Область техники, к которой относится изобретение
Настоящее изобретение относится к новым аминопиридиновым производным, которые применимы в фармацевтической области, и более конкретно, к аминопиридиновым производным, которые ингибируют рост опухолевых клеток на основе селективной ингибирующей активности в отношении киназы Аврора А и обладают противоопухолевым эффектом, а также к селективному ингибитору киназы Аврора А и противоопухолевому средству, содержащему такие производные.
Уровень техники
Киназа Аврора представляет собой серин/треонинкиназу, участвующую в делении клеток. В настоящее время известны три подтипа киназы Аврора: А, В и С, которые высокогомологичны друг другу. Аврора А участвует в созревании и распределении центросомы или в формировании тела веретена. С другой стороны, считается, что Аврора В участвует в агрегации и конъюгации хромосомы, сверочной (контрольной) точке веретена и делении цитоплазмы [Nat. Rev. Mol. Cell Biol., No.4, pp.842-854]. Также считается, что Аврора С действует аналогичным образом в результате взаимодействия с Авророй В [J. Biol. Chem., Epub ahead (2004)]. На основании того факта, что ранее в большинстве раковых клеток была подтверждена высокая экспрессия киназы Аврора А; что высокая экспрессия киназы Аврора А в нормальных клетках приводит к трансформации линий нормальных клеток грызунов и т.п., считается, что Аврора A, являющаяся одним из онкогенов, должна быть подходящей мишенью для противоопухолевого средства [EMBO J., № 17, стр. 3052-3065 (1998)].
Согласно другому сообщению раковые клетки, где Аврора A экспрессируется в высокой степени, обладают резистентностью к паклитакселу [Cancer Cell, Vol. 3, pp. 51-62 (2003)]. В то же время в отношении ингибитора киназы Аврора считается, что разработка лекарственных средств, селективных к определенному ее подтипу, является трудной задачей, если учесть высокую гомологию среди подтипов, структурный анализ белка и т.п.; и хотя известны сообщения о лекарственных средствах, таких как ZM447439, которые одновременно ингибируют как Аврору A, так и Аврору B [J. Cell Biol., № 161, стр. 267-280 (2003); J. Cell Biol., № 161, стр. 281-294, (2003); Nat. Med., № 10, стр. 262-267, (2004)], сообщений по поводу лекарственных средств, селективных в отношении Авроры A, неизвестно. Таким образом, в указанных сообщениях противоопухолевый эффект описан лишь для того случая, когда вводится только одно лекарственное средство, которое одновременно ингибирует как Аврору A, так и Аврору B. Кроме того, сообщалось также, что в лекарственном средстве, которое одновременно ингибирует как Аврору A, так и Аврору B, ингибирующее действие в отношении киназы Аврора ослабляет активность паклитаксела [J. Cell Biol., № 161, стр. 281-294, (2003)].
В настоящее время были поданы патентные заявки, относящиеся к соединениям, обладающим ингибирующей активностью в отношении киназы Аврора (WO 02/057259, патент США № 6664247 и т.д.), и патентные заявки, относящиеся к аминопиридиновым производным, были поданы также (патент США № 6586424 и т.д). При данных обстоятельствах авторы настоящего изобретения подали патентную заявку, относящуюся к аминопиридиновому производному, обладающему превосходной селективной ингибирующей активностью в отношении Авроры A (WO 2006/046734).
Описание изобретения
Проблемы, которые должно решить настоящее изобретение, состоят в создании новых аминопиридиновых производных, которые обладают превосходной селективной ингибирующей активностью в отношении Авроры A и ингибирующей активностью в отношении роста клеток на основании вышеизложенного, а также в достижении синергического действия путем комбинированного применения с другим противоопухолевым средством(ми). Кроме того, проблемы, которые должно решить настоящее изобретение, также состоят в создании, в случае перорального введения, новых аминопиридиновых производных, которые обладают превосходной селективной ингибирующей активностью в отношении Авроры A.
Для решения указанных выше проблем авторы настоящего изобретения синтезировали ряд новых аминопиридиновых производных и обнаружили, что соединение, представленное следующей формулой (I), на основании вышеизложенного обладает превосходной селективной ингибирующей активностью в отношении Авроры A и ингибирующей активностью в отношении роста клеток, а также достигает синергического действия путем комбинированного применения с другими противоопухолевыми средствами, тем самым осуществляя изобретение. Что касается тех типов рака, которые нельзя полностью вылечить с помощью известных противоопухолевых средств, таких как паклитаксел, поскольку из-за побочных эффектов или резистентности к лекарственному средству невозможно применять достаточное количество таких средств, ожидается, что пероральное введение соединения по изобретению или комбинированное введение соединения по изобретению с другим противоопухолевым средством даст превосходный противоопухолевый эффект (включая усиление активности, обусловленное другим противоопухолевым средством) и эффект по ослаблению побочных эффектов.
Таким образом, изобретение относится к соединению общей формулы I:

где:
R1 представляет собой атом водорода, F, CN, COORa1, CONRa2Ra2', NRa3CORa3', CONRa4ORa4', NRa5CONRa5'Ra5", NRa6COORa6', SO2NRa7Ra7', NRa8SO2Ra8', CORa9, SO2Ra10, NO2, ORa11 или NRa12Ra12',
где:
каждый из Ra1, Ra3, Ra4, Ra5, Ra6 и Ra8 независимо представляет собой атом водорода или низший алкил;
каждый из Ra2, Ra2', Ra5', Ra5", Ra7, Ra7', Ra12 и Ra12' независимо представляет собой атом водорода или низший алкил, который может быть замещен одним или несколькими одинаковыми или разными заместителями, выбранными из <группы-заместителя L1>, где <группа-заместитель L1> представляет собой атом галогена, гидрокси, нитро, циано, амино, карбамоил, аминосульфонил, имино, низшую алкиламиногруппу, ди(низший)алкиламиногруппу, низший алкилсульфонил, низшую алкилсульфониламиногруппу, низший алкокси, низший алкоксикарбонил, низшую алкоксикарбониламиногруппу, низший алканоил, низший алканоилокси, низший алкилтио и карбоксил; однако при условии, что каждые из Ra2 и Ra2'; Ra5' и Ra5"; Ra7 и Ra7'; Ra12 и Ra12' независимо, вместе с атомом азота, к которому они присоединены, могут образовывать 5-членную или 6-членную ароматическую или алифатическую гетероциклическая группу, которая может быть замещена одним или несколькими одинаковыми или разными заместителями, выбранными из <группы-заместителя L2>, где <группа-заместитель L2> представляет собой атом галогена, гидрокси, амино и гидроксиметил;
каждый из Ra3', Ra4', Ra6', Ra8', Ra9, Ra10 и Ra11 независимо представляет собой атом водорода или низший алкил, который может быть замещен одним или несколькими одинаковыми или разными заместителями, выбранными из <группы-заместителя L1>; или
R1 представляет собой низший алкил, который может быть замещен одним или несколькими одинаковыми или разными заместителями, выбранными из <группы-заместителя M>, где <группа-заместитель M> представляет собой атом галогена, гидрокси, нитро, циано, амино, карбамоил, аминосульфонил, имино, низшую алкиламиногруппу, ди(низший)алкиламиногруппу, низший алкилсульфонил, низшую алкилсульфониламиногруппу, низший алкокси, низший алкоксикарбонил, низшую алкоксикарбониламиногруппу, низший алканоил, низший алканоилокси, низший алкилтио и карбоксил; или
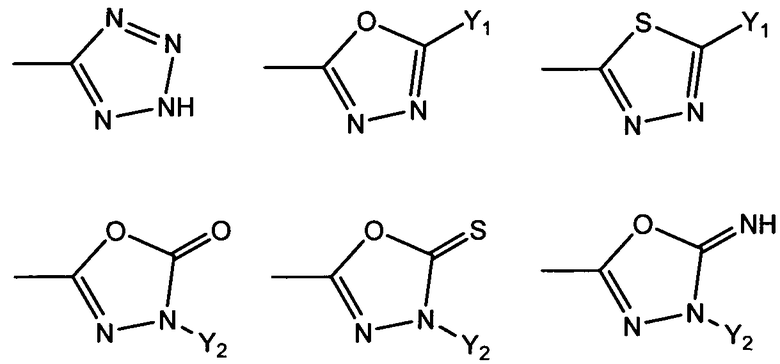
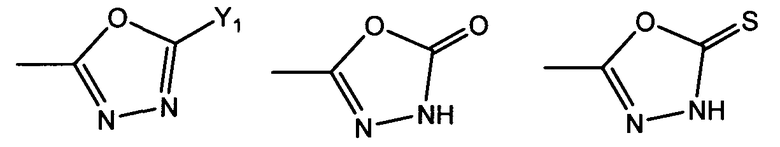
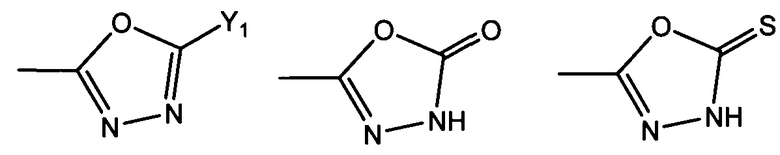
R1 представляет собой гетероциклическую группу, выбранную из следующих групп, где Y1 и Y2 являются одинаковыми или разными, и каждый из них представляет собой атом водорода или низший алкил, который может быть замещен:
R2 представляет собой O, S, SO, SO2, NH, NRb или CRc1Rc2, где Rb представляет собой низший алкил, который может быть замещен, и Rc1 и Rc2, которые могут быть одинаковыми или разными, представляют собой атом водорода или низший алкил;
R3 представляет собой фенил, который может быть замещен;
X2 представляет собой CH, CX2a или N, где:
X2a представляет собой низший алкил; или
X2a представляет собой заместитель, выбранный из <группы-заместителя A1>, или низший алкил, который замещен одним или несколькими одинаковыми или разными заместителями, выбранными из <группы-заместителя A1>, где <группа-заместитель A1> представляет собой атом галогена; циано; гидрокси; низшую алкиламиногруппу; ди(низший)алкиламиногруппу; низший алкокси, который может быть замещен одной или несколькими гидроксигруппами; низший алкилтио; и низший алкилсульфонил; или
X2a представляет собой COORx1, CONRx2Rx3, NHCORx1, NHCONRx2Rx3, NHSO2NRx2Rx3, NRx4Rx5 или CH2NRx4Rx5, где:
Rx1 представляет собой атом водорода или низший алкил, который может быть замещен;
каждый из Rx2 и Rx3, которые могут быть одинаковыми или разными, представляет собой атом водорода, низший алкил, который может быть замещен, или циклоалкил, который может быть замещен; или, альтернативно, Rx2 и Rx3 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную алифатическую гетероциклическую группу, которая содержит, по меньшей мере, один атом, выбранный из N, O и S, и которая может быть замещена; и
Rx4 и Rx5, которые могут быть одинаковыми или разными, представляют собой атом водорода, низший алкил, который может быть замещен, или циклоалкил, который может быть замещен; или
X2a представляет собой 5- или 6-членную алифатическую гетероциклическую группу, которая содержит, по меньшей мере, один атом, выбранный из N, O и S, и которая может быть замещена, в которой два атома водорода, которые присоединены к одному и тому же атому углерода алифатической гетероциклической группы, могут быть замещены оксогруппой, а соседние два атома углерода, составляющие алифатическое гетероциклическое кольцо, могут образовывать двойную связь; или низший алкил, который замещен алифатической гетероциклической группой; или
X2a представляет собой 5- или 6-членную ароматическую гетероциклическую группу, которая содержит, по меньшей мере, один атом, выбранный из N, O и S, и которая может быть замещена; или низший алкил, который замещен ароматической гетероциклической группой;
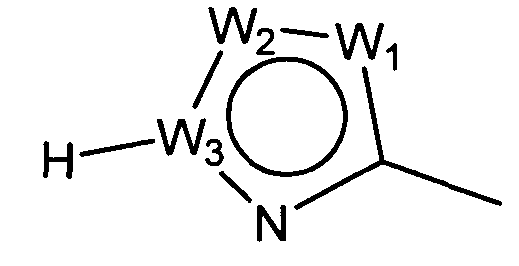
W представляет собой следующий остаток:
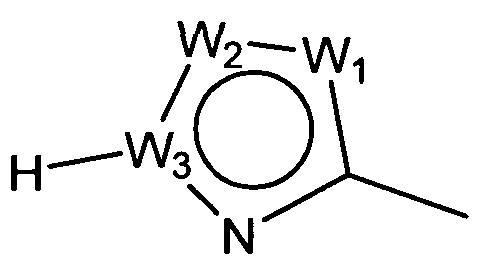
где:
W1 представляет собой CH, N, NH, O или S;
W2 представляет собой CH, CW2a, N, NW2b, O или S, где каждый W2a и W2b независимо представляет собой атом водорода, атом галогена, циано, низший алкил, содержащий от одного до двух атомов углерода, циклоалкил, содержащий от трех до пяти атомов углерода, или низший алкил, содержащий от одного до двух атомов углерода, который может быть замещен одним или несколькими атомами галогена;
W3 представляет собой C или N; и,
по меньшей мере, один из W1, W2 и W3 представляет собой атом углерода; однако два из W1, W2 и W3 одновременно не могут представлять собой O и S,
или к его фармацевтически приемлемой соли или сложному эфиру.
Изобретение также относится к комбинированному препарату для одновременного, отдельного или последовательного введения при лечении рака, содержащему два отдельных препарата, которые представляют собой:
* препарат, содержащий вместе с фармацевтически приемлемым носителем или разбавителем соединение, представленное описанной выше формулой (I), или его фармацевтически приемлемую соль или сложный эфир; и
* препарат, содержащий вместе с фармацевтически приемлемым носителем или разбавителем одно противоопухолевое средство, выбранное из группы, состоящей из противоопухолевых алкилирующих средств, противоопухолевых антиметаболитов, противоопухолевых антибиотиков, противоопухолевых средств растительного происхождения, противоопухолевых координационных соединений платины, противоопухолевых производных камптотецина, противоопухолевых ингибиторов тирозинкиназы, моноклональных антител, интерферонов, модификаторов биологического ответа и других противоопухолевых средств, а также их фармацевтически приемлемой соли(ей) или сложного эфира(ов), где
противоопухолевое алкилирующее средство представляет собой N-оксид азотистого иприта, циклофосфамид, ифосфамид, мелфалан, бусульфан, митобронитол, карбоквон, тиотепа, ранимустин, нимустин, темозоломид или кармустин;
противоопухолевый антиметаболит представляет собой метотрексат, 6-меркаптопуринрибозид, меркаптопурин, 5-фторурацил, тегафур, доксифлуридин, кармофур, цитарабин, окфосфат цитарабина, эноцитабин, S-1, гемцитабин, флударабин или динатрия пеметрексед;
противоопухолевый антибиотик представляет собой актиномицин D, доксорубицин, даунорубицин, неокарциностатин, блеомицин, пепломицин, митомицин C, акларубицин, пирарубицин, эпирубицин, зиностатин стималамер, идарубицин, сиролимус или валрубицин;
противоопухолевое средство растительного происхождения представляет собой винкристин, винбластин, виндезин, этопозид, собузоксан, доцетаксел, паклитаксел или винорелбин;
противоопухолевое координационное соединение платины представляет собой цисплатин, карбоплатин, недаплатин или оксалиплатин;
противоопухолевое производное камптотецина представляет собой иринотекан, топотекан или камптотецин;
противоопухолевый ингибитор тирозинкиназы представляет собой гефитиниб, иматиниб, сорафениб, сунитиниб, дазатиниб или эрлотиниб;
моноклональное антитело представляет собой цетуксимаб, ритуксимаб, бевацизумаб, алемтузумаб или трастузумаб;
интерферон представляет собой интерферон α, интерферон α-2a, интерферон α-2b, интерферон β, интерферон γ-1a или интерферон γ-n1;
модификатор биологического ответа представляет собой крестин, лентинан, сизофиран, пицибанил или убенимекс; и
другое противоопухолевое средство представляет собой митоксантрон, L-аспарагиназу, прокарбазин, дакарбазин, гидроксикарбамид, пентостатин, третиноин, алефацепт, дарбепоэтин-альфа, анастрозол, экземестан, бикалутамид, леупролелин, флутамид, фулвестрант, октанатрия пегаптаниб, денилейкин дифтитокс, алдеслейкин, тиротропин альфа, триоксид мышьяка, бортезомиб, капецитабин или гозерелин.
Изобретение дополнительно относится к фармацевтической композиции, содержащей вместе с фармацевтически приемлемым носителем или разбавителем соединение, представленное описанной выше формулой (I), или его фармацевтически приемлемую соль или сложный эфир, и противоопухолевое средство, выбранное из группы, состоящей из противоопухолевых алкилирующих средств, противоопухолевых антиметаболитов, противоопухолевых антибиотиков, противоопухолевых средств растительного происхождения, противоопухолевых координационных соединений платины, противоопухолевых производных камптотецина, противоопухолевых ингибиторов тирозинкиназы, моноклональных антител, модификаторов биологического ответа и других противоопухолевых средств (здесь определение каждого противоопухолевого средства является таким же, как в приведенном выше определении) или их фармацевтически приемлемой соли или сложного эфира.
Кроме того, изобретение дополнительно относится к способу лечения рака, включающему одновременное, отдельное или последовательное введение терапевтически эффективного количества соединения, представленного описанной выше формулой (I), или его фармацевтически приемлемой соли или сложного эфира в комбинации с терапевтически эффективным количеством противоопухолевого средства, выбранного из группы, состоящей из противоопухолевых алкилирующих средств, противоопухолевых антиметаболитов, противоопухолевых антибиотиков, противоопухолевых средств растительного происхождения, противоопухолевых координационных соединений платины, противоопухолевых производных камптотецина, противоопухолевых ингибиторов тирозинкиназы, моноклональных антител, интерферонов, модификаторов биологического ответа и других противоопухолевых средств (здесь определение каждого противоопухолевого средства является таким же, как в приведенном выше определении) или их фармацевтически приемлемой соли или сложного эфира.
Кроме того, изобретение относится к применению селективного ингибитора Авроры A, для производства лекарственного средства, предназначенного для лечения рака; и к применению селективного ингибитора Авроры A, в комбинации с противоопухолевым средством для производства лекарственного средства, предназначенного для лечения рака; и также относится к способу лечения рака у млекопитающего (в частности, у человека), который включает введение упомянутому млекопитающему терапевтически эффективного количества селективного ингибитора Авроры A; и к способу лечения рака у млекопитающего (в частности, человека), который включает введение упомянутому млекопитающему терапевтически эффективного количества селективного ингибитора Авроры A в комбинации с терапевтически эффективным количеством противоопухолевого средства.
Изобретение относится к фармацевтической композиции, содержащей в качестве активного ингредиента селективный ингибитор Авроры A; и к фармацевтической композиции, содержащей в качестве активного ингредиента селективный ингибитор Авроры A, вместе с противоопухолевым средством.
Далее будут объяснены символы и термины, применяемые в настоящем описании изобретения.
Термин "низший алкил" в описанной выше формуле (I) обозначает алкильную группу с линейной или разветвленной цепью, содержащую от 1 до 6 атомов углерода, и примеры такой группы включают, например, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил и гексил, среди которых предпочтительным является метил.
Термин "циклоалкил" в описанной выше формуле (I) обозначает 3-8-членную алифатическую циклическую группу, такую как, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Термин "гетероциклическая группа" в формуле (I) относится к "ароматической гетероциклической группе" или "алифатической гетероциклической группе". В данном описании термин "ароматическая гетероциклическая группа" относится к ароматической гетероциклической группе, содержащей в дополнение к атому(ам) углерода, по меньшей мере, один гетероатом, такой как атом азота, атом кислорода или т.п., и примеры такой группы включают 5-7-членную моноциклическую гетероциклическую группу, гетероциклическую группу с конденсированным циклом, образованную путем конденсации 3-8-членного цикла с моноциклической гетероциклической группой и т.п. В частности, можно упомянуть тиенильную группу, пирролильную группу, тиазолильную группу, имидазолильную группу, пиразолильную группу, оксазолильную группу, пиридильную группу, пиразинильную группу, пиримидинильную группу, пиридазинильную группу, изоксазолильную группу, изохинолильную группу, изоиндолильную группу, индазолильную группу, индолильную группу, хиноксалинильную группу, хинолильную группу, бензоимидазолильную группу, бензофуранильную группу и т.п. С другой стороны, термин "алифатическая гетероциклическая группа" относится к насыщенной или ненасыщенной алифатической гетероциклической группе, содержащей в добавление к атому(ам) углерода, по меньшей мере, один атом, выбранный из атома азота, атома кислорода и атома серы, и содержащей моноциклический цикл или бициклический или трициклический конденсированный цикл. Примеры такой группы включают азетидильную группу, пирролидинильную группу, пиперидинильную группу, пиперазинильную группу, морфолиногруппу, тетрагидрофуранильную группу, имидазолидинильную группу, тиоморфолиногруппу, тетрагидрохинолильную группу, тетрагидроизохинолильную группу и т.п.
Термин "5- или 6-членная алифатическая гетероциклическая группа" в описанной выше формуле (I) обозначает 5- или 6-членную алифатическую циклическую группу, содержащую, по меньшей мере, один атом, выбранный из атома азота, атома кислорода и атома серы в добавление к атомам углерода, и примеры такой группы включают пирролидинил, пиперидинил, пиперазинил, морфолино, тетрагидрофуранил, имидазолидинил и тиоморфолино. Кроме того, в алифатической гетероциклической группе два атома водорода, которые присоединены к одному и тому же атому углерода, могут быть замещены оксогруппой, а также соседние атомы углерода, составляющие цикл алифатической гетероциклической группы, могут быть соединены двойной связью.
Термин "5- или 6-членная ароматическая гетероциклическая группа" в описанной выше формуле (I) обозначает 5- или 6-членную ароматическую циклическую группу, содержащую, по меньшей мере, один атом, выбранный из атома азота, атома кислорода и атома серы в дополнение к атомам углерода, и примеры такой группы включают тиенил, пирролил, фурил, тиазолил, имидазолил и оксазолил.
Термин "атом галогена" в описанной выше формуле (I) представляет собой, например, атом фтора, атом хлора, атом брома или атом иода. Среди них предпочтительным является, например, атом фтора, атом хлора или атом брома.
Термин "низшая алкиламиногруппа" в описанной выше формуле (I) обозначает группу, в которой аминогруппа является N-замещенной описанным выше "низшим алкилом", и примеры такой группы включают N-метиламино, N-этиламино, N-пропиламино, N-изопропиламино, N-бутиламино, N-изобутиламино, N-трет-бутиламино, N-пентиламино и N-гексиламино.
Термин "ди(низший)алкиламиногруппа" в описанной выше формуле (I) обозначает группу, в которой аминогруппа является N,N-двузамещенной описанным выше "низшим алкилом", и примеры такой группы включают N,N-диметиламино, N,N-диэтиламино, N,N-дипропиламино, N,N-диизопропиламино, N,N-дибутиламино, N,N-диизобутиламино, N,N-ди-трет-бутиламино, N,N-дипентиламино, N,N-дигексиламино, N-этил-N-метиламино и N-метил-N-пропиламино.
Термин "низший алкилсульфонил" в описанной выше формуле (I) обозначает группу, в которой описанный выше "низший алкил" присоединен к сульфонилу, и примеры такой группы включают метилсульфонил, этилсульфонил и бутилсульфонил.
Термин "низшая алкилсульфониламиногруппа" в описанной выше формуле (I) обозначает группу, в которой описанный выше "низший алкилсульфонил" присоединен к аминогруппе, и примеры такой группы включают метилсульфониламино, этилсульфониламино и бутилсульфониламино.
Термин "низший алкокси" в описанной выше формуле (I) обозначает группу, в которой "низший алкил" присоединен к атому кислорода, и примеры такой группы включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентилокси, неопентилокси, гексилокси и изогексилокси.
Термин "низший алкоксикарбонил" в описанной выше формуле (I) обозначает группу, в которой "низший алкокси" присоединен к карбонилу, и примеры такой группы включают метоксикарбонил, этоксикарбонил, пропоксикарбонил, изопропоксикарбонил, бутоксикарбонил, изобутоксикарбонил, втор-бутоксикарбонил, трет-бутоксикарбонил, пентилоксикарбонил, неопентилоксикарбонил, гексилоксикарбонил и изогексилоксикарбонил.
Термин "низшая алкоксикарбониламиногруппа" в описанной выше формуле (I) обозначает группу, в которой "низший алкоксикарбонил" присоединен к аминогруппе, и примеры такой группы включают метоксикарбониламино, этоксикарбониламино, пропоксикарбониламино, изопропоксикарбониламино, бутоксикарбониламино, изобутоксикарбониламино, втор-бутоксикарбониламино, трет-бутоксикарбониламино, пентилоксикарбониламино, неопентилоксикарбониламино, гексилоксикарбониламино и изогексилоксикарбониламино.
Термин "низший алканоил" в описанной выше формуле (I) обозначает группу, в которой описанный выше "низший алкил" присоединен к карбонилу, и примеры такой группы включают ацетил, пропионил, бутирил, изобутирил, валерил, изовалерил, пивалоил и пентаноил.
Термин "низший алканоилокси" в описанной выше формуле (I) обозначает группу, в которой описанный выше "низший алканоил" присоединен к атому кислорода, и примеры такой группы включают ацетилокси, пропионилокси, бутирилокси, изобутирилокси, валерилокси, изовалерилокси, пивалоилокси и пентаноилокси.
Термин "низший алкилтио" в описанной выше формуле (I) обозначает заместитель, где описанный выше "низший алкил" присоединен к атому серы, и примеры такой группы включают метилтио, этилтио и бутилтио.
Термин "селективный ингибитор Авроры A", применяемый в настоящем описании изобретения, представляет собой соединение или лекарственное средство, которое селективно ингибирует Аврору A по сравнению с Авророй B. "Селективный ингибитор Авроры A" предпочтительно представляет собой соединение или лекарственное средство, ингибирующие активности которого в отношении Авроры A, по меньшей мере, в десять раз больше активностей в отношении Авроры B; и более предпочтительно, соединение или лекарственное средство, ингибирующие активности которого в отношении Авроры A, по меньшей мере, в сто раз выше активностей в отношении Авроры B.
Объяснение термина "его фармацевтически приемлемая соль сложного эфира" или термин "фармацевтически приемлемый носитель или разбавитель", применяемые в описании изобретения, будут даны позже.
Термин "лечение рака", применяемый в описании изобретения, означает ингибирование роста раковых клеток путем введения противоопухолевого средства пациенту с раковым заболеванием. Предпочтительно такое лечение делает возможным ретрогрессию роста ракового новообразования, то есть уменьшение поддающегося измерению размера ракового новообразования. Более предпочтительно, такое лечение полностью устраняет раковое новообразование.
Термин "рак", применяемый в описании изобретения, относится к солидному раку и злокачественному заболеванию кроветворной системы. В настоящем описании примеры солидного рака включают опухоль мозга, рак головы и шеи, рак пищевода, рак щитовидной железы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, рак молочной железы, рак желудка, рак желчного пузыря и желчного протока, рак печени, рак поджелудочной железы, рак ободочной кишки, рак прямой кишки, рак яичника, хорионэпителиому, рак матки, рак шейки матки, рак почечной лоханки и мочеточника, рак мочевого пузыря, рак предстательной железы, рак полового члена, рак яичка, эмбриональный рак, опухоль Вильмса, рак кожи, злокачественную меланому, нейробластому, остеосаркому, опухоль Юинга и саркому мягких тканей. С другой стороны, примеры злокачественного заболевания кроветворной системы включают острый лейкоз, хронический лимфолейкоз, хронический миелоцитарный лейкоз, истинную полицитемию, злокачественную лимфому, множественную миелому и неходжкинскую лимфому.
Термин "препарат", применяемый в описании изобретения, включает препараты для перорального введения и препараты для парентерального введения. Примеры препаратов для перорального введения включают таблетки, капсулы, порошки и гранулы, в то время как примеры парентеральных препаратов включают стерилизованные жидкие препараты, такие как растворы или суспензии, в частности, инъекции или вливания с помощью капельницы. Предпочтительно они представляют собой внутривенные инъекции или внутривенные вливания с помощью капельницы и более предпочтительно внутривенные вливания с помощью капельницы.
Термин "комбинированный препарат", применяемый в описании изобретения, относится к препаратам, содержащим два или более препаратов для одновременного, отдельного или последовательного введения при лечении, и такой препарат может представлять собой препарат или фармацевтическую композицию так называемого наборного типа. Термин "комбинированный препарат" также включает препарат, содержащий один или несколько препаратов, дополнительно объединенных с комбинированным препаратом, содержащим два отдельных препарата, применяемых при лечении рака.
Описанные выше два отдельных препарата можно дополнительно объединять в комбинации с фармацевтически приемлемым носителем или разбавителем, по меньшей мере, с одним препаратом, содержащим, по меньшей мере, одно противоопухолевое средство, выбранное из группы, состоящей из противоопухолевых алкилирующих средств, противоопухолевых антиметаболитов, противоопухолевых антибиотиков, противоопухолевых средств растительного происхождения, противоопухолевых координационных соединений платины, противоопухолевых производных камптотецина, противоопухолевых ингибиторов тирозинкиназы, моноклональных антител, интерферонов, модификаторов биологического ответа и других противоопухолевых средств (здесь определение каждого противоопухолевого средства является таким же, как в приведенном выше определении), или их фармацевтически приемлемой соли или сложного эфира. В таком случае упомянутый выше, по меньшей мере, один препарат, который объединен дополнительно, можно вводить одновременно, отдельно или последовательно по отношению к двум отдельным препаратам. Например, комбинированный препарат, содержащий три препарата, может включать тот, который состоит из препарата, включающего препарат, содержащий соединение, представленное описанной выше формулой (I), препарат, содержащий 5-фторурацил, и препарат, содержащий лейковорин.
Здесь в упомянутом выше комбинированном препарате любой из двух или оба вместе отдельных препарата могут представлять собой препарат для перорального введения; а также один может представлять собой препарат для перорального введения, в то время как другой может представлять собой препарат для парентерального введения (инъекции или вливания с помощью капельницы).
Препарат согласно изобретению обычно может содержать терапевтически эффективное количество соединения по изобретению вместе с фармацевтически приемлемым носителем или разбавителем. Считается, что такая методика приготовления лекарственного средства хорошо известна и должна представлять собой технические сведения, общеизвестные для специалистов в данной области. Предпочтительно препараты для перорального введения, внутривенного вливания с помощью капельницы или инъекции можно получать в комбинации с фармацевтически приемлемым носителем или разбавителем различными способами, которые хорошо известны в данной области.
В случае применения комбинированного препарата по изобретению термин "введение", применяемый в настоящем описании изобретения, относится к парентеральному введению и/или пероральному введению и предпочтительно - к пероральному введению. Таким образом, при введении комбинированного препарата оба введения могут быть парентеральными; одно введение может быть парентеральным, в то время как другое может быть пероральным; или оба введения могут быть пероральными. Предпочтительно оба препарата в комбинированном препарате вводятся перорально. Здесь термин "парентеральное введение", например, представляет собой внутривенное введение, подкожное введение или внутримышечное введение, и предпочтительно он представляет собой внутривенное введение. Даже когда объединяются и вводятся три или более препаратов, каждый препарат можно вводить перорально.
В одном из вариантов осуществления настоящего изобретения соединение, представленное описанной выше формулой (I), можно вводить одновременно с другим противоопухолевым средством(ами). Кроме того, сначала можно вводить соединение, представленное описанной выше формулой (I), а затем последовательно другое противоопухолевое средство; или, альтернативно, сначала можно вводить другое противоопухолевое средство, а затем последовательно соединение, представленное описанной выше формулой (I). Также сначала можно вводить соединение, представленное описанной выше формулой (I), а затем через некоторое время отдельно вводить другое противоопухолевое средство; или, альтернативно, сначала можно вводить другое противоопухолевое средство, а затем через некоторое время отдельно вводить соединение, представленное описанной выше формулой (I). Порядок и временной интервал введения соответствующим образом может быть выбран специалистом в данной области в соответствии, например, с применяемым препаратом, содержащим соединение, представленное описанной выше формулой (I), и препаратом, содержащим противоопухолевое средство, которое применяется в комбинации с ним, типом раковых клеток, подвергаемых лечению, и состоянием пациента. Например, в случае введения соединения, представленного описанной выше формулой (I), и паклитаксела или доцетаксела предпочтительно сначала вводить паклитаксел или доцетаксел, а затем через некоторое время последовательно или отдельно вводить соединение, представленное описанной выше формулой (I).
Термин "одновременно", применяемый в описании изобретения, относится к применению препаратов для лечения в значительной степени в одно и то же время, тогда как термин "отдельно" относится к отдельному применению препаратов для лечения в разное время, так что, например, одно средство применяют в первый день, а другое средство применяют на второй день лечения. Термин "последовательно" относится к применению препаратов в таком порядке, что, например, сначала применяют одно средство, а через заданный период времени применяют другое средство для лечения.
Термин "противоопухолевое алкилирующее средство", применяемый в настоящем описании изобретения, относится к алкилирующему средству, обладающему противоопухолевой активностью, и здесь термин "алкилирующее средство" обычно относится к средству, отдающему алкильную группу в реакции алкилирования, при которой атом водорода органического соединения замещается алкильной группой. Термин "противоопухолевое алкилирующее средство" можно проиллюстрировать примерами в виде N-оксида азотистого иприта, циклофосфамида, ифосфамида, мелфалана, бусульфана, митобронитола, карбоквона, тиотепы, ранимустина, нимустина, темозоломида или кармустина.
Термин "противоопухолевый антиметаболит", применяемый в описании изобретения, относится к антиметаболиту, обладающему противоопухолевой активностью, и здесь термин "антиметаболит" включает, в широком смысле, вещества, которые нарушают нормальный обмен веществ, и вещества, которые ингибируют систему переноса электронов для предотвращения образования богатых энергией промежуточных продуктов, благодаря их структурной или функциональной схожести с метаболитами, которые важны для живых организмов (такими как витамины, коферменты, аминокислоты и сахариды). Термин "противоопухолевые антиметаболиты" можно проиллюстрировать примерами в виде метотрексата, 6-меркаптопуринрибозида, меркаптопурина, 5-фторурацила, тегафура, доксифлуридина, кармофура, цитарабина, окфосфата цитарабина, эноцитабина, S-1, гемцитабина, флударабина или динатрия пеметрекседа, а предпочтительными являются 5-фторурацил, S-1, гемцитабин и т.п.
Термин "противоопухолевый антибиотик", применяемый в описании изобретения, относится к антибиотику, обладающему противоопухолевой активностью, и здесь "антибиотик" включает вещества, которые продуцируются микроорганизмами или получаются путем органического синтеза и ингибируют рост клеток и другие функции микроорганизмов и других живых организмов. Термин "противоопухолевый антибиотик" можно проиллюстрировать примерами в виде актиномицина D, доксорубицина, даунорубицина, неокарциностатина, блеомицина, пепломицина, митомицина C, акларубицина, пирарубицина, эпирубицина, зиностатин стималамера, идарубицина, сиролимуса или валрубицина.
Термин "противоопухолевое средство растительного происхождения", применяемый в описании изобретения, включает соединения, обладающие противоопухолевыми активностями, которые происходят из растений, или соединения, получаемые путем применения в отношении вышеупомянутых соединений химической модификации. Термин "противоопухолевое средство растительного происхождения" можно проиллюстрировать примерами в виде винкристина, винбластина, виндезина, этопозида, собузоксана, доцетаксела, паклитаксела и винорелбина, и предпочтительными являются доцетаксел и паклитаксел.
Термин "противоопухолевое производное камптотецина", применяемый в описании изобретения, относится к соединениям, которые являются структурно родственными камптотецину и ингибируют рост раковых клеток, включая камптотецин сам по себе. Термин "противоопухолевое производное камптотецина" специально не ограничивается, хотя его можно проиллюстрировать примерами в виде камптотецина, 10-гидроксикамптотецина, топотекана, иринотекана или 9-аминокамптотецина, с тем, что предпочтительными являются камптотецин, топотекан и иринотекан. Кроме того, иринотекан подвергается превращению в процессе обмена веществ in vivo и обладает противоопухолевым эффектом как SN-38. Считается, что механизм действия и активность производных камптотецина практически должны быть такими же, как у камптотецина (см., например, публикацию Nitta и др., Gan to Kagaku Ryoho, 14, 850-857 (1987)).
Термин "противоопухолевое координационное соединение платины (платиновый комплекс)", применяемый в описании изобретения, относится к координационному соединению платины, обладающему противоопухолевой активностью, и термин "координационное соединение платины" относится здесь к координационному соединению платины, которое обеспечивает платину в ионной форме. Предпочтительные соединения платины включают цисплатин; цис-диамминдиаквоплатина(II)-ион; хлорид хлор(диэтилентриамин)платины (II); дихлор(этилендиамин)платину (II); диамин-(1,1-циклобутандикарбоксилато)платину (II) (карбоплатин); спироплатин; ипроплатин; диамин-(2-этилмалонато)платину (II); этилендиаминмалонатоплатину (II); аква-(1,2-диаминодициклогексан)сульфатоплатину (II); аква-(1,2-диаминодициклогексан)малонатоплатину (II); (1,2-диаминоциклогексан)малонатоплатину (II); (4-карбоксифталато)(1,2-диаминоциклогексан)платину (II); (1,2-диаминоциклогексан)-(изоцитрато)платину(II); (1,2-диаминоциклогексан)оксалатоплатину (II); ормаплатин; тетраплатин; карбоплатин, недаплатин и оксалиплатин, и предпочтительными являются карбоплатин или оксалиплатин. Кроме того, известны и имеются в продаже и/или могут быть получены специалистом в данной области с помощью традиционных методик другие противоопухолевые координационные соединения платины, упомянутые в описании изобретения.
Термин "противоопухолевый ингибитор тирозинкиназы", применяемый в описании изобретения, относится к ингибитору тирозинкиназы, обладающему противоопухолевой активностью, и термин "ингибитор тирозинкиназы" относится здесь к химическому веществу, ингибирующему "тирозинкиназу", которое переносит γ-фосфатную группу АТФ к гидроксигруппе специфического тирозина в белке. Термин "противоопухолевый ингибитор тирозинкиназы" можно проиллюстрировать примерами в виде гефитиниба, иматиниба, сорафениба, сунитиниба, дазатиниба или эрлотиниба.
Термин "моноклональное антитело", применяемый в описании изобретения, который также известен как одноклональное антитело, относится к антителу, продуцируемому клеткой, продуцирующей моноклональное антитело, и их примеры включают цетуксимаб, бевацизумаб, ритуксимаб, алемтузумаб и трастузумаб.
Термин "интерферон", применяемый в описании изобретения, относится к интерферону, обладающему противоопухолевой активностью, и представляющему собой гликопротеин с молекулярной массой приблизительно 20000, который продуцируется и выделяется большинством животных клеток при вирусной инфекции. Он обладает не только эффектом ингибирования роста вируса, но также различными иммуноэффекторными механизмами, включая ингибирование роста клеток (в частности, опухолевых клеток) и повышение активности природных клеток-киллеров, поэтому определяется как один из типов цитокинов. Примеры "интерферона" включают интерферон α, интерферон α-2a, интерферон α-2b, интерферон β, интерферон γ-1a и интерферон γ-n1.
Термин "модификатор биологического ответа", применяемый в описании изобретения, обозначает так называемый модификатор биологического ответа или BRM и обычно представляет собой общее обозначение веществ или лекарственных средств, предназначенных для модификации защитных механизмов живых организмов или биологических ответов, таких как выживание, рост или дифференцировка клеток тканей, для того, чтобы направить их с пользой для индивида против опухоли, инфекции или других заболеваний. Примеры "модификатора биологического ответа" включают крестин, лентинан, сизофиран, пицибанил и убенимекс.
Термин "другое противоопухолевое средство", применяемый в описании изобретения, относится к противоопухолевому средству, которое не принадлежит ни к какому из описанных выше средств, обладающему противоопухолевыми активностями. Примеры "другого противоопухолевого средства" включают митоксантрон, L-аспарагиназу, прокарбазин, дакарбазин, гидроксикарбамид, пентостатин, третиноин, алефацепт, дарбепоэтин альфа, анастрозол, экземестан, бикалутамид, лейпрорелин, флутамид, фулвестрант, октанатрия пегаптаниб, денилейкин дифтитокс, алдеслейкин, тиротропин альфа, триоксид мышьяка, бортезомиб, капецитабин и гозерелин.
Все описанные выше термины "противоопухолевое алкилирующее средство", "противоопухолевый антиметаболит", "противоопухолевый антибиотик", "противоопухолевое средство растительного происхождения", "противоопухолевое координационное соединение платины", "противоопухолевое производное камптотецина", "противоопухолевый ингибитор тирозинкиназы", "моноклональное антитело", "интерферон", "модификатор биологического ответа" и "другое противоопухолевое средство" известны и либо имеются в продаже, либо могут быть получены специалистом в данной области с помощью способов, которые известны по сути или хорошо известны, или с помощью традиционных способов. Способ получения гефитиниба описан, например, в патенте США № 5770599; способ получения цетуксимаба описан, например, в заявке WO 96/40210; способ получения бевацизумаба описан, например, в заявке WO 94/10202; способ получения оксалиплатина описан, например, в патентах США №№ 5420319 и 5959133; способ получения гемцитабина описан, например, в патентах США №№ 5434254 и 5223608; и способ получения камптотецина описан в патентах США №№ 5162532, 5247089, 5191082, 5200524, 5243050 и 5321140; способ получения иринотекана описан, например, в патенте США № 4604463; способ получения топотекана описан, например, в патенте США № 5734056; способ получения темозоломида описан, например, в патенте JP-B № 4-5029; и способ получения ритуксимаба описан, например, в патенте JP-W № 2-503143.
Упомянутые выше противоопухолевые алкилирующие средства имеются в продаже, что проиллюстрировано следующими примерами: N-оксид азотистого иприта от компании Mitsubishi Pharma Corp. в виде препарата Нитромин (торговая марка Nitromin); циклофосфамид от компании Shionogi & Co., Ltd. в виде препарата Эндоксан (торговая марка Endoxan); ифосфамид от компании Shionogi & Co., Ltd. в виде препарата Ифомид (торговая марка Ifomide); мелфалан от компании GlaxoSmithKline Corp. в виде препарата Алкеран (торговая марка Alkeran); бусульфан от компании Takeda Pharmaceutical Co., Ltd. в виде препарата Маблин (торговая марка Mablin); митобронитол от компании Kyorin Pharmaceutical Co., Ltd. в виде препарата Миеброл (торговая марка Myebrol); карбоквон от компании Sankyo Co., Ltd. в виде препарата Эсквинон (торговая марка Esquinon); тиотепа от компании Sumitomo Pharmaceutical Co., Ltd. в виде препарата Теспамин (торговая марка Tespamin); ранимустин от компании Mitsubishi Pharma Corp. в виде препарата Цимерин (торговая марка Cymerin); нимустин от компании Sankyo Co., Ltd. в виде препарата Нидран (торговая марка Nidran); темозоломид от компании Schering Corp. в виде препарата Темодар (торговая марка Temodar); и кармустин от компании Guilford Pharmaceuticals Inc. в виде препарата Глиадел Вафер (торговая марка Gliadel Wafer).
Упомянутые выше противоопухолевые антиметаболиты имеются в продаже, что проиллюстрировано в виде следующих примеров: метотрексат от компании Takeda Pharmaceutical Co., Ltd. в виде препарата Метотрексат (торговая марка Methotrexate); 6-меркаптопурин рибозид от компании Aventis Corp. в виде препарата Тиоинозин (торговая марка Thioinosine); меркаптопурин от компании Takeda Pharmaceutical Co., Ltd. в виде препарата Лейкерин (торговая марка Leukerin); 5-фторурацил от компании Kyowa Hakko Kogyo Co., Ltd. в виде препарата 5-FU (торговая марка); тегафур от компании Taiho Pharmaceutical Co., Ltd. в виде препарата Футрафул (торговая марка Futraful); доксифлуридин от компании Nippon Roche Co., Ltd. в виде препарата Фурутулон (торговая марка Furutulon); кармофур от компании Yamanouchi Pharmaceutical Co., Ltd. в виде препарата Ямафур (торговая марка Yamafur); цитарабин от компании Nippon Shinyaku Co., Ltd. в виде препарата Цилокид (торговая марка Cylocide); окфосфат цитарабина от компании Nippon Kayaku Co., Ltd. в виде препарата Страсид (торговая марка Strasid); эноцитабин от компании Asahi Kasei Corp. в виде препарата Санрабин (торговая марка Sanrabin); S-1 от компании Taiho Pharmaceutical Co., Ltd. в виде препарата TS-1 (торговая марка); гемцитабин от компании Eli Lilly & Co. в виде препарата Гемзар (торговая марка Gemzar); флударабин от компании Nippon Schering Co., Ltd. в виде препарата Флудара (торговая марка Fludara); и динатрия пеметрексед от компании Eli Lilly & Co. в виде препарата Алимта (торговая марка Alimta).
Упомянутые выше противоопухолевые антибиотики имеются в продаже, что проиллюстрировано следующими примерами: актиномицин D от компании Banyu Pharmaceutical Co., Ltd. в виде препарата Космеген (торговая марка Cosmegen); доксорубицин от компании Kyowa Hakko Kogyo Co., Ltd. в виде препарата Адриацин (торговая марка Adriacin); даунорубицин от компании Meiji Seika Kaisha Ltd. в виде препарата Дауномицин (Daunomycin); неокарциностатин от компании Yamanouchi Pharmaceutical Co., Ltd. в виде препарата Неокарциностатин (торговая марка Neocarzinostatin); блеомицин от компании Nippon Kayaku Co., Ltd. в виде препарата Блео (торговая марка Bleo); пепромицин от компании Nippon Kayaku Co, Ltd. в виде препарата Пепро (торговая марка Pepro); митомицин C от компании Kyowa Hakko Kogyo Co., Ltd. в виде препарата Митомицин (торговая марка Mitomycin); акларубицин от компании Yamanouchi Pharmaceutical Co., Ltd. в виде препарата Аклацинон (торговая марка Aclacinon); пирарубицин от компании Nippon Kayaku Co., Ltd. в виде препарата Пинорубицин (торговая марка Pinorubicin); эпирубицин от компании Pharmacia Corp. в виде препарата Фарморубицин (торговая марка Pharmorubicin); зиностатин стималамер от компании Yamanouchi Pharmaceutical Co., Ltd. в виде препарата Сманкс (торговая марка Smancs); идарубицин от компании Pharmacia Corp. в виде препарата Идамицин (торговая марка Idamycin); сиролимус от компании Wyeth Corp. в виде препарата Рапамун (торговая марка Rapamune); и валрубицин от компании Anthra Pharmaceuticals Inc. в виде препарата Валстар (торговая марка Valstar).
Упомянутые выше противоопухолевые средства растительного происхождения имеются в продаже, что проиллюстрировано следующими примерами: винкристин от компании Shionogi & Co., Ltd. в виде препарата Онковин (торговая марка Oncovin); винбластин от компании Kyorin Pharmaceutical Co., Ltd. в виде препарата Винбластин (торговая марка Vinblastine); виндезин от компании Shionogi & Co., Ltd. в виде препарата Филдезин (торговая марка Fildesin); этопозид от компании Nippon Kayaku Co., Ltd. в виде препарата Ластет (торговая марка Lastet); собузоксан от компании Zenyaku Kogyo Co., Ltd. в виде препарата Перазолин (торговая марка Perazolin); доцетаксел от компании Aventis Corp. в виде препарата Таксзотер (торговая марка Taxsotere); паклитаксел от компании Bristol-Myers Squibb Co. в виде препарата Таксол (торговая марка Taxol); и винорелбин от компании Kyowa Hakko Kogyo Co., Ltd. в виде препарата Навелбин (торговая марка Navelbine).
Упомянутые выше противоопухолевые координационные соединения платины имеются в продаже, что проиллюстрировано следующими примерами: цисплатин от компании Nippon Kayaku Co., Ltd. в виде препарата Ранда (торговая марка Randa); карбоплатин от компании Bristol-Myers Squibb Co. в виде препарата Параплатин (торговая марка Paraplatin); недаплатин от компании Shionogi & Co., Ltd. в виде препарата Аквапла (торговая марка Aqupla); и оксалиплатин от компании Sanofi-Synthelabo Co. в виде препарата Элоксатин (торговая марка Eloxatin).
Упомянутые выше противоопухолевые производные камптотецина имеются в продаже, что проиллюстрировано следующими примерами: иринотекан от компании Yakult Honsha Co., Ltd. в виде препарата Кампто (торговая марка Campto); топотекан от компании GlaxoSmithKline Corp. в виде препарата Гикамтин (торговая марка Hycamtin); и камптотецина от компании Aldrich Chemical Co., Inc., U.S.A.
Упомянутые выше противоопухолевые ингибиторы тирозинкиназы имеются в продаже, что проиллюстрировано следующими примерами: гефитиниб от компании AstraZeneca Corp. в виде препарата Иресса (торговая марка Iressa); иматиниб от компании Novartis AG в виде препарата Гливек (торговая марка Gleevec); сорафениб от компании Bayer в виде препарата Нексавар (торговая марка Nexavar); сунитиниб от компании Pfizer в виде препарата Сутент (торговая марка Sutent); дазатиниб от компании Bristol Myers Squibb в виде препарата Спруцел (торговая марка Sprycel); и эрлотиниб от компании OSI Pharmaceuticals Inc. в виде препарата Тарцева (торговая марка Tarceva).
Упомянутые выше моноклональные антитела имеются в продаже, что проиллюстрировано следующими примерами: цетуксимаб от компании Bristol-Myers Squibb Co. в виде препарата Эрбитукс (торговая марка Erbitux); бевацизумаб от компании Genentech, Inc. в виде препарата Авастин (торговая марка Avastin); ритуксимаб от компании Biogen Idee Inc. в виде препарата Ритуксан (торговая марка Rituxan); алемтузумаб от компании Berlex Inc. в виде препарата Кампат (торговая марка Campath); и трастузумаб от компании Chugai Pharmaceutical Co., Ltd. в виде препарата Герцептин (торговая марка Herceptin).
Упомянутые выше интерфероны имеются в продаже, что проиллюстрировано следующими примерами: интерферон-α от компании Sumitomo Pharmaceutical Co., Ltd. в виде препарата Сумиферон (торговая марка Sumiferon); интерферон α-2a от компании Takeda Pharmaceutical Co., Ltd. в виде препарата Канферон-А (торговая марка Canferon-A); интерферон α-2b от компании Schering-Plough Corp. в виде препарата Интрон А (торговая марка Intron A); интерферон β от компании Mochida Pharmaceutical Co., Ltd. в виде препарата IFNβ (торговая марка); интерферон γ-1a от компании Shionogi & Co., Ltd. в виде препарата Имуномакс-γ (торговая марка Imunomax-γ); и интерферон γ-n1 от компании Otsuka Pharmaceutical Co., Ltd. в виде препарата Огамма (торговая марка Ogamma).
Упомянутые выше модификаторы биологического ответа имеются в продаже, что проиллюстрировано следующими примерами: крестин от компании Sankyo Co., Ltd. в виде препарата Крестин (торговая марка Krestin); лентинан от компании Aventis Corp. в виде препарата Лентинан (торговая марка Lentinan); сизофиран от компании Kaken Seiyaku Co., Ltd. в виде препарата Сонифиран (торговая марка Sonifiran); пицибанил от компании Chugai Pharmaceutical Co., Ltd. в виде препарата Пицибанил (торговая марка Picibanil); и убенимекс от компании Nippon Kayaku Co., Ltd. в виде препарата Бестатин (торговая марка Bestatin).
Упомянутые выше другие противоопухолевые средства имеются в продаже, что проиллюстрировано следующими примерами: митоксантрон от компании Wyeth Lederle Japan, Ltd. в виде препарата Новантрон (торговая марка Novantrone); L-аспарагиназа от компании Kyowa Hakko Kogyo Co., Ltd. в виде препарата Леуназ (торговая марка Leunase); прокарбазин от компании Nippon Roche Co., Ltd. в виде препарата Натулан (торговая марка Natulan); дакарбазин от компании Kyowa Hakko Kogyo Co., Ltd. в виде препарата Дакарбазин (торговая марка Dacarbazine); гидроксикарбамид от компании Bristol-Myers Squibb Co. в виде препарата Гидреа (торговая марка Hydrea); пентостатин от компании Kagaku Oyobi Kessei Ryoho Kenkyusho в виде препарата Кофорин (торговая марка Coforin); третиноин от компании Nippon Roche Co., Ltd. в виде препарата Весаноид (торговая марка Vesanoid); алефацепт от компании Biogen Idee Inc. в виде препарата Амевив (торговая марка Amevive); дарбепоэтин-альфа от компании Amgen Inc. в виде препарата Аранесп (торговая марка Aranesp); анастрозол от компании AstraZeneca Corp. в виде препарата Аримидекс (торговая марка Arimidex); экземестан от компании Pfizer Inc. в виде препарата Аромазин (торговая марка Aromasin); бикалутамид от компании AstraZeneca Corp. в виде препарата Казодекс (торговая марка Casodex); лейпрорелин от компании Takeda Pharmaceutical Co., Ltd. в виде препарата Лейплин (торговая марка Leuplin); флутамид от компании Schering-Plough Corp. в виде препарата Эйлексин (торговая марка Eulexin); фулвестрант от компании AstraZeneca Corp. в виде препарата Фаслодекс (торговая марка Faslodex); октанатрия пегаптаниб от компании Gilead Sciences, Inc. в виде препарата Макуген (торговая марка Macugen); денилейкин дифтитокс от компании Ligand Pharmaceuticals Inc. в виде препарата Онтак (торговая марка Ontak); алдеслейкин от компании Chiron Corp. в виде препарата Пролейкин (торговая марка Proleukin); тиротропин-альфа от компании Genzyme Corp. в виде препарата Тироген (торговая марка Thyrogen); триоксид мышьяка от компании Cell Therapeutics, Inc. в виде препарата Триценокс (торговая марка Trisenox); бортезомиб от компании Millennium Pharmaceuticals, Inc. в виде препарата Велкад (торговая марка Velcade); капецитабин от компании Hoffmann-La Roche, Ltd. в виде препарата Кселода (торговая марка Xeloda); и гозерелин от компании AstraZeneca Corp. в виде препарата Золадекс (торговая марка Zoladex).
Термин "противоопухолевое средство", применяемый в описании изобретения, включает описанное выше "противоопухолевое алкилирующее средство", "противоопухолевый антиметаболит", "противоопухолевый антибиотик", "противоопухолевое средство растительного происхождения", "противоопухолевое координационное соединение платины", "противоопухолевое производное камптотецина", "противоопухолевый ингибитор тирозинкиназы", "моноклональное антитело", "интерферон", "модификатор биологического ответа " и "другое противоопухолевое средство".
Термин "аминопиридиновое производное", применяемый в описании изобретения, включает, но не ограничивается перечисленным, любое соединение, содержащее пиридильную группу или группу, аналогичную пиридиновой группе, любая из которых замещена аминогруппой. Сказанное иллюстрируется соединением описанной выше общей формулы (I) и предпочтительно любым из упомянутых ниже соединений от (a) до (e): соединение, которое представляет собой:
(a) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту (пример 1 и 2);
(b) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту (пример 4);
(c) транс-4-(2,3-дихлорфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту (пример 5);
(d) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту (пример 7);
(e) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексанкарбоксамид (пример 11);
(f) 5-(транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он (пример 13);
(g) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он (пример 14);
(h) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он (пример 17); или
(i) 5-(транс-4-((2,3-дихлорфенил)сульфонил)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он (пример 24),
или его фармацевтически приемлемую соль или сложный эфир.
Варианты осуществления соединения, представленного описанной выше общей формулой (I), будут проиллюстрированы более подробно.
R1 представляет собой атом водорода, F, CN, COORa1, CONRa2Ra2', NRa3CORa3', CONRa4ORa4', NRa5CONRa5'Ra5", NRa6COORa6', SO2NRa7Ra7', NRa8SO2Ra8', CORa9, SO2Ra10, NO2, ORa11 или NRa12Ra12',
где:
каждый из Ra1, Ra3, Ra4, Ra5, Ra6 и Ra8 независимо представляет собой атом водорода или низший алкил;
каждый из Ra2, Ra2', Ra5', Ra5", Ra7, Ra7', Ra12 и Ra12' независимо представляет собой атом водорода или низший алкил, который может быть замещен одним или несколькими, одинаковыми или разными заместителями, выбранными из <группы-заместителя L1>, в которой <группа-заместитель L1> представляет собой атом галогена, гидрокси, нитро, циано, амино, карбамоил, аминосульфонил, имино, низшую алкиламиногруппу, ди(низший)алкиламиногруппу, низший алкилсульфонил, низшую алкилсульфониламиногруппу, низший алкокси, низший алкоксикарбонил, низшую алкоксикарбониламиногруппу, низший алканоил, низший алканоилокси, низший алкилтио и карбоксил; однако при условии, что каждые из Ra2 и Ra2'; Ra5' и Ra5"; Ra7 и Ra7'; Ra12 и Ra12' независимо, вместе с атомом азота, к которому они присоединены, могут образовывать 5-членную или 6-членную ароматическую или алифатическую гетероциклическую группу, которая может быть замещена одним или несколькими, одинаковыми или разными заместителями, выбранными из <группы-заместителя L2>, в которой <группа-заместитель L2> представляет собой атом галогена, гидрокси, амино, и гидроксиметил;
каждый из Ra3', Ra4', Ra6', Ra8', Ra9, Ra10 и Ra11 независимо представляет собой атом водорода или низший алкил, который может быть замещен одним или несколькими, одинаковыми или разными заместителями, выбранными из <группы-заместителя L1>; или
R1 представляет собой низший алкил, который может быть замещен одним или несколькими, одинаковыми или разными заместителями, выбранными из <группы-заместителя M>, в которой <группа-заместитель M> представляет собой атом галогена, гидрокси, нитро, циано, амино, карбамоил, аминосульфонил, имино, низшую алкиламиногруппу, ди(низший)алкиламиногруппу, низший алкилсульфонил, низшую алкилсульфониламиногруппу, низший алкокси, низший алкоксикарбонил, низшую алкоксикарбониламиногруппу, низший алканоил, низший алканоилокси, низший алкилтио и карбоксил; или
R1 представляет собой гетероциклическую группу, выбранную из следующих групп, где Y1 и Y2 являются одинаковыми или разными, и каждый представляет собой атом водорода или низший алкил, который может быть замещен:
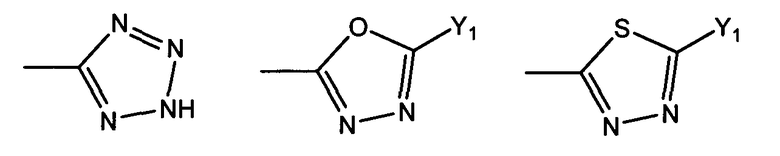
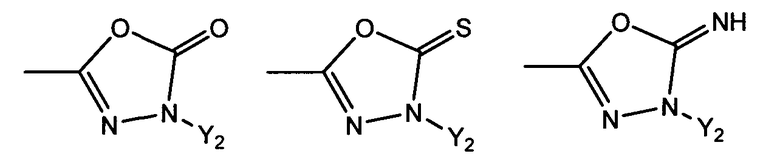
Предпочтительно R1 представляет собой OH, COOH или CONRa2Ra2', в которой Ra2 и Ra2' являются одинаковыми или разными, и каждый представляет собой атом водорода или низший алкил, содержащий от одного до трех атомов углерода; или R1 выбран из следующих групп:
<группа-заместитель L1> представляет собой атом галогена, гидрокси, нитро, циано, амино, карбамоил, аминосульфонил, имино, низшую алкиламиногруппу, ди(низший)алкиламиногруппу, низший алкилсульфонил, низшую алкилсульфониламиногруппу, низший алкокси, низший алкоксикарбонил, низшую алкоксикарбониламиногруппу, низший алканоил, низший алканоилокси, низший алкилтио и карбоксил; предпочтительно, представляет собой атом водорода, гидрокси, амино, карбамоил, низшую алкиламиногруппу, низшую алкиламиногруппу и низший алкокси.
<группа-заместитель L2> представляет собой атом галогена, гидрокси, амино и гидроксиметил; предпочтительно гидрокси и гидроксиметил.
<группа-заместитель M> представляет собой атом галогена, гидрокси, нитро, циано, амино, карбамоил, аминосульфонил, имино, низшую алкиламиногруппу, ди(низший)алкиламиногруппу, низший алкилсульфонил, низшую алкилсульфониламиногруппу, низший алкокси, низший алкоксикарбонил, низшую алкоксикарбониламиногруппу, низший алканоил, низший алканоилокси, низший алкилтио и карбоксил; предпочтительно гидрокси, карбамоил, аминосульфонил, низшую алкилсульфониламиногруппу и карбоксил.
R2 представляет собой O, S, SO, SO2, NH, NRb или CRc1Rc2, в которой Rb представляет собой низший алкил, который может быть замещен, и Rc1 и Rc2, которые могут быть одинаковыми или разными, представляют собой атом водорода или низший алкил, который может быть замещен.
Предпочтительно R2 представляет собой O, S, SO или SO2; более предпочтительно O.
R3 представляет собой фенил, который может быть замещен; предпочтительно R3 представляет собой фенил, который замещен; более предпочтительно R3 представляет собой фенил, который во 2-ом и 3-ем положениях замещен двумя одинаковыми или разными заместителями, выбранными из F, Cl, CF3 и CN.
X2 представляет собой CH, CX2a или N, где:
X2a представляет собой низший алкил; или
X2a представляет собой заместитель, выбранный из <группы-заместителя A1>, или низший алкил, который замещен одним или несколькими, одинаковыми или разными заместителями, выбранными из <группы-заместителя A1>, где <группа-заместитель A1> представляет собой атом галогена; циано; гидрокси; низшую алкиламиногруппу; ди(низший)алкиламиногруппу; низший алкокси, который может быть замещен одной или несколькими гидроксигруппами; низший алкилтио; и низший алкилсульфонил; или
X2a представляет собой COORx1, CONRx2Rx3, NHCORx1, NHCONRx2Rx3, NHSO2NRx2Rx3, NRx4Rx5 или CH2NRx4Rx5, где:
Rx1 представляет собой атом водорода или низший алкил, который может быть замещен;
каждый из Rx2 и Rx3, которые могут быть одинаковыми или разными, представляет собой атом водорода, низший алкил, который может быть замещен, или циклоалкил, который может быть замещен; или, альтернативно, Rx2 и Rx3 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную алифатическую гетероциклическую группу, которая содержит, по меньшей мере, один атом, выбранный из N, O и S, и которая может быть замещена; и
Rx4 и Rx5, которые могут быть одинаковыми или разными, представляют собой атом водорода, низший алкил, который может быть замещен, или циклоалкил, который может быть замещен; или
X2a представляет собой 5- или 6-членную алифатическую гетероциклическую группу, которая содержит, по меньшей мере, один атом, выбранный из N, O и S, и которая может быть замещена, в которой два атома водорода, которые присоединены к одному и тому же атому углерода алифатической гетероциклической группы, могут быть замещены оксогруппой, а соседние два атома углерода, составляющие алифатическое гетероциклическое кольцо, могут образовывать двойную связь; или низший алкил, который замещен алифатической гетероциклической группой; или
X2a представляет собой 5- или 6-членную ароматическую гетероциклическую группу, которая содержит, по меньшей мере, один атом, выбранный из N, O и S, и которая может быть замещена; или низший алкил, который замещен ароматической гетероциклической группой.
Предпочтительно X2 представляет собой CH, CX2a или N, где X2a представляет собой низший алкил.
Более предпочтительно X2 представляет собой CH или N.
<группа-заместитель A1> представляет собой атом галогена; циано; гидрокси; низшую алкиламиногруппу; ди(низший)алкиламиногруппу; низший алкокси, который может быть замещен одной или несколькими гидроксигруппами; низший алкилтио; и низший алкилсульфонил; предпочтительно атом галогена, гидрокси, низшую алкиламиногруппу и низший алкилсульфонил.
W представляет собой следующий остаток:
где:
W1 представляет собой CH, N, NH, O или S;
W2 представляет собой CH, CW2a, N, NW2b, O или S, где каждый из W2a и W2b независимо представляет собой атом водорода, атом галогена, циано, низший алкил, содержащий от одного до двух атомов углерода, циклоалкил, содержащий от трех до пяти атомов углерода, или низший алкил, содержащий от одного до двух атомов углерода, который может быть замещен одним или несколькими атомами галогена;
W3 представляет собой C или N; и
по меньшей мере, один из W1, W2 и W3 представляет собой атом углерода; однако два из W1, W2 и W3 одновременно не могут представлять собой O и S.
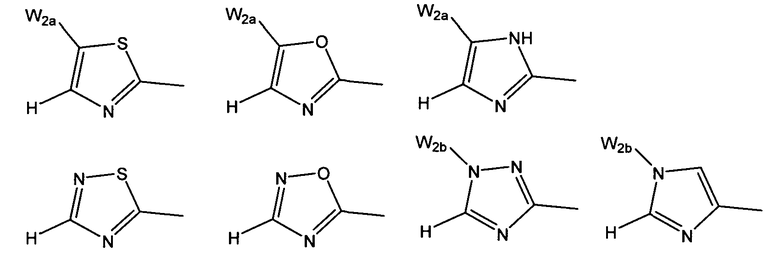
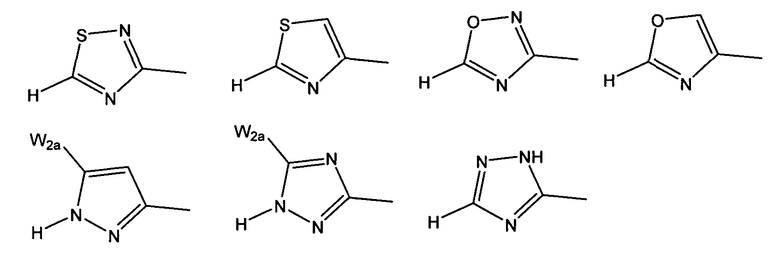
W предпочтительно выбран из:
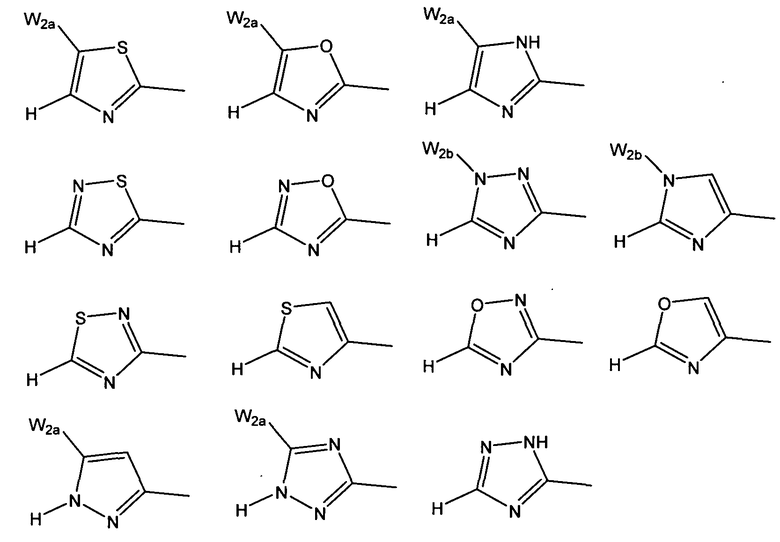
W более предпочтительно выбран из:
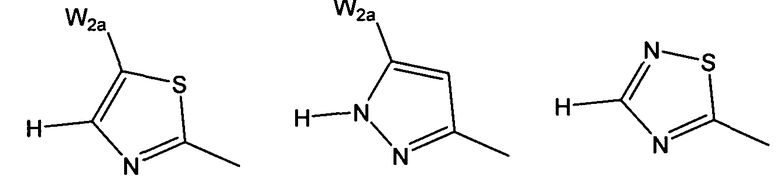
где W2a представляет собой атом водорода, атом галогена, циано или метил, который может быть замещен одним-тремя атомами фтора.
В частности, W предпочтительно выбран из:
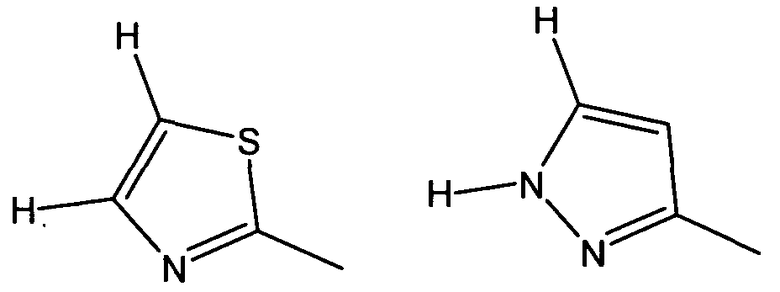
Еще более предпочтительно W выбран из:
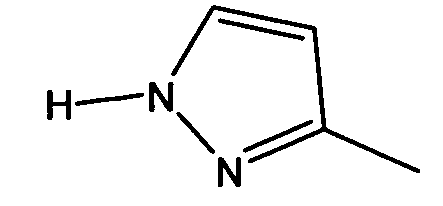
Предпочтительный вариант осуществления соединения, представленного описанной выше общей формулой (I), также можно выразить следующим образом:
(1) Соединение описанной выше формулы (I) или его фармацевтически приемлемая соль или сложный эфир, где W выбран из:
(2) Соединение, описанное в п. (1), или его фармацевтически приемлемая соль или сложный эфир, где R3 представляет собой фенил, который во 2-ом и 3-ем положениях замещен двумя одинаковыми или разными заместителями, выбранными из F, Cl, CF3 и CN.
(3) Соединение, описанное в п. (2), или его фармацевтически приемлемая соль или сложный эфир, где <группа-заместитель L1> представляет собой атом галогена, гидрокси, амино, карбамоил, низшую алкиламиногруппу, ди(низший)алкиламиногруппу и низший алкокси; и <группа-заместитель M> представляет собой гидрокси, карбамоил, аминосульфонил, низшую алкилсульфониламиногруппу и карбоксил.
(4) Соединение, описанное в п. (3), или его фармацевтически приемлемая соль или сложный эфир, где X2 представляет собой CH или N.
(5) Соединение, описанное в п. (4), или его фармацевтически приемлемая соль или сложный эфир, где R1 представляет собой OH, COOH или CONRa2Ra2', где Ra2 и Ra2' являются одинаковыми или разными, и каждый из них представляет собой атом водорода или низший алкил, содержащий от одного до трех атомов углерода; или R1 выбран из следующих групп:
и R2 представляет собой O, S, SO или SO2.
(6) Соединение, описанное в п. (5), или его фармацевтически приемлемая соль или сложный эфир, где:
W выбран из:
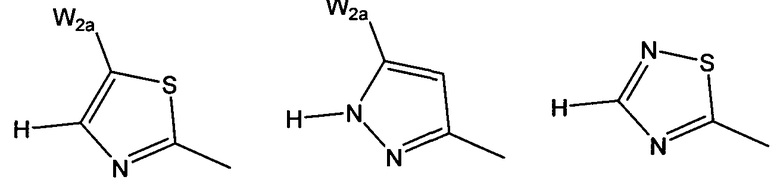
где W2a представляет собой атом водорода, атом галогена, циано или метил, который может быть замещен одним-тремя атомами фтора.
(7) Соединение, описанное в п. (6), или его фармацевтически приемлемая соль или сложный эфир, где W представляет собой любой из следующих остатков:
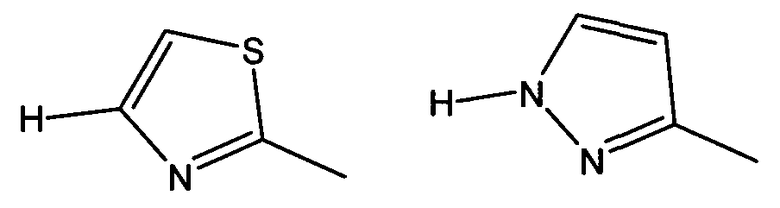
(8) Соединение, которое представляет собой:
(a) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(b) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(c) транс-4-(2,3-дихлорфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(d) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(e) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексанкарбоксамид;
(f) 5-(транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он;
(g) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он;
(h) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он; или
(i) 5-(транс-4-((2,3-дихлорфенил)сульфонил)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он,
или их фармацевтически приемлемую соль или сложный эфир.
Также при еще одном варианте осуществления изобретение относится к соединению общей формулы (I0):
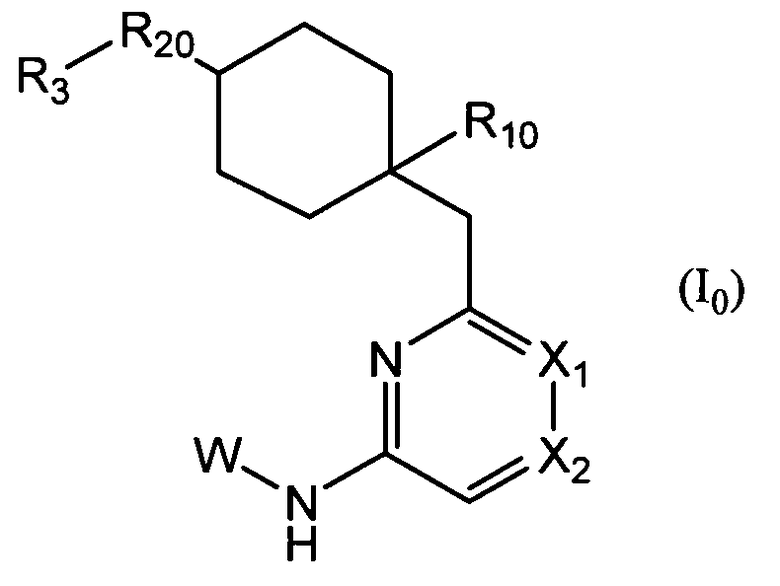
где:
R10 представляет собой атом водорода, F, CN, OH, CH2OH, COOH или CONRa10Ra20, где Ra10 и Ra20, которые могут быть одинаковыми или разными, представляют собой атом водорода или низший алкил;
R20 представляет собой O, S, NH, NRb или CRc1Rc2, где Rb представляет собой низший алкил, и Rc1 и Rc2, которые могут быть одинаковыми или разными, представляют собой атом водорода или низший алкил;
R3 представляет собой фенил, который может быть замещен;
X1 представляет собой CH, CX1a или N, где X1a представляет собой низший алкил, который может быть замещен;
X2 представляет собой CH, CX2a или N, где:
X2a представляет собой низший алкил; или
X2a представляет собой заместитель, выбранный из <группы-заместителя A1>, или низший алкил, который замещен одним или несколькими, одинаковыми или разными заместителями, выбранными из <группы-заместителя A1>, где <группа-заместитель A1> представляет собой атом галогена; циано; гидрокси; низшую алкиламиногруппу; ди(низший)алкиламиногруппу; низший алкокси, который может быть замещен одной или несколькими гидроксигруппами; низший алкилтио; и низший алкилсульфонил; или
X2a представляет собой COORx1, CONRx2Rx3, NHCORx1, NHCONRx2Rx3, NHSO2NRx2Rx3, NRx4Rx5 или CH2NRx4Rx5, где:
Rx1 представляет собой атом водорода или низший алкил, который может быть замещен;
каждый из Rx2 и Rx3, которые могут быть одинаковыми или разными, представляет собой атом водорода, низший алкил, который может быть замещен, или циклоалкил, который может быть замещен; или, альтернативно, Rx2 и Rx3 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную алифатическую гетероциклическую группу, которая содержит, по меньшей мере, один атом, выбранный из N, O и S, и которая может быть замещена; и
Rx4 и Rx5, которые могут быть одинаковыми или разными, представляют собой атом водорода, низший алкил, который может быть замещен, или циклоалкил, который может быть замещен; или
X2a представляет собой 5- или 6-членную алифатическую гетероциклическую группу, которая содержит, по меньшей мере, один атом, выбранный из N, O и S, и которая может быть замещена, в которой два атома водорода, которые присоединены к одному и тому же атому углерода алифатической гетероциклической группы, могут быть замещены оксогруппой, а соседние два атома углерода, составляющие алифатическое гетероциклическое кольцо, могут образовывать двойную связь; или низший алкил, который замещен алифатической гетероциклической группой; или
X2a представляет собой 5- или 6-членную ароматическую гетероциклическую группу, которая содержит, по меньшей мере, один атом, выбранный из N, O и S, и которая может быть замещена; или низший алкил, который замещен ароматической гетероциклической группой;
однако при условии, что среди X1 и X2 количество атомов азота равно 0 или 1;
W представляет собой следующий остаток:
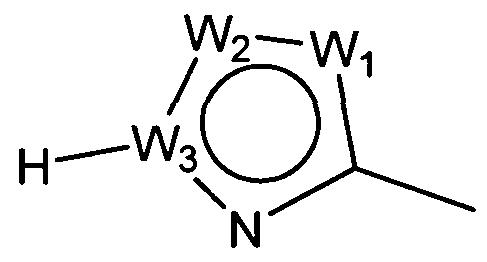
где:
W1 представляет собой CH, N, NH, O или S;
W2 представляет собой CH, CW2a, N, NW2b, O или S, где каждый из W2a и W2b независимо представляет собой атом водорода, атом галогена, циано, низший алкил, содержащий от одного до двух атомов углерода, циклоалкил, содержащий от трех до пяти атомов углерода, или низший алкил, содержащий от одного до двух атомов углерода, который может быть замещен одним или несколькими атомами галогена;
W3 представляет собой C или N; и
по меньшей мере, один из W1, W2 и W3 представляет собой атом углерода; однако два из W1, W2 и W3 одновременно не могут представлять собой O и S,
или к его фармацевтически приемлемой соли или сложному эфиру.
Кроме того, в комбинированном препарате по изобретению, содержащем два отдельных препарата, предпочтительно любой из двух отдельных препаратов или оба являются препаратами для перорального введения.
Комбинированный препарат по изобретению, содержащий два отдельных препарата, предпочтительно является таким препаратом, где один из препаратов представляет собой препарат, содержащий вместе с фармацевтически приемлемым носителем или разбавителем следующие соединения:
(a) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(b) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(c) транс-4-(2,3-дихлорфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(d) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(e) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексанкарбоксамид;
(f) 5-(транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он;
(g) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он;
(h) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он; или
(i) 5-(транс-4-((2,3-дихлорфенил)сульфонил)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он,
или их фармацевтически приемлемую соль или сложный эфир; и
другой препарат представляет собой препарат, содержащий паклитаксел или доцетаксел, или их фармацевтически приемлемую соль или сложный эфир вместе с фармацевтически приемлемым носителем или разбавителем.
Кроме того, комбинированный препарат, содержащий вместе с фармацевтически приемлемым носителем или разбавителем два отдельных препарата, согласно изобретению можно дополнительно объединять, по меньшей мере, с одним препаратом, содержащим противоопухолевое средство, выбранное из группы, состоящей из противоопухолевых алкилирующих средств, противоопухолевых антиметаболитов, противоопухолевых антибиотиков, противоопухолевых средств растительного происхождения, противоопухолевых координационных соединений платины, противоопухолевых производных камптотецина, противоопухолевых ингибиторов тирозинкиназы, моноклональных антител, интерферонов, модификаторов биологического ответа и других противоопухолевых средств (здесь определение каждого противоопухолевого средства является таким же, как в приведенном выше определении) или их фармацевтически приемлемой соли или сложного эфира.
Фармацевтическая композиция по изобретению вместе с фармацевтически приемлемым носителем или разбавителем также предпочтительно содержит следующие соединения:
(a) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(b) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(c) транс-4-(2,3-дихлорфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(d) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(e) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексанкарбоксамид;
(f) 5-(транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он;
(g) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он;
(h) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он; или
(i) 5-(транс-4-((2,3-дихлорфенил)сульфонил)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-он,
или их фармацевтически приемлемую соль или сложный эфир; и паклитаксел или доцетаксел, или их фармацевтически приемлемую соль или сложный эфир вместе с фармацевтически приемлемым носителем или разбавителем.
Далее будут объяснены ингибирующие активности соединения общей формулы (I) по изобретению в отношении Авроры A и Авроры B.
Ингибирующая активность в отношении Авроры A
(1) Очистка Авроры A
кДНК His-меченной по N-концу человеческой киназы Аврора A интегрировали в экспрессирующий вектор, который затем в высокой степени экспрессировался в клетках Escherichia coli BL21-CodonPlus(DE3)-RIL. Клетки Escherichia coli собирали и подвергали лизису, а затем His-меченный человеческий белок Аврора A наносили на никель-хелатную колонку и элюировали с колонки имидазолом. Активные фракции обессоливали на колонке для гель-фильтрации (обессоливания), получая при этом очищенный фермент.
(2) Измерение активности Авроры A
Субстрат, применяемый для измерения активности Авроры A, представлял собой синтетический пептид (5-FAM (5-карбоксифлуоресцеин)-γ-аминомасляная кислота-Ala-Leu-Arg-Arg-Ala-Ser-Leu-Gly-NH2)(SEQ ID NO:1), который был получен от компании Toray Research Center, Inc.
Для реакции фосфорилирования ссылаются на способ Upstate, Inc. [Kinase ProfilerTM протоколы испытаний], и детектировали фосфорилирование субстрата, применяя IMAP-методику (Molecular Devices, Co. Ltd.)(Gaudet EW. и др, J.Biomol.Screen, 8, 164-175(2003)). Более конкретно, реакцию фосфорилирования и детектирование осуществляли следующим образом.
Реакцию фосфорилирования проводили с использованием 384-луночного планшета, реакционный объем составлял 10 мкл/ячейку. Реакционный буфер состоял из 50 мМ Трис-хлоридного буфера (pH 7,4), 15 мМ ацетата магния и 0,2 мМ этилендиамин-N,N,N',N'-тетрауксусной кислоты (EDTA). К нему добавляли очищенный белок Аврора A, 100 нМ пептидного субстрата и 20 мкМ аденозин-5'-трифосфата (АТФ), и затем осуществляли реакцию при 30°C в течение 120 минут.
После этого, для того чтобы завершить и детектировать реакцию, в каждую лунку добавляли по 30 мкл связывающего реагента (IMAP Progressive Binding Reagent, R7284) IMAP (зарегистрированная торговая марка), который был разбавлен (1:400) 1×MAP-связывающим буфером A (IMAP Progressive Binding Buffer A, 5×сток-раствор, R7282). Раствор выдерживали в темноте еще в течение 60 минут и затем измеряли поляризацию флуоресценции, используя высокопроизводительное считывающее устройство для микропланшетов (длина волны возбуждения: 485 нм; длина волны излучения: 520 нм).
В реакционную систему добавляли соединение, подвергаемое тестированию, при этом готовили серии разведений соединения в диметилсульфоксиде (ДМСО) и затем добавляли 0,5 мкл такого раствора для тестирования в каждую лунку. Для каждого раствора обеспечивали контрольную лунку путем добавления в лунку 0,5 мкл ДМСО вместо раствора ДМСО, содержащего подвергаемое тестированию соединение.
Ингибирующая активность в отношении Авроры B
(1) Измерение активности Авроры B (Способ A)
Для реакции фосфорилирования применяли набор реактивов для проведения анализа IMAP (зарегистрированная торговая марка) (Аврора B), предоставленный компанией Carna Biosciences, Inc., и детектировали фосфорилирование субстрата с использованием IMAP-технологии. Применяемый набор реактивов для анализа состоял из буфера для анализа, комплекса, состоящего из GST-меченной человеческой Авроры B (AurB)/комплекса His-меченных человеческих белков INCENP (аминокислотная последовательность: 803-916, AAU04398.1) и раствора АТФ/субстрат. Используя те же средства, проводили реакцию фосфорилирования согласно частично измененной инструкции, прилагаемой к набору, и затем детектировали фосфорилирование субстрата, используя IMAP-технологию.
Для реакции фосфорилирования использовали 384-луночный планшет, а реакционный объем составлял 10 мкл/лунку. Состав реакционного буфера (буфер для анализа) состоял из 20 мМ буфера HEPES (pH 7,4), 0,01% Tween-20 и 2 мМ дитиотрейтола (DTT). К буферу добавляли комплекс AurB/комплекс белка INCENP, 100 нМ субстрата, 40 мкМ АТФ и 1 мМ магниевой соли и затем проводили реакцию при 25°C в течение 45 минут. После этого для завершения и детектирования реакции в каждую лунку добавляли по 30 мкл связывающего реагента (IMAP Progressive Binding Reagent, R7284) IMAP (зарегистрированная торговая марка), который разбавляли (1:400) 1×IMAP-связывающим буфером A (IMAP Progressive Binding Buffer A, 5×сток-раствор, R7282). Раствор выдерживали в темноте еще в течение 60 минут и затем измеряли поляризацию флуоресценции, используя высокопроизводительное считывающее устройство для микропланшетов (длина волны возбуждения: 485 нм; длина волны излучения: 520 нм).
В реакционную систему добавляли тестируемое соединение, при этом готовили серии разведений соединения в ДМСО и затем по 0,5 мкл такого раствора добавляли для тестирования в каждую лунку. Для каждого раствора создавали контрольную лунку, добавляя в нее 0,5 мкл ДМСО вместо раствора ДМСО, содержащего подвергаемое тестированию соединение.
(2) Измерение активности Авроры B (Способ B)
(a) Очистка Авроры B
кДНК человеческой Авроры B, содержащей His-метку, соединенную с N-концом, интегрировали в экспрессирующий вектор, который затем с высокой степенью экспрессировался в клетках бактерии Escherichia coli BL21-CodonPlus(DE3)-RIL. Клетки Escherichia coli собирали, подвергали солюбилизации и затем His-меченный белок Аврора B адсорбировали на никель-хелатной колонке и элюировали с колонки имидазолом. Активные фракции обессоливали на колонке для гель-фильтрации (обессоливания), получая при этом очищенный фермент.
(b) Измерение активности Авроры B
При измерении активности Авроры B используемый субстрат представлял собой Kemptide (Leu-Arg-Arg-Ala-Ser-Leu-Gly) (SEQ ID NO:2), a синтетический пептид был предоставлен компанией Sigma-Aldrich, Inc.
Реакцию проводили с использованием частично модифицированного способа измерения активности Авроры A. Количество реакционной жидкости составляло 21,1 мкл, а в состав реакционного буфера (буфер R2) входило 50 мМ Трис-гидрохлоридного буфера (pH 7,4), 15 мМ ацетата магния и 0,2 мМ этилендиамин-N,N,N',N'-тетраацетата (EDTA). К нему добавляли очищенную Аврору B, 100 мкМ субстратного пептида, 100 мкМ не содержащего метки аденозинтрифосфата (АТФ) и 1 мкКи АТФ, меченного γ-33P (2500 Ки/ммоль или более), и давали смеси возможность взаимодействовать при 30°C в течение 20 минут. Затем к реакционной системе добавляли 10 мкл 350 мМ фосфатного буфера, чтобы остановить реакцию. Субстратный пептид адсорбировали на 96-луночном планшете из бумажного фильтра P81 и затем несколько раз промывали 130 мМ фосфатным буфером. Радиоактивность пептида измеряли с помощью жидкостного сцинтилляционного счетчика. АТФ, меченный γ-33P, был предоставлен компанией Amersham Biosciences Co., Ltd.
Соединение, подвергаемое тестированию, добавляли в реакционную систему, при этом сначала готовили серии разведений соединения в ДМСО и 1,1 мкл такого раствора добавляли в реакционную систему. Контрольный образец обеспечивали путем добавления к реакционной системе 1,1 мкл ДМСО.
С помощью упомянутого выше способа (при измерении активности Авроры B применяли способ A) получены результаты измерения активностей Авроры A и Авроры B, приведенные в таблице 1. Соединение согласно изобретению обладало превосходной селективной ингибирующей активностью в отношении Авроры A. Сходные результаты получали, если при измерении активности Авроры B использовали способ B.
Далее будет выяснена активность соединения общей формулы (I) по изобретению по подавлению роста клеток.
Способ оценки фармацевтического эффекта с применением клеток
a) Реагент
Фетальная телячья сыворотка (FCS) была предоставлена компанией Moregate Biotech, питательная среда DMEM была предоставлена компанией Invitrogen Corp. Реагент WST-8 был предоставлен компанией Kishida Chemical Co., Ltd.
b) Клетки
Раковые клетки шейки матки человека (HeLa S3) были получены от компании American Type Culture Collection (ATCC).
c) Способ оценки эффекта
Готовили суспензию клеток в среде DMEM, содержащей 10% FCS, и распределяли суспензию клеток на 96-луночном пластиковом планшете со степенью загрузки 750 клеток/100 микролитров на лунку. Планшет инкубировали при 37°C в течение ночи в атмосфере, состоящей из 95% воздуха и 5% CO2. Готовили калибровочные растворы лекарственного средства путем разведения в диметилсульфоксиде и дополнительно разбавляли средой DMEM, содержащей 10% FCS. Затем растворы каждого из разведений наносили на отдельный планшет, в лунки которого вносили по 100 микролитров клеточной суспензии на лунку. Планшет дополнительно инкубировали при 37°C в течение трех дней в атмосфере, содержащей 5% CO2 и 95% воздуха. Рост клеток после инкубирования измеряли способом WST-8 (H. Tominaga и др., Anal. Commun., 36, 47-50 (1999)). В данном описании способ WST-8 относится к способу, при котором в каждую лунку добавляют по 20 микролитров раствора реагента WST-8, инкубирование проводят при 37°C в течение 60 минут, планшет встряхивают и колориметрическим способом измеряют количество продуцируемого формазана, чтобы определить процент ингибирования для лекарственного средства. Определяли концентрацию соединения, необходимую для 50% ингибирования роста (IC50, мкМ).
Как видно из таблицы 2, соединение по изобретению обладало превосходным ингибирующим эффектом в отношении роста раковых клеток, полученных от человека (HeLa S3).
Способ оценки эффекта применения в клетках комбинированных лекарственных средств
a) Реагент
Фетальная телячья сыворотка (FCS) была предоставлена компанией Moregate Biotech, питательная среда DMEM была предоставлена компанией Invitrogen Corp., доцетаксел (торговая марка: Taxere) от компании Sigma-Aldrich, Inc., и реагент WST-8 от компании Kishida Chemical Co., Ltd.
b) Клетки
Клетки человеческой злокачественной опухоли шейки матки (HeLa S3) были получены от компании American Type Culture Collection (ATCC).
c) Способ оценки эффекта
Готовили суспензию клеток в среде DMEM, содержащей 10% FCS, и распределяли суспензию клеток на двух 96-луночных пластиковых планшетах со степенью загрузки 750 клеток/100 микролитров на лунку. Планшеты инкубировали при 37°C в течение ночи в атмосфере, состоящей из 95% воздуха и 5% CO2. Готовили калибровочные растворы в диметилсульфоксиде и дополнительно разбавляли их ДМСО или средой DMEM, содержащей 10% FCS, а также содержащей 0,6 нМ доцетаксела. Затем каждый из разбавленных растворов добавляли к лункам одного из планшетов, в которые было внесено по 100 микролитров клеток на лунку. Конечная концентрация доцетаксела на данной стадии составляла 0,3 нМ. Также в случае введения только одного соединения по изобретению концентрации составляли 0,003, 0,01, 0,03, 0,1, 0,3, 1, 3 и 10 мкМ. Планшеты дополнительно инкубировали при 37°C в течение трех дней в атмосфере, состоящей из 95% воздуха и 5% CO2. После инкубации способом WST-8 (H. Tominaga и др., Anal. Commun., 36, 47-50 (1999)) измеряли рост клеток. В данном описании способ WST-8 относится к способу, при котором в каждую лунку добавляют по 20 микролитров раствора реагента WST-8, инкубирование проводят при 37°C в течение 60 минут, планшет встряхивают и колориметрическим способом измеряют количество продуцируемого формазана, чтобы определить процент ингибирования для лекарственного средства. Определяли эффекты ингибирования роста клеток, вызываемые доцетакселом и соединением по изобретению, принимая за 0% значение, полученное при обработке одним только ДМСО.
Как видно из таблицы 3, соединение по изобретению обладало превосходным ингибирующим эффектом в отношении роста клеток, а также синергическим действием с противоопухолевым средством типа таксана, таким как доцетаксел, в отношении раковых клеток, полученных от человека (HeLa S3).
1,0
26,9
90,4
1,0
52,9
93,8
1,0
25,6
87,0
1,0
49,0
95,0
3,0
59,0
95,0
1,0
25,4
84,5
1,0
42,4
90,8
0,3
21,3
77,4
1,0
49,8
94,4
0,3
36,0
87,4
0,3
25,4
87,8
1,0
18,2
88,5
0,3
39,9
92,9
0,3
53,4
92,0
3,0
38,5
89,0
0,3
40,8
94,0
1,0
23,4
91,3
0,3
40,8
93,9
0,1
42,7
89,9
0,3
32,0
88,9
На основе приведенных выше данных предполагается, что соединение по изобретению должно быть применимо в качестве противоопухолевого средства, поскольку оно обладает не только превосходной ингибирующей активностью в отношении роста клеток, основанной на селективной ингибирующей активности в отношении Авроры A, но также обладает синергическим действием при комбинированном применении с другим противоопухолевым средством. Поэтому предполагается, что фармацевтическая композиция или селективный ингибитор Авроры A, содержащие новое аминопиридиновое производное по изобретению, или его фармацевтически приемлемую соль или сложный эфир, или противоопухолевое средство, содержащее соединение по изобретению или его фармацевтически приемлемую соль или сложный эфир, эффективны при лечении пациентов с раковым заболеванием.
Упомянутые выше фармацевтическая композиция и ингибитор и упомянутое выше противоопухолевое средство могут содержать фармацевтически приемлемый носитель или разбавитель. В настоящем описании термин "фармацевтически приемлемый носитель или разбавитель" относится к эксципиентам (например, жирам, воску, полутвердым и жидким полиолам, природным или гидрогенизированным маслам и т.д.); воде (например, дистиллированной воде, в частности дистиллированной воде для инъекции и т.д.), физиологическому солевому раствору, спирту (например, этанолу), глицерину, полиолам, водному раствору глюкозы, манниту, растительным маслам т.д.); добавкам (например, наполнителю, дезинтегрирующему средству, связующему, лубриканту, увлажняющему средству, стабилизатору, эмульгатору, диспергирующей добавке, консерванту, подсластителю, красителю, средству для улучшения вкуса или ароматизатору, концентрирующему средству, разбавителю, буферному веществу, растворителю или солюбилизирующему средству, химическому веществу для обеспечения эффекта хранения, соли для модификации осмотического давления, средству для покрытия или антиоксиданту) и т.п.
Считается, что примером подходящей опухоли, для которой может быть проиллюстрирован терапевтический эффект соединения по изобретению, является солидный рак человека. Примеры солидного рака человека включают рак мозга, рак головы и шеи, рак пищевода, рак щитовидной железы, мелкоклеточную карциному, немелкоклеточную карциному, рак молочной железы, рак желудка, рак желчного пузыря и желчного протока, рак печени, рак поджелудочной железы, рак ободочной кишки, рак прямой кишки, рак яичника, хорионэпителиому, рак матки, рак шейки матки, рак почечной лоханки и мочеточника, рак мочевого пузыря, рак предстательной железы, рак полового члена, рак яичка, эмбриональный рак, опухоль Вильмса, рак кожи, злокачественную меланому, нейробластому, остеосаркому, опухоль Юинга, саркому мягких тканей и т.п.
Далее будет объяснен описанный выше термин "фармацевтически приемлемая соль или сложный эфир". Когда соединение по изобретению применяют в качестве противоопухолевого средства или т.п., его можно также применять в форме фармацевтически приемлемой соли. Типичные примеры фармацевтически приемлемой соли включают соль щелочного металла, такого как натрий и калий; соль неорганической кислоты, такой как гидрохлорид, сульфат, нитрат, фосфат, карбонат, гидрокарбонат и перхлорат; соль органической кислоты, такой как ацетат, пропионат, лактат, малеат, фумарат, тартат, малат, цитрат и аскорбат; соль сульфоновой кислоты, такой как метансульфонат, изотионат, бензолсульфонат и толуолсульфонат; кислую соль двухосновной аминокислоты, такой как аспартат и глутамат; и т.п. Фармацевтически приемлемая соль соединения (I) предпочтительно представляет собой соль неорганической кислоты, такой как гидрохлорид, сульфат, нитрат, фосфат, карбонат, гидрокарбонат и перхлорат; более предпочтительно - гидрохлорид.
Способ получения фармацевтически приемлемой соли соединения по изобретению можно осуществлять с помощью соответствующей комбинации тех способов, которые традиционно применяются в области органической синтетической химии. Конкретным примером такого способа является способ, при котором раствор соединения по изобретению в его свободной форме подвергается титрованию для нейтрализации раствором щелочи или раствором кислоты.
Примеры сложного эфира соединения по изобретению включают сложный метиловый эфир и сложный этиловый эфир. Такие сложные эфиры можно получать этерификацией свободной карбоксильной группы согласно традиционному способу.
Что касается каждого комбинированного препарата по изобретению, можно выбрать различные формы препарата, и такие примеры включают препараты для перорального введения, такие как таблетки, капсулы, порошки, гранулы или жидкости, или стерилизованные жидкие препараты для парентерального введения, такие как растворы или суспензии, суппозитории, мази и т.п.
Твердые препараты можно получать в форме таблетки, капсулы, гранулы и порошка без каких-либо добавок или получать, применяя соответствующие носители (добавки). Примеры таких носителей (добавок) могут включать сахариды, такие как лактоза или глюкоза; кукурузный, пшеничный или рисовый крахмал; жирные кислоты, такие как стеариновая кислота; неорганические соли, такие как метаалюмосиликат магния или безводный фосфат кальция; синтетические полимеры, такие как поливинилпирролидон или полиалкиленгликоль; спирты, такие как стеариловый спирт или бензиловый спирт; синтетические производные целлюлозы, такие как метилцеллюлоза, карбоксиметилцеллюлоза, этилцеллюлоза или гидроксипропилметилцеллюлоза; и другие традиционно применяемые добавки, такие как желатин, тальк, растительные масла и гуммиарабик.
Указанные твердые препараты, такие как таблетки, капсулы, гранулы и порошки, обычно могут содержать в качестве активного ингредиента, например, от 0,1 до 100% по массе и предпочтительно от 5 до 98% по массе соединения описанной выше формулы (I), в расчете на общую массу препарата.
Жидкие препараты производят в форме суспензии, сиропа, инъекции и вливания с помощью капельницы (внутривенная жидкость), применяя соответствующие добавки, которые традиционно применяются в жидких препаратах, такие как вода, спирт или масла растительного происхождения, такие как соевое масло, арахисовое масло и кунжутное масло.
В частности, когда препарат вводится парентерально в форме внутримышечной инъекции, внутривенной инъекции или подкожной инъекции, соответствующий растворитель или разбавитель можно проиллюстрировать примером в виде дистиллированной воды для инъекции, водного раствора гидрохлорида лидокаина (для внутримышечной инъекции), физиологического солевого раствора, водного раствора глюкозы, этанола, полиэтиленгликоля, пропиленгликоля, жидкости для внутривенной инъекции (например, водный раствор лимонной кислоты, цитрата натрия и т.п.) или электролитического раствора (для внутривенного вливания с помощью капельницы и внутривенной инъекции) или их смешанного раствора.
Такая инъекция может быть в форме предварительно приготовленного раствора или в форме порошка как такового или порошка, связанного с подходящим носителем (добавкой), который растворяется во время применения. Жидкость для инъекции может содержать, например, от 0,1 до 10% по массе активного ингредиента, в расчете на общую массу препарата.
Жидкие препараты, такие как суспензия или сироп для перорального введения, могут содержать, например, от 0,1 до 10% по массе активного ингредиента, в расчете на общую массу препарата.
Каждый препарат в комбинированном препарате по изобретению может быть получен специалистом в данной области согласно традиционным способам или общеизвестным методикам. Например, препарат, содержащий другое противоопухолевое средство, который применяют в комбинации с соединением, представленным описанной выше общей формулой (I), можно получить в том случае, если препарат является препаратом для перорального введения, например, путем смешения соответствующего количества противоопухолевого средства с соответствующим количеством лактозы и заполнения полученной смесью твердых желатиновых капсул, которые подходят для перорального введения. С другой стороны, получение можно осуществлять в том случае, если препарат, содержащий противоопухолевое средство, представляет собой инъекцию, например, путем смешения соответствующего количества противоопухолевого средства с соответствующим количеством 0,9%-ного физиологического солевого раствора и заполнения полученной смесью ампул для инъекции.
Также в случае комбинированного препарата, содержащего соединение по изобретению, представленное описанной выше общей формулой (I), и другое противоопухолевое средство, специалист в данной области может легко получить препарат согласно традиционным способам или общеизвестным методикам.
В способе по изобретению предпочтительная терапевтическая доза может изменяться, например, в зависимости от способа введения соединения, представленного общей формулой (I), типа применяемого соединения, представленного общей формулой (I), и дозированной формы применяемого соединения, представленного общей формулой (I); типа, способа введения и дозированной формы другого противоопухолевого средства, применяемого в комбинации; и типа клеток, подвергаемых лечению, состояния пациента и т.п. Оптимальное лечение в данных условиях может определить специалист в данной области, основываясь на установленной традиционной терапевтической дозе и/или основываясь на содержании настоящего описания изобретения.
В способе по изобретению терапевтическая доза соединения, представленного описанной выше общей формулой (I), может изменяться, особенно в зависимости от типа применяемого соединения, типа комбинированной композиции, частоты применения и конкретного места, подлежащего лечению, тяжести заболевания, возраста пациента, диагноза врача, типа рака или т.п. Однако, в качестве типичной рекомендации, в случае перорального введения суточная доза для взрослого человека может находиться, например, в диапазоне от 1 до 1000 мг. В случае парентерального введения, предпочтительно внутривенного введения, и более предпочтительно внутривенного вливания с помощью капельницы, суточная доза может находиться, например, в диапазоне от 1 до 100 мг/м2 (площадь поверхности тела). Здесь в случае внутривенного вливания с помощью капельницы введение можно осуществлять непрерывно, например в течение времени от 1 до 48 часов. Кроме того, частоту введения можно изменять в зависимости от способа введения и симптомов, например, от одного до пяти раз в день. Альтернативно при определенном способе введения также можно использовать введение через определенные промежутки времени, такое как введение через день, введение каждые два дня или т.п. Период прекращения введения лекарственного средства в случае парентерального введения составляет, например, от 1 до 6 недель.
Хотя терапевтическая доза другого противоопухолевого средства, применяемого в комбинации с соединением, представленным общей формулой (I), специально не ограничивается, если необходимо, она может определяться специалистом в данной области согласно известным литературным данным. Примеры могут быть следующими.
Терапевтическая доза 5-фторурацила (5-FU) такова, что в случае перорального введения, например, ежедневно вводят от 200 до 300 мг последовательно от одного до трех раз в день, и в случае инъекции, например, путем внутривенной инъекции или внутривенного вливания с помощью капельницы ежедневно вводят от 5 до 15 мг/кг в день в течение первых 5 дней подряд и затем путем внутривенной инъекции или внутривенного вливания с помощью капельницы один раз в день вводят от 5 до 7,5 мг/кг через день (дозу можно соответствующим образом увеличивать или уменьшать).
Терапевтическая доза S-1 (Тегафур, Гиместат и Остат калия) такова, что, например, исходная доза (разовая доза) устанавливается, исходя из стандартного количества в зависимости от площади поверхности тела, и ее вводят перорально два раза в день после завтрака и после обеда в течение 28 дней подряд с последующим прекращением введения лекарственного средства в течение 14 дней. Это соответствует одному курсу введения, который повторяется. Исходное стандартное количество на единицу площади поверхности тела (эквивалент Тегафура) составляет 40 мг при одном введении для площади менее 1,25 м2; 50 мг при одном введении для площади от 1,25 м2 до площади менее 1,5 м2; 60 мг при одном введении для площади 1,5 м2 или более. Указанная доза соответствующим образом увеличивается или уменьшается в зависимости от состояния пациента.
Терапевтическая доза гемцитабина составляет, например, 1 г гемцитабина/м2 за одно введение, который вводится путем внутривенного вливания с помощью капельницы в течение 30 минут, и одно введение в неделю продолжается в течение 3 недель с последующим прекращением введения лекарственного средства в течение четвертой недели. Это соответствует одному курсу введения, который повторяется. Указанная доза соответствующим образом уменьшается в соответствии с возрастом, симптомом или развитием побочных эффектов.
Терапевтическая доза доксорубицина (например, гидрохлорида доксорубицина), например, такова, что в случае внутривенной инъекции один раз в день путем внутривенного однократного введения вводят 10 мг (0,2 мг/кг) (титр) в течение 4-6 дней подряд с последующим прекращением введения лекарственного средства в течение 7-10 дней. Это соответствует одному курсу введения, который повторяется два или три раза. Здесь суммарная доза предпочтительно составляет 500 мг (титр)/м2 (площадь поверхности тела) или менее, и ее можно соответствующим образом увеличивать или уменьшать внутри диапазона.
Терапевтическая доза этопозида, например, такова, что в случае внутривенной инъекции в течение 5 дней подряд вводят от 60 до 100 мг/м2 (площадь поверхности тела) в день с последующим прекращением введения лекарственного средства в течение трех недель (дозу можно соответствующим образом увеличивать или уменьшать). Это соответствует одному курсу введения, который повторяется. При этом, в случае перорального введения, например, в течение 5 дней подряд вводят от 175 до 200 мг в день с последующим прекращением введения лекарственного средства в течение трех недель (дозу можно соответствующим образом увеличивать или уменьшать). Это соответствует одному курсу введения, который повторяется.
Терапевтическая доза доцетаксела (гидрат доцетаксела), например, такова, что один раз в день путем внутривенного вливания с помощью капельницы вводят 60 мг доцетаксела/м2 (площадь поверхности тела) в течение 1 часа или более с интервалом от 3 до 4 недель (дозу можно соответствующим образом увеличивать или уменьшать).
Терапевтическая доза паклитаксела, например, такова, что один раз в день путем внутривенного вливания с помощью капельницы вводят 210 мг/м2 (площадь поверхности тела) в течение 3 часов, с последующим прекращением введения лекарственного средства в течение, по меньшей мере, 3 недель. Это соответствует одному курсу введения, который повторяется. Дозу можно соответствующим образом увеличивать или уменьшать.
Терапевтическая доза цисплатина, например, такова, что в случае внутривенной инъекции один раз в день вводят от 50 до 70 мг/м2 (площадь поверхности тела) с последующим прекращением введения лекарственного средства в течение 3 недель или более (дозу можно соответствующим образом увеличивать или уменьшать). Это соответствует одному курсу введения, который повторяется.
Терапевтическая доза карбоплатина, например, такова, что один раз в день путем внутривенного вливания с помощью капельницы вводят от 300 до 400 мг/м2 в течение 30 минут или более с последующим прекращением введения лекарственного средства в течение, по меньшей мере, 4 недель (дозу можно соответствующим образом увеличивать или уменьшать). Это соответствует одному курсу введения, который повторяется.
Терапевтическая доза оксалиплатина такова, что один раз в день путем внутривенной инъекции вводят 85 мг/м2 с последующим прекращением введения лекарственного средства в течение двух недель. Это соответствует одному курсу введения, который повторяется.
Терапевтическая доза иринотекана (например, гидрохлорид иринотекана), например, такова, что один раз в день путем внутривенного вливания с помощью капельницы вводят 100 мг/м2 за 3 или 4 приема с интервалом в одну неделю с последующим прекращением введения лекарственного средства в течение, по меньшей мере, двух недель.
Терапевтическая доза топотекана, например, такова, что один раз в день путем внутривенного вливания с помощью капельницы вводят 1,5 мг/м2 в течение 5 дней с последующим прекращением введения лекарственного средства в течение, по меньшей мере, 3 недель.
Терапевтическая доза циклофосфамида, например, такова, что в случае внутривенной инъекции один раз в день путем внутривенной инъекции вводят по 100 мг в течение нескольких дней подряд. Если пациент может терпеть, суточную дозу можно увеличивать до 200 мг. Суммарная доза, которую можно соответствующим образом увеличивать или уменьшать, составляет от 3000 до 8000 мг. Если необходимо, можно вводить путем внутримышечной, интраторакальной или внутриопухолевой инъекции или вливания. С другой стороны, в случае перорального введения, например, ежедневно вводят от 100 до 200 мг.
Терапевтическая доза гефитиниба такова, что вводят перорально по 250 мг один раз день.
Терапевтическая доза цетуксимаба, например, такова, что в первый день путем внутривенного вливания с помощью капельницы вводят 400 мг/м2 и затем каждую неделю путем внутривенного вливания с помощью капельницы вводят по 250 мг/м2.
Терапевтическая доза бевацизумаба, например, такова, что каждую неделю путем внутривенного вливания с помощью капельницы вводят 3 мг/кг.
Терапевтическая доза трастузумаба, например, такова, что обычно взрослому человеку первоначально вводят один раз в день 4 мг трастузумаба/кг (вес тела) с последующим внутривенным вливанием с помощью капельницы 2 мг/кг в течение 90 минут или более каждую неделю со второго введения.
Терапевтическая доза экземестана, например, такова, что обычно взрослому человеку перорально вводят 25 мг один раз в день после еды.
Терапевтическая доза лейпрорелина (например, ацетата лейпрорелина), например, такова, что обычно взрослому человеку подкожно вводят 11,25 мг один раз в 12 недель.
Терапевтическая доза иматиниба, например, такова, что обычно взрослому человеку в хронической фазе хронической миелогенной лейкемии перорально вводят 400 мг один раз в день после еды.
Терапевтическая доза комбинации 5-FU и лейковорина, например, такова, что с первого дня до пятого дня путем внутривенного вливания с помощью капельницы вводят 425 мг/м2 5-FU и 200 мг/м2 лейковорина и повторяют такой курс с интервалом 4 недели.
Терапевтическая доза сорафениба, например, такова, что два раза в день перорально вводят по 200 мг (400 мг в день), по меньшей мере, за 1 час до еды или спустя 2 часа после еды.
Терапевтическая доза сунитиниба, например, такова, что один раз в день перорально вводят 50 мг в течение четырех недель, за которыми следует пропуск 2 недели.
Экспериментальные примеры
В примерах и справочных примерах при проведении тонкослойной хроматографии применяли силикагель Silica gel60F254 (Merck) в виде пластины, а в качестве способа детектирования применялся УФ-детектор. В качестве силикагеля для колонки применяли Biotage FLASH column (SI, NH). При препаративной жидкостной хроматографии с обращенной фазой в качестве колонки применяли колонку XBridge Prep C18 (Waters), а для подвижной фазы применяли 0,1% водный раствор трифторуксусной кислоты и 0,1% раствор трифторуксусной кислоты в ацетонитриле. МС-спектры измеряли, применяя Waters micromass ZQ2000 (ESI, ESCi). Спектры ЯМР измеряли с помощью спектрометра типа JEOL JNM-AL400 (400 МГц) или Varian MERCURY400 (400 МГц), и все значения δ представлены в м.д. Температуры плавления измеряли в условиях повышения температуры со скоростью 1°C/мин, применяя комбинацию Mettler Toledo FP82HT Hot Stage и NIKON Eclipse E600 POL.
Значения аббревиатур следующие.
с: синглет
д: дуплет
дд: двойной дуплет
т: триплет
дт: двойной триплет
кв: квартет
квин: квинтет
м: мультиплет
уш.: уширение
J: константа взаимодействия
Гц: Герц
ДМСО-d6: диметилсульфоксид-d6
TBS: трет-бутил(диметил)силильная группа
MOM: метоксиметильная группа
TBDPS: трет-бутил(дифенил)силильная группа
TsOH: п-толуолсульфоновая кислота
SEM: (2-(триметилсилил)этокси)метильная группа
Пример 1
Синтез гидрохлорида транс-4-(3-хлор-2-фторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
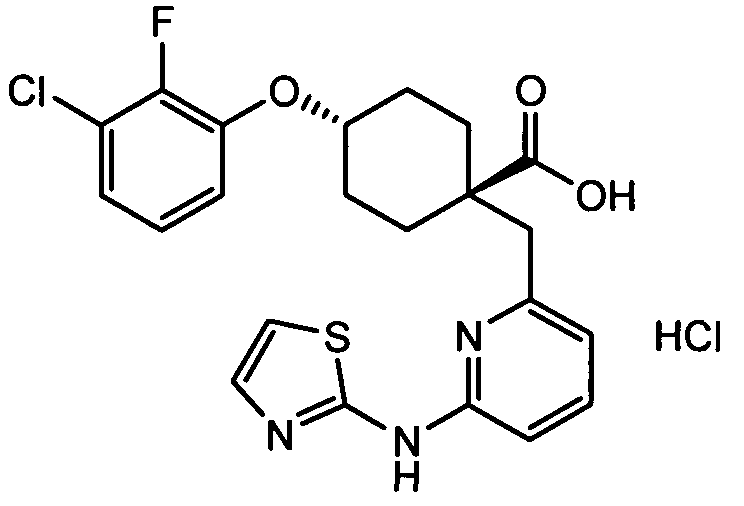
(1) Синтез 2-бром-6-(((трет-бутил(диметил)силил)окси)метил)пиридина
К раствору 10 г (6-бромпиридин-2-ил)метанола в 50 мл N,N-диметилформамида последовательно добавляли 4 г имидазола и 8,4 г трет-бутилдиметилсилилхлорида при комнатной температуре с последующим перемешиванием реакционной смеси при комнатной температуре в течение 2 часов. После добавления к реакционной смеси воды смесь экстрагировали н-гексаном. Полученный гексановый раствор сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме, получая при этом указанное в заголовке соединение в виде бесцветного масла.
(2) Синтез 6-(((трет-бутил(диметил)силил)окси)метил)-N-((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)пиридин-2-амина
Смесь 15,92 г 2-бром-6-(((трет-бутил(диметил)силил)окси)метил)пиридина, 5,53 г 2-аминотиазола, 3,04 г 9,9-диметил-4,5-бис(дифенилфосфино)ксантена, 2,72 г комплекса трис(дибензилиденацетон)дипалладий(0)-хлороформ, 18,86 г карбоната цезия и 100 мл толуола перемешивали при 120°C в течение ночи с последующим охлаждением до комнатной температуры и отфильтровывали нерастворимое вещество с помощью целита. Полученный толуольный раствор промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме, получая при этом сырой (неочищенный) продукт.
Готовили суспензию полученного сырого (неочищенного) продукта в 100 мл хлороформа, и затем при охлаждении льдом последовательно добавляли 13,7 мл N,N-диизопропилэтиламина и 4,8 мл простого хлорметилметилового эфира с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. Хлороформ удаляли в вакууме и к остатку добавляли воду с последующей экстракцией этилацетатом. Полученный этилацетатный раствор сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 5/1), получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
(3) Синтез (6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метанола
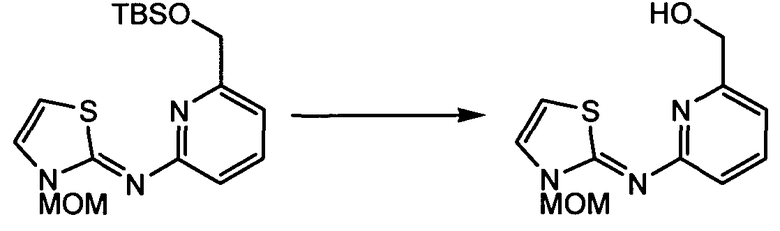
К раствору 14,28 г 6-(((трет-бутил(диметил)силил)окси)метил)-N-((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)пиридин-2-амина в 30 мл хлороформа и 30 мл метанола добавляли 30 мл трифторуксусной кислоты при охлаждении льдом с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. Реакционную смесь упаривали в вакууме. Полученный остаток нейтрализовали насыщенным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Полученный этилацетатный раствор сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 1/1), получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
(4) Синтез 6-(бромметил)-N-((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)пиридин-2-амина
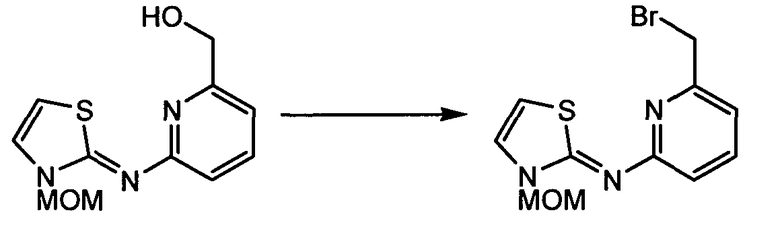
К раствору 4,43 г (6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метанола в 40 мл тетрагидрофурана последовательно добавляли 3,2 мл триэтиламина и 1,5 мл метилсульфонилхлорида при охлаждении льдом с последующим перемешиванием реакционной смеси при комнатной температуре в течение 1 часа. При комнатной температуре последовательно добавляли 0,74 мл триэтиламина и 0,27 мл метилсульфонилхлорида с последующим перемешиванием реакционной смеси при комнатной температуре в течение 1 часа. Осадок отфильтровывали, промывали тетрагидрофураном и затем концентрировали фильтрат в вакууме. К раствору полученного остатка в 30 мл N,N-диметилформамида добавляли 4,58 г бромида лития при охлаждении льдом с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Полученный этилацетатный раствор последовательно промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от хлороформа до смеси хлороформ/этилацетат = 10/1), получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
(5) Синтез трет-бутил-цис-4-((трет-бутил(дифенил)силил)окси)-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата
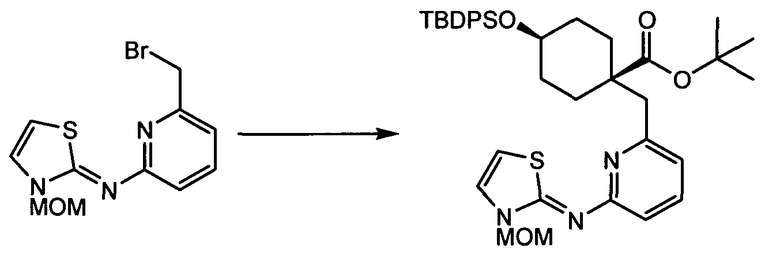
К раствору 3,8 мл диизопропиламина в тетрагидрофуране добавляли 17,3 мл гексанового раствора, содержащего 1,58M н-бутиллития при охлаждении льдом с последующим перемешиванием реакционной смеси в течение 30 минут. После охлаждения до -78°C к раствору добавляли 12 г трет-бутил-4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксилата, полученного по ссылке 1, в 30 мл тетрагидрофурана и перемешивали полученный раствор в течение 2 часов при -78°C. К реакционной смеси добавляли раствор 2,87 г 6-(бромметил)-N-((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)пиридин-2-амина и 7,9 мл гексаметилфосфорамида в 20 мл тетрагидрофурана с последующим постепенным нагреванием реакционной смеси до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Полученный этилацетатный слой промывали насыщенным раствором соли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 10/1-4/1), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
(6) Синтез трет-бутил-цис-4-гидрокси-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата
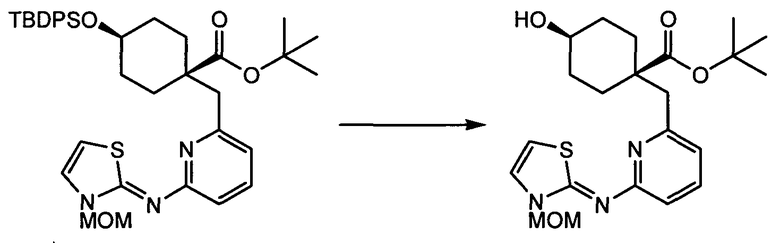
К раствору 5,56 г трет-бутил-цис-4-((трет-бутил(дифенил)силил)окси)-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата в 100 мл тетрагидрофурана добавляли 49,6 мл 1M раствора фторида тетрабутиламмония в тетрагидрофуране при комнатной температуре с последующим перемешиванием реакционной смеси при 60°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры с последующим разбавлением этилацетатом. Полученный раствор последовательно промывали раствором фосфатного буфера с pH 6,8 и насыщенным раствором соли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 1/2), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
(7) Синтез трет-бутил-транс-4-(3-хлор-2-фторфенокси)-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата
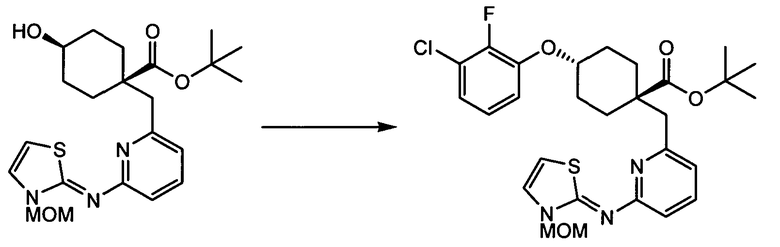
К раствору смеси, состоящей из 4,34 г трет-бутил-цис-4-гидрокси-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата, 2,93 г 3-хлор-2-фторфенола и 5,24 г трифенилфосфина, в 70 мл тетрагидрофурана добавляли 3,94 мл диизопропилазодикарбоксилата при охлаждении льдом с последующим перемешиванием реакционной смеси при комнатной температуре в течение 1 часа. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Полученный этилацетатный слой промывали насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 3/1), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
(8) Синтез гидрохлорида транс-4-(3-хлор-2-фторфенокси)-1-(6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
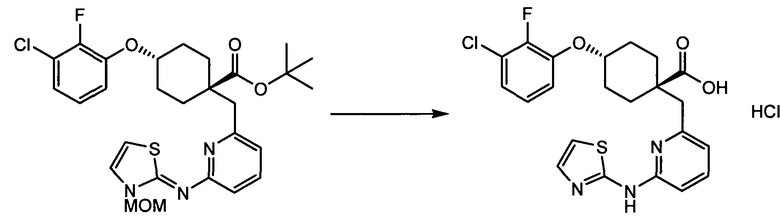
К 3,9 г трет-бутил-транс-4-(3-хлор-2-фторфенокси)-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата добавляли 100 мл 4M раствора хлористого водорода в 1,4-диоксане с последующим перемешиванием реакционной смеси при 90°C в течение 5 часов. После охлаждения реакционной смеси до комнатной температуры к смеси добавляли 100 мл простого трет-бутилметилового эфира. Полученный осадок собирали фильтрованием и промывали простым трет-бутилметиловым эфиром, получая при этом бесцветное твердое вещество.
Полученное бесцветное твердое вещество растворяли в 1,2 л этанола при 80°C. Этанол удаляли дистилляцией, чтобы уменьшить объем раствора приблизительно до одной трети. Полученный раствор охлаждали до комнатной температуры с последующим перемешиванием при комнатной температуре в течение ночи. Полученное твердое вещество собирали фильтрованием и промывали охлажденным этанолом, получая при этом указанное в заголовке соединение в виде бесцветного кристаллического вещества.
1H-ЯМР (ДМСО-d6) δ: 1,60-1,92 (8H, м), 3,03 (2H, c), 4,62 (1H, ушир.c), 6,90 (1H, д, J=7,4 Гц), 7,05-7,22 (5H, м), 7,53 (1H, д, J=4,1 Гц), 7,74 (1H, т, J=7,8 Гц).
Масс: 462,464 (М+1)+
Пример 2
Синтез транс-4-(3-хлор-2-фторфенокси)-1-(6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
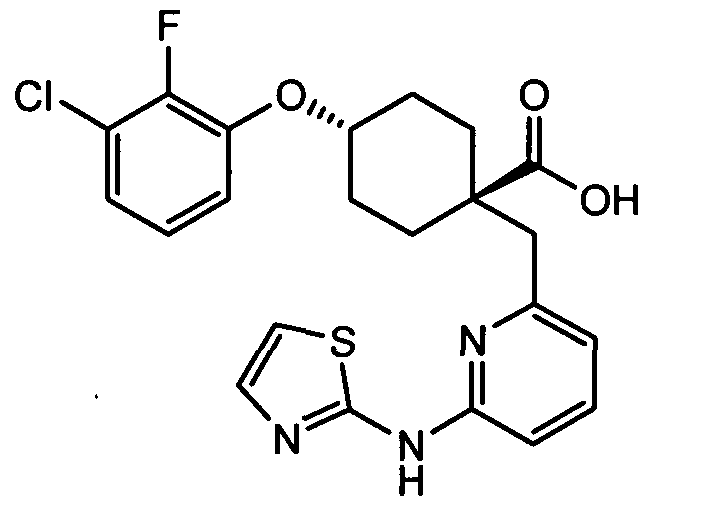
Способ A
К 47,9 мг гидрохлорида транс-4-(3-хлор-2-фторфенокси)-1-(6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, полученного по примеру 1, последовательно добавляли 4 мл воды и 4 мл этанола с последующим перемешиванием реакционной смеси при комнатной температуре в течение 12 часов. Полученный осадок собирали фильтрованием и промывали водой, получая при этом указанное в заголовке соединение в виде бесцветных игольчатых кристаллов (т.пл.: 202-222°C).
1H-ЯМР (ДМСО-d6) δ: 1,60-1,92 (8H, м), 2,98 (2H, c), 4,61 (1H, ушир.c), 6,71 (1H, д, J=7,2 Гц), 6,90 (1Н, д, J=8,2 Гц), 6,98 (1H, д, J=3,5 Гц), 7,10-7,22 (3H, м), 7,38 (1H, д, J=3,5 Гц), 7,60 (1H, т, J=7,6 Гц).
Масс: 462,464 (М+1)+
Способ B
К 460 мг гидрохлорида транс-4-(3-хлор-2-фторфенокси)-1-(6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, полученного по примеру 1, последовательно добавляли 40 мл воды и 40 мл этанола с последующим перемешиванием реакционной смеси при комнатной температуре в течение 4 дней. Полученный осадок собирали фильтрованием и промывали водой, получая при этом указанное в заголовке соединение в виде бесцветных пластинок (т.пл.: 224-242°C).
1H-ЯМР (ДМСО-d6) δ: 1,60-1,92 (8H, м), 2,98 (2H, c), 4,61 (1H, ушир.c), 6,71 (1H, д, J=7,2 Гц), 6,90 (1H, д, J=8,2 Гц), 6,98 (1H, д, J=3,5 Гц), 7,10-7,22 (3H, м), 7,38 (1H, д, J=3,5 Гц), 7,60 (1H, т, J=7,6 Гц).
Масс: 462,464 (М+1)+
Пример 3
Синтез гидрохлорида транс-4-(2,3-дифторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
Указанное в заголовке соединение получали в виде белого твердого вещества по примеру 1, применяя 2,3-дифторфенол вместо 3-хлор-2-фторфенола, который применялся на стадии 1(7) примера 1.
1H-ЯМР (ДМСО-d6) δ: 1,60-1,92 (8H, м), 3,02 (2H, c), 4,62 (1H, ушир.c), 6,84 (1H, д, J=8,4 Гц), 6,97-7,15 (5H, м), 7,49 (1H, д, J=3,7 Гц), 7,70 (1H, т, J=7,8 Гц).
Масс: 446 (М+1)+
Пример 4
Синтез гидрохлорида транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
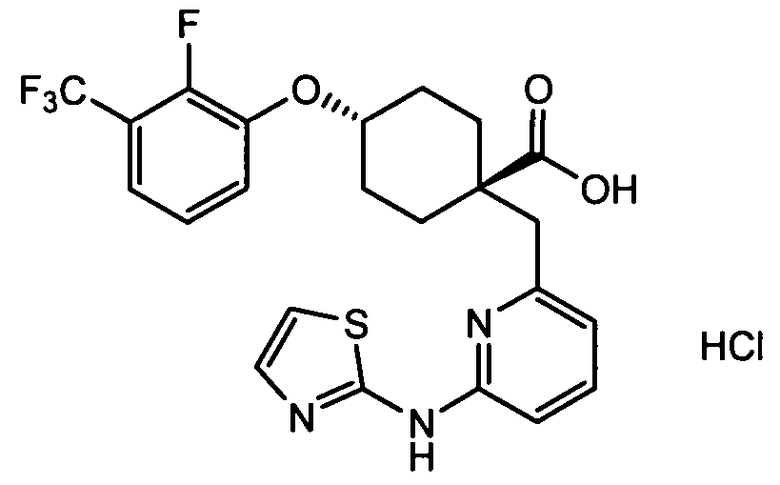
Указанное в заголовке соединение получали в виде белого твердого вещества по примеру 1, применяя 2-фтор-3-(трифторметил)фенол вместо 3-хлор-2-фторфенола, который применялся на стадии 1(7) примера 1.
1H-ЯМР (ДМСО-d6) δ: 1,62-1,95 (8H, м), 3,03 (2H, c), 4,68 (1H, ушир.c), 6,89 (1H, д, J=7,2 Гц), 7,07 (1H, д, J=8,4 Гц), 7,15 (1H, д, J=3,7 Гц), 7,28-7,35 (2H, м), 7,52 (1H, д, J=3,7 Гц), 7,56 (1H, т, J=6,8 Гц), 7,73 (1H, т, J=7,8 Гц).
масс: 496 (М+1)+
Пример 5
Синтез гидрохлорида транс-4-(2,3-дихлорфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
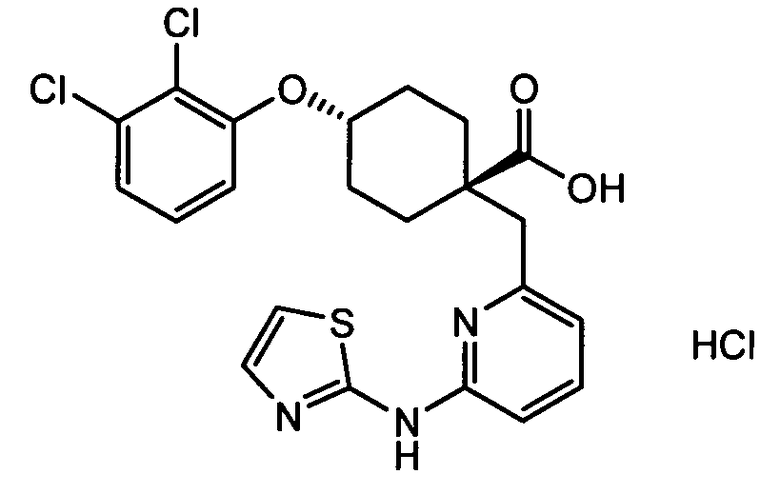
Указанное в заголовке соединение получали в виде белого твердого вещества по примеру 1, применяя 2,3-дихлорфенол вместо 3-хлор-2-фторфенола, который применялся на стадии 1(7) примера 1.
1H-ЯМР (ДМСО-d6) δ: 1,51-1,66 (2H, м), 1,69-1,89 (6H, м), 2,97 (2H, c), 4,72 (1H, ушир.c), 6,75-6,85 (1H, м), 6,95-7,10 (2H, м), 7,10-7,16 (2H, м), 7,25 (1H, т, J=8,2 Гц), 7,42-7,49 (1H, м), 7,62-7,72 (1H, м).
масс: 478,480 (М+1)+
Пример 6
Синтез транс-4-(3-хлор-2-фторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоксамида
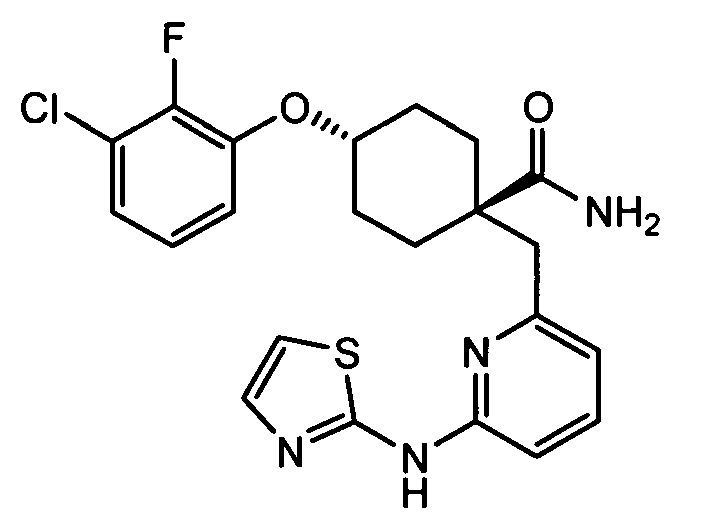
К раствору 20 мг гидрохлорида транс-4-(3-хлор-2-фторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, полученного по примеру 1, в 3 мл хлороформа при комнатной температуре последовательно добавляли 21 мг хлорида аммония, 0,056 мл триэтиламина, 31 мг гидрата гидроксибензотриазола и 38 мг гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. После добавления к реакционной смеси насыщенного водного раствора бикарбоната натрия смесь экстрагировали этилацетатом. Полученный этилацетатный раствор сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью препаративной тонкослойной хроматографии (KieselgelTM60F254, Art5744 (Merck), хлороформ/метанол = 10/1), получая при этом указанное в заголовке соединение в виде белого твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,63-1,93 (8H, м), 2,92 (2H, c), 4,55 (1H, ушир. c), 6,68 (1H, д, J=7,4 Гц), 6,85 (1H, д, J=8,4 Гц), 6,94 (2H, д, J=3,1 Гц), 7,05-7,20 (3H, м), 7,25 (1H, c), 7,35 (1H, д, J=3,5 Гц), 7,55 (1H, т, J=7,8 Гц), 11,13 (1H, c).
масс: 461,463 (М+1)+
Пример 7
Синтез трифторацетата транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
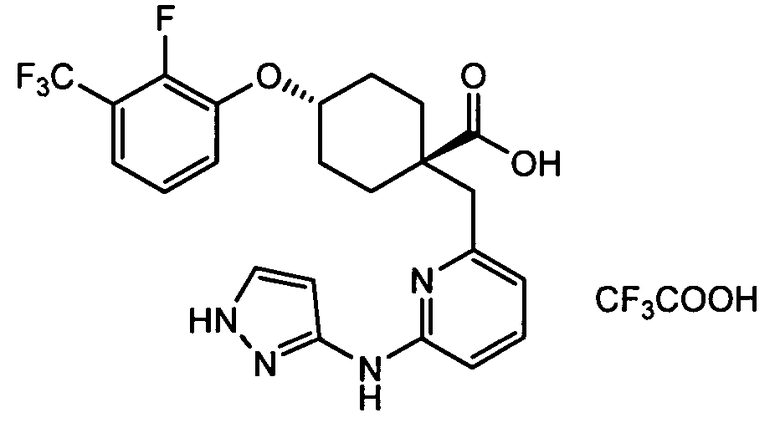
(1) Синтез 2-бром-6-(бромметил)пиридина

К раствору 498 мг (6-бромпиридин-2-ил)метанола в 6 мл N,N- диметилформамида при охлаждении льдом последовательно добавляли 1,15 мл диизопропилэтиламина и раствор 695 мг метансульфонового ангидрида в 2 мл N,N-диметилформамида с последующим перемешиванием реакционной смеси при комнатной температуре в течение 20 минут. Затем к раствору добавляли 693 мг бромида лития с последующим перемешиванием реакционной смеси при комнатной температуре в течение 1 часа. После добавления к реакционной смеси насыщенного водного раствора бикарбоната натрия смесь экстрагировали этилацетатом. Полученный этилацетатный раствор сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан/этилацетат = 20/1-3/2), получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
(2) Синтез трет-бутил-цис-1-((6-бромпиридин-2-ил)метил)-4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксилата
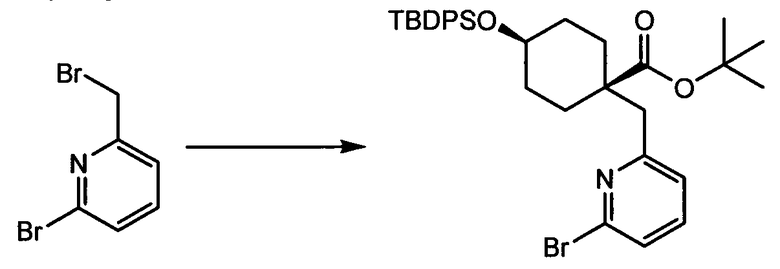
К раствору 0,82 мл диизопропиламина в 20 мл тетрагидрофурана при охлаждении льдом добавляли 3,7 мл гексанового раствора, содержащего 1,58M н-бутиллития, с последующим перемешиванием реакционной смеси в течение 30 минут. После охлаждения реакционной смеси до -78°C к раствору добавляли раствор 2,67 г трет-бутил-4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксилата, полученного по ссылке 1, в 10 мл тетрагидрофурана и полученный раствор перемешивали в течение 1 часа при -78°C. К реакционной смеси добавляли раствор 980 мг 2-бром-6-(бромметил)пиридина и 2,7 мл гексаметилфосфорамида в 5 мл тетрагидрофурана с последующим постепенным нагреванием реакционной смеси до комнатной температуры и затем перемешивали реакционную смесь при комнатной температуре в течение ночи. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией хлороформом. Полученный хлороформный раствор сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан/этилацетат = 100/1-9/1), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
(3) Синтез трет-бутил-цис-1-((6-бромпиридин-2-ил)метил)-4-гидроксициклогексанкарбоксилата
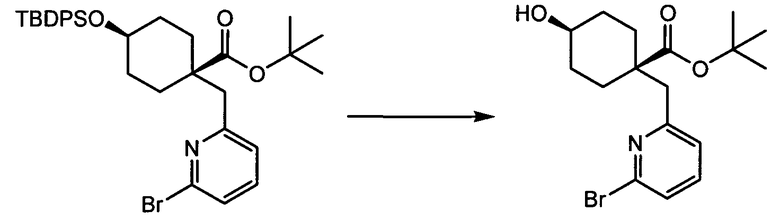
К раствору 1,6 г трет-бутил-цис-1-((6-бромпиридин-2-ил)метил)-4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксилата в 30 мл тетрагидрофурана при комнатной температуре добавляли 16 мл тетрагидрофуранового раствора, содержащего 1M фторида тетрабутиламмония, с последующим перемешиванием реакционной смеси при 60°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры с последующим разбавлением хлороформом. Полученный раствор последовательно промывали раствором фосфатного буфера с pH 6,8 и насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от смеси гексан/этилацетат = 8/1 до этилацетата), получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
(4) Синтез трет-бутил-транс-1-((6-бромпиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоксилата
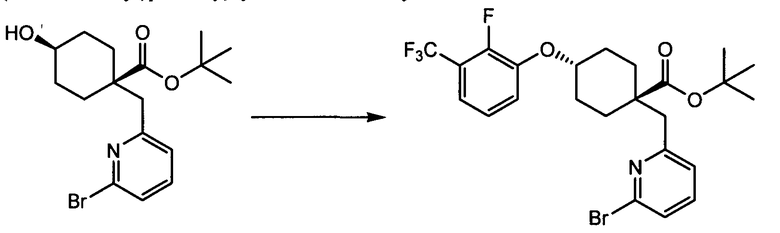
К раствору 150 мг трет-бутил-цис-1-((6-бромпиридин-2-ил)метил)-4-гидроксициклогексанкарбоксилата, 219 мг 2-фтор-3-(трифторметил)фенола и 320 мг трифенилфосфина в 2,5 мл тетрагидрофурана при охлаждении льдом добавляли 0,24 мл диизопропилазодикарбоксилата с последующим перемешиванием реакционной смеси при комнатной температуре в течение 15 минут. Реакционную смесь концентрировали в вакууме и очищали полученный остаток с помощью колоночной хроматографии на силикагеле (элюент: смесь гексан/этилацетат = 50/1-4/1), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
(5) Синтез трет-бутил-транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоксилата
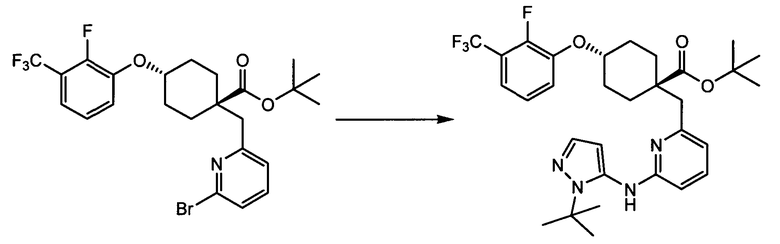
Смесь, состоящую из 140 мг трет-бутил-транс-1-((6-бромпиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоксилата, 102 мг 1-трет-бутил-1H-пиразол-5-амин-п-толуолсульфоната, полученного по ссылке 4, 25,4 мг 9,9-диметил-4,5-бис(дифенилфосфино)ксантена, 21,6 мг комплекса трис(дибензилиденацетон)дипалладий(0)-хлороформа, 139 мг фосфата калия и 4 мл 1,4-диоксана перемешивали при 100°C в течение ночи с последующим охлаждением до комнатной температуры. Нерастворимое вещество отфильтровывали с помощью целита и промывали этилацетатом. Полученный этилацетатный раствор промывали водой, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан/этилацетат = 20/1-3/2), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
(6) Синтез трифторацетата транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
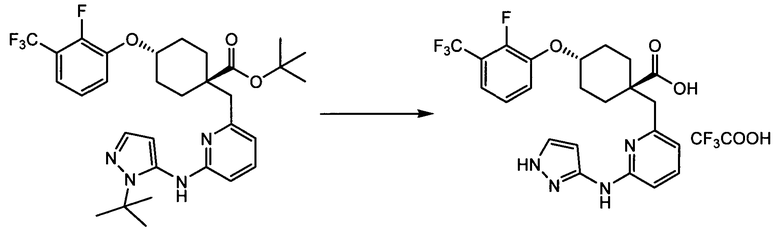
Раствор 88,8 мг трет-бутил-транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоксилата в 1 мл муравьиной кислоты перемешивали при 110°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры с последующим концентрированием в вакууме. Полученный остаток очищали с помощью препаративной жидкостной хроматографии с обращенной фазой с последующим концентрированием полученной фракции в вакууме, получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
1H-ЯМР (CD3OD) δ: 1,82-2,17 (8H, м), 3,24 (2H, c), 4,70 (1H, c), 6,18 (1H, д, J=2,8 Гц), 7,05 (1H, д, J=7,2 Гц), 7,19-7,31 (3H, м), 7,45 (1H, дт, J=8,4, 2,8 Гц), 7,78 (1H, д, J=2,4 Гц), 8,06 (1H, дд, J=8,8, 7,2 Гц).
масс: 479 (М+1)+
Пример 8
Синтез трифторацетата транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
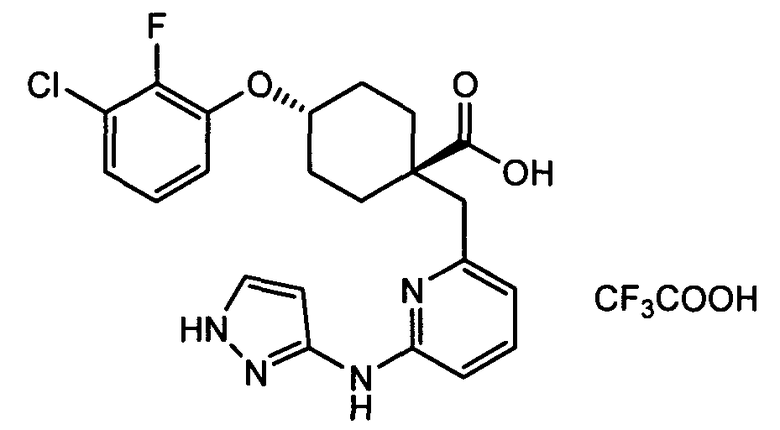
Указанное в заголовке соединение получали в виде белого твердого вещества по примеру 7, применяя 3-хлор-2-фторфенол вместо 2-фтор-3-(трифторметил)фенола, который применялся на стадии 7(4) примера 7.
1H-ЯМР (CD3OD) δ: 1,79-2,15 (8H, м), 3,20 (2H, c), 4,62 (1H, c), 6,18 (1H, д, J=2,4 Гц), 7,01-7,15 (4H, м), 7,20 (1H, д, J=8,8 Гц), 7,79 (1H, д, J=2,4 Гц), 8,04 (1H, дд, J=8,8, 7,2 Гц).
масс: 445,447 (М+1)+
Пример 9
Синтез транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоксамида
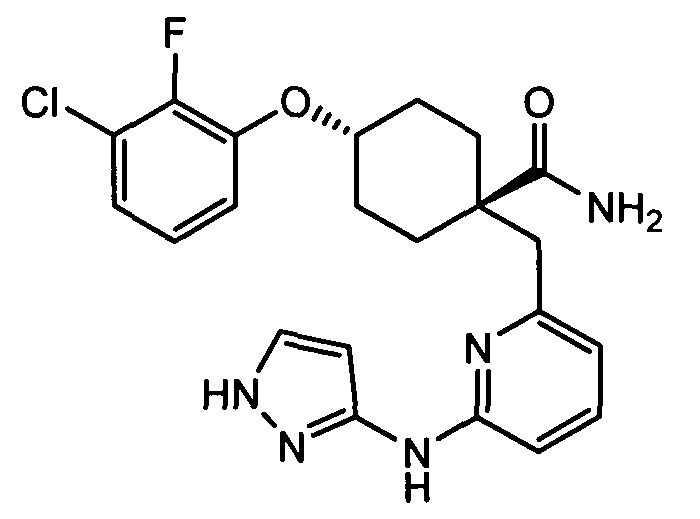
К раствору 32,1 мг трифторацетата транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, полученного по примеру 8, в 1 мл диметилсульфоксида при комнатной температуре последовательно добавляли 7,5 мг хлорида аммония, 0,038 мл триэтиламина, 20,2 мг гидрата гидроксибензотриазола и 24,6 мг гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. Реакционную смесь очищали с помощью препаративной жидкостной хроматографии с обращенной фазой с последующим применением препаративной тонкослойной хроматографии (NH-PLC05 (FUJI SILYSIA CHEMICAL), хлороформ/метанол = 20/1), получая при этом указанное в заголовке соединение в виде белого твердого вещества.
1H-ЯМР (CD3OD) δ: 1,82-2,00 (8H, м),3,04 (2H, c), 4,55 (1H, c), 5,75 (1H, ушир.c), 6,60-6,80 (2H, м), 7,00-7,10 (3H, м), 7,44 (1H, ушир.c), 7,50 (1H, т, J=8,0 Гц).
масс: 444,446 (М+1)+
Пример 10
Синтез трифторацетата транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексанкарбоновой кислоты
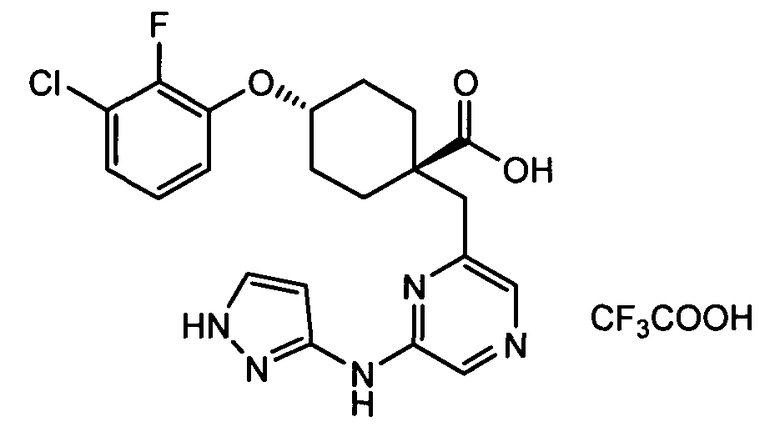
(1) Синтез N-(1-трет-бутил-1H-пиразол-5-ил)-6-хлорпиразин-2-амина
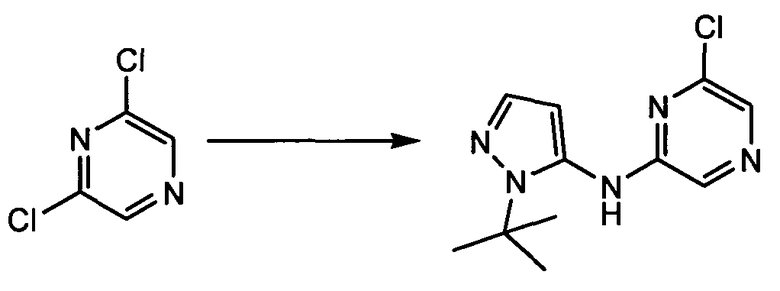
Смесь 60,6 г 2,6-дихлорпиразина, 62,2 г 1-трет-бутил-1H-пиразол-5-амина, полученного по ссылке 3, 23,5 г 9,9-диметил-4,5-бис(дифенилфосфино)ксантена, 21,0 г комплекса трис(дибензилиденацетон)дипалладий(0)-хлороформа, 172,6 г фосфата калия и 1,17 л 1,4-диоксана перемешивали при 100°C в течение ночи с последующим охлаждением до комнатной температуры. Нерастворимое вещество отфильтровывали с помощью целита и промывали этилацетатом. Полученный этилацетатный раствор промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан/этилацетат = 4/1-2/1), получая при этом указанное в заголовке соединение в виде желтого твердого вещества.
(2) Синтез N-(1-трет-бутил-1H-пиразол-5-ил)-6-винилпиразин-2-амина
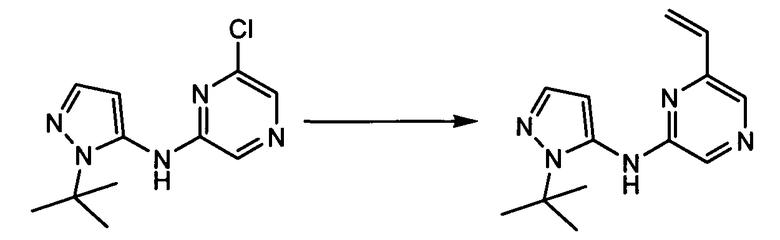
Смесь, состоящую из 65,04 г N-(1-трет-бутил-1H-пиразол-5-ил)-6-хлорпиразин-2-амина, 41,6 г винилтрифторбората калия, 4,22 г комплекса (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладий(II) в дихлорметане, 72 мл триэтиламина и 685 мл 1-пропанола, перемешивали при 110°C в течение ночи с последующим охлаждением до комнатной температуры и концентрировали в вакууме. Полученный остаток разбавляли этилацетатом и отфильтровывали нерастворимое вещество с помощью целита. Полученный этилацетатный раствор промывали водой, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Из полученного остатка готовили суспензию в 100 мл этилацетата и добавляли к смеси 400 мл простого диизопропилового эфира. Полученный осадок собирали, получая при этом указанное в заголовке соединение в виде бледно-коричневого твердого вещества.
(3) Синтез 6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-карбальдегида
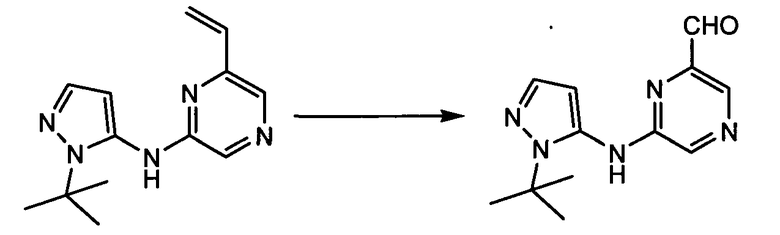
К раствору 56,36 г N-(1-трет-бутил-1H-пиразол-5-ил)-6-винилпиразин-2-амина в 570 мл ацетонитрила при комнатной температуре последовательно добавляли 48,9 г N-оксида N-метилморфолина и 215 мл 0,1M водного раствора тетраоксида осмия с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. После добавления к реакционной смеси 73 г сульфита натрия и 580 мл воды смесь экстрагировали этилацетатом. Полученный этилацетатный раствор промывали насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме, получая при этом сырой (неочищенный) продукт.
К раствору полученного остатка в 572 мл ацетонитрила и 858 мл воды при охлаждении льдом добавляли 62,8 г периодата натрия с последующим перемешиванием реакционной смеси при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Полученный этилацетатный раствор промывали насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от хлороформа до смеси хлороформ/метанол = 20/1), получая при этом указанное в заголовке соединение в виде темно-коричневого масла.
(4) Синтез (6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-ил)метанола
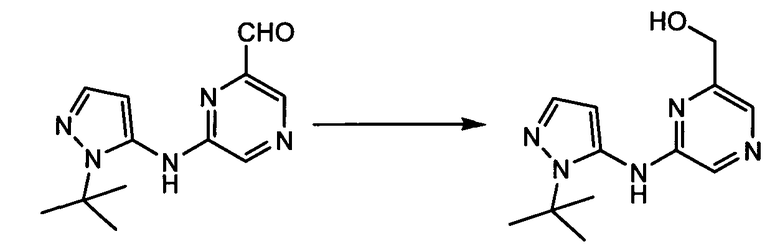
К раствору 14,99 г 6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-карбальдегида в 235 мл этанола при охлаждении льдом добавляли 2,31 г борогидрида натрия с последующим перемешиванием реакционной смеси в течение 1 часа. После медленного добавления к реакционной смеси при охлаждении льдом 61 мл 1M раствора хлористоводородной кислоты этанол отгоняли в вакууме. Полученный остаток разбавляли водой и экстрагировали хлороформом. Полученный хлороформный раствор промывали насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от хлороформа до смеси хлороформ/метанол = 20/1), получая при этом указанное в заголовке соединение в виде коричневого твердого вещества.
(5) Синтез N-(1-трет-бутил-1H-пиразол-5-ил)-6-(хлорметил)пиразин-2-амина
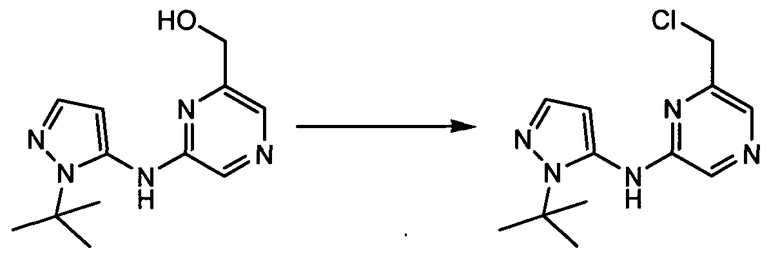
К раствору 507,3 мг (6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-ил)метанола в 6,8 мл хлороформа при охлаждении льдом последовательно добавляли 1,08 мл диизопропилэтиламина и 0,24 мл метилсульфонилхлорида с последующим перемешиванием реакционной смеси при комнатной температуре в течение 1,5 часов. К реакционной смеси последовательно добавляли 442,9 мг хлорида лития и 6,8 мл N,N-диметилформамида с последующим перемешиванием реакционной смеси при комнатной температуре в течение 2 часов. После разбавления реакционной смеси этилацетатом этилацетатный раствор последовательно промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан/этилацетат = 20/1-1/4), получая при этом указанное в заголовке соединение в виде желтого твердого вещества.
(6) Синтез трет-бутил-цис-4-((трет-бутил(дифенил)силил)окси)-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-ил)метил)циклогексанкарбоксилата
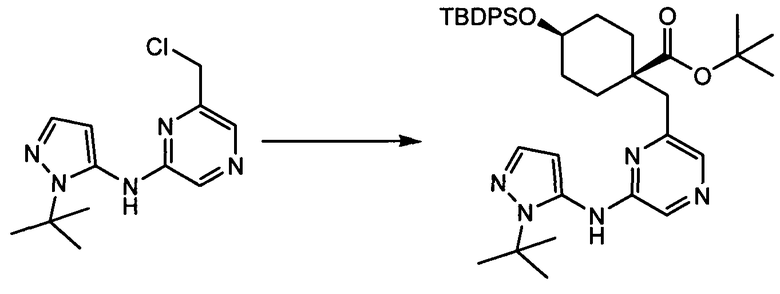
К раствору 0,15 мл диизопропиламина в 4,2 мл тетрагидрофурана при охлаждении льдом добавляли 0,7 мл гексанового раствора, содержащего 1,58 M н-бутиллития с последующим перемешиванием реакционной смеси в течение 30 минут. После охлаждения до -78°C к раствору добавляли раствор 488 мг трет-бутил-4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксилата, полученного по ссылке 1, в 2 мл тетрагидрофурана. Полученный раствор перемешивали в течение 1 часа при -78°C. К реакционной смеси добавляли раствор 115 мг N-(1-трет-бутил-1H-пиразол-5-ил)-6-(хлорметил)пиразин-2-амина и 0,5 мл гексаметилфосфорамида в 1,5 мл тетрагидрофурана с последующим постепенным нагреванием реакционной смеси до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией хлороформом. Полученный хлороформный раствор сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 1/4), получая при этом указанное в заголовке соединение в виде желтого масла.
(7) Синтез трет-бутил-цис-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-ил)метил)-4-гидроксициклогексанкарбоксилата
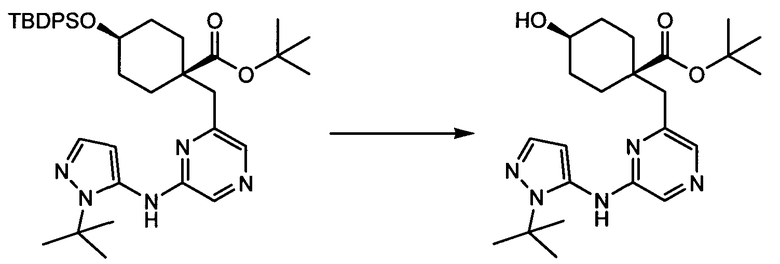
К раствору 60,5 мг трет-бутил-цис-4-((трет-бутил(дифенил)силил)окси)-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-ил)метил)циклогексанкарбоксилата в 1 мл тетрагидрофурана при комнатной температуре добавляли 0,36 мл тетрагидрофуранового раствора, содержащего 1M фторида тетрабутиламмония, с последующим перемешиванием реакционной смеси при 60°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры с последующим разбавлением хлороформом. Полученный раствор последовательно промывали раствором фосфатного буфера с pH 6,8 и насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: хлороформ/метанол = 50/1-4/1), получая при этом указанное в заголовке соединение в виде желтого масла.
(8) Синтез трет-бутил-транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-ил)метил)-4-(3-хлор-2-фторфенокси)циклогексанкарбоксилата
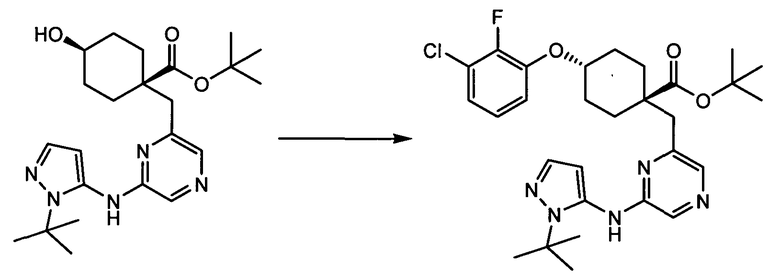
К раствору 28,9 мг трет-бутил-цис-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-ил)метил)-4-гидроксициклогексанкарбоксилата, 30 мг 3-хлор-2-фторфенола и 52,2 мг трифенилфосфина в 0,5 мл тетрагидрофурана при охлаждении льдом добавляли 0,04 мл диизопропилазодикарбоксилата с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. Реакционную смесь концентрировали в вакууме и полученный остаток очищали с помощью препаративной тонкослойной хроматографии (NH-PLC05 (FUJI SILYSIA CHEMICAL), гексан/этилацетат = 1/1), получая при этом указанное в заголовке соединение в виде желтого масла.
(9) Синтез трифторацетата транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексанкарбоновой кислоты
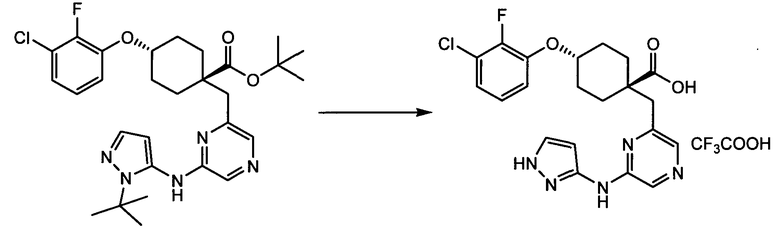
Раствор 16,2 мг трет-бутил-транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-ил)метил)-4-(3-хлор-2-фторфенокси)циклогексанкарбоксилата в 0,5 мл муравьиной кислоты перемешивали при 100°C в течение 1,5 часов. Реакционную смесь охлаждали до комнатной температуры с последующим концентрированием в вакууме. Полученный остаток очищали с помощью препаративной жидкостной хроматографии с обращенной фазой с последующим концентрированием полученной фракции в вакууме, получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
1H-ЯМР (CD3OD) δ: 1,75-2,04 (8H, м), 3,05 (2H, c), 4,53-4,59 (1H, м), 6,39 (1H, д, J=2,4 Гц), 6,98-7,09 (3H, м), 7,62 (1H, д, J=2,4 Гц), 7,84 (1H, c), 8,17 (1H, c).
масс: 446,448 (М+1)+
Пример 11
Синтез транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексанкарбоксамида
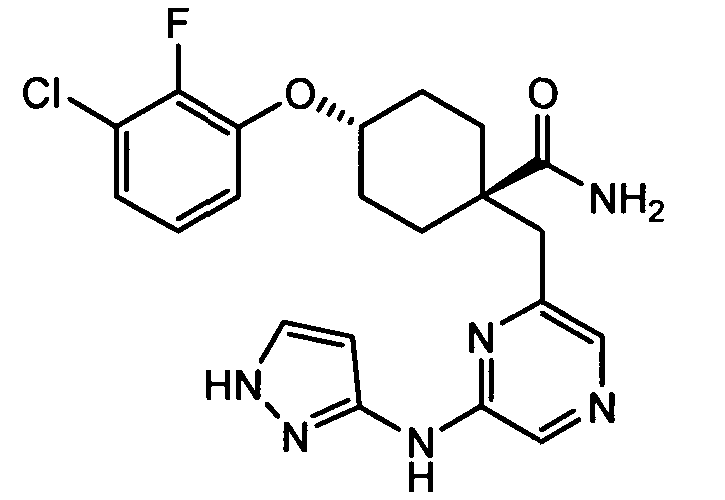
Указанное в заголовке соединение получали в виде бледно-желтого твердого вещества по примеру 9, применяя трифторацетат транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексанкарбоновой кислоты, который получен по примеру 10, вместо трифторацетата транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, который применялся в примере 9.
1H-ЯМР (ДМСО-d6) δ: 1,62-1,93 (8H, м), 2,82 (2H, c), 4,54 (1H, ушир.c), 6,50 (1H, ушир.c), 6,93 (1H, ушир.c), 7,06-7,22 (3H, м), 7,26 (1H, ушир.c), 7,55 (1H, ушир.c), 7,67 (1H, c), 8,28 (1H, ушир.c), 9,60 (1H, ушир.c).
масс: 445,447 (М+1)+
Пример 12
Синтез транс-4-(3-хлор-2-фторфенокси)-N-метокси-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоксамида

Указанное в заголовке соединение получали в виде бледно-желтого твердого вещества по примеру 6, применяя гидрохлорид О-метилгидроксиламина вместо хлорида аммония, который применялся в примере 6.
1H-ЯМР (CDCl3) δ: 1,80-2,15 (8H, м), 3,07 (2H, c), 3,64 (3H, c), 4,46 (1H, ушир.c), 6,60 (1H, c), 6,70-7,03 (5H, м), 7,31 (1H, c), 7,47 (1H, т, J=7,6 Гц), 9,39 (1H, ушир.c).
масс: 491,493 (М+1)+
Пример 13
Синтез 5-(транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-она
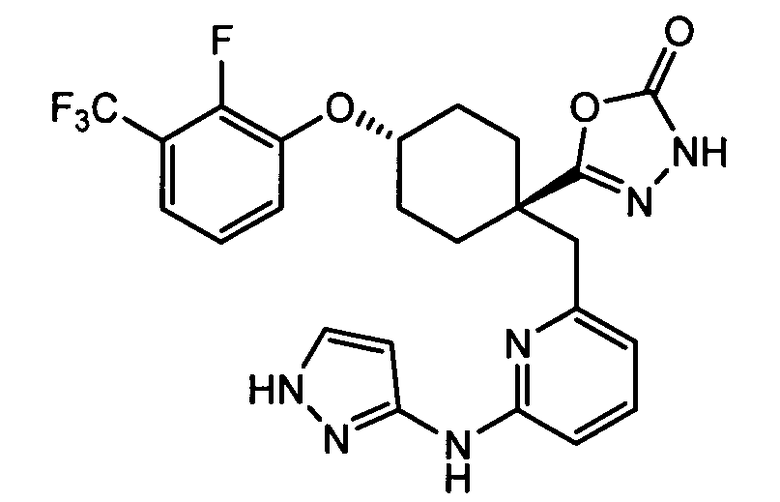
(1) Синтез трифторацетата транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоновой кислоты
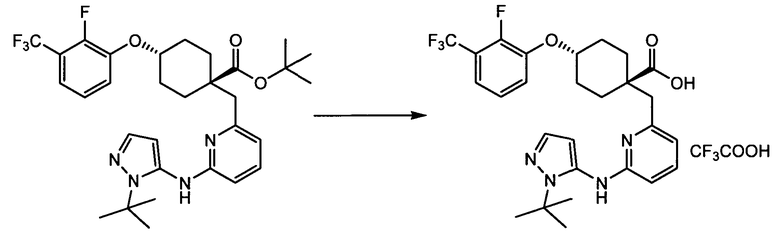
К раствору 2,51 г трет-бутил-транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоксилата, полученного на стадии 7(5) примера 7, в 39 мл хлороформа при 0°C добавляли 19 мл трифторуксусной кислоты с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. Полученный раствор концентрировали в вакууме, получая при этом указанное в заголовке соединение в виде желтого масла.
(2) Синтез трет-бутил 2-((транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексил)карбонил)гидразинкарбоксилата
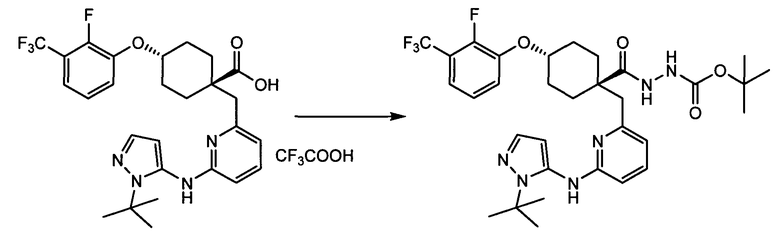
К раствору 3,2 г трифторацетата транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоновой кислоты в 14,1 мл хлороформа при комнатной температуре последовательно добавляли 5,62 г трет-бутилкарбазата, 3,27 г гидрата 1-гидроксибензотриазола и 4,13 г гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида с последующим перемешиванием при комнатной температуре в течение 8 часов. После добавления к реакционной смеси этилацетата органический слой последовательно промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 1/4), получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
(3) Синтез транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбогидразида
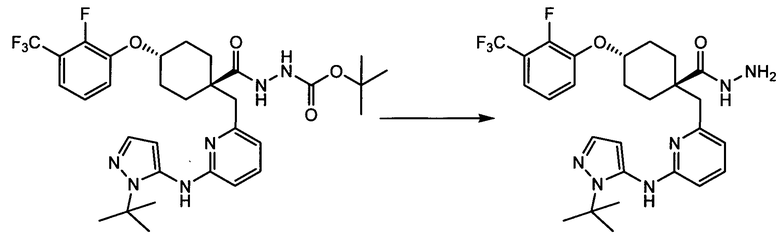
К раствору 2,93 г трет-бутил-2-((транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексил)карбонил)гидразинкарбоксилата в 30 мл хлороформа при комнатной температуре добавляли 15 мл трифторуксусной кислоты с последующим перемешиванием при комнатной температуре в течение 1 часа. После концентрирования реакционной смеси в вакууме полученный остаток растворяли в хлороформе. Хлороформный раствор последовательно промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от хлороформа до смеси хлороформ/метанол = 10/1), получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
(4) Синтез 5-(транс-1-((6-((1-трет-бутил-1Н-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексил)-1,3,4-оксадиазол-2(3H)-она

К раствору 1,9 г транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбогидразида в 35 мл тетрагидрофурана при комнатной температуре добавляли 3,05 мл N,N'-диизопропилэтиламина и 1,70 г 1,1'-карбонилдиимидазола. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов с последующим концентрированием полученного раствора в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от хлороформа до смеси хлороформ/метанол = 10/1), получая при этом указанное в заголовке соединение в виде бледно-желтого твердого вещества.
(5) Синтез 5-(транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-она
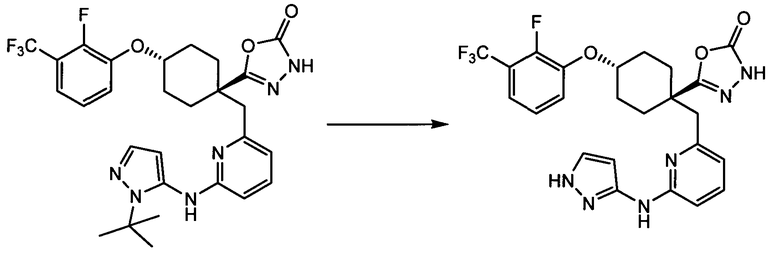
Раствор 2,11 г 5-(транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексил)-1,3,4-оксадиазол-2(3H)-она в 37 мл муравьиной кислоты перемешивали при 95°C в течение 1,5 часов. После концентрирования реакционной смеси в вакууме полученный остаток подщелачивали насыщенным раствором бикарбоната натрия и экстрагировали хлороформом. Хлороформный раствор последовательно промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от хлороформа до смеси хлороформ/метанол = 4/1), получая при этом указанное в заголовке соединение в виде белого твердого вещества.
1H-ЯМР (CDCl3) δ: 1,63-1,80 (2H, м), 1,89-2,07 (6H, м), 3,02 (2H, c), 4,47-4,53 (1H, м), 6,23 (1H, ушир.c), 6,50 (1H, д, J=8,0 Гц), 6,61 (1H, д, J=7,2 Гц), 7,06 (1H, ушир.c), 7,10-7,22 (3H, м), 7,35-7,42 (2H, м).
масс: 519 (М+1)+
Пример 14
Синтез 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-она

(1) Синтез транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(3-хлор-2-фторфенокси)циклогексанкарбогидразида
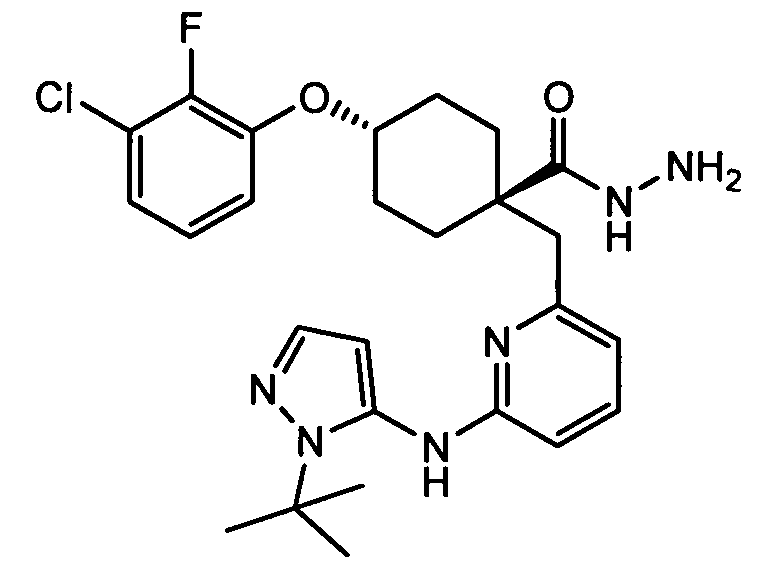
Указанное в заголовке соединение получали в виде бледно-желтого масла таким же способом, как на стадиях 7(4),(5) примера 7 и 13(1)-13(3) примера 13, применяя 3-хлор-2-фторфенол вместо 2-фтор-3-(трифторметил)фенола, который применялся в примере 7(4).
(2) Синтез 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-она
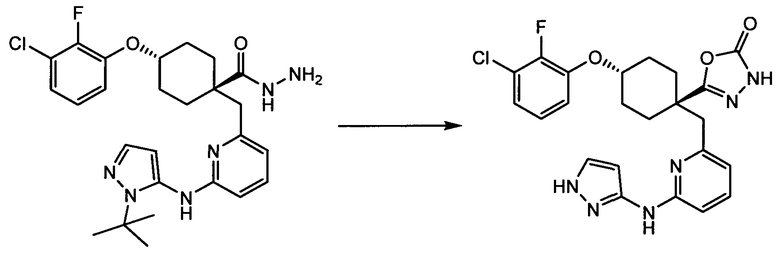
Указанное в заголовке соединение получали в виде твердого белого вещества таким же способом, как на стадиях 13(4) и 13(5) примера 13, применяя транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(3-хлор-2-фторфенокси)циклогексанкарбогидразид вместо транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбогидразида, который применялся в примере 13(4).
1H-ЯМР (ДМСО-d6) δ: 1,65-2,00 (8H, м), 2,97 (2H, c), 4,63 (1H, ушир.c), 6,30-6,45 (2H, м), 6,95-7,30 (4H, м), 7,46 (1H, т, J=8,0 Гц), 7,48-7,56 (1H, м), 9,15 (1H, c), 12,02 (1H, c), 12,07 (1H, ушир.c).
масс: 485,487 (М+1)+
Пример 15
Синтез 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-тиона
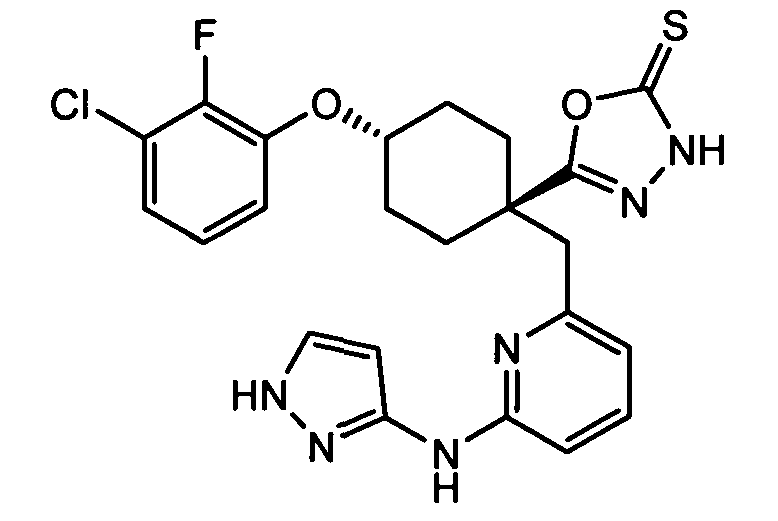
(1) Синтез 5-(транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(3-хлор-2-фторфенокси)циклогексил)-1,3,4-оксадиазол-2(3H)-тиона
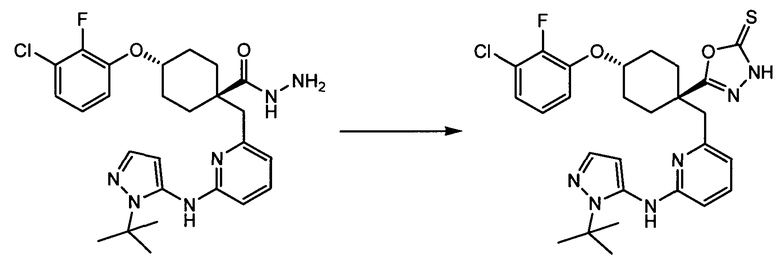
К раствору 97 мг транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(3-хлор-2-фторфенокси)циклогексанкарбогидразида, полученного на стадии примера 14(1), в 3 мл этанола при комнатной температуре добавляли 0,078 мл сероуглерода и 0,432 мл этанольного раствора, содержащего 0,87M гидроксида калия. Реакционную смесь перемешивали при 80°C в течение 3 часов, подкисляли 2M раствором хлористоводородной кислоты и экстрагировали этилацетатом. Этилацетатный раствор сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 2/3), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
(2) Синтез 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1Н-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-тиона
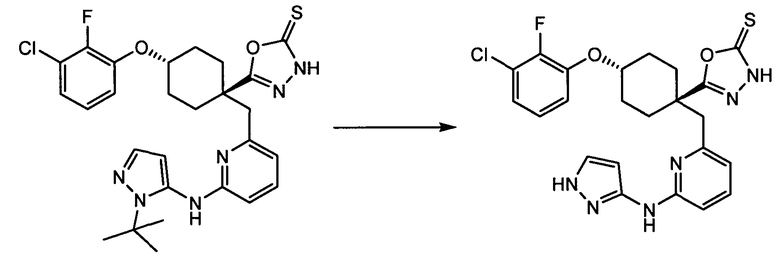
Указанное в заголовке соединение получали в виде твердого бледно-желтого вещества таким же способом, как на стадии 13(5) примера 13, применяя 5-(транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(3-хлор-2-фторфенокси)циклогексил)-1,3,4-оксадиазол-2(3H)-тион вместо 5-(транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексил)-1,3,4-оксадиазол-2(3H)-она, который применялся в примере 13(5).
1H-ЯМР (ДМСО-d6) δ: 1,61-1,74 (2H, м), l,85-2,01 (6H, м), 2,96 (2H, c), 4,56 (1H, ушир.c), 6,24 (1H, c), 6,33 (1H, д, J=7,2 Гц), 6,90-7,00 (1H, м), 7,09-7,24 (3H, м), 7,37 (1H, т, J=7,6 Гц), 7,48 (1H, c), 9,01 (1H, ушир.c).
масс: 501,503 (М+1)+
Пример 16
Синтез 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-3-метил-1,3,4-оксадиазол-2(3Н)-она
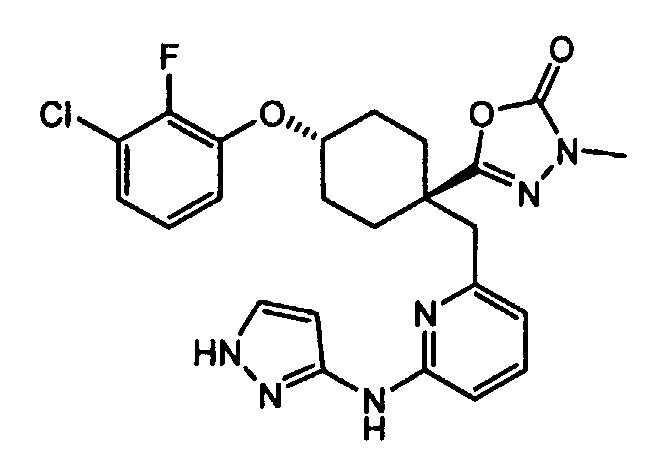
Указанное в заголовке соединение получали по примеру 14 в виде твердого бледно-желтого вещества, применяя трет-бутил-1-метилгидразинкарбоксилат вместо трет-бутилкарбазата, который применялся в примере 14.
1H-ЯМР (CDCl3) δ: 1,58-1,75 (2Н, м), 1,96-2,05 (6Н, м), 2,99 (2Н, с), 3,20 (3Н, с), 4,46 (1Н, ушир.с), 6,09 (1Н, с), 6,55 (1Н, д, J=6,8 Гц), 6,77 (1Н, д, J=8,0 Гц), 6,80-7,20 (4Н, м), 7,42 (1Н, т, J=7,6 Гц), 7,46 (1Н, с).
масс: 499,501 (M+1)+
Пример 17
Синтез 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1H-пиразол-3-иламино)пиразин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3Н)-она
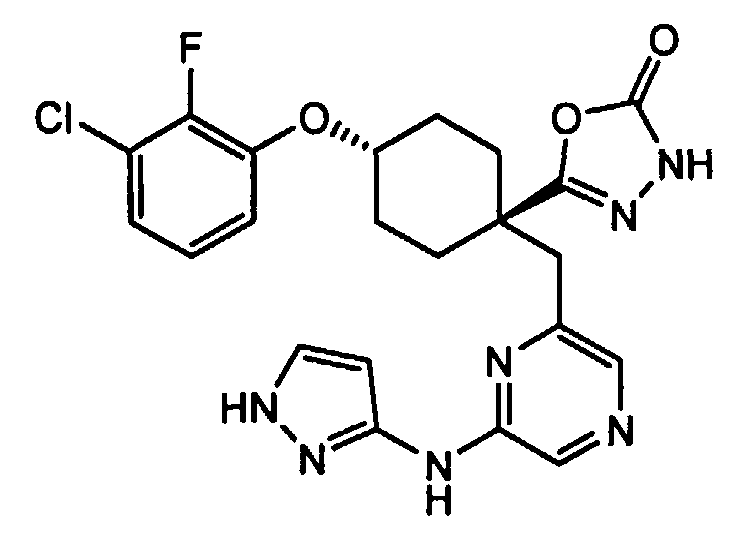
Указанное в заголовке соединение получали по примеру 13 в виде твердого белого вещества, применяя трет-бутил-транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиразин-2-ил)метил)-4-(3-хлор-2-фторфенокси)циклогексанкарбоксилат, который получен на стадии 10(8) примера 10.
1H-ЯМР (ДМСО-d6) δ: 1,60-1,95 (8H, м), 2,92 (2H, c), 4,57 (1Н, ушир.c), 6,38 (1Н, c), 7,10-7,25 (3H, м), 7,52 (1Н, c), 7,62 (1Н, c), 8,27 (1Н, c), 9,66 (1Н, c), 11,98 (1Н, c), 12,16 (1H, c).
масс: 486,488 (М+1)+
Пример 18
Синтез трифторацетата транс-4-((2,3-дихлорфенил)тио)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
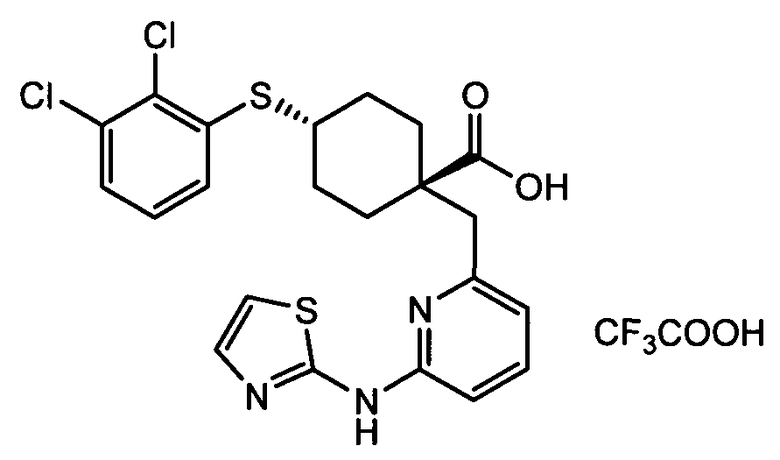
(1) Синтез трет-бутил-транс-4-((2,3-дихлорфенил)тио)-1-(((6-((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата
К раствору 107,4 мг трет-бутил-цис-4-гидрокси-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата, полученного на стадии 1(6) примера 1, в 0,83 мл тетрагидрофурана при 0°C добавляли 0,070 мл триэтиламина и 0,029 мл метансульфонилхлорида с последующим перемешиванием реакционной смеси при комнатной температуре в течение 30 минут. Осадок отфильтровывали и промывали тетрагидрофураном и концентрировали фильтрат в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 4/1), получая при этом трет-бутил-цис-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3Н)-илиден)амино)пиридин-2-ил)метил)-4-((метилсульфонил)окси)циклогексанкарбоксилат.
К раствору 110 мг трет-бутил-цис-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)-4-((метилсульфонил)окси)циклогексанкарбоксилата в 0,72 мл N-метил-2-пирролидинона при комнатной температуре добавляли 62,8 мг карбоната калия и 78,0 мг 2,3-дихлорбензолтиола с последующим перемешиванием реакционной смеси при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, добавляли к реакционной смеси воду и экстрагировали этилацетатом. Этилацетатный слой промывали насыщенным раствором соли, сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 1/4), получая при этом указанное в заголовке соединение в виде желтого масла.
(2) Синтез трифторацетата транс-4-((2,3-дихлорфенил)тио)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
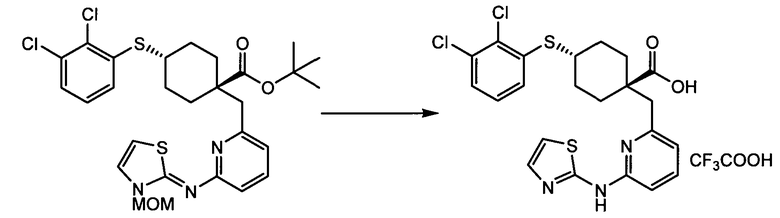
К 39,0 мг трет-бутил-транс-4-((2,3-дихлорфенил)тио)-1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата добавляли 1 мл 4M раствора хлористого водорода в 1,4-диоксане с последующим перемешиванием реакционной смеси при 95°C в течение 2,5 часов. После охлаждения реакционной смеси до комнатной температуры реакционную смесь концентрировали в вакууме. Полученный остаток очищали с помощью препаративной жидкостной хроматографии с обращенной фазой с последующим концентрированием полученной фракции в вакууме, получая при этом указанное в заголовке соединение в виде белого твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,71-1,96 (8H, м), 3,00 (2H, c), 3,64 (1H, ушир.c), 6,74 (1H, д, J=7,6 Гц), 6,91 (1H, д, J=8,0 Гц), 7,03 (1H, д, J=4,0 Гц), 7,31 (1H, т, J=8,0 Гц), 7,40 (1H, д, J=4,0 Гц), 7,41-7,47 (2H, м), 7,60 (1H, дд, J=8,0, 7,6 Гц), 11,40 (1H, ушир.c).
масс: 494,496 (М+1)+
Пример 19
Синтез трифторацетата транс-4-((2,3-дихлорфенил)сульфинил)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
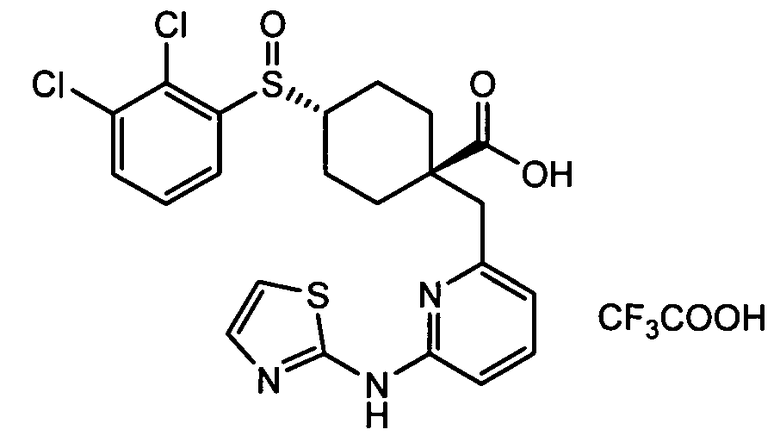
К суспензии 8,0 мг трифторацетата транс-4-((2,3-дихлорфенил)тио)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, полученного по примеру 18, в 0,24 мл ацетонитрила и 0,12 мл воды при комнатной температуре добавляли раствор 8,6 мг препарата OXONE® (пероксимоносульфат калия, предоставленный компанией Aldrich) в 0,12 мл воды с последующим перемешиванием реакционной смеси при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали в вакууме. Полученный остаток очищали с помощью препаративной жидкостной хроматографии с обращенной фазой с последующим концентрированием полученной фракции в вакууме, получая при этом указанное в заголовке соединение в виде белого твердого вещества.
1H-ЯМР (CD3OD) δ: 1,26-1,41 (1H, м), 1,73-1,82 (1H, м), 1,85-2,21 (6H, м), 3,05-3,15 (1H, м), 3,29 (2H, с), 7,07 (1H, д, J=8,0 Гц), 7,11 (1H, д, J=7,6 Гц), 7,28 (1H, д, J=4,0 Гц), 7,59 (1H, д, J=4,0 Гц), 7,59 (1H, т, J=8,0 Гц), 7,71-7,78 (2H, м), 7,83 (1H, т, J=8,0 Гц).
масс: 510,512 (M+1)+
Пример 20
Синтез трифторацетата транс-4-((2,3-дихлорфенил)сульфонил)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
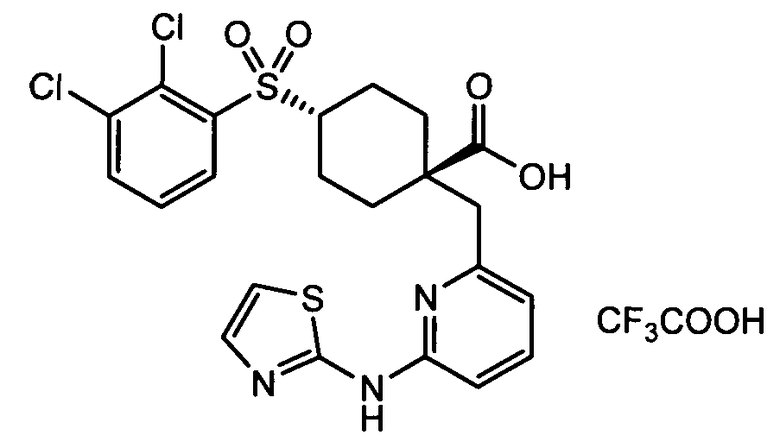
К суспензии 6,60 мг трифторацетата транс-4-((2,3-дихлорфенил)тио)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, полученного по примеру 18, в 0,24 мл ацетонитрила и 0,12 мл воды при комнатной температуре добавляли раствор 14,7 мг препарата OXONE® (пероксимоносульфат калия) в 0,12 мл воды с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. Реакционную смесь концентрировали в вакууме. Полученный остаток очищали с помощью препаративной жидкостной хроматографии с обращенной фазой с последующим концентрированием полученной фракции в вакууме, получая при этом указанное в заголовке соединение в виде белого твердого вещества.
1H-ЯМР (ДМСО-d6) δ: 1,59-1,76 (4H, м), 1,85-1,92 (2H, м), 1,98-2,11 (2H, м), 3,02 (2H, c), 3,61-3,70 (1H, м), 6,65 (1H, д, J=8,0 Гц), 6,86 (1H, д, J=8,0 Гц), 7,01 (1H, J=3,6 Гц), 7,38 (1H, д, J=3,6 Гц), 7,56 (1H, т, J=8,0 Гц), 7,65 (1H, т, J=8,0 Гц), 8,03 (2H, д, J=8,0 Гц), 11,15 (1H, ушир.c).
масс: 526,528 (М+1)+
Пример 21
Синтез трифторацетата транс-4-((2,3-дихлорфенил)тио)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
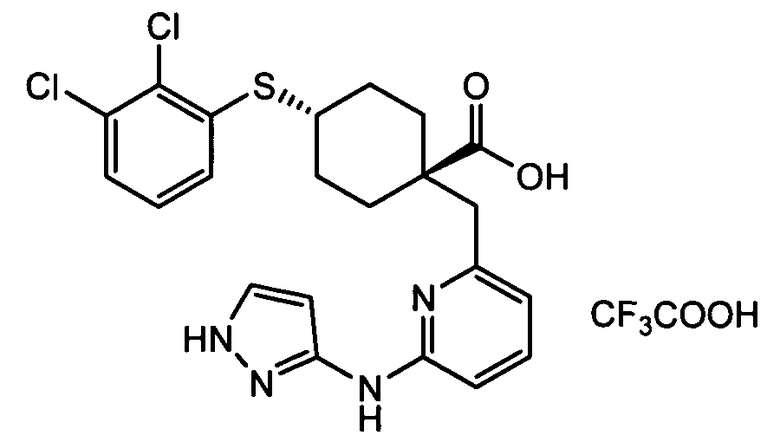
(1) Синтез трет-бутил-цис-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-гидроксициклогексанкарбоксилата
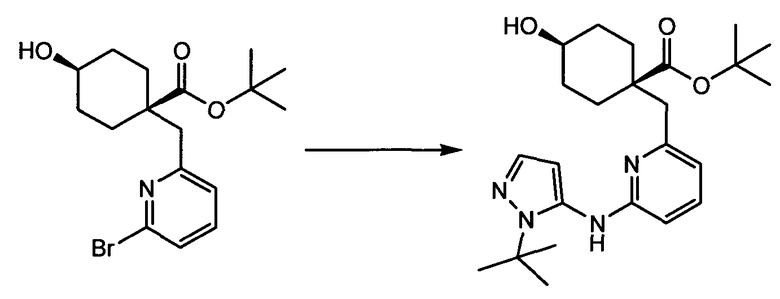
Указанное в заголовке соединение получали в виде твердого желтого вещества таким же способом, как на стадии 7(5) примера 7, применяя трет-бутил-цис-1-((6-бромпиридин-2-ил)метил)-4-гидроксициклогексанкарбоксилат, полученный на стадии 7(3) примера 7, вместо трет-бутил-транс-1-((6-бромпиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоксилата, который применялся в примере 7(5).
(2) Синтез трет-бутил-транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-((2,3-дихлорфенил)тио)циклогексанкарбоксилата
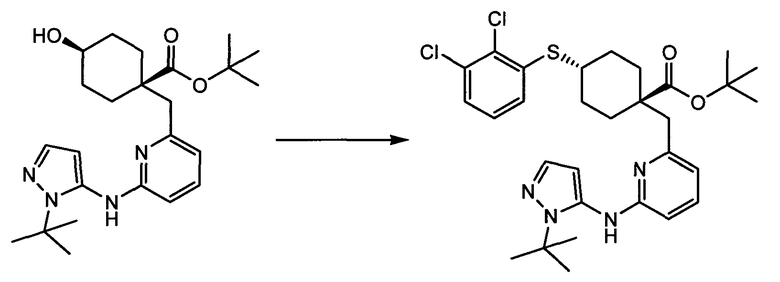
Указанное в заголовке соединение получали в виде не совсем белого твердого вещества таким же способом, как на стадии 18(1) примера 18, применяя трет-бутил-цис-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-гидроксициклогексанкарбоксилат вместо трет-бутил-цис-4-гидрокси- 1-((6-(((2Z)-3-(метоксиметил)-1,3-тиазол-2(3H)-илиден)амино)пиридин-2-ил)метил)циклогексанкарбоксилата, который применялся на стадии 18(1) примера 18.
(3) Синтез трифторацетата транс-4-((2,3-дихлорфенил)тио)-1-((6-(1Н-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты
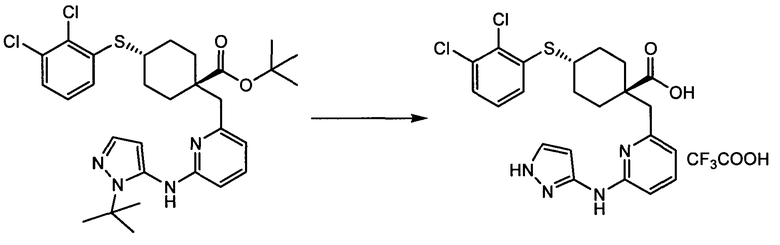
Указанное в заголовке соединение получали в виде твердого белого вещества таким же способом, как на стадии 7(6) примера 7, применяя трет-бутил-транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-((2,3-дихлорфенил)тио)циклогексанкарбоксилат вместо трет-бутил-транс-1 -((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоксилата, который применялся на стадии 7(6) примера 7.
1H-ЯМР (CD3OD) δ: 1,77-2,12 (8H, м), 3,22 (2H, c), 3,60-3,70 (1H, м), 6,13 (1H, д, J=2,4 Гц), 6,99 (1Н, д, J=8,0 Гц), 7,14 (1Н, д, J=8,0 Гц), 7,24 (1Н, т, J=8,0 Гц), 7,37 (1H, дд, J=8,0, 1,2 Гц), 7,40 (1H, дд, J=7,6, 1,2 Гц), 7,75 (1H, д, J=2,4 Гц), 8,01 (1H, дд, J=8,0, 7,6 Гц).
масс: 477,479 (М+1)+
Пример 22
Синтез трифторацетата 5-(транс-4-((2,3-дихлорфенил)тио)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-она
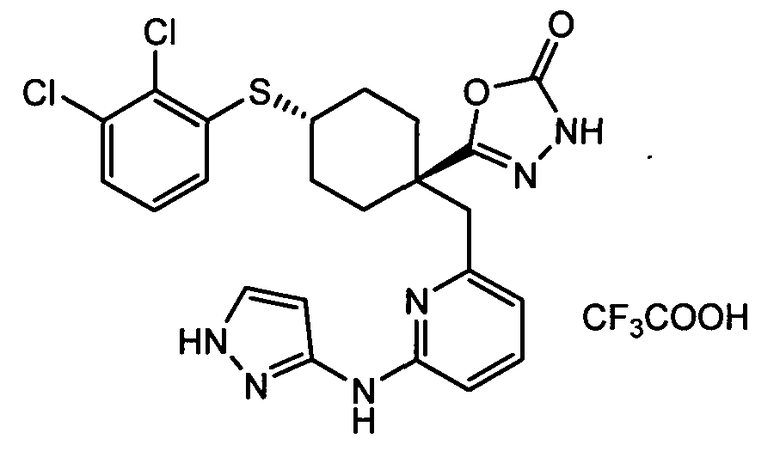
Указанное в заголовке соединение получали в виде твердого белого вещества таким же способом, как на стадиях 13(2)-13(4) примера 13, применяя трифторацетат транс-4-((2,3-дихлорфенил)тио)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, полученный по примеру 21, вместо трифторацетата транс-1-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-4-(2-фтор-3-(трифторметил)фенокси)циклогексанкарбоновой кислоты, который применялся в примере 13(2).
1H-ЯМР (CD3OD) δ: 1,82-1,92 (2H, м), 1,96-2,17 (6H, м), 3,29 (2H, c), 3,66-3,72 (1H, м), 6,14 (1H, д, J=2,8 Гц), 6,99 (1H, д, J=7,6 Гц), 7,17 (1H, д, J=8,8 Гц), 7,25 (1H, т, J=8,0 Гц), 7,38 (1H, дд, J=8,0, 1,6 Гц), 7,41 (1H, дд, J=8,0, 1,6 Гц), 7,75 (1H, д, J=2,8 Гц), 8,02 (1H, дд, J=8,8, 7,6 Гц).
масс: 517,519 (М+1)+
Пример 23
Синтез трифторацетата 5-(транс-4-((2,3-дихлорфенил)сульфинил)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-она
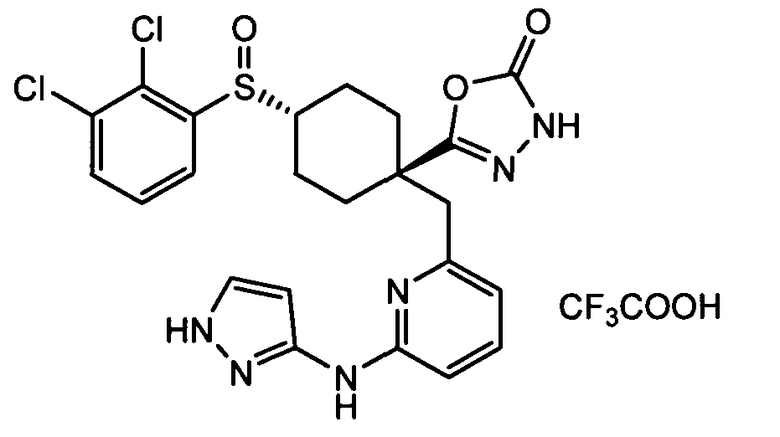
Указанное в заголовке соединение получали по примеру 19 в виде твердого белого вещества, применяя трифторацетат 5-(транс-4-((2,3-дихлорфенил)тио)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-она, полученный по примеру 22, вместо трифторацетата транс-4-((2,3-дихлорфенил)тио)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, который применялся в примере 19.
1H-ЯМР (CD3OD) δ: 1,45-1,56 (1H, м), 1,86-1,99 (1H, м), 2,00-2,23 (6H, м), 3,12-3,23 (1H, м), 3,36 (1H, д, J=14,4 Гц), 3,42 (1H, д, J=14,4 Гц), 6,15 (1H, д, J=2,8 Гц), 6,97 (1H, д, J=7,2 Гц), 7,17 (1H, д, J=8,8 Гц), 7,62 (1H, дд, J=8,0, 7,2 Гц), 7,76-7,80 (3H, м), 8,03 (1H, дд, J=8,8, 7,2 Гц).
масс: 533,535 (М+1)+
Пример 24
Синтез трифторацетата 5-(транс-4-((2,3-дихлорфенил)сульфонил)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-она
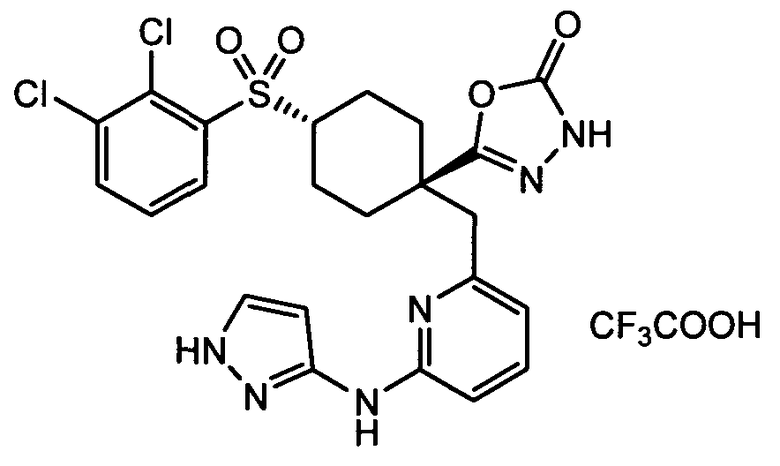
Указанное в заголовке соединение получали по примеру 20 в виде твердого желтого вещества, применяя трифторацетат 5-(транс-4-((2,3-дихлорфенил)тио)-1-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3H)-она, полученный по примеру 22, вместо трифторацетата транс-4-((2,3-дихлорфенил)тио)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновой кислоты, который применялся в примере 20.
1H-ЯМР (CD3OD) δ: 1,90-2,25 (8H, м), 3,34 (2H, c), 3,73-3,83 (1H, м), 6,15 (1H, д, J=2,8 Гц), 6,97 (1H, д, J=7,2 Гц), 7,17 (1H, д, J=8,8 Гц), 7,58 (1H, т, J=8,0 Гц), 7,77 (1H, д, J=2,8 Гц), 7,93 (1H, дд, J=8,0, 1,6 Гц), 8,02 (1H, дд, J=8,8, 7,2 Гц), 8,09 (1H, дд, J=8,0, 1,6 Гц).
масс: 549,551 (М+1)
Ссылка 1
Синтез трет-бутил-4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксилата
(1) Синтез этил-4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксилата
К раствору 25 г 4-гидроксициклогексанкарбоновой кислоты в 125 мл N,N-диметилформамида при охлаждении льдом последовательно добавляли 21,7 г имидазола и 39,6 мл трет-бутил(дифенил)силилхлорида с последующим перемешиванием реакционной смеси при комнатной температуре в течение 3 часов. К реакционной смеси добавляли воду и экстрагировали гексаном. Полученный гексановый раствор промывали насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме, получая при этом указанное в заголовке соединение.
(2) Синтез 4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоновой кислоты
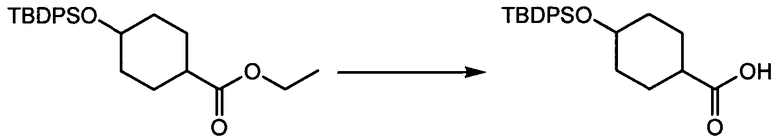
К раствору 64,2 г этил-4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксилата в 200 мл метанола и 200 мл тетрагидрофурана добавляли 58 мл 5M водного раствора гидроксида натрия с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную смесь нейтрализовали 5M водным раствором хлористоводородной кислоты (гидрохлорида) с последующим удалением метанола и тетрагидрофурана в вакууме и полученный остаток экстрагировали этилацетатом. Полученный этилацетатный раствор промывали насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме, получая при этом указанное в заголовке соединение.
(3) Синтез трет-бутил-4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксилата
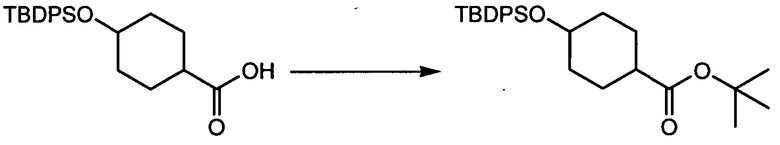
К раствору 62,8 г 4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоновой кислоты в 270 мл трет-бутилового спирта при комнатной температуре последовательно добавляли 63,3 г ди-трет-бутилдикарбоната и 5,31 г 4-диметиламинопиридина с последующим перемешиванием реакционной смеси при комнатной температуре в течение 3 часов. Для удаления трет-бутилового спирта реакционную смесь концентрировали в вакууме и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: от гексана до смеси гексан/этилацетат = 19/1), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
Ссылка 2
Синтез 4-((трет-бутил(дифенил)силил)окси)циклогексанкарбонитрила
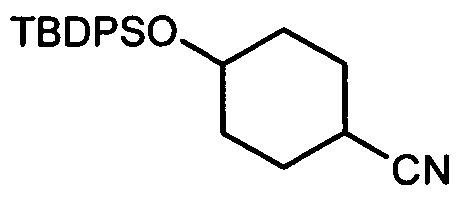
(1) Синтез 4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксамида

К раствору 6,64 г 4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоновой кислоты, полученной на стадии 1(2) ссылки 1, в 100 мл хлороформа при комнатной температуре последовательно добавляли 4,65 г хлорида аммония, 30,3 мл диизопропилэтиламина, 8,0 г гидрата гидроксибензотриазола и 10,0 г гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида с последующим перемешиванием реакционной смеси при комнатной температуре в течение ночи. Реакционную смесь промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан/этилацетат = 10/1 до этилацетата), получая при этом указанное в заголовке соединение.
2) Синтез 4-((трет-бутил(дифенил)силил)окси)циклогексанкарбонитрила

К раствору из 6,42 г 4-((трет-бутил(дифенил)силил)окси)циклогексанкарбоксамида и 2,39 мл диметилсульфоксида в 90 мл метиленхлорида при -78°C добавляли раствор 2,06 мл оксалилхлорида в 10 мл метиленхлорида с последующим перемешиванием реакционной смеси при -78°C в течение 15 минут. К реакционной смеси при -78°C добавляли 7,05 мл триэтиламина с последующим перемешиванием реакционной смеси при -78°C в течение 30 минут и дополнительным перемешиванием в течение 1,5 часов при комнатной температуре. Реакционную смесь промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан до смеси гексан/этилацетат = 4/1), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
Ссылка 3
Синтез 1-трет-бутил-1H-пиразол-5-амина
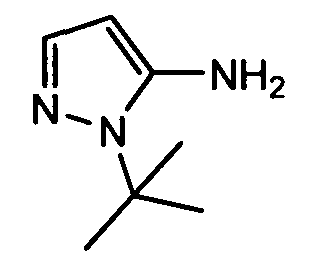
К 600 мл этанола при комнатной температуре последовательно добавляли 59,94 г гидрохлорида трет-бутилгидразина, 79,3 г ацетата натрия и 50 мл 2-хлоракрилонитрила с последующим перемешиванием реакционной смеси при 80°C в течение 12 часов. После удаления растворителя в вакууме к остатку добавляли воду. Смесь нейтрализовали гидрокарбонатом натрия и экстрагировали этилацетатом. Полученный этилацетатный раствор промывали насыщенным раствором соли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан/этилацетат = 2/1-1/2), получая при этом указанное в заголовке соединение в виде бледно-желтого масла.
Ссылка 4
Синтез 1-трет-бутил-1H-пиразол-5-амин-п-толуолсульфоната
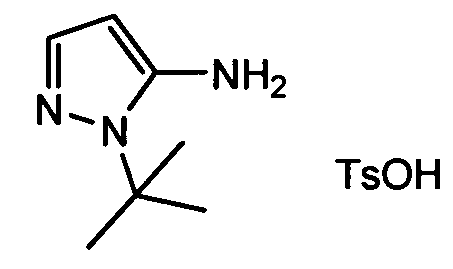
К 850 мл этанола при комнатной температуре последовательно добавляли 85,64 г гидрохлорида трет-бутилгидразина, 112,54 г ацетата натрия и 72 мл 2-хлоракрилонитрила с последующим перемешиванием реакционной смеси при 80°C в течение 12 часов. После удаления растворителя в вакууме к остатку добавляли воду. Смесь нейтрализовали гидрокарбонатом натрия и экстрагировали этилацетатом. Полученный этилацетатный раствор промывали насыщенным раствором соли, сушили над безводным сульфатом магния, фильтровали и концентрировали фильтрат в вакууме. К раствору полученного остатка в 700 мл этилацетата при перемешивании добавляли раствор 96,16 г гидрата п-толуолсульфоновой кислоты в 140 мл этанола с последующим оставлением полученной смеси, как она есть, на ночь. Полученный осадок собирали и промывали этилацетатом, получая при этом указанное в заголовке соединение в виде твердого белого вещества.
Промышленная применимость
Соединение по изобретению характеризуется тем, что оно обладает ингибирующей активностью в отношении роста клеток, а также синергическим действием при применении с другими противоопухолевыми средствами, основанной на превосходной селективной ингибирующей активности в отношении Авроры A, и благодаря этому ожидается, что оно применимо в качестве противоопухолевого средства в области фармацевтических препаратов.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПРОИЗВОДНОЕ АМИНОПИРИДИНА С ИНГИБИТОРНОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ SYK. | 2006 |
|
RU2363699C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ PLK1 | 2007 |
|
RU2458062C2 |
| НОВОЕ ПИПЕРИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2581834C1 |
| ПРОИЗВОДНЫЕ ДИГИДРОПИРАЗОЛОПИРИМИДИНОНА | 2007 |
|
RU2437885C2 |
| СОЕДИНЕНИЯ ДИБЕНЗИЛАМИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ТЕРАПЕВТИЧЕСКИЙ ИЛИ ПРОФИЛАКТИЧЕСКИЙ АГЕНТ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ГИПЕРЛИПИДЕМИИ ИЛИ АРТЕРИОСКЛЕРОЗА | 2003 |
|
RU2293078C2 |
| ЗАМЕЩЕННЫЕ ПИРИМИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ JNK | 2007 |
|
RU2493155C2 |
| 6, 6-БИЦИКЛИЧЕСКИЕ КОЛЬЦЕВЫЕ ЗАМЕЩЕННЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2005 |
|
RU2379308C2 |
| ДИАМИНОГЕТЕРОЦИКЛИЧЕСКОЕ КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2010 |
|
RU2526253C2 |
| ИНГИБИТОРЫ АВРОРА-КИНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2822464C2 |
| ПРОИЗВОДНЫЕ 1,5-НАФТИРИДИНА И ИНГИБИТОРЫ MELK, СОДЕРЖАЩИЕ ИХ | 2012 |
|
RU2645339C1 |
Настоящее изобретение относится к соединению общей формулы I, где: R1 представляет собой COORa1, CONRa2Ra2', CONRa4ORa4', где: каждый из Ra1 и Ra4 представляет собой атом водорода; каждый из Ra2 и Ra2' представляет собой атом водорода; Ra4' представляет собой низший алкил; или R1 представляет собой гетероциклическую группу, выбранную из следующих групп, где Y2 представляет собой атом водорода или низший алкил:
R2 представляет собой О, S, SO, SO2; R3 представляет собой фенил, который замещен 2 заместителями, выбранными из галогена, CF3; Х2 представляет собой СН или N; W представляет собой следующий остаток:
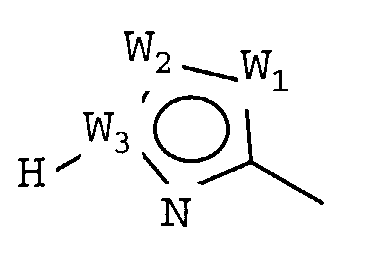
где: W1 представляет собой СН или S; W2 представляет собой СН; W3 представляет собой С или N; и, по меньшей мере, один из W1, W2 и W3 представляет собой атом углерода; или его фармацевтически приемлемая соль или сложный эфир. Изобретение также относится к фармацевтической композиции, обладающей селективной ингибирующей активностью в отношении Авроры А, содержащей вместе с фармацевтически приемлемым носителем или разбавителем, по меньшей мере, одно соединение формулы I в качестве активного ингредиента. Технический результат - аминопиридиновые или аминопиразиновые производные, ингибирующие рост опухолевых клеток на основе селективной ингибирующей активности в отношении киназы Аврора А. 6 н. и 5 з.п. ф-лы, 3 табл.
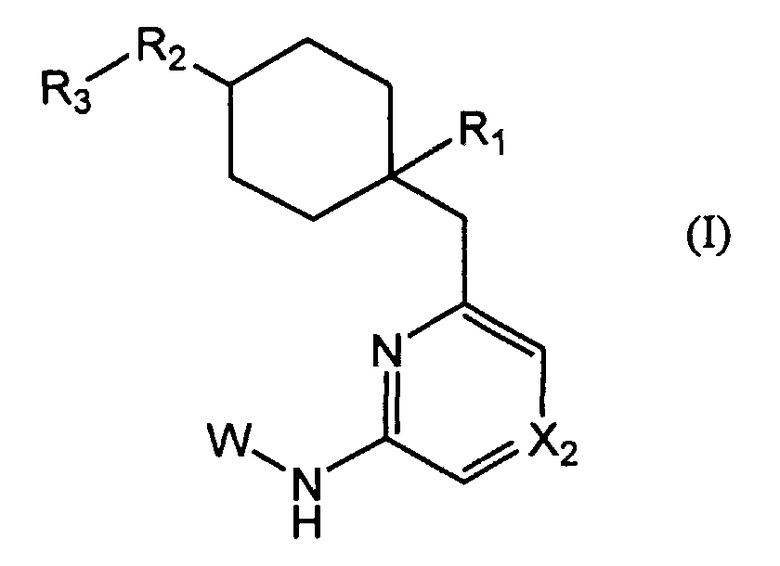
1. Соединение общей формулы I:
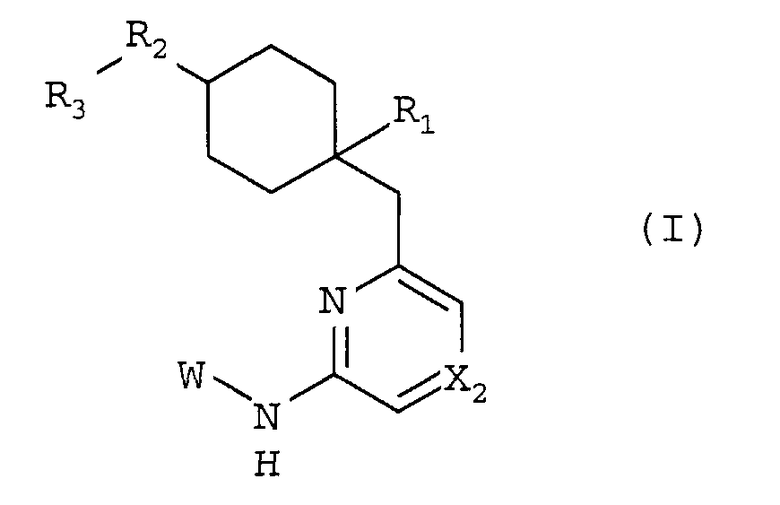
где R1 представляет собой COORa1, CONRa2Ra2', CONRa4ORa4',
где каждый из Ra1 и Ra4 представляет собой атом водорода;
каждый из Ra2 и Ra2' представляет собой атом водорода;
Ra4' представляет собой низший алкил; или
R1 представляет собой гетероциклическую группу, выбранную из следующих групп, где Y2 представляет собой атом водорода или низший алкил:
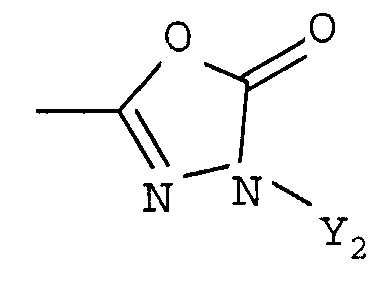
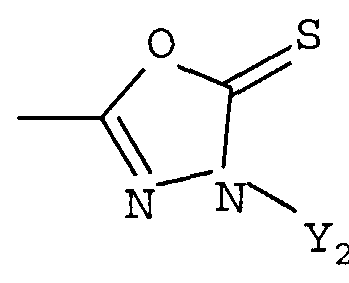
R2 представляет собой О, S, SO, SO2;
R3 представляет собой фенил, который замещен 2 заместителями, выбранными из галогена, CF3;
Х2 представляет собой СН или N;
W представляет собой следующий остаток:
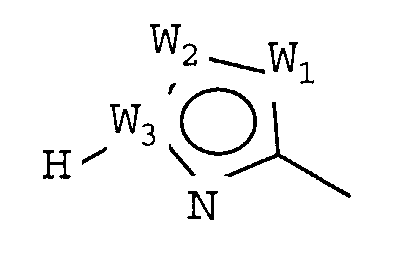
где W1 представляет собой СН или S;
W2 представляет собой СН;
W3 представляет собой С или N; и,
по меньшей мере, один из W1, W2 и W3 представляет собой атом углерода; или его фармацевтически приемлемая соль или сложный эфир.
2. Соединение по п.1 или его фармацевтически приемлемая соль или сложный эфир, где W выбран из:
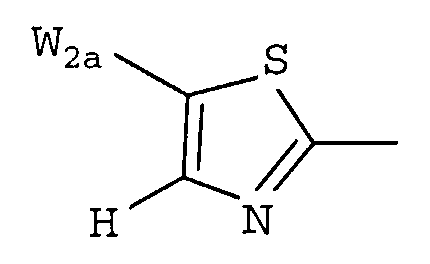 ,
, 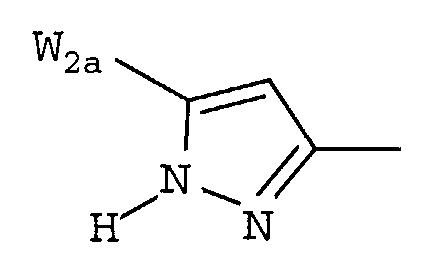
где W2a представляет собой водород.
3. Соединение по п.2 или его фармацевтически приемлемая соль или сложный эфир, где R3 представляет собой фенил, 2-е и 3-е положения которого замещены двумя заместителями, выбранными из F, Cl и CF3.
4. Соединение по п.3 или его фармацевтически приемлемая соль или сложный эфир, где X2 представляет собой СН.
5. Соединение по п.4 или его фармацевтически приемлемая соль или сложный эфир, где R1 представляет собой СООН или CONRa2Ra2', где Ra2 и Ra2' каждый представляет собой атом водорода; или R1 выбран из следующего:
 ,
, 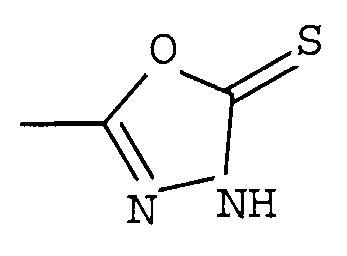 ,
,
и R2 представляет собой О, S, SO или SO2.
6. Соединение по п.5 или его фармацевтически приемлемая соль или сложный эфир, где W представляет собой любой из следующих остатков:
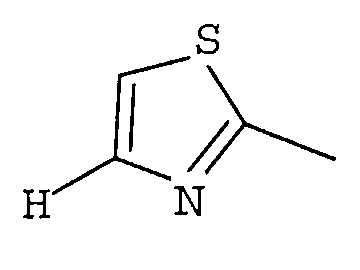 ,
, 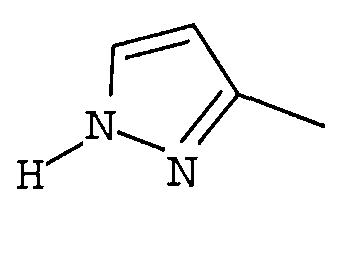
7. Соединение, которое представляет собой:
(a) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(b) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(c) транс-4-(2,3-дихлорфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(d) транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1Н-пиразол-3-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту;
(e) транс-4-(3-хлор-2-фторфенокси)-1-((6-(1Н-пиразол-3-иламино)пиразин-2-ил)метил)циклогексанкарбоксамид;
(f) 5-(транс-4-(2-фтор-3-(трифторметил)фенокси)-1-((6-(1Н-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3Н)-он;
(g) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1Н-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3Н)-он;
(h) 5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1Н-пиразол-3-иламино)пиразин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3Н)-он, или
(i) 5-(транс-4-((2,3-дихлорфенил)сульфонил)-1-((6-(1Н-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3Н)-он,
или его фармацевтически приемлемую соль или сложный эфир.
8. Соединение, представляющее собой
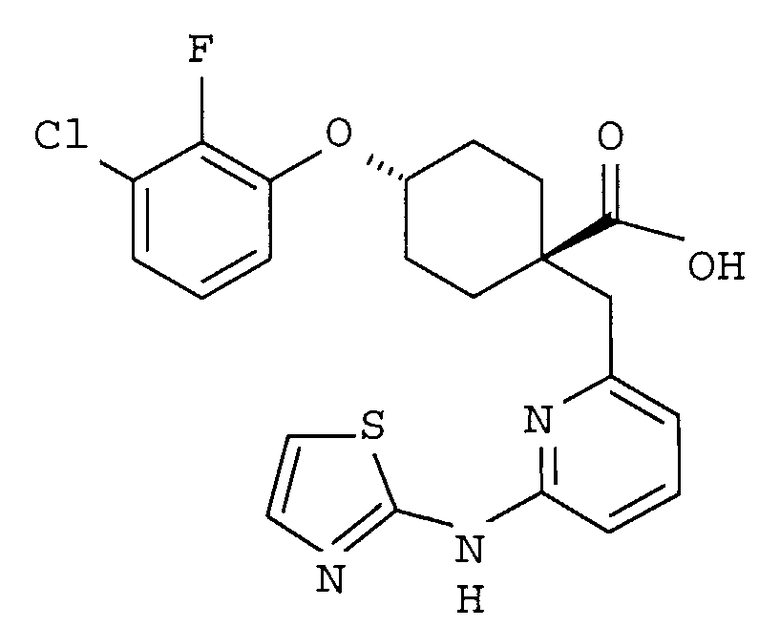
транс-4-(3-хлор-2-фторфенокси)-1-((6-(1,3-тиазол-2-иламино)пиридин-2-ил)метил)циклогексанкарбоновую кислоту,
или его фармацевтически приемлемая соль или сложный эфир.
9. Соединение, представляющее собой
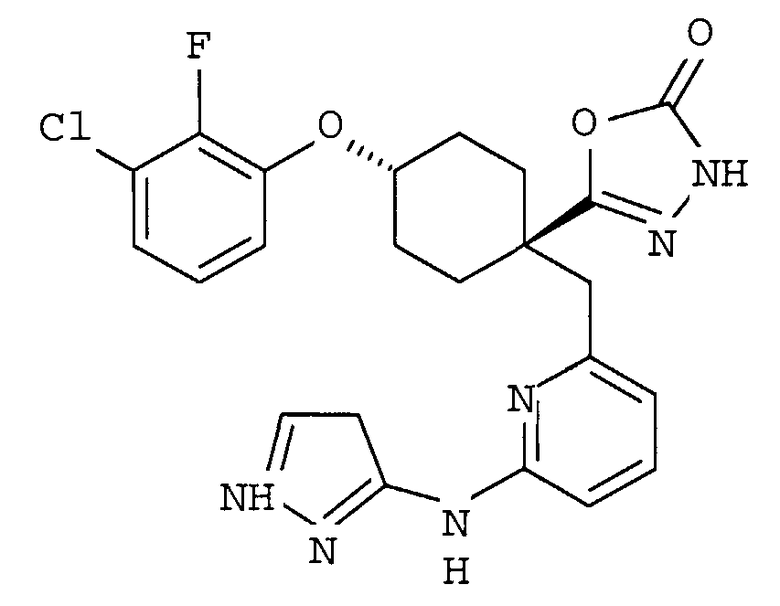
5-(транс-4-(3-хлор-2-фторфенокси)-1-((6-(1Н-пиразол-3-иламино)пиридин-2-ил)метил)циклогексил)-1,3,4-оксадиазол-2(3Н)-он,
или его фармацевтически приемлемая соль или сложный эфир.
10. Фармацевтическая композиция, обладающая селективной ингибирующей активностью в отношении Авроры А, содержащая вместе с фармацевтически приемлемым носителем или разбавителем, по меньшей мере, одно соединение по п.1 в качестве активного ингредиента.
11. Селективный ингибитор Авроры А, содержащий вместе с фармацевтически приемлемым носителем или разбавителем, по меньшей мере, одно соединение по п.1 в качестве активного ингредиента.
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| ЭПОКСИДНОЕ СВЯЗУЮЩЕЕ, ПРЕПРЕГ НА ЕГО ОСНОВЕ И ИЗДЕЛИЕ, ВЫПОЛНЕННОЕ ИЗ НЕГО | 2014 |
|
RU2585638C1 |
| RU 2003122209 A, 10.02.2005 | |||
| RU 2003122210 A, 27.12.2004 | |||
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| СПОСОБ СООРУЖЕНИЯ НАПРЯЖЕННЫХ ЖЕЛЕЗОБЕТОННЫХ КОНСТРУКЦИЙ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2014 |
|
RU2567569C2 |
| ЭПОКСИДНОЕ СВЯЗУЮЩЕЕ, ПРЕПРЕГ НА ЕГО ОСНОВЕ И ИЗДЕЛИЕ, ВЫПОЛНЕННОЕ ИЗ НЕГО | 2014 |
|
RU2585638C1 |

