Настоящее изобретение относится к новым производным бензазепина, обладающим фармакологической активностью, к способам их получения, к композициям, содержащим данные соединения, и к их применению для лечения неврологических и психиатрических нарушений.
В JP 2001226269 и WO 00/23437 (Takeda Chem. Ind. Ltd) описывается ряд производных бензазепина, которые заявляются как средства для лечения ожирения. В DE 2207430, US 4210749, FR 2171879 (Pennwalt Corp.) и GB 1268243 (Wallace and Tiernan Inc.) описывается ряд производных бензазепина в качестве антагонистов наркотиков (таких как морфин или кодеин), а также антигистаминные и антихолинергические средства. В WO 02/14513 (Takeda Chem. Ind. Ltd) описывается ряд производных бензазепина с активностью GPR12 как средства для лечения синдрома нарушения внимания, нарколепсии или беспокойства. В WO 02/02530 (Takeda Chem. Ind. Ltd) описывается ряд производных бензазепина как антагонистов GPR14, как средства для лечения гипертонии, атеросклероза и инфаркта миокарда. В WO 01/03680 (Isis Innovation Ltd) описывается ряд производных бензазепина, которые заявляются как эффективные средства, использующиеся при получении клеток для трансплантации, а также для подавления таких заболеваний, как диабет. В WO 00/21951 (SmithKline Beecham pic) раскрывается ряд производных тетрагидробензазепина как модуляторов рецепторов допамина D3 в качестве антипсихотических средств. В WO 01/87834 (Takeda Chem. Ind Ltd) описывается ряд производных бензазепина как антагонистов MCH, которые заявляются как средства для лечения ожирения. В WO 02/15934 (Takeda Chem. Ind. Ltd) описывается ряд производных бензазепина как антагонистов рецепторов уротензина II, которые заявляются как средства для лечения нейродегенеративных заболеваний.
Рецептор гистамина H3 преимущественно экспрессируется в центральной нервной системе млекопитающих (CNS), в периферических тканях он экспрессируется на минимальном уровне, за исключением некоторых симпатических нервов (Leurs et al. (1998), Trends Pharmacol. Sci. 19, 177-183). Активация рецепторов H3 под действием селективных агонистов или гистамина приводит к ингибированию высвобождения нейромедиаторов из ряда различных популяций нервных клеток, в том числе из гистаминергических и холинергических нейронов (Schlicker et al. (1994), Fundam. Clin. Pharmacol. 8, 128-137). Кроме того, исследования in vitro и in vivo показали, что антагонисты H3 могут способствовать высвобождению нейромедиаторов из таких участков мозга, как кора головного мозга и гиппокамп, которые имеют отношение к познавательной способности (Onodera et al. (1998), In: The Histamine H3 receptor, ed Leurs and Timmerman, pp. 255-267, Elsevier Science B.V.). Кроме того, в ряде публикаций было продемонстрировано, что антагонисты H3 (например, тиоперамид, клобенпропит, ципроксифан и GT-2331) улучшают познавательную способность у кроличьих моделей, в том числе выполнение следующих тестов: задачи пятикратного выбора, распознавания объекта, приподнятого крестообразного лабиринта, обнаружения новой задачи и пассивного уклонения (Giovanni et al. (1999), Behav. Brain Res. 104, 147-155). Приведенные данные позволяют предположить, что новые антагонисты H3 и/или обратные агонисты, например ряд соединений настоящего изобретения, можно использовать для лечения ухудшения познавательной способности при таких неврологических заболеваниях, как болезнь Альцгеймера и родственные нейродегенеративные нарушения.
В первом аспекте настоящее изобретение предлагает соединение, которое представляет собой 6-(3-Циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)-N-метилникотинамид
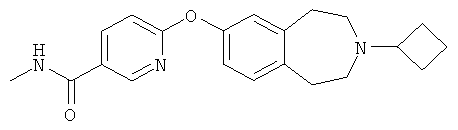
или его фармацевтически приемлемую соль.
Соединение или его фармацевтически приемлемая соль по изобретению представляет собой соединение формулы (I) или его фармацевтически приемлемую соль:
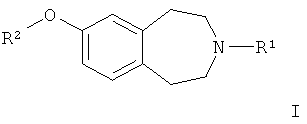
где R1 представляет собой незамещенный циклобутил;
R2 представляет собой 2-пиридинил, замещенный CON(H)(Me), и представляет собой 4-метиламинокарбонилпиридин-2-ил или его сольват.
Алкильные группы, присутствующие отдельно или в составе другой группы, могут быть линейными или разветвленными, группы алкокси и алканоил интерпретируют так же. Более предпочтительно, алкильные фрагменты представляют собой C1-4алкил, например метил или этил. Термин 'галоген' используется в описании, если не указано иначе, для определения группы, выбранной из фтора, хлора, брома или иода.
Термин 'арил' включает моноциклические карбоциклические ароматические кольца (например, фенил) и бициклические карбоциклические ароматические кольца (например, нафтил) или карбоциклические бензоконденсированные кольца (например, C3-8циклоалкил, конденсированный с фенильным циклом, такой как дигидроинденил или тетрагидронафталинил).
Подразумевается, что термин "гетероциклил" обозначает 4-7-членный моноциклический насыщенный или частично ненасыщенный алифатический цикл, или 4-7-членный насыщенный или частично ненасыщенный алифатический цикл, конденсированный с бензольным циклом, содержащим от 1 до 3 гетероатомов, выбранных из кислорода, азота или серы. Подходящие примеры таких моноциклических колец включают в себя пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, тетрагидрофуранил, тетрагидропиранил, диазепанил, азепанил, имидазолидинил, изотиазолидинил, оксазолидинил, пирролидинон и тетрагидрооксазепинил. Подходящие примеры бензоконденсированных гетероциклических колец включают в себя индолинил, изоиндолинил, бензодиоксолил, дигидроизоиндол, дигидробензофуранил, дигидробензотиопиранил и дигидроизохинолинил.
Подразумевается, что термин "гетероциклил, содержащий атом азота" относится к любой гетероциклической группе, как определено выше, которая содержит атом азота.
Подразумевается, что термин "гетероарил" обозначает 5-7-членное моноциклическое ароматическое или конденсированное 8-11-членное бициклическое ароматическое кольцо, содержащее от 1 до 3 гетероатомов, выбранных из кислорода, азота и серы. Подходящие примеры таких моноциклических ароматических циклов включают в себя тиенил, фурил, пирролил, триазолил, имидазолил, оксазолил, тиазолил, оксадиазолил, изотиазолил, изоксазолил, тиадиазолил, пиразолил, пиримидил, пиридазинил, пиразинил, пиридил и тетрагидропиранил.
Подходящие примеры таких конденсированных ароматических циклов включают в себя бензоконденсированные ароматические циклы, такие как хинолинил, изохинолинил, хиназолинил, хиноксалинил, циннолинил, нафтиридинил, индолил, индазолил, фуропиридинил, пирролопиридинил, бензофуранил, бензотиенил, бензимидазолил, бензоксазолил, бензизоксазолил, бензотиазолил, бензизотиазолил, бензоксадиазолил, бензотиадиазолил и т.п.
Соединения формулы (I) могут образовывать кислотно-аддитивные соли с традиционными фармацевтически приемлемыми кислотами, такими как малеиновая, хлористоводородная, бромистоводородная, фосфорная, уксусная, фумаровая, салициловая, серная, лимонная, молочная, миндальная, виннокаменная и метансульфоновая. Таким образом, соли, сольваты и гидраты соединений формулы (I) составляют аспект данного изобретения.
Таутомеры также составляют аспект данного изобретения.
Настоящее изобретение также предлагает способ получения соединения формулы (I) или его фармацевтически приемлемой соли, данный способ включает в себя
(a) взаимодействие соединения формулы (II)
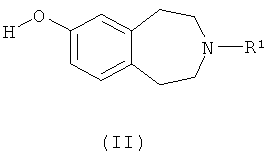
где R1 такой, как определено выше, с соединением формулы R2'-L1, где R2' такой, как определено выше для R2, или представляет собой группу, которая способна превратиться в R2, а L1 обозначает подходящую уходящую группу, такую как атом галогена (например, бром или иод) или необязательно активированная гидроксильная группа;
(b) взаимодействие соединения формулы (III)
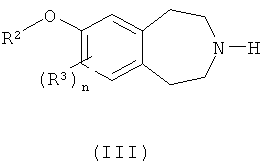
где R2, R3 и n такие, как определено выше, с соединением формулы R1'-L2, где R1' такой, как определено выше для R1, или представляет собой группу, которая способна превратиться в R1, а L2 обозначает подходящую уходящую группу, такую как атом галогена (например, бром, иод) или тозилат; или
(c) взаимодействие соединения формулы (III), как определено выше, с кетоном формулы R1'=O, где R1' такой, как определено выше для R1, или представляет собой группу, которая способна превратиться в R1; или
(d) удаление защитной группы с защищенного соединения формулы (I); и
(e) взаимопревращение в другие соединения формулы(I).
Если уходящая группа L1 присоединена к атому углерода в состоянии sp3-гибридизации, например, если R2'-L1 обозначает алкилгалогенид, процесс (a), как правило, включает в себя применение подходящего основания, такого как карбонат калия, в подходящем растворителе, таком как 2-бутанон, необязательно в присутствии катализатора, такого как иодид калия, при подходящей температуре, такой как температура кипения с обратным холодильником.
Если уходящая группа L1 присоединена к атому углерода в состоянии sp2-гибридизации, например, если R2'-L1 обозначает арилгалогенид, процесс (a), как правило, включает в себя применение соли меди (I), такой как иодид меди (I), в присутствии такого основания, как гидрид натрия, в подходящем растворителе, таком как пиридин, при подходящей температуре, такой как температура кипения с обратным холодильником.
Если уходящая группа L1 присоединена к активированному атому углерода в состоянии sp2-гибридизации, например, если R2'-L1 обозначает гетероарилгалогенид, такой как 2-хлорпиридин или 2-хлорпиразин, процесс (a), как правило, включает в себя применение подходящего основания, такого как гидрид натрия, в подходящем растворителе, таком как диметилформамид или диметилсульфоксид, при подходящей температуре. Альтернативно можно использовать трет-бутоксид калия в трет-бутаноле при подходящей температуре.
Если уходящая группа L1 присоединена к активированному атому углерода в состоянии sp2-гибридизации, например, если R2'-L1 обозначает арилгалогенид, такой как 3,4-дифторбензонитрил, процесс (a), как правило, включает в себя применение подходящего основания, карбоната калия, в подходящем растворителе, таком как диметилсульфоксид, при подходящей температуре.
Если L1 обозначает гидроксильную группу, присоединенную к атому углерода в состоянии sp3-гибридизации, например, если R2'-L1 обозначает спирт, процесс (a), как правило, включает в себя применение фосфина, такого как трифенилфосфин, в подходящем растворителе, таком как тетрагидрофуран, с последующим добавлением азодикарбоксилата, такого как диэтилазодикарбоксилат, при подходящей температуре, такой как комнатная температура.
Процесс (b), как правило, включает применение подходящего основания, такого как карбонат калия, в подходящем растворителе, таком как 2-бутанон, необязательно в присутствии катализатора, такого как иодид калия, при подходящей температуре, такой как температура кипения с обратным холодильником.
Процесс (c), как правило, включает применение восстанавливающих условий (таких как обработка боргидридом, например триацетоксиборгидридом натрия), необязательно в присутствии кислоты, такой как уксусная кислота, в подходящем растворителе, таком как дихлорметан, при подходящей температуре, такой как комнатная температура.
Что касается процесса (d), примеры защитных групп и способы их удаления можно найти в T. W. Greene 'Protective Groups in Organic Synthesis' (J. Wiley and Sons, 1991). Подходящие защитные группы для аминогруппы включают в себя сульфонил (например, тозил), ацил (например, ацетил, 2',2',2'-трихлорэтоксикарбонил, бензилоксикарбонил или трет-бутоксикарбонил) и арилалкил (например, бензил), которые можно удалить гидролизом (например, используя такую кислоту, как хлористоводородная в диоксане, или трифторуксусная в дихлорметане) или восстановлением (например, гидрированием бензильной группы, или, в случае 2',2',2'-трихлорэтоксикарбонильной группы, восстановлением цинком в уксусной кислоте), соответственно. Другие подходящие защитные группы для аминогруппы включают в себя трифторацетил (-COCF3), который можно удалить гидролизом, катализируемым основанием или с использованием в качестве твердой фазы смолы с иммобилизованной бензильной группой, такой как смола Merrifield, связанная с 2,6-диметоксибензильной группой (линкер Ellman), которую можно удалить кислым гидролизом, например, с помощью трифторуксусной кислоты.
Процесс (e) можно проводить с использованием традиционных методов взаимопревращения, таких как эпимеризация, окисление, восстановление, алкилирование, нуклеофильное или электрофильное замещение в ароматическом ядре, гидролиз сложного эфира, образование амидной связи или реакции сочетания, опосредованные переходным металлом. Примеры реакций сочетания, катализируемых переходными металлами и используемых в качестве способов взаимопревращения, включают в себя следующие: катализируемые палладием реакции сочетания электрофилов, таких как арилгалогениды, с металлорганическими реагентами, например борными кислотами (реакции перекрестного сочетания Сузуки (Suzuki)); катализируемые палладием реакции аминирования и амидирования органических электрофилов, таких как арилгалогениды, проводимые с использованием таких нуклеофилов, как амины и амиды; катализируемые медью реакции амидирования органических электрофилов (таких как арилгалогениды), проводимые с использованием таких нуклеофилов, как амиды; и катализируемые медью реакции сочетания фенолов с борными кислотами.
Соединения формул (II) и (III) можно получить по следующей схеме
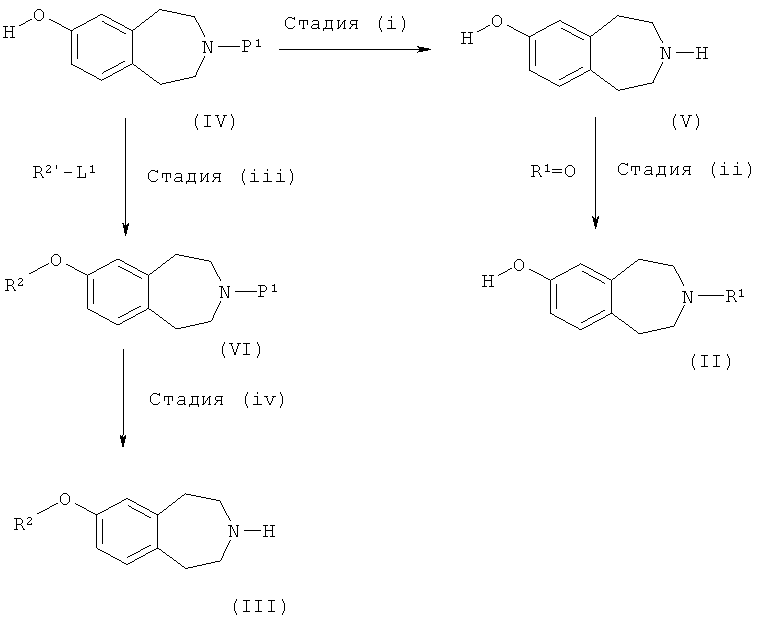
где R1, R2, R2' и L1 такие, как определено выше, а P1 обозначает подходящую защитную группу, такую как Boc.
Стадия (i), как правило, включает в себя реакцию удаления защитной группы, например, если P1 обозначает Boc, реакция удаления защитной группы включает в себя взаимодействие соединения формулы (IV) с кислотой, например хлористоводородной кислотой в диоксане или трифторуксусной кислотой в дихлорметане.
Стадию (ii) можно проводить в восстанавливающих условиях, как описано для процесса (c).
Стадию (iii) можно проводить по способу, описанному для процесса (a).
Стадия (iv), как правило, включает в себя реакцию удаления защитной группы с получением соединения формулы (III) и может проводиться в соответствии со способом стадии (i).
Соединения формулы (VI), где R2 обозначает -2-пиридинил, замещенный CON(H)(Me) и представляет собой 4-метиламинокарбонилпиридин-2-ил, могут быть также получены по следующей схеме
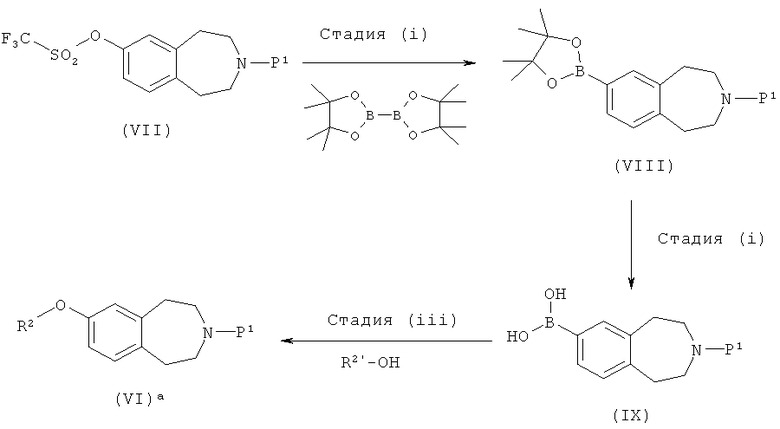
где R2 и R2' такие, как определено выше, а P1 обозначает подходящую защитную группу, такую как Boc.
Стадию (i) можно проводить в условиях перекрестного сочетания, катализируемого палладием, например, с использованием в качестве каталитической системы комплекса бис(дифенилфосфино)ферроцендихлорпалладия(II) и
1,1'-бис(дифенилфосфино)ферроцена, в сочетании с подходящим основанием, таким как ацетат калия, в подходящем растворителе, например в диоксане, при подходящей температуре, например, при температуре кипения с обратным холодильником.
Стадию (ii) можно проводить в окисляющих условиях, например, с использованием периодата натрия в присутствии ацетата аммония, в подходящей системе растворителей, такой как ацетон и вода, при подходящей температуре, например, при комнатной температуре.
Стадию (iii) можно проводить в присутствии соли меди, например ацетата меди, в сочетании с подходящим основанием, таким как триэтиламин, с использованием молекулярных сит, в подходящем растворителе, например дихлорметане, при подходящей температуре, например при комнатной температуре.
Соединения формулы (IV) можно получить по способу, приведенному в описании 3 WO 02/40471.
Соединения формулы (VII) можно получить по способу, описанному в Bioorg. Med. Chem. Lett.; 10; 22; 2000; 2553-2556.
Соединения формулы (I) и их фармацевтически приемлемые соли обладают сродством к рецептору гистамина H3, являются антагонистами и/или обратными агонистами указанного рецептора и предположительно могут применяться для лечения неврологических заболеваний, включающих в себя болезнь Альцгеймера, деменцию, старческое расстройство памяти, легкое ухудшение познавательной способности, нарушение познавательной способности, эпилепсию, невропатическую боль, воспалительную боль, мигрень, болезнь Паркинсона, рассеянный склероз, удар и нарушение сна, включая нарколепсию; психиатрических заболеваний, включающих шизофрению (особенно, нарушение познавательной способности при шизофрении), синдром дефицита внимания с гиперактивностью, депрессию и привыкание; а также других заболеваний, включающих ожирение, астму, аллергический ринит, заложенность носа, хроническое обструктивное заболевание легких и желудочно-кишечные заболевания.
Следовательно, данное изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения в качестве терапевтического средства для лечения или профилактики вышеуказанных заболеваний, особенно нарушения познавательной способности при таких заболеваниях, как болезнь Альцгеймера и родственные нейродегенеративные нарушения.
Далее, данное изобретение относится к способу лечения или профилактики вышеуказанных заболеваний у млекопитающих, в том числе у людей, который включает в себя введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В другом аспекте данное изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного средства для лечения вышеуказанных заболеваний.
Для применения в терапевтических целях соединения формулы (I) обычно вводят в состав стандартной фармацевтической композиции. Такие композиции можно получить с помощью стандартных методов.
Следовательно, настоящее изобретение также относится к фармацевтической композиции для лечения вышеуказанных заболеваний, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Далее, настоящее изобретение относится к фармацевтической композиции, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Соединения формулы (I) можно использовать в комбинации с другими терапевтическими средствами, например с антагонистами гистамина H1 или с медикаментами, заявленными как средства для облегчения или симптоматического лечения болезни Альцгеймера. Подходящими примерами упомянутых других терапевтических средств могут служить средства, которые, как известно, изменяют холинергическую передачу, такие как антагонисты 5-HT6, мускариновые агонисты M1, мускариновые антагонисты M2 или ингибиторы ацетилхолинестеразы. Если соединения используются в комбинации с другими терапевтическими средствами, их можно вводить последовательно или одновременно любым подходящим способом.
Таким образом, в следующем аспекте данное изобретение относится к комбинации соединения формулы (I), или его фармацевтически приемлемого производного, с другим терапевтическим средством или другими терапевтическими средствами.
Упомянутые выше комбинации удобно использовать в виде фармацевтической композиции и, следовательно, фармацевтические композиции, содержащие указанную выше комбинацию вместе с фармацевтически приемлемым носителем или наполнителем, составляют следующий аспект данного изобретения. Отдельные компоненты таких сочетаний можно вводить последовательно или одновременно в разных фармацевтических композициях или в одной фармацевтической композиции.
Если соединение формулы (I) или его фармацевтически приемлемое производное используется в сочетании с другим терапевтическим средством, активным против того же заболевания, доза каждого соединения может отличаться от дозы, используемой в случае применения одного соединения. Специалисты в данной области могут легко определить нужную дозу.
Фармацевтическую композицию данного изобретения, которую можно получить путем смешивания, обычно при температуре окружающей среды и атмосферном давлении, как правило, модифицируют для перорального, парентерального или ректального введения, и, как таковая, она может находиться в виде таблеток, капсул, жидких препаратов для перорального применения, порошков, гранул, пастилок, порошков для разбавления, растворов или суспензий для инъекций или инфузий, или свечей. Как правило, предпочтительными являются композиции для перорального введения.
Таблетки и капсулы для перорального введения могут находиться в виде стандартной лекарственной формы и могут содержать традиционные наполнители, такие как связующие средства, наполнители, смазывающие средства, используемые для получения таблеток, дезинтегрирующие средства и приемлемые увлажняющие средства. Таблетки могут быть покрыты в соответствии со способами, хорошо известными в обычной фармацевтической практике.
Жидкие препараты для перорального применения могут находиться в виде, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров, или они могут находиться в виде сухого продукта для разбавления водой или другой подходящей средой перед применением. Такие жидкие препараты могут содержать традиционные добавки, такие как суспендирующие средства, эмульгирующие средства, неводные среды (которые могут содержать пищевые масла), консерванты и, при желании, традиционные ароматизаторы или красители.
Жидкие стандартные лекарственные формы для парентерального введения получают, используя соединение данного изобретения, или его фармацевтически приемлемую соль, и стерильную среду. В зависимости от используемых среды и концентрации соединение может быть либо суспендированным, либо растворенным в среде. При получении растворов соединение можно растворить с получением раствора для инъекции, который стерилизуют с помощью фильтра, затем наполняют им подходящие флаконы или ампулы и герметично закрывают их. Предпочтительно в среде растворяют вспомогательные средства, такие как местные анестезирующие средства, консерванты и забуферивающие средства. Для увеличения стабильности после заполнения флаконов и удаления воды под вакуумом композицию можно заморозить. Суспензии для парентерального введения получают по существу таким же способом, за исключением того, что соединение не растворяют, а суспендируют в среде, и стерилизацию нельзя проводить путем фильтрации. Соединение можно стерилизовать путем воздействия этиленоксида перед суспендированием в стерильной среде. Чтобы обеспечить однородное распределение соединения, предпочтительно, в состав композиции вводят поверхностно-активное вещество или увлажняющее средство.
Композиция может содержать от 0,1% до 99% по массе, предпочтительно от 10 до 60% по массе, активного вещества, в зависимости от способа введения. Доза соединения, используемого для лечения вышеупомянутых заболеваний, как правило, варьирует в зависимости от тяжести заболевания, массы пациента и других факторов. Однако, в общем случае, подходящие разовые дозы могут составлять от 0,05 до 1000 мг, более предпочтительно - от 1,0 до 200 мг, причем такие разовые дозы можно вводить более одного раза в день, например, два или три раза в день. Такое лечение может продолжаться несколько недель или месяцев.
Нижеследующие описания и примеры иллюстрируют получение соединений данного изобретения.
Описание 1
Трет-бутиловый эфир 7-бензилокси-1,2,4,5-тетрагидробензо[d]азепин-3-карбоновой кислоты (D1)
Трет-бутиловый эфир 7-гидрокси-1,2,4,5-тетрагидробензо[d]азепин-3-карбоновой кислоты (PCT Int. Appl. (2002), WO 02/40471) (790 мг, 3 ммоль), карбонат калия (1,24 г, 9 ммоль) и катализатор иодид калия суспендируют в 2-бутаноне (20 мл). Добавляют бензилбромид (536 мкл, 4,5 ммоль) и смесь нагревают с обратным холодильником 24 часа. Твердые вещества фильтруют и затем промывают ацетоном. Фильтрат концентрируют в вакууме, и неочищенное масло очищают колоночной хроматографией, элюируя смесью этилацетата и гексана (1:4), получая указанное в заголовке соединение (D1) (1,06 г, 100%), 1H ЯМР (CDCl3) 7,44 (5H, м), 7,03 (1H, д, J=8,1 Гц), 6,77 (1H, с), 6,74 (1H, дд, J=8,1 и 2,4 Гц), 3,49 (4H, м), 2,84 (4H, м), 1,48 (9H, с).
Описание 2
7-Бензилокси-1,2,4,5-тетрагидробензо[d]азепин (D2)
Трет-бутиловый эфир 7-бензилокси-1,2,4,5-тетрагидробензо[d]азепин-3-карбоновой кислоты (D1) (1,06 г, 3 ммоль) растворяют в дихлорметане (15 мл) и обрабатывают трифторуксусной кислотой (15 мл). Раствор перемешивают при комнатной температуре 2 часа, концентрируют в вакууме и затем дважды упаривают с дихлорметаном. Остаток растворяют в метаноле, наносят на ионообменную колонку SCX (Varian bond-elute, 10 г) и промывают сначала метанолом, затем смесью 0,880 аммиак/метанол. Объединенные основные фракции упаривают в вакууме и остаток очищают колоночной хроматографией, элюируя смесью 0,880 аммиак:этанол:дихлорметан (1:9:90), получая указанное в заголовке соединение (D2) (702 мг, 93%), МС (ES+) m/e 254 [M+H]+.
Описание 3
1-(6-Хлорпиридин-3-ил)-1-морфолин-4-илметанон (D3)
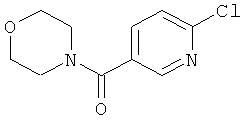
Морфолин (0,2 мл, 2,2 ммоль) добавляют к перемешиваемому раствору 6-хлорникотиноилхлорида (250 мг, 1,4 ммоль) в дихлорметане (10 мл). Через 2 часа реакционную смесь оставляют охлаждаться, неочищенную смесь наносят на ионообменный картридж SCX (Varian bond-elute, 10 г) и промывают метанолом. Метанольные фракции концентрируют в вакууме, получая указанное в заголовке соединение (D3).
Описания 4-31
Соединения описаний 4-31 (D4-D31) получают по способу описания 3 (D3) из соответствующих арилгалогенидов и аминов, указанных в таблице, и используют их без дополнительной характеристики:
Описание 30
1,1-Диметилэтил 7-({5-[(метиламино)карбонил]-2-пиридинил}окси)-1,2,4,5-тетрагидро-3H-3-бензазепин-3-карбоксилат (D30)
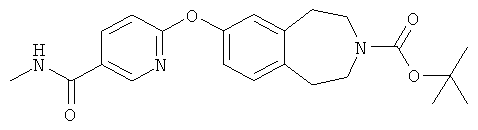
Трет-бутиловый эфир 7-гидрокси-1,2,4,5-тетрагидробензо[d]азепин-3-карбоновой кислоты (PCT Int. Appl. (2002), WO 02/40471) (8,7 г, 33 ммоль) растворяют в трет-бутаноле и обрабатывают трет-бутоксидом калия (4 г, 36 ммоль). После перемешивания в течение 30 минут при комнатной температуре добавляют 6-хлор-N-метилникотинамид (D8) (5,1 г, 30 ммоль) и реакционную смесь перемешивают при температуре кипения с обратным холодильником 20 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. К неочищенному остатку добавляют воду со льдом, что приводит к выпадению осадка, который собирают фильтрацией. Твердый осадок растворяют в этилацетате, промывают насыщенным соляным раствором и сушат (сульфат магния). Органический слой фильтруют, концентрируют в вакууме, полученный остаток очищают колоночной хроматографией, элюируя смесью этилацетат:гексан (1:1), и получают указанное в заголовке соединение (D30). 1H ЯМР (CDCl3) 8,52 (1H, д, J=2,4), 8,12 (1H, дд, J=8,8), 7,16 (1H, м), 6,95-6,81 (3H, м), 6,02 (1H, ушир.), 3,57 (4H, ушир.), 3,02 (3H, д, J=2,4), 2,89 (4H, ушир.), 1,49 (9H, с).
Описание 31
N-Метил-6-(2,3,4,5-тетрагидро-1H-3-бензазепин-7-илокси)-3-пиридинкарбоксамид (D31)
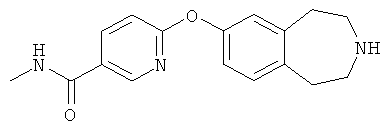
1,1-Диметилэтил 7-({5-[(метиламино)карбонил]-2-пиридинил}окси)-1,2,4,5-тетрагидро-3H-3-бензазепин-3-карбоксилат (D30) (3,98 г, 10 ммоль) растворяют в диоксане (40 мл) и обрабатывают 4M раствором хлористого водорода в диоксане (35 мл). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 6 часов и затем концентрируют в вакууме, получая указанное в заголовке соединение (D31); MS (ES+) m/e 298 [M+H]+.
Пример 1
7-Бензилокси-3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин (Е1)
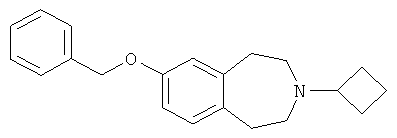
7-Бензилокси-1,2,4,5-тетрагидробензо[d]азепин (D2) (25,3 г, 100 ммоль) растворяют в 2,5% растворе уксусной кислоты в дихлорметане (400 мл) при 0°C и обрабатывают по каплям циклобутаноном (11,2 мл, 150 ммоль). Смесь перемешивают 30 минут и затем порциями добавляют триацетоксиборгидрид натрия (31,8 г, 150 ммоль). Реакционную смесь перемешивают при комнатной температуре 4 часа, подщелачивают насыщенным раствором карбоната натрия и экстрагируют дихлорметаном. Объединенные экстракты промывают водой, насыщенным соляным раствором, сушат над безводным сульфатом натрия и концентрируют в вакууме.
Неочищенный остаток перетирают с гексаном и фильтруют, получая указанный в заголовке продукт (E1).
МС (ES+) m/e 308 [M+H]+.
Пример 2
3-Циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ол (E2)
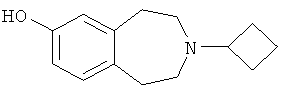
7-Бензилокси-3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин (E1) (9,22 г, 30 ммоль) растворяют в этаноле (150 мл) и тетрагидрофуране (50 мл). Добавляют палладий (1,5 г, 10% на угольной пасте) и реакционную смесь перемешивают при комнатной температуре в атмосфере водорода (1 атмосфера) в течение 5 часов. Реакционную смесь фильтруют через целит, и фильтрат концентрируют в вакууме. Неочищенный остаток перетирают с диэтиловым эфиром и фильтруют, получая указанный в заголовке продукт (E2); МС (ES+) m/e 218 [M+H]+.
Пример 3
6-(3-Циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)-N-метилникотинамид (E3)
Гидрид натрия (60% дисперсия в минеральном масле, 60 мг, 1,5 ммоль) добавляют к перемешиваемому раствору 3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ола (E2) (200 мг, 0,9 ммоль) в диметилсульфоксиде (10 мл). Через 0,5 часа добавляют 6-хлор-N-метилникотинамид (D8) (400 мг, 2,5 ммоль) и реакционную смесь нагревают при 120°C в течение 6 часов. Реакционную смесь оставляют охлаждаться, затем неочищенную смесь наносят на ионообменный картридж SCX (Varian bond-elute, 10 г) и промывают сначала метанолом, затем смесью .880 аммиак:метанол (1:9). Объединенные основные фракции упаривают в вакууме, получая указанное в заголовке соединение (E3). 1H ЯМР (ДМСО-d6) δ 8,56 (1H, дд, J=2,4, 0,4 Гц), 8,48 (1H, ушир.м), 8,20 (1H, дд, J=8,4, 2,4 Гц), 7,16 (1H, д, J=8,0 Гц), 7,03 (1H, дд, J=8,4, 0,4 Гц), 6,91 (1H, д, J=2,4 Гц), 6,86 (1H, дд, J=8,0, 2,4 Гц), 2,87-2,77 (8H, м), 2,36 (4H, м), 2,00 (2H, м), 1,78 (2H, м), 1,58 (2H, м); МС (ES+) m/e 352 [M+H]+.
Пример 3 (Альтернативный способ 1)
6-(3-Циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)-N-метилникотинамид (E3)
Гидрид натрия (0,331 г, 8,28 ммоль, 60% дисперсия в минеральном масле) добавляют к перемешиваемому раствору 3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ола (E3) (1,5 г, 6,9 ммоль) в диметилсульфоксиде (15 мл). Через 0,5 часа добавляют 6-хлор-N-метилникотинамид (D8) (2,34 г, 13,8 ммоль) и реакционную смесь нагревают при 100°C в течение 18 часов. Реакционную смесь оставляют охлаждаться до комнатной температуры и затем распределяют между этилацетатом и водой. Этилацетатный слой отделяют, а водный слой промывают дополнительным объемом этилацетата. Затем объединенные органические слои промывают водой, насыщенным соляным раствором, сушат (Na2SO4) и фильтруют. Смесь концентрируют в вакууме, и полученный остаток очищают колоночной хроматографией, элюируя смесью .880 аммиак:этанол:дихлорметан (0,5:4,5:95, затем 1:9:90), и получают указанное в заголовке соединение (E3), которое затем перекристаллизовывают из толуола. 1H ЯМР (ДМСО-d6) δ 8,56 (1H, дд, J=2,4, 0,4 Гц), 8,48 (1H, ушир.м), 8,20 (1H, дд, J=8,4, 2,4 Гц), 7,16 (1H, д, J=8,0 Гц), 7,03 (1H, дд, J=8,4, 0,4 Гц), 6,91 (1H, д, J=2,4 Гц), 6,86 (1H, дд, J=8,0, 2,4 Гц), 2,87-2,77 (8H, м), 2,36 (4H, м), 2,00 (2H, м), 1,78 (2H, м), 1,58 (2H, м); МС (ES+) m/e 352 [M+H]+.
Пример 3 (Альтернативный способ 2)
6-(3-Циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)-N-метилникотинамид (E3)
Смесь N-метил-6-(2,3,4,5-тетрагидро-1H-3-бензазепин-7-илокси)-3-пиридинкарбоксамида (D31) (1,04 г, 3,5 ммоль) в дихлорметане (12 мл), содержащем уксусную кислоту (240 мкл) при 0°C обрабатывают по каплям циклобутаноном (400 мкл, 5,3 ммоль) и затем перемешивают при комнатной температуре в течение 1 часа. Затем смесь охлаждают до 0°C, порциями добавляют триацетоксиборгидрид натрия (1,11 г, 5,3 ммоль) и перемешивают при комнатной температуре 16 часов. Раствор осторожно подщелачивают 2N раствором гидроксида натрия, перемешивают 30 минут и затем экстрагируют дихлорметаном. Объединенные экстракты промывают насыщенным соляным раствором, сушат над безводным сульфатом натрия и концентрируют в вакууме. Неочищенное вещество очищают колоночной хроматографией, элюируя сначала дихлорметаном, затем смесью .880 аммиак:метанол:дихлорметан (1:9:90), и получают указанное в заголовке соединение (E3); МС (ES+) m/e 352[M+H]+.
Пример 4
Метиловый эфир 5-(3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)пиразин-2-карбоновой кислоты (E4)
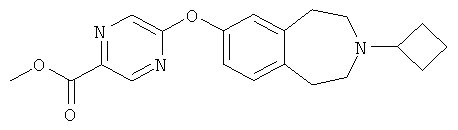
Гидрид натрия (60% дисперсия в минеральном масле, 332 мг, 8,3 ммоль) добавляют к перемешиваемому раствору 3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ола (E2) (1,64 г, 7,5 ммоль) в диметилформамиде (4 мл). Через 0,5 часа добавляют раствор метилового эфира 5-хлорпиразин-2-карбоновой кислоты (1,95 г, 11,3 ммоль) в диметилформамиде (8 мл) и реакционную смесь перемешивают при комнатной температуре 1 час. Реакционную смесь разбавляют дихлорметаном, и органический слой промывают водой, насыщенным соляным раствором, и сушат над сульфатом магния. Органический слой фильтруют, концентрируют в вакууме, и полученный остаток очищают колоночной хроматографией, элюируя смесью .880 аммиак:этанол:дихлорметан (1:9:90), и получают указанное в заголовке соединение (E4). МС (ES+) m/e 354 [M+H]+.
Пример 5a
5-(3-Циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)пиразин-2-карбоновая кислота (E5a)
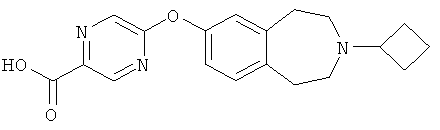
Метиловый эфир 5-(3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)пиразин-2-карбоновой кислоты (E4) (880 мг, 2,5 ммоль) растворяют в смеси этанола (15 мл) и 2N раствора гидроксида натрия (4 мл). Полученную смесь перемешивают при комнатной температуре 0,5 часа и затем концентрируют в вакууме, чтобы удалить органические растворители. Затем реакционную смесь подкисляют до pH 5 (2N раствором хлористоводородной кислоты) и полученные осадки фильтруют, промывают водой и сушат в вакууме, получая указанное в заголовке соединение (E5a); МС (ES+) m/e 340 [M+H]+.
Пример 5
1-[5-(3-Циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)пиразин-2-ил]-1-морфолин-4-илметанон (E5)
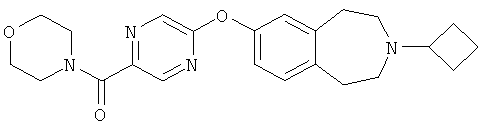
Стадия 1: 5-(3-Циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)пиразин-2-карбонилхлорид
Тионилхлорид (5 мл) медленно добавляют к 5-(3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)пиразин-2-карбоновой кислоте (E5a) (485 мг). Полученную реакционную смесь перемешивают при комнатной температуре 1 час и затем нагревают с обратным холодильником еще 1 час. Реакционную смесь охлаждают, разбавляют толуолом и концентрируют в вакууме, получая указанное в заголовке соединение, которое используют без дополнительной характеристики.
Стадия 2: 1-[5-(3-Циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-илокси)пиразин-2-ил]-1-морфолин-4-илметанон
Морфолин (0,17 мл, 2,0 ммоль) добавляют к перемешиваемому раствору продукта стадии 1 (394 мг, 1 ммоль) и диэтиламинометилполистирола (1,88 г, 3,2 ммоль/г, 6 ммоль) в дихлорметане (10 мл). Полученную смесь оставляют перемешиваться при комнатной температуре в течение 24 часов и фильтруют. Фильтрат концентрируют в вакууме, полученный неочищенный остаток очищают колоночной хроматографией, элюируя смесью .880 аммиак:этанол:дихлорметан (1:9:90), и получают указанное в заголовке соединение (E5). МС (ES+) m/e 409 [M+H]+.
Пример 6a
Метиловый эфир 6-[(3-Циклобутил-2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)окси]-3-пиридинкарбоновой кислоты
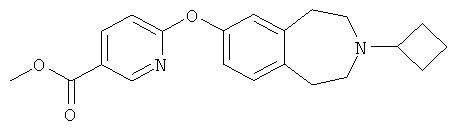
Указанное в заголовке соединение получают из 3-циклобутил-2,3,4,5-тетрагидро-1H-бензо[d]азепин-7-ола (E2) и метил 6-хлор-3-пиридинкарбоксилата по способу, описанному для соединения E4; МС (ES+) m/e 353 [M+H]+.
Пример 6b
6-[(3-Циклобутил-2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)окси]-3-пиридинкарбоновая кислота (E6b)
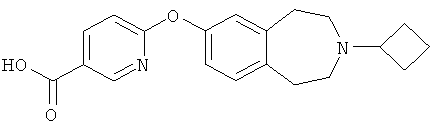
Указанное в заголовке соединение получают из соединения примера 6a (E6a) по способу примера 5a (E5a); МС (ES+) m/e 339 [M+H]+.
Пример 6
6-[(3-Циклобутил-2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)окси]-N-циклопропил-3-пиридинкарбоксамид (E6)
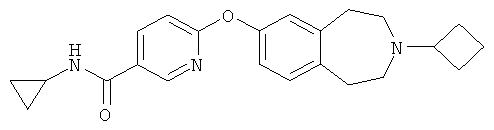
Карбонилдиимидазол (142 мг, 0,88 ммоль) добавляют к перемешиваемому раствору 6-[(3-циклобутил-2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)окси]-3-пиридинкарбоновой кислоты (E6b) (150 мг, 0,44 ммоль) в дихлорметане (5 мл). После перемешивания при комнатной температуре в течение 3 часов добавляют циклопропиламин (0,15 мл, 2,2 ммоль) и смесь оставляют перемешиваться еще 18 часов. Реакционную смесь наносят на ионообменный картридж SCX (Varian bond-elute, 10 г) и промывают сначала метанолом, затем смесью .880 аммиак:метанол (1:9). Объединенные основные фракции концентрируют в вакууме, получая указанное в заголовке соединение (E6). МС (ES+) m/e 378 [M+H]+.
Биологические данные
Мембранный препарат, содержащий рецепторы гистамина Н3, можно получить следующим способом:
(i) Получение клеточной линии, содержащей гистамин H3
ДНК, содержащую ген человеческого гистамина H3 (Huvar, A. et al. (1999) Mol. Pharmacol. 55(6), 1101-1107), клонируют в несущем векторе pCDNA3.1 TOPO (InVitrogen), кДНК выделяют из данного вектора путем рестрикционного расщепления плазмидной ДНК ферментами BamH1 и Not-1 и лигируют в индуцируемый вектор экспрессии pGene (InVitrogen), расщепляемый такими же ферментами. С системой GeneSwitchTM (система, где экспрессия трансгена прекращается в отсутствие индуцирующего фактора и запускается в присутствии индуцирующего фактора) работают по методу, описанному в патентах №№: 5364791; 5874534; и 5935934. Лигированную ДНК переносят в компетентные бактериальные клетки хозяина DH5α E. coli и помещают их на агар с бульоном Луриа (Luria Broth (LB)), содержащим ZeocinTM (антибиотик, который позволяет осуществить селекцию клеток, экспрессирующих ген sh ble, который присутствует в pGene и pSwitch) в концентрации 50 мкг/мл. Колонии, содержащие заново лигированную плазмиду, идентифицируют с помощью рестрикционного анализа. ДНК для трансфекции в клетки млекопитающих получают из 250 мл культуры бактериального хозяина, содержащего плазмиду pGeneH3, и выделяют с помощью набора для получения ДНК (Qiagen Midi-Prep), следуя инструкциям производителя (Qiagen). Клетки CHO K1, заранее трансфицированные регуляторной плазмидой pSwitch (InVitrogen), за 24 часа до использования высевают в концентрации 2×10e6 клеток на колбу T75 в полной среде, содержащей среду HamsF12 (GIBCOBRL, Life Technologies), дополненную 10% об./об. диализированной фетальной бычьей сывороткой, L-глутамином и гидромицином (100 г/мл). Плазмидную ДНК трансфицируют в клетки, используя липофектамин, в соответствии с инструкциями производителя (InVitrogen). Через 48 часов после трансфекции клетки помещают в полную среду, дополненную 500 мкг/мл ZeocinTM.
Чтобы индуцировать экспрессию рецептора, через 10-14 дней после селекции к культуральной среде добавляют 10 нМ мифепристона (InVitrogen). Через 18 часов после индуцирования клетки отсоединяют от колбы, используя этилендиаминтетрауксусную кислоту (EDTA; 1:5000; InVitrogen), затем их несколько раз промывают забуференным фосфатом физиологическим раствором pH 7,4 и повторно суспендируют в среде для сортинга, содержащей минимальную поддерживающую среду (MEM) без фенолового красного, дополненную солями Эрла и 3% Foetal Clone II (Hyclone). Приблизительно 1×10e7 клеток анализируют на экспрессию рецептора путем окрашивания кроличьими поликлональными антителами, 4a, против N-концевого домена рецептора гистамина H3, затем их инкубируют на льду в течение 60 минут и дважды промывают средой для сортинга. Антитела, связанные с рецептором, детектируют путем инкубации клеток в течение 60 минут на льду с козлиными антителами против кроличьих антител, конъюгированными с флуоресцентным маркером Alexa 488 (Molecular Probes). После двух дополнительных промываний средой для сортинга клетки фильтруют через 50 мкм FilconTM (BD Biosciences) и затем анализируют на проточном цитометре FACS Vantage SE, оборудованном автоматической приставкой для приготовления клеточных препаратов. В качестве контроля используют неиндуцированные клетки, обработанные подобным образом. Окрашенные клетки помещают в виде отдельных клеток в 96-луночные планшеты, в которые добавлена полная среда, содержащая 500 мкг/мл ZeocinTM, и оставляют расти перед повторным анализом экспрессии рецептора по связыванию антител и лигандов. Клон 3H3 отбирают для получения мембран.
(ii) Получение мембран из культивированных клеток
Все стадии метода проводят при 4°C с использованием предварительно охлажденных реагентов. Клеточный осадок ресуспендируют в 10 объемах буфера A2, содержащего 50 мМ N-2-гидроксиэтилпиперазин-N'-2-этансульфоновую кислоту (HEPES) (pH 7,40), дополненного 10-4 M лейпептина (ацетил-лейцил-лейцил-аргинал; Sigma L2884), 25 мкг/мл бацитрацина (Sigma B0125), 1 мМ этилендиаминтетрауксусной кислоты (EDTA), 1 мМ фенилметилсульфонилфторида (PMSF) и 2×10-6 M пепстатина A (Sigma). Затем клетки гомогенизируют импульсами 2×15 секунд в 1-литровом стеклянном смесителе Waring и центрифугируют 20 минут при 500g. После чего супернатант центрифугируют 30 минут при 48000g. Осадок снова суспендируют в 4 объемах буфера A2 путем интенсивного перемешивания в течение 5 секунд, затем гомогенизируют в гомогенизаторе Dounce (10-15 ударов). В этот момент берут аликвоты препарата, помещают их в полипропиленовые пробирки и хранят при -70°C.
Биологическую активность соединений данного изобретения можно анализировать in vitro следующими методами:
(I) Анализ связывания гистамина H3
Для каждого анализируемого соединения в 96-луночный планшет, лунки которого имеют прозрачное дно и белые стенки, добавляют:
(a) 10 мкл анализируемого соединения (или 10 мкл иодофенпропита (известного антагониста гистамина H3) с конечной концентрацией 10 мМ), разбавленного до нужной концентрации 10% ДМСО;
(b) 10 мкл 125I 4-[3-(4-иодфенилметокси)пропил]-1H-имидазолия (иодпроксифан) (Amersham; 1,85 мБк/мкл или 50 мкКи/мл; удельная активность ~2000 Ки/ммоль) разбавляют до 200 пМ буфером для анализа (50 мМ трис(гидроксиметил)аминометановый буфер (TRIS) pH 7,4, 0,5 мМ этилендиаминтетрауксусная кислота (EDTA)), получая конечную концентрацию 20 пМ;
(c) 80 мкл смеси шарики/мембраны, полученной следующим образом: шарики для сцинтилляционного проксимального анализа (SPA) типа WGA-PVT суспендируют в буфере для анализа в концентрации 100 мг/мл, затем смешивают с мембранами (полученными по описанному выше способу) и разбавляют в буфере для анализа, получая конечный объем 80 мкл, который содержит 7,5 мкг белка и 0,25 мг шариков на лунку - смесь предварительно смешивают при комнатной температуре в течение 60 минут на роллере. Планшет встряхивают 5 минут и затем оставляют стоять при комнатной температуре 3-4 часа перед прочтением на счетчике Wallac Microbeta с помощью программы, нормализованной в течение 1 минуты для счета по тритию. Данные анализируют, используя 4-параметрическое логистическое уравнение.
(II) Функциональный антагонистический анализ гистамина H3
Для каждого анализируемого соединения в 96-луночный планшет, лунки которого имеют прозрачное дно и белые стенки, добавляют:
(a) 10 мкл анализируемого соединения (или 10 мкл гуанозин-5'-трифосфата (ГТФ) (Sigma) в качестве контроля неспецифического связывания), разбавленного до нужной концентрации буфером для анализа (20 мМ N-2-гидроксиэтилпиперазин-N'-2-этансульфоновая кислота (HEPES) + 100 мМ NaCl +10 мМ MgCl2, pH 7,4 NaOH);
(b) 60 мкл смеси шарики/мембраны/ГДФ, полученной следующим образом: шарики для сцинтилляционного проксимального анализа (SPA) агглютинин зародышей пшеницы-поливинилтолуол (WGA-PVT) суспендируют в буфере для анализа в концентрации 100 мг/мл, затем смешивают с мембранами (полученными по описанному выше способу) и разбавляют буфером для анализа, получая конечный объем 60 мкл, который содержит 10 мкг белка и 0,5 мг шариков на лунку - смесь предварительно смешивают при 4°С в течение 30 минут на роллере и непосредственно перед внесением в планшет добавляют гуанозин 5'-дифосфат (ГДФ) (Sigma; разбавленный аналитическим буфером) с получением конечной концентрации 10 мкМ. Чтобы уравновесить антагонист и рецептор/шарики, планшет инкубируют при комнатной температуре и при встряхивании в течение 30 минут, после чего добавляют:
(c) 10 мкл гистамина (Tocris) с конечной концентрацией 0,3 мкМ;
(d) 20 мкл соли триэтиламина и гуанозин 5'[y35-S]тиотрифосфата (Amersham; концентрация радиоактивности = 37 кВк/мкл или 1 мКи/мл; удельная активность 1160 Ки/ммоль), разбавленного буфером для анализа до концентрации 1,9 нМ, с получением конечной концентрации 0,38 нМ.
Затем планшет инкубируют на шейкере при комнатной температуре в течение 30 минут, после чего центрифугируют 5 минут при 1500 об/мин. Через 3-6 часов после завершения центрифугирования планшет прочитывают на счетчике Wallac Microbeta с помощью программы, нормализованной в течение 1 минуты для счета по тритию. Данные анализируют, используя 4-параметрическое логистическое уравнение. Базальную активность, когда в лунки не добавляют гистамин, используют как минимальную.
Результаты
Соединения примеров E1-3, E5-149, E151-230, E233-235, E237-256, E258, E260-270, E273 и E275-288 тестируют с помощью функционального антагонистического анализа гистамина H3 и обнаруживают, что они проявляют антагонизм в следующем интервале: 6,5-10,5 pKb. Более конкретно, соединения примеров 1, 52, 121, 125 и 217 проявляют антагонизм в следующем интервале: 9,0-10,5 pKb. Еще более конкретно, соединение примера 121 проявляет антагонизм >9,5 pKb.
| название | год | авторы | номер документа |
|---|---|---|---|
| (3-ЦИКЛОАЛКИЛ-2,3,4,5-ТЕТРАГИДРО-1Н-БЕНЗО[d]АЗЕПИН-7-ИЛОКСИ)ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ Н3 РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ | 2003 |
|
RU2388752C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ АЗЕПИНА И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ АНТИДИУРЕТИЧЕСКИХ АГЕНТОВ | 2001 |
|
RU2269517C2 |
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНА, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ ВАЗОПРЕССИНА | 2008 |
|
RU2471784C2 |
| ПРОИЗВОДНЫЕ 1,2,4,5-ТЕТРАГИДРОБЕНЗО[D]АЗЕПИНОВ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2000 |
|
RU2240317C2 |
| КОНДЕНСИРОВАННЫЕ АЗЕПИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ, СПОСОБ ЛЕЧЕНИЯ С ИСПОЛЬЗОВАНИЕМ УКАЗАННОЙ КОМПОЗИЦИИ | 2001 |
|
RU2259367C2 |
| СОЕДИНЕНИЯ | 2003 |
|
RU2327690C2 |
| БИЦИКЛИЧЕСКИЕ АГОНИСТЫ ВАЗОПРЕССИНА | 2000 |
|
RU2254332C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ИНГИБИТОРЫ НЭП (НЕЙТРАЛЬНОЙ ЭНДОПЕПТИДАЗЫ), ИНГИБИТОРЫ ЭНДОГЕННОЙ ПРОДУЦИРУЮЩЕЙ ЭНДОТЕЛИН СИСТЕМЫ И ИНГИБИТОРЫ ГМГ (ГИДРОКСИМЕТИЛГЛУТАРИЛ)СоА РЕДУКТАЗЫ | 2005 |
|
RU2410118C2 |
| ТИОФЕНИЛЬНЫЕ И ПИРРОЛИЛЬНЫЕ АЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ СЕРОТОНИНОВОГО 5-HT РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2434872C2 |
| МОДУЛЯТОРЫ 5HT РЕЦЕПТОРА | 2003 |
|
RU2317982C2 |
Настоящее изобретение относится к соединению, которое представляет собой 6-(3-Циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамид
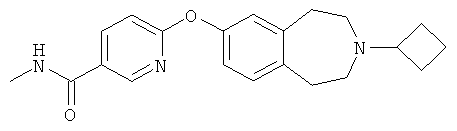
или к его фармацевтически приемлемой соли. Это соединение и его фармацевтически приемлемые соли обладают сродством к рецептору гистамина НЗ и являются антагонистами и/или обратными агонистами указанного рецептора. Настоящее изобретение также относится к способам получения производного бензазепина, к фармацевтическим композициям, содержащим его, и к применению производного бензазепина для лечения неврологических и психиатрических нарушений. 6 н. и 12 з.п. ф-лы, 1 табл.
1. Соединение общей формулы (I), которое представляет собой 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамид
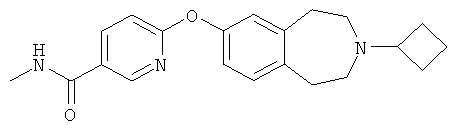
или его фармацевтически приемлемая соль.
2. Фармацевтически приемлемая соль соединения по п.1, которая представляет собой кислотно-аддитивную соль, образованную 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамидом и кислотой.
3. Фармацевтически приемлемая соль соединения по п.2, где кислота представляет собой малеиновую, хлористоводородную, бромистоводородную, фосфорную, уксусную, фумаровую, салициловую, серную, лимонную, молочную, миндальную, виннокаменную и метансульфоновую кислоту.
4. Фармацевтическая композиция, являющаяся антагонистом рецептора гистамина Н3, включающая 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамид
или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксцепиент.
5. Соединение по п.1, используемое в терапии.
6. Применение 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамид или его фармацевтически приемлемой соли в получении лекарственного средства для лечения нейрологического заболевания или психиатрического расстройства.
7. Применение по п.6, где лекарственное средство предназначено для лечения: нейрологического заболевания, такого как болезнь Альцгеймера, деменцию, старческое расстройство памяти, легкое ухудшение познавательной способности, нарушение познавательной способности, эпилепсию, невропатическую боль, болезнь Паркинсона, рассеянный склероз или удар; или психиатрических расстройств, таких как нарушение познавательной способности при шизофрении, синдром дефицита внимания с гиперактивностью, депрессию или привыкание.
8. Применение по п.6, где лекарственное средство используют для лечения ухудшения познавательной способности, вызванной заболеванием.
9. Применение по п.8, где лекарственное средство используют для лечения ухудшения познавательской способности, вызванной болезнью Альцгеймера или родственного нейродегенеративного нарушения.
10. 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамид или его фармацевтически приемлемая соль в качестве терапевтического вещества используемого при лечении нейрологического заболевания или психиатрического расстройства.
11. 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамид или его фармацевтически приемлемая соль по п.10 в качестве терапевтического вещества, используемого при лечении: нейрологического заболевания, такого как болезнь Альцгеймера, деменцию, старческое расстройство памяти, легкое ухудшение познавательной способности, нарушение познавательной способности, эпилепсию, невропатическую боль, болезнь Паркинсона, рассеянный склероз или удар; или психиатрических заболеваний, таких как нарушение познавательной способности при шизофрении, синдром дефицита внимания с гиперактивностью, депрессию и привыкание.
12. 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамид или его фармацевтически приемлемая соль по п.10 в качестве терапевтического вещества, используемого для лечения ухудшения нарушения познавательной способности, вызванной заболеванием.
13. 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамид по п.12 или его фармацевтически приемлемая соль в качестве терапевтического вещества, используемого для лечения ухудшения познавательной способности, вызванной болезнью Альцгеймера или родственным нейродегенеративным нарушением.
14. 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метилникотинамид по п.13 или его фармацевтически приемлемая соль в качестве терапевтического вещества, используемого для лечения ухудшения познавательной способности, вызванной болезнью Альцгеймера.
15. Фармацевтическая композиция, предназначенная для лечения неврологического заболевания, включающая 6-(3-циклобутил-2,3,4,5-тетрагидро-1Н-бензо[d]азепин-7-илокси)-N-метил-никотинамид или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксцепиент.
16. Фармацевтическая композиция по п.15, где неврологическим заболеванием является болезнь Альцгеймера, деменция, старческое расстройство памяти, легкое ухудшение познавательной способности, нарушение познавательной способности, эпилепсия, невропатическая боль, болезнь Паркинсона, рассеянный склероз или удар.
17. Способ получения соединения формулы (I) по п.1 или его фармацевтически приемлемой соли
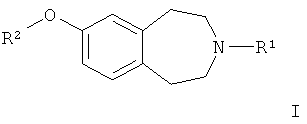
где R1 представляет собой незамещенный циклобутил;
R2 представляет собой 2-пиридинил, замещенный CON(H)(Me), и представляет собой 4-метиламинокарбонилпиридин-2-ил, который включает
а) взаимодействие соединения формулы (II)
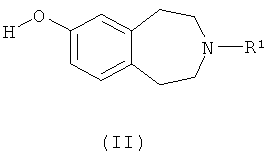
где R1 определен выше,
с соединением формулы R2'-L1, где R2' такой, как определен выше для R2 или группа, превращаемая в него, a L1 обозначает подходящую уходящую группу, или
в) взаимодействие соединения формулы (III)
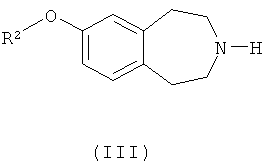
где R2 такой, как определено выше,
с соединением формулы R1'-L2, где R1' такой, как определено выше для R1 или группа, превращаемая в него, а L2 обозначает подходящую уходящую группу, или
c) взаимодействие соединения формулы (III), как определено выше, с кетоном формулы R1'=O, где R1' такой, как определен для R1, или группа, превращаемая в него, или
d) удаление защитной группы с защищенного соединения формулы (I).
18. Способ по п.17, где
на стадии а) R2' определен выше для R2 и L1 обозначает подходящую уходящую группу, такую как атом галогена или активированная гидроксильная группа, и
на стадии в) R1' определен выше для R1 и L2 обозначает подходящую уходящую группу, такую как атом галогена.
| US 2003158177 A1, 21.08.2001 | |||
| Устройство для предупреждения столкновений поездов | 1925 |
|
SU6254A1 |
| 2,3,4,5-ТЕТРАГИДРО-1Н-3-БЕНЗАЗЕПИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ | 1993 |
|
RU2114839C1 |
| Pillot С et al, The Journal of Neuroscience, 2002, 22(16), 7272-7280 | |||
| Ligneau X et al, The Journal of Pharmacology and Experimental Therapeutics, 1998, 287(2), 658-666. | |||

