Область изобретения
Настоящее изобретение относится к новым пролекарствам оптически чистой бензохинолизин-2-карбоновой кислоты и к фармацевтическим композициям, которые включают эти пролекарства. Соединения и композиции по изобретению могут использоваться для лечения инфекций, вызываемых грамположительными, грамотрицательными и анаэробными бактериями, особенно инфекций, вызываемых резистентным грамположительным организмом и грамотрицательным организмом, микобактериальных инфекций и инфекций, вызываемых внутрибольничными патогенами.
Уровень техники
Пролекарства представляют собой терапевтические средства, которые являются неактивными per se, но in vivo они трансформируются в терапевтически активную родительскую молекулу. Пролекарства имеют оптимальные физико-химические, фармакокинетические и фармакодинамические свойства. Они могут быть предназначены, чтобы преодолеть фармацевтические, фармакокинетические или фармакодинамические барьеры, такие как недостаточная пероральная абсорбция, недостаточная растворимость, недостаточная химическая стабильность, неприемлемый вкус или запах, раздражение или боль, неадекватная проходимость через гематоэнцефалический барьер, токсичность и выраженный пресистемный метаболизм.
Для удобства пациентов большинство лекарственных средств вводят пероральным путем. Существуют значительные проблемы, препятствующие доставке лекарственного средства из перорального пути, что часто означает, что не все введенное соединение достигает его предполагаемого места действия. То, в какой степени соединение может преодолеть препятствия при пероральном использовании лекарственного средства и достигнуть большого круга кровообращения, определяют количественно термином пероральная биодоступность.
Хиральный фторхинолон S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновая кислота, также известная как S-(-)-надифлоксацин, описана в японских патентах JP 63,192,753A и JP 05,339,238A.
S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновая кислота имеет потенциал для использования в качестве коммерческого антибактериального агента вследствие ее профиля антибактериальной активности. Однако натриевая соль S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты имеет тенденцию вызывать флебит у крыс при введении внутривенным путем. Также S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновая кислота имеет растворимость в воде 0,8-2,0 мг/мл в диапазоне рН 8,0-9,5 при 28°C, таким образом создавая проблемы в плане необходимости составлять лекарственное средство в форме таблетки или капсулы или в создании составов для искусственного питания и парентеральной инъекции. Таким образом, желаемый агент, который мог бы преодолеть указанные выше проблемы, может помочь в разработке лекарственной формы, приемлемой для системного использования у человека и животных.
В этом направлении авторами было показано, что соль S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и L-аргинина представляет собой антибиотик широкого спектра действия, который обладает более низкой склонностью вызвать флебит у крыс при введении внутривенным путем, как раскрыто в заявке РСТ WO00/68229. Наиболее стабильная форма соли, тетрагидрат соли S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и L-аргинина, раскрыт в заявке РСТ WO05/023805A1 (и в соответствующем американском патенте US 7 164 023). Полезно получать стабильные фармацевтические лекарственные формы, включая водный раствор. Форма для инъекций является особенно подходящей для долгосрочной внутривенной терапии для лечения заболеваний, вызываемых бактериальными инфекциями, ввиду ее благоприятной растворимости в воде при рН 9,5, ее идеальной пригодности в силу того, что она не вызывает венозного воспаления при повторном внутривенном введении, и ее профиле безопасности.
Однако было обнаружено, что пероральная биодоступность S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и тетрагидрата соли S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и L-аргинина диметилбензойной кислоты является недостаточной при использовании у животных. Недостаточная пероральная биодоступность S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты, вероятно, является следствием неадекватной растворимости, и, следовательно, недостаточной абсорбции in vivo. В то же время недостаточная пероральная биодоступность соли L-аргинина и S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты, вероятно, является следствием склонности соединения к осаждению при кислом рН при транзите через желудочную область, где рН~1-2.
Таким образом, для улучшения пероральной биодоступности S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты был предусмотрен подход пролекарств. Пролекарства S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты раскрыты в патенте США 6 750 224 на имя заявителя. В патенте '224 раскрыты два типа пролекарств - пролекарства в 2-карбоксильной группе и в 4-гидроксильной группе боковой цепи пиперидина. Пролекарства, раскрытые в 4-гидроксипиперидине, включают аминокислотные пролекарства. Эти пролекарства были изучены с целью разработать пероральную фармацевтическую композицию. Пролекарства S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты, раскрытые там, или не обладали оптимальными характеристиками, такими как адекватная растворимость в широком диапазоне рН, который, вероятно, будет присутствовать в пищеварительном тракте, для того, чтобы продемонстрировать реальный эффект пролекарства, или пролекарства были не в состоянии улучшить пероральную биодоступность S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты по сравнению с тетрагидратом соли S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и L-аргинина. Среди аминокислотных пролекарств были также раскрыты пролекарства L-аланина и L-валина. Оба пролекарства имели ограниченную водорастворимость (<10 мг/мл). В целом предполагается, что неадекватная растворимость в воде представляет собой важный фактор, ограничивающий пероральную биодоступность.
Цель настоящего изобретения состоит в получении соединения с улучшенной растворимостью в воде в широком диапазоне рН, который, вероятно, будет иметь место в пищеварительном тракте, и с увеличенной пероральной биодоступностью.
Сущность изобретения
Согласно настоящему изобретению описаны соли пролекарств S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты Формулы I
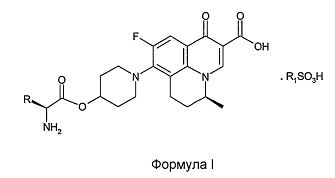
и их фармацевтически приемлемые сольваты, полиморфы или гидраты,
в которой
R обозначает CH3 или CH(CH3)2 и
R1 обозначает C1-C6 алкил или фенил, в случае необходимости замещенный одним или более заместителями, выбранными из C1-C6 алкила, галогена, нитро, гидрокси или C1-C6 алкокси.
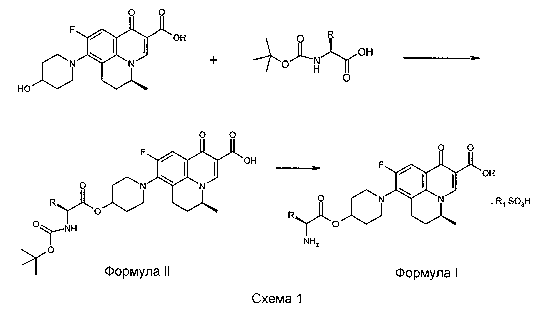
Настоящее изобретение также относится к способу получения соединения формулы I, включающему стадии сочетания амин-защищенного L-аланина или L-валина с S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислотой с получением соединения формулы II, удаления защитной группы от соединения формулы II и выделения соединения по изобретению формулы I или его фармацевтически приемлемой соли.
Дополнительно, это изобретение относится к способу или способам получения соединений по изобретению формулы I и их полиморфов и гидратов.
Изобретение также относится к фармацевтическим композициям, содержащим соединения по изобретению, и к способу лечения или профилактики бактериальных инфекций с использованием соединений и композиций по изобретению.
Детали одного или более вариантов осуществления изобретения представлены ниже в описании. Другие признаки, объекты и преимущества изобретения будут очевидны из описания и формулы изобретения.
Подробное описание изобретения
Согласно настоящему изобретению описаны пролекарства S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты Формулы I
и их фармацевтически приемлемые сольваты, полиморфы или гидраты,
в которой
R обозначает CH3 или CH(CH3)2 и
R1 обозначает C1-C6 алкил или фенил, в случае необходимости замещенный одним или более заместителями, выбранными из C1-C6 алкила, галогена, нитро, гидрокси или C1-C6 алкокси.
Растворимость в воде является важным параметром для пероральной биодоступности лекарственного средства. Было обнаружено, что растворимость в воде соединений по изобретению составляет >200 мг/мл при рН 7, что в основном больше, чем у свободного основания или гидрохлорида.
Было показано, что соединения формулы I обеспечивают особенно хорошую абсорбцию, отраженную в увеличенном AUC и Cmax при введении пероральным путем у крыс по сравнению с родительским соединением, то есть S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислотой и ее солью с L-аргинином. Также по сравнению с гидрохлоридом пролекарства, S-(-)-9-фтор-6,7-дигидро-8-(4-L-аланинилоксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты, соединения по изобретению показали увеличенный AUC и Cmax при введении пероральным путем у крыс. Кроме того, более низкая растворимость гидрохлорида S-(-)-9-фтор-6,7-дигидро-8-(4-L-аланинилоксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]-хинолизин-2-карбоновой кислоты может страдать от общего ионного эффекта при прохождении через желудочную область. Также было обнаружено, что соединения по изобретению формулы I хорошо переносятся в больших дозах у грызунов и собак.
Другой вариант осуществления настоящего изобретения относится к способу получения соединений формулы I. Соединения формулы I могут быть получены путем сочетания амин-защищенной аминокислоты с S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислотой в присутствии агента сочетания. Используемая аминокислота представляет собой L-аланин или L-валин. Соединение формулы подвергают удалению защитной группы, чтобы получить соединение по изобретению формулы I.
Соединения по изобретению могут быть получены из S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты путем этерификации 4-гидроксипиперидина N-трет-бутоксикарбонил-L-аланином или N-трет-бутоксикарбонил-L-валином в присутствии агента сочетания при использовании методик, известных из уровня техники. Обычно реакцию сочетания осуществляют в присутствии агента сочетания в подходящем растворителе и в присутствии одного или более основания при температуре, составляющей от -30°C до +150°C, получая соединение формулы II. Предпочтительные агенты сочетания включают карбодиимиды, 2,4,6-трихлорбензоилхлорид, метансульфонилхлорид и т.п. Карбодиимиды, такие как дициклогексилкарбодиимид или N-(3-диметиламинопропил)-N-этилкарбодиимид, могут использоваться в качестве агентов сочетания. Подходящий растворитель выбирают из галогенированных растворителей, таких как дихлорметан, хлороформ, или биполярных апротонных растворителей, таких как тетрагидрофуран, N,N-диметилформамид, или их смесей. Одно или более оснований выбирают из триэтиламина, N,N-диметиламинопиридина, основания Ханига (N,N-диизопропилэтиламин).
Реакцию сочетания осуществляют, обрабатывая S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновую кислоту N-трет-бутоксикарбонил-L-аланином или N-трет-бутоксикарбонил-L-валином в галогенированном углеводороде, таком как дихлорметан, хлороформ или этилендихлорид и в присутствии N,N-диметиламинопиридина и агента сочетания, дициклогексилкарбодиимида, при температуре от -10 до 0°C. Или, в альтернативном варианте, реакция сочетания может быть выполнена обработкой S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты N-трет-бутоксикарбонил-L-аланином или N-трет-бутоксикарбонил-L-валином в тетрагидрофуране и диметилформамиде с использованием в качестве агента сочетания трихлорбензоилхлорида в присутствии N,N-диметиламинопиридина при температуре от -10 до 0°C. Реакция сочетания может также быть выполнена путем обработки S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты N-трет-бутоксикарбонил-L-аланином или N-трет-бутоксикарбонил-L-валином в тетрагидрофуране и диметилформамиде с использованием метансульфонил хлорида в качестве агента сочетания в присутствии триэтиламина при температуре от -10 до 0°C.
Стадия удаления защитной группы N-трет-бутоксикарбонила (ВОС) может быть выполнена при использовании способов, описанных в T. W. Greene and P. G. M. Wuts, Protective groups in organic synthesis, 3rd edition, John Wiley & Sons, New York (1999). Соответственно удаление защитной группы может быть выполнено путем обработки соединения формулы II с использованием умеренно кислых условий в водных или неводных растворах. Растворители, которые могут использоваться для стадии удаления защитной группы в растворе, представляют собой водные растворы кислоты с добавлением или без добавления органических растворителей, или неводные органические растворители. Органические растворители выбирают из группы, включающей ацетон, ацетонитрил, этанол, изопропанол или их смеси. Кислые условия могут использоваться при использовании неорганической кислоты, такой как соляная кислота, или, в качестве альтернативы, при использовании любой другой органической кислоты, такой как метансульфоновая кислота, толуолсульфоновая кислота, бензолсульфоновая кислота, п-нитробензолсульфоновая кислота, трифторуксусная кислота, уксусная кислота, муравьиная кислота или их смеси. Предпочтительно, соединение формулы II обрабатывают метансульфоновой кислотой, п-толуолсульфоновой кислотой, бензолсульфоновой кислотой, п-бромбензолсульфоновой кислотой или п-нитробензолсульфоновой кислотой при температуре в пределах от -10°C до 100°C в ацетоне или ацетонитриле, получая соединение по изобретению формулы I.
Другой аспект настоящего изобретения относится к очистке сырого продукта соединения по изобретению формулы I. Очистку соединений по изобретению осуществляют удалением примесей, растворяя примеси в органическом растворителе. Используемыми растворителями являются ацетон, простой диэтиловый эфир или их смеси.
Полиморфы соединений формулы I могут быть получены кристаллизацией соединения формулы I в различных условиях, например температуры, времени и/или использования специфических растворителей. Гидраты соединений формулы I могут быть получены способами, известными специалисту.
Термин “C1-C6 алкил” относится к насыщенным углеводородным радикалам с прямой или разветвленной цепью, содержащим от одного до шести атомов углерода. Примеры C1-C6 алкильных радикалов включают, но не ограничены ими, метил, этил, пропил, бутил, пентил, гексил и их разветвленные изомеры, такие как изопропил, изобутил, трет-бутил.
Термин “C1-C6 алкокси” относится к радикалу кислорода, имеющему заместитель в виде углеводородной цепи, где углеводородная цепь представляет собой алкил (т.e. -O-алкил). Примерами C1-C6 алкокси являются метокси, этокси, пропилокси, изопропилокси, бутилокси, пентилокси, гексилокси.
Термин "галоген" в рамках изобретения относится к атому, выбранному из фтора, хлора, брома и йода.
Термин "нитро" относится к группе формулы -NO2-.
Термин “алкилсульфокислота” относится к формуле (C1-C6 алкил)-SO3H; например, метансульфокислота метана (CH3SO3H), этансульфокислота (CH3CH2SO3H).
Изобретение также относится к жидким и твердым фармацевтическим составам, которые содержат пролекарство по изобретению, таким как, например, инъецируемые растворы, суспензии, эмульсии, таблетки, таблетки, покрытые оболочкой, сердцевины таблеток, покрытых оболочкой, капсулы, растворы, пастилки, дисперсии, пилюли, гранулы, суппозитории, твердые или мягкие желатиновые капсулы и т.п.
Фармацевтические композиции получают согласно обычным процедурам, используемым специалистами для получения стабильных и эффективных композиций. Такие способы включают смешивание, перемешивания, суспендирование, диспергирование, эмульгирование, растворение и т.п. активных соединений с фармацевтическими вспомогательными средствами или в фармацевтических вспомогательных средствах, таких как носитель, разбавитель, растворитель или эксципиент, и переработку компонентов в фармацевтически подходящие формы для парентерального, перорального, внутриносового, щечного или ректального введения и т.п. В твердых, жидких, парентеральных лекарственных формах эффективное количество активного соединения или активного ингредиента представляет собой любое количество, которое приводит к желательным результатам. В соответствии с настоящим изобретением было обнаружено, что выгодные свойства растворимости пролекарства по изобретению могут быть приданы составу фармацевтических лекарственных форм. Это может также использоваться для получения таблеток мокрой грануляцией или обычной сухой грануляцией. Это может также использоваться для получения водных лекарственных форм.
Лекарственные формы могут быть получены любыми обычными методиками, известными из уровня техники, но предпочтительно их составляют, смешивая пролекарство по изобретению с другими ингредиентами. Другие ингредиенты, используемые для составления твердых пероральных лекарственных форм, включают обычные инертные ингредиенты, такие как микрокристаллическая целлюлоза, метилцеллюлоза и т.п., подходящие подсластители, красители и/или ароматизаторы, и, в случае необходимости, их консерванты. Такие твердые пероральные лекарственные формы или сухие составы, подходящие для получения суспензий, составляют таким образом, что они содержат эффективную дозу соединения по изобретению. В целом рассматриваются твердые лекарственные формы, содержащие от 100 мг до 1500 мг соединения по изобретению. Препараты, подходящие для пероральной суспензии, содержат подобную дозу.
Фармацевтические составы могут быть составлены вместе со вспомогательными средствами и добавками, обычно используемыми в фармации, такими как связующие таблеток, наполнители, консерванты, дезинтеграторы таблеток, регуляторы потока, мягчители, смачивающие вещества, диспергирующие агенты, эмульгаторы, растворители, модификаторы рН, ароматизаторы и т.п. Вторым предпочтительным способом является парентеральное внутримышечное, внутривенное или подкожное введение.
Когда фармацевтическую композицию составляют в форме препарата для инъекций, при составлении фармацевтической композиции в форму раствора или суспензии могут использоваться все растворители, обычно используемые в данной области техники. Примерами подходящих растворителей являются вода, этанол, полипропиленгликоль, этоксилированный изостеариловый спирт, полиоксиэтиленсорбит и сложные эфиры сорбитана. В терапевтическое средство могут быть включены хлорид натрия, глюкоза или глицерин. Предпочтительно, чтобы концентрация активного ингредиента в препарате для инъекций находилась в диапазоне от 0,1 мг/мл до 100 мг/мл.
В дополнение к обычным лекарственным формам, описанным выше, соединения, согласно настоящему изобретению, могут также вводиться с помощью средств контролируемого высвобождения и/или устройствами для доставки, такими как описанные в патентах США 3 845 770; 3 916 899; 3 536 809; 3 598 123 и 4 008 719, раскрытия которых тем самым включены в настоящее описание путем ссылки.
Диапазон полной суточной дозы обычно составляет от приблизительно 200 мг до приблизительно 5000 мг пролекарства по изобретению. Предпочтительно, диапазон полной суточной дозы составляет от 300 мг до 3000 мг пролекарства по изобретению. Однако доза может быть более высокой или более низкой в зависимости от потребностей и состояний пациента.
Антибактериальные соединения и фармацевтические композиции по изобретению могут быть использованы в лечении человека и животных, имеющих широкий спектр бактериальных инфекций, таких как импетиго, пневмония, бронхит, фарингит, эндокардит, инфекции мочевых путей, язвы диабетической стопы, желудочно-кишечные инфекции и бактериемия. Эти бактериальные инфекции могут быть вызваны любой из следующих бактерий - Staphylococcus aureus, коагулаза-негативный стафилококк, метицилин-резистентный Staphylococcus aureus, метицилин-резистентный коагулаза-негативный стафилококк, энтерококки, бета-гемолитические стрептококки, стрептококки группы viridans, микобактериальные инфекции, вызванные M. tuberculosis с множественной лекарственной резистентностью и другими атипичными микобактериями, такими как M. intracellulare и M. avium, а также как новыми грамотрицательными патогенами, такими как Chryseobacterium meningosepticum, Chryseobacterium indologense, и другими грамотрицательными патогенами, такими как E. coli, Klebsiella, Proteus, Serratia, Citrobacter и Pseudomonas.
Настоящее изобретение также охватывает противоинфекционную композицию для лечения человека и животных в целях профилактики и/или терапии системных инфекций, особенно инфекций, вызываемых резистентными грамположительными организмами, инфекций, вызываемых грамотрицательными организмами, микобактериальных инфекций и инфекций, вызываемых внутрибольничными патогенами, которая содержит некоторое количество соединения по изобретению, в основном достаточное, чтобы ликвидировать указанную инфекцию, но которое не вызывает каких бы то ни было нежелательных побочных эффектов. Соединение и композиции по изобретению могут вводиться человеку и животным, которые подвергаются риску инфицирования, например соединение или композиция по изобретению могут вводиться пациенту до и/или после хирургии, работникам сферы здравоохранения или другим лицам, которые подвергаются риску инфицирования.
Настоящее изобретение охватывает введение соединений животным или человеку. Соединение и композиции, которые используются в изобретении, должны соответственно быть фармацевтически приемлемыми. В рамках изобретения такой "фармацевтически приемлемый" компонент представляет собой компонент, который является подходящим для использования у человека и/или животных без нежелательных неблагоприятных побочных эффектов (таких как токсичность, раздражение и аллергическая реакция), соразмерным с приемлемым отношением выгоды/риска. Животные, которые могут быть подвергнуты лечению при использовании соединений по изобретению, включают, но не ограничены ими, млекопитающих, рыб, птиц.
Следующие детализированные примеры служат, чтобы более полно иллюстрировать изобретение, не ограничивая его объем. Следует понимать, что различные другие варианты осуществления и модификации в практике изобретения будут очевидны для специалиста и могут быть легко им осуществлены без отхода от объема и духа изобретения, как описано выше. Соответственно объем приложенной формулы изобретения не ограничен точным описанием, приведенным выше, а должен рассматриваться как включающий все признаки патентоспособной новизны, которые включены в настоящее изобретение, включая все признаки и варианты осуществления, которые специалист мог бы трактовать как эквиваленты этого изобретения. Изобретение далее описано в отношении следующей экспериментальной работы.
Следующие детализированные примеры служат, чтобы более полно иллюстрировать изобретение, не ограничивая его объем.
Экспериментальная часть:
(S)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]-хинолизин-2-карбоновую кислоту получали согласно процедуре, описанной в Chem. Pharm. Bull. 1996, 44(4), 642-645.
Пример 1
Получение (2'S,5S)-9-фтор-6,7-дигидро-8-(4-(N-трет-бутоксикарбонил-L-аланинил-окси)-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты
Способ 1: К смеси N-трет-бутоксикарбонил-L-аланина (473 г) в дихлорметане (2 л) при температуре от -10 до 0°C добавляли дициклогексилкарбодиимид (515 г), растворенный в дихлорметане (2 л), получая мутную суспензию. К этой мутной суспензии добавляли 300 г (S)-9-фтор-6,7-дигидро-8-(4-гидрокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты, затем 4-N,N-диметиламинопиридин (58 г), и реакционную смесь перемешивали при температуре от -10 до 5°C в течение 2 часов. Суспензию фильтровали, и твердое вещество промывали 500 мл дихлорметана. Фильтрат промывали водой. Фильтрат высушивали над безводным сульфатом натрия. Высушенный органический слой затем концентрировали до половины его объема, после чего осаждали твердое вещество. Твердое вещество отфильтровывали и промывали 300 мл дихлорметана. Чистый органический фильтрат концентрировали досуха, получая масляную массу. Масляную массу растирали с простым диэтиловым эфиром (4 л), получая твердое вещество белого цвета. Твердое вещество отфильтровывали с отсасыванием и промывали простым диэтиловым эфиром (1 л), получая целевое соединение в количестве 415 г (94 %).
Способ 2: К смеси триэтиламина (98,0 мл) и N-трет-бутоксикарбонил-L-аланина (110 г) в смеси тетрагидрофурана (1050 мл) и N,N-диметилформамида (350 мл) добавляли 2,4,6-трихлорбензоил хлорид (100 мл). Полученную смесь перемешивали при температуре от -5 до 0°C в течение 5 часов. К реакционной смеси добавляли 4-N,N-диметиламинопиридин (24 г) и (S)-9-фтор-6,7-дигидро-8-(4-гидрокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновую кислоту (70 г). Реакционную смесь перемешивали еще 7 часов при температуре от -5 до 0°C. Суспензию фильтровали при комнатной температуре и фильтрат экстрагировали этилацетатом после добавления воды. Упариванием органического слоя при пониженном давлении получали липкое твердое вещество, которое после растирания с простым диэтиловым эфиром приводило к твердому веществу белого цвета в количестве 85 г.
Способ 3: К раствору N-трет-бутоксикарбонил-L-аланина (7,9 г) в смеси тетрагидрофурана (75 мл) и N,N-диметилформамида (25 мл) при температуре от -10 до 0°C добавляли по каплям метансульфонилхлорид (2,42 мл). К указанному раствору добавляли по каплям за 5 мин триэтиламин (8,7 мл). Реакционную смесь перемешивали в течение 1,5 часов, поддерживая температуру от -10 до 0°C. К реакционной смеси добавляли (S)-9-фтор-6,7-дигидро-8-(4-гидрокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновую кислоту (5,01 г) и 4-N,N-диметиламинопиридин (1,70 г). Реакционную смесь перемешивали в течение дополнительного 1 часа при температуре от -5 до 0°C. Суспензию фильтровали при комнатной температуре и фильтрат разбавляли водой (300 мл) и экстрагировали этилацетатом (150 мл×2). Упариванием органического слоя при пониженном давлении получали липкое твердое вещество, которое после растирания с простым диэтиловым эфиром приводило к твердому веществу белого цвета в количестве 6,38 г (86%).
Пример 2
Получение соли (2'S,5S)-9-фтор-6,7-дигидро-8-(4-L-аланинил-окси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты с метансульфоновой кислотой
К смеси (2'S,5S)-9-фтор-6,7-дигидро-8-(4-N-трет-бутоксикарбонил-L-аланинилокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты (415 г) в ацетоне (4,5 л) добавляли метансульфоновую кислоту (66 мл). Реакционную смесь перемешивали при температуре 65-67°C в течение ночи. Суспензию фильтровали при 40-45°C. Твердое вещество промывали ацетоном (1,5 л), затем простым диэтиловым эфиром (1,5 л). Твердое вещество грязно-белого цвета высушивали в вакууме (40-45 мм) при температуре 55-60°C за период 3-4 часов. Целевое соединение получали в форме свободно текучего грязно-белого материала 383,0 г (93 %).
Для MF: C23H30FN3O8S, MS (ES+) m/z 432 (полученный как свободное основание для MF: C22H26FN3O5); M.P. 278,50°C DSC.
Пример 3
Получение соли (2'S,5S)-9-фтор-6,7-дигидро-8-(4-L-валинилокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и метансульфоновой кислоты
Стадия 1: Получение (2'S,5S)-9-фтор-6,7-дигидро-8-{4-[N-трет-бутилоксикарбонил-L-валинилокси]-пиперидин-1-ил}-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты
К смеси триэтиламина (14 мл) и N-трет-бутилоксикарбонил-L-валина (18 г) в смеси тетрагидрофурана (50 мл) и N,N-диметилформамида (15 мл) добавляли 2,4,6-трихлорбензоилхлорид (14,3 мл). Полученную смесь перемешивали при температуре от -5 до 0°C в течение 5 часов. К реакционной смеси добавляли 4-N,N-диметиламинопиридин (3,4 г) и (S)-9-фтор-6,7-дигидро-8-{4-гидрокси-пиперидин-1-ил}-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновую кислоту (10 г). Реакционную смесь перемешивали еще 7 часов при температуре от -5 до 0°C. Суспензию фильтровали при комнатной температуре и фильтрат экстрагировали этилацетатом после добавления воды. Упариванием органического слоя при пониженном давлении получали липкое твердое вещество, которое после растирания с простым диэтиловым эфиром приводило к твердому веществу белого цвета в количестве 12,5 г.
Стадия 2: Получение соли (2'S,5S)-9-фтор-6,7-дигидро-8-{4-L-валинилокси-пиперидин-1-ил}-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и метансульфоновой кислоты
Смесь соединения, полученного на стадии 1 (12 г), и метансульфокислоты (1,81 мл) в ацетоне (150 мл) перемешивали при температуре 63-65°C в течение 10 часов. Суспензию фильтровали при комнатной температуре и твердое вещество промывали ацетоном, получая твердое вещество грязно-белого цвета в количестве 9,8 г.
Пример 4
Получение соли (2'S,5S)-9-фтор-6,7-дигидро-8-{4-(L-аланинилокси)-пиперидин-1-ил}-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и толуолсульфоновой кислоты
К смеси (2'S,5S)-9-фтор-6,7-дигидро-8-(4-N-трет-бутоксикарбонил-L-аланинилокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты (3,0 грамма, 5,64 моль) в ацетоне (35 мл) добавляли моногидрат п-толуолсульфоновой кислоты (1,61 грамма, 8,47 моль). Реакционную смесь нагревали с обратным холодильником в течение ночи. Суспензию фильтровали при 40-45°C и полученное твердое вещество промывали ацетоном, затем простым диэтиловым эфиром. Твердое вещество высушивали в вакууме (40-45 мм) при температурах 55-60°C за период 3-4 часов, получая целевое соединение в форме твердого вещества грязно-белого цвета в количестве 3,0 граммов (выход 88,23%). Для MF: C29H34FN3O8S, MS (M+H) m/z 432 (полученный как свободное основание для MF: C22H26FN3O5); M.P. 232°C.
Пример 5
Получение соли (2'S,5S)-9-фтор-6,7-дигидро-8-{4-(L-аланинилокси)-пиперидин-1-ил}-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и бензолсульфоновой кислоты
К смеси (2'S,5S)-9-фтор-6,7-дигидро-8-(4-N-трет-бутоксикарбонил-L-аланинилокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты (3,0 грамма, 5,64 моль) в ацетоне (35 мл) добавляли бензолсульфоновую кислоту (1,33 грамма, 8,41 моль). Реакционную смесь нагревали с обратным холодильником в течение ночи. Суспензию фильтровали при 40-45°C и полученное твердое вещество промывали ацетоном, затем простым диэтиловым эфиром. Твердое вещество высушивали в вакууме (40-45 мм) при температурах 55-60°C за период 3-4 часов, получая целевое соединение в форме твердого вещества светло-желтого цвета в количестве 3,1 граммов (выход 90,0%). Для MF: C28H32FN3O8S, MS (M+H) m/z 432 (полученный как свободное основание для MF: C22H26FN3O5); M.P. 229°C.
Пример 6
Получение соли (2'S,5S)-9-фтор-6,7-дигидро-8-{4-(L-аланинилокси)-пиперидин-1-ил}-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и нитробензолсульфоновой кислоты
К смеси (2'S,5S)-9-фтор-6,7-дигидро-8-(4-N-трет-бутоксикарбонил-L-аланинилокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты (3,0 грамма, 5,64 моль) в ацетоне (35 мл) добавляли п-нитробензолсульфоновую кислоту (1,73 грамма, 8,47 моль). Реакционную смесь нагревали с обратным холодильником в течение ночи. Суспензию фильтровали при 40-45°C и полученное твердое вещество промывали ацетоном, затем простым диэтиловым эфиром. Твердое вещество высушивали в вакууме (40-45 мм) при температурах 55-60°C за период 3-4 часов, получая целевое соединение в форме твердого вещества светло-желтого цвета в количестве 3,3 граммов (выход 92,43%). Для MF: C28H31FN4O10S, MS (M+H) m/z 432 (полученный как свободное основание для MF: C22H26FN3O5); M.P. 240°C.
Пример тестирования 1
Анализ растворимости: Способ для определения растворимости в воде при различных физиологических значениях рН при температурах 25-30°C. К точно взвешенному количеству тестируемого вещества добавляли частями специфический буферный раствор (части приблизительно по 50 микролитров) при температурах 25-30°C, до полного растворения твердого вещества и получения прозрачного раствора.
pH 7
Биологический Пример
Способ изучения фармакокинетики (PK) на крысах Sprague Dawley: фармакокинетическое исследование проводили на самцах крыс Sprague Dawley весом 200-220 г, которых выдерживали натощак в течение ночи. Соль (2'S,5S)-9-фтор-6,7-дигидро-8-(4-L-аланинилокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]-хинолизин-2-карбоновой кислоты и метансульфоновой кислоты (A), гидрохлорид (2'S,5S)-9-фтор-6,7-дигидро-8-(4-L-аланинилокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты (B) и соль (2'S,5S)-9-фтор-6,7-дигидро-8-(4-L-валинилокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и метансульфоновой кислоты (C) растворяли в воде Milli Q. Соединения A, B и C вводили в дозе 50 мг/кг, перорально, количества в пересчете на активный ингредиент. Соль (5S)-9-фтор-6,7-дигидро-8-(4-гидрокси-пиперидин-1-ил)-5-метил-1-оксо-1H,5H-бензо[i,j]хинолизин-2-карбоновой кислоты и L-аргинина (D) растворяли в 1%-ном Tween перед использованием. Соединения A и D вводили в дозе 200 мг/кг, перорально, количества в пересчете на активный ингредиент. Объем дозы составлял 0,5 мл/200 г. Пробы крови собирали через предопределенные временные интервалы, то есть 0, 0,5, 1, 2, 4, 6 и 8 часов в ампулы eppendorff, содержащие 10 мкл насыщенного раствора фторида натрия. Плазму собирали после центрифугирования пробы крови при 10000 об/мин в течение 10 мин. Образцы плазмы анализировали с помощью ВЭЖХ для того, чтобы определить уровни лекарственного средства. Параметры PK AUC, Cmax вычисляли некомпартментным анализом при использовании программного обеспечения Winnonlin.
при пероральном введении у крыс
50 мг/кг per os
200 мг/кг per os
50 мг/кг per os
Настоящее изобретение относится к новым производным бензохинолизин-2-карбоновой кислоты формулы I и их фармацевтически приемлемым сольватам, в которой R обозначает СН3 или СН(СН3)2 и R1 обозначает С1-С6 алкил или фенил, в случае необходимости замещенный одним или более заместителями, выбранными из C1-С6 алкила или нитро. Также изобретение относится к способу получения соединения формулы I, к фармацевтической композиции на основе соединения формулы I и применению соединения формулы I. Технический результат: получены новые производные бензохинолизин-2-карбоновой кислоты, обладающие улучшенной растворимостью в воде широком диапазоне рН, которые могут использоваться для лечения бактериальных грамположительных, грамотрицательных и анаэробных инфекций. 4 н. и 14 з.п. ф-лы, 2 табл.
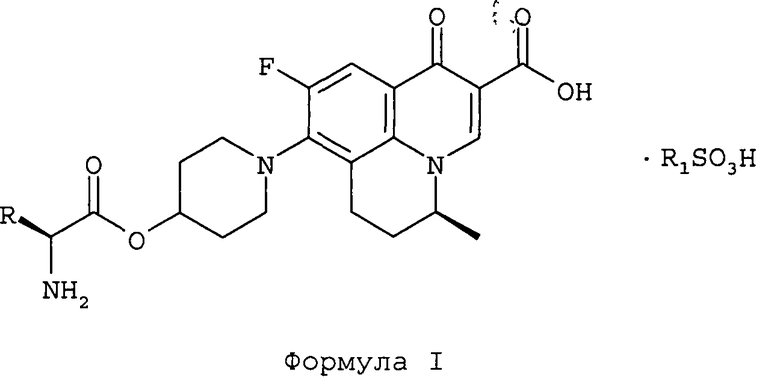
1. Соединения, имеющие структуру Формулы I
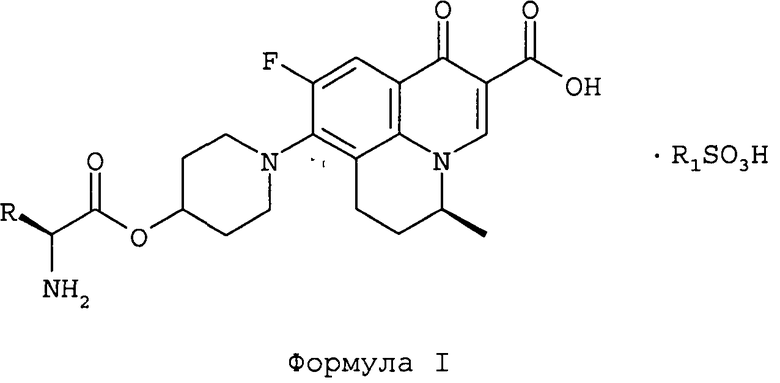
и их фармацевтически приемлемые сольваты,
в которой R обозначает СН3 или СН(СН3)2; и
R1 обозначает C1-С6 алкил или фенил, в случае необходимости замещенный одним или более заместителями, выбранными из С1-С6 алкила или нитро.
2. Соединения по п.1, в которых R1 обозначает C1-С6 алкил.
3. Соединения по п.1, в которых R1 обозначает фенил, в случае необходимости замещенный одним или более заместителями, выбранными из C1-С6 алкила или нитро.
4. Соединения по п.1, в которых R обозначает СН3 или СН(СН3)2 и R1 обозначает метил, этил, фенил, п-нитрофенил, толил.
5. Способ получения соединения Формулы (I) и его фармацевтически приемлемых сольватов, включающий:
а) сочетание S-(-)-9-фтор-6,7-дигидро-8-(4-гидроксипиперидин-1-ил)-5-метил-1-оксо-1Н,5Н-бензо[i,j]хинолизин-2-карбоновой кислоты с амин-защищенным L-аланином или L-валином в одном или более растворителях с получением защищенного соединения Формулы II;
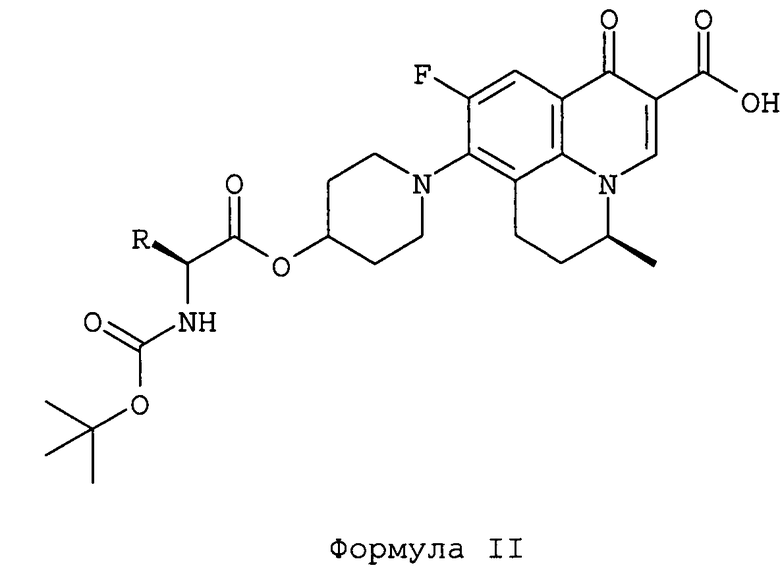
b) удаление защитной группы от соединения Формулы II с помощью сульфокислоты общей Формулы R1-SO3H, в которой R1 имеет значение, определенное в п.1;
c) выделение соединения Формулы I.
6. Способ по п.5, в котором реакцию сочетания осуществляют в присутствии агента сочетания.
7. Способ по п.6, в котором агент сочетания включает одно или более соединений из числа дициклокарбодиимида, 2,4,6-трихлорбензоилхлорида и метансульфонилхлорида.
8. Способ по п.5, дополнительно включающий осуществление реакции сочетания в присутствии основания.
9. Способ по п.8, в котором основание включает одно или более соединений из числа триэтиламина, N,N-диметиламинопиридина и диизопропилэтиламина.
10. Способ по п.5, в котором растворитель включает одно или более соединений из числа дихлорметана, хлороформа, этиленхлорида, тетрагидрофурана, N,N-диметилформамида и их смесей.
11. Способ по п.5, в котором реакцию сочетания осуществляют при температуре от приблизительно -10°С до приблизительно 0°С.
12. Способ по п.5, в котором используемой сульфокислотой является алкилсульфокислота.
13. Способ по п.12, в котором сульфокислотой является метансульфокислота.
14. Фармацевтическая композиция, обладающая антибактериальной активностью, содержащая терапевтически эффективное количество одного или более соединений по п.1 вместе с фармацевтически приемлемыми носителями, эксципиентами или разбавителями.
15. Применение соединений по п.1 для получения лекарственного средства для лечения или профилактики состояния, вызванного или обусловленного бактериальными инфекциями, включающее введение млекопитающему терапевтически эффективных количеств одного или более соединений по п.1.
16. Применение соединений по 15, в котором состояние выбрано из импетиго, пневмонии, бронхита, фарингита, эндокардита, инфекций мочевых путей, язв диабетической стопы, желудочно-кишечных инфекций или бактериемии.
17. Применение соединений по п.15, в котором бактериальная инфекция вызвана грамположительными, грамотрицательными или анаэробными бактериями.
18. Применение по п.17, в котором грамположительные, грамотрицательные или анаэробные бактерии выбраны из Staphylococcus aureus, коагулаза-негативных стафилококков, метицилин-резистентного Staphylococcus aureus, метицилин-резистентных коагулаза-негативных стафилококков, энтерококков, бета-гемолитических стрептококков, стрептококков группы viridans, микобактериальных инфекций, вызванных М. tuberculosis с множественной лекарственной резистентностью и другими атипичными микобактериями, такими как М. intracellulare и М. avium, а также новыми грамотрицательными патогенами, такими как Chryseobacterium meningosepticum, Chryseobacterium indologense, и другими грамотрицательными патогенами, такими как Е. coli, Klebsiella, Proteus, Serratia, Citrobacter и Pseudomonas.
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ получения производных пиперазинилбензогетероциклических соединений или их кислотно-аддитивных солей | 1979 |
|
SU993818A3 |
| Замещенные бензо( )хинолизин2-карбоновые кислоты или их производные по карбоксильной группе | 1974 |
|
SU522183A1 |

