Область техники, к которой относится изобретение
Изобретение относится к соединению 2Н-хромена и его производному, которые являются полезными в качестве активного ингредиента для фармацевтической композиции, в частности, фармацевтической композиции для профилактики или лечения заболеваний, вызванных нежелательной инфильтрацией лимфоцитов, или заболеваний, вызванных аномальной пролиферацией или аккумуляцией клеток.
Предшествующий уровень техники
Сфингозин-1-фосфат является метаболитом сфинголипида, который представляет собой физиологически активное вещество, секретируемое из активированных тромбоцитов (Annual Review Biochemistry, 2004, Vol. 73, pp. 321-354). Рецептор сфингозин-1-фосфата представляет собой связывающийся с G-белком тип рецептора и относится к Edg-семейству, которое представляет собой ген дифференциации эндотелиальных клеток. До сих пор были обнаружены пять рецепторов S1P1 (Edg1), S1P2 (Edg5), S1P3 (Edg3), S1P4 (Edg6) и S1P5 (Edg8). Все эти рецепторы широко распространены в клетках и тканях по всему организму, но S1P1, S1P3 и S1P4 преимущественно экспрессируются в лимфоцитах и эндотелиальных клетках, S1P2 преимущественно экспрессируется в клетках гладких мышц сосудов, S1P5 преимущественно экспрессируется в головном мозге и селезенке, и его аминокислотные последовательности хорошо сохранены у людей и грызунов (Annual Review Biochemistry 2004, Vol. 73, pp. 321-354).
Многие рецепторы связываются с G-белками в результате стимуляции сфингозин-1-фосфата. S1P1 связывается с Gi/0, S1P2 и S1P3 связываются с Gi/0, Gq, G12/13 и Gs, S1P4 связывается с Gi/0, G12/13 и Gs, S1P5 связывается с Gi/0 и G12/13, и индуцируются клеточная пролиферация, вызываемая активацией MAPK, изменения в цитоскелетной системе и клеточная инфильтрация, вызываемые активацией Rac (и/или Rho), и продукция цитокинов и медиаторов, вызываемая активацией PLC и притоком кальция в клетку, и т.п. (Annual Review Biochemistry, 2004, Vol. 73, pp. 321-354).
Известно, что через стимуляцию действия S1P1 сфингозин-1-фосфата индуцируются миграция лимфоцитов, ингибирование апоптоза, продукция цитокинов и секвестрация лимфоцитов в тимусе и других вторичных лимфоидных тканях, и это способствует ангиопластике в эндотелиальных клетках сосудов (Nature Review Immunology, 2005, Vol. 5, pp. 560-570). С другой стороны, экспрессия S1P3 также обнаружена на кардиомиоцитах, и наблюдается временное снижение частоты сердечных сокращений (редкий пульс) или кровяного давления через стимуляцию сфингозин-1-фосфата (Japanese Journal of Pharmacology, 2000, Vol. 82, pp. 338-342). Редкий пульс не наблюдается при стимуляции сфингозин-1-фосфата у мышей с “нокаутом”, где имеется генетическая недостаточность S1P3 (Journal of Pharmacology and Experimental Therapeutics, 2004, Vol. 309, pp. 758-768).
Известно, что FTY720 и FTY720 фосфат, который является его активным основным членом, обладают отличным действием агониста S1P1 и, таким образом, индуцируют секвестрацию лимфоцитов, и сообщалось об их эффектах на кожный трансплантат или рассеянный склероз, которые являются аутоиммунными заболеваниями (Cellular & Molecular Immunology, 2005, Vol. 2, No. 6, pp. 439-448; и The New England Journal of Medicine, 2006, Vol. 355, pp. 1124-40). Однако также были сообщения о побочных эффектах, таких как редкий пульс, пониженная функция легких (Transplantation, 2006, 82, pp. 1689-1967). Было сообщение о том, что FTY720 фосфат обладает неселективным агонистическим действием на S1P3, S1P4 и S1P5 (Science, 2002, Vol. 296, pp. 346-349), и сообщалось о результатах клинического испытания, выявивших в качестве нежелательного побочного эффекта очень частые случаи редкого пульса, индуцируемого стимулирующим действием через S1P3 (Journal of American Society of Nephrology, 2002, Vol. 13, pp. 1073-1083).
В качестве соединения, обладающего действием агониста S1P1, Патентный документ 1 раскрывает соединение общей формулы (A):
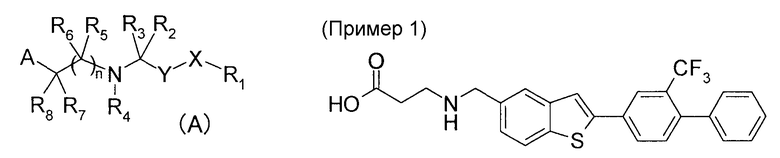
[где n имеет значение 1 или 2; A представляет собой -C(O)OR9 или т.п.; R9 представляет собой водород или алкил; X представляет собой связь, C1-4 алкилен, -X1OX2- или т.п., где X1 и X2 независимо выбраны из связи и C1-3 алкилена; Y представляет собой конденсированную 5,6- или 6,6-гетеробициклическую кольцевую систему, содержащую, по меньшей мере, одно ароматическое кольцо, где конденсированная бициклическая кольцевая система Y может быть замещена, если это желательно; R1 выбран из C6-10 арила и C2-9 гетероарила, где любой арил или гетероарил замещен C6-10 арил C0-4 алкилом, C2-9 гетероарилом, C0-4 алкилом, C1-6 алкилом или т.п., если это желательно, R2, R3, R5, R6, R7 и R8, независимо, представляют собой водород, C1-6 алкил, галоген или т.п.; R4 представляет собой водород или C1-6 алкил; или R7 и любой из R2, R4 или R5 объединены с атомом, с которым они связаны, с образованием 4-7-членного кольца; где 4-7-членное кольцо является насыщенным или частично ненасыщенным] и его фармацевтически приемлемую соль, гидрат, сольват, изомер и пролекарство (подробнее см. в Патентном документе 1), и в качестве конкретного соединения, например, описанное выше бензотиенильное соединение раскрыто в качестве Примера 1.
Кроме того, Патентный документ 2 раскрывает, что соединение общей формулы (B):
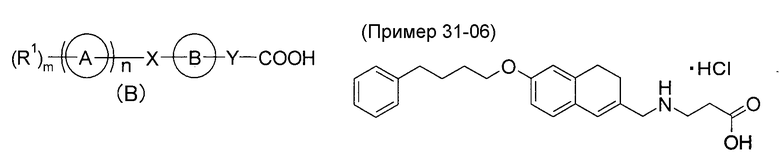
[в общей формуле, Кольцо A представляет собой циклическую группу; Кольцо B представляет собой циклическую группу, которая может содержать заместитель; X представляет собой спейсер, содержащий от одного до восьми атомов в основной цепи, или т.п.; Y представляет собой спейсер, содержащий от одного до десяти атомов в основной цепи, или т.п.; n имеет значение 0 или 1; в случае, когда n имеет значение 0, m имеет значение 1 и более, R1 представляет собой атом водорода или заместитель; в случае, когда n имеет значение 1, m имеет значение 0 или целое число от 1 до 7, и далее, R1 представляет собой заместитель (когда m имеет значение 2 или более, несколько групп R1 может быть одинаковыми или отличными друг от друга)], его соль, его сольват или его пролекарство (подробнее см. в Патентном документе 2) обладают способностью связывания с рецептором S1P, и в качестве такого конкретного соединения, например, раскрыто тетрагидронафталиновое производное в качестве Примера 31-06.
Кроме того, Патентный документ 3 раскрывает, что соединение общей формулы (C):
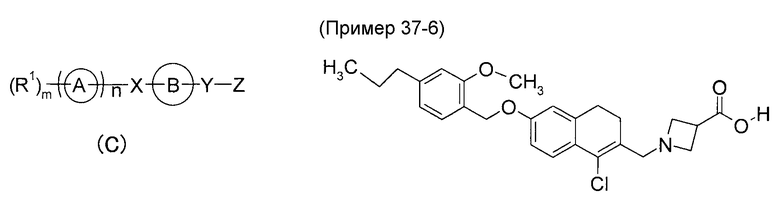
[где Кольцо A представляет собой циклическую группу, Кольцо B представляет собой циклическую группу, которая может дополнительно содержать заместитель, X представляет собой связывающее плечо или спейсер, содержащий от одного до восьми атомов в основной цепи, где один атом спейсера может быть объединен с заместителем Кольца B с образованием кольца, которое может содержать заместитель, Y представляет собой связывающее плечо или спейсер, содержащий от одного до десяти атомов в основной цепи, где один атом спейсера может быть объединен с заместителем Кольца B с образованием кольца, которое может содержать заместитель, Z представляет собой кислотную группу, которая может быть защищена, и n имеет значение 0 или 1, при условии, что в случае, когда n имеет значение 0, m имеет значение 1 и далее, R1 представляет собой атом водорода или заместитель, в случае, когда n имеет значение 1, m имеет значение 0 или целое число от 1 до 7 и далее, R1 представляет собой заместитель (когда m имеет значение 2 или более, несколько групп R1 могут быть одинаковыми или отличными друг от друга)], его соль, его N-оксид, его сольват или его пролекарство обладают способностью связывания с рецептором S1P. В качестве такого конкретного соединения раскрыто, например, тетрагидронафталиновое производное, представленное Примером 37-6.
Однако вплоть до настоящего времени оставалась потребность в новом и высокостабильном S1P1 агонисте, обладающем сильным действием S1P1 агониста сфингозин-1-фосфата и, соответственно, обладающего превосходным действием секвестрации лимфоцитов и, кроме того, не имеющего нежелательных эффектов, таких как редкий пульс, ослабление функции легких и подобные, о которых сообщалось в связи с обычными S1P1 агонистами.
Документы предшествующего уровня техники
Патентные документы
[Патентный документ 1] Брошюра международной публикации WO 2005/000833
[Патентный документ 2] Брошюра международной публикации WO 2005/020882
[Патентный документ 3] Брошюра международной публикации WO 2006/064757
Раскрытие изобретения
Задачи, решаемые настоящим изобретением
Представлено соединение, которое является полезным в качестве активного ингредиента фармацевтической композиции, в частности, фармацевтической композиции для профилактики или лечения заболеваний, вызванных нежелательной инфильтрацией лимфоцитов, или заболеваний, вызванных аномальной пролиферацией или аккумуляцией клеток, на основании агонистического действия в отношении S1P1.
Средства решения задач
Авторы настоящего изобретения провели всесторонние исследования соединения, обладающего действием агониста S1P1, и, как результат, было обнаружено, что соединение 2Н-хромена, представленное ниже формулой (I), или его производное обладает превосходным действием агониста S1P1 и является полезным в качестве активного ингредиента фармацевтической композиции для профилактики или лечения заболеваний, вызванных нежелательной инфильтрацией лимфоцитов, или заболеваний, вызванных аномальной пролиферацией или аккумуляцией клеток, завершив, таким образом, настоящее изобретение.
Таким образом, настоящее изобретение относится к соединению 2Н-хромена, представленному следующей формулой (1):
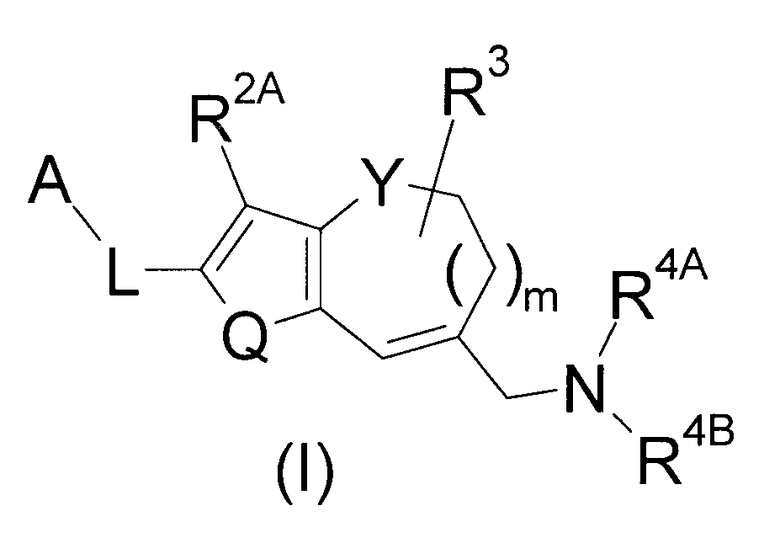
(где
A представляет собой низший алкил, циклоалкил, арил или гетероарил,
где арил и гетероарил могут быть, соответственно, замещены одним-пятью заместителями R1, которые являются одинаковыми или отличными друг от друга,
R1 представляет собой галоген, -CN, -NO2, низший алкил, низший алкенил, низший алкинил, галоген-низший алкил, арил, гетероарил, циклоалкил, -OH, -O-(низший алкил), -O-(галоген-низший алкил), -O-(арил), -O-(циклоалкил), -O-(гетероарил), -NH2, -NH(низший алкил), -NH(галоген-низший алкил), -N(низший алкил)2 или циклический амино,
где арил, гетероарил, циклоалкил и циклический амино могут быть, соответственно, замещены одним-пятью заместителями, которые являются одинаковыми или отличными друг от друга и выбраны из группы, включающей галоген, -CN, низший алкил и галоген-низший алкил,
L представляет собой низший алкилен, низший алкенилен, низший алкинилен, -(низший алкилен)-O-, -O-(низший алкилен)- или -(низший алкилен)-O-(низший алкилен)-,
Q представляет собой S или -C(R2B)=C(R2C)-,
R2A, R2B и R2C являются одинаковыми или отличными друг от друга и представляют собой -H, галоген, низший алкил, галоген-низший алкил, -O-(низший алкил) или -O-(галоген-низший алкил),
Y представляет собой O, S или -CH2-, при условии, что когда Y представляет собой -CH2-, Q представляет собой S,
m имеет значение 0 или 1,
R3 представляет собой -H, галоген, низший алкил или арил,
R4A представляет собой -H или низший алкил,
R4B представляет собой низший алкил, замещенный группой, выбранной из Группы G, или циклоалкил, замещенный группой, выбранной из Группы G,
или R4A и R4B объединены с атомом N, с которым они связаны, с образованием циклического амино, замещенного группой, выбранной из Группы G, где циклический амино может дополнительно содержать от одного до четырех заместителей, которые являются одинаковыми или отличными друг от друга и выбраны из группы, включающей галоген, низший алкил и галоген-низший алкил, и
Группа G представляет собой, -C(=O)OH, тетразолил, -C(=O)NHS(=O)2(низший алкил), -(низший алкилен)-C(=O)OH, или
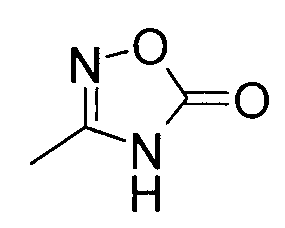 ) или его производному, или его соли.
) или его производному, или его соли.
В этой связи, в случае когда символы в любой из химических формул в настоящем описании используются также и в других химических формулах, одни и те же символы имеют одинаковые значения, если конкретно не описано иным образом.
Кроме того, настоящее изобретение относится к фармацевтической композиции, которая содержит соединение 2Н-хромена формулы (I) или его производное или его соль и фармацевтически приемлемый эксципиент, в частности, к (1) S1P1 агонисту, (2) фармацевтической композиции для профилактики или лечения заболеваний, вызванных нежелательной инфильтрацией лимфоцитов, связанной с S1P1, (3) фармацевтической композиции для профилактики или лечения реакций отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей, аутоиммунных заболеваний или воспалительных заболеваний у человека или животных, (4) фармацевтической композиции для профилактики или лечения реакций отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей у человека или животных, (5) фармацевтической композиции для профилактики или лечения рассеянного склероза, (6) фармацевтической композиции для профилактики или лечения заболеваний, вызванных аномальной пролиферацией или аккумуляцией клеток, связанной с S1P1, и (7) фармацевтической композиции для профилактики или лечения рака или лейкоза.
Кроме того, настоящее изобретение относится к способу профилактики или лечения заболеваний, вызванных нежелательной инфильтрацией лимфоцитов, связанной с S1P1, в частности, реакций отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей, или рассеянного склероза у человека или животных, который включает введение пациенту эффективного количества соединения 2Н-хромена формулы (I) или его производного или его соли. Кроме того, настоящее изобретение включает применение соединения 2Н-хромена формулы (I) или его производного или его соли для профилактики или лечения заболеваний, вызванных нежелательной инфильтрацией лимфоцитов, связанной с S1P1, в частности, реакций отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей, или рассеянного склероза у человека или животных, и соединение 2Н-хромена формулы (I) или его производное или его соль для применения для профилактики или лечения заболеваний, вызванных нежелательной инфильтрацией лимфоцитов, связанной с S1P1, в частности, реакций отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей, или рассеянного склероза у человека или животных.
Эффекты настоящего изобретения
Соединение формулы (I) или его соль по настоящему изобретению обладает действием агониста S1P1 и может быть использовано для профилактики или лечения заболеваний, вызванных нежелательной инфильтрацией лимфоцитов, например, аутоиммунных заболеваний или воспалительных заболеваний, таких как реакции отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей, ревматоидный артрит, рассеянный склероз, системная красная волчанка, нефротический синдром, менингоэнцефалит, тяжелая миастения, панкреатит, гепатит, нефрит, диабет, легочные заболевания, бронхиальная астма, атопический дерматит, воспалительные заболевания кишечника, атеросклероз, ишемическое реперфузионное расстройство, и заболеваний, вызванных аномальной пролиферацией или аккумуляцией клеток, например, рака, лейкоза и подобных.
Лучший вариант осуществления настоящего изобретения
Далее представлено подробное объяснение настоящего изобретения.
В описании “галоген” означает F, Cl, Br или I. Предпочтительно, его примеры включают F и Cl.
В настоящем описании “низший алкил” представляет собой линейный или разветвленный алкил, содержащий от одного до шести атомов углерода (далее называемый просто как C1-6), и его примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и подобные, в другом варианте воплощения, C1-4 алкил, и в следующем варианте воплощения, метил, этил и изопропил.
“Низший алкенил” представляет собой линейный или разветвленный C2-6 алкенил, и его примеры включают винил, пропенил, бутенил, пентенил, 1-метилвинил, 1-метил-2-пропенил, 1,3-бутадиенил, 1,3-пентадиенил и подобные, и в другом варианте воплощения, C2-4 алкенил.
“Низший алкилен” представляет собой линейный или разветвленный C1-6 алкилен, и его примеры включают метилен, этилен, триметилен, тетраметилен, пентаметилен, гексаметилен, пропилен, метилметилен, этилэтилен, 1,2-диметилэтилен, 1,1,2,2-тетраметилэтилен и подобные, в другом варианте воплощения, C1-4 алкилен, и в следующем варианте воплощения, метилен и этилен.
“Низший алкенилен” представляет собой линейный или разветвленный C2-6 алкенилен, и его примеры включают винилен, этилиден, пропенилен, бутенилен, пентенилен, гексенилен, 1,3-бутадиенилен, 1,3-пентадиенилен и подобные, в другом варианте воплощения, C2-4 алкенилен, и в следующем варианте воплощения, винилен и этилиден.
“Низший алкинилен” представляет собой линейный или разветвленный C2-6 алкинилен, и его примеры включают этинилен, пропинилен, бутинилен, пентинилен, гексинилен, 1,3-бутадиинилен, 1,3-пентадиинилен и подобные, в другом варианте воплощения, C2-4 алкинилен, и в следующем варианте воплощения, этинилен, пропинилен, бутинилен и пентинилен.
“Галоген-низший алкил” представляет собой C1-6 алкил, замещенный одним или несколькими атомами галогена, в другом варианте воплощения, низший алкил, замещенный одним-пятью атомами галогена, в следующем варианте воплощения, C1-3 низший алкил, замещенный одним-пятью атомами галогена, и еще в одном варианте воплощения его примеры включают -CF3, -CH2CF3, -CH(CH3)CF3 и -CH(CH2F)2.
“Циклоалкил” представляет собой C3-10 насыщенную углеводородную кольцевую группу, которая может содержать мостик. Его примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, адамантил и подобные, в другом варианте воплощения, C3-8 циклоалкил, в следующем варианте воплощения, C3-6 циклоалкил, и еще в одном варианте воплощения, циклопропил, циклопентил и циклогексил.
“Арил” представляет собой C6-14 моноциклическую-трициклическую ароматическую углеводородную кольцевую группу, и его примеры включают фенил и нафтил, и в другом варианте воплощения, фенил.
“Гетероарил” представляет собой 5-6-членный моноциклический гетероарил, содержащий от одного до четырех гетероатомов, выбранных из N, S и O, и бициклический гетероарил, образованный путем его конденсации с бензольным кольцом или 5-6-членным моноциклическим гетероарилом, и может быть частично насыщенным. В другом варианте воплощения, его примеры включают пиридил, пирролил, пиразинил, пиримидинил, пиридазинил, имидазолил, триазолил, триазинил, тетразолил, тиазолил, пиразолил, изотиазолил, оксазолил, изоксазолил, тиадиазолил, оксадиазолил, тиенил, фурил, бензотиазолил и индолил, в другом варианте воплощения, гетероарил, состоящий из 5-членного кольца, которое может быть конденсировано с бензольным кольцом, и еще в одном варианте воплощения, пирролил, имидазолил, тиазолил, тиенил, бензотиазолил и индолил.
“Азот-содержащий моноциклический гетероарил” означает моноциклический гетероарил, в котором один из составляющих кольцо атомов обязательно представляет собой N, и может содержать один или два гетероатома, выбранных из N, S и O, в качестве составляющих кольцо атомов, и в другом варианте воплощения, его примеры включают 5-6-членное кольцо, в следующем варианте воплощения, пиридил, пирролил, пиразинил, пиримидинил, пиридазинил, имидазолил, триазолил, тетразолил, тиазолил, пиразолил, изотиазолил, оксазолил, изоксазолил, тиадиазолил, оксадиазолил и подобные, еще в одном варианте воплощения, 5-членное кольцо, и еще в одном варианте воплощения, пирролил и имидазолил.
“Циклический амино” означает моноциклический-трициклический гетероциклоалкил, в котором один из составляющих кольцо атомов обязательно представляет собой N, может содержать один или два гетероатома, выбранных из N, S и O, в качестве составляющих кольцо атомов, и может содержать частично ненасыщенную связь. В другом варианте воплощения, он представляет собой кольцо, со степенью восстановления от 4 до 9, в следующем варианте воплощения, его примеры включают азетидинил, пирролидинил, пиперидинил, морфолинил, гомопиперидинил, 3-азабицикло[3,1,0]гексанил, тетрагидропиридил, октагидроциклопента[c]пирролил, хинуклидинил и подобные, еще в одном варианте воплощения, его примеры включают циклический амино, состоящий из 6-членного кольца, еще в одном варианте воплощения, его примеры включают пиперидинил, пиперазинил, морфолинил и тетрагидропиридил, и еще в одном варианте воплощения, его примеры включают азетидинил, пирролидинил, пиперидинил и тетрагидропиридил.
В настоящем описании, выражение "который может быть замещен одним-пятью заместителями R1, которые являются одинаковыми или отличными друг от друга” означает незамещенный или содержащий от одной до пяти групп R1 в качестве заместителей. Кроме того, в случае, когда присутствуют несколько заместителей R1, заместители R1 могут быть как одинаковыми, так и отличными друг от друга.
Варианты воплощения настоящего изобретения будут описаны ниже.
(1) Соединение 2Н-хромена или его соль, где Y представляет собой O, Q представляет собой -C(R2B)=C(R2C)-, и m имеет значение 0.
(2) Соединение 2Н-хромена или его соль, где R4A и R4B объединены с атомом N, с которым они связаны, с образованием циклического амино, выбранного из азетидинила, пирролидинила, пиперидинила и тетрагидропиридила, который замещен группой(группами), выбранными из Группы G, и может быть замещен низшим алкилом или галогеном.
(3) Соединение 2Н-хромена или его соль, где группа, представленная Группой G, представляет собой -C(=O)OH или -C(=O) NHS(=O)2CH3.
(4) Соединение 2Н-хромена или его соль, где A представляет собой фенил, пиридил или тиенил, который может быть замещен одним-тремя заместителями R1, которые могут быть как одинаковыми, так и отличными друг от друга.
(5) Соединение 2Н-хромена или его соль, где L представляет собой -(низший алкилен)-O-, низший алкенилен или низший алкинилен.
(6) Соединение 2Н-хромена или его соль, где R2A представляет собой -H или низший алкил, R2B представляет собой -H, R2C представляет собой -H или галоген, R3 представляет собой -H или галоген, R1 представляет собой галоген, низший алкил, галоген-низший алкил, фенил, пирролил, циклоалкил, -O-(низший алкил) или -O-(галоген-низший алкил) и далее, L представляет собой -CH2-O-, -CH=CH- или 3-бутинилен.
(7) Соединение 2Н-хромена или его соль, где R4A и R4B объединены с атомом N, с которым они связаны, с образованием пиперидинила или тетрагидропиридила, который замещен группой -C(=O)OH, L представляет собой -CH2-O-, R2A представляет собой -H, R2B представляет собой -H, R2C представляет собой -H или галоген, R3 представляет собой -H, и A представляет собой фенил или пиридил, который замещен двумя заместителями R1, которые являются одинаковыми или отличными друг от друга, где R1 представляет собой галоген, галоген-низший алкил, -O-(низший алкил) или -O-(галоген-низший алкил).
(8) Соединение 2Н-хромена или его соль, где R4A и R4B объединены с атомом N, с которым они связаны, с образованием пиперидинила, который замещен группой -C(=O)OH, и A представляет собой фенил, который замещен двумя заместителями R1, которые являются одинаковыми или отличными друг от друга.
(9) Соединение 2Н-хромена или его соль, где R4A и R4B объединены с атомом N, с которым они связаны, с образованием тетрагидропиридила, который замещен группой -C(=O)OH, A представляет собой пиридил, который замещен двумя заместителями R1, которые являются одинаковыми или отличными друг от друга.
Примеры конкретных соединений, включенных в настоящее изобретение, включают следующие соединения или их соли:
1-{[7-({3-хлор-4-[(1S)-2,2,2-трифтор-1-метилэтокси]бензил}окси)-2H-хромен-3-ил]метил}-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
1-({7-[(3-хлор-4-изопропилбензил)окси]-2H-хромен-3-ил}метил)-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
1-[(7-{[4-изопропокси-3-(трифторметил)бензил]окси}-2H-хромен-3-ил)метил]-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
1-{[7-({3-хлор-4-[2-фтор-1-(фторметил)этокси]бензил}окси)-2H-хромен-3-ил]метил}-1,2,3,6-тетрагидропиридин-4-карбоновая кислота,
1-{[7-({5-хлор-6-[(1S)-2,2,2-трифтор-1-метилэтокси]пиридин-3-ил}метокси)-2H-хромен-3-ил]метил}-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
(3R)-1-{[7-({4-[(1,3-дифторпропан-2-ил)окси]-3-(трифторметил)бензил}окси)-5-фтор-2H-хромен-3-ил]метил}пиперидин-3-карбоновая кислота,
1-[(7-{[4-циклопентил-3-(трифторметил)бензил]окси}-2H-хромен-3-ил)метил]-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
(3R)-1-{[7-({3-хлор-4-[(1,3-дифторпропан-2-ил)окси]бензил}окси)-5-фтор-2H-хромен-3-ил]метил}пиперидин-3-карбоновая кислота,
(3S)-1-{[7-({4-[(1,3-дифторпропан-2-ил)окси]-3-(трифторметил)бензил}окси)-5-фтор-2H-хромен-3-ил]метил}пиперидин-3-карбоновая кислота,
(3R)-1-[(7-{[4-(2,2,2-трифторэтокси)-3-(трифторметил)бензил]окси}-2H-хромен-3-ил)метил]пиперидин-3-карбоновая кислота,
(3R)-1-[(7-{[3-(трифторметил)-4-{[(2S)-1,1,1-трифторпропан-2-ил]окси}бензил]окси}-2H-хромен-3-ил)метил]пиперидин-3-карбоновая кислота,
(3S)-1-[(7-{[4-(2,2,2-трифторэтокси)-3-(трифторметил)бензил]окси}-5-фтор-2H-хромен-3-ил)метил]пиперидин-3-карбоновая кислота,
(3R)-1-{[7-({4-[(1,3-дифторпропан-2-ил)окси]-3-(трифторметил)бензил}окси)-5-фтор-2H-хромен-3-ил]метил}-N-(метилсульфонил)пиперидин-3-карбоксамид или
1-[(7-{[4-(2,2,2-трифторэтокси)-3-(трифторметил)бензил]окси}-2H-хромен-3-ил)метил]пиперидин-4-карбоновая кислота.
Соединение формулы (I) может существовать в виде таутомеров или геометрических изомеров, в зависимости от типа заместителей. В настоящем описании соединение формулы (I) будет описано только в одной форме изомера, хотя настоящее изобретение включает другие изомеры, выделенные формы изомеров или их смеси.
Кроме того, соединение формулы (I) может содержать ассиметричные атомы углерода или осевую хиральность, в некоторых случаях, и, соответственно, оно может существовать в форме оптических изомеров. Настоящее изобретение включает как отдельные формы оптических изомеров соединения формулы (I), так и их смеси.
Кроме того, настоящее изобретение также включает фармацевтически приемлемое пролекарство соединения, представленного формулой (I). Фармацевтически приемлемое пролекарство представляет собой соединение, содержащее группу, которая может быть преобразована в аминогруппу, гидроксильную группу, карбоксильную группу или т.п. в результате сольволиза или в физиологических условиях. Примеры группы, образующей пролекарство, включают группы, описанные в Prog. Med., 5, 2157-2161 (1985) and Pharmaceutical Research and Development, Drug Design, Hirokawa Publishing Company (1990), Vol. 7, 163-198.
Кроме того, соль соединения формулы (I) представляет собой фармацевтически приемлемую соль соединения формулы (I) и может образовывать кислотно-аддитивную соль или соль с основанием, в зависимости от вида заместителей. Конкретные примеры включают кислотно-аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные, и с органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, миндальная кислота, винная кислота, дибензоилвинная кислота, дитолилвинная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, аспарагиновая кислота, глутаминовая кислота и подобные, и соли с неорганическими основаниями, такими как натрий, калий, магний, кальций, алюминий и подобные, или с органическими основаниями, такими как метиламин, этиламин, этаноламин, лизин, орнитин и подобные, соли с различными аминокислотами или аминокислотными производными, такими как ацетиллейцин и подобные, аммониевые соли и т.д.
Кроме того, настоящее изобретение также включает различные гидраты или сольваты и полиморфные кристаллические вещества соединения формулы (I) и его соли. Кроме того, настоящее изобретение также включает соединения, меченные различными радиоактивными или нерадиоактивными изотопами.
В настоящем описании в некоторых случаях могут быть использованы следующие сокращения.
ADDP=1,1'-(азодикарбонил)дипиперидин, AIBN=2,2'-азобисизобутиронитрил, AcOH=уксусная кислота, CDI=1,1'-карбонилбис-1H-имидазол, DAST=(диэтиламино)сера трифторид, DBU=1,8-диазабицикло[5,4,0]ундец-7-ен, DCC=дициклогексилкарбодиимид, DCE=дихлорэтан, ДХМ=дихлорметан, DIBAL=диизобутилалюмогидрид, DIBOC=ди-трет-бутилдикарбонат, DIC=N,N'-диизопропилкарбодиимид, DIPEA=диизопропилэтиламин, DMA=N,N'-диметилацетамид, DMAP=4-(N,N'-диметиламино)пиридин, DME=диметоксиэтан, DMF=N,N'-диметилформамид (ДМФА), ДМСО=диметилсульфоксид (ДМСО), DPPA=дифенилфосфорилазид, DPPP=1,3-бис(дифенилфосфино)пропан, EDCI·HCl=N-[3-(диметиламино)пропил]-N'-этилкарбоксамидгидрохлорид, Et=этил, Et2O=диэтиловый эфир, TEA=триэтиламин, EtOAc=этилацетат, EtOH=этанол, HOBt=1-гидрокси-1H-бензотриазол, IPE=диизопропиловый эфир, t-BuOK=трет-бутоксид калия, LAH= литийалюмогидрид, MS4 Angstrom=молекулярные сита 4 Ангстрема, MeCN=ацетонитрил, MeOH=метанол, MgSO4=безводный сульфат магния, NBS=N-бромсукцинимид, NCS=N-хлорсукцинимид, NMP=N-метилпирролидон, NT = не испытывали, Na2SO4= безводный сульфат натрия, NaBH(OAc)3=триацетоксиборгидрид натрия, NaBH4= боргидрид натрия, NaOEt=этоксид натрия, NaOH=гидроксид натрия, NaOMe=метоксид натрия, TBP=три-нормальный бутилфосфин, PDC=пиридинийдихромат, POCl3=оксихлорид фосфора, PPh3=трифенилфосфин, Pd(OAc)2=ацетат палладия (II), Pd(PPh3)4=тетракис(трифенилфосфин)палладий (0), TEA=триэтиламин, TFA=трифторуксусная кислота, ТГФ=тетрагидрофуран, TMEDA=N,N,N'N'-тетраметилэтилендиамин, Tf=CF3S(=O)2-, насыщенный солевой раствор=насыщенный солевой раствор, i-PrOH=2-пропанол, н-BuLi=нормальный бутиллитий, н-BuOH=нормальный бутиловый спирт, t-BuOH=третичный бутиловый спирт и tert= третичный.
(Способы получения)
Соединение формулы (I) и его соль можно получить с использованием характеристик, основанных на основной структуре или типе заместителей, и путем применения различных известных способов синтеза. В процессе получения, замена соответствующей функциональной группы подходящей защитной группой (группой, которую можно легко преобразовать в функциональную группу) на стадии от исходного вещества до промежуточного соединения в некоторых случаях может быть эффективным, в зависимости от типа функциональной группы в используемом способе получения. Защитная группа для такой функциональной группы может включать, например, защитные группы, описанные в “Greene's Protective Groups in Organic Synthesis (4th Ed., 2006)” авторами P. G. M. Wuts и T. W. Greene, и можно выбрать одну из таких групп и использовать, как это необходимо, в зависимости от реакционных условий. В таком способе, желаемое соединение можно получить путем введения защитной группы, путем осуществления реакции и путем удаления защитной группы, как это необходимо.
Кроме того, пролекарство соединения формулы (I) можно получить путем введения специфической группы или путем осуществления реакции с использованием полученного соединения формулы (I) на стадии от исходного вещества до промежуточного соединения, как в случае с вышеуказанной защитной группой. Реакцию можно осуществить с использованием способов, известных специалистам в данной области, таких как обычная этерификация, амидирование, дегидратация и подобные.
Ниже описаны репрезентативные способы получения соединения формулы (I). Каждый из способов получения также можно осуществить со ссылкой на Ссылочные документы, указанные в настоящем описании. Кроме того, способы получения по настоящему изобретению не ограничены примерами, представленными ниже.
<Способ получения 1>
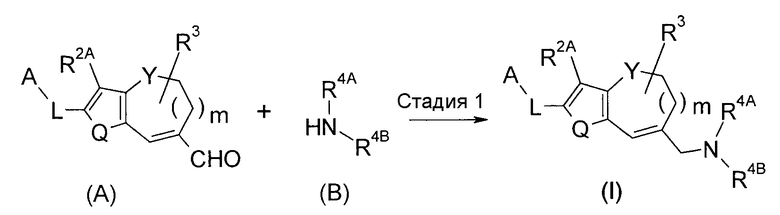
Соединение (I) по настоящему изобретению можно получить, подвергая соединение (A) и соединение (B) восстановительному аминированию.
Способ на стадии 1 представляет собой восстановительное аминирование. Соединение (A) и соединение (B) используют в эквивалентных количествах, или любое из них используют в избыточном количестве, и смесь перемешивают в любых условиях, включающих температуру от -45°C до температуры кипения с обратным холодильником, в частности, от 0°C до комнатной температуры, обычно в течение времени от 0,1 часа до 5 дней, в растворителе, который является инертным для реакции, в присутствии восстановителя. Примеры растворителя включают спирты, такие как MeOH, EtOH и подобные; простые эфиры, такие как Et2O, ТГФ, диоксан, DME и подобные; галогенированные углеводороды, такие как ДХМ, DCE, хлороформ и подобные; и состоящий из них смешанный растворитель. Примеры восстановителя включают NaBH3CN, NaBH(OAc)3, NaBH4 и подобные. Может быть предпочтительным в некоторых случаях осуществлять реакцию в присутствии агента дегидратации, такого как молекулярные сита и подобные, или кислоты, такой как уксусная кислота, хлористоводородная кислота, комплекс изопропоксида титана (IV) и подобные. Имин, который является промежуточным соединением реакции, можно выделить в виде стабильного промежуточного соединения и путем восстановления иминового промежуточного соединения можно получить соединение (I). Кроме того, реакцию можно осуществить в растворителе, таком как MeOH, EtOH, EtOAc и подобные, в присутствии или в отсутствие кислоты, такой как уксусная кислота, хлористоводородная кислота и подобные, с использованием катализатора восстановления (например, палладия на углероде, никеля Ренея и подобных), вместо восстановителя. В этом случае, реакцию осуществляют в атмосфере водорода при давлении от нормального до 50 атмосфер, в температурных условиях от охлаждения до нагревания.
[Ссылочные документы] (1) “Comprehensive Organic Functional Group Transformations II” written by A. R. Katritzky and R. J. K. Taylor, Vol. 2, Elsevier Pergamon, 2005, (2) “Jikken Kagaku Koza (Courses in Experimental Chemistry) (5th Edition)” edited by The Chemical Society of Japan, Vol. 14 (2005) (Maruzen)
<Способ получения 2>
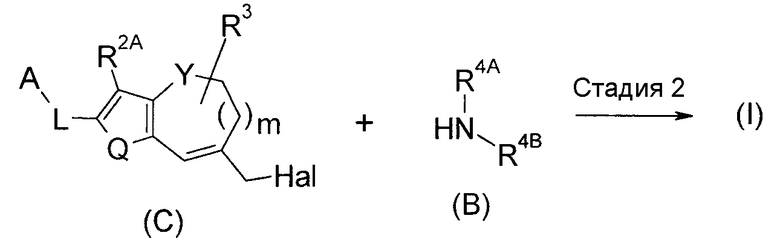
(где Hal представляет собой галоген).
Соединение (I) по настоящему изобретению можно получить путем алкилирования соединения (C) соединением (B).
Способ на стадии 2 представляет собой алкилирование. Соединение (B) и соединение (C) используют в эквивалентных количествах, или любое из них используют в избыточном количестве, и смесь перемешивают в температурных условиях от охлаждения до нагревания и температуры кипения с обратным холодильником, предпочтительно от 0°C до 80°C, обычно в течение времени от 0,1 часа до 5 дней, в растворителе, который является инертным для реакции, или без растворителя. Примеры растворителя включают ароматические углеводороды; простые эфиры; галогенированные углеводороды; ДМФА, ДМСО, EtOAc и MeCN; и состоящий из них смешанный растворитель. Может быть выгодным в некоторых случаях для ровного протекания реакции осуществлять реакцию в присутствии органического основания, такого как TEA, DIPEA или N-метилморфолин и подобные, или неорганического основания, такого как K2CO3, Na2CO3 или KOH и подобные. Может быть выгодным в некоторых случаях для ровного протекания реакции добавить неорганическую соль, такую как NaI и подобные, в систему реакции.
[Ссылочный документ] “Jikken Kagaku Koza (Courses in Experimental Chemistry) (5th Edition)” edited by The Chemical Society of Japan, Vol. 14 (2005) (Maruzen)
<Промежуточный процесс получения 1>
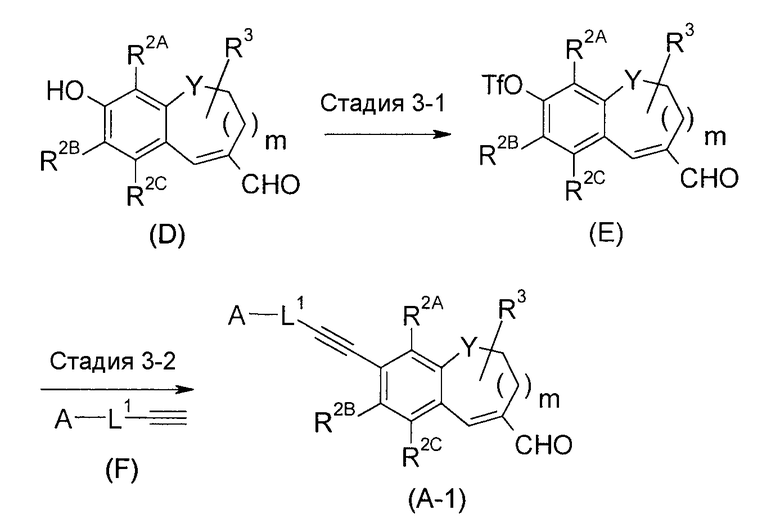
(где Tf представляет собой CF3S(=O)2-, и L1 представляет собой низший алкилен или низший алкенилен).
Соединение (A-1) можно получить с использованием реакции Соногашира, исходя из соединения (D).
Стадия 3-1 представляет собой стадию получения трифлата. Соединение (E) можно получить, подвергая соединение (D) взаимодействию с трифторметансульфоновым ангидридом. В качестве растворителя, который обычно не мешает реакции, используют растворитель, выбранный из галогенированных углеводородов, реакцию осуществляют в присутствии органических оснований, такие как пиридин, TEA, DIPEA и подобные, в температурных условиях от -10°C до охлаждения льдом. Кроме того, органическое основание можно использовать в комбинации с растворителем.
Стадия 3-2 представляет собой так называемую реакцию Соногашира. Соединение (A-1) можно получить путем добавления каталитического количества Pd(0) катализатора и основания к соединению (E), давая возможность прореагировать концевому ацетилену. Может быть выгодным в некоторых случаях для ровного протекания реакции добавить йодид меди в систему реакции. Примеры растворителя включают простые эфиры; ароматические углеводороды, такие как толуол, ксилол и подобные; ДМФА, ДМСО, EtOAc; и состоящий из них смешанный растворитель. Например, основание, такое как TEA, пирролидин и подобные, можно использовать в комбинации с растворителем. Что касается температуры реакции, реакцию можно осуществить в температурных условиях от комнатной температуры до температуры кипения с обратным холодильником.
[Ссылочный документ] K. Sonogashira, Тетраhedron Letters, 1975, 50, pp. 4467.
<Промежуточный процесс получения 2>
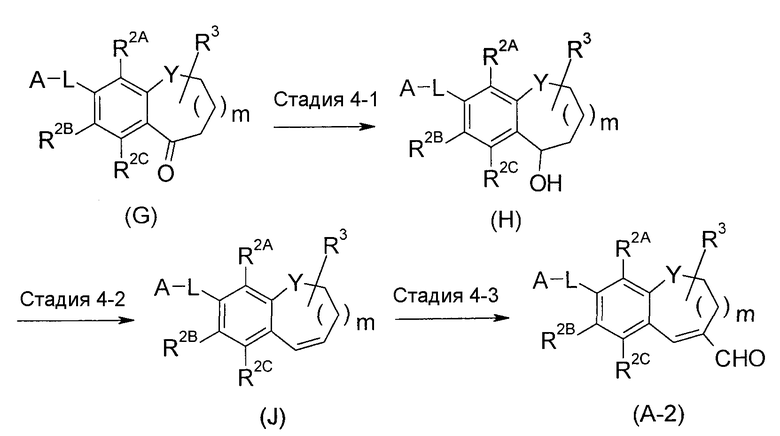
соединение (A-2) можно получить путем восстановления соединения (G) и его дегидратации и формилирования полученного соединения (J).
Стадия 4-1 представляет собой реакцию восстановления кетона. Соединение (G) обрабатывают эквивалентным количеством или избыточным количеством восстановителя в температурных условиях от охлаждения до нагревания, предпочтительно от -20°C до 80°C, обычно в течение времени от 0,1 часа до 3 дней, в растворителе, который является инертным для реакции. Примеры растворителя включают простые эфиры; спирты; ароматические углеводороды; ДМФА, ДМСО, EtOAc и состоящий из них смешанный растворитель. В качестве восстановителя, подходящими для использования являются гидридные восстановители, такие как NaBH4, DIBAL и подобные, металлические восстановители, такие как натрий, цинк, железо и подобные, а также восстановители, указанные в следующих Ссылочных документах.
[Ссылочные документы] (1) “Reductions in Organic Chemistry, 2nd ed. (ACS Monograph: 188)” written by M. Hudlicky, ACS, 1996, (2) “Comprehensive Organic Transformations” written by R. C. Larock, 2nd ed., VCH Publishers, Inc., 1999, (3) “Oxidation and Reduction in Organic Synthesis (Oxford Chemistry Primers 6)” written by T. J. Donohoe, Oxford Science Publications, 2000, (4) “Jikken Kagaku Koza (Courses in Experimental Chemistry) (5th Edition)” edited by The Chemical Society of Japan, Vol. 14 (2005) (Maruzen)
Стадия 4-2 представляет собой реакцию дегидратации. Обычно исходное вещество перемешивают в концентрированной серной кислоте в условиях нагревания и затем продолжают дистилляцию вплоть до полного исчезновения элюента.
Стадия 4-3 представляет собой формилирование. Соединение (A-2) получают путем взаимодействия соединения (J) с формамидным производным. В данном случае, формамидное производное означает формамидное соединение, в котором низшие алкилы или арилы, которые являются одинаковыми или отличными друг от друга, связываются с атомами азота формамида. Что касается комплекса Вилсмейера, полученного путем взаимодействия формамидного производного с POCl3, ароматическое кольцо подвергают нуклеофильному замещению с получением аммониевой соли. Ее можно гидролизовать в щелочных условиях с получением формильного продукта. В этой реакции, соединение (J) и ДМФА эквивалент используют в эквивалентных количествах, или любое из них используют в избыточном количестве, и смесь перемешивают в растворителе, который является инертным для реакции, или без растворителя, в присутствии агента галогенирования. Эту реакцию осуществляют в температурных условиях от комнатной температуры до нагревания и температуры кипения с обратным холодильником, обычно в течение времени от 0,1 часа до 5 дней. Примеры растворителя включают галогенированные углеводороды; простые эфиры; или MeCN. Агент галогенирования используют таким образом, чтобы преобразовать ДМФА производное в комплекс Вилсмейера, и обычно оно конкретно не ограничено, при условии, что это реагент, используемый для галогенирования спиртов, но подходящими для использования являются пентахлорид фосфора, POCl3 или т.п.
[Ссылочный документ] (1) “Strategic Applications of Named Reactions in Organic Synthesis” written by L. Kurti and B. Czako, Elsevier Inc, 2005, pp. 468-469
<Промежуточный процесс получения 3>
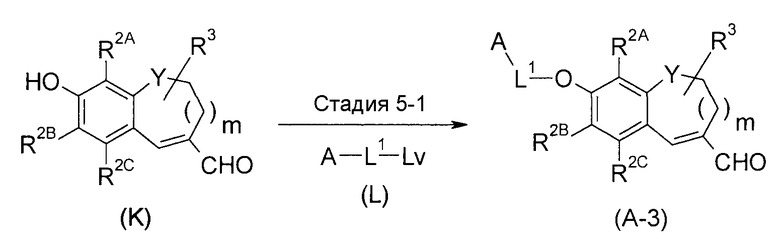
(где Lv представляет собой удаляемую группу).
Соединение (A-3) получают путем взаимодействия соединения (K) с соединением (L).
Стадия 5-1 представляет собой алкилирование. Примеры удаляемой группы Lv включают галоген, метансульфонилокси, п-толуолсульфонилокси группы и подобные.
Соединение (K) и соединение (L) используют в эквивалентных количествах, или любое из них используют в избыточном количестве, и смесь перемешивают в растворителе, который является инертным для реакции, или без растворителя, в температурных условиях от охлаждения до нагревания и температуры кипения с обратным холодильником, предпочтительно от 0°C до 80°C, обычно в течение времени от 0,1 часа до 5 дней. Примеры растворителя включают ароматические углеводороды; простые эфиры; галогенированные углеводороды; ДМФА, ДМСО, EtOAc, MeCN; и состоящий из них смешанный растворитель. Может быть выгодным в некоторых случаях для ровного протекания реакции осуществлять реакцию в присутствии органических оснований, таких как TEA, DIPEA, N-метилморфолин и подобные, или неорганических оснований, таких как K2CO3, Na2CO3, KOH и подобные. Может быть выгодным в некоторых случаях для ровного протекания реакции добавить неорганические соли, такие как NaI и подобные, в систему реакции.
[Ссылочный документ] “Jikken Kagaku Koza (Courses in Experimental Chemistry) (5th Edition)” edited by The Chemical Society of Japan, Vol. 14 (2005) (Maruzen)
<Промежуточный процесс получения 4>
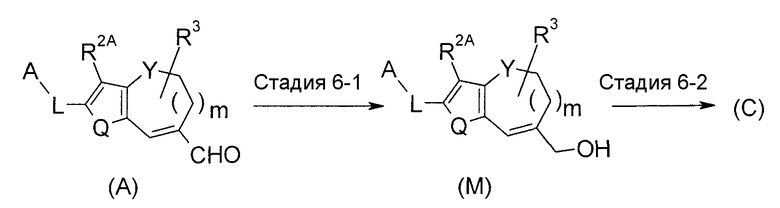
Соединение (C) можно получить из соединения (A) через соединение (M).
Стадия 6-1 представляет собой восстановление. Соединение (M) можно получить путем перемешивания соединения (A) с эквивалентным количеством или избыточным количеством восстановителя в растворителе, который является инертным для реакции, в температурных условиях от охлаждения до нагревания, предпочтительно от -20°C до 80°C, обычно в течение времени от 0,1 часа до 3 дней. Примеры используемого растворителя конкретно не ограничены, но включают простые эфиры, такие как диэтиловый эфир, ТГФ, диоксан и диметоксиэтан, спирты, такие как MeOH, EtOH, 2-пропанол и подобные, ароматические углеводороды, такие как бензол, толуол, ксилол и подобные, ДМФА, ДМСО, EtOAc и состоящий из них смешанный растворитель. В качестве восстановителя, подходящими для использования являются гидридные восстановители, такие как NaBH4, DIBAL и подобные, металлические восстановители, такие как натрий, цинк, железо и подобные, и восстановители, указанные в следующих Ссылочных документах.
[Ссылочные документы]
“Reductions in Organic Chemistry, 2nd ed. (ACS Monograph: 188)” written by M. Hudlicky, ACS, 1996
“Comprehensive Organic Transformations” written by R. C. Larock, 2nd ed., VCH Publishers, Inc., 1999
“Oxidation and Reduction in Organic Synthesis (Oxford Chemistry Primers 6)” written by T. J. Donohoe, Oxford Science Publications, 2000
“Jikken Kagaku Koza (Courses in Experimental Chemistry) (5th Edition)” edited by The Chemical Society of Japan, Vol. 14 (2005) (Maruzen)
Стадия 6-2 представляет собой галогенирование. Соединение (C) можно получить, подвергая соединение (M) галогенированию. В качестве агента галогенирования используют агент галогенирования для преобразования гидроксильной группы в галоген. Агент галогенирования конкретно не ограничен, но, например, используют PBr3, HBr, BBr3, PCl3, PCl5 или т.п. В качестве растворителя простые эфиры являются предпочтительными, и например, используют ТГФ, диэтиловый эфир, диметоксиэтан, метил-трет-бутиловый эфир, диоксан, 2-метилтетрагидрофуран или т.п.
<Промежуточный процесс получения 5>
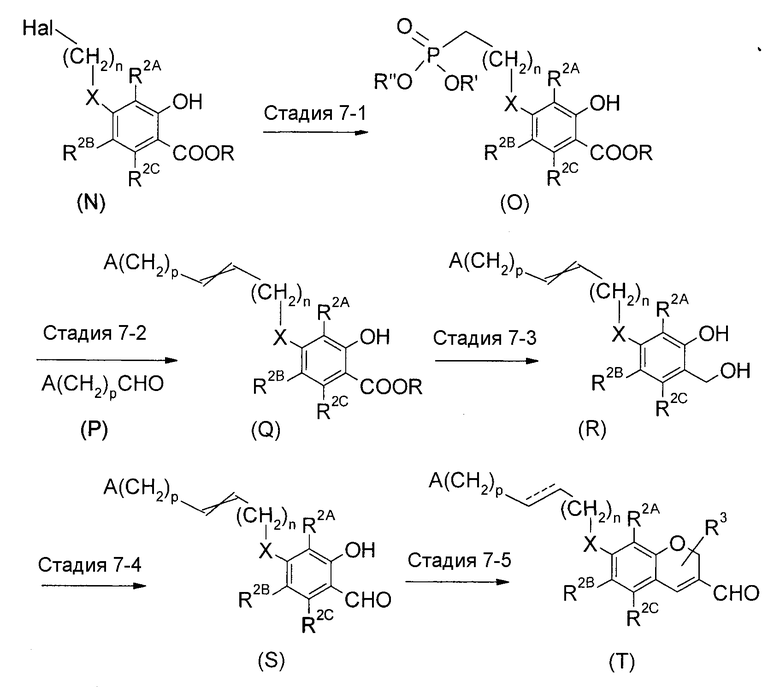
(где X представляет собой -O- или связь, R представляет собой защитную группу для карбоксильной группы, R' и R” представляют собой низший алкил, n и p каждый представляют собой целое число от 0 до 4, которые являются одинаковыми или отличными друг от друга, и далее, сумма n и p равна 4 или меньше. ---- представляет собой простую связь или двойную связь).
Соединение (T) можно получить путем последовательного осуществления реакции Виттига, восстановления, окисления и построения хроменового скелета из соединения (N). Соединение (T), в котором ---- представляет собой простую связь, получают путем осуществления реакции восстановления на стадии, которая не мешает реакции.
Стадия 7-1 представляет собой реакцию образования илида форсфора. Соединение (O) получают путем взаимодействия соединения (N), например, с триэтилфосфитом или т.п., обычно в растворителе, который не мешает реакции. Примеры растворителя включают ароматические углеводороды; простые эфиры; галогенированные углеводороды; кетоны, такие как ацетон, этилметилкетон и подобные; ДМФА, ДМСО, EtOAc, MeCN; и состоящий из них смешанный растворитель. Что касается температуры реакции, реакцию можно осуществить в температурных условиях от -20°C до нагревания.
Стадия 7-2 представляет собой так называемую реакцию Виттига. Соединение (O) можно подвергнуть взаимодействию с альдегидным соединением (P) с получением соединения (Q). Путем альдегидного присоединения замещающих фосфорильные группы карбанионов, можно получить олефины через Виттиг-подобный механизм. Температура реакции представляет собой любую, выбранную из диапазона от 0°C до нагревания.
[Ссылочные документы] (1) J. Boutagy CRV, 79, 87, 1974, (2) W. S. Wadsworth Jr OR, 25, 73, 1977.
Стадия 7-3 представляет собой реакцию восстановления. В качестве восстановителя используют LiAlH4, LiAlH(OMe)3 или DIBAL, и реакцию можно осуществить в растворителе, который является инертным для реакции, таком как ТГФ, простые эфиры и подобные, обычно в температурных условиях от охлаждения до нагревания.
Стадия 7-4 представляет собой реакцию окисления. В качестве окислителя используют диоксид марганца или PDC. Примеры растворителя обычно включают галогенированные углеводороды и подобные. Что касается температуры реакции, реакцию осуществляют в температурных условиях от 0°C до нагревания, обычно при комнатной температуре. Что касается других способов, существует способ с использованием реагента на основе ДМСО-POCl3. Способ с использованием реагента, такого как DCC, ангидриды кислот, хлор или реагенты на основе Me2S-NCS (Corey-Kim окисления) или с использованием перйодинана Dess-Martin, вместо POCl3, также можно использовать. Реакцию обычно осуществляют в температурных условиях от комнатной температуры до нагревания. Примеры растворителя конкретно не ограничены, но включают ароматические углеводороды; простые эфиры; галогенированные углеводороды; MeCN и состоящий из них смешанный растворитель.
Стадия 7-5 представляет собой реакцию образования хроменового кольца. Соединение (T) можно получить путем добавления акролеинового производного к соединению (S) с последующим перемешиванием в температурных условиях от комнатной температуры до нагревания в присутствии неорганического основания, такого как K2CO3 и подобные. Примеры растворителя включают ароматические углеводороды; простые эфиры; галогенированные углеводороды; MeCN и состоящий из них смешанный растворитель. Обычно используют растворители на основе простого эфира, такие как ТГФ, DME, диоксан и подобные.
Соединение, в котором --- в соединении (T) представляет собой простую связь, получают путем восстановления некоторых соединений из соединений (P) через соединение (S). Это так называемая реакция восстановления олефинов. Обычно соединение перемешивают в растворителе, который является инертным для реакции, в присутствии металлического катализатора, обычно в течение от 1 часа до 5 дней, в атмосфере водорода. Эту реакцию обычно осуществляют в температурных условиях от охлаждения до нагревания, предпочтительно при комнатной температуре. Примеры растворителя конкретно не ограничены, но включают спирты, такие как MeOH, EtOH, i-PrOH и подобные; простые эфиры; воду, EtOAc, ДМФА, ДМСО; и состоящий из них смешанный растворитель. В качестве металлического катализатора, подходящими для использования являются палладиевые катализаторы, такие как палладий на углероде, палладиевая чернь, гидроксид палладия и подобные, платиновые катализаторы, такие как платиновая пластина, оксид платины и подобные, никелевые катализаторы, такие как восстановленный никель, никеля Ренея и подобные, родиевые катализаторы, катализаторы на основе железа, такие как восстановленное железо и подобные, и т.д. Вместо газообразного водорода также можно использовать муравьиную кислоту или формиат аммония в эквивалентом количестве или в избыточном количестве, в качестве источника водорода для соединения.
[Ссылочные документы] (1) “Reductions un Organic Chemistry, 2nd ed. (ACS Monograph: 188)” written by M. Hudlicky, ACS, 1996, (2) “Jikken Kagaku Koza (Courses in Experimental Chemistry) (5th Edition)” edited by The Chemical Society of Japan, Vol. 19 (2005) (Maruzen).
Кроме того, некоторые соединения, представленные формулой (I), также можно получить путем комбинирования стадий, которые обычно может использовать специалист в данной области, таких, которые представляют собой известные стадии алкилирования, ацилирования, реакцию замещения, окисления, восстановления, гидролиза, удаления защиты, галогенирования и подобные, из соединения по настоящему изобретению, полученного, как описано выше.
Например, для алкилирования можно использовать реакцию алкилирования, которую обычно используют специалисты в данной области, и алкилирование можно осуществить в органическом растворителе, который является инертным для реакции, таком как простые эфиры; ароматические углеводороды; галогенированные углеводороды; ДМФА, MeCN; апротонных полярных растворителях и подобных, в условиях охлаждения, от охлаждения до комнатной температуры, или от комнатной температуры до нагревания, в присутствии оснований, таких как NaH; щелочного соединения угольной кислоты; гидрокарбонатного щелочного соединения; алкоксида; третичного амина; органических оснований и т.п.
Кроме того, например, для ацилирования можно использовать реакцию ацилирования, которую обычно используют специалисты в данной области, но ацилирование осуществляют в органическом растворителе, который является инертным для реакции, таком как простые эфиры; ароматические углеводороды; галогенированные углеводороды; сложные эфиры, такие как EtOAc и подобные; MeCN; апротонные растворители и подобные, с использованием агента конденсации, такого как EDCI·HCl, CDI, дифенилфосфориланид и подобные, в зависимости от реакционных условий, но обычно в условиях охлаждения, в температурных условиях от охлаждения до комнатной температуры, или от комнатной температуры до нагревания, в частности, в присутствии HOBt.
Соединения формулы (I) могут быть выделены и очищены в форме их свободных соединений, солей, гидратов, сольватов или полиморфных кристаллических веществ. Соли соединения формулы (I) можно получить путем осуществления традиционной реакции солеобразования.
Выделение и очистку осуществляют с использованием обычных химических процедур, таких как экстракция, фракционированная кристаллизация, различные типы фракционной хроматографии и подобные.
Различные изомеры можно получить путем выбора подходящего исходного соединения или разделения с использованием разницы в физико-химических свойствах между изомерами. Например, оптические изомеры можно получить с использованием общего способа оптического разделения рацемических продуктов (например, фракционированная кристаллизация для индукции диастереомерных солей оптически активными основаниями или кислотами, хроматография с использованием хиральной колонки или т.п. и другие), и, кроме того, изомеры также можно получить из подходящего оптически активного исходного соединения.
Фармакологическую активность соединения формулы (I) подтверждали путем испытания, описанного ниже.
Пример испытания 1: Оценка in vitro агонистической активности в отношении S1P1 рецептора в биологическом организме
(Метод 1) Метод оценки агонистического действия на рецепторе при помощи GTP[γ-35S] анализа связывания с использованием мембран клеток, экспрессирующих человеческий S1P1
In vitro агонистическое действие соединения по настоящему изобретению в отношении S1P1 оценивали по повышению функциональной связывающей активности GTP[γ-35S] с G-белком с использованием мембран клеток, экспрессирующих человеческий S1P1. кДНК, кодирующую человеческий S1P1, клонировали из библиотеки кДНК колоректальных клеток человека и вводили в экспрессирующий вектор pcDNA3.1 для получения конструкции S1P1-pcDNA3.1. Затем, при помощи Липофектамина 2000 (GIBCO) S1P1-pcDNA3.1 трансфицировали в CHO клетки и культивировали в F-12 культуральной среде Хэма, содержащей 10% фетальную бычью сыворотку, 100 Ед./мл пенициллина, 100 мкг/мл стрептомицина и 1 мг/мл G418 дисульфата, с получением стабильного G418-резистентного штамма. Культивированные клетки, экспрессирующие человеческий S1P1, изолировали в 1 мМ EDTA·2Na-содержащего PBS и разрывали при охлаждении льдом с использованием гомогенизатора, сделанного из стекла, в буферном растворе 1 мМ Tris HCl (pH 7,4), содержащем 0,1 мМ EDTA и ингибитор белка. Осуществляли центрифугирование при 1400×10 мин и супенатант далее центрифугировали при 4°C в течение 60 мин при 100000×g и суспендировали в буферном растворе 10 мМ Tris HCl (pH 7,4), содержащем 1 мМ EDTA, для очистки мембран. Осуществляли взаимодействие полученных мембран (0,13 мг/мл) и 50 пМ GTP[γ-35S] (NEN; неактивный 1250 Ки/ммоль) в буферном растворе 20 мМ HEPES (pH 7,0) (общее количество: 150 мкл), содержащем 100 мМ NaCl, 10 мМ MgCl2, 0,1% не содержащего жирных кислот BSA и 5 мкМ GDP, в течение 1 часа вместе с соединением по настоящему изобретению (10−12-10−5 M) и затем мембраны выделали на GF-C фильтровальной пластине с использованием клеточного харвестера (Packard, FilterMate). Фильтровальную пластину сушили при 50°C в течение 60 мин и добавляли Microscinti-o (Packard) для измерения при помощи жидкостного сцинтилляционного счетчика для микропланшета (Packard, TOP count). Для оценки агонистического действия в отношении человеческого S1P1 соединения по настоящему изобретению и сравнительного соединения, использовали процентное значение, установленное как 100% для максимальной скорости реакции, делающей GTP[γ-35S] связи насыщенными, в присутствии соединения, и установленное как 0% для скорости реакции образования GTP[γ-35S] связей в отсутствие соединения, строили кривую нелинейной регрессии и концентрацию, вызывающую агонистическое действие, которое составляет 50% от максимальной реакции, определяли как значение EC50 (нМ).
(Метод 2) Метод оценки агонистического действия в отношении рецептора при помощи анализа притока Ca2+ с использованием клеток, экспрессирующих человеческий S1P1
In vitro агонистическое действие соединения по настоящему изобретению в отношении S1P1 оценивали по повышению концентрации Ca2+ в клетках, экспрессирующих человеческий S1P1. кДНК, кодирующую человеческий S1P1, клонировали из библиотеки кДНК колоректальных клеток человека и вводили в экспрессирующий вектор pcDNA3.1 для получения конструкции S1P1-pcDNA3.1. Затем, при помощи Липофектамина 2000 (GIBCO) S1P1-pcDNA3.1 трансфицировали в CHO клетки и культивировали в F-12 культуральной среде Хэма, содержащей 10% фетальную бычью сыворотку, 100 Ед./мл пенициллина, 100 мкг/мл стрептомицина и 1 мг/мл G418 дисульфата, с получением стабильного G418-резистентного штамма. Культивированные клетки, экспрессирующие человеческий S1P1, изолировали в 1 мМ EDTA·2Na-содержащего PBS и суспендировали в F-12 культуральной среде Хэма, содержащей 10% фетальную бычью сыворотку, 100 Ед./мл пенициллина, 100 мкг/мл стрептомицина. Эту клеточную суспензию распределяли в 96-луночный планшет при плотности 50000 клеток/лунка и культивировали в CO2 инкубаторе (5% CO2, 37°C) в течение ночи. Культуральную среду заменяли содержащим кальций-чувствительный флуоресцентный реагент (FLIPR (зарегистрированная торговая марка) набор для анализа кальция 3, molecular device) нагружающим буфером (сбалансированный солевой раствор Хэнка, 20 мМ HEPES, 2,5 мМ пробенецида) и оставляли выстаиваться в CO2 инкубаторе (5% CO2, 37°C) в течение 1 часа. Планшет устанавливали на систему для скрининга функционального лекарственного средства FDSS6000 (Hamamatsu Photonics K. K.) и постоянно осуществляли измерения 124 раза через каждые 1,02 сек при длине волны возбуждения 480 нм. Испытываемое соединение (конечная концентрация 10-12-10-5 M) добавляли одновременно с 12-м измерением и изменение Ca2+ концентрации в клетках оценивали по изменению интенсивности флуоресценции. Для оценки агонистического действия в отношении человеческого S1P1 соединения по настоящему изобретению и сравнительного соединения использовали процентное значение, установленное как 100% для максимальной скорости реакции, делающей повышение Ca2+ концентрации в клетках насыщенным после добавления соединения, и установленное как 0% для скорости повышения Ca2+ концентрации в клетках при добавлении только носителя, строили кривую нелинейной регрессии и концентрацию, вызывающую агонистическое действие, которое составляет 50% от максимальной реакции, определяли как значение EC50 (нМ).
Пример испытания 2: Оценка уменьшения количества лимфоцитов в периферической крови у крыс
Действие на лимфоциты периферической крови оценивали с использованием крыс. 6-10-недельных самцов крыс Lewis (Japan Charles River Laboratories Japan, Inc.) рандомизированно разделяли на группы (n=3) и соединение по настоящему изобретению суспендировали в содержащей 0,5% метилцеллюлозы дистиллированной воде и перорально вводили через зонд. В момент времени 4 часа или 24 часа после введения, собирали 0,2 мл крови из глазного дна под эфирной анестезией. К образцу крови сразе же добавляли EDTA·4K и гепарин для предотвращения свертывания и количество лимфоцитов в крови измеряли при помощи автоматического анализатора форменных элементов крови (Sysmex Corp.; XT-2000i). Для определения уменьшения количества лимфоцитов в периферической крови соединением по настоящему изобретению использовали процентное значение, установленное как 100% для количества лимфоцитов в группах, которым осуществляли введение с содержащей 0,5% метилцеллюлозы дистиллированной водой, и дозу, вызывающую 50% уменьшение количества лимфоцитов в периферической крови путем введения соединения по настоящему изобретению, определяли как значение ED50 (мг/кг).
Результаты Примера испытания 1 и Примера испытания 2 для некоторых соединений формулы (I) представлены в Таблицах 1 и 2. В таблицах, колонка A показывает in vitro действие агониста S1P1, EC50 значения (нМ), полученные методом 1 Примера испытания 1, при этом значение с * показывает EC50 значения, измеренные методом 2. Кроме того, колонка B показывает действие по уменьшению числа лимфоцитов в периферической крови в момент времени 4 часа или 24 часа после введения лекарственного средства Примера испытания 2, при этом представлены значения ED504 час (мг/кг) или ED5024 час (мг/кг), соответственно.
Как показано в Таблице 1 и 2, было подтверждено, что соединение формулы (I) по настоящему изобретению обладает отличным действием агониста S1P1 и обладает сильным действием по снижению числа лимфоцитов в периферической крови, даже через 4 часа или 24 часа после введения, в фармакологическом испытании с использованием крыс.
(мг/кг)
(мг/кг)
(мг/кг)
(мг/кг)
Пример испытания 3: Оценка увеличения массы легкого у крыс
Оценивали увеличенную массу легкого у крыс, один из нежелательных эффектов, наблюдаемых для традиционных S1P1 агонистов. 6-10-недельных самцов крыс Lewis или крыс SD (Japan Charles River Laboratories Japan, Inc.) рандомизированно разделяли на группы (n=3 до 4) и соединение по настоящему изобретению суспендировали в содержащей 0,5% метилцеллюлозы дистиллированной воде и перорально вводили через зонд. Для однократного введения, в момент времени 24 часа после введения массу крысы измеряли, кровь удаляли под анестезией пентобарбиталом и легкое извлекали и измеряли его массу. Для повторного введения, введение осуществляли раз в день в течение 7 дней и в момент времени 24 часа после конечного введения измеряли массу тела и массу легкого. Для увеличенной массы легкого, показатель увеличения среднего значения соответствующих массовых значений группы, которой вводили суспензию соединения по настоящему изобретению в содержащей 0,5% метилцеллюлозы дистиллированной воде относительно среднего значения соответствующих массовых значений группы, которой вводили содержащую 0,5% метилцеллюлозу дистиллированную воду определяли в виде процента, и вводимое количество, показывающее 10% или больше увеличение массы, определяли как положительное.
Было подтверждено, что из соединений по настоящему изобретению соединения Примеров 31, 43, 44, 45, 56, 62, 66, 69, 74, 81, 85, 89, 109, 116, 137, 143, 149, 151, 152, 160, 171, 178, 181, 183, 212, 216, 223, 230 и 236 дают увеличение массы легкого менее 10%, даже при дозе 1 мг/кг, и оказывают слабое действие на легкие.
Пример испытания 4: Оценка ингибирующего действия на отторжение в гетеротопной крысиной модели абдоминального сердечного трансплантата
Гетеротопную крысиную модель абдоминального сердечного трансплантата можно осуществить в соответствии со способом Ono and Lindsey (Transplantation, 1969, 517, pp. 225-229). В качестве донора использовали 6-8-недельных самцов крыс ACI (CLEA Japan, Inc.) и сердца обнажали под анестезией пентобарбиталом. Правую и левую полую вену, отличную от аорты, и легочную артерию, легочные вены и нижнюю полую вену сразу лигировали и аорту и легочную вену отделяли и удаляли в качестве трансплантата. 6-8-недельных самцов крыс Lewis (Japan Charles River Laboratories Japan, Inc.) использовали в качестве реципиентов. Под анестезией пентобарбиталом, конец легочной артерии трансплантата и абдоминальную аорту реципиента соединяли при помощи анастомоза и конец легочной артерии трансплантата и полую вену реципиента соединяли при помощи анастомоза с получением модели (разделяли по группам по 6-10 примеров на группу). Определению отторжения трансплантированного сердца помогает абдоминальная пальпация реципиента через каждые 29 дней после трансплантации, и присутствие или отсутствие пульсирования трансплантата определяют по отторжению. Соединение по настоящему изобретению суспендировали в содержащей 0,5% метилцеллюлозы дистиллированной воде и перорально вводили один или два раза в день в течение 14 дней с момента трансплантации. В качестве контроля, перорально вводили содержащую 0,5% метилцеллюлозы дистиллированную воду такое же количество раз в течение того же периода времени. Одновременно вводили внутримышечным путем 0,02 мг/мл/кг такролимуса всем животным группы. При помощи этого испытания определяли действие соединения по настоящему изобретению по ингибированию отторжения при использовании такролимуса в сочетании.
Пример испытания 5: Оценка выраженного редкого пульса с использованием бодрствующих крыс
Самцов крыс Lewis анестезировали при помощи ингаляции изофлураном и полиэтиленовую трубку вводили в бедренную артерию и вену. Трубку соединяли с установкой для измерения кровяного давления•частоты сердечных сокращений через датчик давления от артериальной линии и измеряли артериальное давление и частоту сердечных сокращений. Постоянно вводили внутривенную инфузию, включающую Носитель (10% HCO40/tween80/PEG, 90% физиологического раствора) и соединения по настоящему изобретению, со скоростью 1 мл/кг/мин в течение 10 минут. Показатели измерений считывали (общее время оценки 20 минут) из таблицы значений перед введением, в точках времени 1, 2, 5 и 10 минут после начала постоянной инфузии и 1, 2, 5 и 10 минут после завершения инфузии и, таким образом, с учетом частоты сердечных сокращений и кровяного давления до введения, рассчитывали скорости снижения (%) до и после инфузии.
Было подтверждено, что из соединений по настоящему изобретению, например, соединение Примера 230 не оказывает влияние на частоту сердечных сокращений и кровяное давление при дозе введения 1 мг/кг, согласно данной оценке, и редкий пульс не наблюдается.
В качестве результатов описанных выше испытаний, было подтверждено, что соединение формулы (I) по настоящему изобретению обладает отличным действием агониста S1P1 и обладает действием ингибирования инфильтрации лимфоцитов. Кроме того, как показано в Примерах испытаний 3 и 4 выше, иллюстративные соединения некоторых вариантов воплощения настоящего изобретения могут обладать действием агониста S1P1, который имеет слабые нежелательные эффекты, такие, как наблюдаемые у традиционных S1P1 агонистов, такие как увеличенная масса легких, редкий пульс и подобные, и незначительные побочные эффекты.
Соответственно, соединение формулы (I) по настоящему изобретению является полезным для профилактики или лечения заболеваний, индуцируемых нежелательной инфильтрацией лимфоцитов, например, реакций отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей, аутоиммунных заболеваний или воспалительных заболеваний, таких как ревматоидный артрит, рассеянный склероз, системная красная волчанка, нефротический синдром, менингоэнцефалит, тяжелая миастения, панкреатит, гепатит, нефрит, диабет, легочные заболевания, бронхиальная астма, атопический дерматит, воспалительные заболевания кишечника, атеросклероз, ишемическое реперфузионное расстройство и подобные, и заболеваний, вызванных аномальной пролиферацией или аккумуляцией клеток, таких как рак, лейкемия и подобные, в частности, для профилактики или лечения реакций отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей и рассеянного склероза.
Кроме того, соединение по настоящему изобретению можно вводить в качестве S1P1 агониста отдельно или в комбинации с, по меньшей мере, одним средством, в одинаковых или разных дозах, одинаковым или разным путем введения. Примеры средства для использования в комбинации включают, но не ограничиваются этим, циклоспорин A, такролимус, сиролимус, эверолимус, микофенолят, азатиоприне, бреквинар, Лефлуномид, финголимод, антитело против IL-2 рецептора (например, даклизумаб и подобные), анти-CD3 антитело (например, OKT3), анти-T-клеточный иммуноглобулин (например, AtGam и подобные), белатасепт, абатасепт, циклофосфамид, β-интерферон, аспирин, ацетаминофен, ибупрофен, напроксен, пироксикам, противовоспалительное стероидное средство (например, преднизолон и дексаметазон) и подобные.
Фармацевтическую композицию, содержащую один или два или более типов соединения формулы (I) или его соли в качестве активного ингредиента, можно получить с использованием эксципиентов, которые обычно используют в данной области, то есть эксципиентов для фармацевтических препаратов, носителей для фармацевтических препаратов и т.п., в соответствии со способами, которые обычно используют.
Введение можно осуществить либо путем перорального введения через таблетки, пилюли, капсулы, гранулы, порошки, растворы и подобные, либо путем парентерального введения через инъекции, такие как внутрисосудистые, внутривенные или внутримышечные инъекции и подобные, суппозитории, глазные растворы, глазные мази, чрескожные жидкие препараты, мази, чрескожные пластыри, жидкие препараты для введения через слизистую оболочку, пластыри для введения через слизистую оболочку, препараты для ингаляций и подобные.
Твердую композицию для перорального введения используют в форме таблеток, порошков, гранул или т.п. В такой твердой композиции один или несколько активных ингредиентов смешивают с, по меньшей мере, одним неактивным эксципиентом, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон, алюмометасиликат магния и/или т.п. В соответствии с обычным способом, композиция может содержать неактивные добавки, включая лубриканты, такие как стеарат магния и т.п., дезинтегранты, такие как натрий карбоксиметилкрахмал, стабилизаторы и вещества, способствующие солюбилизации. Если необходимо, таблетки или пилюли могут иметь сахарное покрытие или пленочное покрытие из гастро- или энтеросолюбильного вещества.
Жидкая композиция для перорального введения содержит фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры или т.п., а также содержит обычно используемые инертные разбавители, например, дистиллированную воду или этанол. Помимо инертного разбавителя, жидкая композиция также может содержать вспомогательные вещества, такие как вещество, способствующее солюбилизации, смачивающее вещество и суспендирующее вещество, а также подсластители, отдушки, ароматизаторы и антисептики.
Препараты для инъекций для парентерального введения включают стерильные водные или не-водные растворы, суспензии или эмульсии. В качестве водного растворителя, например, включены дистиллированная вода для инъекций или физиологический солевой раствор. Примеры не-водного растворителя включают пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и подобные, спирты, такие как этанол и подобные, полисорбат 80 (pharmacopeia) и т.п. такая композиция может дополнительно содержать агент тоничности, антисептик, смачивающее вещество, эмульгатор, диспергирующее вещество, стабилизатор или вещества, способствующие солюбилизации. Композиции стерилизуют, например, при помощи фильтрации через удерживающий бактерии фильтр, смешивания с бактерицидами или облучения. Кроме того, их также можно использовать путем получения стерильной твердой композиции и растворения или суспендирования такой композиции в стерильной воде или стерильном растворителе для инъекций перед использованием.
Примеры препаратов для наружного применения включают мази, пластыри, кремы, желе, пластыри, спреи, лосьоны, глазные капли, глазные мази и подобные. Лекарственное средство содержит обычно используемые основы для мазей, основы для лосьонов, водные или не-водные жидкие препараты, суспензии, эмульсии или т.п. Примеры основы для мазей или основы для лосьонов включают полиэтиленгликоль, пропиленгликоль, белый вазелин, отбеленный пчелиный воск, полиоксиэтилен-гидрированное касторовое масло, глицерилмоностеарат, стеариловый спирт, цетиловый спирт, лауромакроголь, сорбитансесквиолеат и подобные вещества.
Что касается средства для введения через слизистую оболочку, такого как препарат для ингаляций, средство для введения через нос и т.п., используют средства для введения через слизистую оболочку в твердой, жидкой или полутвердой форме, и их можно получить в соответствии с широко известным способом. Например, можно подходящим образом добавить известный эксципиент, а также агент регулирования pH, антисептик, поверхностно-активное вещество, смазывающее вещество, стабилизатор, загуститель или т.п. Для их введения, можно использовать подходящее устройство для ингаляции или инсуффляции. Например, соединение можно вводить отдельно или в виде смеси, сформулированной в порошок, или в виде раствора или суспензии, путем объединения с фармацевтически приемлемым носителем, с использованием широко известного устройства или распылителя, такого как устройство для введения отмеренных доз путем ингаляции и подобные. Ингалятор сухого порошка или т.п. может быть предназначен для однократного или многократного введения, и можно использовать сухой порошок или содержащую порошок капсулу. Альтернативно, это может быть в форме находящегося под давлением аэрозольного спрея, в котором используют подходящий пропеллент, например, хлорфторалкан, гидрофторалкан, или подходящий газ, такой как диоксид углерода и подобные.
Обычно, в случае перорального введения, подходящей является суточная доза, составляющая от 0,001 до 100 мг/кг массы тела, предпочтительно от 0,1 до 30 мг/кг, и более предпочтительно от 0,1 до 10 мг/кг, и ее вводят в виде одной дозы или разделяют на 2-4 части. В случае внутривенного введения, подходящей является суточная доза, составляющая от около 0,0001 до 10 мг/кг массы тела, и ее вводят раз в день или два или более раз в день. Кроме того, средства для введения через слизистую оболочку вводят при дозе от около 0,001 до 100 мг/кг массы тела, и его вводят раз в день или два или более раз в день. Дозу подходящим образом определяют с учетом конкретного случая, принимая во внимание симптомы, возраст, пол и подобные факторы.
Соединение формулы (I) можно использовать в комбинации с различными средствами для лечения или профилактики заболеваний, где соединение формулы (I), описанное выше, считается эффективным. Комбинированный препарат можно вводить одновременно или по отдельности, и непрерывно или с желаемыми интервалами. Препараты для одновременного введения могут представлять собой смесь или могут быть получены отдельно.
ПРИМЕРЫ
Кроме того, следующие аббревиатуры можно использовать в некоторых случаях в Примерах, Примерах получения и Таблицах, описанных ниже.
Пр.пол. = № Примера получения, Пр. = № Примера, Ссыл.пр. = № Ссылочного Примера, Стр. = Структурная формула, MS = Масс-спектрометрические данные, ESI (EI) = Данные анализа ионизации электрораспылением, FAB = Масс-спектрометрические данные в соответствии с методом ионизации путем бомбардировки быстрыми атомами, Гц = Герц, CDCl3 = дейтерированный хлороформ, ДМСО-d6 = диметилсульфоксид d6.
Также, поперечные двойные связи в структурных формулах означают смесь цис-формы и транс-формы. В данных 1H-ЯМР, тетраметилсилан используют в качестве внутреннего стандарта, если специально не указано иное, и δ (м.д.) (интегрированное значение, картина расщепления) сигналов в 1H-ЯМР, где ДМСО-d6 используют в качестве растворителя для измерений. В настоящем описании, ЯМР представляет собой 1H-ЯМР: Протонный Ядерный Магнитный Резонанс. Кроме того, знаки + и - в данных MS и ESI (EI), каждый, представляет собой положительные и отрицательные массовые значения.
Пример получения 1
7-[(5-Бром-4-фенил-2-тиенил)метокси]-2H-хромен-3-карбальдегид (120 мг) растворяли в ДМФА (2,4 мл). К этой реакционной жидкости добавляли Zn(CN)2 (65 мл) и Pd(PPh3)4 (65 мг) при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 5 часов и затем выливали в 1:1 смешанный растворитель, состоящий из водного раствора NaHCO3 и EtOAc, с последующим перемешиванием в течение 1 часа. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с последующей очисткой при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=100:0 до 70:30) с получением 5-{[(3-формил-2H-хромен-7-ил)окси]метил}-3-фенилтиофен-2-карбонитрила (83 мг) в виде бледно-желтого твердого вещества.
Пример получения 2
К раствору метил 5-бром-4-фенилтиофен-2-карбоксилата в диоксане добавляли 2-изопропенил-4,4,5,5-тетраметил 1,3,2-диоксаборолан и 2 M водный раствор Na2CO3. К реакционной смеси добавляли ацетат палладия и PPh3 с последующим перемешиванием при 100°C в течение 5 часов. После того, как оставляли охлаждаться, к этой смеси добавляли насыщенный водный раствор NH4Cl с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с последующей очисткой при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=95:5 до 80:20) с получением метил 5-изопропенил-4-фенилтиофен-2-карбоксилата в виде бесцветной жидкости.
Таким же образом, как и в Примере получения 2, получали соединения Примера получения 2-1 - Примера получения 2-4, приведенные в описанных далее таблицах.
Пример получения 3
К раствору ДМФА (2, мл) в ДХМ (3 мл) по каплям добавляли POCl3 (2 мл) при 0°C с последующим перемешиванием при комнатной температуре в течение 30 минут. Затем к реакционной жидкости добавляли по каплям 8-(бензилокси-3,4-дигидро-1-бензоксепин-5(2H)-он в ДХМ (4 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа и при 50°C в течение 3 часов. К реакционной жидкости добавляли воду с последующей экстракцией два раза при помощи EtOAc. Органический слой объединяли, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки; смесь гексан:EtOAc=97:3 до 90:10) с получением 8-(бензилокси)-5-хлор-2,3-дигидро-1-бензоксепин-4-карбальдегида (445 мг).
Пример получения 4
К раствору ДМФА (2 мл) в ДХМ (7,5 мл) добавляли по каплям POCl3 (1,39 мл) при 0°C с последующим перемешиванием при комнатной температуре в течение 30 минут. Затем к реакционной жидкости добавляли по каплям раствор 7-{[трет-бутил(дифенил)силил]окси}-2,3-дигидро-4H-хромен-4-она (2,00 г) в ДХМ (11 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа и при 50°C в течение 3 часов. К реакционной жидкости добавляли воду с последующей экстракцией при помощи EtOAc два раза. Органический слой объединяли, промывали водой и насыщенным солевым раствором и сушили над MgSO4 и жидкость концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 80:20) с получением 4-хлор-7-гидрокси-2H-хромен-3-карбальдегида (720 мг).
Пример получения 5
7-(бензилокси)-2,3-дигидро-4H-хромен-4-он растворяли в ТГФ, к этой смеси по каплям добавляли раствор (0,97 M, 5 мл) метилмагнийбромида в ТГФ при 0°C с последующим перемешиванием при комнатной температуре в течение 1 часа и затем по каплям добавляли раствор (0,97 M, 5 мл) метилмагнийбромида в ТГФ с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной жидкости добавляли насыщенный водный раствор NH4Cl и затем 2 M раствор хлористоводородной кислоты (20 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов и затем экстрагировали при помощи EtOAc три раза. Органический слой объединяли, промывали водой и насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=95:5 до 90:10) с получением 7-(бензилокси)-4-метил 2H-хромена (445 мг) в виде бесцветной прозрачной жидкости.
Таким же образом, как и в Примере получения 5, получали соединение Примера получения 5-1, представленное в описанных далее таблицах.
Пример получения 6
К раствору 2-гидрокси-4-[(2-метокси-4-пропилфенокси)метил]бензальдегида (120 мг) в диоксане (2,4 мл) при 25°C добавляли K2CO3 (55,2 мг) и акролеин (0,267 мл). Реакционную смесь нагревали до 100°C с последующим перемешиванием при 100°C в течение 15 часов. Реакционную смесь оставляли нагреваться до 25°C и затем фильтровали через целит и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 80:20) с получением 7-[(2-метокси-4-пропилфенокси)метил]-2H-хромен-3-карбальдегида (104,2 мг) в виде бесцветной жидкости.
Таким же образом, как и в Примере получения 6, получали соединения Примера получения 6-1 - Примера получения 6-9 и Примера получения 6-11, приведенные в описанных далее таблицах.
Пример получения 6-10
K2CO3 (835 мг) суспендировали в диоксане (40 мл) и к этой смеси добавляли 2-гидрокси-4-(метоксиметокси)бензальдегид (1 г) и 3-метил 2-бутаналь (0,787 мл) с последующим перемешиванием при 110°C в течение ночи. К этой смеси добавляли EtOAc, нерастворимые вещества удаляли при помощи фильтрации через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=95:5 до 70:30) с получением 7-(метоксиметокси)-2,2-диметил-2H-хромен-3-карбальдегида (320 мг) в виде желтого масла.
Пример получения 7
При 0°C к перемешиваемому раствору концентрированной HCl (8 мл) и AcOH (1,6 мл) добавляли трет-бутил 3-циано-3-(фторметил)азетидин-1-карбоксилат (800 мг). Жидкость нагревали до 25°C с последующим перемешиванием при 25°C в течение 1 часа и затем при 100°C в течение 5 часов. Реакционную жидкость концентрировали при пониженном давлении, с последующей азеотропной перегонкой с толуолом (30 мл) три раза. Остаток растворяли в смешанном растворителе ацетона (4,8 мл) и воды (8,0 мл) и при 0°C к этой смеси добавляли Na2CO3 (593,7 мг) и DIBOC (1223 мг). Реакционную жидкость нагревали до 25°C с последующим перемешиванием при 25°C в течение 15 часов. Через пятнадцать часов реакционный раствор концентрировали и ацетон выпаривали. Остаток экстрагировали три раза (50 мл ×3) путем добавления эфира (50 мл). Водный слой объединяли и охлаждали до 0°C и при 0°C к этой смеси добавляли 2 M раствор HCl (10 мл) до получения раствора при pH=2-3. Осажденное твердое вещество белого цвета собирали при помощи фильтрации и промывали гексаном (50 мл) с получением 1-(трет-бутоксикарбонил)-3-(фторметил)азетидин 3-карбоноой кислоты (801,2 мг) в виде белого твердого вещества.
Пример получения 8
трет-Бутил 3-циано-3-(гидроксиметил)азетидин-1-карбоксилат (5,0 г) растворяли в ДХМ (100 мл). К этой смеси добавляли при 0°C DAST (3,74 мл) с последующим перемешиванием при 0°C в течение 3 часов. Через три часа к реакционной жидкости добавляли водный раствор NaHCO3 (100 мл) с последующей экстракцией при помощи ДХМ (50 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 50:50) с получением трет-бутил 3-циано-3-(фторметил)азетидин-1-карбоксилата (1,24 г) в виде коричневого твердого вещества.
Пример получения 9
2-Фтор-4,6-дигидроксибензальдегид (12 г) растворяли в MeCN (250 мл) и к этой смеси добавляли карбонат цезия (25,1 г) и хлорметилметиловый эфир (6,95 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. Нерастворимые вещества удаляли при помощи фильтрации через целит и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 94:6) с получением 2-фтор-6-гидрокси-4-(метоксиметокси)бензальдегида (11,89 г) в виде белого порошка.
Таким же образом, как и в Примере получения 9, получали соединения Примера получения 9-1 - Примера получения 9-4, приведенные в описанных далее таблицах.
Пример получения 10
7-Гидрокси-2,3-дигидро-4H-хромен-4-он (900 мг) растворяли в ДМФА (10 мл) и к этой смеси добавляли трет-бутил(хлор)дифенилсилан (1,711 мл) и 1H-имидазол (448 мг) с последующим перемешиванием при комнатной температуре в течение ночи. К реакционной жидкости добавляли воду с последующей экстракцией при помощи EtOAc три раза. Органический слой объединяли, промывали водой и насыщенным солевым раствором в указанном порядке, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=90:10 до 80:20) с получением 7-{[трет-бутил(дифенил)силил]окси}-2,3-дигидро-4H-хромен-4-она (2,08 г) в виде бесцветного прозрачного сиропа.
Таким же образом, как и в Примере получения 10, получали соединение Примера получения 10-1, представленное в описанных далее таблицах.
Пример получения 11
К раствору 7-гидрокси-2H-хромен-3-карбальдегида в ДХМ при 0°C добавляли пиридин. К реакционной жидкости при 0°C добавляли по каплям трифторметансульфоновый ангидрид. После перемешивания при комнатной температуре в течение 1 часа к этой смеси при 0°C добавляли воду. Смесь экстрагировали при помощи EtOAc. Органический слой промывали 1 M раствором HCl, водой и насыщенным солевым раствором, в указанном порядке, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=95:5 до 80:20) с получением 3-формил-2H-хромен-7-ил трифторметансульфоната в виде желтого масляного вещества.
Таким же образом, как и в Примере получения 11, получали соединение Примера получения 11-1, представленное в описанных далее таблицах.
Пример получения 12
К ДМФА (1 мл) по каплям добавляли POCl3 (0,25 мл) при 0°C с последующим перемешиванием при комнатной температуре в течение 30 минут. К реакционной смеси добавляли по каплям раствор 7-(бензилокси)-4-метил 2H-хромена (280 мг) в ДХМ (1 мл) с последующим перемешиванием при комнатной температуре в течение 3 часов. Реакционную жидкость выливали в смесь льда с водой с последующей экстракцией при помощи EtOAc три раза. Органический слой объединяли, промывали водой и насыщенным солевым раствором, в указанном порядке, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=85:15 до 70:30) с получением 7-(бензилокси)-4-метил-2H-хромен-3-карбальдегида (234 мг) в виде бледно-желтого порошка.
Таким же образом, как и в Примере получения 12, соединение Примера получения 12-4 получали из соединения Примера получения 12-1, представленного в описанных далее таблицах.
Пример получения 13
Раствор NaH (105,63 мг) в ДМФА (5,5 мл) охлаждали до 0°C и к этой смеси добавляли метил 2-{[трет-бутил(диметил)силил]окси}-4-[(диэтоксифосфорил)метил]бензоат (550 мг). Реакционную смесь нагревали до 25°C, затем перемешивали в течение 1 часа и снова охлаждали до 0°C и к этой смеси добавляли 2-метокси-4-пропилбензальдегид (235,34 мг). Реакционную смесь нагревали до 25°C и затем перемешивали в течение 15 часов. К реакционной жидкости добавляли насыщенный водный раствор NH4Cl (50 мл) с последующей экстракцией при помощи EtOAc (50 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 70:30) с получением метил 2-гидрокси-4-[(E)-2-(2-метокси-4-пропилфенил)винил]бензоата (304,2 мг) в виде белого твердого вещества.
Таким же образом, как и в Примере получения 13, получали соединение Примера получения 13-1, представленное в описанных далее таблицах.
Пример получения 14
К ДМФА (40 мл) при охлаждении льдом добавляли 60% раствор NaH (634 мг) и к этой смеси медленно добавляли раствор 4-фтор-3-(трифторметил)бензонитрила (2 г) в ДМФА (20 мл). После перемешивания при комнатной температуре в течение 5 часов, реакцию гасили насыщенным раствором NH4Cl с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем фильтровали. Фильтрат концентрировали с получением 4-изопропокси-3-(трифторметил)бензонитрила (2,4 г) в виде бледно-желтого твердого вещества.
Таким же образом, как и в Примере получения 14, получали соединения Примера получения 14-1 - Примера получения 14-16, приведенные в описанных далее таблицах.
Пример получения 15
К раствору метил 4-фтор-2-(трифторметил)бензоата в ДМФА добавляли K2CO3 и пиперидин с последующим перемешиванием при 100°C в течение 3 часов. Реакционную смесь охлаждали до 0°C и к этой смеси добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=100:0 до 90:10) с получением метил 4-пиперидин-1-ил-2-(трифторметил)бензоата в виде бесцветного маслянистого вещества.
Таким же образом, как и в Примере получения 15, получали соединения Примера получения 15-1 - Примера получения 15-4, приведенные в описанных далее таблицах.
Пример получения 16
К раствору метил 1H-индол-5-карбоксилата (1,5 г) в ДМФА (30 мл) при 0°C добавляли NaH (410 мг). Реакционную смесь нагревали до 25°C с последующим перемешиванием в течение 0,5 часа. Затем реакционную смесь снова охлаждали до 0°C и далее к этой смеси добавляли метилйодид (1,38 мл). Реакционную смесь нагревали до 25°C с последующим перемешиванием в течение 3 часов. К реакционной жидкости добавляли воду (50 мл) с последующей экстракцией при помощи EtOAc (50 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь CHCl3:MeOH=100:0 до 98:2) с получением метил 1-этил-1H-индол-5-карбоксилата (1465 мг) в виде белого твердого вещества.
Пример получения 17
К раствору 4-фтор-2-(трифторметил)бензойной кислоты в MeOH при 0°C добавляли концентрированную серную кислоту. Реакционную смесь нагревали и кипятили с обратным холодильником в течение 2 дней. Реакционную смесь концентрировали при пониженном давлении и остаток разбавляли при помощи EtOAc. Органический слой промывали насыщенным водным раствором NaHCO3, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением метил 4-фтор-2-(трифторметил)бензоата в виде бесцветного маслянистого вещества.
Пример получения 18
К суспензии 4-бром-5-этилтиофен-2-карбоновой кислоты (800 мг) в MeOH (4 мл) при 0°C добавляли по каплям SOCl2 (0,50 мл). Реакционную смесь перемешивали при 0°C в течение 1 часа, нагревали до 60°C и затем перемешивали в течение 15 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=98:2 до 70:30) с получением метил 4-бром-5-этилтиофен-2-карбоксилата (765,0 мг) в виде бесцветной жидкости.
Таким же образом, как и в Примере получения 18, получали соединения Примера получения 18-1 - Примера получения 18-6, приведенные в описанных далее таблицах.
Пример получения 19
К раствору N-изопропилпропан-2-амина (165,5 мг) в ТГФ (1 мл) при -78°C по каплям добавляли раствор н-бутиллития в гексане (1,6 M, 0,98 мл) с последующим нагреванием до 25°C и последующим перемешиванием в течение 30 минут. После повторного охлаждения до -78°C к этой смеси по каплям добавляли раствор 1-трет-бутил-3-метилпирролидин-1,3-дикарбоксилата (300 мг) в ТГФ (1 мл). Реакционную смесь нагревали до -40°C и затем перемешивали в течение 1 часа. Реакционную смесь снова охлаждали до -78°C и к этой смеси по каплям добавляли раствор N-фтор-N-(фенилсульфонил)бензолсульфонамида (495,1 мг) в ТГФ (1 мл). Реакционную смесь перемешивали при -78°C в течение 1 часа, затем нагревали до 25°C и перемешивали в течение 15 часов. Через пятнадцать часов к реакционной жидкости добавляли насыщенный водный раствор NH4Cl (30 мл) с последующей экстракцией при помощи EtOAc (30 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 80:20) с получением 1-трет-бутил 3-метил-3-фторпирролидин-1,3-дикарбоксилата (154,3 мг) в виде желтой жидкости.
Пример получения 20
К раствору метил 4-фенилтиофен-2-карбоксилата (1,8 г) в ДХМ (18 мл) при 0°C порциями добавляли пиридинийтрибромид (13,2 г). Реакционную жидкость нагревали до 25°C и затем перемешивали в течение 45 часов. Реакционную смесь охлаждали до 0°C и медленно по каплям добавляли насыщенный водный раствор Na2S2O3 (100 мл). Реакционную смесь экстрагировали при помощи ДХМ (50 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 90:10) с получением метил 5-бром-4-фенилтиофен-2-карбоксилата (2,06 г) в виде бесцветной жидкости.
Пример получения 21
К раствору 4-хлор-5,5,5-трифтор-3-фенилпент-3-ен-2-она (950 мг) и метилсульфанилацетата (446 мг) в MeCN (23,8 мл) при 25°C по каплям добавляли DBU (0,63 мл) с последующим перемешиванием при той же температуре в течение 15 часов. К реакционной жидкости добавляли насыщенный водный раствор NH4Cl (50 мл) с последующей экстракцией диэтиловым эфиром (50 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=98:2 до 90:0) с получением метил 3-метил-4-фенил-5-(трифторметил)тиофен-2-карбоксилата (1,08 г) в виде бесцветной жидкости.
Пример получения 22
К раствору бензил 3-цианопирролидин-1-карбоксилата (1,0 г) и TEA гидрохлорида (2,99 г) в толуоле при 25°C добавляли азид натрия (1,41 г) с последующим перемешиванием при 115°C в течение 5 часов. Реакционную жидкость оставляли охлаждаться и к этой смеси добавляли ДХМ (10 мл). Затем к 5% водному раствору салициловой кислоты (100 мл) по каплям добавляли реакционную жидкость с последующим перемешиванием при 25°C в течение 1 часа. Реакционную жидкость экстрагировали при помощи EtOAc (30 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 80:20) с получением бензил 3-(1H-тетразол-5-ил)пирролидин-1-карбоксилата (10,8 г) в виде бесцветной жидкости.
Пример получения 23
К раствору метил 2-{[трет-бутил(диметил)силил]окси}-4-метилбензоата (3,4 г) в тетрахлориде углерода (68 мл) добавляли NBS (2,16 г) и к этой смеси при комнатной температуре добавляли AIBN (398 мг) с последующим перемешиванием при 80°C в течение 1 часа. Завершение реакции подтверждали при помощи показателей ТСХ и для остановки реакции к реакционной жидкости добавляли воду с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили, используя MgSO4, и затем концентрировали при пониженном давлении с последующей очисткой при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 95:5) с получением метил 4-(бромметил)-2-{[трет-бутил(диметил)силил]окси}бензоата (3,79 г) в виде бесцветной жидкости.
Пример получения 23-1
К раствору (2S)-3-(4-хлорфенил)-2-метилпропан-1-ола (300 мг) в ДХМ (20 мл) при охлаждении льдом добавляли N-бромсукцинимид (347 мг) и трифенилфосфин (511 мг). Реакционную жидкость перемешивали при комнатной температуре в течение 2 часов и затем реакционную жидкость выливали в воду с последующей экстракцией при помощи хлороформа. Органический слой промывали насыщенным солевым раствором и сушили над MgSO4 и затем растворитель выпаривали при пониженном давлении. Остаток подвергали очистке при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=100:0 до 90:10) с получением 1-[(2S)-3-бром-2-метилпропил]-4-хлорбензола (373 мг) в виде бесцветной жидкости.
Пример получения 24
К раствору N-изопропилпропан-2-амина (11,54 мл) в ТГФ (50 мл) при -78°C добавляли по каплям раствор н-бутиллития в гексане (1,6 M, 51,45 мл). Реакционную смесь нагревали до 0°C и затем перемешивали в течение 30 минут. Реакционную смесь снова охлаждали до -78°C и затем по каплям добавляли раствор трет-бутил 3-цианоазетидин-1-карбоксилата (5,0 г) в ТГФ (30 мл) с последующим перемешиванием при -78°C в течение 1 часа. К реакционной смеси при -78°C по каплям добавляли раствор 1H-бензотриазол-1-ил-метанола (8,19 г) в ТГФ (20 мл) с последующим перемешиванием при -78°C в течение 3 часов. К реакционной смеси добавляли насыщенный водный раствор NH4Cl (100 мл) с последующей экстракцией при помощи EtOAc (50 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 50:50) с получением трет-бутил 3-циано-3-(гидроксиметил)азетидин-1-карбоксилата (5,68 г) в виде белого твердого вещества.
Пример получения 25
Раствор метил 4-амино-(2-трифторметил)бензоатгидрохлорида (1,24 г) и 2,5-диметокситетрагидрофурана (773 мг) в AcOH (20 мл) перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и подвергали азеотропной перегонке с толуолом, и AcOH выпаривали. Полученное желтовато-коричневое маслянистое вещество растворяли в хлороформе и к этой смеси добавляли насыщенный водный раствор NaHCO3. Органический слой промывали насыщенным водным раствором NaHCO3, водой и насыщенным солевым раствором, в указанном порядке, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь CHCl3:MeOH=97:3) с получением метил 4-(1H-пирроло-1-ил)-2-(трифторметил)бензоата (11,8 г).
Таким же образом, как и в Примере получения 25, получали соединение Примера получения 25-1, представленное в описанных далее таблицах.
Пример получения 26
К раствору триметил(про-1-пин-1-ил)силана (877 мг) в ТГФ (60 мл) при -78°C добавляли раствор н-BuLi в гексане (1,58 M, 4,5 мл). Реакционную смесь перемешивали при -78°C в течение 3 часов и затем к этой смеси по каплям добавляли раствор 1-(бромметил)-2,4-бис(трифторметил)бензола (2 г) в ТГФ (10 мл) с последующим перемешиванием в течение 1 часа. К реакционной жидкости добавляли водный раствор NH4Cl с последующей экстракцией при помощи эфира. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=100:0) с получением {4-[2,4-бис(трифторметил)фенил]бут-1-ин-1-ил}(триметил)силана (1,8 г) в виде бесцветной жидкости.
Пример получения 27
1-(хлорметил)-2-метокси-4-пропилбензол (1,1 г) растворяли в ДМФА (20 мл) и к этой смеси добавляли 7-гидрокси-2H-хромен-3-карбальдегид (975 мг) и K2CO3 (1,15 г) с последующим перемешиванием при 80°C в течение 1 часа. Далее к этой смеси добавляли йодид натрия (416 мг) с последующим перемешиванием при 80°C в течение 1 часа. После подтверждения завершения реакции к реакционной жидкости добавляли воду, чтобы остановить реакцию, с последующей экстракцией при помощи EtOAc три раза. Органический слой объединяли, промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=95:5 до 80:20) с получением 7-[(2-метокси-4-пропилбензил)окси]-2H-хромен-3-карбальдегида (1,21 г) в виде желтого порошка.
Таким же образом, как и в Примере получения 27, получали соединения Примера получения 27-1 - Примера получения 27-6, приведенные в описанных далее таблицах.
Пример получения 28
7-Гидрокси-2H-хромен-3-карбальдегид (200 мг) растворяли в ДМФА (5 мл) и к этой смеси добавляли K2CO3 (235 мг) и 1-(бромметил)-2,4-бис(трифторметил)бензол (0,234 мл) с последующим перемешиванием при 80°C в течение 30 минут. Реакционную жидкость выливали в воду и получившийся порошок собирали при помощи фильтрации и сушили при пониженном давлении с получением 7-{[2,4-бис(трифторметил)бензил]окси}-2H-хромен-3-карбальдегида (455 мг) в виде бледно-желтого порошка.
Таким же образом, как и в Примере получения 28, получали соединения Примера получения 28-1 - Примера получения 28-27, приведенные в описанных далее таблицах.
Пример получения 29
К раствору 3-формил-2H-хромен-7-илтрифторметансульфоната (520 мг) в ДМФА (10,4 мл) при комнатной температуре добавляли 1-этинил-4-(трифторметил)бензол (330 мкл), дихлорид бис(трифенилфосфин)палладия(II) (355 мг) и йодид меди(I) (161 мг) и TEA (470 мкл). Реакционную смесь перемешивали при 100°C в течение 5 часов. К реакционной смеси добавляли воду при охлаждении льдом, нерастворенные вещества отделяли при помощи фильтрации и фильтрат экстрагировали при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc) с получением 7-{[4-(трифторметил)фенил]этинил}-2H-хромен-3-карбальдегида (189 мг).
Таким же образом, как и в Примере получения 29, получали соединения с Примера получения 29-1 до Примера получения 29-15, приведенные в описанных далее таблицах.
Пример получения 30
К раствору Pd(PPh3)4 (542 мг) и TEA (4 мл) в ДМФА (16 мл) при комнатной температуре добавляли 1-бром-4-изобутилбензол (1 г) и этинил(триметил)силан (553 мг) с последующим перемешиванием при 60°C в течение 4 часов. К реакционной жидкости добавляли 1 M раствор хлористоводородной кислоты с последующей экстракцией при помощи эфира. Нерастворенные вещества фильтровали через целит. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан) с получением [(4-изобутилфенил)этинил](триметил)силана (459 мг) в виде желтой жидкости.
Таким же образом, как и в Примере получения 30, получали соединение Примера получения 30-1, представленное в описанных далее таблицах.
Пример получения 31
К раствору хлорида меди (10 мг) и Pd(PPh3)4 (60 мг) в ДМФА (2 мл) при комнатной температуре добавляли [(4-изобутилфенил)этинил](триметил)силан (288 мг) и 5-фтор-3-формил-2H-хромен-7-илтрифторметансульфонат (340 мг) с последующим перемешиванием при 80°C в течение 12 часов. Реакционную жидкость концентрировали и остаток очищали при помощи колоночной хроматографии на силикагеле (CHCl3) с получением 5-фтор-7-[(4-изобутилфенил)этинил]-2H-хромен-3-карбальдегида (83 мг) в виде желтого твердого вещества.
Таким же образом, как и в Примере получения 31, получали соединение Примера получения 31-1, представленное в описанных далее таблицах.
Пример получения 32
К раствору 1-[4-фенил-5-(трифторметил)-2-тиенил]этанона (1,0 г) в ТГФ (20 мл) при -78°C добавляли по каплям DIBAL (0,99 M раствор в толуоле, 9,34 мл). Реакционную смесь нагревали до 25°C и перемешивали в течение 3 часов и к этой смеси добавляли насыщенный водный раствор сегнетовой соли (50 мл) с последующей экстракцией при помощи EtOAc (50 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=98:2 до 90:10) с получением 1-[4-фенил-5-(трифторметил)-2-тиенил]этанола (0,99 г) в виде бесцветной жидкости.
Пример получения 33
К раствору 3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]бензойной кислоты (1,085 г) в ТГФ (43 мл) при 0°C добавляли по каплям раствор BH3·ТГФ в ТГФ (1 M, 14 мл). Реакционную смесь нагревали до комнатной температуры и затем перемешивали в течение 15 часов. К реакционной жидкости при 0°C добавляли 1 M раствор хлористоводородной кислоты, чтобы остановить реакцию, с последующим перемешиванием в течение 30 минут и экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением {3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}метанола (570 мг) в виде белого маслянистого вещества.
Таким же образом, как и в Примере получения 33, получали соединения Примера получения 33-1 - Примера получения 33-21, приведенные в описанных далее таблицах.
Пример получения 34
К раствору метил 4-пиперидин-1-ил-2-(трифторметил)бензоата (955 мг) в ТГФ (19 мл) при охлаждении льдом добавляли по каплям раствор DIBAL в гексане (1 M, 10,0 мл) с последующим перемешиванием при той же температуре в течение 2 часов. К реакционной жидкости добавляли по каплям MeOH и затем к этой смеси добавляли насыщенный водный раствор сегнетовой соли с последующим перемешиванием при комнатной температуре в течение 1 часа. Смесь экстрагировали при помощи EtOAc и органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc) с получением[4-пиперидин-1-ил-2-(трифторметил)фенил]метанола (846 мг).
Таким же образом, как и в Примере получения 34, получали соединения Примера получения 34-1 - Примера получения 34-30, приведенные в описанных далее таблицах.
Пример получения 35
Метил 2-гидрокси-4-[(2-метокси-4-пропилфенокси)метил]бензоат (300 мг) растворяли в ТГФ (15 мл). К реакционной жидкости при 0°C добавляли LAH (103,4 мг) с последующим нагреванием от 0°C до 25°C и перемешиванием в течение 3 часов. К реакционной жидкости добавляли насыщенный водный раствор сегнетовой соли (30 мл) с последующей экстракцией при помощи EtOAc (30 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 80:20) с получением 2-(гидроксиметил)-5-[(2-метокси-4-пропилфенокси)метил]фенола (245,2 мг) в виде белого твердого вещества.
Таким же образом, как и в Примере получения 35, получали соединения Примера получения 35-1 - Примера получения 35-3, приведенные в описанных далее таблицах.
Пример получения 36
К раствору NaBH4 (93,1 мг) в EtOH (15 мл) добавляли по каплям раствор 7-{[2,4-бис(трифторметил)бензил]окси}-2,3-дигидро-4H-тиохромен-4-она (1,0 г) в EtOH (5 мл) при 0°C. Реакционную смесь нагревали до 25°C с последующим перемешиванием в течение 3 часов. Реакционную жидкость концентрировали при пониженном давлении и к остатку при 0°C добавляли ДХМ (20 мл) и затем насыщенный водный раствор NH4Cl (30 мл) с последующим перемешиванием в течение 1 часа и экстракцией при помощи ДХМ три раза (30 мл ×3). Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 60:40) с получением 7-{[2,4-бис(трифторметил)бензил]окси}тиохроман-4-ола (845 мг) в виде белого твердого вещества.
Таким же образом, как и в Примере получения 36, получали соединения Примера получения 36-1 - Примера получения 36-2, приведенные в описанных далее таблицах.
Пример получения 37
При нормальном давлении в атмосфере газообразного водорода к раствору метил 4-фенил-5-винилтиофен-2-карбоксилата (250 мг) в EtOH (5 мл) при 25°C добавляли Pd/C (50% влажность) (50 мг) с последующим перемешиванием в течение 5 часов. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением метил 5-этил-4-фенилтиофен-2-карбоксилата (247,3 мг) в виде бесцветной жидкости.
Таким же образом, как и в Примере получения 37, получали соединения Примера получения 37-1 - Примера получения 37-3, приведенные в описанных далее таблицах.
Пример получения 38
К раствору 5-фтор-7-гидрокси-2H-хромен-3-карбальдегида (275 мг) и 2-(гидроксиметил)-5-метил-4-фенил-тиазола (436 мг) в толуоле (8,2 мл) при охлаждении льдом добавляли ADDP (393 мг) и TBP (315 мг). Реакционную жидкость перемешивали при комнатной температуре в течение 15 часов, затем к этой смеси добавляли IPE и твердое вещество удаляли при помощи фильтрации. Фильтрат концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=90:20 до 70:30) с получением 5-фтор-7-[(5-5-метил-4-фенил-1,3-тиазол-2-ил)метокси]-2H-2H-хромен-3-карбальдегида (381 мг) в виде бледно-желтого твердого вещества.
Таким же образом, как и в Примере получения 38, получали соединения Примера получения 38-1 - Примера получения 38-61, приведенные в описанных далее таблицах.
Пример получения 39
К раствору 5-({[2'-фтор-2-(трифторметил)бифенил-4-ил]окси}метил)-2-(гидроксиметил)фенола (520 мг) в хлороформе (10 мл) при комнатной температуре добавляли диоксид марганца (1 г). Реакционную жидкость перемешивали при комнатной температуре в течение 16 часов и затем фильтровали через целит. Фильтрат концентрировали при пониженном давлении и очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=95:5 до 80:20) с получением 4-({[2'-фтор-2-(трифторметил)бифенил-4-ил]окси}метил)-2-гидроксибензальдегида (180 мг) в виде белого твердого вещества.
Таким же образом, как и в Примере получения 39, получали соединения Примера получения 39-1 - Примера получения 39-6, приведенные в описанных далее таблицах.
Пример получения 40
К раствору [2-({[2-(трифторметил)бифенил-4-ил]окси}метил)-4,5-дигидро-1-бензотиен-6-ил]метанола (1,0 г) в ДХМ (20 мл) при 25°C добавляли PDC (1,36 г) и MS4 Ангстрем (1,36 г). Реакционную жидкость перемешивали в течение 3 часов и затем фильтровали через целит. Фильтрат концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 80:20) с получением 2-({[2-(трифторметил)бифенил-4-ил]окси}метил)-4,5-дигидро-1-бензотиофен-6-карбальдегида (345 мг) в виде бесцветной жидкости.
Пример получения 41
К раствору [1-(трет-бутоксикарбонил)пиперидин-4-ил]уксусной кислоты (200 мг) в диоксане (1 мл) добавляли 4 M раствор хлорида водорода в диоксане (1 мл). Реакционную жидкость перемешивали при комнатной температуре в течение 15 часов и затем концентрировали при пониженном давлении с получением гидрохлорида пиперидин-4-илуксусной кислоты (140 мг) в виде белого твердого вещества.
Таким же образом, как и в Примере получения 41, получали соединения Примера получения 41-1 - Примера получения 41-5, приведенные в описанных далее таблицах.
Пример получения 42
Бензил 3-(1H-тетразол-5-ил)пирролидин-1-карбоксилат (300 мг) добавляли к смешанному раствору концентрированной хлористоводородной кислоты (3 мл) и AcOH (0,6 мл) с последующим перемешиванием при 100°C в течение 5 часов. Реакционную жидкость концентрировали и затем подвергали азеотропной перегонке с толуолом три раза (30 мл ×3). Остаток растворяли в смешанном растворе ацетона (0,9 мл) и воды (1,5 мл) и затем охлаждали до 0°C и к этой смеси добавляли Na2CO3 (174,5 мг) и DIBOC (359,4 мг). Реакционную смесь нагревали до 25°C и затем перемешивали в течение 15 часов. Реакционную жидкость концентрировали. К остатку добавляли диэтиловый эфир (30 мл) для экстракции (три раза). Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением трет-бутил 3-(1H-тетразол-5-ил)пирролидин-1-карбоксилата (102,7 мг) в виде бесцветной жидкости.
Пример получения 43
К 1-трет-бутил 3-метил 3-фторпирролидин-1,3-дикарбоксилату (100 мг) при 0°C добавляли смешанный раствор концентрированной хлористоводородной кислоты (1 мл) и AcOH (0,2 мл). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 1 часа и затем перемешивали при 100°C в течение 5 часов. После подтверждения того, что исходные материалы исчезли, полученную смесь концентрировали при пониженном давлении и затем подвергали азеотропной перегонке с толуолом три раза. Остаток растворяли в смешанном жидком растворе ацетона (0,6 мл) и воды (1 мл) и к этой смеси добавляли при 0°C Na2CO3 (64 мг) и DIBOC (132 мг) с последующим нагреванием до комнатной температуры и перемешиванием в течение 15 часов. Реакционную смесь концентрировали при пониженном давлении и ацетон выпаривали. К остатку добавляли диэтиловый эфир для сепарации жидкости. Водный слой объединяли, охлаждали до 0°C и доводили до pH=2-3 при помощи 2 M раствора хлористоводородной кислоты. К этой смеси добавляли EtOAc для экстракции. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением 1-(трет-бутоксикарбонил)-3-фторпирролидин-3-карбоновой кислоты (70 мг) в виде белого твердого вещества.
Пример получения 44
5-Фтор-7-(метоксиметокси)-2H-хромен-3-карбальдегид (1,5 г) растворяли в ацетоне (25 мл) и к этой смеси добавляли 1 M раствор HCl (20 мл) с последующим нагреванием и кипячением с обратным холодильником в течение 5 часов. Реакционную жидкость концентрировали и остаток растворяли в EtOAc, промывали водой и насыщенным солевым раствором и сушили над MgSO4 и фильтрат концентрировали. Остаток промывали хлороформом с получением 5-фтор-7-гидрокси-2H-хромен-3-карбальдегида (0,95 г) в виде желтого порошка.
Таким же образом, как и в Примере получения 44, получали соединения Примера получения 44-1 - Примера получения 44-6, приведенные в описанных далее таблицах.
Пример получения 45
7-(Метоксиметокси)-2,2-диметил-2H-хромен-3-карбальдегид (300 мг) растворяли в EtOH (10 мл) и к этой смеси добавляли (1S)-(+)-10-камфорсульфоновую кислоту (421 мг) с последующим перемешиванием при 80°C в течение ночи. К реакционной жидкости добавляли силикагель с последующим концентрированием. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=90:10 до 70:30) с получением 7-гидрокси-2,2-диметил-2H-хромен-3-карбальдегида (175 мг) в виде красного порошка.
Пример получения 46
К раствору KOH (358 мг) в MeOH (30 мл) добавляли {4-[2,4-бис(трифторметил)фенил]бут-1-ин-1-ил}(триметил)силан (1,8 г) с последующим перемешиванием при комнатной температуре в течение 18 часов. Реакционную жидкость нейтрализовали при помощи 1 M раствора хлористоводородной кислоты и экстрагировали эфиром. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=100:0) с получением 1-бут-3-ин-1-ил-2,4-бис(трифторметил)бензола (426 мг) в виде бесцветной жидкости.
Пример получения 47
К 7-(бензилокси)-4-метил-2H-хромен-3-карбальдегиду (230 мг) и 1,2,3,4,5-пентаметилбензолу (608 мг) добавляли TFA (3 мл) с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную жидкость выливали в водный раствор NaHCO3 с последующей экстракцией при помощи EtOAc три раза. Органический слой объединяли, промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=80:20 до 20:80) с получением 7-гидрокси-4-метил-2H-хромен-3-карбальдегида (130 мг) в виде бледно-желтого порошка.
Таким же образом, как и в Примере получения 47, получали соединения Примера получения 47-1 - Примера получения 47-2, приведенные в описанных далее таблицах.
Пример получения 48
2-Фтор-4,6-диметоксибензальдегид (22 г) растворяли в ДХМ (110 мл) и к этой смеси при охлаждении льдом добавляли по каплям раствор BBr3 в ДХМ (1 M, 300 мл) с последующим перемешиванием при комнатной температуре в течение ночи. После подтверждения завершения реакции реакционную жидкость выливали в смесь льда с водой (100 мл) с последующим перемешиванием в течение 1 часа и затем экстракцией при помощи EtOAc три раза. Органический слой объединяли, промывали водой и насыщенным солевым раствором, в указанном порядке, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=80:20 до 60:40) с получением 2-фтор-4,6-дигидроксибензальдегида (12 г) в виде белого порошка.
Пример получения 49
К раствору 7-{[2,4-бис(трифторметил)бензил]окси}тиохроман-4-ола (800 мг) в толуоле (16 мл) добавляли 4-метилбензолсульфоновую кислоту (33,7 мг) с последующим перемешиванием при 120°C в течение 3 часов. К реакционной жидкости добавляли насыщенный водный раствор NaHCO3 (50 мл) с последующей экстракцией при помощи EtOAc (50 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=100:0 до 80:20) с получением 2,4-бис(трифторметил)бензил 2H-тиохроман-7-илового эфира (753,2 мг) в виде бесцветной жидкости.
Пример получения 50
Смесь метил 4-(бромметил)-2-{[трет-бутил(диметил)силил]окси}бензоата (0,30 г) и триэтилфосфита (0,17 г) перемешивали при 25°C и затем перемешивали при 130°C в течение 24 часов. Реакционную жидкость концентрировали при пониженном давлении и дважды подвергали азеотропной перегонке с толуолом (30 мл ×2). Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=40:60 до 10:90) с получением метил 2-{[трет-бутил(диметил)силил]окси}-4-[(диэтоксифосфорил)метил]бензоата (0,23 г) в виде бесцветной жидкости.
Пример получения 51
К раствору 7-[(4-бром-5-этил-2-тиенил)метокси]-2H-хромен-3-карбальдегида (150 мг) в диоксане (4,5 мл) при 25°C добавляли [2-(трифторметил)фенил]борную кислоту и 2 M водный раствор Na2CO3. Затем к реакционной смеси добавляли ацетат палладия (4,44 мг) и PPh3 (20,75 мг) с последующим нагреванием до 100°C и перемешиванием в течение 5 часов. К реакционной жидкости добавляли насыщенный водный раствор NH4Cl (30 мл) с последующей экстракцией при помощи EtOAc (30 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=95:5 до 80:20) с получением 7-({5-этил-4-[2-(трифторметил)фенил]-2-тиенил}метокси)-2H-хромен-3-карбальдегида (134,8 мг) в виде бледно-желтой жидкости.
Таким же образом, как и в Примере получения 51, получали соединения Примера получения 51-1 - Примера получения 51-5, приведенные в описанных далее таблицах.
Пример получения 52
В атмосфере азота к раствору [3-хлор-4-(трифторметил)фенил]метанола (800 мг) и фенилборной кислоты (1,90 г) в толуоле (16 мл) при 25°C добавляли фосфат калия (1,61 г), ацетат палладия (42,6 мг) и дициклогексил(2',6'-диметоксибифенил-2-ил)фосфин (195,0 мг). Реакционную смесь нагревали до 100°C и затем перемешивали в течение 15 часов. К реакционной жидкости добавляли воду (30 мл) с последующей экстракцией при помощи EtOAc (30 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, смесь гексан:EtOAc=70:30 до 50:50) с получением [6-(трифторметил)бифенил-3-ил]метанола (678,3 мг) в виде желтого твердого вещества.
Таким же образом, как и в Примере получения 52, получали соединения Примера получения 52-1 - Примера получения 52-4, приведенные в описанных далее таблицах.
Пример получения 53
Раствор {3-хлор-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}метанола (284 мг) и SOCl2 (179 мкл) в ДХМ (7 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционную жидкость выливали в воду с последующей экстракцией при помощи хлороформа. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем концентрировали при пониженном давлении с получением 2-хлор-4-(хлорметил)-1-[(1S)-2,2,2-трифтор-1-метилэтокси]бензола (285 мг) в виде бесцветной жидкости.
Таким же образом, как и в Примере получения 53, получали соединения Примера получения 53-1 - Примера получения 53-4, приведенные в описанных далее таблицах.
Пример получения 54
К раствору метил 4-бром-3-бензоата (300 мг) в ТГФ (1 мл) добавляли бромид циклопентилцинка (9,6 мл) и палладий-три-трет-бутилфосфин (1:2) (123 мг). Реакционную жидкость перемешивали при комнатной температуре в течение 20 часов. К этой смеси при 0°C добавляли насыщенный водный раствор NH4Cl, с последующей фильтрацией через целит и экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и сушили над MgSO4. Растворитель выпаривали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=100:0 до 90:10) с получением метил 3-хлор-4-циклопентилбензоата (280 мг) в виде желтого твердого вещества.
Пример получения 55
К раствору (7-{[2,4-бис(трифторметил)бензил]окси}-5-фтор-2H-хромен-3-ил)метанола (200 мг) в MeCN (5 мл) добавляли трифенилфосфиндибромид (240 мг). Реакционную жидкость перемешивали при комнатной температуре в течение 2 часов и концентрировали при пониженном давлении. К остатку добавляли EtOAc и IPE, полученное твердое вещество удаляли при помощи фильтрации и фильтрат концентрировали с получением 7-{[2,4-бис(трифторметил)бензил]окси}-3-(бромметил)-5-фтор-2H-хромена (230 мг) в виде коричневой жидкости. 60% Гидрид натрия (24 мг) добавляли к раствору ДМФА (5 мл) при 0°C и далее к этой смеси добавляли этил 1H-пиразол-4-карбоксилат (76 мг). Смесь перемешивали при комнатной температуре в течение 0,5 часа и при 0°C к ней добавляли раствор 7-{[2,4-бис(трифторметил)бензил]окси}-3-(бромметил)-5-фтор-2H-хромена (220 мг) в ДМФА (5 мл). Реакционную жидкость перемешивали при комнатной температуре в течение 13 часов, гасили насыщенным раствором NH4Cl и затем экстрагировали при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=100:0 до 70:30) с получением этил 1-[(7-{[2,4-бис(трифторметил)бензил]окси}-5-фтор-2H-хромен-3-ил)метил]-1H-пиразол-4-карбоксилата (111 мг) в виде бледно-желтого твердого вещества.
Таким же образом, как и в Примере получения 55, получали соединение Примера получения 55-1, представленное в описанных далее таблицах.
Пример получения 56
Раствор 4-изопропокси-3-(трифторметил)бензонитрила (2,4 г) и 5 M раствор NaOH (50 мл) в EtOH (50 мл) нагревали и кипятили с обратным холодильником в течение 18 часов. Раствор охлаждали до комнатной температуры, подкисляли при помощи хлористоводородной кислоты и затем экстрагировали хлороформом. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь хлороформ:MeOH=100:0 до 95:5). Продукт промывали гексаном с получением 4-изопропокси-3-(трифторметил)бензойной кислоты (2,2 г) в виде белого твердого вещества.
Таким же образом, как и в Примере получения 56, получали соединения Примера получения 56-1 - Примера получения 56-6, приведенные в описанных далее таблицах.
Пример получения 57
5-Бром-3-(трифторметил)-2-{[(2S)-1,1,1-трифторпропан-2-ил]окси}пиридин (500 мг) растворяли в смешанном растворителе ДМСО (5 мл) и MeOH (5 мл). Затем к этой смеси при 25°C добавляли TEA (0,42 мл) и далее при 25°C в нее добавляли Pd(OAc)2 (17 мг) и DPPP (60 мг) с последующим перемешиванием при 70°C в течение 15 часов в атмосфере CO. К реакционному раствору добавляли воду (30 мл) с последующей экстракцией при помощи EtOAc (20 мл) три раза. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем фильтровали и фильтрат концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (устройство автоматической очистки, проявляющий раствор; смесь гексан:EtOAc=100:0 до 80:20) с получением метил 5-(трифторметил)-6-{[(2S)-1,1,1-трифторпропан-2-ил]окси}никотината (403 мг) в виде желтого твердого вещества.
Пример получения 58
К раствору этил (1-метил-1,2,3,6-тетрагидропиридин-4-ил)ацетата (2,05 г) в дихлорэтане (14 мл) при 0°C добавляли 1-хлорэтил-хлорформиат (1,5 мл). Реакционную жидкость нагревали и кипятили с обратным холодильником в течение 2,5 часов и затем концентрировали при пониженном давлении. Остаток растворяли в MeOH (14 мл) и нагревали и кипятили с обратным холодильником в течение 1 часа. После концентрирования при пониженном давлении остаток очищали при помощи колоночной хроматографии на аминосиликагеле (смесь хлороформ:метанол=100:0 до 80:20) с получением этил 1,2,3,6-тетрагидропиридин-4-илацетата (130 мг) в виде коричневой жидкости.
Таким же образом, как и в Примере получения 58, получали соединение Примера получения 58-1, представленное в описанных далее таблицах.
Пример получения 59
Метил 5,6-дихлорникотинат (1,5 г) и 60% раствор гидрида натрия (640 мг) растворяли в ТГФ (45 мл). К этой смеси добавляли при 0°C 1,3-дифторпропан-2-ол (1,5 г) с последующим перемешиванием при 0°C в течение 3 часов и реакционный раствор гасили водным раствором NH4Cl. После эстракции при помощи EtOAc органический слой сушили над MgSO4 и затем фильтровали и осушитель удаляли. Растворитель выпаривали при пониженном давлении с последующей очисткой при помощи колоночной хроматографии на силикагеле (смесь гексан:AcOEt=100:0 до 50:50) с получением метил 5-хлор-6-[(1,3-дифторпропан-2-ил)окси]никотината (1,56 г) в виде бесцветной жидкости.
К раствору метил 5-хлор-6-[(1,3-дифторпропан-2-ил)окси]никотината (1 г) в ТГФ (20 мл) при 0°C добавляли по каплям 0,99 M раствор DIBAL в толуоле (11,3 мл) с последующим перемешиванием при 0°C в течение 2 часов. Затем реакционный раствор выливали в водный раствор сегнетовой соли с последующим перемешиванием при комнатной температуре в течение 1 часа. После экстракции с системой EtOAc-вода органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и затем фильтровали и осушитель удаляли. Растворитель выпаривали при пониженном давлении с последующей очисткой при помощи колоночной хроматографии на силикагеле (смесь Hex:AcOEt=98:2 до 70:30) с получением {5-хлор-6-[(1,3-дифторпропан-2-ил)окси]пиридин-3-ил}метанола (650 мг) в виде бесцветной жидкости.
Пример получения 60
К 5,6-дихлорникотиновой кислоте (2,2 г) добавляли 1,1,1-триметоксиэтан (4,3 мл) с последующим облучением в микроволновой печи при 120°C в течение 15 минут. Реакционную смесь растворяли в EtOAc и промывали водой. Органический слой сушили над MgSO4 и растворитель выпаривали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=85:15 до 80:20) с получением метил 5,6-дихлорникотината (2,2 г) в виде белого твердого вещества.
Пример получения 61
К раствору этил 4-пиридилацетата (2 г) в MeCN (20 мл) добавляли метилйодид (2,3 мл). Реакционную жидкость перемешивали при комнатной температуре в течение ночи и затем концентрировали при пониженном давлении. К остатку добавляли IPE и полученное твердое вещество собирали при помощи фильтрации. Твердое вещество растворяли в MeOH и туда добавляли боргидрид натрия (916 мг) при 15°C или ниже.
Реакционную жидкость перемешивали при комнатной температуре в течение 6 часов и затем к этой смеси добавляли воду с последующей экстракцией при помощи хлороформа. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии (смесь хлороформ:MeOH=100:0 до 90:10) с получением этил (1-метил-1,2,3,6-тетрагидропиридин-4-ил)ацетата (2,08 г) в виде бледно-желтой жидкости.
Пример получения 62
К раствору {3-(трифторметил)-4-[(1S)-2,2,2-трифтор-1-метилэтокси]фенил}метанола (200 мг) в дихлорэтане (5 мл) добавляли тионилхлорид (111 мкл) и каталитическое количество ДМФА с последующим перемешиванием при 60°C в течение 2 часов. Реакционную жидкость концентрировали при пониженном давлении и затем к остатку добавляли раствор этил (3R)-1-[(7-гидрокси-2H-хромен-3-ил)метил]пиперидин-3-карбоксилата (175 мг) в ДМФА (8,75 мл) и карбонат калия (152 мг), в указанном порядке, с последующим перемешиванием при 80°C в течение 2 часов. Реакционную жидкость охлаждали до комнатной температуры и выливали в воду с последующей экстракцией при помощи EtOAc. Органический слой промывали водой и насыщенным солевым раствором, в указанном порядке, и затем сушили над безводным сульфатом натрия и растворитель выпаривали. Остаток очищали при помощи колоночной хроматографии на силикагеле с получением этил (3R)-1-[(7-{[3-(трифторметил)-4-{[(2S)-1,1,1-трифторпропан-2-ил]окси}бензил]окси}-2H-хромен-3-ил)метил]пиперидин-3-карбоксилата (271 мг) в виде желтого маслянистого вещества.
Таким же образом, как и в Примере получения 62, получали соединения Примера получения 62-1 - Примера получения 62-19, приведенные в описанных далее таблицах.
Пример получения 63
К раствору 4-хлор-3-(трифторметил)бензонитрила (1,5 г), ацетилацетоната железа(III) (130 мг) и 1-метилпирролидин-2-она (4 мл) в ТГФ (45 мл) при 5°C добавляли 1 M раствор циклопентилмагнийбромида в ТГФ (8,8 мл) с последующим перемешиванием при комнатной температуре в течение 0,5 часа и разбавлением диэтиловым эфиром. К этой смеси медленно добавляли 1 M раствор хлористоводородной кислоты с последующей экстракцией при помощи EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=100:0 до 95:5) с получением 4-циклопентил-3-(трифторметил)бензонитрила (367 мг) в виде белого твердого вещества.
Пример получения 64
К раствору 7-(метоксиметокси)-2H-хромен-3-карбальдегида (5,00 г) и этил (3R)-пиперидин-3-карбоксилата (4,20 мл) в дихлорэтане (150 мл) добавляли триацетоксиборгидрид натрия (12,0 г) с последующим перемешиванием при 80°C в течение 4 часов. Реакционную жидкость охлаждали до комнатной температуры и затем к этой смеси добавляли насыщенный водный раствор NaHCO3 с последующей экстракцией при помощи хлороформа. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель выпаривали. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь гексан:EtOAc=4:1) с получением этил (3R)-1-{[7-(метоксиметокси)-2H-хромен-3-ил]метил}пиперидин-3-карбоксилата (7,30 г) в виде желтого маслянистого вещества.
Таким же образом, как и в Примере получения 64, получали соединения Примера получения 64-1 - Примера получения 64-7, приведенные в описанных далее таблицах.
Для соединений Примеров получения структуры представлены в Таблицах 3-57, а физико-химические данные и способы получения представлены в Таблицах 99-107.
Пример 1
К раствору 1-[(7-{[2,4-бис(трифторметил)бензил]окси}-5-фтор-2H-хромен-3-ил)метил]пирролидин-3-карбоновой кислоты (98 мг) в ДМФА (2 мл) добавляли CDI (46 мг) с последующим перемешиванием при 70°C в течение 12 часов. К реакционной жидкости добавляли метансульфонамид (27 мг) и DBU (43 мг), в указанном порядке, с последующим перемешиванием в течение 12 часов. К реакционной жидкости добавляли AcOH с последующим концентрированием при пониженном давлении и остаток очищали при помощи обращенно-фазовой колоночной хроматографии (смесь H2O:MeCN=100:0 до 90:10) с получением желтого аморфного вещества (70 мг). Это желтое аморфное вещество растворяли в диоксане (1 мл) и к этой смеси добавляли 4 M раствор HCl/диоксан (1 мл) с последующим перемешиванием и затем концентрированием при пониженном давлении. Остаток промывали гексаном с получением 1-[(7-{[2,4-бис(трифторметил)бензил]окси}-5-фтор-2H-хромен-3-ил)метил]-N-(метилсульфонил)пирролидин-3-карбоксамидгидрохлорида (60 мг) в виде бледно-желтого твердого вещества.
Пример 2
К раствору гидрохлорида пирролидин-3-карбоновой кислоты в MeOH добавляли TEA с последующим перемешиванием при комнатной температуре в течение 10 минут. Реакционную смесь концентрировали при пониженном давлении и к этой смеси при комнатной температуре добавляли раствор 7-{[4-фенил-5-(трифторметил)-2-тиенил]метокси}-2H-хромен-3-карбальдегида в MeOH и AcOH. Реакционную смесь нагревали до 70°C, перемешивали в течение 2 часов и оставляли охлаждаться до 25°C и к ней при комнатной температуре добавляли NaBH3CN с последующим перемешиванием при 70°C в течение 5 часов. Реакционную жидкость очищали при помощи обращенно-фазовой колоночной хроматографии (смесь MeCN:H2O=20:80 до 50:50) и полученное белое твердое вещество промывали диизопропиовым эфиром с получением 1-[(7-{[4-фенил-5-(трифторметил)-2-тиенил]метокси}-2H-хромен-3-ил)метил]пирролидин-3-карбоновой кислоты в виде белого твердого вещества.
Пример 3
Гидрохлорид пирролидин-3-карбоновой кислоты (165 мг) растворяли в MeOH и к этой смеси добавляли TEA с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и затем к этой смеси при комнатной температуре добавляли раствор 7-{[2,4-бис(трифторметил)фенил]этинил}-5-фтор-2H-хромен-3-карбальдегида в MeOH (8 мл) и AcOH (0,5 мл) с последующим перемешиванием при 70°C в течение 0,5 часа. После того, как раствор оставляли охлаждаться до комнатной температуры, к реакционной смеси при комнатной температуре добавляли NaBH3CN (57 мг) с последующим перемешиванием при 50°C в течение 2 часов. После подтверждения завершения реакции при помощи показателей ЖХ реакционную жидкость очищали при помощи обращенно-фазовой колоночной хроматографии (смесь MeCN:H2O=20:80 до 50:50), полученное аморфное вещество (233 мг) растворяли в диоксане (1 мл) и к этой смеси добавляли 4 M раствор HCl/диоксан (1 мл). Реакционную жидкость концентрировали при пониженном давлении и остаток промывали при помощи MeCN с получением гидрохлорида 1-[(7-{[2,4-бис(трифторметил)фенил]этинил}-5-фтор-2H-хромен-3-ил)метил]пирролидин-3-карбоновой кислоты (185 мг) в виде бледно-желтого твердого вещества.
Пример 154
Раствор этил 1-[(7-{[2,4-бис(трифторметил)бензил]окси}-5-фтор-2H-хромен-3-ил)метил]-1H-пиразол-4-карбоксилата (100 мг) и 1 M водный раствор NaOH (0,55 мл) в смеси EtOH/ТГФ (3 мл/1 мл) перемешивали при 100°C в течение 2 часов, нейтрализовали при помощи 1 M раствора HCl и экстрагировали хлороформом. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь хлороформ:метанол=100:0 до 90:10) и полученное твердое вещество промывали при помощи IPE с получением 1-[(7-{[2,4-бис(трифторметил)бензил]окси}-5-фтор-2H-хромен-3-ил)метил]-1H-пиразол-4-карбоновой кислоты (71 мг) в виде белого твердого вещества.
Пример 156
К раствору этил (3R)-1-[(7-{[3-(трифторметил)-4-{[(2S)-1,1,1-трифторпропан-2-ил]окси}бензил]окси}-2H-хромен-3-ил)метил]пиперидин-3-карбоксилата (271 мг) в смеси EtOH (5,4 мл) - ТГФ (2,7 мл) добавляли 1 M водный раствор NaOH (923 мкл) с последующим перемешиванием при 50°C в течение 2 часов. Реакционную жидкость охлаждали до комнатной температуры, затем к этой смеси добавляли 1 M раствор хлористоводородной кислоты (923 мкл) и растворитель выпаривали. Остаток очищали при помощи обращенно-фазовой колоночной хроматографии (смесь H2O:MeCN=100:0 до 30:70) с получением желтого маслянистого вещества, которое растворяли в диоксане (3 мл), обрабатывали 4 N раствором HCl/диоксан (1 мл) и промывали при помощи IPE с получением гидрохлорида (3R)-1-[(7-{[3-(трифторметил)-4-{[(2S)-1,1,1-трифторпропан-2-ил]окси}бензил]окси}-2H-хромен-3-ил)метил]пиперидин-3-карбоновой кислоты (215 мг) в виде белого порошка.
Таким же образом, как в способах Примеров 1-3, 154 или 156, получали соединения Примеров, приведенные в описанных далее таблицах. Для соединений Примеров структуры представлены в Таблицах 58-98, а физико-химические данные и способы получения представлены в Таблицах 108-131.
Таблицы 3-131 приведены в графической части.
Промышленная применимость
Соединение по настоящему изобретению обладает действием агониста S1P1, и его можно использовать для профилактики или лечения заболеваний, вызванных нежелательной инфильтрацией лимфоцитов, например, аутоиммунных заболеваний или воспалительных заболеваний, таких как реакции отторжения трансплантата или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей, ревматоидный артрит, рассеянный склероз, системная красная волчанка, нефротический синдром, менингоэнцефалит, тяжелая миастения, панкреатит, гепатит, нефрит, диабет, легочные заболевания, бронхиальная астма, атопический дерматит, воспалительные заболевания кишечника, атеросклероз, ишемическое реперфузионное расстройство, и заболеваний, вызванных аномальной пролиферацией или аккумуляцией клеток, например, рак, лейкемия и подобные.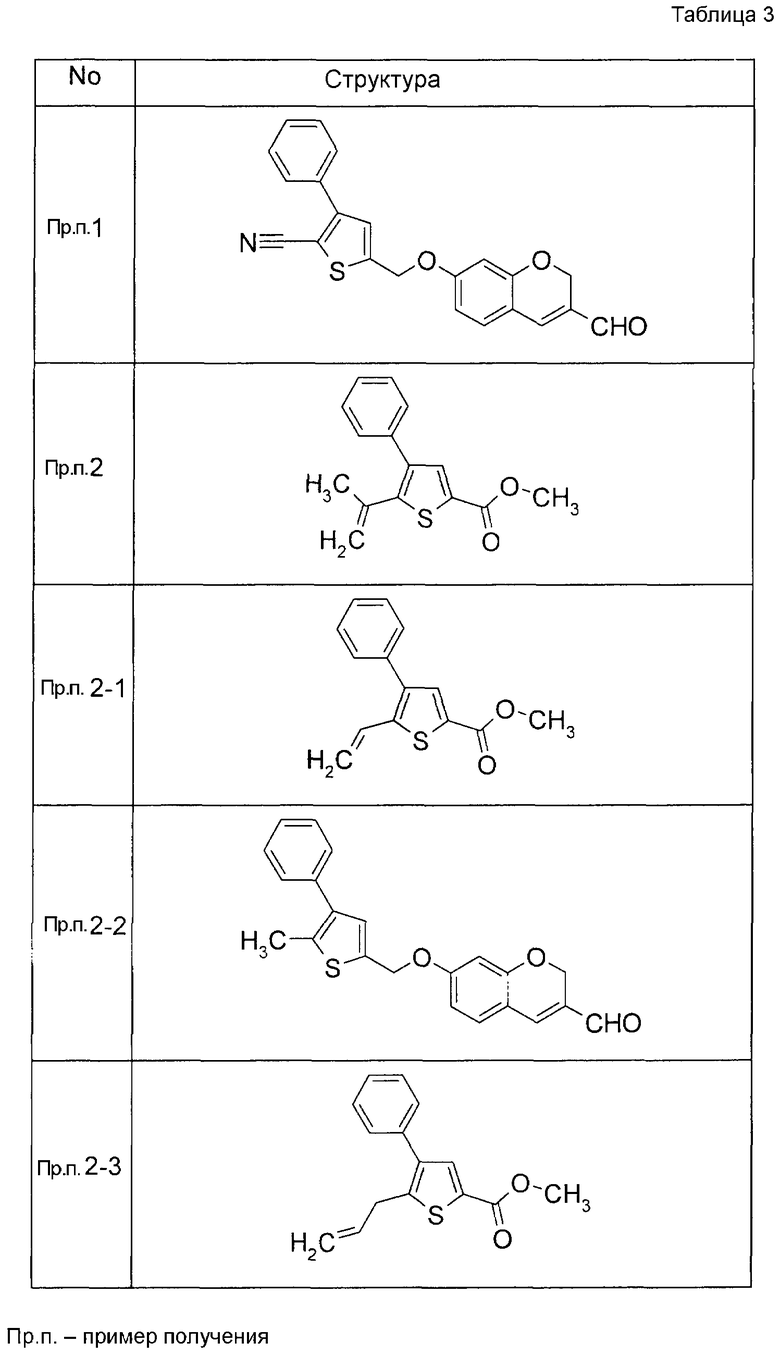
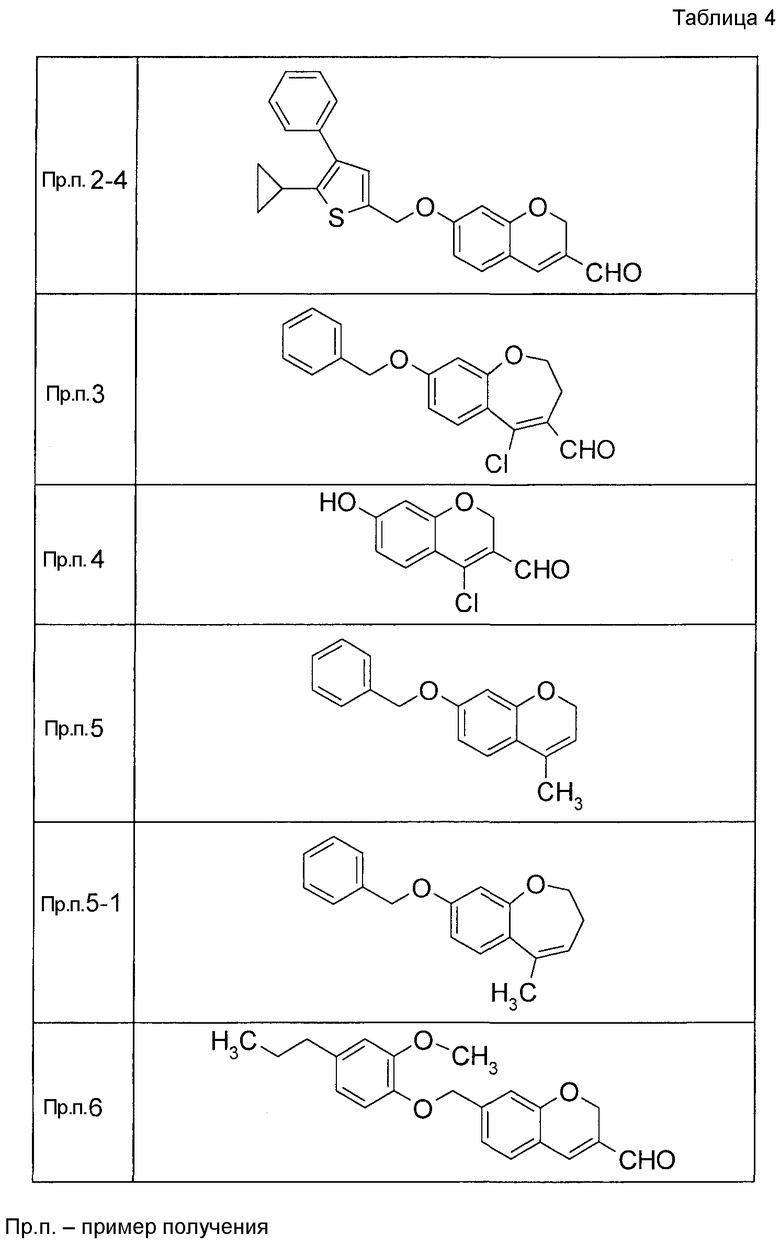
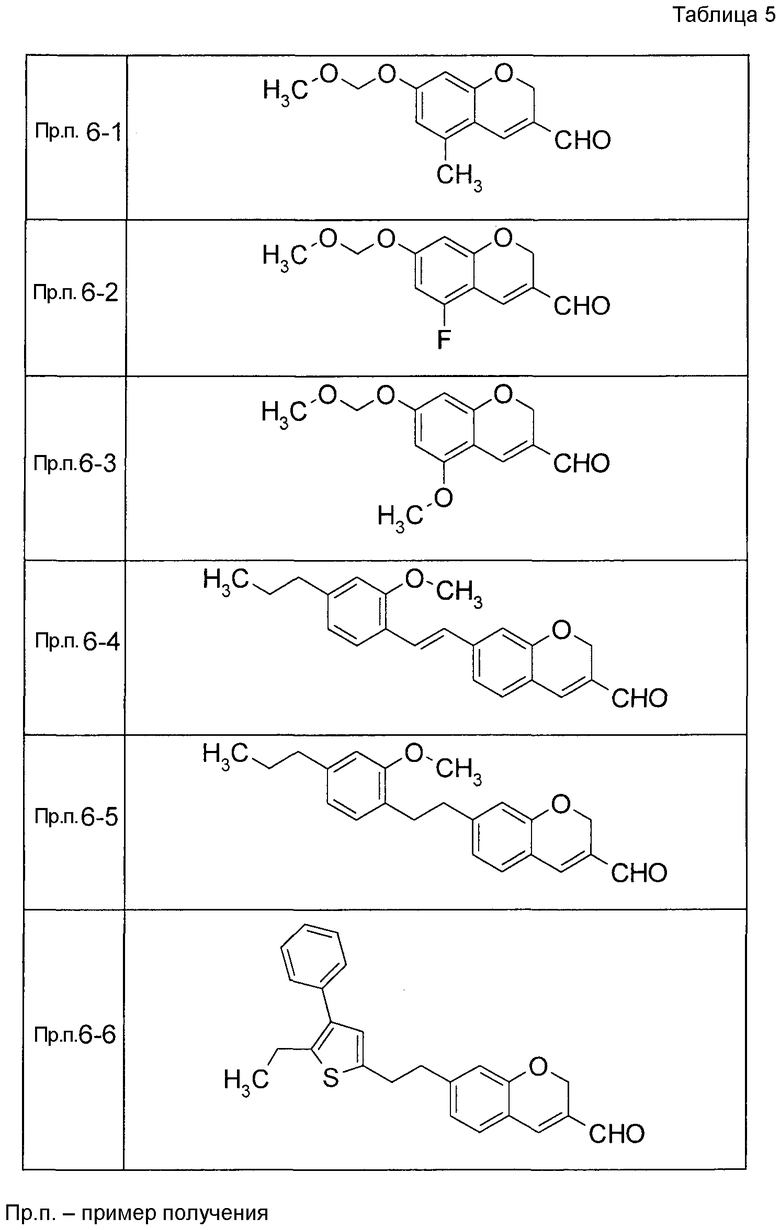
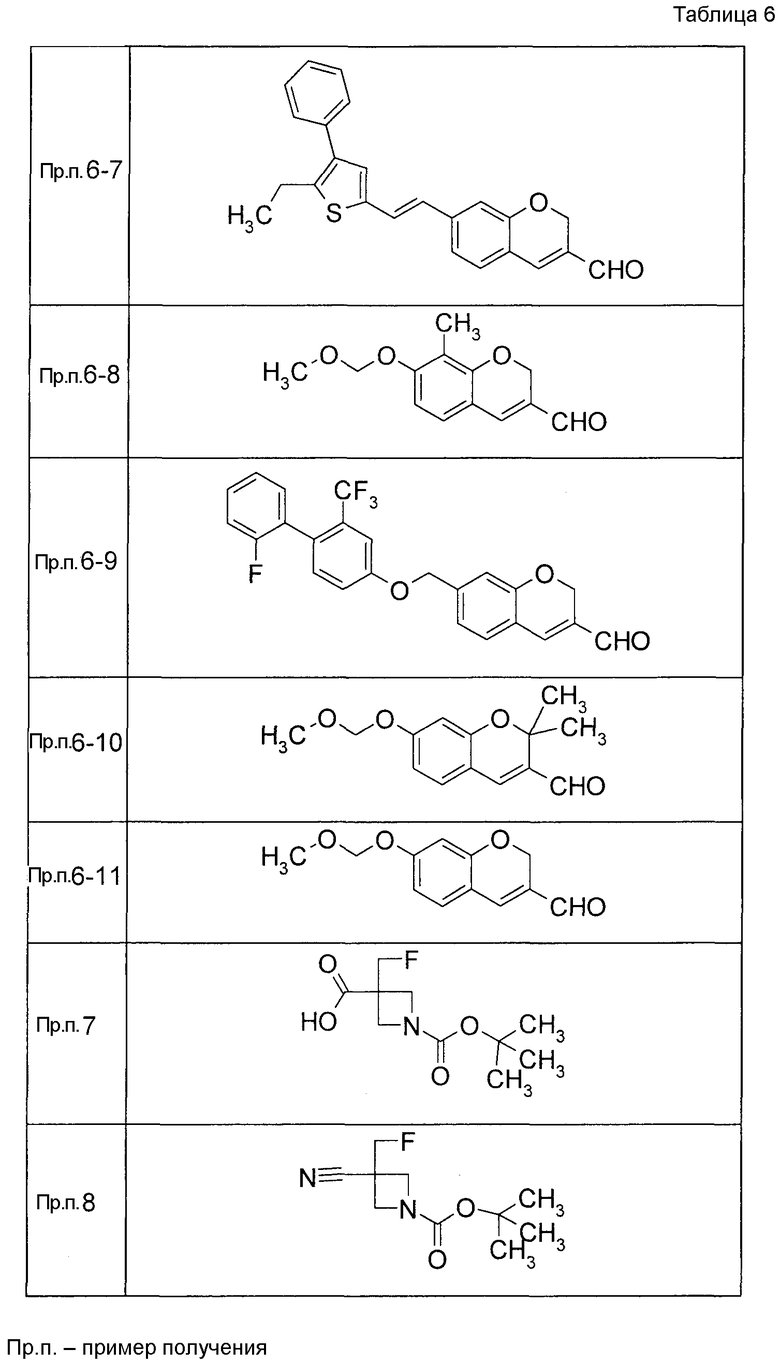
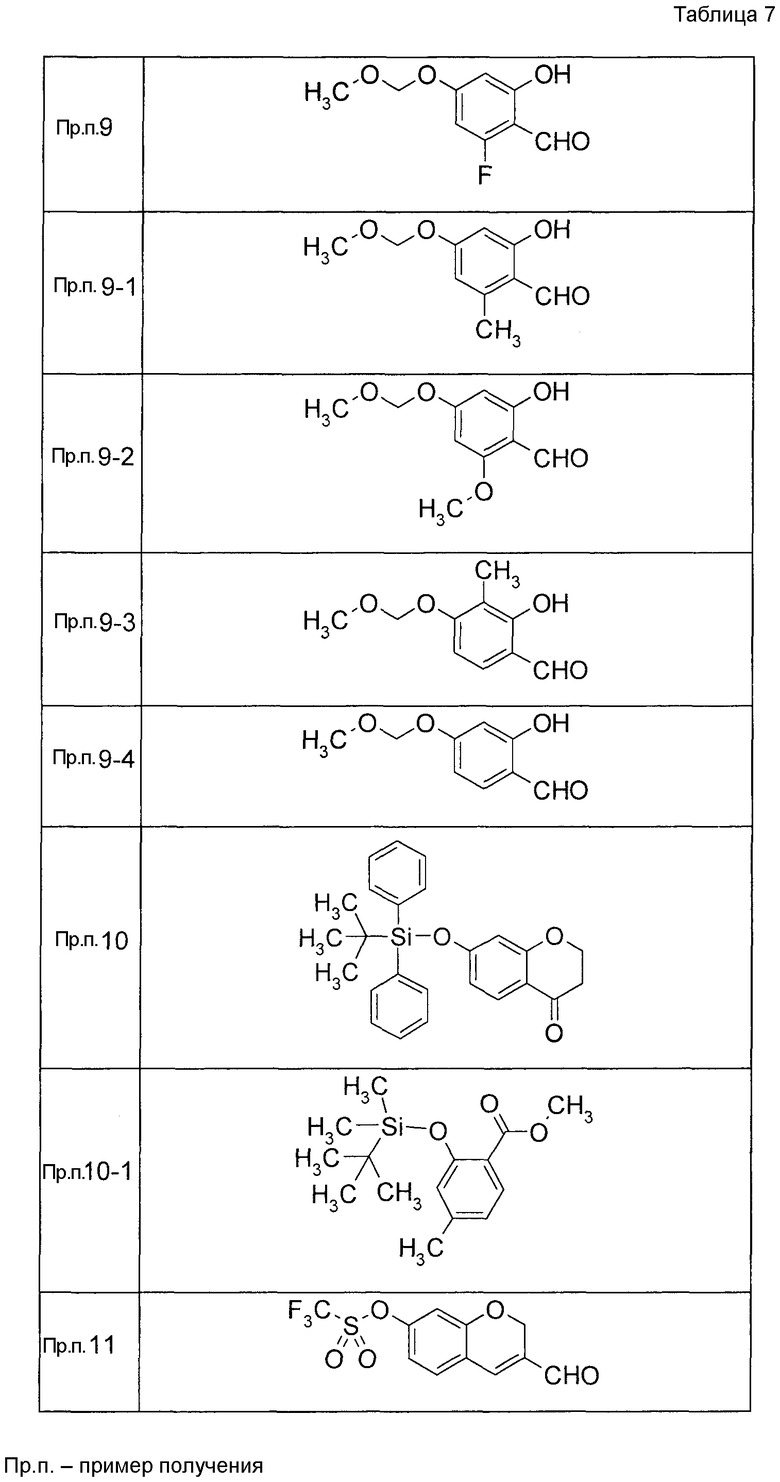
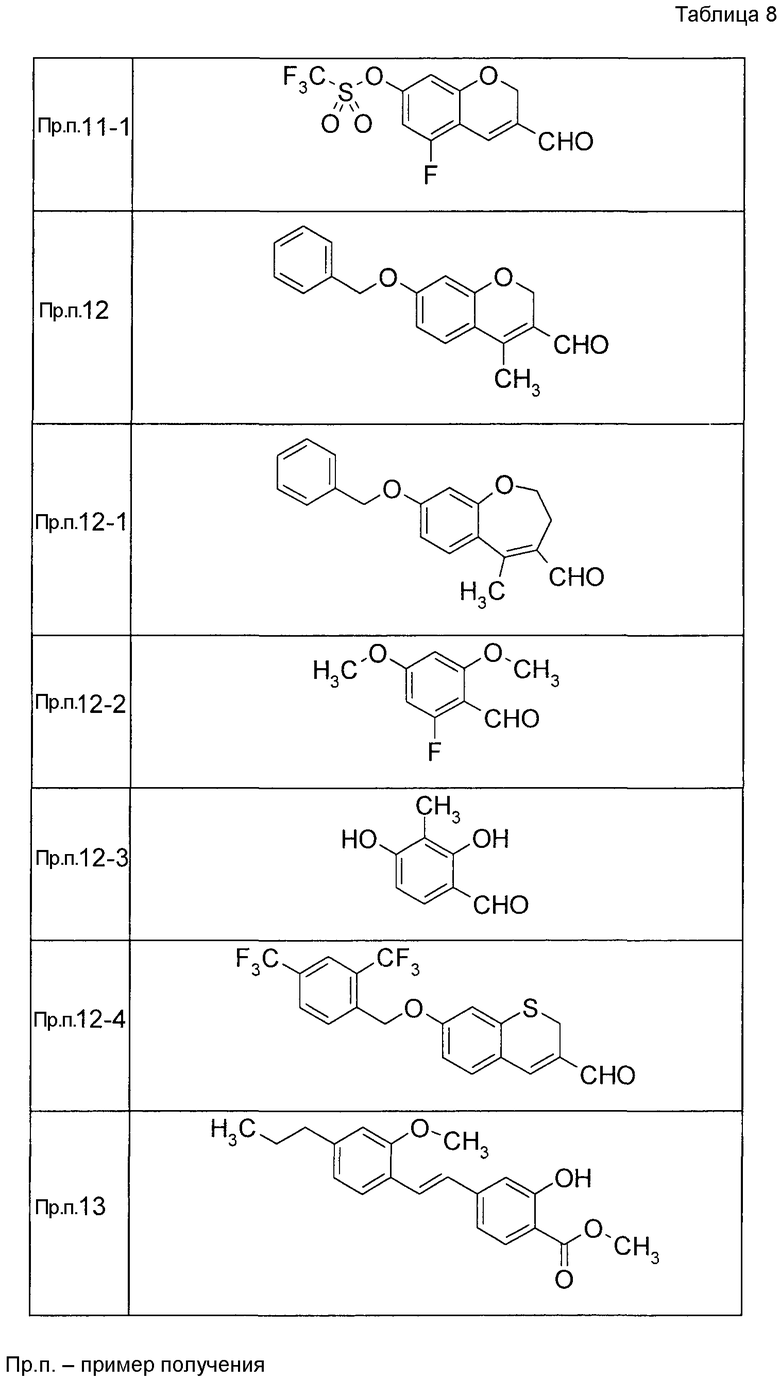
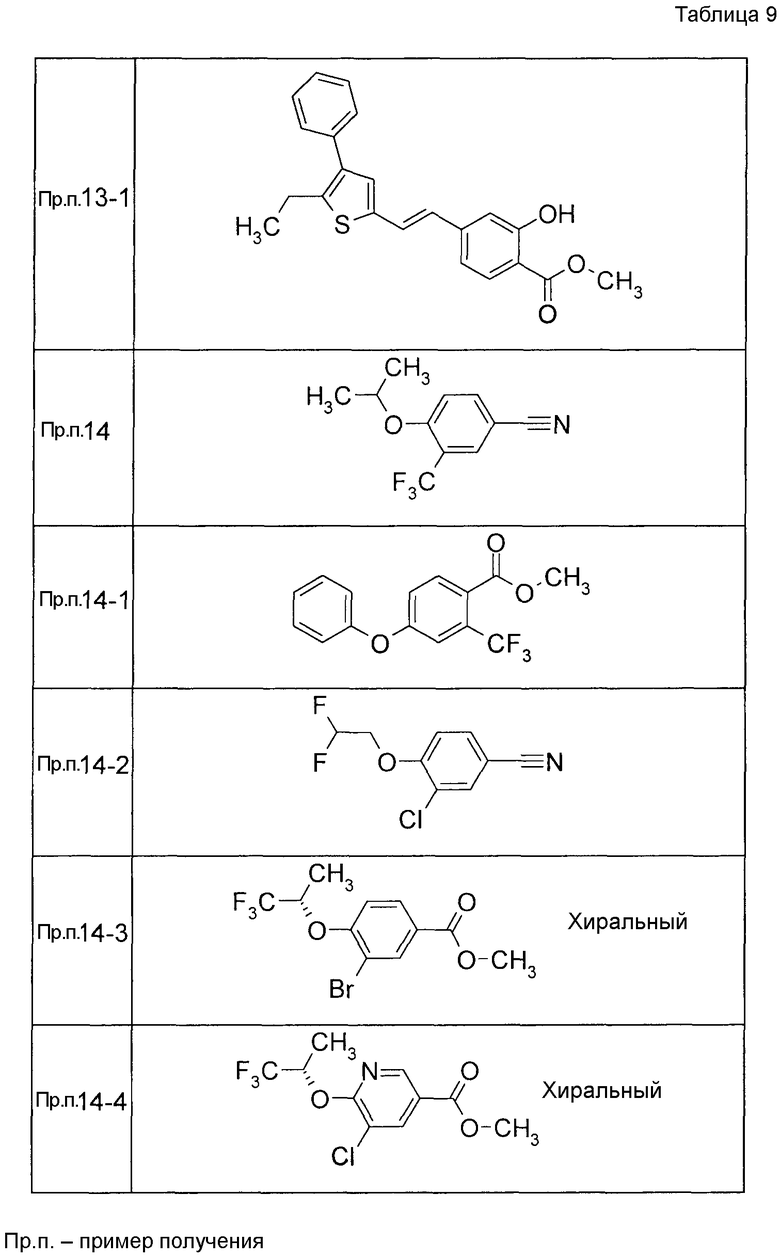
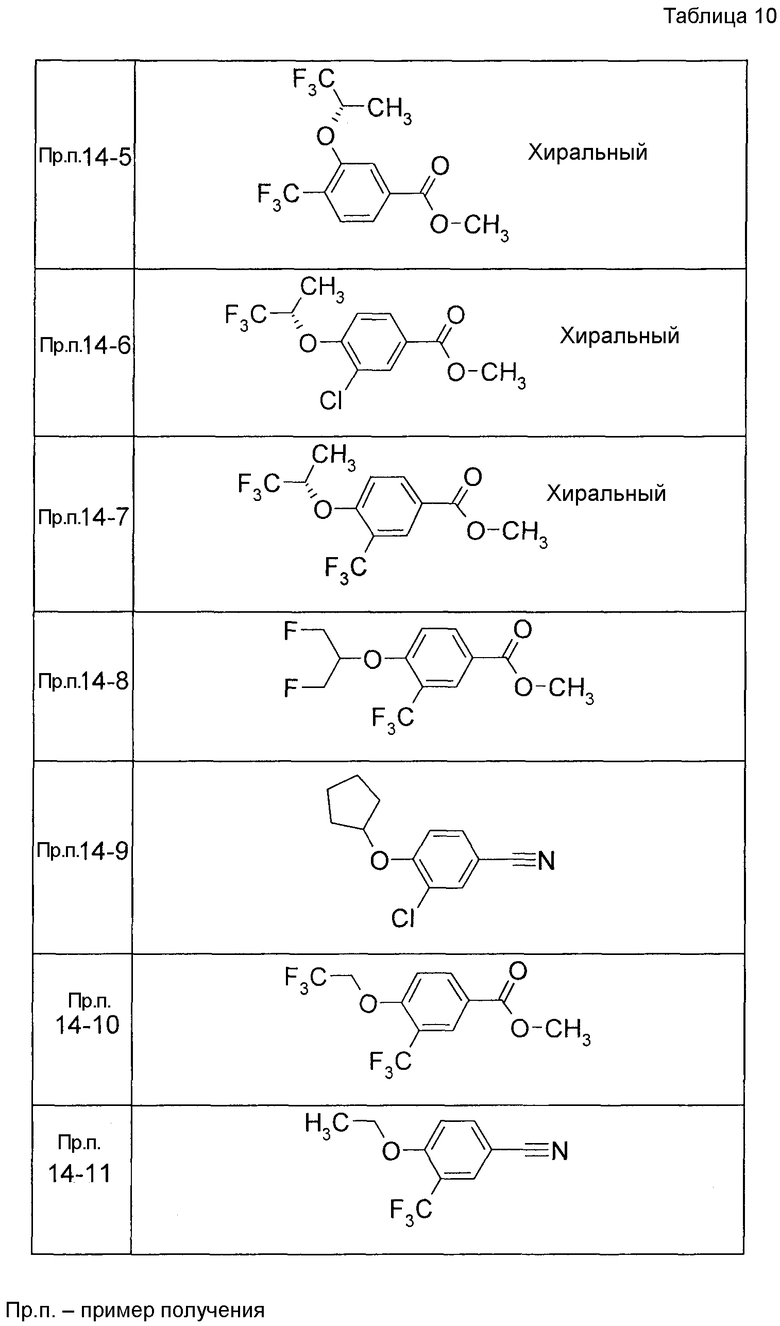
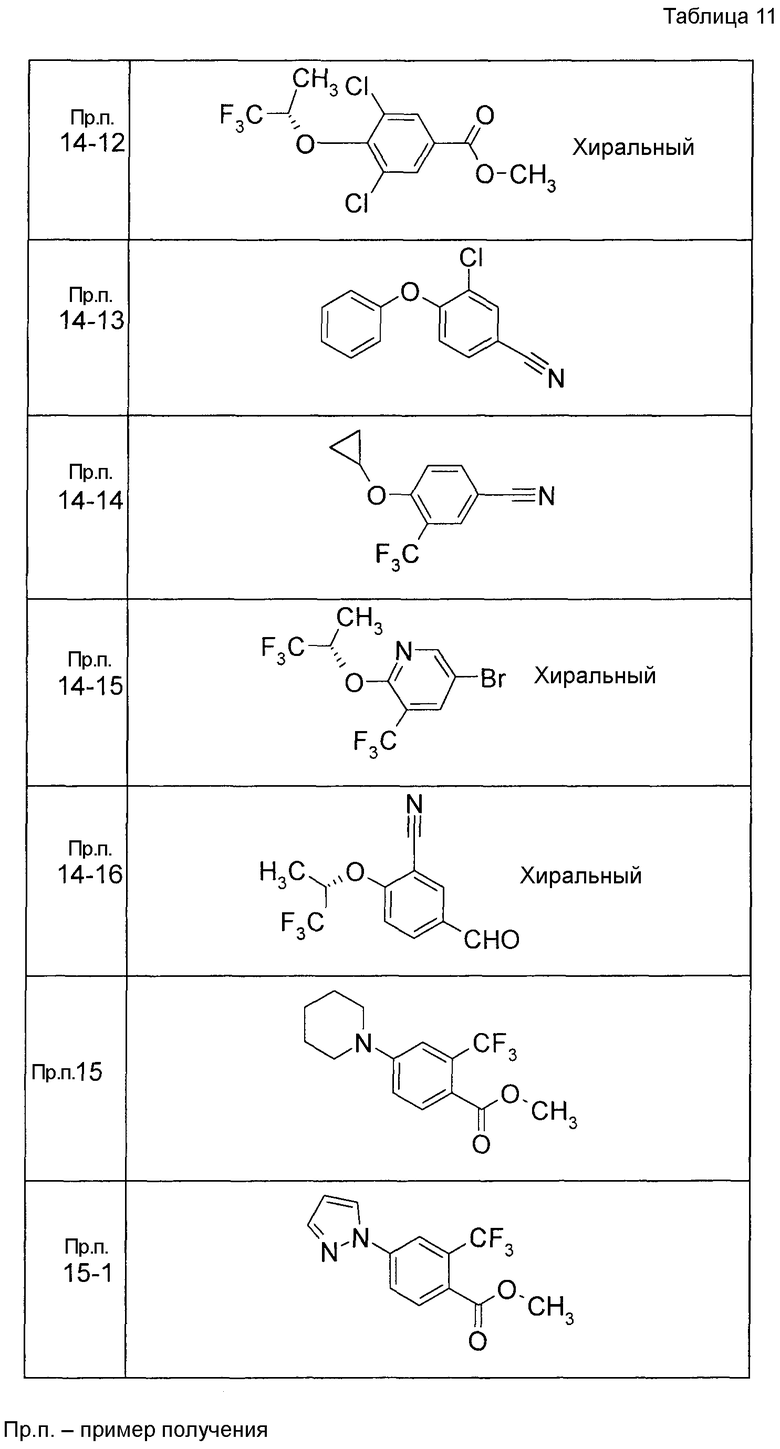
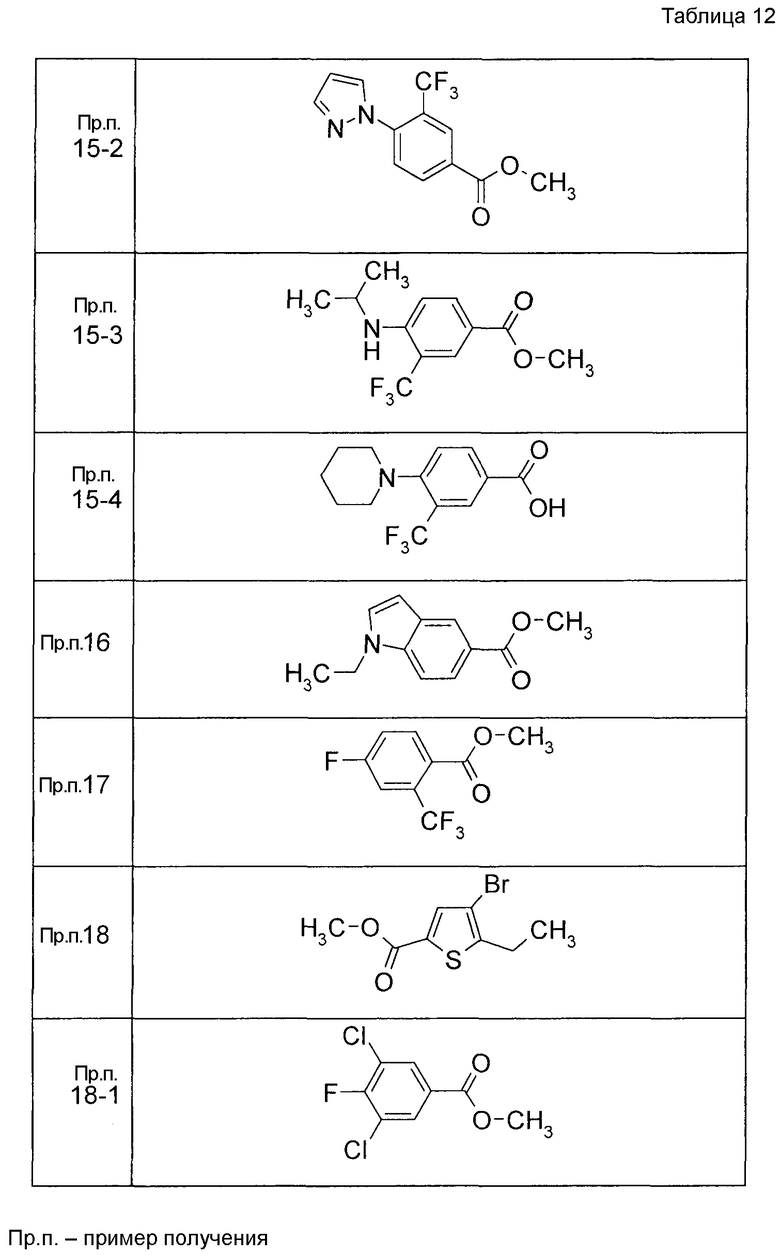
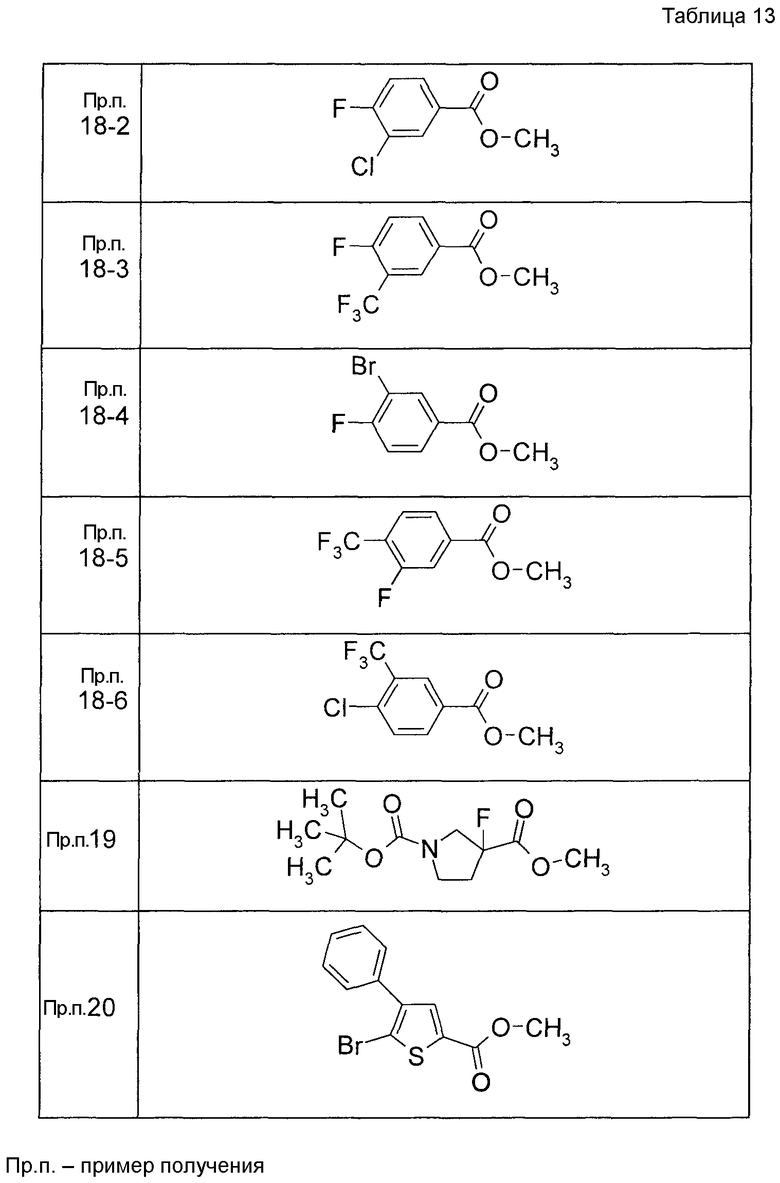
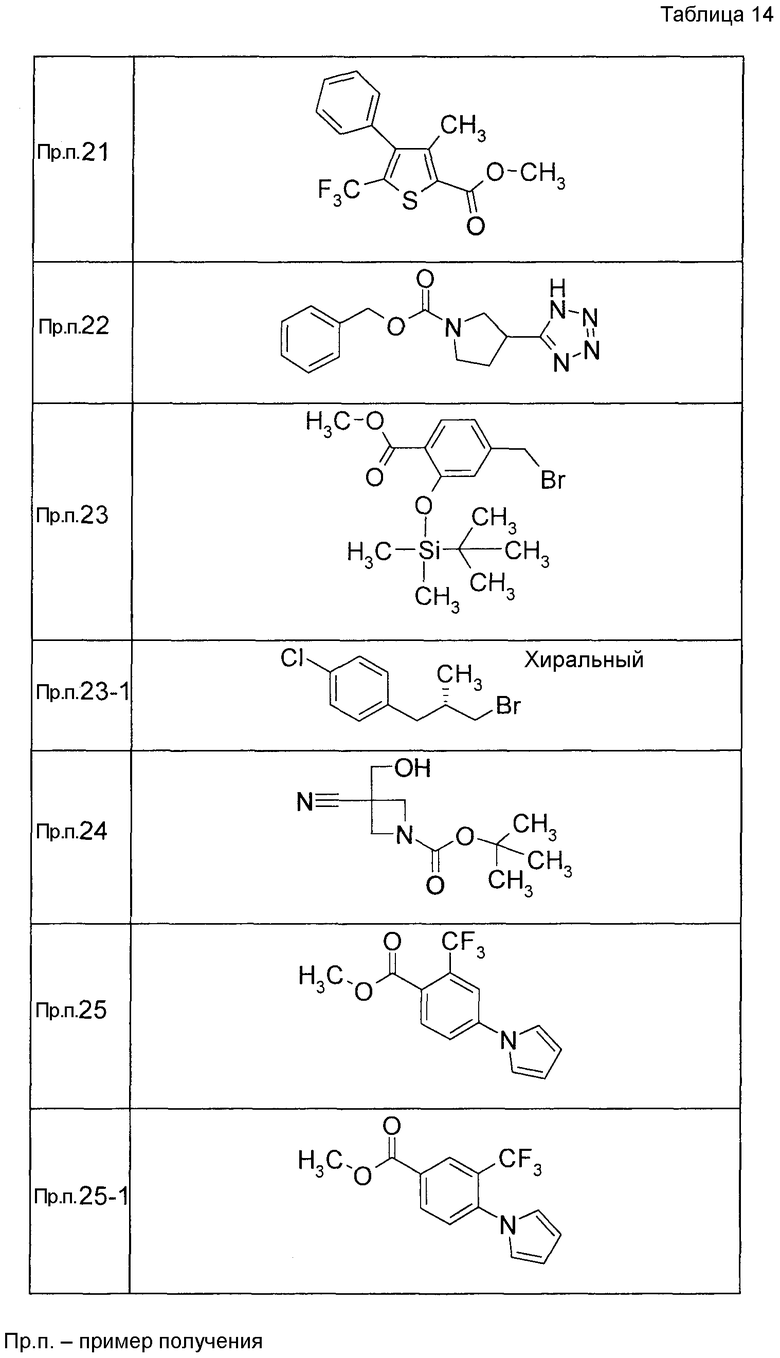
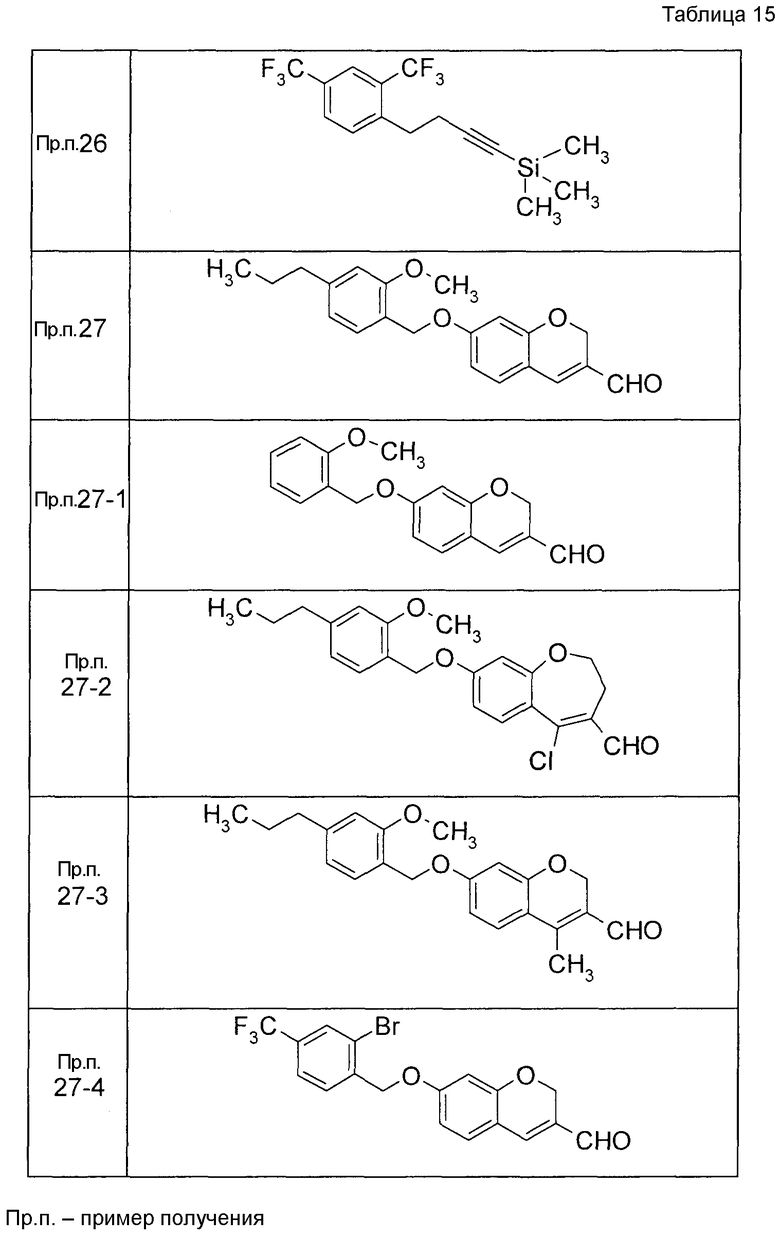
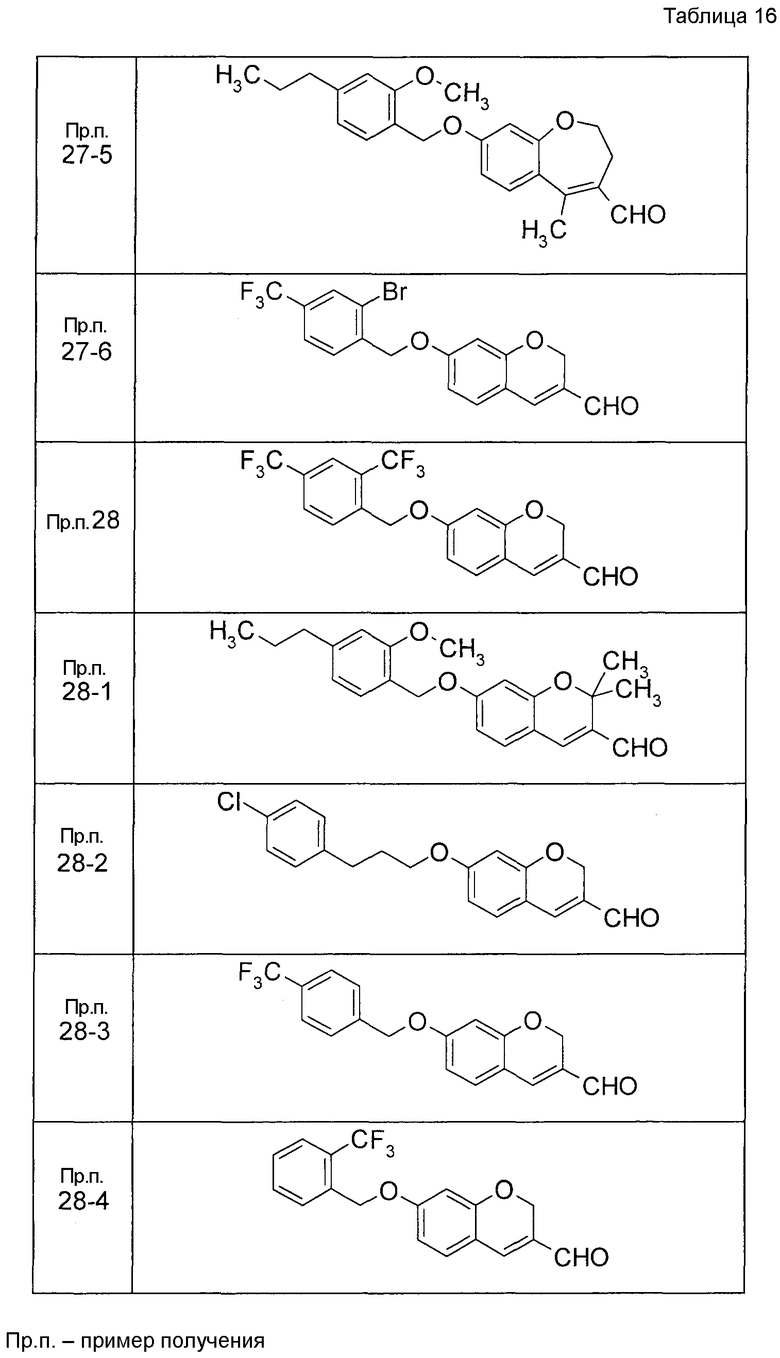
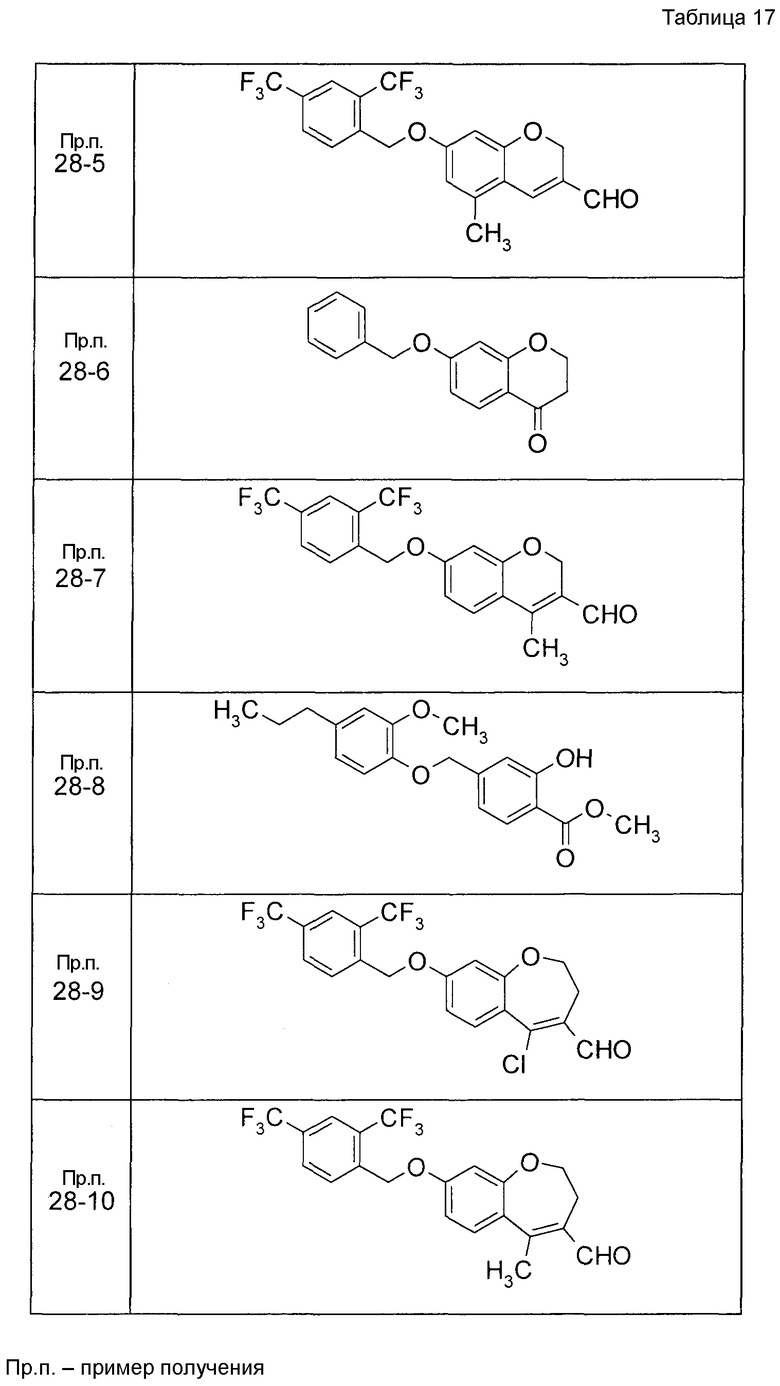
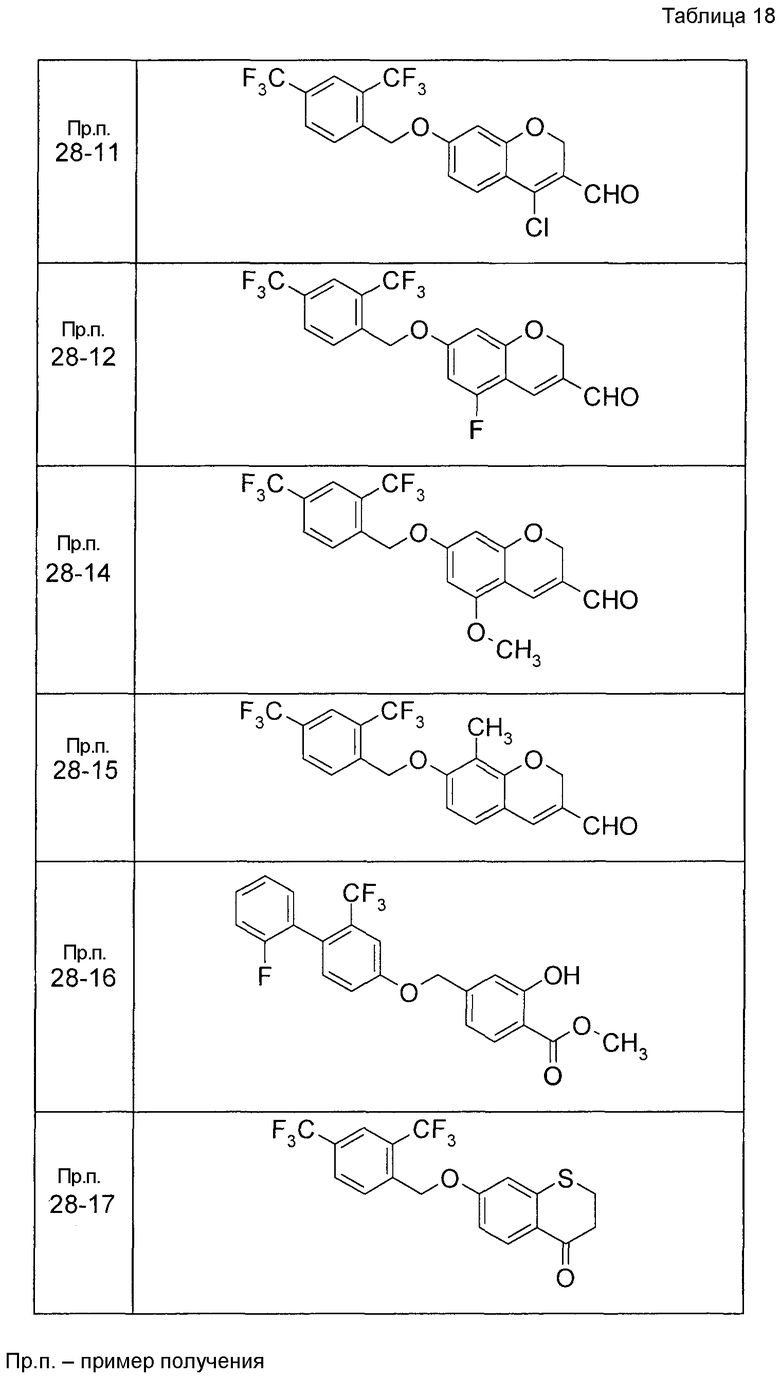
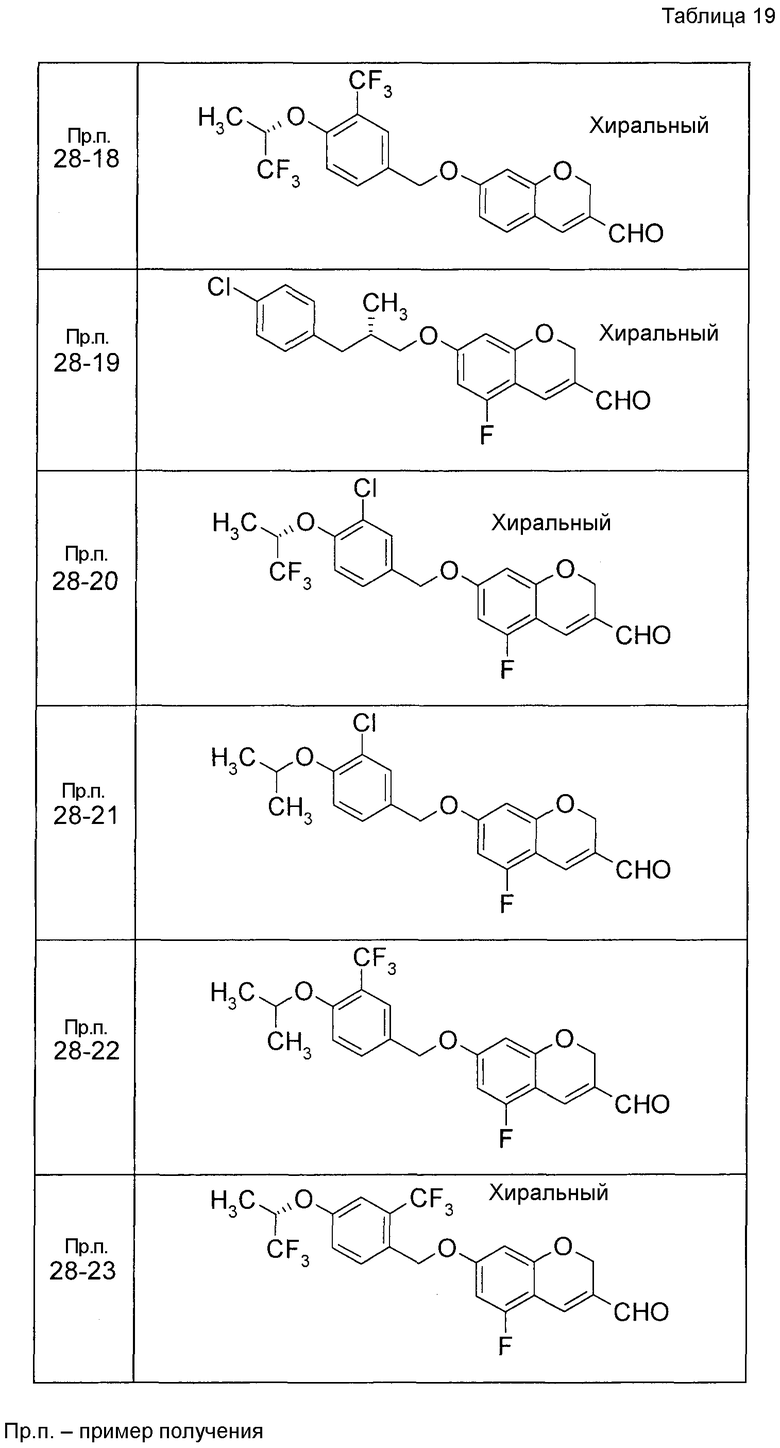
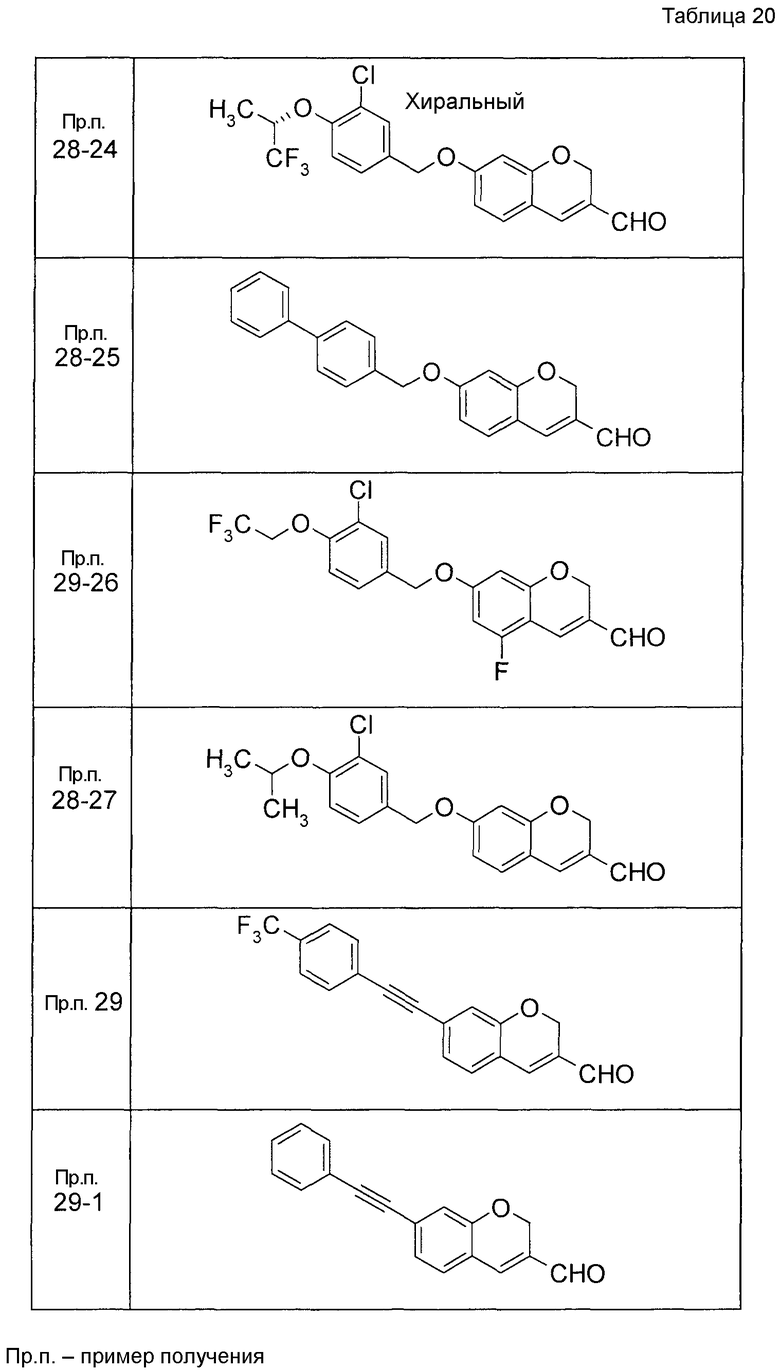
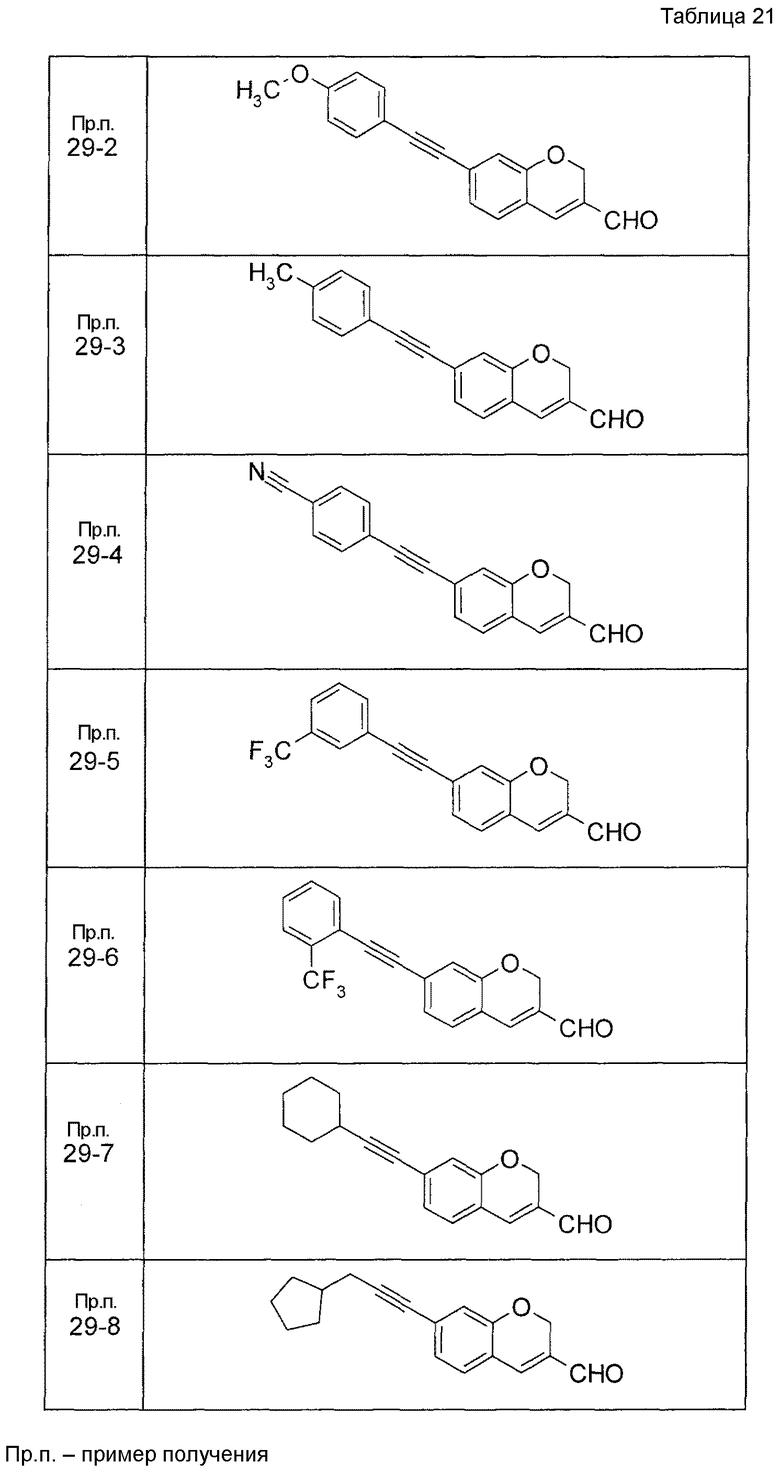
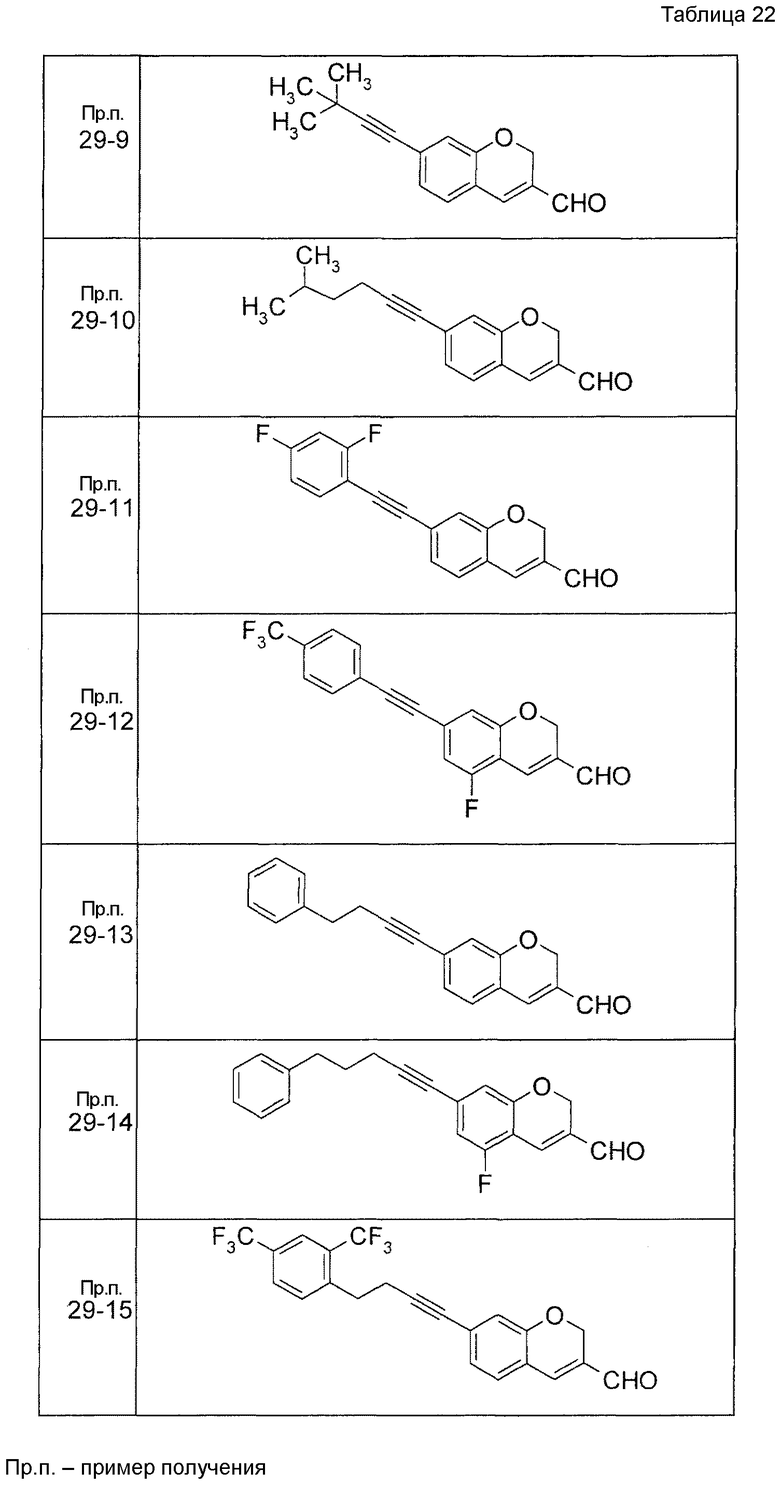
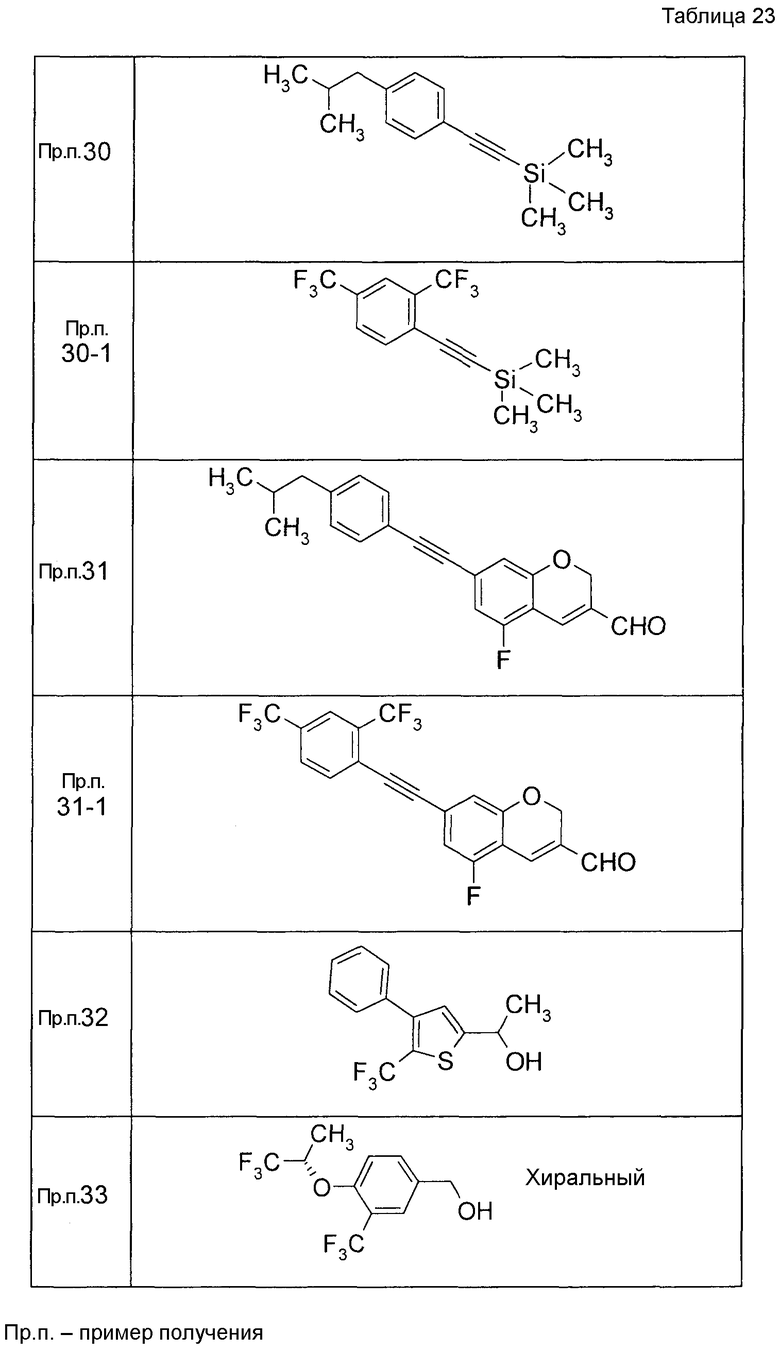
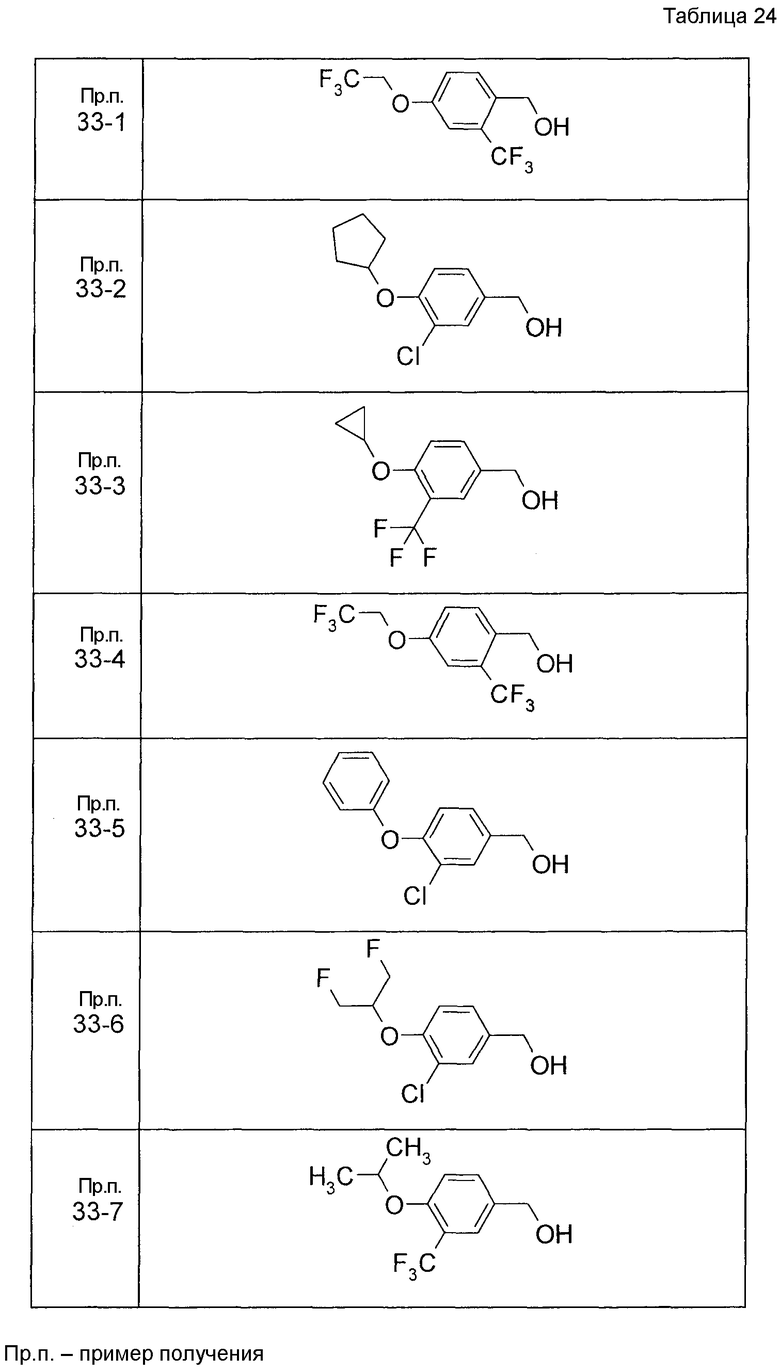
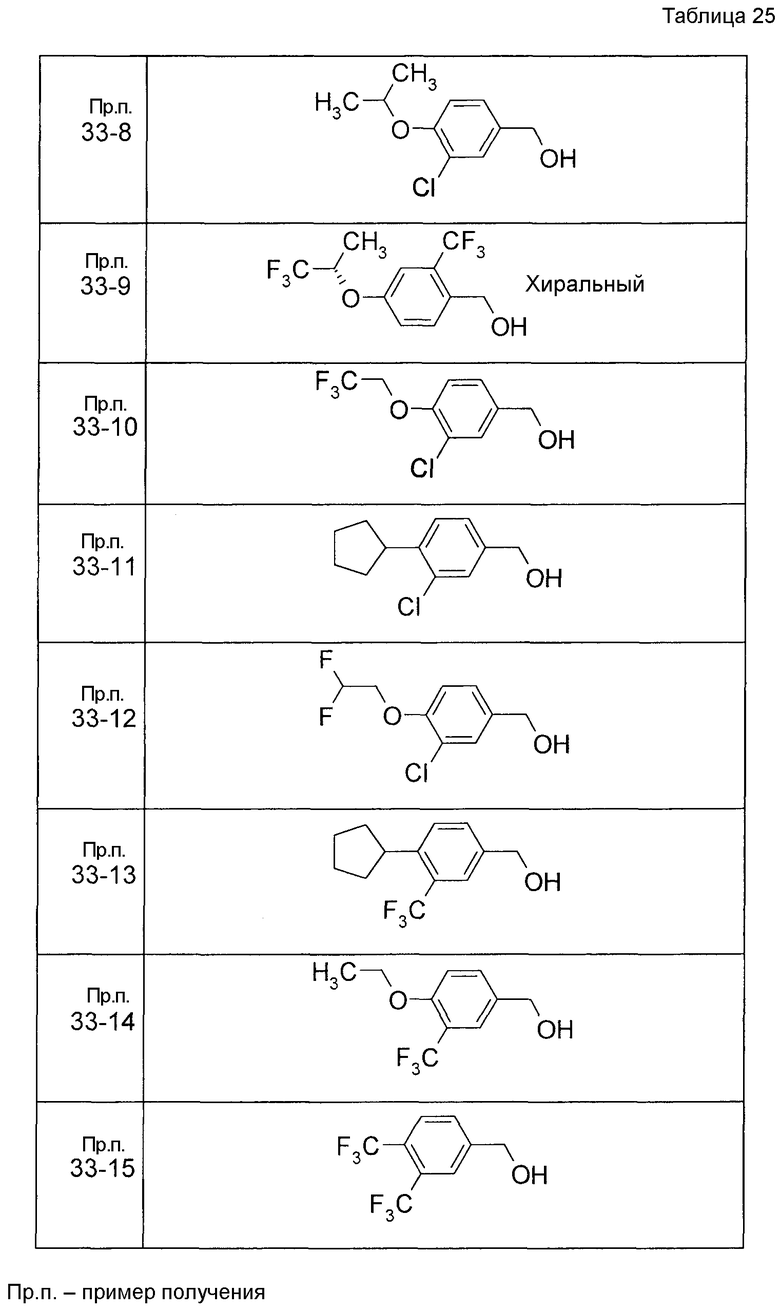
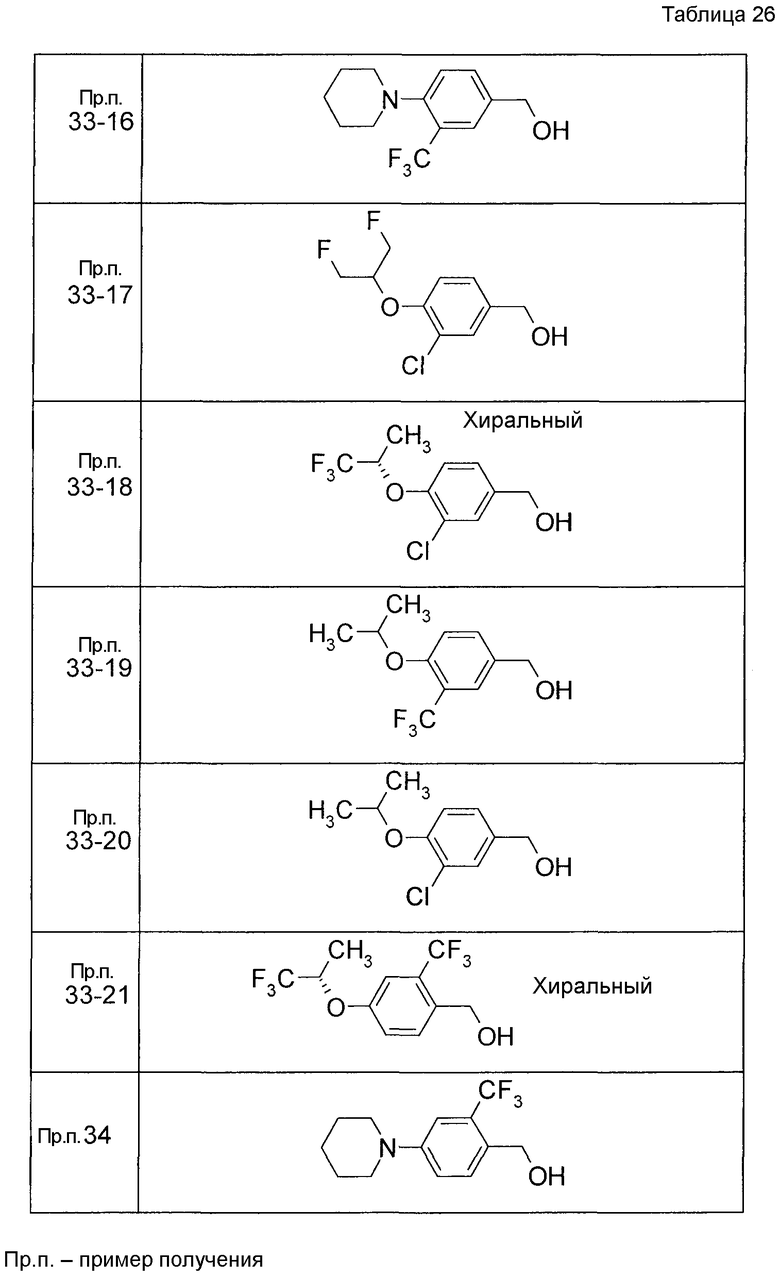
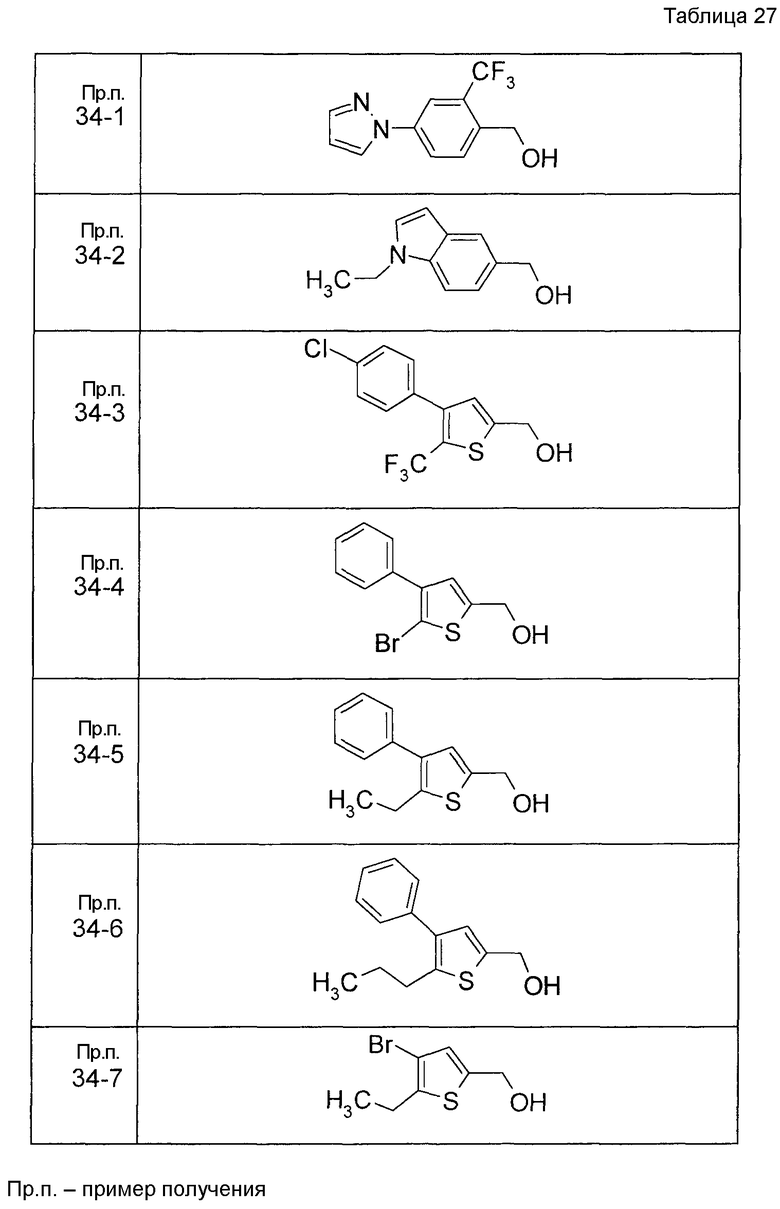
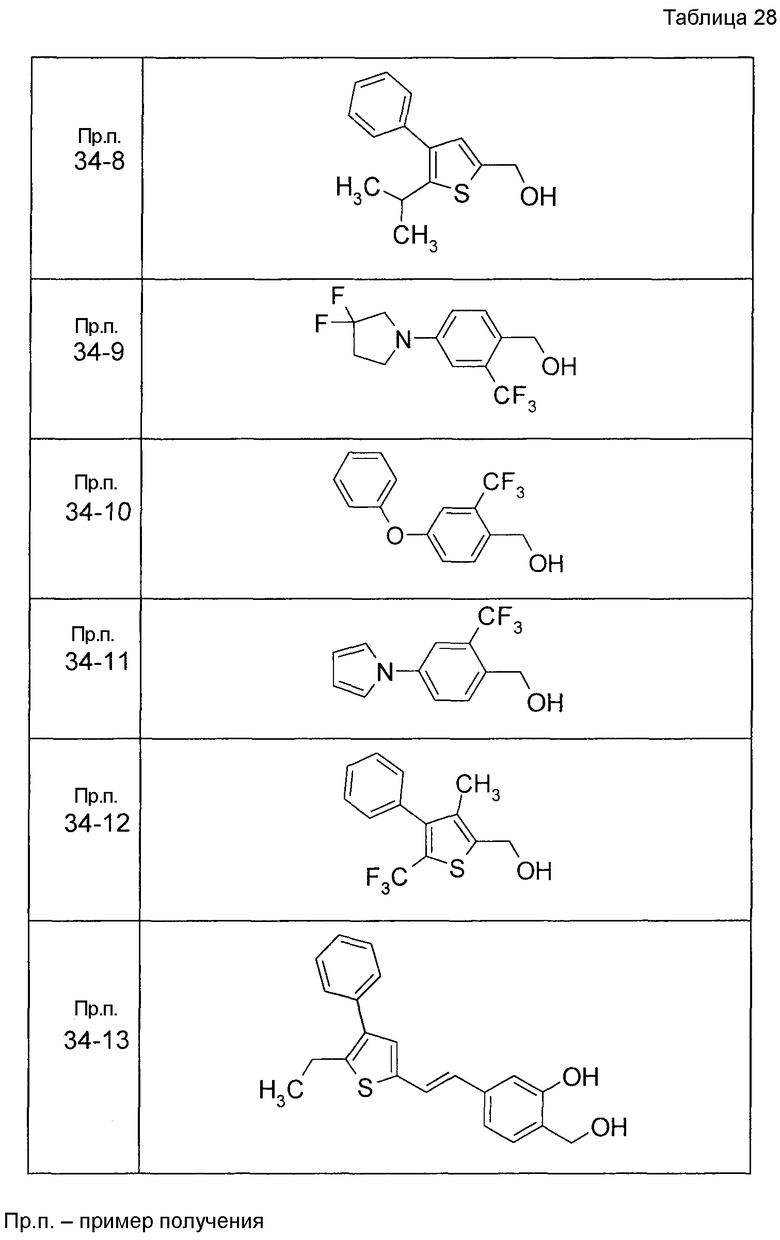
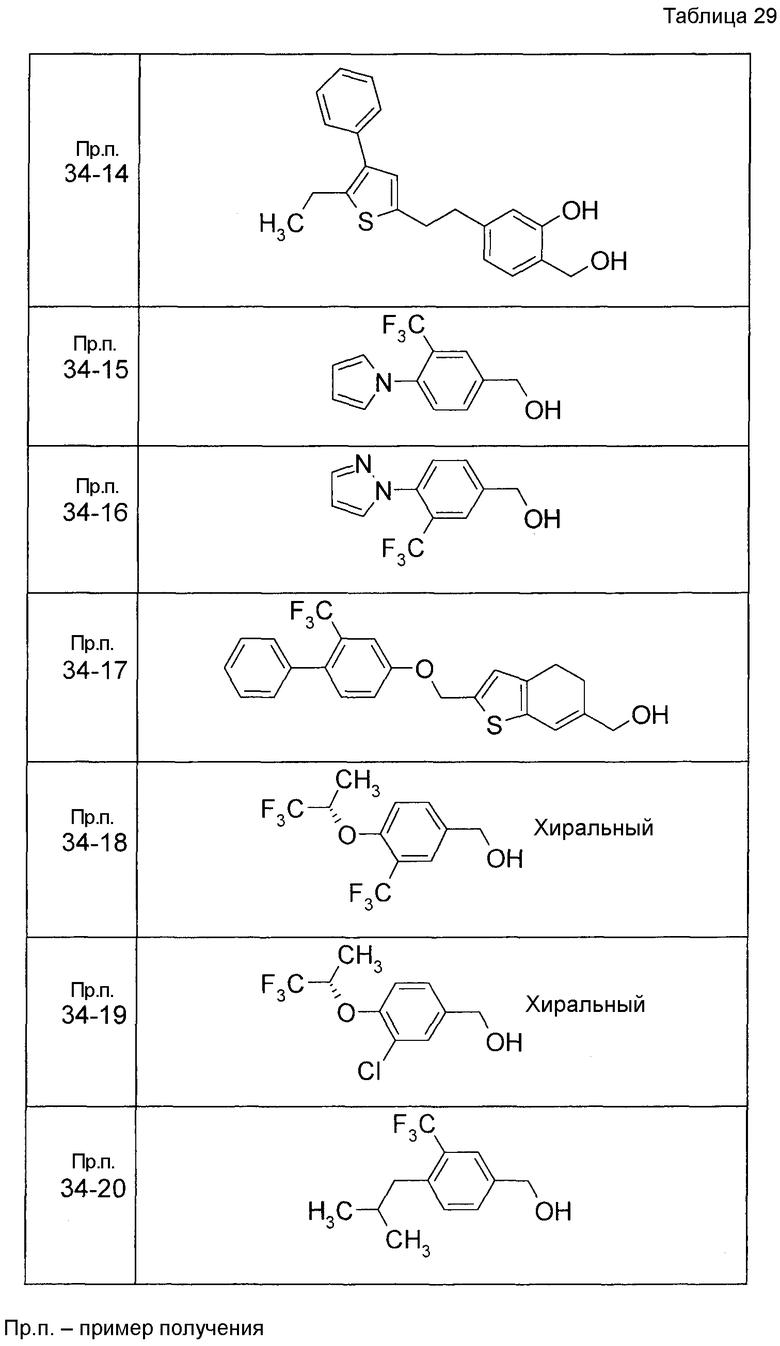
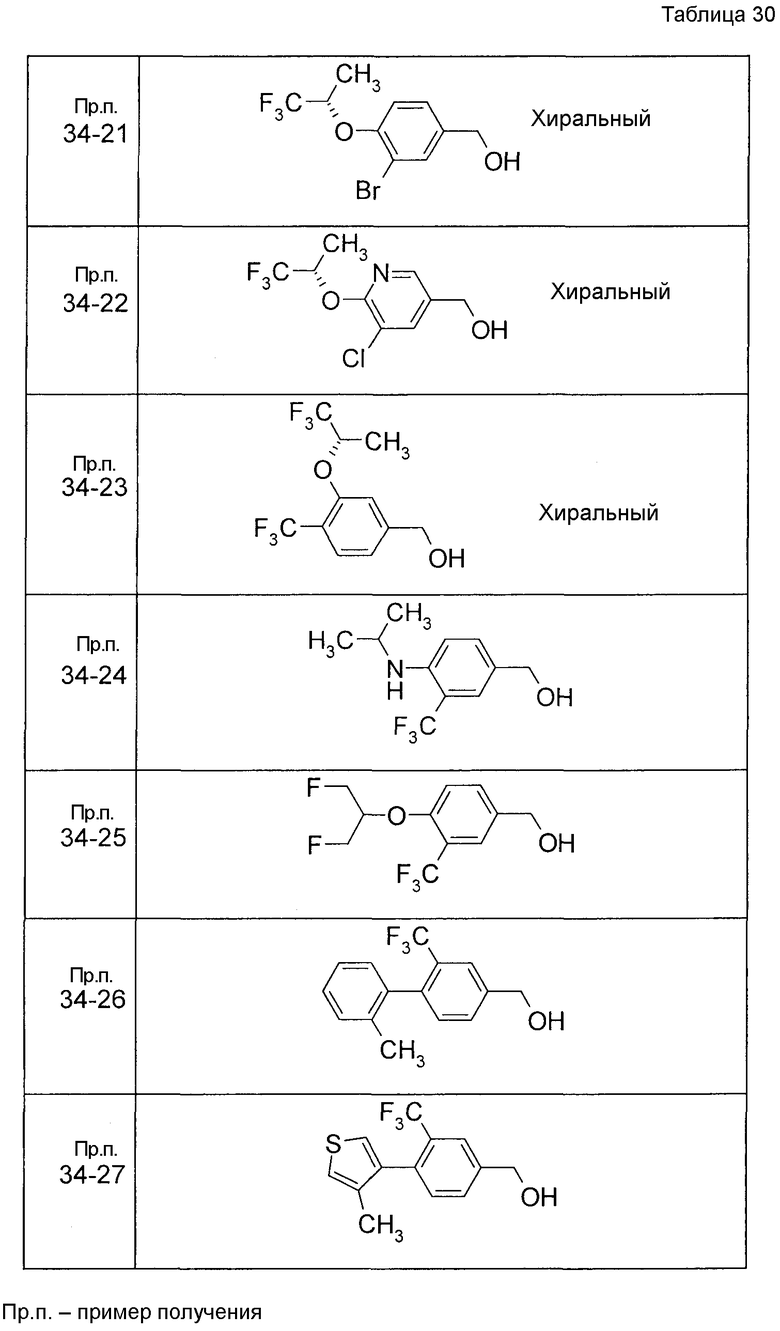
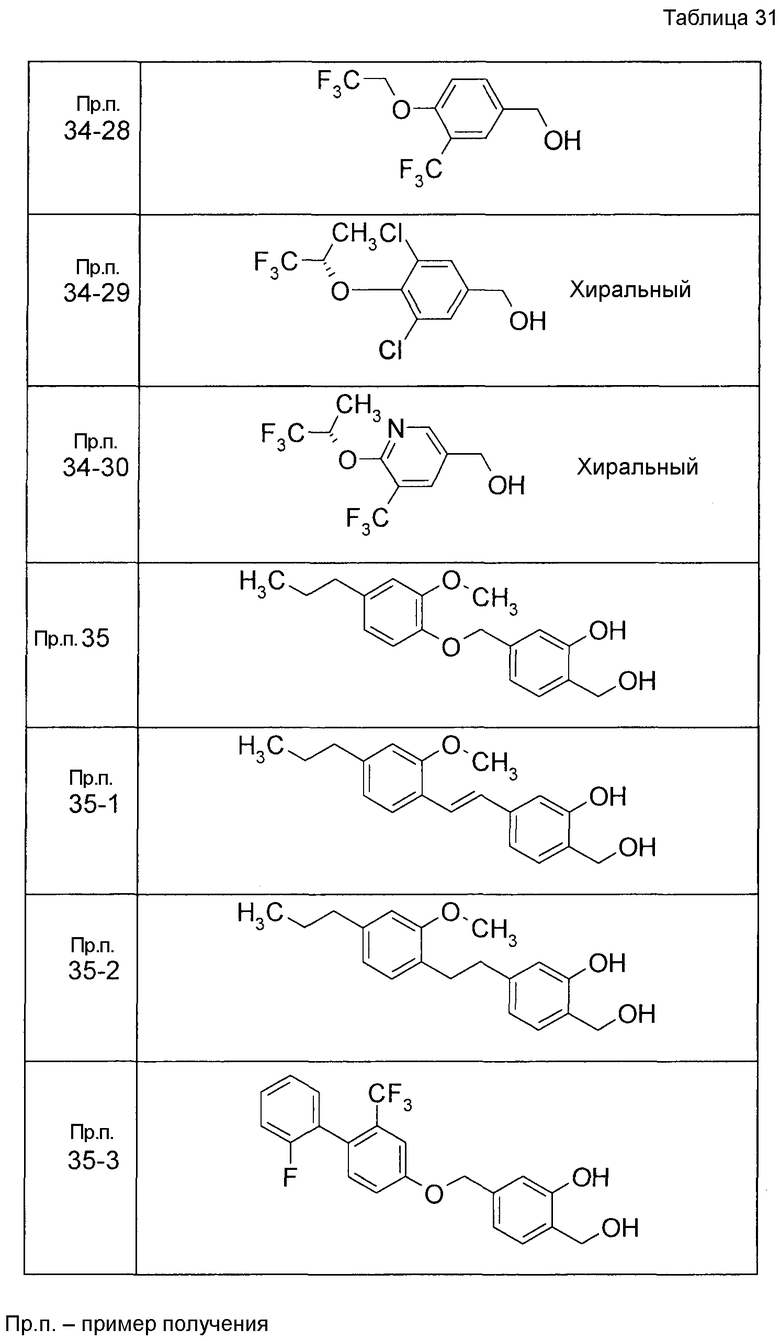
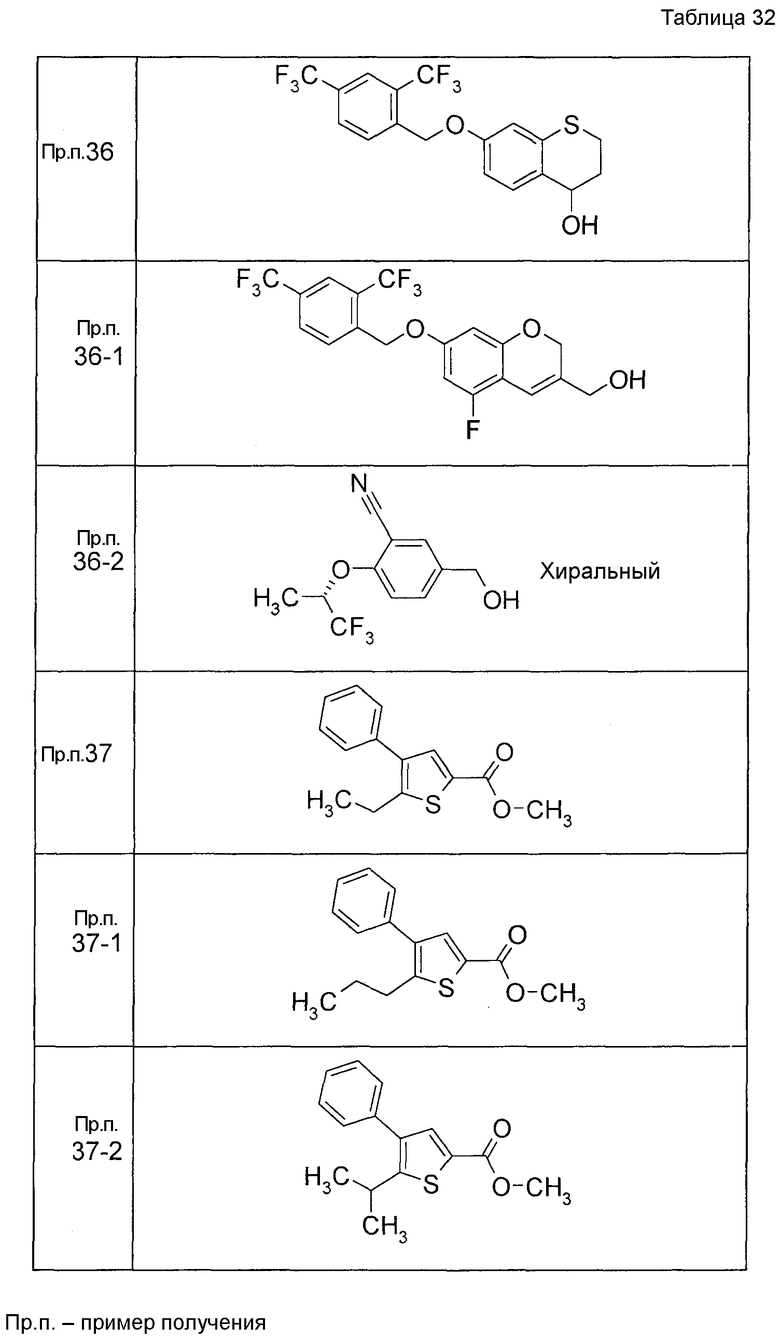
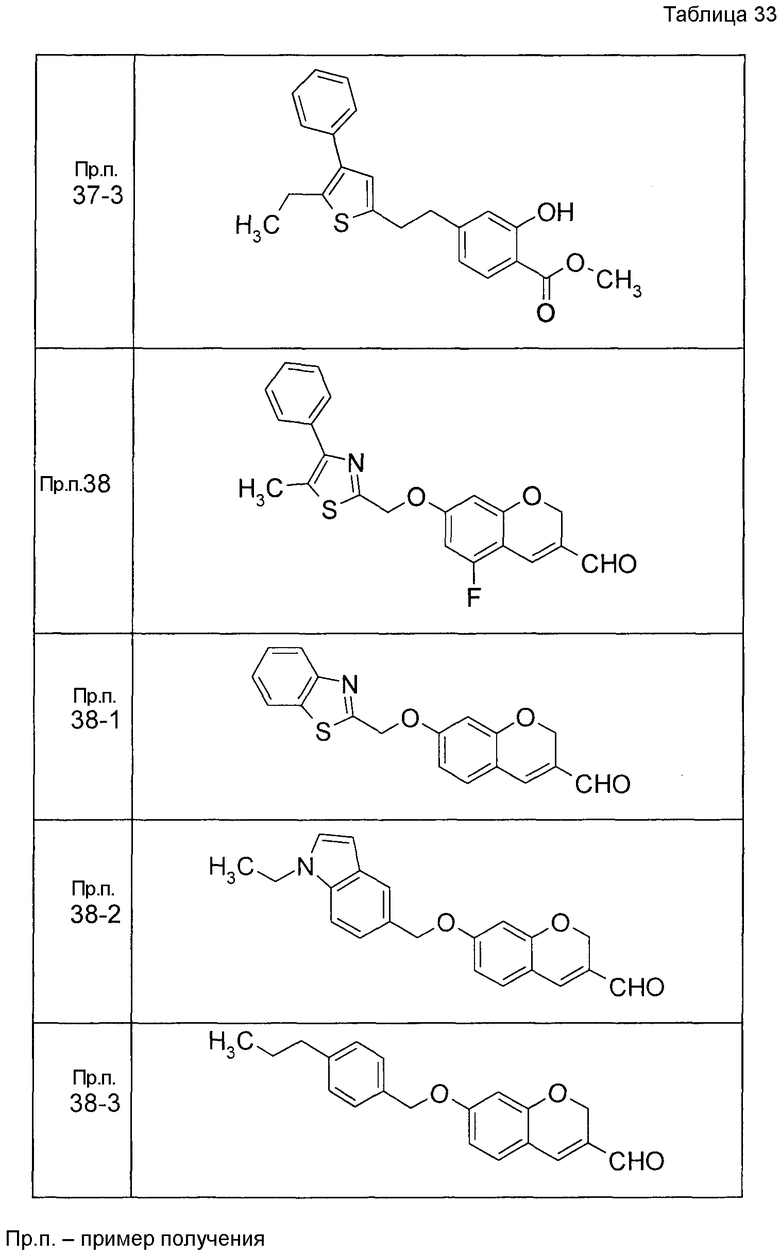
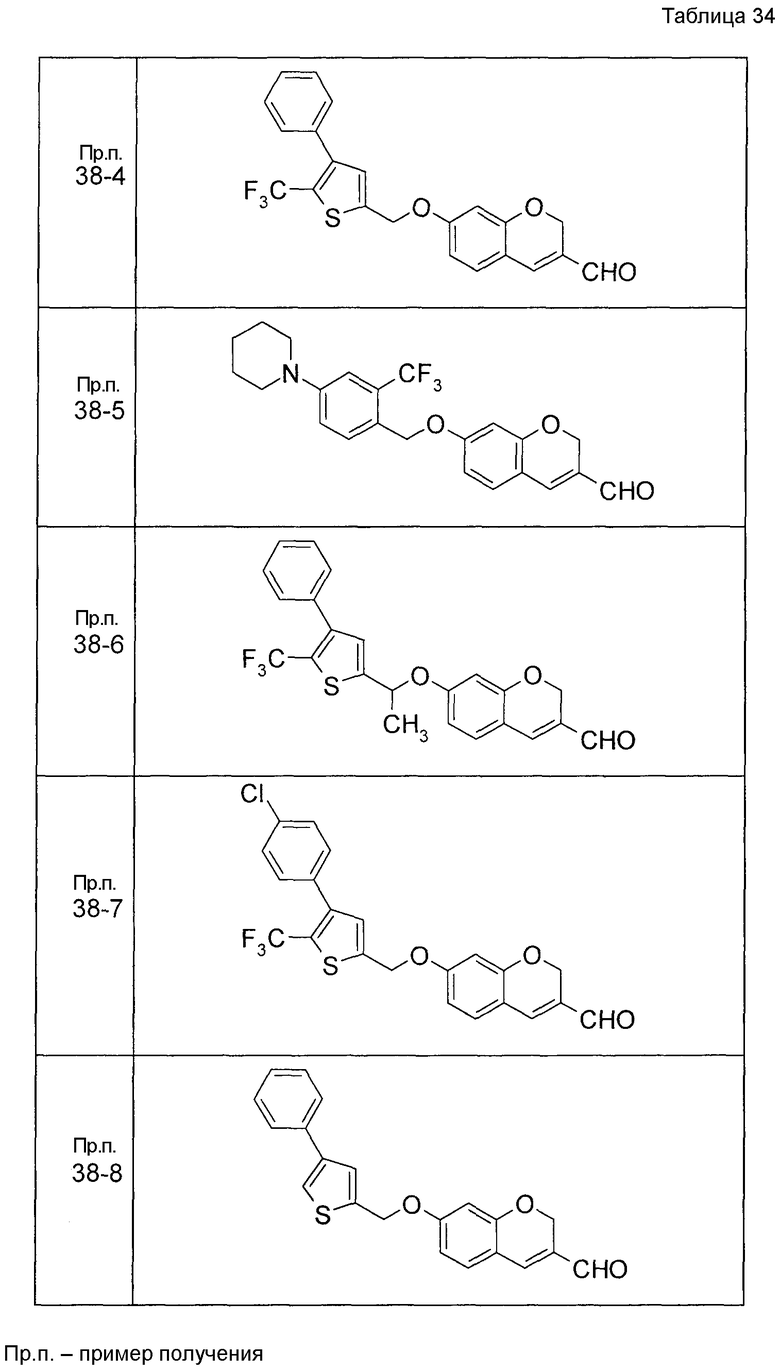
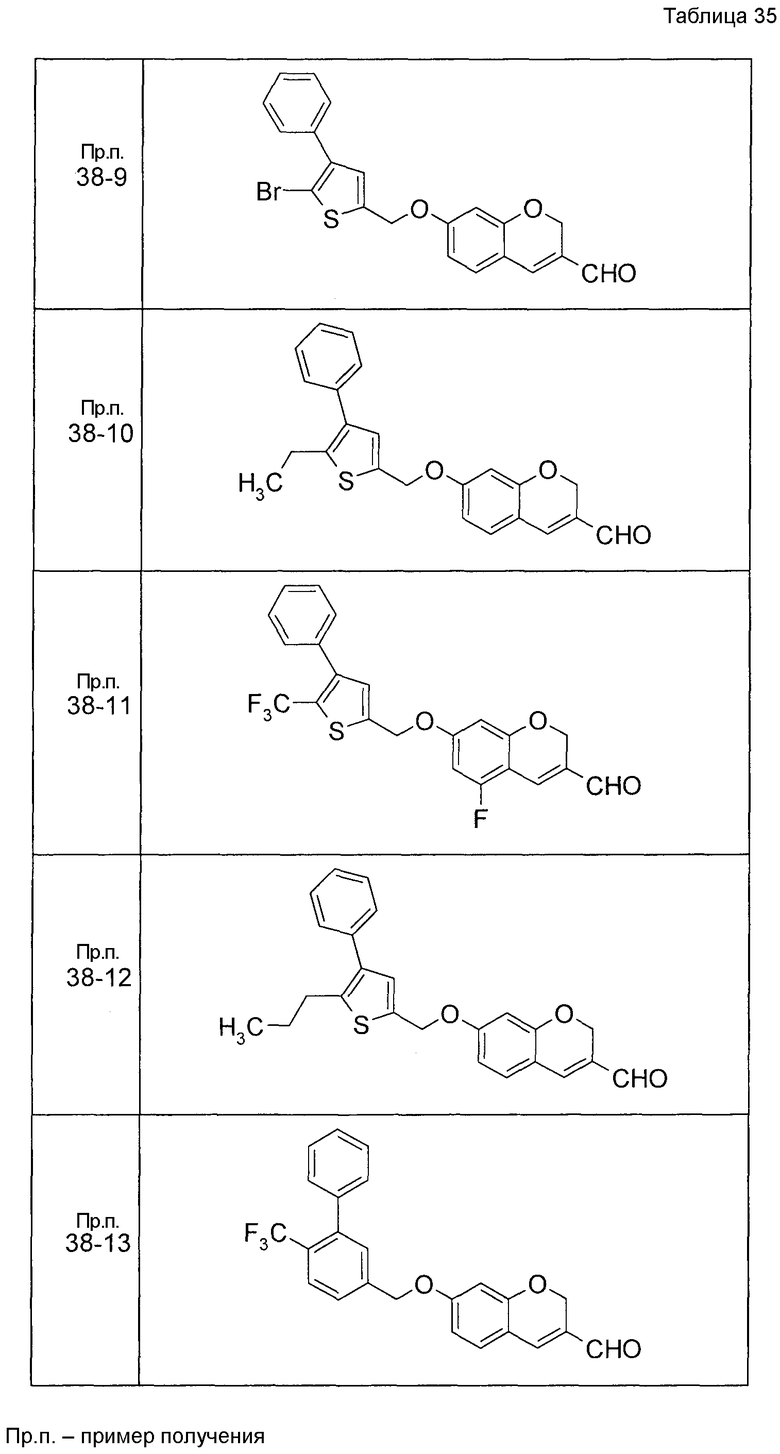
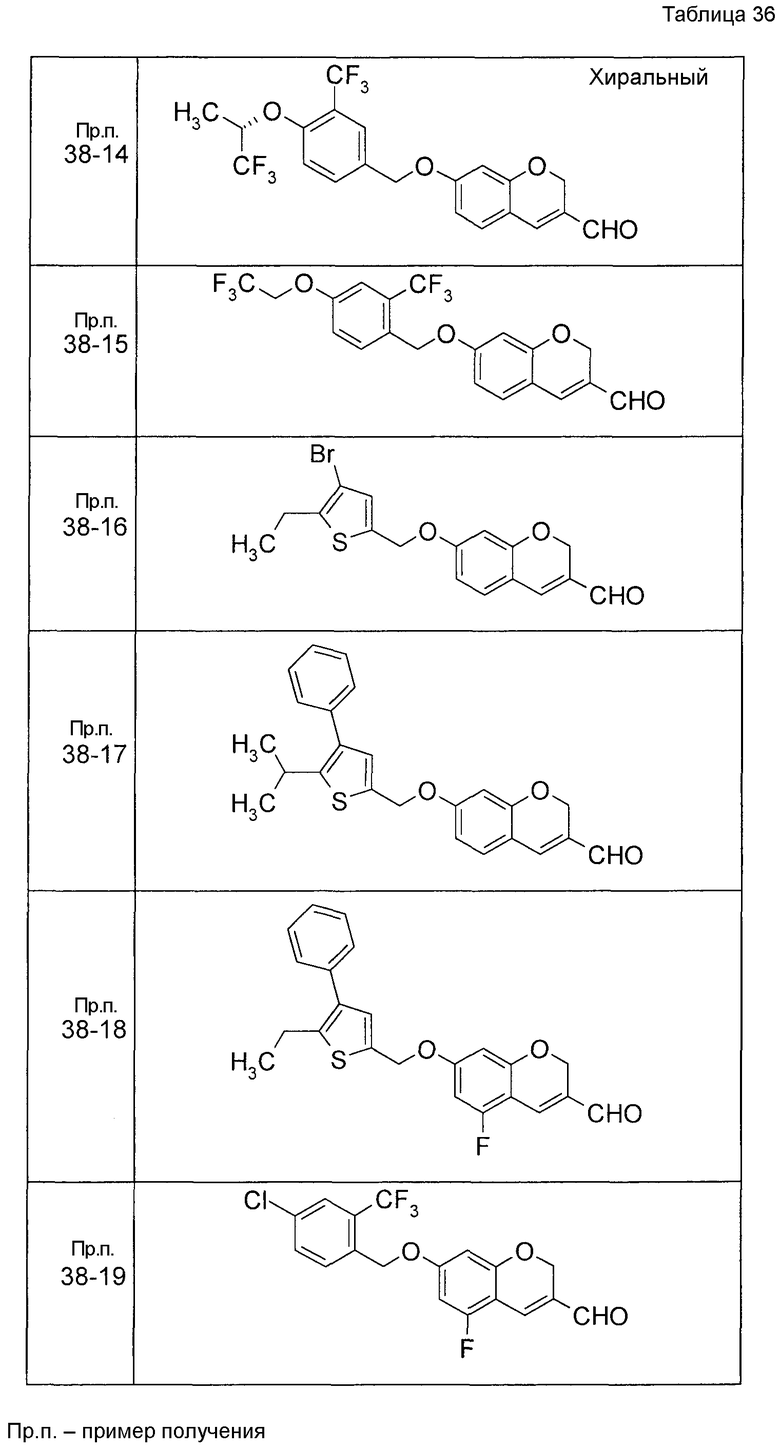
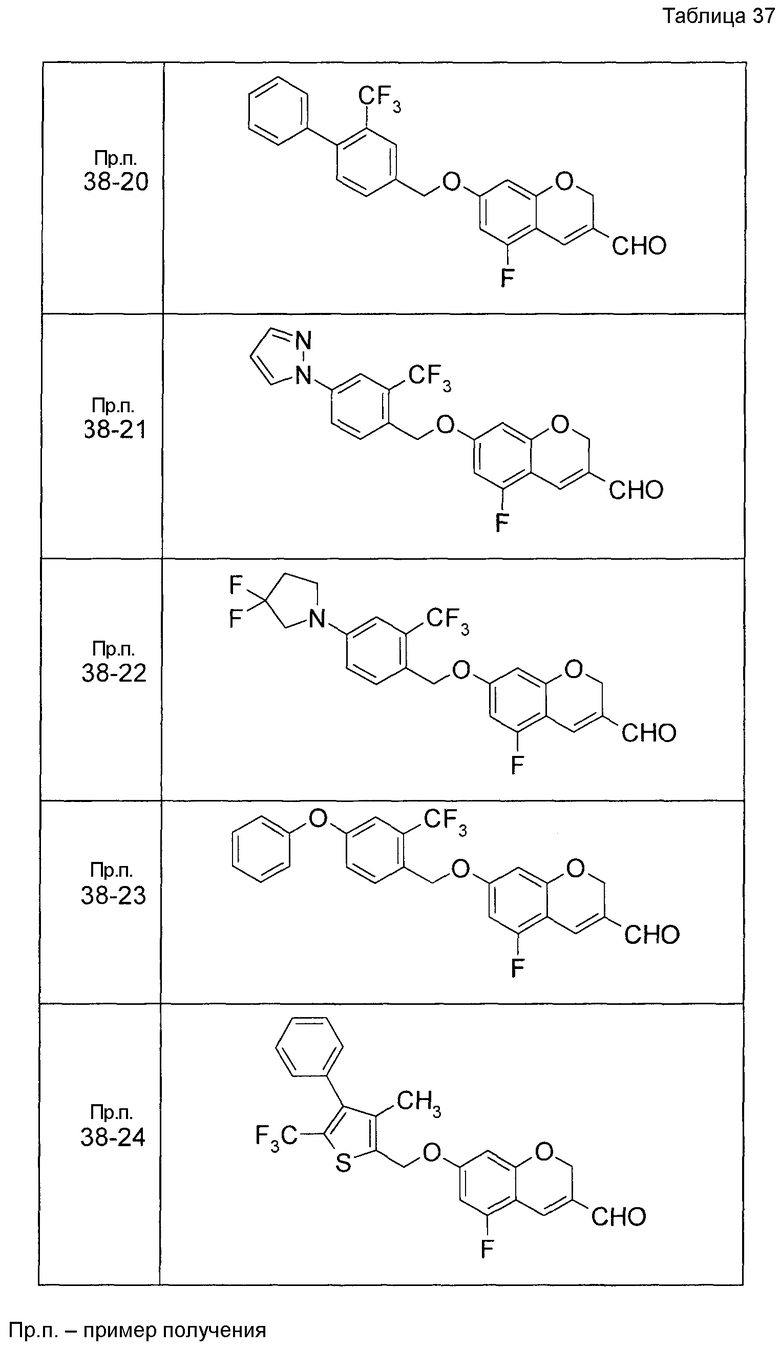
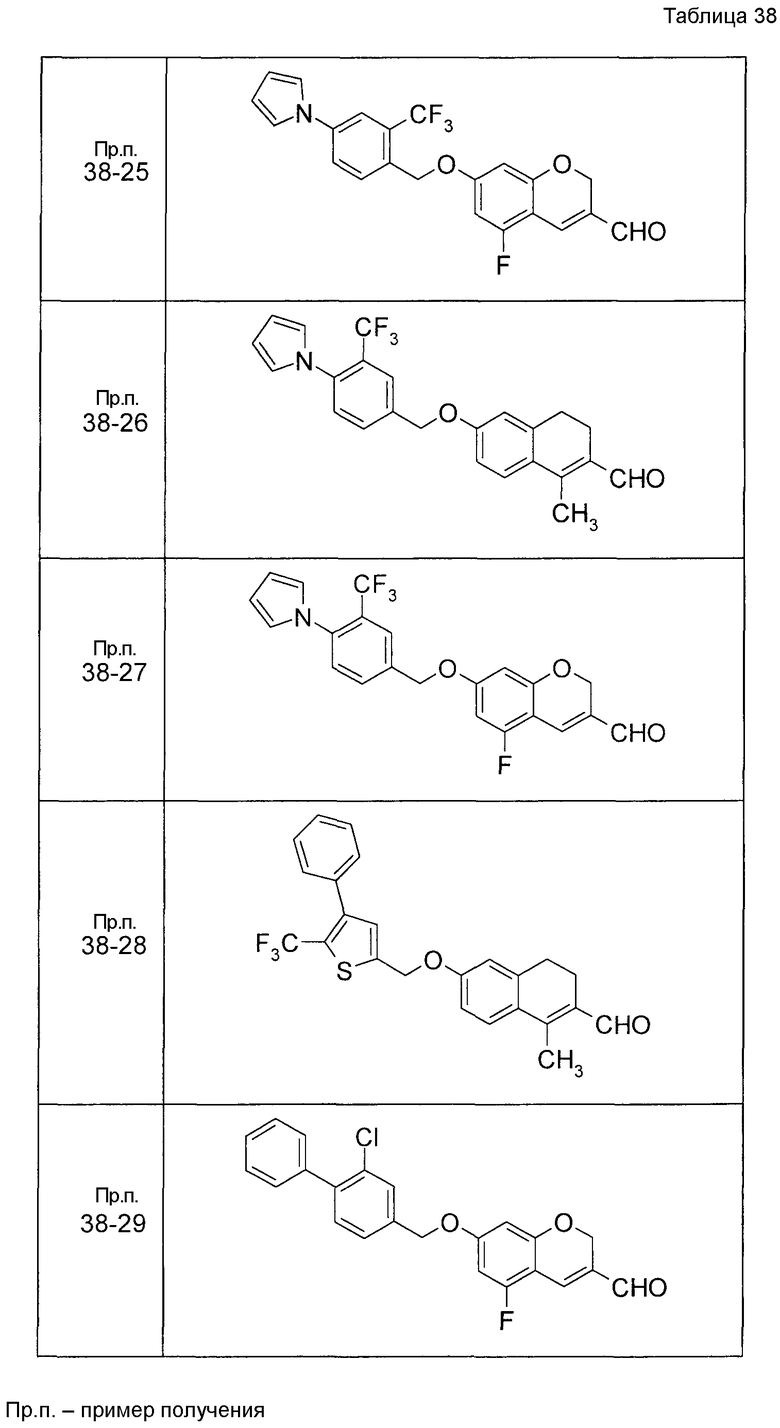
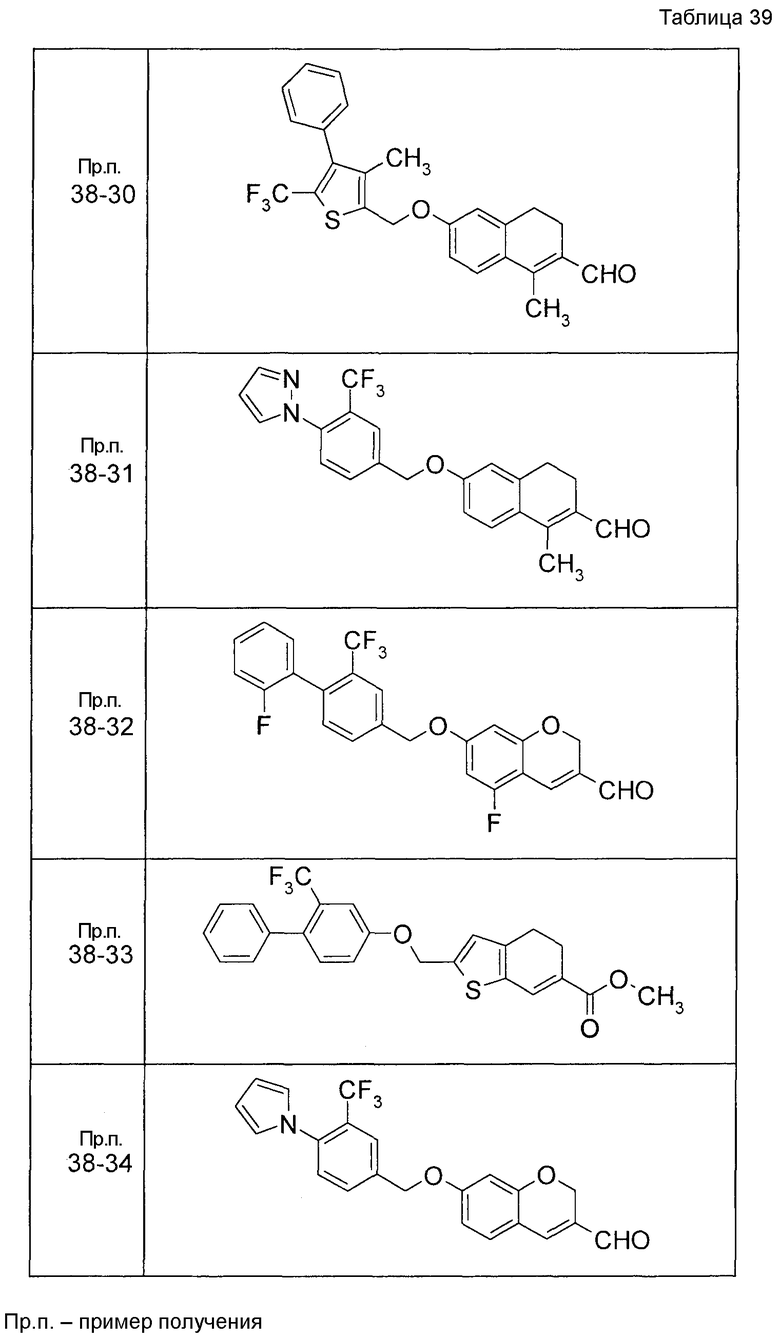
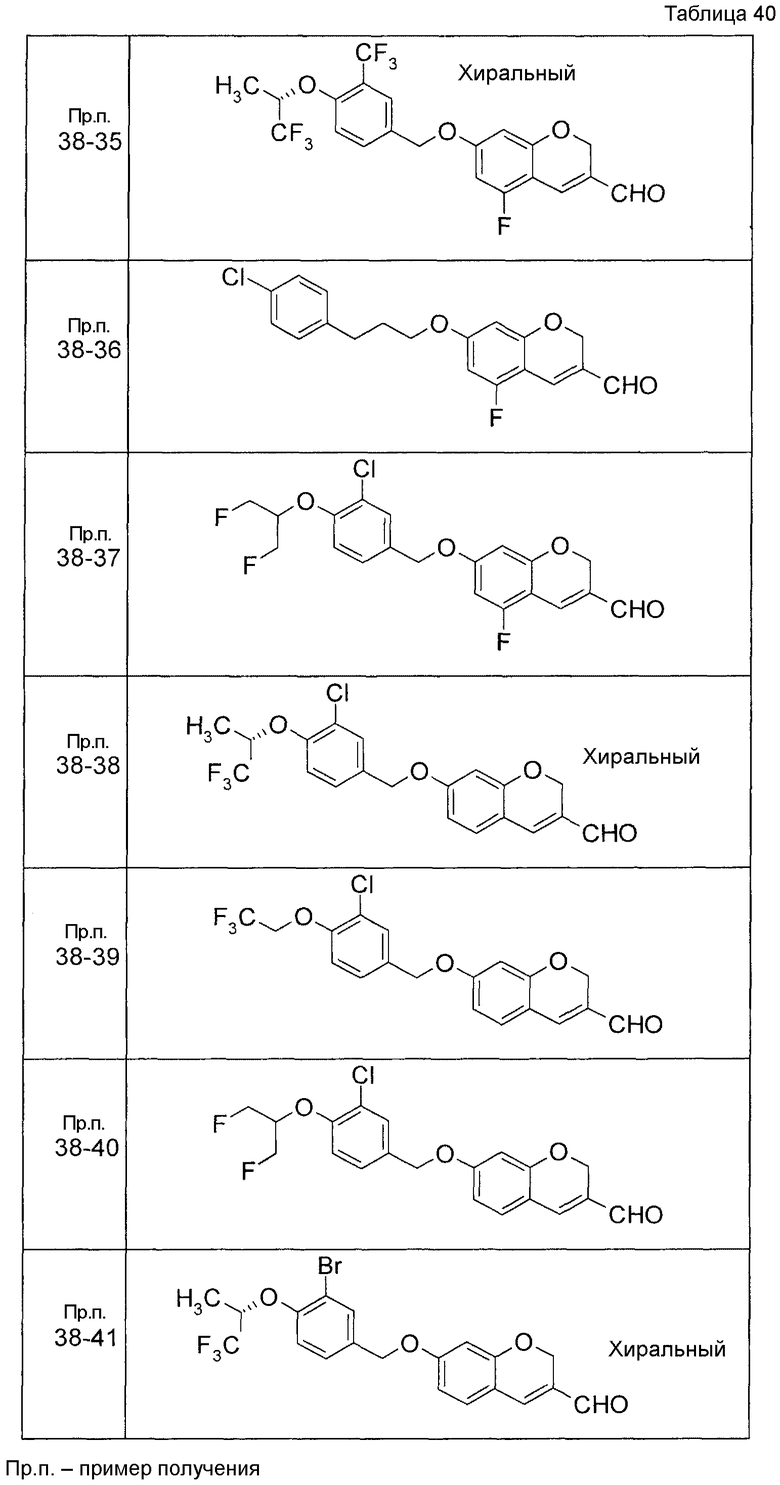
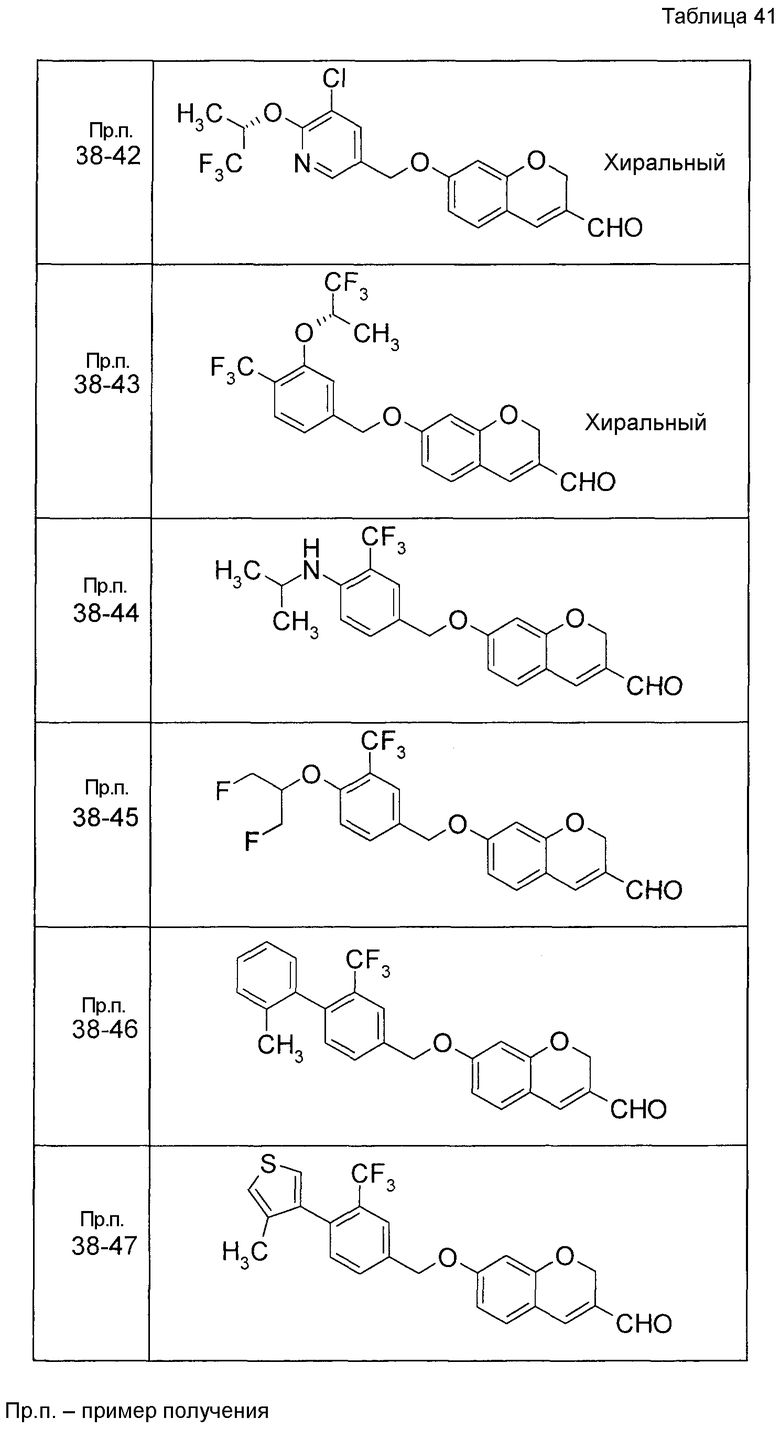
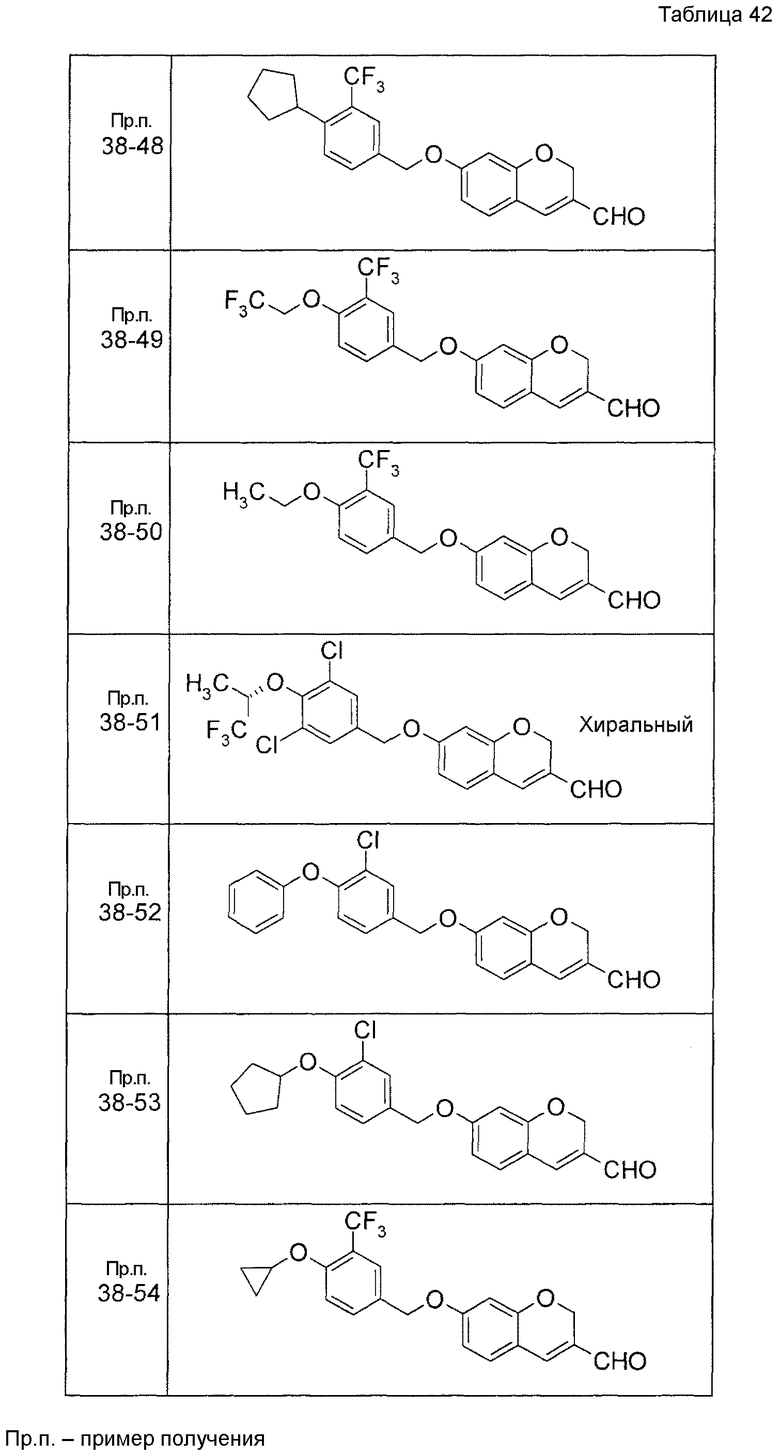
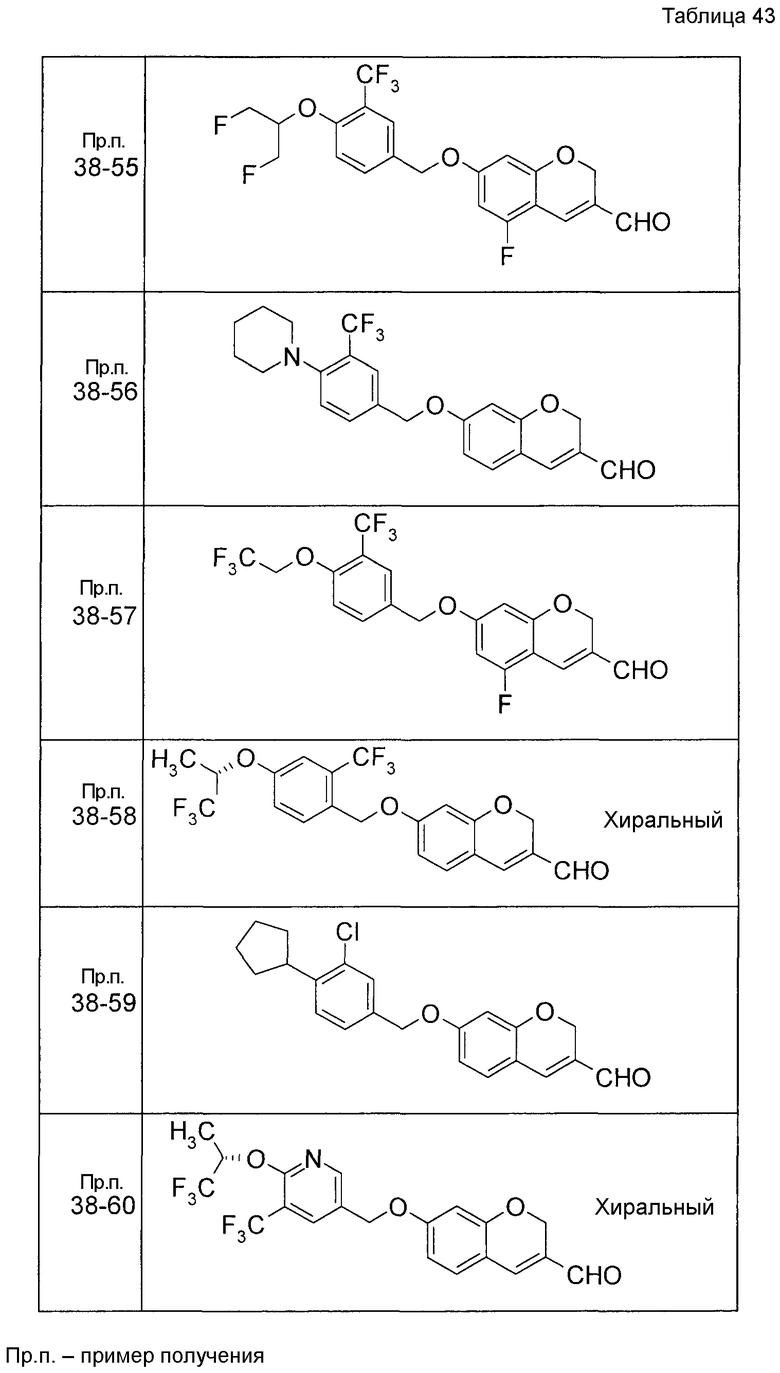
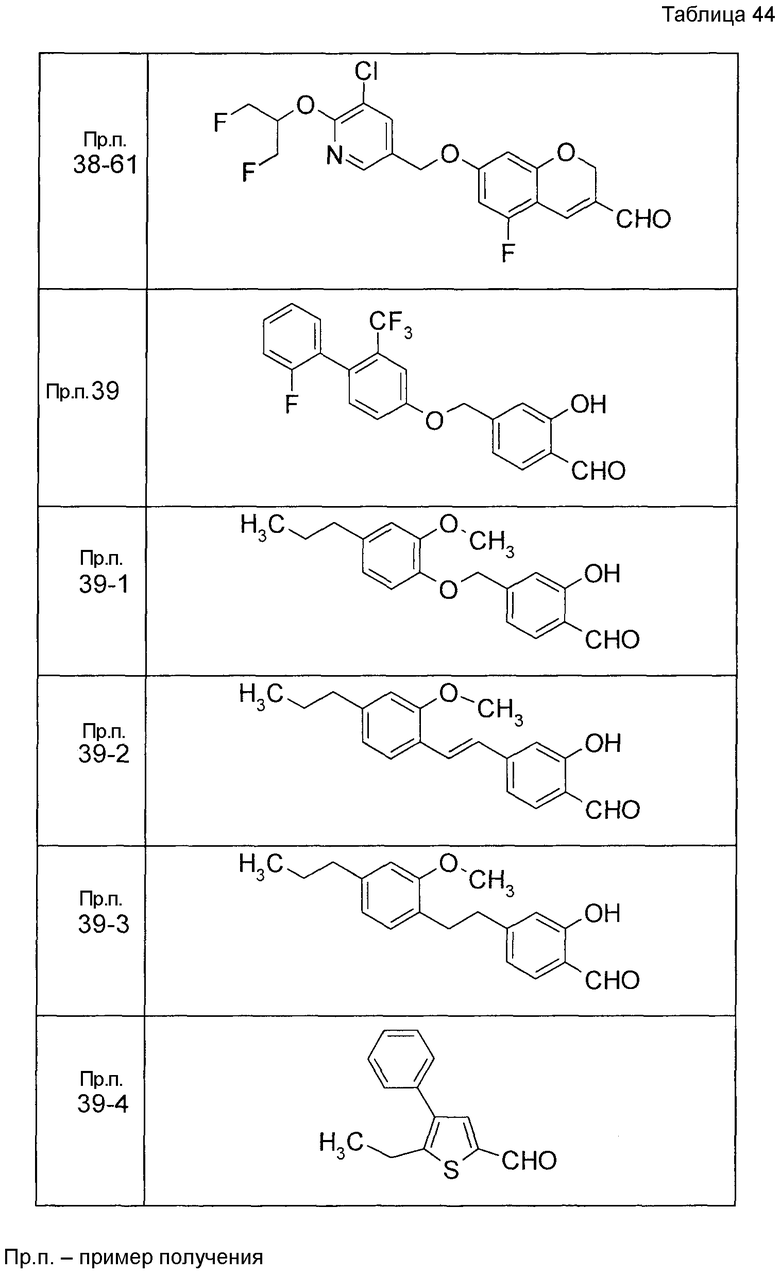
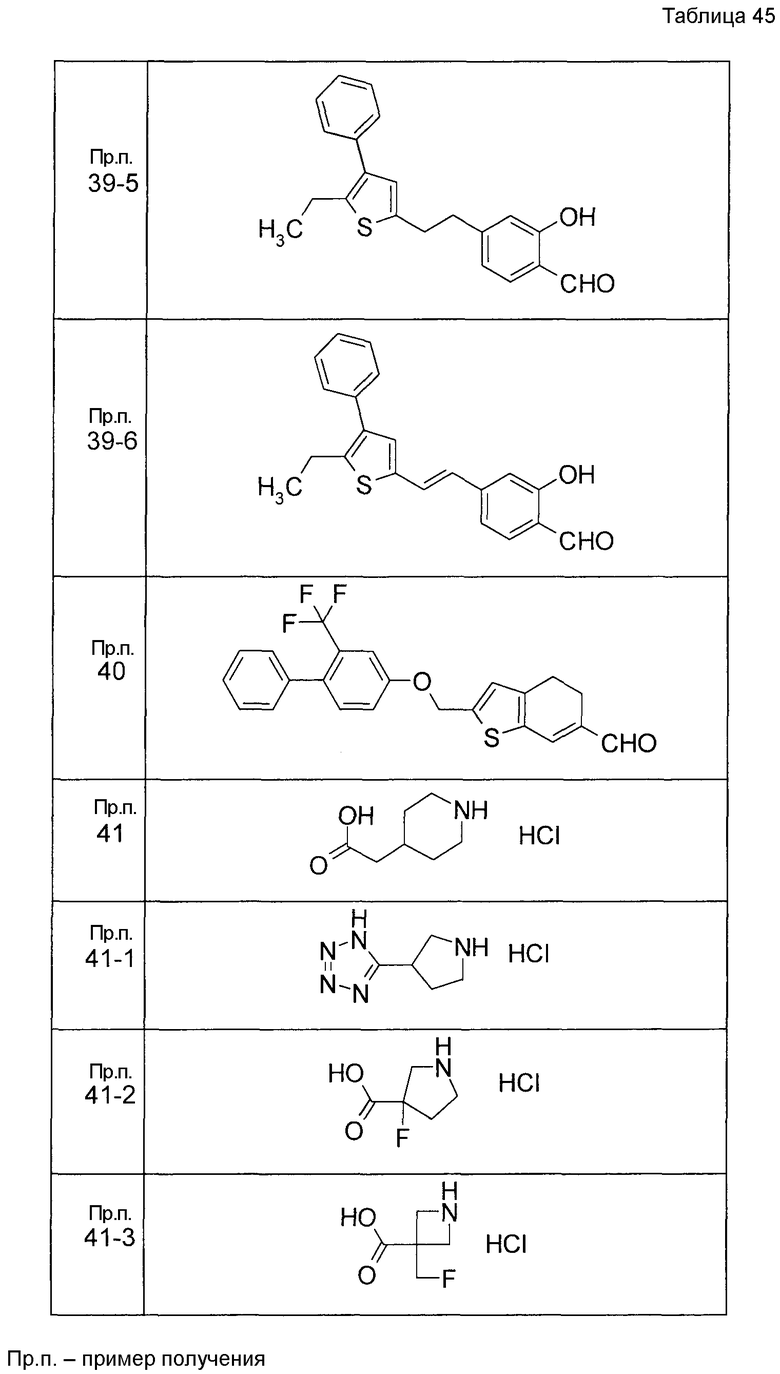
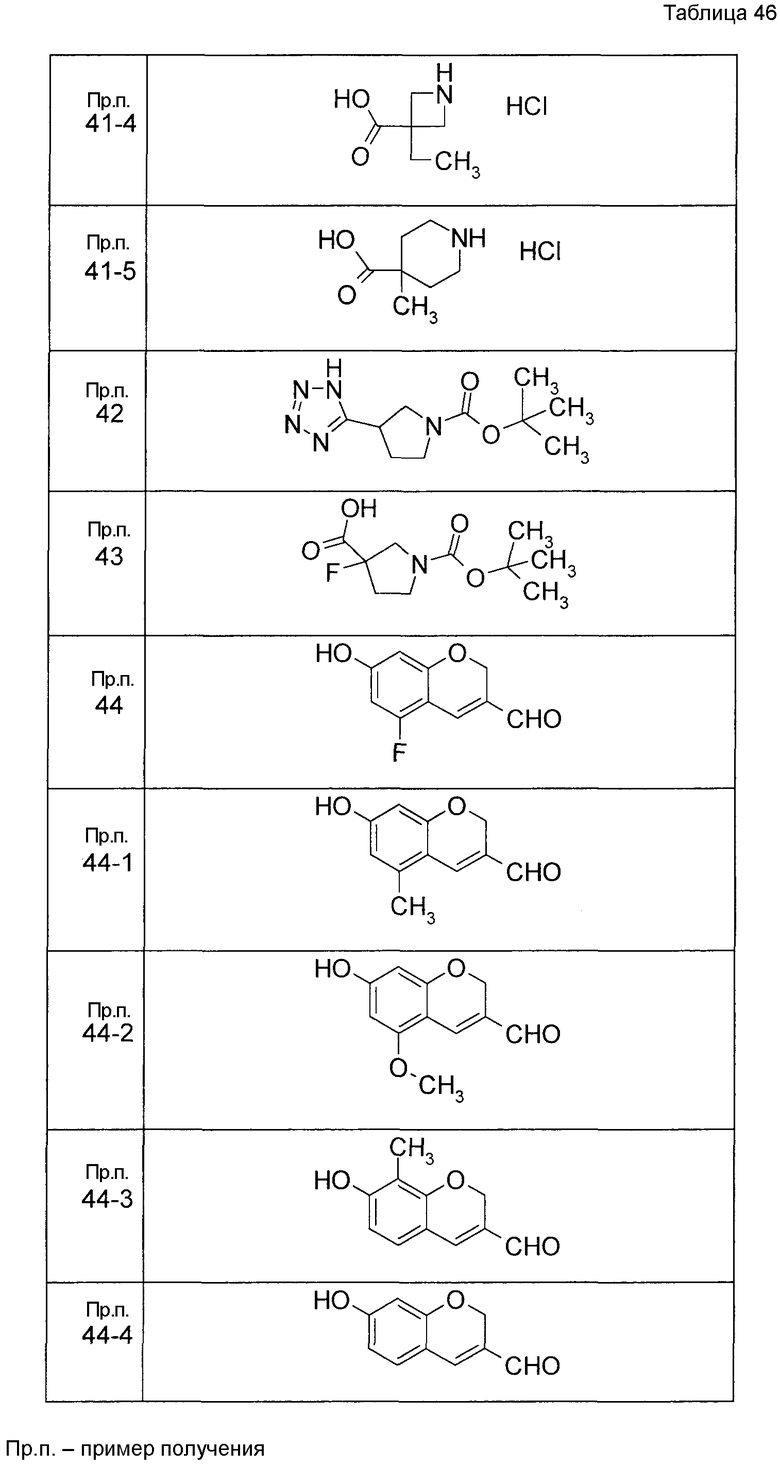
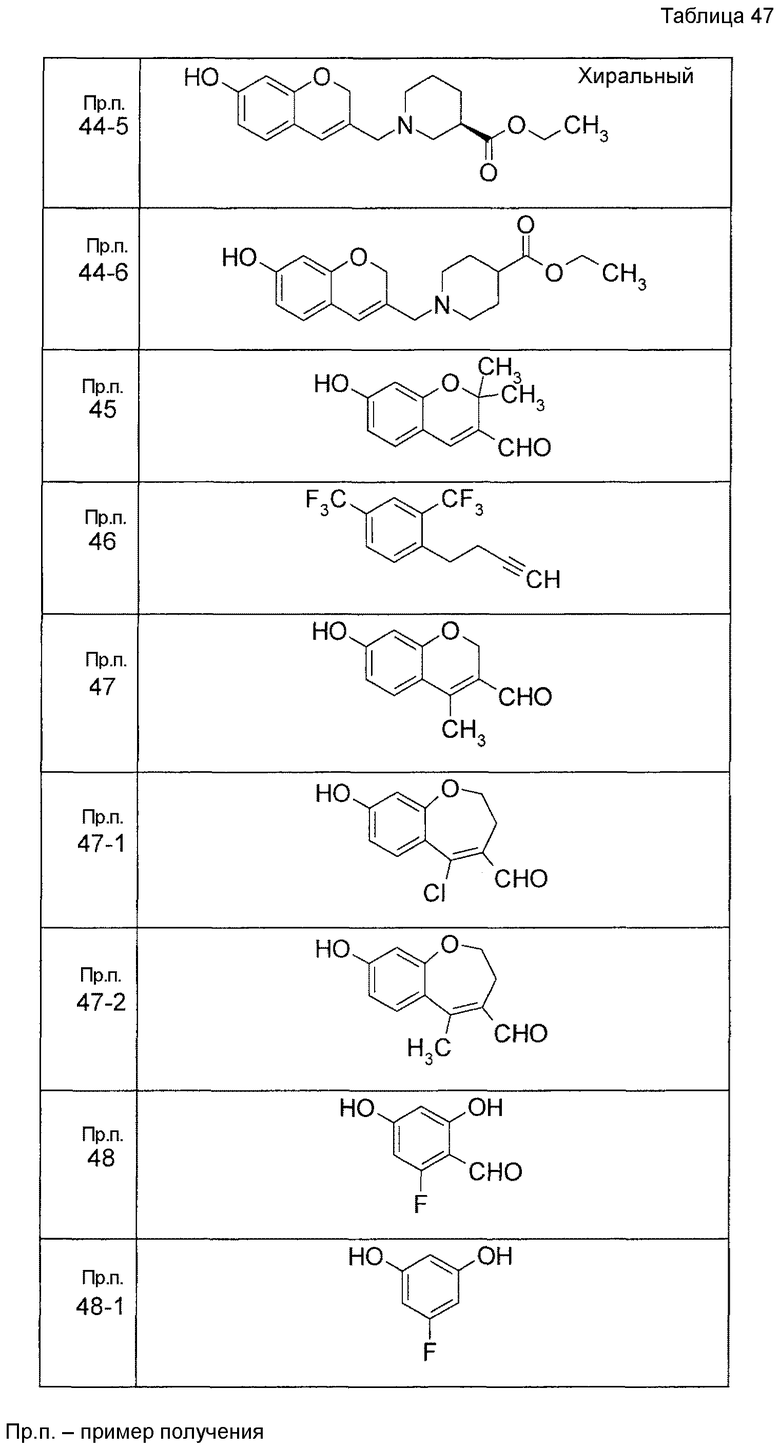
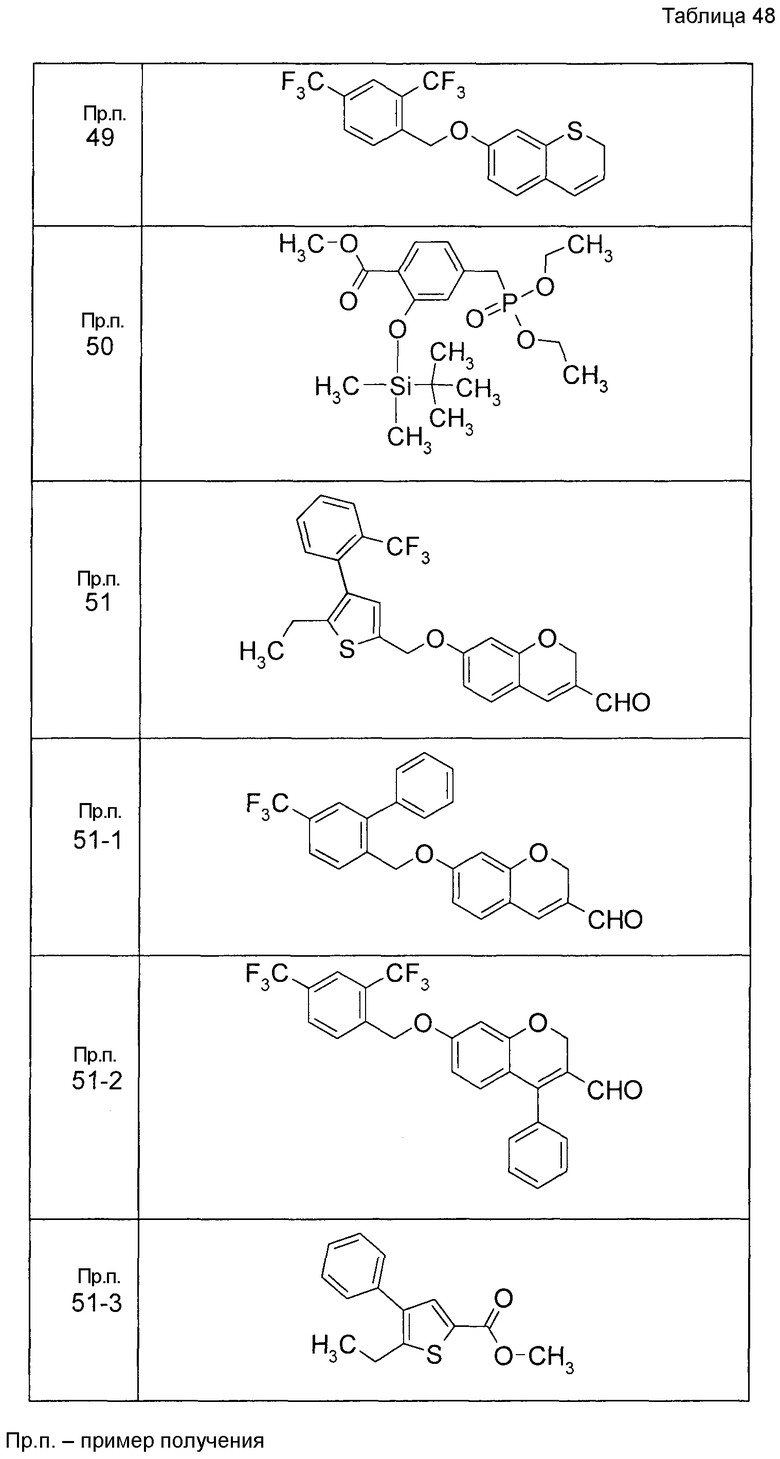
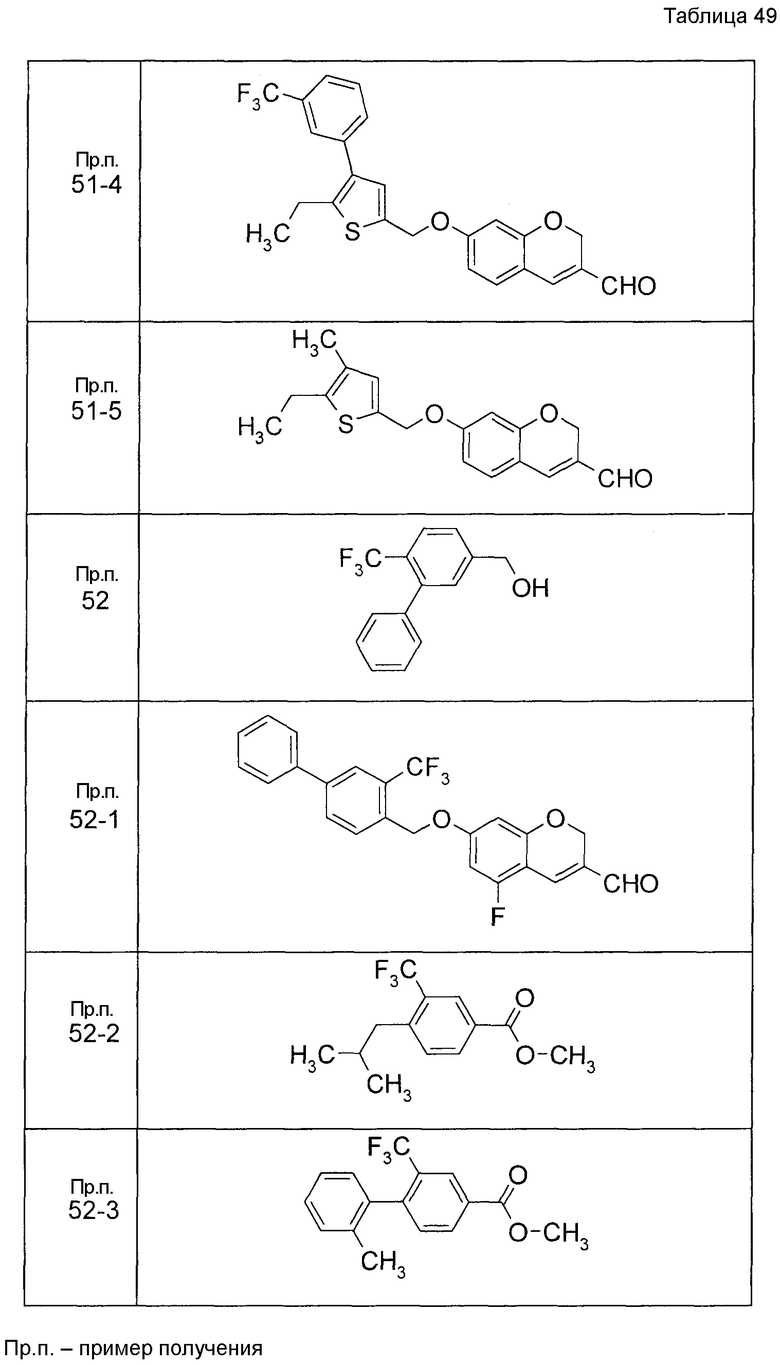
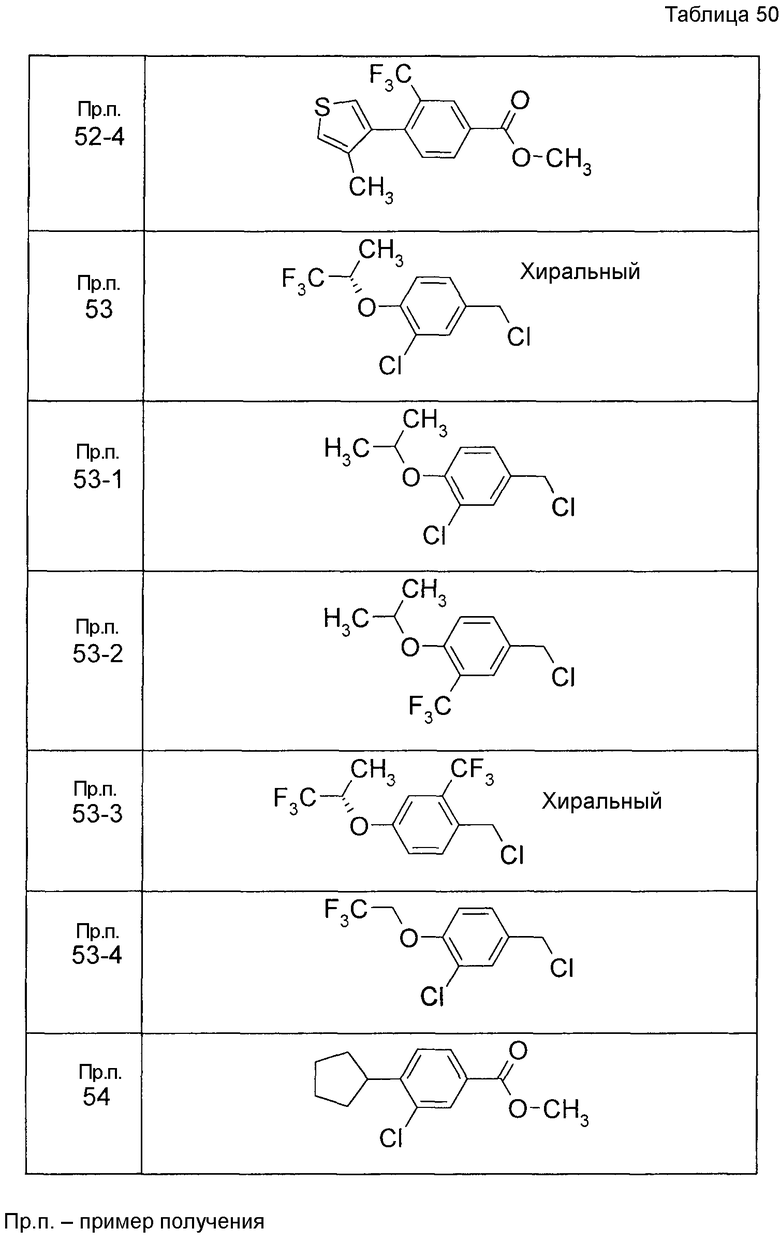
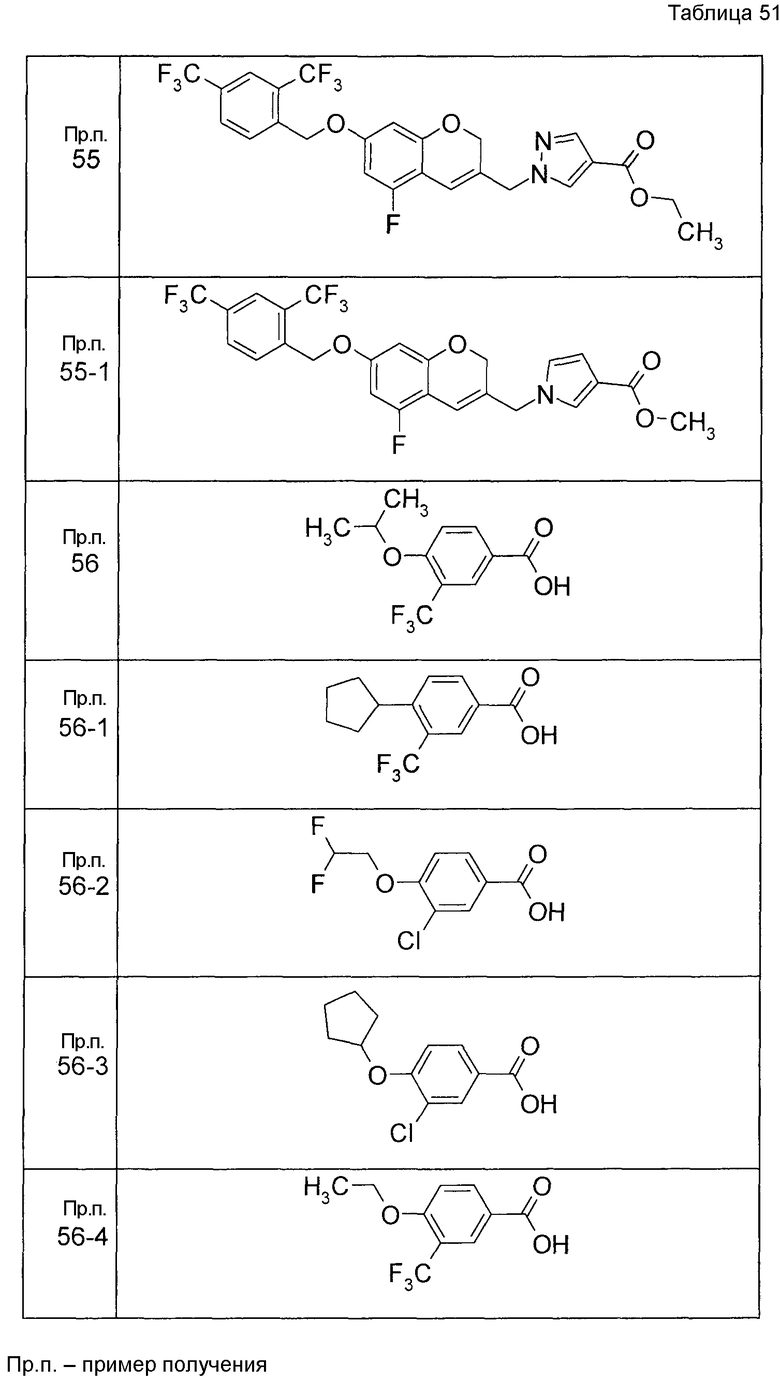
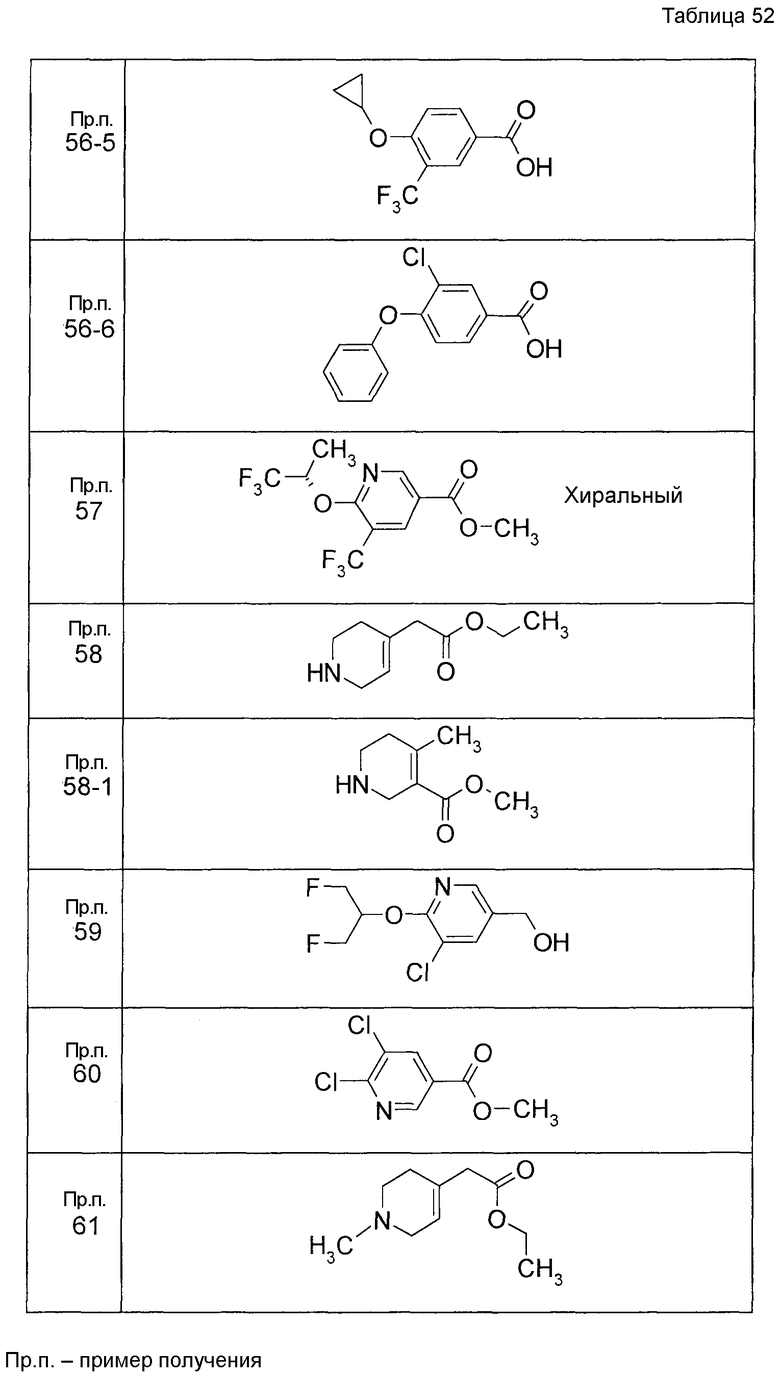
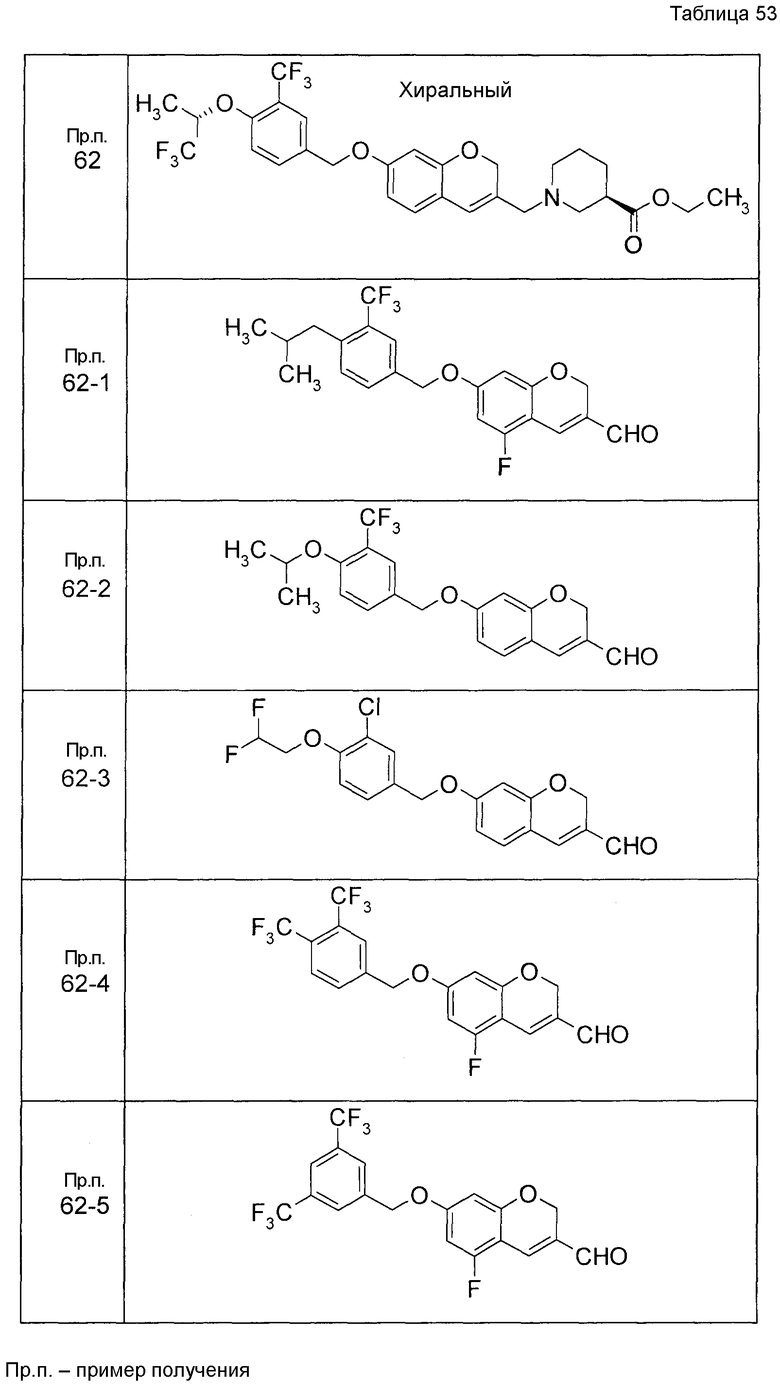
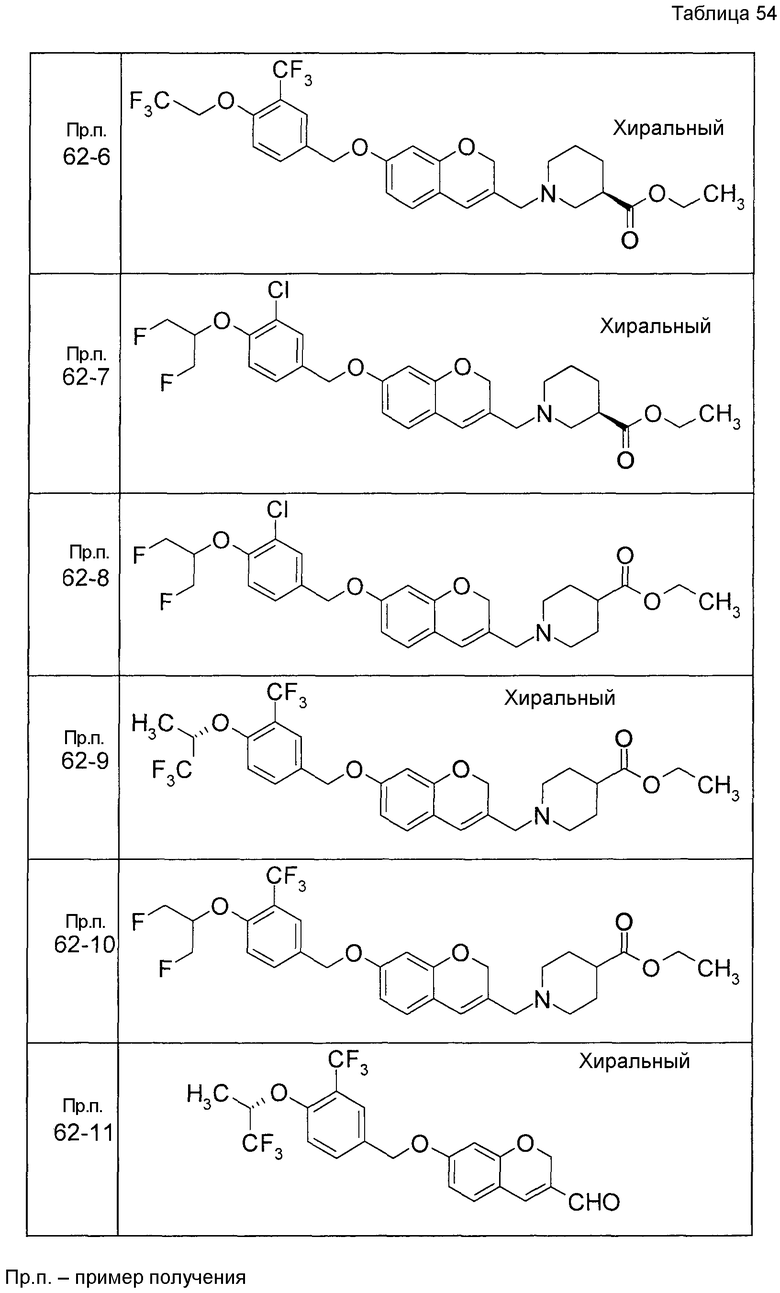
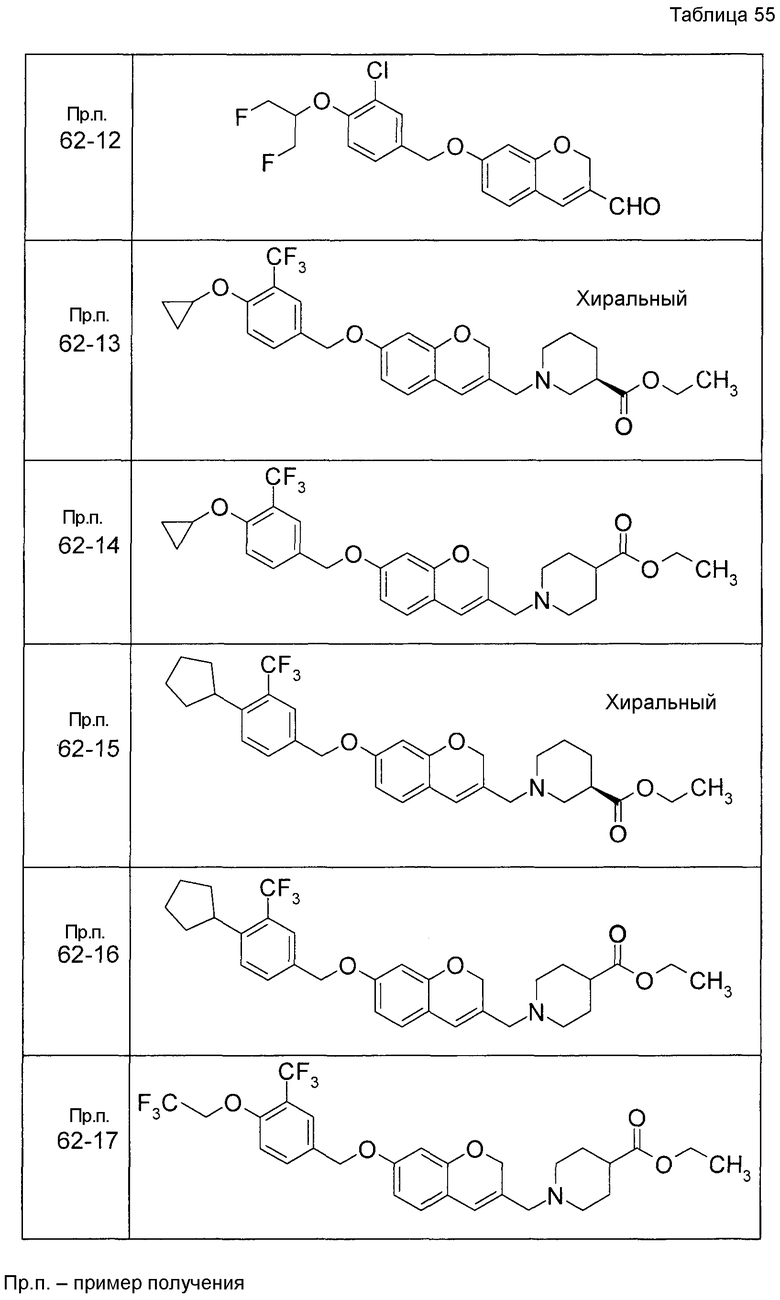
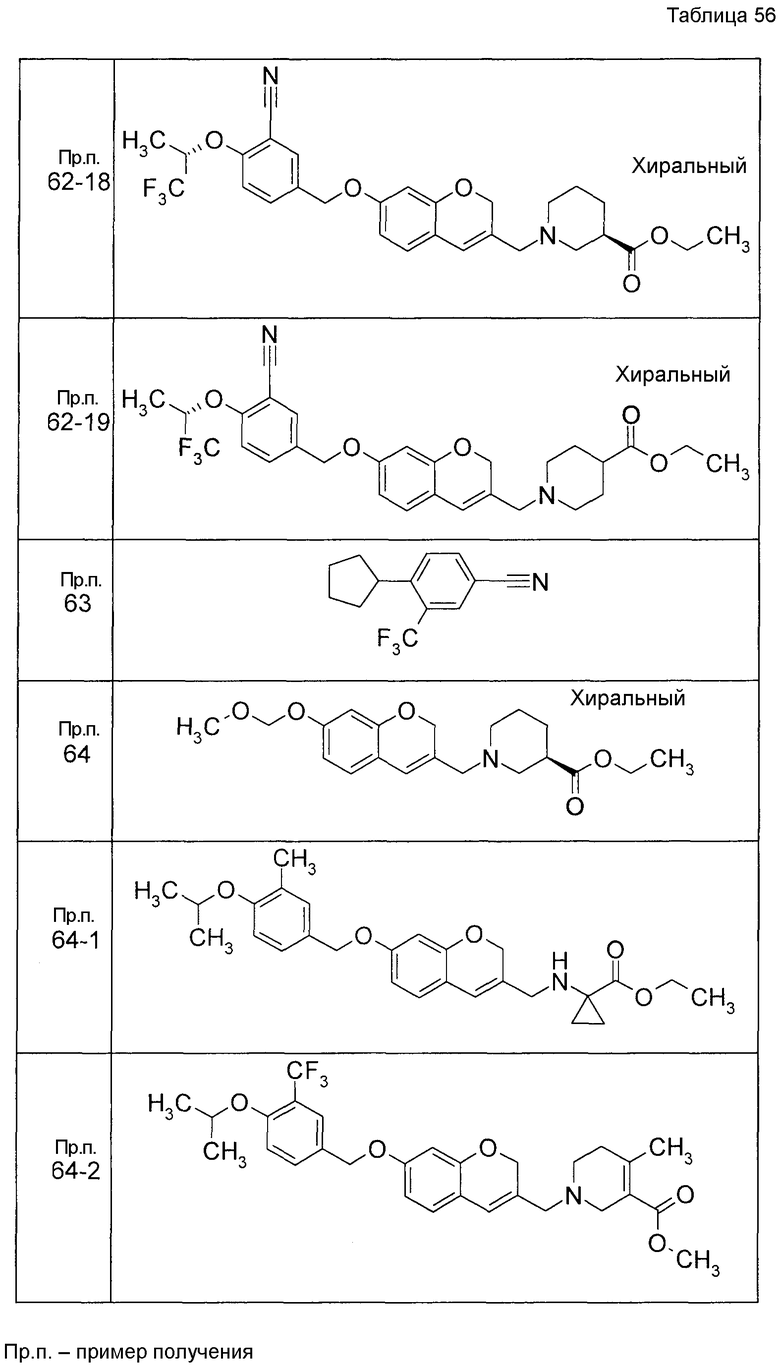
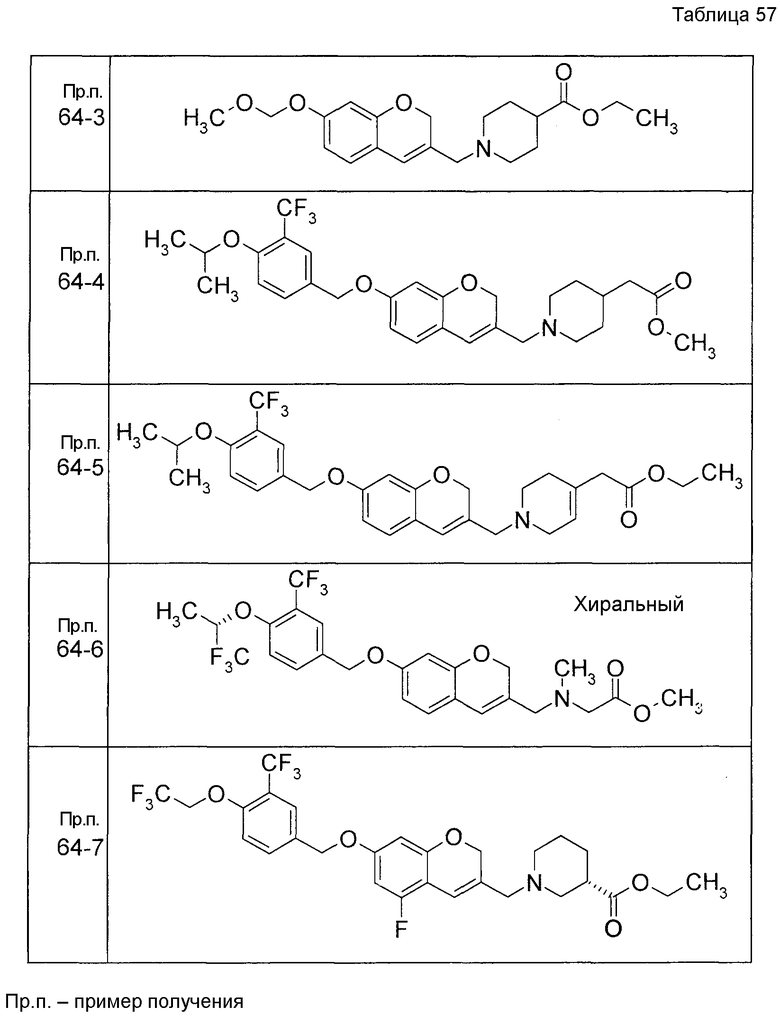
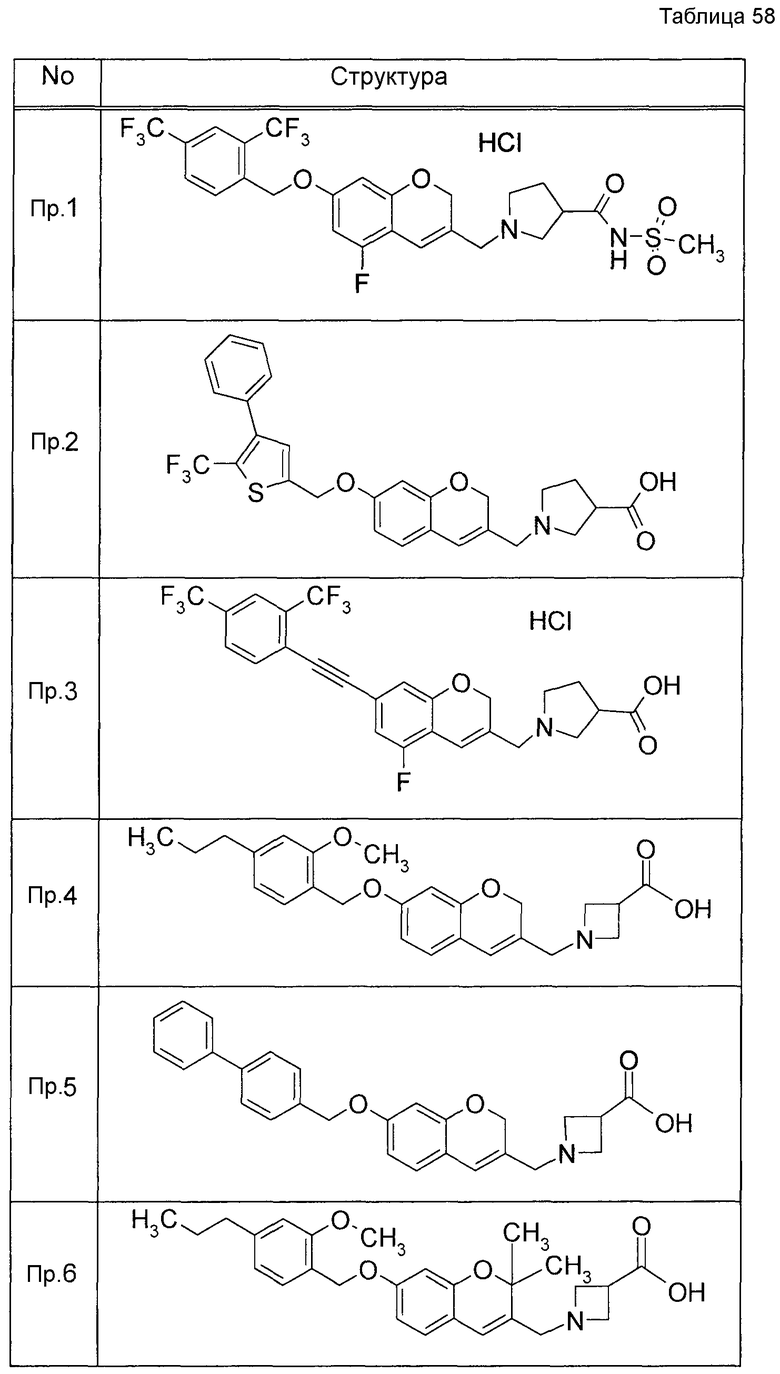
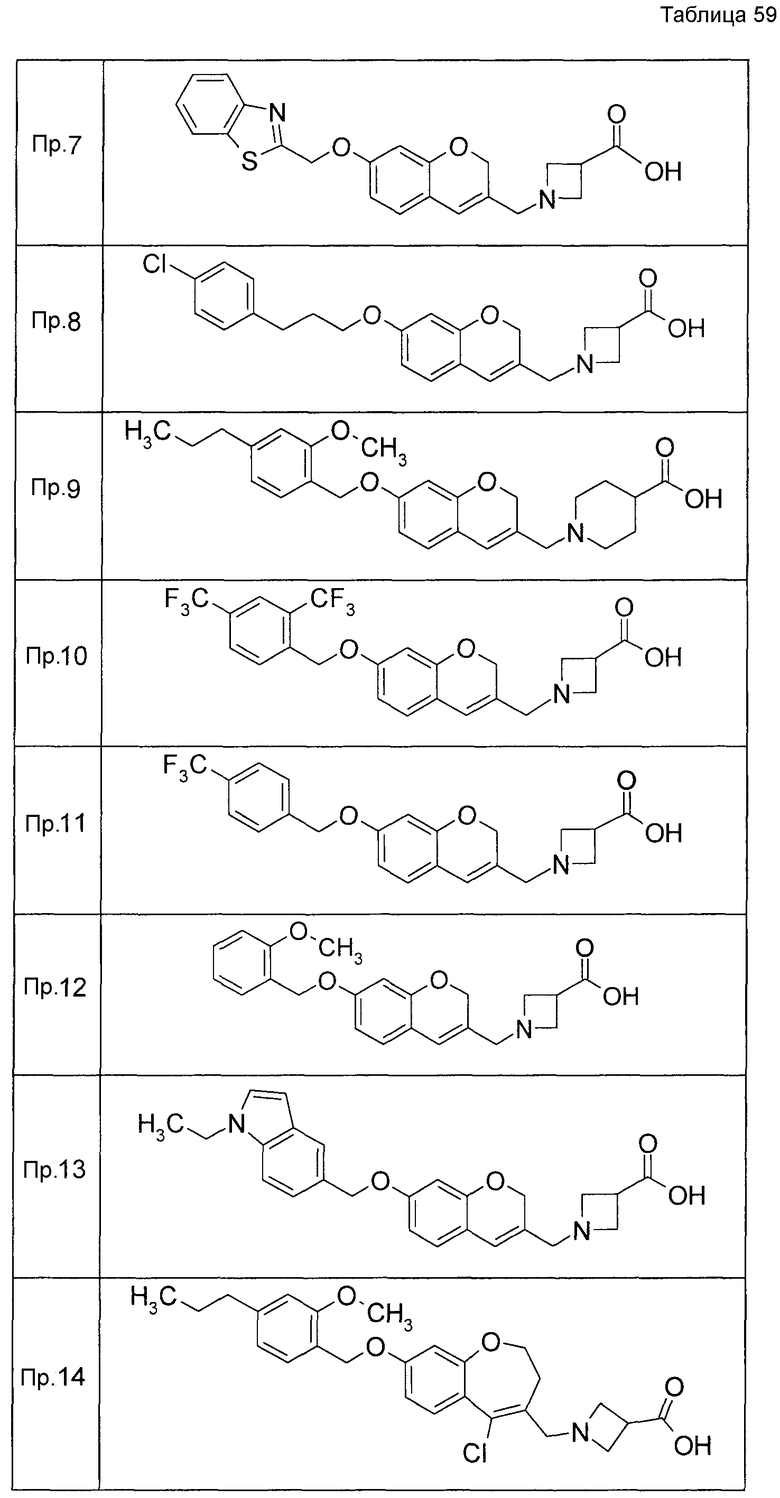
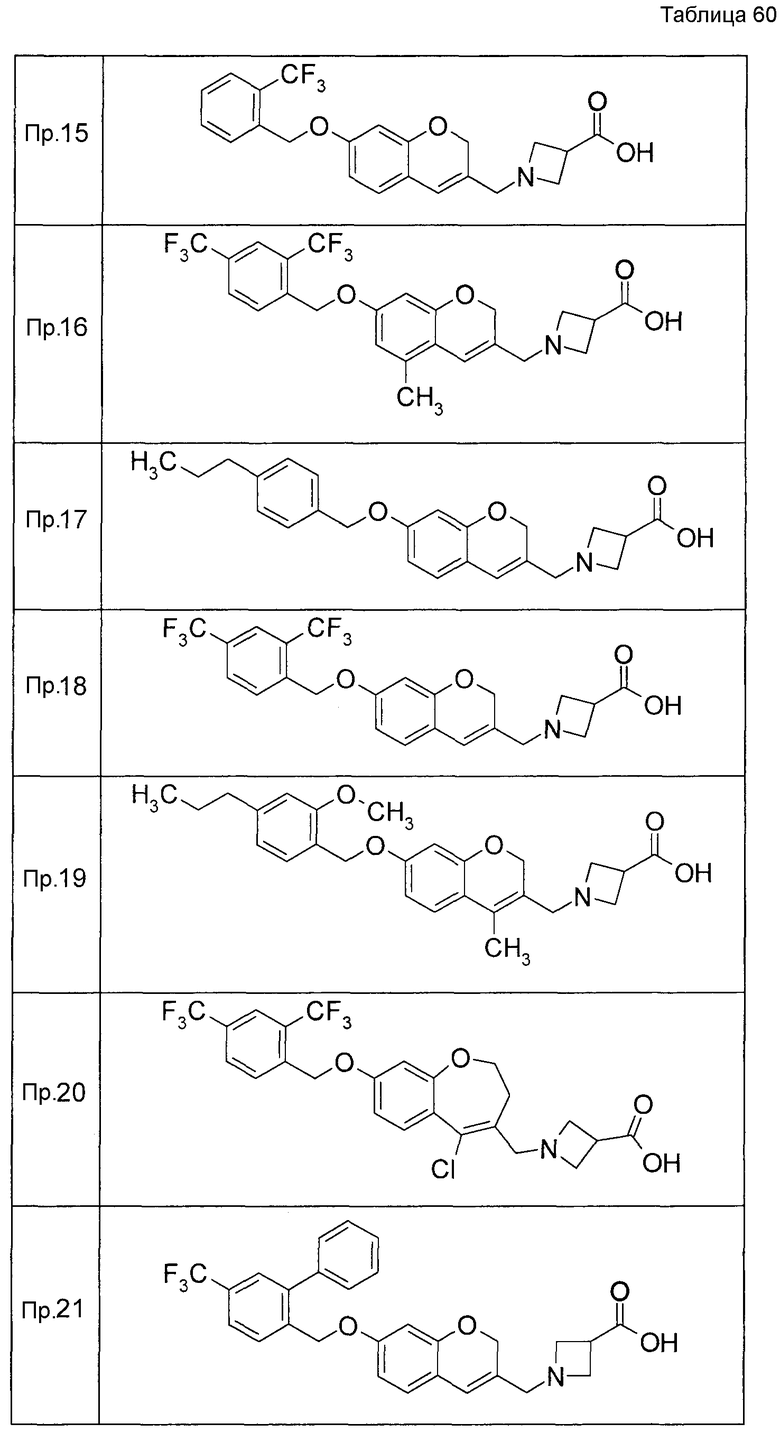
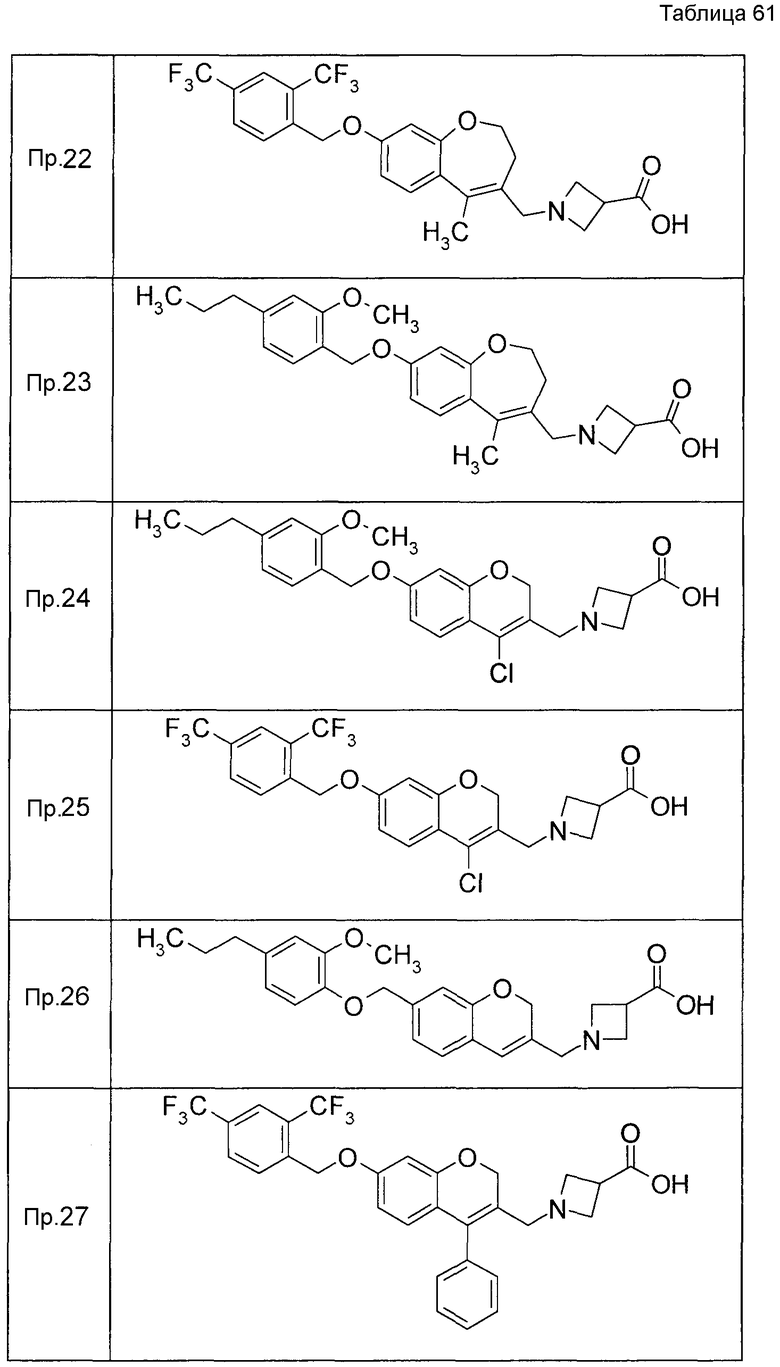
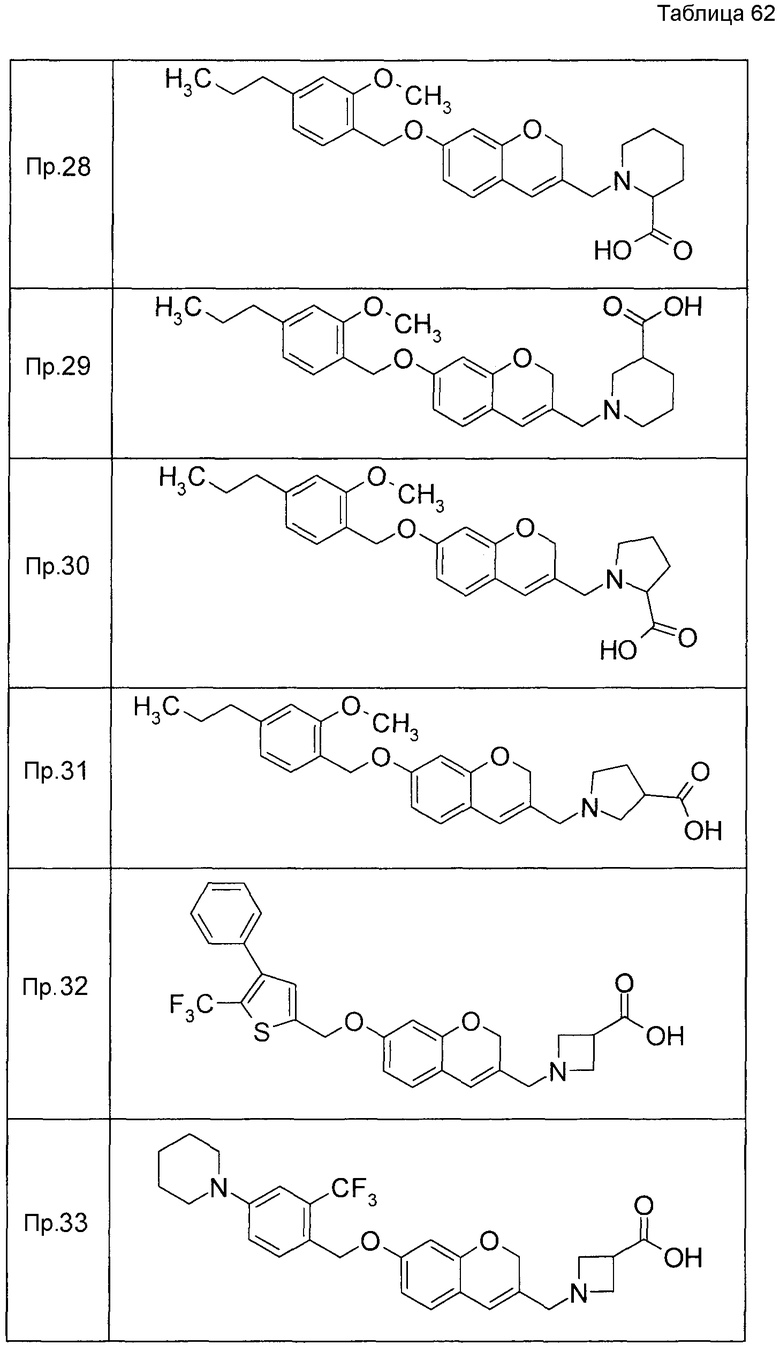
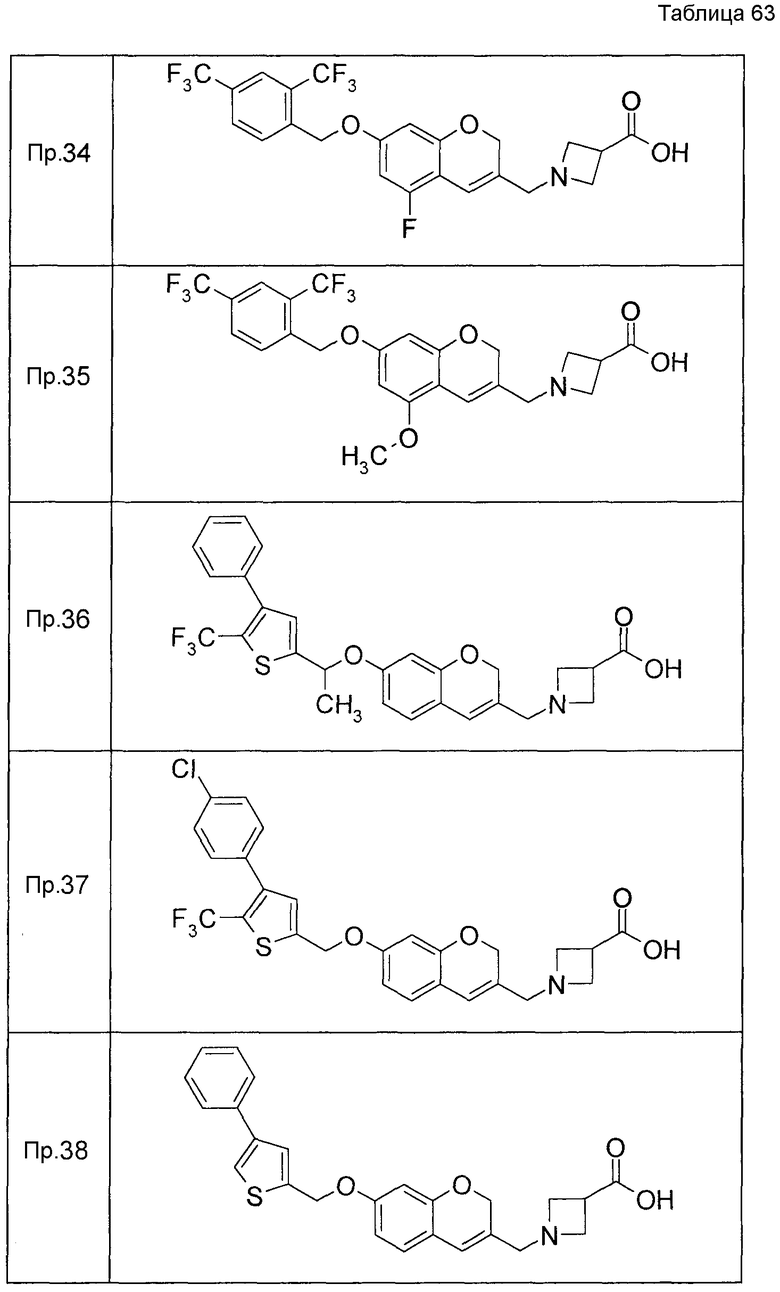
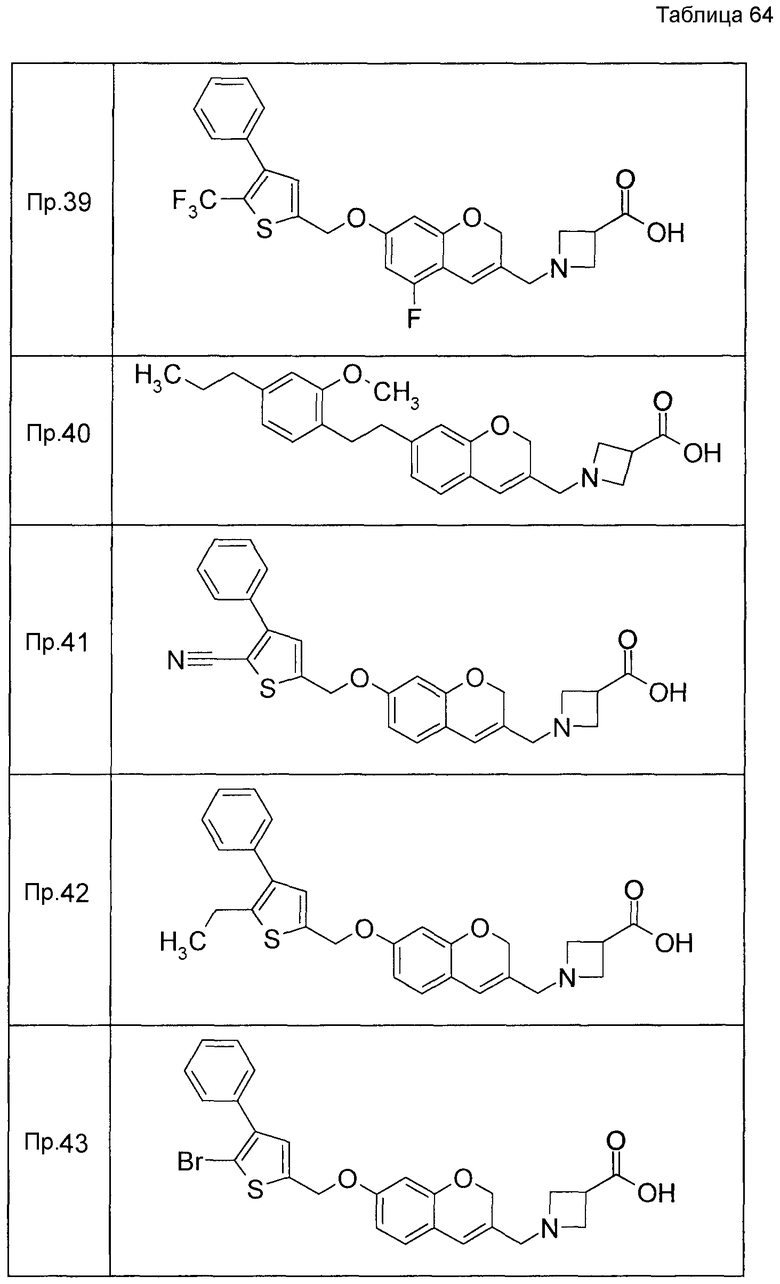
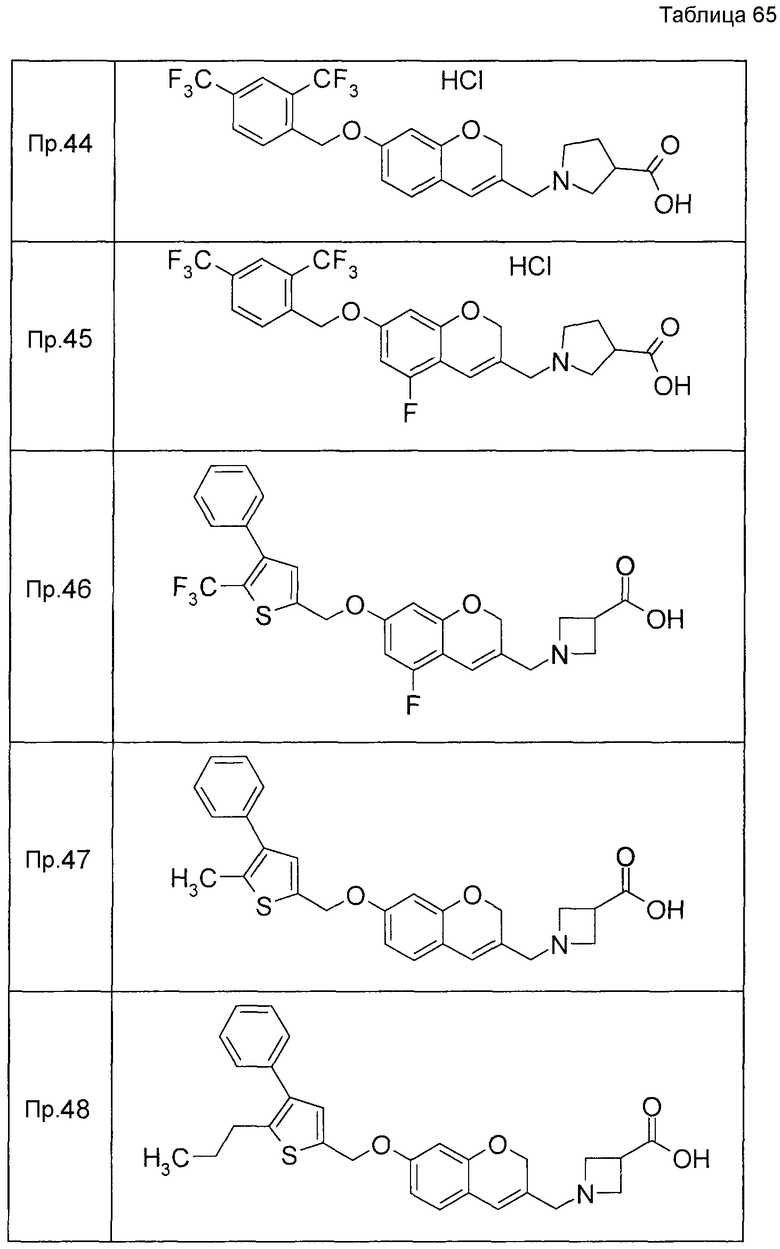
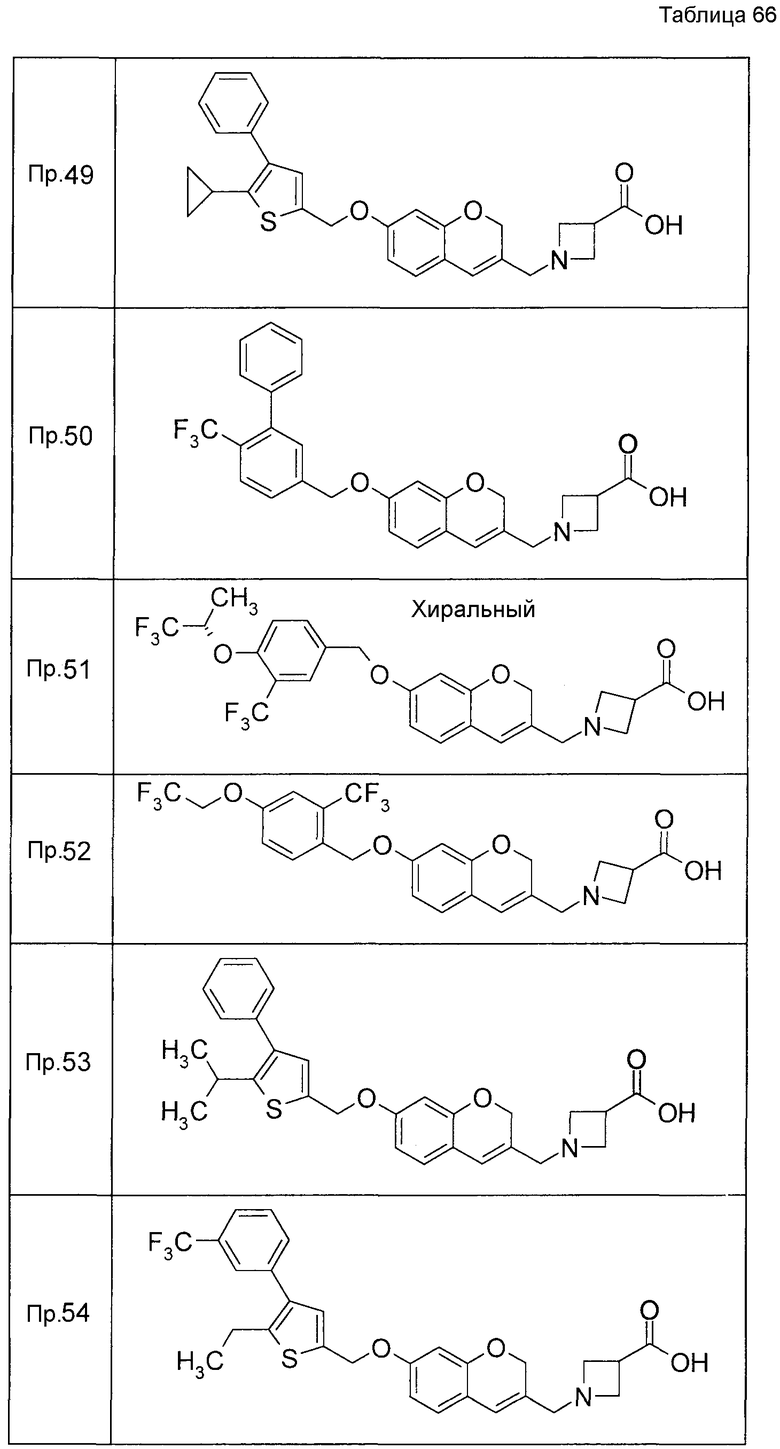
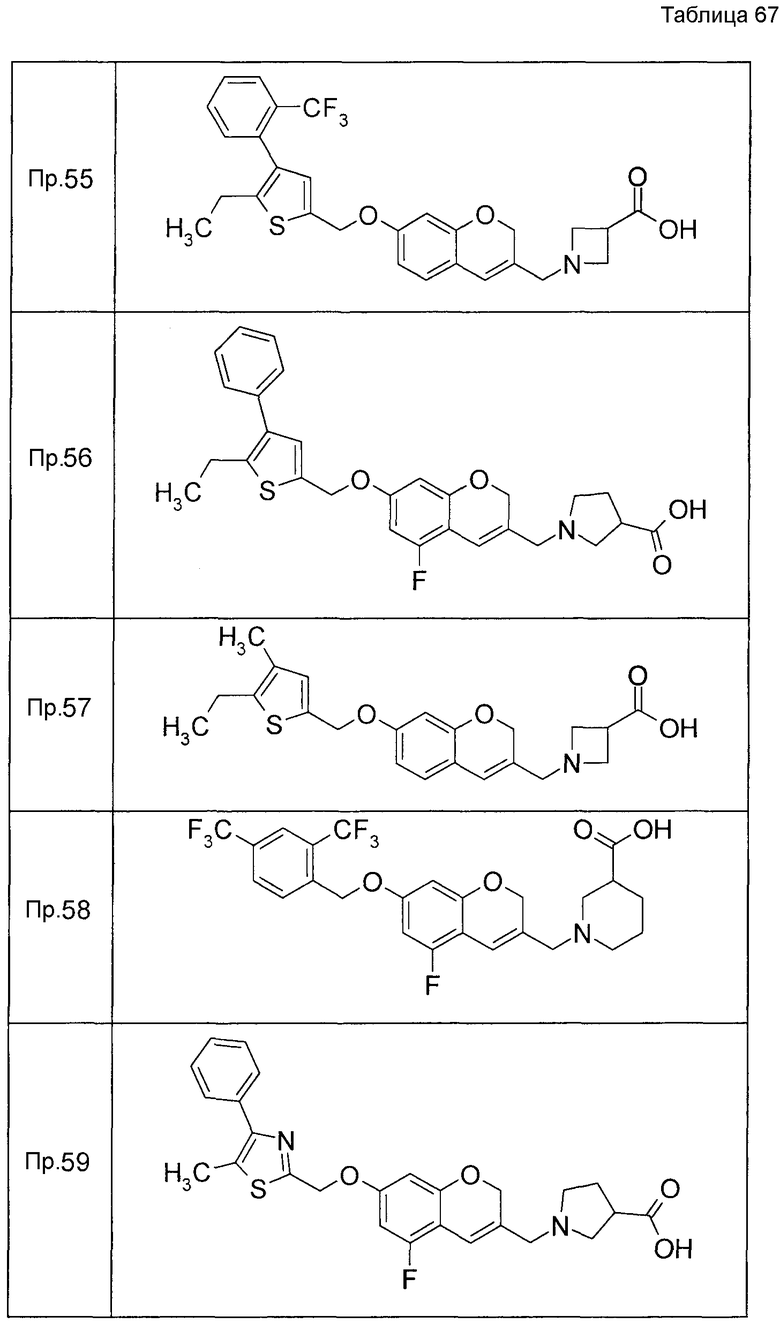
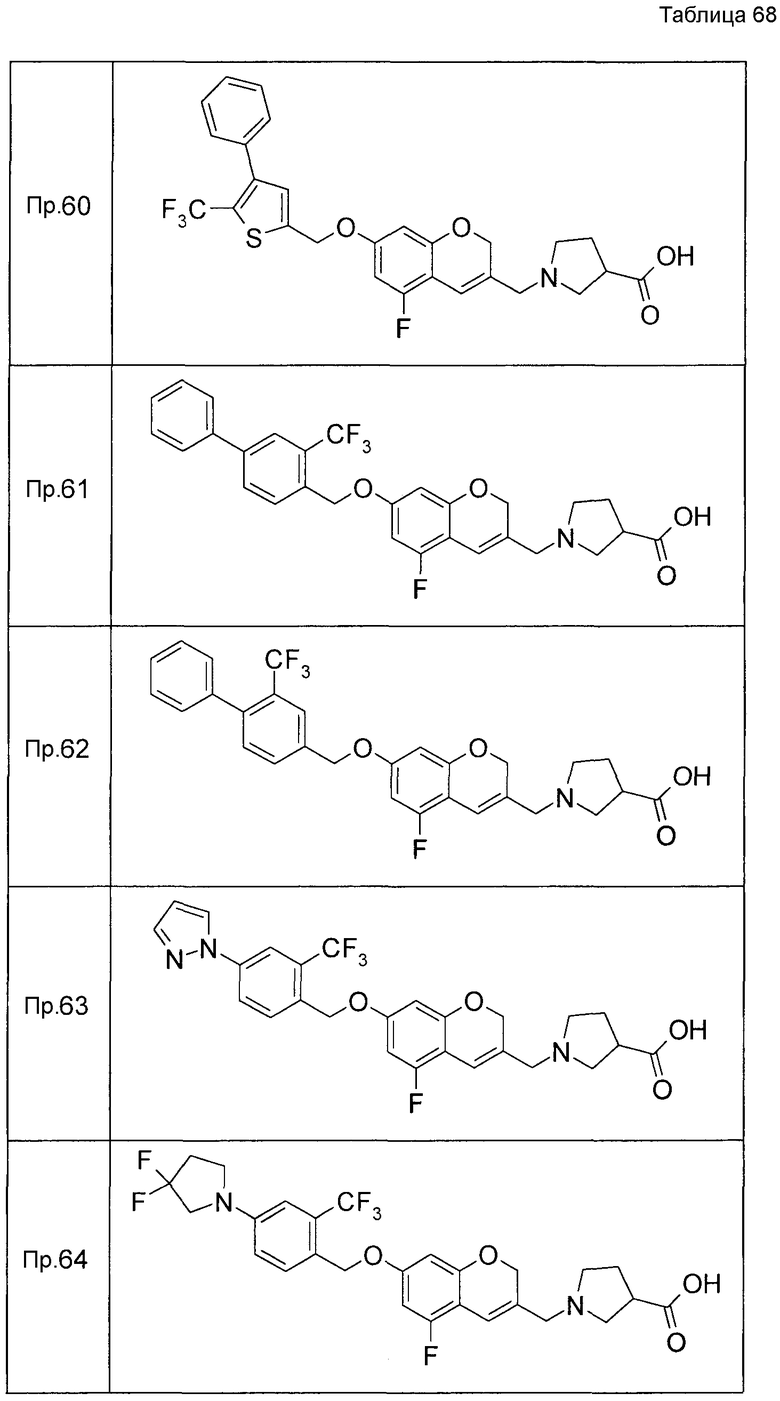
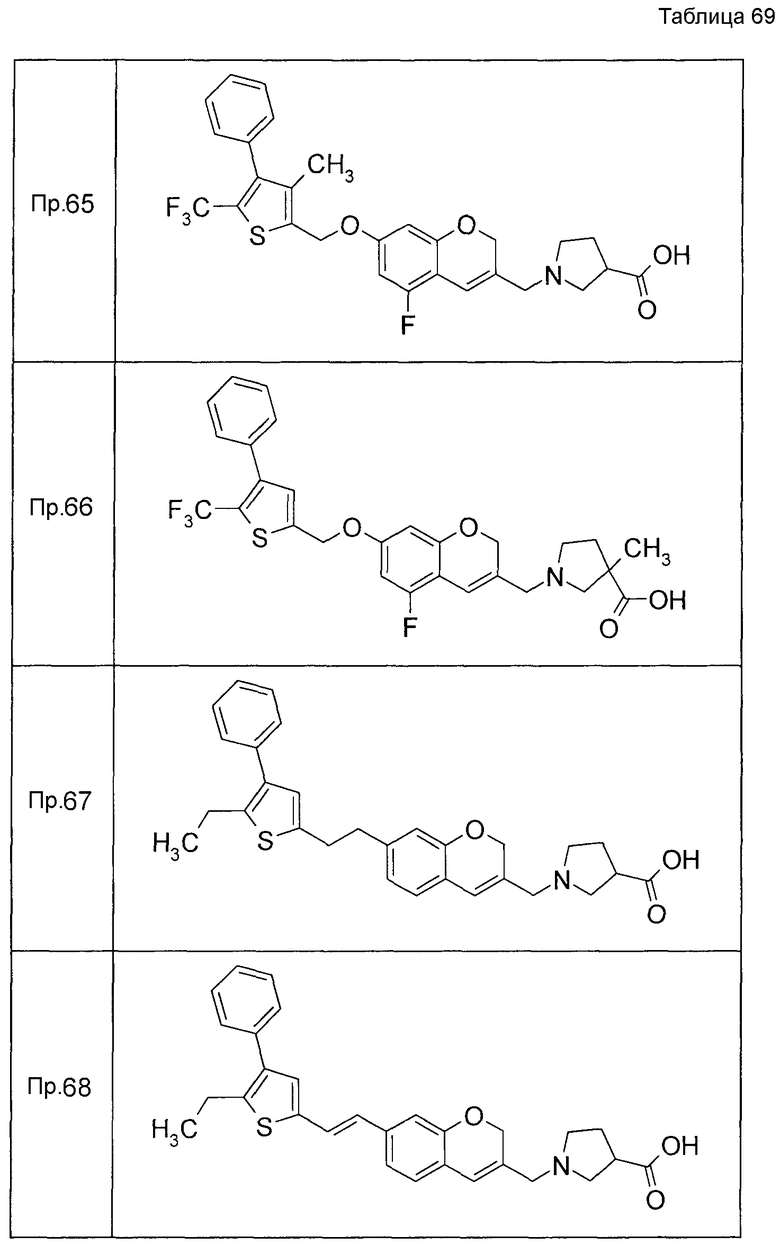
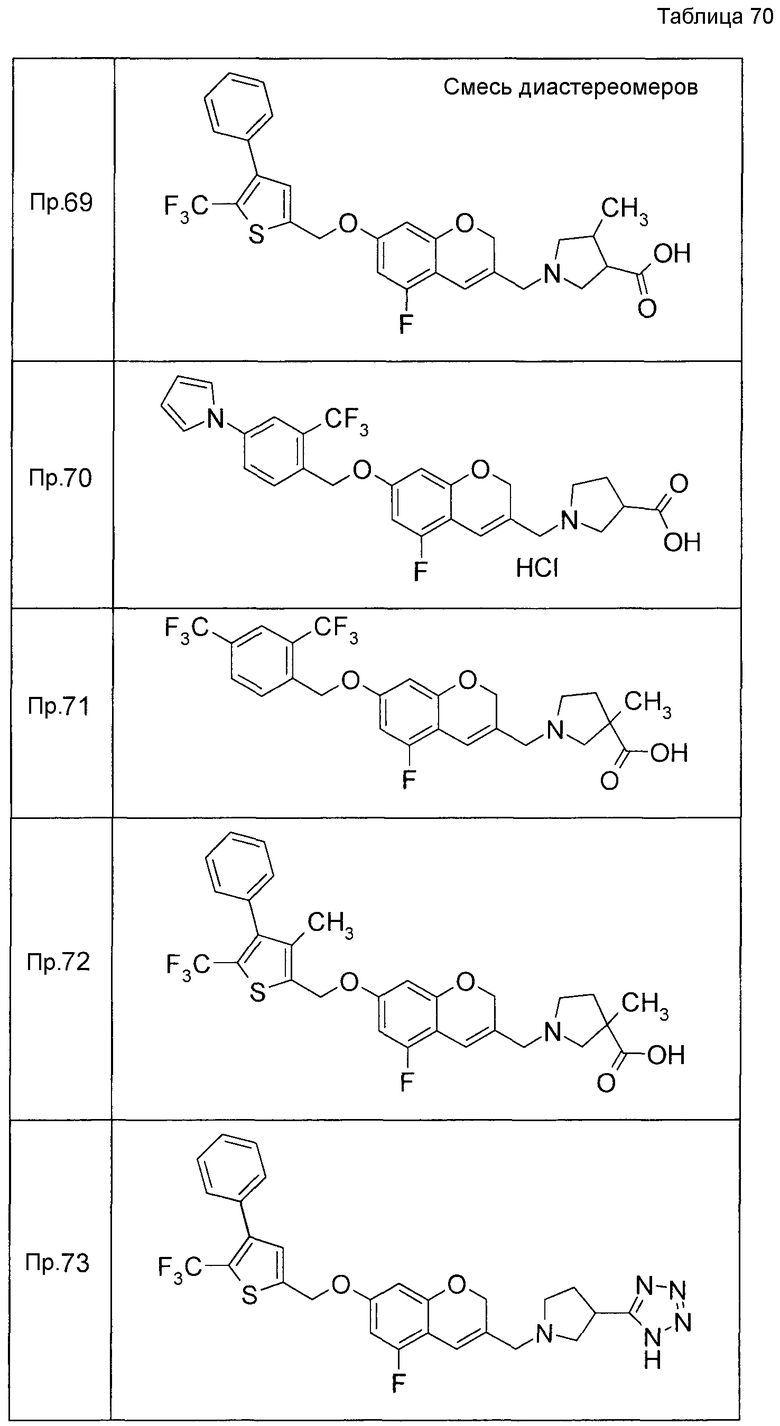
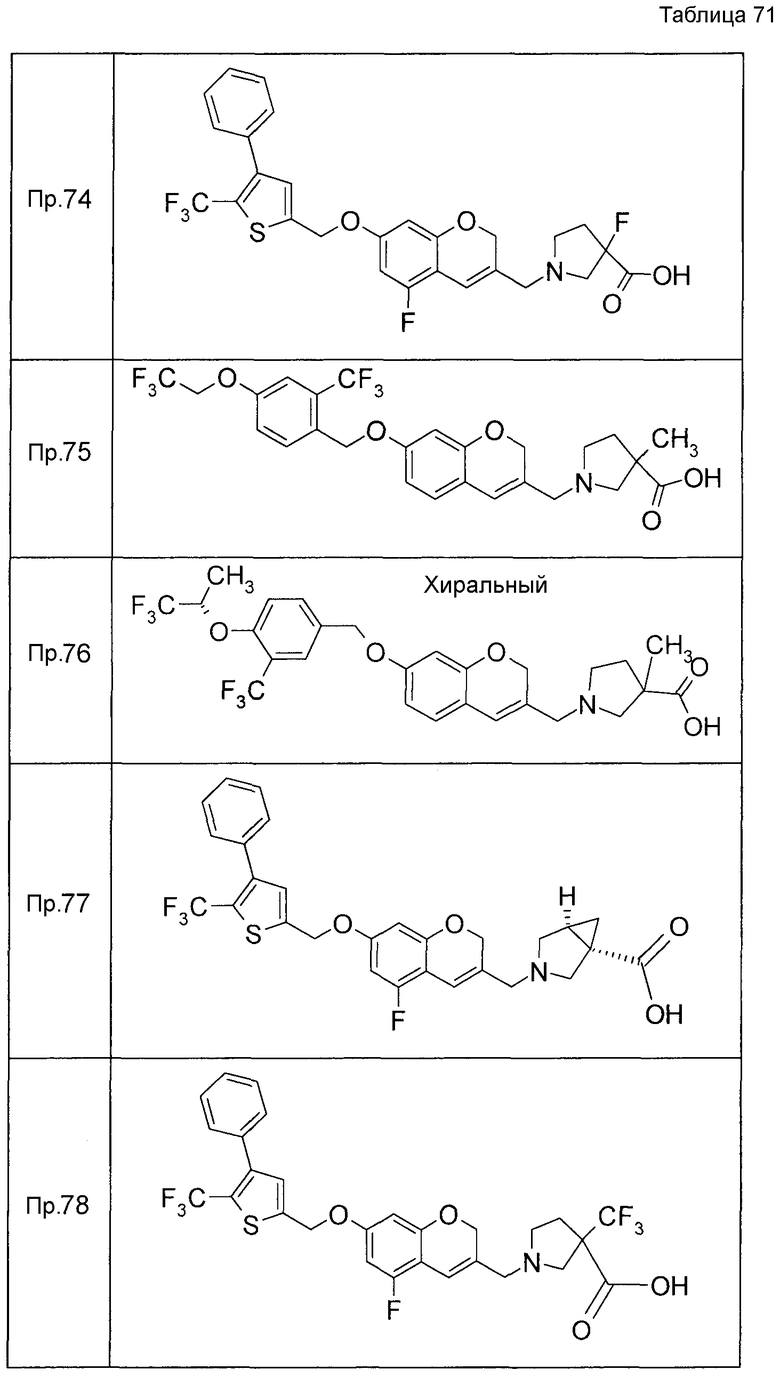
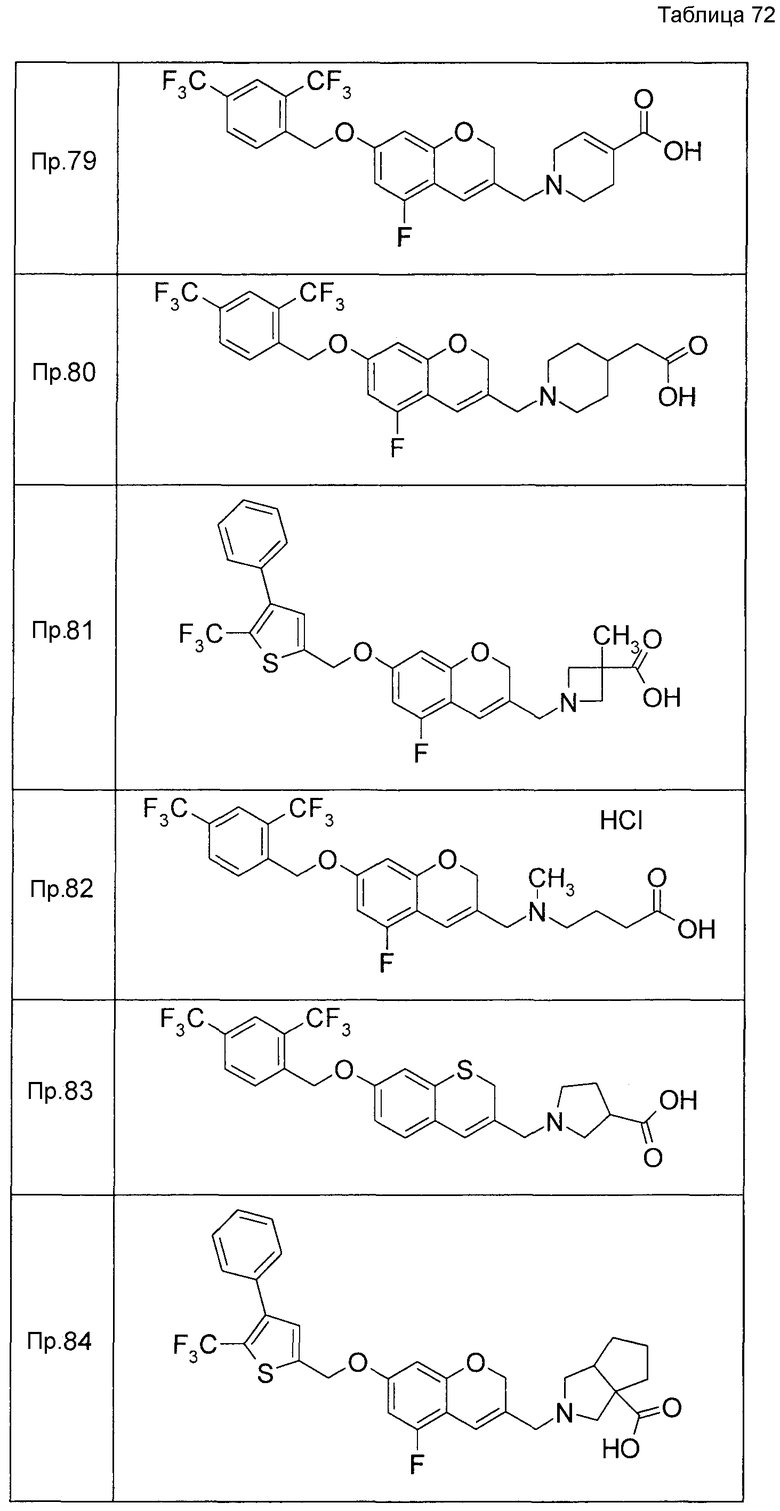
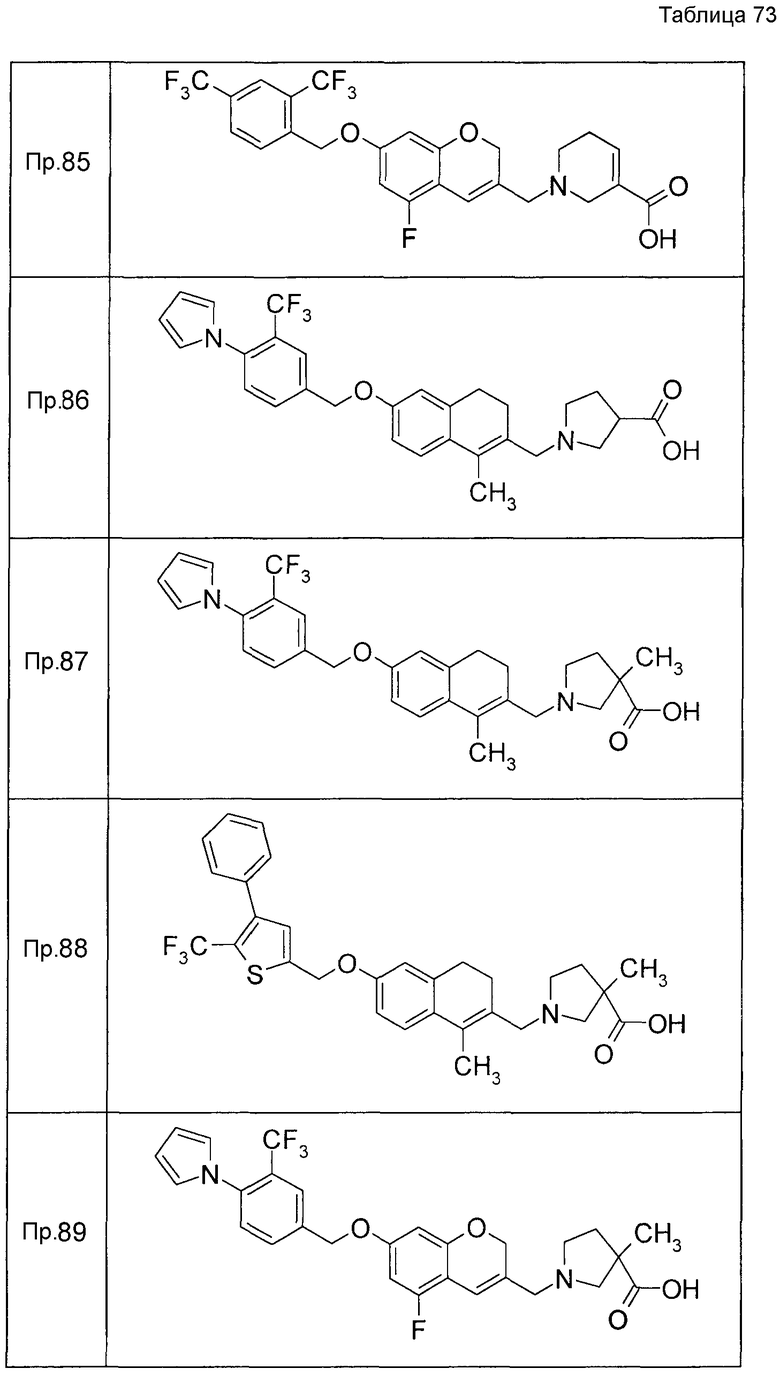
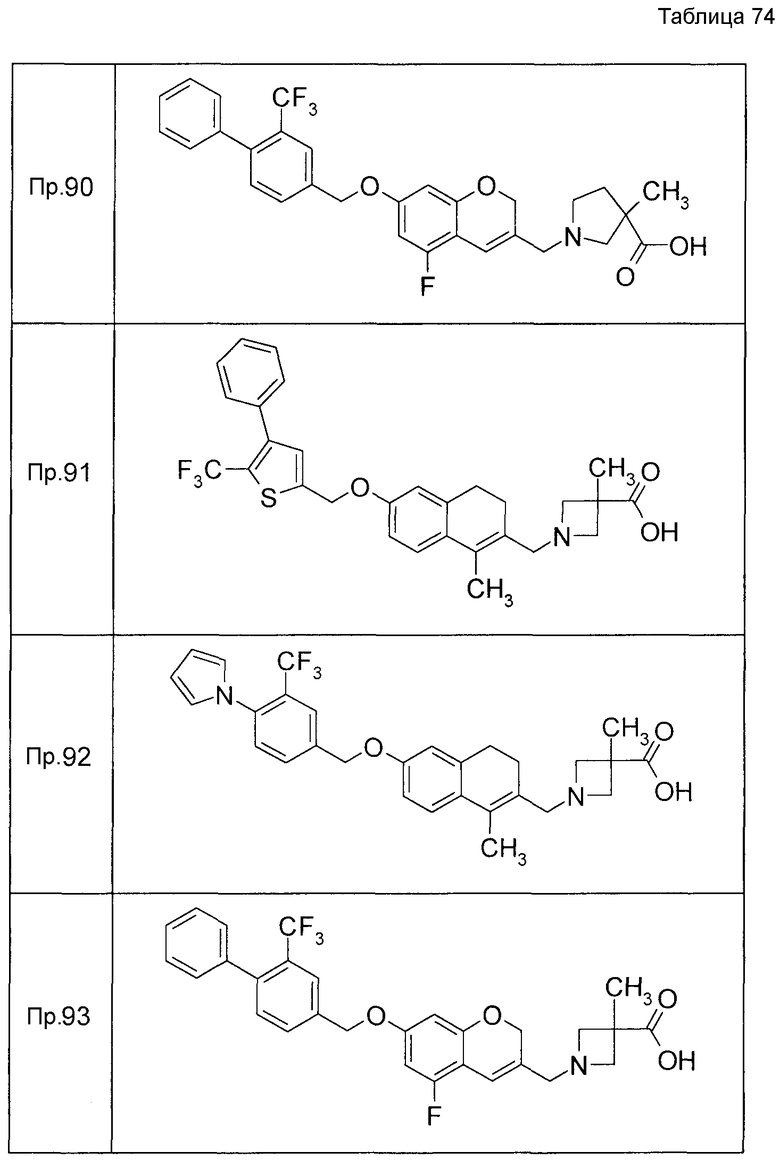
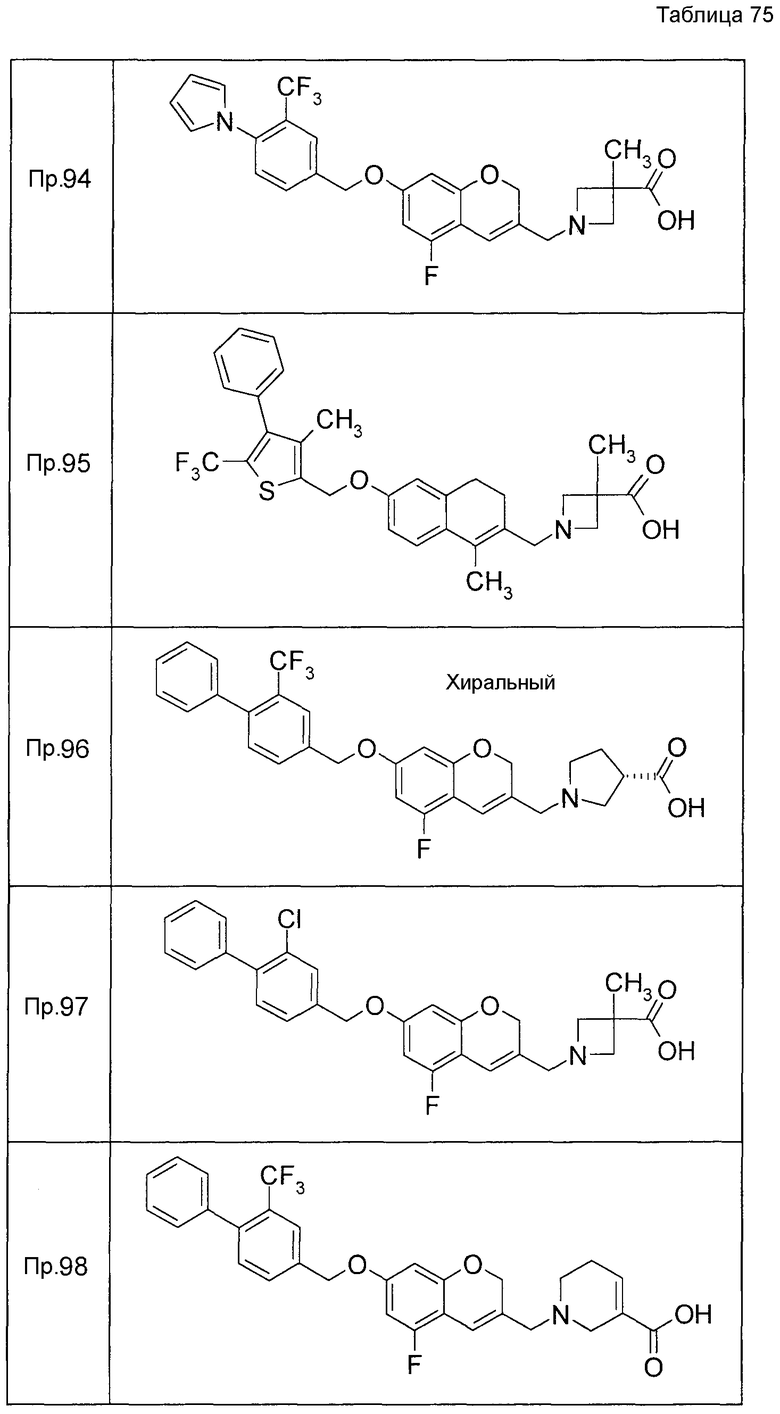
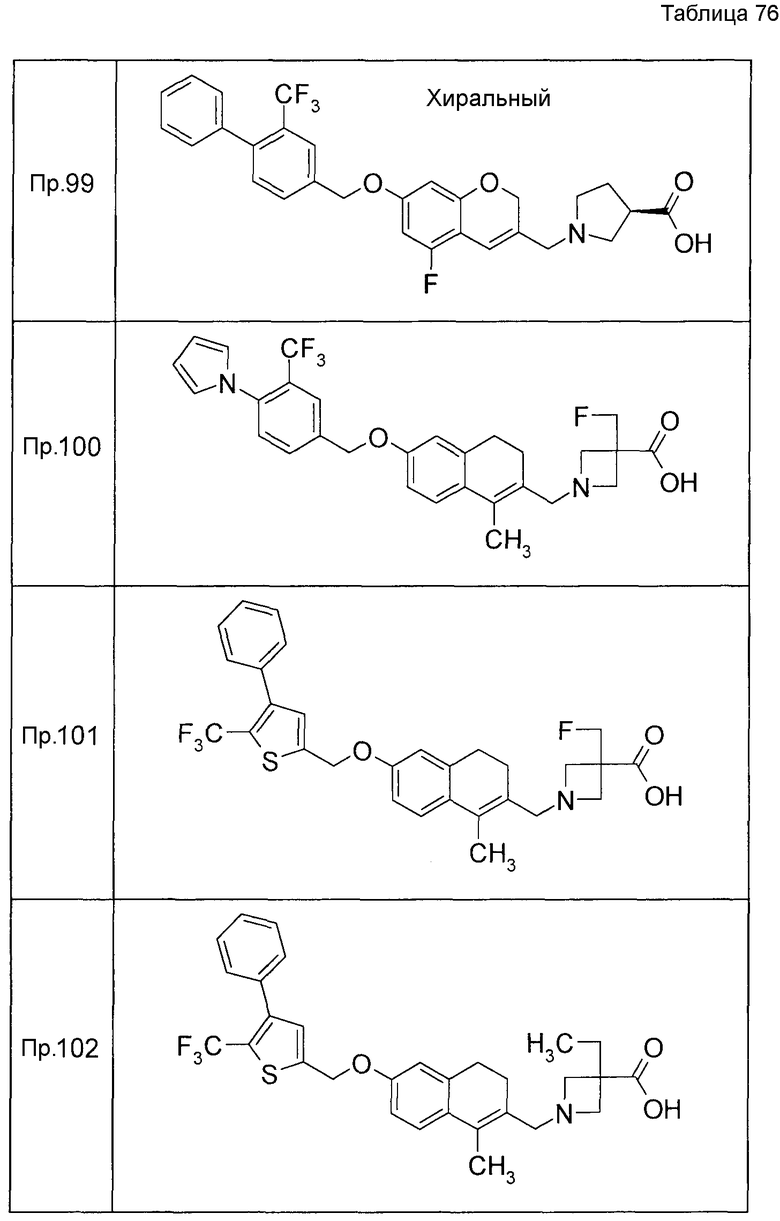
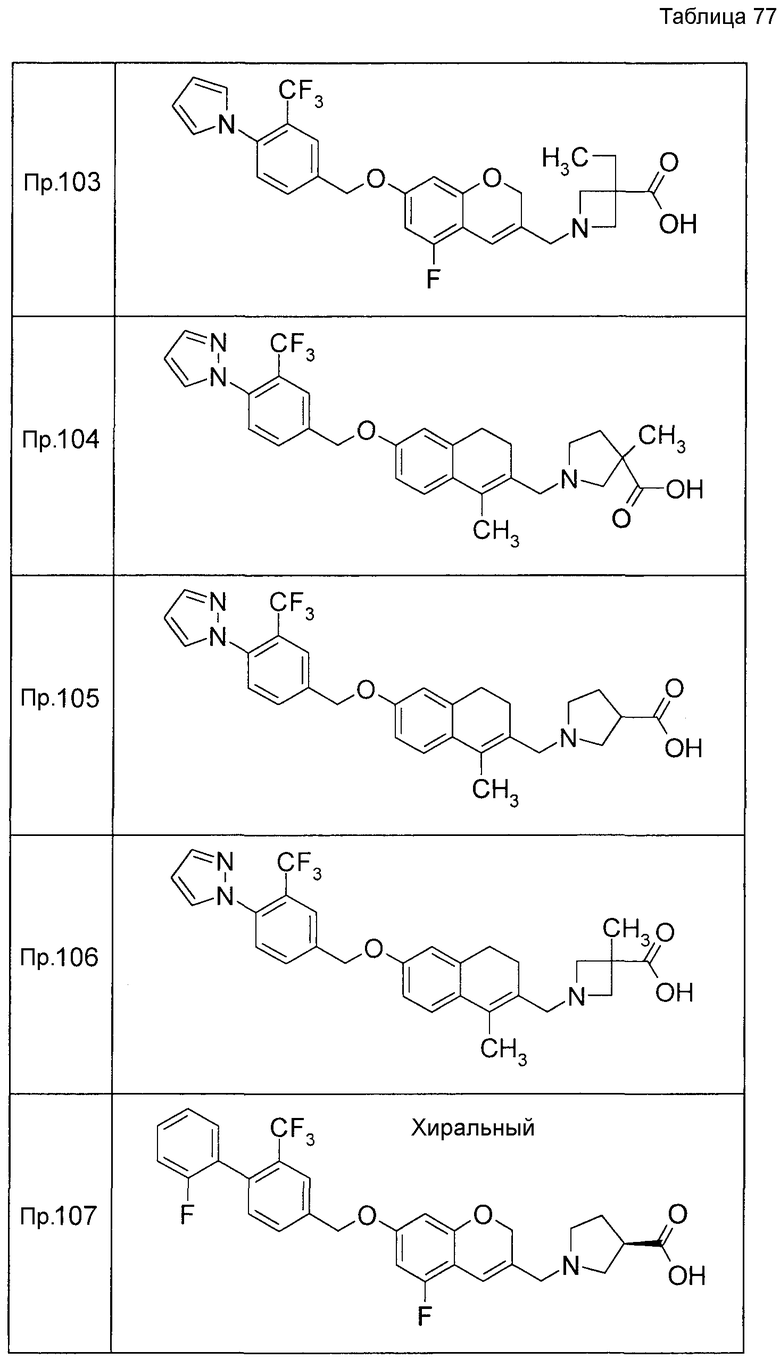
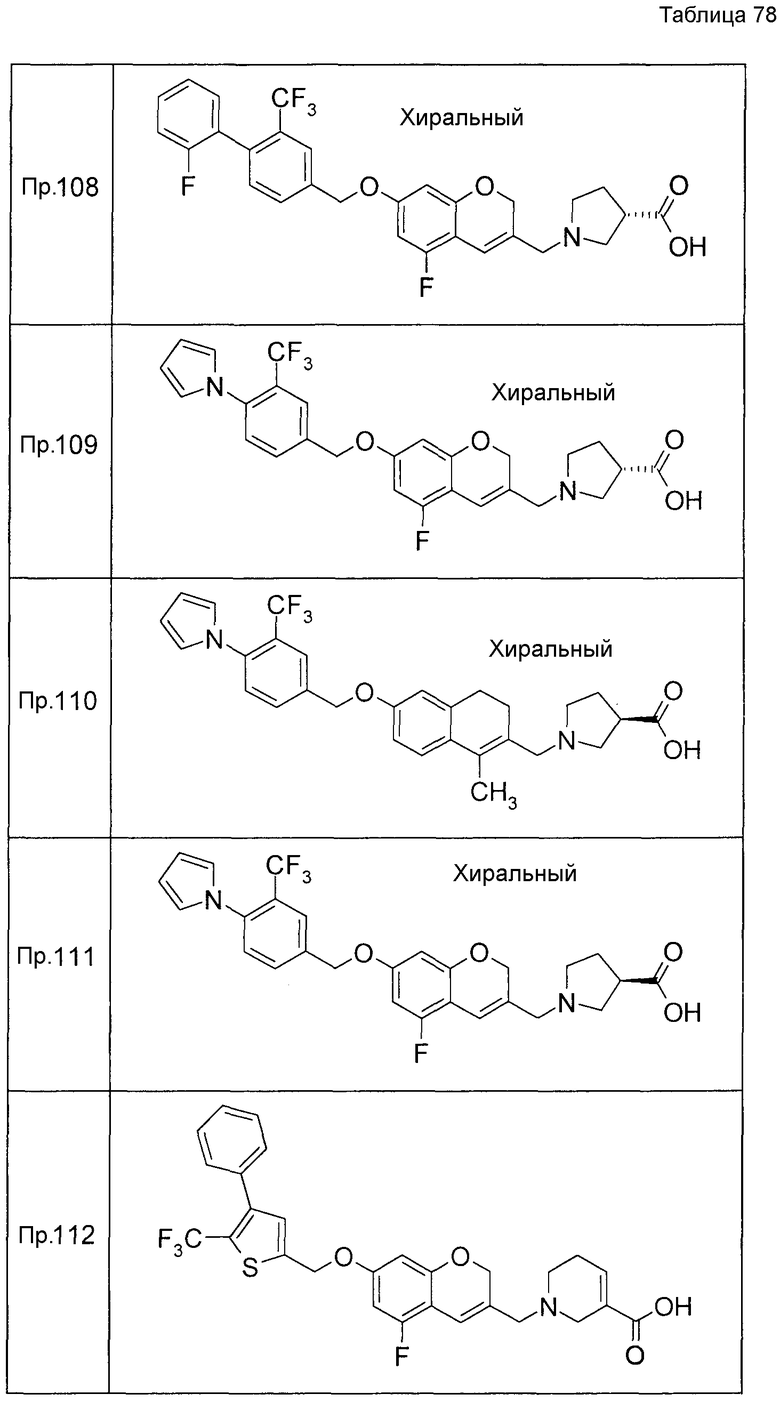
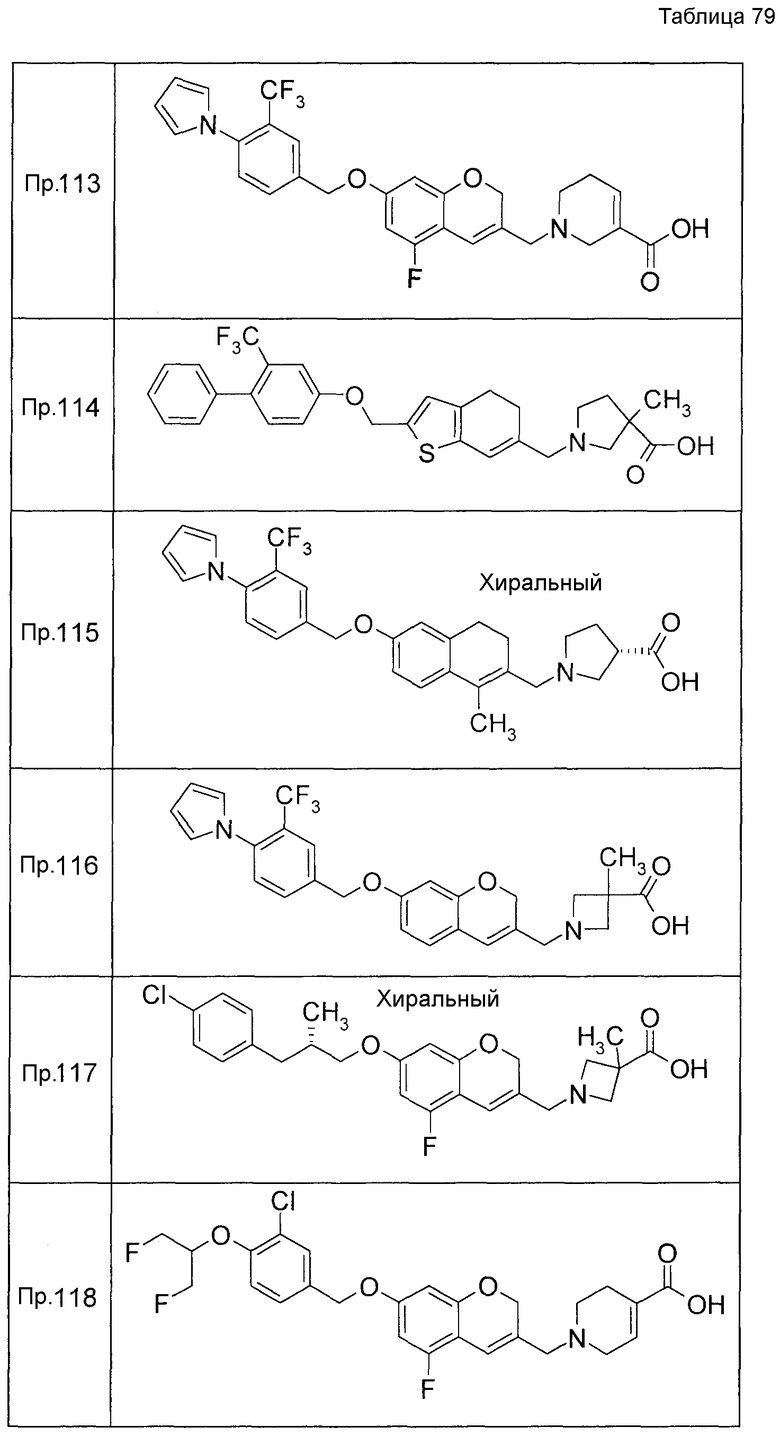
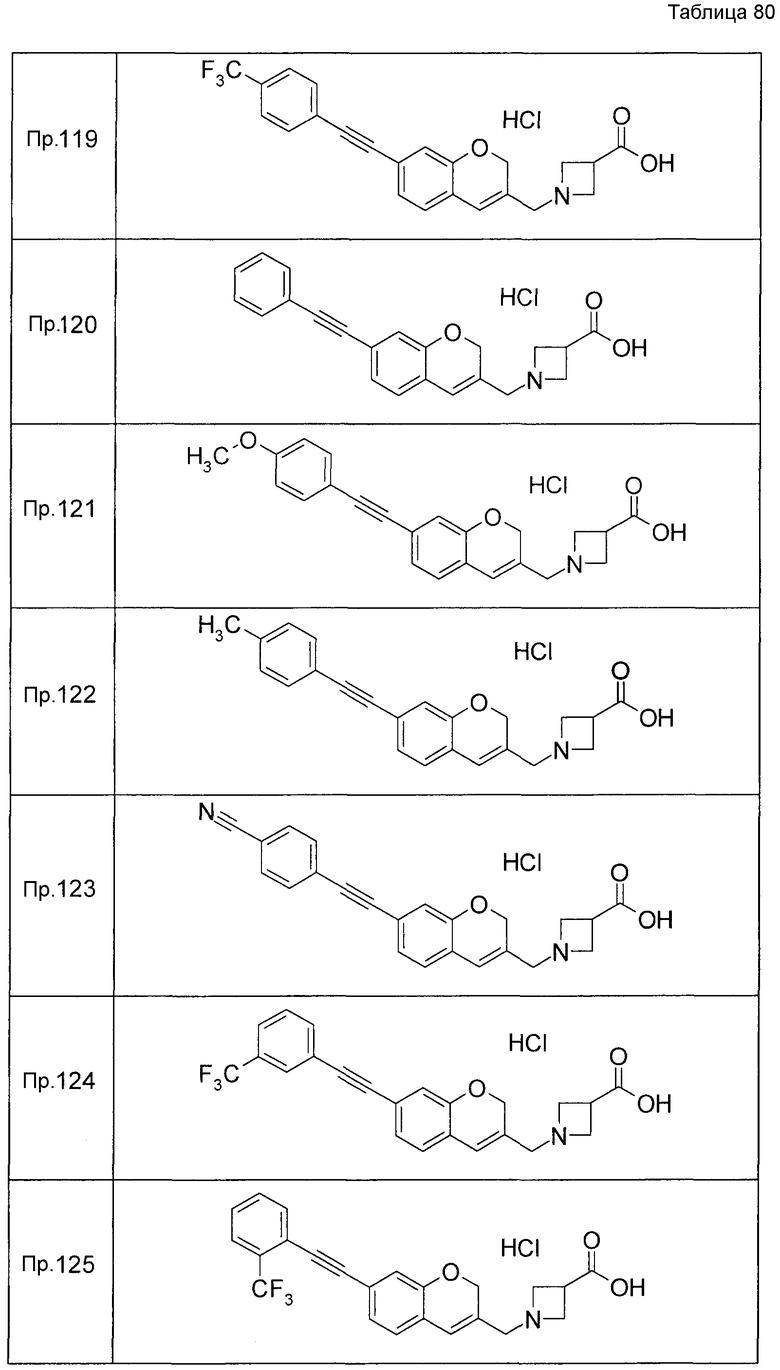
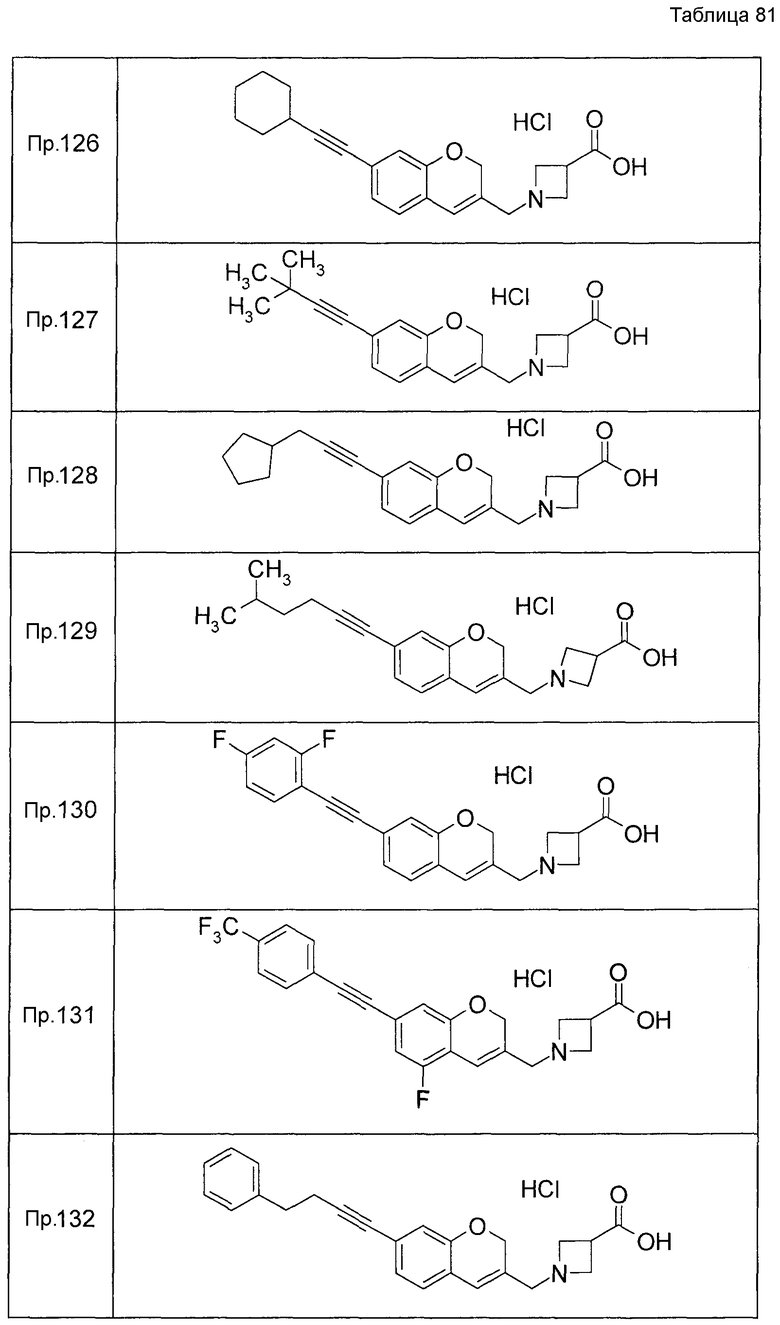
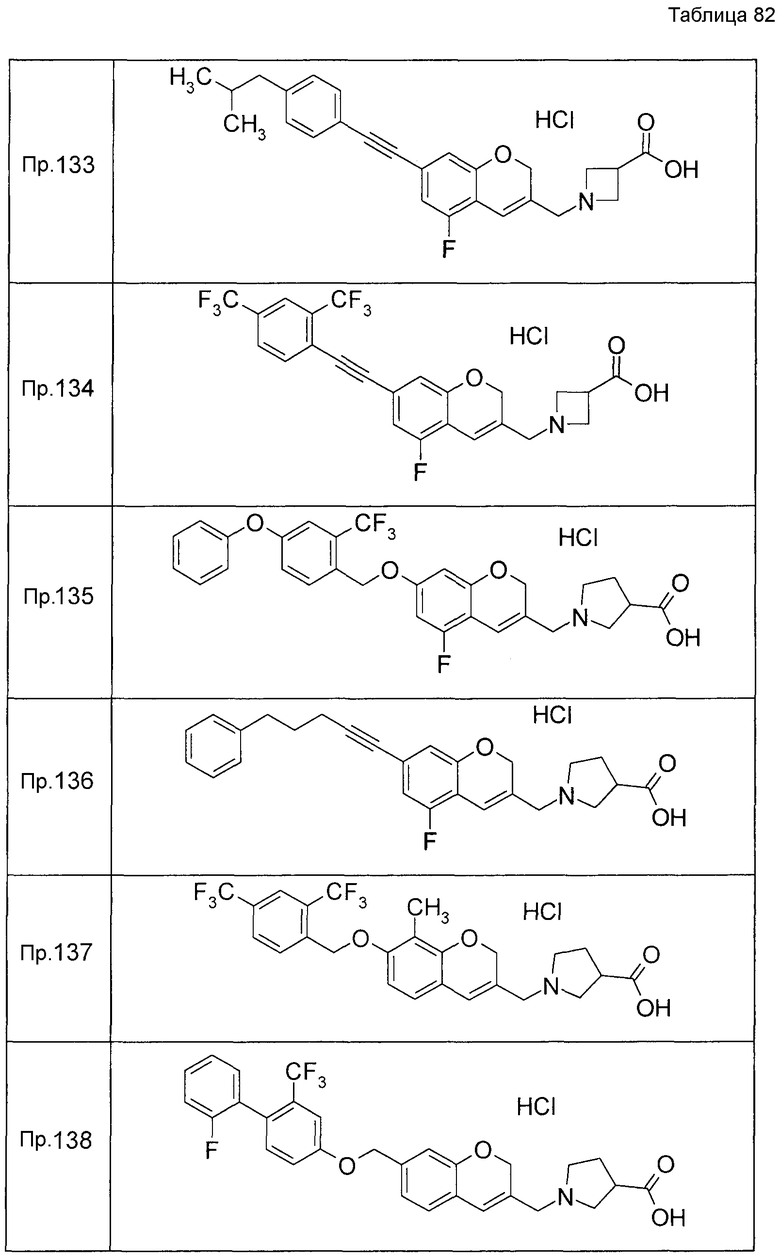
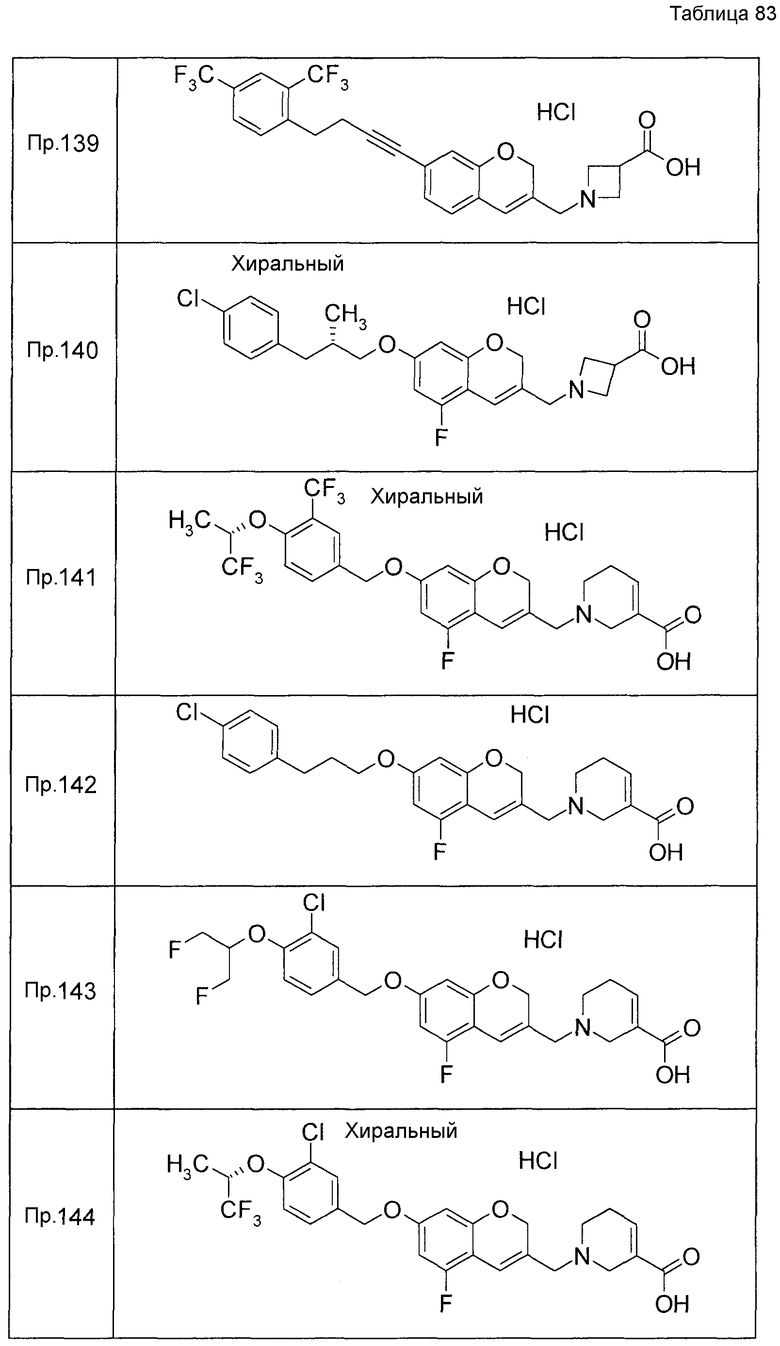
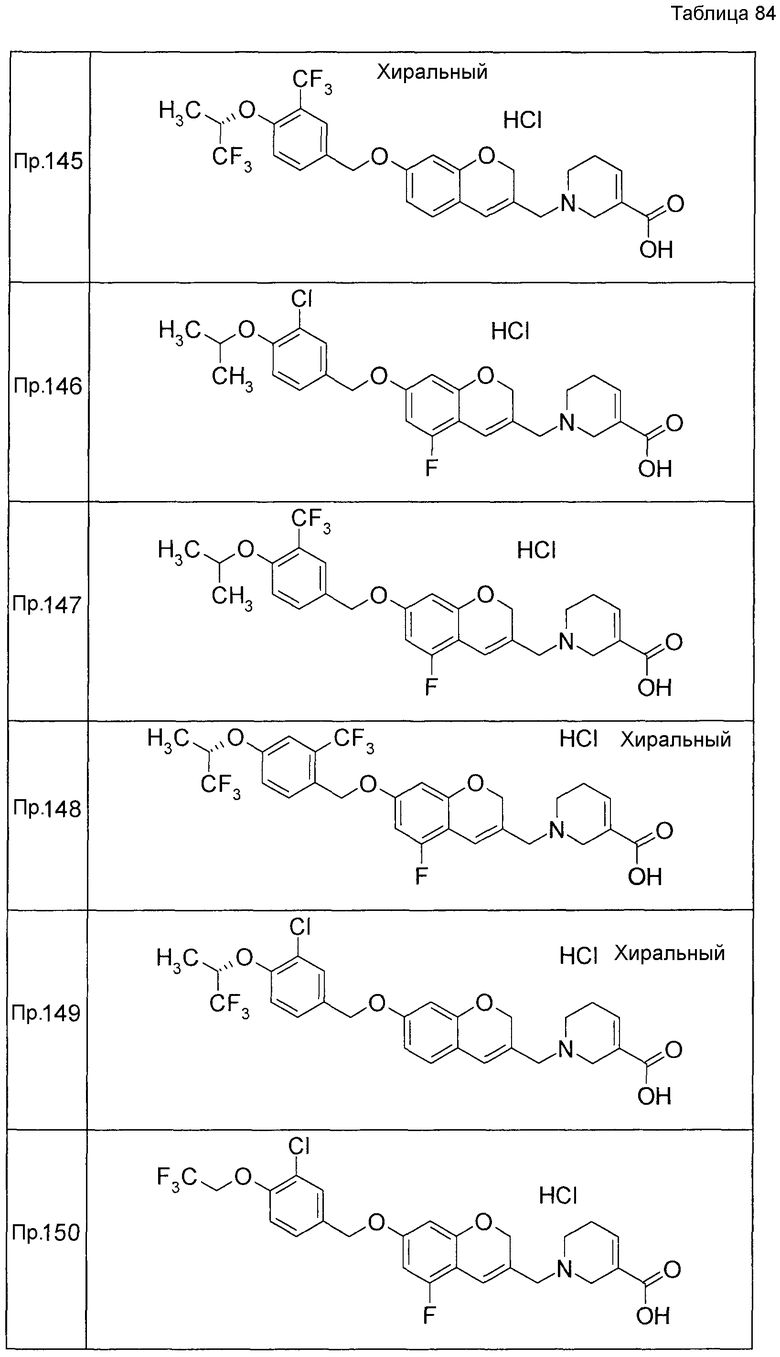
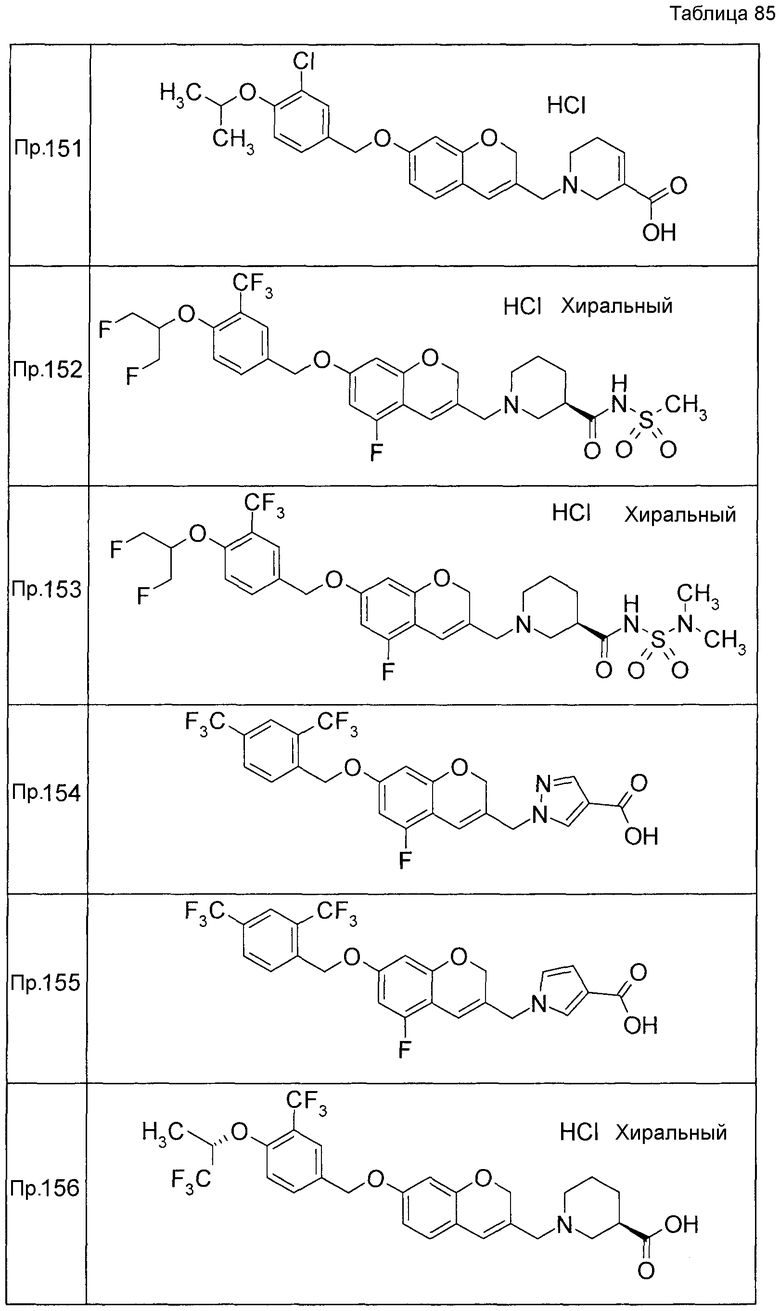
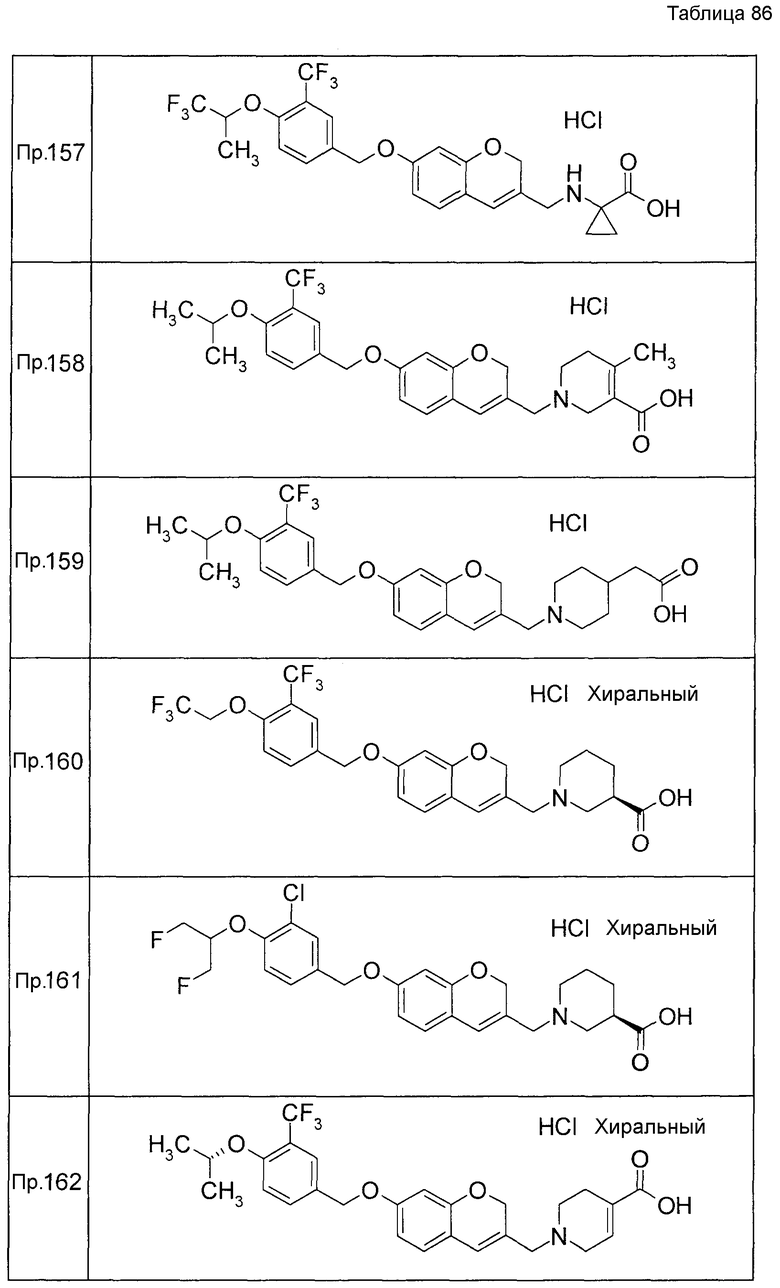
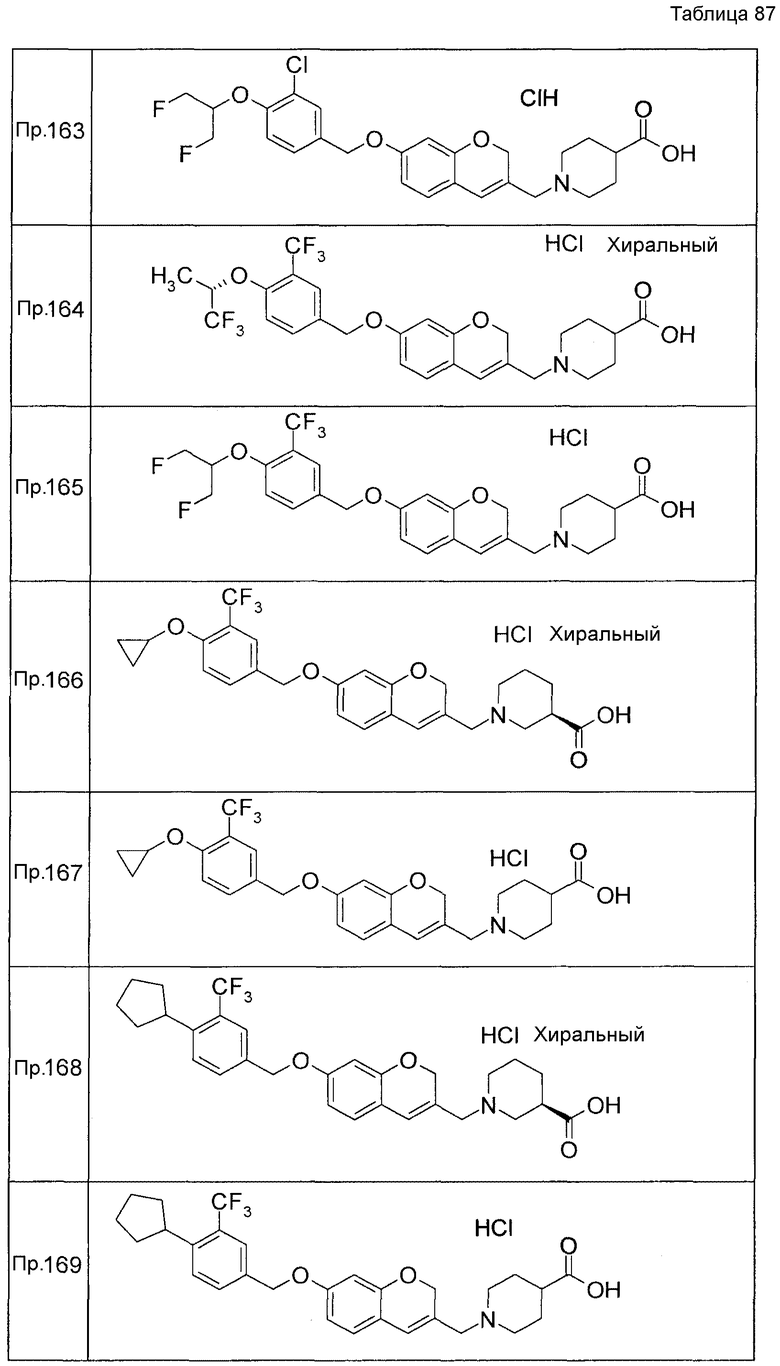
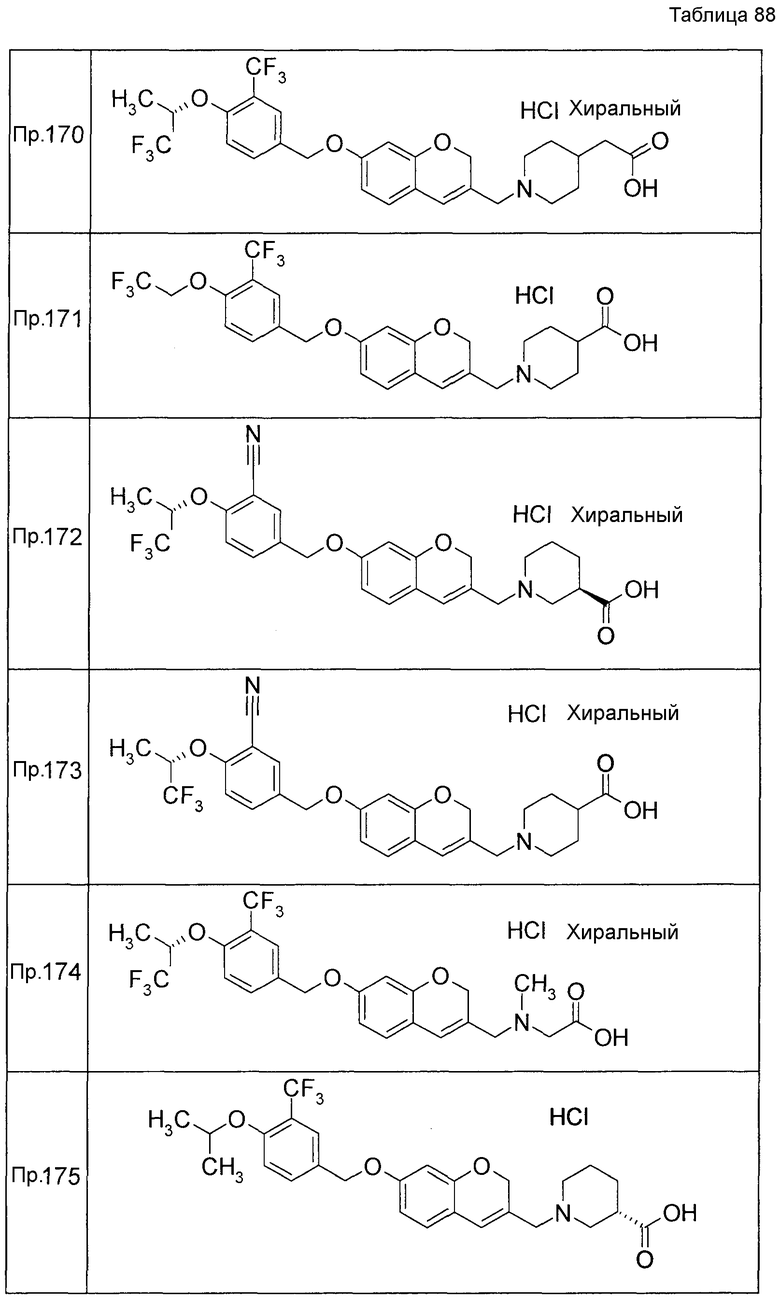
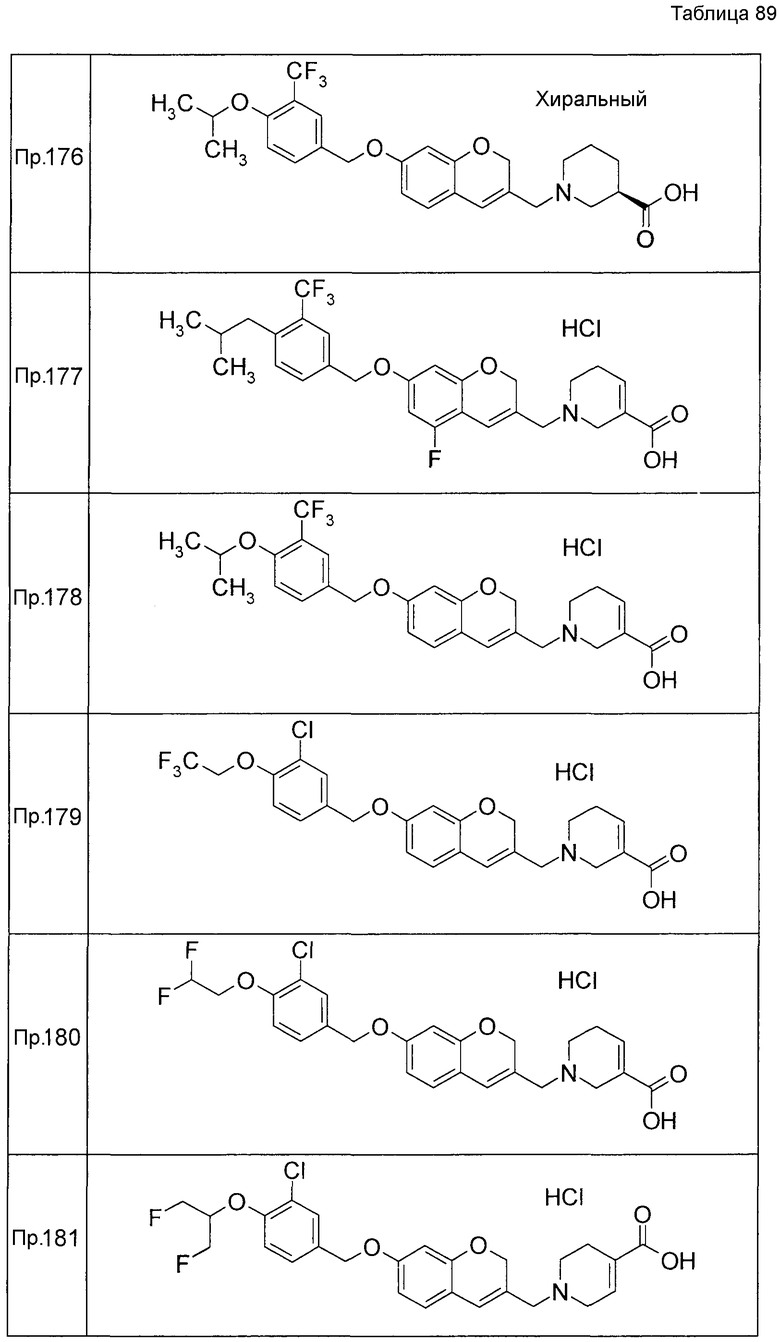
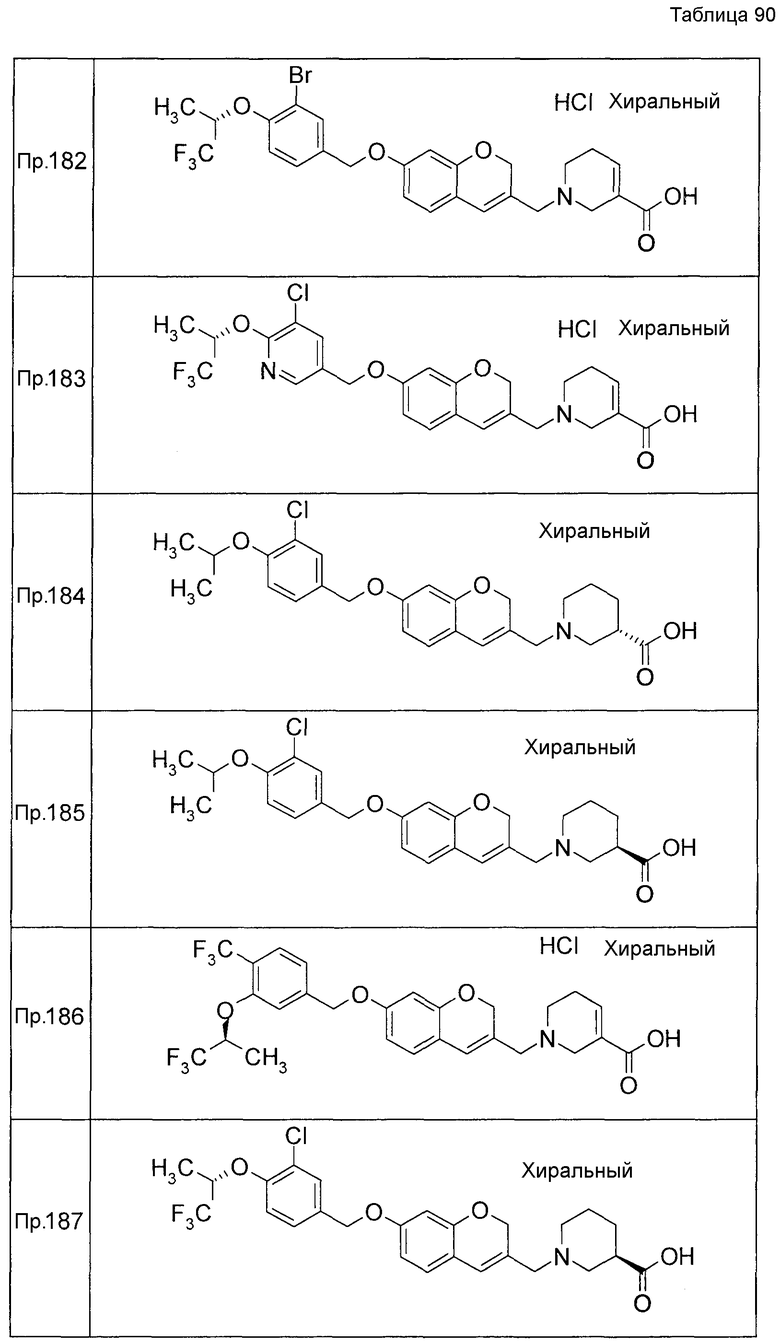
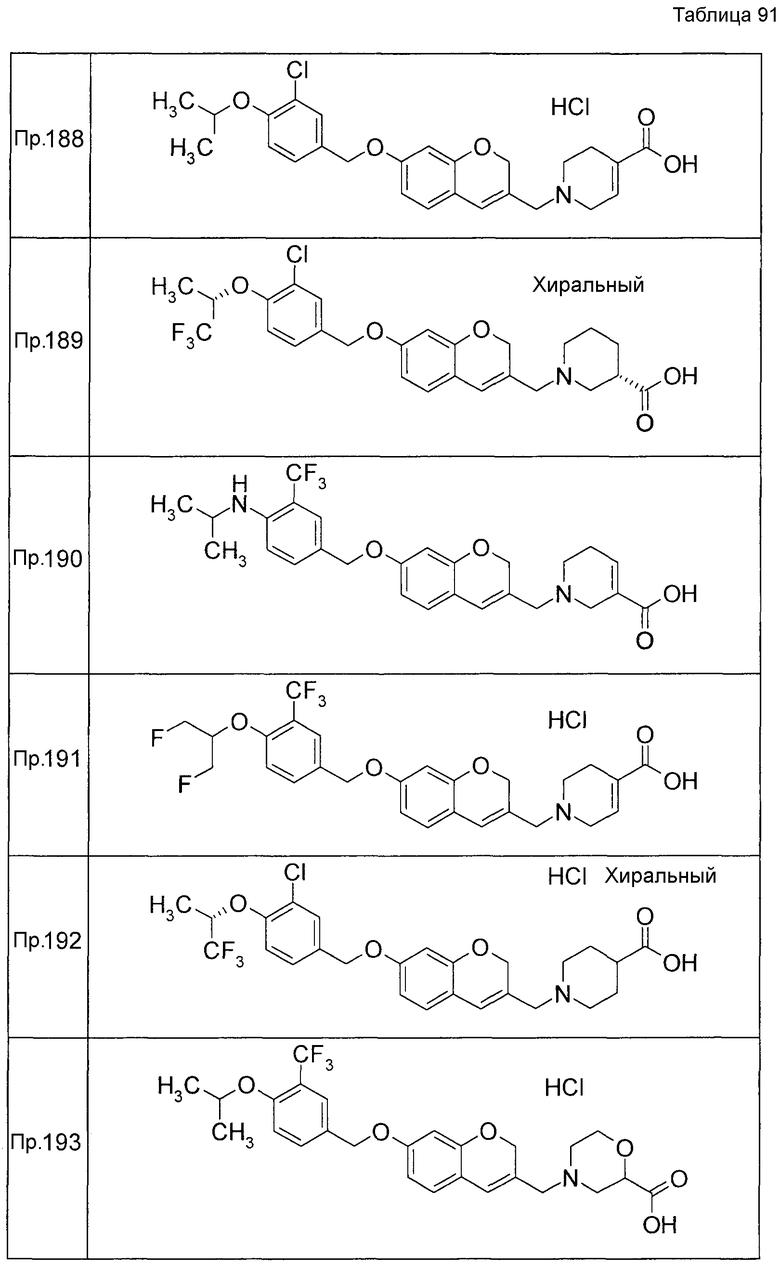
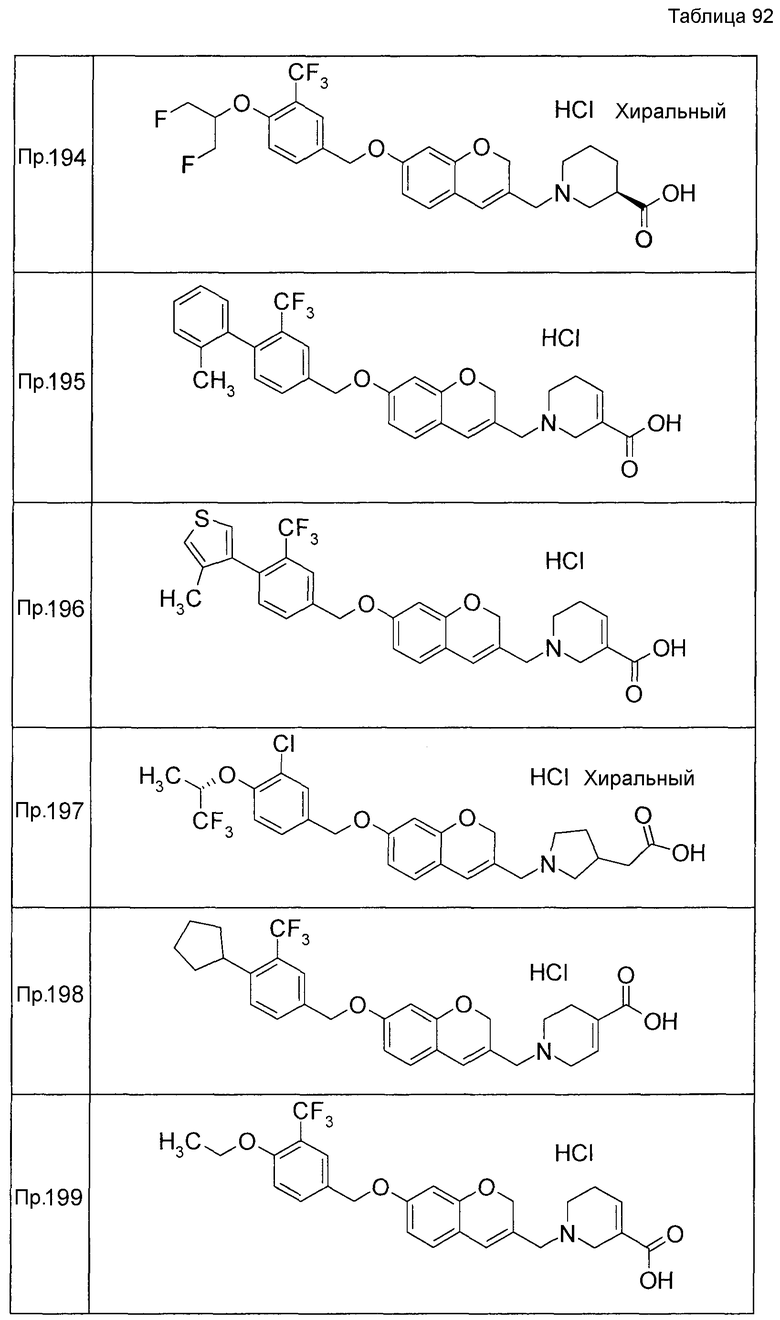
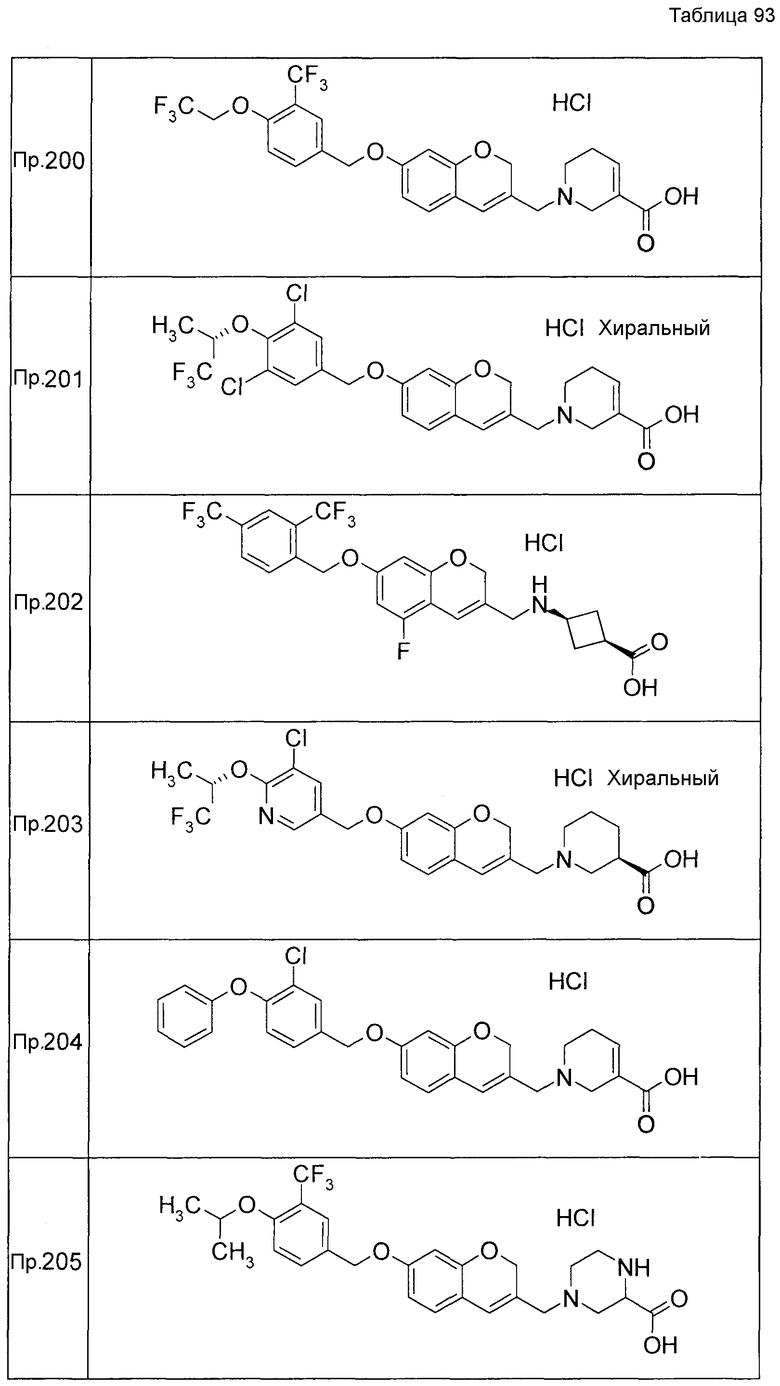
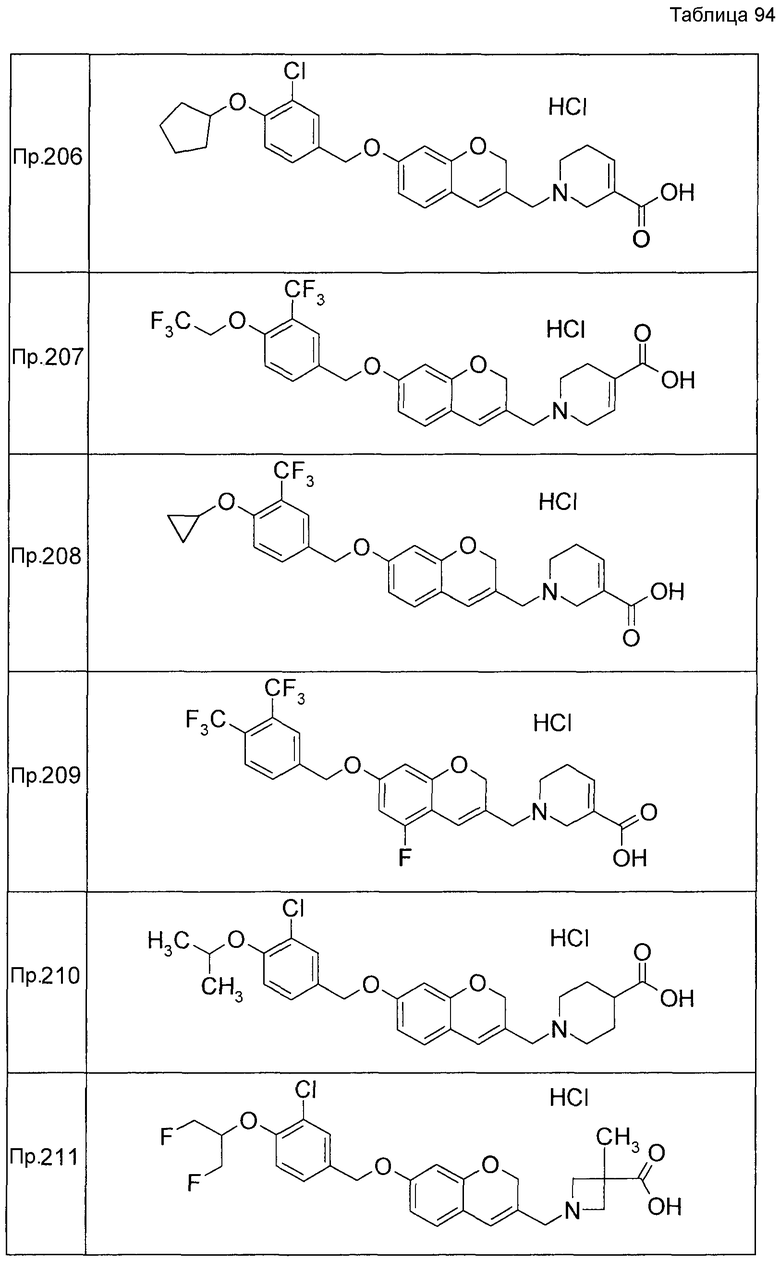
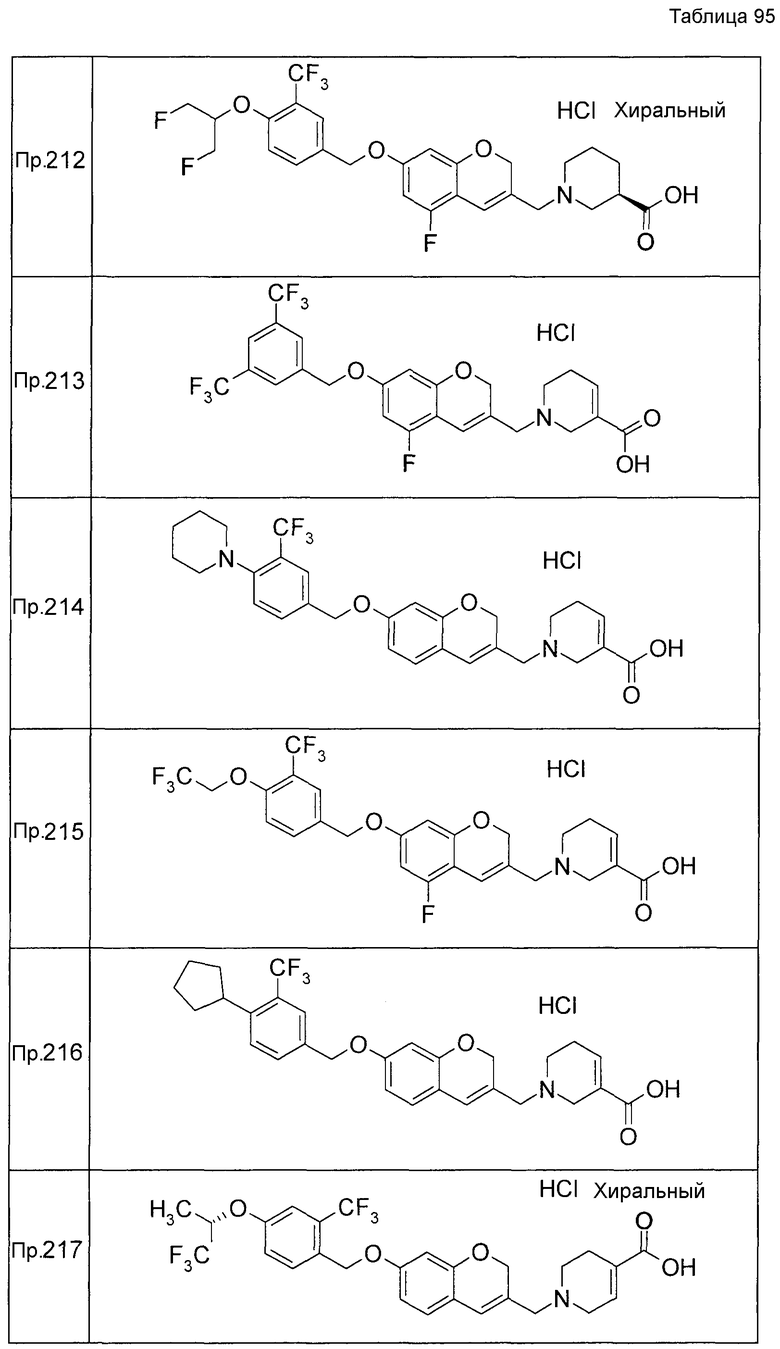
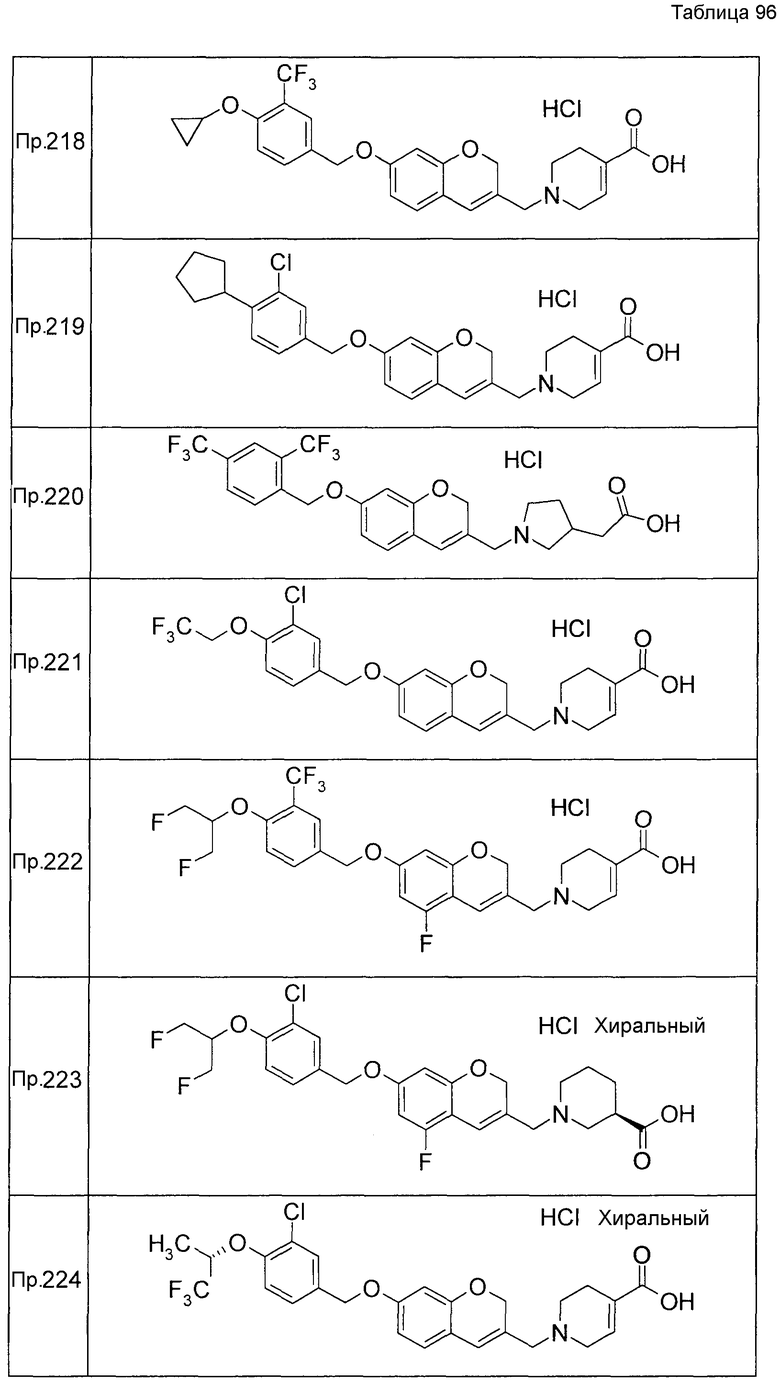
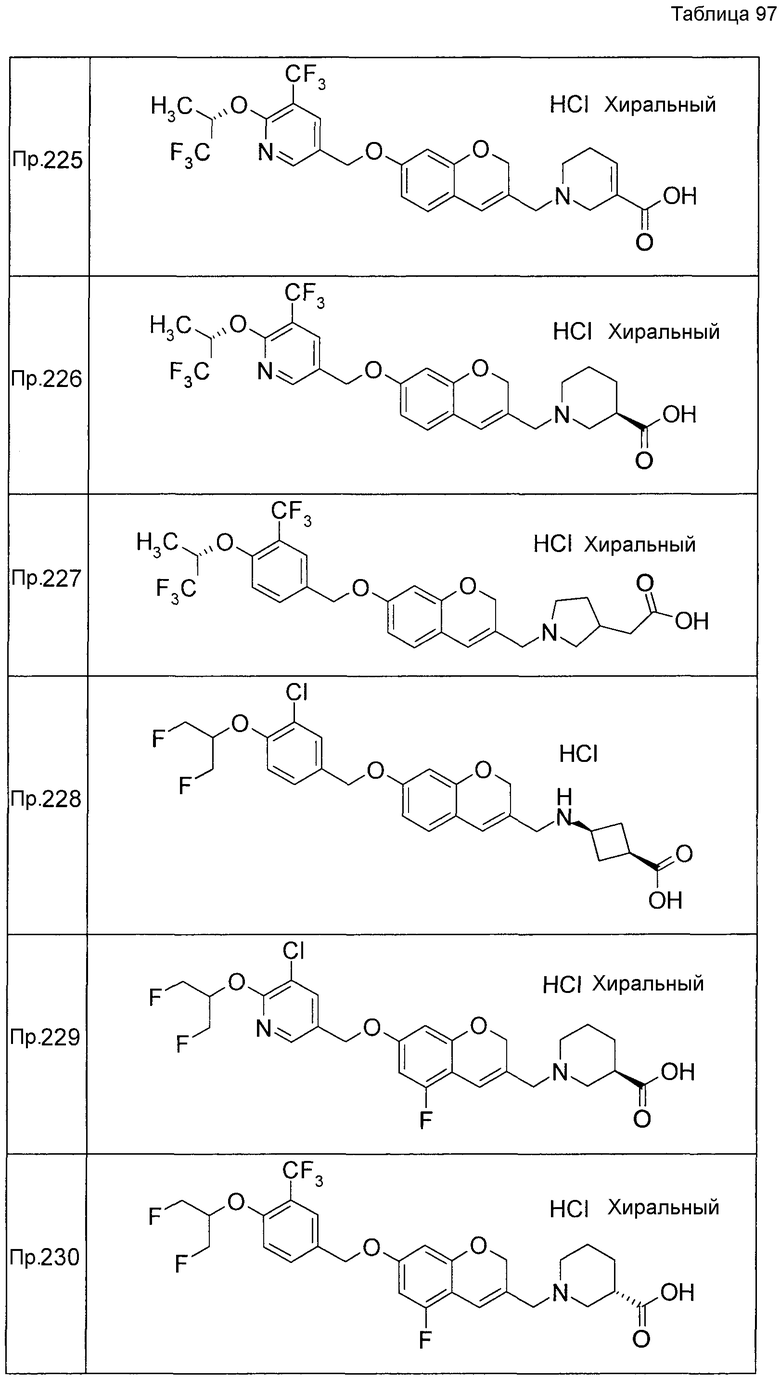
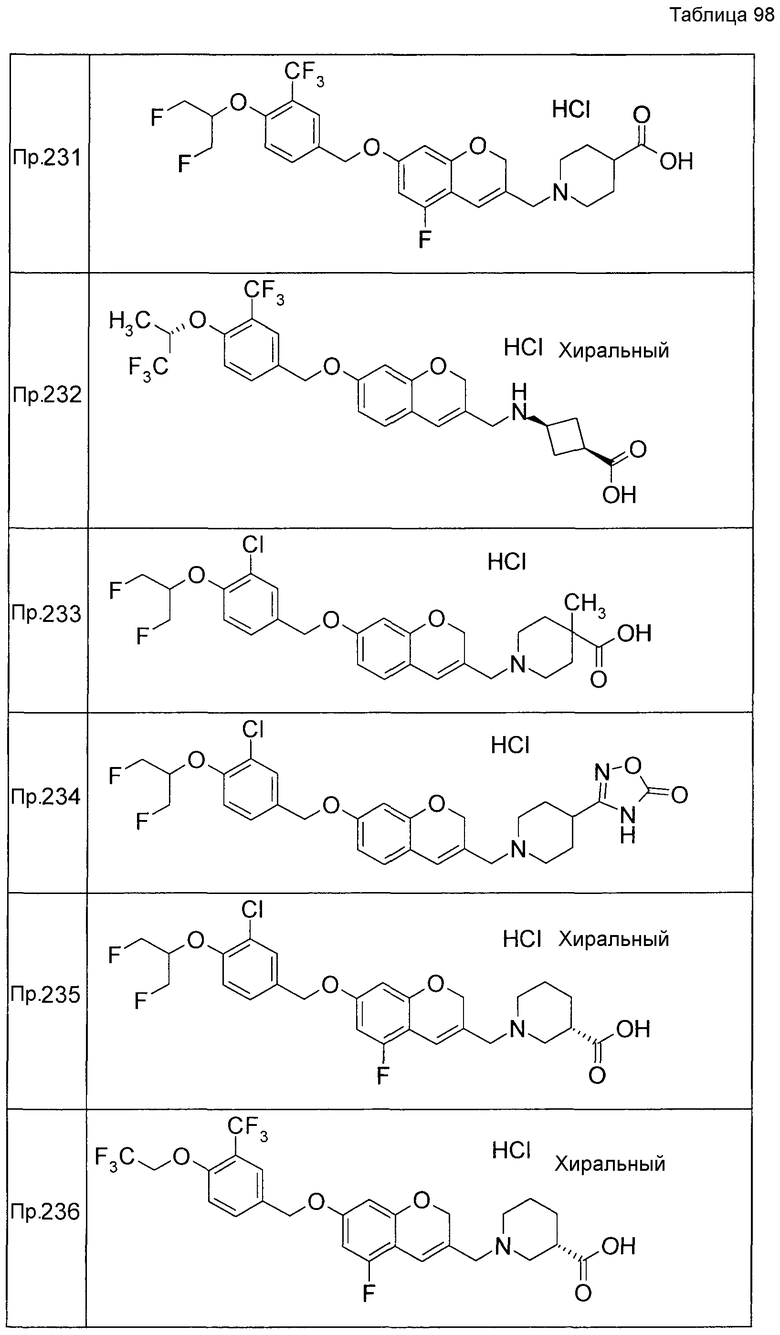

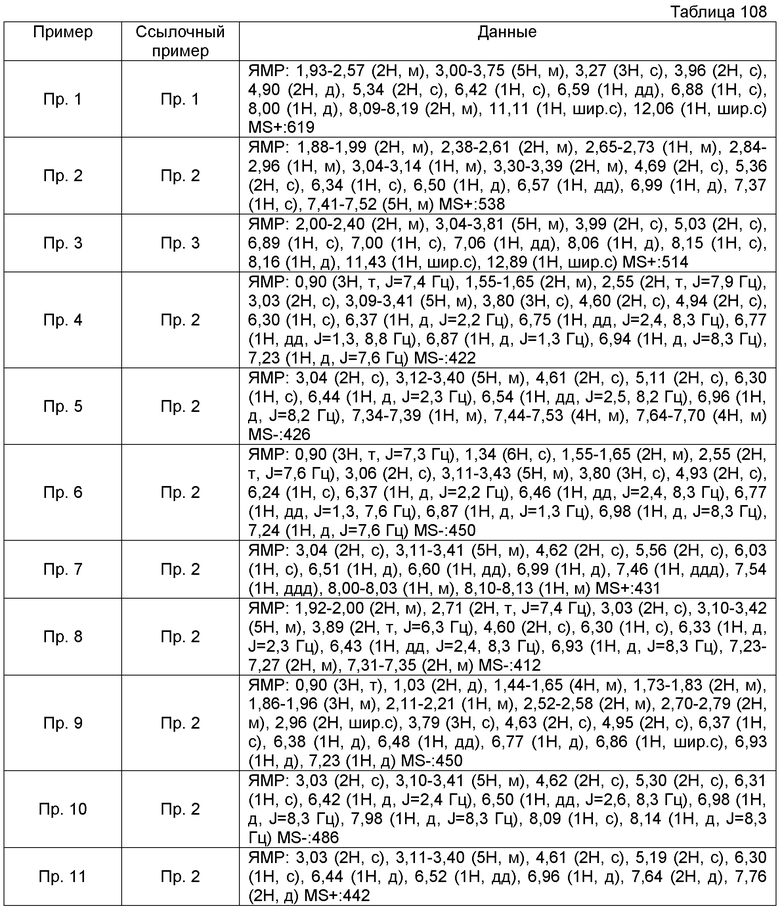
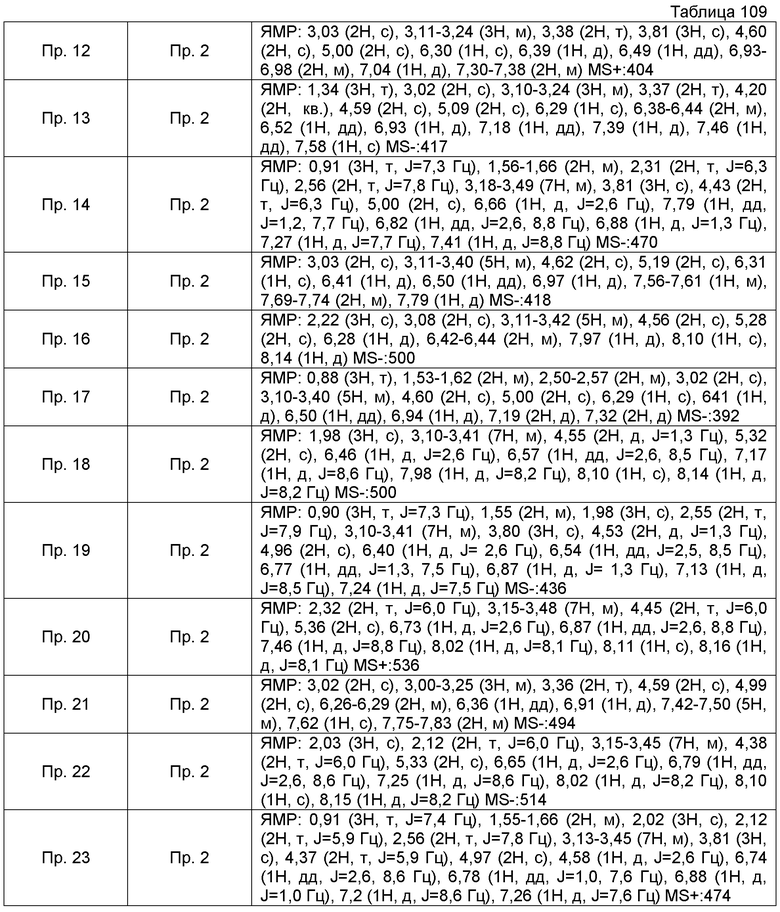
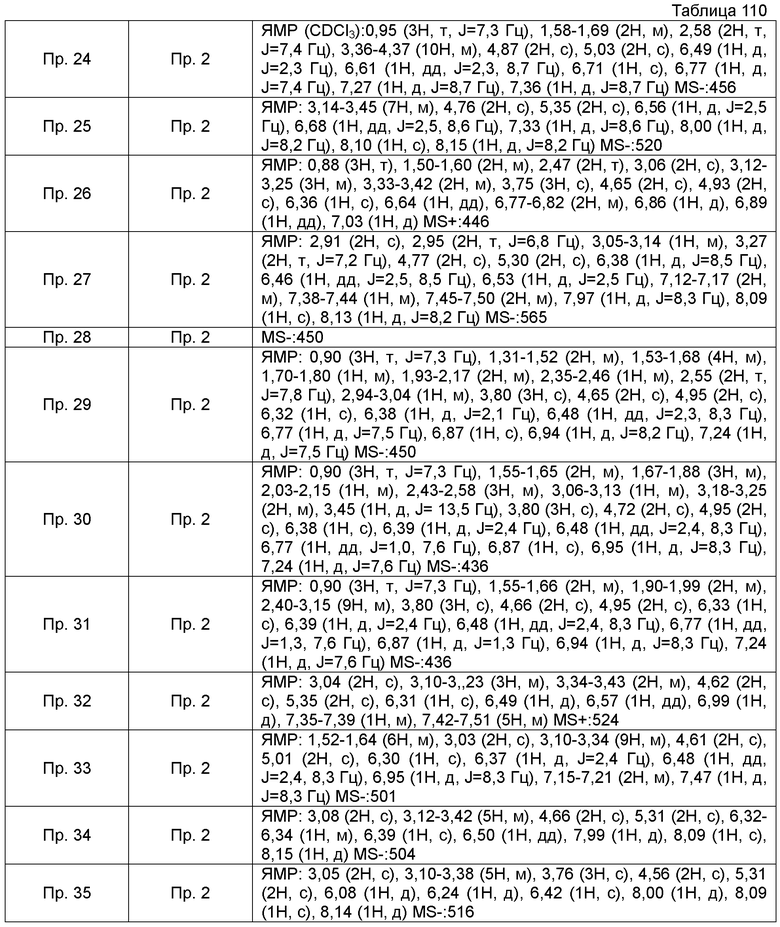
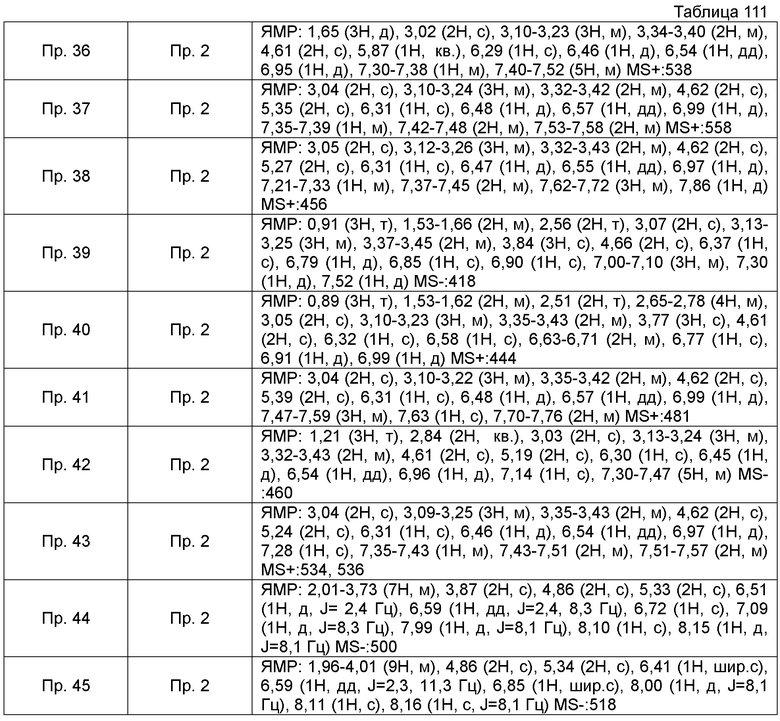
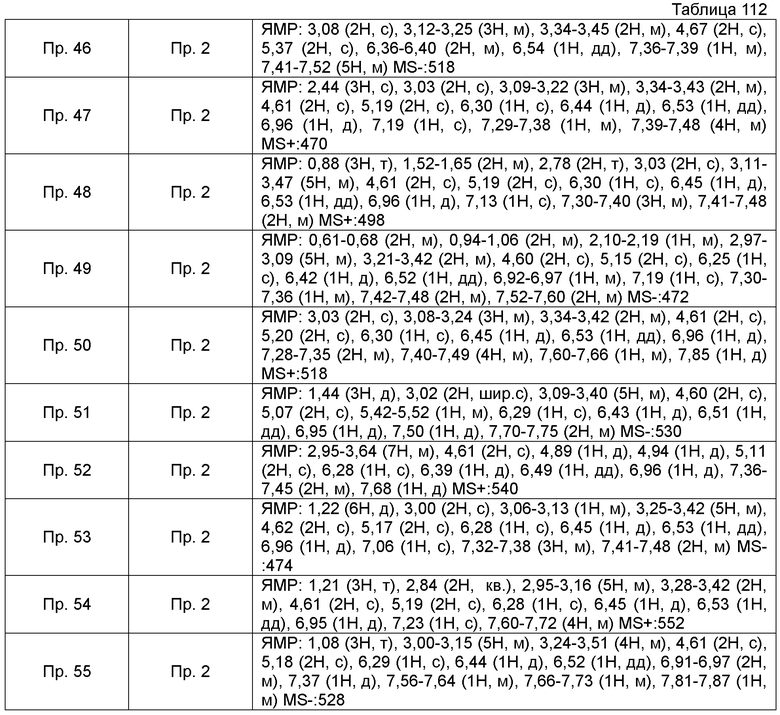
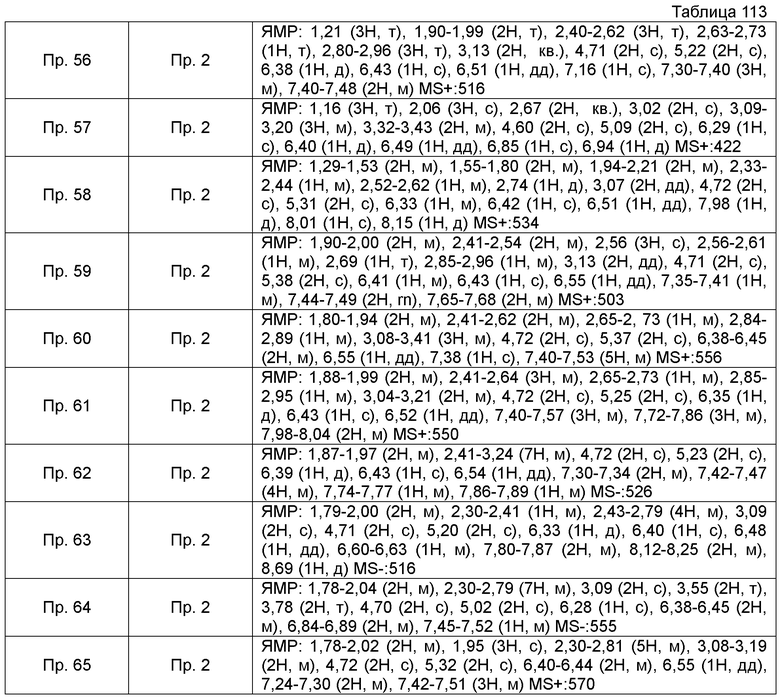
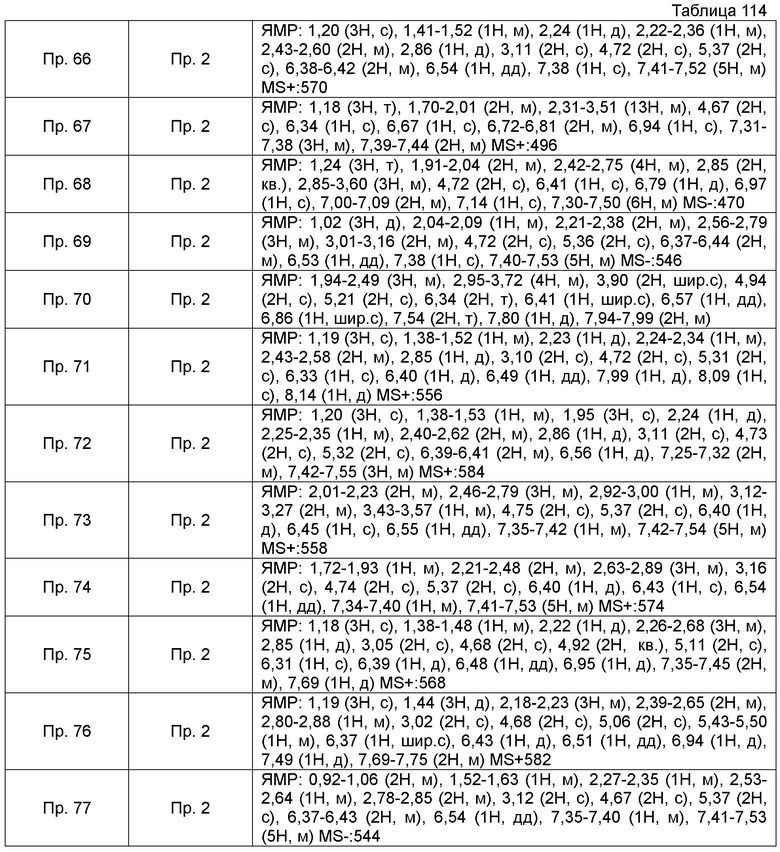
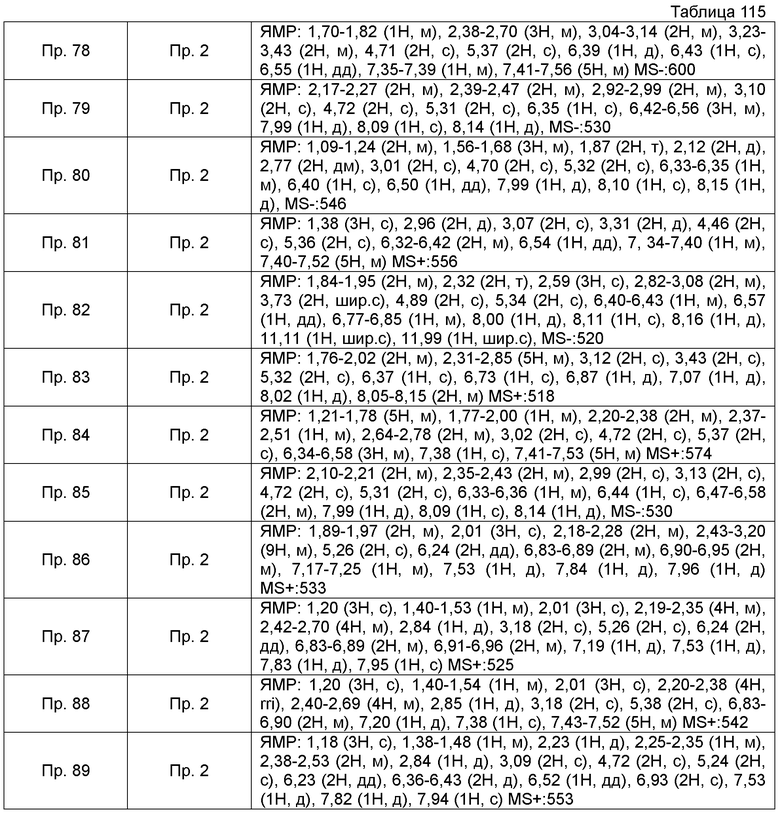
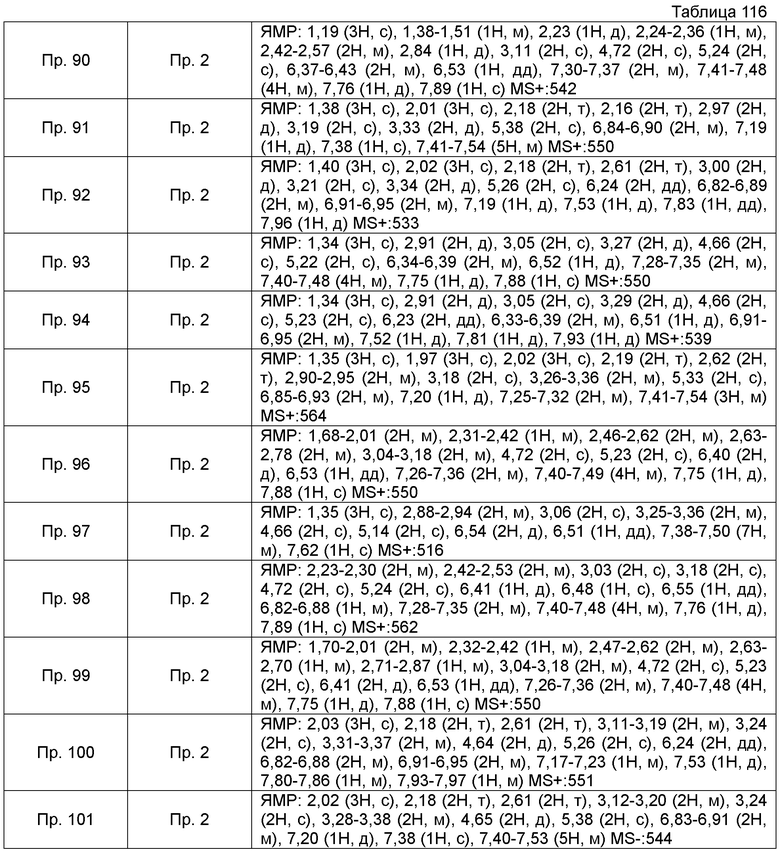
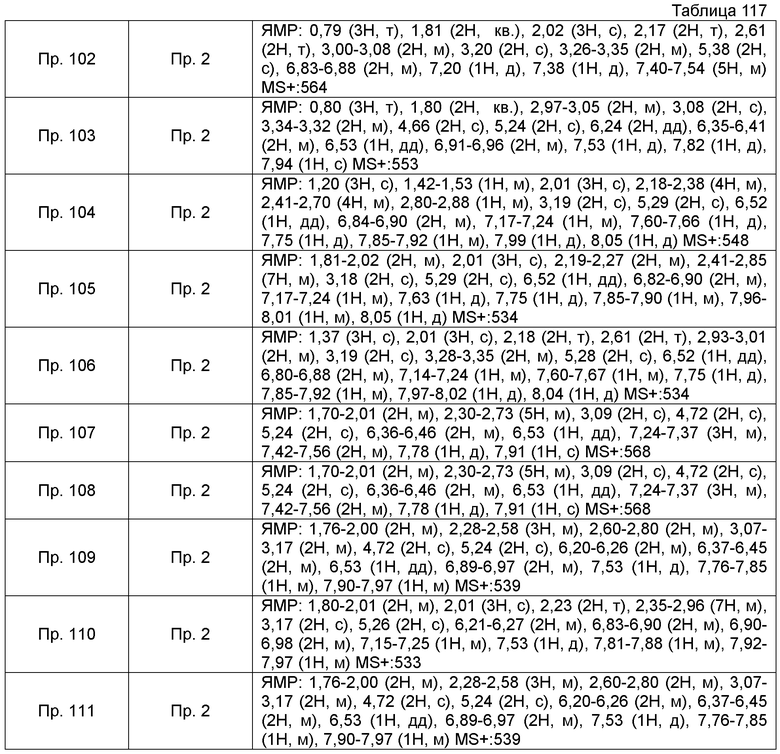
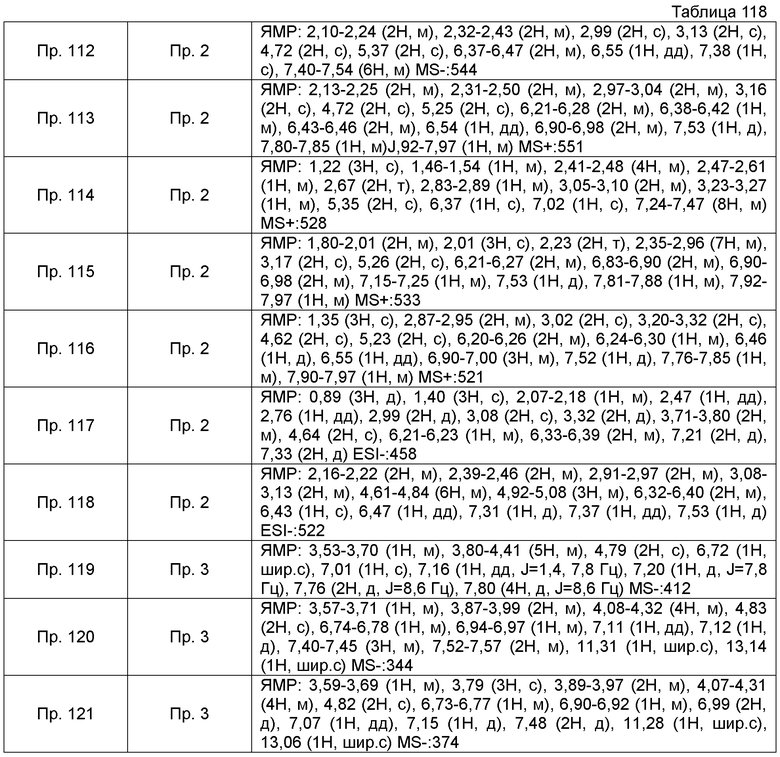
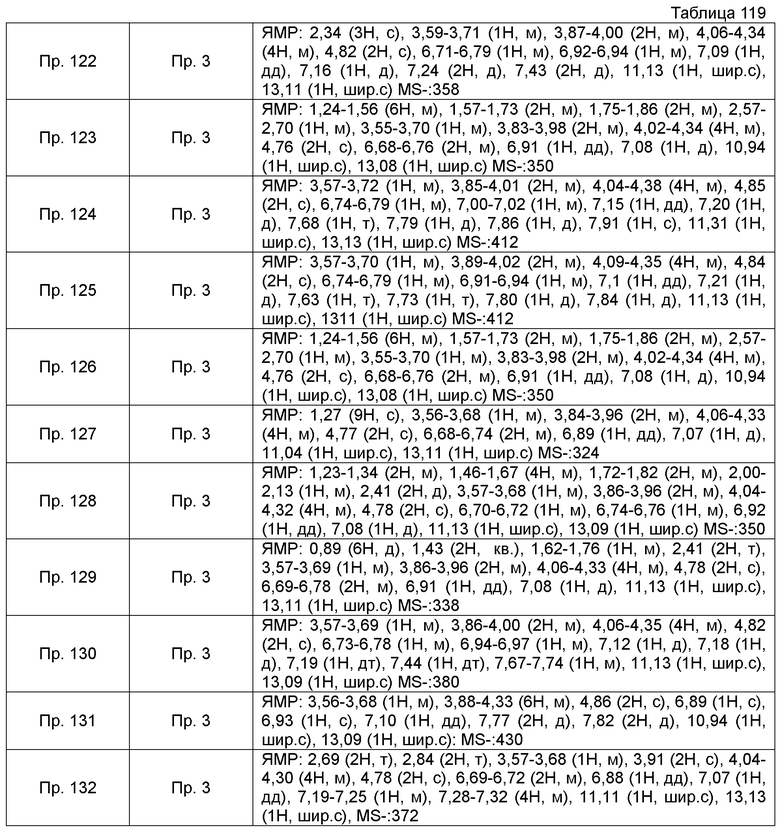
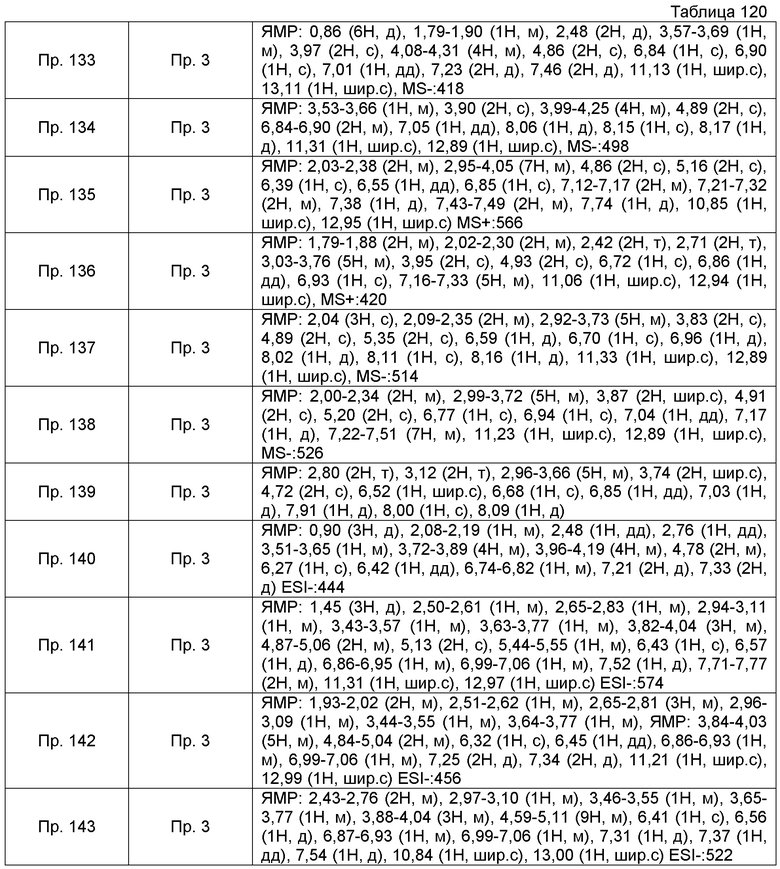
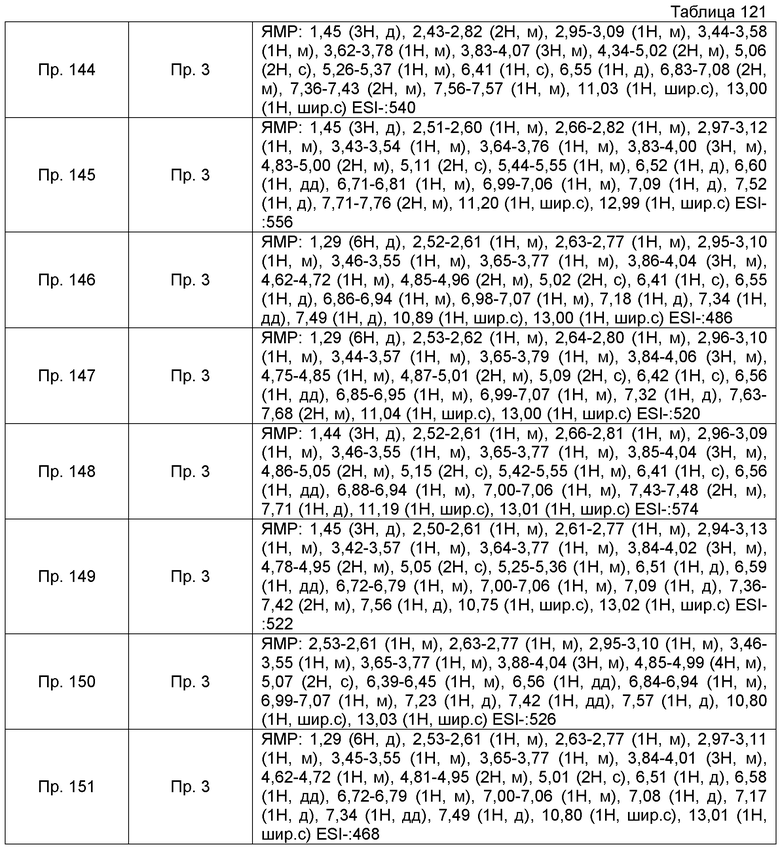
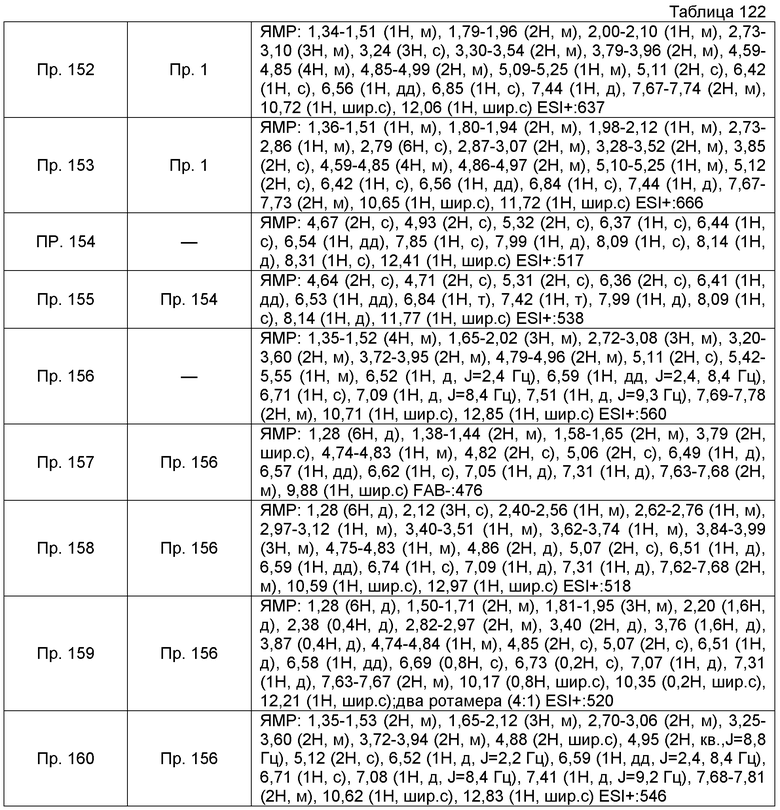
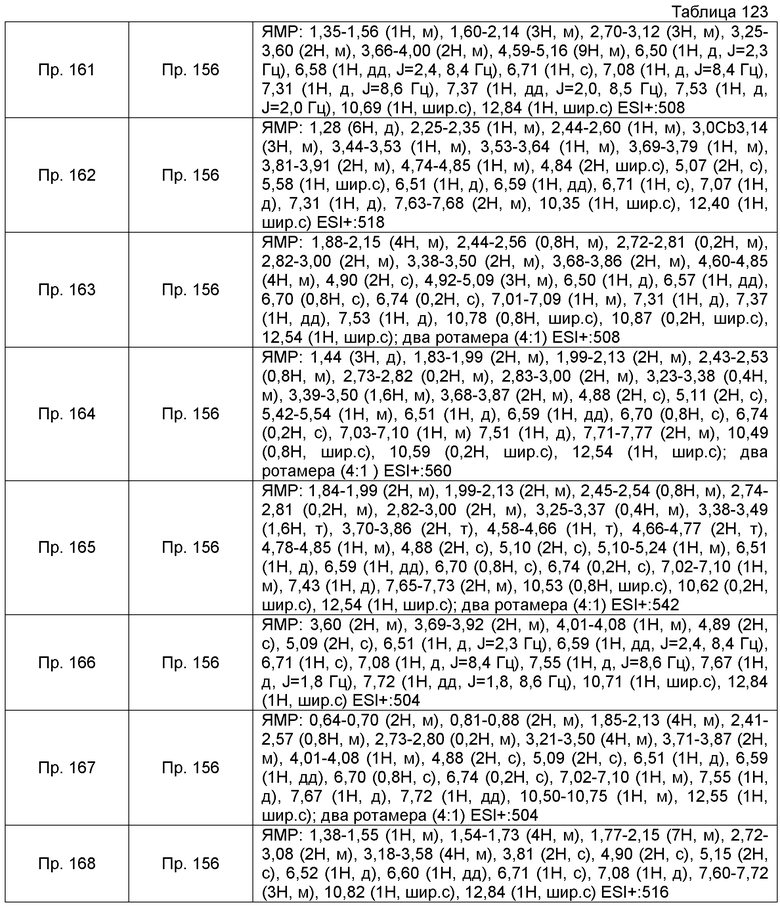
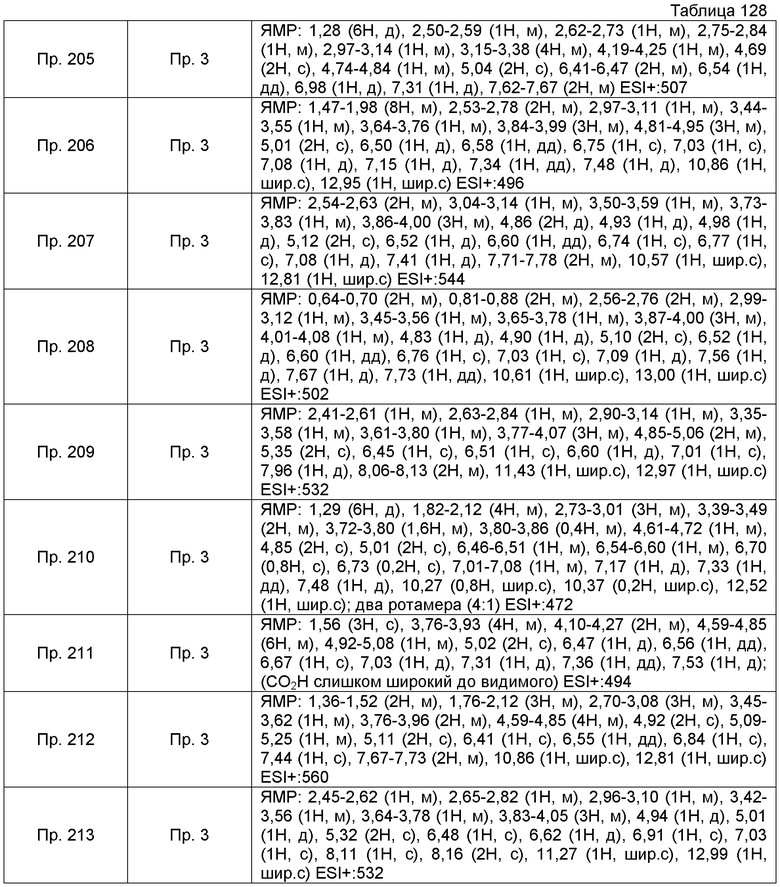
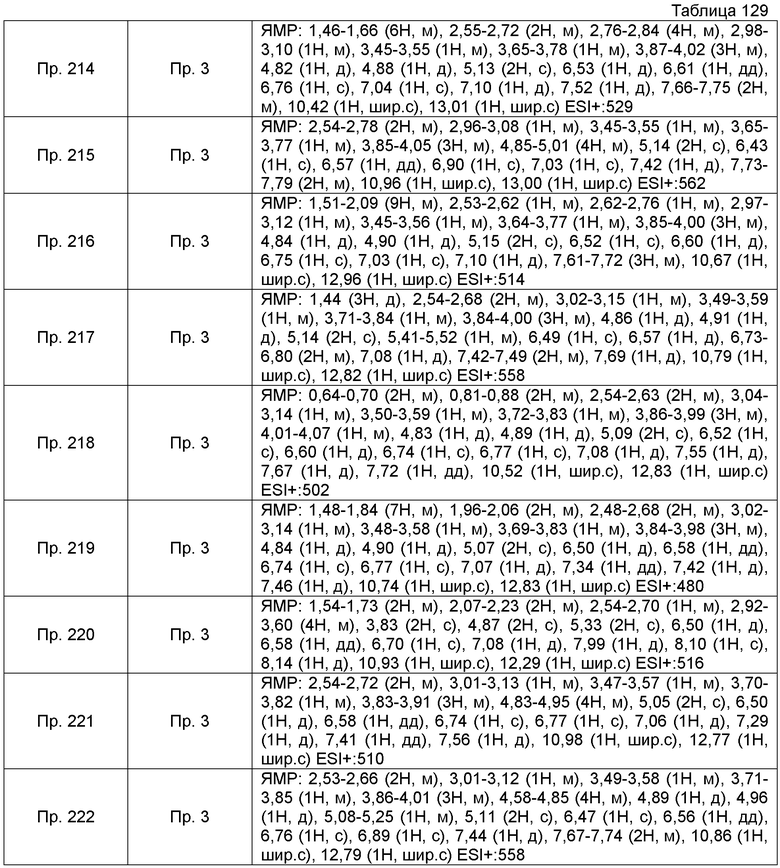
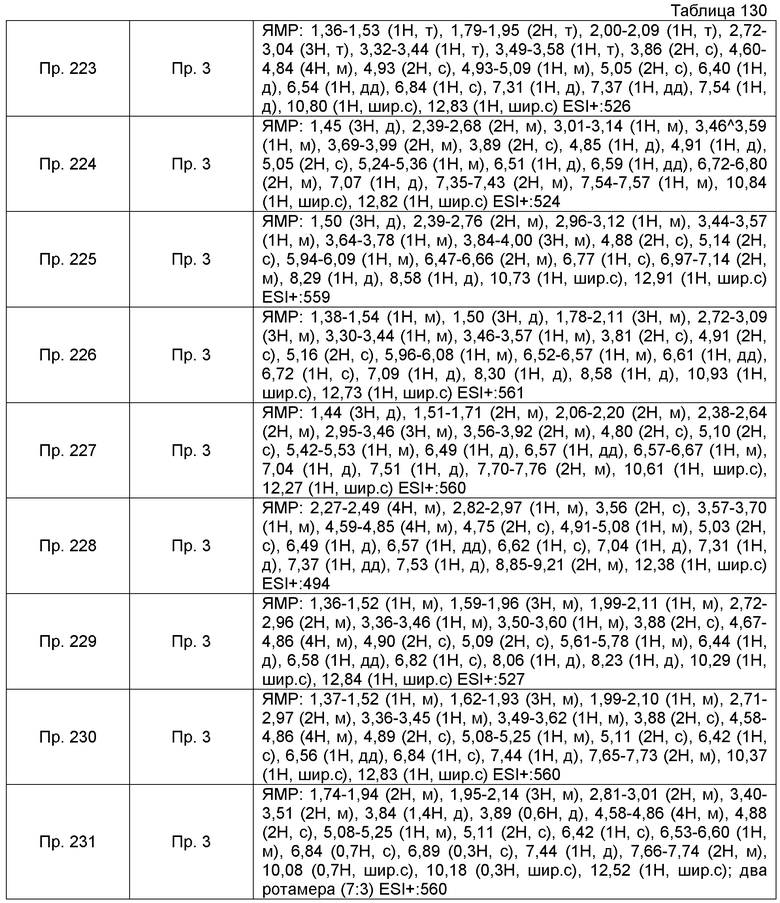
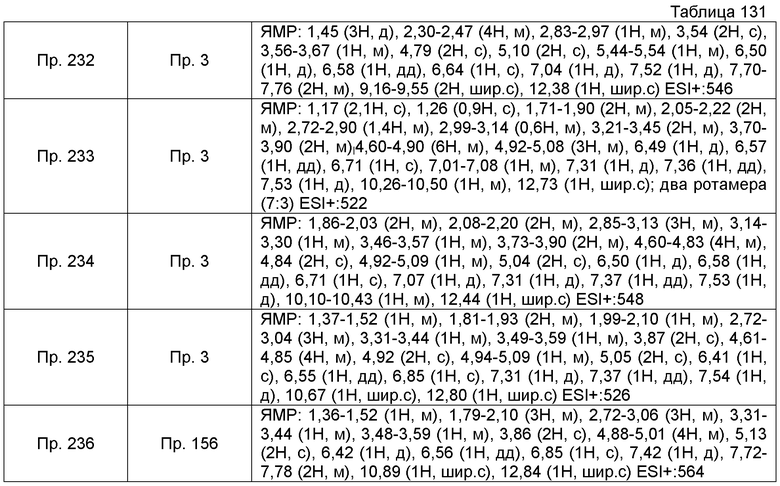
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОСОЕДИНЕНИЕ | 2007 |
|
RU2425832C2 |
| АЗА-КОЛЬЦЕВОЕ СОЕДИНЕНИЕ С ВНУТРЕННИМ МОСТИКОМ | 2008 |
|
RU2441868C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| ПРОЛЕКАРСТВЕННЫЕ СРЕДСТВА ИНГИБИТОРОВ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИЧ | 2015 |
|
RU2693622C2 |
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2629118C2 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| ЛЕЧЕНИЕ ИЛИ ПРОФИЛАКТИКА ПРОЛИФЕРАТИВНЫХ СОСТОЯНИЙ | 2010 |
|
RU2705548C2 |
| МОДУЛЯТОРЫ CCR2 | 2016 |
|
RU2726206C2 |
Изобретение относится к соединению 2H-хромена или его производному, которые обладают действием агониста S1P1. Их можно использовать для профилактики и/или лечения заболевания, вызванного нежелательной инфильтрацией лимфоцитов, или заболевания, вызванного аномальной пролиферацией или аккумуляцией клеток. 3 н. и 5 з.п. ф-лы, 131 табл., 156 пр.
1. Соединение 2H-хромена или его соль, выбранное из
следующих:
1-{[7-({3-хлор-4-(1S)-2,2,2-трифтор-1-метилэтокси]бензил}окси)-2H-хромен-3-ил]метил}-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
1-({7-[(3-хлор-4-изопропилбензил)окси|-2H-хромен-3-ил} метил)-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
1-[(7-{[4-изопропокси-3-(трифторметил)бензил]окси}-2H-хромен-3-ил)метил]-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
1-{[7-({3-хлор-4-[2-фтор-1-(фторметил)этокси]бензил}окси)-2H-хромен-3-ил]метил}-1,2,3,6-тетрагидропиридин-4-карбоновая кислота,
1-{[7-({5-хлор-6-[(1S)-2,2,2-трифтор-1-метилэтокси]пиридин-3-ил}метокси)-2Н-хромен-3-ил]метил}-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
(3R)-1-{[7-({4-[(1,3-дифторпропан-2-ил)окси]-3-(трифторметил)бензил}окси)-5-фтор-2H-хромен-3-ил]метил} пиперидин-3-карбоновая кислота,
1-[(7-{[4-циклопентил-3-(трифторметил)бензил]окси}-2H-хромен-3-ил)метил]-1,2,5,6-тетрагидропиридин-3-карбоновая кислота,
(3R)-1-{[7-({3-хлор-4-[(1,3-дифторпропан-2-ил)окси]бензил}окси)-5-фтор-2H-хромен-3-ил]метил}пиперидин-3-карбоновая кислота,
(3S)-1-{[7-({4-[(1,3-дифторпропан-2-ил)окси]-3-(трифторметил)бензил}окси)-5-фтор-2H-хромен-3-ил]метил}пиперидин-3-карбоновая кислота,
(3R)-1-[(7-{[4-(2,2,2-трифторэтокси)-3-трифторметил)бензил]окси}-2H-хромен-3-ил)метил]пиперидин-3-карбоновая кислота,
(3R)-1-[(7-{[3-(трифторметил)-4-{[(2S)-1,1,1-трифторпропан-2-ил]окси}бензил]окси}-2H-хромен-3-ил)метил]пиперидин-3-карбоновая кислота,
(3S)-1-[(7-{[4-(2,2,2-трифторэтокси)-3-(трифторметил)бензил]окси}-5-фтор-2H-хромен-3-ил)метил]пиперидин-3-карбоновая кислота,
(3R)-1-{[7-({4-[(1,3-дифторпропан-2-ил)окси]-3-(трифторметил)бензил}окси)-5-фтор-2H-хромен-3-ил]метил}-N-(метилсульфонил)пиперидин-3-карбоксамид, или
1-[(7-{[4-(2,2,2-трифторэтокси)-3-(трифторметил)бензил]окси}-2H-хромен-3-ил)метил]пиперидин-4-карбоновая кислота.
2. Соединение, представленное следующей формулой:
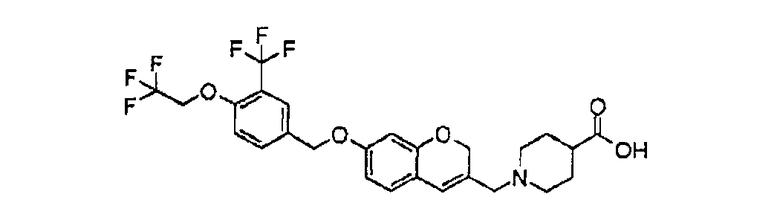
или его соль.
3. Фармацевтическая композиция, обладающая S1P1 агонистической активностью, содержащая эффективное количество соединения 2H-хромена, или его производного, или его соли по п.1 и фармацевтически приемлемый носитель или эксципиент.
4. Фармацевтическая композиция по п.3, которая является агонистом S1P1.
5. Фармацевтическая композиция по п.4, которая представляет собой фармацевтическую композицию для профилактики или лечения заболевания, вызванного нежелательной инфильтрацией лимфоцитов, связанной с S1P1.
6. Фармацевтическая композиция по п.5, где заболевание, вызванное нежелательной инфильтрацией лимфоцитов, связанной с S1P1, представляет собой реакции отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей, аутоиммунные заболевания или воспалительные заболевания у человека или животного.
7. Фармацевтическая композиция по п.6, которая представляет собой фармацевтическую композицию для профилактики или лечения реакций отторжения или «трансплантат против хозяина» при трансплантации органов, костного мозга или тканей у человека или животного.
8. Фармацевтическая композиция по п.6, которая представляет собой фармацевтическую композицию для профилактики или лечения рассеянного склероза.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| EP 0754455 A1, 08.02.1996 | |||
| ГРУНТОВОЧНЫЙ РАСТВОР ДЛЯ ПОДГОТОВКИ К ОТДЕЛКЕ ДРЕВЕСНО-СТРУЖЕЧНЫХ И ДРУГИХ ПЛИТ | 0 |
|
SU332064A1 |
| RU 2007109207 A, 20.09.2008 | |||
| RU 2006110027 A, 10.10.2007. | |||

