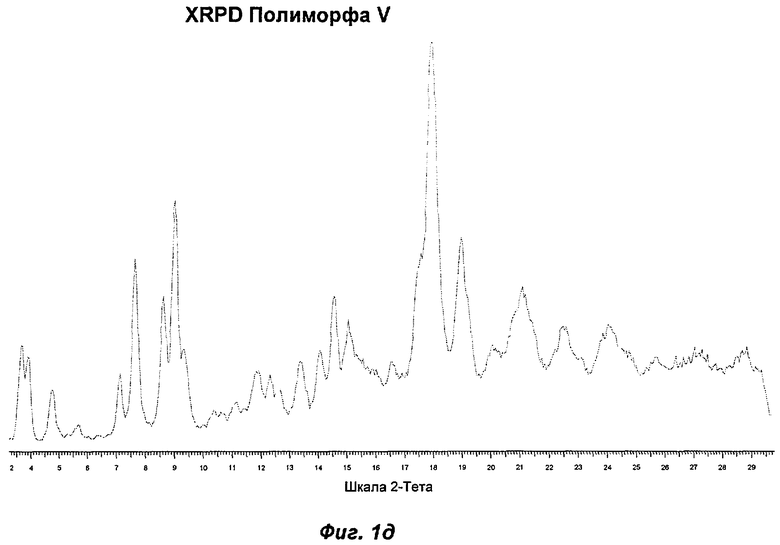
Полиморф V получали созреванием в трет-бутил метиловом эфире ("ТВМЕ"). Он был охарактеризован как белый порошок с частицами небольшого размера (<10 мкм) и без какой-либо различимой морфологии. Он был кристаллическим по данным XRPD, его растворимость в воде составляла 0,72 мг/мл. Термический анализ показал переход при плавлении с началом при 100°С. Это коррелировало с потерей массы на 8,7% по TGA (термогравиметрический анализ), эквивалентной 1 моль ТВМЕ, указывая на то, что Полиморф V представляет собой моносольват ТВМЕ. Кристалличность Полиморфа V уменьшилась через одну неделю хранения при 40°С и RH 75%, и он превращался в Полиморф II при проведении анализа растворимости. Он является сольватом, и это свидетельствует о том, что он нежелателен в качестве кандидата для создания композиций. Репрезентативные данные XRPD и DSC для Полиморфа V показаны на Фиг.1д и 2г соответственно.
Полиморф VI представляет собой частично кристаллический полиморф, полученный из этилацетата, изопропилацетата или анизола. Небольшие ступенчато изменяющиеся потери массы наблюдали в процессе проведения термогравиметрического анализа (TGA), что указывает на то, что он относится к семейству изоморфных сольватов. Начало эндотерма для формы, полученной из этилацетата, составляло 107°С; соответствующее начало для изопропилацетатной формы составляло 90°С. Анизольная форма имела два эндотерма с началом при 98 и 110°С. Полиморф VI превращался в Полиморф IV в процессе хранения при 40°С и RH 75% или в Полиморф IV или II при проведении анализа растворимости. Его превращение в Полиморф IV указывает на то, что он недостаточно стабилен, чтобы быть желательным кандидатом для создания композиций.
Репрезентативные данные XRPD, DSC и GVS для Полиморфа VI (этилацетатная форма) показаны на Фиг.1е, 2д и 3г соответственно.
Полиморф VII получали после созревания в толуоле. Он был охарактеризован как белый порошок с частицами небольшого размера (<20 мкм) без какой-либо различимой морфологии. Он был частично кристаллическим по данным XRPD. Его растворимость в воде составила 0,75 мг/мл. Он демонстрировал постоянную потерю массы в гравиметрическом анализе сорбции пара ("GVS") с соответствующей потерей кристалличности по данным повторного анализа с использованием XRPD в условиях окружающей среды. Термический анализ показал переход при плавлении с началом при 103°С, сопровождающийся потерей массы на 4,7% по данным TGA, эквивалентной 0,5 моль толуола. Таким образом, по-видимому, Полиморф VII представляет собой геми-толуол-сольват. Полиморф VII терял кристалличность через одну неделю хранения при 40°С и RH 75% и превращался в смесь Полиморфов II и IV при проведении анализа растворимости. То, что он представляет собой сольват, и его нестабильность делают его менее желательным в качестве кандидата для создания композиций.
Репрезентативные данные XRPD, DSC и GVS для Полиморфа VII показаны на Фиг.1ж, 2е и 3д соответственно.
На Фиг.16 показана репрезентативная картина XRPD для Полиморфа II. В Таблице 1 представлена табличная сводка главных пиков, приведенных на Фиг.16. Таким образом, в одном аспекте Полиморф II может быть определен по его характеристическим пикам XRPD при 3,5±0,1; 6,9±0,1; 9,2±0,1; 9,6±0,1 и 10,4±0,1 градусах 2θ или по его характеристическим пикам XRPD при 3,5±0,1; 6,9±0,1; 9,2±0,1; 10,4±0,1 и 18,0±0,1 градусах 2θ.
На Фиг.1 г показана репрезентативная картина XRPD для Полиморфа IV. В Таблице 2 представлена табличная сводка главных пиков, приведенных на Фиг.1 г. Таким образом, в одном аспекте Полиморф IV может быть определен по его характеристическим пикам XRPD при 3,8±0,1; 7,5±0,1; 8,1±0,1; 9,6±0,1 и 11,0±0,1 градусах 2θ или по его характеристическим пикам XRPD при 3,8±0,1; 7,5±0,1; 16,1±0,1; 16,5±0,1 и 17,1±0,1 градусах 2θ.
На Фиг.2в показан репрезентативный полученный сканированием результат DSC Полиморфа IV. (В этом случае образец Полиморфа IV получали с использованием DIPE в соответствии с Примером 4.) Полиморф IV демонстрирует широкий эндотерм при температуре в диапазоне от температуры окружающей среды до 110°С, обусловленный потерей растворителя, за которым следует эндотерм плавления с началом при 143-156°С и минимумом при 149-161°С. Такой эндотерм отсутствует у других полиморфов Соединения Ia, идентифицированных авторами изобретения. Таким образом, в одном аспекте Полиморф IV может быть охарактеризован как имеющий эндотерм плавления с температурой начала плавления от примерно 143 до примерно 156°С, отличающий его от других полиморфов Соединения Ia.
На Фиг.3в показан репрезентативный полученный сканированием результат GVS Полиморфа IV при постоянной температуре 25°С. Полиморф IV демонстрирует увеличение массы на 3,5% в диапазоне RH 0-90%. Прирост/потеря массы происходят очень равномерно в процессе многочисленных циклов сорбции и десорбции. Полиморфы I (Фиг.3а), II (Фиг.3б) и VI (Фиг.3г) демонстрировали увеличение массы на 6-10% в диапазоне RH 0-90%, и для них прирост/потеря массы радикально изменялся в процессе многочисленных циклов сорбции и десорбции. Полиморф VII (Фиг.3д) демонстрировал увеличение массы на 3% в диапазоне RH 0-90%, но его прирост/потеря массы также радикально изменялись в процессе многочисленных циклов сорбции и десорбции. Таким образом, в одном аспекте Полиморф IV может быть охарактеризован как имеющий увеличение массы на 3,5% в диапазоне RH 0-90% (при 25°С) и равномерный прирост/потерю массы в процессе многочисленных циклов сорбции и десорбции.
На Фиг.5 показан репрезентативный FT-IR-спектр Полиморфа IV. Могут быть отмечены следующие основные полосы поглощения (см-1) (s = сильное, m = среднее, w = слабое, ошибка эксперимента составляет +/- 2 см-1): 3381 (m), 2973(m), 2936(m), 1721(m), 1674(m), 1558(w), 1450(m), 1408(w), 1375(m), 1347(m), 1325(w), 1272(w), 1250(w), 1176(s), 1167(s), 1130(w), 1108(s), 1080(w), 1053(w), 1038(w), 1029(w), 993(s), 982(w), 958(m), 930(w), 898(m), 864(w), 844(w), 833(w), 804(w), 778(w), 753(w), 724(w), 701 (w) и 668(w). Наиболее характерными являются следующие пики: 1558(w), 1347(m), 1130(w), 1108(s) и 993(s).
На Фиг.6 показан репрезентативный FT-Raman-спектр Полиморфа IV. Могут быть отмечены следующие основные сдвиги частот комбинационного рассеяния (см-1) (vs = очень сильный, s = сильный, m = средний, w = слабый, ошибка эксперимента составляет +/- 2 см-1): 2977(vs), 2940(vs), 2916(m), 2848(s), 2719(m), 1726(w), 1662(w), 1463(s), 1412(w), 1374(w), 1356(m), 1330(w), 1282(w), 1249(w), 1208(w), 1160(m), 1130(w), 1109(w), 1058(w), 1037(w), 1000(w), 983(w), 960(w), 933(w), 900(w), 865(m), 829(w), 812(w), 773(w), 753(w), 736(w), 670(w), 615(w), 527(w), 486(w), 460(w), 433(w), 407(w), 346(w), 279(w) и 226(w). Наиболее характерными являются следующие сдвиги: 1463(s), 933(w), 736(w) и 615(w).
На Фиг.7 показан репрезентативный спектр твердотельного 13С-ЯМР Полиморфа IV. Наблюдаются следующие химические сдвиги (млн-1 относительно внешнего образца адамантина при 29,5 млн-1; интенсивности эквивалентны высотам пиков в скобках): 177.6 (4.68), 177.3 (3.6), 171.7 (1.18), 170.8 (2.68), 103.2 (5.08), 101.2 (5.08), 97.1 (5.09), 95.7 (6.76), 85.6 (2.27), 80.3 (2.72), 78.2 (6.35), 77.4 (5.09), 77.1 (5.42), 76.4 (11.6), 74.7 (7.69), 74.1 (9.97), 73.9 (10.11), 73.4 (4.39), 72.1 (2.62), 71.6 (6.35), 71.2 (5.61), 69.8 (1.75), 69.5 (4.22), 68.8 (5.34), 68.4 (4.79), 66.0 (5.13), 65.3 (5.72), 62.0 (2.31), 52.9 (2.59), 51.2 (5.06), 49.5 (5.74), 45.7 (12), 44.4 (5.26), 39.9 (3.58), 36.6 (3.32), 35.6 (3.82), 35.5 (3.41), 34.6 (3.29), 34.0 (2.48), 33.5 (5.01), 32.9 (2.86), 32.8 (7.31), 32.2 (5.15), 29.4 (1.69), 28.4 (6.71), 27.1 (5.53), 26.2 (3.22), 23.6 (7.16), 23.3 (1.67), 22.6 (5.05), 22.3 (10.17), 22.1 (6.25), 21.9 (4.88), 21.4 (7.3), 21.2 (6.22), 20.6 (7.42), 20.5 (8.01), 19.9 (9.82), 19.5 (2.79), 19.2 (6.23), 18.9 (7.85), 18.4 (2.93), 17.8 (5.67), 12.7 (6.44), 11.6 (4.1), 11.3 (5.13), 9.6 (6.09) и 7.7 (7.11). Наиболее характерными являются следующие химические сдвиги: 177.6, 170.8, 45.7, 28.4, 12.7 и 7.7 млн-1.
На Фиг.8 показан репрезентативный спектр твердотельного 15N-ЯМР Полиморфа IV. Наблюдают следующие химические сдвиги (млн-1 относительно внешнего образца DL-аланина при -331.5 млн-1; интенсивности эквивалентны высотам пиков в скобках): -270.8 (4.29), -273.4 (12), -342.4 (8.16) и -345.1 (9.27).
Полиморфы по изобретению могут быть использованы в композициях Соединения Ia в комбинации с обычными нетоксичными фармацевтически приемлемыми носителями для таблеток, пилюль, капсул, суппозиториев, пессариев, растворов, эмульсий, суспензий и любой другой формы, подходящей для применения. Для целей обработки как лекарственное вещество и для применения в твердых композициях особенно предпочтителен Полиморф IV.
Эксципиенты, которые могут быть использованы, включают носители, поверхностно-активные агенты, загустители или эмульгаторы, твердые связывающие вещества, вспомогательные вещества для образования дисперсий или суспензий, солюбилизаторы, окрашивающие вещества, корригенты, образующие покрытия вещества (coatings), разрыхлители, смазывающие вещества, подсластители, консерванты, изотонические агенты и их комбинации. Выбор и использование подходящих эксципиентов даны в книге Gennaro, ed., Remington: The Science and Practice of Pharmacy, 20th Ed. (Lippincott Williams & Wilkins 2003), описание которой включено в данное изобретение посредством ссылки.
Полиморфы по изобретению могут быть введены перорально. Пероральное введение может включать глотание, чтобы соединение поступало в желудочно-кишечный тракт, или может быть использовано трансбуккальное или сублингвальное введение, посредством чего соединение поступает в кровоток непосредственно изо рта. Композиции, подходящие для перорального введения, включают твердые композиции, такие как таблетки, капсулы, содержащие частицы, жидкости или порошки, пастилки (включая заполненные жидкостью), жевательные конфеты, мульти- и наночастицы, гели, твердый раствор, липосому, пленки, овули, спреи и жидкие композиции.
Жидкие композиции включают суспензии, растворы, сиропы и эликсиры. Такие композиции можно использовать в качестве наполнителей в мягких или твердых капсулах, и они обычно содержат носитель, например воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло и один или более эмульгаторов и/или суспендирующих агентов. Жидкие композиции также могут быть изготовлены путем растворения твердого соединения, например из саше.
Что касается таблетированных лекарственных форм, то в зависимости от дозы лекарственное вещество может составлять от 1 до 80 мас.% лекарственной формы, в более типичном случае от 5 до 60 мас.% лекарственной формы. В дополнение к лекарственному веществу таблетки обычно содержат разрыхлитель. Примеры разрыхлителей включают крахмал-гликолят натрия, натриевую соль карбоксиметилцеллюлозы, кальциевую соль карбоксиметилцеллюлозы, натриевую соль кроскармелозы, кросповидон, поливинилпирролидон, метил целлюлозу, микрокристаллическую целлюлозу, замещенную низшим алкилом гидроксипропилцеллюлозу, крахмал, прежелатинизированный крахмал и альгинат натрия. Как правило, разрыхлитель будет составлять от 1 до 25 мас.%. В одном из воплощений настоящего изобретения разрыхлитель будет составлять от 5 до 20 мас.% лекарственной формы. Связывающие вещества обычно используют для придания таблеточной композиции когезионных свойств. Подходящие связывающие вещества включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, природные и синтетические камеди, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки могут также содержать разбавители, такие как лактоза (моногидрат, высушенный с использованием распылительной сушки моногидрат, безводная и тому подобное), маннит, ксилит, декстрозу, сахарозу, сорбит, микрокристаллическую целлюлозу, крахмал и дигидрат гидрофосфата кальция. Возможно таблетки также могут содержать поверхностно-активные агенты, такие как лаурилсульфат натрия и полисорбат 80, и скользящие вещества, такие как диоксид кремния и тальк. Если присутствуют, то поверхностно-активные агенты могут составлять от 0,2 до 5 мас.% таблетки, а скользящие вещества могут составлять от 0,2 до 1 мас.% таблетки. Обычно таблетки также содержат смазывающие вещества, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Обычно смазывающие вещества составляют от 0,25 до 10 мас.% таблетки. В одном из воплощений настоящего изобретения смазывающие вещества составляют от 0,5 до 3 мас.% таблетки. Другие возможные ингредиенты включают антиоксиданты, окрашивающие вещества, корригенты, консерванты и маскирующие вкус агенты.
Типичные таблетки содержат примерно до 80% лекарственного вещества, от примерно 10 мас.% до примерно 90 мас.% связывающего вещества, от примерно 0 мас.% до примерно 85 мас.% разбавителя, от примерно 2 мас.% до примерно 10 мас.% разрыхлителя и от примерно 0,25 мас.% до примерно 10 мас.% смазывающего вещества.
Для образования таблеток таблеточные смеси могут быть спрессованы непосредственно или с использованием ролика. Альтернативно, перед таблетированием таблеточные смеси или части смесей могут быть подвергнуты влажной или сухой грануляции либо грануляции плавлением, застыванию путем плавления или экструзии. Конечная композиция может содержать один или более слоев и может иметь покрытие или не иметь его; она даже может быть инкапсулирована. Составы таблеток обсуждены в Н. Lieberman and L. Lachman, Pharmaceutical Dosage Forms: Tablets, Vol.1 (Marcel Dekker, New York, 1980).
В общем случае Полиморф IV очищают в результате применения методики получения, с помощью которой в него превращается другой полиморф Соединения la. В таком случае количество Полиморфа IV в образце увеличивается по отношению к его количеству (которое могло быть нулевым) в образце до применения методики получения. К тому же, в результате такой очистки могут быть удалены другие примеси. Предпочтительно очищенный Полиморф IV содержит преобладающее количество Полиморфа IV, и тем самым исключены другие полиморфы Соединения I.
Предпочтительный способ получения очищенного Полиморфа IV состоит в растворении Соединения Ia в этилацетате и затем добавлении С5-С7алкана или -алкена для того, чтобы вызвать кристаллизацию Полиморфа IV. Алкан или алкен должны содержать небольшое количество воды, предпочтительно ниже 0,005% (об./об.). Эта методика до некоторой степени чувствительна к содержанию воды в этилацетатном растворе Соединения Ia и температуре кристаллизации. Вода может попасть в этот раствор двумя путями. Используемое Соединение Ia может быть в форме полиморфа с некоторым содержанием воды (например, дигидрат Полиморфа II). Или этилацетат может содержать следовые количества воды. Предпочтительно содержание воды в этилацетатном растворе Соединения Ia составляет менее 3,6%, более предпочтительно менее 1,9% и наиболее предпочтительно от примерно 1,1 до примерно 1,9% (объем/объем или об./об.). Содержание воды можно поддерживать на желаемом низком уровне с помощью различных методик, используемых индивидуально или в комбинации:
(а) путем использования полиморфа Соединения Iа, который не является гидратом;
(б) путем предварительной сушки используемого Соединения Iа, например, при 40°С в течение 17 ч под вакуумом;
(в) путем использования этилацетата высокой чистоты с низким содержанием воды или предварительно высушенного этилацетата;
(г) путем сушки этилацетатного раствора перед добавлением С5-С7алкана или -алкена, например с использованием безводного сульфата натрия.
Ввиду наличия чувствительности к содержанию воды в этилацетатном растворе рекомендуется перед добавлением С5-С7алкана или -алкена рассчитывать или измерять содержание воды в нем, и если оно превышает 3,6%, то содержание воды необходимо уменьшить перед добавлением C5-С7алкана или -алкена.
Температура кристаллизации может колебаться от примерно 20°С до примерно 36°С. В общем случае, если содержание воды в этилацетатном растворе равно или ниже 1,9%, для получения Полиморфа IV рекомендуется температура выше 25°С (например, 25-36°С).
Примеры подходящих С5-С7алканов или -алкенов, которые могут быть использованы в приведенной выше методике (или в альтернативной методике созревания), включают: н-пентан, циклопентан, 1-пентен, 2-пентен, изопентан, неопентан, н-гексан, 1-гексен, циклогексан, н-гептан, 1-гептен и тому подобное. Предпочтителен н-гептан.
Практическое применение этого изобретения может быть дополнительно понято посредством ссылки на следующие далее примеры, которые приведены только для иллюстрации, а не для ограничения.
Пример 1. Общие аналитические методики
Картины XRPD получали на дифрактометре Bruker AXS C2 GADDS, используя СuКα-излучение (40 кВ, 40 мА), автоматизированную XYZ-платформу, видеомикроскоп с лазерным источником для автоматического расположения образца и 2-мерный детектор (2-dimensional area detector) HiStar. Рентгеновская оптика состояла из одиночного многослойного зеркала Гебеля, сопряженного с точечным коллиматором 0,3 мм.
Расходимость пучка, то есть действительный размер рентгеновского пучка, направляемого на образец, составляла приблизительно 4 мм. Использовали режим постоянного сканирования θ-θ с расстоянием между образцом и детектором 20 см, который давал эффективный диапазон 2θ от 3,2° до 29,7°. Обычно образец подвергали воздействию рентгеновского пучка в течение 120 секунд.
Картины XRPD получали посредством Pharmorphix Ltd. (Cambridge, United Kingdom). Картины дифракции рентгеновских лучей на порошке для образцов получали на дифрактометре Siemens D5000, используя СuКα-излучение (40 кВ, 40 мА), θ-θ гониометр, автоматические отклоняющую и приемную щели, графитовый вторичный монохроматор и сцинтилляционный счетчик. Данные собирали в диапазоне углов от 2° до 42° 2θ в режиме постоянного сканирования, используя размер шага 2θ 0,02° и продолжительность шага 1 с. Перед проведением анализа образцы сушили под вакуумом при 30°С в течение 24 ч, хотя приемлемы и другие режимы сушки.
XRPD-образцы для работы в условиях окружающей среды готовили в виде образцов в форме плоских пластинок, используя порошок в таком виде, в каком его получали, без измельчения. Приблизительно 25-50 мг образца осторожно помещали в полости диаметром 12 мм, глубиной 0,5 мм, вырезанные в полированных кремниевых пластинах с нулевым фоном (zero-background) (510) (The Gem Dugout, 1652 Princeton Drive, Pennsylvania State College, PA 16803, USA). Все образцы измеряли в стационарном режиме.
Данные GVS также получали посредством Pharmorphix, Ltd. Все образцы измеряли на анализаторе сорбции влаги (moisture sorption analyzer) Hiden IGASorp, работающем с программным обеспечением CFRSorp. Размеры образцов обычно составляли 10 мг. Изотерму адсорбции-десорбции влаги выполняли, как изложено ниже, с двумя сканированиями, составляющими один полный цикл. Все образцы загружали и выгружали при типичной влажности окружающей среды (комнатной влажности) и температуре (RH 40%; 25°С). Все образцы после GVS-анализа анализировали с использованием XRPD. Стандартную изотерму выполняли при 25°С с интервалами RH 10% в диапазоне RH 0-90%.
Содержание воды в этилацетате, н-гептане и соединении Ia определяли методом Карла Фишера. Содержание воды в этилацетатных растворах соединения Ia рассчитывали исходя из баланса масс и результаты выражали в виде % об./об.
Данные FT-IR получали, используя спектрометр ThermoNicolet Avatar 360 FTIR, оборудованный ATR-приставкой для однократного отражения (ATR=Attenuated Total Reflectance; нарушенное полное внутреннее отражение) (алмазным ATR-кристаллом с оптикой из селенида цинка) от Smart Golden Gate™ и КВr-детектором d-TGS (Deuterated Triglycine Sulphate). Спектр получали при разрешении 2 см-1 и суммировании результатов 256 сканирований. Использовали аподизацию функции Хаппа-Гензеля (Нарр-Genzel). Поскольку FT-IR-спектр регистрировали, используя приставку для однократного ATR, никакой подготовки образцов не требовалось. Использование ATR FT-IR будет приводить к получению относительных интенсивностей инфракрасных полос, отличных от таковых, наблюдающихся в спектре поглощения FT-IR, получаемом с использованием таблеток КВr, или для образцов, приготовленных в виде нуйол-суспензий. Обусловленные природой ATR FT-IR полосы с меньшим волновым числом характеризуются большей интенсивностью, чем полосы с большим волновым числом. Ошибка эксперимента, если не указано иное, составляла ±2 см-1. Пики выбирали с использованием программного обеспечения ThermoNicolet Omnic 6.1 а. Интерпретации интенсивностей производили относительно основной полосы в спектре, так что они не основывались на абсолютных значениях, измеряемых от базовой линии. При оценке расщепленных пиков величину интенсивности брали от базовой линии, но опять же интенсивность задавали относительно наиболее сильной полосы в спектре.
Данные FT-Raman получали, используя спектрометр комбинационного рассеяния Bruker Vertex70 FT-IR с модулем Ramll FT-Raman, оборудованный 1064 нм МаYАG (алюмо-иттриевый гранат с добавкой неодима)-лазером и детектором LN-Germanium. Все спектры регистрировали, используя разрешение 2 см-1 и аподизацию с помощью четырехзвенного окна Блэкмана-Харриса (Blackman-Harris 4-term apodisation), мощность лазера 300 мВт и 4096 сканирований. Образец измеряли непосредственно в стеклянном флаконе и подвергали воздействию лазерного излучения. Данные представляют в виде интенсивности как функции от сдвига частот комбинационного рассеяния и корректируют на ответ прибора и зависящее от частоты рассеяние, используя спектр белого света от эталонной лампы с применением корректирующей функции Bruker-Raman (Bruker Raman Correct function) (программное обеспечение Bruker - OPUS 6.0). Ошибка эксперимента, если не указано иное, составляла ±2 см-1. Пики выбирали, используя программное обеспечение ThermoNicolet Omnic 6.1 а. Интерпретации интенсивностей производили относительно основной полосы в спектре, так что они не основывались на абсолютных значениях, измеряемых от базовой линии. При оценке расщепленных пиков величину интенсивности брали от базовой линии, но опять же интенсивность задавали относительно наиболее сильной полосы в спектре.
Данные твердотельного 13С- и 15N-ЯМР получали в условиях окружающей среды на 4 мм датчике CPMAS Bruker-Biospin, расположенном на ЯМР-спектрометре Bruker-Biospin Avance 500 МГц со стандартным отверстием. Спектр азота получали с использованием 7 мм датчика BL CPMAS. Образец помещали в 4 и 7 мм ZrO2 роторы, помещали на магический угол и вращали при 7,0 кГц. Спектры углерода и азота получали с использованием протонного расщепления в эксперименте с кросс-поляризацией и вращением под магическим углом (cross-polarization magic angle spinning (CPMAS)). Продолжительность кросс-поляризации устанавливали на 2,5 мс. Прикладывали протон-расщепляющее поле приблизительно 90 кГц (4 мм датчик) и 70 кГц (7 мм датчик). Проводили 5120 (13С) и 30000 (15N) сканирований. Задержки рециклов (recycle delays) подводили приблизительно до 1,5*T1H (где T1H обозначает время продольной релаксации протонов, рассчитанное на основании эксперимента с протон-детектируемой релаксацией по методу "инверсии-восстановления" протонов (proton detected proton inversion recovery relaxation)). Углеродный спектр сравнивали с эталонным, используя внешний стандарт кристаллического адамантана, устанавливая его резонанс в сильных полях (upfield resonance) к 29,5 млн-1. Азотный спектр сравнивали с эталонным, используя внешний стандарт кристаллического 98%-ного 15N-меченного D,L-аланина, устанавливая его резонанс к -331,5 млн-1.
Пример 2. Общая методика получения Соединения Ia
Соединение Ia получали, как описано в заявке Liu '616, включенной в данное описание посредством ссылки. На Фиг.7 суммирована используемая схема синтеза. Эритромицин А (1) восстанавливали борогидридом натрия, получая промежуточный (9S)-дигидроэритромицин А (7). После деметилирования (9S)-дигидроэритромицина А (7) йодом в присутствии основания, такого как ацетат натрия или трис(гидроксиметил)аминометан ("ТРИС"), получали N-дезметил-(9S)-дигидроэритромицин А (8), алкилирование которого 2-йодпропаном в свою очередь позволило получить промежуточное соединение 9. После алкилирования промежуточного соединения 9 N-метилбромацетамидом получали соединение Ia. То, каким будет получен полиморф соединения Ia, будет зависеть от стадий выделения и очистки после синтеза.
Получение промежуточного соединения 9 также описано в Santi и др., US 6946482 В2 (2005), включенном в данное описание посредством ссылки. Стадия деметилирования также описана в Liu, заявка США №11/591726, поданная 1 ноября 2006 года, описание которой включено в данное изобретение посредством ссылки.
Пример 3. Получение Соединения Ia и выделение в виде Полиморфа I
В 5-литровую трехгорлую круглодонную колбу, оборудованную механической мешалкой и внутренним датчиком температуры на основе термопары, загружали раствор соединения 9 (156,7 г; 205 ммоль), N-метил-бромацетамида (37,4 г; 246 ммоль) в безводном тетрагидрофуране ("THF", 1800 мл) с охлаждением до 0°С на ледяной бане. Добавляли твердый трет-бутилат калия (25,3 г; 226 ммоль, 1,1 экв.) в виде одной порции с перемешиванием в атмосфере азота. Реакционную смесь перемешивали при 0°С в течение 1 ч. Тонкослойная хроматография (элюент гексан-ацетон (1:2)) показала, что реакция прошла полностью. Реакцию гасили добавлением насыщенного раствора NaHCO3 (300 мл). Смесь распределяли между разбавленным NaHCO3 (2500 мл) и этилацетатом ("ЕtOАс," 1500 мл). Водный слой экстрагировали ЕtOАс (2×1500 мл). Объединенные органические слои сушили над Na2SO4. Неочищенное соединение Ia (178,1 г) получали в виде слегка желтого твердого вещества, которое затем очищали на колонке с силикагелем (2800 г силикагеля, градиент элюирования от 20 до 40% ацетона в гексане, 1% триэтиламина), получая чистое соединение Ia (135 г; выход 79%).
Для удаления следовых количеств растворителей и триэтиламина указанный выше продукт неоднократно растворяли в дихлорметане и подвергали четырем циклам упаривания на роторном испарителе и затем сушили под высоким вакуумом. Затем его лиофилизировали из смеси ацетонитрил-вода (1:1 об./об., 4 мл/г), сушили в вакуумной печи (16 ч, 50°С), получая конечный продукт (т.пл. 106-108°С, по результатам измерений с использованием капиллярного прибора для измерения точки плавления). В результате применения такой методики обработки получали соединение Ia в виде Полиморфа I (отметим слабый эндотерм при приблизительно 110°С в DSC-картине Полиморфа I на Фиг.2а). В заявке Liu '616 сообщалось об аналогичной точке плавления, таким образом, по-видимому, это есть описанный там полиморф.
Пример 4. Получение Соединения Ia и выделение в виде Полиморфа II
Соединение 9 (светло-оранжевое вещество; 353 г; 462 ммоль) и N-бромацетамид (84 г; 600 ммоль; 1,3 экв.) растворяли в THF (3,9 л, безводном и без антиокислителей). Желтый раствор охлаждали до 0±2°С, разбавляли 1 М трет-бутилатом калия в THF (549 мл; 549 ммоль; 1,2 экв.) в течение 20 мин, поддерживая температуру от 0 до 3°С. Перемешивание продолжали при 0±2°С, регистрируя прохождение реакции с помощью HPLC по расходованию исходного вещества. Через 15 мин оставалось только примерно 0,34% исходного вещества. Реакцию гасили 5%-ным NаНСО3 (2,6 л). Слои разделяли и водную фазу экстрагировали ЕtOАс (2,9 л). Объединенные органические слои промывали водой (1,2 л) и затем рассолом (1,2 л). Органическую фазу сушили над MgSO4 (75 г). Осушающий агент удаляли фильтрованием и промывали ЕtOАс (200 мл). Объединенные фильтраты концентрировали, получая соединение Ia в виде светло-желтого остатка (392 г).
Этот остаток растворяли в ацетоне (3,1 л; 8 мл/г) и светло-желтый раствор разбавляли деионизированной водой (3,1 л). Слегка мутный раствор охлаждали до диапазона 0-5°С в течение 20 мин, что приводило к выпадению осадка (кристаллы заметны при приблизительно 10°С). Суспензию перемешивали в течение 15 мин при 0-5°С и разбавляли дополнительным количеством деионизированной воды (3,1 л) в течение 30 мин. Смесь перемешивали в течение дополнительных 30 мин при 0-5°С. Твердые вещества выделяли фильтрованием и затем промывали смесью ацетона (0,15 л) и деионизированной воды (0,30 л). Твердые вещества сушили на воздухе в течение ночи (приблизительно 16 ч) и затем сушили дополнительно (30°С; 29 дюйм Нg (98 кПа)) в течение 64 ч, получая Соединение Ia (322 г) в виде кремового твердого вещества.
Пример 5. Получение Полиморфа IV
DIPE (1,0 мл) добавляли к полиморфу II соединения (Ia) (250 мг) в небольшом флаконе с завинчивающейся крышкой. Флакон и его содержимое подвергали трем циклам нагревания и охлаждения в диапазоне от температуры окружающей среды до 50°С в течение периода времени 24 ч. Полученное твердое вещество отфильтровывали и анализировали с использованием XRPD после сушки при 30°С в течение 24 ч, который показал, что имело место превращение в Полиморф IV.
1H-ЯМР-анализ полученного таким образом Полиморфа IV показал следовые количества (0,9%; 0,07 эквивалента) присутствующего DIPE. DIPE удаляли посредством суспендирования в воде следующим образом: к образцу Полиморфа IV (30 мг) в небольшом флаконе с завинчивающейся крышкой добавляли воду (1,0 мл) и встряхивали при 25°С в течение 72 ч. Полученное твердое вещество отфильтровывали и сушили. Анализ с использованием XRPD и 1H-ЯМР показал, что DIPE удален без изменения формы образца.
Пример 6. Альтернативное получение Полиморфа IV Соединение Ia (2,0 г) растворяли в этилацетате (12,0 мл) при температуре окружающей среды. Содержание воды в этилацетатном растворе составляло 1,1% (об./об.). Светло-желтый раствор помещали в трехгорлую круглодонную колбу емкостью 500 мл, оборудованную подвесной мешалкой (1КА RW16 basic). Раствор перемешивали при 32°С на 180-185 об/мин и добавляли н-гептан (80 мл) со скоростью 0,8 мл/мин, используя шприцевой насос (KdScientific). Добавление гептана было прервано на 4 мин после того, как было добавлено 50 мл гептана, для повторного заполнения шприца. После добавления следующих 30 мл гептана (для общего количества 80 мл) полученную суспензию перемешивали в течение следующих 2,5 ч при 185 об/мин и 32°С. Суспендированные кристаллы Полиморфа IV собирали фильтрованием с использованием керамической воронки Бюхнера (5 см) и фильтровальной бумаги Ватман №4. Кристаллы промывали смесью гептан:этилацетат (90:10 об./об.; 20 мл) и сушили на воздухе в течение 10 мин. Кристаллы дополнительно сушили при 40°С под вакуумом (29,5 дюйм Нg (99,9 кПа)) в течение 16 ч, получая 1,62 г Полиморфа IV. Идентичность продукта Полиморфу IV была подтверждена с использованием DSC и XRPD.
Эксперимент повторяли при 25°С, в результате чего также получили Полиморф IV (хотя и с немного меньшим выходом).
Пример 7. Другое альтернативное получение Полиморфа IV
В этом примере описано получение Полиморфа IV посредством созревания в н-гептане. н-Гептан (500 мкл) добавляли к Полиморфу I в небольшом флаконе с завинчивающейся крышкой. Флакон подвергали 12 циклам нагревания/охлаждения в диапазоне от 5 до 40°С в течение периода времени 24 ч с перемешиванием. XRPD-анализ подтвердил получение Полиморфа IV. Эта же методика может быть использована с DIPE.
Предшествующее подробное описание изобретения включает отрывки, которые в основном или исключительно касаются конкретных частей или аспектов изобретения. Следует понимать, что это сделано для ясности и удобства, что конкретный признак может быть релевантным в отношении нечто большего, чем просто отрывок, в котором он описан, и что представленное в данном изобретении описание включает все соответствующие комбинации информации, имеющиеся в различных отрывках. Подобным образом, несмотря на то что различные графические материалы и описания в данном изобретении касаются конкретных воплощений изобретения, следует понимать, что там, где раскрыт конкретный признак в контексте конкретного графического материала или воплощения, такой признак также может быть использован, насколько это возможно, в контексте другого графического материала или воплощения, в комбинации с другим признаком или в изобретении в целом.
Далее, несмотря на то что настоящее изобретение конкретно описано в терминах некоторых предпочтительных воплощений, изобретение не ограничено такими предпочтительными воплощениями. В значительной степени объем изобретения определяется прилагаемой формулой изобретения.
Настоящее изобретение относится к очищенному полиморфу IV соединения формулы Iа, характеризующемуся пиками дифракции рентгеновских лучей на порошке (XRPD) при 3,8, 7,5, 16,1, 16,5 и 17,1 градусах 2θ (±0,1), полученными с использованием рентгеновского медного К-альфа1-излучения (длина волны равна 1,5406 ангстрем). Кроме того, изобретение относится к двум способам получения указанного полиморфа IV. Первый включает подвергание полиморфа II соединения формулы Iа по меньшей мере двум циклам нагревания и охлаждения в температурном диапазоне от 5 до 50°С в присутствии среды, выбранной из диизопропилового эфира и С5-С7алкана или -алкена, где указанный Полиморф II соединения формулы Iа характеризуется пиками XRPD при 3,5, 6,9, 9,2, 10,4 и 18,0 градусах 2θ (±0,1), полученными с использованием рентгеновского медного К-альфа1-излучения (длина волны равна 1,5406 ангстрем). Второй способ включает получение этилацетатного раствора соединения формулы Iа, где содержание воды составляет менее 3,6% об./об., и добавление к этому раствору С5-С7алкана или -алкена при температуре от 20 до 36°С. Изобретение относится также к твердой фармацевтической композиции на основе полиморфа IV, имеющей агонистическую активность в отношении мотилина, а также к способу лечения с помощью полиморфа IV заболевания нарушенной двигательной функции желудка. 5 н. и 8 з.п. ф-лы, 2 табл., 23 ил.
1. Очищенный полиморф IV соединения, имеющего структуру, представленную формулой Iа
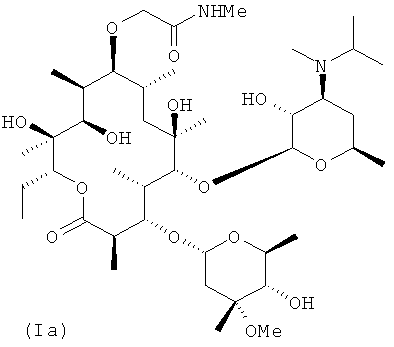
характеризующийся пиками дифракции рентгеновских лучей на порошке (XRPD) при 3,8, 7,5, 16,1, 16,5 и 17,1 градусах 2θ (±0,1), полученными с использованием рентгеновского медного К-альфа1-излучения (длина волны равна 1,5406 ангстрем).
2. Очищенный полиморф IV соединения, имеющего структуру, представленную формулой Iа, как определено в п.1, для применения в качестве лекарственного средства для лечения заболевания нарушенной двигательной функции желудка.
3. Очищенный полиморф IV соединения, имеющего структуру, представленную формулой Iа, как определено в п.1, для применения в лечении гастроэзофагеального рефлюкса ("GERD").
4. Способ получения очищенного полиморфа IV соединения, имеющего структуру, представленную формулой Iа, как определено в п.1, включающий подвергание полиморфа II соединения формулы Iа по меньшей мере двум циклам нагревания и охлаждения в присутствии среды, выбранной из диизопропилового эфира и С5-C7алкана или -алкена, где указанный полиморф II соединения формулы Iа характеризуется пиками XRPD при 3,5, 6,9, 9,2, 10,4 и 18,0 градусах 2θ (±0,1), полученными с использованием рентгеновского медного К-альфа1-излучения (длина волны равна 1,5406 ангстрем), и температурный диапазон для циклов нагревания и охлаждения составляет от 5 до 50°С.
5. Способ по п.4, где среда представляет собой гептан.
6. Способ получения очищенного полиморфа IV соединения, имеющего структуру, представленную формулой Iа, как определено в п.1, включающий получение этилацетатного раствора соединения формулы Iа и добавление к этому раствору С5-С7алкана или -алкена для того, чтобы вызвать кристаллизацию соединения формулы Iа в виде очищенного полиморфа IV, где содержание воды в этилацетатном растворе соединения формулы 1а составляет менее 3,6% об./об. и добавление С5-С7алкана или -алкена для того, чтобы вызвать кристаллизацию, осуществляют при температуре от 20 до 36°С.
7. Способ по п.6, где С5-С7алкан или -алкен представляет собой гептан.
8. Способ по п.6, где добавление С5-С7алкана или -алкена для того, чтобы вызвать кристаллизацию, осуществляют при температуре от примерно 25 до примерно 36°С.
9. Способ по п.6, где содержание воды в растворе составляет менее 1,9% об./об.
10. Способ по любому из пп.6-9, дополнительно включающий стадию анализа содержания воды раствора и, если содержание воды превышает 3,6% об./об., снижение содержания воды перед добавлением С5-С7алкана или -алкена.
11. Твердая фармацевтическая композиция, имеющая агонистическую активность в отношении мотилина, содержащая эффективное количество очищенного Полиморфа IV соединения, имеющего структуру, представленную формулой Iа, как определено в п.1, и фармацевтически приемлемый эксципиент.
12. Способ лечения заболевания нарушенной двигательной функции желудка, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества очищенного полиморфа IV соединения, имеющего структуру, представленную формулой Iа, как определено в п.1.
13. Способ по п.12, где заболевание нарушенная двигательная функция желудка представляет собой гастроэзофагеальный рефлюкс ("GERD").
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ПРОИЗВОДНЫЕ [2R,3R (2'R,3'R,), 6R,7S,8S,9R,10R] -3-(2',3'-ДИГИДРОКСИПЕНТ-2'-ИЛ)-2,6,8,10,12-ПЕНТАМЕТИЛ -4,13-ДИОКСАБИЦИКЛО [8,2,1]-ТРИДЕЦ-12-ЕН-5-ОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО | 1992 |
|
RU2107070C1 |
| ПРОИЗВОДНЫЕ 10,13,15-ТРИОКСАТРИЦИКЛО (9.2.1.1.)-ПЕНТАДЕКАНОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 1997 |
|
RU2181727C2 |

