Область техники, к которой относится изобретение
Данное изобретение относится к улучшенному способу получения атенолола высокой степени чистоты, по существу, свободного от примесей.
Уровень техники
Бета-адренергические антагонисты, называемые бета-блокаторами, представляют собой класс соединений, которые используются для лечения стенокардии, гипертензии и аритмии. Основной функцией бета-блокаторов является снижение частоты приступов стенокардии и повышение ангинального порога путем ослабления хронотропных и инотропных реакций на адренергическую стимуляцию.
Рацемический атенолол, химически известный как 4-[2-гидрокси-3-[(1-метилэтил)амино]пропокси]бензолацетамид и представленный формулой (I-A), принадлежит к классу бета-блокаторов, проявляющих свое действие блокированием бета-рецепторов в организме, и существует в виде рацемической смеси оптических (R)- и (S)-изомеров из-за присутствия асимметричного центра у атома углерода с присоединенной гидроксигруппой 1-арилокси-3-аминопропан-2-ольной группы.
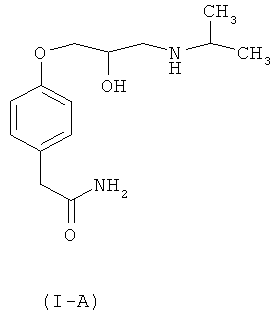
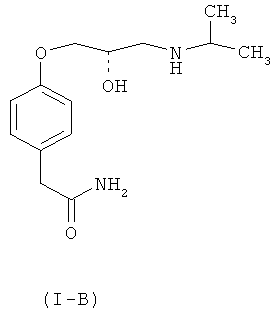
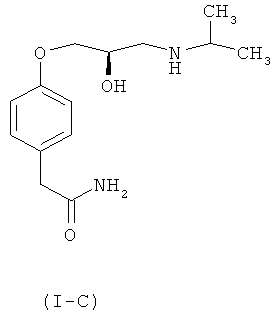
Достаточно хорошо известно, что гипотензивная активность рацемического атенолола присуща (S)-изомеру (1-В) атенолола, в то время как (R)-изомер лишен гипотензивной активности. Кроме того, (S)-изомер обладает дополнительным преимуществом, которое проявляется в отсутствии побочного действия снижения частоты сердечных сокращений, иногда встречающегося в связи с рацемической смесью атенолола формулы (I-A).
В известном уровне техники имеется несколько описанных способов получения как рацемического атенолола (I-A), так и его оптически активных изомеров (S)-атенолола (I-В) и (R)-атенолола (I-C).
В US 3663607 (переданном Imperial Chemical Industries) раскрывается способ получения рацемического атенолола, кратко отображенный на схеме I, который включает взаимодействие фенольного соединения формулы (II) с эпихлоргидрином формулы (III-A) в присутствии каталитического количества пиперидина при температуре 95-100°С в течение нескольких часов с образованием простого глицидилового эфира (IV), который при последующем взаимодействии с низшим алкиламином, подобным изопропиламину, дает атенолол формулы (I-A).
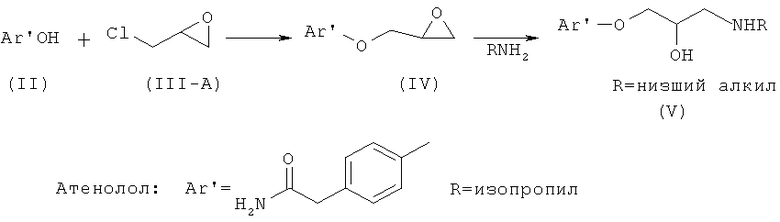
Схема I. Способ получения атенолола, раскрытый в US 3663607.
В US 4182911 (переданном Imperial Chemical Industries) раскрывается способ получения оптически активного (S)-атенолола, кратко отображенный на схеме II.
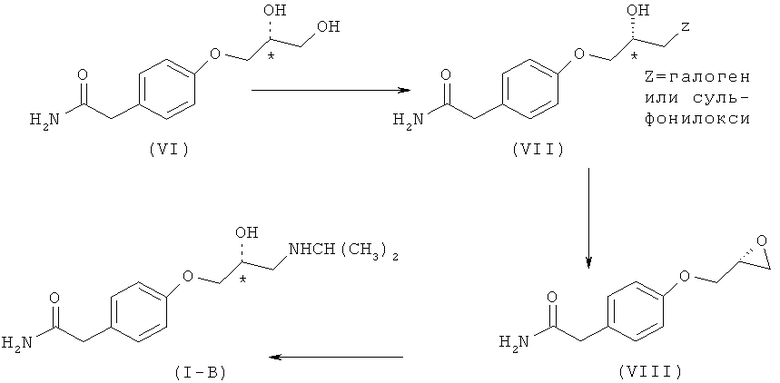
Схема II. Способ получения (S)-атенолола, раскрытый в US 4182911.
Данный способ имеет несколько недостатков, ограничивающих его промышленное масштабирование, которыми являются
а) необходимость осуществления нескольких стадий для получения соединения (VI) из D-маннита, что увеличивает продолжительность каждого рабочего цикла,
b) присутствие в исходном соединении (VI) вторичной, а также первичной гидроксигруппы, и во время конверсии первичной гидроксигруппы соединения (VI) в соединение формулы (VIII) карбамоилметильная группа соединения (VII) взаимодействует с функциональной группой (Z), причем таким образом появляются примеси, которые значительно снижают выход атенолола,
c) кроме того, полученное таким образом промежуточное соединение простой глицидиловый эфир (VIII) имеет низкую оптическую чистоту (80% эи).
В US 5223646 (переданном Daiso Company Ltd) описывается способ получения оптически активного промежуточного соединения для атенолола, т.е. оптически активного простого глицидилового эфира (VIII), взаимодействием фенола и эпихлоргидрина в смешивающемся с водой растворителе или воде при температуре в интервале 0°С-35°С. Полученное таким образом оптически активное промежуточное глицидилсодержащее соединение (VIII) имеет невысокую чистоту - 90-96% и при последующем взаимодействии с изопропиламином дает (S)-атенолол с низкой оптической чистотой - 93-94%.
Кроме того, в данном патенте также описывается способ очистки с использованием кислоты Бронстеда, такой как неорганическая кислота или органическая кислота. Неорганическую кислоту выбирают из числа галогенводородов, серной кислоты и фосфорной кислоты, в то время как органические кислоты выбирают из числа моно- и дикарбоновых кислот, таких как муравьиная кислота, уксусная кислота, органосульфоновых кислот, таких как метансульфоновая кислота, пара-толуолсульфоновая кислота, или фенола, которые можно использовать одни или в сочетании друг с другом. (S)-Атенолол превращают в его соль присоединения кислоты нагреванием в органическом растворителе, таком как ацетон, оставляя ее постепенно кристаллизоваться в течение ночи. Свободное основание оптически чистого (S)-изомера получают из его солей присоединения кислот обработкой ионообменной смолой.
Данный способ имеет недостатком необходимость жестких условий взаимодействия, так как существует опасность рацемизации глицидилового эфира (VIII) при температуре выше 35°С, что снижает его оптическую чистоту. Кроме того, когда температура реакции превышает 35°С, глицидиловый эфир (VIII) взаимодействует с фенолом с образованием ассоциированных примесей, очистку от которых трудно осуществить с использованием обычных способов.
Поэтому можно легко заметить, что процесс, используемый для получения (S)-атенолола по такому способу, является действительно слишком длительным, утомительным и требующим времени и не подходит для получения его в промышленном масштабе.
В US 5290958 (переданном Industrial Technology Research Institute) раскрывается способ получения рацемического атенолола (I-A), показанный на схеме III, в котором используют межфазный катализатор, такой как соль четвертичного аммония с высшими алкильными группами или соли четвертичного аммония с низшими алкильными группами в качестве межфазного катализатора (РТС) при взаимодействии п-гидроксифенилацетамида и эпихлоргидрина с образованием промежуточных эпоксидных соединений или галогенгидринов.
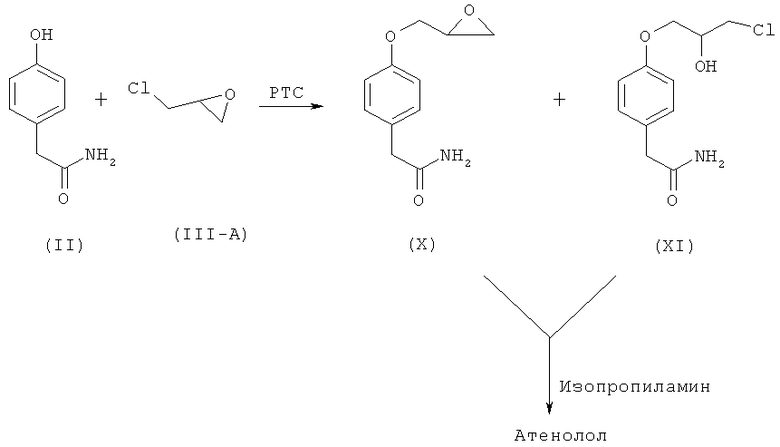
Схема III. Способ получения атенолола, описанный в US 5290958
Уместно отметить, что во всех вышеописанных способах для получения атенолола используют фенол или его производные, и также следует указать, что продукт, полученный вышеописанными способами известного уровня техники, генерирует ассоциированные примеси, которые раскрываются в British Pharmacopoeia 2003, Vol.1, page 173.
Ассоциированными примесями, как показано на схеме IV, являются 2,2'-[(1-метилэтил)иминобис(2-гидроксипропан-3,1-диилокси-4,1-фенилен)]диацетамид (XII), 2-(4-гидроксифенил)ацетамид (XIII), 2-[4-[[(2RS)-оксиран-2-ил]метокси]фенил]ацетамид (XIV), 2-[4-[(2RS)-3-хлор-2-гидроксипропокси]фенил]ацетамид (XV), 2,2'-[2-гидроксипропан-1,3-диилбис(окси-4,1-фенилен)]диацетамид (XVI), 2-[4-[[(2RS)-2-гидрокси-3-[(1-метилэтил)амино]пропокси]фенил]уксусная кислота (XVII), 2-[4-[(2RS)-2-гидрокси-3-[(1-метилэтил)амино]пропокси]фенил]ацетонитрил (XVIII) и 2-[4-[(2RS)-2,3-дигидроксипропокси]фенил]ацетамид (XIX), которые с трудом удаляются с использованием обычных способов очистки, и если удаляются, существенно уменьшается выход нужного продукта.
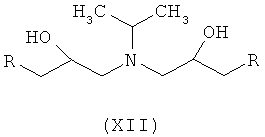
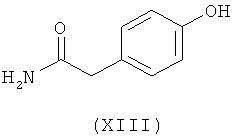
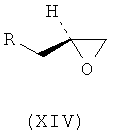
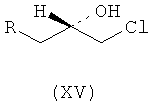
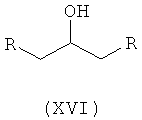
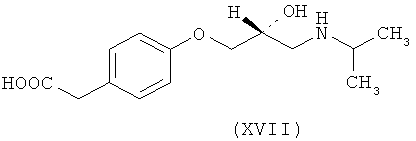
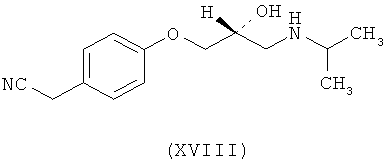
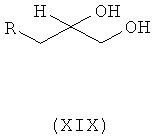
где R представляет собой 
Схема IV. Примеси, ассоциированные с получением атенолола
Органы власти во всем мире становятся весьма строгими в отношении уровня примесей в одобренных лекарственных средствах. Однако большинство способов известного уровня техники не дают продукт, удовлетворяющий требованиям фармакопеи.
В Chemical and Pharmaceutical Bulletin, 1997, 45 (2), 412-414, раскрывается способ очистки (S)-атенолола перекристаллизацией загрязненного (S)-атенолола из органического растворителя, такого как метанол. Такой способ получения (S)-атенолола не подходит для коммерческой цели, так как процедура длительная, поскольку большинство стадий, раскрытых в данной работе, требуют стадии очистки.
В Chemical and Pharmaceutical Bulletin, 1998, 46 (3), 505-507 раскрывается способ очистки (S)-атенолола, включающий получение солей присоединения кислот к (S)-атенололу с использованием различных кислот, подобных бензойной кислоте, п-толуиловой кислоте, п-толуолсульфоновой кислоте и п-трет-бутилбензойной кислоте. (S)-Атенолол получают из таких солей присоединения кислот обработкой ионообменной смолой.
Недостатком данного способа является использование ионообменной смолы для выделения в свободном состоянии атенолола из его соли присоединения кислоты. Использование ионообменных смол требует больших капиталовложений, и также время, требуемое для очистки таким способом, является очень длительным, что существенно увеличивает длительность каждого рабочего цикла и снижает производительность.
В свете вышеуказанных недостатков необходимо разрабатывать другие пути синтеза, которые позволят не только минимизировать содержание всех примесей до уровней ниже фармакопейных пределов, но также получать соединение формулы (I-A), или (I-B), или (I-C) высокой чистоты и с хорошим выходом.
Авторы настоящего изобретения разработали новый способ синтеза для получения рацемического атенолола (I-A), (S)-атенолола (I-B) и (R)-атенолола (I-C) высокой степени чистоты и, по существу, свободных от примесей.
В настоящем изобретении соединения формулы (I-A), (I-B) или (I-C) очищают способом, который включает обработку свободного основания атенолола [рацемического или его (S)-изомера] органической кислотой с получением соответствующей соли присоединения кислоты, которую затем дополнительно обрабатывают водным раствором неорганического основания, выделяя свободный амин, т.е. чистый атенолол [рацемический или его (S)-изомер] высокой степени чистоты и с хорошим выходом.
В способе по настоящему изобретению для очистки атенолола вместо ионообменных смол используют неорганическое основание, такое как легкодоступный гидроксид натрия, что приводит к значительной экономии, причем посредством этого весь процесс становится рентабельным и пригодным для промышленного производства.
Цель изобретения
Целью настоящего изобретения является улучшенный способ получения рацемического атенолола, т.е. 4-[2-гидрокси-3-[(1-метилэтил)амино]пропокси]бензолацетамида формулы (I-A), и его энантиомеров синтезом, который отличается от способов известного уровня техники.
Раскрытие изобретения
Один аспект изобретения относится к улучшенному способу получения рацемического атенолола формулы (I-A), (S)-атенолола формулы (I-B) и (R)-атенолола формулы (I-C) высокой степени чистоты, по существу, свободных от примесей.
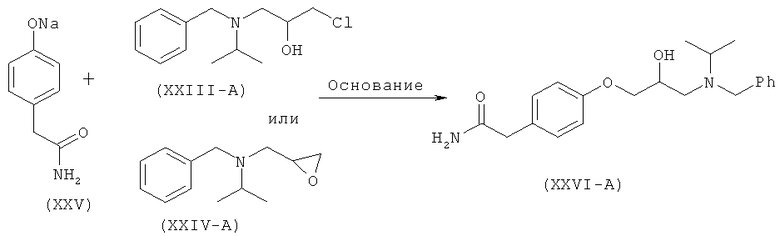
Другой аспект изобретения относится к улучшенному способу получения рацемического атенолола формулы (I-A), включающему взаимодействие хлорметилбензола (XX) и изопропиламина (XXI) с образованием N-бензил-N-изопропиламина (XXII), который после взаимодействия с эпихлоргидрином (IIIA) дает 1-[бензил(изопропил)амино]-3-хлорпропан-2-ол формулы (XXIII-A). Данное соединение после обработки неорганическим основанием дает N-бензил-N-[(2)-оксиран-2-илметил]пропан-2-амин (XXIV-A), который после взаимодействия с натриевой солью 2-(4-гидроксифенил)ацетамида формулы (XXV) дает М-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}фенил)ацетамид (XXVI-A), который затем восстанавливают, и получают рацемический атенолол формулы (I-A).
Еще один аспект изобретения относится к улучшенному способу получения (S)-атенолола формулы (I-B), включающему взаимодействие хлорметилбензола (XX) и изопропиламина (XXI) с образованием N-бензил-N-изопропиламина (XXII), который после последующего взаимодействия с S-эпихлоргидрином (IIIB) дает (2S)-1-[бензил(изопропил)амино]-3-хлорпропан-2-ол формулы (XXIII-B). Данное соединение после обработки неорганическим основанием дает N-бензил-N-[(2S)-оксиран-2-илметил]пропан-2-амин (XXIV-B), который после взаимодействия с 2-(4-гидроксифенил)ацетамидом (XXV) дает N-бензил-2-(4-{[(2S)-2-гидрокси-3-(изопропиламино)пропил]окси}фенил)ацетамид (XXVI-B), который затем восстанавливают и получают (S)-атенолол формулы (I-B).
Другой аспект изобретения относится к улучшенному способу получения (R)-атенолола формулы (I-C), включающему взаимодействие хлорметилбензола (XX) и изопропиламина (XXI) с образованием N-бензил-N-изопропиламина (XXII), который после последующего взаимодействия с R-эпихлоргидрином (IIIC) дает (2R)-1-[бензил(изопропил)амино]-3-хлорпропан-2-ол формулы (XXIII-C). Данное соединение после обработки неорганическим основанием дает N-бензил-N-[(2R)-оксиран-2-илметил]пропан-2-амин (XXIV-C), который после взаимодействия с 2-(4-гидроксифенил)ацетамидом (XXV) дает N-бензил-2-(4-{[(2R)-2-гидрокси-3-(изопропиламино)пропил]окси}фенил)ацетамид (XXVI-C), который затем восстанавливают и получают (R)-атенолол формулы (I-C).
Еще один аспект изобретения относится к способу получения атенолола формулы или (I-A), или (I-B), или (I-C), включающему взаимодействие хлорметилбензола (XX) и изопропиламина (XXI) с образованием R-бензил-N-изопропиламина (XXII), который после последующего взаимодействия с 2-{4-[(2S)-оксиран-2-илметокси]фенил}ацетамидом формулы (XXVII) дает N-бензил-2-(4-{[(2R)-2-гидрокси-3-(изопропиламино)пропил]окси}фенил)ацетамид (XXVI-C), который затем восстанавливают и получают атенолол любой формулы (I-A), (I-B) или (I-C).
Еще один аспект изобретения относится к простому рентабельному способу очистки рацемического атенолола формулы (I-A), (S)-атенолола формулы (I-B) и (R)-атенолола формулы (I-C), включающему обработку атенолола [рацемического или (S)-изомера или (R)-изомера] органической кислотой или неорганической кислотой с образованием соответствующей соли присоединения кислоты, которую затем нейтрализуют водным раствором неорганического основания и получают атенолол высокой степени чистоты, по существу, свободный от примесей.
Осуществление изобретения
Атенолол (I-A), (S)-атенолол (I-B) и (R)-атенолол (I-C) получают способом, отображенным на схеме V, схеме VA и схеме VB соответственно. Кроме того, атенолол формулы (I-A), (I-B) или (I-C) можно получить способом, отображенным на схеме (V-C).
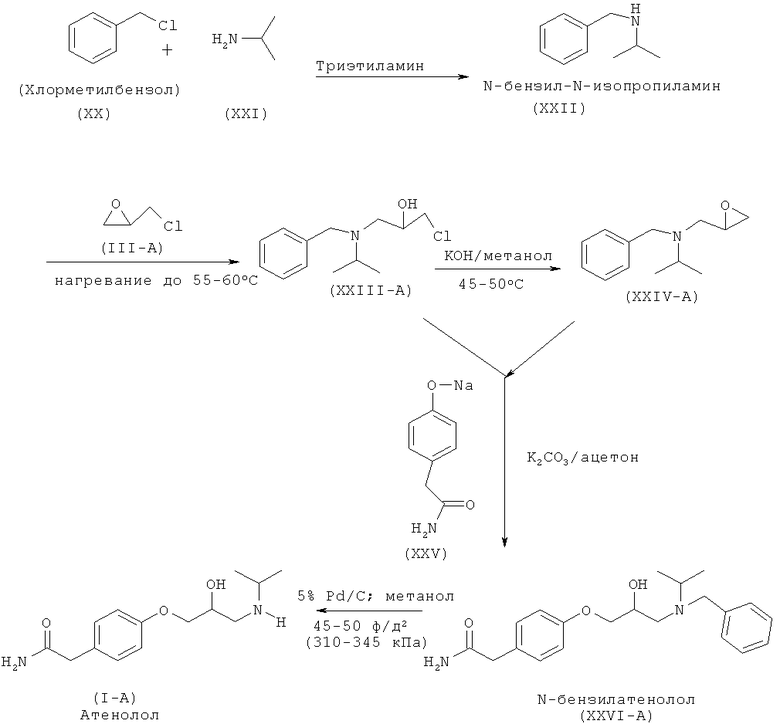
Схема V. Способ получения рацемического атенолола по настоящему изобретению
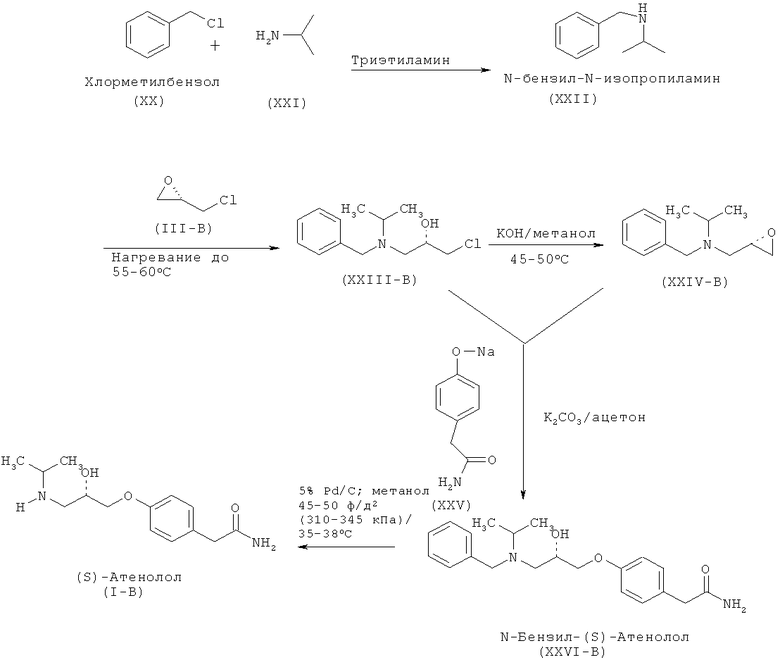
Схема VA. Способ получения (S)-атенолола по настоящему изобретению
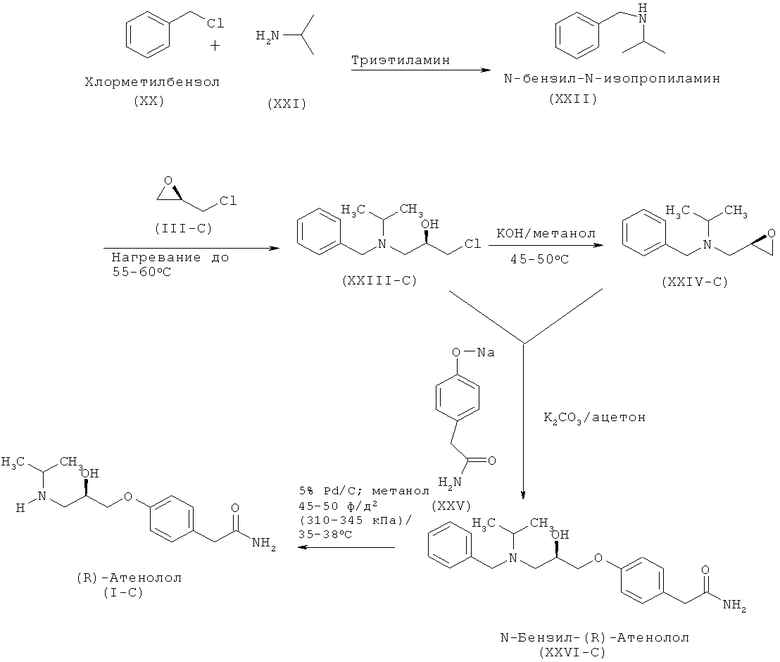
Схема VB. Способ получения (R)-атенолола по настоящему изобретению
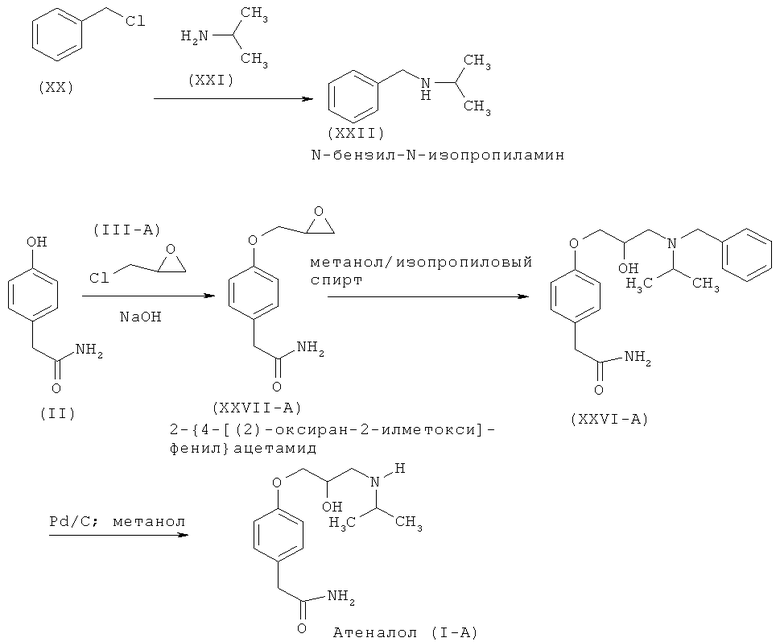
Схема VC. Способ получения рацемического атенолола или конкретно его (R)- или (S)-изомеров.
Настоящее изобретение относится к способу получения атенолола (отображенному на схеме V), включающему взаимодействие хлорметилбензола (XX) и изопропиламина (XXI) с образованием N-бензил-N-изопропиламина (XXII). Данное промежуточное соединение после взаимодействия или с рацемическим эпихлоргидрином (III-A) или S-эпихлоргидрином (III-B) или R-эпихлоргидрином (III-C) дает галогенгидрин (XXIII-A, или В, или -С) и эпоксид (XXFV-A, или В, или -С). Интервал молярного количества эпихлоргидрина 0,25-5 молей. Данное взаимодействие осуществляют при температуре 40-70°С. Такое взаимодействие также можно осуществить в спиртовом растворителе. Галогенгидрин (XXIII-A, или В, или -С) после обработки неорганическим основанием в органическом растворителе превращается в эпоксид (XXIV-A, или В, или -С). Неорганическое основание выбирают из группы, состоящей из гидроксидов, карбонатов, бикарбонатов и т.д. щелочных или щелочноземельных металлов. Предпочтительными неорганическими основаниями являются гидроксиды щелочных или щелочноземельных металлов. Предпочтительнее, неорганическое основание находится в группе, состоящей из NaOH, КОН и т.д. Интервал молярного количества используемого неорганического основания 1,0-3,5 молей. Данное взаимодействие осуществляют при температуре 30-60°С. Полученное соединение (XXIV-A) при взаимодействии с соединением формулы (XXV) в присутствии основания, выбранного из группы, состоящей из гидроксидов, карбонатов, бикарбонатов и т.д. щелочных или щелочноземельных металлов, дает (N)-бензилированный атенолол (XXVI-A, или В, или -С), который после восстановления дает рацемический атенолол (I-A), или S-атенолол (I-B), или R-атенолол. Такое взаимодействие также катализируют с использованием межфазного катализатора, такого как иодид тетрабутиламмония, бромид тетрабутиламмония и т.д. Предпочтительные неорганические основания выбирают из группы, состоящей из карбонатов щелочных или щелочноземельных металлов. Предпочтительнее, неорганическим основанием является К2СО3, Na2CO3 и т.д. Интервал молярного количества неорганического основания 1-3 моля. Данное взаимодействие осуществляют при 40-85°С.
Кроме того, настоящее изобретение также относится к способу, включающему взаимодействие N-бензил-N-изопропиламина с 2-{4-[(2)-оксиран-2-илметокси]фенил}ацетамидом (XXVII-A) с образованием N-бензилированного атенолола, который после восстановления с палладием на угле в спиртовом растворителе дает атенолол. Спиртовый растворитель выбирают из группы, состоящей из метанола, этанола, н-пропанола, изопропанола и т.д. Такой способ, как отображено на схеме VC, можно использовать для получения рацемического атенолола, а также конкретно его (S)- или (R)-изомеров с использованием соответственно (S)- или (R)-изомеров эпихлоргидрина.
Настоящее изобретение превосходит способы известного уровня техники, так как образование примесей является минимальным. Это происходит в силу того факта, что в способах известного уровня техники конечный продукт атенолол (I-A, или I-B, или I-C), полученный взаимодействием глицидилового эфира (VIII) с изопропиламином (XXI), имеет склонность к дальнейшему взаимодействию, так как весьма вероятно, что атенолол (I-A, или I-B, или I-C), содержащий вторичную функциональную аминогруппу, снова взаимодействует с глицидиловым эфиром (VIII), присутствующим в реакционной смеси, с образованием ассоциированной примеси (XII), как отображено на схеме VI, которую трудно удалить обычными способами очистки.
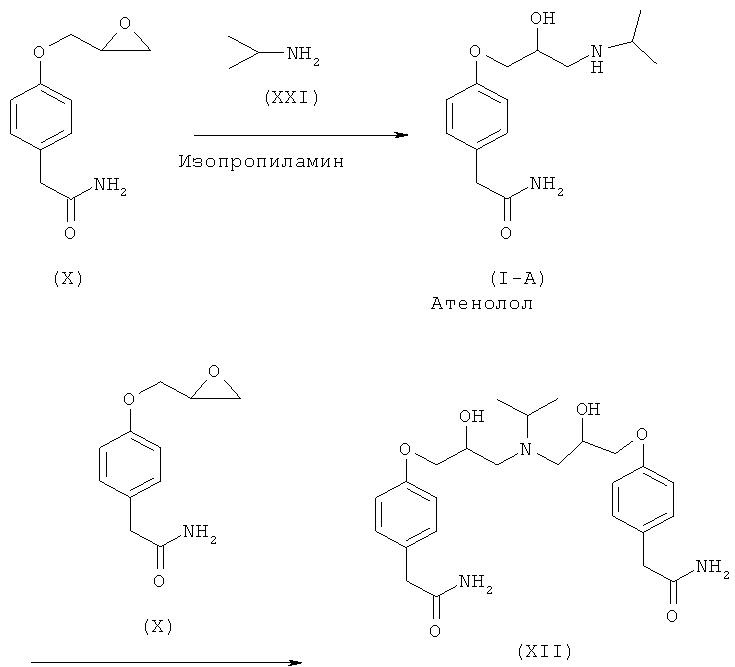
Схема VI. Примеси, образующиеся во время получения атенолола способами известного уровня техники.
Способ, воплощенный в данном изобретении, не может привести к образованию примеси (XII), так как после взаимодействия с эпихлоргидрином (IIIA), (IIIB) или (IIIC) образуется третичный амин (XXIII) или (XXIV), который не может взаимодействовать далее с образованием ассоциированной примеси (XII), образующейся в способах известного уровня техники, из-за полного блокирования атома азота.
Очистка атенолола
Рацемический атенолол формулы (I-А), (S)-атенолол формулы (I-B) и (R)-атенолол формулы (I-C), полученные разными способами, раскрытыми на известном уровне техники, ассоциируются с различными примесями. Как уже указывалось ранее, органы власти во всем мире весьма строги в отношении уровня примесей.
Следовательно, существует потребность в очистке для удаления ассоциированных примесей. Указанное соединение очищают согласно схеме VII, которая описывает способ очистки (S)-атенолола (I-B), который схож со способами очистки рацемического атенолола (I-А) и (R)-атенолола.
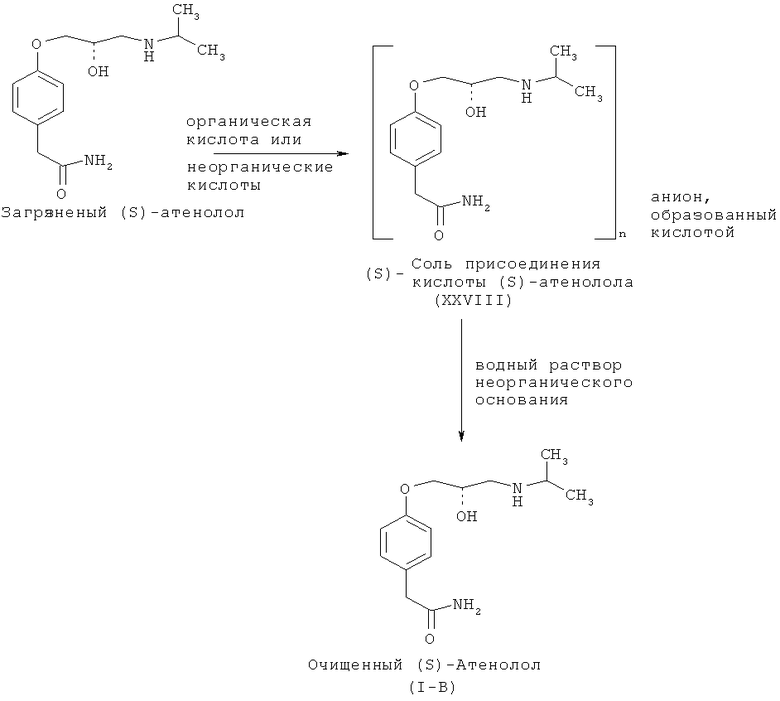
Схема VII. Способ очистки атенолола (I-B), воплощенный в настоящем изобретении.
Типично (S)-атенолол (I-B) обрабатывают органической или неорганической кислотой с использованием органического растворителя, подобного кетону, при температуре окружающей среды и получают соль присоединения кислоты (S)-атенолола (XXVIII), которая выделяется из смеси в виде твердого вещества.
Органические кислоты, используемые для получения соли присоединения кислоты, выбирают из числа таких органических кислот, как уксусная кислота, метансульфоновая кислота, бензолсульфоновая кислота, трифторуксусная кислота, L-винная кислота, малеиновая кислота, фумаровая кислота и т.д. Неорганические кислоты, используемые для получения соли, выбирают из группы, состоящей из кислот, таких как HNO3, HCl, H2SO4, HBr и т.д.
Соль присоединения кислоты, полученную таким образом, растворяют в воде и обрабатывают разбавленным раствором неорганического основания для достижения рН между 12,0 и 12,5, при котором оптически чистый (S)-изомер (I-B) выделяется в виде белого твердого вещества.
Неорганическое основание, используемое для выделения в свободном состоянии основания из его соли присоединения кислоты, выбирают из числа гидроксидов, карбонатов и бикарбонатов щелочных или щелочноземельных металлов. Предпочтительные основания можно выбрать из группы, состоящей из NaOH, KOH, Na2CO3, NaHCO3, K2CO3, LiOH, Са(ОН)2 и т.д.
Преимущество изобретения состоит в том, что рацемический атенолол формулы (I-А), (S)-атенолол формулы (I-B) или (R)-атенолол формулы (I-C), полученные таким образом по способу изобретения, имеют высокую степень чистоты и, по существу, свободны от примесей. Еще одним преимуществом является то, что полученный атенолол имеет высокую степень оптической чистоты.
Данное изобретение описано подробно ниже со ссылкой на приведенные далее примеры, которые приводятся только для пояснения и не предназначены для ограничения объема изобретения каким-либо образом. В случае получения атенолола или его энантиомеров стратегия синтеза остается одной и той же, но с применением соответствующего энантиомера эпихлоргидрина.
Пример 1
Получение N-бензил-N-изопропиламина формулы (XXII)
При перемешивании при температуре окружающей среды к изопропиламину (XXI; 1000 мл) добавляют триэтиламин (119,9 г). Реакционную смесь охлаждают до 10-15°C и перемешивают в течение 10 мин. При 10-15°С постепенно добавляют хлорметилбензол (XX; 100 г), и смесь перемешивают в течение 24 час при 10-15°С. Реакционную смесь концентрируют и к остатку добавляют воду (500 мл). Водный слой экстрагируют метилендихлоридом (2×500 мл). Органический слой отделяют, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, получают густую маслянистую жидкость. Выход 126,7 г (90,5%).
Пример 2
Получение (2S)-1-[бензил(изопропил)амино]-3-хлорпропан-2-ола(XXIII-В)
К N-бензил-N-изопропиламину (XXII; 100,5 г) добавляют (2S)-2-(хлорметил)оксиран (III-B; 310 г), и реакционную смесь перемешивают при 55-60°С в течение 6 час. Реакционную смесь концентрируют, добавляют толуол (300 мл) и снова концентрируют, получают маслянистый остаток продукт реакции (2S)-1-[бензил(изопропил)амино]-3-хлорпропан-2-ол. Выход 158,4 г (% выход 97,3).
Пример 3
Получение N-бензил-N-[(2S)-оксиран-2-илметил]пропан-2-амина (XXIV-B)
К раствору КОН в метаноле (1,66 г в 25 мл метанола) добавляют (2S)-1-[бензил(изопропил)амино]-3-хлорпропан-2-ол (2,42 г), и смесь перемешивают до завершения реакции при 45-50°C. Затем реакционную смесь концентрируют и к остатку добавляют воду (100 мл), а затем метиленхлорид (50 мл). Органический слой, содержащий продукт реакции, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, получают N-бензил-N-[(2S)-оксиран-2-илметил]пропан-2-амин (XXIV-B) в виде густой маслянистой жидкости. Выход 1,3 г (% выход 63,41).
Пример 4
Получение N-бензил-2-(4-{[(2S)-2-гидрокси-3-изопропиламино)пропил]окси}-фенил)ацетамида (XXVIB)
Добавляют 2-(4-гидроксифенил)ацетамид (XXV; 0,75 г) к ацетону (30 мл), а затем добавляют К2СО3 (1,035 г), смесь кипятят с обратным холодильником в течение одного часа при перемешивании. Добавляют (2S)-1-[бензил(изопропил)амино]-3-хлорпропан-2-ол (XXIII-B; 3,26 г), и реакционную смесь кипятят с обратным холодильником в течение 48 час. Реакционную смесь охлаждают до 30°C, фильтруют, промывают ацетоном и сушат в вакууме. Выход 1,70 г (% выход 96,0).
Пример 5
Получение 2-(4-{[(2S-2-гидрокси-3-изопропиламино)пропил]окси}фенил)-ацетамида S(-)-атенолола (IB)
N-Бензил-N-[(2S)-оксиран-2-илметил]пропан-2-амин (10 г, XXVI-B), метанол (400 мл) и 5% Pd/C (2 г) загружают в автоклав и продувают газом N2, после чего пропускают газ H2 до получения давления 310-345 кПА (45-50 ф/д2). Смесь перемешивают при 35-38°C в течение 24 час и фильтруют. Фильтрат концентрируют, к остатку добавляют холодную воду для отделения белого твердого вещества, которое затем отфильтровывают и сушат в печи при 55-60°C. Выход 7,2 г (% выход 96,5).
Пример 6
Получение ацетата 2-{4-[2-гидрокси-3-изопропиламино)пропокси]фенил}-ацетамида - ацетата S-(-)-атенолола
Неочищенный S-(-)-2-{4-[2-гидрокси-3-изопропиламино)пропокси]фенил}-ацетамид (255,0 г) при перемешивании добавляют к ацетону (5,1 л, 20 об.). Реакционную смесь охлаждают до 15-20°C и добавляют ледяную уксусную кислоту (102,0 г) в течение 10-15 мин. Смесь перемешивают в течение 2,0 час при 15-20°C и фильтруют. Смесь промывают холодным ацетоном (2 об.). Влажный ацетат S-(-)-атенолола 5-7 раз промывают ацетоном (7 об.).
Чистота неочищенного S-(-)-атенолола по ВЭЖХ = 97,5%.
Чистота ацетата S-(-)-атенолола по ВЭЖХ = 99,58%.
Пример 7
Получение (S)-атенолола (IB) из ацетата (S)-атенолола (XXVII)
Влажный ацетат S-(-)-атенолола растворяют в воде (1,53 л) (6,0 об.) и при 10°C доводят раствор до рН 12,5 с использованием 30% раствора NaOH (95,0 мл). Раствор перемешивают в течение 1,0 часа. Выпавшее в осадок вещество отфильтровывают, промывают холодной водой (510,0 мл, 2,0 об.) и сушат вещество при 50-55°C (70,27%).
Чистота чистого S-(-)-атенолола по ВЭЖХ = 100,0%.
Пример 8
Получение (2R)-1-[бензил(изопропил)амино]-3-хлорпропан-2-ола(XXVI-С)
В 1 л-круглодонную колбу загружают N-бензил-N-изопропиламин (100,5 г) и (2R)-2-(хлорметил)оксиран (III-C; 310 г). Реакционную смесь нагревают до 55-60°С и при 55-60°С перемешивают в течение 6 час. Сначала из реакционной смеси отгоняют избыток непрореагировавшего (2R)-2-(хлорметил)оксирана, затем добавляют 300 мл толуола и отгоняют следовые количества непрореагировавшего (2R)-2-(хлорметил)оксирана при пониженном давлении. Этот процесс повторяют до полного удаления непрореагировавшего (2R)-2-(хлорметил)оксирана. Масса полученного (2R)-1-[бензил(изопропил)амино]-3-хлорпропан-2-ола составляет 158,4 г (97,3%).
Пример 9
Получение N-бензил-2-(4-{[(2R)-2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-C)
В 1 л-круглодонную колбу загружают 2-(4-гидроксифенил)ацетамид (0,75 г) и ацетон (30 мл) и смесь перемешивают в течение 15 мин при 30°С. Добавляют К2СО3 (1,035 г) и смесь перемешивают в течение одного часа при температуре образования флегмы. Добавляют (2R)-1-[бензил(изопропил)амино]-3-хлорпропан-2-ол (3,626 г) и продолжают кипячение с обратным холодильником в течение 48 час. Через 48 час реакционную смесь охлаждают до 30°С, фильтруют, промывают ацетоном и фильтрат собирают и концентрируют при пониженном давлении.
Масса полученного N-бензил-2-(4-{[(2R)-2-гидрокси-3-(изопропиламино)-пропил]окси}фенил)ацетамида составляет 1,70 г (96,0%).
Пример 10
Получение 2-(4-{[(2R)-2-гидрокси-3-(изопропиламино)пропил]окси}фенил)-ацетамида, т.е. R-(+)-атенолола (I-C)
В 1 л-реактор - автоклав для высоких давлений загружают N-бензил-N-[(2R)-оксиран-2-илметил]пропан-2-амин (10 г), метанол (400 мл) и 5% Pd/C (2 г), реактор продувают N2 и создают повышенное давление газа H2 (310-345 кПа (45-50 ф/д2)). Смесь перемешивают при 35-38°C в течение 24 час. Через 24 часа реакционную смесь выгружают, катализатор отфильтровывают и фильтрат концентрируют. Остаток обрабатывают холодной водой и извлекают белое твердое вещество, которое сушат в сушильной печи при 55-60°C.
Масса полученного 2-(4-{[(2R)-2-гидрокси-3-(изопропиламино)пропил]-окси}фенил)ацетамида, т.е. R-(+)-атенолола, составляет 7,2 г (96,5%).
Пример 11
Получение N-бензил-2-(4-{[(2R)-2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-C)
В 100 мл-круглодонную колбу загружают 2-(4-{[(2R)-оксиран-2-илметокси]фенил}ацетамид (5 г), метанол (50 мл) и N-бензилизопропиламин (5,04 г), и реакционную смесь перемешивают при 60-65°С в течение 24 час. Из реакционной смеси удаляют метанол отгонкой при пониженном давлении. К остатку добавляют метилендихлорид (100 мл), выпавшее в осадок твердое вещество отфильтровывают и фильтрат концентрируют. Получают густую маслянистую жидкость.
Масса полученного N-бензил-2-(4-{[(2R)-2-гидрокси-3-(изопропиламино)пропил]-окси)фенил)ацетамида составляет 8,0 г (93,13%).
Пример 12
Получение 1-[бензил(изопропил)амино]-3-хлорпропан-2-ола (XXIII-A)
В 1 л-круглодонную колбу загружают N-бензил-N-изопропиламин (100,5 г) и 2-(хлорметил)оксиран (310 г). Реакционную смесь нагревают до 55-60°С и при 55-60°С перемешивают в течение 6 час. Сначала отгоняют избыток непрореагировавшего 2-(хлорметил)оксирана (III-A) из реакционной смеси, затем добавляют 300 мл толуола и отгоняют следовые количества непрореагировавшего 2-(хлорметил)оксирана при пониженном давлении. Этот процесс повторяют до полного удаления непрореагировавшего 2-(хлорметил)оксирана.
Масса полученного 1-[бензил(изопропил)амино]-3-хлорпропан-2-ола составляет 158,4 г (97,3%).
Пример 13
Получение N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-A)
В 1 л-круглодонную колбу загружают 2-(4-гидроксифенил)ацетамид (0,75 г) и ацетон (30 мл), и смесь перемешивают в течение 15 мин при 30°С. Добавляют К2СО3 (1,035 г) и смесь перемешивают в течение одного часа при температуре образования флегмы. Добавляют 1-[бензил(изопропил)амино]-3-хлорпропан-2-ол (3,626 г) и продолжают кипячение с обратным холодильником в течение 48 час. Через 48 час реакционную смесь охлаждают до 30°С, фильтруют, промывают ацетоном и фильтрат собирают и концентрируют при пониженном давлении.
Масса полученного N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]-окси}фенил)ацетамида составляет 1,70 г (96,0%).
Пример 14
Получение 2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}фенил)-ацетамида, т.е. атенолола (I-A)
В 1 л-автоклав загружают N-бензил-N-[оксиран-2-илметил]пропан-2-амин (10 г), метанол (400 мл) и 5% Pd/C (2 г), реактор продувают N2 и создают повышенное давление газа H2 (310-345 кПа (45-50 ф/д2)). Реакционную смесь перемешивают при 35-38°С в течение 24 час. Через 24 часа реакционную смесь выгружают, катализатор отфильтровывают и фильтрат концентрируют. Остаток обрабатывают холодной водой и получают продукт реакции - белое твердое вещество, которое сушат в сушильной печи при 55-60°С.
Масса полученного 2-(4-{[2-гидрокси-3-(изопропиламино)пропил]-окси}фенил)ацетамида составляет 7,2 г (96,5%).
Пример 15
Получение N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-A)
В 100 мл-круглодонную колбу загружают 2-{4-[оксиран-2-илметокси]фенил}ацетамид (5 г), метанол (50 мл) и N-бензилизопропиламин (5,04 г), и реакционную смесь перемешивают при 60-65°С в течение 24 час. Через 24 часа из реакционной смеси удаляют метанол отгонкой при пониженном давлении. К остатку добавляют метилендихлорид (100 мл), выпавшее в осадок твердое вещество отфильтровывают и фильтрат концентрируют. Получают густую маслянистую жидкость.
Масса полученного N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида составляет 8,0 г (93,13%).
Пример 16
Получение 2-{4-[(2S)-оксиран-2-илметокси]фенил}ацетамида (XXVII)
В 2 л-круглодонную колбу загружают 2-(4-гидроксифенил)ацетамид (100 г) и воду (400 мл) и смесь перемешивают в течение получаса. Добавляют предварительно охлажденный раствор NaOH [31,78 г + вода DM, 200 мл] и смесь охлаждают до 0°С. В течение 1 часа добавляют (2R)-2-(хлорметил)оксиран (91,88 г). Реакционную смесь перемешивают в течение 25 час при 0°С. Через 25 час реакционную смесь фильтруют и полученное твердое вещество сушат при 50°С.
Масса полученного 2-{4-[(2S)-оксиран-2-илметокси]фенил}ацетамида составляет 137,08 г (73,24%).
Пример 17
Получение N-бензил-2-(4-{[(2S)-2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида
В 100 мл-круглодонную колбу загружают 2-(4-{[(2S)-оксиран-2-илметокси]фенил}ацетамид (5 г), метанол (50 мл) и N-бензилизопропиламин (5,04 г) и реакционную смесь перемешивают при 60-65°С в течение 43 час. Через 43 часа из реакционной смеси отгоняют метанол при пониженном давлении. К остатку добавляют метилендихлорид (100 мл), выпавшее в осадок твердое вещество отфильтровывают и фильтрат концентрируют. Получают густую маслянистую жидкость.
Масса полученного N-бензил-2-(4-{[(2S)-2-гидрокси-3-(изопропиламино)пропил]-окси}фенил)ацетамида составляет 8,0 г (93,13%).
Пример 18
Получение N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-A)
В 1 л-круглодонную колбу загружают 2-(4-гидроксифенил)ацетамид (5 г), ацетон (200,0 мл) и К2СО3 (4,57 г). Смесь нагревают до 55°С и перемешивают в течение одного часа при температуре образования флегмы. Добавляют N-бензил-N-изопропиламинохлоргидрин (16,0 г) и продолжают кипячение с обратным холодильником в течение 15 суток. Смесь охлаждают до 30°С, фильтруют, промывают ацетоном и собирают фильтрат. Фильтрат концентрируют. Добавляют воду. Доводят рН реакционной смеси до 2,0 соляной кислотой и смесь экстрагируют дихлорметаном. Доводят рН водного слоя до 12,2 30%-раствором гидроксида натрия и экстрагируют его дихлорметаном. Органический слой концентрируют и получают маслянистый продукт реакции.
Масса полученного N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-A) составляет 8,5 г (72,1%).
Пример 19
Получение N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-A)
В 1 л-круглодонную колбу загружают 2-(4-гидроксифенил)ацетамид (10 г), ацетонитрил (100 мл) и К2СО3 (18,31 г) и смесь нагревают до 80°С. Смесь перемешивают при температуре образования флегмы в течение 1 часа. Добавляют каталитическое количество иодида тетрабутиламмония. Добавляют N-бензил-N-изопропиламинохлоргидрин (31,96 г) и продолжают кипячение с обратным холодильником в течение 28 часов. Через 28 часов реакционную смесь охлаждают до 30°С и фильтруют. Фильтрат собирают и концентрируют при пониженном давлении. Добавляют воду. Доводят рН смеси до 1,5 соляной кислотой и смесь экстрагируют дихлорметаном. Доводят рН водного слоя до 12,2 30%-раствором гидроксида натрия и экстрагируют его дихлорметаном. Органический слой концентрируют и получают маслянистый продукт реакции.
Масса полученного N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-A) составляет 15 г (63,64%).
Пример 20
Получение N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-A)
В 1 л-круглодонную колбу загружают 2-(4-гидроксифенил)ацетамид (5 г) и ацетонитрил (50 мл) и смесь перемешивают в течение 15 мин при 30°С. Добавляют К2СО3 (9,2 г) и смесь при перемешивании нагревают до 80°С. Добавляют каталитическое количество иодида тетрабутиламмония. Добавляют N-бензил-N-изопропиламинохлоргидрин (9 г) и продолжают кипячение с обратным холодильником в течение 46 часов. Реакционную смесь охлаждают до 30°С, фильтруют и промывают ацетонитрилом и концентрируют при пониженном давлении. К концентрированному маслу добавляют воду и дихлорметан, доводят рН смеси до 0,7 соляной кислотой и смесь экстрагируют дихлорметаном. Доводят рН водного слоя до 12,2 30%-раствором гидроксида натрия и экстрагируют его дихлорметаном. Органический слой концентрируют и получают маслянистый продукт реакции. Масса полученного N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси} фенил)ацетамида (XXVI-A) составляет 11,2 г (95,07%).
Пример 21
Получение N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-A)
В 1 л-круглодонную колбу загружают 2-(4-гидроксифенил)ацетамид (5 г) и ацетонитрил (50 мл) и смесь перемешивают в течение 15 мин при 28°С. Добавляют К2СО3 (9,15 г) и смесь нагревают до 80°С. Перемешивают смесь в течение 1 часа при температуре образования флегмы. Добавляют каталитическое количество иодида тетрабутиламмония. Добавляют N-бензил-N-изопропиламинохлоргидрин (9 г) и продолжают кипячение с обратным холодильником в течение 58 часов. Реакционную смесь охлаждают до 30°С, фильтруют и промывают ацетонитрилом. Смесь концентрируют при пониженном давлении. К концентрированному маслу добавляют воду и дихлорметан, доводят рН смеси до 1,0 соляной кислотой и смесь экстрагируют дихлорметаном. Доводят рН водного слоя до 12,2 30%-раствором гидроксида натрия и экстрагируют его дихлорметаном. Органический слой концентрируют и получают маслянистый продукт реакции. Масса полученного N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}фенил)ацетамида (XXVI-A) составляет 11,5 г (97,62%).
Пример 22
Получение N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}-фенил)ацетамида (XXVI-A)
В 1 л-круглодонную колбу загружают 2-(4-гидроксифенил)ацетамид (5 г) и ацетонитрил (50 мл) и смесь перемешивают в течение 15 мин при 27°С. Добавляют К2СО3 (9,15 г) и смесь нагревают при перемешивании до 80°С. Добавляют каталитическое количество иодида тетрабутиламмония. Добавляют N-бензил-N-изопропиламинохлоргидрин (9,6 г) и смесь кипятят с обратным холодильником в течение 24 часов. Реакционную смесь охлаждают до 30°С, фильтруют и промывают ацетонитрилом. Реакционную смесь концентрируют при пониженном давлении. Масса полученного N-бензил-2-(4-{[2-гидрокси-3-(изопропиламино)пропил]окси}фенил)-ацетамида (XXVI-A) составляет 10,2 г (86,58%).
Пример 23
Получение N-бензил-N-[(2S)-оксиран-2-илметил]пропан-2-амина (XXIV-A)
К раствору КОН в метаноле (8,192 г в 100,0 мл метанола) добавляют 1-[бензил(изопропил)амино]-3-хлорпропан-2-ол (10,0 г) и смесь перемешивают до завершения реакции при 45-50°С. Реакционную смесь концентрируют и к остатку добавляют воду (250 мл), а затем метиленхлорид (250 мл). Органический слой, содержащий продукт реакции, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, получают N-бензил-N-[(2S)-оксиран-2-илметил]пропан-2-амин (XXIV-A) в виде густой маслянистой жидкости. Выход 8,7 г (выход 98%).
Пример 24
Получение рацемического ацетата 2-{4-(2-гидрокси-3-(изопропиламино)-пропокси)фенил}ацетамида
Неочищенный рацемический 2-гидрокси-3-изопропиламино)пропокси)ацетамид (25,0 г) при перемешивании в течение 15 минут добавляют к ацетону (0,5 л, 20 об.). Реакционную смесь охлаждают до 15-20°С и добавляют ледяную уксусную кислоту (10,0 г) в течение 10-15 мин. Смесь перемешивают в течение 2,0 час при 15-20°С. Смесь фильтруют и промывают холодным ацетоном (2 об.). Влажный рацемический ацетат атенолола 5-7 раз промывают ацетоном (7 об.). Растворяют влажный рацемический ацетат атенолола в воде (150 мл, 6 об.) и при 10°С доводят раствор до рН 12,5 с использованием 30%-раствора NaOH (11,0 мл). Смесь перемешивают в течение 1,0 часа, фильтруют и промывают холодной водой (50,0 мл, 2,0 об.). Полученное вещество сушат при 50-55°С. Выход 75,04%.
Чистота неочищенного атенолола по ВЭЖХ = 96,96%.
Чистота ацетата атенолола по ВЭЖХ = 99,32%.
Чистота очищенного атенолола по ВЭЖХ = 99,77%.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗАЦИКЛИЧЕСКИЕ СПИРО-СОЕДИНЕНИЯ | 2010 |
|
RU2550495C2 |
| АНТИМИКРОБНЫЕ ФЕНИЛОКСАЗОЛИДИНОНЫ | 1995 |
|
RU2134692C1 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ И АГОНИСТОВ БЕТА-2-АДРЕНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2013 |
|
RU2661877C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛАЛКИЛПИПЕРАЗИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2243970C1 |
| ПРОИЗВОДНЫЕ 4-ГИДРОКСИПИПЕРИДИНА | 1997 |
|
RU2178412C2 |
| ПРОИЗВОДНЫЕ ДИФЕНИЛАЗЕТИДИНОНА, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ЛЕКАРСТВЕННЫЕ СРЕДСТВА И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2275370C2 |
| НИТРАТНЫЕ СОЛИ ГИПОТЕНЗИВНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 1999 |
|
RU2235097C2 |
| ПРОИЗВОДНЫЕ 4-ФЕНИЛ-5-ОКСО-1, 4, 5, 6, 7, 8-ГЕКСАГИДРОХИНОЛИНА В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ БЕСПЛОДИЯ | 2006 |
|
RU2403243C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ | 2006 |
|
RU2408584C2 |
| БИЦИКЛИЧЕСКИЕ ОКСАЗИН- ИЛИ ТИАЗИН-ОКСАЗОЛИДИНОНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБЫ ЛЕЧЕНИЯ МИКРОБНЫХ ИНФЕКЦИЙ | 1995 |
|
RU2128660C1 |
Настоящее изобретение относится к новому улучшенному способу получения атенолола, включающему стадии а) сочетания хлорметилбензола (XX) с изопропиламином с образованием N-бензил-N-изопропиламина (XXII); b) взаимодействия полученного N-бензил-N-изопропиламина с эпихлоргидрином (IIIA); с) превращения галогенгидрина (XXIII-А), полученного на стадии b) в присутствии основания в эпоксид; d) взаимодействия соли фенилацетамида (XXV) с галогенгидрином (XXIII-A) или эпоксидом (XXIV-A) в присутствии основания; е) дебензилирования N-бензелированного атенолола (XXVI-A) до рацемического атенолола (I-A). Техническим результатом является возможность получения атенолола высокой чистоты в промышленном масштабе. 10 з.п. ф-лы.
1. Способ получения атенолола, включающий стадии
а) сочетание хлорметилбензола (XX) с изопропиламином с образованием N-бензил-N-изопропиламина
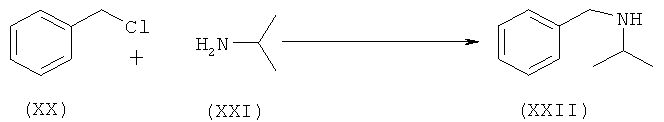
b) взаимодействие N-бензил-N-изопропиламина (XXII) с эпихлоргидрином (IIIA)
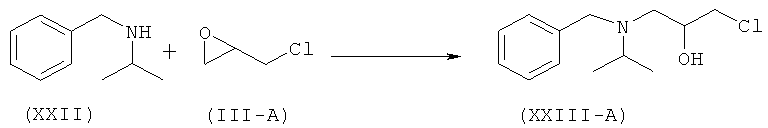
c) превращение галогенгидрина (XXIII-A) в присутствии основания в эпоксид
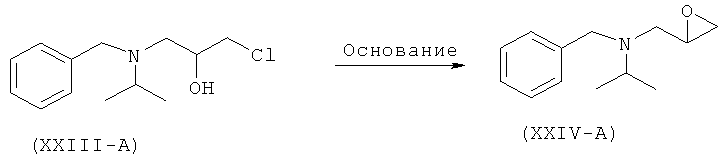
d) взаимодействие соли фенилацетамида (XXV) с галогенгидрином (XXIII-А) или эпоксидом (XXIV-A) в присутствии основания
е) дебензилирование N-бензелированного атенолола (XXVI-A) до рацемического атенолола (I-A)
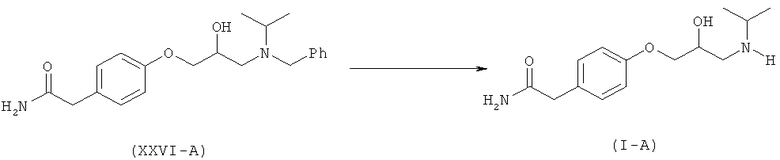
2. Способ по п.1, где полученный атенолол представляет собой (S)-атенолол при условии, что используемый эпихлоргидрин представляет собой (S)-эпихлоргидрин.
3. Способ по п.1, где полученный атенолол представляет собой (R)-атенолол при условии, что используемый эпихлоргидрин представляет собой (R)-эпихлоргидрин.
4. Способ по п.1 (с), где используемое основание выбирают из группы, состоящей из гидроксидов, карбонатов, бикарбонатов щелочных и щелочноземельных металлов.
5. Способ по п.1 (с), где используемое основание выбирают из группы, состоящей из гидроксидов щелочных и щелочноземельных металлов.
6. Способ по п.1 (с), где используемое основание выбирают из группы, состоящей из КОН и NaOH.
7. Способ по п.1 (d), где используемое основание выбирают из группы, состоящей из гидроксидов, карбонатов, бикарбонатов щелочных и щелочноземельных металлов.
8. Способ по п.1 (d), где используемое основание выбирают из группы, состоящей из карбонатов щелочных и щелочноземельных металлов.
9. Способ по п.1 (d), где используемое основание выбирают из группы, состоящей из K2CO3 и Na2CO3.
10. Способ по п.1 (d), где основание используют вместе с межфазным катализатором.
11. Способ по п.10, где межфазный катализатор выбирают из числа иодида тетрабутиламмония или бромида тетрабутиламмония.
| ФИКСАТОР СТРОЯ МЕХАНИЗМА ТРЕМОЛО СТРУННОГО МУЗЫКАЛЬНОГО ИНСТРУМЕНТА | 1992 |
|
RU2008725C1 |
| US 6982349 B1, 03.01.2006 | |||
| ТЕРМОПЛАСТИЧНАЯ ЭЛАСТОМЕРНАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2276167C1 |
| Способ прокатки слябов на листовых станах | 1981 |
|
SU990352A1 |
| Способ получения 1-изопропиламино-3-фенокси-2-пропанола | 1983 |
|
SU1321371A3 |
| Kitaori, K | |||
| et al | |||
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |
| Kitaori, K | |||
| et al | |||
| "Convenient preparation of | |||

