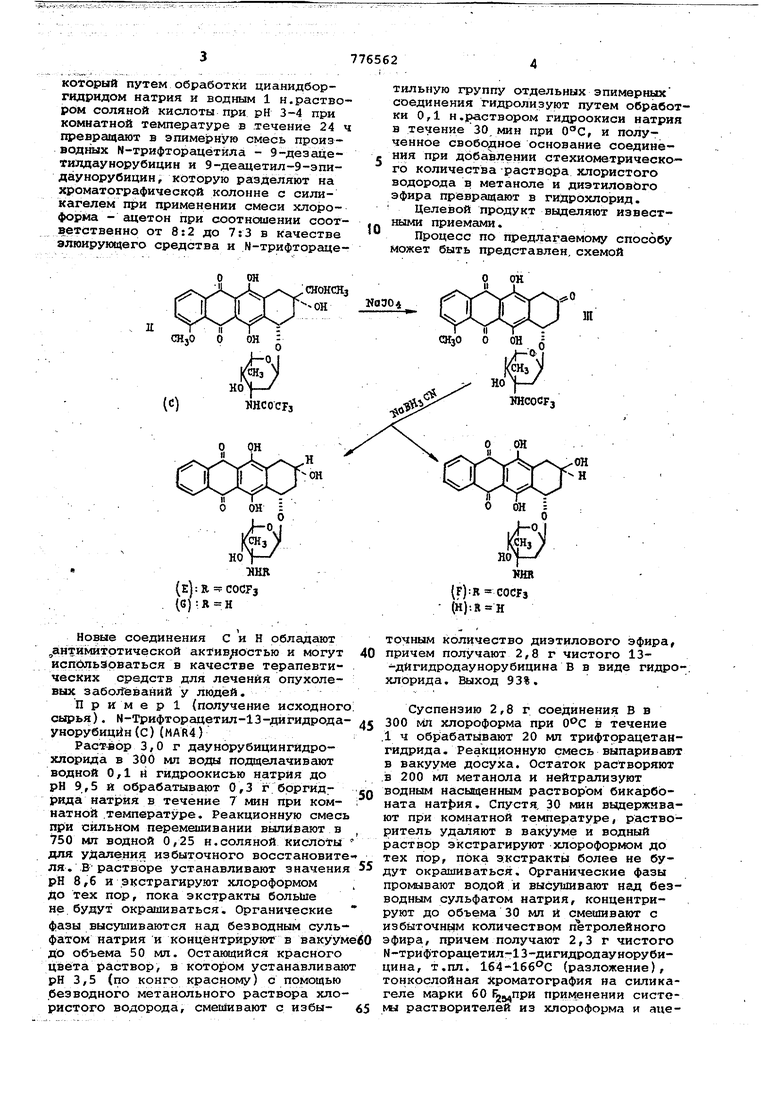
1 Изобретение относится к способу получения новых производных антрапиклина, обладаняцих ценными фармакологическими свойствами. Цель изобретения - получение новых полезных соединений, расширяющих, арсенал средств воздействия на живой организм, достигается путем синтеза последних, основанного на известных реакциях р1сисления и вос становления Описывается, согласно изобретению, способ получения антрациклинов Общей формулы I; где X означает отличающийся тем, что М-Тоифторацетил-13-дигидр6даунорубиции общей (/ла II QJt ,СНОНСНз МНСОСРз в трет-бутаноле вприсутствии двух эквивалентов иодата натрия окисляют при комнатной температуре в течение 2 ч с получением 9-деза1(етил-9-оксо-М-трифторааетилдаунорубицина общей формулы III о ОН ттсосрз
кофорый путем обработки цианидборгидридом натрия и водным 1 н.раствором соляной кислоты при рН 3-4 при комнатной температуре в течение 24 ч превращают в эпимерную смесь производных N-трифторацётйла - 9-деэацетилдаунорубицин и 9-деацетил-9-эпидаунорубицин, которую разделяют на хроматографической колонне с силикагелем при применении смеси хлороформа - ацетон при соотнесении соответственно от 8:2 до 7:3 в качестве элюирующего средства и N-трифтораце:снонскз
-.он Уозо-i
. 31
рз Новые соединения с и Н обладают ,,антймитртической актив/юстью и могут исп1бльзрваться в качестве терапевтических средств для лечения опухолевых забол1еваний у людей. Пример 1 (получение исходного сырья). М-Трифторацетил-13-дигидродаунорубицйн{С) (MAR4} Раствор 3,0 г даунорубицингйдрохлсчрида в 300 мл воды подщелачивают водной 0/1 н гидроокисью натрия до рН 9,5 и обрабатывают 0,3 г боргидрида натрия в течение 7 мин при комнатной .температуре. Реакционную смесь при сильном перемешивании вьшйвают в 750 мл водной 0,25 н.соляной кислоты для удаления избыточного восстановите ля. В растворе устанавливают значения рН 8,6 и экстрагируют хлороформом До тех пор, пока экстракты болыае не будут окрашиваться. Органические фазы высушиваются над безводным сульфатом натрия и концентрируют в вакуум До объема 50 мп. Остающийся красного цвета раствор, в котором устанавливаю рН 3,5 (по конго красному) с помощью безводного метанольного раствора хлористого водорода, смешивают с избытильную группу отдельных эпимерных соединения гидролизуют путем обработки 0,1 н.раствором гидроокиси натрия в течение 30 мин при , и полученное свободное основание соединения при добавлении стехиометрическо го количества раствора хлористого водорода в метаноле и диэтиловЬго эфира превращают в гидрохлорид.
Целевой продукт выделяют известными приемами. .
Процесс по тфедлагаемому способу может быть представлен, схемой
.
смзо о он
О точным количество диэтилового эфира, причем получают 2,8 г чистого 13-дйгидродаунорубицина В в виде гидрохлорида. Выход 93%. Суспензию 2,8 г соединения В в 300 МП хлороформа при в течение .1 ч обрабатывают 20 мл трифторацетангидрида. Реакционную смесь выпаривают в вакууме досуха. Остаток растворяют в 200 мл метанола и нейтрализуют водным насыщенным раствором бикарбоната нат|)ия, Спустя, 30 мин выдерживают при комнатной температуре, растворитель удаляют в вакууме и водный раствор экстрагируют хлороформом до тех пор, пока экстракты более не будут окрашиваться. Органические фазы промавают водой и высушивают над безводным сульфатом натрия, концентрируют до объема 30 мл и смешивают с избыточным количеством пЪтролейного эфира, причем получают 2,3 г чистого М-трифторацетил-13-дигидродаунорубицина, т.пл. 1б4-1б6°С (разложение), тонкослойная хроматография на силикагеле марки 60 применении систоN« растворителей из хлороформа и ацетона (2:1 по объему) дает R 0,25. Выход 70%. П р и м е р 2. 9-Дезрцетил-9-оксо -N-тpифтopaцeтилдayнopyбицинa(д)(MARl Раствор 2,3 г полученного соедине ния С в 120 мл треттбутанола обрабаты вают 1,53 г йодата натрия, растворен ных в 120 мл воды. Реакционную смесь в течение 2 ч перемешивают при комна ной температуре. Выпавший в осадок (соединение Д) отфильтровывают, промы вают водой и высушивают в вакууме. Получают 1,34 г чистого соединения Д, т.пл. 200°С (разложение); тонкослойная хроматография на силикагеле 60 р25.при использовании системы растворителей из хлороформа и аце тона (2:1 по объему) дает R 0,57. Выход 64%. Найдено, %: Н 4,26, С 55,6. C57H24FNO,o, Вычислено, %: Н 4,18, С 55,9. Пример 3. 9-Дезацетилдаунсрубицин(С)(MAR29) и 9-дезацетил-9-эпидаунорубицин(Н) (MAR 30).Раствор 1,34 г соединения Д в 250 мл диоксана и 50 мл воды подкисляют водной 1 Н.соляной кислотой до рН 3 и обрабатывают 0,78 г цианборгидрида натрия в течение 24 ч при комнатной температуре, причем поддер живают кислые условия путем добавки 1 Н.соляной кислоты. Реакционную смесь смешивают с 300 мл воды и пять раз экстрагируют по 200мл хлороформа Промытую водой и высушенную нйд безводным сульфатом натрия органическую фазу выпаривают в вакууме досуха. Остаток (1 г) хроматографируют на колонне с силикагелем при использовании сначала смеси хлороформ ацетон 8:2 V/V и затем смеси хлороформ - ацетон 7:3 V/V в качестве элю ирукядего средства, причем получают 0,67 г чистого соединения Е, т.пл. 204-2060С (разложение). Выход 50%. Тонкослойная хроматография на Силикагеле марки 60 применении системы растворителей из хлороформа и ацетона (2:1 по объему) дает R 0,44 и получают 0,078 г чистого соединения F, т.пл. 180-182°С (разложение). Выход 6%, R.p определенным при таких же условиях, что и для соединения Е, 0,3. Чтобы гидролизовать М-трифто гщётильную группу, соединения Вир обрабатывают следующим образом. К 0,67 г соединения Е добавляют 50 мл 0,1 Н.гидроокиси натрия, спустя 30 мин при устанавливают с помощью 0,1 Н,соляной кислоты рН 8,4 и раствор повторно промывают хлороформом. Объединенные экстракты сушат над безводным сульфатом натрия и концентрируют в вакууме до объема 20 мл. Раствор при добавке стехиомет рического количества метанольного ряоткора хлористого водорода и диэтилового эфира дает осадок красного цвета, который отделяют, промывают диэтиловым эфиром и высушивают в вакууме. Соединение С имеет т.пл. 166-1670С (разложение) , 282° (с 0,15, метанол). Тонкослойная хроматография на силикагелевых пластинах F2S4 (марка) с системой растворителей из хлороформа и ащетона. (13:6:1 по объему) дает R 0,55. Получено-0,56 г соединения G, Выход 84%. Общий выход 9-дезацетил-9-кето-М-трифторацетилдаунорубицина(MAR-18) 27%. Найдено, %: Н 5,45/ С 57,16 N 2,68. . C25Hf N09 HCe . Bычиcлieн6, Н 5,42J С 57,52; N 2,68. Соединение Н 0,065 г имеет т.пл. 176°с (разложение), R при таких же УСЛОВИЯХ, что и для соединения G равно 0,4. Найдено, %: Н 5,52, С 57,61, N 2,63. C5jaH27N09-HC1. Шчислено, %: Н 5,42; С 57,52/ N 2,68. (с 0,148 МеОН). Общий выход 9--дезацетил-9-оксо-N-тpифтopalteтилдayн6pyбицинa(MAR 18) 3%. Формула изобретения. Способ получения антрациклинов общей формулы 1 х. где X означает ,с или ;с отличающийся тем, что М-трифторацётил-13-дигидродаунорубицин общей формулы 11 СЯОНСНз ОН ТШСОСРз в трет-бутаноле в присутствии двух
э.квйвс|лёнтов йрдата натрия окисляют при комнатной температуре в течение 2 ч с получением 9--дезацетил-9-оксо-М-трйфторацетилдаунорубицина общей формула 111
KOifopau йутём обработки цйанилЬор гидридом натрия, и водным 1н.раГЬтвором соляной кислоты при рН 3-4 при комнатной температуре в течение 24 ч itpespiattiaiet в эпимерную смесь производных N-трифторацетила - 9-дезацетилдаунорубицин и 9-гдезацетил-9-эпидаунорубицин, которую разделяют на хроматографической колонне с силикагелемпри применении сначала смеси хлороформ - ацетон при соотношении 8:2, затем 7:3 соответственно в качестве элюйруквдего средства к Nr-трифторацетильную группу отдельных эпимерных (Соединений гидролизуют путем обработки 0,1 и.раствором гидроокиси натрия в течение 30 мин при и полученное свободное основание соединения при добавлении стехиометрического количества раствора хлористого вЬйо)Ьда в Метаноле и лового эфира превраща от в гидрохло. ,рид. :: :: / : -V .. -.-;.
Источники информации, принятые во внимание при экспертизе 1. Общий практикум по органической химии. Под ред. А.Н.Коста,М,, 1У1ир, с. 332, 484, 1965.


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения гликозидов | 1979 |
|
SU963471A3 |
| Способ получения гидрохлорида 4-деоксидауномицина | 1976 |
|
SU670226A3 |
| Способ получения гликозидов антрациклина | 1976 |
|
SU1014477A3 |
| Способ получения гидрохлорида 4 -эпи-6оксидауномицина | 1975 |
|
SU646913A3 |
| Способ получения гликозидов антрациклина | 1976 |
|
SU728719A3 |
| Способ получения гидрохлорида оптически активных дауносаминилпроизводных антрациклинонов | 1977 |
|
SU724087A3 |
| Способ получения производных антрациклина | 1977 |
|
SU897111A3 |
| Способ получения гликозидных антибиотиков или их гидрохлоридов | 1975 |
|
SU650507A3 |
| Способ получения гликозида | 1987 |
|
SU1590045A3 |
| Способ получения хлоргидрата 4 -эпи-6 оксиадриамицина | 1976 |
|
SU628822A3 |
