Область техники, к которой относится изобретение
Настоящее изобретение относится к новому гетеросоединению и лекарственному средству, содержащему его в качестве активного ингредиента, а именно как средство для лечения иммунологических заболеваний.
Уровень техники изобретения
Сфингозин-1-фосфат представляет собой метаболит сфинголипида, который является физиологически активным веществом, выделяющимся из активированного тромбоцита (Непатентный документ 1). Рецептор сфингозин-1-фосфата - это G протеинсвязывающего типа и принадлежит к Edg-семейству, который представляет собой ген эндотелиальной дифференциации. В настоящее время было найдено пять рецепторов S1P1(Edg1), S1P2(Edg5), S1P3(Edg3), S1P4(Edg6) и S1P5(Edg8). Все эти рецепторы широко представлены в клетках и тканях во всем теле, однако S1P1, S1P3 и S1P4 главным образом выражены в лимфоците и эндотелиальной клетке, S1P2 главным образом выражены в васкулярной гладкомышечной клетке и S1P5 главным образом выражены в мозге и селезенке, и их аминокислотные последовательности хорошо сохраняются у человека и грызунов (Непатентный документ 1). Многие рецепторы связаны с G протеинами путем стимуляции сфингозин-1-фосфата. S1P1 связан с Gi/0, S1P2 и S1P3 связан с Gi/0, Gq, G12/13 и Gs, S1P4 связан с Gi/0, G12/13 и Gs, S1P5 - это комбинация Gi/0 и G12/13 и вызывают рост клеток? обусловленный активацией МАПК, изменение цитоскелетной системы и инфильтрацию клеток, обусловленую активацией Rac (и/или Rho), и генерацию цитокина и медиатора, обусловленую активацией PLC и притоком кальция в клетку и т.п. (Непатентный документ 1). Было известно, что при стимулировании действия S1P1 сфингозин-1-фосфатом вызывается миграция лимфоцитов, ингибирование апоптоза, генерация цитокина, секвестирование лимфоцитов в тимусе и других вторичных лимфоидных тканях и ангиопластика в васкулярных эндотелиальных клетках (Непатентный документ 2). С другой стороны, S1P3 также найдены в кардиомиоците и обнаружено случайное снижение частоты сердцебиения (брадисфигмия) и кровяного давления при стимуляции сфингозин-1-фосфата (Непатентный документ 3), в то время как брадисфигмия не обнаружена при стимуляции сфингозин-1-фосфата у нокаутной мыши, у которой S1P3 генетически удален (Непатентный документ 4). Сообщалось что фосфатный эфир FTY720, который представляет собой активное вещество FTY720, в настоящее время проходит клинические испытания имеет неселективную агонистическую активность по отношению S1P1, S1P3, S1P4 и S1P5 (Непатентный документ 5), и, в особенности, брадисфигмия, вызванная эффектом стимуляции S1P3, зачастую выражается как нежелательный побочный эффект при клинических испытаниях (Непатентный документ 6). Таким образом, считается, что для секвестирования лимфоцита посредством рецептора сфингозин-1-фосфата необходима стимуляция S1P1 (Непатентный документ 7), в то время как стимуляция S1P3 не нужна, и она скорее рассматривается как причина нежелательного побочного эффекта. Следовательно, для разработки имуносупрессирующего средства с меньшими побочными эффектами желательно разработать агонист, обладающий слабым действием на S1P3 и селективно воздействующий на S1P1.
Например, в качестве соединения, обладающего активностью как агонист S1P1, было известно производное карбоновой кислоты, изображаемое следующей формулой (Патентный документ 1):
[Хим.1]
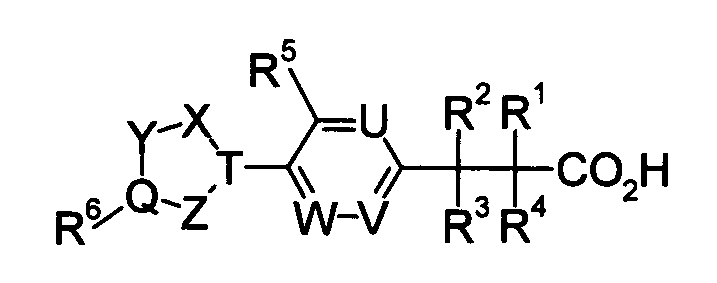
[по поводу символа в формуле обращайтесь к публикации]
В качестве соединения, обладающего активностью как агонист S1P1, известно производное индана, изображенное следующей формулой (Патентный документ 2):
[Хим.2]
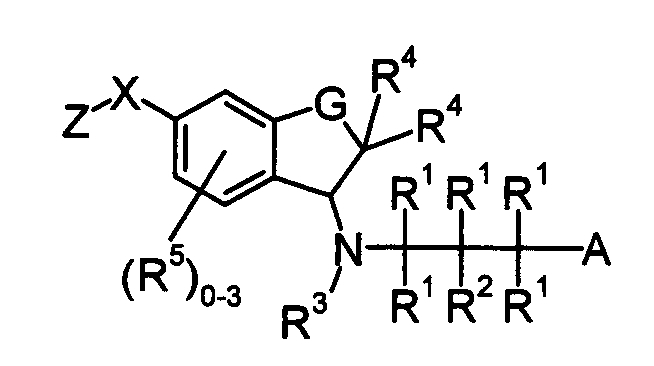
[по поводу символа в формуле обращайтесь к публикации]
В качестве соединения, обладающего активностью как агонист S1P1, известно производное оксадиазола, изображаемое следующей формулой (последующие фигуры, Патентные документы 3, 4, 5, и 6):
[Хим.3]
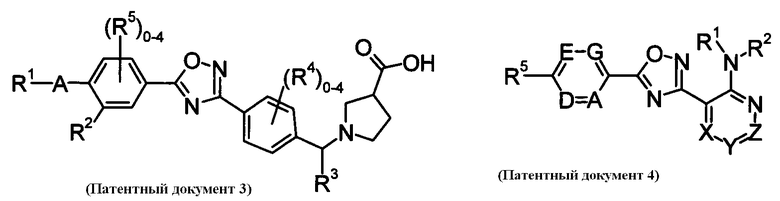
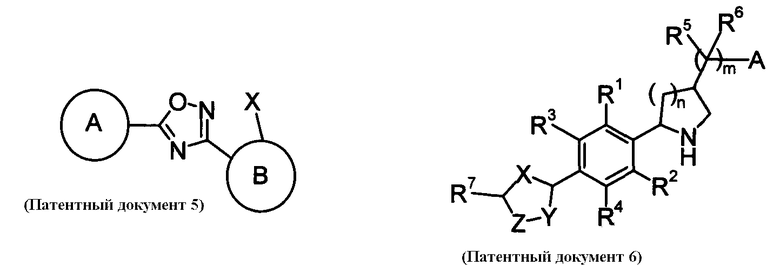
[по поводу символа в формуле обращайтесь к публикации]
В качестве соединения, обладающего активностью как агонист S1P1, известно производное, изображенное следующей формулой (следующая фигура, Патентный документ 7):
[Хим.4]
[по поводу символа в формуле обращайтесь к публикации]
Однако соединение по настоящему изобретению не было описано ни в одном документе.
Непатентный документ 1: Annulan Review Biochemistry, 204, 73, 321-354
Непатентный документ 2: Nature Review Immunology, 2005, 5, 560-570
Непатентный документ 3: Japanese Journal of Pharmacology, 2000, 82, 328-342
Непатентный документ 4: Journal of Pharmacology и Experimental Therapeutics, 2004, 309, 758-768
Непатентный документ 5: Science, 2002, 296, 346-349
Непатентный документ 6: Journal of American Society of Nephrology, 2002, 13, 1073-1083
Непатентный документ 7: Nature, 2004, 427, 355-360
Патентный документ 1: Международная заявка на патент WO 2005/058848 брошюра
Патентный документ 2: Международная заявка на патент WO 2004/058149 брошюра
Патентный документ 3: Международная заявка на патент WO 2003/105771 брошюра
Патентный документ 4: Международная заявка на патент WO 2004/103279 брошюра
Патентный документ 5: Международная заявка на патент WO 2005/032465 брошюра
Патентный документ 6: Международная заявка на патент WO 2006/047195 брошюра
Патентный документ 7: Международная заявка на патент WO 2006/001463 брошюра
Описание изобретения
Проблема, которую изобретение призвано решить
Авторы настоящего изобретения провели исследование с целью обнаружения вещества, применимого для профилактики и/или лечения отторжения при трансплантации органа/костного мозга/ткани или аутоиммунных заболеваний, основываясь на агонистической активности по отношению к S1P1, и, в дальнейшем, предоставить лекарственный препарат, содержащий это вещество.
Средства решения задачи
Разработчики данного изобретения провели широкие исследования по отношению к веществу, имеющему S1P1 агностическую активность, и в результате они обнаружили, что новое гетеросоединение может быть использовано в качестве агониста S1P1, завершив, таким образом, настоящее изобретение. Иными словами, исходя из данного изобретения, можно предусмотреть новое гетеросоединение, представленное нижеследующей формулой (I), или его фармацевтически приемлемую соль.
Соединение, представленное формулой (I):
[Хим. 5]
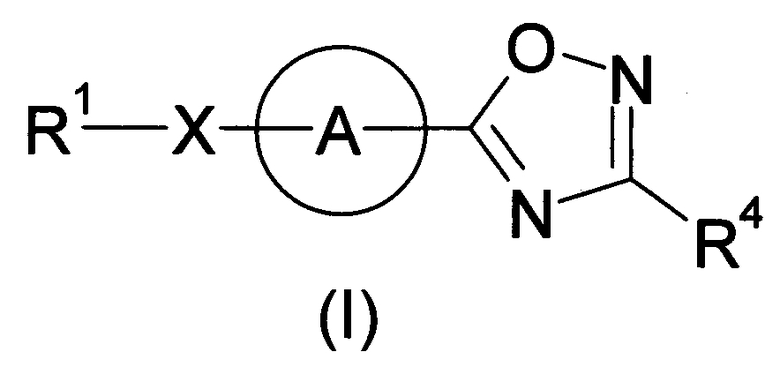
или его фармацевтически приемлемая соль
[В формуле символы означают, как следует ниже.]
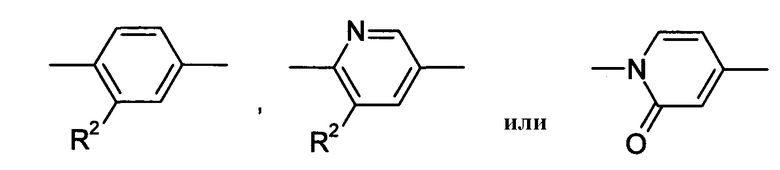
Кольцо представляет собой:
[Хим. 6]
Х представляет собой простую связь, -СН2-, -NR3-, -O-, -S-, -S(=O)- или -S(=O2)-,
R1 представляет собой -Н, галоген, арил, гетероарил, (С3-С8)циклоалкил, С3-С8)циклоалкенил, (С3-С8)гетероциклоалкил; или (С1-С6)алкил или (С2-С6)алкенил, каждый из которых может содержать галоген, -CONH2, арил, или (С3-С8)циклоалкил, как заместитель,
R2 представляет собой -CN, -O-(C1-C6)алкил, -С(=О)Н, галоген или (С1-С6)алкил, который может быть замещен с помощью галогена или -ОН,
R3 представляет собой -Н, где R3 может образовывать морфолино, 1-пирролидинил или 3,4-дегидропиперидин-1-ил вместе с R1 и азотом,
где когда -Х- представляет собой простую связь, R1 и R2 могут в комбинации образовывать 5-членное кольцо и, кроме того, содержать (С1-С6)алкил, как заместитель.
R4 представляет собой следующее ниже кольцо:
где любая одна из связей от кольца связана с оксадиазольным кольцом,
[Хим.7]
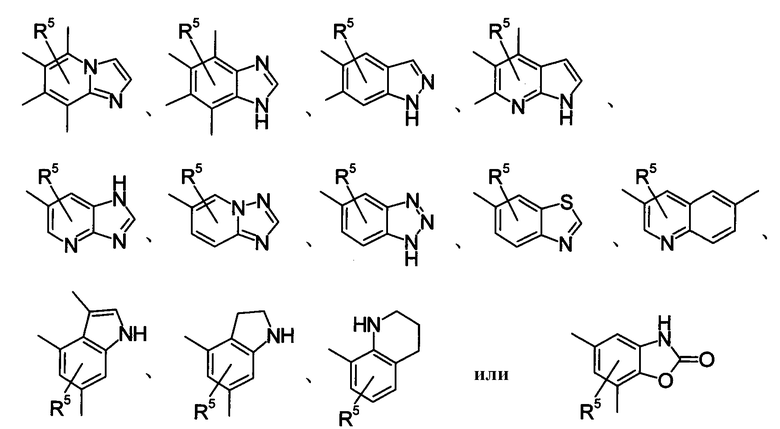
R5 представляет собой -Н; (С1-С6)алкил, который может быть замещен не менее одной группой, выбранной из ряда, содержащего: -CN, -C(=O)NRXRY, -NHRX, -SRX, -S(=O)2RX и -ORX (данный определен как R0-(C1-C6)алкил); R0-(C1-C6)алкил-О-; R0-(C1-C6)алкил-C(=O)-; R0-(C1-C6)алкил-S(=О)2-, R0-O-(C1-C6)алкил; R0-C(=O)(C1-C6)алкил-; R0-S(=O)2-(C1-C6)алкил-; (C2-C6)алкенил-; -С(=О)Н; -ORX; -S(=O)2RX; галоген; =О; NRXRY; -C(=O)NRXRY;
RX и RY могут быть одинаковыми или различными друг с другом и представлять собой -Н; или (С1-С6)алкил, который может быть замещен -ОН, NH2, которые могут быть защищены защитной группой, или гетероарил, в котором RX могут образовывать (С3-С8)гетероциклоалкил вместе с RY и азотом.
Что касается -Х-, он в формуле (I) предпочтительно представляет собой простую связь или -О- и, более предпочтительно, представляет собой -О-. Что касается R1, он предпочтительно представляет собой (С1-С4)алкил или (С2-С4)алкенил, каждый из которых может быть замещен галогеном или (С3-С8)циклоалкилом и, кроме того, более предпочтительно представляет собой (С1-С4)алкил, который может быть замещен с помощью F. Что касается кольца А, оно предпочтительно представляет собой:
[Хим.8]
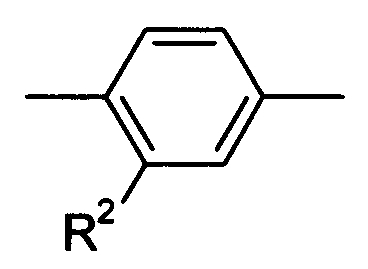
Что касается R2, он предпочтительно представляет собой галоген, -CN, (C1-C4)алкил, который может быть замещен галогеном, и, более предпочтительно, он представляет собой Cl, CF3. Что касается R4, он предпочтительно представляет собой:
[Хим.9]
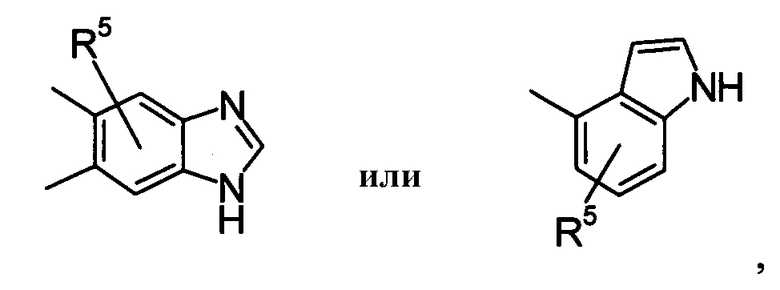
и более предпочтительно он представляет собой:
[Хим.10]
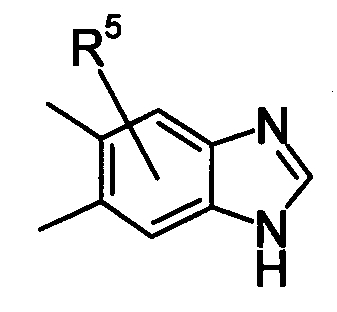
Что касается R5, он предпочтительно представляет собой -Н, (С1-С6)алкил, который может быть замещен с помощью -С(=О)NRXRY. Что касается RX, он предпочтительно представляет собой -Н, (С1-С6)алкил, который может быть замещен с помощью -ОН. Что касается RY, он предпочтительно представляет собой -Н; (С1-С6)алкил, который может быть замещен с помощью -ОН.
Соединение по настоящему изобретению, изображенное формулой (I), охарактеризовано химической структурой с точки зрения того, что бициклический азотсодержащий ненасыщенный гетероцикл или бициклический азотсодержащий частично ненасыщенный гетроцикл связан с 3-позицией оксазола и обладает фармакологическими характеристиками с точки зрения того, что соединение обладает S1P1 агонистической активностью.
Эффекты изобретения
Поскольку соединение по изобретению обладает S1P1 агонистической активностью, оно может быть использовано в качестве активного ингредиента средства для лечения или в качестве средства для профилактики заболеваний, вызванных неблагоприятной лимфоцитарной инфильтрацией, например отторжения трансплантата при пересадке органа, костного мозга или ткани, или реакции "трансплантат против хозяина", аутоиммунных заболеваний или воспалительных заболеваний, таких как ревматический артрит, рассеянный склероз, системная красная волчанка, нефротический синдром, энцефаломенингит, бульбоспинальный паралич, панкреатит, гепатит, нефрит, диабет, легочные расстройства, астма, атопический дерматит, воспалительные кишечные заболевания, атеросклероз или ишемическо-реперфузионное повреждение, и, кроме того, заболеваний, вызванных аномальным ростом или аккумуляцией клеток, таких как рак или лейкемия.
Лучший вариант осуществления изобретения
В дальнейшем в этом документе настоящее изобретение будет описано более детально.
В описании «алкил» означает линейную или разветвленную одновалентную группу. «С1-С6алкил» означает С1-С6 линейную или разветвленную группу, и ее специфические примеры включают: метил, этил, н-пропил, изопропил, н-бутил, изобутил, т-бутил, н-пропил и н-гексил, предпочтительны С1-С4алкил и особенно предпочтительны метил, этил, н-пропил и изопропил.
В описании «галоген» означает F, Cl, Br и I и предпочтительные примеры включают F и Сl.
В описании «С2-С6алкенил» означает С2-С6 линейную или разветвленную группу, которая имеет двойную связь в данном месте, и их специфические примеры включают: этинил (винил), 1-пропенил, 2-пропенил, 1-метилэтин-1-ил, 1-бутен-1-ил, 2-бутен-1-ил, 3-бутен-1-ил, 1-метил-1-пропен-1-ил, 2-метил-1-пропен-1-ил, 1-метил-2-пропен-1-ил и 2-метил-2-пропен-1-ил и предпочтительно, 1-метил-2-пропен-1-ил или 1-пентинил.
В описании «С3-С8циклоалкил» означает одновалентную группу неароматического углеродного цикла, имеющего степень восстановления 3-8, который частично может иметь ненасыщенные связи. Таким образом, специфические примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
В описании «С3-С8 гетероциклоалкил» означает одновалентную неароматическую группу имеющую степень восстановления от 4 до 9, содержащую один или более гетероатомов, одинаковых или различных друг с другом, выбранные из ряда, содержащего азот, кислород и, по желанию, окисленную серу, которая может быть частично ненасыщенной. Их специфические примеры включают азиридинил, азетидинил, пирролидинил, пиперидинил, гомопиперидинил, морфолил, тиоморфолил, тетрагидропиранил и тетрагидротиопиранил.
В описании «арил» означает ароматическую углеводородную группу, но предпочтительными являются арильные группы, имеющие 6-14 атомов углерода. Их специфические примеры включают фенил, нафтил и антранил, и более предпочтительным является фенил.
В описании «гетероарил» означает 5- или 6-членный цикл ароматического гетероцикла, содержащий один или более гетероатомов, которые различны или отличаются друг от друга, выбранные из группы, содержащей азот, кислород и серу. Их специфические примеры включают пиридил, пиразил, пиримидинил, пирадизинил, пирролил, пиразолил, имидазолил, оксазолил, тиазолил, тиенил, фурил, аксадиазолил и тиадиазолил. Предпочтительным является 6-членный гетероарил и, в частности, предпочтительным является пиридил.
Соединение по настоящему изобретению в некоторых случаях может существовать в виде геометрического изомера или таутомера, в зависимости от типа заместителя. Более того, данное соединение может иметь асимметрический углерод. Настоящее изобретение включает все изолированные виды этих изомеров, а также их смесь. Кроме того, меченые соединения, т.е. соединения, в которых по меньшей мере один элемент в соединении по настоящему изобретению замещен радиоактивными или нерадиоактивными изотопами, также включаются в настоящее изобретение.
Более того, фармацевтически применимые, так называемые пролекарства, содержащие соединение по настоящему изобретению, также включены в настоящее изобретение. Фармацевтически допустимое пролекарство - это соединения, имеющие группу, которая может быть превращена в аминогруппу, гидроксильную группу, карбоксильную, или подобие соединения по настоящему изобретению, посредством сольволиза или в результате физиологического условия. Примеры группы, способной образовывать пролекарства, включают группы, описанные в "Prog. Med., vol. 5, 2157-2161 (1985) и "Iyakuhin no Kaihatsu (Development of Medicines) (Hirokawa Shoten, 1990), vol. 7, Bunshi Sekkei (Molecular Design)", 163-198.
Соединение, представленное формулой (I), может образовывать соли с кислотами или основаниями. Это могут быть любые фармацевтически приемлемые соли, и их специфические примеры включают соли присоединения кислоты неорганических кислот, такие как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота, и органических кислот, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, аспаргиновая кислота и глутаминовая кислота; и соли с неорганическими основаниями, такими как натрий, калий, магний, кальций и алюминий, и с органическими основаниями, таким как метиламин, этиламин, этаноламин, лизин, орнитин и аммонийные соли.
Дополнительно настоящее изобретение также включает различные гидраты и сольваты и полиморфные соединения соединения, представленного формулой (I), и их соли.
В описании использовали следующие аббревиатуры.
Pr: метод получения, АсОН: уксусная кислота, n-BuLi: нормальный бутиллитий, t-BuOH: третичный бутанол, n-BuOH: нормальный бутанол, BrCN: бромистый цианид, CDI: 1,1'-карбонилбис-1Н-имидазол, DBU: диазобицикло[5.4.0]ундека-7-ен, DMAP: 4(N,N-диметиламино)пиридин, DIC: N,N'-диизопропилкарбодиимид, DMF: N,N-диметилформамид, DMSO: диметилсульфоксид, DPPA: дифенилфосфорилазид, Et: этил, EDCI/HCl: N[3-(диметиламино)пропил]-N'-этилкарбоксамида гидрохлорид, EtOH: этанол, Et3N: триэтиламин, EtOAc: этилацетат, HOBt: 1-гидрокси-1Н-бензотриазол, HPLC: высокоэффективная жидкостная хроматография, IPE: диизопропиловый эфир, i-PrOH: 2-пропанол, K2CO3: карбонат калия, КСN: цианид калия, KHCO3: гидрокарбонат калия, KOtBu: третичный бутоксид калия, LC-MS: жидкостная хроматография-масс-спектроскопия, LiH: гидрид лития, MeOH: метанол, NaH: гидрид натрия, NaOH: гидроксид натрия, NaBH4: боргидрид натрия, NaCN: цианид натрия, NaHCO3: гидрокарбонат натрия, Na2CO3: карбонат натрия, NaOMe: метоксид натрия, NaOEt: этоксид натрия, NCS: N-хлорсукцинимид, NH4Cl: хлорид аммония, NMP: N-метилпирролидон, POCl3: оксохлорид фосфора, Р2О5: пентаоксид фосфора, THF: тетрагидрофуран, TLC: тонкослойная хроматография, TMEDA: N,N,N',N'-тетраметилэтилендиамин, Zn(CN)2: цианид цинка.
(Метод Получения)
Соединение (I) по настоящему изобретению и его фармацевтически приемлемая соль могут быть получены путем применения различных известных синтетических методов, используя преимущества, даваемые их основным каркасом или типом заместителей. Здесь, в зависимости от типа функциональных групп, в некоторых случаях, с точки зрения методики приготовления, эффективно защитить функциональную группу подходящей защитной группой либо заменить ее группой, которая может быть легко превращена в функциональную группу, на протяжении стадий от исходных веществ до промежуточных соединений. Примеры таких функциональных групп включают аминогруппу, гидроксильную группу, карбоксильную группу, и примеры защитных групп для них включают защитные группы, которые описаны в «Protective Groups in Organic Synthesis» edited T.W. Green and P.G.M. Wuts, (USA) (3rd edition, 1999), которые могут быть выбраны по желанию, и их использование обуславливается условиями реакции. При помощи таких методов требуемое соединение можно получить путем введения защитной группы с целью проведения реакции, и затем, если требуется, удаления защитной группы или превращения в требуемую группу.
Дополнительно, пролекартво соединения (I) по настоящему изобретению может быть приготовлено путем введения специфических групп на протяжении стадий от исходных веществ до промежуточных соединений, сходной с вышеописанными защитными группами, или при проведении реакции, используя полученное соединение (I) по настоящему изобретению. Реакция может быть проведена при применении обычных методов этерефикации, аминирования, дегидратирования или методом, хорошо известным специалисту в данной области техники.
<Первый метод получения промежуточного вещества>
[Хим.11]
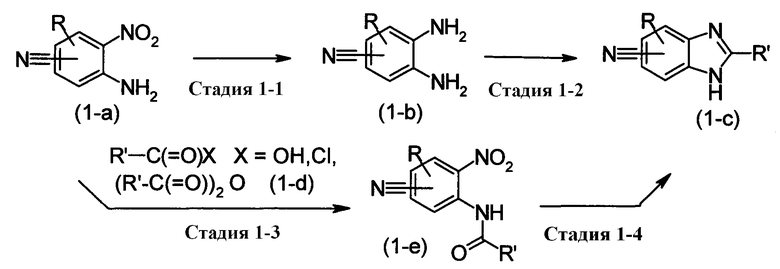
[где R или R' означает низший алкил, который может быть замещен не менее чем одним заместителем, выбранным из ряда, содержащего -CN, -C(=O)OH, -C(=O)ORX, -C(=O)NRXRY, -C(=O)NHSO2RX, -C(=O)-(С1-С8 гетероциклоалкил), -NHRX, -OH, SRX, -S(=O2)RX, галоген и -ORX (который определен как RZ-низший алкил); R0-(C1-C6)алкил-О-, R0-(C1-C6)алкил-С(=О)-; R0-(C1-C6)алкил-S(=O)2-; R0-O-(C1-C6)алкил-; R0-C(=O)-(C1-C6)алкил; R0-S(=O)2-(C1-C6)алкил-; (С2-С6)алкенил, -С(=О)Н, -ORX; -S(=O2)RX; галоген; =О; -NRXRY или -С(=О)NRXRY;
RX и RY одинаковы или отличаются друг от друга и каждый означает -Н; (С1-С6)алкил, который может быть замещен с помощью -ОН или пиридила. Также RX может быть связан с RY и атомом азота, образуя (C3-C8)гетероциклоалкил.
Этот метод получения представляет собой метод получения соединения безимидазола, изображенного формулой (1-с), при вступлении альдегидного соединения в реакцию с соединением 1,2-диаминобензола, изображенного формулой (1-b), которое может быть получено при восстановлении соединения, изображенного формулой (1-а).
Стадия, изображенная Стадией 1-1, представляет собой стадию восстановления нитрогруппы соединения, изображенного формулой (1-а), в аминогруппу, которая может быть проведена при нормальном давлении или при повышенном давлении в растворителе, инертном к реакции.
На стадии, изображенной Стадией 1-2, где R' представляет собой Н, можно создать имидазольное кольцо, например, при вступлении ортомуравьиного эфира, такого как этилортоформиат, в реакцию с соединением, изображенным формулой (1-b), в присутствии кислого катализатора.
Кроме того, на стадии, изображенной Стадией 1-2, где R' не является Н, например метод в котором аминогруппа соединения, изображенного формулой (1-а), предварительно проацилирована, используя карбоновую кислоту, хлорангидрид карбоновой кислоты, ангидрид карбоновой кислоты или подобные и циклизована при нагревании или в присутствии кислоты, метод, при котором тетраалакилортокарбонат, СDI и BrCN используют вместо ортомуравьиного эфира, или другие методы можно привести в качестве примера.
Кроме того, наряду с другими методами, можно привести в качестве примера и метод, в котором аминную часть соединения нитробензола (1-а) подвергают карбомилированию, приводящему к ациламинному соединению (1-е), подвергаемому восстановлению его нитрогруппы и циклизации при нагревании (Стадия 1-3, Стадия 1-4).
Все эти реакции могут быть проведены в растворителе, инертном к реакции, или без растворителя, от при комнатной температуре до при нагревании или при нагревании с кипячением.
<Второй метод получения промежуточного соединения>
[Хим.12]
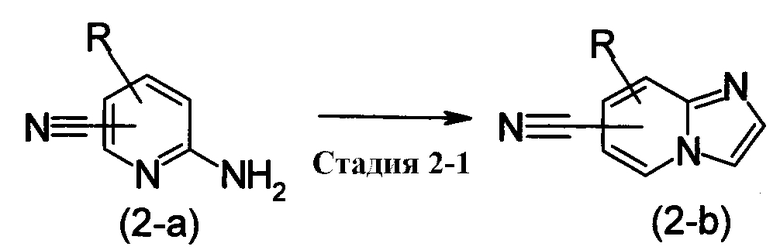
[где R имеет те же самые значения, как определено выше]
Этот метод получения представляет собой метод получения имидазо[1,2-а]пиридина, замещенного нитрильной группой, изображенного при помощи формулы (2-b), при использовании соединения 2-аминопиридина, изображенного формулой (2-а), в качестве исходного соединения.
Стадия, изображенная Стадией 2-1, представляет собой реакцию сборки кольца имидазо[1,2-а]пиридина при вступлении в реакцию хлорацетальдегида или α-хлоркетона с соединением, изображенным формулой (2-а).
Ее предпочтительно проводить в присутствии основания, и специфические примеры оснований включают карбонаты щелочных металлов, таких как Na2CO3 и K2CO3; гидрокарбонаты щелочных металлов, таких как NaHCO3 и KHCO3; алкоксидов, таких как NaOMe, NaOEt и KOtBu; третичных аминов, таких как Et3N и DIPEA; и органических аминов, таких как DBU, пиридин и лутидин.
Все эти реакции могут быть проведены в растворителе, инертном к реакции, или без растворителя от при комнатной температуре до при нагревании или при нагревании с кипячением.
<Третий метод получения промежуточного соединения>
[Хим. 13]
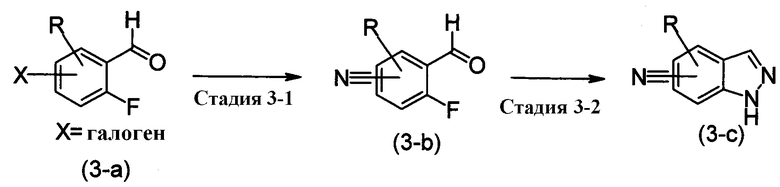
[где R имеет те же самые значения, как определено выше]
Этот метод получения представляет собой метод получения соединения имидазола, изображенного при помощи формулы (3-с), при проведении реакции гидразингидрата с нитрильной группой соединения цианобензола, изображенного формулой (3-b), полученного путем замещения галогена соединения, изображенного формулой (3-а).
Стадия, изображенная Стадией 3-1, представляет собой реакцию галогена, связанного ароматическим кольцом с нитрильной группой. Примером этого могут служить реакция Zn(CN)2 в присутствии тетракисфенилфосфин палладия (0), реакция TMEDA и Pd катализатора в присутствии Na2CO3 в DMA, и реакции, в которых KCN, NaCN или им подобные вступают в реакцию вместо Zn(CN)2. Как правило, соединение, изображенное формулой (3-b), можно получить при реакции соединения, изображенного формулой (3-а), с трис(дибензилдиацетон)дипалладий (0),
1'-бис(дифенилфосфино)ферроценом и Zn(CN)2.
Здесь, примеры уходящей группы включают галоген, такие как Br и Сl; метансульфонилокси, этансульфонилокси, бензолсульфонилокси, п-толуилсульфонилокси и трифторметлисульфонилокси.
Стадия, изображенная Стадией 3-2, представляет собой реакцию создания имидазольного цикла из соединения цианобензальдегида, изображенного формулой (3-b). Как правило, в этой реакции используют гидразингидрат, эта реакция может быть проведена без растворителя или в растворителе, инертном к реакции, таком как МеОН и толуол, от при комнатной температуре до при нагревании или при нагревании при кипении. Дополнительно, также в качестве примера можно привести метод с использованием цианида меди и может быть добавлено основание, такое как пиридин. Также эту реакцию предпочтительно поводят в атмосфере азота.
<Четвертый метод получения промежуточного соединения>
[Хим.14]
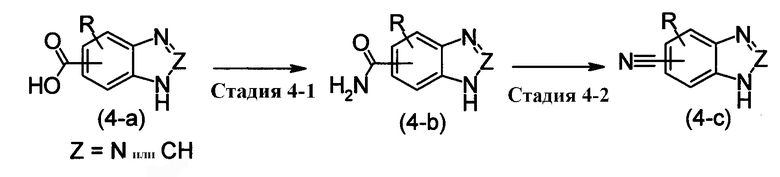
[где R имеет те же самые значения, как определено выше. Z обозначает -СН= или -N=].
Этот метод получения представляет собой метод получения соединения бензотриазола или бензимидазола, изображенного формулой (4-с), путем дегидратации амидного соединения, изображенного формулой (4-b), полученного, используя в качестве исходного соединения карбоновую кислоту, изображенную формулой (4-а).
Стадия, изображенная стадией 4-1, представляет собой реакцию конденсации карбоновой кислоты, изображенной формулой (4-а), с аммиаком и создания функциональной группы амида карбоновой кислоты, изображенной формулой (4-b). Соединение, изображенное формулой (4-а), может быть использовано в виде свободной кислоты, но также могут быть использованы ее реакционноспособные производные. Примеры таких реакционноспособных производных, исходя из соединения, изображенного формулой (4-b), включают галогенангидриды карбоновых кислот, такие как хлорангидрид кислоты и бромангидридкислоты; обыкновенные сложные эфиры, такие как метиловый эфир, этиловый эфир и бензиловый эфир; азиды карбоновых кислот; активированные эфиры, такие как НОBt, п-нитрофениловый и N-гидроскисукцинимидный; симметричные ангидриды карбоновых кислот; смешанные ангидриды карбоновых кислот алкиловых эфиров галогенкарбоновых кислот, таких как хлорангидрид алкилугольной кислоты, пивалоилгалогенид и хлорангидрид п-толуолсульфоновой кислоты; смешанные ангидриды карбоновых кислот, такие как смешанные ангидриды фосфорной кислоты, такие как те, что получаются по реакции дифенилфсфорилхлорида с N-метилморфолином.
В случае, когда соединение, изображенное формулой (4-а), вступает в реакцию в виде свободной кислоты, или без выделения активированного эфира, или нечто подобное, предпочтительно используют конденсирующий агент, такой как DCC, CDI, DPPA, диэтилфосфорилцианид и EDCI/HCl.
Растворитель для реакции варьируется в зависимости от реакционной способности производного или конденсирующего агента, которые используют, но реакцию проводят в органическом растворителе, инертном к реакции, таких как галогенпроизводные углеводородов, ароматические углеводороды, простые эфиры, сложные эфиры, такие как EtOAc, ацетонитрил, DMF и DMSO или в их смесях. Также реакцию проводят при охлаждении, от при охлаждении до при комнатной температуре или от при комнатной температуре до при нагревании.
Кроме того, в реакции, для того чтобы реакция прошла хорошо, желательно проводить реакцию с избыточным количеством аммиака или в присутствии основания, такого как N-метилморфолин, триметиламин, Et3N, DIPEA, N,N-диметиланилин, пиридин, DMAP, пиколин и лутидин. Пиридин может быть использован в комбинации с растворителем.
Стадия, изображенная Стадией 4-2, представляет собой дегидратацию, для которой основание может или не может использоваться и могут быть использованы такие дегидратирующие агенты, как трифторуксуный ангидрид, POCl3 и Р2О5.
Кроме того, в случае синтеза конденсированных, промежуточного гетероциклического соединения, отличного от описанного, в вышеописанных методах получения промежуточных веществ, можно применять методы, описанные в Справочных Примерах или в Примерах в данном описании, или равноценные методы, или дополнительно, для получения следует использовать хорошо известные методы или методы, очевидные для специалистов в данной области техники.
<Первый метод получения>
[Хим.15]
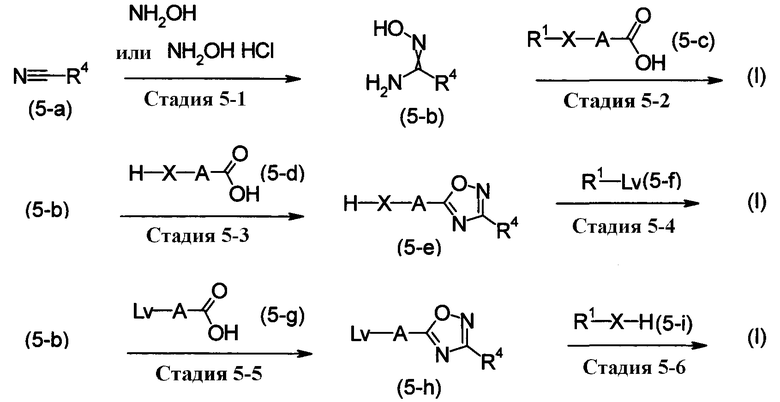
[где А, Х, R1 и R4, как описаны выше. Lv обозначает уходящую группу. Карбоновая кислота, изображенная формулами (5-с), (5-d) и (5-g), можно приобрести как коммерчески доступный продукт, или приготовлен так же, как коммерчески доступный продукт].
Этот метод получения представляет собой метод получения соединения по настоящему изобретению, изображенного формулой (I), при помощи реакции гидроксиамида, изображенного формулой (5-b), полученного при вступлении в реакцию гидроксиламина с ароматическим нитрилом, изображенным формулой (5-а), с карбоновой кислотой, изображенной формулой (5-с).
На стадии, изображенной Стадией 5-1, гидроксиамидин, изображенный формулой (5-b), может быть получен при вступлении в реакцию свободного гидроксиламина или гидрохлорида гидроксиламина в присутствии основания.
Эту реакцию следует проводить в растворителе, инертном к реакции. Специфические примеры растворителей включают спирты, такие как МеОН, EtOH и i-PrOH; ароматические углеводороды, такие как толуол и ксилол; простые эфиры, такие как THF, диоксан и диэтоксиэтан; галогенуглеводороды, такие как дихлорметан, 1,2-дихлорэтан, хлороформ и четыреххлористый углерод; ацетонитрилы; апротонные полярные растворители, такие как DMF, 1,3-димитил-2-имидазолидинон и DMSO; воду или смеси таких растворителей. Как правило, в реакции используют спирты. Как описано выше, в случае, когда используют гидрохлорид гидроксиламина, реакцию проводят предпочтительно в присутствии основания, и специфические примеры оснований включают карбонаты щелочных металлов, такие как Na2CO3 и K2CO3; гидрокарбонаты щелочных металлов, такие как NaHCO3 и КНСО3; алкоксиды, такие как NaOMe, NaOEt и KOtBu; третичные амины, такие как Et3N и DIPEA; органические амины, такие как DBU, пиридин и лутидин. Температура реакции варьируется в зависимости от типа исходных веществ, условий реакции и т.п., но реакцию следует проводить, как правило, от при комнатной температуры до около температуры кипения растворителя. Как правило, в присутствии снования, такого как Na2CO3, реакцию следует проводить в органическом растворителе, инертном к реакции, таком как МеОН, от при комнатной температуры до при нагревании.
Стадия, изображенная Стадией 5-2, состоит из двух стадий, т.е. из стадии ацилирования гидроксиамидина и стадии реакции цикизации в соответствующем порядке. Стадию ацилирования можно провести следующим образом. Соединения, изображенное формулой (5-с), может быть использовано в реакции в виде свободной кислоты, но его реакционноспособное производное также может использоватся в реакции. Примеры таких реакционноспособных производных включают хлорангидрид кислоты и бромангидрид кислоты; обычные простые эфиры, такие как метиловый эфир, этиловый эфир и бензиловый эфир; азиды кислоты; активированные эфиры, такие как HOBt, п-нитрофениловый, N-гидроксисукцинимидный; симметричный ангидрид кислоты; смешанный ангидрид кислоты с алкиловыми эфирами галогенкарбоновых кислот, такими как алкиловый эфир галогенугольной кислоты, смешанный ангидрид смешанной фосфорной кислоты, такие как полученные по реакции дифенилфософорилхлорида с N-метилморфолином.
В случае, когда соединение, изображенное формулой (5-с), реагирует в виде свободной кислоты, или без выделения активированного эфира или нечто подобное, предпочтительно используют конденсирующий агент, такой как DCC, CDI, DPPA, диэтилфосфорилцианид и EDCI/HCl.
Растворитель для реакции варьируется в зависимости от реакционной способности производного и конденсирующего агента, который используют, но реакцию проводят в органическом растворителе, инертном к реакции, таких как галогенуглеводороды, ароматические углеводороды, простые эфиры, сложные эфиры, как, например, EtOAC, ацетонитрил, DMF и DMSO, или смеси таких растворителей, при охлаждении, от при охлаждении до при комнатной температуре или от при комнатой температуре до при нагревании.
Для того чтобы хорошо провести реакцию, в некоторых случаях предпочтительно ее проводить в присутствии таких оснований, как N-метилморфолин, триметиламин, Et3N, DIPEA, N,N-диметиланилин, пиридин, DMAP, пиколин и лутидин. Также пиридин может использоваться в комбинации с растворителем. Ацилированный продукт как промежуточное соединение может быть очищен путем выделения и нагрет в органическом растворителе, инертном к реакции, таком как EtOH, диоксан, толуол и вода. Как правило, эту двухстадийную реакцию следует проводить в одну стадию, при нагревании или микроволновом облучении продукта в том виде, в котором он есть, или в виде реакционной смеси после ацилирования.
Специфические примеры растворителей выключают ароматические, такие как толуол, ксилол и пиридин; простые эфиры, такие как диэтиловый эфир, THF, диоксан и диэтоксиэтан; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан, хлороформ и четыреххлористый углерод; ацетонитрилы; апротонные полярные растворители, такие как DMF, DMA, 1,3-диметил-2-имидазолидинон, NMP и DMSO; воду или смеси этих растворителей. Температура реакции варьируется в зависимости от типа исходных веществ, условий реакции или нечто подобное, но реакцию следует проводить обычно от при комнатной температуры до при нагревании.
В случае, когда Х представляет собой -О- или -NH-, синтез может быть осуществлен нижеследующим методом приготовления.
Стадии, изображенные Стадией 5-3 и Стадией 5-5, могут быть проведены таким же образом, как и стадия, изображенная Стадией 5-2.
Стадии, изображенные Стадией 5-4 и Стадией 5-6, представляют собой стадии приготовления соединения по настоящему изобретению, изображенного формулой (I), осуществляемые при вступлении в реакцию фенола, анилина, спирта или амина, изображенных формулой (5-е) или формулой (5-i), с соединением, имеющим уходящую группу, изображенным формулой (5-f) или формулой (5-h). Здесь, примеры уходящих групп включают галогены, такие как хлор или бром; и сульфонилокси, такие как матансульфонилокси, этансульфонилокси, бензолсульфонилокси, п-толуолсульфонилокси, п-нитробензолсулфанилокси и трифотрметансульфонилокси.
Реакцию проводят при нормальном давлении или при повышенном давлении, без растворителя или в подходящем растворителе.
Специфические примеры растворителей включают ароматические углеводороды, такие как толуол или ксилол; кетоны, такие как ацетон и метилэтилкетон; простые эфиры, такие как эфир, THF, диоксан и диэтоксиэтан; спирты, такие как МеОН, EtOH, i-PrOH и n-BuOH; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлоэтан, хлороформ и четыреххлористый углерод; ацетонитрилы; апротонные полярные растворители, такие как DMF, 1,3-диметил-2-имидазолидинон, NMP и DMSO; воду или смеси таковых растворителей. В этой реакции предпочтительно используют основание, и специфические примеры оснований включают NaH; карбонаты щелочных металлов, такие как Na2CO3 и K2CO3, гидрокарбонаты щелочных металлов, такие как NaНCO3 и KНCO3, алкоксиды, такие как NaOMe, NaOEt и KOtBu, третичные амины, такие как Et3N, трибутиламин и DIPEA; органические амины, такие как DBU, пиридин и лутидин, но избыточное количество может быть объединено с амином, изображенным формулой (5-е) или (5-i). Температура реакции варьируется в зависимости от типа исходных веществ, условий реакции или тому подобного, но, как правило, реакцию следует проводить от при комнатной температуре до при около температуры кипения растворителя. Как правило, реакцию следует проводить в присутствии основания, такого как NaH и Na2CO3, в органическом растворителе, инертном к реакции, таком как DMF и DMA, от при -10°С до при нагревании. Также амин, изображенный формулой (5-е) или формулой (5-i), может быть представлен в виде его соли. Дополнительно, может быть осуществлено микроволновое облучение при нагревании при приготовлении.
Кроме того, некоторые соединения, изображенные формулой (I), могут быть приготовлены путем сочетания хорошо известных процессов, которые обычно могут использоваться квалифицированными специалистами в области техники, такие как хорошо известные алкилирование, ацилирование, реакция замещения, окисление, восстановление, гидролиз, снятие защитной группы и галогенирование, исходя из соединений по настоящему изобретению, как приготовленные вышеописанным методом.
Например, относительно алкилирования, реакция алкилирования, которая обычно используется квалифицированными специалистами в области техники, может быть применена, которую следует проводить в органическом растворителе, инертном к реакции, таком как простые эфиры; ароматические углеводороды; галогенированные углеводороды, такие как дихлорметан, дихлорэтан и хлороформ; DMF; ацетонитрилы; и полярные апротонные растворители, при охлаждении, от при охлаждении до комнатной температуры или от комнатной температуры до при нагревании, в присутствии основания, такого как NaH; карбонаты щелочных металлов; гидрокарбонаты щелочных металлов; алкоксиды; третичные амины и органические основания.
Кроме того, например относительно ацилирования, реакция ацилирования, которую, как правило, используют квалифицированные специалисты в области техники, может быть использована, в частности, та, которая может быть проведена в присутствии HOBt, в растворителе, варьируемом в зависимости от конденсирующего агента, такого как EDCI/HCl или CDI и дифенилфосфорилцианид, в растворителе, варьирующемся от условий реакции, но, как правило, в органическом, инертном растворителе, таком как простые эфиры; ароматические углеводороды; галогенированные углеводороды, такие как дихлорметан, дихлорэтан и хлороформ; сложные эфиры, такие как EtOAc; ацетонитрилы; и апротонные растворители, при охлаждении, от при охлаждении до при комнатной температуры или от при комнатной температуры до при нагревании.
Приготовленное таким образом соединение очищается путем выделения, как оно если или в виде его соли после солеобразующей обработки при помощи соответствующего метода. Очищение при помощи выделения проводится при применении общеизвестных химических операций, таких как экстракция, концентрирование, удаление отгонкой, кристаллизация, фильтрация, перекристаллизация и различные методы хроматографии.
Различные типы изомеров могут быть выделены соответствующими методами, использующими разницу физико-химических свойств между изомерами. Например, рацемическая смесь может быть превращена в оптически чистые изомеры, например, при помощи обычного метода расщепления рацемических продуктов, таких как метод расщепления оптических изомеров в виде диастереомерных солей с общеизвестными оптически активными кислотами, такими как винная кислота. Также диастереомерная смесь может быть разделена, например, при помощи дробной кристаллизации или различных методов хроматографии. Кроме того, оптически активные соединения также могут быть приготовлены, используя соответствующие оптически активные исходные вещества.
Действие соединений данного изобретения подтвердили при помощи следующих фармакологических тестов.
Экспериментальный Пример 1: Тест для подтверждения агонистической S1P 1 активности
1) Оценка активности агониста рецептора путем анализа связывания GTP[γ-35S], используя мембрану клетки человека, экспрессирующую S1P1.
In vitro S1P1 агонистическую активность соединения данного изобретения оценивали при помощи возрастания функциональной связывающей активности в G-протеине GTP[γ-35S], используя мембрану человеческой S1P1 выраженной клетки. Кодировку кДНК человеческого S1P1 клонировали из библиотеки человеческой колоректальной кДНК и вводили в вектор экспрессии pcDNA3.1 с целью создания S1P1-pcDNA3.1. Затем при помощи Lipofectamine 2000 (GIBCO) S1P1-pcDNA3.1 трансфектировали в клеточную линию СНО и культивировали в Ham's F-12 питательной среде, содержащей 10% фетальной бычьей сыворотки, 100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина и 1 мг/мл G418 дисульфата, получая G418-резистентный штамм. Культивированные человеческие S1P1 экрпессированные клетки выделяли в чистом виде в 1 мМоль ЕДТА/2Na-содержащем ФСБ и разрушали при охлаждении льдом в гомогенизаторе, сделанном из стекла, в 1 мМоль Трис-НCl (рН 7,4) буферном растворе, содержащем 0,1 мМоль ЕДТА и ингибитор протеина. Его центрифуговали при 1400×10 мин и надосадочную жидкость в дальнейшем центрифуговали при 4°С в течение 60 мин при 100000хг и суспендировали в 10 мМоль в 1 мМоль Трис-НCl (рН 7,4) буферном растворе, содержащем 1 мМоль ЕДТА. Полученные мембраны (0,13 мг/мл) и 50 пМоль GTP[γ-35S] (NEN; неактивен 1250 Сi/mmol) вводили в реакцию в 20 мМоль HEPES (рН 7,0) буферном растворе (общее количество 150 мкл), содержащем 100 мМоль NaCl, 10 мМоль MgCl2, 0,1% свободный от жирных кислот бычий сывороточный альбумин и 5 мкМоль цитиндисфосфата в течение 1 часа вместе с соединением по настоящему изобретению (10-12 до 10-15 Моль) и затем мембраны отделяли в GF-С планшет с халвестором клеток (Packard, FilterMate). FilterMate сушили при 50°С в течение 60 мин и в дополнение добавляли Microscinti-o (Packard) для вычисления при помощи детектора сцинтилляции липидов для микропланшета (Packard, TOP count). Для оценки агонистической активности по отношению к человеческому S1P1 соединения по настоящему изобретению и сравнительного соединения процент со значением максимальной реакции, делающей GTP[γ-35S] связи насыщенными, в присутствии соединения было принято за 100% и значении реакции GTP[γ-35S] связей в отсутствии соединения было принято за 0%, создали нелинейную кривую регрессии, и концентрацию, вызывающую возникновение агонистической активности на уровне 50% от максимальной реакции, определили как значение ЕС50 (нМоль).
№
EC
50
№
EC
50
№
EC
50
В результате подтвердилось, что соединения по настоящему изобретению обладают S1P1 агонистической активностью.
2) Оценка лимфоцитопении периферической крови у крыс
Лимфоцитопению периферической крови у крыс определяли спустя 24 часа после перорального введения нижеследующим образом. Самцов крысы Льюиса возраста шесть-десять недель (Japan Charles речная лаборатория) произвольно распределяли по группам (n=3) и соединение по настоящему изобретению суспендировали в содержащей 0,5% метилцеллюлозы дистиллированной воде и перорально вводили с помощью зонда. Спустя 24 часа после введения под эфирным наркозом отбирали 0,2 мл крови из глазного дна. В образец крови сразу же добавляли ЭДТА/4К и гепарин для предотвращения свертывания и число лимфоцитов в крови подсчитывали с помощью автоматического гематологического анализатора (Sysmex Corp.; XT-2000i).
Для снижения числа лимфоцитов в периферической крови под действием соединения по настоящему изобретению процент с числом лимфоцитов в группах, которым назначали содержащую 0,5% метилцеллюлозы дистиллированную воду, был принят за 100%, как делали в прошлый раз, и дозу, которая обуславливает снижение на 50% числа лимфоцитов в периферической крови при назначении соединения по настоящему изобретению, определяли как значение ED50 (мкг/кг).
Для сравнительных соединений 1 и 2, которые описаны в пояснительной записке Международной Заявки №WO2004/103279, сравнительное соединение 3, которое, как считается, точно описано в пояснительной записке Международной Заявки №WO2005/032465, и соединение Примера 119, сравнивали значение ED50 спустя 24 часа после назначения для снижения числа лимфоцитов в периферической крови у крыс.
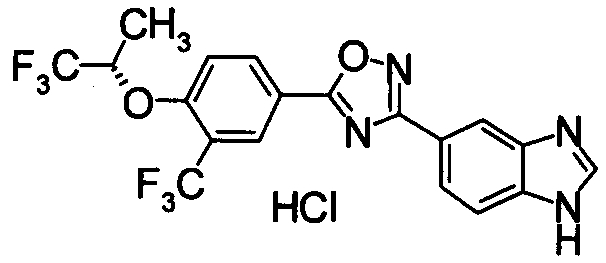
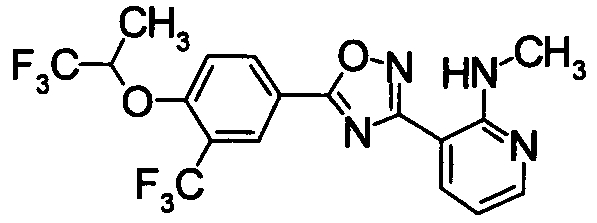
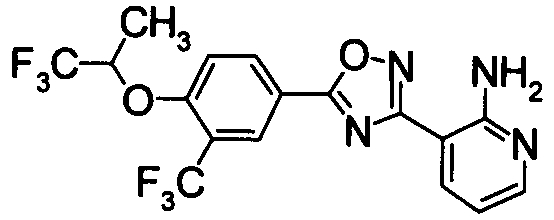
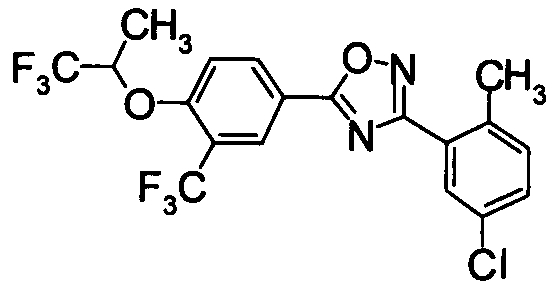
В результате видно, что соединение Примера 119 показывает высокое значение ED50 даже после 24 часов, что указывает на сохранение эффекта.
В связи с тем, что соединение по настоящему изобретению обладает S1P1 агонистической активностью, оно применимо в качестве активного ингредиента средства для лечения или средства для профилактики заболевания, обусловленного неблагоприятной лимфоцитарной инфильтрацией, например аутоиммунного заболевания, такого как отторжение трансплантата при пересадке органов, костного мозга или тканей или реакция «трансплантат против хозяина», ревматические артриты, рассеянный склероз, системная красная волчанка, нефротический синдром, энцефаломинигит, злокачественная миастения, панкреатит, гепатит, легочные заболевания, астма, атопический дерматит, воспалительное заболевание кишечника, атеросклероз, ишемическое реперфузионное повреждение или воспалительное заболевание, и кроме того, заболевания, обусловленного ненормальным ростом или аккумуляцией клеток, таких как рак или лейкемия.
Кроме того, соединение по настоящему изобретению применимо для лечения и/или профилактики следующих заболеваний, базирующиеся на агонитсической активности по отношению к S1P1.
Оно применимо для лечения и/или профилактики воспалительных или гиперпластических кожных заболеваний, таких как псориаз, контактный дерматит, экзематоидный дерматит, себорейный дерматит, красный плоский лишай, вульгарная пузырчатка, пемфигоид, булелезный эпидермолиз, крапивница, васкулярная водянка, окклюзия сосудов, эритема, эозинофильный лейкоцитоз кожи, красная волчанка, акне и круговая алопеция или проявления кожных заболеваний, вызванных иммунной системой; аутоиммунных заболеваний или аллергических заболеваний глаз, таких как кератоконъюктивит, весенний конъюктивит, аллергические конъюктивиты, уевиты, связанные с болезнью Бехчета, кератит, герпетический кератит, кератоконусный кератит, корнеальная эпителиальная дистрофия, бельмо роговицы, атрофический мукосинехилиальный буллезный дерматит, разъедающая язва роговицы, воспаление склеры, офтальмопатия Грейвса, болезнь Фогта-Коянаги-Харада, сухой кератоконъюктивит (сухие глаза), пузырьки, иридоциклит, саркоидоз и офтальмические заболевания гланд; обратимые обструкционные заболевания легких (астма, например бронхиальная астма, аллергическая астма, эндогенная бронхиальная астма, экзогенная бронхиальная астма и астма на грязь), в частности хроническая или тяжелая астма (например, астма с поздним началом или заболевания дыхательных путей); мукоз или ангинит (например, язва желудка, ишемическое или тромбозное сосудистое повреждение, возрастная макуляропатия, диабетическая мукуляропатия, ишемическая болезнь кишечника, болезнь кишечника, некротический энтерит, поражение кишечника термическим ожогом и заболевания, обусловленные нейромедиатором лекотриена В4) воспаление кишечника или аллергическое заболевание кишечника, включающие, например, проктит, эозинофильный энтерит, мастоцитоз, глютеиновую болезнь, болезнь Крона и неспецифический язвенный колит; связанные с пищей аллергические заболевания, проявляющие симптомы на участке, удаленном от желудочно-кишечного тракта, включающие, например, мигрень, ринит и экзему; аутоиммунные заболевания или воспалительные заболевания, включающие, например, первичный отек слизистой, аутоиммунный атрофический гастрит, преждевременный климактерический период, ювенильный диабет, обыкновенная пузырчатка, пемфигоид, симпатическое разрушение глазного яблока, факоанафилактичсекий эндофтальмит, паркоксимзальная лейкопения, хронический активный гепатит, пракосизмальный цирроз печени, дискоидная красная волчанка, синдром Шегрена, аутоиммунное воспаление яичек, артрит (например, модифицированный артрит) и полихандрия; ренальные заболевания, например мембранозная нефропатия, мембранозно-пролиферативный нефрит, фокальный глобальный гломерусклероз, серповидный нефрит, гломерулярный нефрит, первичная нефропатия IgA-типа, тублопатия межуточного нефрита и диабетическая нефропатия. Кроме того, соединение по настоящему изобретению также применимо для лечения и/или профилактики заболеваний печени (например, иммуногенных заболеваний (например, аутоиммунных заболеваний печени, хронических аутоиммунных заболеваний печени, таких как биллиарный цирроз печени, склерозирующий холангит и подобные), частичное расслоение печени, острый некротический гепатит (например, некроз, обусловленный токсином, вирусным гепатитом, шоком, аноксией или подобным), гепатит В, не-типа А гепатит, цирроз, печеночная недостаточность (например, молниеносный гепатит, гепатит с поздним началом, печеночная недостаточность (острая печеночная недостаточность или хроническая болезнь печени)) и подобных.
Дополнительно, соединение по настоящему изобретению может назначаться, как агонист S1P1 самостоятельно иди в комбинации с не менее чем одним средством, в той же доже или разных дозах, посредством такого же или отличного пути введения. Примеры средства, которое можно комбинировать, включают, но не ограничиваются им, циклоспорин А, тарколимус, сиролимус, эверолимус, микофенолат, азатиофен, бреквинар, Лефлуномид, финголимод, и антитела анти-IL-2 рецептора (например, даклизумаб) и анти-CD3 антитела (например, ОКТ3), и атни-Т клеточный иммуноглобулин (например, AtGam), белатацепт, абатацепт, циклофосамид, β-интерферон, аспирин, ацетаминофен, ибупрофен, напроксен, пироксикам и противовоспалительный стероид (например, преднизолон и дексаметазон).
Препарат, содержащий соединение, изображенное формулой (I), либо одну, две или более его солей в качестве активных ингредиентов, изготавливаются с использованием носителя, эксципиента или других добавок, обычно используемых при изготовлении медикаментов.
Прием может осуществляться в любой форме, как в виде таблеток, гранул, порошков и растворов, так и в виде парентеральных инъекций, как внутривенных, так и внутримышечных, суппозиториев, подкожных препаратов, трансназальных препаратов, ингаляций и т.п. Доза рассчитывается в каждом индивидуальном случае с учетом симптомов, возраста, пола и т.п.пациента, однако обычно она варьируется от 0,001 мг/кг до 100 мг/кг в день для взрослого в случае перорального приема и может приниматься одной порцией или разделяться на 2-4 порции. Так же, в случае внутривенного введения, в соответствии с симптомами, препарат принимается из расчета от 0,0001 мг/кг до 10 мг/кг в день для взрослого, один или два или более раз в день. Дополнительно, в случае ингаляций, прием осуществляется из расчета 0,0001-1 мг/кг для взрослого человека, один или два или более раз в день.
Что касается твердых композиций по настоящему изобретению для орального введения, используются таблетки, порошки, гранулы и т.п. В таких твердых композициях одно или более активное вещество смешиваются с по меньшей мере одним неактивным наполнителем, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон и алюминия-магния силикат. В стандартном методе композиция может содержать неактивные добавки, например смазывающие вещество, такое как стеарат магния, дезинтегратор, такой как натрия какрбоксиметилкрахмал или солюбилизирующий компонент. При необходимости таблетки или пилюли могут быть покрыты оболочкой из сахара, или растворимым в желудке, или энтеросолюбильным покрытием.
Жидкая композиция для орального приема включает фармацевтически применимые эмульсии, растворы, суспензии, сиропы, эликсиры и т.п. и содержит обычно используемые инертные растворители, такие как очищенная вода и этанол. Вдобавок к инертному растворителю композиция может содержать вспомогательное средство, такое как солюбилизирующий агент, увлажняющий агент или суспендирующий агент, подсластитель, нейтрализующее средство, ароматизатор и антисептик.
Инъекции для парентерального введения включают стерильные водные или неводные растворы, суспензии и эмульсии. Примеры водных растворителей включают дистиллированную воду для инъекций и физиологический раствор. Примеры неводного растворителя включают пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое, спирты, такие как этанол, и Polysorbate 80 (Pharmacopeia). Такая композиция может также содержать средство для поддержания тонуса мышц, антисептик, увлажняющее средство, эмульгирующее средство, диспергирующее средство, стабилизирующее средство и солюбилизирующее средство. Эти вещества стерилизуются, например, с помощью фильтрации через фильтр, задерживающий бактерии, смешивания бактерицидных препаратов или с помощью облучения. Кроме того, их можно использовать при изготовлении стерильной твердой композиции и растворении или суспендировании ее в стерильной воде или стерильном растворителе для инъекции непосредственно перед ее введением.
Что касается трансмукозального средства, как, например, ингаляции или трансназального средства, то здесь применяются твердые, жидкие или полужидкие формы, которые могут быть изготовлены в соответствии с общепринятым известным методом. Например, при необходимости могут быть добавлены наполнители, такие как лактоза или крахмал, рН-контролирующие средства, антисептик, ПАВ, смазывающее средство, стабилизатор, загуститель. Для их приема могут быть использованы соответствующие устройства для ингаляций или впрыскивания. К примеру, соединение может приниматься посредством традиционного устройства или пульверизатора, такого как устройство для дозированной ингаляции, в чистом виде, либо в виде порошка, изготовленного по рецепту, либо в виде раствора или суспензии, в которых он объединяется с фармацевтически применимым носителем. Ингалятор для вдыхания сухого порошка может быть как одноразовым, так и многоразовым, с применением сухого порошка либо капсулы, содержащей порошок. В качестве альтернативы может использоваться аэрозольный пульверизатор с высоким давлением, в котором применяется соответствующий газ-вытеснитель, к примеру хлорфторалкан, гидрофторалкан и диоксид углерода.
Наружное средство включает мази, пластыри, кремы, желе, пасты, пульверизаторы, лосьоны, глазные капли, глазные мази и т.п. Наружное средство может содержать мазевые основы, лосьонные основы, водные и неводные жидкости, суспензии, эмульсии и т.п., применяемые в подобных случаях. Примеры основ мазей или лосьонов включают полиэтиленгликоль, пропиленгликоль, белый вазелин, осветленный пчелиный воск, полиэтоксиэтиленовое твердое касторовое масло, глицеринмоностеарат, стеариловый спирт, цетиловый спирт, лаурилмакроголь и сорбитансексвиолеат.
Пример
В дальнейшем в этом документе соединения по настоящему изобретению будут описаны более подробно с ссылкой на Примеры. Настоящее изобретение не ограничивается изобретением, как описано в следующих примерах. Методы приготовления исходных веществ показаны в Примерах Получения.
[В последующих таблицах Pr означает Пример Получения №., и Структура означает структурную формулу. Как сокращенные символы в структурной формуле Ме обозначает метильную группу и Et обозначает этильную группу. Перекрещивающаяся двойная связь обозначает смесь цис/транс, и если в секции Данных описаны только номера, это показывает МS-данные. MS обозначает масс-спектрометрические данные. В таблицах RT обозначает время удерживания в высокоэффективной жидкостной хроматографии (ВЭЖХ) и М обозначает минуты. Условия выполнения ВЭЖХ следующие: колонка: Intertsil ODS-34,6×150 мм, элюент 0,01M KH2PO4 водн./MeCN(3:7), расход элюента 1,0 мл/мин, длина волны детектирования: 254 нм. Если данные 1Н-ЯМР описаны в таблицах, в качестве внутреннего стандарта использовали тетраметилсилан, и если не сказано что-либо иное, δ (ppm) (интегрированное значение, схема измерения) сигналов в 1Н-ЯМР показан в DMSO-d6 как в растворителе, используемом для регистрации. Сокращенные символы имеют те же самые значение, как в следующем: s: синглет, d: дублет, t: триплет, q: квартет, dd: дублет дублетов, ddd: дублет дублета дублетов, dt: дублет триплета, dm: дублет мультиплета, br: уширенный, brs: уширенный синглет, Hz: Герц, CDCl3: дейтерированный хлороформ, DMSO-d6: диметилсульфоксид-d6, и в данном описании ЯМР обозначает 1Н-ЯМР: протонный ядерный магнитный резонанс. Подобные будут применяться здесь в этом документе].
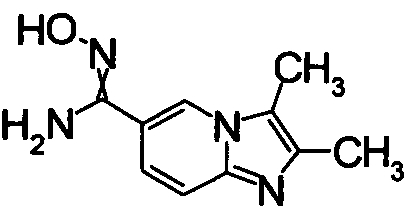
Пример Получения 1
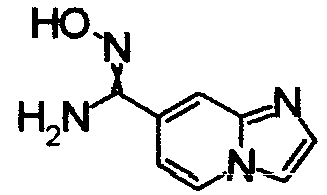
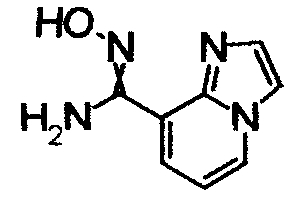
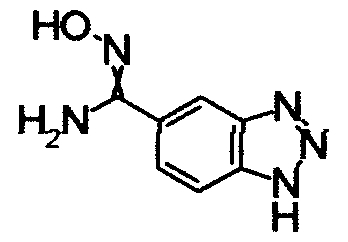
Имидазо[1,2-а]пиридин-7-карбонитрил гидрохлорид (1,5 г), гидрохлорид гидроксиламина (301 мг) и Na2CO3 (3,5 г) перемешивали при 60°С в течение 6 часов в СН3ОН (57 мл). Реакционный раствор охлаждали и концентрировали и завершение реакции определяли при помощи ЖХ-МС. К остатку добавляли воду, затем экстрагировали EtOAC. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным MgSO4 и, затем, отфильтровывали и фильтрат концентрировали, получая N'-гидроксиимидазо[1,2-a]пиридин-7-карбоксамид (850 мг) в виде твердого вещества.
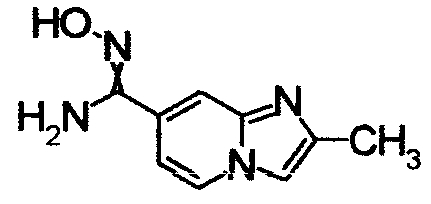
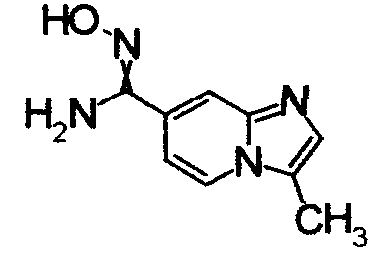
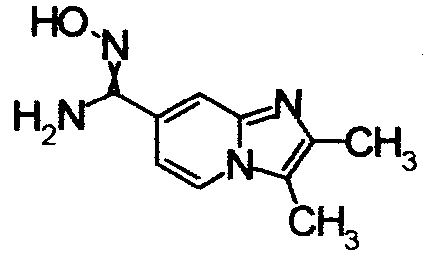
Соединения, показанные в Pr 1-1 до Pr 1-17, получали таким же образом, как в Примере Получения 1.
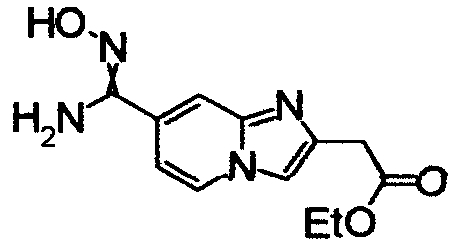
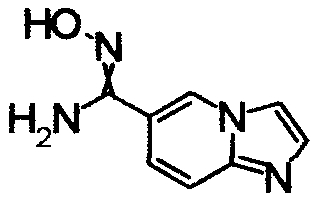
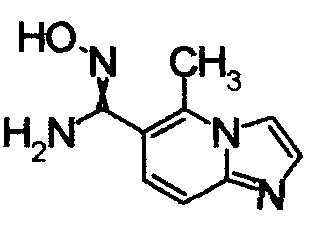
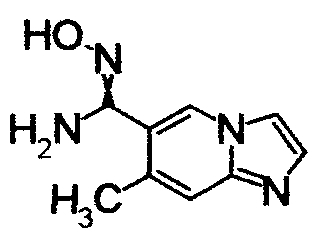
1,79
M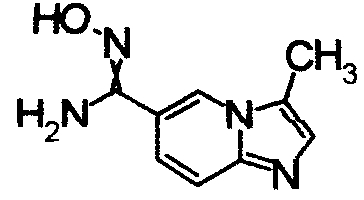
1,64M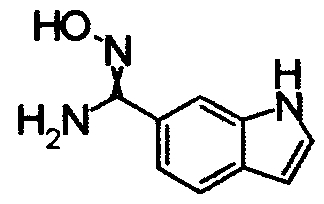
1,60
M
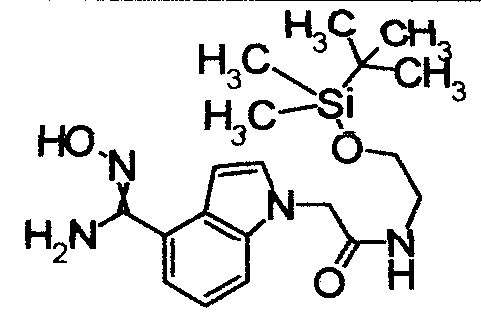
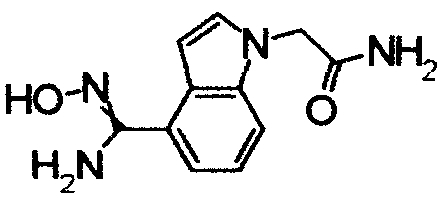
Пример Получения 2
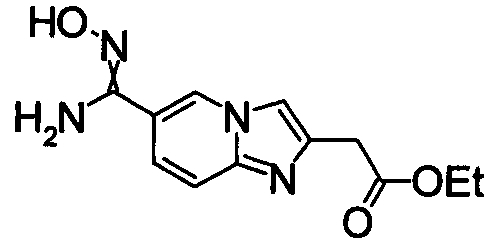
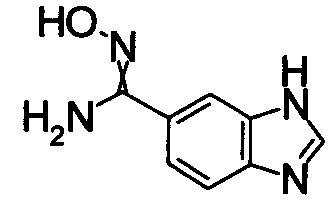
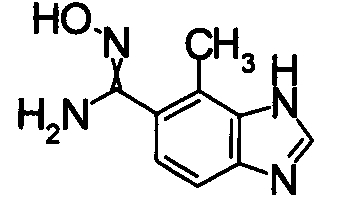
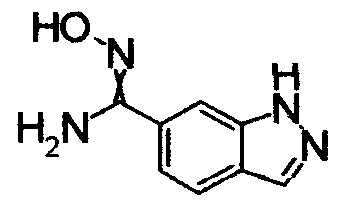
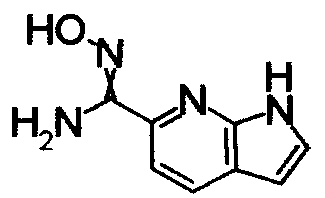
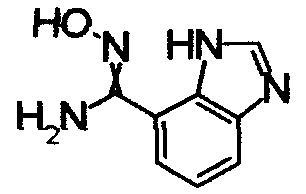
В реакционном сосуде на 50 мл к раствору 1Н-индол-4-карбонитлира (5,00 г) в СН3ОН (100 мл) добавляли гидроксиламин (50% водный раствор) при комнатной температуре, затем кипятили в течение 15 часов (завершение реакции подтверждали при помощи ТСХ). Реакционный раствор концентрировали при уменьшенном давлении и сушили азеотропной отгонкой с толуолом три раза. Полученное твердое вещество промывали IPE. Получили N'-гидрокси-1Н-индол-4-карбоксимдиамид (6,12 г) в виде твердого белого вещества.
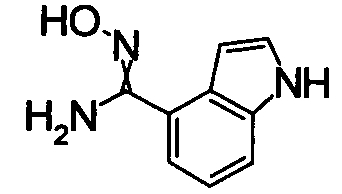
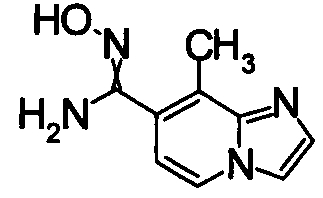
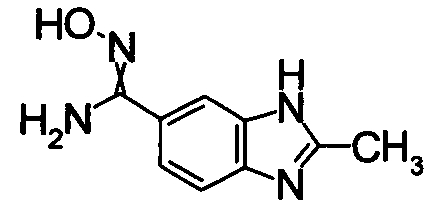
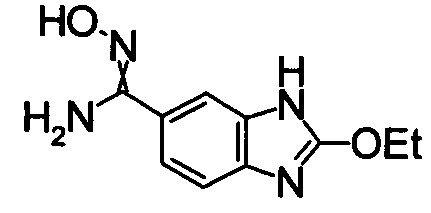
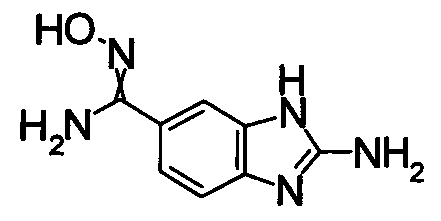
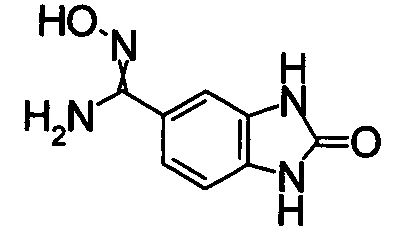
Соединения, показанные в Pr 2-1 до Pr 2-26, получали тем же самым образом, как и в Примере Получения 2.
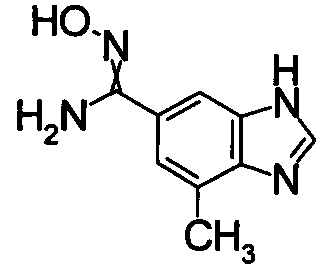
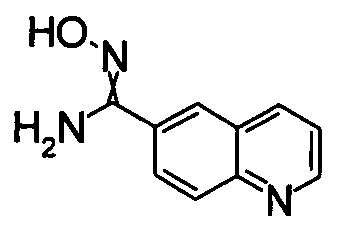
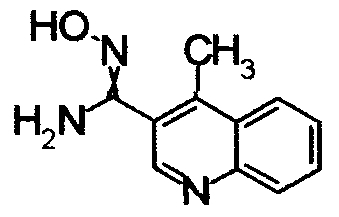
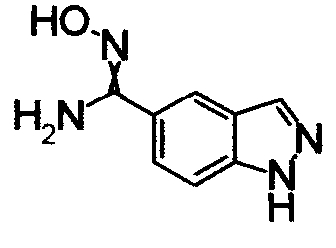
1,68
M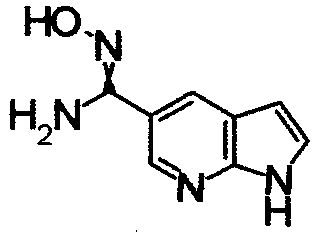
ниже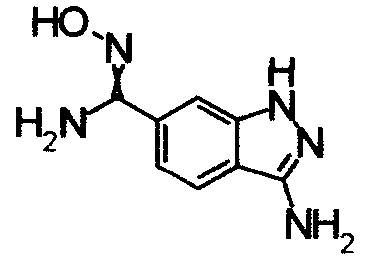
1,62
M
1,69
M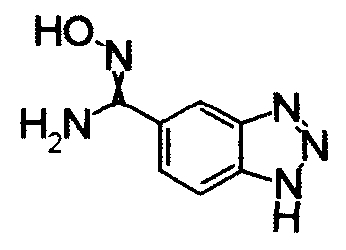
1,62M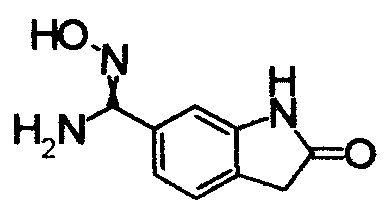
1,62M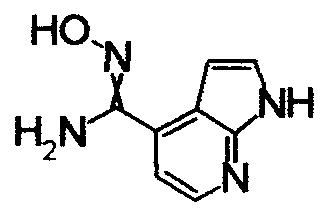
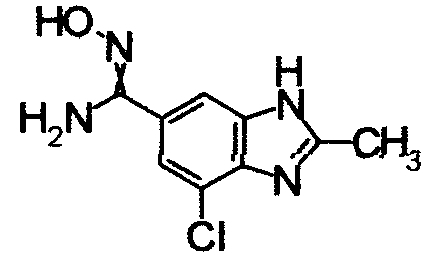
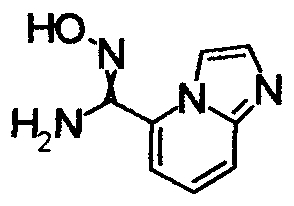
Пример Получения 3
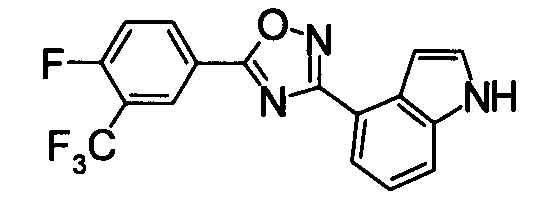
Суспензию N2-гидрокси-1Н-индол-4-карбоксамида (1,00 г), 4-фтор-3-(трифторметил)бензойной кислоты (1,19 г) и EDCI/HCl (1,32 г) в диоксане (30 мл) перемешивали при комнатной температуре в течение 1 часа, и затем нагревали при кипении в течение 18 часов. Реакционную смесь концентрировали при уменьшенном давлении, добавляли хлороформ и воду, затем перемешивали. Нерастворившуюся часть отделяли фильтрованием. Органический слой маточной жидкости промывали водой, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали при уменьшенном давлении. Нерастворимые примеси отделяли фильтрованием, вместе с маточной жидкостью очищали при помощи хроматографии на силикагеле (н-гексан:EtOAc=80:20). К целевому веществу добавляли ацетон, затем растворяли при нагревании и добавляли н-гексан, выпавший осадок отделяли фильтрованием, получая 4-{5-[4-фтор-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол (391 мг) в виде белого твердого вещества.
Пример Получения 4
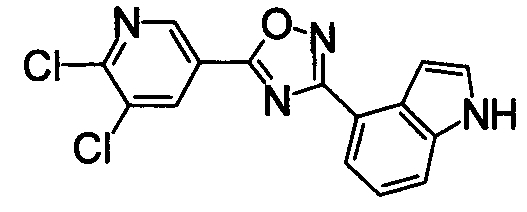
N2-{[(5,6-Дихлорпиридин-3-ил)карбонил]окси}-1H-индол-4-карбоксамид (1,91 г) добавляли к диоксану (40 мл), затем нагревали при кипении в течение 5 часов. Реакционную смесь концентрировали при уменьшенном давлении и затем очищали при помощи колоночной хроматографии на силикагеле (EtOAc). К полученному твердому веществу добавляли ацетон, затем суспендировали при нагревании. После того как раствору позволили охладиться, нерастворимые частицы удаляли фильтрованием, получая 4-[5-(5,6-дихлорпиридин-3-ил)-1,2,4-оксадиазол-3-ил]-1H-индол (1,44 г) в виде бледно-желтого порошка.
329
Пример Получения 5
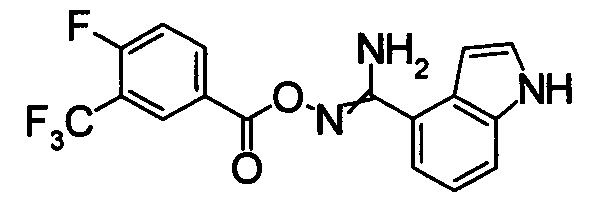
Раствор N2-гидрокси-1Н-индол-4-карбоксамида (3,42 г) и 4-фтор-3-(трифторметил)бензойной кислоты (4,07 г) в ТГФ (70 мл) охлаждали до -10°С или ниже и добавляли DIC (3,7 мл). После перемешивания от при -15 до -5°С в течение 3 часов реакционную смесь концентрировали при уменьшенном давлении. Остаток суспендировали в хлороформе и затем нерастворившуюся часть отделяли фильтрованием. Полученный порошок очищали при помощи хроматографии на силикагеле (н-гексан:EtOAc=50:50), получая N2-{[4-фтор-3-(трифторметил)бензоил]окси}-1H-индол-4-карбоксамид (8,40 г) в виде белого твердого вещества.
Соединение, показанное в Pr 5-1, получали тем же самым образом, как в Примере Получения 5.
366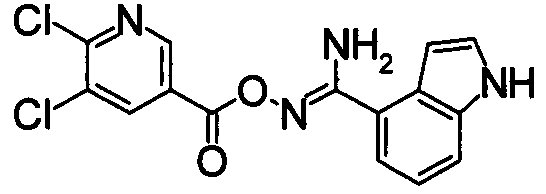
Пример Получения 6
К раствору 6-амино-2-метилникотинонитрила (960 мг) в этаноле (34 мл) добавляли 40% водный раствор хлорацетальдегида (2,36 мл) при 60°С. Реакционную смесь кипятили в течение 8 часов. Образующийся осадок отделяли фильтрованием, получая 5-метилимидазо[1,2-а]пиридин-6-карбонитрил гидрохлорид (580 мг) в виде белого твердого вещества.
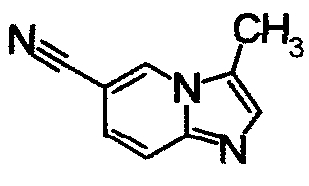
Соединения, показанные в Pr 6-1 до Pr 6-11, получали тем же самым образом, как в Примере Получения 6.
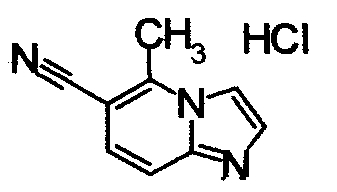
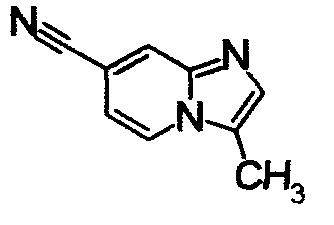
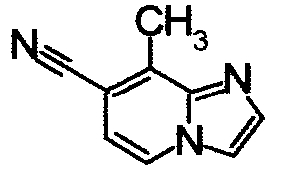
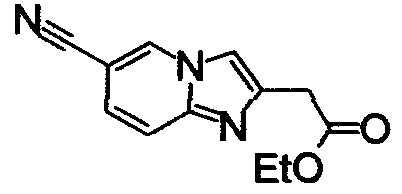
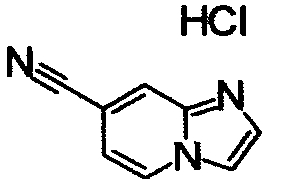
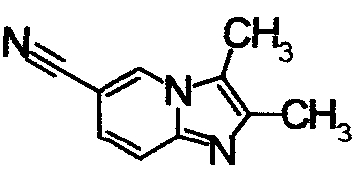
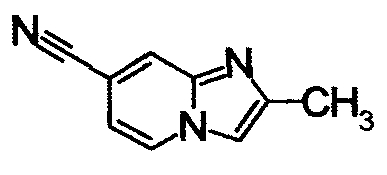
ниже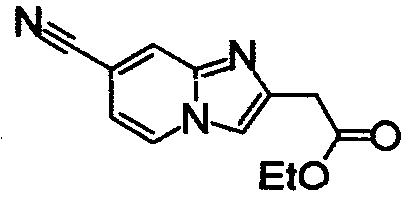
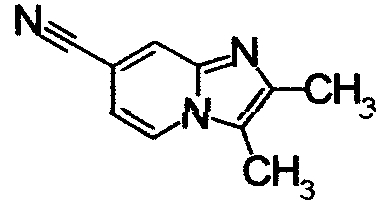
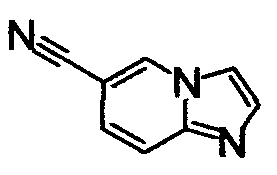
ниже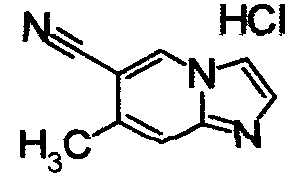
Пример Получения 7
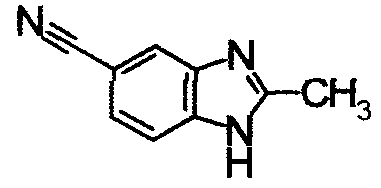
К раствору 3,4-диаминобензонитрила (500 мг) в AcOH (10 мл) добавляли Ас2О (372 мкл) при комнатной температуре. Реакционную смесь кипятили в течение 15 часов (масляная баня при 150°С). Реакционную смесь охлаждали до комнатной температуры и концентрировали до тех пор, пока количество АсОН не уменьшилось до половины. Реакционную смесь нейтрализовали водным раствором Na2CO3 и экстрагировали EtOAc. Органический слой промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали колоночной хроматографией на силикагеле, получая 2-метил-1Н-бензимидазол-5-карбонитрил (390 мг) в виде бледно-красного вещества.
Пример Получения 8
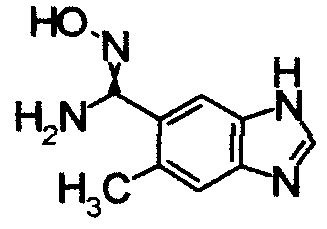
Реакционную смесь 4,5-диамино-2-метилбензонитрила (20 мг) и муравьиной кислоты (6 мл) кипятили в течение 3 часов. Реакционный раствор охлаждали и концентрировали. К остатку добавляли 1М водный раствор NaOH и экстрагировали EtOAc. Органический слой сушили над безводным MgSO4 затем фильтровали и фильтрат концентрировали, получая 5-метил-1Н-бензимидазол-6-карбонитрил в виде окрашенного порошка.
Соединения, показанные в Pr 8-1, получали тем же самым образом, как в Примере Получения 8.
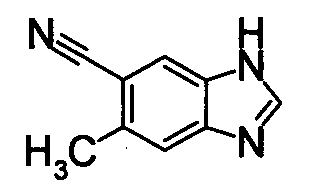
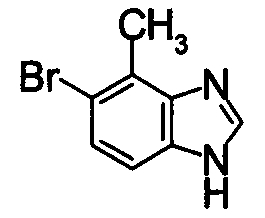
213
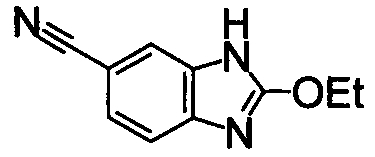
Пример Получения 9
В раствор 3,4-диаминобензонитрила (400 мг) в этилортоформиате (6,48 г) добавляли АсОн (238 мг), затем перемешивали при 80°С в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры и разделяли между EtOAc и 1М водным раствором NaOH. Органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO3 и затем отделяли фильтрованием и фильтрат концентрировали и очищали при помощи колоночной хроматографии на силикагеле, получая 2-этокси-1Н-бензимидазол-6-карбонитрил (164 мг) в виде окрашенного порошка.
Пример Получения 10
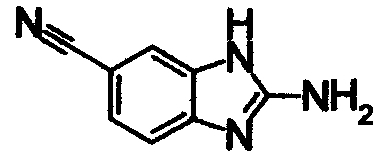
К суспензии 3,4-диаминобензонитрила (400 мг) в СН3ОН (4 мл) добавляли BrCN (477 мг), затем перемешивали при 20°С в течение 14 часов. К реакционной смеси добавляли 1М водный раствор NaOH (0,117 мл), затем концентрировали. К остатку добавляли хлороформ:СН3ОН=10:1 (10 мл) и образовавшиеся нерастворимые примеси удаляли фильтрованием. Фильтрат концентрировали и полученный остаток очищали при помощи колоночной хроматографии на силикагеле, получая 2-амино-1Н-бензимидазол-6-карбонитрил (311 мг) в виде светло-оранжевого порошка.
Пример Получения 11
К раствору 3,4-диаминобензонитрила (350 мг) в толуоле (5,5 мл) добавляли CDI (554 мг), затем перемешивали при 125°С в течение 2 часов. К реакционной смеси добавляли 1М водный раствор NaOH (0,117 мл) и затем экстрагировали EtOAc. Органически слой сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток перетирали в порошок/промывали IPE/IPA, получая 2-оксо-2,3-дигидро-1Н-бензимидазол-5-карбонитрил (423 мг) в виде окрашенного порошка.
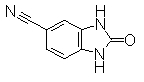
8,2Гц), 11,12(2H, ушир.)
Пример Получения 12
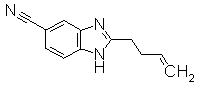
К смешанному раствору N-(4-циано-2-нитрфенил)пента-4-енамида (1,0 г) в АсОН)/этанол (1:1,20 мл) добавляли порошок железа (710 мг). Реакционный раствор нагревали при 110°С в течение 3 часов и затем концентрировали. К остатку добавляли хлороформ, затем нейтрализовали насыщенным водным раствором NaHCO3. Органический слой сушили безводным MgSO4 и затем фильтровали для удаления осушителя и растворитель концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле, получая 3-бутенил-1Н-бензимидазол-5-карбонитрил (405 мг) в виде окрашенной жидкости.
Соединения, показанные в Pr 12-1 до Pr 12-2, получали тем же самым образом, как и в Примере Получения 12.
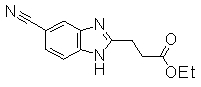
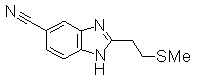
Пример Получения 13
К раствору 2-фтортерефталонитрила (500 мг) и Et3N (572 мкл) в EtOH (20 мл) добавляли гидразин (моногидрат), затем смеси давали прореагировать при 60°С в течение 16 часов и концентрировали. Остаток промывали диэтиловым эфиром, получая 3-амино-1Н-имидазол-6-карбонитрил (488 мг) в виде желтого твердого вещества.
Соединение, показанное в Pr 13-1, получали таким же образом, как в Примере Получения 13.
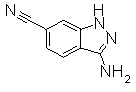
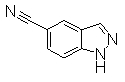
Пример Получения 14
К суспензии 3-амино-1Н-имидазол-6-карбонитрила (345 мг) в АсОН медленно добавляли водный раствор NaNO2 (301 мг) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2,5 дней и остаток отделяли фильтрованием и промывали холодной водой. К остатку добавляли 0,1М HCl и DME, затем перемешивали при 80°С в течение 2 часов. Реакционную смесь нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали EtOAc. Органический слой сушили над безводным MgSO4 и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (н-гексан:EtOAc=80:20 до 50:50), получая 1Н-индазол-6-карбонитрил (175 мг) в виде желтого твердого вещества.
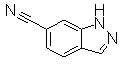
Пример Получения 15
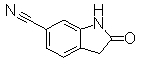
Метил(4-циано-2-нитрофенил)ацетат (128 мг) растворяли в АсОН (3,0 мл), затем добавляли порошок железа (129 мг) и реакционный раствор перемешивали на масляной бане при 100°С в течение 1,5 часов. Реакционный раствор концентрировали для того чтобы удалить АсОн, затем добавляли EtOAc. Коричневое твердое вещество отделяли фильтрованием и органический слой промывали 1М HCl и насыщенным раствором соли, сушили над безводным MgSO4 и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=99:1 до 95:5), получая 2-оксоиндолин-6-карбонитрил (52,0 мг) в виде светло-желтого твердого вещества.
Пример Получения 16
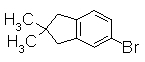
К раствору 6-бром-2,2-диметилинданметилиндан-1-она (124 мг) в TFA (4,44 г, 3,0 мл) добавляли триэтилсилан (150 мг, 207 мкл) при комнатной температуре. После перемешивания при комнатной температуре в течение 2,5 дней к реакционному раствору добавляли воду для остановки реакции, затем промывали водой и насыщенным раствором NaHCO3. Реакционную смесь экстрагировали EtOAc. Органический слой сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии (только н-гексан), получая 5-бром-2,2-диметилиндан (114 мг) в виде окрашенного масла.
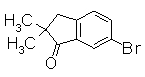
Пример Получения 17
В THF (30 мл) к смеси 6-бром-1-инданона (300 мг) и диазометана (504 мг, 221 мкл) добавляли 60% NaH (125 мг) при 0°С. Смесь перемешивали при комнатной температуре в течение 3,5 часов и промывали насыщенным раствором NH4Cl. Смесь экстрагировали EtOAc и органический слой сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали колоночной хроматографией на силикагеле (н-гексан:EtOAc=100:0 до 80:20), получая 6-бром-2,2-диметил-1-инданон (158 мг) в виде светло-желтого масла.
263
Пример Получения 18
К раствору 3-амино-4-гидроксибензонитрила (730 мг) в DMF (10 мл) добавляли CDI (1,06 г) при 0°С, затем перемешивали при комнатной температуре в течение 3,5 часов. Реакционный раствор разбавляли водой (10 мл) и экстрагировали EtOAc (200 мл). Органический слой сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=98:2 до 97:3), получая 2-оксо-2,3-дигидро-1,3-бензоксазол-5-карбонитрил (647 мг) в виде светло-желтого твердого вещества.
Соединения Pr 18-1 получали тем же самым образом, как и Примере Получения 18.
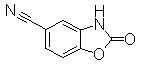
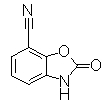
Пример Получения 19
К раствору 60% NaH (12,38 г) в DMF (480 мл) добавляли раствор 1Н-индол-4-карбонитрила (40,0 г) в DMF (80 мл) при 0°С. После перемешивания при 0°С в течение 30 мин раствор перемешивали при комнатной температуре в течение 0,5 часов. Впоследствии раствор 2-бромацетамида (40,76 г) в DMF (80 мл) добавляли по каплям при 0°С. Раствор нагревали от 0°С до комнатной температуры и перемешивали в течение 12 часов. К реакционному раствору добавляли воду (1200 мл) и выпавший твердый осадок отделяли фильтрованием. Раствор промывали горячей водой (300 мл) и диизопропиловым эфиром (200 мл), получая 2-(4-циано-1Н-индол-1-ил)ацетамид (5,21 г) в виде белого твердого вещества.
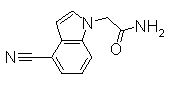
Пример Получения 20
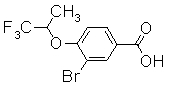
К раствору 3-хлор-4-(2,2,2-трифтор-1-метилэтокси)бензонитрила (430 мг) в IPA (3 мл) добавляли 5М водный раствор NaOH (1,37 мл), затем перемешивали при 80°С в течение 24 часов и в дальнейшем добавляли 5М водный раствор NaOH (1,37 мл), далее перемешивали при 95°С в течение 24 часов. Реакционный раствор концентрировали до тех пор, пока его количество не уменьшилось до половины. К остатку добавляли 12М HCl и образовавшийся осадок отделяли фильтрованием и затем сушили, получая 3-хлор-4-(2,2,2-трифтор-1-метилэтокси)бензойную кислоту в виде желтого твердого вещества.
Соединение, показанное в Pr 20-1 до Pr 20-3, получали тем же образом, как и в Примере Получения 20.
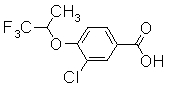
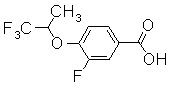
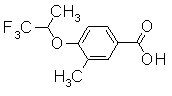
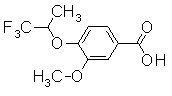
Пример Получения 21
К 3-(дифторметил)-4-(2,2,2-трифтор-1-метилэтокси)бензонитрил (234 мг) добавляли воду (2 мл) и серную кислоту (2 мл), затем кипятили течение 24 часов. После охлаждения до комнатной температуры реакционный раствор подщелачивали с помощью 5М водного раствора NaOH и экстрагировали диэтиловым эфиром (30 мл). Водный слой подкисляли 1М HCl и экстрагировали EtOAc. Органический слой сушили безводным MgSO4 и затем фильтровали и фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии (хлороформ:СН3ОН=97:3 до 90:10), получая 3-формил-4-(2,2,2-трифтор-1-метилэтокси)бензойную кислоту (151 мг) в виде белого твердого вещества.
Соединения, показанные в Pr 21-1 до Pr 21-6, получали тем же самым образом, как и в Примере Получения 21.
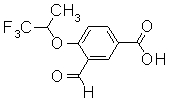
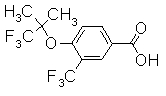
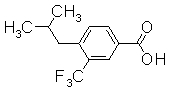
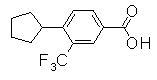
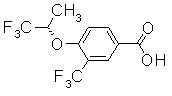
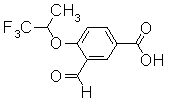
313
Пример Получения 22
К 5-бром-2-(2,2,2-трифтор-1-метилэтокси)бензонитрилу в смеси растворителей толуол/ТГФ (4:1) добавляли раствор n-BuLi в н-гексане при -78°С. Раствор перемешивали в течение 0,5 часов, пропуская через него газообразный СО2. К реакционному раствору добавляли 1М водный раствор NaOH для завершения реакции и затем экстрагировали диэтиловым эфиром. Органический слой подкисляли, добавляя 1М HCl, экстрагировали EtOAc, сушили над безводным MgSO4 и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=97:3 до 90:10), получая 3-циано-4-(2,2,2-трифтор-1-метилэтокси)бензойную кислоту в виде белого твердого вещества.
Соединение, показанное в Pr 22-1, получали тем же самым образом, как и в Примере Получения 22.
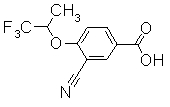
Пример Получения 23
К перемешиваемому раствору метил 1-изобутил-2-оксо-1,2-дигидропиридин-4-карбоксилата (430 мг) в CH3OH-THF (4 мл-3 мл) добавляли водный раствор 1М NaOH. Раствор перемешивали при комнатной температуре в течение 10 часов и затем концентрировали при уменьшенном давлении и добавляли воду (10 мл) и далее добавляли 1М HCl до тех пор, пока рН не достиг значения 3. Образовавшиеся твердое вещество отделяли фильтрованием, промывали водой и сушили при уменьшенном давлении, получая 1-изобутил-2-оксо-1,2-дигидроксипиридин-4-карбоновую кислоту (235 мг) в виде белого порошка.
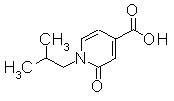
Пример Получения 24
Раствор 1Н-бензимидазол-5-карбоновой кислоты (75,0 г) в дихлорметане (750 мл) реагировал с оксалил хлоридом (76,3 г, 52,4 мл) при комнатной температуре в течение 1 часа, и затем реакционный раствор концентрировали. К раствору остатка в THF (750 мл) добавляли 28% водный раствор NH3 (5 мл) при охлаждении льдом. Эту реакционную смесь перемешивали при той же температуре и реакционный раствор концентрировали. Пурпурный остаток растирали в порошок/промывали IPE/IPA и отделяли фильтрованием, получая 1Н-бензимидазол-6-карбоксамид (129 г) (включая неорганические соли).
Соединение, показанное в Pr 24-1, получали тем же самым образом, как и в Примере Получения 24.
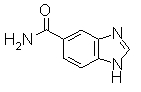
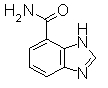
Пример Получения 25
Пример Получения 25-1
К раствору 1Н-1,2,3-бензотриазол-5-карбоновой кислоты (2 г), EDCI/HCl (2,82 г) и HOBt в DMF (70 мл) добавляли водный раствор NH3 (5,1 мл), затем смесь реагировала при комнатной температуре в течение 2 часов. Смесь концентрировали и остаток промывали насыщенным водным раствор NaHCO3, отделяли фильтрованием и сушили, получая 1Н-1,2,3-бензотриазол-5-карбоксамид (1,98 г) в виде черного твердого вещества.
Пример Получения 25-2
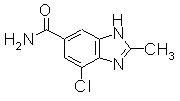
В реакционном сосуде на 50 мл к раствору метил 4-хлор-2-метил-1Н-бензимидазол-6-карбоксилат эфиру (300 мг) в формамиде (2,65 мл) добавляли NaОСН3 (288 мг) при комнатной температуре. Раствор перемешивали при 80°С в течение 3 часов. Завершение реакции определяли по ТСХ и ЖХ и затем реакционный раствор концентрировали и добавляли воду для завершения реакции. Реакционную смесь экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (автоматический очиститель, хлороформ:СН3ОН=100:0 до 90:10), получая 4-хлор-2-метил-1Н-бензимидазол-6-карбоксамид (257 мг) в виде белого твердого вещества.
Соединения, показанные в Pr 25-2, получали тем же образом, как и в Примере Получения 25-1.
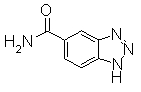
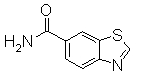
234
Пример Получения 26
К раствору 3-формил-4-(2,2,2-трифтор-1-метиэтокси)бензойной кислоты (490 мг) и К2СО3 (387 мг) в ацетоне (10 мл) добавляли йодметан (350 мкл) при комнатной температуре и затем перемешивали в течение 2 часов. Реакционную смесь разбавляли водой (15 мл) и экстрагировали EtOAc (30 мл). Органический слой сушили над безводным MgSO4 и фильтровали для удаления осушителя и растворитель концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (н-гексан:EtOAC=95:5 до 80:20), получая метил 3-формил-4-(2,2,2-трифтор-1-метилэтокси)бензоат (122 мг) в виде твердого вещества.
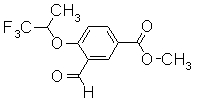
Пример Получения 27
При охлаждении льдом к DMF (30 мл) медленно добавляли по каплям РОСl3 (6,68 г, 4,06 мл), далее смесь реагировала при комнатной температуре в течение 2 часов и затем добавляли раствор 1Н-бензимдазол-6-карбоксамида (2,38 г) в DMF (47,6 мл) и затем смесь перемешивали при комнатной температуре в течение 2 часов. К раствору добавляли 1М водный раствор NaOH (рН 6-7), далее перемешивали при комнатной температуре в течение 0,5 часов. Раствор экстрагировали EtOAc и органические слои объединяли и промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии и растирали в порошок/промывали IPE, получая 1Н-бензимидазол-6-карбонитрил (0,58 г) в виде бледно-красных кристаллов.
Соединения, показанные в Pr 27-1 до Pr 27-2, получали тем же самым образом, как и в Примере Получения 27.
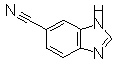
Пример Получения 28
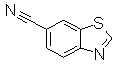
Раствор 1,3-бензотиазол-6-карбоксамида (1,96 г) в POCl3 (10 мл) кипятили в течение 4 часов. Реакционный раствор концентрировали и медленно добавляли воду при 0°С. Смесь экстрагировали EtOAc и органический слой сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии (н-гексан:EtOAc=80:20 до 60:40), получая 1,3-бензотиазол-6-карбонитрил в виде светло-желтого твердого вещества.
Следующий Pr 28-1 получали тем же самым образом, как и в Примере Получения 28
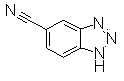
Пример Получения 29
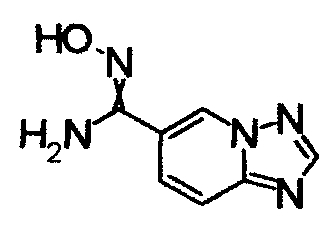
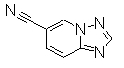
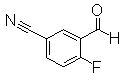
К раствору 6-бром[1,2,4]триазоло[1,5-а]пиридина (400 мг) в DMF добавляли трис(дибензилдиацетно)дипалладий (0), 1'-бис(дифенилфосфино)ферроцен и Zn(CN)2 в атмосфере азота, далее перемешивали при 110°С в течение 23 часов. Смесь охлаждали до комнатной температуры и добавляли насыщенный NH4Cl (2 мл), насыщенный раствор NH3 (6 мл) и Н2О (12 мл). Реакционную смесь трижды экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли и сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи хроматографии на силикагеле (0-5% СН3ОН/хлороформ), получая [1,2,4]триазоло[1,5-а]пиридин-6-карбнитрил в виде темно-красного твердого вещества.
Пример Получения 30
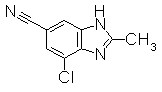
В реакционном сосуде на 100 мл к раствору 6-бром-7-метил-1Н-бензимидазола (500 мг) в DMF добавляли Zn(CN)2 (834 мг) и Pd(PPh)4 (547 мг) при комнатной температуре, далее перемешивали при 150°С в течение 5 часов. Реакционный раствор выливали в смесь 1:1 насыщенного раствора NaHCO3 и EtOAc и перемешивали 1 час. Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем отфильтровывали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии (автоматический очиститель, хлороформ:СН3ОН=98:2 до 90:10), получая 7-метил-1Н-бензимидазол-6-карбонитрил (161,8 мг) в виде коричневого твердого вещества.
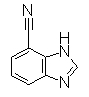
Следующие Pr 30-1 до Pr 30-7 получали тем же самым образом, как и в Примере Получения 30. Они также были получены при помощи метода Примера Получения 29.
(1H, д), 8,04
(1H, д), 8,74
(1H, с), 9,90-
9,87(1H, м)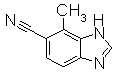
(1H,дд), 8,25
(1H, ддд),
8,34(1H, дд), 10,17(1H, с)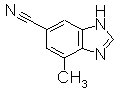
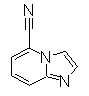
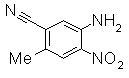
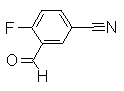
(1H, с), 8,34
(1H, дд), 8,25
(1H, ддд),
7,67(1H, дд)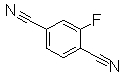
7,95(1H,
дд), 8,19
(1H, дд),
8,23(1H,дд)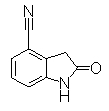
Пример Получения 31
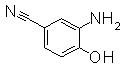
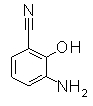
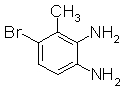
К смешанному раствору 4-гидрокси-3-нитробензонитрила (1 г) и NH4Cl (163 мг) в этаноле (20 мл), THF (10 мл) и в воде (10 мл) добавляли Целит (5 г) и восстановленное железо (1,7 г) далее нагревали при кипении при 70°С в течение 30 мин. Реакционный раствор охлаждали до комнатной температуры, разбавляли EtOAc (200 мл) и затем фильтровали через целит. Раствор промывали насыщенным раствором соли, органический слой сушили над безводным MgSO4 и фильтровали и фильтрат упаривали при уменьшенном давлении, получая 3-амино-4-гидроксибензонитрил (740 мг) в виде светло-коричневого твердого вещества.
Следующие Pr 31-1 до Pr 31-3 были приготовлены тем же самым образом, как и в Примере Приготовления 31.
227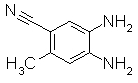
Пример Получения 32
К раствору 4-амино-3-нитробензонтрила (8 г) в смеси растворителей EtOH/THF (40 мл/40 мл) добавляли Pd-C (50% по весу) (0,8 г), далее перемешивали в атмосфере H2 в течение 12 часов. Реакционный раствор отфильтровывали через целит и концентрировали. Остаток растирали в порошок/промывали смесью растворителей IPE и IPA и отделяли фильтрованием, получая 3,4-диаминобензонитрил (6,3 г) в виде оранжевого порошка.
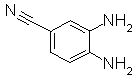
Пример Получения 33
К раствору 2-амино-3-нитробензонитрила (2 г) в THF (30 мл) добавляли 4-петиноилхлорид (2,90 г) и диизопропилэтиламин (4,27 г) далее перемешивали при 80°С в течение 12 часов. Реакционный раствор выливали в воду и экстрагировали EtOAc. Органический слой сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:CH3OH), получая N-(4-циано-2-нитрофенил)пент-4-енамид (174 мг) в виде окрашенной жидкости.
Следующие Pr 33-1 до Pr 33-2 получали тем же самым образом, как и в Примере Получения 33.
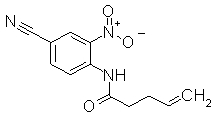
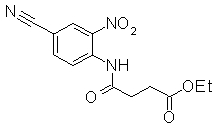
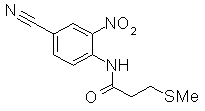
Пример Получения 34
К раствору 3-хлор-4-фторбензонитрила (300 мг) и 1,1,1-трифтор-2-пропанола в THF (15 мл) добавляли 60% NaH (92,5 мг) при 5°С, далее перемешивали при комнатной температуре в течение 2 часов, добавляли насыщенный раствор NH4Cl для завершения реакции и экстрагировали EtOAc. Полученный органический слой сушили над безводным MgSO4 и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (н-гексан:EtOAc=97:3 до 85:15, получая 3-хлор-4-(2,2,2-трифтор-1-метилэтокси)бензонитрил (435 мг) в виде окрашенного маслянистого вещества.
Следующие Pr 34-1 до Pr 34-6 получали тем же самым образом, как и в Примере Получения 34.
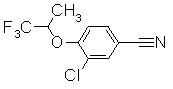
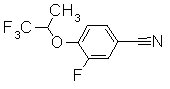
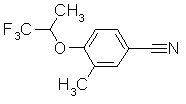
252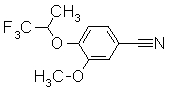
268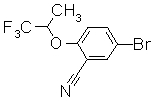
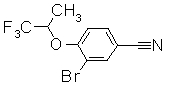
316,
318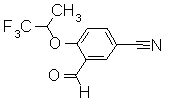
242
Пример Получения 35
В реакционном сосуде на 50 мл к раствору 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол (100 мг) в DMF (1 мл) добавляли 60% NaH (10,9 мг) при 0°С. Далее трет-бутил(2-йодэтокси)диметилсилан добавляли к смеси при 0°С, затем перемешивали при комнатной температуре в течение 15 часов. Завершение реакции подтверждали при помощи ХЖ-МС и затем к реакционному раствору добавляли воду (30 мл). Смесь экстрагировали трижды EtOAC (20 мл). Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (автоматический очиститель, (н-гексан:EtOAC=100:0 до 90:10), получая 1-(2-{[трет-бутил(диметил)силил]окси}этил)-4-{5-[3-(трифторметил)-4-2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол (86,4 мг) в виде белого твердого вещества.
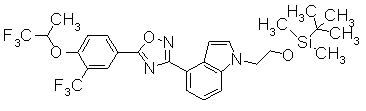
Пример Получения 36
LiH суспензировали в DMF (5 мл) и к той суспензии добавляли по каплям суспензию метил-2-оксо-1,2-дигидропиридин-4-карбоксилата (500 мг) в DMF (5 мл) при комнатной температуре. Суспензию перемешивали и добавляли раствор 1-йод-2-метилпропана (506 мкл) в DMF (5 мл) по каплям в течение 10 мин, далее перемешивали при 50°С в течение 15 часов. К реакционному раствору добавляли 1М HCl при 0°С и далее экстрагировали EtOAc и органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и фильтровали и растворитель упаривали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (н-гексан:EtOAc=90:10 до 50:50), получая метил-1-изобутил-2-оксо-1,2-дигидропиридин-4-карбоксилат (440 мг) в виде белого порошка.
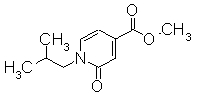
Пример Получения 37
К раствору 4-фтор-3-нитробенонитрила (300 мг) и диметилмалоната (286 мг) в DMF добавляли 60% NaH при 0°С, далее смесь реагировала при комнатной температуре, образуя диметил(4-циано-2-нитрофенил)малонат (198 мг).
Пример Получения 38
К раствору диметил(4-циано-2-нитрофенил)малоната (198 мг) в DMSO (5 мл) добавляли LiCl (60,3 мг) и Н2О (12 мкл), далее перемешивали при 100°С в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и выливали в EtOAc и насыщенный раствор соли порциями. Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (н-гексан:EtOAc=90:10 до 75:25), получая метил(4-циано-2-нитрофенил)ацетат (128 мг) в виде желтого твердого вещества.
Пример Получения 39
К раствору 4-хлор-3-(трифторметил)бенонитрила (1 г) и железа(3+)трис[(2Z)-4-окскопент-2-ен-2-олеата] (86 мг) и 1-метилпирролидин-2-она (2,8 мл) в THF (30 мл) добавляли раствор 3М бром(изобутил)магния в диэтиловом эфире (2,9 мл) при охлаждении льдом. Раствор перемешивали при комнатной температуре в течение 30 минут и разбавляли диэтиловым эфиром (30 мл) и затем осторожно добавляли 1М HCl для завершения реакции. Реакционный раствор экстрагировали EtOAc (100 мл) и органический слой сушили над безводным MgSO4 и затем фильтровали и фильтрат упаривали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (н-гексан:EtOAc=100:5 до 95:5), получая 4-изобутил-3-(трифторметил)бензонитрил (320 мг) в виде светло-желтой жидкости.
Следующий Pr 39-1 получали тем же самым образом, как и в Примере Получения 39.
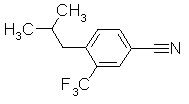
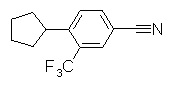
Пример Получения 40
К раствору 4-фтор-3-формилбензонитрила (300 мг) в дихлорметане (7 мл) добавляли 2-меткоси-N-(2-метоксиэтил)-N-(трифтор-λ4-сульфанил)этанамин (757 мг) при комнатной температуре, далее перемешивали в течение 6 часов и добавляли насыщенный водный раствор NaHCO3 (15 мл). После экстракции хлороформом (30 мл) органический слой сушили над безводным MgSO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (н-гексан:EtOAc=95:5 до 80:20), получая 3-(дифторметил)-4-фторбензонитрил (174 мг) в виде окрашенной жидкости.
Следующий Pr 40-1 получали таким же образом, как и в Примере Получения 40.
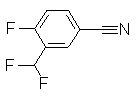
т), 7,66(1H, дд), 8,14-8,20(1H,м), 8,22(1H, дм)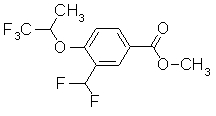
321
Пример Получения 41
Следующие Pr 41-1 до Pr 41-10 получали тем же самым образом, как и в Примере 2.
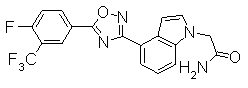
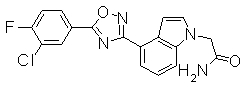
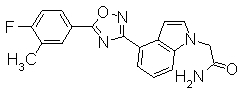
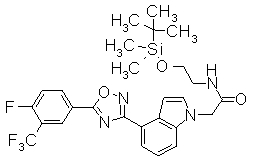
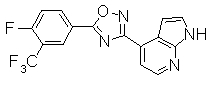
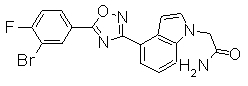
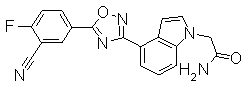
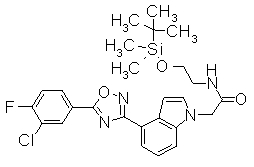
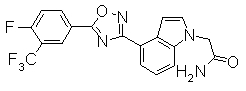
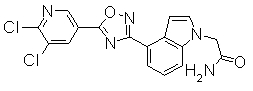
Пример Получения 42
Следующие Pr 42-1 до Pr 42-3 получали тем же самым образом, как и в Примере 5.
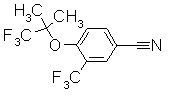
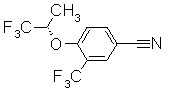
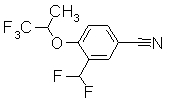
MS:288
Пример Получения 43
Следующий Pr 43 получали таким же образом, как и в Примере 6.
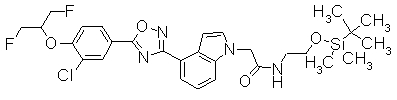
Пример Получения 44
Следующие Pr 44-1 до Pr 44-3 получали тем же самым образом, как и в Примере 12.
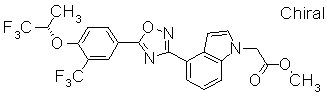
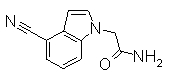
Пример Получения 45
Следующий Pr 45 получали тем же самым образом, как и в Примере Получения 47.
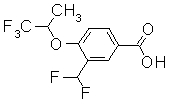
8,07-8,13(2H, м)
Пример Получения 46
Следующие Pr 46-1 до Pr 46-5 получали тем же самым образом, как и в Примере 19.
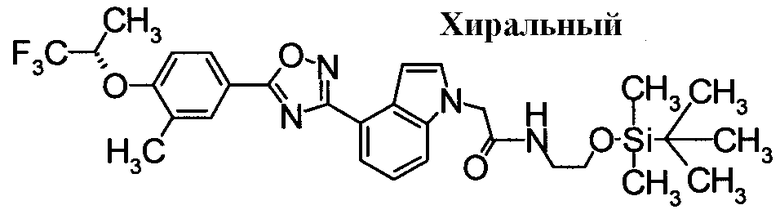
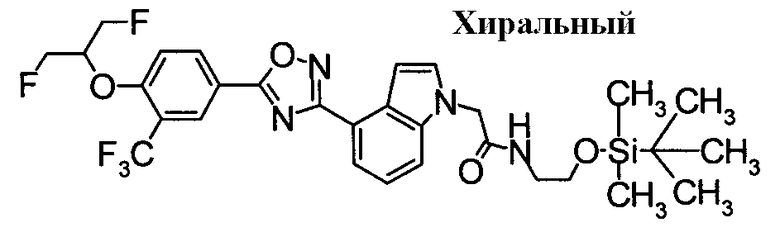
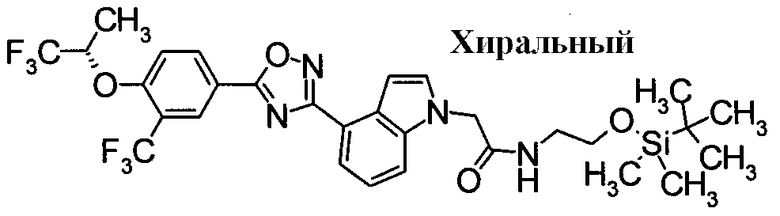
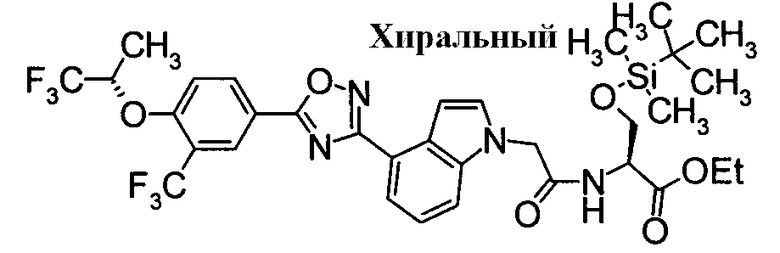
Пример Получения 47
К раствору этил(7-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксаиазол-3-ил}имидазо[1,2-a]пиридин-2-ил)ацетата (230 мг) в THF (2,0 мл) добавляли водный раствор NaOH (1 мл), далее перемешивали при 80°С в течение 2 часов. После охлаждения до комнатной температуры добавляли 1М HCl, затем экстрагировали хлороформом. Органический слой сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=10:1 до 5:1), получая окрашенный порошок. К раствору этого окрашенного порошка в EtOAc добавляли раствор 4М HCl/EtOAc, затем концентрировали. Образовавшийся окрашенный порошок растирали в порошок/промывали IPE, получая (7-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксаиазол-3-ил}имидазо[1,2-a]пиридин-2-ил)уксусной кислоты гидрохлорид (180,4 мг) в виде окрашенного порошка.
Следующие Pr 47-1 до Pr 46-13 получали тем же самым образом, как и в Примере Получения 47.
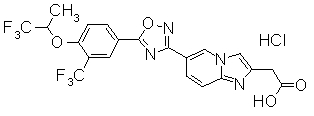
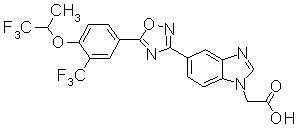
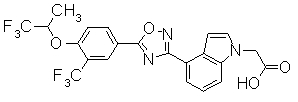
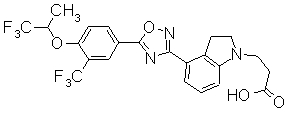
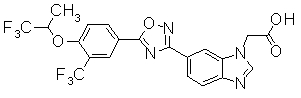
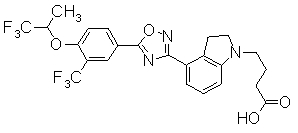
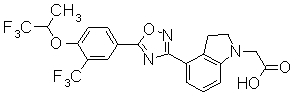
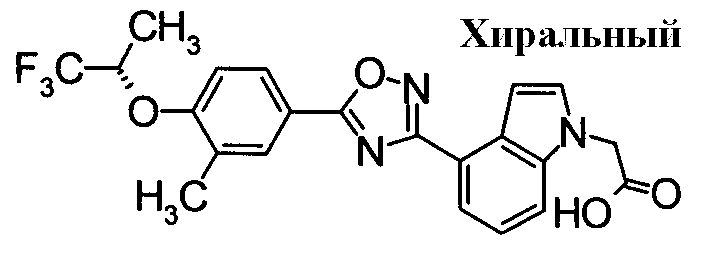
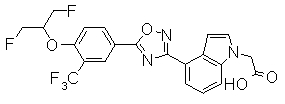
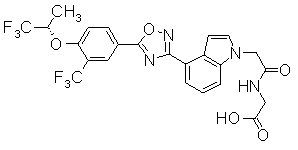
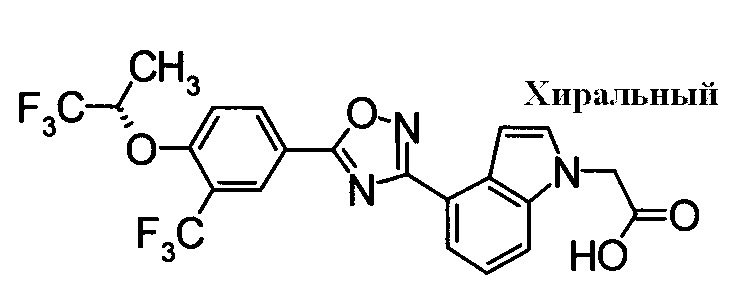
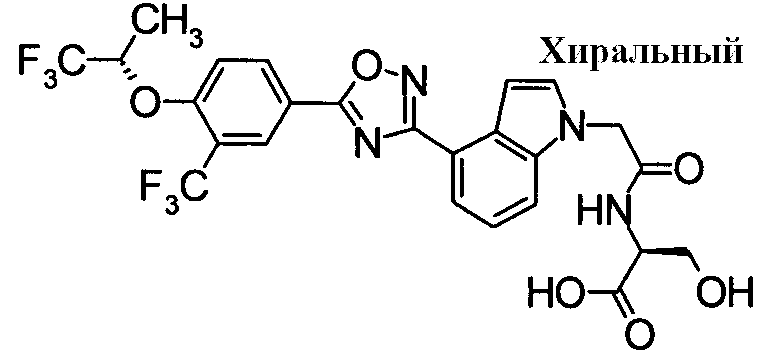
Пример 1
Раствор 3-(трифторметил)-4-(2,2,2-трифтор-1-меткосиметил)бензойной кислоты (810 мг), EDCI/HCl (616 мг) и N'-гидрокси-7-метилимидазо[1,2-а]пиридин-6-карбоксамида (510 мг) в диоксане перемешивали при 115°С в течение 60 часов. Реакционный раствор концентрировали и остаток распределяли между водой и хлороформом. Органический слой сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали колоночной хроматографией на силикагеле (СН3ОН/хлороформ=0 до 5%) и перекристаллизовывали из EtOH, получая 7-метил-6-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}имидазо[1,2-a]пиридин (60 мг) в виде белого твердого вещества.
Пример 2
К раствору 3-(трифторметил)-4-(2,2,2-трифтор-1-меткосиметил)бензойной кислоты (349 мг) в дихлорметане (6 мл) добавляли оксалил хлорид (333 мг) и каталитическое количество DMF при охлаждении льдом, далее перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали и сушили азеотропной отгонкой с толуолом. К раствору остатка в THF добавляли N'-гидрокси-2-метил-1Н-бензимидазол-6-карбоксиимидамид (200 мг), N-этил-N-изопропил-2-пропаненамин (543 мг). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. К реакционному раствору добавляли воду, далее экстрагировали трижды EtOAc. Органические слои объединяли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток растворяли в диоксане, далее перемешивали при 100°С в течение 3 часов. После охлаждения до комнатной температуры раствор концентрировали при уменьшенном давлении для удаления растворителя и очищали при помощи колоночной хроматографии на силикагеле, получая окрашенное маслянистое вещество. К раствору этого маслянистого вещества в EtOAc добавляли раствор 4М HCl/EtOAc, далее перемешивали в течение нескольких минут и концентрировали, получая 2-метил-5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазола гидрохлорид (239 мг) в виде окрашенных кристаллов.
Пример 3
К раствору 4-(2,2,2-трифтор-1,1-диметилэтокси)-3-(трифторметил)бензойной кислоты (118 мг) и 2-{4-[амино(гидроксиимино)метил]-1H-индол-1-ил}ацетамида (104 мг) в диоксане (5 мл) добавляли DIC (69 мкл) далее перемешивали при комнатной температуре в течение 3 часов и затем нагревали при кипении в течение 20 часов. Реакционный раствор концентрировали и затем к остатку добавляли воду (15 мл), затем экстрагировали хлороформом (15 мл). Органический слой промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи препаративной ВЖХ (колонка: CAPCEL PAK, C18, MG, S-5, 30×50 мм; растворитель: 50-90% ацетонитрил/10 мМоль карбонат аммония-аммиак (рН 9,2); 40 мл/мин) и перекристаллизовывали из диизопропилового эфира, получая 2-(4{5-[4-(2,2,2-трифтор-1,1-диметлиэтокси)-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-ил)ацетамид (40 мг) в виде белого твердого вещества.
Пример 4
К суспензии 60% NaH (68,0 мг) в DMF добавляли циклопропилметанол (99 мг) при 0°С, далее перемешивали при той же температуре затем добавляли 5-{5-[4-фтор-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазол (120 мг). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем добавляли воду. Смесь экстрагировали EtOAc и органический слой концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (СН3ОН/хлороформ=0 до 5%), получая маслянистое вещество. В раствор маслянистого вещества в хлороформе-СН3ОН добавляли раствор 4М HCl/диоксан (0,5 мл) и концентрировали получая 5-{5-[4-(циклопропилметокси)-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазол гидрохлорид (20 мг) в виде белого вещества.
ВЭЖХ анализ: Условия (TSK-GEL (TOSOH) ODS-80TM 4,6×150 мм, MeCN: 0,01M KH2PO4 (7:3), 1,0 мл/мин, 254 нм)[RT: 7,90 мин]
Пример 5
К раствору 2-(4-{5-[4-фтор-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-ил)ацетамида (100 мг) и 2-пропанола (35 мкл) в DMF (3 мл) добавляли 60% NaH (12 мг) при 0°С далее перемешивали при комнатной температуре в течение 9 часов. К реакционному раствору добавляли воду (5 мл) для завершения реакции и экстрагировали смесью растворителей хлороформ:СН3ОН (8:2). Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=98:2 до 93:7) и кристаллизовали из диэтилового эфира, получая 2-(4-{5-[4-изопропокси-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-ил)ацетамид (25 мг) в виде светло-желтого твердого вещества.
Пример 6
К раствору 1,3-дифторпропанола (62 мг) в DMF (2,4 мл) добавляли 60% NaH (19 мг) при -10°С, далее перемешивали при -10°С, в течение 0,5 часов. К этой реакционной смеси добавляли 2-{4-[5-(3-хлор-4-фторфенил)-1,2,4-оксадиазол-3-ил]-1H-индол-1-ил}ацетамид (120 мг) при -10°С и далее перемешивали при -10°С в течение 3 часов. После добавления воды к реакционному раствору реакционную смесь экстрагировали EtOAc, органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=100:0 до 95:5), получая 2-[4-(5-{3-хлор-4-[2-фтор-1-(фторметил)этокси]фенил}-1,2,4-оксадиазол-3-ил)-1H-индол-1-ил]ацетамид (76,9 мг) в виде белого вещества.
Пример 7
К раствору 2-{4-[5-(4-фтор-3-метилфенил)-1,2,4-оксадиазол-3-ил]-1H-индол-1-ил}ацетамида (100 мг) и (2R)-1,1,1-трифторпропан-2-ола (109 мг) в DMF (3 мл) добавляли 60% NaH (17 мг) при 0°С далее перемешивали при 80°С в течение 4 часов. В реакционный раствор добавляли воду (15 мл) для завершения реакции, фильтровали и затем сушили. Полученный порошок очищали при помощи колоночной хроматографии (хлороформ:СН3ОН=100:0 до 95:5) и кристаллизовали из диизопропилового эфира, получая 2-[4-(5-{3-метил-4-[(1R)-2,2,2-трифтор-1-метилэтокси]фенил}-1,2,4-оксадиазол-3-ил)-1H-индол-1-ил]ацетамид (70 мг) в виде светло-желтого твердого вещества.
Пример 8
К суспензии 60% NaH (43 мг) в DMF (4 мл) добавляли 2-пропанол (65 мг) при 0°С, далее перемешивали при комнатной температуре в течение 20 мин. После охлаждения до 0°С добавляли 2-{4-[5-(3-хлор-4-фторфенил)-1,2,4-оксадиазол-3-ил]-1H-индол-1-ил-ацетамид (200 мг). Реакционную смесь облучали микроволновым излучением при 60°С в течение 50 мин. Реакционную смесь добавляли к водному раствору NH4Cl, далее перемешивали и затем растворитель упаривали. После добавления смеси растворителей (4:1) хлороформ-СН3ОН и суспендирования твердые частицы отделяли, добавляли силикагель и растворитель упаривали. Остаток очищали при помощи колоночной хроматографии (хлороформ:СН3ОН=100:0 до 98:2; н-гексан:EtOAc=0:100), получая 2-{4-[5-(3-хлор-4-изопропилфенил)-1,2,4-оксадиазол-3-ил]-1H-индол-1-ил}ацетамид (17,5 мг) в виде белого твердого вещества.
Пример 9
К раствору 4-{5-[4-фтор-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1H-индола (300 мг) в THF (1,5 мл) добавляли пропан-2-амин (0,75 мл) и после закупоривания в трубку раствор перемешивали при 50°-55°С в течение 40 часов. Раствор концентрировали при уменьшенном давлении и затем очищали при помощи колоночной хроматографии (н-гексан:EtOAc). Полученное твердое вещество растворяли в ацетоне при нагревании и добавляли н-гексан и осадок отфильтровывали, получая 4-[3-(1H-индол-4-ил)-1,2,4-оксадиазол-5-ил]-N-изопропил-2-(трифторметил)анилин (295 мг).
Пример 10
К смешанному раствору 2-{4-[5-(5,6-дихлорпиридин-3-ил)-1,2,4-оксадиазол-3-ил]-1H-индол-1-ил}ацетамида (100 мг) в диоксане (2 мл) и NMP (2 мл) добавляли изопропиламин (220 мкл), далее перемешивали при 150°С в течение 1 часа в микроволновом реакционном сосуде. Реакционную смесь концентрировали при уменьшенном давлении и затем остаток очищали при помощи колоночной хроматографии на силикагеле (н-гескан:EtOAc=40:60 до 0:100) и полученный остаток суспендировали в диизопропиловом эфире при нагревании и отделяли фильтрованием, получая 2-(4-{5-[5-хлор-6-(изопропиламино)пиридин-3-ил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-ил)ацетамид (62 мг) в виде белого порошка.
Пример 11 (11-1 и 11-2)
К раствору 5-{5-[3-(трифтормтил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазола (105 мг) в DMF (3,15 мл) добавляли 60% NaH (31 мг) при охлаждении льдом, далее перемешивали при той же самой температуре в течение 15 мин и добавляли метилйодид (0,22 мл), затем перемешивали при комнатной температуре в течение 5 часов. К реакционной смеси добавляли воду, далее экстрагировали EtOAc, органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии (автоматический процессор, хлороформ:СН3ОН=10:1). Целевое соединение растворяли в EtOAc (5 мл), добавляли раствор 4М HCl/EtOAc (5 мл) и концентрировали, получая приблизительно 1:1 два региоизомера. Смесь кристаллизовали из ацетонитрила, получая 1-метил-5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазол гидрохлорид (12,1 мг). Маточную жидкость концентрировали, получая 1-метил-5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазол гидрохлорид и 1-метил-6-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазол гидрохлорид (70,2 мг) в виде окрашенного порошка.
Пример 12
К раствору 4-(5-{3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}-1,2,4-оксазол-3-ил)-1H-индола (150 мг) в DMF (1,5 мл) добавляли 60% NaH (16 мг) при 0°С и далее перемешивали при комнатной температуре в течение 0,5 часов. Затем к реакционной смеси добавляли 2-бромацетамид (70 мг) также при 0°С, далее перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли воду, затем экстрагировали EtOAc, органический слой отделяли, промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии (хлороформ:СН3ОН=100:0 до 90:10), получая 2-[4-(5-{3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилфенокси]фенил}-оксадиазол-3-ил)-1H-индол-1-ил]ацетамид (145 мг) в виде белого твердого вещества.
Пример 13
К раствору 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси]фенил]-1,2,4-оксадиазол-3-ил)индолина (100 мг) в ацетонитриле (2,5 мл) добавляли К2СО3 (46 мг) и 3-йодпропанамид (124 мг) при комнатной температуре, далее перемешивали при 80°С в течение 15 часов. К реакционному раствору добавляли насыщенный водный раствор NaHCO3, далее экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии (хлороформ:СН3ОН=100:0 до 90:10), получая 3-(4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-2,3-дигидро-1H-индол-1-ил)пропанамид (23,6 мг) в виде белого твердого вещества.
Пример 14
К раствору 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-3-карбальдегида (150,0 мг) в СН3ОН (1,5 мл) добавляли 40% раствор СН3ОН в СН3NH2 (74,5 мг) при 0°С. После нагревания до комнатной температуры раствор перемешивали при комнатной температуре в течение 4 часов. После подтверждения образования иминиевой соли органический растворитель упаривали при уменьшенном давлении. Остаток растворяли в EtOH (1,5 мл). К этому раствору добавляли NaBH4 (12,09 мг). После нагревания до комнатной температуры раствор перемешивали при комнатной температуре в течение 15 часов. К реакционному раствору добавляли воду (30 мл), далее экстрагировали трижды EtOAc (20 мл). Органический слой объединяли, промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (автоматический очиститель, хлороформ:СН3ОН=100:0 до 90:10), получая N-метил-1-(4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-3-ил)метанамин (87,4 мг) в виде белого вещества.
Пример 15
К раствору 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}индлина (100 мг) в DMF (1,0 мл) добавляли 69% NaH (10,9 мг) при 0°С, затем перемешивали при комнатной температуре в течение 0,5 часов. Алкилхлорид (24,1 мкл) прибавляли при 0°С, далее перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду (30 мл), далее экстрагировали трижды EtOAc (20 мл). Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (автоматический очиститель, н-гесан: EtOAc=90:10 до 60:40), получая 1-ацетил-4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}индолин (56,8 мг) в виде белых кристаллов.
Пример 16
К раствору 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индола (80 мг) в DMF (0,80 мл) добавляли 60% NaH (8,7 мг) при 0°С, затем перемешивали при комнатной температуре в течение 0,5 часа. Добавляли метансульфонилхлорид (21,1 мкл) при 0°С, далее перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду (30 мл), далее экстрагировали трижды EtOAc (20 мл). Органический слой объединяли, промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Фильтрат очищали при помощи колоночной хроматографии на силикагеле (автоматический очиститель, хлороформ:СН3ОН=100:0 до 98:2), получая 1-(метилсульфонил)-4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метиэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол (15,6 мг) в виде белого твердого вещества.
Пример 17
4-{5-[3-(Трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол (100 мг) растворяли в DMF (1,0 мл), затем добавляли 60% NaH (10,9 мг) при 0°С и перемешивали при комнатной температуре в течение 0,5 часа. Метилхлорид карбонат (26,3 мкл) прибавляли при 0°С, далее перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду (30 мл), далее экстрагировали трижды EtOAc (20 мл), органический слой объединяли, промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (автоматический очиститель, хлороформ:СН3ОН=100:0 до 98:2), получая метил 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-карбоксилат (98,6 мг) в виде белого твердого вещества.
Пример 18
К раствору 3-(5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазол-2-ил)пропановой кислоты (23,5 мг) в дихлорметане добавляли оксалил хлорид (0,01 мл) и несколько капель DMF, далее перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали для удаления растворителя и реагента. К раствору остатка в THF добавляли NH4ОН, далее перемешивали в течение 1 часа. К реакционному раствору добавляли насыщенный водный раствор NH4Cl, затем экстрагировали EtOAc. Органический слой сушили над безводным MgSO4 и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=10:1), получая окрашенный порошок. К раствору окрашенного порошка в EtOAc добавляли раствор 4N-HCl в EtOAc. Реакционную смесь концентрировали и остаток растирали в порошок и промывали IPE, получая 3-(5-{5-[3-(трифтометил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазол-2-ил)пропанамид гидрохлорид в виде светло-желтого порошка.
Пример 19
К раствору (4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-ил)уксусной кислоты (150 мг) и HOBt (65 мг) в DMF (1,5 мл) добавляли EDCI/HCl(69 мг) при 0°С, далее перемешивали при комнатной температуре в течение 1 часа. После охлаждения до 0°С добавляли 1-пиридин-2-илметанамин (39 мг) затем перемешивали при комнатной температуре в течение 15 часов. К реакционному раствору добавляли насыщенный водный раствор NaHCO3 для завершения реакции. Реакционный раствор экстрагировали EtOAc, органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=100:0 до 95:5), получая N-(пиридин-2-илметил)-2-(4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-ил)ацетамид (157,8 мг) в виде твердого белого вещества. К раствору N-(пиридин-2-илметил)-2-(4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-ил)ацетамида (120 мг) в метиленхлорид (2,2 мл) добавляли по каплям 10 эквивалентов 4М НС/диоксан, далее перемешивая 1 час. Реакционную смесь концентрировали при уменьшенном давлении и растирали в порошок/промывали в IPE. Полученное твердое вещество отделяли фильтрованием и сушили, получая N-(пиридин-2-илметил)-2-(4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-ил)ацетамид гидрохлорид (126 мг) в виде белого твердого вещества.
Пример 20
К раствору [4-(5-{3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метиэтокси]фенил}-1,2,4-оксадиазол-3-ил)-1H-индол-1-ил]уксусной кислоты (100 мг) в DMF (1 мл) добавляли CDI (39 мг) и через 30 минут добавляли метансульфонамид (23 мг) и 2,3,4,6,7,8,9,10-октагидрпиримид[1,2-а]азепин (37 мг), далее перемешивали при комнатной температуре в течение 15 часов. К реакционной смеси добавляли воду для завершения реакции. Реакционный раствор экстрагировали EtOAc, полученный органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=100:0 до 90:10), получая N-(метилсульфонил)-2-[4-(5-{3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}-1,2,4-оксадиазол-3-ил)-1H-индол-1-ил]ацетамид (56,4 мг) в виде белого твердого вещества.
Пример 21
РОCl3 (158,4 мкл) добавляли по каплям к раствору DMF (4 мл) при 0°С. После нагревания до комнатной температуры раствор перемешивали в течение 0,5 часов. Затем раствор 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индола (500,0 мг) в DMF (1 мл) добавляли при 0°С, далее перемешивали при комнатной температуре в течение 15 часов. После охлаждения до 0°С к реакционному раствору добавляли 1М водный раствор NaOH для доведения рН до 9-10. Этот раствор перемешивали при 100°С в течение 1 часа. Затем оставляли для охлаждения, к реакционному раствору добавляли воду (30 мл), далее экстрагировали трижды EtOAc (20 мл). Органический слой объединяли, промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (автоматический очиститель, н-гексан: EtOAc =90:10 до 70:30), получая 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-3-карбальдегид (456,7 мг) в виде белого твердого вещества.
Пример 22
К смеси раствора 5-[3-(1H-бензимидазол-6-ил)-1,2,4-оксадиазол-5-ил]-2-(2,2,2-трифтор-1-метилэтокси)бензальдегида (83 мг), калия дигидрофосфата (421 мг) и 2-метил-2-бутена (0,5 мл) в tBuOH (2 мл) и воды (0,5 мл) добавляли хлорид натрия (187 мг) при комнатной температуре. Смешанный реакционный раствор перемешивали при комнатной температуре в течение 3 часов, затем разбавляли водой (10 мл) и экстрагировали EtOAc (20 мл). Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. К раствору остатка в диоксане добавляли 4 н. HCl-диоксан раствор, далее концентрировали. Образовавшийся остаток перекристаллизовывали из IPA (10 мл), получая 5-[3-(1H-бензимидазол-5-ил)-1,2,4-оксадиазол-5-ил]-2-(2,2,2-трифтор-1-метилэтокси)бензойной кислоты гидрохлорид (80 мг) в виде белого порошка.
Пример 23
К раствору 2-[2-(метилтио)этил]-5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазола (400 мг) в дихлорметане (8,0 мл) добавляли mCBA (534 мг), затем перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли водный раствор Na2S2O4, далее перемешивали в течение 1 часа. Реакционный раствор экстрагировали трижды EtOAc и органический слой объединяли, сушили над безводным MgSO4, фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=10:1), получая желтое маслянистое вещество. Это вещество растворяли в EtOAc и добавляли 4М-HCl/EtOAc затем концентрировали. Остаток промывали IPE, получая 2-[2-(метилсульфанил)этил]-5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазол гидрохлорид (146 мг) в виде светло-желтого порошка.
Пример 24
К реакционной смеси 2-бут-3-ен-1-ил-5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазола (200 мг) в ацетоне и воде (1 мл) добавляли тетраоксосмий (51 мг) и 4-метилморфолин-4-оксид (94 мг), далее перемешивали в течение ночи. Реакционную смесь фильтровали и к фильтрату добавляли водный раствор тиосульфата натрия, затем перемешивали в течение 1 часа. Раствор экстрагировали хлороформом и органические слои концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ-СН3ОН) и концентрировали и полученный окрашенный порошок добавляли к раствору HCl в этаноле и растворяли в нем, после чего концентрировали. Остаток растирали в порошок/промывали диизопропиловым эфиром, получая 4-(5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-бензимидазол-2-ил)бутан-1,2-диол гидрохлорид (102,3 мг).
Пример 25
К раствору 3-(4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-2,3-дигидро-1H-индол-1-ил)пропанамида (100 мг) в хлороформе (1 мл) добавляли диоксид марганца (67,7 мг) после чего кипятили в течение 15 часов. Реакционный раствор оставляли для охлаждения до комнатной температуры и фильтровали через Целит для удаления диоксида марганца. Фильтрат концентрировали при уменьшенном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=100:0 до 90:10), получая 3-(4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол-1-ил)пропанамид (46,3 мг).
Пример 26
К раствору 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индола (100 мг) в АсОН (3 мл) добавляли порциями цианоборат натрия (29 мг). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли водой, подщелачивали 1М водным раствором NaОН и экстрагировали EtOAc.
Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали и остаток очищали при помощи колоночной хроматографии на силикагеле (н-гесан: EtOAc =90:10 до 75:25) и промывали н-гексаном, получая 4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}индолин (90 мг) в виде светло-желтого твердого вещества.
Пример 27
К раствору 5-[3-(1H-бензимидазол-6-ил)-1,2,4-оксадиазол-5-ил]-2-(2,2,2-трифтор-1-метилэтокси)бензальдегида (80 мг) в этаноле (3 мл) добавляли NaBH4 (9 мг) при 0°С. После перемешивания при комнатной температуре в течение 0,5 часов добавляли насыщенный раствор NH4Cl (10 мл), после чего экстрагировали EtOAc (20 мл). Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. К раствору остатка в диоксане добавляли 4М HCl/диоксан после чего концентрировали. Образовавшийся порошок перекристаллизовывали из IPA (10 мл), получая [5-[3-(1H-бензимидазол-5-ил)-1,2,4-оксадиазол-5-ил]-2-(2,2,2-трифтор-1-метилэтокси)фенил]метанол гидрохлорид (70 мг) в виде белого порошка.
Пример 28
Раствор 5-метил-6-{5-[3-трифтор)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}имидазо[1,2-a]пиридина (120 мг) и NCS в THF/EtOH (1/1) перемешивали при 80°С в течение ночи. Раствор концентрировали и полученный остаток очищали при помощи колоночной хроматографии на силикагеле (СН3ОН/хлороформ 0-5%), получая 3-хлор-5-метил-6-{5-[3-(трифтор)-4-(2,2,2-трифтор-1-метоксиэтил)фенил]-1,2,4-оксадиазол-3-ил}имидазо[1,2-a]пиридин (45 мг) в виде светло-желтого твердого вещества.
Пример 29
К раствору 5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}имидазо[1,2-a]пиридин (150 мг) в этаноле добавляли NCS (67 мг) при комнатной температуре, после чего перемешивали при 80°С в течение 15 часов. К реакционному раствору добавляли воду, после чего экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (н-гесан: EtOAc=90:10 до 75:25), получая 3-хлор-5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}имидазо[1,2-a]пиридин (110,4 мг) в виде белого твердого вещества. К раствору 3-хлор-5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}имидазо[1,2-a]пиридина (100 мг) в метиленхлориде (2 мл) добавляли по каплям 10 эквивалентов 4М HCl/диоксана при комнатной температуре. После перемешивания при комнатной температуре в том виде, в котором она есть, в течение 1 часа смесь концентрировали при уменьшенном давлении. Остаток растирали в порошок/промывали диизопропиловым эфиром и затем отделяли фильтрованием, получая 3-хлор-5-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}имидазо[1,2-a]пиридин гидрохлорид (102,6 мг) в виде белого твердого вещества.
Пример 30
1-(2-{[Трет-бутилдиметилсилил]окси}этил)-4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1H-индол (60 мг) растворяли в THF (1,2 мл), и TBAF (150 мкл) к нему добавляли при 0°С, после чего перемешивали при комнатной температуре в течение 3,0 часов. К реакционной смеси добавляли воду (30 мл), после чего трижды экстрагировали EtOAc (20 мл). Органический слой объединяли, промывали насыщенным раствором соли, сушили над безводным MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ:СН3ОН=100:0 до 90:10), получая 2-(4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил-1H-индол-1-ил)этанол (36,5 мг) в виде белого твердого вещества.
Пример 31
К раствору трет-бутил 4-{[4-(5-{3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}-1,2,4-оксадиазол-3-ил)-1H-индол-1-ил]ацетил}пиперазин-1-карбоксилата (70,7 мг) в метиленхлориде (1 мл) добавляли по каплям 10 эквивалентов 4М HCl/диоксана, после чего перемешивали при комнатной температуре в течение 3 часов. Спустя 5 часов реакционный раствор концентрировали. При добавлении диизопропилового эфира выпадало белое твердое вещество. Белое твердое вещество промывали IPE, получая 1-(2-оксо-2-пиперазин-1-илэтил)-4-(5-{3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}-1,2,4-оксадиазол-3-ил)-1H-индол гидрохлорид (58,9 мг) в виде светло-красного твердого вещества.
В следующих таблицах показаны структурные формулы соединений Примеров. Ex: Пример №.
№
№
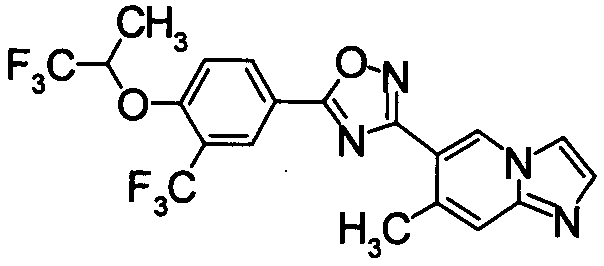
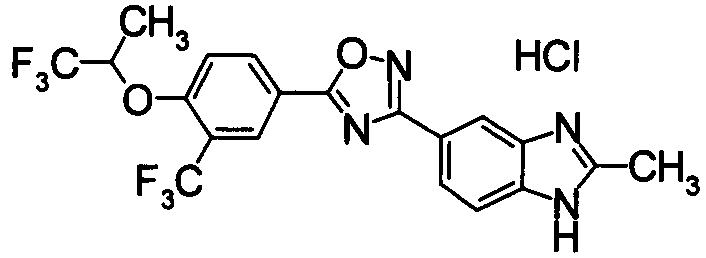
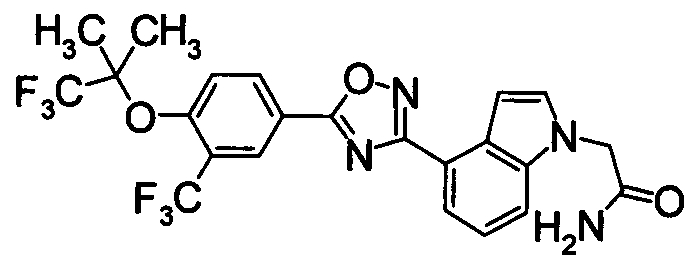
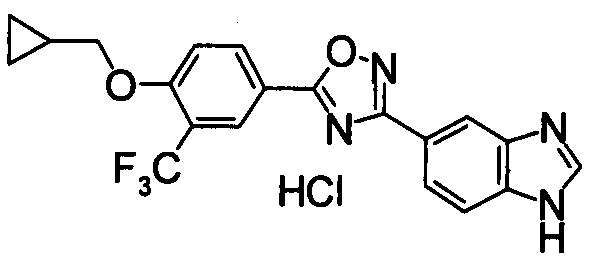
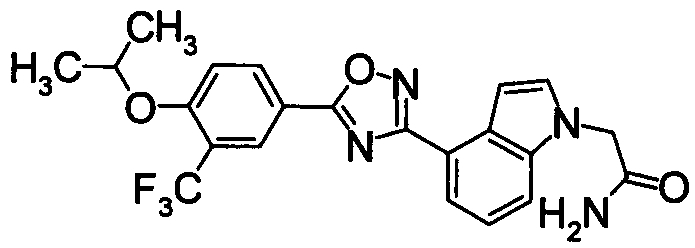
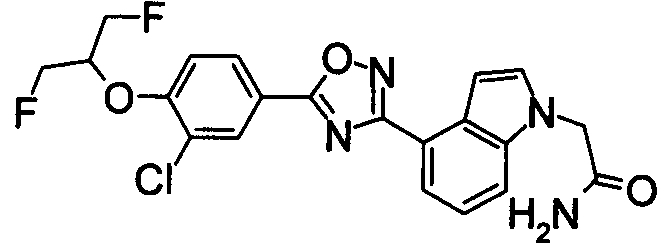
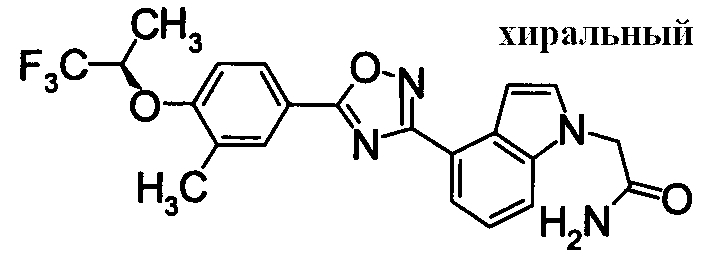
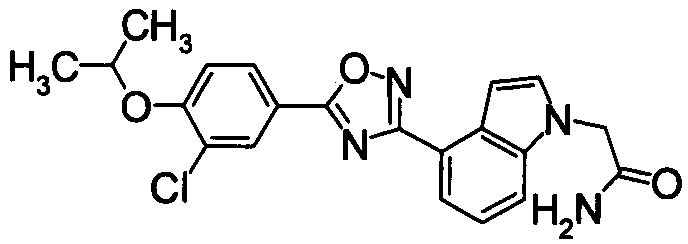
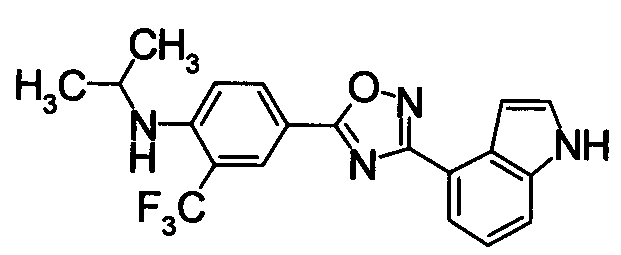
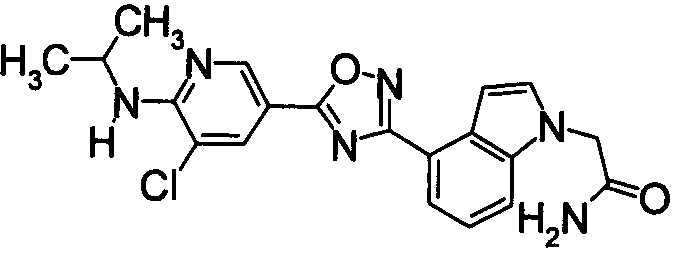
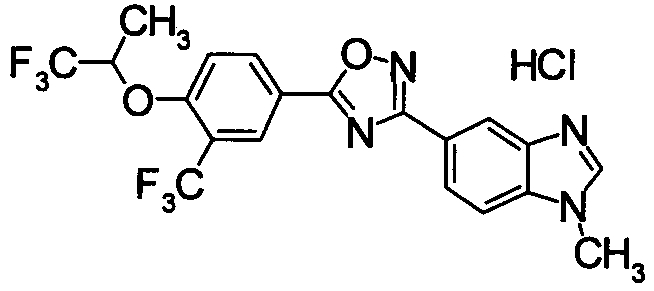
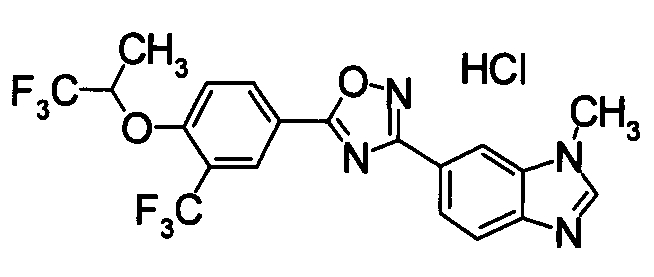
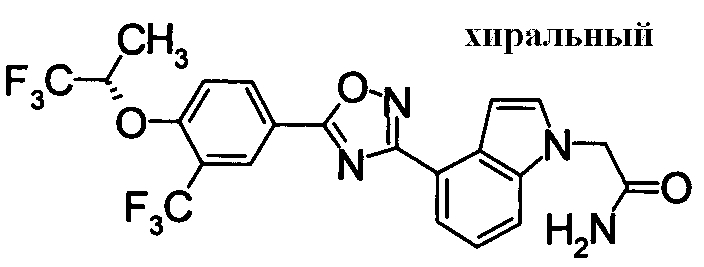
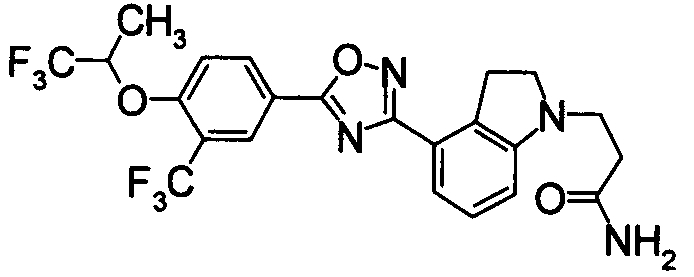
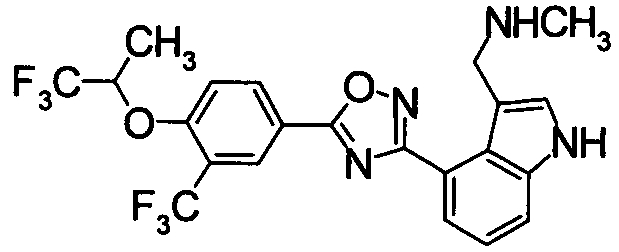
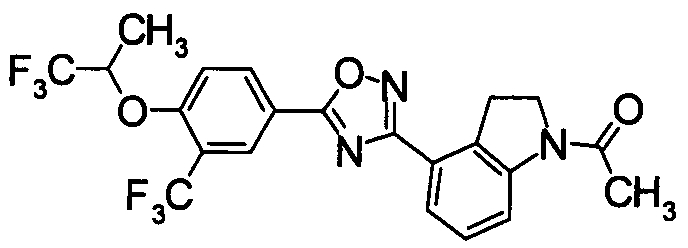
№
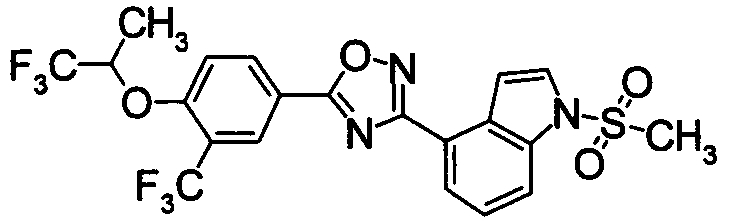
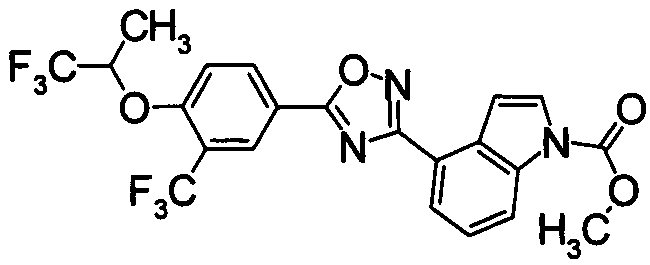
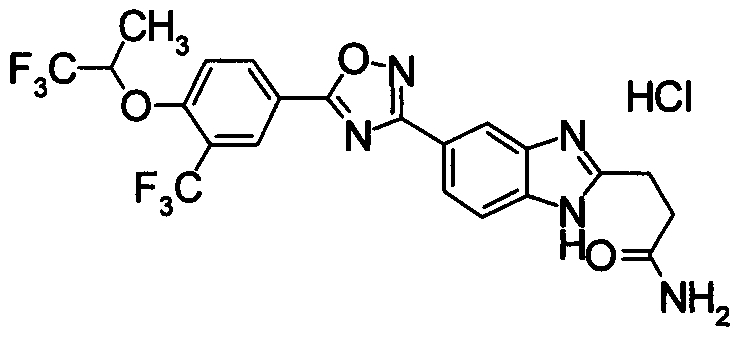
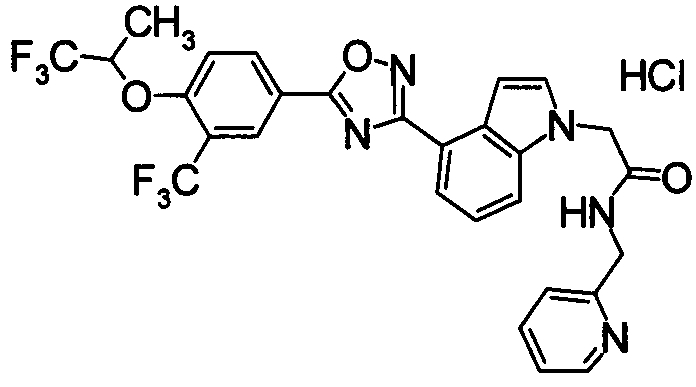
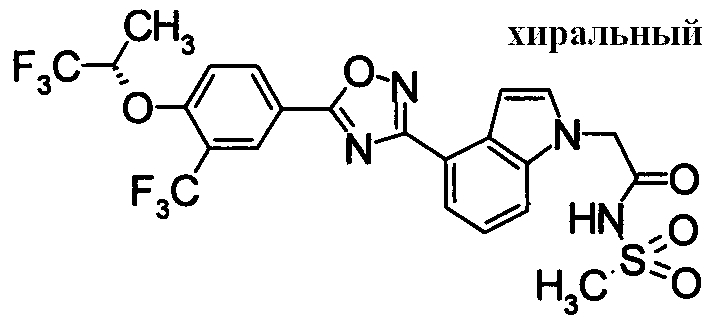
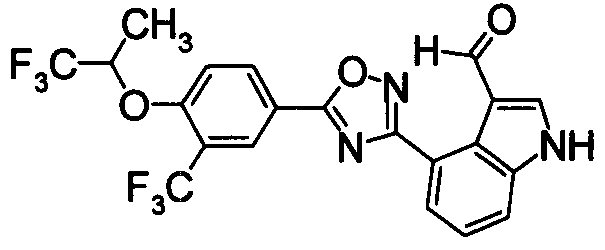
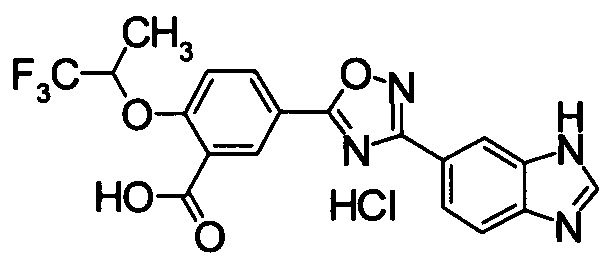
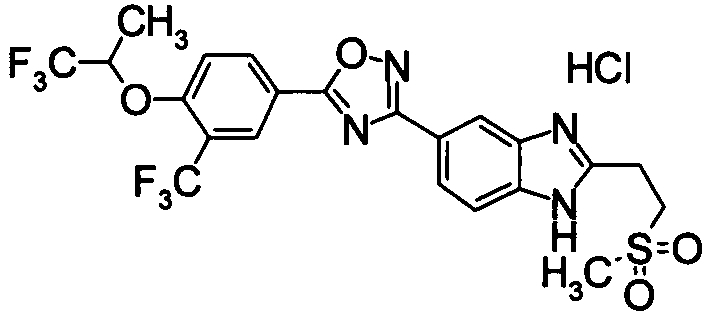
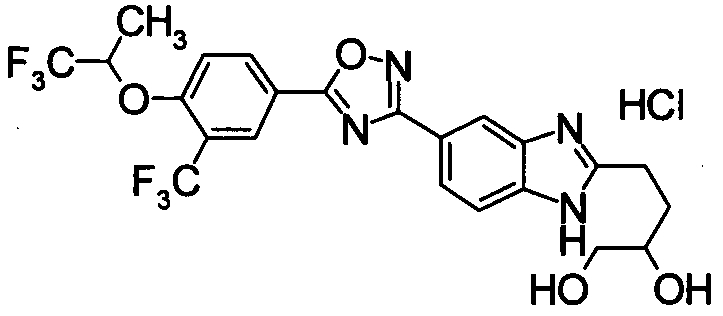
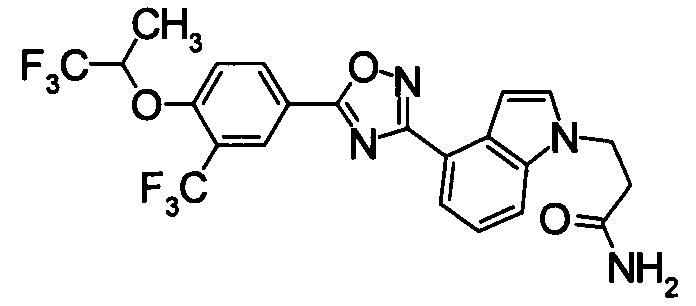
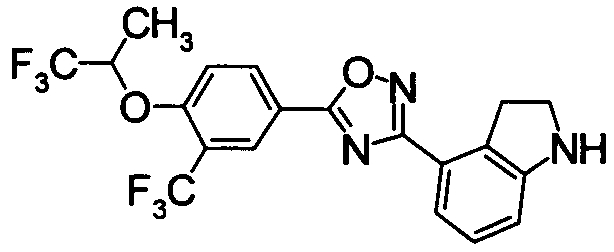
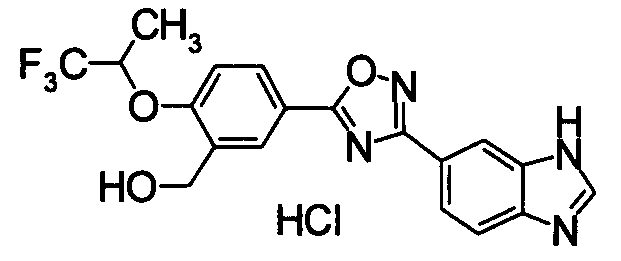
№
№
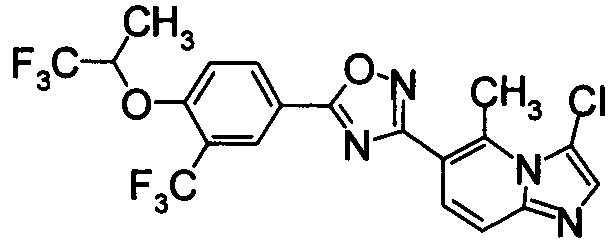
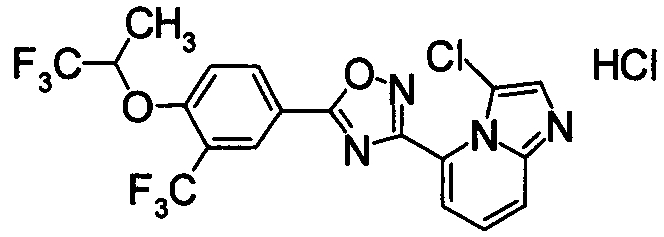
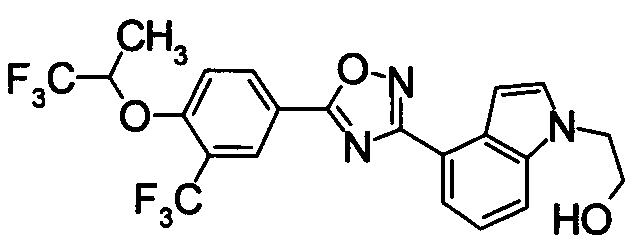
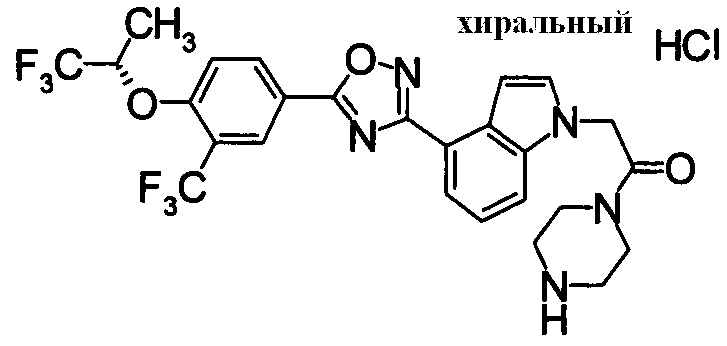
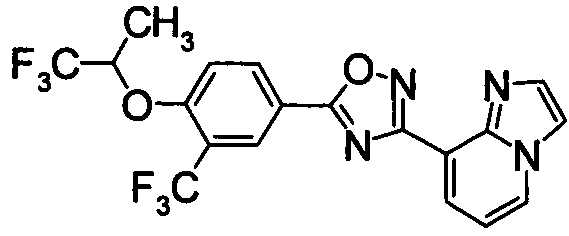
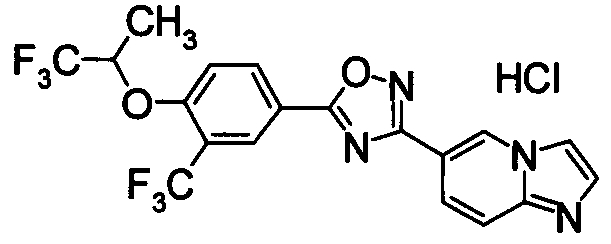
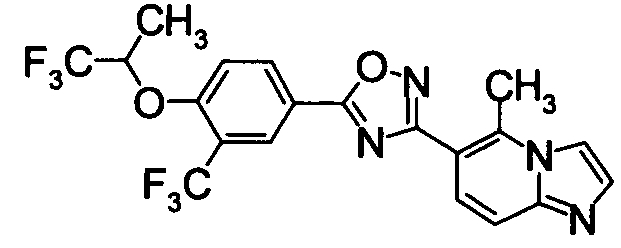
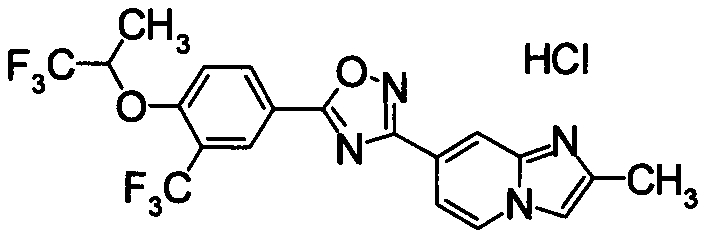
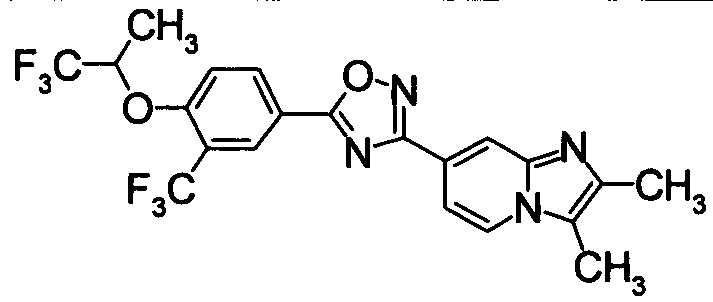
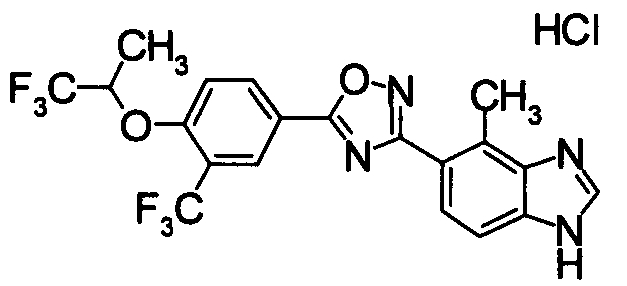
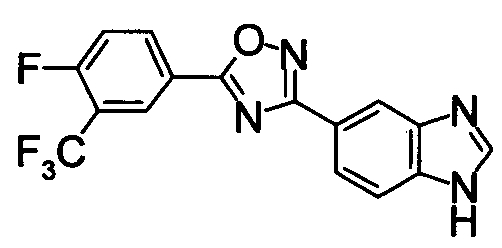
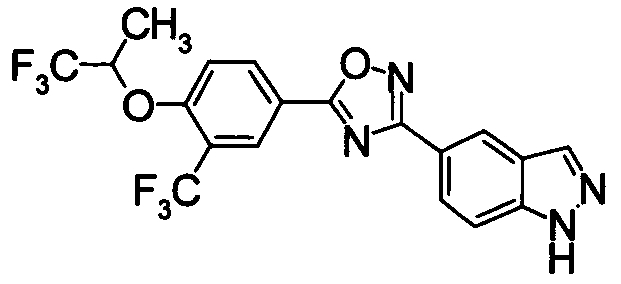
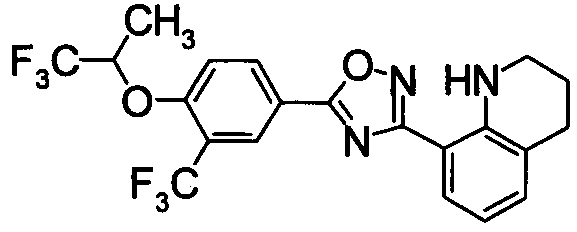
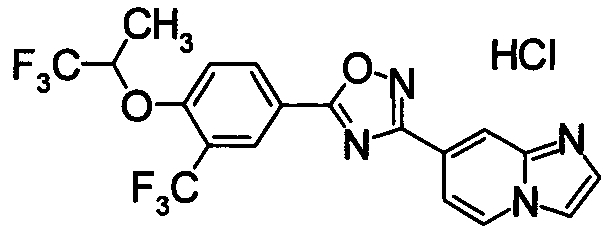
№
№
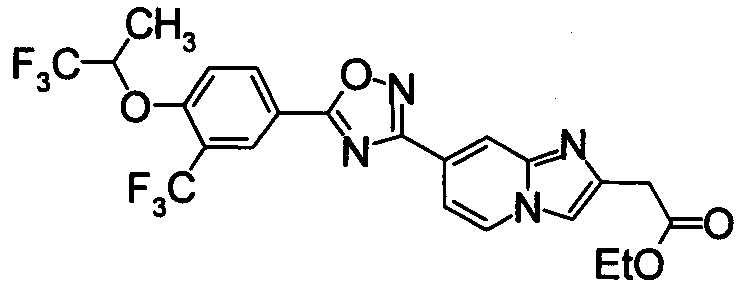
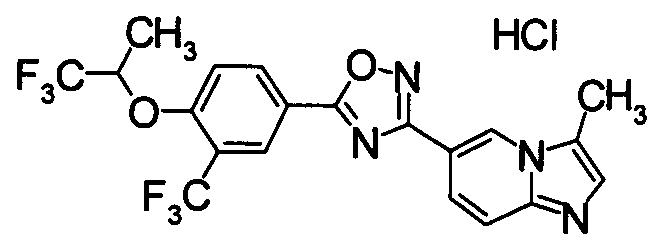
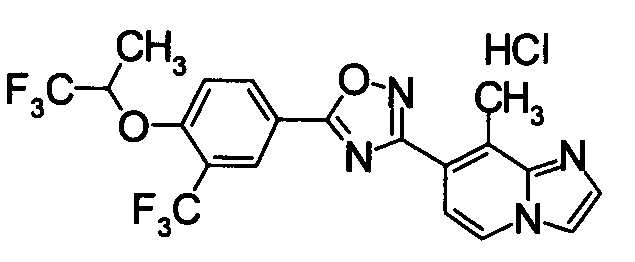
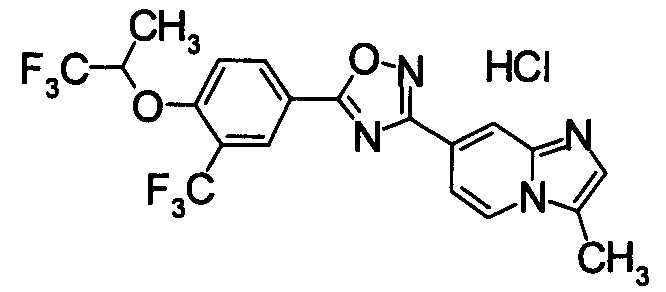
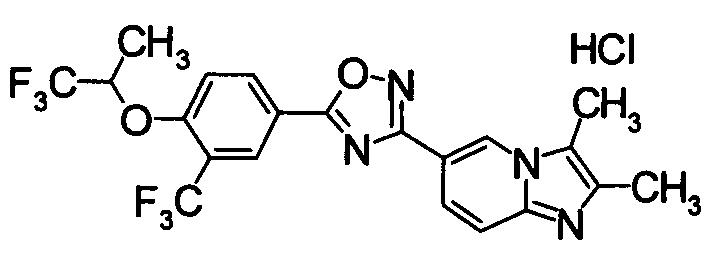
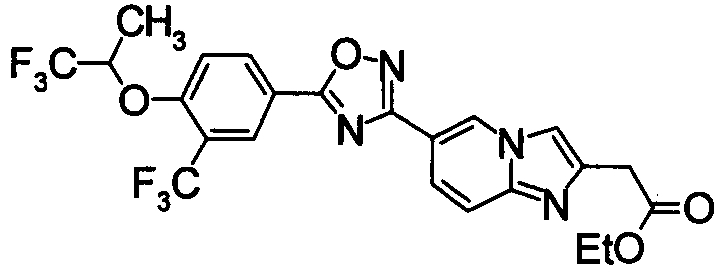
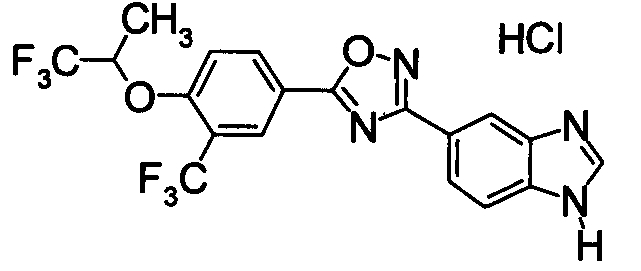
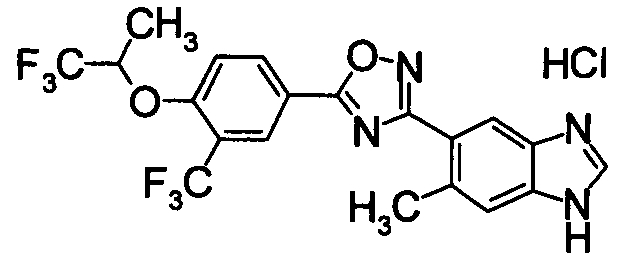
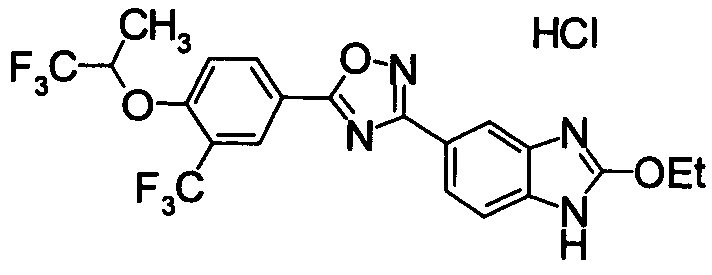
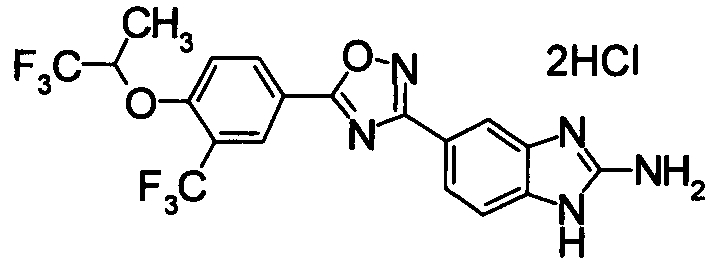
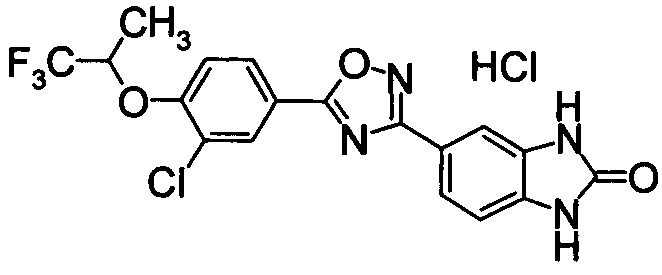
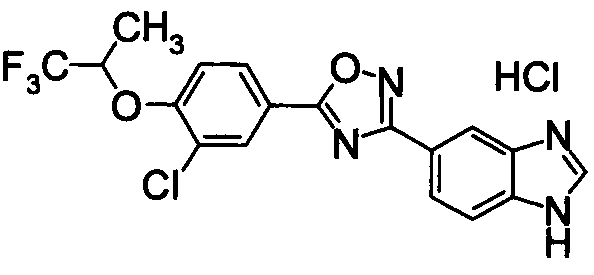
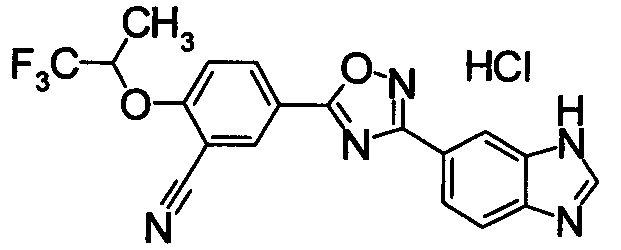
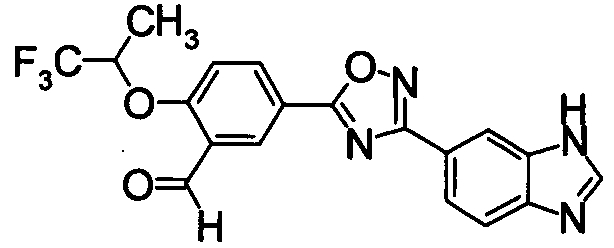
№
№
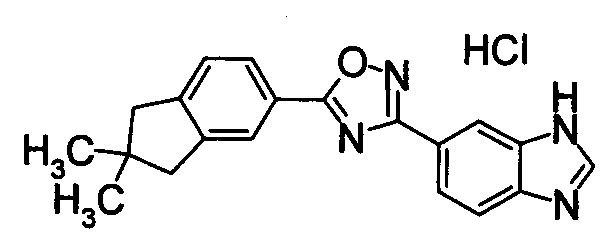
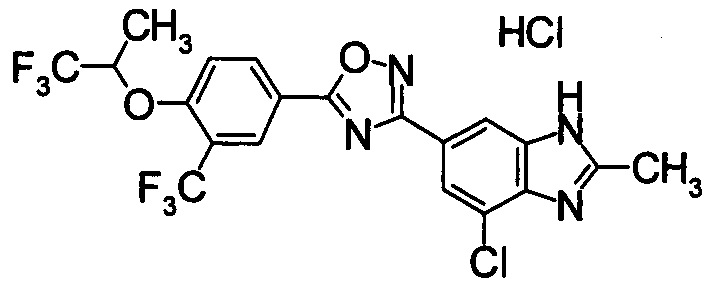
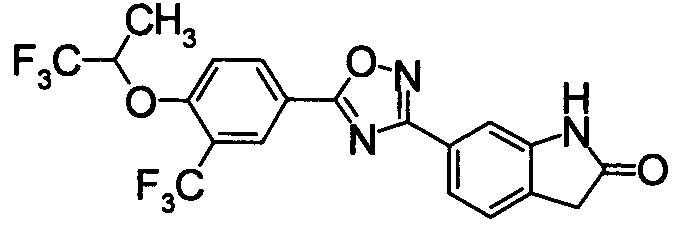
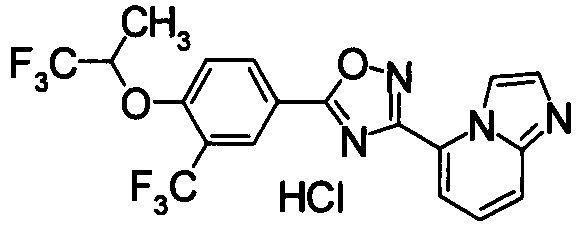
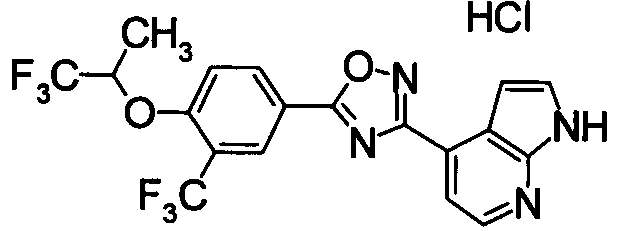
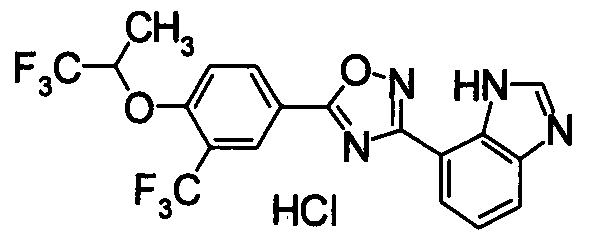
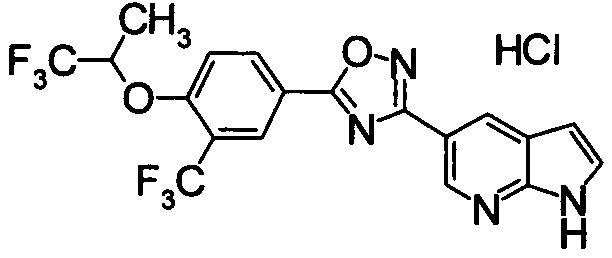
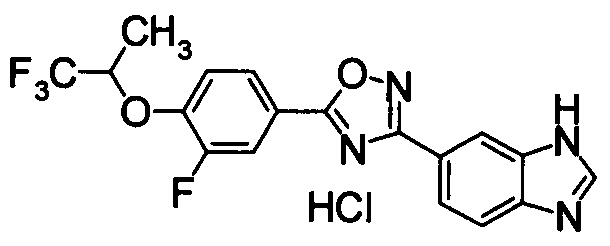
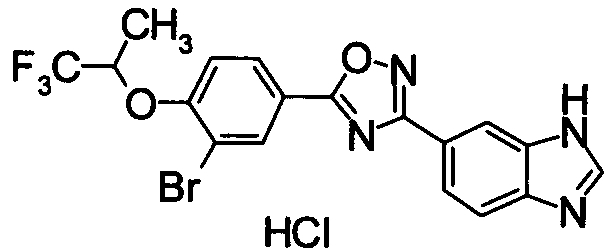
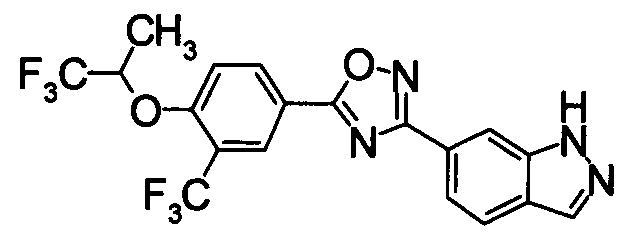
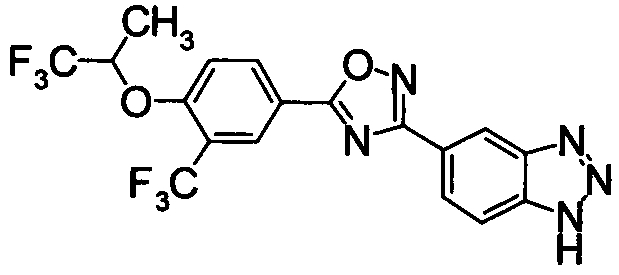
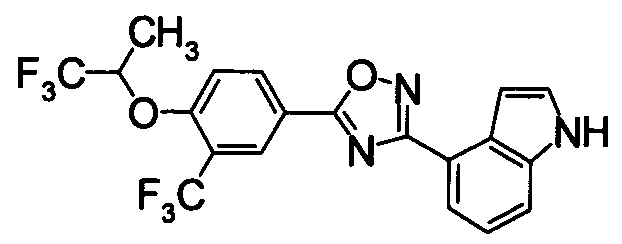
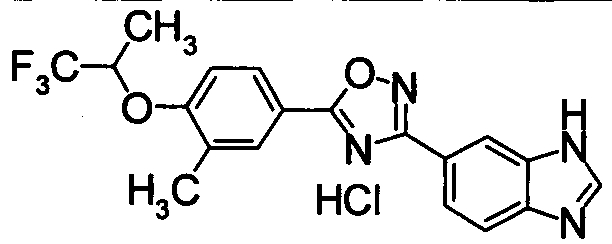
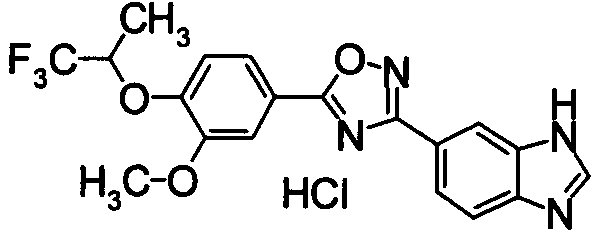
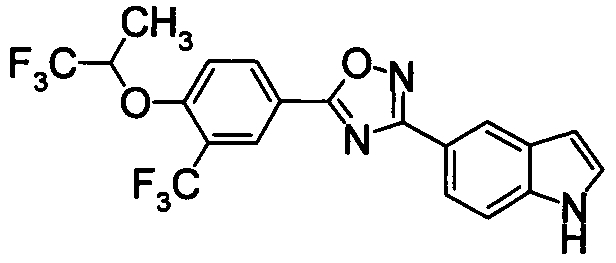
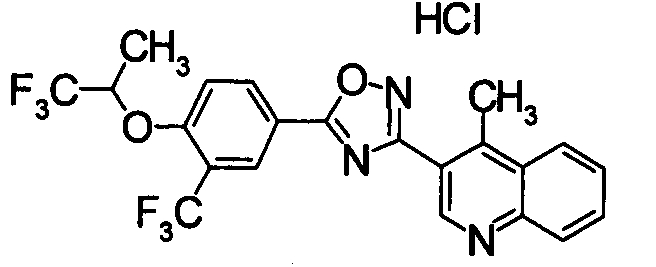
№
№
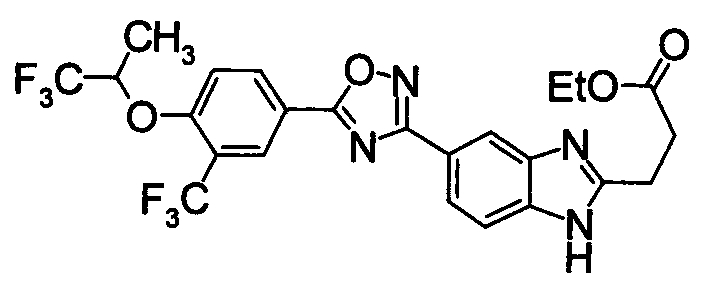
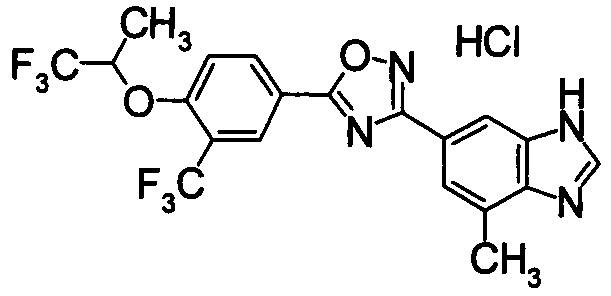
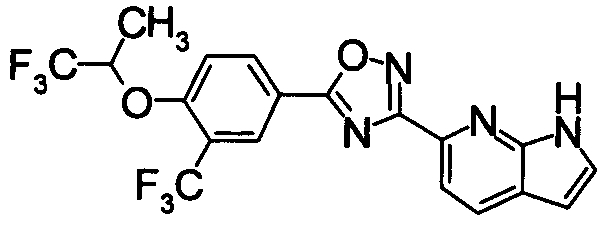
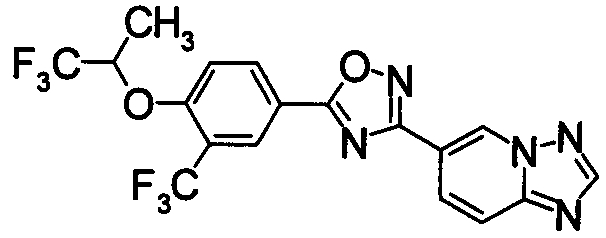
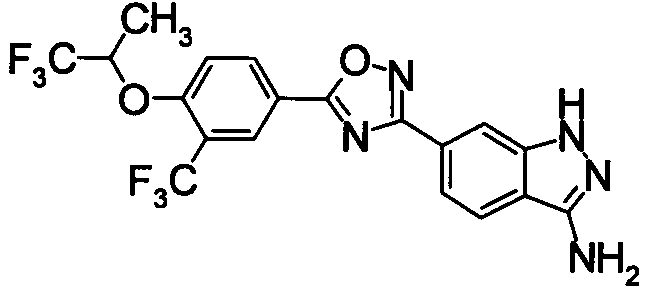
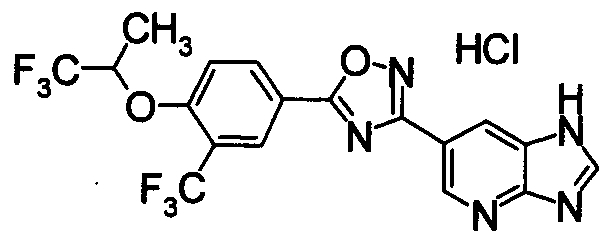
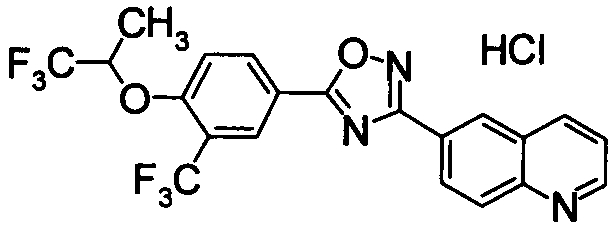
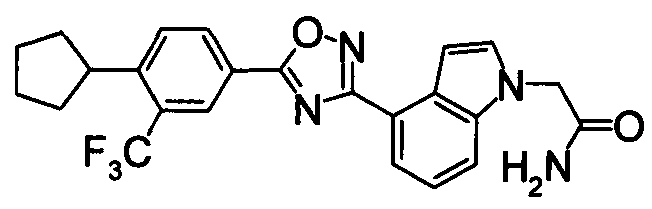
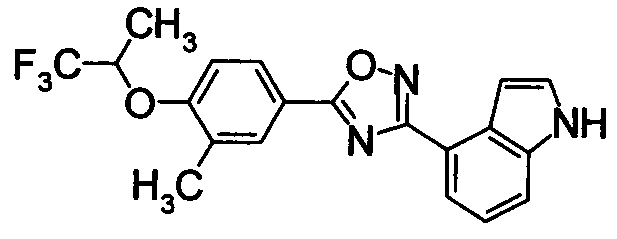
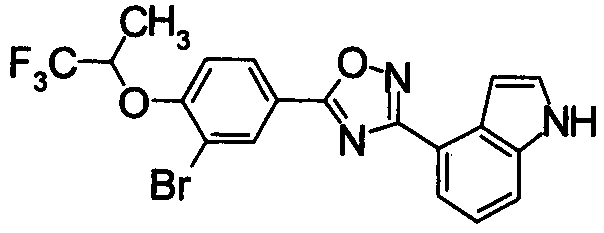
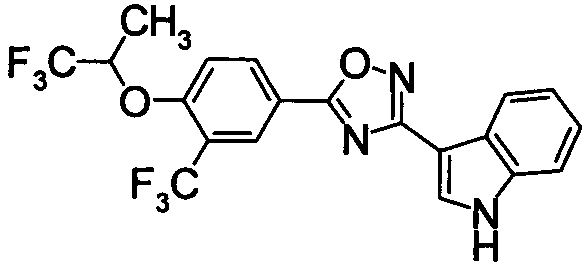
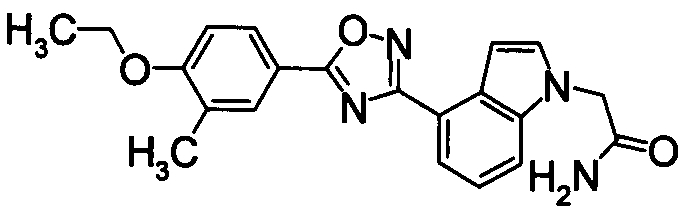
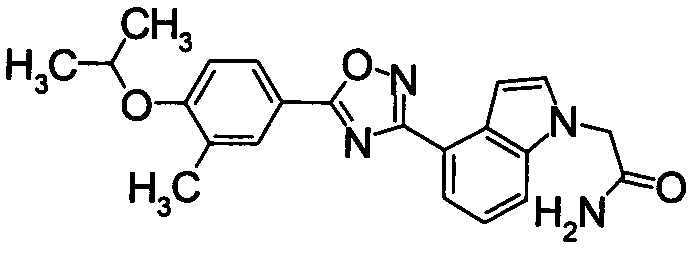
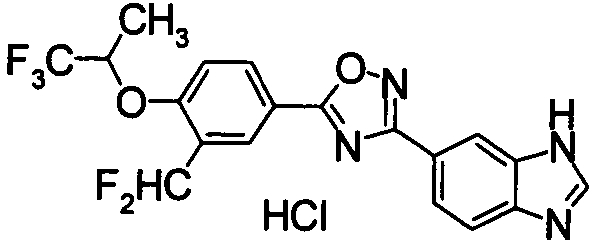
№
№
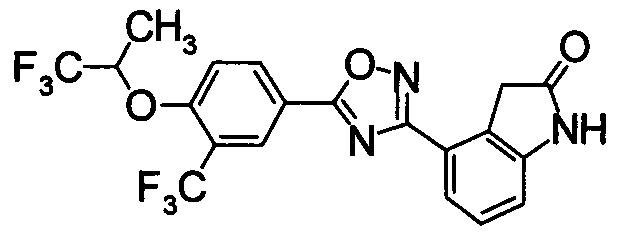
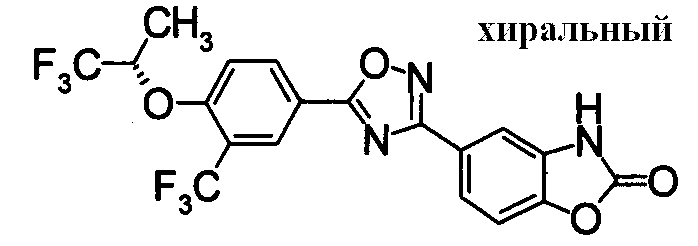
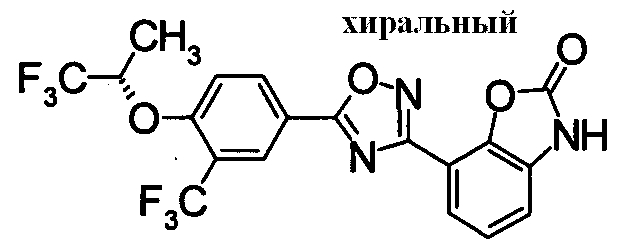
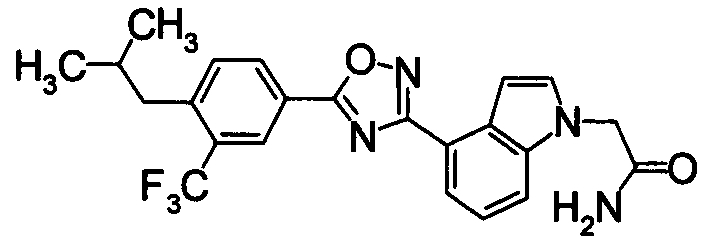
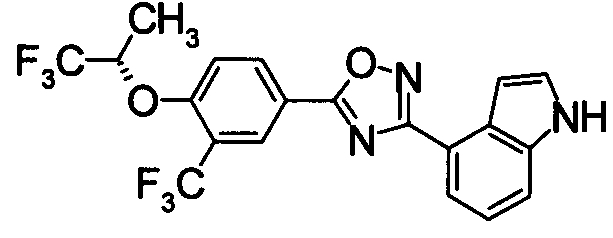
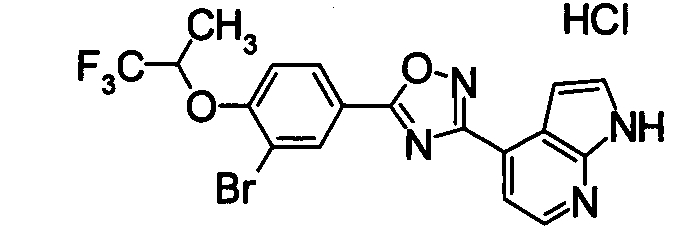
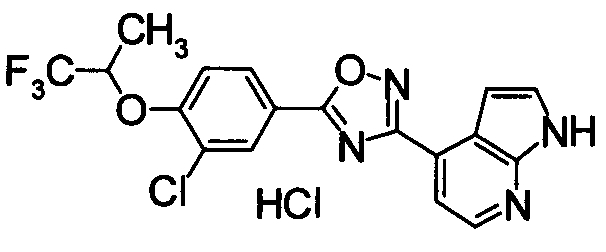
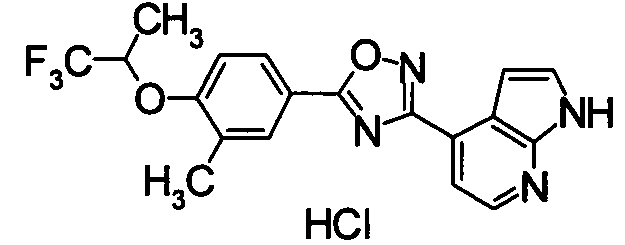
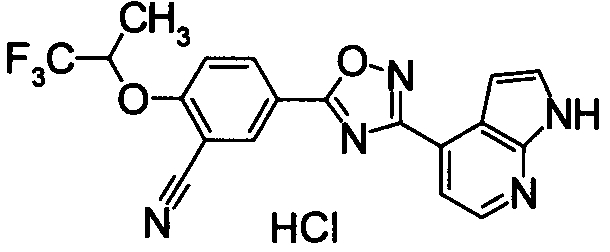
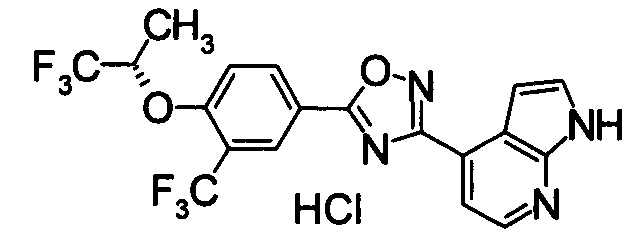
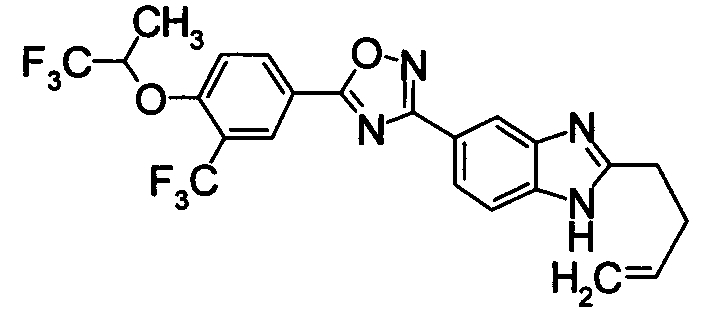
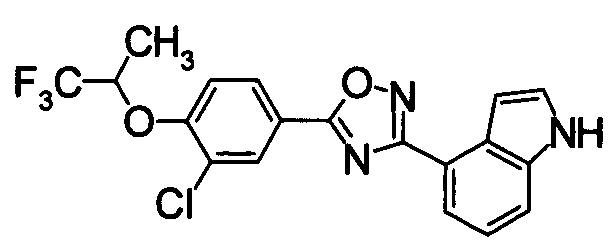
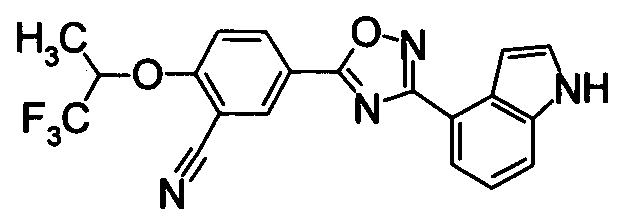
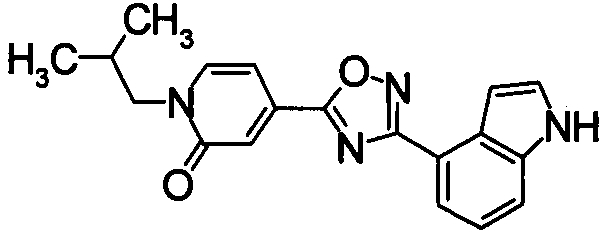
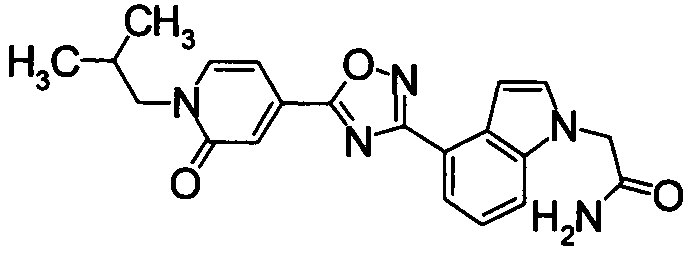
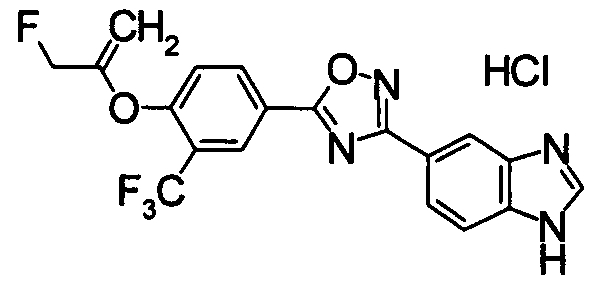
№
№
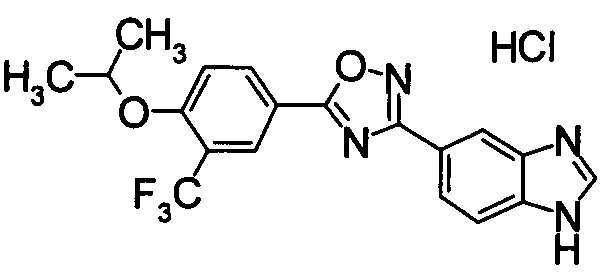
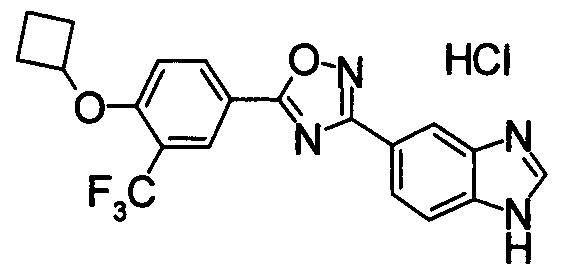
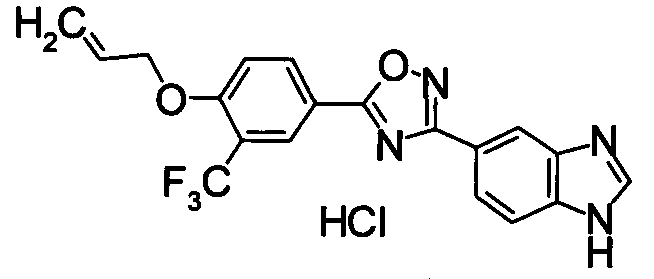
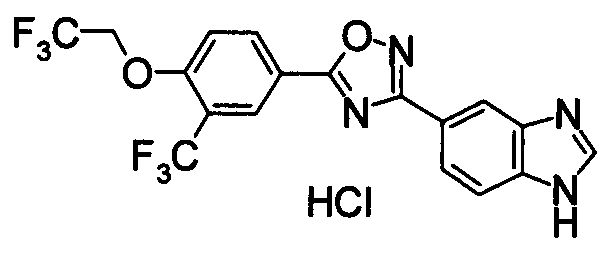
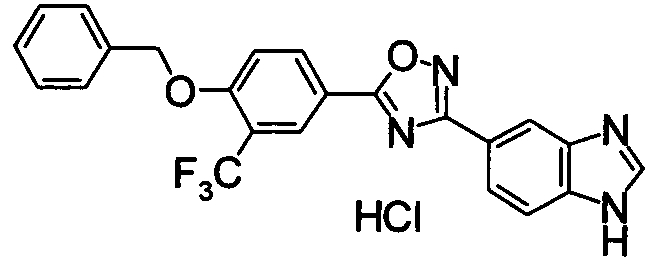
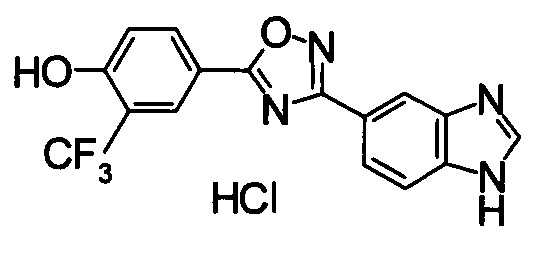
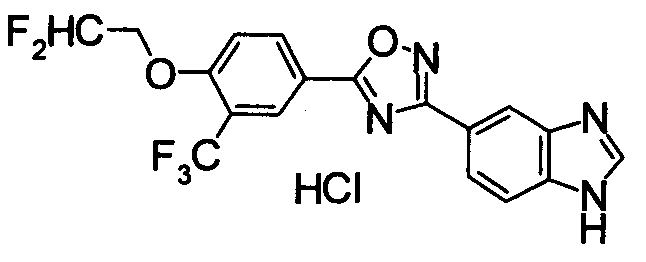
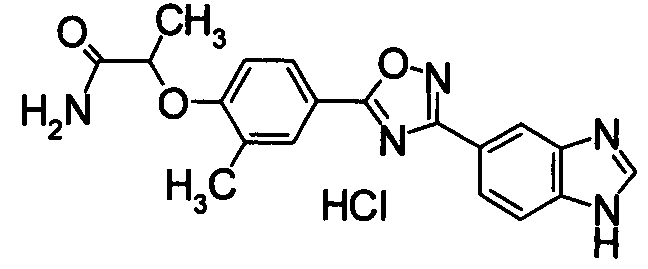
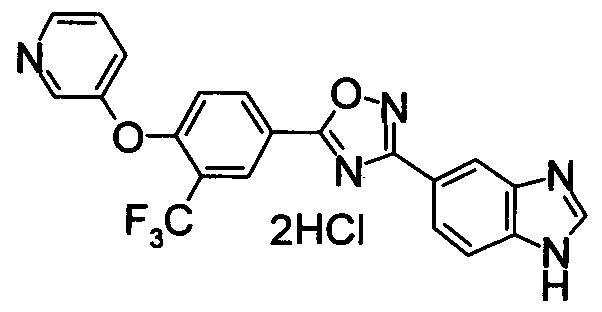
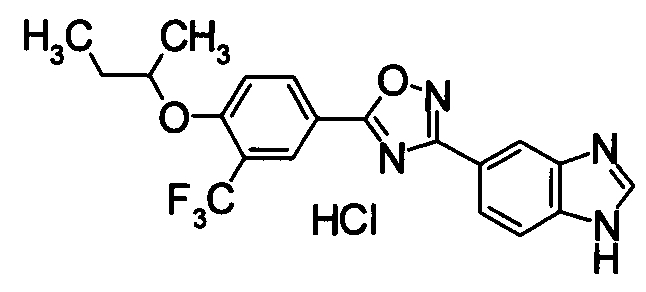
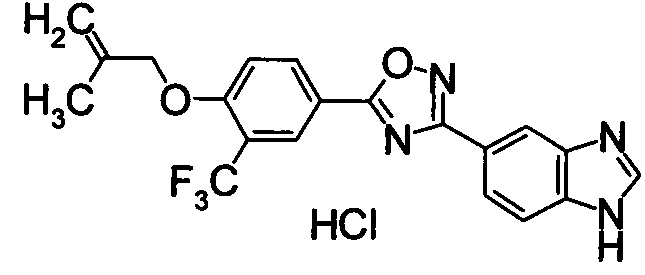
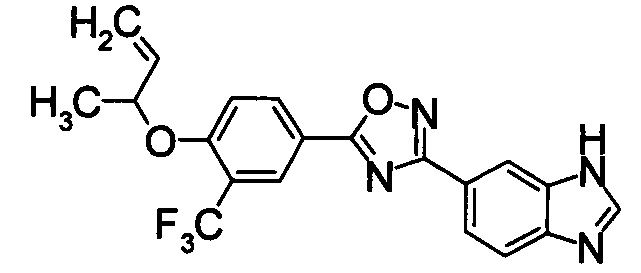
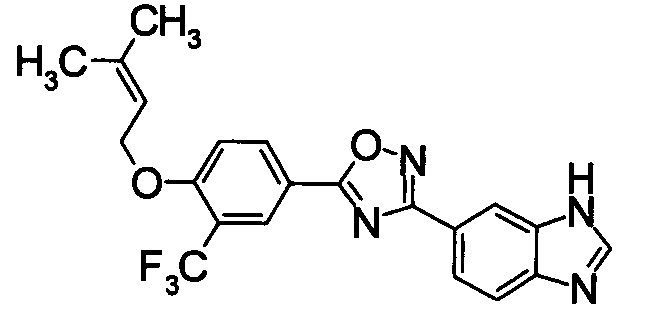
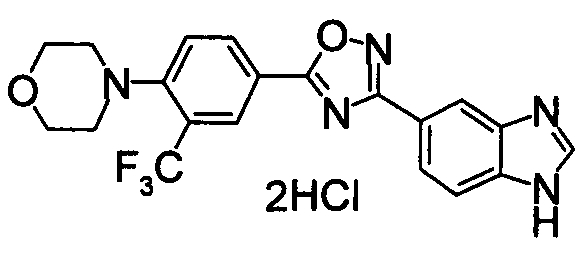
№
№
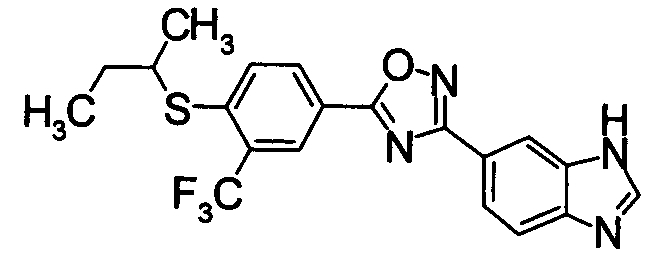
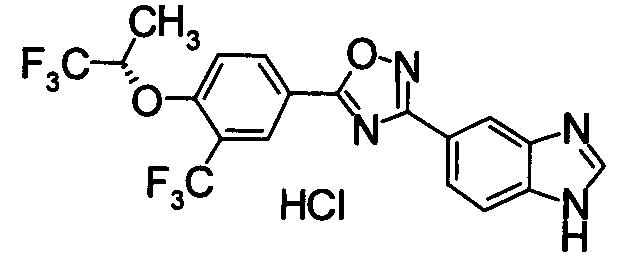
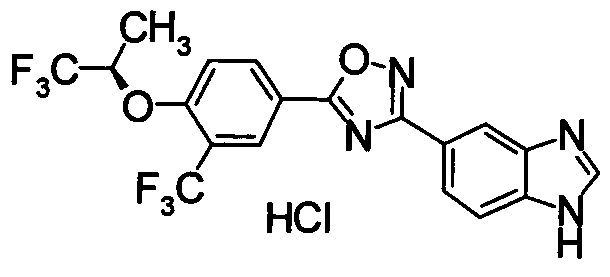
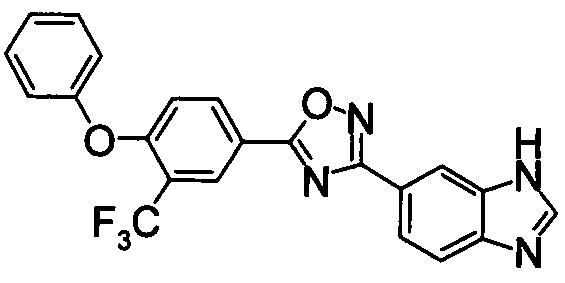
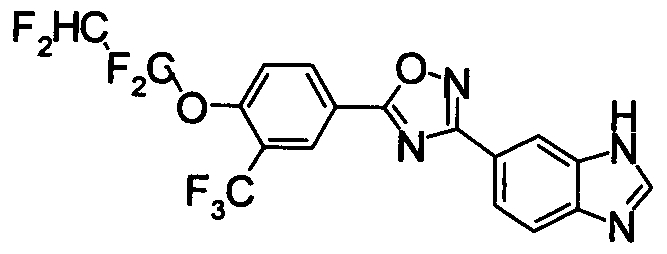
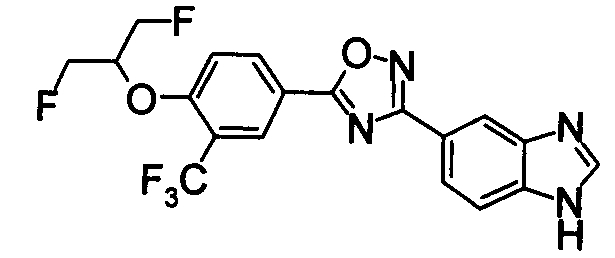
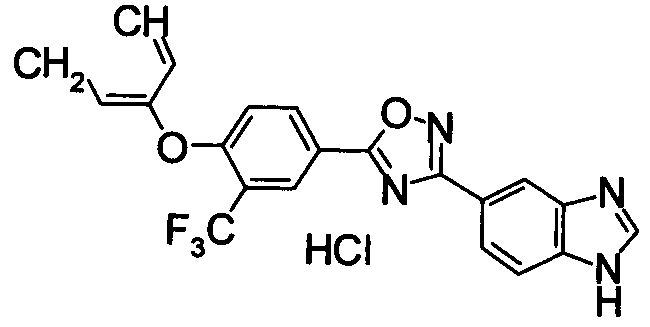
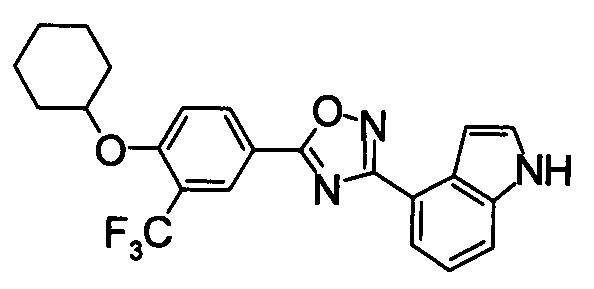
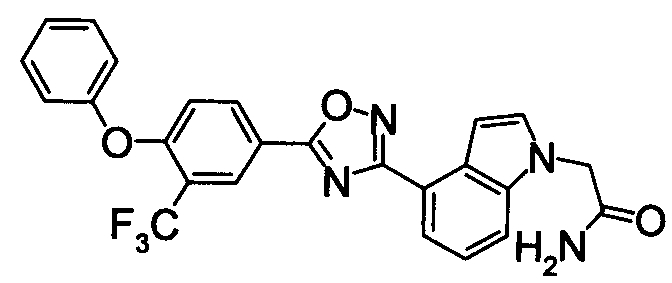
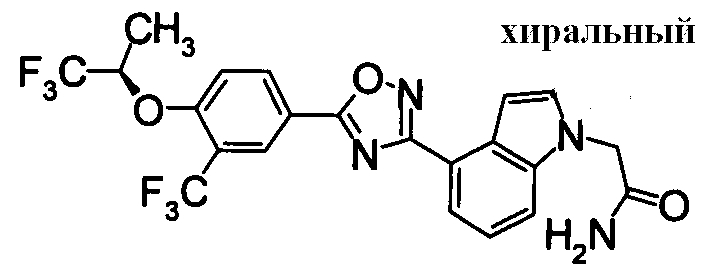
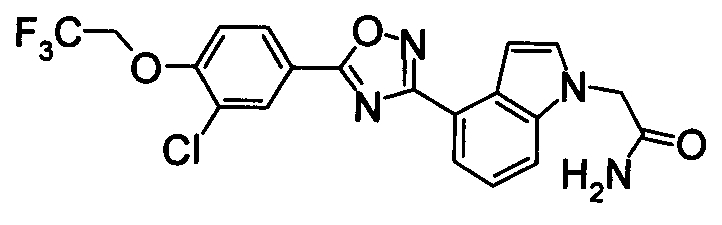
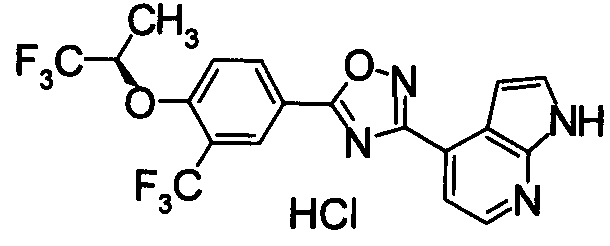
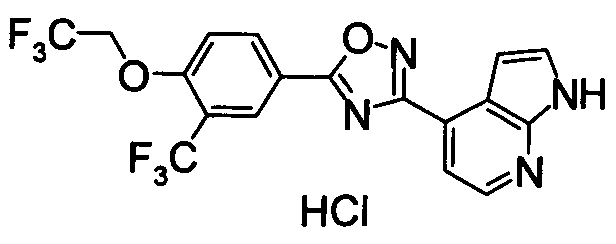
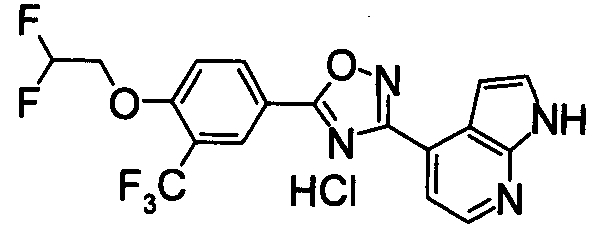
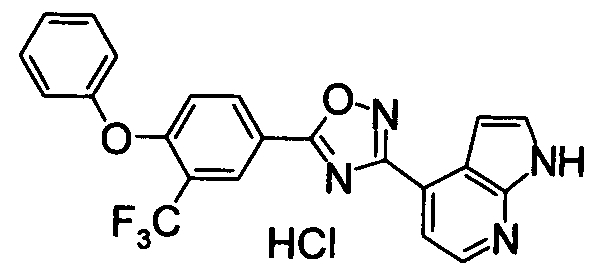
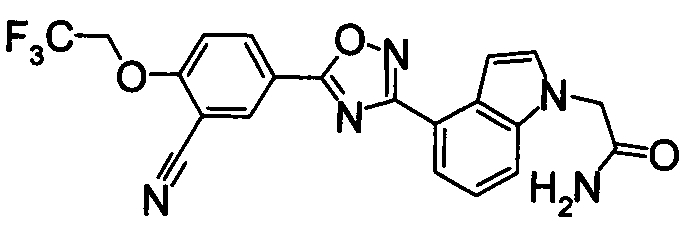
№
№
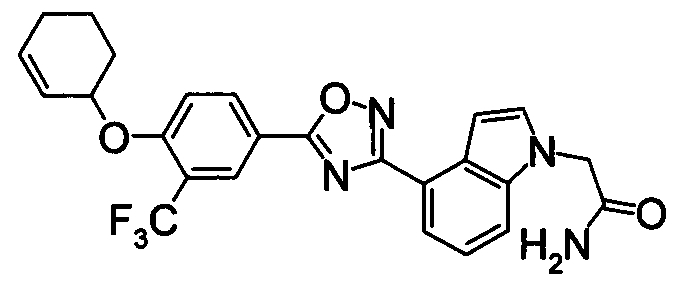
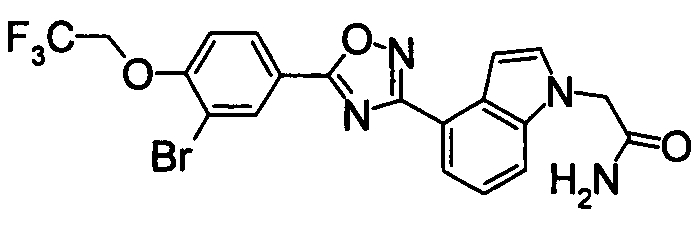
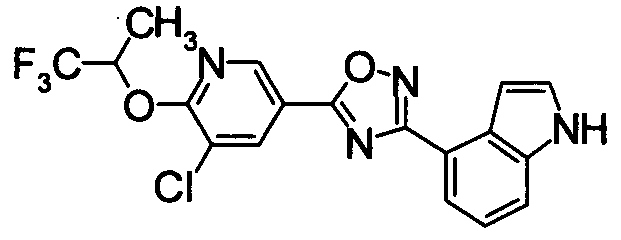
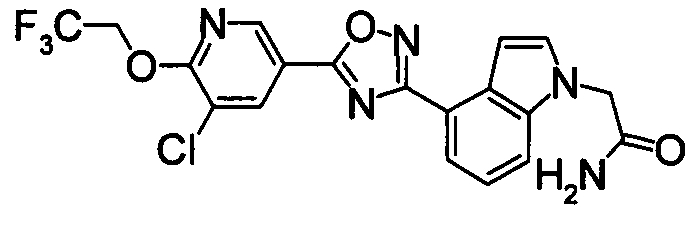
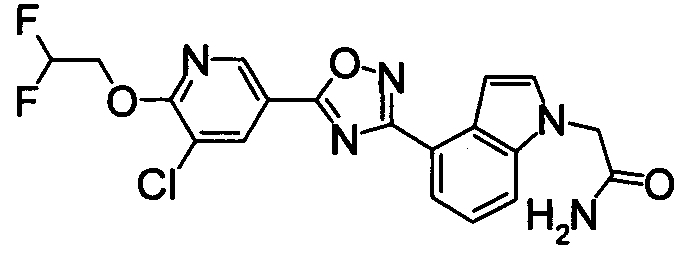
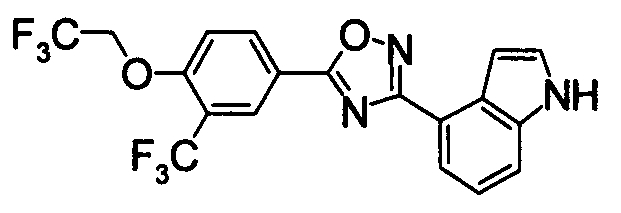
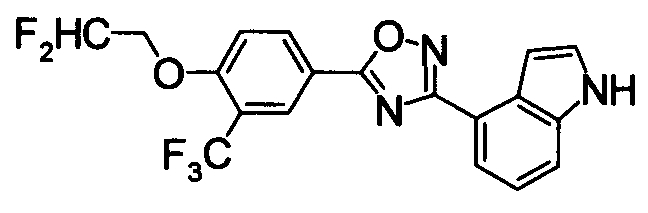
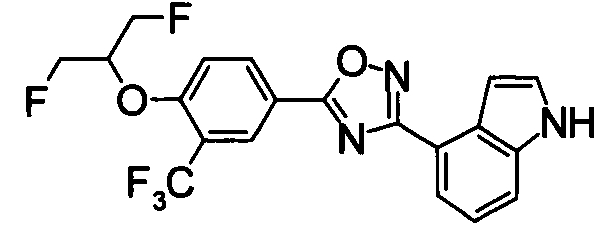
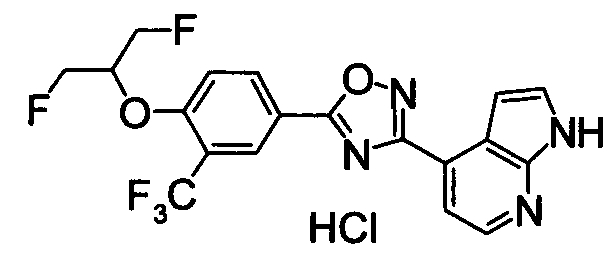
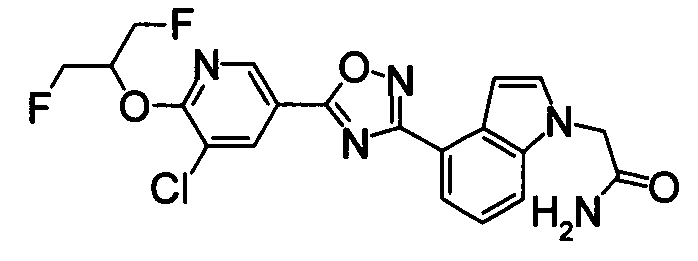
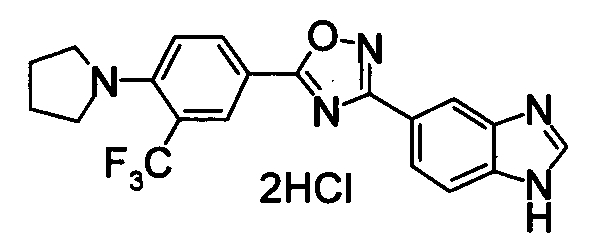
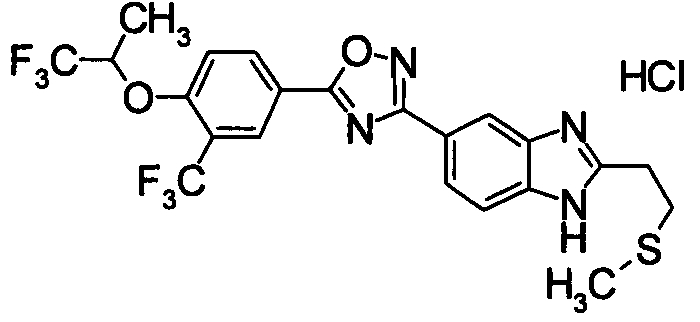
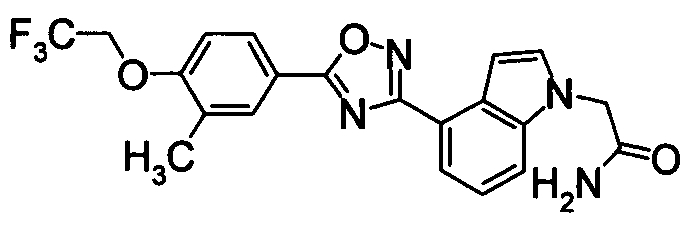
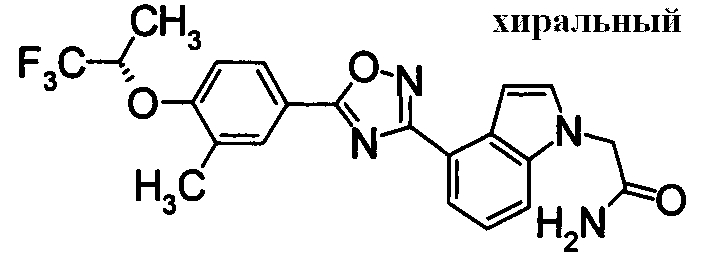
№
№
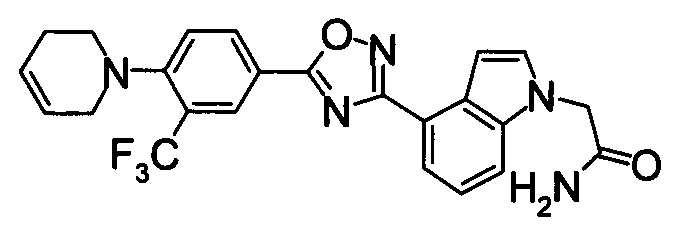
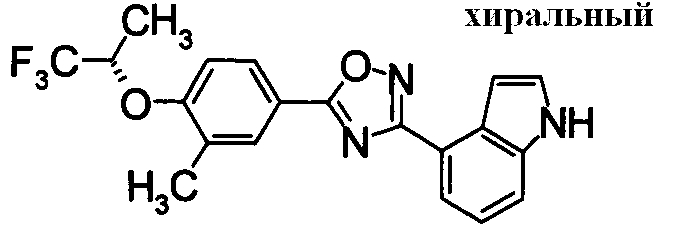
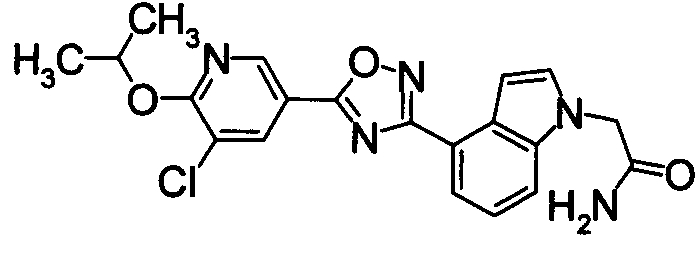
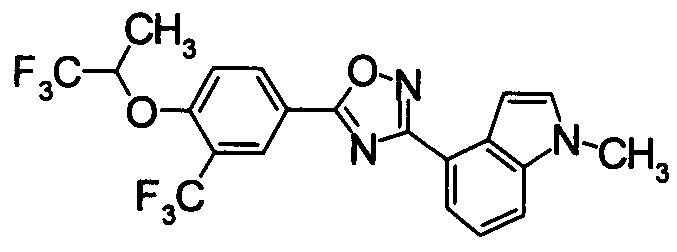
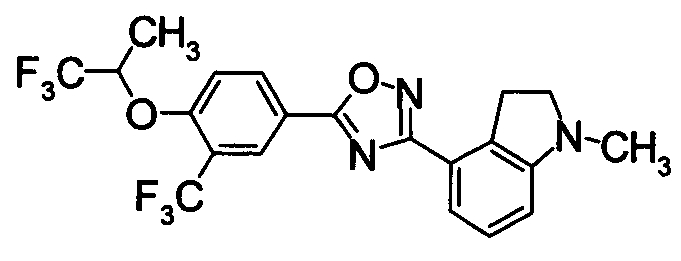
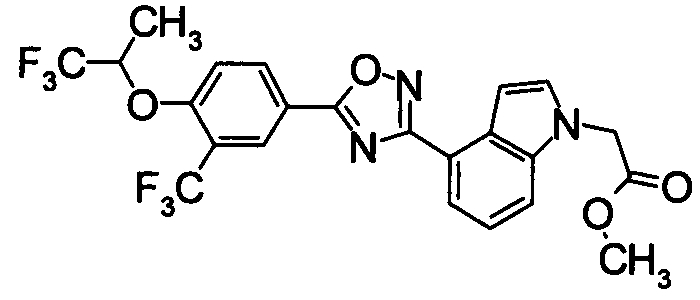
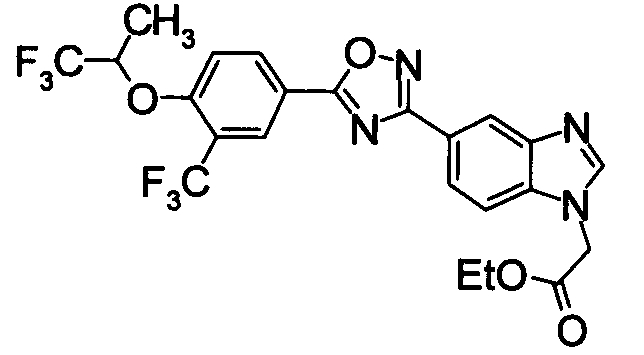
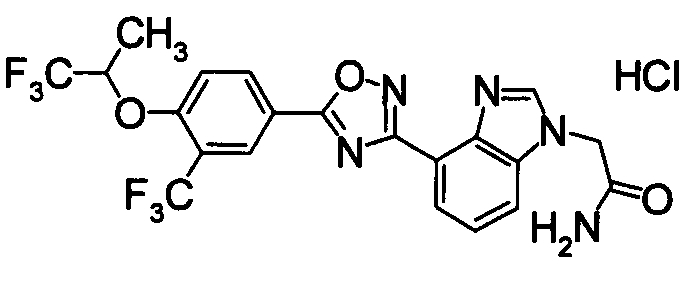
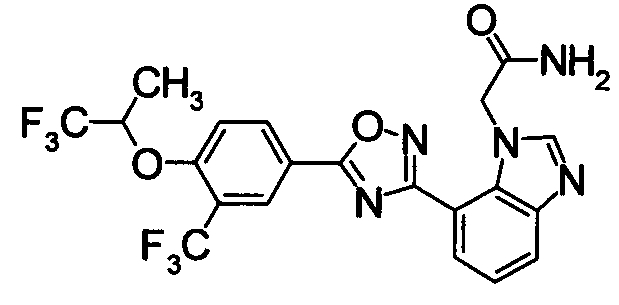
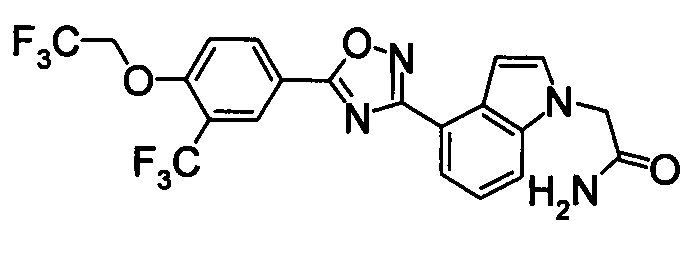
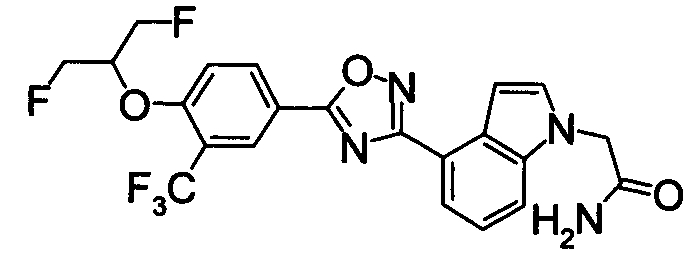
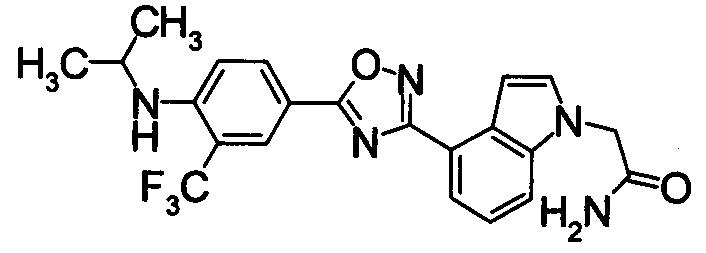
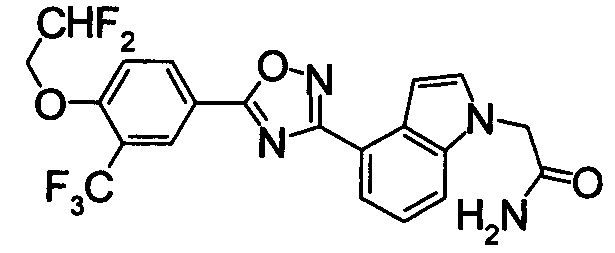
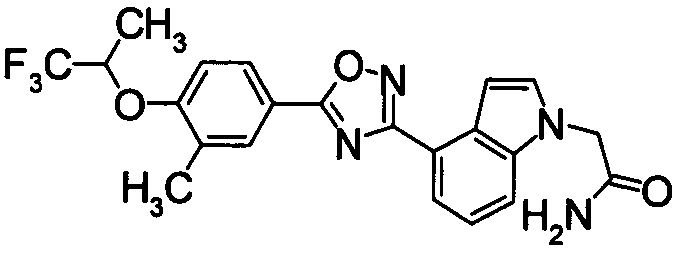
№
№
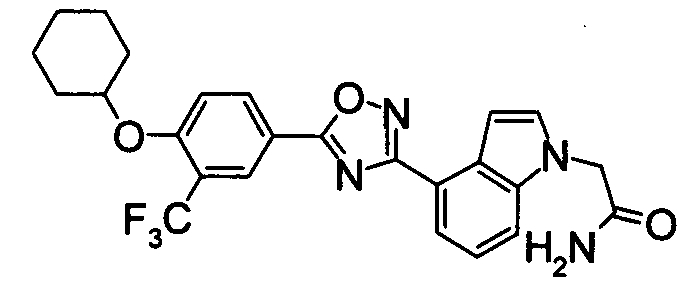
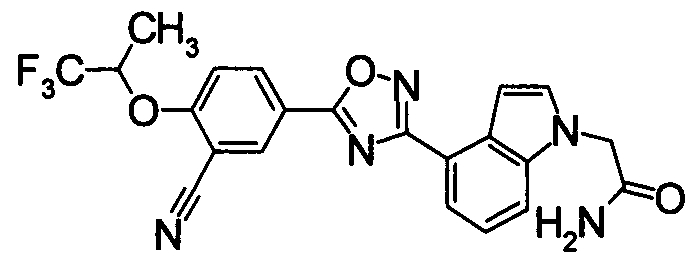
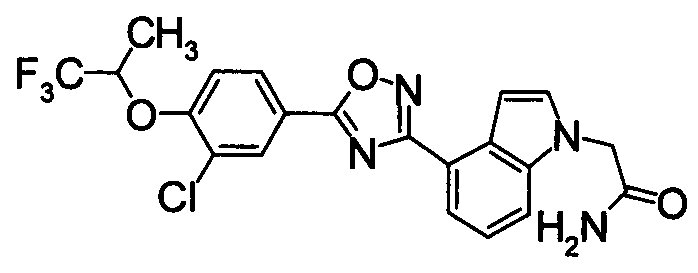
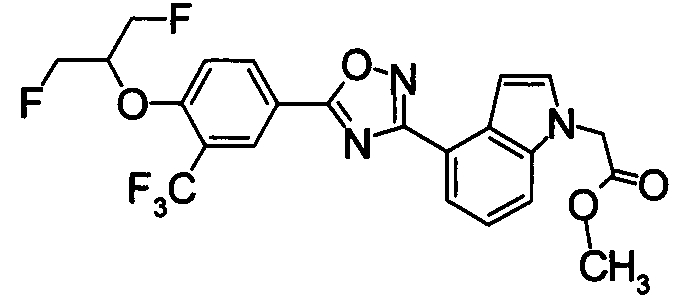
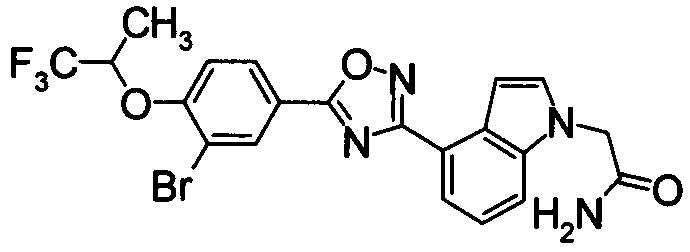
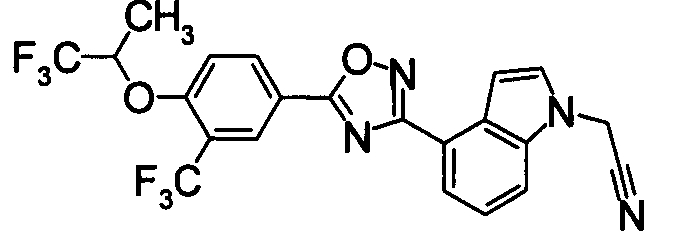
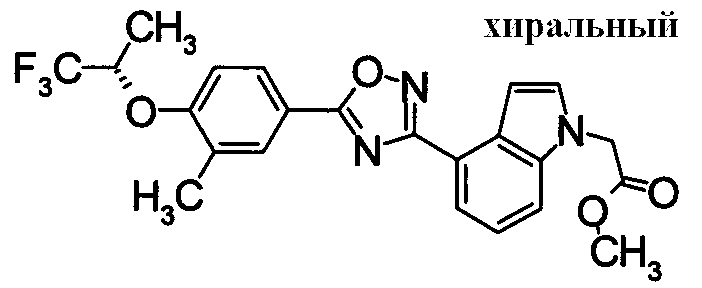
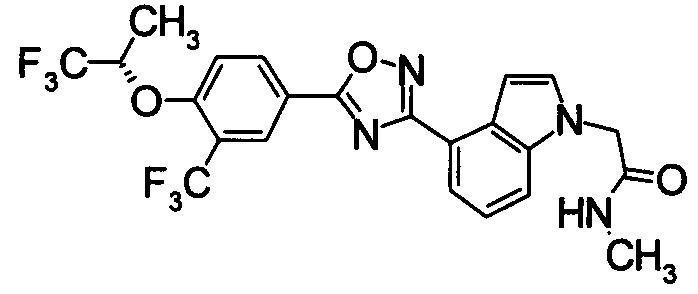
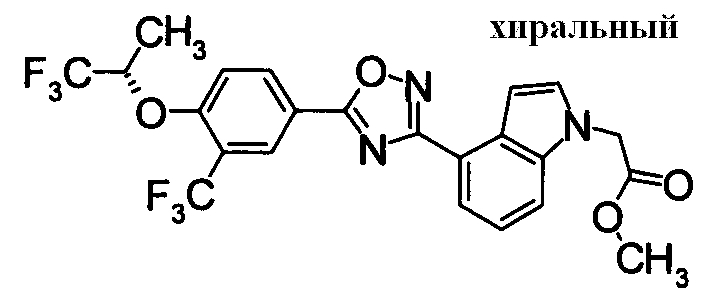
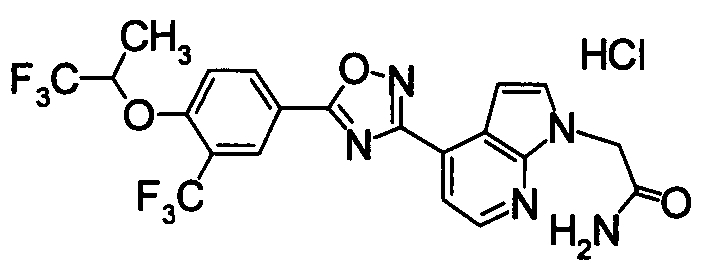
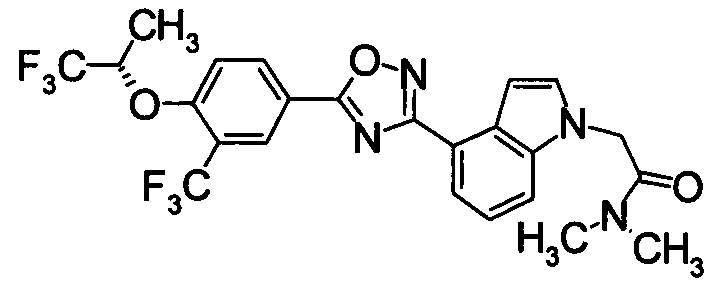
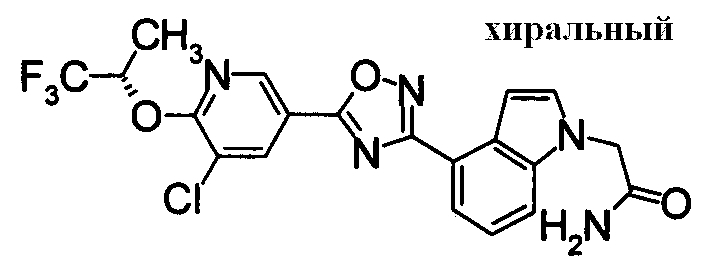
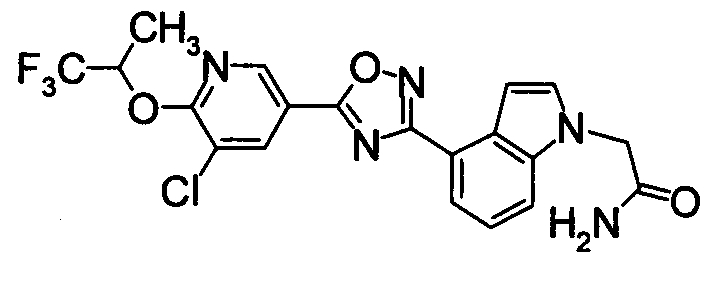
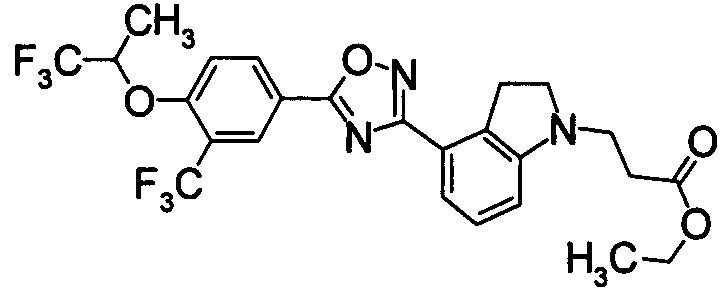
№
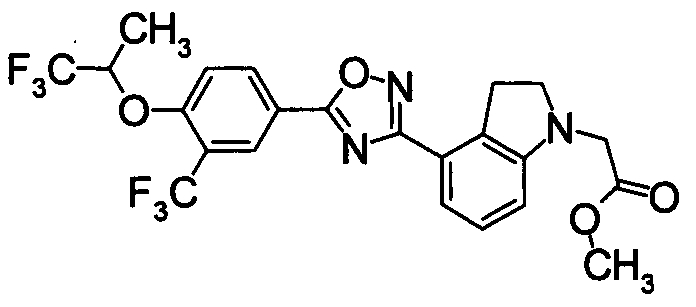
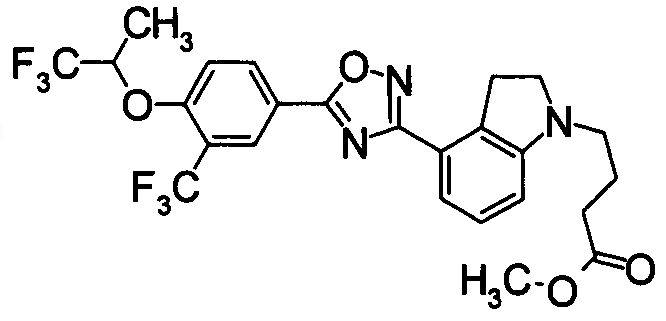
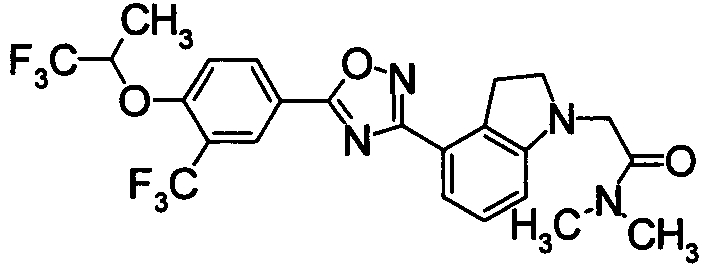
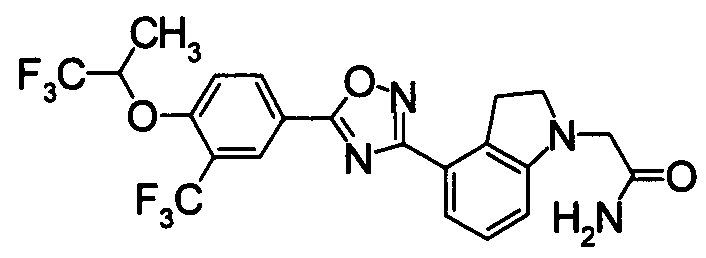
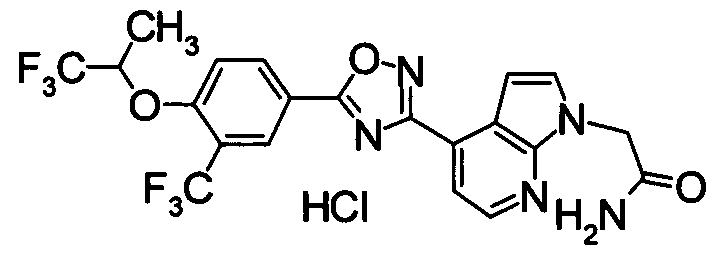
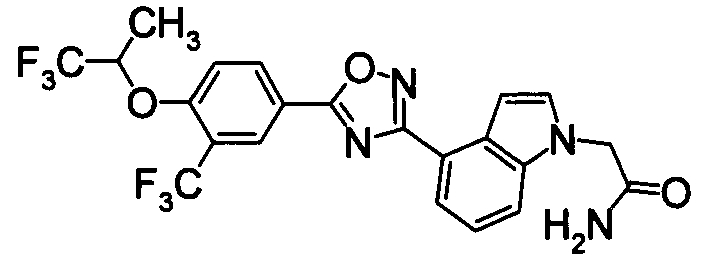
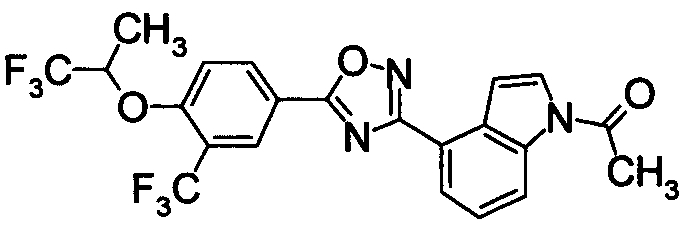
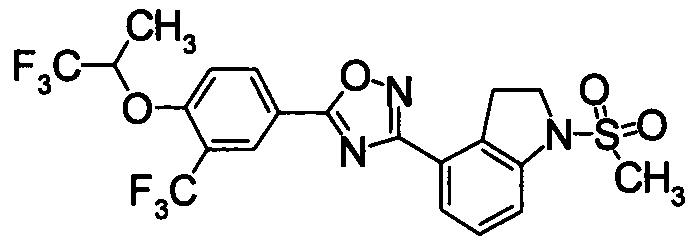
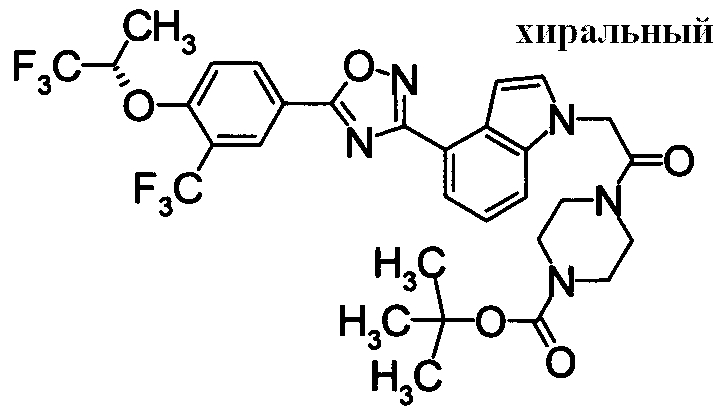
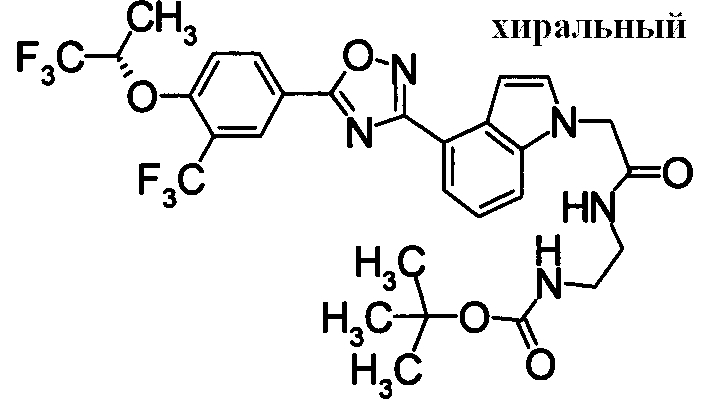
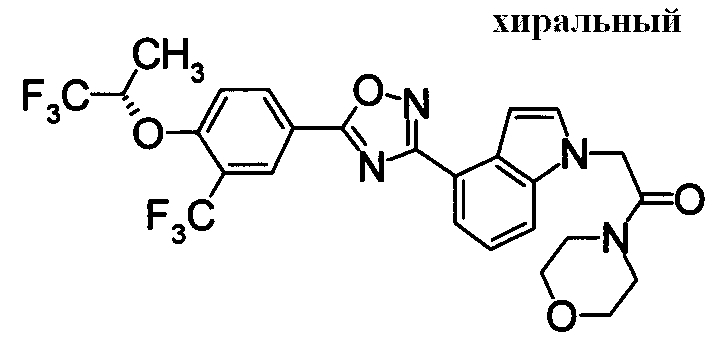
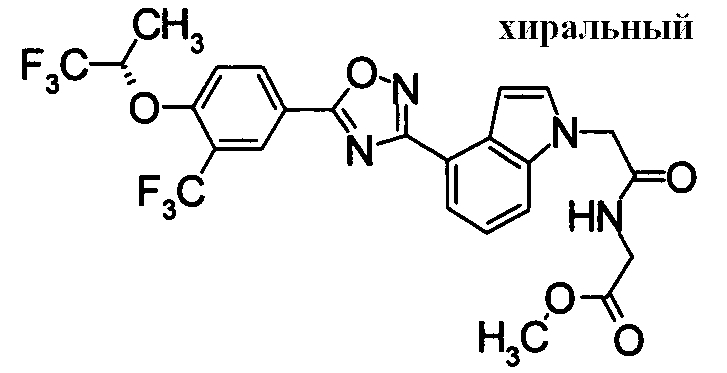
№
№
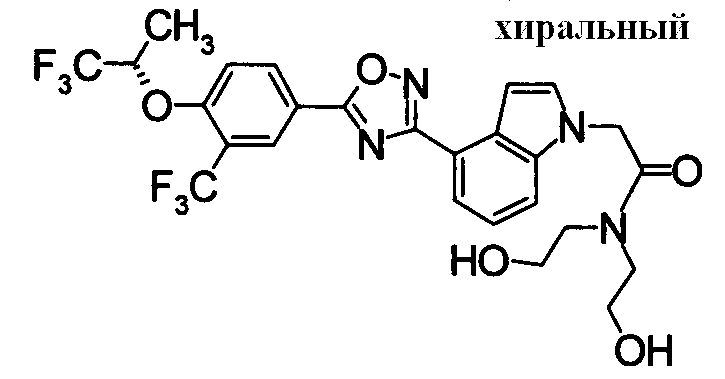
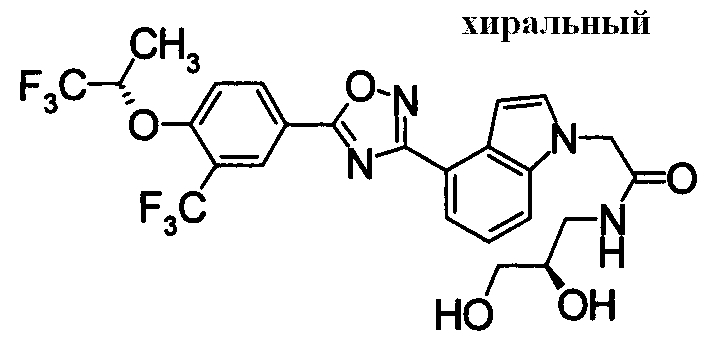
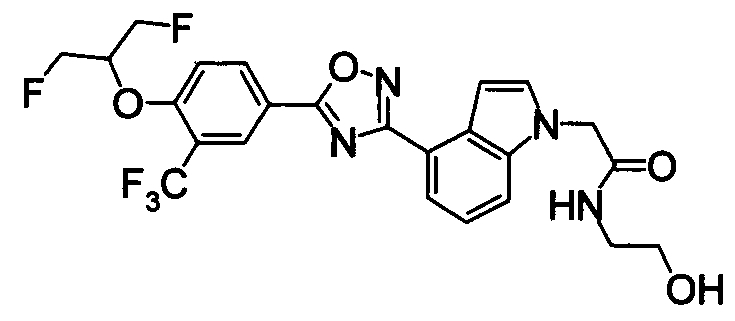
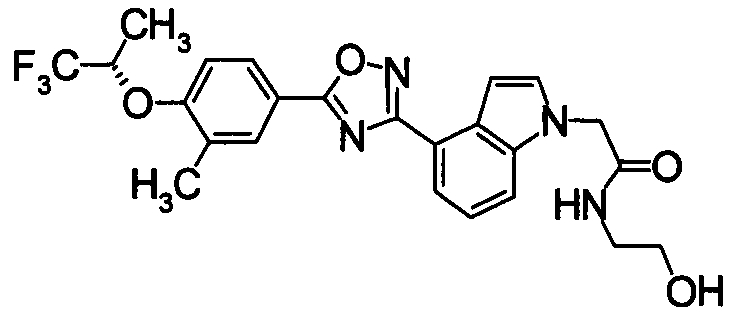
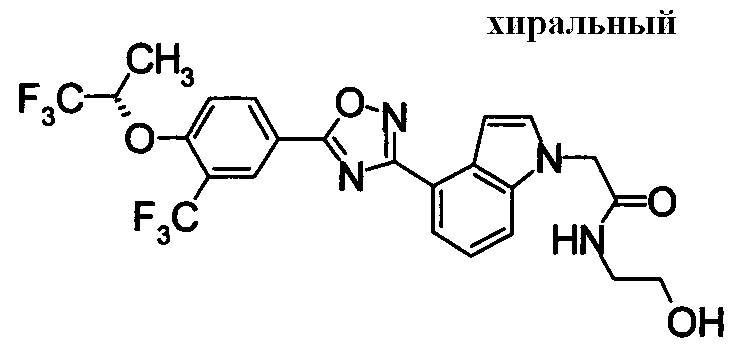
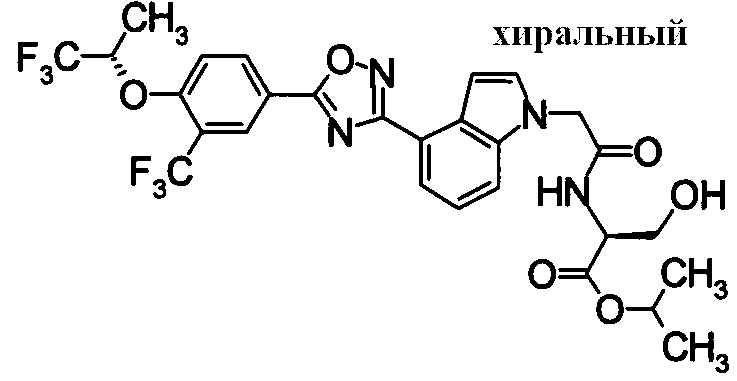
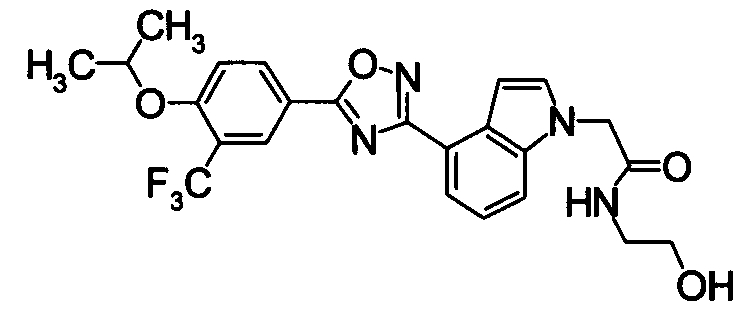
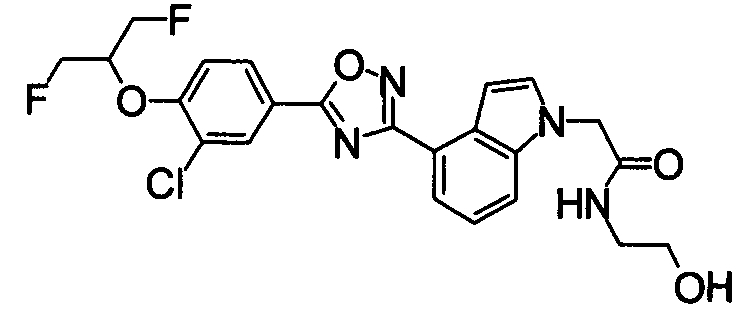
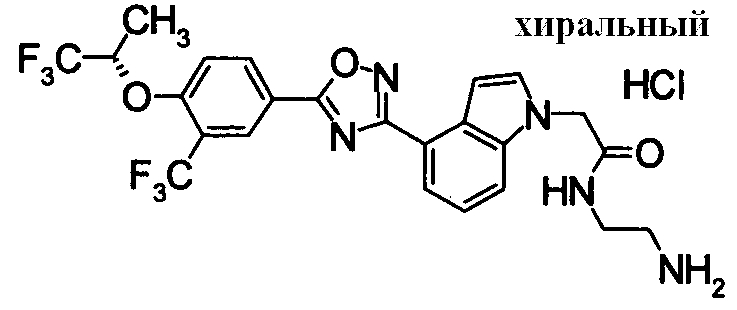
В следующих таблицах показаны данные ЯМР соединений Примеров. В качестве внутреннего стандарта использовался тетраметилсилан и, если не сказано что-либо иное, δ (ppm) сигнала 1H-ЯМР показаны как измеренные в DMSO-d6, используемом как в растворитель. Ref-Ex обозначает Пример №, который следует относить к получению.
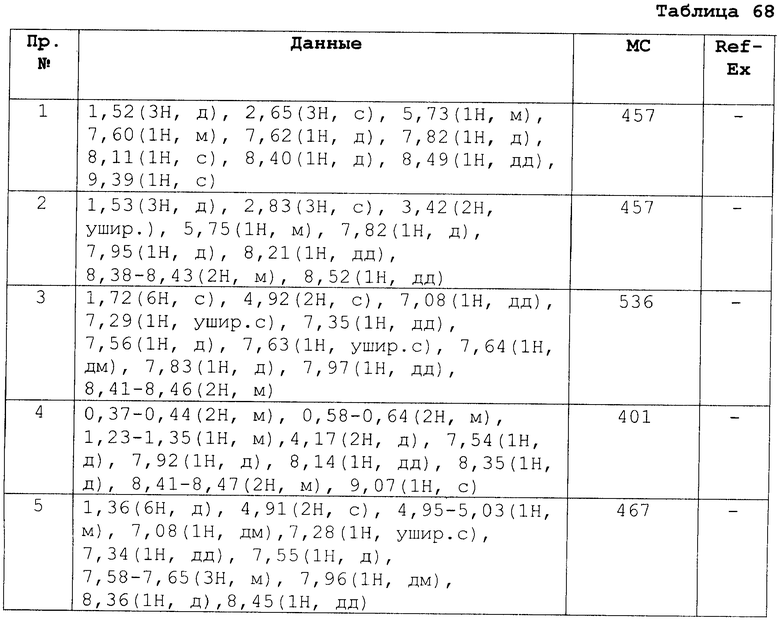
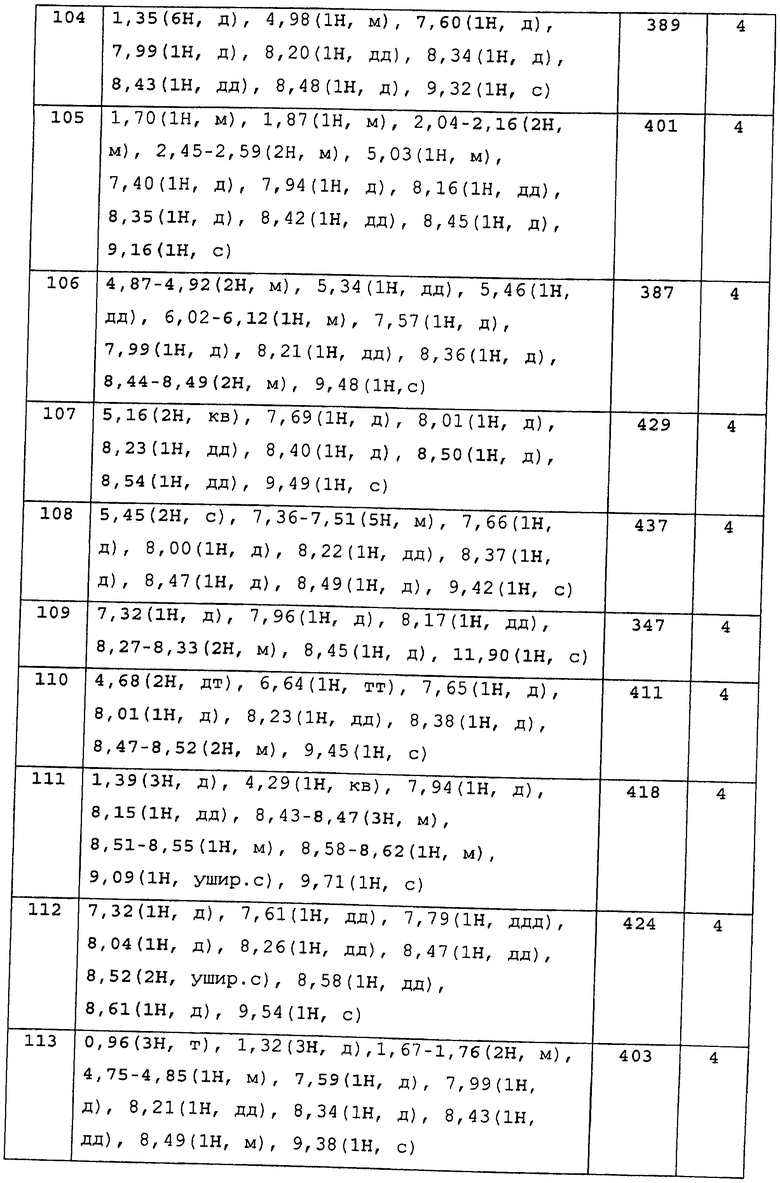
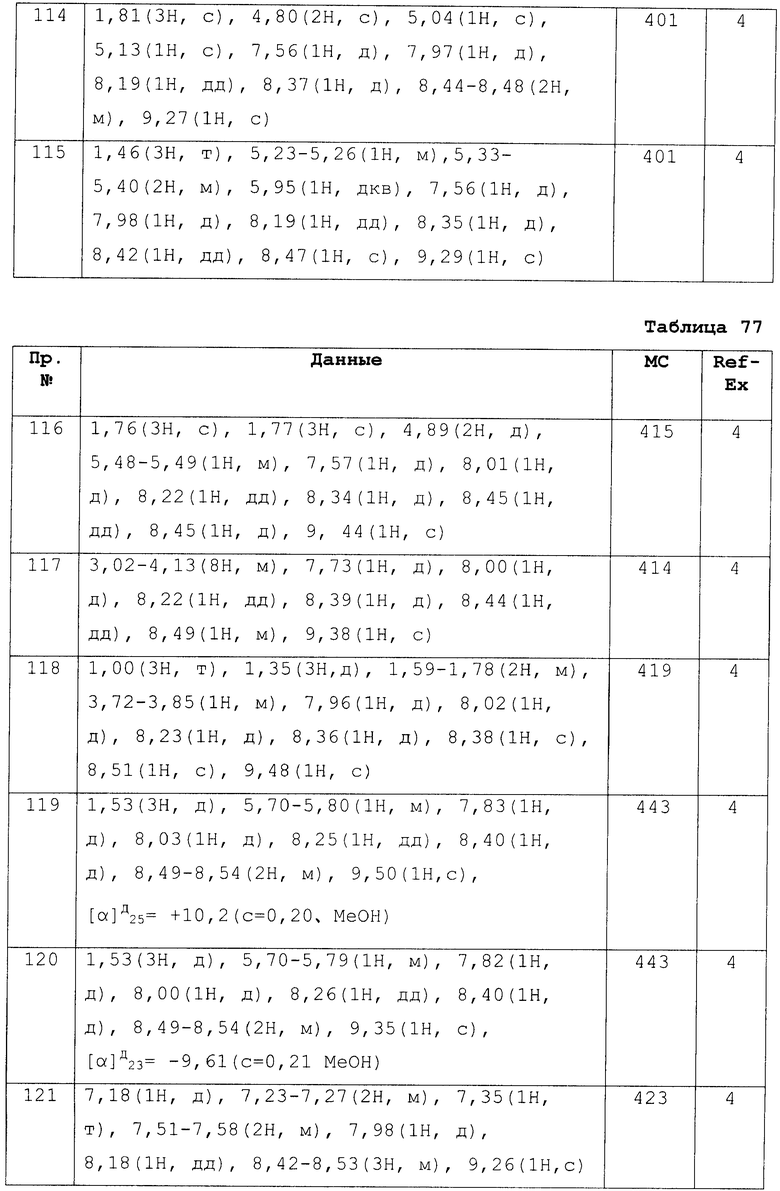
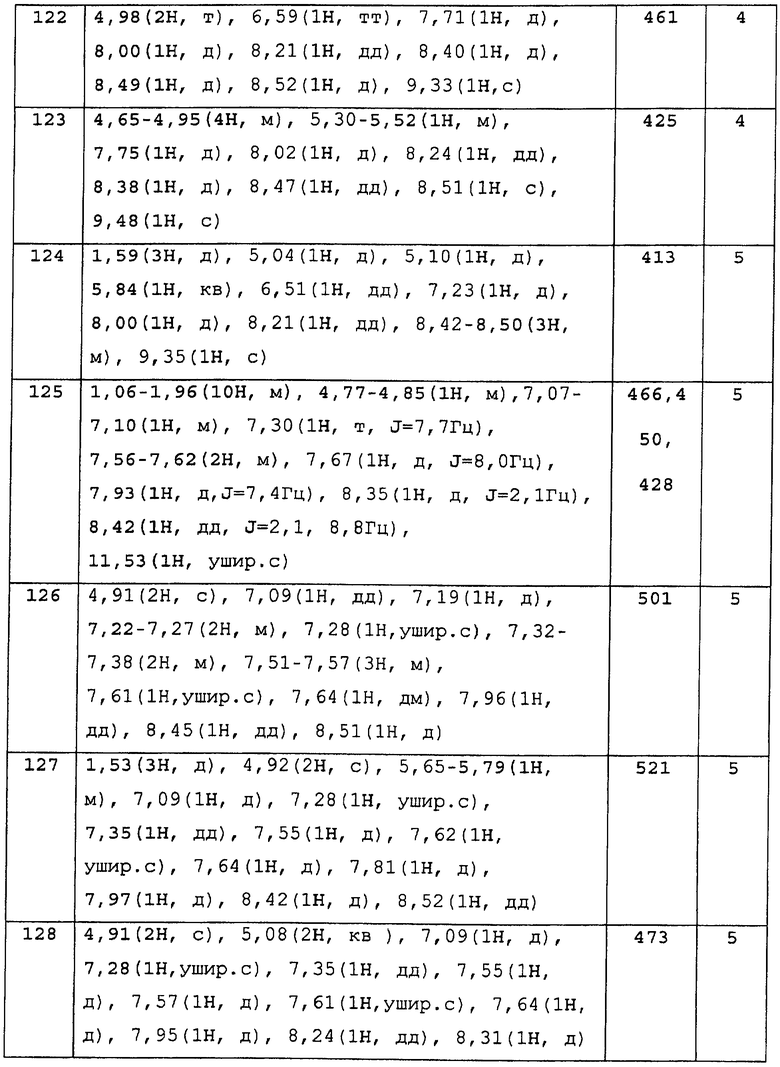
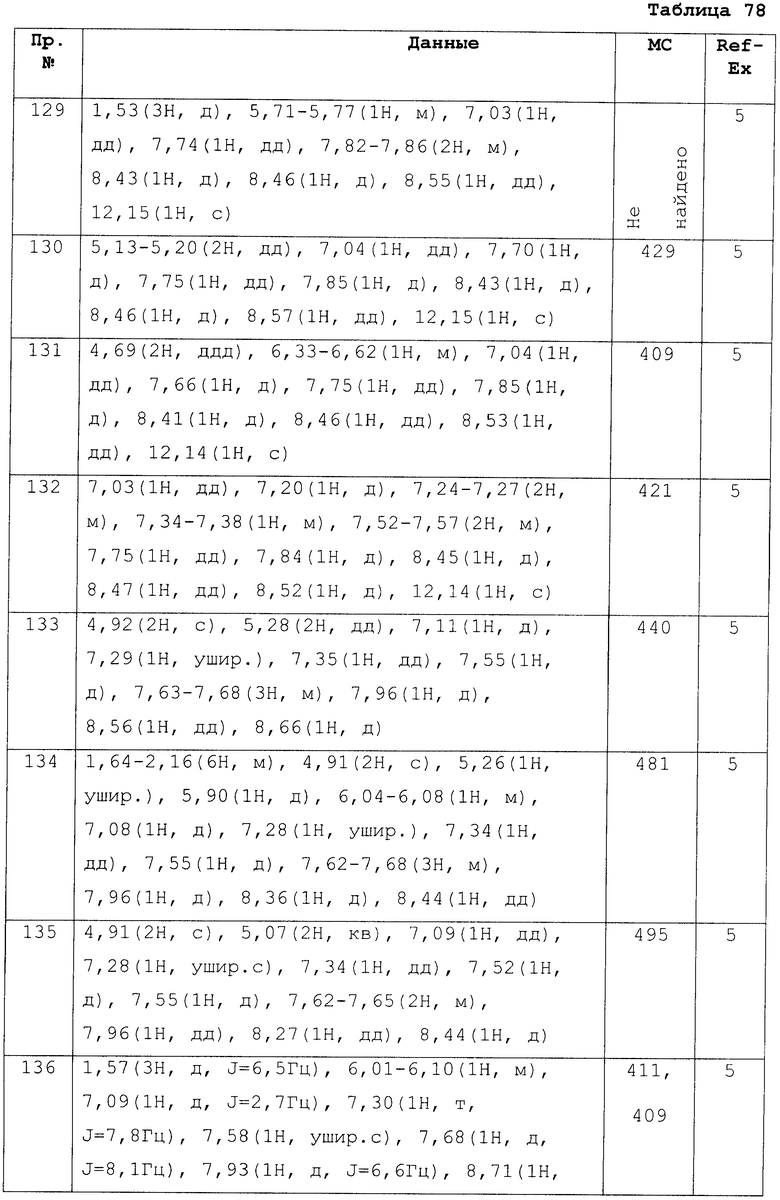
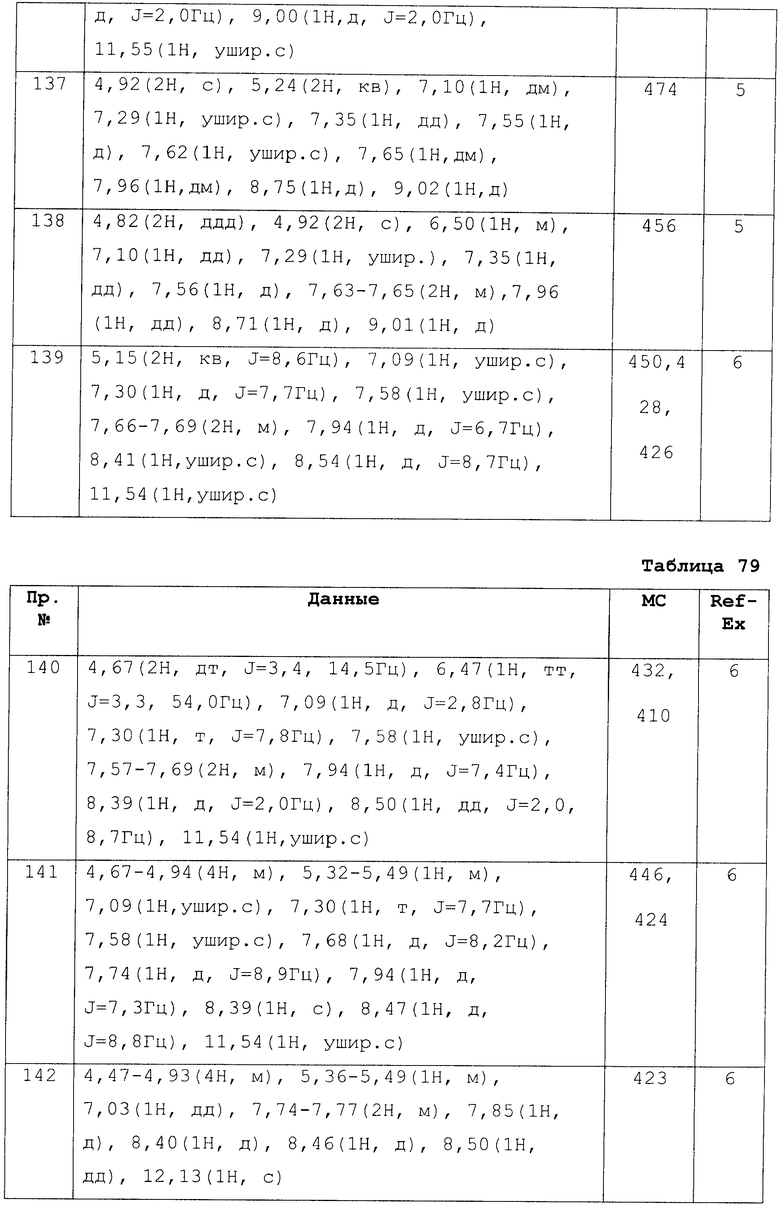
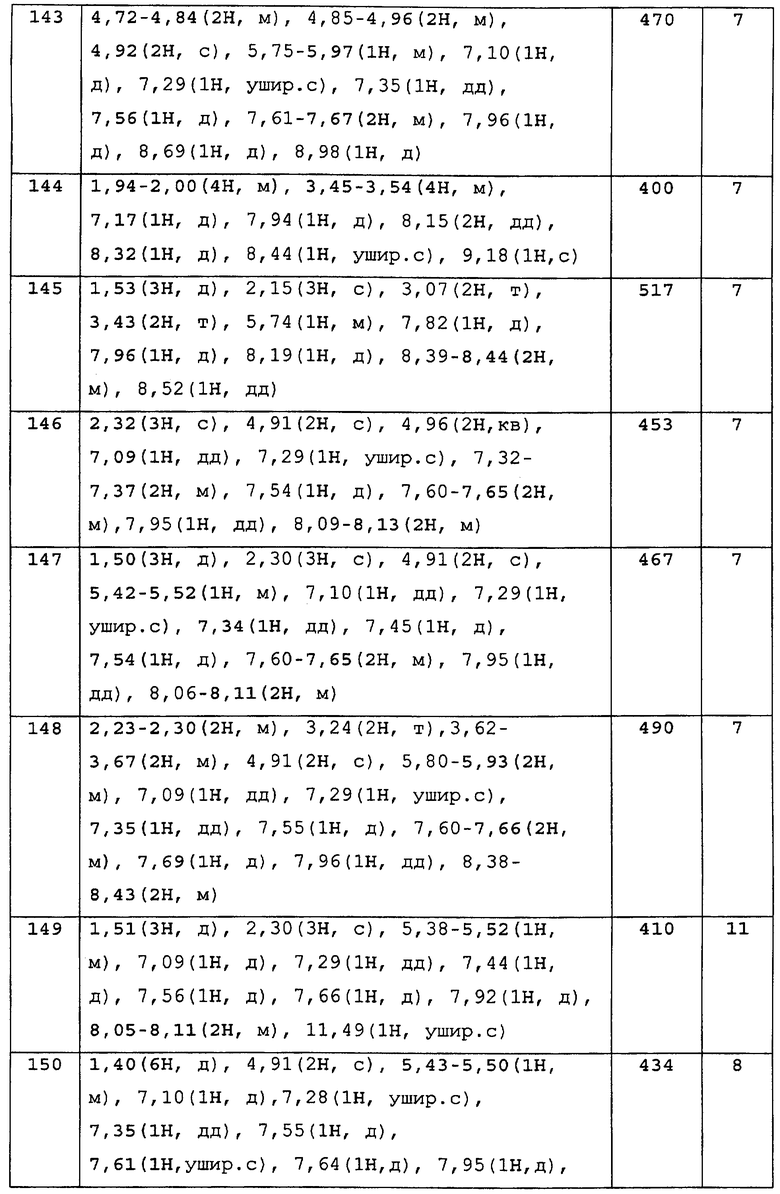
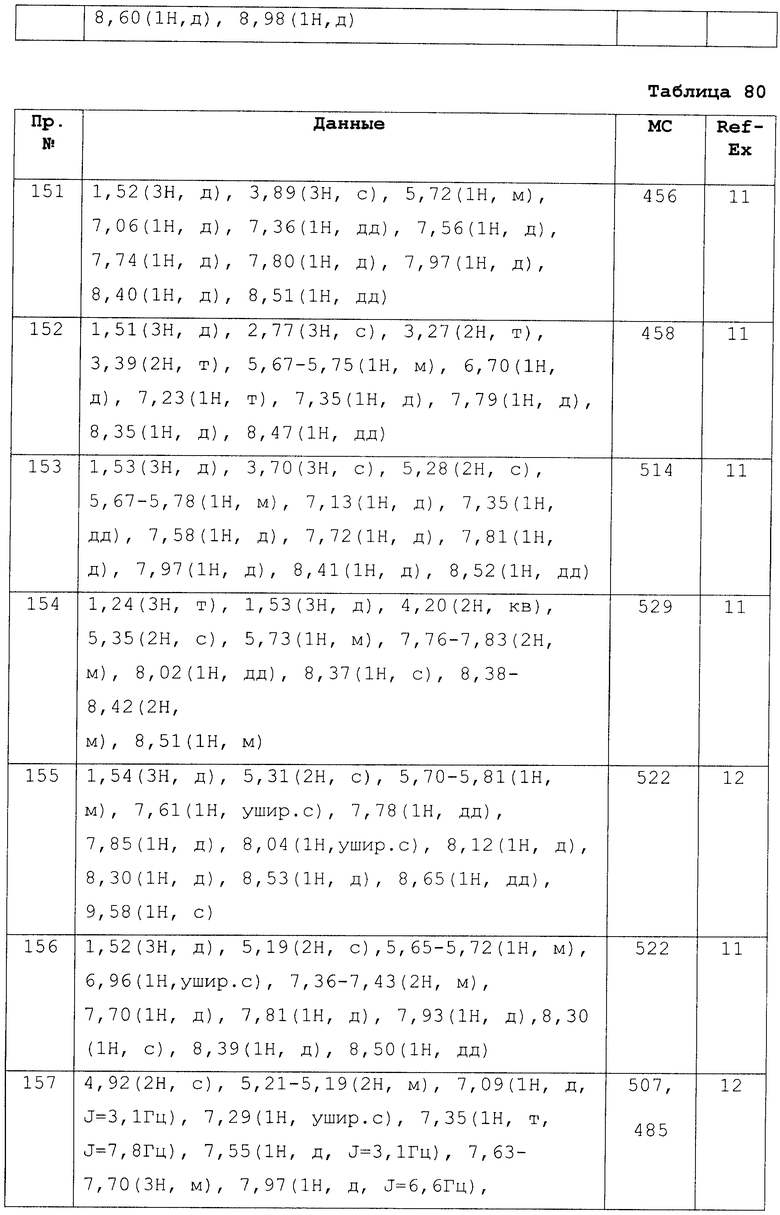
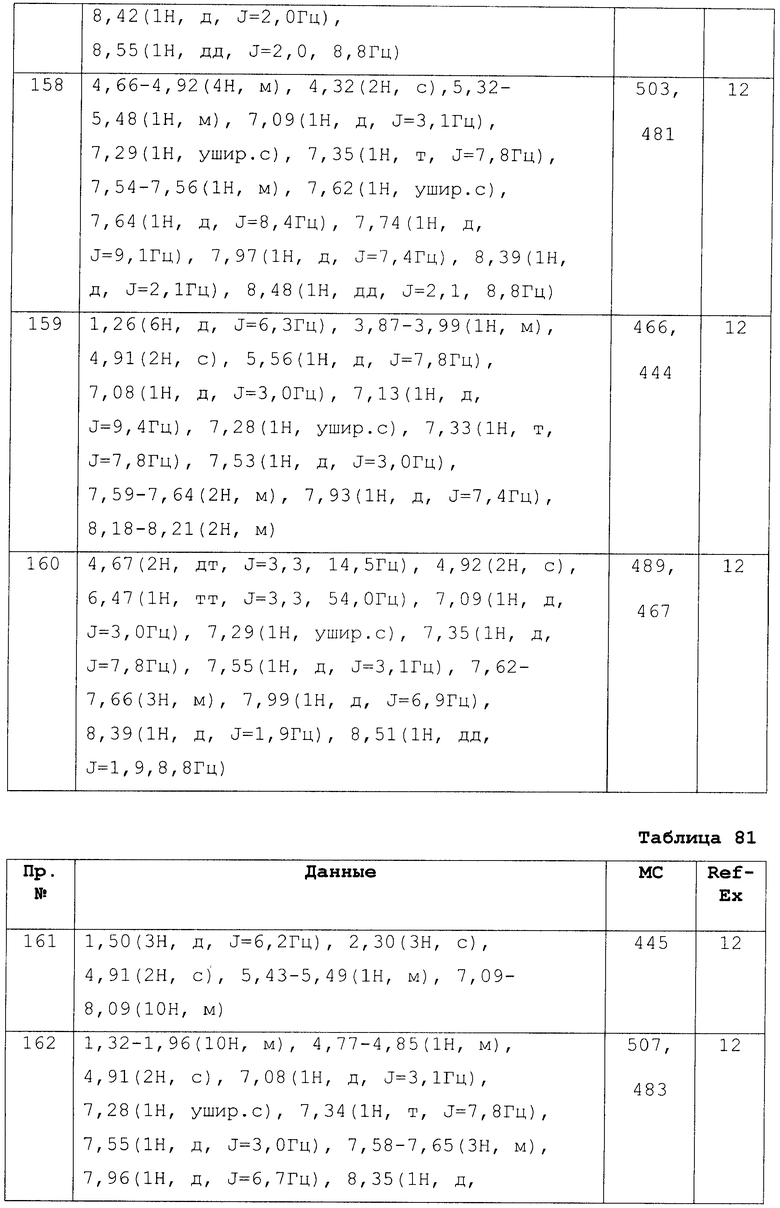
Применение в промышленных условиях
Соединение по настоящему изобретению применимо в качестве лекарственного средства и, в частности, для профилактики и/или лечения отторжения органов при трансплантации, костного мозга или тканей, аутоиммунных заболеваний и т.п., так как оно обладает S1P1 агонистической активностью.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ 2Н-ХРОМЕНА И ЕГО ПРОИЗВОДНОЕ | 2009 |
|
RU2490266C2 |
| ИНГИБИТОРЫ ОКСАЗИНМОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ (MAGL) | 2019 |
|
RU2794334C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ГИСТОНДЕЗАЦЕТИЛАЗЫ 6 | 2018 |
|
RU2797613C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА МИНЕРАЛОКОРТИКОИДОВ | 2012 |
|
RU2598842C2 |
| ЧЕТЫРЕХЗАМЕЩЕННЫЕ БЕНЗОЛЫ | 2008 |
|
RU2527177C2 |
| ПРОИЗВОДНЫЕ ТИАЗОЛОПИРИДИНА КАК АГОНИСТЫ GPR119 | 2017 |
|
RU2749111C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| ТИОФЕНИЛЬНЫЕ И ПИРРОЛИЛЬНЫЕ АЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ СЕРОТОНИНОВОГО 5-HT РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2434872C2 |
| СПИРО-КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА В КАЧЕСТВЕ ИНГИБИТОРОВ HSL, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА | 2010 |
|
RU2607080C2 |
Изобретение относится к соединению формулы (I)
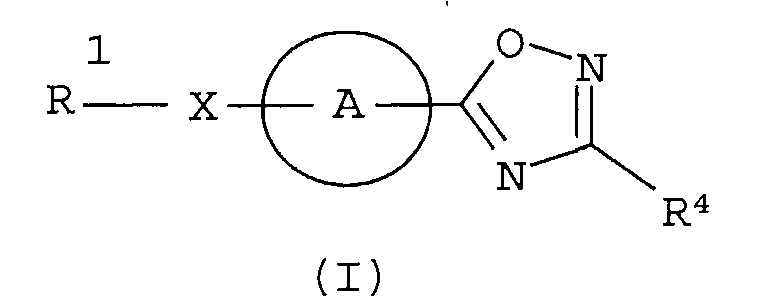
или к его фармацевтически приемлемой соли, где символы имеют следующие значения: кольцо А представляет собой 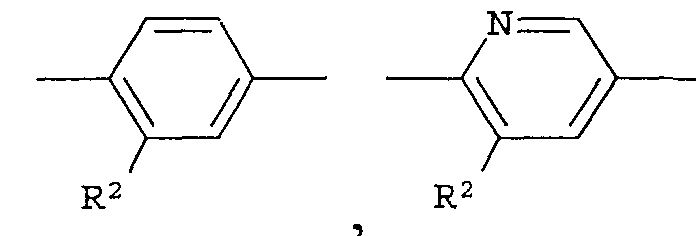
или X представляет собой простую связь, -CH2-, -NR3-, -O-, -S-, R1 представляет собой галоген; фенил; пиридил; (С3-C8) циклоалкил, или (C1-С6) алкил, или (С2-С6) алкенил, каждый из которых может содержать галоген, -CONH2, фенил или (С3-С8)циклоалкил в качестве заместителя, R2 представляет собой -CN, -O-(С1-С6)алкил, -C(=O)H, галоген или (С1-С6)алкил, который может быть замещен с помощью галогена или -ОН, R3 может образовывать морфолино или 1-пирролидинил вместе с R1 и азотом, и, когда -Х- представляет собой простую связь, R1 и R2 могут в комбинации образовывать 5-членное кольцо и дополнительно содержать (C1-С6)алкил в качестве заместителя, R4 представляет собой следующее кольцо:
 ,
,  ,
,  ,
,
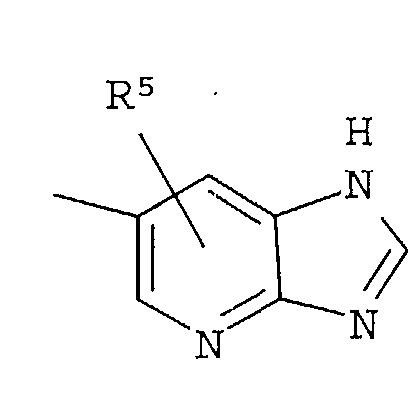 ,
, 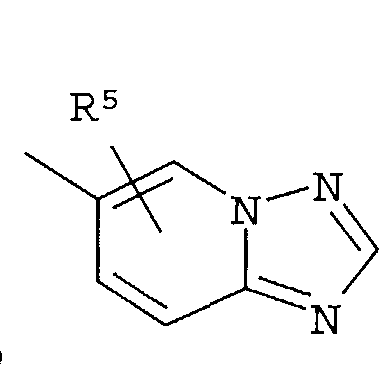 ,
, 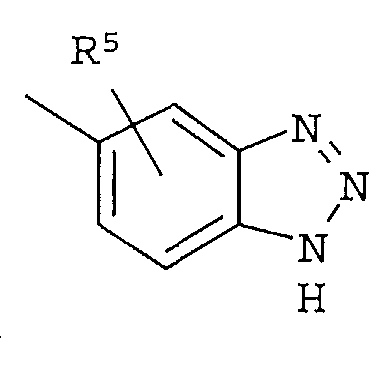 ,
, 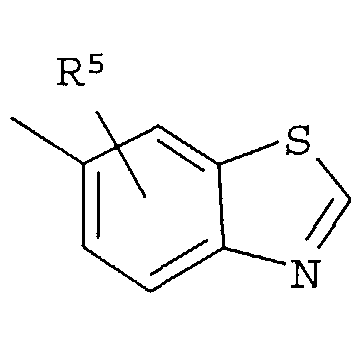 ,
,
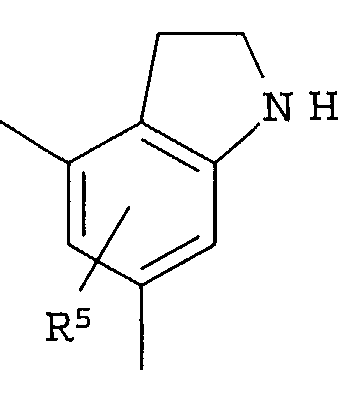 ,
, 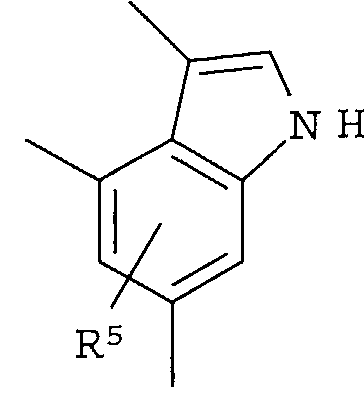 ,
, 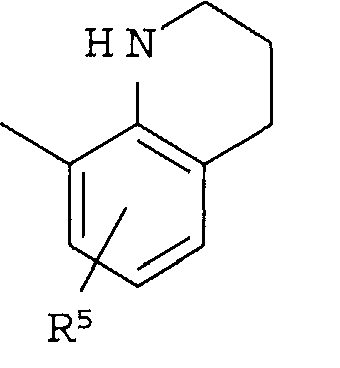 или
или 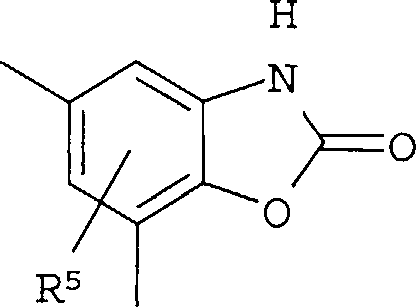
где любая одна из связей от кольца связана с оксазольным кольцом, R5 представляет собой -Н; (С1-С6)алкил, который может быть замещен не менее одной группой, выбранной из ряда, содержащего: -C(=O)NRXRY -NHRX и -ORX-(С2-С6)алкенил-; -С(=O)H; -C(=O)NRXRY, RX и RY могут быть одинаковыми или отличатся друг от друга и представлять собой -H или (C1-С6) алкил. Изобретение также относится к фармацевтической композиции, обладающей активностью агониста SlP1, на основе указанных соединений. Соединения и композиции могут найти свое применение в медицине для профилактики и/или лечения отторжения при трансплантации органа, костного мозга или тканей, аутоиммунных заболеваний. 9 н. и 7 з.п. ф-лы, 84 табл.
1. Соединение формулы (I)
[Хим. 16]
или его фармацевтически приемлемая соль,
где символы имеют следующие значения:
кольцо А представляет собой
[Хим. 17]
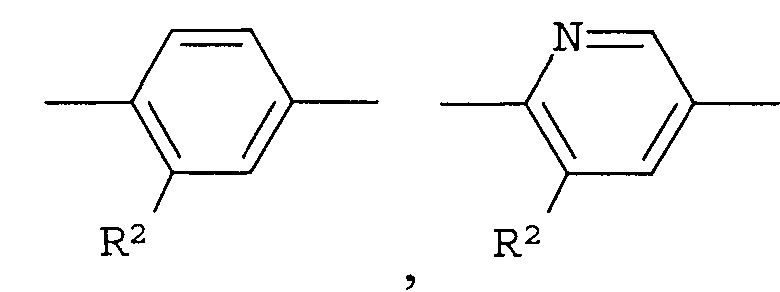 или
или 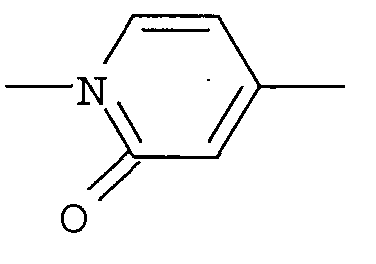
Х представляет собой простую связь, -СН2-, -NR3-, -O-, -S-,
R1 представляет собой галоген, фенил, пиридил, (С3-C8) циклоалкил, или (C1-С6) алкил, или (C2-C6) алкенил, каждый из которых может содержать галоген, -CONH2, фенил или (С3-С8)циклоалкил в качестве заместителя,
R2 представляет собой -CN, -O-(С1-С6)алкил, -С(=O)H, галоген, или (C1-С6)алкил, который может быть замещен с помощью галогена или -ОН,
R3 может образовывать морфолино или 1-пирролидинил вместе с R1 и азотом, и, когда -Х- представляет собой простую связь, R1 и R2 могут в комбинации образовывать 5-членное кольцо и дополнительно содержать (С1-С6)алкил в качестве заместителя,
R4 представляет собой следующее кольцо:
[Хим. 18]
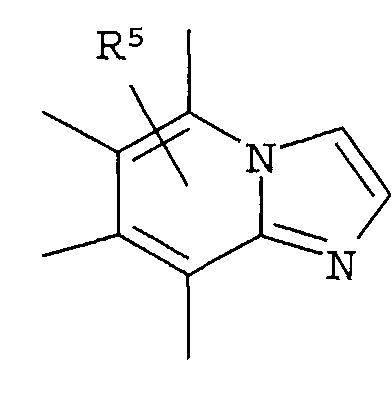 ,
, 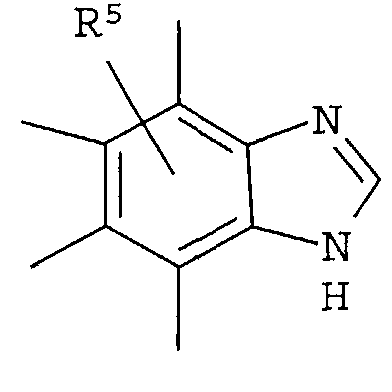 ,
, 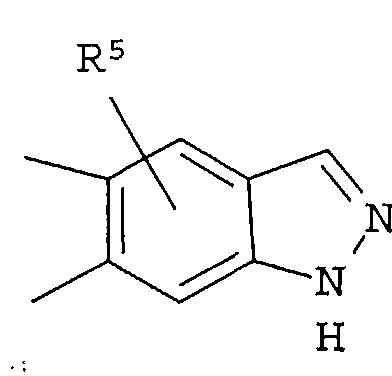 ,
,
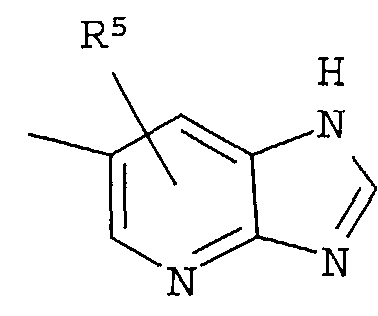 ,
, 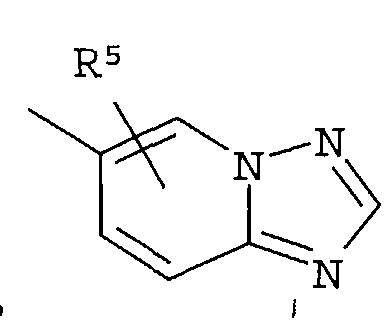 ,
, 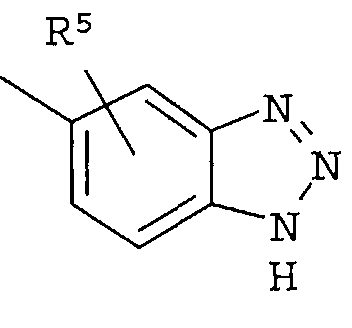 ,
, 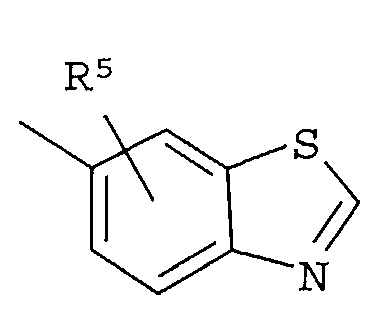 ,
,
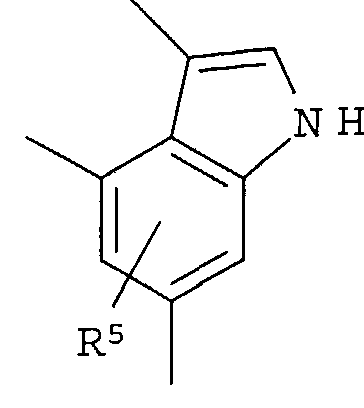 ,
, 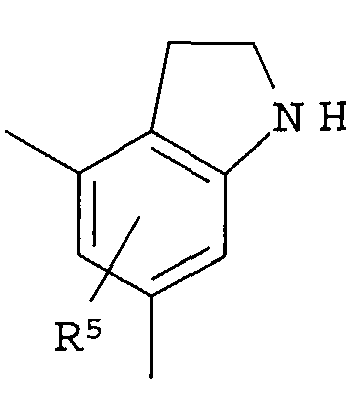 ,
, 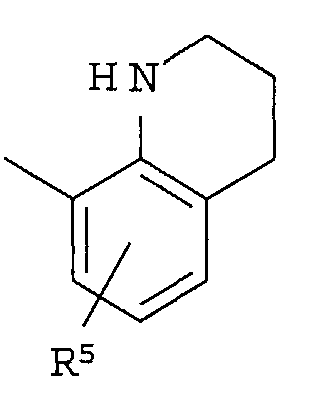 или
или 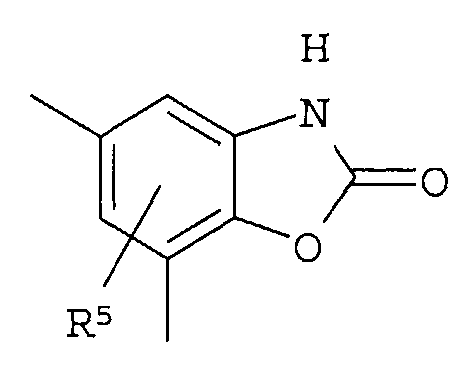
где любая одна из связей от кольца связана с оксазольным кольцом
R5 представляет собой -Н, (С1-С6)алкил, который может быть замещен не менее одной группой, выбранной из ряда, содержащего -С(=O)NRXRY, -NHRX и -ORX- (С2-С6)алкенил-, -С(=O)H, -C(=O)NRXRY,
RX и RY могут быть одинаковыми или отличаться друг от друга и представлять собой -Н или (С1-С6) алкил.
2. Соединение по п.1, которое изображено при помощи формулы (II):
[Хим. 19]
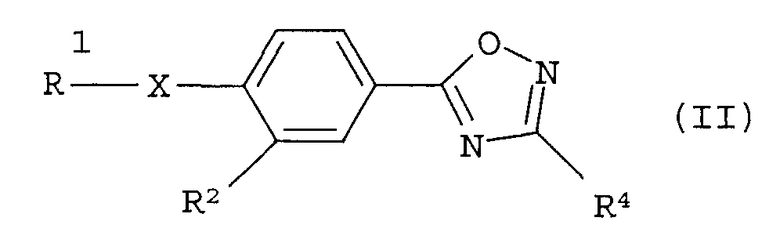
или его фармацевтически приемлемая соль.
3. Соединение по п.2 или его фармацевтически приемлемая соль, в котором R1 представляет собой (C1-С6) алкил, который может содержать галоген в качестве заместителя.
4. Соединение по п.2 или его фармацевтически приемлемая соль, в котором R2 представляет собой -CN, галоген или (С1-С6)алкил, который может быть замещен с помощью галогена или -ОН.
5. Соединение по п.2 или его фармацевтически приемлемая соль, в котором R5 представляет собой -Н; или (С1-С6)алкил, который может быть замещен по меньшей мере одним -C(=O)NRXRY.
6. Соединение по п.2 или его фармацевтически приемлемая соль, в котором Х представляет собой простую связь, -СН2- или -O-.
7. Соединение по п.2 или его фармацевтически приемлемая соль, в котором R4 представляет собой следующее кольцо:
[Хим.20]
 или
или 
где любая одна из связей от кольца R4 связана с оксадиазольным кольцом.
8. Соединение формулы (I)
[Хим. 16]
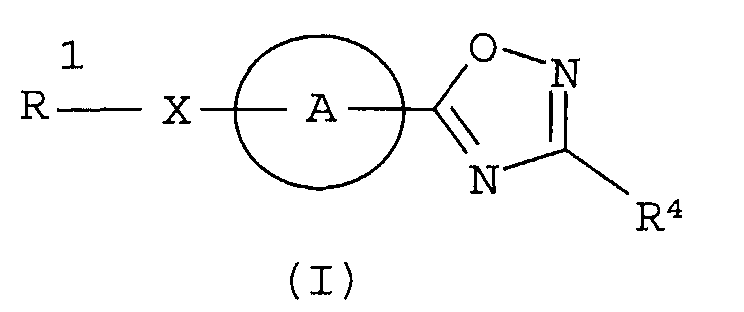
или его фармацевтически приемлемая соль, где символы имеют следующие значения:
кольцо А представляет собой
Х представляет собой -O-,
R1 представляет собой (С1-C4) алкил, необязательно замещенный F,
R2 представляет собой галоген, -CN, или (С1-С6)алкил, необязательно замещенный галогеном,
R4 представляет собой следующее кольцо:
[Хим. 18]
где любая одна из связей от кольца связана с оксазольным кольцом,
R5 представляет собой -Н; (C1-С6) алкил, необязательно замещенный -C(=O)NRXRY,
RX представляет собой -Н; (С1-С6)алкил,
RY представляет собой -Н.
9. Соединение, выбранное из следующих:
5-(5-[3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил)-1,2,4-оксадиазол-3-ил)-1Н-бензимидазол гидрохлорид;
5-(5-[3-(трифторметил)-4-[(1R)-2,2,2-трифтор-1-метилэтокси]фенил)-1,2,4-оксадиазол-3-ил)-1Н-бензимидазол гидрохлорид;
6-{5-[3-бром-4-(2,2,2-трифтор-1-метилэтокси]фенил)-1,2,4-оксадиазол-3-ил}-1Н-бензимидазол гидрохлорид;
5-(5-[4-фенокси-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил)-1Н-бензимидазол гидрохлорид;
2-(4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси)фенил)-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)ацетамид;
4-{5-[3-(трифторметил)-4-(2,2,2-трифтор-1-метилэтокси]фенил)-1,2,4-оксадиазол-3-ил)-1Н-пирроло[2,3-b]пиридин гидрохлорид;
6-{5-[3-(дифторметил)-4-(2,2,2-трифтор-1-метилэтокси]фенил)-1,2,4-оксадиазол-3-ил)-1Н-бензимидазол гидрохлорид;
2-(4-{5-[3-хлор-4-(2,2,2-трифтор-1-метилэтокси)фенил)-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)ацетамид;
2-(4-{5-[4-циклопентил-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
2-[4-(5-{3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил)-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
2-[4-(5-{4-[2-фтор-1-(фторметил)этокси]-3-(трифторметил)фенил)-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
2-(4-{5-[3-циано-4-(2,2,2-трифтор-1-метилэтокси)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)ацетамид;
N-(2-гидроксиэтил)-2-[4-(5-{3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
2-[4-(5-{5-хлор-6-[(1S)-2,2,2-трифтор-1-метилэтокси]пиридин-3-ил]-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
2-[4-(5-{5-хлор-6-[2-фтор-1-(фторметил)этокси]пиридин-3-ил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
2-[4-(5-{3-хлор-4-[2-фтор-1-(фторметил)этокси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
2-[4-(5-{3-метил-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
2-(4-{5-[4-изопропил-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
2-{4-[5-(3-хлор-4-изопропоксифенил)-1,2,4-оксадиазол-3-ил]-1Н-индол-1-ил)ацетамид;
N-(2-гидроксиэтил)-2-[4-(5-{3-метил-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид;
N-(2-гидроксиэтил)-2-(4-{5-[4-изопропокси-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил)ацетамид; и
2-[4-(5-{3-хлор-4-[2-фтор-1-(фторметил)этокси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]-N-(2-гидроксиэтил)ацетамид.
10. Фармацевтическая композиция, обладающая активностью агониста S1P1, содержащая эффективное количество соединения по любому из пп.1-9, или его фармацевтически приемлемой соли, в качестве активного ингредиента, и фармацевтически приемлемый носитель или эксципиент.
11. Соединение по любому из пп.1-9 или его фармацевтически приемлемая соль, применяемое для лечения и/или профилактики отторжения при пересадке органа/ткани у человека или животного, аутоиммунного заболевания или рассеянного склероза.
12. Средство, обладающее активностью агониста S1P1, содержащее соединение по любому из пп.1-9 или его фармацевтически приемлемую соль, в качестве активного ингредиента и фармацевтически приемлемый носитель или эксципиент.
13. Способ лечения и/или профилактики отторжения при трансплантации органа/костного мозга/ткани у человека или животного, реакции «трансплантат против хозяина» или рассеянного склероза, причем указанный способ включает введение соединения, или его фармацевтически приемлемой соли по любому из пп.1-9 пациенту, страдающему вышеупомянутым заболеванием.
14. Применение соединения по любому из пп.1-9 или его фармацевтически приемлемой соли для получения средства для лечения и/или профилактики отторжения при трансплантации органа/костного мозга/ткани у человека или животного, реакции «трансплантат против хозяина» или рассеянного склероза.
15. Применение соединения по любому из пп.1-9 или его фармацевтически приемлемой соли для лечения и/или профилактики отторжения при трансплантации органа/костного мозга/ткани у человека или животного, реакции «трансплантат против хозяина» или рассеянного склероза.
16. Фармацевтическая композиция, содержащая соединение по любому из пп.1-9 или его фармацевтически приемлемую соль, для применения для лечения и/или профилактики отторжения при трансплантации органа/костного мозга/ткани у человека или животного, реакции «трансплантат против хозяина» или рассеянного склероза.
| JP 11279176 A, 12.10.1999 | |||
| WO 2006001463 A1, 05.01.2006 | |||
| WO 2004113330 A1, 29.12.2004 | |||
| RU 2005108070 A, 27.08.2005. |

