Уровень техники
Настоящее изобретение относится к усовершенствованию композиций, содержащих пептидные аналоги глюкагоноподобного пептида-1 и/или их фармацевтически приемлемые соли, к способам получения таких композиций, к фармацевтическим композициям и к способам получения таких композиций для лечения млекопитающих.
Глюкагоноподобный пептид-1(7-36)амид (GLP-1) синтезируется в L-клетках кишечника при тканеспецифичном посттрансляционном процессинге предшественника глюкагона предпроглюкагона (Varndell, J.M. et al., J. Histochem Cytochem, 1985:33:1080-6) и высвобождается в кровь в ответ на прием пищи. Концентрация GLP-1 в плазме повышается от уровня приблизительно 15 пмоль/л натощак до максимального уровня 40 пмоль/л после приема пищи. Было показано, что, при таком повышении концентрации глюкозы в плазме, происходит приблизительно трехкратное увеличение инсулина в плазме, если глюкоза поступает перорально по сравнению с внутривенным введением (Kreymann B. et al., Lancet 1987:2, 1300-4). Такое алиментарное увеличение секреции инсулина, известное как "инкретиновый эффект", является прежде всего гуморальным, и полагают, что GLP-1 является наиболее сильным физиологическим инкретином человека. Кроме инсулинотропного эффекта, GLP-1 подавляет секрецию глюкагона, задерживает опорожнение желудка (Wettergren A. et al., Dig Dis Sci 1993:38:665-73) и может повышать периферическую утилизацию глюкозы (D'Alessio D.A. et al., J. Clin Invest 1994:93:2293-6).
В 1994 году в результате наблюдения было сделано предположение, что GLP-1 обладает терапевтической эффективностью, что единичная подкожная (п/к) доза GLP-1 может полностью нормализовать уровни глюкозы после приема пищи у пациентов с инсулинонезависимым сахарным диабетом (NIDDM) (Gutniak M.K. et al., Diabetes Care 1994:17:1039-44). Этот эффект, как полагают, опосредуется как усилением секреции инсулина, так и ослаблением секреции глюкагона. Кроме того, было показано, что внутривенное введение GLP-1 задерживает опорожнение желудка после приема пищи у пациентов с NIDDM (Williams B. et al., J. Clin Endo Metab 1996:81:327-32). В отличие от производных сульфонилмочевины, инсулинотропный эффект GLP-1 зависит от концентрации глюкозы в плазме (Holz G.G. 4th, et al., Nature 1993:361:362-5). Таким образом, ослабление секреции инсулина, опосредованное GLP-1 при низкой концентрации глюкозы в плазме, защищает от тяжелой гипогликемии. Такое сочетание эффектов дает GLP-1 исключительные преимущества над другими средствами, в настоящее время используемыми для лечения NIDDM.
Многочисленные исследования показали, что прием здоровыми индивидами GLP-1 значительно влияет на гликемические уровни, а также концентрацию инсулина и глюкагона (Orskov C, Diabetologia 35:701-711, 1992; Holst J.J. et al., Potential of GLP-1 in diabetes management in Glucagon III, Handbook of Experimental Pharmacology, Lefevbre P.J., Ed. Berlin, Springer Verlag, 1996, p. 311-326), а эти эффекты зависят от глюкозы (Kreymann B. et al., Lancet ii: 1300-1304, 1987; Weir G.C. et al., Diabetes 38:338-342, 1989). Более того, GLP-1 также эффективен у пациентов с диабетом (Gutniak M., N. Engl J Med 226:1316-1322, 1992; Nathan D.M. et al., Diabetes Care 15:270-276, 1992), так как нормализует уровни глюкозы крови у больных диабетом типа 2 (Nauck M.A. et al., Diabetologia 36:741-744, 1993) и улучшает гликемический контроль у больных диабетом типа 1 (Creutzfeldt W.O. et al., Diabetes Care 19:580-586, 1996), что указывает на его способность, в числе прочего, повышать чувствительность к инсулину/снижать резистентность к инсулину. GLP-1 и его агонисты были предложены для использования у индивидов с риском развития инсулинонезависимого диабета (смотрите WO 00/07617), а также для лечения гестационного диабета (патент США № 20040266670).
Кроме указанного выше, GLP-1 и его агонисты были предложены для использования в ряде терапевтических схем у млекопитающих, например людей, включая, но ими не ограничиваясь: повышение обучаемости, усиление нейропротективного действия и/или уменьшение симптома заболевания или расстройства центральной нервной системы, например, за счет регулирования нейрогенеза, и, например, при болезни Паркинсона, болезни Альцгеймера, болезни Хантингтона, боковом амиотрофическиом склерозе (ALS), инсульте, синдроме дефицита внимания (ADD) и психоневрологических синдромах (патентные публикации США №№ 20050009742 и 20020115605); дифференцировка стволовых клеток/клеток-предшественников печени в функциональные клетки поджелудочной железы (WO03/033697); профилактика разрушения бета-клеток (патентные публикации США №№ 20040053819 и 20030220251) и стимуляция пролиферации бета-клеток (патентная публикация США № 20030224983); лечение ожирения (патентная публикация США № 20040018975; WO98/19698); подавление аппетита и стимуляцию чувства насыщения (патентная публикация США № 20030232754); лечение синдрома раздраженного кишечника (WO 99/64060); снижение заболеваемости и/или смертности при инфаркте миокарда (патентная публикация США № 20040162241, WO98/08531) и инсульте (смотрите WO 00/16797); лечение острого коронарного синдрома, проявляющегося инфарктом миокарда с отсутствием зубца Q на ЭКГ (патентная публикация США № 20040002454); ослабление послеоперационных катаболических изменений (патент США № 6006753); лечение гибернации миокарда или диабетической кардиомиопатии (патентная публикация США № 20050096276); снижение уровней норэпинефрина в плазме (патентная публикация США № 20050096276); усиление выведения натрия с мочой, снижение концентрации калия в моче (патентная публикация США № 20050037958); лечение состояний или расстройств, связанных с токсической гиперволемией, например, при почечной недостаточности, застойной сердечной недостаточности, нефротическом синдроме, циррозе, отеке легких и гипертензии (патентная публикация США № 20050037958); индукцию инотропного эффекта и усиление сокращаемости сердечной мышцы (патентная публикация США № 20050037958); лечение синдрома поликистоза яичников (патентные публикации США №№ 20040266678 и 20040029784); лечение дыхательной недостаточности (патентная публикация США № 20040235726); улучшение питания при неалиментарном пути введения, а именно при внутривенной, подкожной, внутримышечной, перитонеальной или другой инъекции или инфузии (патентная публикация США № 20040209814); лечение нефропатии (патентная публикация США № 20040209803); лечение систолической дисфункции левого желудочка, например, при патологической фракции выброса левого желудочка (патентная публикация США № 20040097411); ингибирование антро-дуоденальной моторики, например, при лечении или профилактики желудочно-кишечных расстройств, таких как диарея, послеоперационный демпинг-синдром и синдром раздраженного кишечника, и в качестве медикаментозной подготовки при эндоскопических процедурах (патентная публикация США № 20030216292); лечение полиневропатии критического состояния (CIPN) и синдрома системной воспалительной реакции (SIRS) (патентная публикация США № 20030199445); регулирование уровней триглицеридов и лечение дислипидемии (публикации патентов США №№ 20030036504 и 20030143183); лечение поражения ткани органа, вызванного реперфузией кровотока после ишемии (патентная публикация США № 20020147131); уменьшение фактора риска развития коронарной болезни сердца (CHDRF) (патентная публикация США № 20020045636) и другие.
Однако GLP-1 является метаболически нестабильным, его период полувыведения из плазмы крови (t1/2) составляет лишь 1-2 минуты in vivo. Экзогенно введенный GLP-1 также быстро деградирует (Deacon C.F. et al., Diabetes 44:1126-1131, 1995). Такая метаболическая нестабильность ограничивает терапевтические возможности природного GLP-1. Были сделаны попытки улучшить терапевтические возможности GLP-1 и его аналогов путем улучшения его лекарственных форм. Например, в международной патентной публикации № WO01/57084 описан процесс получения кристаллов аналогов GLP-1, которые, как указывается, используются при получении фармацевтических композиций, таких как лекарственные средства, вводимые инъекцией, содержащие кристаллы и фармацевтически приемлемый носитель. Неоднородные микрокристаллические кластеры GLP-1(7-37)OH выращивали из солевых растворов и анализировали после пропитки цинком и/или м-крезолом (Kim and Haren, Pharma. Res. Vol. 12 No. 11 (1995)). Из фосфатных растворов, содержащих цинк или протамин, были получены сырые суспензии кристаллов GLP(7-36)NH2, содержащие игольчатые кристаллы и аморфный осадок (Pridal et. al., International Journal of Pharmaceutics Vol. 136, pp. 53-59 (1996)). В европейской патентной публикации № EP0619322A2 описано получение микрокристаллических форм GLP-1(7-37)OH путем смешивания белковых растворов в буфере pH 7-8,5 с определенными комбинациями солей и низкомолекулярных полиэтиленгликолей (ПЭГ). В патенте США № 6566490 описано введение затравки микрокристаллов, и в числе прочего, GLP-1, которое, как указано, способствует получению очищенных пептидных продуктов. В патенте США 6555521 (США '521) описаны кристаллы GLP-1, имеющие форму тетрагонального плоского стержня или пластинчатую форму, которые, как указывается, обладают повышенной чистотой и демонстрируют пролонгированную активность in vivo. В США '521 сообщается, что такие кристаллы являются относительно однородными и сохраняются в суспензии в течение более длительного периода времени, чем предшествующие кристаллические кластеры и аморфные кристаллические суспензии, которые, как указано, быстро оседают, агрегируют или слипаются, забивают иглы для шприцев и, как правило, осложняют непредсказуемое дозирование.
Биодеградируемый триблоксополимер поли[(dl-лактид-co-гликолид)-β-этиленгликоль-β-(лактид-co-гликолид)] был предложен для использования в лекарственных формах с контролируемым высвобождением GLP-1. Однако, как и в случае других полимерных систем, получение триблоксополимера предусматривает сложные методики и формирование несоответствующих частиц.
Аналогично, также были предложены биодеградируемые полимеры, например, поли(сополимер молочной и гликолевой кислоты) (PLGA), для использования в лекарственных формах пептидов с замедленным высвобождением. Однако использование таких биодеградируемых полимеров, не принятых в области техники, поскольку эти полимеры в основном плохо растворимы в воде, и при их получении требуются не смешивающиеся с водой органические растворители, например, метиленхлорид, и/или жесткие условия получения. Такие органические растворители и/или жесткие условия получения, как полагают, повышают риск индукции конформационного превращения интересующего пептида или белка, что приводит к снижению структурной целостности и нарушению биологической активности (Choi et al., Pharm. Research, Vol. 21, No. 5, (2004)). Полоксамеры имеют аналогичные проблемы.
Композиции GLP-1, описанные в указанных выше ссылках, не являются оптимальными для получения фармацевтических композиций GLP, так как задерживают примеси, и/или существует другая трудность, чтобы осуществлять воспроизводимо производство и введение. Также известно, что аналоги GLP вызывают тошноту при повышенных концентрациях, таким образом, существует необходимость обеспечить пролонгированный эффект лекарственного средства при пониженных начальных концентрациях в плазме (Ritzel et al., Diabetologia, 38: 720-725 (1995); Gutniak et al., Diabetes Care, 17(9): 1039-1044 (1994); Deacon et al., Diabetes, 44: 1126-1131 (1995)). Следовательно, существует необходимость в лекарственных формах GLP-1 с надежным и простым производством, которые можно легко и воспроизводимо вводить пациенту и которые обеспечивают пониженные начальные концентрации в плазме для того, чтобы снизить или исключить нежелательные побочные эффекты.
Сущность изобретения
Сущность изобретения приведена в абзацах ниже, а также в формуле изобретения. Таким образом, изобретение относится к фармацевтической композиции, содержащей аналог GLP-1. Особенно предпочтительным является аналог GLP-1 следующей формулы (I):
(Aib8,35)hGLP-1(7-36)NH2
(I)
или его фармацевтически приемлемая соль, где состав указанной композиции позволяет упростить получение, введение, улучшить фармакокинетические и фармакодинамические свойства, а также минимизировать отрицательные побочные эффекты. Предпочтительно, фармацевтическая композиция по изобретению не имеет в своем составе прозрачного водного раствора ZnCl2 с pH 4, в котором указанный [Aib8,35]hGLP-1(7-36)NH2 находится в концентрации 4 мг/мл и указанный ZnCl2 присутствует в концентрации 0,5 мг/мл.
В одном из предпочтительных вариантов осуществления изобретение относится к фармацевтической композиции с улучшенными характеристиками высвобождения лекарственного средства, предпочтительно, с пониженной начальной концентрацией.
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формулы (I) с пролонгированным действием.
В другом варианте осуществления изобретение также относится к фармацевтической композиции, которая выпадает в осадок in vivo при физиологическом значении pH с образованием in situ депо для обеспечения замедленного высвобождения лекарственного средства.
В еще одном варианте осуществления изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель или разбавитель. Предпочтительно, указанный носитель или разбавитель содержит воду.
В предпочтительных вариантах изобретение относится к фармацевтической композиции, содержащей соединение или аналог пептида GLP-1, полученный в виде соли пептида или в виде смеси пептида и его соли.
Предпочтительно, соль аналога пептида GLP-1 в указанной фармацевтической композиции выбрана из перечня фармацевтически приемлемых солей органических кислот, таких как уксусная, молочная, яблочная, аскорбиновая, янтарная, бензойная, лимонная, метансульфоновая или толуолсульфоновая кислоты, или фармацевтически приемлемых солей неорганических кислот, таких как хлористоводородная, бромистоводородная, йодистоводородная, серная или фосфорная кислоты. Фармацевтически приемлемые соли сильных кислот, таких как хлористоводородная кислота, являются особенно предпочтительными. Под сильной кислотой понимают кислоту с pKA, равной менее 4,5. Другими предпочтительными солями пептида в указанной фармацевтической композиции являются соли органических кислот, таких как уксусная кислота или трифторуксусная кислота, молочная, яблочная, аскорбиновая, янтарная, бензойная или лимонная кислота.
В одном из предпочтительных вариантов осуществления растворимость, pH и характер высвобождения фармацевтической композиции могут регулироваться путем подбора молярного отношения аналога GLP-1 в форме соли и аналога GLP-1 не в форме соли для улучшения характеристик высвобождения и уменьшения начального пика концентрации аналога GLP-1.
В предпочтительном варианте осуществления фармацевтическая композиция, кроме того, содержит двухвалентный металл, для снижения растворимости композиции в воде, и, таким образом, улучшения характеристик высвобождения, наряду с уменьшением начального выброса или пика концентраций в плазме. Предпочтительные двухвалентные металлы включают в себя цинк и медь. Формы солей двухвалентных металлов являются особенно предпочтительными, включая, но ими не ограничиваясь, хлоридные и ацетатные соли двухвалентных металлов. Наиболее предпочтительными являются CuAc2, CuCl2, ZnAc2 и/или ZnCl2. Предпочтительно, двухвалентный металл и/или соли двухвалентного металла находятся в указанной фармацевтической композиции в концентрации от приблизительно 0,0005 до приблизительно 50 мг/мл. Даже более предпочтительно, двухвалентный металл и/или соли двухвалентного металла находятся в указанной фармацевтической композиции в концентрации от приблизительно 0,01 до приблизительно 0,50 мг/мл. Более предпочтительно, указанная фармацевтическая композиция содержит разбавитель, где указанный разбавитель содержит фармацевтически приемлемый водный раствор. Разбавитель может содержать стерильную воду.
В другом варианте осуществления указанная фармацевтическая композиция дополнительно содержит двухвалентный металл и/или соль двухвалентного металла, где молярное соотношение указанного аналога GLP-1 и указанного двухвалентного металла и/или соли двухвалентного металла в указанной фармацевтической композиции составляет от приблизительно 6:1 до приблизительно 1:1. Предпочтительно, указанное соотношение составляет от приблизительно 5,5:1 до приблизительно 1:1. Более предпочтительно, указанное соотношение составляет от приблизительно 5,4:1 до приблизительно 1,5:1. Однако еще более предпочтительно, указанное соотношение составляет приблизительно 5,4:1, 4,0:1 или 1,5:1. Наиболее предпочтительно, указанное соотношение составляет приблизительно 1,5:1. В данном аспекте изобретения "приблизительно" означает соотношение 1,5:1±10% каждой указанной величины, таким образом, предполагаемое соотношение включает в себя соотношения, включающие, например, 1,35-1,65:0,85-1,15.
Предпочтительно, указанная фармацевтическая композиция содержит водную смесь, суспензию или раствор, где указанный аналог GLP-1, соединение формулы (I) или его соль находятся в концентрации приблизительно 0,5-30% (масс./масс.). Более предпочтительно, концентрация указанного аналога GLP-1 и/или его соли в указанной водной смеси, суспензии или растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30% (масс./масс.). Более предпочтительно, концентрация указанного аналога GLP-1 и/или его соли в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 14, 15, 16, 19, 20, 21, 22, 23, 24, 25, 26, 29 или 30% (масс./масс.). Еще более предпочтительно, концентрация указанного аналога GLP-1 и/или его соли в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 9, 10, 11, 22, 23, 24, 25 или 26% (масс./масс.). Еще более предпочтительно, концентрация указанного аналога GLP-1 и/или его соли в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 10, 22, 23, 24, 25 или 26% (масс./масс.). Еще более предпочтительно, концентрация указанного аналога GLP-1 и/или его соли в указанном водном растворе составляет приблизительно 1, 2, 5, 10, 23 или 25% (масс./масс.). Под "приблизительно" понимают следующее: для концентраций от приблизительно 0,5 до приблизительно 4% необходимый диапазон составляет ±0,5% указанной величины (например, 0,5-1,5% составляет приблизительно 1%); для указанных концентраций приблизительно 5% и выше, желательный диапазон составляет 20% указанной величины (например, 8-12% соответствует приблизительно 10%).
Предпочтительно, концентрация [Aib8,35]hGLP-1(7-36)NH2, аналога GLP-1 или его соли в фармацевтической композиции составляет приблизительно 1% (масса/объем), а молярное соотношение [Aib8,35]hGLP-1(7-36)NH2 и указанного двухвалентного металла и/или соли двухвалентного металла составляет приблизительно 1,5:1. Более предпочтительно, концентрация [Aib8,35]hGLP-1(7-36)NH2 или его соли в указанной фармацевтической композиции составляет приблизительно 2% (масса/объем), а молярное соотношение [Aib8,35]hGLP-1(7-36)NH2 и указанного двухвалентного металла и/или соли двухвалентного металла составляет приблизительно 1,5:1. Еще более предпочтительно, концентрация [Aib8,35]hGLP-1(7-36)NH2 или его соли в указанной фармацевтической композиции составляет приблизительно 10% (масса/объем), а молярное соотношение [Aib8,35]hGLP-1(7-36)NH2 и указанного двухвалентного металла и/или соли двухвалентного металла составляет приблизительно 1,5:1. Наиболее предпочтительно, концентрация [Aib8,35]hGLP-1(7-36)NH2 или его соли в указанной фармацевтической композиции составляет приблизительно 23 или приблизительно 25% (масса/объем), а молярное соотношение [Aib8,35]hGLP-1(7-36)NH2 и указанного двухвалентного металла и/или соли двухвалентного металла составляет приблизительно 1,5:1.
В предпочтительном варианте осуществления концентрация аналога GLP-1, [Aib8,35]hGLP-1(7-36)NH2 или его солей в фармацевтической композиции составляет приблизительно 5% (масса/объем), а молярное соотношение пептида и двухвалентного металла и/или соли двухвалентного металла составляет приблизительно 5,4:1. Более предпочтительно, концентрация [Aib8,35]hGLP-1(7-36)NH2 или его соли в указанной композиции составляет приблизительно 5% (масса/объем), а указанное соотношение составляет приблизительно 4,0:1. Однако более предпочтительно, концентрация [Aib8,35]hGLP-1(7-36)NH2 или его соли в указанной композиции составляет приблизительно 10% (масса/объем), а указанное соотношение составляет приблизительно 5,4:1. Еще более предпочтительно, концентрация [Aib8,35]hGLP-1(7-36)NH2 или его соли в указанной композиции составляет приблизительно 10% (масса/объем), а указанное соотношение составляет приблизительно 4,0:1.
Предпочтительно, указанный двухвалентный металл и/или соль двухвалентного металла находятся в виде хлорида цинка или ацетата цинка. Более предпочтительно, указанный ацетат цинка находится в виде ZnAc2·2H2O.
В другом варианте осуществления указанный двухвалентный металл и/или соль двухвалентного металла находятся в виде хлорида меди или ацетата меди.
В одном из вариантов осуществления значение pH указанной фармацевтической композиции увеличивают с помощью основания. Более предпочтительно, регулирование указанного значения pH осуществляют с помощью NaOH. Еще более предпочтительно, значение pH указанной фармацевтической композиции регулируют с помощью NaOH, и если разбавляют до приблизительно 1/2 от начальной концентрации, используя 0,9% NaCl, то получают значение pH, равное приблизительно 5,0-5,5, используя непосредственно потенциометрическое титрование.
В предпочтительном варианте осуществления изобретение относится к фармацевтической композиции, которая составлена так, что пептидный аналог GLP-1 или его соль, например соединение формулы (I) или его соль, высвобождаются в организме индивида, например, млекопитающего, предпочтительно, человека, в течение продолжительного периода времени. Предпочтительно, указанное высвобождение указанного соединения продолжается по меньшей мере час, более предпочтительно, по меньшей мере 4, 6, 12 или 24 часа. Еще более предпочтительно, указанная композиция составлена так, что соединение формулы (I) высвобождается в организме индивида в течение по меньшей мере 36, 48, 60, 72, 84 или 96 часов. Более предпочтительно, указанная композиция составлена так, что соединение формулы высвобождается в организме индивида в течение по меньшей мере приблизительно 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 дней. Еще более предпочтительно, указанная композиция составлена так, что соединение формулы (I) высвобождается в организме индивида в течение по меньшей мере приблизительно 2, 3 или 4 недель. Еще более предпочтительно, указанная композиция составлена так, что соединение формулы (I) высвобождается в организме индивида в течение не менее приблизительно 1, 1,5, 2 или 3 месяцев или дольше.
В одном из аспектов изобретения регулирование состава солей пептидного аналога GLP-1 в указанной фармацевтической композиции улучшает растворимость и стабильность пептидного аналога GLP-1 в фармацевтической композиции и, кроме того, обеспечивает улучшение характера высвобождения in vivo за счет снижения начального выброса высвобождения.
Термин "регулирование" в данном аспекте изобретения означает изменение состава соли за счет регулирования молярного соотношения аналога GLP-1, находящегося в форме соли, и аналога GLP-1 не в виде соли.
Еще более предпочтительно, соль пептида в указанной фармацевтической композиции представляет собой соль хлористоводородной или уксусной кислоты, или хлориды или ацетаты указанного пептида формулы (I). В указанной фармацевтической композиции ацетат или хлорид находится в конечном молярном соотношении ацетата или хлорида и указанного соединения формулы (I) в диапазоне от приблизительно 0,5:1 до приблизительно 10:1. Более предпочтительно, указанное соотношение изменяется от приблизительно 0,8:1 до приблизительно 9:1. Даже более предпочтительно, указанное соотношение составляет от приблизительно 1:1 до приблизительно 6:1. Наиболее предпочтительно, указанное соотношение составляет приблизительно 3,0:1, в частности, 3,2:1.
В этом аспекте изобретения молярное соотношение ацетата или хлорида и пептида обозначает молярную пропорцию ацетата (CH3COO-) или хлорида (Cl-) в фармацевтической композиции к молярной пропорции пептида в фармацевтической композиции. Например, при молярном соотношении 3:1 в фармацевтической композиции ацетата, молярное содержание в три раза больше молярного содержания пептида в пропорции. Это является стехиометрическим соотношением соединения по сравнению с другим соединением.
В данном аспекте изобретения термин "приблизительно" означает соотношение 1,5:1±10% каждой указанной величины, таким образом, предполагаемое соотношение включает в себя соотношения, включающие, например, 1,35-1,65:0,85-1,15.
В других предпочтительных аспектах изобретения значение pH фармацевтической композиции устанавливают путем регулирования содержания ацетата в композиции. Предпочтительно, диапазон значений pH указанной фармацевтической композиции составляет pH от 3 до 6. Более предпочтительно, указанный диапазон значений pH указанной фармацевтической композиции составляет pH от 3,5 до 5,5. Наиболее предпочтительно, указанный диапазон значений pH указанной фармацевтической композиции составляет pH от 4,2 до 4,6.
Предпочтительно, для подкисления фармацевтической композиции содержание ацетата может увеличиваться при добавлении уксусной кислоты.
В одном из вариантов осуществления значение pH указанной фармацевтической композиции может быть увеличено исходя из пептидной соли аналога GLP-1, имеющей низкое содержание ацетата или не содержащей ацетат, путем регулирования содержания ацетата.
В предпочтительных вариантах осуществления установление значения pH в конечной фармацевтической композиции путем регулирования содержания ацетата и хлорида делает возможным регулирование таких параметров, как концентрация пептида, концентрация цинка, химическая стабильность, физическая стабильность и характер высвобождения in vivo путем снижения начального выброса высвобождения.
В одном из аспектов изобретения содержание Zn или Cu является фиксированным, а значение pH контролируется путем регулирования содержания ацетата. Повышенное содержание ацетата приводит к улучшению растворимости и физической стабильности, а пониженное содержание ацетата приводит к увеличению влияния на значение pH и снижению влияния на Cмакс.
В предпочтительных вариантах осуществления указанная фармацевтическая композиция содержит водную смесь, суспензию или раствор.
Настоящее изобретение также относится к способу индукции эффекта агониста GLP-1, где указанный способ включает приведение рецептора лиганда GLP-1(7-36)NH2 в контакт с аналогом GLP-1 или его солью, напрямую или опосредованно.
В указанном выше способе указанный рецептор лиганда GLP-1(7-36)NH2 находится у животного, предпочтительно, примата, более предпочтительно, человека. Таким образом, в этом варианте осуществления настоящее изобретение относится к способу индукции эффекта агониста рецептора GLP-1 у индивида, который включает введение указанному индивиду композиции по настоящему изобретению, где указанная композиция содержит эффективное количество аналога GLP-1 или его фармацевтически приемлемую соль.
В предпочтительном аспекте указанного выше способа указанный индивид является человеком, страдающим или имеющим риск развития заболевания или состояния, выбранного из группы, состоящей из диабета типа I, диабета типа II, гестационного диабета, ожирения, булимии, отсутствия чувства насыщения и нарушения обмена веществ. Предпочтительно, указанное заболевание представляет собой диабет типа I или диабет типа II.
В еще одном более предпочтительном аспекте указанного выше способа указанный индивид является человеком, страдающим или имеющим риск развития заболевания, выбранного из группы, состоящей из диабета типа I, диабета типа II, ожирения, глюкагономы, секреторных расстройств дыхательных путей, артрита, остеопороза, растройств центральной нервной системы, рестеноза, нейродегенеративного заболевания, почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза, отека легких, гипертензии и расстройств, при которых желательно снизить потребление пищи, заболевания или нарушения центральной нервной системы (например, за счет регулирования нейрогенеза и, например, болезнь Паркинсона, болезнь Альцгеймера, болезнь Хантингтона, ALS, инсульт, ADD и психоневрологические синдромы), синдрома раздраженного кишечника, инфаркта миокарда (например, снижая связанные с ним заболеваемость и/или смертность), инсульта, острого коронарного синдрома (например, отличающегося отсутствием зубца Q) инфаркта миокарда; послеоперационных катаболических изменений, гибернации миокарда или диабетической кардиомиопатии, недостаточного выведения натрия с мочой, избыточной концентрации калия в моче, состояний или расстройств, связанных с токсической гиперволемией (например, при почечной недостаточности, застойной сердечной недостаточности, нефротическом синдроме, циррозе, отеке легких и гипертензии), синдрома поликистоза яичников, дыхательной недостаточности, нефропатии, систолической дисфункции левого желудочка (например, при патологической фракции выброса левого желудочка), желудочно-кишечных расстройств, таких как диарея, послеоперационный демпинг-синдром и синдром раздраженной кишки (а именно, за счет ингибирования антро-дуоденальной моторики), полиневропатии критического состояния (CIPN), синдрома системной воспалительной реакции (SIRS), дислипидемии, поражения ткани органа, вызванного реперфузией кровотока после ишемии, фактора риска развития ишемической болезни сердца (CHDRF).
В дополнительном аспекте изобретения изобретение относится к способу дифференцировки стволовых клеток/клеток-предшественников печени в функциональные клетки поджелудочной железы, предотвращения разрушения бета-клеток и стимуляции пролиферации бета-клеток, снижению уровней норэпинефрина в плазме крови, индукции инотропного эффекта и усиления сокращаемости сердечной мышцы, улучшения питания при неалиментарном пути (например, за счет внутривенной, подкожной, внутримышечной, перитонеальной или другой инъекции или инфузии), медикаментозной подготовки индивида к эндоскопическим процедурам, и регулированию уровней триглицеридов у больных, где указанный способ включает введение указанному индивиду композиции по настоящему изобретению, содержащей эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли. Предпочтительно, указанный индивид является млекопитающим, более предпочтительно, приматом, еще более предпочтительно, человеком.
Краткое описание чертежей
На фиг.1 представлены параметры плазмы (медиана), полученные после однократного подкожного (s.с.) введения собакам приблизительно 1 мг [Aib8,35]hGLP-1(7-36)NH2. В каждом случае пептид вводили в виде водной цинксодержащей композиции, содержащей приблизительно 1% (масс./об.) пептида и имеющей молярное соотношение пептид:Zn, равное приблизительно 1,5. Закрашенные квадраты и пустые квадраты соответствуют композициям, в которых значение pH регулируется с помощью NaOH, как описано в описании; закрашенные треугольники соответствуют композиции, в которой рН не регулировали с помощью NaOH; закрашенные круги соответствуют композиции, забуференной AcOH/AcO-.
На фиг.2 представлены параметры плазмы (медиана), полученные после однократного подкожного (s.с.) введения собакам приблизительно 15 мг [Aib8,35]hGLP-1(7-36)NH2. В каждом случае пептид вводили в виде водной цинксодержащей композиции, содержащей приблизительно 10% (масс./об.) пептида и имеющей молярное соотношение пептид:Zn, равное приблизительно 1,5. Закрашенные квадраты и пустые квадраты соответствуют композициям, в которых значение pH регулируется с помощью NaOH, как описано в описании; закрашенные треугольники соответствуют композиции, в которой значение pH не регулировали с помощью NaOH; закрашенные круги соответствуют композициии, забуференной AcOH/AcO-.
На фиг.3 представлены параметры плазмы (медиана), полученные после однократного подкожного (s.с.) введения собакам приблизительно 1 мг [Aib8,35]hGLP-1(7-36)NH2. В каждом случае пептид вводили в виде полутвердой водной цинксодержащей композиции следующим образом: черные круги: приблизительно 5% (масс./об.) пептида, молярное соотношение пептид:Zn составляет приблизительно 5,4:1, без регулирования значения pH; белые круги: приблизительно 10% (масс./об.) пептида, молярное соотношение пептид:Zn составляет приблизительно 5,4:1, без регулирования значения pH; белые квадраты: приблизительно 10% (масс./об.) пептида, молярное соотношение пептид:Zn составляет приблизительно 5,4:1, pH регулировали с помощью NaOH; черные квадраты: приблизительно 10% (масс./об.) пептида, молярное соотношение пептид:Zn составляет приблизительно 4:1, pH регулировали с помощью NaOH.
На фиг.4 схематично представлены различные устройства, которые можно использовать для получения некоторых композиций по настоящему изобретению.
На фиг.5 представлены параметры плазмы (медиана), полученные после однократного подкожного (s.c.) введения собакам приблизительно 1 мг [Aib8,35]hGLP-1(7-36)NH2. Пептид вводили в виде водной цинксодержащей композиции с концентрацией пептида, равной приблизительно 2%, и молярным соотношением пептид:Zn, равным приблизительно 1,5:1.
На фиг.6 представлены параметры плазмы (медиана), полученные после однократного подкожного (s.c.) введения собакам приблизительно 15 мг [Aib8,35]hGLP-1(7-36)NH2. Пептид вводили в виде полутвердой цинксодержащей композиции с концентрацией пептида, равной приблизительно 25%, и молярным соотношением пептид:Zn, равным приблизительно 4:1.
На фиг.7 представлены параметры плазмы (медиана), полученные после однократного подкожного (s.c.) введения собакам приблизительно 15 мг [Aib8,35]hGLP-1(7-36)NH2. Пептид вводили в виде полутвердой цинксодержащей композиции с концентрацией пептида, равной приблизительно 23%, и молярным соотношением пептид:Zn, равным приблизительно 1,5:1.
На фиг.8 представлен полный цикл параметров плазмы (медиана), полученных после однократного подкожного (s.c.) введения крысам 0,3 мг (3 мкл 10% раствора) анализируемых композиций HCl-соли аналога GLP-1:
(1) HCl-соль [Aib8,35]hGLP-1(7-36)NH2 с CuCl2: молярное соотношение [Aib8,35]hGLP-1(7-36)NH2/CuCl2 составляет 1,5:1. Концентрация пептида составляет 10% (30 мМ) в воде (масс./масс.) с приблизительно pH 5,5.
(2) HCl-соль [Aib8,35]hGLP-1(7-36)NH2 с ZnCl2: молярное соотношение ([Aib8,35]hGLP-1(7-36)NH2/ZnCl2 составляет 1,5:1. Концентрация пептида составляет 10% (30 мМ) в воде (масс./масс.) с приблизительно pH 5,5.
На фиг.9 представлен полный цикл параметров плазмы (медиана), полученных после однократного подкожного (s.c.) введения крысам 0,3 мг (3 мкл 10% раствора) анализируемых композиций ацетатной соли аналога GLP-1:
Ацетатная соль [Aib8,35]hGLP-1(7-36)NH2 с ZnCl2: молярное соотношение [Aib8,35]hGLP-1(7-36)NH2/ZnCl2 составляет 1,5:1. Концентрация пептида составляет 10% (30 мМ) в воде (масс./масс.) с pH приблизительно 5,5.
На фиг.10 представлена начальная часть параметров плазмы (медиана), полученных после однократного подкожного (s.c.) введения крысам 0,3 мг (3 мкл 10% раствора) анализируемых композиций, представленных на фиг.8.
На фиг.11 представлена начальная часть параметров плазмы (медиана), полученных после однократного подкожного (s.c.) введения крысам 0,3 мг (3 мкл 10% раствора) анализируемых композиций, представленных на фиг.9.
На фиг.12 представлен рассчитанный процент [Aib8,35]hGLP-1(7-36)NH2, оставшегося на месте инъекции у крыс после однократного подкожного (s.c.) введения 0,3 мг (3 мкл 10% раствора) трех анализируемых композиций, представленных на фиг.8.
Подробное описание изобретения
Предпочтительный пептид GLP-1, который может быть использован в виде соли пептида по изобретению, обозначен в описании следующим образом, например, [Aib8,35]hGLP-1(7-36)NH2, с аминокислотными заменами в природной последовательности, расположенными в первых скобках (например, Aib8,35 обозначает, что Ala8 и Gly35 в hGLP-1 заменены на Aib). Aib является аббревиатурой α-аминоизомасляной кислоты. Аббревиатура GLP-1 обозначает глюкагоноподобный пептид-1; hGLP-1 обозначает глюкагоноподобный пептид-1 человека. Цифры между вторыми скобками обозначают число аминокислот в пептиде (например, hGLP-1(7-36) обозначает аминокислоты 7-36 пептидной последовательности GLP-1 человека). Последовательность hGLP-1(7-37) приведена в статье Mojsov S., Int. J. Peptide Protein Res., 40, 1992, pp. 333-342. Обозначение "NH2" в hGLP-1(7-36)NH2 указывает на то, что C-конец пептида является амидированным. hGLP-1(7-36) означает, что C-конец является свободной кислотой. В hGLP-1(7-38) остатки в положениях 37 и 38 являются Gly и Arg, соответственно, если не указано иное.
Особенно предпочтительные аналоги пептида GLP-1, используемые в настоящем изобретении, находятся в форме фармацевтически приемлемых солей. Примеры таких солей включают, но ими не ограничиваются, соли органических кислот (например, уксусной, молочной, малеиновой, лимонной, яблочной, аскорбиновой, янтарной, бензойной, метансульфоновой, толуолсульфоновой или памовой кислот), неорганических кислот (например, хлористоводородной кислоты, серной кислоты или фосфорной кислоты) и полимерных кислот (например, дигалловой кислоты, карбоксиметилцеллюлозы, полимолочной, полигликолевой или сополимеров поли(молочной-гликолевой) кислот). Обычный способ получения соли пептида по настоящему изобретению хорошо известен в данной области и может проводиться стандартными способами обмена солей. Таким образом, соль ТФУ пептида по настоящему изобретению (соль ТФУ получают после очистки пептида путем использования препаративной ВЭЖХ, элюируя ТФУ-содержащими буферными растворами) может быть преобразована в другую соль, такую как ацетатная соль, путем растворения пептида в небольшом количестве 0,25 н. водного раствора уксусной кислоты. Полученный раствор наносят на полупрепаративную колонку ВЭЖХ (Zorbax, 300 SB, C-8). Колонку элюируют (1) 0,1 н. водным раствором ацетата аммония в течение 0,5 часа, (2) 0,25 н. водным раствором уксусной кислоты в течение 0,5 часа и (3) в линейном градиенте (20-100% раствора B в течение 30 минут) со скоростью потока 4 мл/мин (раствор A представляет собой 0,25 н. водный раствор уксусной кислоты; раствор B представляет собой 0,25 н. уксусную кислоту в ацетонитриле/воде 80:20). Фракции, содержащие пептид, собирают и лиофилизуют досуха.
Специалистам в данной области хорошо известно, что известное и возможное применение GLP-1 различно и многочисленно (смотрите Todd J.F. et al., Clinical Science, 1998, 95, pp. 325-329; и Todd J.F. et al., European Journal of Clinical Investigation, 1997, 27, pp.533-536). Таким образом, введение соединений по изобретению для индукции эффекта агониста может иметь те же эффекты и применение, что и собственно GLP-1. Эти разнообразные пути применения GLP-1 могут быть суммированы в зависимости от лечения следующим образом: диабет типа I, диабет типа II, ожирение, глюкагонома, секреторные расстройства дыхательных путей, нарушение обмена веществ, артрит, остеопороз, расстройства центральной нервной системы, рестеноз, нейродегенеративное заболевание, почечная недостаточность, застойная сердечная недостаточность, нефротический синдром, цирроз, отек легких, гипертензия и расстройства, при которых желательно снизить потребление пищи, а также различные другие обсуждаемые в описании состояния или нарушения. Таким образом, объемом настоящего изобретения предусмотрены описанные в настоящем описании фармацевтические композиции, содержащие в качестве активного компонента соединение формулы (I).
Доза активного компонента в композициях по настоящему изобретению может применяться, однако необходимо, чтобы количество активного компонента было достаточным для получения подходящей дозы. Выбранная доза зависит от желаемого терапевтического эффекта, способа введения и длительности лечения и, как правило, определяется лечащим врачом. Обычно эффективная доза для осуществления изобретения находится в диапазоне от 1×10-7 до 200 мг/кг/день, предпочтительно от 1×10-4 до 100 мг/кг/день и может быть введена в виде единичной дозы или разделена на множество доз.
Композиции по изобретению предпочтительно вводят парентерально, например внутримышечно, внутрибрюшинно, внутривенно, подкожно и тому подобное.
Препараты по изобретению для парентерального введения включают стерильные водные или неводные растворы, суспензии, гели или эмульсии при условии достижения желаемых параметров высвобождения in vivo. Примерами неводных растворов или везикул являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло, желатин и инъецируемые сложные органические эфиры, такие как этилолеат. Такие лекарственные формы могут также содержать вспомогательные вещества, такие как консерванты, увлажнители, эмульгаторы и диспергирующие агенты. Эти формы могут быть стерилизованы, например, путем фильтрования через задерживающий бактерии фильтр, путем добавления стерилизующих агентов в композиции, путем облучения композиций или путем нагрева композиций. Также они могут быть получены в виде твердых стерильных композиций, которые могут быть растворены в стерильной воде или некоторой другой стерильной инъецируемой среде непосредственно перед использованием.
Синтез пептидов
Пептиды, которые могут быть использованы в практике настоящего изобретения, могут быть получены и были получены с помощью стандартного твердофазного синтеза пептидов. Смотрите, например, Stewart J.M. et al., Solid Phase Synthesis (Pierce Chemical Co., 2d ed. 1984).
Следующие примеры описывают способы синтеза, которые могут быть и были использованы для получения пептидов, с которыми настоящее изобретение может быть эффективно использовано на практике, при этом способы синтеза хорошо известны специалистам в данной области. Другие способы также хорошо известны специалистам в данной области. Примеры приведены с целью иллюстрации и не предназначены для ограничения объема настоящего изобретения.
Указанные пептиды, такие как аналог GLP-1, могут быть получены различными способами синтеза, хорошо известными специалистам в данной области, которые могут включать окончательное осаждение пептида, процесс лиофилизации, сушку в вакууме или другие известные в данной области способы сушки. Ионообменная хроматография, осмотический обмен буфера и дифильтрация могут быть подходящими способами в настоящем изобретении для очистки или выделения пептида в виде различных солей.
Boc-βAla-OH, Boc-D-Arg(Tos)-OH и Boc-D-Asp(OcHex) были приобретены у Nova Biochem, Сан-Диего, Калифорния. Boc-Aun-OH был приобретен у Bachem, Кинг-оф-Пруссия, Пенсильвания. Boc-Ava-OH и Boc-Ado-OH были приобретены у Chem-Impex International, Вуд-Дейл, Иллинойс. Boc-2Nal-OH был приобретен у Synthetech, Inc., Олбани, Орегон.
Далее приведены расшифровки других используемых в настоящем описании аббревиатур: Boc - трет-бутилоксикарбонил, HF - фтороводород, Fm - формил, Xan - ксантил, Bzl - бензил, Tos - тозил, DNP - 2,4-динитрофенил, ДМФА (DMF) - диметилформамид, ДХМ (DCM) - дихлорметан, HBTU - 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат, DIEA - диизопропилэтиламин, HOAc - уксусная кислота, ТФУ - трифторуксусная кислота, 2ClZ - 2-хлорбензилоксикарбонил, 2BrZ - 2-бромбензилоксикарбонил, OcHex - O-циклогексил, Fmoc - 9-флуоренилметоксикарбонил, HOBt - N-гидроксибензотриазол, PAM-смола - 4-гидроксиметилфенилацетамидометильная смола, Трис (Tris) - трис(гидроксиметил)аминометан и Бис-Трис (Bis-Tris) - бис(2-гидроксиэтил)амино-трис(гидроксиметил)метан (а именно, 2-бис(2-гидроксиэтил)амино-2-(гидроксиметил)-1,3-пропандиол). Термин "галоген" включает фтор, хлор, бром и йод.
Если не указано иначе, все используемые технические и научные термины имеют те же значения, которые известны среднему специалисту в данной области, к которой относится настоящее изобретение. Кроме того, все публикации, патентные заявки и патенты, а также другие указанные в настоящем описании ссылки включены в качестве ссылки.
Пример 1
[Aib 8,35 ]hGLP-1(7-36)NH 2
Подробное описание способа синтеза [Aib8,35]hGLP-1(7-36)NH2 дано в международной патентной публикации № WO00/34331 (PCT/EP99/09660), содержание которой приведено в настоящем описании в полном объеме. Кратко, соединение синтезировали с использованием пептидного синтезатора Applied Biosystems (Фостер Сити, Калифорния), модель 430A, который был модифицирован для ускорения твердофазного синтеза пептидов с использованием Boc-химии. Смотрите Schnolzer et al., Int. J. Peptide Protein Res., 40:180 (1992). Использовали 4-метилбензгидриламиновую (MBHA) смолу (Peninsula, Бельмонт, Калифорния) с замещением 0,91 ммоль/г. Использовали Boc-аминокислоты (Bachem, Калифорния, Torrance, Калифорния; Nova Biochem., Лайола, Калифорния) со следующей защитой боковых цепей: Boc-Ala-OH, Boc-Arg(Tos)-OH, Boc-Asp(OcHex)-OH, Boc-Tyr(2BrZ)-OH, Boc-His(DNP)-OH, Boc-Val-OH, Boc-Leu-OH, Boc-Gly-OH, Boc-Gln-OH, Boc-Ile-OH, Boc-Lys(2ClZ)-OH, Boc-Thr(Bzl)-OH, Boc-Ser(Bzl)-OH, Boc-Phe-OH, Boc-Aib-OH, Boc-Glu(OcHex)-OH и Boc-Trp(Fm)-OH. Boc-группы удаляли путем обработки 100% ТФУ в течение 2×1 минуту. Boc-аминокислоты (2,5 ммоль) предварительно активировали с помощью HBTU (2,0 ммоль) и DIEA (1,0 мл) в 4 мл ДМФА и проводили реакцию присоединения без предшествующей нейтрализации соли ТФУ пептида на смоле. Время реакции присоединения составило 5 минут, за исключением остатков Boc-Aib-OH и следующих остатков, Boc-Lyys(2ClZ)-OH и Boc-His(DNP)-OH, для которых время реакции присоединения составило 2 часа.
В конце сборки пептидной цепи смолу обрабатывали раствором 20% меркаптоэтанол/10% DIEA в ДМФА в течение 2×30 минут для удаления DNP-групп боковой цепи His. N-концевую Boc-группу затем удаляли обработкой 100% ТФУ в течение 2×2 минуты. После нейтрализации пептида-смолы с помощью 10% DIEA в ДМФА (1×1 минуту), формильную группу боковой цепи Trp удаляли с помощью раствора 15% этаноламин/15% вода/70% ДМФА в течение 2×30 минут. Пептид-смолу промывали ДМФА и ДХМ и сушили при пониженном давлении. Конечное расщепление проводили при перемешивании пептида-смолы в 10 мл HF, содержащего 1 мл анизола и дитиотреитола (24 мг) при 0°C в течение 75 минут. HF удаляли в потоке азота. Остаток промывали простым эфиром (6×10 мл) и экстрагировали 4 н. HOAc (6×10 мл).
Пептидную смесь в водном экстракте очищали с помощью препаративной высокоэффективной жидкостной хроматографии с обращенной фазой (ВЭЖХ), используя колонку с обращенной фазой VYDAC® C18 (Nest Group, Southborough, Массачусетс). Колонку элюировали в линейном градиенте (20-50% раствора B в течение 105 минут) со скоростью потока 10 мл/мин (раствор А = вода, содержащая 0,1% ТФУ; раствор B = ацетонитрил, содержащий 0,1% ТФУ). Фракции собирали и проверяли с помощью аналитической ВЭЖХ. Содержащие чистый продукт фракции объединяли и лиофилизовали досуха. В одном из примеров синтеза соединения было получено 135 мг белого твердого вещества. Чистота составила 98,6% на основании анализа с помощью аналитической ВЭЖХ. Анализ масс-спектрометрии с ионизацией электроспреем (MS(ES))S дал молекулярную массу 3339,7 (в полном соответствии с вычисленной молекулярной массой 3339,7).
Пример 2
Методика получения лекарственной формы I
2.1. Материалы, базовые растворы, расчеты
A) Материалы: ZnCl2, гранулы NaOH и хлористоводородную кислоту, 35%, получали от Panreac Quimica, Барселона, Испания. WFI (стерильную воду для инъекций/промывания) получали от B. Braun Medical, Барселона, Испания.
B) Базовые растворы
(i) ZnCl 2 , pH=3:
1. При перемешивании добавляли 35% HCl в WFI до достижения pH=3.
2. В мерную колбу переносили взвешенное количество ZnCl2. При перемешивании добавляли pH=3 HCl до достижения конечной концентрации приблизительно 1-4 мг ZnCl2/мл.
(ii) ZnCl 2 , pH=2:
1. При перемешивании добавляли 35% HCl в WFI до достижения pH=2.
2. В мерную колбу переносили взвешенное количество ZnCl2. При перемешивании добавляли pH=2 HCl до достижения конечной концентрации приблизительно 4-12 мг ZnCl2/мл.
(iii) NaOH, 0,1-10 мг/мл:
1. В мерную колбу переносили взвешенное количество NaOH. При перемешивании добавляли WFI до достижения конечной концентрации приблизительно 0,1-10 мг NaOH/мл.
(iv) Лиофилизованные 20-мг аликвоты (Aib 8,35 )hGLP-1(7-36)NH 2 /флакон:
1. Получали 0,04% (об./об.) раствор уксусной кислоты и WFI.
2. В мерную колбу переносили взвешенное количество (Aib8,35)hGLP-1(7-36)NH2 (ацетатная соль). При перемешивании добавляли достаточное количество 0,04% уксусной кислоты до достижения конечной концентрации 20 мг (Aib8,35)hGLP-1(7-36)NH2/мл. После стерилизации фильтрованием на фильтрах размером 0,45 микрон аликвоты по 1 мл раствора переносили во флаконы для лиофилизации, растворы лиофилизовали, и высушенный продукт хранили при -22ºС.
(v) Лиофилизованные 50-мг аликвоты (Aib 8,35 )hGLP-1(7-36)NH 2 /флакон:
1. Получали 0,1% (об./об.) раствор уксусной кислоты и WFI.
2. В мерную колбу переносили взвешенное количество (Aib8,35)hGLP-1(7-36)NH2 (ацетатная соль). При перемешивании добавляли достаточное количество 0,1% уксусной кислоты до достижения конечной концентрации 50 мг (Aib8,35)hGLP-1(7-36)NH2/мл. После стерилизации фильтрованием аликвоты по 1 мл раствора переносили во флаконы для лиофилизации и лиофилизовали.
C) Расчеты
(i) Определение общей массы/объема наполнителя (Е) для композиции:
Е=(A×100/T)-(A/P),
где
Е = наполнитель в мг;
A = содержание чистого пептида (мг);
T = целевая концентрация композиции; например 2, если целью является 2%; и
P = концентрация чистого пептида (мг пептида/100 мг состава).
Что касается общего объема наполнителя, принимается допущение, что 1 мл = 1 г.
(ii) Определение объема/массы (W) ZnCl2 для добавления в каждый мл или г раствора композиции:
a) W=100% E для композиции, в которой регулирование pH не осуществляется;
b) W=80% E для жидких композиций, в которых пептид составляет приблизительно 1% или приблизительно 2%, или приблизительно до 10%, и pH регулируют с помощью основания;
c) W=50% E для полутвердых или гелевых композиций, в которых пептид составляет приблизительно 1% или приблизительно 2%, или приблизительно до 10%, и pH регулируют с помощью основания;
d) W=66,66% E для полутвердых или гелевых композиций, в которых пептид составляет приблизительно 25%, и pH регулируют с помощью основания;
e) W=90% E для составов, в которых пептид восстанавливают из лиофилизованного препарата, и pH регулируют с помощью основания.
(iii) Определение объема/массы (W) NaOH для добавления в каждый мл или г раствора композиции:
a) W=20% E для композиций, в которых пептид составляет приблизительно 1% или приблизительно 2%, или приблизительно до 10%, и pH регулируют с помощью основания;
b) W=50% E для полутвердых или гелевых композиций, в которых пептид составляет приблизительно 1% или приблизительно 2%, или приблизительно до 10%, и pH регулируют с помощью основания;
c) W=33,33% E для полутвердых или гелевых композиций, в которых пептид составляет приблизительно 25%, и pH регулируют с помощью основания;
d) W=10% E для композиций, в которых пептид восстанавливают из лиофилизованного препарата, и pH регулируют с помощью основания.
(iv) Определение концентрации ZnCl2 (мг/мл или мг/г), которую используют в каждой композиции:
[ZnCl2]=(136,29×A)/(W×3339,76×R),
где
A = содержание чистого пептида (мг);
R = молярное соотношение пептид/Zn;
R=1,5 для композиций, в которых пептид составляет приблизительно 1% или приблизительно 2%, или приблизительно 10%, или приблизительно до 23%;
R=4,0 для композиций, в которых пептид составляет приблизительно 25%; и
W = масса (г) или объем (мл) раствора ZnCl2, которые добавляют к каждому г или мл раствора композиции.
2.2. Получение композиций с 1-10% лиофилизованного пептида и ZnCl 2 , без регулирования pH
Как используется в описании, композиция с процентным содержанием пептида соответствует композиции, содержащей количество пептида по массе на общую массу композиции, например, 1% пептида описывает состав, содержащий 1 г пептида на 100 г всей композиции. Композиции, содержащие приблизительно 1% или приблизительно 2%, приблизительно до 10% пептида получали следующим образом. Лиофилизованные образцы (Aib8,35)hGLP-1(7-36)NH2, полученные как описано, тщательно смешивали с базовым раствором ZnCl2 pH 3 при 100% общем объеме наполнителя и [пептид:Zn]=1,5:1.
A) 1% композиции получали путем смешивания 20 мг лиофилизованного (Aib8,35)hGLP-1(7-36)NH2 (смотрите 2.1 B (iv) выше) с 2 мл раствора ZnCl2 (0,272 мг/мл; смотрите 2.1 B (i) выше).
B) 2% композиции получали путем смешивания 20 мг лиофилизованного (Aib8,35)hGLP-1(7-36)NH2 (смотрите 2.1 B (iv) выше) с 1 мл раствора ZnCl2 (0,544 мг/мл; смотрите 2.1 B (i) выше).
C) 10% композиции получали путем смешивания 50 мг лиофилизованного (Aib8,35)hGLP-1(7-36)NH2 (смотрите 2.1 B (v) выше) с 0,45 мл раствора ZnCl2 (3,023 мг/мл; смотрите 2.1 B (i) выше).
Лиофилизованные пептиды и растворы оставляли уравновешиваться при комнатной температуре. Необходимый объем раствора ZnCl2 вливали во флакон, содержащий лиофилизованный пептид, и позволяли протекать гидратации в течение приблизительно 2 минут для 1% или 2% композиций пептида, приблизительно до 60 минут для 10% композиции пептида или до тех пор, пока весь лиофилизованный пептид полностью гидратируется и раствор не будет содержать комочки пептида. После гидратации растворенный пептид встряхивали в течение приблизительно 1 минуты.
Соответствующее количество пептида может быть отобрано для дозирования, например, 100 мкл 1% раствора пептида, полученного согласно A выше, соответствует дозе 1 мг, 50 мкл 2% раствора пептида, полученного согласно B выше, соответствует дозе 1 мг, 150 мкл 10% раствора пептида, полученного согласно C выше, соответствует дозе 15 мг, и т.д.
Используя сведения рассматриваемой заявки, специалист в данной области может легко варьировать количества пептида и ZnCl2 для получения композиций, отличных от 1%, 2% и 10% композиций, подробно описанных ниже, а также желаемых доз.
2.3. Получение композиций с 1-10% лиофилизованного пептида и ZnCl 2 , с регулированием pH
Композиции, содержащие приблизительно 1% или приблизительно 2%, приблизительно до 10% пептида, были получены следующим образом. Лиофилизованные образцы (Aib8,35)hGLP-1(7-36)NH2, полученные как описано, тщательно смешивали с базовым раствором ZnCl2 pH 3 при 90% общего объема наполнителя. Необходимого значения pH достигали при добавлении разбавленного раствора NaOH.
A) 1% композиции получали путем смешивания 20 мг лиофилизованного (Aib8,35)hGLP-1(7-36)NH2 (смотрите 2.1 B (iv) выше) с 1,8 мл раствора ZnCl2 (смотрите 2.1 B (i) выше).
B) 2% композиции получали путем смешивания 20 мг лиофилизованного (Aib8,35)hGLP-1(7-36)NH2 (смотрите 2.1 B (iv) выше) с 0,9 мл раствора ZnCl2 (смотрите 2.1 B (i) выше).
C) 10% композиции получали путем смешивания 50 мг лиофилизованного (Aib8,35)hGLP-1(7-36)NH2 (смотрите 2.1 B (v) выше) с 0,40 мл раствора ZnCl2 (смотрите 2.1 B (i) выше).
К полученным выше растворам добавляли необходимый объем (10% общего объема наполнителя) разбавленного раствора NaOH для достижения целевой концентрации и pH. Например, для каждой:
1% композиции: добавляли 0,2 мл раствора NaOH с соответствующей концентрацией;
2% композиции: добавляли 0,1 мл раствора NaOH с соответствующей концентрацией;
10% композиции: добавляли 0,05 мл раствора NaOH с соответствующей концентрацией.
Используя сведения рассматриваемой заявки, специалист в данной области может варьировать количества пептида и ZnCl2 для получения композиций, отличных от 1%, 2% и 10% композиций, подробно описанных ниже.
2.4. Получение жидких композиций с 1-10% пептида и ZnCl 2 , без регулирования pH
Жидкие композиции, содержащие приблизительно 1% или приблизительно 2%, приблизительно до 10% пептида, получали следующим образом. Образцы (Aib8,35)hGLP-1(7-36)NH2 взвешивали и смешивали с базовым раствором ZnCl2 pH 3 для достижения целевой концентрации пептида 1%, 2%, до 10%. После смешивания композицию стерилизовали фильтрованием и хранили до использования.
2.5. Получение жидких композиций с 1-10% пептида и ZnCl 2 , с регулированием pH
Жидкие композиции, содержащие приблизительно 1% или приблизительно 2%, приблизительно до 10% пептида получали следующим образом. Образцы (Aib8,35)hGLP-1(7-36)NH2 взвешивали и тщательно смешивали с базовым раствором ZnCl2 pH 3 при 80% общего объема наполнителя. Раствор цинка может быть либо ZnCl2, либо ZnAc2·2H2O. Необходимого значения pH раствора достигали путем добавления разбавленного раствора NaOH. Препараты C5-C13 были получены с применением данного способа.
Используя сведения рассматриваемой заявки, специалист в данной области может варьировать количества пептида и ZnCl2 для получения композиций, отличных от 1%, 2% и 10% композиций, подробно описанных ниже.
2.6. Получение полутвердых/гелевых композиций с 25% пептида и ZnCl2, без регулирования pH
Полутвердые или гелевые композиции, содержащие приблизительно 25% пептида, были получены следующим образом. Образцы (Aib8,35)hGLP-1(7-36)NH2 взвешивали и тщательно смешивали с базовым раствором ZnCl2 pH 2 при 66,66% общего объема наполнителя. Раствор цинка может быть либо ZnCl2, либо ZnAc2·2H2O. Препараты C1-C2 были получены с применением данного способа.
В частности, полутвердые или гелевые композиции были получены с применением метода смешивания «к себе-от себя»:
a) желаемое количество пептида взвешивали в цилиндре одноразового шприца S1, предварительно оборудованном специальным ручным клапаном двухстороннего действия HV (В.Д.=0,5 мм), и трубку помещали внутрь отверстия шприца Люэра;
b) поршень шприца закрепляли штоком из нержавеющей стали SR;
c) HV в S1 присоединяли к источнику вакуума, и HV открывали. Через 10 минут HV закрывали;
d) раствор цинка аккуратно взвешивали в цилиндре второго одноразового шприца S2;
e) затем S2 соединяли со свободной частью HV;
f) HV открывали, и растворитель вытягивали с помощью вакуума в цилиндр, содержащий порошок пептида S1;
g) HV закрывали, и шприц с растворителем S2 удаляли, таким образом, гидратируя порошок пептида в S1;
h) SR удаляли, и поршень шприца медленно высвобождали;
i) поршень шприца двигали (к себе-от себя), без открытия HV, так что порошковая масса полностью смачивалась растворителем;
j) двухстороннюю соединительную вставку из нержавеющей стали SC (В.Д.=1,0 мм) помещали в шприц S2 с трубкой, помещенной внутрь отверстия шприца Люэра, и его поршень толкали до конца;
k) HV в S1 открывали, чтобы выпустить вакуум, и затем отделяли HV. Поршень шприца двигали так, чтобы минимизировать воздух в цилиндре; и
i) S1 и S2 соединяли с помощью SC, и композицию проталкивали из S1 в S2 через SC.
Используя сведения рассматриваемой заявки, специалист в данной области может варьировать количества пептида и ZnCl2 для получения композиций, отличных от 25% композиции, описанной в описании.
2.7. Получение полутвердых/гелевых композиций с 25% пептида и ZnCl 2 , с регулированием pH
Полутвердые или гелевые композиции, содержащие приблизительно 25% пептида, получали следующим образом. Образцы (Aib8,35)hGLP-1(7-36)NH2 взвешивали и тщательно смешивали с базовым раствором ZnCl2 pH 2 при 66,66% общего объема наполнителя. Раствор цинка может быть либо ZnCl2, либо ZnAc2·2H2O. Необходимого значения pH раствора достигали путем добавления разбавленного раствора NaOH. В данном примере общий объем жидкой фазы, добавленный к порошку, должен быть разделен между растворами цинка и NaOH. По этой причине концентрацию раствора цинка устанавливали так, чтобы общий объем необходимого раствора цинка был доведен до 50% общего объема жидкой фазы, добавляемой к порошку пептида (этап d). Остальные 50% общей жидкой фазы, добавляемые к порошку пептида, добавляли в виде раствора NaOH, как подробно описано ниже. Препараты C3 и C4 были получены с применением данного способа.
pH-отрегулированные полутвердые или гелевые композиции были получены с применением метода смешивания «к себе-от себя»:
a) желаемое количество пептида взвешивали в цилиндре одноразового шприца S1, предварительно оборудованном специальным ручным клапаном двухстороннего действия HV (В.Д.=0,5 мм), и трубку помещали внутрь отверстия шприца Люэра;
b) поршень шприца закрепляли штоком из нержавеющей стали SR;
c) HV в S1 присоединяли к источнику вакуума, и HV открывали. Через 10 минут HV закрывали;
d) раствор цинка аккуратно взвешивали в цилиндре второго одноразового шприца S2;
e) затем S2 соединяли со свободной частью HV;
f) HV открывали, и растворитель вытягивали с помощью вакуума в цилиндр, содержащий порошок пептида S1;
g) HV закрывали, и шприц с растворителем S2 отделяли, таким образом, гидратируя порошок пептида в S1;
h) SR отделяли, и поршень шприца медленно высвобождали;
i) поршень шприца двигали (к себе-от себя), без открытия HV, так что порошковая масса полностью смачивалась растворителем;
j) двухстороннюю соединительную вставку из нержавеющей стали SC (В.Д.=1,0 мм) помещали в шприц S2 с трубкой, помещенной внутрь отверстия шприца Люэра, и его поршень толкали до конца;
k) HV в S1 открывали, чтобы выпустить вакуум, и затем отделяли HV. Поршень шприца двигали так, чтобы минимизировать воздух в цилиндре; и
i) S1 и S2 соединяли с помощью SC, и композицию проталкивали из S1 в S2 через SC;
m) после гомогенизации отбирали аликвоту смешанного продукта для определения концентрации пептида;
n) оставшуюся промежуточную массу продукта аккуратно взвешивали, и рассчитывали необходимое для достижения желаемого значения рН количество раствора NaOH;
o) раствор NaOH аккуратно взвешивали в цилиндре третьего одноразового шприца S3; и
p) поршни шприцев медленно сжимали для минимизации воздуха в камерах шприцев. Оба шприца соединяли SC, и композицию проталкивали через SC.
Используя сведения рассматриваемой заявки, специалист в данной области может варьировать количества пептида и ZnCl2 для получения композиций, отличных от 25% композиции, описанной в описании.
**Показано целевое значение. Действительные значения находились в пределах 10% от целевого во всех случаях
3.0. Определение аффинности рецептора GLP-1
Соединение для применения на практике настоящего изобретения может быть тестировано на способность связываться с рецептором GLP-1 с применением следующей методики.
Культура клеток
Клетки инсулиномы крысы RIN 5F (номер ATCC CRL-2058, Американская коллекция типовых культур, Манассас, Вирджиния), экспрессирующие рецептор GLP-1, культивировали в модифицированной по Дульбекко среде Игла (DMEM), содержащей 10% фетальной телячьей сыворотки, и выдерживали при приблизительно 37ºC во влажной атмосфере с 5% CO2/95% воздуха.
Радиолигандное связывание
Получали мембраны для исследования радиолигандного связывания путем гомогенизации клеток RIN в 20 мл ледяного 50 мМ Трис-HCl с помощью Политрона Бринкмана (Brinkman Polytron, Вестбери, Нью-Йорк) (установка 6, 15 секунд). Гомогенаты промывали дважды с помощью центрифугирования (39000 g/10 минут), и конечные гранулы ресуспендировали в 50 мМ Tris-HCl, содержащем 2,5 мМ MgCl2, 0,1 мг/мл бацитрацина (Sigma Chemical, Сант Луис, Миссури) и 0,1% BSA. В течение анализа аликвоты (0,4 мл) инкубировали с 0,05 нМ (125I)GLP-1(7-36) (~2200 Ки/ммоль, New England Nuclear, Бостон, Массачусетс), в присутствии и в отсутствие 0,05 мл немеченых конкурирующих тестируемых пептидов. Через 100 минут инкубации (25ºC) связанный (125I)GLP-1(7-36) отделяли от несвязанного путем быстрого фильтрования через фильтры GF/C (Brandel, Гейтесбург, Мериленд), которые предварительно замачивали в 0,5% полиэтиленимине. Фильтры промывали три раза 5-мл аликвотами ледяного 50 мМ Трис-HCl, и задержанную на фильтрах соответствующую радиоактивность подсчитывали с помощью гамма-спектрометрии (Wallac LKB, Гейтесбург, Мериленд). Специфическое связывание определяли как общее связывание (125I)GLP-1(7-36) минус связывание в присутствии 1000 нМ GLP-1(7-36) (Bachem, Торренс, Калифорния).
4. Определение зависимости растворимости от значения pH
4.1. Определение зависимости растворимости соединения от значения pH в фосфатно-солевом буферном растворе (PBS)
Соединение, которое может быть предпочтительно применено для практического использования изобретения, может быть тестировано для определения его растворимости в PBS при различных значениях pH и температурах, используя следующую методику.
Базовый буферный раствор PBS получали путем растворения одной упаковки предварительно смешанного порошка (SIGMA, Продукт № P-3813) в одном литре деионизированной воды с получением 10 мМ фосфатно-солевого буферного раствора с 138 мМ NaCl, 2,7 мМ KCl и pH 7,4. Буферы PBS с различными значениями pH получали, регулируя значение pH данного базового раствора с помощью фосфорной кислоты и/или гидроксида натрия.
Два мг образцов соединения, которое тестируется, например, 2 мг соединения примера 1, взвешивали в стеклянных сосудах. В каждый сосуд добавляли 50-мкл аликвоты буфера PBS при определенном значении pH. Раствор перемешивали на вортексе и, если необходимо, обрабатывали ультразвуком до получения прозрачного раствора. Записывали для каждого тестируемого pH общий объем буфера, необходимый для растворения 2 мг соединения, и рассчитывали растворимость.
Растворы пептидов, которые являлись прозрачными при комнатной температуре (20-25°C), помещали в холодильник (4°C) на ночь, и проверяли растворимость пептида при 4°C.
4.2. Определение зависимости растворимости соединения от значения pH в солевом растворе
Соединение, которое может быть предпочтительно применено для практического использования изобретения, может быть тестировано для определения его растворимости в солевом растворе при различных значениях pH и температурах, используя следующую методику.
Базовый солевой раствор получали путем растворения 9 грамм NaCl в одном литре деионизированной воды. Солевые растворы с различными значениями pH получали путем регулирования значения pH данного базового раствора с HCl и/или NaOH.
Два мг образцов соединения, которое тестируется, например, 2 мг соединения примера 1, взвешивали в стеклянных сосудах. В каждый сосуд добавляли 50-мкл аликвоты солевого раствора при определенном значении pH. Раствор перемешивали на вортексе и, если необходимо, обрабатывали ультразвуком до получения прозрачного раствора. Записывали для каждого тестируемого pH общий объем солевого раствора, необходимый для растворения 2 мг соединения, и рассчитывали растворимость.
Растворы, которые являлись прозрачными при комнатной температуре (20-25°C), помещали в холодильник (4°C) на ночь, и проверяли растворимость пептида при 4°C.
4.3. Определение растворимости соединения в солевом растворе при pH 7,0
Соединения, которые могут быть предпочтительно применены для практического использования изобретения, могут быть тестированы для определения их растворимости при комнатной температуре в солевом растворе со значением pH=7, используя следующую методику.
Солевой раствор получали путем растворения 9 грамм NaCl в одном литре деионизированной воды. Взвешивали 2 мг образца тестируемого соединения, например, 2 мг соединения примера 1, в стеклянных сосудах и добавляли 1-мл аликвот солевого раствора, перемешивали на вортексе и обрабатывали ультразвуком до получения прозрачного раствора. Записывали общий объем солевого раствора, необходимый для растворения 2 мг пептида, и рассчитывали растворимость при комнатной температуре.
4.4. Определение растворимости соединения в солевом растворе при различных значениях pH
Соединение, которое может быть предпочтительно применено для практического использования изобретения, может быть тестировано для определения его растворимости при комнатной температуре в солевых растворах с различными значениями pH, используя следующую методику.
Базовый солевой раствор получали путем растворения 9 грамм NaCl в одном литре деионизированной воды. Солевые растворы с различными значениями pH получали путем обработки аликвот базового раствора HCl и NaOH.
Взвешивали 2 мг образца соединения, которое тестируется, например, соединения примера 1, в стеклянных сосудах. Добавляли 50-мкл аликвоты солевого буферного раствора при определенном значении pH. Раствор перемешивали на вортексе и обрабатывали ультразвуком до получения прозрачного раствора. Записывали общий объем буфера, использованного для растворения 2 мг пептида, и рассчитывали растворимость.
5. Определение растворимости соединения в воде в зависимости от концентрации цинка
Соединение, которое может быть предпочтительно применено для практического использования изобретения, может быть тестировано для определения его растворимости в воде с pH 7 при различных концентрациях цинка, используя следующую методику.
Базовый раствор цинка получали путем растворения ZnCl2 в деионизированной воде при концентрации 100 мг/мл и доводили pH до 2,7, используя HCl. Растворы с различными концентрациями ZnCl2 ("тестовые растворы Zn") получали путем соответствующих разбавлений базового раствора.
Один мг соединения, которое тестируется, например, 1 мг соединений примера 1, растворяли в 250 мкл каждого тестового раствора Zn с получением раствора, имеющего 4 мг/мл соединения. Затем значение pH данного раствора регулировали с помощью 0,2 н. NaOH до тех пор, пока не наблюдали образование белого осадка. Раствор с выпавшим осадком центрифугировали, и маточный раствор анализировали с помощью ВЭЖХ. Измеряли площадь УФ-поглощения пика тестируемого соединения и определяли концентрацию тестируемого соединения в маточном растворе посредством сравнения с калибровочной кривой.
В качестве иллюстративного примера соединения, которое может быть применено для практического использования изобретения, тестировали соединение примера 1 непосредственно в вышеупомянутом анализе, и были получены следующие результаты (водный, pH 7,0, комнатная температура):
6. Определение изоэлектрической точки (pI) с помощью ИЭФ-гелей
Для измерения pI пептидов GLP-1, например, соединения примера 1, использовали гели Invitrogen's Novex ИЭФ pH3-10. Тестируемые пептидные соединения растворяли в воде при концентрации 0,5 мг/мл. В случае каждого такого раствора, 5 мкл полученного раствора смешивали с 5 мкл 2× буфера для образца Novex® (содержащего 20 мМ свободного основания аргинина, 20 мМ свободного основания лизина и 15% глицерина), и полученные 10 мкл раствора образца вносили в гель вместе с образцом белковых стандартов.
Подвижные буферы также получали от Invitrogen, и гель прогоняли в соответствии с инструкциями производителей, обычно следующим образом: постоянное значение 100 В в течение 1 часа, с последующим постоянным значением 200 В в течение 1 часа, с последующим постоянным значением 500 В в течение 30 минут.
Затем гель фиксировали в 12% ТХУ (TCA, трихлоруксусная кислота), содержащей 3,5% сульфосалициловой кислоты, в течение 30 минут и затем оставляли на 2 часа с коллоидным Кумасси синим, в соответствии с инструкциями набора Novex® Colloidal Blue, затем удаляли краситель в воде в течение ночи.
Гель сканировали и анализировали с помощью программы Fragment Analysis 1.2. Значения pI неизвестных пептидов рассчитывали относительно pI стандартных соединений, обладающих значениями pI: 10,7, 9,5, 8,3, 8,0, 7,8, 7,4, 6,9, 6,0, 5,3, 5,2, 4,5, 4,2 и 3,5.
Измеренное значение pI соединения примера 1 составило 7,60.
7. Исследования in vivo на крысах
Композиции по настоящему изобретению могут тестироваться для определения их способности вызывать и усиливать эффект in vivo, используя следующие испытания.
7.1. Экспериментальная методика
За день до эксперимента взрослым самцам крыс Sprague-Dawley (Taconic, Германтаун, Нью-Йорк), которые весили приблизительно 300-350 г, имплантировали правую предсердную яремную канюлю под анестезией хлоргидратом. Затем крыс не кормили в течение 18 часов до инъекции соответствующего тестируемого соединения или наполнителя в качестве контроля в момент времени 0. Крыс не кормили на протяжении всего эксперимента.
Получали раствор 0,5 мг/мл ZnCl2 путем разбавления раствора 100 мг/мл ZnCl2 в растворе HCl, имеющем pH 2,7 воды. Растворяли 1 мг соединения формулы (I) ((Aib8,35)hGLP-1(7-36)NH2) в 250 мкл данного раствора с получением прозрачного раствора, имеющего 4 мг/мл соединения и 0,5 мг/мл Zn при pH 4.
В нулевой момент времени крысам подкожно (s.c.) инъецировали либо (a) упомянутый в описании выше раствор (Aib8,35)hGLP-1(7-36)NH2, либо носитель-контроль. В обоих случаях инъецируемый раствор был очень мал (4-6 мкл), а доза введенного особи соединения GLP-1 составляла 75 мкг/кг. В соответствующее время после s.c. инъекций забирали 500 мкл образца крови посредством внутривенной (i.v.) канюли, и крыс подвергали i.v. глюкозной нагрузке, чтобы тестировать на наличие усиления секреции инсулина. Время глюкозной нагрузки составляло 0,25, 1, 6, 12 и 24 часа после инъекции соединения. После забора первого образца крови инъецировали i.v. глюкозу (1 г/кг) и промывали 500 мкл гепаринизированного солевого раствора (10 Ед./мл). После этого на 2,5, 5, 10 и 20 минуты после инъекции глюкозы забирали образцы крови по 500 мкл. За каждым немедленно следовала i.v. инъекция 500 мкл гепаринизированного солевого раствора (10 Ед./мл) через канюлю. Образцы крови центрифугировали, из каждого образца собирали плазму, и образцы хранили при -20°C до их использования в анализе на содержание инсулина. Количество инсулина в каждом образце определяли, используя набор для твердофазного иммуноферментного анализа (ELISA) инсулина крысы (American Laboratory Products Co., Виндхам, Нью-Гэмпшир).
7.1.1. Результаты
Наблюдали стабильную инсулинстимулирующую активность, которую индуцировала инъекция глюкозы, в течение всех 24 часов эксперимента.
8. Исследования in vivo на собаках
Существует ряд исследований in vivo, известных в области техники, которые дают возможность специалистам в данной области определять способность композиции поддерживать пролонгированное высвобождение активного компонента in vivo.
8.1. 1% композиция пептида
В качестве примера были получены водные тестовые составы, содержащие 1% (масс./масс.) соединения формулы (I) в буферном растворе ZnCl2 (соотношение пептид:Zn=1,5:1,0).
Всего 6 самцов собак породы Бигль (Beagle) возрастом 42-78 месяцев и массой тела 14-21 кг содержали со свободным доступом к воде и питанием один раз в день (приблизительно 400 г сухого стандартного питания (SAFE 125)). Собак не кормили в течение 18 часов до введения тестируемой композиции.
Тестируемую композицию вводили подкожно в межлопаточную область. Объем введения (приблизительно 20 микролитров на животное) формировали посредством шприцев Terumo 0,3 мл с 0,33-12 мм (BS=30M2913). Таким образом достигали теоретической дозы приблизительно 0,2 мг пептида.
Через определенные промежутки времени забирали образцы крови, приблизительное время = 0, 8, 15, 30, 45 минут и 1, 2, 4, 8 и 12 часов и 1, 2, 3, 4, 5 и 6 дней после введения. Кровь быстро охлаждали после отбора образца, прежде чем центрифугировать, и плазму декантировали и быстро замораживали в ожидании анализа. Определение концентрации пептида в плазме осуществляли после произвольной твердофазной экстрации, с последующей оперативной фазовой экстракцией, связанной с ЖХ-МС/МС, и полученные данные обрабатывали с помощью программного обеспечения Analyst v1.2.
Композиции демонстрировали пролонгированное высвобождение активного пептида в течение 2 дней.
8.2. 1% раствор (Aib 8,35 )hGLP-1(7-36)NH 2 :
Используя по существу ту же самую методику анализа in vivo, которая описана в разделе 8.1 выше, исследовали следующие композиции на их способность высвобождать испытуемый пептид на протяжении длительного периода времени. Для каждой из следующих четырех композиций концентрация пептида составляла приблизительно 1% (масс./масс.), соотношение пептида и цинка составляло приблизительно 1,5:1, и вводимая доза пептида была приблизительно 1 мг.
Раствор 8.2.A: (Aib8,35)hGLP-1(7-36)NH2 в растворе, содержащем (i) 90% ZnCl2 (0,298 мг/мл) и (ii) 10% NaOH (0,975 мг/мл).
Раствор 8.2.B: (Aib8,35)hGLP-1(7-36)NH2 в растворе ZnCl2 (0,286 мг/мл).
Раствор 8.2.C: по существу аналогичен раствору 8.2.B и буферизован с применением AcOH/AcO-.
Раствор 8.2.D: по существу аналогичен раствору 8.2.A.
Композиции обеспечивали пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как представлено на фиг.1.
8.3. 1% раствор (Aib8,35)hGLP-1(7-36)NH2
Используя по существу ту же самую методику анализа in vivo, которая описана в разделе 8.1 выше, исследовали следующую композицию на способность высвобождать испытуемый пептид на протяжении длительного периода времени. Для следующей композиции концентрация пептида составляла приблизительно 2% (масс./масс.), соотношение пептида и цинка составляло приблизительно 1,5:1, и вводимая доза пептида была приблизительно 1 мг.
Раствор 8.3: (Aib8,35)hGLP-1(7-36)NH2 в растворе, содержащем (i) 80% ZnCl2 (0,695 мг/мл) и (ii) 20% NaOH (1,75 мг/мл).
Композиция обеспечивала пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как представлено на фиг.5.
8.4. 10% растворы пептида
Используя по существу ту же самую методику анализа in vivo, которая описана в разделе 8.1 выше, исследовали следующие композиции на их способность высвобождать испытуемый пептид на протяжении длительного периода времени. Для каждой из следующих четырех композиций концентрация пептида составляла приблизительно 10% (масс./масс.), соотношение пептида и цинка составляло приблизительно 1,5:1, и вводимая доза пептида была приблизительно 15 мг.
Раствор 8.4.A: (Aib8,35)hGLP-1(7-36)NH2 в растворе, содержащем (i) 90% ZnCl2 (3,367 мг/мл) и (ii) 10% NaOH (5,01 мг/мл).
Раствор 8.4.B: (Aib8,35)hGLP-1(7-36)NH2 в растворе ZnCl2 (2,993 мг/мл).
Раствор 8.4.C: по существу аналогичен раствору 8.4.B и буферизован с применением AcOH/AcO-.
Раствор 8.4.D: по существу аналогичен раствору 8.4.A.
Композиции обеспечивали пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как представлено на фиг.2.
8.5. Полутвердые композиции
Используя по существу ту же самую методику анализа in vivo, которая описана в разделе 8.1 выше, исследовали следующие полутвердые композиции на их способность высвобождать испытуемый пептид на протяжении длительного периода времени. Для композиции 8.5.A концентрация пептида была приблизительно 5%, в то время как для композиций 8.5.B, 8.4.C и 8.5.D концентрация пептида была приблизительно 10% (масс./масс.). Соотношение пептида и цинка для композиций 8.5.A, 8.5.B и 8.5.C было приблизительно 5,4:1, в то время как для композиции 8.5.D соотношение составляло приблизительно 4,0:1. Для всех четырех композиций вводимая доза пептида была приблизительно 1 мг.
Композиция 8.5.A: (Aib8,35)hGLP-1(7-36)NH2 в полутвердой композиции, содержащей ZnCl2 (0,40 мг/мл) в WFI.
Композиция 8.5.B: по существу аналогична композиции 8.5.A, где концентрацию ZnCl2 устанавливали выше, чтобы удерживать соотношение пептид:Zn приблизительно 5,4:1.
Композиция 8.5.C: (Aib8,35)hGLP-1(7-36)NH2 в полутвердой композиции, содержащей (i) 50% ZnCl2 (1,69 мг/мл) и (ii) 50% NaOH (1 мг/мл).
Композиция 8.5.D: (Aib8,35)hGLP-1(7-36)NH2 в полутвердой композиции, содержащей (i) 50% ZnCl2 (2,28 мг/мл) и (ii) 50% NaOH (1 мг/мл).
Композиции обеспечивали пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как представлено на фиг.3.
8.6. Полутвердые композиции
Используя по существу ту же самую методику анализа in vivo, которая описана в разделе 8.1 выше, исследовали следующую полутвердую композицию на способность высвобождать испытуемый пептид на протяжении длительного периода времени. Данную композицию получали с применением 5,22 мг/мл раствора ZnCl2, при pH=2,0. Обеспечивали достаточное количество пептида, чтобы в результате получить 25% полутвердую композицию пептида, имеющую соотношение пептида и цинка приблизительно 4:1. Значение pH композиции регулировали в соответствии с предусмотренной в описании процедурой, используя 10 мг/мл NaOH. Доза введенного пептида составляла приблизительно 15 мг.
Композиция 8.6 обеспечивала пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как представлено на фиг.6.
8.7. Полутвердые композиции
Используя по существу ту же самую методику анализа in vivo, которая описана в разделе 8.1 выше, исследовали следующую полутвердую композицию на способность высвобождать испытуемый пептид на протяжении длительного периода времени. Данную композицию получали с применением 8,5 мг/мл раствора ZnCl2, при pH=2,0. Обеспечивали достаточное количество пептида, чтобы в результате получить 23% полутвердую композицию пептида, имеющую соотношение пептида и цинка приблизительно 1,5:1. Композицию получали в соответствии с методикой, подробно описанной в разделе 2.6 выше. Доза введенного пептида составляла приблизительно 15 мг (соответствует приблизительно 65 микролитрам композиции).
Композиция 8.7 обеспечивала пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как представлено на фиг.7.
Дополнительные исследования с различными изменениями в раскрытом изготовлении состава подвергались таким же образом анализу in vivo и подтверждали, что композиции по настоящему изобретению обеспечивают полезную платформу для доставки лекарственного средства соединения формулы (I). Используя сведения рассматриваемой заявки, специалист в данной области техники может варьировать количества пептида и ZnCl2 и значения рН для получения композиций по настоящему изобретению, как описано в описании.
Пример 9
1. Регулирование ФК-профиля путем изменения содержания ацетата в 10% пептидных растворах
В данном примере представлено фармакокинетическое исследование (Aib8,35)hGLP-1(7-36)NH2 у самцов собак породы бигль после однократного подкожного введения двух импровизированных композиций, содержащих 10% (Aib8,35)hGLP-1(7-36)NH2 и хлорид цинка [(Aib8,35)hGLP-1(7-36)NH2:Zn=1,5:1], с уровнем дозы 15 мг/собаку.
Метод проведения анализа in vivo аналогичен описанному в разделе 8.1.
Данный пример иллюстрирует регулирование ФК-профиля с помощью содержания ацетата в фармацевтической композиции и, таким образом, влияние соотношения [ацетат/пептид] в фармацевтической композиции на значение pH.
Регулирование значения pH контролировали путем регулирования содержания ацетата, снижение содержания ацетата обнаруживало увеличение влияния на значение pH.
Варьирование ацетата также демонстрировало влияние на Cмакс. Обычно снижение содержания ацетата уменьшало величину Cмакс.
Увеличение содержания ацетата демонстрировало улучшение растворимости и физической стабильности.
Согласно выбранному составу, улучшение путем регулирования соотношения ацетат/пептид растворимости или стабильности компенсируется путем регулирования соотношения пептид/Zn, например, Смакс. Можно видеть, как в системе с тремя возможными переменными регулируется стабильность, растворимость, значение рН или Cмакс.
В данном примере аббревиатура SD означает стандартное отклонение. AUC означает площадь под кривой зависимости концентрации в плазме от времени Артемизинина.
Значением аббревиатуры MRT является среднее время удержания (MRT) - параметр для оценки скорости биодоступности в сравнении с MRT с tмакс, которое представляет собой время достижения максимальной концентрации лекарственного средства. MRTt рассчитывали, используя данные от нулевого момента времени до времени последнего забора образца крови.
В таблице 3 собраны результаты по партиям 10% композиции пептида с различными соотношениями [ацетат/пептид] и подкожным введением собакам породы Бигль. Значения максимума концентрации лекарственного средства в плазме (Cмакс) составило 8,10 нг/мл (SD=1,80 нг/мл) в случае молярного соотношения [ацетат/пептид], равного [3,7:1], где партия, имеющая меньшее соотношение [3,2:1], обеспечивала значение Cмакс 5,65 нг/мл (SD=2,61 нг/мл).
10. Составы соль пептида GLP-1/двухвалентный металл
10.1. Способы
Получали растворы 1 мг/мл (Aib8,35)hGLP-1(7-36)NH2 в воде и PBS, и доводили значение pH до 7,0. Получали базовые растворы 10 мг/мл CaCl2, CuCl2, MgCl2 и ZnCl2 в воде. Доводили значение рН растворов CaCl2, MgCl2 и ZnCl2 до 7,0. pH раствора CuCl2 не может быть изменено в сторону повышения основности, поскольку выпадает в осадок Cu. Поэтому использовали раствор CuCl2 с pH 4,4.
Добавляли 4 мкл растворов ионов металлов в воде или PBS к 200 мкл 1 мг/мл раствора (Aib8,35)hGLP-1(7-36)NH2 с получением конечной концентрации иона металла 200 мкг/мл. Конечный раствор перемешивали и проверяли на образование осадка. Если осадок формировался, суспензию центрифугировали. Определяли концентрацию (Aib8,35)hGLP-1(7-36)NH2 в надосадочной жидкости методом ВЭЖХ.
10.2. Результаты
Растворимость (Aib
8,35
)hGLP-1(7-36)NH
2
в присутствии ионов двухвалентных металлов
10.3. Фармакокинетические исследования прозрачных составов (Aib 8,35 )hGLP-1(7-36)NH 2 /двухвалентный металл с pH 5,5
Получали три различных состава (Aib8,35)hGLP-1(7-36)NH2, используя следующие методики:
(1) HCl-соль (Aib8,35)hGLP-1(7-36)NH2 с CuCl2,
(2) HCl-соль (Aib8,35)hGLP-1(7-36)NH2 с ZnCl2,
(3) Ацетатная соль (Aib8,35)hGLP-1(7-36)NH2 с ZnCl2.
Соль ТФУ аналога GLP-1 (ТФУ-соль получается в результате очистки пептида с применением препаративной ВЭЖХ при элюировании буферными растворами, содержащими ТФУ) может быть превращена в другую соль, такую как ацетатная соль, путем растворения пептида в небольшом количестве 0,25 н. водного раствора уксусной кислоты. Полученный раствор используют на полупрепаративной колонке ВЭЖХ (Zorbax, 300 SB, C.8). Колонку элюируют (1) 0,1 н. водным раствором ацетата аммония в течение 0,5 часа, (2) 0,25 н. водным раствором уксусной кислоты в течение 0,5 часа и (3) в линейном градиенте (20-100% раствора B на протяжении 30 минут) со скоростью потока 4 мл/мин (раствор A представляет собой 0,25 н. водный раствор уксусной кислоты; раствор B представляет собой 0,25 н. уксусную кислоту в ацетонитриле/воде 80:20). Фракции, содержащие пептид, собирают и лиофилизуют досуха.
HCl-соль (Aib8,35)hGLP-1(7-36)NH2 получали с помощью метода лиофилизации. Растворяли 20 мг ацетата (Aib8,35)hGLP-1(7-36)NH2 в 4 мл 20 мМ водного раствора HCl и выдерживали при комнатной температуре в течение 10 минут. Образец замораживали и лиофилизовали в течение ночи. Лиофилизацию осуществляли еще два раза и определяли содержание хлорида в конечном продукте. Измеренное содержание хлорида составило 5,38%.
(1) HCl-соль (Aib 8,35 )hGLP-1(7-36)NH 2 с CuCl 2
Растворяли 5,3 мг HCl-соли (Aib8,35)hGLP-1(7-36)NH2 (содержание пептида составляет 95%) в 50 мкл 20 мМ водного раствора CuCl2. pH регулировали с помощью приблизительно 2 мкл 1 н. NaOH приблизительно до 5,5. Молярное соотношение (Aib8,35)hGLP-1(7-36)NH2/CuCl2 составляло 1,5:1. Концентрация пептида в воде (масс./масс.) была 10% (30 мМ) с pH приблизительно 5,5.
(2) HCl-соль (Aib 8,35 )hGLP-1(7-36)NH 2 с ZnCl 2
Растворяли 5,3 мг HCl-соли (Aib8,35)hGLP-1(7-36)NH2 (содержание пептида составляет 95%) в 50 мкл 20 мМ водного раствора ZnCl2. pH регулировали с помощью приблизительно 2 мкл 1 н. NaOH приблизительно до 5,5. Молярное соотношение (Aib8,35)hGLP-1(7-36)NH2/ZnCl2 составляло 1,5:1. Концентрация пептида в воде (масс./масс.) была 10% (30 мМ) с pH приблизительно 5,5.
(3) Ацетатная соль (Aib 8,35 )hGLP-1(7-36)NH 2 с ZnCl 2
Растворяли 5,5 мг ацетатной соли (Aib8,35)hGLP-1(7-36)NH2 (содержание пептида составляет 92%) в 50 мкл 20 мМ водного раствора ZnCl2. Полученный раствор лиофилизовали в течение ночи и повторно растворяли в 50 мкл воды. pH регулировали с помощью приблизительно 1 мкл 1 н. NaOH приблизительно до 5,5. Молярное соотношение (Aib8,35)hGLP-1(7-36)NH2/ZnCl2 составляло 1,5:1. Концентрация пептида в воде (масс./масс.) была 10% (30 мМ) с pH приблизительно 5,5.
10.4. Дозирование и сбор образцов крови
Крысам вводили подкожно данные три состава (Aib8,35)hGLP-1(7-36)NH2 в дозе 0,3 мг/крысу (3 мкл 10% раствора). Образцы крови забирали на 5, 10, 15, 30 минут, 1, 2, 4, 8 часов и 1, 2, 3, 4, 7, 10 дней. Плазму крови собирали после центрифугирования и хранили при -80°C. Ткань на месте инъекции также собирали, гомогенизировали в 5x метаноле и хранили при -80°C.
Двух крыс использовали для экспериментальных точек 5, 10, 15, 30 минут и 1, 2, 4, 8 часов. Одну крысу использовали для экспериментальных точек 1, 2, 3, 4, 7, 10 дней.
10.5. Подготовка образца для ЖХ-МС/МС
Плазму (200 мкл) подкисляли 10 мкл муравьиной кислоты и высаживали с помощью 600 мкл ацетонитрила. Супернатант собирали путем центрифугирования и концентрировали досуха в вакууме. Остаток растворяли в 150 мкл 30% ацетонитрила в воде и центрифугировали. 50 мкл супернатанта вводили для ЖХ-МС/МС анализа.
Экстракт ткани в метаноле (10 мкл) разводили до 1 мл 30% ацетонитрилом в воде и 50 мкл вводили для ЖХ-МС/МС анализа.
10.6. ЖХ-МС/МС анализ
ЖХ-МС/МС анализ осуществляли с помощью системы масс-спектрометра API4000, оборудованной ионным источником Turbo Ionspray. Использовали метод детектирования молекулярных ионов MRM (мониторинг множественных ионов) с парой ионов 668,9 и 136,1.
Разделение ВЭЖХ осуществляли с помощью колонки Luna C8(2) 2×30 мм 3 мкм прогоняя от 10 до 90% B за 10 минут при скорости течения 0,30 мл/минуту. Буфер A представляет собой 1% муравьиную кислоту в воде, а буфер B представляет собой 1% муравьиную кислоту в ацетонитриле.
LOQ (чувствительность метода) составляла 0,5 нг/мл.
10.7. Результаты и выводы
Концентрации пептида в плазме вычисляли с помощью его стандартной калибровочной кривой. Для расчета, сколько процентов осталось в месте введения, за 100% принимали 0,06 мг/мл (Aib8,35)hGLP-1(7-36)NH2 (0,3 мг/крысу в 5 мл метанольного экстракта).
Концентрации в плазме (Aib
8,35
)hGLP-1(7-36)NH
2
На фиг.8 представлена кривая полного цикла фармакокинетического профиля составов HCl-соли (Aib8,35)hGLP-1(7-36)NH2. Кривая более ранней части цикла фармакокинетического профиля составов HCl-соли (Aib8,35)hGLP-1(7-36)NH2 представлена на фиг.9. На фиг.10 представлена кривая полного цикла фармакокинетического профиля составов ацетатной соли (Aib8,35)hGLP-1(7-36)NH2. Кривая более ранней части цикла фармакокинетического профиля составов ацетатной соли (Aib8,35)hGLP-1(7-36)NH2 представлена на фиг.11.
Оцененные проценты оставшегося на месте инъекции (Aib
8,35
)hGLP-1(7-36)NH
2
Профиль аккумуляции в ткани (Aib8,35)hGLP-1(7-36)NH2 на месте инъекции дополнительно представлен на фиг.12.
ФК параметры
Результаты указывают, что солевые формы аналогов GLP-1, в частности, в комбинации с солями двухвалентных металлов, обеспечивают приемлемые составы с замедленным высвобождением с пониженными начальными концентрациями в плазме, которые снижают или исключают нежелательные побочные эффекты.
Данные указывают, что соли с сильными кислотами, например, HCl-соли аналога GLP-1, показывают дополнительное понижение начальных концентраций в плазме. Без связи с данной теорией полагают, что исключительное понижение начальных концентраций в плазме HCl-солей аналогов GLP-1 связано с процессом нейтрализации in vivo. В композициях (1) и (2) выше при pH 5,5, 100% кислоты находится в виде хлоридной соли, и свободная кислота отсутствует. Соответственно, после подкожного введения жидкая среда организма способна нейтрализовать раствор более быстро, таким образом, приводя к более быстрому осаждению раствора. Данное снижение времени нейтрализации приводит к меньшей, менее заявленной, начальной концентрации в плазме или пику.
Цитируемые выше публикации включены в настоящее описание посредством ссылки. Дополнительные примеры осуществления настоящего изобретения будут очевидны из предшествующего раскрытия и входят в объем изобретения, которое полностью описано в описании и определено в последующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ hGLP-1, ЭКСЕНДИНА-4 И ИХ АНАЛОГОВ | 2007 |
|
RU2419452C2 |
| АНАЛОГИ GLP-1 | 1999 |
|
RU2288232C9 |
| GLP-1-МОЛЕКУЛЯРНЫЙ КОМПЛЕКС, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ КОМПЛЕКСА | 1995 |
|
RU2147588C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ЛИГАНДОВ РЕЦЕПТОРОВ МЕЛАНОКОРТИНОВ | 2010 |
|
RU2548753C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЬЮГАТ ГИАЛУРОНОВОЙ КИСЛОТЫ, ЛИНКЕРА И ДВОЙНОГО АГОНИСТА GLP-1/ГЛЮКАГОНА | 2016 |
|
RU2719482C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ НАЗАЛЬНОГО ВСАСЫВАНИЯ | 2002 |
|
RU2327484C2 |
| СОСТАВЫ АНАЛОГОВ ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-2 (GLP-2) | 2019 |
|
RU2833459C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТ ЭКСЕНДИН-ЛИНКЕР | 2011 |
|
RU2593774C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ АГОНИСТ GLP-1, ИНСУЛИН И МЕТИОНИН | 2010 |
|
RU2537239C2 |
| ГЛИКОЗИЛИРОВАННЫЙ ПЕПТИД GLP-1 | 2009 |
|
RU2543157C2 |
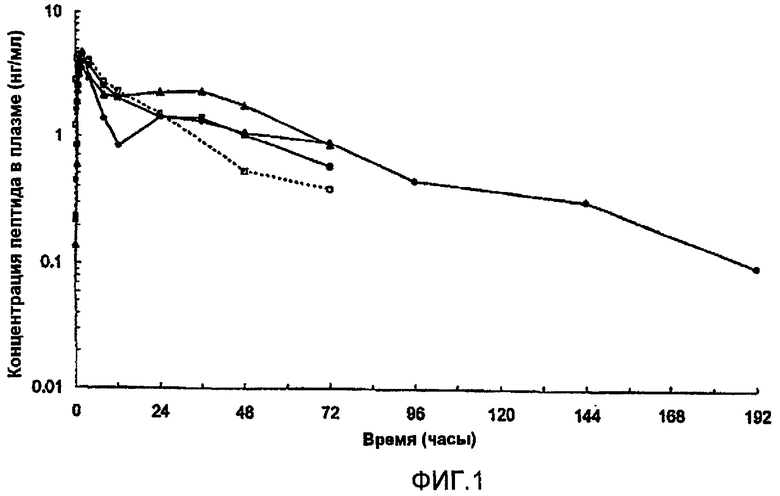
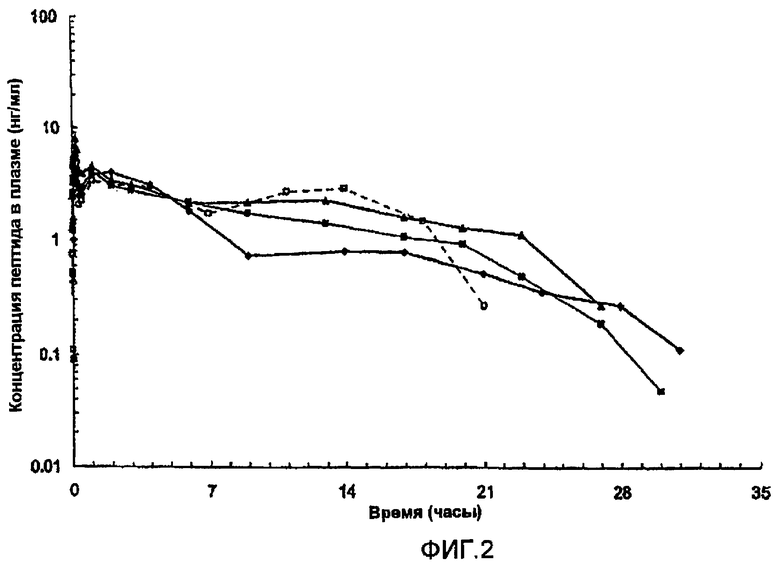
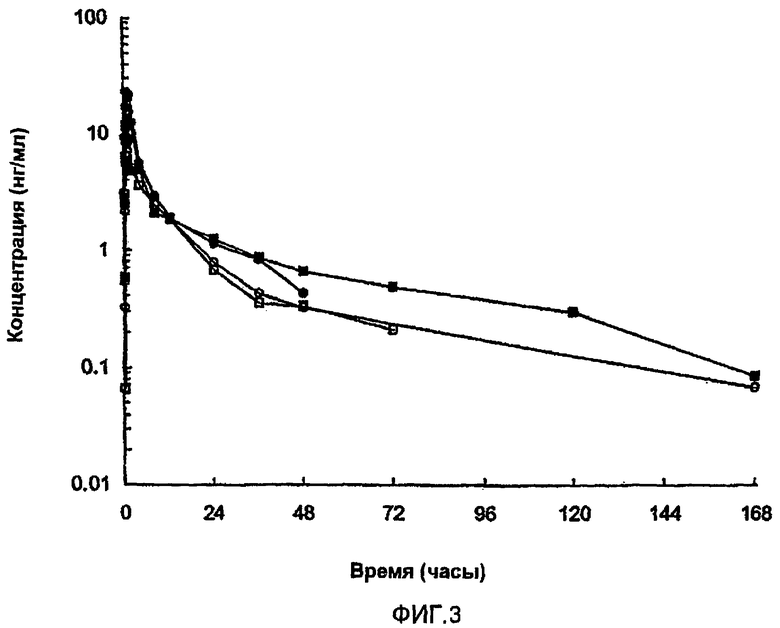
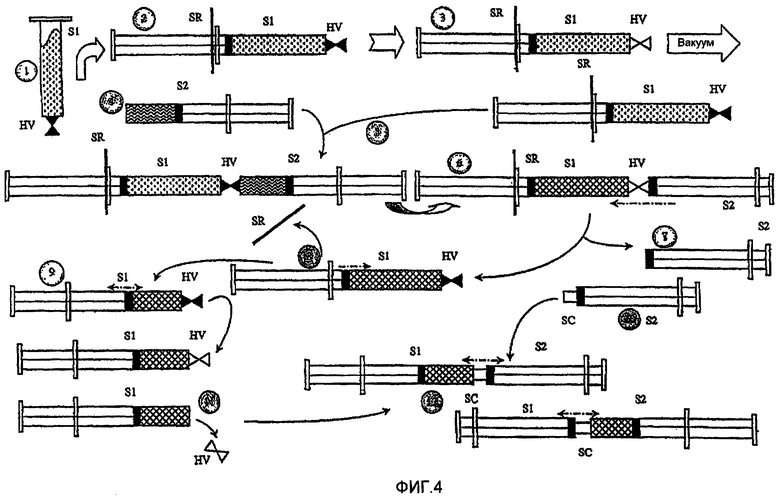
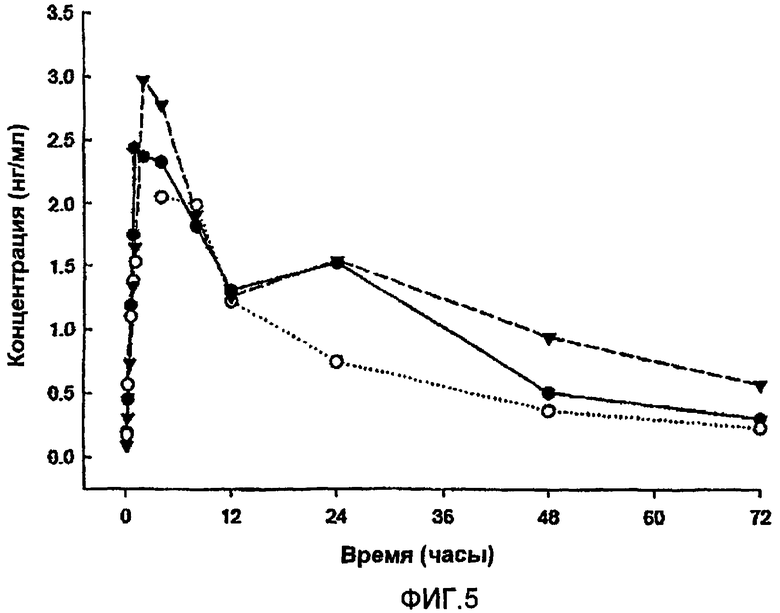
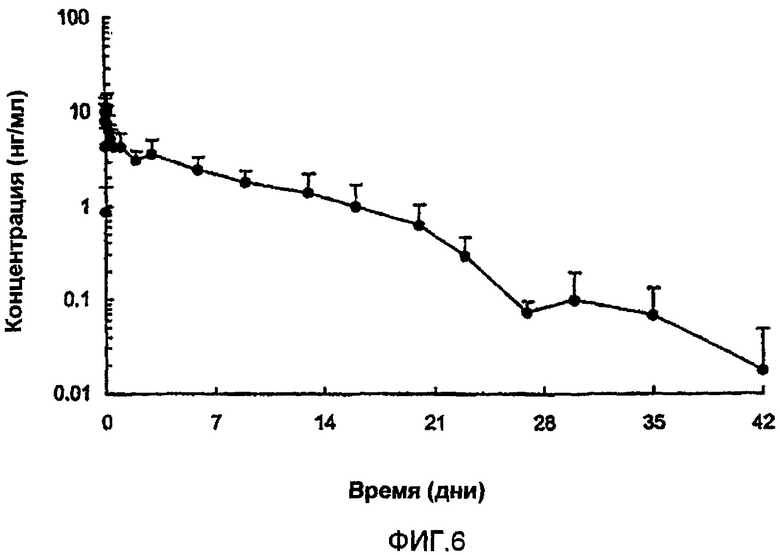
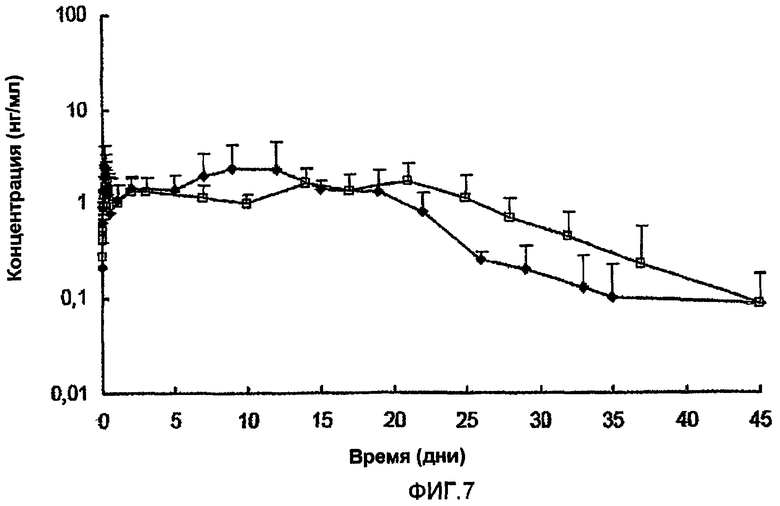
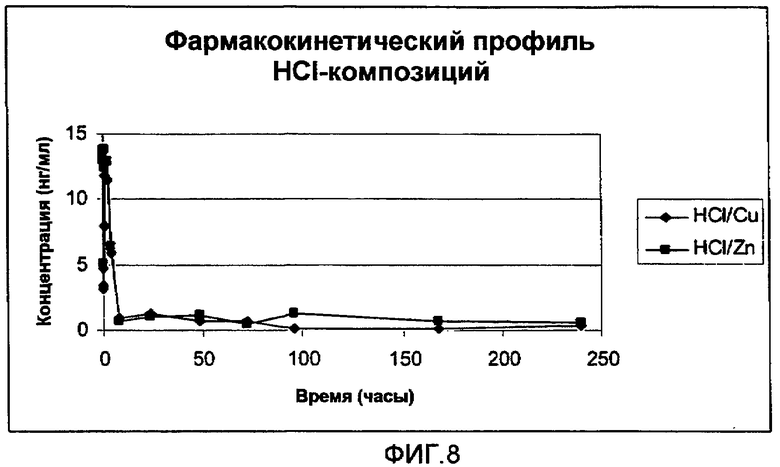
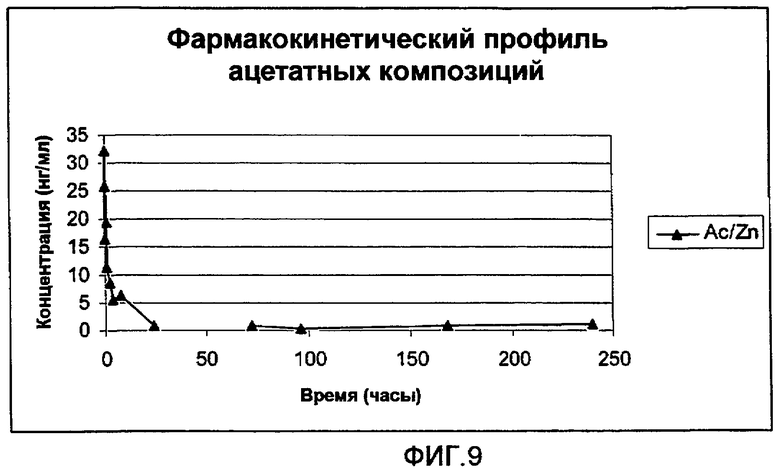
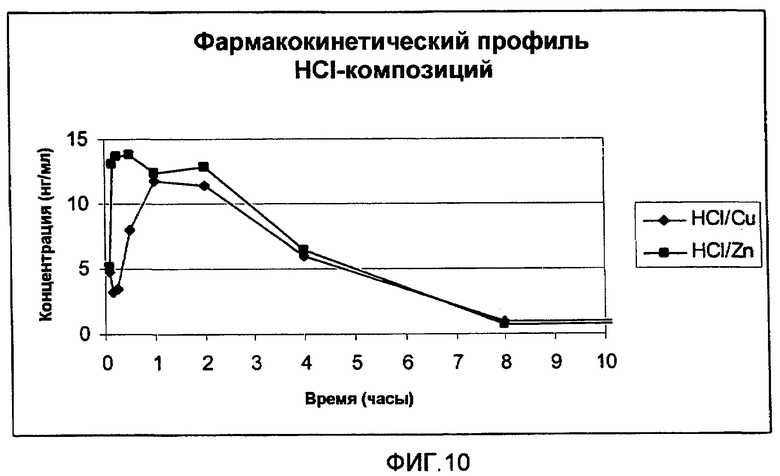
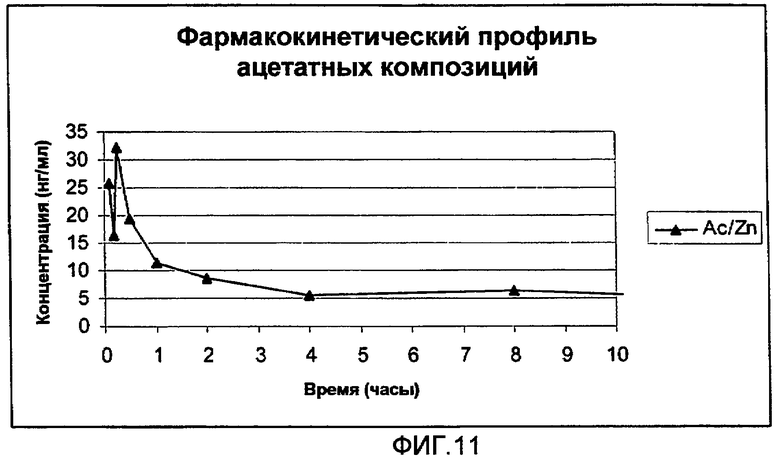
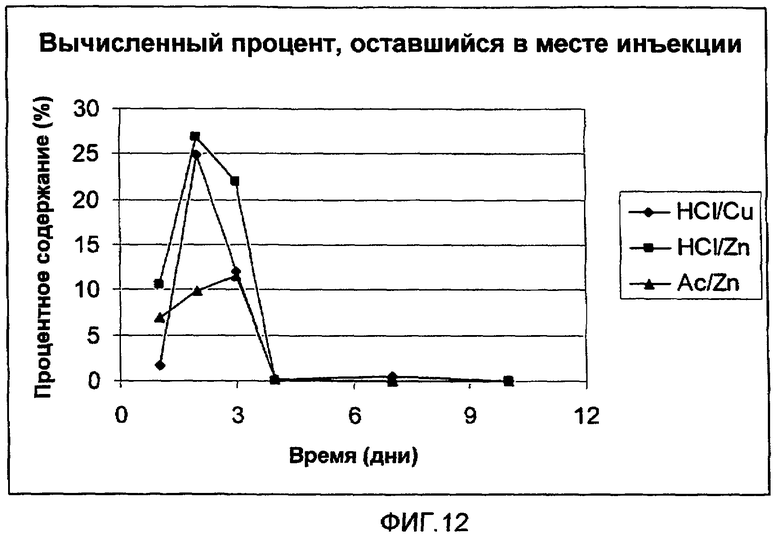
Изобретение относится к области фармакологии и представляет собой фармацевтическую композицию, содержащую аналог GLP-1 [Aib8,35]hGLP-1(7-36)NH2, полученную вместе с фармацевтически приемлемой солью указанного аналога или смесью соли указанного аналога и аналога, указанная композиция дополнительно содержит двухвалентный метал и/или соль двухвалентного металла, причем молярное соотношение указанного аналога GLP-1 и указанного двухвалентного металла и/или соли двухвалентного металла в указанной фармацевтической композиции составляет приблизительно от 5,4:1 до приблизительно 1,5:1; где указанный выбранный двухвалентный металл представляет собой цинк или медь и где диапазон значений молярного соотношения указанной соли (Aib8,35)hGLP-1(7-36)NH2 и аналога пептида GLP-1 составляет от приблизительно 0,5:1 до приблизительно 10:1. Изобретение обеспечивает легкое и воспроизводимое введение пациенту композиции, а также снижение или исключение нежелательных побочных эффектов. 10 з.п. ф-лы, 9 пр., 7 табл., 12 ил.
1. Фармацевтическая композиция, содержащая аналог GLP-1 [Aib8,35]hGLP-1(7-36)NH2, полученная вместе с фармацевтически приемлемой солью указанного аналога или смесью соли указанного аналога и аналога, указанная композиция дополнительно содержит двухвалентный металл и/или соль двухвалентного металла, причем молярное соотношение указанного аналога GLP-1 и указанного двухвалентного металла и/или соли двухвалентного металла в указанной фармацевтической композиции составляет приблизительно от 5,4:1 до приблизительно 1,5:1; где указанный выбранный двухвалентный металл представляет собой цинк или медь, и где диапазон значений молярного соотношения указанной соли (Aib8,35)hGLP-1(7-36)NH2 и аналога пептида GLP-1 составляет от приблизительно 0,5:1 до приблизительно 10:1.
2. Фармацевтическая композиция по п.1, где указанная композиция содержит соли двухвалентных металлов, выбранные из группы, состоящей из СuАс2, CuCl2, ZnAc2 и/или ZnCl2.
3. Фармацевтическая композиция по п.1 или 2, где указанная соль (Aib8,35)hGLP-1 (7-36)NH2 является фармацевтически приемлемой солью органической кислоты.
4. Фармацевтическая композиция по п.3, где указанная органическая кислота выбрана из группы, состоящей из уксусной, трифторуксусной, молочной, яблочной, аскорбиновой, янтарной, бензойной, лимонной, метансульфоновой и толуолсульфоновой кислот.
5. Фармацевтическая композиция по п.3, где указанная кислота представляет собой уксусную кислоту.
6. Фармацевтическая композиция по п.1 или 2, где указанная соль (Aib8,35)hGLP-1(7-36)NH2 является фармацевтически приемлемой солью неорганической кислоты.
7. Фармацевтическая композиция по п.6, где указанная неорганическая кислота выбрана из группы, состоящей из хлористоводородной, бромистоводородной, йодистоводородной, серной и фосфорной кислот.
8. Фармацевтическая композиция по п.6, где указанная кислота представляет собой хлористоводородную кислоту.
9. Фармацевтическая композиция по п.1, где указанная фармацевтически приемлемая соль представляет собой (Aib8,35)hGLP-1(7-36)NH2·HCl·Zn.
10. Фармацевтическая композиция по п.1, где указанная фармацевтически приемлемая соль представляет собой (Aib8,35)hGLP-1(7-36)NH2·ацетат·Zn.
11. Фармацевтическая композиция по п.1, где указанная фармацевтически приемлемая соль представляет собой (Aib8,35)hGLP-1(7-36)NH2·HCl·медь.
| US 20050159356 А, 21.07.2005 | |||
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ФУНКЦИОНАЛЬНОЙ ДИСПЕПСИИ И/ИЛИ СИНДРОМА РАЗДРАЖЕННОЙ ТОЛСТОЙ КИШКИ И НОВОЕ ПРИМЕНЕНИЕ ВЕЩЕСТВ В НЕЙ | 1999 |
|
RU2226402C2 |
| АНАЛОГИ GLP-1 | 1999 |
|
RU2214418C2 |

