ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение касается способа получения катализатора реформинга, который преобразует высокотемпературный смолосодержащий газ, выделяющийся во время термического разложения углеродистого сырья, и превращает его в газ, состоящий, главным образом, из водорода, моноксида углерода и метана, способа газификации смолы (способ реформинга), который использует данный катализатор, и способа регенерации, используемого, когда данный катализатор для реформинга смолосодержащего газа ухудшается.
Настоящее изобретение имеет приоритет на основании японской патентной заявки №2008-244852, зарегистрированной в Японии 24 сентября 2008 г., содержание которой включено сюда посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
Хотя сталелитейная промышленность является высокоэнергопотребляющей промышленностью, которая потребляет приблизительно 10% всего количества энергии, потребляемой в Японии, приблизительно 40% энергии процесса непрерывного производства стали в доменной печи представляет собой неиспользованное отходящее тепло. Оно включает в себя теплосодержание высокотемпературного неочищенного газа коксовой печи (ГКП) (называемого "сырой ГКП"), выделяющегося из коксовых печей, в качестве источника тепла, которое легко извлекать, но обычно не используют. Способы, состоящие, главным образом, из непрямого извлечения тепла, ранее были предложены в патентном документе 1 и патентном документе 2 как технологии для извлечения теплосодержания сырого ГКП, и был описан способ, который состоит из обеспечения теплопереносящей трубы внутри стоячей трубы коксовой печи или между частью стоячей трубы и частью газосборной трубы и извлечения теплосодержания путем предоставления возможности теплоносителю циркулировать и течь сквозь внутреннюю часть данной теплопереносящей трубы. В этих способах, однако, адгезия смолы, светлых нефтепродуктов и подобного и уплотнение, относящееся к процессу зауглероживания и агрегации, присущему образовавшемуся ГКП, на внешней поверхности теплопереносящей трубы, приводит к неизбежным проблемам снижения эффективности теплопереноса и эффективности теплообмена со временем. В патентном документе 3 описан способ в качестве технологии для решения этих проблем, который состоит из нанесения катализатора, такого как кристаллический силикат алюминия или кристаллический оксид кремния, на внешнюю поверхность теплопереносящей трубы и разложения смолы и других приставших веществ на углеводороды с низкой молекулярной массой с помощью катализатора, чтобы поддерживать стабильную эффективность теплопереноса. Однако этот способ также не покидает области технологии непрямого извлечения тепла для теплосодержания сырого ГКП, и нет какого-либо рассмотрения того, становятся ли продукты разложения смолы и других тяжелых углеводородов легкими углеводородами, которые легко утилизировать в качестве газового топлива и подобного. Кроме того, эффекты ухудшения активности разложения со временем, вызываемые отравлением катализатора сернистыми соединениями, такими как высококонцентрированный сероводород, содержащийся в сыром ГКП, также не были рассмотрены.
С другой стороны, составная система, объединяющая электростанцию газотурбинного объединенного цикла (ГТОЦ) и другую установку, была предложена в отношении так называемой технологии преобразования тепловая энергия - химическая энергия, с помощью которой тепловая энергия, качество которой сильно изменяется соответственно температуре, превращается в химическую энергию, и примеры этого нерегулярно наблюдали, включая объединение с производством кислорода, используя высокотемпературный кислород-переносящий твердый электролит (патентный документ 4), и реформинг водяного пара и получение водорода из природного газа, используя теплосодержание отходящего газа газовой турбины, и его применение в качестве топлива (патентный документ 5). В каждой из этих технологий тепловая энергия превращается в химическую энергию в форме кислорода или водорода путем предоставления возможности воздуху или природному газу действовать через функциональный материал в форме твердого электролита или катализатора.
Есть только несколько технологий для превращения в химическую энергию путем прямого проведения химической реакции в реакционном газе, образованном при высоких температурах в присутствии катализатора с использованием его теплосодержания, и почти во всех случаях предшествующего уровня техники теплосодержание высокотемпературного газа либо извлекается непрямым образом, либо не используется совсем, а используется только охлажденный газ после воздействия различных обработок. Однако, даже хотя сырой ГКП имеет теплосодержание, так как содержание сернистых соединений превышает 2000 ч./млн, считается, что его очень трудно реализовать с точки зрения организации каталитической реакции термического разложения тяжелых углеводородов, таких как смола, и хотя в прошлом проводили исследования, описанные в патентном документе 6, активность реформинга необязательно была надлежащей. Кроме того, хотя катализаторы превращения энергии обычно получают методом нанесения, в котором частицы активного металла наносят извне на пористый керамический носитель, такой как оксид кремния или оксид алюминия, в случае этих методов трудно увеличить дисперсность нанесенного металлического компонента, а также существует чувствительность к отравлению серой и отложению углерода, и трудно получить катализатор, подходящий для реакций разложения смолы, состоящей, главным образом, из конденсированных полициклических ароматических соединений, которые поддаются отложению углерода в атмосфере, содержащей высококонцентрированные сернистые соединения, как описано выше. Кроме того, так как результатом сжигания в воздухе с целью регенерации после работы является ухудшение последующей реакции, спекание (укрупнение) нанесенных металлических гранул легко происходит, затрудняя выполнение восстановления активности путем регенерации.
Кроме того, что касается способов получения смесей никель-магниевых оксидных соединений и оксида алюминия, если порошок никель-магниевого оксидного соединения и порошок оксида алюминия просто смешивают с последующим формованием и прокаливанием, состояние, в котором каждый элемент присутствует в катализаторе, не является однородным, и активные частицы никеля, в частности, претерпевают коагуляцию без достижения высокой площади поверхности, что вызывает такие проблемы, как ненадлежащая активность реформинга и большие количества осажденного углерода, тем самым мешая этим способам достичь уровня, пригодного для промышленного применения.
С другой стороны, хотя способы получения оксидов, содержащих никель, магний и алюминий, описаны в публикациях, таких как непатентный документ 1 и патентный документ 8, в отношении материалов, которые прокаливают после образования осадка (главным образом, образование структуры гидротальцита) с осаждающим агентом из водного раствора, в котором растворен каждый металлический компонент, эти способы имеют проблемы с практическим применением из-за ненадлежащей активности реформинга и больших количеств осажденного углерода.
В результате проведения обширных исследований ввиду вышесказанного, изобретатели настоящего изобретения установили, что катализатор, полученный согласно способу, состоящему из образования совместного осадка с осаждающим агентом из водного раствора, содержащего никелевый компонент и магниевый компонент, сушки и прокаливания совместного осадка, и затем сушки и обжига или сушки, прокаливания, формования и обжига смеси, полученной путем добавления порошка оксида алюминия и воды или золя оксида алюминия, демонстрирует высокую активность реформинга и демонстрирует сравнительно низкое отложение углерода, тем самым приведя к регистрации патента (японская патентная заявка №2008-155887). Однако считалось необходимым дополнительно уменьшить количество осажденного углерода, чтобы разработать катализатор, который демонстрирует устойчивую активность на протяжении длительного периода времени в целях практического применения.
Кроме того, недавно внимание было сфокусировано на использовании биомассы, которая является видом углеродистого сырого материала, в качестве эффективного средства снижения уровней эмиссии диоксида углерода из-за проблемы глобального потепления, и исследования, касающиеся высокоэффективного преобразования энергии биомассы, проводят на различном оборудовании. Кроме того, с современной точки зрения сохранения источников энергии, исследования, касающиеся эффективного использования угля, которые интенсивно проводили в прошлом, также повторно рассматривают для практического применения. Среди этих исследований, хотя проводили различные изучения, включая описанные в патентном документе 7, способов генерации сырого газа (неочищенного газа) путем газификации смолы, образованной при сухой перегонки биомассы, и затем утилизации ее теплосодержания, обращаясь, в частности, к каталитическому реформингу смолы с использованием катализатора, данный подход не всегда является адекватным с точки зрения активности катализатора и регенерации катализатора так же, как реакции разложения угольной смолы, описанные ранее.
Документы предшествующего уровня техники
Патентные документы
[Патентный документ 1] японская рассмотренная патентная заявка, вторая публикация № S59-44346.
[Патентный документ 2] японская нерассмотренная патентная заявка, первая публикация № S58-76487.
[Патентный документ 3] японская нерассмотренная патентная заявка, первая публикация № Н8-134456.
[Патентный документ 4] патент США №5516359.
[Патентный документ 5] японская нерассмотренная патентная заявка, первая публикация №2000-54852.
[Патентный документ 6] японская нерассмотренная патентная заявка, первая публикация №2003-55671.
[Патентный документ 7] японская нерассмотренная патентная заявка, первая публикация №2005-53972.
[Патентный документ 8] японская нерассмотренная патентная заявка, первая публикация №S50-4001.
Непатентные документы
[Непатентный документ 1] F. Basile, et al., Stud. Surf. Sci. Catal., Vol. 119 (1998).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Проблемы, решаемые изобретением
Целью настоящего изобретения является обеспечить способ получения катализатора реформинга и способ реформинга смолы, который используют для обработки сырого газа для преобразования химической энергии, состоящий из превращения легких химических веществ в присутствии катализатора и превращения в топливную композицию, состоящую, главным образом, из метана, моноксида углерода и водорода, фокусируясь на теплосодержании, которым обладает сырой газ (необработанный газ), образовавшийся во время термического разложения угля, биомассы и другого углеродистого сырья, и использования высокой активности химической реакции высокотемпературной смолы, содержащейся в данном сыром газе и присущей ему. Катализатор реформинга, полученный в способе получения настоящего изобретения, демонстрирует высокую производительность и высокую устойчивость к отложению углерода даже в отношении смолосодержащего газа, содержащего высокие концентрации сероводорода, такого как сырой ГКП, в частности.
Кроме того, целью настоящего изобретения является разработка способа регенерации катализатора реформинга смолосодержащего газа, в котором удаляют осажденный углерод и поглощенную на катализаторе серу с восстановлением активности катализатора, и обеспечение стабильной работы путем введения водяного пара или воздуха для создания окислительной атмосферы для катализатора, который ухудшился из-за протекания реакции газификации с использованием данного катализатора.
Средство решения проблем
В ходе разработки катализатора, обращая внимание на элементы, которые образуют катализатор и каталитическую композицию, и выполняя обширные исследования способа получения этого катализатора, изобретатели настоящего изобретения обращали внимание на способ кристаллизации твердой фазы для катализатора настоящего изобретения, который превращает смолу, содержащуюся в сыром газе и присущую ему, в легкие химические вещества, состоящие, главным образом, из метана, моноксида углерода и водорода, во время термического разложения углеродистого сырья. Этот способ кристаллизации твердой фазы отличается от обычных способов нанесения и имеет различные отличия, такие как возможность быстрой реакции, позволяющей тонкое осаждение активных частиц металла, возможность ингибирования уменьшения активности вследствие устойчивости к спеканию (укрупнению), так как осажденный активный металл сильно связан с матрицей (материнской фазой), и возможность регенерации, что позволяет ингибировать спекание путем предоставления возможности осажденным частицам активного металла возвращаться в твердый раствор в матрице путем прокаливания. Более конкретно, изобретатели настоящего изобретения обнаружили способ получения, допускающий превращение тяжелых углеводородов в легкие химические вещества, такие как метан, моноксид углерода и водород, путем увеличения площади поверхности активного металла и возможности осаждения нового активного металла, даже при воздействии отравления серой, даже в жестких условиях, когда компоненты, которые легко вызывают осаждение углерода, такие как смола и другие тяжелые углеводороды, содержатся в больших количествах в атмосфере, имеющей высокие концентрации сернистых компонентов, способных вызывать отравление серой, путем предварительного соединения активных частиц в форме никеля с оксидом алюминия или оксидом магния, и это служит в качестве матрицы, и использования тонкого осаждения металлического никеля из оксидной матрицы в кластерах на оксидной поверхности во время восстановительной обработки до реакции.
Кроме того, путем смешения со связующим из оксида алюминия, добавляемого для формования соединения никеля и оксида магния, описанного выше, во влажных условиях, было обнаружено, что смешанное состояние соединения никеля и оксида магния с оксидом алюминия становится очень однородным, и формованный катализатор, полученный с помощью последующей сушки и дробления с последующим формованием и обжигом или сушкой, прокаливанием, дроблением, формованием и обжигом, способен демонстрировать высокую активность реформинга при снижении количества осажденного углерода.
Последующее указывает отличия настоящего изобретения:
(1) способ получения катализатора для реформинга смолосодержащего газа, содержащий получение катализатора путем добавления осаждающего агента к смешанному раствору соединения никеля и соединения магния, образование осадка путем соосаждения никеля и магния, образование смеси путем добавления порошка оксида алюминия и воды или золя оксида алюминия к данному осадку и перемешивание, и, по меньшей мере, сушку и прокаливание данной смеси;
(2) способ получения катализатора для реформинга смолосодержащего газа, описанный в (1), где данный катализатор получают с помощью сушки и обжига или с помощью сушки, прокаливания, дробления, формования и обжига данной смеси;
(3) способ получения катализатора для реформинга смолосодержащего газа, содержащий получение катализатора путем добавления осаждающего агента к смешанному раствору соединения никеля и соединения магния, образование осадка путем соосаждения никеля и магния, образование смеси путем добавления порошка оксида алюминия и воды или золя оксида алюминия к данному осадку и перемешивание, сушку и прокаливание данной смеси, и дополнительное формование второй смеси путем смешения порошка оксида алюминия и воды или золя оксида алюминия, и, по меньшей мере, сушку и обжиг данной второй смеси;
(4) способ получения катализатора для реформинга смолосодержащего газа, описанный в (3), где данный катализатор получают с помощью сушки и обжига или с помощью сушки, прокаливания, дробления, формования и обжига данной второй смеси;
(5) способ получения катализатора для реформинга смолосодержащего газа, описанный в любом из пунктов (1)-(4), где данный катализатор для реформинга смолосодержащего газа получают так, что содержание никеля составляет от 1 до 50% масс., содержание магния составляет от 5 до 45% масс. и содержание оксида алюминия составляет от 20 до 80% масс.;
(6) способ получения катализатора для реформинга смолосодержащего газа, описанный в (5), где данный катализатор для реформинга смолосодержащего газа получают так, что содержание никеля составляет от 1 до 35% масс., содержание магния составляет от 10 до 25% масс. и содержание оксида алюминия составляет от 20 до 80% масс.;
(7) способ реформинга смолосодержащего газа, который использует катализатор для реформинга смолосодержащего газа, полученный согласно любому из способов получения, описанных в (1)-(6);
(8) способ реформинга смолосодержащего газа, описанный в (7), где смола, содержащаяся в смолосодержащем газе, превращается и газифицируется путем контакта с водородом, диоксидом углерода и водяным паром в смолосодержащем газе, образованном во время термического разложения углеродистого сырья, в присутствии катализатора для реформинга смолосодержащего газа или в присутствии данного катализатора после восстановления;
(9) способ реформинга смолосодержащего газа, описанный в (8), где смолосодержащий газ превращается и газифицируется путем контакта любого из, по меньшей мере, водорода, диоксида углерода и водяного пара извне со смолосодержащим газом, образованным во время термического разложения;
(10) способ реформинга смолосодержащего газа, описанный в любом из пунктов (7)-(9), где смолосодержащий газ представляет собой смолосодержащий газ, содержащий 20 ч./млн или более сероводорода;
(11) способ реформинга смолосодержащего газа, описанный в любом из пунктов (7)-(10), где кислородсодержащий газ добавляют к реформированному газу и приводят в контакт со смолосодержащим газом, и часть водорода или углеводородов в смолосодержащем газе сгорает;
(12) способ реформинга смолосодержащего газа, описанный в любом из пунктов (7)-(11), где смолосодержащий газ представляет собой газ сухой перегонки, образованный во время сухой перегонки угля;
(13) способ реформинга смолосодержащего газа, описанный в любом из пунктов (7)-(11), где смолосодержащий газ представляет собой газ коксовой печи, выпускаемый из коксовой печи;
(14) способ реформинга смолосодержащего газа, описанный в любом из пунктов (7)-(11), где смолосодержащий газ представляет собой газ сухой перегонки, образованный во время сухой перегонки, по меньшей мере, или древесной биомассы, или биомассы пищевых отходов;
(15) способ реформинга смолосодержащего газа, описанный в любом из пунктов (7)-(14), где смолосодержащий газ контактирует с катализатором для реформинга смолосодержащего газа при 600-1000°С; и
(16) способ регенерации катализатора для реформинга смолосодержащего газа, содержащий регенерацию катализатора путем контакта, по меньшей мере, водяного пара или воздуха с данным катализатором в случае, если производительность катализатора ухудшилась из-за, по меньшей мере, осаждения углерода или отравления серой в результате осуществления способа реформинга смолосодержащего газа, описанного в любом из пунктов (7)-(15).
Действие изобретения
Согласно настоящему изобретению может быть получен катализатор реформинга, который имеет высокую активность, высокую устойчивость к осаждению углерода, и способен к превращению в топливную композицию, состоящую, главным образом, из метана, моноксида углерода и водорода, путем превращения смолы, содержащейся в сыром газе, образованном во время термического разложения углеродистого сырья, и присущего ему, в легкие химические вещества в присутствии катализатора. В частности, может быть получен катализатор реформинга, который демонстрирует высокую производительность и имеет высокую устойчивость к отложению углерода даже для смолосодержащего газа, содержащего высокие концентрации сероводорода, такого как газ газификации биомассы или сырой ГКП. Кроме того, согласно одному аспекту настоящего изобретения смола может превращаться в легкие химические вещества путем использования теплосодержания смолосодержащего газа.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг.1 представляет собой чертеж, показывающий часть, где газ коксовой печи выпускается из коксовой печи.
Фиг.2 представляет собой чертеж для объяснения способа сухой перегонки угля в примере и способа реформинга смолосодержащего газа, образованного в нем, с использованием катализатора.
Фиг.3 представляет собой широкоугольную рентгеновскую дифрактограмму в части, эквивалентной пику Ni (200), катализатора после реакции в примере 2.
Фиг.4 представляет собой широкоугольную рентгеновскую дифрактограмму в части, эквивалентной пику Ni (200), катализатора после реакции в сравнительном примере 5.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Последующее обеспечивает более подробное объяснение настоящего изобретения путем приведения его конкретных примеров.
Катализатор для реформинга смолосодержащего газа, полученный согласно способу получения настоящего изобретения, функционирует как основной активный компонент, который позволяет никелю (Ni) вызывать реакцию реформинга между тяжелыми углеводородами и водяным паром, водородом и диоксидом углерода, присутствующими или вводимыми извне в газ. Даже в случаях, когда сероводород присутствует в высокой концентрации в смолосодержащем газе, данный катализатор считается устойчивым к снижению активности, вызванному отравлением серой, так как металлический никель тонко диспергирован в кластерах на поверхности катализатора, что приводит к увеличению площади поверхности, и в восстановительной атмосфере новые активные металлические гранулы тонко осаждаются из матрицы, даже если активные металлические гранулы подвергаются отравлению во время реакции. Активные металлические гранулы могут осаждаться из этого матричного соединения в форме мелких кластеров в восстановительной атмосфере. Кроме того, смола, которая, главным образом, образована из конденсированных полициклических ароматических соединений, находится в очень реакционноспособном состоянии при высоких температурах сразу после сухой перегонки, и считается, что в результате контакта с высокоактивным металлическим никелем, который тонко диспергирован и имеет высокую удельную площадь поверхности, превращается с высокой эффективностью и разлагается в легкие углеводороды. Кроме того, среди компонентов, которые соединены с элементарным никелем, оксид магния является основным оксидом, который имеет функцию адсорбции диоксида углерода, и так как он играет роль в реакции с осажденным углеродом на основном активном составляющем элементе, приводя к его удалению путем окисления в виде моноксида углерода, считается, что он способен поддерживать поверхность катализатора чистой и поддерживать стабильную производительность катализатора в течение длительного периода времени. В добавлении к демонстрации связующей функции, которая сохраняет стабильную матрицу соединения, оксид алюминия тонко разделяет кристаллические фазы, содержащие никель и магний, и, в результате, делает эти материалы высокодисперсными в оксидной твердой фазе, выполняя функцию, которая заставляет никелевые зерна активных частиц, которые осаждаются из каждой кристаллической фазы на поверхности, быть маленькими и в высокодисперсном состоянии.
Упоминаемое здесь "углеродистое сырье" означает сырой материал, который содержит углерод, который образует смолу при термическом разложении, и относится к широкому диапазону материалов, содержащих углерод в качестве составляющего элемента, таких как уголь, биомасса и пластиковые контейнеры и упаковки. В частности, термин "биомасса" относится к древесной биомассе, такой как остатки от рубки леса, древесина после уборки, неиспользованные деревья, остатки лесопильных заводов, строительные отходы или вторичные продукты, такие как древесная щепа и гранулы, которые используют их в качестве исходных материалов; бумажная биомасса, такая как бумажные отходы, которые не могут больше повторно использоваться в качестве вторичной бумаги; травянистая биомасса, такая как бамбуковая зелень, серебристая трава и другие сорняки, скошенные в парках или вдоль рек и дорог; биомасса на основе пищевых отходов, таких как кухонные отходы, сельскохозяйственные остатки, такие как рисовая солома, пшеничная солома или рисовая шелуха; целевые культуры, такие как сахарный тростник и другие источники сахаридов, зерно и другие источники крахмала, или рапсовое и другие масла; ил; и экскременты скота.
Кроме того, хотя ее свойства отличаются от сырого материала, который термически разлагается, "смолой" является то, что образуется во время термического разложения углеродистого сырья, относимое к органическому соединению, которое является жидкостью при комнатной температуре, в которой присутствует 5 или более атомов углерода, и представляет собой смесь, образованную из линейных углеводородов и циклических углеводородов. Конкретные примеры включают в себя, но не ограничиваются ими, смеси, образованные, главным образом, из конденсированных полициклических ароматических соединений, таких как нафталин, фенантрен, пирен или антрацен в случае термического разложения угля, соединения, образованные, главным образом, из бензола, толуола, нафталина, индена, антрацена или фенола в случае термического разложения древесных отходов, и гетероциклические соединения, содержащие разные элементы, такие как элементарный азот, в шестичленном или пятичленном кольце, такие как индол или пиррол в добавление к указанным примерам в случае термического разложения пищевых отходов. Смола термического разложения существует в газообразной форме, когда находится в высокотемпературном состоянии сразу после термического разложения.
Кроме того, реакция реформинга смолы, в которой смола газифицируется путем контактного разложения, представляет собой реакцию, которая превращает смолу, состоящую, главным образом, из тяжелых углеводородов, в легкие химические вещества, такие как метан, моноксид углерода и водород. Хотя маршрут реакции является сложным и не обязательно полностью понятным, считается, что эта реакция протекает в виде реакции гидрирования, реакции парового реформинга или реакции сухого реформинга и подобных, которые могут происходить между водородом, водяным паром, диоксидом углерода и подобным, присутствующими в смолосодержащем газе или вводимыми извне. Так как этот ряд реакций является эндотермическим, хотя высокотемпературный газ, имеющий теплосодержание, присутствующий в реакционном резервуаре, реформируется внутри слоя катализатора и снижается по температуре на выходе в случае фактического применения, в случае реформинга тяжелых углеводородных компонентов, таких как смола, более эффективно введение воздуха или кислорода в слой катализатора по необходимости, что позволяет реакции протекать дополнительно, поддерживая температуру слоя катализатора в определенной степени за счет теплоты сгорания, возникающей от сгорания части углеводородных компонентов.
Катализатор реформинга настоящего изобретения получают путем образования осадка, используя осаждающий агент, в смешанном растворе соединения никеля и соединения магния, образования смеси путем добавления порошка оксида алюминия и воды или золя оксида алюминия к данному осадку и смешения без сушки или прокаливания осадка, и, по меньшей мере, сушки или прокаливания данной смеси. Альтернативно, катализатор реформинга настоящего изобретения получают путем добавления осаждающего агента к смешанному раствору соединения никеля и соединения магния, образования осадка путем соосаждения никеля и магния, образования смеси путем добавления порошка оксида алюминия и воды или золя оксида алюминия к данному осадку и смешения без сушки или прокаливания осадка, образования второй смеси путем дополнительного подмешивания порошка оксида алюминия и воды или золя оксида алюминия после сушки и прокаливания данной смеси, и, по меньшей мере, сушки и прокаливания второй смеси.
Кроме того, примеры способов для, по меньшей мере, сушки и обжига смеси или второй смеси включают в себя сушку и обжиг; сушку, дробление и обжиг; сушку, дробление, формование и обжиг; сушку, прокаливание, дробление, формование и обжиг; и сушку, дробление, прокаливание, дробление, формование и обжиг.
Здесь нет особых ограничений на температуру и способ сушки при сушке смеси, и обычный способ сушки может быть использован. Высушенную смесь можно грубо дробить по необходимости с последующим обжигом (грубое дробление не требуется в случае, когда осадок сохраняет зернистую форму после сушки в результате сушки в псевдоожиженном слое).
Кроме того, смесь предпочтительно фильтруют перед сушкой, так как хлопоты, связанные с сушкой, могут быть снижены, и количество энергии, требуемой для сушки, может быть уменьшено. Кроме того, осадок более предпочтительно промывают чистой водой и подобным после фильтрования, так как это позволяет снизить количество примесей.
Кроме того, обжиг смеси можно выполнять на воздухе, и температура предпочтительно находится в диапазоне от 700 до 1300°С и более предпочтительно в диапазоне от 900 до 1150°С. Хотя высокая температура обжига вызывает спекание смеси, приводящее к увеличению прочности, с другой стороны, так как уменьшение удельной площади поверхности вызывает снижение каталитической активности, желательно определять температуру обжига соответственно балансу между ними. После обжига, хотя данная смесь может непосредственно использоваться в качестве катализатора, ее можно использовать как продукт для формования путем формования в пресс-форме и подобном. Кроме того, этапы прокаливания и формования также могут быть добавлены между сушкой и обжигом, и, если необходимо приводить в гранулированную форму перед формованием, между стадиями прокаливания и формования, формование можно выполнять после дробления. В этом случае прокаливание выполняют на воздухе при температуре приблизительно от 400 до 800°С, а формование выполняют путем пресс-формования и подобного.
Путем использования катализатора, полученного согласно такому способу получения, присущие тяжелые углеводороды, такие как смола, могут быть реформированы с высокой эффективностью и превращены в легкие химические вещества, главным образом, состоящие из водорода, моноксида углерода и метана, даже в случае смолосодержащего газа, состоящего, главным образом, из конденсированных полициклических ароматических соединений, который содержит большие количества сероводорода, образовавшегося во время термического разложения углеродистого сырья, и подверженного возникновению отложения углерода. Кроме того, путем контакта, по меньшей мере, водяного пара или воздуха с катализатором при высокой температуре, когда производительность катализатора ухудшилась, осажденный углерод и адсорбированная сера на катализаторе могут удаляться, тем самым, позволяя восстанавливать производительность катализатора и делать возможной стабильную работу в течение длительного периода времени.
Катализатор для реформинга смолосодержащего газа, полученный согласно настоящему способу получения, отличается от катализатора, полученного просто путем образования совместного осадка никеля и магния с последующим физическим подмешиванием порошка оксида алюминия в обожженный порошок и затем формования и обжига, тем, что путем мокрого подмешивания порошка оксида алюминия и воды или золя оксида алюминия в осадок никеля и магния водяной компонент, содержащий компонент из оксида алюминия, способен образовывать высокооднородную смесь с совместным осадком никеля и магния. Затем, в результате сушки и обжига или сушки, прокаливания, дробления, формования и обжига смеси, образуется спеченное тело, в котором соединения никеля и магния и оксид алюминия равномерно распределены, никель-магниевая кристаллическая фаза становится тоньше, и, так как осажденные из нее зерна Ni очень тонко диспергированы, считается, что может быть получен формованный продукт, который имеет высокую активность и низкое количество углеродных отложений.
Действительно, оценка размера зерен Ni, полученная из пика Ni (200) с помощью широкоугольной рентгеновской дифракции катализатора после реакции, показала, что зерна Ni осаждались в состоянии, имеющем малый средний размер зерна.
Более конкретно, можно использовать различные соединения металлов, имеющие высокую растворимость в воде, при приготовлении смешанного раствора соединения никеля и соединения магния, и примеры соединений, которые могут быть использованы, предпочтительно включают не только неорганические соли, такие как нитраты, карбонаты, сульфаты или хлориды, но также органические соли, такие как ацетаты. Нитраты, карбонаты или ацетаты особенно предпочтительны, так как считается, что они с меньшей вероятностью оставляют примеси, которые могут вызывать отравление катализатора после обжига. Кроме того, любой осаждающий агент может использоваться в качестве осаждающего агента, применяемого при образовании осадка из этих растворов, при условии, что он заставляет рН раствора изменяться в нейтральный-основный рН, при котором никель и магний осаждаются, главным образом, как гидроксиды. В частности, предпочтительно используются осаждающие агенты, такие как водный раствор карбоната калия, водный раствор карбоната натрия, водный раствор аммиака или раствор мочевины.
Кроме того, катализатор реформинга настоящего изобретения предпочтительно имеет содержание никеля от 1 до 50% масс., который служит в качестве основного активного компонента. Если содержание никеля меньше чем 1% масс., производительность реформинга никеля не демонстрируется в надлежащей степени, тем самым, делая это нежелательным. В случае, когда содержание никеля превышает 50% масс., так как содержание магния и алюминия, которые образуют матрицу, снижается, концентрация металлического никеля, который осаждается на катализаторе, увеличивается, и он становится очень грубым, что приводит к риску ухудшения производительности со временем в условиях настоящей реакции.
Кроме того, содержание магния предпочтительно составляет от 5 до 45% масс. Если содержание магния меньше чем 5% масс., это приводит к затруднению поддержания стабильной производительности катализатора на протяжении длительного периода времени путем ингибирования отложения углерода из углеводородов за счет преимущества свойств основного оксида, которыми обладает оксид магния, поэтому содержание магния предпочтительно составляет 5% или более. В случае, когда содержание магния превышает 45% масс., так как содержание других компонентов - никеля и алюминия - уменьшается, существует опасность невозможности адекватно демонстрировать активность реформинга катализатора.
Кроме того, содержание оксида алюминия предпочтительно составляет от 20 до 80% масс. Если содержание оксида алюминия меньше чем 20% масс., образуется керамика, состоящая, главным образом, из никеля-оксида магния, что вызывает заметное снижение прочности при формовании, что делает это нежелательным. В случае, когда содержание оксида алюминия превышает 80% масс., так как доли никеля, который служит в качестве основного активного компонента, и оксида магния, который ингибирует отложение углерода, снижаются, существует опасность неспособности адекватно демонстрировать активность реформинга катализатора.
Кроме того, катализатор реформинга настоящего изобретения более предпочтительно получают так, что содержание никеля составляет от 1 до 35% масс., содержание магния составляет от 10 до 25% масс., а содержание оксида алюминия составляет от 20 до 80% масс. Кроме того, упоминание добавления здесь оксида алюминия означает, что его добавляют к оксидам никеля и магния в форме порошка оксида алюминия или золя оксида алюминия. В случае добавления в виде порошка, предпочтительно использовать насколько возможно тонкий размер зерна, и, например, средний размер зерна 100 микрометров или менее является предпочтительным, и данный порошок используют в форме суспензии путем добавления воды или подобного во время смешения. Кроме того, в случае добавления в форме золя оксида алюминия, предпочтительно использовать золь, в котором гранулы оксида алюминия имеют средний размер зерна 100 нанометров или менее. Кроме того, предпочтительно готовить каждый из исходных материалов после предварительного вычисления, чтобы подгонять содержания всех металлических частиц так, что они находятся внутри указанных диапазонов. Кроме того, когда желаемая композиция катализатора достигнута, катализатор можно затем готовить на основании использованной тогда формулы.
Кроме того, хотя неизбежные примеси, которые входят в процесс получения катализатора, или другие компоненты, которые не изменяют производительность катализатора, также могут содержаться в дополнение к указанным элементам, желательно предотвращать загрязнение примесями насколько это возможно.
Кроме того, метод, называемый методом сканирующей высокочастотной индуктивно связанной плазмы (ICP), может использоваться для измерения содержания каждой из металлических частиц, которые составляют катализатор реформинга. Более конкретно, после дробления образца щелочной флюс (такой как карбонат натрия или борат натрия) добавляют с последующим нагревом и плавлением в платиновом тигле и полном растворении в растворе соляной кислоты с нагревом после охлаждения. Когда данный раствор вводят в ICP анализатор, так как раствор образца атомизируется и термически возбуждается в высокотемпературном плазменном состоянии внутри анализатора, что вызывает генерацию эмиссионного спектра с длиной волны, характерной для данного элемента, когда образец возвращается его основное состояние, типы содержащихся элементов и их количества могут быть качественно и количественно определены из длин волн эмиссии и их интенсивностей.
Катализатор реформинга, полученный в настоящем изобретении, может быть в форме порошка или формованного продукта, и в случае формованного продукта может быть сферическим, цилиндрическим, кольцеобразным, колесообразным или гранулированным и т.п., или может иметь каталитический компонент, нанесенный на металлический или керамический сотообразный базовый материал. Кроме того, в случае использования псевдоожиженного слоя может использоваться катализатор, который был образован путем распылительной сушки и подобного. Кроме того, в случае использования неподвижного слоя или подвижного слоя, примеры предпочтительных способов формования включают в себя гранулирование, экструзионное формование, пресс-формование и таблеточное формование, но не ограничиваются ими.
Согласно способу реформинга смолосодержащего газа, который использует катализатор реформинга, полученный согласно способу получения настоящего изобретения, получаются ранее описанные действия и эффекты. В этом способе реформинга смолосодержащего газа смола, присутствующая в смолосодержащем газе, реформируется и газифицируется путем контакта водорода, диоксида углерода или водяного пара, присутствующего в данном газе или вводимого извне, со смолосодержащим газом, образованным во время термического разложения углеродистого сырья, в присутствии катализатора или после восстановления катализатора. Хотя предпочтительно восстанавливать катализатор реформинга, так как восстановление происходит во время данной реакции, восстановление катализатора не требуется. Таким образом, смола, присутствующая в смолосодержащем газе, реформируется и газифицируется путем контакта смешанного газа, полученного путем добавления водяного пара и воздуха или кислорода, вводимого извне, со смолосодержащим газом, образованным во время термического разложения углеродистого сырья, в присутствии катализатора или после восстановления катализатора.
Хотя нет особых ограничений на условия в случае обеспечения восстановления катализатора, восстановление выполняют при сравнительно высокой температуре и в восстановительной атмосфере, так как зерна никеля, служащие в качестве активного металла, осаждаются в форме мелких кластеров из катализатора настоящего изобретения, данные условия могут состоять, например, из газовой атмосферы, содержащей, по меньшей мере, любой компонент из водорода, моноксида углерода или метана, газовой атмосферы, в которой водяной пар смешан с этими восстанавливающими газами, или атмосферы, в которой азот или другие инертные газы смешаны с этими газами. Кроме того, температура восстановления составляет предпочтительно, например, от 600 до 1000°С, время восстановления зависит от количества имеющегося катализатора, и хотя предпочтительно, например, от 30 минут до 4 часов, требуется только, чтобы за данное количество времени восстанавливалось полное количество имеющегося катализатора, и нет особых ограничений на это условие.
Реактор с неподвижным слоем, псевдоожиженным слоем или подвижным слоем предпочтительно используют в качестве каталитического реактора, и его температура входа слоя катализатора предпочтительно составляет от 600 до 1000°С. В случае, когда температура входа слоя катализатора ниже чем 600°С, трудно демонстрировать какую-либо каталитическую активность при реформинге смолы в легкие углеводороды, состоящие, главным образом, из водорода, моноксида углерода и метана, что делает это нежелательным. С другой стороны, в случае, когда температура входа слоя катализатора превышает 1000°С, требуется термостойкая структура, что увеличивает стоимость устройства реформинга и делает это экономически невыгодным. Кроме того, температура входа слоя катализатора более предпочтительно составляет от 650 до 1000°С. Кроме того, в случае, когда углеродистым сырьем является уголь, реакцию можно проводить при относительно высокой температуре, тогда как в случае древесной биомассы, бумажной биомассы или биомассы на основе пищевых отходов реакцию можно проводить при относительно низкой температуре.
Кроме того, хотя коксовая печь обычно применяется для образования смолосодержащего газа в случае использования угля в качестве сырья, или барабанная печь с внешним нагревом, печь с подвижным дном или печь с псевдоожиженным слоем и подобные применяются в случае использования биомассы в качестве сырья, нет особых ограничений на это.
Смола, присутствующая в газе, может реформироваться и газифицироваться с помощью настоящего изобретения, даже если смолосодержащий газ, образованный путем термического разложения или частичного окисления углеродистого сырья, представляет собой смолосодержащий газ, имеющий очень высокую концентрацию сероводорода, такой как высокотемпературный газ коксовых печей, выпускаемый из коксовой печи. Упомянутые здесь термическое разложение или частичное окисление более конкретно означают получение смолосодержащего газа путем сухой перегонки или путем окисления только части углеродистого сырья в целях газификации. В современных коксовых печах, хотя кокс производят путем заполнения данной печи угольным сырьем с последующим нагревом и сухой перегонкой, как показано на фиг.1, образующийся попутно газ коксовой печи собирают в сухой трубопровод 4, служащий в качестве газосборной трубы, из части, называемой стоячая труба 1, расположенной сверху печи, после охлаждения путем распыления водного аммиака 2. Однако, даже хотя газовый компонент сохраняет теплосодержание приблизительно 800°С в стоячей трубе 1 коксовой печи 3, оно пропадает при быстром охлаждении до 100°С или ниже после распыления водного аммиака 2, тем самым теплосодержание предотвращается от эффективного использования. Следовательно, если можно превращать тяжелые углеводороды, такие как смола, в топливные компоненты в форме легких углеводородов, таких как водород, моноксид углерода и метан, эффективно используя это теплосодержание газа, это не только приведет к приросту энергии, но, в результате существенного увеличения объема полученного здесь восстановительного газа, будет возможно применять способ получения восстановленного железа путем его приложения к железной руде, например, тем самым, позволяя резко уменьшить уровни выброса диоксида углерода, образующегося в современном способе в доменной печи, применяемом для восстановления железной руды коксом. Согласно настоящему изобретению газ коксовой печи, сохраняющий теплосодержание, образованный в коксовой печи, может превращаться в топливные компоненты в виде легких углеводородов, таких как водород, моноксид углерода и метан, путем проведения реформинга при эффективном использовании теплосодержания газа путем контакта газа коксовой печи с катализатором реформинга, полученным согласно способу получения настоящего изобретения.
Кроме того, хотя катализатор реформинга настоящего изобретения позволяет реакции реформинга стабильно протекать даже в атмосфере сероводорода, концентрация сероводорода в газе предпочтительно является насколько возможно низкой, так как это приводит к меньшему отравлению катализатора. В частности, концентрация сероводорода предпочтительно составляет 4000 ч./млн или менее и более предпочтительно 3000 ч./млн или менее.
С другой стороны, катализатор реформинга смолы, введенный в каталитический реактор, подвергается ухудшению каталитической производительности из-за отложения углерода на поверхности катализатора во время превращения смолы в легкие химические вещества, состоящие, главным образом, из водорода, моноксида углерода и метана, или из-за адсорбции сернистых компонентов на катализаторе, которые содержатся в газе термического разложения, полученном на стадии термического разложения, как описано выше. Следовательно, ухудшенный катализатор может регенерироваться путем удаления углерода на поверхности катализатора посредством введения водяного пара в каталитический реактор и предоставления возможности водяному пару реагировать с углеродом, или путем удаления серы, которая адсорбировалась на катализаторе, посредством реакции между водяным паром и серой. Кроме того, путем замены всего или части водяного пара воздухом и его введения, углерод на поверхности катализатора может удаляться посредством реакции горения между кислородом в воздухе и углеродом, или сера, которая адсорбировалась на катализаторе, может удаляться посредством реакции кислорода с серой, затем катализатор может регенерироваться.
ПРИМЕРЫ
Хотя последующее обеспечивает более подробное объяснение настоящего изобретения посредством его примеров, настоящее изобретение не ограничивается этими примерами.
Пример 1
Водный раствор карбоната калия, нагретый до 60°С, добавляли к водному раствору, нагретому до 60°С, приготовленному путем точного взвешивания нитрата никеля и нитрата магния, так что мольное отношение металлических элементов было 1:9, и никель и магний совместно осаждали в виде гидроксида с последующим надлежащим перемешиванием мешалкой. Затем, после старения путем непрерывного перемешивания в течение заданного периода времени, поддерживая при 60°С, данный осадок подвергали вакуумному фильтрованию с последующим надлежащим промыванием чистой водой при 80°С. Затем золь оксида алюминия добавляли к полученному осадку до 50% масс. в виде оксида алюминия с последующим надлежащим перемешиванием в смесителе, оборудованном лопастной мешалкой, переносом смеси в колбу для извлечения, прикреплением данной колбы к роторному испарителю и испарением воды путем аспирации при перемешивании. Смесь никеля, магния и оксида алюминия, приставшую к стенкам колбы для извлечения, переносили в испарительную чашку, сушили при 120°С и дробили с помощью ступки и пестика с последующим пресс-формованием порошка в форме таблеток, имеющих диаметр 3 мм, используя машину компрессионного формования, получая формованную таблетку. Формованную таблетку прокаливали на воздухе при 1100°С, получая формованный катализатор, в котором 50% масс. оксида алюминия были подмешаны к Ni0,1Mg0,9О.
Пробы 60 см3 этого катализатора фиксировали кварцевой ватой так, чтобы он располагался в центре SUS реакционной трубы, термопару вставляли в центр слоя катализатора, и реакционные трубы с неподвижным слоем устанавливали в заданных положениях.
Перед началом реакции реформинга реактор нагревали до 800°С в атмосфере азота, и восстанавливающую обработку проводили в течение 30 минут, позволяя газообразному водороду протекать сквозь реактор при 100 см3/мин. Затем смесь водорода и азота 1:1, используемую в качестве моделированного газа коксовой печи, и H2S в концентрации, показанной в таблице 1, вводили путем регулирования каждого газа до полных 125 см3/мин с последующим проведением реакции при каждой температуре, показанной в таблице 1, при нормальном давлении. Дополнительно, 1-метилнафталин, который действительно содержится в смоле и является жидким веществом, которое имеет низкую вязкость при нормальных температурах, использовали в качестве типичного примера вещества, которое моделирует смолу, образованную во время сухой перегонки угля, и вводили в реакционные трубы прецизионным насосом при скорости потока 0,025 г/мин. Дополнительно, чистую воду вводили в реакционные трубы прецизионным насосом при скорости потока 0,1 г/мин, так что отношение (число молей Н2О)/(число молей углерода в 1-метилнафталине) = 3. После соответствующего удаления нафталина и воды из полученного газа, выпускаемого из выхода, путем пропускания сквозь ловушку при комнатной температуре и ловушку со льдом, полученный газ вводили в газовый хроматограф (Hewlett-Packard HP6890), выполняя TCD и FID анализы. Степень реакции реформинга (скорость разложения метилнафталина) определяли на основании селективности по метану, селективности по СО, селективности по СО2 и величины отложения углерода, отлагающегося на катализаторе. Эти величины вычисляли, используя следующие уравнения, из концентраций каждого компонента в выходящем газе.
Селективность по метану (%) = (величина объема СН4)/(количество поданного С в поданном метилнафталине) × 100.
Селективность по СО (%) = (величина объема СО)/(количество поданного С в поданном метилнафталине) × 100.
Селективность по СО2 (%) = (величина объема СО2)/(количество поданного С в поданном метилнафталине) × 100.
Величина отложения углерода (%) = (масса осажденного углерода)/(количество поданного С в поданном метилнафталине) × 100.
Дополнительно, отношение объема выходящего газообразного водорода к объему входящего газообразного водорода (величина прироста водорода) также показана.
Согласно результатам для №№1-5 в таблице 1, реакция разложения метилнафталина, служащего в качестве модельной смолы, выполняемая даже в атмосфере, содержащей высокую концентрацию H2S 2000 ч./млн, при условии, что катализатор получен согласно настоящему способу получения, очень устойчива к отравлению серой. Кроме того, величины разложения (селективность по метану + селективность по СО + селективность по СО2 + величина отложения углерода) увеличивались с ростом температуры реакции, в частности, и было определено, что реакция разложения метилнафталина протекает даже в жестких условиях с высокими уровнями отравления серой и отложения углерода. Кроме того, влияние отравления серой уменьшалось при уменьшении концентрации H2S, а величина разложения дополнительно увеличивалась. Кроме того, так как отношение прироста водорода также увеличивалось, сопровождая увеличение величины разложения модельной смолы, считается, что водород, связанный с углеродом, образующим метилнафталин, превращался в молекулы водорода, сопровождая разложение с помощью катализатора. Кроме того, величины отложения углерода показали относительно низкие значения, и эти значения уменьшались, когда температура становилась выше. Кроме того, общая реакция реформинга протекала эффективно при высоких температурах 800°С или выше, и было определено, что величина разложения увеличивалась, когда температура становилась выше.
Пример 2
Каталитическую активность определяли в условиях, показанных в таблице 2, используя такой же катализатор, как в примере 1, и такой же метод, как в примере 1, за исключением того, что прокаливали при 950°С и использовали 30 см3 катализатора. Эти результаты показаны в таблице 2.
Согласно результатам №6-9 в таблице 2, активность реформинга модельной смолы увеличивалась и величина разложения улучшалась с ростом температуры до 900°С в атмосфере, имеющей высокую концентрацию H2S 2000 ч./млн. Кроме того, при сравнении результатов №8, 10 и 11, в которых температура реакции была одинаковой, а концентрация H2S разной, хотя величина разложения модельной смолы имела тенденцию к уменьшению, когда концентрация становилась выше, было обнаружено, что величины разложения демонстрировали высокую активность 74% при 800°С даже при концентрации H2S 2000 ч./млн.
Дополнительно, определение размера зерен Ni из пика Ni (200) катализатора после реакции, определяемого с помощью широкоугольной рентгеновской дифракции, проводили описанным ниже образом. Сначала, после размещения образца в держателе порошкового образца, луч CuKα генерировали при мощности 40 кВ и 150 мА, используя Rigaku RINT1500, и измерение выполняли, используя графит в качестве монохроматора в условиях величины 1° для щели расхождения и щели рассеяния, величины 0,15 мм для приемной щели, величины 0,8 мм для монохроматической приемной щели, величины 0,01° для ширины дискретизации и величины 2 град/мин для скорости сканирования. В профилях измерения ширину пика на положении высоты половины вершины пика (полуширина) измеряли для пика Ni (200), в котором 2θ появляется вблизи приблизительно 52 градусов, в частности, и размер зерен Ni вычисляли из этой величины, используя уравнение Шеррера, приведенное ниже
Dhkl=Kλ/βcosθ
Так как Dhkl показывает размер кристаллических зерен, когда определяется на основании линии дифракции Ni (200), он представляет размер кристаллических зерен Ni. Хотя К является константой, так как полуширина используется для β, как показано ниже, величина этой константы становится 0,9. λ представляет собой рентгеновскую длину волны, использованную для измерения, и в выполненном здесь измерении она имеет величину 1,54056 Å. Кроме того, β представляет собой уширение дифракционных линий соответственно размеру кристаллических зерен, и вышеуказанную величину полуширины использовали для этой величины. θ обозначает угол Брегга дифракционной линии Ni (200).
Согласно этому методу размер зерен Ni, определенный из пика Ni (200) с помощью широкоугольной рентгеновской дифракции, показанной на фиг.3, после дробления катализаторов, использованных в этом тесте, вычисляли равным 13 нм, тем самым демонстрируя осаждение очень мелких зерен Ni, что считается причиной возникновения высоких уровней активности реформинга и устойчивости к отложению углерода.
Пример 3
Катализаторы готовили таким же образом, как в примере 1, за исключением использования массовых процентов никеля, магния и оксида алюминия в оксидах никеля и магния, показанных в таблицах 3 и 4. Условия №3 в примере 1 использовали для условий эксперимента при температуре реакции 800°С, концентрации H2S 2000 ч./млн и нормальном давлении. Результаты показаны в таблицах 3 и 4.
Согласно результатам таблиц 3 и 4 величина разложения метилнафталина становится ниже и величина прироста водорода также уменьшается с уменьшением количества никеля, служащего в качестве основного активного компонента, и в случае №12, где содержание Ni было меньше чем 1% масс., результаты были низкими и для величины разложения, и для величины прироста водорода. С другой стороны, величина разложения и величина прироста водорода увеличивались с увеличением содержания Ni. Однако в случае №24, где содержание Ni превышает 50% масс., была большая величина отложения углерода. Кроме того, при сравнении между №13 и 20 и между №14 и 21, даже хотя содержания Ni были приблизительно равными, из-за различия в содержании оксида алюминия большее количество компонента оксида алюминия приводило к более высокой каталитической активности. Считается, что это происходит из-за того, что оксид алюминия тонко расщепляет смешанную никель-магниевую фазу, приводя к снижению размера частиц металлического Ni, осаждающихся во время восстановления, и, в свою очередь, вызывая увеличение реакционной площади поверхности. Кроме того, величины отложения углерода были ниже для более высоких содержаний Mg, и в случае №16, где содержание Mg было меньше чем 5% масс., был высокий уровень отложения углерода.
Пример 4
Катализаторы готовили и оценивали таким же образом, как в примере 1, за исключением того, что использовали температуру реакции 800°С, концентрацию H2S 2000 ч./млн и вводили Н2О, СО2 и О2 в условиях, показанных в таблице 5, во время реакции. Результаты показаны в таблице 5. Кроме того, С в отношениях Н2О/С, СО2/С и О2/С в таблице обозначает подаваемое количество С в подаваемом метилнафталине.
Согласно результатам таблицы 5, по сравнению с результатами для №3 примера 1 было доказано, что реакция реформинга промотируется введением Н2О, СО2 и О2 извне. Кроме того, в случае введения О2, так как поглощаемое тепло парового реформинга при использовании Н2О или сухого реформинга при использовании СО2 может быть компенсировано теплотой сгорания, это является очень эффективной технологией в случае реального реактора. Кроме того, введение О2 также дополнительно снижает величину отложения углерода.
Пример 5
80 кг загрузку угля, используемого в реальных коксовых печах, помещали в печь периодического действия, способную моделировать коксовую печь, с последующим нагревом до 800°С таким же образом, как в реальной коксовой печи, с образованием реального газа коксовой печи и присущей ему реальной смолы. Количество смолы в смолосодержащем газе в это время было приблизительно 0,04 г/л. Данный газ захватывали всасывающим насосом и использовали в эксперименте. Реакционную трубу помещали внутри электрической печи, нагретой до температуры реакции 800°С, оксид, полученный путем подмешивания 50% масс. оксида алюминия в Ni0,1Mg0,9О, формованный в форме кольца с помощью такого же способа получения, как в примере 1, помещали в центре реакционной трубы, и, после восстановления в течение 2 часов при скорости потока водорода 10 нл/мин, газ, захваченный из печи периодического действия, подавали в слой катализатора с последующим непрерывным определением активности каталитического разложения реального газа коксовой печи и реальной, присущей ему смолы в течение 5 часов. Скорость потока входящего газа была приблизительно 10 нл/мин, а количество наполняющего катализатора было приблизительно 1 л. Кроме того, с помощью газовой хроматографии было доказано, что композиция входящего газа была приблизительно такой же, как композиция реального газа коксовой печи. Кроме того, было доказано, что газ содержал от 2400 до 2500 ч./млн сероводорода. Концентрацию смолы в газе определяли, используя описанный ниже способ, а именно каждый газ захватывали путем предварительного прикрепления 1 л вакуумной пробоотборной колбы, сохраняющей вакуум до контрольного крана, прикрепленного ко входу и выходу слоя катализатора, и затем открывания данного крана. Внутренность пробоотборной колбы затем промывали дихлорметаном, и после полного удаления дихлорметана жидкий компонент взвешивали при нормальной температуре. Величину разложения смолы определяли из отношения массы смоляного компонента в выходящем газе слоя катализатора к массе смоляного компонента во входящем газе слоя катализатора, захваченными с использованием описанного выше способа. В результате величина разложения смолы, когда 2 часа протекало от начала реакции, составляла 89%, а величина прироста водорода достигала в среднем 2,3 в течение 5 часов.
Пример 6
После осуществления реакции непрерывно в течение 8 часов в условиях №3 примера 1 подачу сырья прерывали, и углерод и серу, осажденные на катализаторе, удаляли, используя N2 в качестве газа-носителя при 60 см3/мин и Н2О при 60 см3/мин в виде газа, устанавливая температуру слоя катализатора на 800°С и поддерживая ее в течение 5 часов. Затем, когда подачу сырья возобновляли в таких же условиях, как в примере 1, было доказано, что катализатор демонстрировал активность, равную 90% или более от активности до регенерации. Кроме того, было доказано, что концентрация водорода в газе после реформинга в этом тесте была высокой, и было доказано, что газ превращался в газ, состоящий, главным образом, из водорода, моноксида углерода и метана.
Пример 7
После осуществления реакции непрерывно в течение 8 часов в условиях №3 примера 1 таким же образом, как в примере 6, подачу сырья прерывали, и углерод и серу, осажденные на катализаторе, удаляли, используя N2 в качестве газа-носителя при 60 см3/мин и воздух при 60 см3/мин, устанавливая температуру слоя катализатора на 800°С и поддерживая ее в течение 2 часов. Затем, когда подачу сырья возобновляли в таких же условиях, как в примере 1, было доказано, что катализатор демонстрировал активность, равную 90% или более от активности до регенерации. Кроме того, было доказано, что концентрация водорода в газе после реформинга в этом тесте была высокой, и было доказано, что газ превращался в газ, состоящий, главным образом, из водорода, моноксида углерода и метана.
Пример 8
После нагрева барабанной печи 7, показанной на фиг.2, используемой в качестве печи сухой перегонки, до 800°С, куски угля вводили в барабанную печь 7 при скорости подачи 20 кг/ч из бункера 5, заполненного кусками угля (размером 5 см или менее), используя устройство 6 постоянной количественной подачи, для генерации газа сухой перегонки, содержащего смолу. Смолосодержащий газ (газ сухой перегонки) вводили в каталитическую колонну 8, заполненную кольцеобразным катализатором, имеющим такую же композицию, как композиция в примере 1, и имеющим внешний диаметр 15 мм, внутренний диаметр 5 мм и высоту 15 мм, с последующим нагревом до приблизительно 800°С при регулировке скорости потока газа приблизительно 10 нм3/ч вытяжным вентилятором 11, и активность каталитического разложения смолосодержащего газа непрерывно определяли в течение 8 часов, позволяя смолосодержащему газу контактировать с катализатором. Затем реформированный газ охлаждали водой в скруббере 9 и очищали от пыли в масляном барботере 10 с последующим сжиганием для рассевания в факельной стойке 12. Кроме того, восстановительную обработку проводили в течение 30 минут водородом при 5 нм3/ч до подачи сырья. Скорость потока входящего газа была приблизительно 10 нм3/ч, а количество наполняющего катализатора было приблизительно 15 л. Количество смолы в смолосодержащем газе в это время было приблизительно 60 г/нм3. Кроме того, с помощью газовой хроматографии было доказано, что композиция входящего газа была приблизительно такой же, как композиция реального газа коксовой печи. Кроме того, приблизительно 6% воды, содержащейся в угле, используемом в качестве сырья, испарялось и содержалось в газе в виде водяного пара. Кроме того, было доказано, что газ содержал от 2000 до 2500 ч./млн сероводорода. Концентрацию смолы в газе определяли, пропуская газ из входа и выхода слоя катализатора через пятистадийный импинжер, заполненный дихлометаном, путем аспирации в течение заданного периода времени для захвата смоляного компонента в газе, с последующим количественным определением жидкого компонента при нормальной температуре после удаления дихлорметана. Величину разложения смолы определяли из отношения массы смоляного компонента в выходящем газе слоя катализатора к массе смоляного компонента во входящем газе слоя катализатора, захваченными с использованием описанного выше способа. В результате величина разложения смолы, когда 3 часа протекало от начала реакции, составляла 82%, а величина прироста водорода достигала в среднем 2,35 в течение 8 часов, и было подтверждено, что реакция каталитической сухой газификации смолосодержащего газа протекала в масштабе лабораторной установки.
Пример 9
Смолосодержащий газ из биомассы (газ сухой перегонки) получали, используя такое же оборудование, как в примере 8, подавая щепки строительных отходов (размером 5 см или менее) в это оборудование при скорости подачи 10 кг/ч и выполняя сухую перегонку щепок в барабанной печи 7, поддерживаемой при температуре 800°С. Смолосодержащий газ вводили в каталитическую колонну, поддерживаемую при приблизительно 800°С и заполненную таким же формованным катализатором, как в примере 8, за исключением того, что он имел такую же композицию, как №18 примера 3, и активность каталитического разложения смолосодержащего газа непрерывно определяли в течение 8 часов, позволяя смолосодержащему газу контактировать с катализатором. Кроме того, восстановительную обработку выполняли в течение 30 минут водородом при 5 нм3/ч до подачи сырья. Скорость потока входящего газа была приблизительно 10 нм3/ч, а количество наполняющего катализатора было приблизительно 15 л. Количество смолы в смолосодержащем газе из биомассы в это время было приблизительно 10 г/нм3. Кроме того, с помощью газовой хроматографии было доказано, что композиция входящего газа была приблизительно такой же, как композиция газа коксовой печи и была образована, главным образом, из водорода, СО, метана и СО2. Кроме того, приблизительно 16% воды, содержащейся в строительных отходах, используемых в качестве сырья, испарялось и содержалось в виде водяного пара. Кроме того, было доказано, что газ содержал приблизительно 25 ч./млн сероводорода. Кроме того, величину разложения смолы определяли, используя такой же метод, как в примере 8, путем захвата смоляного компонента в смолосодержащем газе из входа и выхода слоя катализатора и количественного определения полученного смоляного компонента. В результате величина разложения смолы, когда 3 часа протекало от начала реакции, составляла 94,4%, величина прироста водорода стабилизировалась при приблизительно 6,5 в течение 8 часов, и было подтверждено, что реакция каталитической сухой газификации смолосодержащего газа из биомассы стабильно протекала в масштабе лабораторной установки.
Пример 10
Смолосодержащий газ из биомассы (газ сухой перегонки) получали, используя такое же оборудование, как в примере 8, подавая сухие куски (размером 5 см или менее) древесных отходов, собираемых из супермаркетов и подобного, в это оборудование при скорости подачи 10 кг/ч и выполняя сухую перегонку древесных отходов в барабанной печи 7, поддерживаемой при температуре 800°С. Смолосодержащий газ вводили в каталитическую колонну, поддерживаемую при приблизительно 800°С и заполненную таким же формованным катализатором, как в примере 8, за исключением того, что он имел такую же композицию, как №18 примера 3, и активность каталитического разложения смолосодержащего газа непрерывно определяли в течение 8 часов, позволяя смолосодержащему газу контактировать с катализатором. Кроме того, восстановительную обработку выполняли в течение 30 минут водородом при 5 нм3/ч до подачи сырья. Скорость потока входящего газа была приблизительно 10 нм3/ч, а количество наполняющего катализатора было приблизительно 15 л. Количество смолы в смолосодержащем газе из биомассы в это время было приблизительно 23 г/нм3. Кроме того, с помощью газовой хроматографии было доказано, что композиция входящего газа была приблизительно такой же, как композиция газа коксовой печи и была образована, главным образом, из водорода, СО, метана и СО2. Кроме того, приблизительно 30% воды, содержащейся в смеси сухих древесных отходов и строительных отходов, используемой в качестве сырья, испарялось и содержалось в виде водяного пара. Кроме того, было доказано, что газ содержал приблизительно 400 ч./млн сероводорода. Кроме того, величину разложения смолы определяли, используя такой же метод, как в примере 8, путем захвата смоляного компонента в смолосодержащем газе из входа и выхода слоя катализатора и количественного определения полученного смоляного компонента. В результате величина разложения смолы, когда 3 часа протекало от начала реакции, составляла 88,5%, величина прироста водорода оставалась стабильной при более чем 4,5 в течение 4 часов после начала реакции, даже хотя она постепенно снижалась в начале в реакции из-за отравления серой, и было подтверждено, что реакция каталитической сухой газификации смолосодержащего газа из биомассы стабильно протекала в масштабе лабораторной установки.
Пример 11
Смолосодержащий газ из биомассы (газ сухой перегонки) получали, используя такое же оборудование, как в примере 8, подавая смесь таких же сухих кусков (размером 5 см или менее) древесных отходов из примера 10 и щепок строительных отходов (размером 5 см или менее), смешанных с массовым отношением 1:2, в это оборудование при скорости подачи 10 кг/ч и выполняя сухую перегонку древесных отходов в барабанной печи 7, поддерживаемой при температуре 800°С. Смолосодержащий газ вводили в каталитическую колонну, поддерживаемую при приблизительно 800°С и заполненную таким же формованным катализатором, как в примере 8, за исключением того, что он имел такую же композицию, как №18 примера 3, и активность каталитического разложения смолосодержащего газа непрерывно определяли в течение 8 часов, позволяя смолосодержащему газу контактировать с катализатором. Кроме того, восстановительную обработку выполняли в течение 30 минут водородом при 5 нм3/ч до подачи сырья. Скорость потока входящего газа была приблизительно 10 нм3/ч, а количество наполняющего катализатора было приблизительно 15 л. Количество смолы в смолосодержащем газе из биомассы в это время было приблизительно 14 г/нм3. Кроме того, с помощью газовой хроматографии было доказано, что композиция входящего газа была приблизительно такой же, как композиция газа коксовой печи и была образована, главным образом, из водорода, СО, метана и СО2. Кроме того, приблизительно 20% воды, содержащейся в смеси сухих древесных отходов и строительных отходов, используемой в качестве сырья, испарялось и содержалось в виде водяного пара. Кроме того, было доказано, что газ содержал приблизительно 200 ч./млн сероводорода. Кроме того, величину разложения смолы определяли, используя такой же метод, как в примере 5, путем захвата смоляного компонента в смолосодержащем газе из входа и выхода слоя катализатора и количественного определения полученного смоляного компонента. В результате величина разложения смолы, когда 3 часа протекало от начала реакции, составляла 87,5%, величина прироста водорода оставалась стабильной при приблизительно 4,6 в течение 4 часов после начала реакции, даже хотя она постепенно снижалась в начале в реакции из-за отравления серой, и было подтверждено, что реакция каталитической сухой газификации смолосодержащего газа из биомассы стабильно протекала в масштабе лабораторной установки.
Пример 12
После реформинга в течение 8 часов в примере 8 подачу угля, служащего в качестве сырья, прерывали, и систему продували азотом. Затем, путем отбора воздуха из порта ввода газа, установленного вблизи входа барабанной печи 7 при 800°С и ввода воздуха, нагретого в барабанной печи, в каталитическую колонну в течение приблизительно 10 часов, удаляли осажденный углерод и адсорбированную серу, которые осели на поверхности катализатора после реформинга, с помощью окисления, регенерируя катализатор. Затем, после продувки системы азотом для удаления кислородного компонента, снова выполняли восстановительную обработку в течение 30 минут водородом при 5 нм3/ч. Затем сырье подавали с такой же скоростью, как в примере 8, и активность каталитического разложения смолосодержащего газа непрерывно определяли в течение 8 часов, позволяя смолосодержащему газу контактировать с катализатором. В результате получали величины разложения смолы и прироста водорода после регенерации, которые были подобны величинам до регенерации, и было подтверждено, что катализатор надлежащим образом регенерировался путем горения в воздухе. Кроме того, хотя каталитическое разложение этого смолосодержащего газа и последующую регенерацию катализатора повторяли пять раз, стабильные результаты получали для величины прироста водорода, которые были подобны результатам до регенерации, и было обнаружено, что продолжительная работа возможна.
Пример 13
После реформинга в течение 8 часов в примере 9 подачу щепок строительных отходов, служащих в качестве сырья, прерывали, и систему продували азотом таким же образом, как в примере 12. Затем, путем отбора воздуха из порта ввода газа, установленного вблизи входа барабанной печи 7 при 800°С и ввода воздуха, нагретого в барабанной печи, в каталитическую колонну в течение приблизительно 10 часов, удаляли осажденный углерод и адсорбированную серу, которые осели на поверхности катализатора после реформинга, с помощью окисления, регенерируя катализатор. Затем, после продувки системы азотом для удаления кислородного компонента, снова выполняли восстановительную обработку в течение 30 минут водородом при 5 нм3/ч. Затем сырье подавали с такой же скоростью, как в примере 9, и активность каталитического разложения смолосодержащего газа непрерывно определяли в течение 8 часов, позволяя смолосодержащему газу контактировать с катализатором. В результате получали величины разложения смолы и прироста водорода после регенерации, которые были подобны величинам до регенерации, и было подтверждено, что катализатор надлежащим образом регенерировался путем горения в воздухе также в случае щепок строительных отходов. Кроме того, хотя каталитическое разложение этого смолосодержащего газа из биомассы и последующую регенерацию катализатора повторяли шесть раз, стабильные результаты получали для величины прироста водорода, которые были подобны результатам до регенерации, и было обнаружено, что продолжительная работа возможна.
Пример 14
После реформинга в течение 8 часов в примере 10 подачу сухих кусков древесных отходов, служащих в качестве сырья, прерывали, и систему продували азотом таким же образом, как в примере 12. Затем, путем отбора воздуха из порта ввода газа, установленного вблизи входа барабанной печи 7 при 800°С и ввода воздуха, нагретого в барабанной печи, в каталитическую колонну в течение приблизительно 10 часов, удаляли осажденный углерод и адсорбированную серу, которые осели на поверхности катализатора после реформинга, с помощью окисления, регенерируя катализатор. Затем, после продувки системы азотом для удаления кислородного компонента, снова выполняли восстановительную обработку в течение 30 минут водородом при 5 нм3/ч. Затем сырье подавали с такой же скоростью, как в примере 10, и активность каталитического разложения смолосодержащего газа непрерывно определяли в течение 8 часов, позволяя смолосодержащему газу контактировать с катализатором. В результате получали величины разложения смолы и прироста водорода после регенерации, которые были подобны величинам до регенерации, и было подтверждено, что катализатор надлежащим образом регенерировался путем горения в воздухе также в случае сухих кусков древесных отходов. Кроме того, хотя каталитическое разложение этого смолосодержащего газа из биомассы и последующую регенерацию катализатора повторяли пять раз, стабильные результаты получали для величины прироста водорода, которые были подобны результатам до регенерации, и было обнаружено, что продолжительная работа возможна.
Пример 15
После реформинга в течение 8 часов в примере 11 подачу смеси сухих кусков древесных отходов и щепок строительных отходов, служащих в качестве сырья, прерывали, и систему продували азотом таким же образом, как в примере 12. Затем, путем отбора воздуха из порта ввода газа, установленного вблизи входа барабанной печи 7 при 800°С и ввода воздуха, нагретого в барабанной печи, в каталитическую колонну в течение приблизительно 10 часов, удаляли осажденный углерод и адсорбированную серу, которые осели на поверхности катализатора после реформинга, с помощью окисления, регенерируя катализатор. Затем, после продувки системы азотом для удаления кислородного компонента, снова выполняли восстановительную обработку в течение 30 минут водородом при 5 нм3/ч. Затем сырье подавали с такой же скоростью, как в примере 11, и активность каталитического разложения смолосодержащего газа непрерывно определяли в течение 8 часов, позволяя смолосодержащему газу контактировать с катализатором. В результате получали величины разложения смолы и прироста водорода после регенерации, которые были подобны величинам до регенерации, и было подтверждено, что катализатор надлежащим образом регенерировался путем горения в воздухе также в случае смеси сухих кусков древесных отходов и щепок строительных отходов. Кроме того, хотя каталитическое разложение этого смолосодержащего газа из биомассы и последующую регенерацию катализатора повторяли пять раз, стабильные результаты получали для величины прироста водорода, которые были подобны результатам до регенерации, и было обнаружено, что продолжительная работа возможна.
Сравнительный пример 1
Когда тест реформинга выполняли, используя один сорт промышленного катализатора в форме катализатора первичного реформинга нафты, изготовленного Sud-Chemie Catalysts Japan (SC11NK: содержащий 20% масс. Ni и имеющий высокую прочность 500 Н), используя такой же экспериментальный метод, как в примере 1, и в условиях №8 примера 2, селективность по метану была 2,5%, селективность по СО была 4,2%, селективность по СО2 была 5,9%, величина отложения углерода была 32,8%, величина разложения была 45,4% и величина прироста водорода была 1,3.
Таким образом, получали результаты, показывающие, что, хотя имела место низкая величина конверсии метилнафталина в газовые компоненты (12,6%), промышленный катализатор имел очень высокую величину отложения углерода. Так как величина отложения углерода очень высока, существует значительный риск короткого срока службы катализатора. Кроме того, даже если проводить регенерирующую обработку после реакции, так как необходимо выполнять окислительную обработку при высокой температуре или в течение длительного периода времени, частицы, имеющие каталитическую активность, подвергаются спеканию из-за большой теплоты сгорания в это время, и предполагается, что производительность после регенерации будет даже ниже.
Сравнительный пример 2
Определение проводили, помещая промышленный катализатор (SC11NK), используемый в сравнительном примере 1, в реакционную трубу, используя такое же тестовое оборудование, как в примере 5, и в таких же условиях, как в примере 5. В результате величина разложения смолы достигала только 22% после 2 часов, прошедших от начала реакции, величина прироста водорода также в среднем составляла только приблизительно 1,5 в течение 5 часов, а также было обнаружено, что величина разложения смолы была еще ниже, когда промышленный катализатор испытывали в присутствии реального газа коксовой печи и реальной смолы.
Сравнительный пример 3
После приготовления осадка никеля и магния таким же образом, как в примере 1, с последующим фильтрованием, промыванием и сушкой, осадок прокаливали в течение 20 часов на воздухе при 950°С, получая соединение никеля и оксида магния. Затем к нему добавляли золь кремнекислоты, так что отношение SiO2 в катализаторе было 50% масс., получая суспензию. Затем распылительную сушку выполняли в таких условиях, что средний размер зерен был 50 мкм, и полученный порошок прокаливали на воздухе при 950°С. Кроме того, после формования и прокаливания полученного твердого раствора оксида, используя такую же процедуру, как в примере 1, определяли активность в таких же условиях, как в №8 примера 2. В результате каталитическая активность была такова, что величина разложения метилнафталина была очень низкой только при приблизительно 15%, величина прироста водорода была 1,0, показывая, что водород не прирастал совсем, и было определено, что активность катализатора была низкой.
Сравнительный пример 4
Осадок никеля и магния готовили таким же образом, как в примере 1, с последующим фильтрованием, промыванием и сушкой. Затем осадок прокаливали на воздухе при 950°С, получая соединение никеля и оксида магния. Затем порошок оксида алюминия взвешивали до 50% масс., и эти два компонента физически смешивали, используя ступку и пестик. После формования и прокаливания полученной смеси, используя такую же процедуру, как в примере 1, определяли активность в таких же условиях, как в №8 примера 2. В результате, хотя каталитическая активность была такова, что величина разложения метилнафталина была приблизительно 66,7% и величина прироста водорода была 1,6, показывая умеренную каталитическую активность, прочность была очень низкой, и была определена трудность практического применения.
Сравнительный пример 5
Осадок получали, используя осаждающий агент, из водного раствора, содержащего никель, оксид магния и оксид алюминия с последующим обжигом согласно способу получения, описанному, например, в непатентном документе 1 или патентном документе 8, а именно смешанный раствор готовили путем вычисления и точного взвешивания нитрата никеля, нитрата магния и нитрата алюминия, так что мольное отношение металлических элементов никеля и магния было 1:9 так же, как в примере 1, и содержание оксида алюминия было 50% масс., и затем смешивания в чистой воде при 60°С, после чего добавляли водный раствор карбоната калия, нагретый до 60°С, таким же образом, как в примере 1, с последующим надлежащим перемешиванием мешалкой. Затем, после старения путем непрерывного перемешивания в течение заданного периода времени при поддержании 60°С, осадок подвергали вакуумному фильтрованию с последующим надлежащим промыванием чистой водой при 80°С. Затем осадок переносили в испарительную чашку, сушили при 120°С и дробили с помощью ступки и пестика с последующим пресс-формованием таким же образом, как в примере 1, используя машину компрессионного формования, получая формованную таблетку. Формованную таблетку прокаливали при 1100°С, получая формованный каталитический продукт. Активность определяли, используя этот формованный продукт, в таких же условиях, как №8 примера 2. В результате катализатор демонстрировал только умеренную каталитическую активность, демонстрируя величину разложения метилнафталина приблизительно 62,6% (величина отложения углерода: 19,9%) и величину прироста водорода 1,6, и было обнаружено, что количество осажденного углерода было очень высоко.
Дополнительно, прореагировавший катализатор дробили, используя такую же процедуру, как в примере 2, вычисляли, что размер зерен Ni, определенный из пика Ni (200) с помощью широкоугольной рентгеновской дифракции, как показано на фиг.4, составлял 35 нм, и, так как Ni не был способен тонко осаждаться из соединения, приготовленного согласно данному способу, считается, что это приводило к большому количеству осажденного углерода и низкой активности реформинга.
КРАТКОЕ ОПИСАНИЕ ЧИСЛЕННЫХ ОБОЗНАЧЕНИЙ
1 Стоячая труба
2 Водный аммиак
3 Коксовая печь
4 Сухой трубопровод
5 Бункер сырья
6 Устройство постоянной количественной подачи
7 Барабанная печь с внешним нагревом
8 Каталитическая колонна
9 Водяной скруббер
10 Масляный барботер
11 Принудительный вытяжной вентилятор
12 Расширяющаяся факельная стойка
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР РЕФОРМИНГА | 2011 |
|
RU2558150C2 |
| СПОСОБ КАТАЛИТИЧЕСКОГО ПРЕДВАРИТЕЛЬНОГО РЕФОРМИНГА УГЛЕВОДОРОДНОГО СЫРЬЯ, СОДЕРЖАЩЕГО ВЫСШИЕ УГЛЕВОДОРОДЫ | 2000 |
|
RU2250873C2 |
| КАТАЛИЗАТОР КОНВЕРСИИ УГЛЕВОДОРОДОВ, СПОСОБ КАТАЛИТИЧЕСКОГО РЕФОРМИНГА ИСХОДНОЙ НАФТЫ С ИСПОЛЬЗОВАНИЕМ КАТАЛИЗАТОРА | 1997 |
|
RU2186622C2 |
| СПОСОБ РЕФОРМИНГА С ИСПОЛЬЗОВАНИЕМ КАТАЛИЗАТОРА ВЫСОКОЙ ПЛОТНОСТИ | 2005 |
|
RU2388534C2 |
| КАТАЛИЗАТОР И СПОСОБ РИФОРМИНГА КИСЛОРОДСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 2006 |
|
RU2438968C2 |
| ВЫХЛОПНАЯ СИСТЕМА С КАТАЛИЗАТОРОМ РЕФОРМИНГА | 2014 |
|
RU2650142C2 |
| КАТАЛИЗАТОР ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА И СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА С ЕГО ИСПОЛЬЗОВАНИЕМ | 2004 |
|
RU2333797C2 |
| КАТАЛИЗАТОР И СПОСОБ РЕФОРМИНГА | 2012 |
|
RU2547466C1 |
| КАТАЛИЗАТОР ДЛЯ ПОЛУЧЕНИЯ ВОДОРОДА | 2011 |
|
RU2585610C2 |
| Катализатор для риформинга лигроинового сырья и способ каталитического риформинга лигроинового сырья | 1987 |
|
SU1797496A3 |
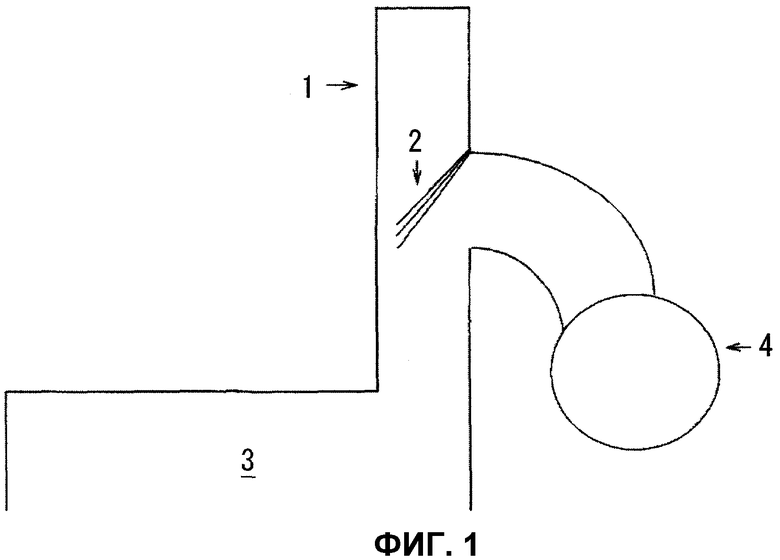
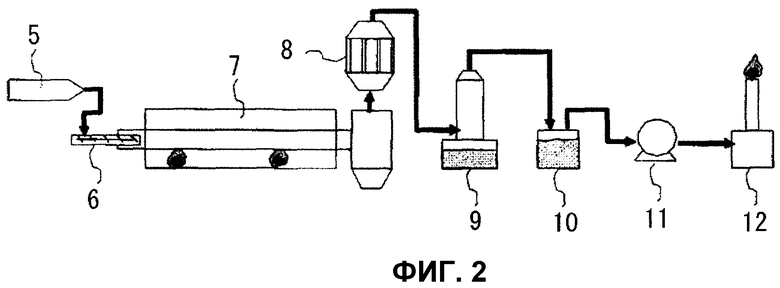
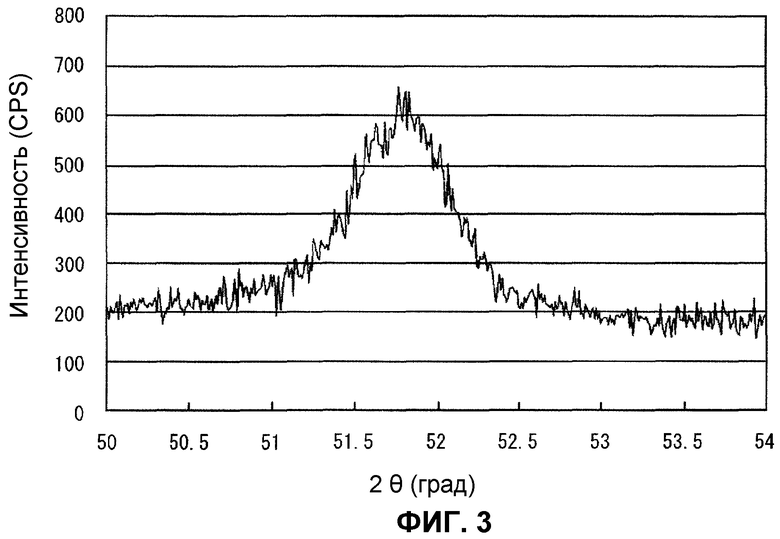
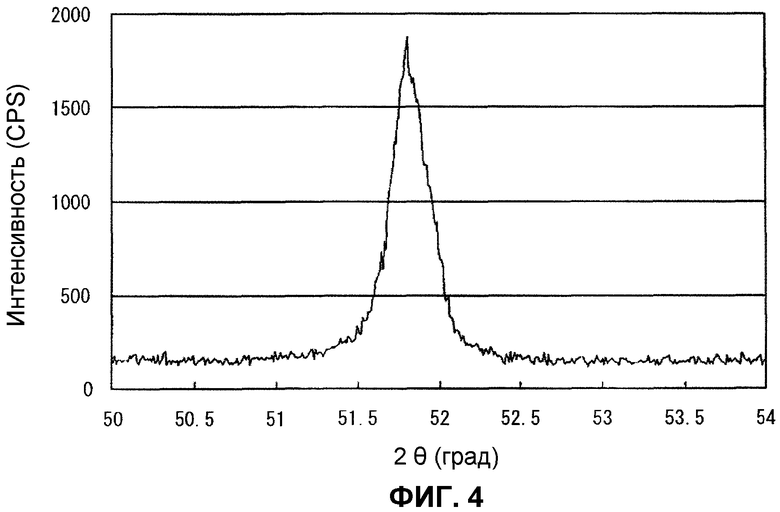
Изобретение предлагает способ получения высокоактивного катализатора для реформинга смолосодержащего газа, смолообразующий газ образуется во время термического разложения углеродистого сырья. Способ включает получение катализатора путем добавления агента осаждения к смешанному раствору соединения никеля и соединения магния, образование осадка путем соосаждения никеля и магния, образование смеси путем добавления порошка оксида алюминия и воды или золя оксида алюминия к данному осадку, перемешивание, и, по меньшей мере, сушку и обжиг данной смеси, где катализатор для реформинга смолосодержащего газа получают так, что содержание никеля составляет от 1 до 50% масс., содержание магния составляет от 5 до 45% масс. и содержание оксида алюминия составляет от 20 до 80% масс., причем смолосодержащий газ содержит 20 ч./млн или более сероводорода. Изобретение включает вариант способа получения катализатора, способы реформинга смолы и способы регенерации катализатора. Технический результат - преобразование химической энергии реформинга в топливную композицию. Катализатор устойчив к отложению углерода даже при реформинге смолосодержащего газа с высоким содержанием сероводорода, катализатор имеет высокую производительность, регенерация обеспечивает стабильность каталитического результата и подобность результату до регенерации. 6 н. и 19 з.п. ф-лы, 4 ил., 5 табл.
1. Способ получения катализатора для реформинга смолосодержащего газа, образованного во время термического разложения углеродистого сырья, причем смолосодержащий газ содержит 20 ч/млн или более сероводорода, включающий
получение катализатора путем добавления агента осаждения к смешанному раствору соединения никеля и соединения магния,
образование осадка путем соосаждения никеля и магния,
образование смеси путем добавления порошка оксида алюминия и воды или золя оксида алюминия к данному осадку и перемешивание, и, по меньшей мере, сушку и обжиг данной смеси, где
катализатор для реформинга смолосодержащего газа получают так, что содержание никеля составляет от 1 до 50 мас.%, содержание магния составляет от 5 до 45 мас.% и содержание оксида алюминия составляет от 20 до 80 мас.%.
2. Способ получения катализатора для реформинга смолосодержащего газа по п.1, где данный катализатор получают с помощью сушки и обжига или с помощью сушки, прокаливания, дробления, формования и обжига данной смеси.
3. Способ получения катализатора для реформинга смолосодержащего газа по п.1, где содержание никеля составляет от 1 до 35 мас.% и содержание магния составляет от 10 до 25 мас.%.
4. Способ получения катализатора для реформинга смолосодержащего газа, образованного во время термического разложения углеродистого сырья, включающий
добавление осаждающего агента к смешанному раствору соединения никеля и соединения магния,
образование осадка путем соосаждения никеля и магния,
образование смеси путем добавления порошка оксида алюминия и воды или золя оксида алюминия к данному осадку и перемешивание, образование второй смеси путем дальнейшего смешения в порошке оксида алюминия и воды или золя оксида алюминия после сушки и обжига смеси и, по меньшей мере, сушку и обжиг второй смеси, где
катализатор получают так, что содержание никеля составляет от 1 до 50 мас.%, содержание магния составляет от 5 до 45 мас.% и содержание оксида алюминия составляет от 20 до 80 мас.%.
5. Способ получения катализатора для реформинга смолосодержащего газа по п.4, где данный катализатор получают с помощью сушки и обжига или с помощью сушки, прокаливания, дробления, формования и обжига данной второй смеси.
6. Способ получения катализатора для реформинга смолосодержащего газа по п.4, где содержание никеля составляет от 1 до 35 мас.%, и содержание магния составляет от 10 до 25 мас.%.
7. Способ реформинга смолосодержащего газа, в котором используют катализатор для реформинга смолосодержащего газа, полученный согласно любому из способов получения по пп.4-6.
8. Способ реформинга смолосодержащего газа по п.7, в котором смолу, содержащуюся в смолосодержащем газе, реформируют и газифицируют путем контакта с водородом, диоксидом углерода и водяным паром в смолосодержащем газе, в присутствии катализатора для реформинга смолосодержащего газа или в присутствии данного катализатора после восстановления.
9. Способ реформинга смолосодержащего газа по п.7, в котором смолосодержащий газ подвергают реформингу и газифицируют путем контакта по меньшей мере водорода, диоксида углерода и водяного пара, введенного извне со смолосодержащим газом, образованным во время термического разложения, в присутствии катализатора реформинга смолосодержащего газа или в присутствии данного катализатора после восстановления.
10. Способ реформинга смолосодержащего газа по п.7, в котором смолосодержащий газ представляет собой смолосодержащий газ, содержащий 20 ч./млн или более сероводорода.
11. Способ реформинга смолосодержащего газа по п.7, в котором кислородсодержащий газ добавляют к реформированному газу и приводят в контакт со смолосодержащим газом, и часть водорода или углеводородов в смолосодержащем газе сжигают.
12. Способ реформинга смолосодержащего газа по п.7, в котором смолосодержащий газ представляет собой газ сухой перегонки, образованный во время сухой перегонки угля.
13. Способ реформинга смолосодержащего газа по п.7, в котором смолосодержащий газ представляет собой газ коксовой печи, выпускаемый из коксовой печи.
14. Способ реформинга смолосодержащего газа по п.7, в котором смолосодержащий газ представляет собой газ сухой перегонки, образованный во время сухой перегонки, по меньшей мере, или древесной биомассы, или биомассы на основе пищевых отходов.
15. Способ реформинга смолосодержащего газа по п.7, в котором смолосодержащий газ приводят в контакт с данным катализатором для реформинга смолосодержащего газа при 600-1000°С.
16. Способ регенерации катализатора для реформинга смолосодержащего газа, в котором регенерируют катализатор путем контакта, по меньшей мере, водяного пара или воздуха с данным катализатором в случае, если производительность катализатора ухудшилась из-за, по меньшей мере, осаждения углерода или отравления серой в результате осуществления способа реформинга смолосодержащего газа по п.7.
17. Способ реформинга смолосодержащего газа, где используют катализатор реформинга смолосодержащего газа, полученный способом по любому из пп.1-3.
18. Способ реформинга смолосодержащего газа по п.17, где смолу, содержащуюся в смолосодержащем газе, реформируют и газифицируют путем контакта с водородом, диоксидом углерода и водяным паром в смолосодержащем газе, в присутствии катализатора для реформинга смолосодержащего газа или в присутствии данного катализатора после восстановления.
19. Способ реформинга смолосодержащего газа по п.17, в котором смолосодержащий газ подвергают реформингу и газифицируют путем контакта по меньшей мере водорода, диоксида углерода и водяного пара, введенного извне со смолосодержащим газом, образованным во время термического разложения, в присутствии катализатора реформинга смолосодержащего газа или в присутствии данного катализатора после восстановления.
20. Способ реформинга смолосодержащего газа по п.17, в котором кислородсодержащий газ добавляют к реформированному газу и приводят в контакт со смолосодержащим газом, и часть водорода или углеводородов в смолосодержащем газе сжигают.
21. Способ реформинга смолосодержащего газа по п.17, в котором смолосодержащий газ представляет собой газ сухой перегонки, образованный во время сухой перегонки угля.
22. Способ реформинга смолосодержащего газа по п.17, в котором смолосодержащий газ представляет собой газ коксовой печи, выпускаемый из коксовой печи.
23. Способ реформинга смолосодержащего газа по п.17, в котором смолосодержащий газ представляет собой газ сухой перегонки, образованный во время сухой перегонки, по меньшей мере, или древесной биомассы, или биомассы на основе пищевых отходов.
24. Способ реформинга смолосодержащего газа по п.17, в котором смолосодержащий газ приводят в контакт с данным катализатором для реформинга смолосодержащего газа при 600-1000°С.
25. Способ регенерации катализатора для реформинга смолосодержащего газа, в котором регенерируют катализатор путем контакта, по меньшей мере, водяного пара или воздуха с данным катализатором в случае, если производительность катализатора ухудшилась из-за, по меньшей мере, осаждения углерода или отравления серой в результате осуществления способа реформинга смолосодержащего газа по п.17.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| WO 2008049266 A1, 02.05.2008 | |||
| WO 2004060557 A2, 22.07.2004 | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| US 3932534 A, 13.01.1976 | |||
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ДЛЯ РИФОРМИНГА УГЛЕВОДОРОДОВ | 0 |
|
SU257371A1 |

