Уровень техники
Фенилэфрин и его фармацевтически приемлемые соли признаются специалистами в данной области техники в качестве назальных деконгестантов, безопасных и эффективных для человека при введении с частыми интервалами. Доступные для приобретения лекарственные формы включают назальный гель, назальные капли и назальный спрей (то есть назальные капли Alconefrin® или назальный гель Neo-Synephrine®), так же как и пероральные таблетки или желатиновые капсулы немедленного высвобождения (то есть Sudafed РЕ™ или жидкие капсулы DayQuil®). Благодаря короткому периоду полураспада in vivo активной формы фенилэфрина в плазме, фенилэфрин и его фармацевтически приемлемые соли, в виде существующих в настоящее время лекарственных форм, как правило, вводятся каждые четыре часа для облегчения заложенности носа.
Таким образом, существует неудовлетворенная потребность в менее частой доставке фенилэфрина для удобства пациента и в продолжительном наличии терапевтически активного фенилэфрина в организме субъекта, нуждающегося в таком введении.
Менее частое введение приводит к более точному соблюдению больными соответствующих режимов введения. Кроме того, постоянные терапевтические уровни активных компонентов в плазме могут быть более эффективными и даже действенными по сравнению с колебаниями, наблюдаемыми при введении многократных доз обычной лекарственной формы немедленного высвобождения, при условии продолжительных эффективных уровней и уменьшения серьезности и частоты побочных эффектов от высоких пиковых уровней в плазме. Таким образом, лекарственные формы, содержащие фенилэфрин, которые могут вводиться менее часто, например один раз за каждые 8, 12, 16, 20 или 24 часа, необходимы.
Краткое изложение сущности изобретения
Данное изобретение предоставляет композиции и способы эффективной доставки фенилэфрина с обеспечением улучшенной и стабильной биодоступности, которые обеспечивают эффективный фенилэфрин в дополнение к повышенному удобству. Изобретение отчасти основано на наблюдении изобретателей, что фенилэфрин эффективно абсорбируется из ободочной кишки, и их оценки, что лекарственные формы, которые позволяют абсорбцию из ободочной кишки, будут обеспечивать более высокую долю фенилэфрина, оставшегося в терапевтически активной несвязанной форме, по сравнению с лекарственными формами, позволяющими абсорбцию из верхних отделов желудочно-кишечного (ЖК) тракта. Введение лекарственной формы, обеспечивающей абсорбцию из ободочной кишки, имеет дополнительное преимущество благодаря продолжительным концентрациям в плазме активного фенилэфрина при однократном болюсном введении препарата.
В одном объекте изобретение предоставляет фармацевтические композиции, подходящие для перорального введения, содержащие фенилэфрин или его фармацевтически приемлемую соль, в которых фенилэфрин сделан доступным для абсорбции из ободочной кишки. В одном варианте выполнения изобретения композиция находится в виде твердой лекарственной формы, содержащей: ядро, содержащее фенилэфрин или его фармацевтически приемлемую соль, и поддающийся эрозии слой, инкапсулирующий ядро, где состав и толщина поддающегося эрозии слоя являются такими, что ядро обнажается, когда композиция поступает в ободочную кишку или приблизительно в то время, когда композиция достигает ободочной кишки.
В определенных вариантах выполнения изобретения поддающийся эрозии слой содержит полимерную матрицу. В других вариантах выполнения изобретения поддающимся эрозии слоем является покрывающий слой. В одном объекте поддающимся эрозии слоем является полимерная матрица определенного состава и толщины, так что слой разрушается через определенное количество времени. В другом объекте поддающимся эрозии слоем является полимерная матрица или покрывающий слой определенного состава, который разрушается при взаимодействии с определенной средой. В определенных вариантах выполнения изобретения поддающийся эрозии слой разрушается при определенном значении рН. В других вариантах выполнения изобретения поддающийся эрозии слой содержит субстраты, специфичные к ободочной кишке, и разрушается в ободочной кишке. В определенных вариантах выполнения изобретения субстрат, специфичный к ободочной кишке, разрушается в присутствии специфичных для ободочной кишки ферментов, но не во время прохода через верхние отделы пищеварительного тракта, включая желудок и/или тонкую кишку. В другом объекте изобретение предоставляет фармацевтические композиции, подходящие для перорального введения, содержащие фенилэфрин или его фармацевтически приемлемую соль, в которых фенилэфрин сделан доступным для абсорбции из всех отделов ЖК тракта, включая двенадцатиперстную кишку, тощую кишку, подвздошную кишку и ободочную кишку. Определенными вариантами выполнения изобретения являются фармацевтические композиции, разработанные в виде единичной лекарственной формы для доставки фенилэфрина или его фармацевтически приемлемой соли в организм субъекта, нуждающегося в этом, с обеспечением максимума концентрации неметаболизированного фенилэфрина в плазме (вышеупомянутого субъекта) в промежутке от около 0.1 до 16 часов после приема композиции внутрь, и где неметаболизированный фенилэфрин поддерживается на уровне, превышающем 0.1 нг/мл, на момент около 6, 8, 12, и/или 24 часов после приема композиции внутрь.
В определенных вариантах выполнения изобретения поддающийся эрозии слой (слои) и/или другая составная часть(и) композиции, отличная(ые) от ядра, содержит(ат) фенилэфрин или его фармацевтически приемлемую соль. Например, помимо ядра, содержащего фенилэфрин или его фармацевтически приемлемую соль, фенилэфрин так же распределен в поддающемся эрозии слое, содержащем полимерную матрицу. Полимерная матрица содержит фенилэфрин или его фармацевтически приемлемую соль, включенные в состав лекарственной формы для их немедленного высвобождения.
В определенных вариантах выполнения изобретения композиция, кроме того, содержит энтеральное покрытие и/или верхние покрытие, где функцией верхнего покрытия является улучшение внешнего вида или вкусовых качеств лекарственной формы.
В одном варианте выполнения изобретения композиция представляет собой лекарственную форму в виде капсулы, где фенилэфрин или его фармацевтически приемлемая соль заключены в капсулу, которая выгружает свое содержимое при поступлении в ободочную кишку или приблизительно в момент достижения ободочной кишки. В одном варианте выполнения изобретения композиция представляет собой лекарственную форму в виде капсулы, где фенилэфрин или его фармацевтически приемлемая соль заключены в капсулу, которая выгружает часть своего содержимого, когда композиция поступает в ободочную кишку.
В одном варианте выполнения изобретения лекарственная форма в виде капсулы, кроме того, включает фенилэфрин или его фармацевтически приемлемую соль для их немедленного высвобождения и/или один или более дополнительных терапевтических агентов для их немедленного или пролонгированного высвобождения.
В еще одном варианте выполнения изобретения фармацевтическая композиция изобретения представляет собой контейнер с механизмом высвобождения. Такая структура состоит из нерастворимого в воде контейнера, содержащего в себе фенилэфрин или его фармацевтически приемлемую соль, и пробки. Пробка удаляется через заданное время запаздывания вследствие набухания, эрозии или растворения.
В еще одном варианте выполнения изобретения фармацевтическая композиция изобретения по своей структуре разработана в виде порошка, перорального геля, эликсира, дисперсных гранул, сиропа, суспензии или тому подобного. В одном варианте выполнения изобретения, в котором фармацевтическая композиция изобретения по своей структуре разработана в виде порошка, суспензия из такого порошка может быть замешена непосредственно перед применением.
В одном варианте выполнения изобретения фармацевтическая композиция изобретения разработана так, что подходит для педиатрического применения.
В одном варианте выполнения изобретения фармацевтическая композиция, кроме того, содержит один или более дополнительных терапевтических агентов. Такой агент или агенты может (могут) быть добавлен(ны) в лекарственную форму для его (их) немедленного высвобождения при принятии внутрь, пролонгированного высвобождения или высвобождения в ободочной кишке одновременно с фенилэфрином, или для любой комбинации вышеперечисленного. Дополнительным терапевтическим агентом может быть деконгестант, жаропонижающее средство, противовоспалительное средство, супрессант кашля, отхаркивающее средство, анальгетик или любой другой терапевтический агент или комбинация таких агентов, полезная для облегчения симптомов простуды, сезонной или другой аллергии, сенной лихорадки или проблем с синусом, любые из которых могут вызвать усиление выделений из носа.
Другим объектом изобретения является метод лечения симптомов простуды, гриппа, аллергии или неаллергического ринита у субъекта, нуждающегося в этом, включающий введение композиции изобретения. В определенных вариантах выполнения изобретения композиция вводится каждые 8, 12, 16, 20 или 24 часа. В одном предпочтительном варианте выполнения изобретения композиция вводится каждые 12 часов.
Другим объектом изобретения являются способы введения фенилэфрина, включающие доставку фенилэфрина в ободочную кишку субъекта. Примерные композиции, полезные для этих способов, описаны выше. В определенных вариантах выполнения изобретения таким способом является способ, в котором максимальная концентрация несвязанного фенилэфрина в плазме субъекта достигается в промежутке от около 5 до около 24 часов после введения, и более предпочтительно, в промежутке от около 6 до около 12 часов после введения.
Определенными вариантами выполнения изобретения являются способы, поддерживающие стабильную биодоступность фенилэфрина в организме субъекта, содержащие пероральное введение в организм субъекта композиции, содержащей фенилэфрин или его фармацевтически приемлемую соль, где, по меньшей мере, часть фенилэфрина абсорбируется из ободочной кишки, и где концентрация несвязанного фенилэфрина в плазме субъекта составляет, по меньшей мере, 0.1, 0.5, 1.0 или 2.5 нг/мл на момент 6 часов после введения композиции. В конкретных вариантах выполнения изобретения концентрация несвязанного фенилэфрина в плазме субъекта составляет, по меньшей мере, 0.1, 0.5, 1.0 или 2.5 нг/мл на момент 12 часов после введения композиции. В конкретных вариантах выполнения изобретения концентрация несвязанного фенилэфрина в плазме субъекта составляет, по меньшей мере, 0.1, 0.5, 1.0 или 2.5 нг/мл на момент 24 часов после введения композиции. Другими определенными вариантами выполнения изобретения являются способы введения фенилэфрина в организм субъекта, содержащие пероральное введение в организм субъекта композиции, содержащей фенилэфрин или его фармацевтически приемлемую соль, причем вышеупомянутая композиция доставляет фенилэфрин в ободочную кишку, где фенилэфрин высвобождается в ободочную кишку и абсорбируется из ободочной кишки, таким образом, достигая относительного AUC0-24 (определяемого как процентное отношение значения AUC0-24 для несвязанного фенилэфрина к значению AUC0-24 для всего (то есть несвязанного + связанного) фенилэфрина в плазме субъекта (смотрите абзац [0049] для примерных способов определения)), по меньшей мере, 1, 2 или 6%. В одном варианте выполнения изобретения процентное отношение значения AUC0-24 для несвязанного фенилэфрина к значению AUC0-24 для всего фенилэфрина в плазме субъекта составляет, по меньшей мере, от около 1 до около 14%. В одном варианте выполнения изобретения процентное отношение значения AUC0-24 для несвязанного фенилэфрина к значению AUC0-24 для всего фенилэфрина в плазме субъекта составляет, по меньшей мере, от около 2 до около 10%.
В еще одном варианте выполнения изобретения композиция включает двухслойную таблетку со слоем немедленного высвобождения и слоем пролонгированного высвобождения.
Как будет понятно специалистам в данной области техники после прочтения данного описания изобретения, поддающийся эрозии слой фармацевтических композиций данного изобретения обеспечивает пролонгированное или контролированное высвобождение фенилэфрина, доставляя в организм субъекта терапевтически эффективное количество в течение 8, 12, или 24 часов. В предпочтительных вариантах выполнения изобретения данные фармацевтические композиции, кроме того, содержат дополнительное количество фенилэфрина в части немедленного высвобождения, доставляющей количество фенилэфрина при введении в организм субъекта.
В определенных вариантах выполнения изобретения предпочтительно иметь оба вида высвобождения, то есть немедленное высвобождение фенилэфрина, которое обеспечивает первоначальный всплеск, сопровождаемое пролонгированным высвобождением фенилэфрина в ободочной кишке в течение 6, 8, 12, 16 или 24 часов. Так же, в определенных вариантах выполнения изобретения, предпочтительно иметь немедленное высвобождение, которое обеспечивает первоначальный всплеск, сопровождаемое пролонгированным высвобождением фенилэфрина в верхних отделах ЖК тракта (то есть тощей кишке и подвздошной кишке) так же как и в ободочной кишке в течение 6, 8, 12, 16 или 24 часов.
Другие объекты изобретения предоставляют способы введения фенилэфрина в организм субъекта, в которых пресистемная трансформация фенилэфрина минимизирована. Любые методы, описанные здесь, могут быть осуществлены на практике, применяя фармацевтическую композицию изобретения.
Данное изобретение может быть более полно понято посредством Фигур, Подробного описания и Примеров, которые следуют ниже.
Краткое описание Фигур
Фиг.1А представляет собой схематическое изображение примерной лекарственной формы с контролируемой продолжительностью эрозии. Фиг.1В представляет собой схематическое изображение гранулированного ядра.
Фиг.2 представляет собой схематическое изображение примерной лекарственной формы с рН-контролируемой эрозией.
Фиг.3 показывает профиль растворения примерных лекарственных форм A-G.
Фиг.4 показывает профиль растворения примерных предпочтительных лекарственных форм, содержащих ядро таблетки с продолжительным высвобождением, компоненты которого включают 22.5 мг гидрохлорида фенилэфрина, и активное покрытие с немедленным высвобождением, инкапсулирующее ядро, компоненты которого включают 7.5 мг гидрохлорида фенилэфрина и 5 мг лоратадина.
Фиг.5 показывает профиль растворения двухслойной таблетки, содержащей слой фенилэфрина, разработанный для пролонгированного высвобождения, и слой фенилэфрина и лоратадина, разработанный для немедленного высвобождения.
Фиг.6 показывает профили значений концентрации в плазме всего, так же как связанного и несвязанного фенилэфрина. В частности, Фиг.6А показывает полулогарифмический график, и Фиг.6В показывает линейный график. Кроме того, Фиг.6С показывает диаграмму значений Cmax всего, так же как связанного и несвязанного фенилэфрина, достигшего уровня дозы 1 мг, после доставки 10 мг и 30 мг фенилэфрина в ободочную кишку.
Фиг.7 показывает профили симулированных концентраций в плазме для вариантов 10 мг дозы фенилэфрина немедленного высвобождения, так же как и 30 мг дозы фенилэфрина (подробно изложенных в Таблице 17), перекрывающих интервалы, подробно изложенные в Таблице 16.
Фиг.8 показывает профили симулированных концентраций в плазме для вариантов 10 мг дозы фенилэфрина немедленного высвобождения, также как и 30 мг дозы фенилэфрина (подробно изложенных в Таблице 18), перекрывающих интервалы, подробно изложенные в Таблице 16.
Подробное описание изобретения
Определения
В качестве применяемого здесь термин "фармацевтически приемлемая соль фенилэфрина" включает, но без ограничения к этому, гидрохлорид фенилэфрина, битартрат фенилэфрина, таннат фенилэфрина и т.д. В одном предпочтительном варианте выполнения изобретения фармацевтически приемлемой солью фенилэфрина является гидрохлорид фенилэфрина.
Термин "неметаболизированный фенилэфрин" означает фенилэфрин, который не был химически изменен с момента поступления в организм субъекта, за исключением высвобождения свободного основания, то есть фенилэфрин, который не был связан сульфотрансферазой или UDP-глюкуронилтрансферазой. Неметаболизированный фенилэфрин проявляет терапевтическую активность (активности). В этом описании изобретения термин "несвязанный фенилэфрин" используется взаимозаменяемо с термином "Неметаболизированный фенилэфрин" и означает терапевтически активную форму фенилэфрина. "Неметаболизированный фенилэфрин" не включает фенилэфрин, который был однажды инактивирован вследствие связывания, но позже стал несвязанным и не является терапевтически активным.
Термин "пресистемная трансформация", в качестве применяемого здесь в связи с фенилэфрином, означает трансформацию фенилэфрина перед тем, как фенилэфрин захватывается кровотоком и таким образом поступает в плазму. Пресистемная трансформация исключает трансформацию фенилэфрина в печени или внутри кровотока.
Термин "среда в ободочной кишке" или "ободочнокишечная среда", в качестве применяемого здесь, означает среду внутри ободочной кишки, в пищеварительном тракте.
Термин "специфичный для ободочной кишки (к ободочной кишке)", в качестве применяемого здесь, означает типично, преимущественно или исключительно обнаруживаемый в ободочной кишке, связанный с ободочной кишкой или относимый к ободочной кишке, но не каким другим частям пищеварительного тракта.
Термин "дозировка" или "доза", в качестве используемого здесь, означает количество фармацевтической композиции, содержащей терапевтически активный(ые) агент(ы), вводимые одновременно. "Дозировка" или "доза" включает введение одной или более единиц фармацевтической композиции, вводимых одновременно.
Термин "AUC", в качестве применяемого здесь, для любого данного лекарственного средства означает "площадь под кривой концентрация - время" в интервале дозирования или активацию лекарственного средства к моменту времени на графике, вычисленную по формуле трапеций. AUC - это параметр, показывающий кумулятивную концентрацию лекарственного средства в плазме по прошествии времени, и является индикатором общего количества и наличия лекарственного средства в плазме. "AUQ0-t" определяется как AUC для любого значения времени (t) вплоть до 24 часов. В предпочтительном варианте выполнения изобретения t составляет 24 часа (обозначается здесь как AUC0-24). "AUC0-∞" определяется как вычисленное значение AUC, экстраполированное до бесконечности. AUC0-∞ вычисляется как равное AUC0-t+Ct/λz, где Ct обозначает концентрацию в момент 24 часов, и λz означает константу скорости конечной стадии или константу скорости элиминации. Константа скорости конечной стадии или элиминации λz определяется из угла наклона кривой "концентрация лекарственного вещества - время", применяя линейную регрессию по конечным измерительным точкам кривой. "Относительное AUC0-24" определяется как процентное отношение значения AUC0-t для несвязанного фенилэфрина к значению AUC0-t для всего фенилэфрина в плазме субъекта в интервале доз.
Данное изобретение предоставляет композиции и способы действенной и эффективной доставки фенилэфрина, обеспечивающие улучшенную биодоступность и удобство.
Фенилэфрин эффективен при введении через короткие интервалы времени для временного облегчения гиперемии и/или заложенности носа, вызванных простудой, сезонной или другой аллергией, сенной лихорадкой, проблемами синуса или аллергическим и неаллергическим ринитом, которые могут вызвать увеличение выделений из носа. В настоящее время фенилэфрин доступен в виде назальных лекарственных форм и пероральных лекарственных форм для лечения этих недомоганий. Однако пероральные лекарственные формы должны приниматься часто и показывают субоптимальную эффективность из-за короткого периода полураспада фенилэфрина в плазме, около 2 часов, вызванного быстрым метаболизмом активного агента. Кроме того, при введении перорально фенилэфрин быстро абсорбируется из ЖК тракта, но подвергается экстенсивной пресистемной трансформации в слизистой кишечника, что приводит к соединению, превращенному в форму, не являющуюся активной. Пресистемная трансформация фенилэфрина в основном имеет место во время абсорбции из верхних отделов кишечного тракта, то есть тощей кишки или подвздошной кишки, благодаря активностям сульфотрансферазы или UDP-глюкуронилтрансферазы, которые находятся там в большом числе и количестве, по сравнению с более низким отделом ЖК тракта, то есть ободочной кишкой. При проникновении в кровоток фенилэфрин выводится из плазмы через печень.
Без какого-либо намерения ограничиваться каким-либо механизмом, изобретатели предполагают, что быстрое и экстенсивное превращение путем пресистемной трансформации уменьшает эффективную концентрацию фенилэфрина в плазме. На самом деле, введение фенилэфрина каждые 4 часа в виде лекарственной формы с немедленным высвобождением, в форме таблетки ли, капсулы или раствора, как оказывается, имеет субоптимальную эффективность в качестве назального деконгестанта.
Данное изобретение отчасти основано на наблюдении, что абсорбция фенилэфрина из нижнего отдела кишечного тракта, где сульфотрансферазы и UDP-глюкуронилтрансферазы обнаруживаются в небольшом числе и количествах, увеличивает биодоступность фенилэфрина на фактор, равный трем, по сравнению с биодоступностью, измеренной для перорального введения фенилэфрина в форме немедленного высвобождения. Известно, что перорально вводимая лекарственная форма достигает илеоцекального соединения в организме здорового взрослого человека в среднем за около 5 часов и поступает в ободочную кишку в среднем через около 6-8 часов. В общем, интервал достижения перорально вводимой лекарственной формой ободочной кишки составляет от около 2 часов до около 8 часов. В среднем время пребывания принятого внутрь вещества в ободочной кишке здорового взрослого человека составляет от 12 до 24 часов. Кроме того, благодаря ограниченной проходимости слизистой мембраны ободочной кишки и времени пребывания проглоченных веществ в ободочной кишке, болюсное введение одного или более дополнительных терапевтических агентов, которые могут быть абсорбированы из ободочной кишки, может привести к продолжительным уровням в плазме такого агента, абсорбированного из ободочной кишки. Весьма увеличенная абсорбция несвязанного фенилэфрин из ободочной кишки намного превышает ту, которая может быть ожидаема при сравнении с абсорбцией из ободочной кишки других активных терапевтических агентов, которые, как известно, являются субъектом для подобной пресистемной трансформации.
Фармацевтические композиции
В одном объекте изобретение предоставляет фармацевтические композиции, подходящие для перорального введения, содержащие фенилэфрин или его фармацевтически приемлемую соль, в которых фенилэфрин сделан доступным для абсорбции из ободочной кишки. Определенными вариантами выполнения изобретения являются фармацевтические композиции, приготовленные в единичной лекарственной форме для доставки фенилэфрина или его фармацевтически приемлемой соли в организм субъекта, нуждающегося в этом, причем такая доставка приводит к обнаруживаемому неметаболизированному фенилэфрину в плазме субъекта через около 5 часов после принятия вышеупомянутым субъектом композиции внутрь. В других вариантах выполнения изобретения неметаболизированный фенилэфрин обнаруживается через около 6, 8, 12, 16, 20 или 24 часа после принятия вышеупомянутым субъектом композиции внутрь. Наличие фенилэфрин в плазме субъекта определяется посредством способов, применяемых специалистами в данной области техники для определения содержания всего фенилэфрина и несвязанного фенилэфрина в плазме.
Примерные способы для определения общего фенилэфрина и несвязанного фенилэфрина описываются в следующих работах: K.Gumbhir. An Investigation of Pharmacokinetics of Phenylephrine and its Metaboiites in Humans, PhD-диссертация по фармацевтическим наукам Миссурийского университета в Канзас-Сити (1993); K. Gumbhir и W.D. Mason. Determination of m-hydroxymandelic acid, m-hydroxyphenyiglycol and their conjugates in human plasma using liquid chromatography with electrochemical detection. Журнал «Journal of Pharmaceutical and Biomedical Analysis» 12: 943-949 (1994); J.H.Hengstmannand J.Goronzy. Pharmacokinetics of 3H-Phenylephrine in Man. Журнал «European Journal of Clinical Pharmacology» 21: 335-341 (1982); V.Vumaand I.Kanfer. High-performance liquid chromatographic determination of phenylephrine in human serum with coulometric detection. Журнал «Journal of Chromatography» 678: 245-252 (1996); M.Yamaguchi, H.Monji, I.Aoki, and T.Yashiki. High Performance liquid chromatographic determination of phenylephrine in human serum using coulometric switching flourescence detection. Журнал «Journal of Chromatography B.» 661: 93-99 (1994); A.Stockis, X.Deroubaix, B.Jeanbaptiste, R.Lins, A.M.Allemon, and H.Laufen. Relative Bioavailability of Carbinoxamine and Phenylephrine from a Retard Capsule after Single and Repeated Dose Adminstration in Healthy Subjects. Arzneim. - Forsch./Drug Res. 45: 1009-1012 (1995); A.Martinsson, S.Bevegard, and P.Hjemdahl. Analysis of Phenylephrine in Plasma: Initial Data About Concentration-Effect Relationship. Журнал «European Journal of Clinical Pharmacology» 30: 427-431 (1986); все из которых включены здесь путем ссылки. Количество связанного фенилэфрина может быть вычислено путем вычитания оцененного количества несвязанного фенилэфрина из оцененного количества всего фенилэфрина.
Определенными вариантами выполнения изобретения являются фармацевтические композиции, созданные в виде единичной лекарственной формы для доставки фенилэфрина или его фармацевтически приемлемой соли в организм субъекта, нуждающегося в этом, так что несвязанный фенилэфрин обнаруживается в плазме субъекта в течение, по меньшей мере, 4.5 часов после принятия вышеупомянутым субъектом композиции внутрь. В других вариантах выполнения изобретения неметаболизированный фенилэфрин обнаруживается в течение, по меньшей мере, около 5, 6, 8, 12, 16, 20 или 24 часов после принятия композиции внутрь.
В некоторых вариантах выполнения изобретения композиция создается в виде лекарственной формы для доставки, по меньшей мере, части фенилэфрина в ободочную кишку субъекта. В других вариантах выполнения изобретения помимо доставки части фенилэфрина в ободочную кишку субъекта композиция выполнена в виде такой лекарственной формы, что, кроме того, пролонгированное высвобождение фенилэфрина достигается в верхних отделах ЖК тракта субъекта. В других вариантах выполнения изобретения, помимо доставки части фенилэфрина в ободочную кишку субъекта, композиция выполнена в виде такой лекарственной формы, что, кроме того, немедленное высвобождение фенилэфрина достигается в верхних отделах ЖК тракта субъекта. В еще одних вариантах выполнения изобретения помимо доставки части фенилэфрина в ободочную кишку субъекта композиция выполнена в виде такой лекарственной формы, что, кроме того, как немедленное высвобождение, так и пролонгированное высвобождение фенилэфрина достигается в верхних отделах ЖК тракта субъекта. Во всех вариантах выполнения изобретения по доли это составляет около 5-95% от количества введенного фенилэфрина или его фармацевтически приемлемой соли. В конкретных вариантах выполнения изобретения доля составляет около 5, 10, 20, 25, 30, 50, 67, 75, 90 или 95% от количества введенного фенилэфрина или его фармацевтически приемлемой соли. В определенных вариантах выполнения изобретения доля составляет около 1/4, 1/5, 3/4 или 9/10 количества введенного фенилэфрина или его фармацевтически приемлемой соли. В других вариантах выполнения изобретения доля составляет около 17, 42, 45, 50, 54 или 61% от количества введенного фенилэфрина или его фармацевтически приемлемой соли. Предпочтительно доля составляет около 5-20% от количества введенного фенилэфрин или его фармацевтически приемлемой соли. Более предпочтительно доля составляет около 10-15% от количества введенного фенилэфрина или его фармацевтически приемлемой соли.
В одном объекте изобретение предоставляет фармацевтические композиции, подходящие для перорального введения, содержащие фенилэфрин или его фармацевтически приемлемую соль, в которых фенилэфрин сделан доступным для абсорбции из всех отделов ЖК тракта, включая двенадцатиперстную кишку, тощую кишку, подвздошную кишку и ободочную кишку. Определенными вариантами выполнения изобретения являются фармацевтические композиции, созданные в виде единичной лекарственной формы для доставки фенилэфрина или его фармацевтически приемлемой соли в организм субъекта, нуждающегося в этом, причем такая доставка приводит к тому, что субъект показывает максимум концентрации неметаболизированного фенилэфрина в плазме в промежутке от около 0.1 до 16 часов после принятия композиции внутрь, и неметаболизированный фенилэфрин поддерживается на уровне, превышающем 0.1 нг/мл, в течение около 6, 8, 12 и/или 24 часов после принятия композиции внутрь. Например, в определенных вариантах выполнения изобретения субъект показывает максимум концентрации неметаболизированного фенилэфрина в плазме между около 0.1 и 14 часами, 0.1 и 12 часами, 0.1 и 10 часами, 0.1 и 8 часами, 0.1 и 6 часами, 0.1 и 4 часами, 0.1 и 2 часами после принятия композиции внутрь, и несвязанный фенилэфрин поддерживается на уровне, превышающем 0.1 нг/мл (например, 0.5 нг/мл, 1 нг/мл или 2.5 нг/мл) в течение около 6, 8, 12 и/или 24 часов после принятия композиции внутрь. В одном предпочтительном варианте выполнения изобретения субъект показывает максимум концентрации неметаболизированного фенилэфрина в плазме между около 0.75 и 2 часами после принятия композиции внутрь, и несвязанный фенилэфрин поддерживается на уровне, превышающем 0.1 нг/мл (например, 0.5 нг/мл, 1 нг/мл или 2.5 нг/мл), на момент около 6, 8, 12 и/или 24 часа после принятия композиции внутрь.
В одном предпочтительном варианте выполнения изобретения композиция обеспечивает 12-часовое пролонгированное высвобождение фенилэфрина или его фармацевтически приемлемой соли. В одном варианте выполнения изобретения композиция находится в виде твердой лекарственной формы, содержащей ядро с пролонгированным высвобождением, содержащее фенилэфрин или его фармацевтически приемлемую соль, и покрывающий ядро слой, содержащий фенилэфрин или его фармацевтически приемлемую соль, с немедленным или пролонгированным высвобождением.
В одном варианте выполнения изобретения композиция находится в виде лекарственной формы для доставки более чем 40% от всего фенилэфрина или его фармацевтически приемлемой соли до входа в ободочную кишку. В другом варианте выполнения изобретения композиция находится в виде лекарственной формы для доставки, по меньшей мере, части всего фенилэфрина или его фармацевтически приемлемой соли, предпочтительно более чем 20%, немедленно или в течение 1 часа после принятия внутрь. Более предпочтительно более чем 40% всего фенилэфрина доставляется посредством пролонгированного высвобождения в верхние отделы ЖК тракта субъекта до входа в ободочную кишку. Смотрите, например, варианты доз в Таблицах 16 и 17, где около 33% доставляется в ободочную кишку, конкретно Таблица 17 (вариант дозы: 10 мг IR+10 мг SR+10 мг ободочнокишечного) и Таблица 18 (вариант дозы: 20 мг IR+20 мг SR+20 мг ободочнокишечного). Смотрите так же предпочтительный вариант выполнения изобретения для дозы, содержащей 30 мг фенилэфрина, где около 17% доставляется в ободочную кишку, конкретно 5 мг из 30 мг дозы доставляется в ободочную кишку.
В другом варианте выполнения изобретения композиция находится в виде лекарственной формы для доставки 25% от всего фенилэфрина или его фармацевтически приемлемой соли в форме немедленного высвобождения и 75% всего фенилэфрина или его фармацевтически приемлемой соли в течение 6-8 часов. Смотрите, например, Таблицы 8 и 9. В предпочтительном варианте выполнения изобретения около 10-15% от введенного количества фенилэфрина или его фармацевтически приемлемой соли доставляется в ободочную кишку.
В одном варианте выполнения изобретения композиция содержит ядро, содержащее фенилэфрин или его фармацевтически приемлемую соль, и один или более поддающихся эрозии слоев, которые разрушаются до обнажения ядра и высвобождения фенилэфрина или его фармацевтически приемлемой соли для абсорбции в ободочной кишке. В определенных вариантах выполнения изобретения поддающийся эрозии слой (слои) содержит(ат) покрытие(ия), полимерную(ые) матрицу(ы) и/или оболочку(и), инкапсулирующую(ие) такое ядро. В других вариантах выполнения изобретения поддающийся эрозии слой (слои) включает(ют) матрицу, в которую вставлено ядро.
Понятно, что дополнительные компоненты для содействия фармацевтической композиции и ее улучшения, такие как один или более вязкость модифицирующий агентов, стабилизирующих агентов и суспендирующих агентов, и буферов для поддержания соответствующего рН, известных в данной области техники в качестве фармацевтически приемлемых и обычно применяемых в фармацевтической композиции, добавляются по желанию. Дополнительные фармацевтические эксципиенты, общепринятые и применяемые, обнаруживаются, например, в Remington's Pharmaceutical Sciences (Gennaro, A., ed.), Mack Pub., 1990. Так же, один или более подсластителей, таких как сахароза, сахарин, Сукралоза и т.д., для улучшения вкуса, один или более консервантов, таких как бензоат натрия, и/или пищевой краситель, возможно, но необязательно, добавляются по желанию. Фармацевтические композиции изобретения могут так же содержать любую другую, одну или более добавку, обычно применяемую в лекарственных формах фармацевтических композиций.
Способы создания матриц и покрытий, с или без терапевтически активных агентов внутри таких матриц и покрытий, известны в данной области техники. Например, способы создания пероральных фармацевтических лекарственных форм с контролируемым высвобождением описываются у Gupta and Robinson, "Oral controlled release delivery," Chapter 6 in Treatise on Controlled Drug Delivery, Editor A.Kydonieus, Dekker, N.Y., 1992; и в патенте США №7,163,696 (например, смотрите от раздела 3, строка 22, до раздела 4, строка 53), включенных здесь путем ссылки.
Иллюстративные примеры лекарственных форм или композиций данного изобретения
1) Лекарственная форма с зависимой от времени или контролируемой по продолжительности эрозией.
У воздерживавшегося от пищи здорового взрослого человека желудок опорожняется каждые 45-80 минут, и время прохода от рта до илеоцекального соединения составляет около 5 часов. Поэтому фармацевтическая композиция, содержащая поддающийся эрозии слой, который полностью разрушается через около 5 часов - около 12 часов, предпочтительно через около 6 часов - около 8 часов после принятия внутрь, будет защищать ядро, содержащее фенилэфрин, до тех пор, пока не будет достигнут целевой участок абсорбции в ободочной кишке для высвобождения фенилэфрина.
В одном варианте выполнения изобретения композиция представляет собой твердую лекарственную форму, содержащую: ядро, содержащее фенилэфрин или его фармацевтически приемлемую соль, и поддающийся эрозии слой, инкапсулирующий ядро, и, возможно, но не обязательно, содержащий фенилэфрин или его фармацевтически приемлемую соль, где состав и толщина поддающегося эрозии слоя являются такими, что ядро обнажается, когда композиция поступает в ободочную кишку, или приблизительно в то время, когда композиция достигает синапса ободочной кишки.
В определенных вариантах выполнения изобретения, где поддающийся эрозии слой содержит фенилэфрин или его фармацевтически приемлемую соль, поддающийся эрозии слой высвобождает фенилэфрин или его фармацевтически приемлемую соль для абсорбции на протяжении всего желудочно-кишечного тракта. Например, включение фенилэфрина или его фармацевтически приемлемой соли в один или более поддающихся эрозии слоев и/или внешнее покрытие обеспечивает немедленное высвобождение и/или пролонгированное высвобождение фенилэфрина или его фармацевтически приемлемой соли в верхних отделах желудочно-кишечного тракта, при приеме композиции внутрь, помимо высвобождения фенилэфрина из обнаженного ядра, когда композиция достигает ободочной кишки.
В определенных вариантах выполнения изобретения композиция, кроме того, включает внешний слой, возможно, но не обязательно, содержащий фенилэфрин или его фармацевтически приемлемую соль в форме немедленного высвобождения, в дополнение к фенилэфрину или его фармацевтически приемлемой соли для абсорбции в ободочной кишке. Внешним покрытием может быть энтеральное покрытие. Схема, изображающая пересечение частей такой лекарственной формы, представлена на Фиг.1А. Лекарственная форма может, кроме того, включать второе внешнее покрытие или верхнее покрытие для улучшения вкусовой привлекательности композиции (не показано). Включение фенилэфрина в поддающийся эрозии слой и/или внешнее покрытие обеспечивает пролонгированное или немедленное высвобождение фенилэфрина в верхних отделах желудочно-кишечного тракта при принятии внутрь.
В одном варианте выполнения изобретения ядро окружено поддающимся эрозии слоем или слоями, разработанное в виде обычной твердой лекарственной формы с немедленным высвобождением. В другом варианте выполнения изобретения ядром является инкапсулированная жидкость. В другом варианте выполнения изобретения ядро является полутвердым или находится в форме жидкого геля. При поступлении ядра в среду ободочной кишки, фенилэфрин или его фармацевтически приемлемая соль высвобождаются и доставляются.
В другом варианте выполнения изобретения ядром является гранулированное ядро (мультичастичное ядро). С помощью термина "гранулированное ядро" обозначают, что участок ядра композиции не является однородной композицией, но включает небольшие гранулы, содержащие фенилэфрин или его фармацевтически приемлемую соль, гранулы включаются в вещество матрицы с формированием ядра. Каждая гранула, возможно, но не обязательно, включена в поддающийся эрозии слой для дополнительного контроля высвобождения фенилэфрина ("поддающееся эрозии покрытие гранул"). В одном варианте выполнения изобретения такое поддающееся эрозии покрытие гранул включает рН-чувствительный полимер. В другом варианте выполнения изобретения поддающееся эрозии покрытие гранул включает полимер, специфичный к ободочной кишке (смотрите ниже Часть 3 для подходящих, специфичных к ободочной кишке полимеров, полезных для применения в поддающемся эрозии покрытии гранулы). Гранулы являются сферическими и эллиптическими и имеют размер диаметра или оси от 0.1 мм до 5 мм, предпочтительно от 0.2 до 2 мм и более предпочтительно от 0.5 до 1.5 мм. Схематическое изображение такого гранулированного ядра показано на Фиг.1В. Общая структура фармацевтической композиции является такой, как показано на Фиг.1А. Маленькие гранулы, как было показано, имеют более долгое удерживание в ободочной кишке, чем таблетки стандартной дозы.
В одном варианте выполнения изобретения общая лекарственная форма содержит 1-150 мг (например, 1 мг, 2.5 мг, 5 мг, 7.5 мг, 10 мг, 12.5 мг, 15 мг, 20 мг, 25 мг, 30 мг, 35 мг, 40 мг, 45 мг, 50 мг, 55 мг, 60 мг, 65 мг, 70 мг, 75 мг, 80 мг, 85 мг, 90 мг, 95 мг, 100 мг, 105 мг, 110 мг, 115 мг, 120 мг, 125 мг, 130 мг, 135 мг, 140 мг, 145 мг или 150 мг) фенилэфрина или его фармацевтически приемлемой соли, что могло бы составлять 0.2-90% в/в по отношению ко всей лекарственной форме.
В одном варианте выполнения изобретения гранулированное ядро содержит 0.2-10% в/в от всего фенилэфрина или его фармацевтически приемлемой соли, содержащихся в ядре; 0-90% в/в микрокристаллической целлюлозы, такой как Avicel® PH 101 (ФМС Биополимер, Филадельфия, Пенсильвания); 0-80% агентов для контроля скорости высвобождения лекарственного средства, включая, но не ограничиваясь к этому, один или более из следующих: гидрофобное основание, такое как глицерилмоностеарат или глицерил бегенат, доступный как Compitrol® 888 АТО (Gattefosse SA), гидрофильное основание, такое как гидроксипропилметилцеллюлоза (ГПМЦ), поливиниловый спирт, привитой сополимер поливинилового спирта и полиэтиленгликоля (например, Kollicoat IR), гидроксиэтилцеллюлоза (ГЭЦ), карбоксиметилцеллюлоза (КМЦ) или соль КМЦ (например, натриевая соль КМЦ, кальциевая соль КМЦ); 1-10% одного или более дезинтегрирующих агентов, таких как кросповидон или L-HPC.
Гранулы изготавливают с помощью способов, известных в данной области техники. В примерном способе гранулы изготавливают в соответствии с вышеуказанным составом, применяя дистиллированную воду в качестве смачивающей жидкости (до 80% от сухого веса композиции). Все компоненты смешиваются, смачиваются и экструдируются через экструдер (такой, как экструдер, доступный для приобретения у фирмы Caleva Process Solutions Ltd., Соединенное королевство), и затем приобретают сферическую или эллиптическую форму на сферонизаторе (Caleva spheronizer). Полученные влажные гранулы высушивают при соответствующей температуре (путем лотковой сушки или сушки в кипящем слое). Полученные гранулы являются эллиптическими или сферическими и имеют размер 0.5-1.3 мм в диаметре.
В определенных вариантах выполнения изобретения гранулированное ядро содержит матрицу и гранулы, содержащие фенилэфрин или его фармацевтически приемлемую соль. В определенных вариантах выполнения изобретения матрицы так же часто содержат фенилэфрин или его фармацевтически приемлемую соль. В отдельных вариантах выполнения изобретения только гранулы содержат фенилэфрин или его фармацевтически приемлемую соль. В других вариантах выполнения изобретения только матрица включает фенилэфрин или его фармацевтически приемлемую соль.
В одном варианте выполнения изобретения помимо доставки фенилэфрина или его фармацевтически приемлемой соли в форме пролонгированного высвобождения гранулированное ядро разработано для доставки фенилэфрина или его фармацевтически приемлемой соли в форме немедленного высвобождения. Подобным образом, в определенных вариантах выполнения изобретения помимо доставки фенилэфрина или его фармацевтически приемлемой соли в форме пролонгированного высвобождения гранулированное ядро разработано для доставки одного или более дополнительных терапевтических агентов в форме немедленного высвобождения и/или в форме пролонгированного высвобождения.
В одном варианте выполнения изобретения ядро содержит отдельные гранулы, на которые наносится поддающийся эрозии слой. В одном варианте выполнения изобретения многочисленные гранулы помещают в капсулы (например, желатиновые капсулы) или прессуют в таблетки для доставки соответствующей дозы фенилэфрина. В одном варианте выполнения изобретения поддающийся эрозии слой содержит полимер, такой как Eudragit® L-30D, который устойчив к разрушению при рН ниже 5.6, Eudragit® L100-55, который устойчив к разрушению при рН ниже 5.5. Другими подходящими веществами являются фталат гидроксипропилметилцеллюлозы (ГПМЦФ), который доступен в формах с рН-порогом эрозии, например, 4.5-4.8, 5.2 или 5.4. Может так же применяться фталат ацетата целлюлозы (ФАЦ).
В одном варианте выполнения изобретения полная лекарственная форма содержит от 1 до 150 мг фенилэфрина или его фармацевтически приемлемой соли на лекарственную форму, 0-90% (в/в по отношению к лекарственной форме) микрокристаллической целлюлозы или других фармацевтически приемлемых разбавителей и 0-5% в/в стеарата магния или другого фармацевтически приемлемого смазочного вещества; поддающийся эрозии слой содержит 20-40% в/в гидроксипропилметилцеллюлозы (ГПМЦ), 0-50% в/в карбоксиметалцеллюлозы (КМЦ) или соли КМЦ (например, натриевой соли КМЦ, кальциевой соли КМЦ), 0-5% диоксида кремния и 0-5% в/в стеарата магния или другого фармацевтически приемлемого смазывающего вещества. Такие лекарственные формы могут, кроме того, содержать поддающийся эрозии слой, содержащий 1-150 мг фенилэфрина или его фармацевтически приемлемой соли на одну лекарственную форму; и верхнее покрытие, где верхнее покрытие предназначено для улучшения внешнего вида лекарственной формы, которое содержит 1-10% в/в низкомолекулярной гидроксипропилметилцеллюлозы (ГПМЦ), поливинилового спирта или Kollicoat IR, включая пластификатор, вплоть до 10% по его весу, и в случае активного покрытия 1-30 мг фенилэфрина или его фармацевтически приемлемой соли в расчете на лекарственную форму.
В одном варианте выполнения изобретения помимо ядра, содержащего фенилэфрин или его фармацевтически приемлемую соль, композиция, кроме того, содержит фенилэфрин или его фармацевтически приемлемую соль для их немедленного высвобождения и/или один или более дополнительных терапевтических агентов для их либо немедленного, либо пролонгированного высвобождения. В одном варианте выполнения изобретения помимо ядра, содержащего фенилэфрин или его фармацевтически приемлемую соль, композиция, кроме того, содержит фенилэфрин или его фармацевтически приемлемую соль для их немедленного высвобождения и антигистамин (например, лоратадин или деслоратадин) для его немедленного высвобождения. В одном варианте выполнения изобретения активный агент (агенты) для немедленного высвобождения находится(ятся) в покрытии, которое разрушается при пероральном введении, таким образом, обнажая внутренний слой (слои) композиции (например, поддающийся эрозии слой).
В одном варианте выполнения изобретения композиция, кроме того, содержит верхнее покрытие, например, для улучшения внешнего вида или вкусовой привлекательности лекарственной формы. В одном варианте выполнения изобретения верхнее покрытие содержит фармацевтически приемлемый покрывающий полимер или краситель. Примеры подходящих фармацевтически приемлемый покрывающих полимеров содержат гидроксипропилметилцеллюлозу (ГПМЦ), карбоксиметилцеллюлозу (КМЦ) или соль КМЦ (например, натриевую соль КМЦ, кальциевую соль КМЦ), гидроксипропилцеллюлозу (ГПЦ), поливиниловый спирт, привитой сополимер поливинилового спирта и полиэтиленгликоля и фармацевтически приемлемые гидрофильные полимеры.
В одном варианте выполнения изобретения верхнее покрытие содержит от 1 до 25 мг (от 0.02% в/в до 5% в/в) поливинилового спирта и от 0.1 до 5.0 мг (от 0.02% в/в до 1% в/в) красителя Синий No. 1 в расчете на лекарственную форму.
В одном варианте выполнения изобретения полная лекарственная форма содержит: 1-150 мг фенилэфрина или его фармацевтически приемлемой соли, что могло бы составлять 0.2-90% в/в от полной лекарственной формы; 0-90% в/в микрокристаллической целлюлозы, такой как Avicel® РН 102 (ФМС Биополимер, Филадельфия, Пенсильвания) или любого фармацевтически приемлемого таблетирующего наполнителя/разбавителя, описанного в справочнике Handbook of Pharmaceutical Excipients, четвертое издание (Row, Shesheky and Walter, Pharmaceutical Press); 0-80% агентов для контроля скорости высвобождения лекарственного средства, включая, но не ограничиваясь к этому, один или более из следующих: гидроксипропилцеллюлоза (ГПЦ), гидроксипропилметилцеллюлоза (ГПМЦ) (например, Methocel® KI5M, Methocel® KI00M, Methocel® K4M (Dow Corning)), карбоксиметилцеллюлоза (КМЦ) или соль КМЦ (например, натриевая соль КМЦ, кальциевая соль КМЦ) и фармацевтически приемлемые гидрофильные полимеры; и 0-15% стеарата магния или другого равноценного смазывающего вещества.
Благодаря высокой растворимости в воде фенилэфрина применение гидрофильных полимеров само по себе приводит к быстрой диффузии и высвобождению активного агента. Чтобы уменьшить взрывной эффект более раннего профиля высвобождения, вышеуказанный фармацевтически приемлемый гидрофильный полимер может быть объединен с одним или более гидрофобным полимером (полимерами) (включая, но не ограничиваясь этим, этилцеллюлозу (Ethocel®) или сополимер акриловой кислоты).
В одном варианте выполнения изобретения предпочтительная комбинация полимеров для пролонгированного высвобождения формирует матрицу с активным агентом (агентами) (то есть фенилэфрином или его фармацевтически приемлемой солью и необязательным одним или более дополнительным терапевтическим агентом (агентами)), распределенным(ми) внутри матрицы, что обеспечивает нулевой или близкий к нулю порядок высвобождения активного агента (агентов).
В одном предпочтительном варианте выполнения изобретения комбинация анионной натриевой соли карбоксиметилцеллюлозы и неионной гидроксипропилцеллюлозы обеспечивает матрицу с сильными поперечными связями, приводя к более высокой вязкости и более низкой скорости диффузии через матрицу для конкретного профиля растворимости фенилэфрина. Комбинация гидроксипропилцеллюлозы и натриевой соли карбоксиметилцеллюлозы позволяет моделирование профиля высвобождения, который является специфичным и особенным для фенилэфрина, так что ядро полностью разрушается через 4-12 часов после введения. В более предпочтительном варианте выполнения изобретения ядро полностью разрушается через около 4-8 часов после принятия внутрь.
В одном варианте выполнения изобретения ядро содержит: фенилэфрин или его фармацевтически приемлемую соль, возможно, но не обязательно, микрокристаллическую целлюлозу, карбоксиметилцеллюлозу (КМЦ) или ее соль (например, натриевая или кальциевая соль КМЦ), гидроксипропилцеллюлозу, возможно, но не обязательно, коллоидный диоксид кремния и стеарат магния. Например, ядро содержит компоненты и интервалы их процентного содержания по весу, подробно изложенные в таблице 1.
Примерные лекарственные формы A-G для ядра с продолжительным высвобождением фенилэфрина подробно описаны в Таблице 2.
В одном предпочтительном варианте выполнения изобретения полная лекарственная форма содержит компоненты и интервалы их процентного содержания (% в/в), подробно описанные ниже в Таблице 3.
Один особенно предпочтительный вариант ядра с продолжительным высвобождением фенилэфрина приводится в Таблице 4.
В определенных предпочтительных вариантах выполнения изобретения один или более полимеров, контролирующих скорость высвобождения, применяются вместо описанных в Таблице 4. Например, подходящие альтернативные гидрофильные полимеры включают гидроксипропилцеллюлозу (ГПЦ), имеющую другую вязкость, гидроксипропилметилцеллюлозу (ГПМЦ), имеющую другую вязкость, натриевую или кальциевую соль карбоксиметилцеллюлозы (КМЦ), имеющую другую вязкость, и ксантановую смолу. Таблица 5 приводит предпочтительные интервалы для примерных альтернативных гидрофильных полимеров.
Подобным образом, подходящие альтернативные гидрофобные полимеры включают этилцеллюлозу (Ethocel®), имеющую различный молекулярный вес, сополимеры акриловой кислоты, фармацевтический воск и метилцеллюлозу, имеющую другую вязкость. Таблица 6 приводит предпочтительные интервалы примерных альтернативных гидрофобных полимеров.
В одном варианте выполнения изобретения композиция, кроме того, содержит от 1 до 25 мг фенилэфрина или его фармацевтически приемлемой соли в форме немедленного высвобождения, 0-90% (в/в относительно лекарственной формы) одного или более фармацевтического разбавителя (разбавителей) или другого фармацевтически приемлемого таблетирующего агента (агентов). Примерами таких таблетирующих агентов является микрокристаллическая целлюлоза, крахмал, пептизированный крахмал, лактоза или другие таблетирующие сахара, фосфат кальция или любые другие фармацевтически приемлемые таблетирующие эксципиенты.
В одном варианте выполнения изобретения композиция, кроме того, содержит поддающееся эрозии покрытие, содержащее от 1 до 25 мг фенилэфрина или его фармацевтически приемлемой соли в форме немедленного высвобождения, 2-20% (в/в относительно лекарственной формы) поливинилового спирта или другого фармацевтически приемлемого агента, образующего покрытие. В другом варианте выполнения изобретения композиция, кроме того, содержит поддающееся эрозии покрытие, содержащее от 1 до 25 мг фенилэфрина или его фармацевтически приемлемой соли в форме немедленного высвобождения, 4-8% (в/в относительно лекарственной формы) поливинилового спирта или другого фармацевтически приемлемого агента, образующего покрытие. Примерами фармацевтически приемлемых агентов, образующих 0покрытие, являются гидроксипропилметилцеллюлоза (ГПМЦ); поливиниловый спирт, Kollicoat IR, карбоксиметилцеллюлоза (КМЦ) или соль КМЦ (например, натриевая соль КМЦ, кальциевая соль КМЦ), гидроксипропилцеллюлоза (ГПЦ), или любые другие фармацевтически приемлемые гидрофильные полимеры. В одном предпочтительном варианте выполнения изобретения фармацевтически приемлемый агент, образующий покрытие, основан на поливиниловом спирте (например, Opadry™ II серия 85, предпочтительно 18422; привитой сополимер поливинилового спирта и полиэтиленгликоля (например, Kollicoat IR)).
В предпочтительном варианте выполнения изобретения часть немедленного высвобождения содержит фенилэфрин или его фармацевтически приемлемую соль, так же как и поливиниловый спирт.
В одном варианте выполнения изобретения композиция кроме того содержит один или более дополнительных терапевтических агентов, включенных в состав лекарственной формы для их немедленного и/или пролонгированного высвобождения. В одном предпочтительном варианте выполнения изобретения одним или более дополнительным терапевтическим агентом (агентами) является антигистамин, предпочтительно, лоратадин или деслоратадин. Соответственно, в одном варианте выполнения изобретения композиция содержит следующую часть пролонгированного высвобождения и часть немедленного высвобождения, подробно изложенные ниже в Таблице 7.
В другом предпочтительном варианте выполнения изобретения композиция содержит следующие часть пролонгированного высвобождения и часть немедленного высвобождения, подробно изложенные ниже в Таблице 8.
В любом из предшествующих вариантов выполнения изобретения ядро, возможно, но не обязательно, содержит вещество, способствующее всасыванию. Примерами веществ, способствующих всасыванию, являются: салицилаты, такие как салицилат натрия, 3-метоксисалицилат, 5-метоксисалицилат и гомованилат; желчные кислоты, такие как таурохолевая, тауродезоксихолевая, дезоксихолевая, холевая, глихолевая, литохолевая, хенодезоксихолевая, урсодезоксихолевая, урсохолевая, дегидрохолевая, фусидовая и т.п.; такие неионные поверхностно-активные вещества, как простые полиоксиэтиленовые эфиры (например, Brij 36T®, Brij 52®, Brij 56®, Brij 76®, Brij 96®, Texaphor® A6, Texaphor® A14, Texaphor® A60 и т.п.), п-трет-октилфенолполиоксиэтилены (Triton® X-45, Triton® X-100, Triton® X-114, Triion® X-305 и т.п.), нонилфеноксиполиоксиэтилены (например, Igepal® серии СО), сложные полиоксиэтиленовые эфиры сорбита (например, Tween®-20, Tween®-80 и т.п.); такие анионные поверхностно-активные вещества, как диоктил сульфосукцинат натрия; такие лизофосфолипиды, как лизолецитин и лизофосфатидилэтаноламин; такие ацилкарнитины, ацилхолины и ациламинокислоты, как лауроилкарнитин, миристоилкарнитин, пальмитоилкарнитин, лауроилхолин, миристоилхолин, пальмитоилхолин, гексадециллизин, N-ацилфенилаланин, N-ацилглицин и т.п.; водно-растворимые фосфолипиды; глицериды со средней длиной цепи, представляющие собой смеси моно-, ди- и триглицеридов, содержащих жирные кислоты со средней длиной цепи (каприловая, каприновая и лауриновая кислоты); этилендиаминтетрауксусная кислота (EDTA); такие катионные поверхностно-активные вещества, как цетилпиридиний хлористый; такие жирно-кислотные производные полиэтиленгликоля, как Labrasol®, Labrafac® и т.п.; и такие алкилсахариды, как лаурил мальтозид, лауроил сахароза, миристоил сахароза и палмитоил сахароза.
В еще одних вариантах выполнения изобретения фармацевтическая композиция изобретения представляет собой контейнер с механизмом высвобождения. Такая структура представляет собой нерастворимый контейнер, содержащий фенилэфрин или его фармацевтически приемлемую соль, и пробку. Пробка удаляется через предопределенное время запаздывания, посредством набухания, эрозии или растворения. В определенных вариантах выполнения изобретения контейнер, как правило, представляет собой капсулу. Доступной для приобретения капсульной системой является Pulsincap® (Scherer DDS, Ltd, Кпайдбанк, Глазго, Соединенное королевство). В этой системе нерастворимая капсула запечатывается гидрогелевой пробкой, которая гидратируется в жидкой среде желудочно-кишечного тракта, зависимым от времени образом, и набухает до той степени, что она вытесняется из тела капсулы, таким образом, высвобождая ее содержимое. В одном варианте выполнения изобретения пробка вытесняется через около 5-12 часов после принятия внутрь. В предпочтительном варианте выполнения изобретения пробка вытесняется через около 6-8 часов после принятия внутрь. В конкретном варианте выполнения изобретения контейнер содержит фенилэфрин или его фармацевтически приемлемую соль и один или более дополнительный терапевтический агент (агенты), так же как и эксципиенты, известные в данной области техники. Примерные варианты выполнения изобретения содержат капсулу, содержащую 1-150 мг фенилэфрина, 0-90% (в/в относительно лекарственной формы) микрокристаллической целлюлозы или другого фармацевтически приемлемого разбавителя и 0-5% в/в стеарата магния или другого фармацевтически приемлемого смазывающего вещества. В другом варианте выполнения изобретения капсула содержит гранулы, как описано выше, гранулы содержат фенилэфрин или его фармацевтически приемлемую соль и один или более дополнительный терапевтический агент (агенты), так же как и эксципиенты, известные в данной области техники.
В одном варианте выполнения изобретения нерастворимый контейнер покрыт колпачком, который немедленно растворяется при пероральном введении, обнажая полимерную пробку, возможно, но не обязательно, содержащую фенилэфрин или его фармацевтически приемлемую соль для немедленного высвобождения. Полимерная пробка состоит из поддающегося эрозии полимера, пробка имеет соответствующую толщину и разрушается в течение желательного промежутка времени. Примерами таких гидрофильных полимеров являются гидроксипропилметилцеллюлоза (ГПМЦ), поливиниловый спирт, Kollicoat IR, карбоксиметилцеллюлоза (КМЦ) или соль КМЦ (например, натриевая соль КМЦ, кальциевая соль КМЦ), гидроксипропилцеллюлоза (ГПЦ), или любые другие фармацевтически приемлемые гидрофильные полимеры. Пробка разрушается через около 5-12 часов после принятия внутрь, более предпочтительно через около 6-8 часов после принятия внутрь.
2) Лекарственная форма с рН-зависимой контролируемой эрозией
В другом варианте выполнения изобретения композиция находится в виде твердой лекарственной формы, где фенилэфрин или его фармацевтически приемлемая соль высвобождаются при условии, которое типично, преимущественно или исключительно существует в ободочной кишке (условие, специфичное для ободочной кишки) и не существует вовремя прохода через верхние отделы пищеварительного тракта, включая желудок и/или тонкий кишечник. рН-Чувствительный полимер, применяемый при соответствующей толщине, может быть использован для предотвращения высвобождения фенилэфрина до тех пор, пока продукт ни достигнет ободочной кишки. Посредством термина "рН-чувствительный" обозначают, что полимер разрушается при рН выше или ниже определенного значения, или внутри определенного интервала значений рН. В предпочтительных вариантах выполнения изобретения слой, поддающийся рН-зависимой эрозии, может иметь увеличенную толщину, чтобы контролировать продолжительность эрозии, и/или учитывать наличие одного или более ферментов, специфичных для ободочной кишки или превалирующих в ободочной кишке, для последующего продолжения эрозии.
Вдоль желудочно-кишечного тракта рН изменяется следующим образом: внутри несодержащего пищу желудка рН находится в пределах от 1.5 до 3 и для наполненного пищей желудка рН 2-5. В тонком кишечнике, внутри несодержащей пищу двенадцатиперстной кишки, рН составляет приблизительно 6.1, тогда как после приема пищи оно падает до 5.4. Подвздошная кишка имеет рН около 7-8. В ободочной кишке: рН внутри слепой кишки и ободочной кишки находится в интервале от 5.5 до 7. Это нейтральное значение рН поддерживается и внутри прямой кишки. Смотрите Patel et al., Drug Delivery Technol, 6(7):62-71 (Июль/Август 2006).
В одном варианте выполнения изобретения композиция изобретения содержит: ядро, содержащее фенилэфрин или его фармацевтически приемлемую соль, и поддающийся эрозии, специфичный к ободочной кишке слой, инкапсулирующий ядро и разрушающийся при рН ободочной кишки, равном около 5.5 или выше, необязательный слой, содержащий фенилэфрин для немедленного высвобождения в верхних отделах кишечного тракта, и необязательное второе покрытие (например, энтеральное покрытие). Предпочтительно специфичный к ободочной кишке слой разрушается при рН ободочной кишки, равном от около 5.7 до около 6.8. Лекарственная форма может, кроме того, содержать верхнее покрытие для улучшения вкусовой привлекательности лекарственной формы, верхние покрытие возможно, но не обязательно, так же может быть активным покрытием, содержащим фенилэфрин для немедленного высвобождения. Такой вариант выполнения изобретения доставляет фенилэфрин посредством от одного до трех импульсных высвобождений, в различные области ЖК тракта. Схема, изображающая поперечное сечение такой лекарственной формы, представлена на Фиг.2. Ядро разработано в виде обычной твердой лекарственной формы немедленного высвобождения, которая позволяет болюсную доставку фенилэфрина при поступлении ядра в среду ободочной кишки. Специфичный к ободочной кишке слой может, кроме того, содержать дополнительное поддающееся эрозии покрытие для защиты от низкого рН желудка. В таком варианте выполнения изобретения ядро и любой необязательный дополнительный слой (слои) инкапсулируются энтеральным покрытием, содержащим композицию, которая устойчива к разрушению при преобладающем значении рН желудка. Обычно применяемое энтеральное покрытие устойчиво к разрушению в желудке, где рН ниже 2. Пример такого энтерального покрытия содержит фталат гидроксипропилметилцеллюлозы, поливинилацетатфталат или сополимеры метакриловой кислоты. Доступные для приобретения препараты включают Eudragit® L-100, который растворяется при рН 6.0, и S-100, который растворяется при рН 7.0, применяемые в виде смеси (Rohm Pharma GmbH, Германия). Другим примером полимеров, подходящих для применения в качестве энтерального покрытия, является ацетат сукцинат гипромеллозы (HPMCAS). Смотрите, например, Tanno et al.. Drug Dev Ind Pharm, 30(1):9-17 (Январь 2004). HPMCAS доступен для приобретения под различными марками, такими как HPMCAS MF (порог рН 6.0) или HPMCAS HF (порог рН 6.8) (Shin-Etsu Chemical Co. Ltd., Токио, Япония) и может применяться в виде комбинации обоих этих полимеров. В состав покрытия входит один или более соответствующих пластификаторов. Способы приготовления желательного покрытия известны в данной области техники и более полно описываются в рассказывающем о товаре информационном материале, предоставляемом производителями этих полимеров.
В примерной лекарственной форме полная лекарственная форма содержит от 1 до 150 мг фенилэфрина или его фармацевтически приемлемой соли в расчете на лекарственную форму, 0-90% (в/в по отношению к лекарственной форме) микрокристаллической целлюлозы или другого фармацевтически приемлемого разбавителя и 0-5% в/в стеарата магния или другого фармацевтически приемлемого смазывающего вещества; слой, специфичный к ободочной кишке, содержит Eudragit L-100, и если дополнительное энтеральное покрытие является полезным, такое энтеральное покрытие достигает 10% в/в от лекарственной формы в целом (5-35% привеса засчет добавления матриц и покрытий) и содержит фталат гидроксипропилметилцеллюлозы, растворимый в среде со значением рН выше 6.8. Кроме того, композиция необязательно содержит верхнее покрытие, содержащее 1-10% в/в низкомолекулярной гидроксипропилметилцеллюлозы, поливинилового спирта или Kollicoat IR, включая пластификатор до 10% от ее веса, и, в случае активного покрытия, 1-30 мг фенилэфрина или его фармацевтически приемлемой соли в расчете на лекарственную форму.
В другом варианте выполнения изобретения ядро фармацевтической композиции изобретения представляет собой гранулированное ядро и содержит гранулы, содержащие фенилэфрин или его фармацевтически приемлемую соль. Гранулированные ядра описываются выше и показаны на Фиг.1B. В другом варианте выполнения изобретения ядро содержит отдельные гранулы, содержащие фенилэфрин или его фармацевтически приемлемую соль, на гранулы наносится специфичное к ободочной кишке покрытие. Многочисленные гранулы загружают в желатиновые капсулы или прессуют в таблетки для доставки соответствующей дозы фенилэфрина. В других вариантах выполнения изобретения многочисленные слои могут быть нанесены на гранулы для обеспечения дополнительных импульсов высвобождения лекарственного средства в различных отделах ЖК тракта.
3) Лекарственная форма со специфичной к ободочной кишке эрозией
В других вариантах выполнения изобретения композиция содержит ядро, содержащее фенилэфрин или его фармацевтически приемлемую соль и специфичный к ободочной кишке слой, разрушаемый посредством действия одного или более ферментов, специфичных для ободочной кишки или преобладающих в ней. Такие ферменты упоминаются в настоящем документе как "специфичные для ободочной кишки ферменты". Ферменты могут производиться клетками ободочной кишки млекопитающих или могут быть выделены популяцией бактерий микрофлоры ободочной кишки. Примером такого фермента является азоредуктаза, которая расщепляет ароматическую азо-связь. Гель, основанный на N,N-диметилакриламиде, N-трет-бутилакриламиде и акриловой кислоте, поперечно связанной с азоароматическими соединениями различной длины, может применяться для получения поддающегося эрозии слоя, разрушающегося при действии такого специфичного для ободочной кишки фермента. Brondsted et al., Pharmaceutical Res, 9(12): 1540-1545 (Декабрь 1992). Аналоги, основанные на уретане, содержащие азоароматическую связь, так же полезны для получения поддающегося эрозии слоя при осуществлении данного изобретения. Дополнительными ферментами, обнаруживаемыми в ободочной кишке, являются нитроредуктаза, N-оксидоредуктаза, сульфоксидредуктаза, гидрогеназа, эстеразы и амидазы, глюкозидаза, глюкуронидаза, сульфатаза и другие. Примеры таких специфичных к ободочной кишке слоев включают, но без ограничения к этому, слои, содержащие полисахариды, такие как хитозан, природный полимер, полученный путем гидролиза хитина, шеллак, и определенные формы крахмала, как, например, гороховый крахмал. Комбинации пектина, хитозана и гидроксипропилметилцеллюлозы, поливиниловый спирт, или Kollicoat IR, так же полезны при реализации этого варианта выполнения изобретения. Пленка, полученная из смеси амилоза/этилцеллюлоза, так же может применяться. Смотрите Siew et al., AAPS PharmSciTech, 1(3): article 22 (2000); Tuleu et al., Alimentary Pharmacol Therapeut, 16(10): 1771 (Октябрь 2002); Chaubal, Drug Delivery Technol, Article 131. Гуаровая смола, метакрилированный инулин и ацетат декстрана являются несколькими дополнительными примерами. Ядро, содержащее фенилэфрин, при необходимости защищено от желудочной кислоты энтеральным покрытием, как, например, когда применяется покрытие, основанное на полисахаридах.
В другом варианте выполнения изобретения ядро фармацевтической композиции изобретения представляет собой гранулированное ядро и содержит гранулы, содержащие фенилэфрин или его фармацевтически приемлемую соль. Гранулированные ядра описываются выше и показаны на Фиг.1B. В другом варианте выполнения изобретения ядро содержит отдельные гранулы, содержащие фенилэфрин или его фармацевтически приемлемую соль, на гранулы наносится специфичный к ободочной кишке слой. Многочисленные гранулы загружают в желатиновые капсулы или прессуют в таблетки для доставки соответствующей дозы фенилэфрина. В другом варианте выполнения изобретения многочисленные слои могут быть нанесены на гранулы для обеспечения дополнительных импульсов высвобождения лекарства в различных областях ЖК тракта. В еще одном варианте выполнения изобретения лекарственные формы изобретения, подходящие для композиций фенилэфрина или его фармацевтически приемлемой соль, имеющих в качестве мишени ободочную кишку, содержат комбинацию двух или более поддающихся эрозии слоев с контролируемой по продолжительности, рН-контролируемой или контролируемой посредством ферментов ободочной кишки эрозией для более точного контроля. Смотрите выше для подробностей и в общем Cheng et al. World J Gastroenterol, 10(12):1769-1774 (2004); Asghar et al., J Pharm PharmaceutSci, (3):327-338 (2006); Li et al., AAPS PharmSciTech, 3(4); article 33 (2002). Например, помимо ядра, содержащего фенилэфрин или его фармацевтически приемлемую соль, предпочтительно количество фенилэфрина также распределено в слое покрытия, содержащем полимерную композицию. Слой покрытия, содержащий часть фенилэфрина, высвобождает содержащийся в нем фенилэфрин немедленно после принятия внутрь, способствуя достижению максимальной концентрации в плазме неметаболизированного или несвязанного фенилэфрина.
4) Лекарственные формы, включающие комбинацию с одним или более дополнительным терапевтическим агентом (агентами).
В другом варианте выполнения изобретения фармацевтическая композиция, кроме того, содержит один или более дополнительных терапевтических агентов. Такой агент или агенты могут быть включены в лекарственную форму для их немедленного высвобождения при принятии внутрь, пролонгированного высвобождения, высвобождения в ободочной кишке или любой комбинации из вышеперечисленного.
В определенных вариантах выполнения изобретения один или более дополнительных терапевтических агентов добавляются в любое из покрытий или в любой из слоев лекарственной формы, описанные выше, в любой подходящей комбинации. В одном варианте выполнения изобретения фармацевтической композицией является лекарственная форма с контролируемой по времени эрозией, имеющая структуру, изображенную на Фиг.1. В одном варианте выполнения изобретения ядро, кроме того, содержит один или более дополнительных терапевтических агентов для высвобождения их в верхних отделах ЖК тракта и/или ободочной кишке, одновременно с фенилэфрином. В конкретном варианте выполнения изобретения один или более дополнительный(ые) терапевтический(ие) агент(ы) включен(ы) в гранулы гранулированного ядра. В другом варианте выполнения изобретения один или более дополнительный(ые) терапевтический(ие) агент(ы) включен(ы) в матрицу, окружающую гранулы в гранулированном ядре. В одном варианте выполнения изобретения один или более дополнительный(ые) терапевтический(ие) агент(ы) включен(ы) в гранулы гранулированного ядра, также как и в матрицу, окружающую гранулы в гранулированном ядре. В другом варианте выполнения изобретения поддающийся эрозии слой содержит один или более дополнительный(ые) терапевтический(ие) агент(ы) для его(их) немедленного высвобождения в тонкой кишке. В другом варианте выполнения изобретения один или более дополнительный(ые) терапевтический(ие) агент(ы) вкпючен(ы) в поддающийся эрозии слой лекарственной формы для его(их) пролонгированного высвобождения. В другом варианте выполнения изобретения активное верхнее покрытие содержит один или более дополнительный(ые) терапевтический(ие) агент(ы) для его(их) немедленного высвобождения после принятия внутрь. В определенных вариантах выполнения изобретения фармацевтические композиции изобретения содержат один или более дополнительный(ые) терапевтический(ие) агент(ы) в одном или более ядре или любом из слоев или покрытий, как описано выше.
В другом варианте выполнения изобретения один или более дополнительный(ые) терапевтический(ие) агент(ы) добавляется(яются) к лекарственной форме с рН-зависимой или специфичной к ободочной кишке эрозией, имеющей структуру, изображенную на Фиг.2. Подобно вышеизложенному описанию, любое ядро или слой лекарственной формы может содержать один или более дополнительный(ые) терапевтический(ие) агент(ы) для его (их) высвобождения в течение требуемого времени.
Дополнительным терапевтическим агентом может быть деконгестант, включая антигистамин, жаропонижающее средство, нестероидное противовоспалительное средство или любой другой терапевтический агент или комбинация двух или более таких агентов для содействия облегчению симптомов простуды, сезонной или несезонной аллергии, сенной лихорадки или проблем синуса. В предпочтительном варианте выполнения изобретения фармацевтические композиции включают антигистамин.
Антигистаминами могут быть H1 или Н2 антагонисты или другие типы ингибиторов высвобождения гистамина. H1 антагонисты могут быть седативными или неседативными, как, например, дифенгидрамин, хлорфенирамин, трипеленнамин, прометазин, клемастин, доксиламин, астемизол, терфенадин и лоратадин, среди прочего. Примеры Н2 антагонистов включают, не без ограничения к этому, циметидин, фамотидин, низатидин и ранитидин. Примеры ингибиторов высвобождения гистамина включают кромолин. Антигистамины долгого действия, выбранные из одного или более членов группы, состоящей из лоратадина, деслоратадина, азатидина, фексофенадина, терфенадина, цетиризина, астемизола и левокабастина, или их фармацевтически приемлемых солей, являются подходящими для фармацевтических композиций изобретения.
Предпочтительные антигистамины включают лоратадин и деслоратадин. Лоратадин раскрывается в патенте США №4,282,233 в качестве неседативного антигистамина, полезного, например, для облегчения симптомов сезонного аллергического ринита, таких как чихание и зуд. Активным метаболитом лоратадина является деслоратадин, который имеет период полураспада (t1/2) приблизительно 15-19 часов. Патент США №5,595,997 раскрывает способы и композиции для лечения симптомов сезонного аллергического ринита посредством применения деслоратадина. Лоратадин и деслоратадин доступны в форме обычных таблеток, которые высвобождают активное вещество обычным образом. Примерная лекарственная форма высвобождает лоратадин посредством процесса разрушения и растворения, так что лоратадин начинает вызывать его антигистаминный эффект в течение от 1 до 3 часов и эффект продолжается в течение более 24 часов. Благодаря продолжительному периоду полураспада лоратадина по сравнению с фенилэфрином, лоратадин в лекарственной форме по изобретению предпочтительно доступен для немедленного высвобождения. Например, лоратадин или деслоратадин может присутствовать в растворе в жидкости-носителе жидкого ядра или может быть инкорпорирован в верхнее покрытие продукта.
Другие антигистамины также полезны для выполнения настоящего изобретения. Азатадин раскрывается в Бельгийском Патенте №647,043 и в соответствующих Патентах США №3,326,924 и №3,419,565. Сообщают, что период полувыведения составляет 9-12 часов. Терфенадин и фексофенадин раскрываются в Патенте США №3,878,217 и имеют продолжительность действия 12-24 часа, и больше чем 24 часа, соответственно. Цетиризин раскрывается в Патенте США №4,525,358 и, как сообщают, имеет продолжительность действия 12-24 часа. Астемизол раскрывается в Патенте США №4,219,559 и, как сообщают, имеет продолжительность действия более 24 часов. Левокабастин раскрывается в Патенте США №4,369,184 и, как сообщают, имеет продолжительность действия 16-24 часа.
Доза антигистамина, такого как лоратадин или деслоратадин, может быть представлена в различных концентрациях, таких как 1-20 мг; предпочтительно 2.5 мг, 5 мг или 10 мг.
Подходящими противовоспалительными и/или жаропонижающими средствами, полезными для данных композиций, могут быть: нестероидные противовоспалительные средства (НСПС), производные аминоарилкарбоновой кислоты, такие как энфенамовая кислота, этофенамат, флуфенамовая кислота, изониксин, меклофенамовая кислота, мефанамовая кислота, нифлумовая кислота, талнифлумат, терофенамат и толфенамовая кислота; производные арилуксусной кислоты, такие как ацеметацин, алклофенак, амфенак, буфексамак, цинметацин, клопирак, диклофенак натрия, этодолак, фельбинак, фенклофенак, фенклорак, фенклозовая кислота, фентиазак, глюкаметацин, ибуфенак, индометацин, изофезолак, изоксепак лоназолак, метиазиновая кислота, оксаметацин, проглюметацин, сулиндак, тиарамид, толметин и зомепирак; производные арилмасляной кислоты, такие как бумадизон, бутибуфен, фенбуфен и ксенбуцин; арилкарбоновые кислоты, такие как клиданак, кеторолак и тиноридин; производные арилпропионовой кислоты, такие как алминопрофен, беноксапрофен, буклоксовая кислота; карпрофен, фенопрофен, флуноксапрофен, флурбипрофен, ибупрофен, ибупроксам, индопрофен, кетопрофен, локсопрофен, миропрофен, напроксен, оксапрозин, пикетопрофен, пирпрофен, пранопрофен, протизиновая кислота, супрофен и тиапрофеновая кислота; пиразолы, такие как дифенамизол и эпиризол; пиразолоны, такие как апазон, бензпиперилон, фепразон, мофебутазон, моразон, оксифенбутазон, фенибутазон, пипебузон, пропифеназон, рамифеназон, суксибузон и тиазолинобутазон; производные салициловой кислоты, такие как ацетаминосалол, аспирин, бенорилат, бромосалигенин, ацетилсалицилат кальция, дифлунизал, этерсалат, фендозал, гентизиновая кислота, гликольсалицилат, имидазола салицилат, ацетилсалицилат лизина, месаламин, морфолин салицилат, 1-нафтилсалицилат, ольсалазин, парсалмид, фенилацетилсалицилат, фенилсалицилат, салацетамид, салициламин, о-уксусная кислота, салицилсерная кислота, сальсалат и сульфасалазин; тиазинкарбоксамиды, такие как дроксикам, изоксикам, пироксикам и теноксикам; другие, такие как у-ацетамидокапроевая кислота, s-аденозилметионин, 3-амино-4-гидроксимасляная кислота, амиксетрин, бендазак, бензидамин, буколом, дифенпирамид, дитазол, эморфазон, гвайазулен, набуметон, нимесулид, орготеин, оксацепрол, паранилин, перизоксал, пифоксим, прогуазон, проксазол и тенидап; и их фармацевтчески приемлемые соли; и другие анальгетики, как, например, ацетаминофен. Доза анальгетика и/или жаропонижающего средства, такого как аспирин, ацетаминофен, и т.д., будет известна специалистам в данной области техники и может находиться в интервале от 80 мг до 250 мг. Доза НСПС будет известна специалистам в данной области техники и может находиться в интервале от 80 мг до 500 мг.
Примерные лекарственные формы, содержащие фенилэфрин в комбинации с лоратадином, описываются ниже.
Предпочтительным вариантом выполнения изобретения является лекарственная форма, содержащая пролонгированное высвобождение фенилэфрина или его фармацевтически приемлемой соли и немедленное высвобождение фенилэфрина или его фармацевтически приемлемой соли и лоратадина. Например, лекарственная форма содержит ядро таблетки продолжительного высвобождения, в котором компоненты содержат 22.5 мг гидрохлорида фенилэфрина, и активное покрытие немедленного высвобождения, инкапсулирующее ядро, компоненты которого содержат 7.5 мг гидрохлорида фенилэфрина и 5 мг лоратадина (Таблица 9). В дополнение к 22.5 мг гидрохлорида фенилэфрина, компоненты ядра таблетки содержат одно или более из следующего: гидроксипрлопилцеллюлоза, натрий-карбоксиметилцеллюлоза, микрокристаллическая целлюлоза и стеарат магния. Компоненты покрытия немедленного высвобождения, кроме того, содержат поливиниловый спирт в качестве полимерной матрицы, образующей пленку, с лоратадином и фенилэфрином или фармацевтически приемлемой солью фенилэфрина. Верхние покрытие может, кроме того, наносится в качестве защитного покрытия, компоненты которого содержат поливиниловый спирт и могут так же содержать краситель для внешнего вида.
Предпочтительным технологическим процессом для одного варианта выполнения изобретения, содержащего лекарственную форму в виде таблетки, такую, как показано в Таблице 9, является прямое прессование для обеспечения ядра и поддающегося эрозии слоя фенилэфрина на поверхности ядра. Для ядра таблетки с продолженным высвобождением гидрохлорид фенилэфрина сначала перемалывают через коническую мельницу лабораторного масштаба, применяя сито 20 меш. Гидроксипропилцеллюлозу klucel EXF, Гидроксипропилцеллюлозу klucel GXF, натрий-карбоксиметилцеллюлозу и одну третью часть микрокристаллической целлюлозы пропускают через ту же коническую мельницу. Перемолотые вещества затем смешивают в РK смесителе подходящего размера в течение 5 минут. Остаток микрокристаллической целлюлозы добавляют к смеси и перемешивают в течение дополнительных 5 минут. Стеарат магния просеивают через сито 30 меш, добавляют к смеси и перемешивают в течение дополнительных 3 минут. Порошкообразную смесь затем прессуют в ядра с продолженным высвобождением, применяя экстраглубокий чашечный круглый резец с резьбой 7/16''. Активный покрывающий слой с немедленным высвобождением, как подробно изложено ниже, является первым из двух слоев покрытия, следующих друг за другом, наносимых на ядро таблетки. Покрытие ядер с продолженным высвобождением может быть выполнено с помощью обычного оборудования для нанесения покрытия, так же как и в лабораторных условиях, с помощью обычного дражировочного котла (O'Hara Labcoat MX), или обычного оборудования промышленного масштаба, такого как Accela-Cota®. В случае как покрытия с немедленным высвобождением, так и покрытия с продолженным высвобождением, соответствующий избыток покрытия должен быть приготовлен. Обычно применяются смесительные сосуды с емкостью, составляющей приблизительно двойной конечный объем.
Активную покрывающую дисперсию приготавливают посредством сначала растворения Фенилэфрина HCl в воде, перемешивая при низкой поперечной силе. Полимер, основанный на поливиниловом спирте (например, Opadry™ II серии 85F (например, 18422 (Белый)), или привитой сополимер поливинилового спирта и полиэтиленгликоля (например, Kollicoat IR)) затем медленно добавляют в раствор, перемешивая, начиная при низкой и продолжая при умеренной поперечной силе, в течение, по меньшей мере, одного часа, или до тех пор, пока не перестанут существовать видимые скопления и/или агломерированные частицы. Затем в дисперсию добавляют лоратадин. Обычно лоратадину позволяют диспергироваться или включаться в дисперсию при постоянной умеренной поперечной силе до тех пор, пока лоратадин не перестает наблюдаться на поверхности дисперсии. Для того чтобы затем диспергировать агломерировавшие частицы лоратадина, требуется применение высокой поперечной силы и мешалки Ultra Turrax с частотой вращения 5000-6200 об/мин в течение 5 минут. Рециркуляционная система с применением высокой поперечной силы может так же применяться. Должна быть образована молочная, белая однородная суспензия с содержанием твердых веществ 11.5-23%. Этот интервал процентного содержания уже был оценен и успешно нанесен на сопоставимые таблетки. Отдельные частицы лекарственного средства лоратадин должны преобладать во всем объеме дисперсии на этой стадии. Затем эту дисперсию перед распылением необходимо перемешивать при низкой поперечной силе для дегазации, в течение по меньшей мере одного часа или до тех пор, пока первоначальный объем раствора не будет достигнут. Покрывающая дисперсия является однородной на этой стадии и на всем протяжении процесса нанесения покрытия. Нанесение покрытия продолжается до тех пор, пока желаемый привес не будет достигнут на основании измерений гравиметрического веса. Дисперсию синего цвета для завершающего покрытия готовят путем медленного добавления красителя Opadry II 85F99O01 (Синий) в воду, перемешивая, начиная при низкой и продолжая при умеренной поперечной силе, в течение, по меньшей мере, одного часа, или до тех пор, пока не перестанут существовать скопления и/или агломераты. Должна быть образована молочная, синяя однородная суспензия с содержанием твердых веществ приблизительно 18%. Нанесение покрытия продолжается до тех пор, пока желаемый привес не будет достигнут на основании измерений гравиметрического веса. Определение соответствующего оборудования, параметры технологического процесса и управление технологическим процессом должны быть организованы для применения этой покрывающей дисперсии, как описано выше. На таблетках ставят торговый знак с помощью Blue Opacode WB NS-78-10526 согласно стандартному методу нанесения отпечатка, так что на поверхности таблетки остается следовое количество. В еще одном варианте выполнения изобретения композиция содержит двухслойную таблетку с слоем немедленного высвобождения и слоем продолженного высвобождения. В одном предпочтительном варианте выполнения изобретения лекарственная форма в виде двухслойной таблетки спрессована вместе со слоем продолженного высвобождения, содержащим 22.5 мг гидрохлорида фенилэфрина, и слоем немедленного высвобождения, содержащим 7.5 мг гидрохлорида фенилэфрина и 5 мг лоратадина (Таблица 10). В дополнение к 22.5 мг гидрохлорида фенилэфрина слой с продолженным высвобождением содержит гидроксипропилцеллюлозу, натрий-карбоксиметилцеллюлозу, микрокристаллическую целлюлозу и стеарат магния. Слой с немедленным высвобождением содержит микрокристаллическую целлюлозу, коллоидный диоксид кремния, Кросповидон и стеарат магния, формирующие слой немедленного высвобождения с лоратадином и фенилэфрином или фармацевтически приемлемой солью фенилэфрина. Краситель может быть добавлен для улучшения внешнего вида или различения двух слоев.
В одном особенно предпочтительном варианте выполнения изобретения компонентами и количествами двухслойной композиции являются те, которые подробно описаны в таблице 10 в качестве примерной Лекарственной формы II. Предпочтительным технологическим процессом является процесс таблетирования путем прямого прессования, применяя двухслойный роторный таблеточный пресс. Предпочтительная композиция представляет собой двухслойную таблетку с двумя различными слоями таблетки; слоем продолжительного высвобождения и слоем немедленного высвобождения. Для слоя таблетки с продолженным высвобождением гидрохлорид фенилэфрина сначала измельчают с помощью конической мельницы лабораторного масштаба, применяя сито 20 меш. Гидроксипропилцеллюлозу Klucel EXF, Гидроксипропилцеллюлозу Klucel GXF, натрий-карбоксиметилцеллюлозу и одну третью часть микрокристаллической целлюлозы пропускают через ту же коническую мельницу. Перемолотые вещества затем смешивают в РK смесители соответствующего размера в течение 5 минут. Остаток микрокристаллической целлюлозы добавляют к смеси и перемешивают в течение дополнительных 5 минут. Стеарат магния просеивают вручную через сито 30 меш, добавляют к смеси и перемешивают в течение дополнительных 3 минут и помещают в контейнер, маркированный соответствующей этикеткой.
Для слоя таблетки с немедленным высвобождением гидрохлорид фенилэфрина и лоратадин сначала измельчают с помощью конической мельницы лабораторного масштаба, применяя сито 20 меш. Синий органический пигмент №1, коллоидный диоксид кремния, кросповидон и одну третью часть микрокристаллической целлюлозы пропускают через ту же коническую мельницу. Перемолотые вещества затем смешивают в РК смесители соответствующего размера в течение 5 минут. Остаток микрокристаллической целлюлозы добавляют к смеси и перемешивают в течение дополнительных 5 минут. Стеарат магния просеивают вручную через сито 30 меш, добавляют к смеси и перемешивают в течение дополнительных 3 минут. Обе порошковые смеси, смесь продолженного высвобождения и смесь немедленного высвобождения затем спрессовывают в двухслойные таблетки с помощью двухслойного роторного таблеточного пресса, применяя экстраглубокий чашечный круглый резец с резьбой 7/16''.
Общий процесс производства лекарственных форм
Другим объектом изобретения является процесс изготовления лекарственных форм, описанных выше. Твердые лекарственные формы получают, применяя способы, в общем известные в данной области техники для приготовления многослойных лекарственных форм. Лекарственные формы находятся в форме таблеток, капсул, гелевых капсул или жидких капсул, среди прочего. Исследования стабильности и деградации могут быть выполнены в соответствии со стандартами Международной Конференцией по Гармонизации (ICH), как описано в руководящих указаниях «Примеси в новых лекарственных веществах», для воспроизведения двух и более лет срока хранения. Например, анализ стабильности может проводиться при 40 градусах Цельсия / 75%-ной относительной влажности в течение 3-месячного периода. Стандартные фармацевтические условия хранения известны в данной области техники. Композиции по изобретению могут быть оценены с удовлетворением всех руководящих указаний ICH для активного фармацевтического анализа со степенями разложения, которые ниже отчетных границ, предпочтительно ниже идентификационных границ, наиболее предпочтительно ниже квалификационных границ.
Способы лечения и введения
Другими объектами изобретения являются способы лечения симптомов простуды, гриппа или аллергии у субъекта, нуждающегося в этом, содержащие введение фармацевтических композиций, описанных здесь. В определенных вариантах выполнения изобретения способы содержат введение фармацевтической композиции каждые 8, 12, 16 или 24 часа. Другим объектом изобретения является способ доставки, по меньшей мере, части фенилэфрина или его фармацевтически приемлемой соли в ободочную кишку субъекта, содержащий введение лекарственной формы или композиции изобретения вышеупомянутому субъекту. Композициями, полезными для способов, являются пероральные лекарственные формы или фармацевтические композиции изобретения, как описано выше. В определенных вариантах выполнения изобретения таким способом является способ, в котором несвязанный фенилэфрин присутствует в плазме субъекта, по меньшей мере, на момент 5 часов после введения и более предпочтительно в промежутке от около 6 до около 12 часов после введения. В определенных вариантах выполнения изобретения таким способом является способ, в котором максимальная концентрация несвязанного фенилэфрина в плазме субъекта достигается в промежутке от около 5 до около 24 часов после введения и более предпочтительно в промежутке от около 6 до около 12 часов после введения. Способ может осуществляться для любого субъекта, в настоящее время считается подходящим для введения фенилэфрина и любого дополнительного фармацевтического агента.
В другом объекте изобретения способами изобретения являются способы для поддержания стабильной биодоступности фенилэфрина в несвязанной терапевтически активной форме, содержащие введение путем перорального введения фармацевтической композиции, описанной здесь, где, по меньшей мере, часть фенилэфрина абсорбируется через ободочную кишку. В определенных вариантах выполнения изобретения несвязанный фенилэфрин биодоступен в плазме в течение более чем 4, 6, 8, 12, 16 или 24 часов после введения. С помощью термина "биодоступный" обозначают, что активная форма фенилэфрина поддается количественному определению в плазме субъекта, и предпочтительно при более чем 0.1 нг/мл, и даже при 0.2, 0.3, 0.4, 0.5, 1.0 или 2.5 нг/мл. Это также является объектом данного изобретения, что концентрация несвязанного фенилэфрина в плазме увеличивается после более чем около получаса от момента введения композиции изобретения, и максимальная концентрация в плазме несвязанного фенилэфрина достигается через 1, 2, 4, 6, 8 или 12 часов после введения фармацевтической композиции.
Кроме того, объектом изобретения является способ введения фенилэфрина путем перорального введения, в котором сохраняется более высокая доля несвязанной формы фенилэфрина по сравнению с обычным введением пероральной лекарственной формы с немедленным высвобождением, приводящий к улучшенной биодоступности и увеличенной эффективности введения фенилэфрина. В определенных вариантах выполнения изобретения способы включают введение фармацевтической композиции изобретения без каких-либо частей немедленного высвобождения, содержащих фенилэфрин, где максимальная концентрация несвязанного фенилэфрина в плазме составляет, по меньшей мере, около 1% от максимальной концентрации в плазме всего фенилэфрина. В определенных вариантах выполнения изобретения процентное содержание составляет, по меньшей мере, 2, 3, 4, 5, 6 или 7%. Для сравнения, максимальная концентрация в плазме несвязанного фенилэфрина составляет менее чем 0.7% от максимальной концентрации всего фенилэфрина в плазме при введении в виде лекарственной формы немедленного высвобождения путем перорального введения. Понятно, что даже при введении фармацевтической композиции изобретения, кроме того, содержащей часть с немедленным высвобождением фенилэфрина, процент активного фенилэфрина в плазме является аддитивным и поэтому всегда более высоким, чем если все количество фенилэфрина было перорально введено в форме немедленного высвобождения.
В другом объекте способы изобретения приводят к более высоким относительным AUC0-t и AUC0-∞ для несвязанного фенилэфрина, отнесенного к общему фенилэфрину в плазме субъекта, которому был введен фенилэфрин. AUC0-24 - это параметр, показывающий кумулятивную концентрацию в плазме в течение 24 после введения дозы, определяемую как площадь под кривой «концентрация-время» в диапазоне доз или активацию к моменту 24 после введения дозы; AUC0-∞ - это параметр, показывающий общую кумулятивную концентрацию в плазме по прошествии длительного времени, определяемую как вычисленная площадь под кривой «концентрация-время» в диапазоне доз или активацию к моменту времени, равному бесконечности. В определенных вариантах выполнения изобретения способы изобретения содержат введения фармацевтической композиции изобретения субъекту, в которых значение AUC0-24 для несвязанного фенилэфрина в плазме субъекта составляет, по меньшей мере, 1% от значения AUC0-24 для всего фенилэфрина в плазме субъекта. В предпочтительном варианте выполнения изобретения относительная величина AUC0-24 для несвязанного фенилэфрина составляет, по меньшей мере, 2, 3, 4, 5, 6 или 7% от значения AUC0-24 для всего фенилэфрина.
Другим объектом изобретения является способ введения фенилэфрина субъекту, содержащий пероральное введение фармацевтической композиции для доставки в ободочную кишку субъекта, содержащей фенилэфрин, где фенилэфрин высвобождается в ободочной кишке, таким образом, достигая минимальной пресистемной трансформации фенилэфрина. С помощью термина "минимальный" определяют, что трансформация значительно меньше, чем такая трансформация, обнаруживаемая при пероральном введении обычной, доступной в настоящее время композиции немедленного высвобождения, содержащей фенилэфрин или его фармацевтически приемлемую соль.
Необходимо так же понимать, что специфическая схема приема и лечения для любого конкретно пациента будет зависеть от многообразия факторов. Такие факторы включают возраст, вес тела, общее состояние здоровья, пол, питание, комбинацию лекарственных средств и оценку лечащего врача и серьезность конкретного состояния, подлежащего лечению. Для любого дополнительного терапевтического агента доза так же будет зависеть от того, какое конкретное соединение (соединения) находится (находятся) в композиции, от активности конкретного применяемого соединения (соединений), биодоступности соединения (соединений) и скорости выделения и химического разложения. Точная дозировка, подходящая для субъекта, может быть легко определена, в качестве установившейся практики, специалистом в данной области техники, например врачом или фармацевтом.
Содержание любых патентов, заявок на патент, публикаций патентов или научных статей, упоминаемых где-либо в этой заявке, включены здесь путем ссылки на их полное содержание.
Следующие экспериментальные примеры предназначаются для иллюстрации раскрытого изобретения. Примеры являются неограничивающими, и специалист определит, что другие варианты выполнения изобретения включены в объем раскрытого изобретения.
Экспериментальные примеры
Пример 1. Биодоступность фенилэфрина
Следующее исследование было проведено, чтобы изучить протекание абсорбции фенилэфрина в ЖК тракте. Субъектам, а именно здоровому мужчине и здоровой женщине, не являющейся беременной и не вскармливающей грудью, ввели дозу, содержащую 10 мг гидрохлорида фенилэфрина, доставляемого в ободочную кишку в капсулах Enterion™ (режим А - 9 субъектов), 10 мг Sudafed РЕ™ (режим В - 8 субъектов) или 30 мг гидрохлорида фенилэфрина, доставляемого в ободочную кишку в капсулах Enterion™ (режим С - 8 субъектов), все вводилось перорально, после воздержания от пищи в течение ночи. Капсулы Enterion™ (Pharmaceutical Profiles Ltd, UK) учитывают доставку лекарственного средства при желательном расположении внутри желудочно-кишечного тракта. Для того чтобы определить, достигли ли капсулы желательного места, препараты режимов А и С водились перорально с 210 мл воды, сопровождающейся радиоактивномеченным напитком, содержащим 4 МБк 99м-Тс-диэтилентриаминпентауксусной кислоты (DTPA) в 30 мл воды, так что расположение проглоченных веществ могло бы быть визуализировано с помощью сцинтиграфического изображения. Капсулы Enterion™ были успешно активированы, и фенилэфрин высвободился у 8 из 9 субъектом в режиме А и 6 из 8 субъектов в режиме С. Концентрации фенилэфрина в плазме, как в несвязанной активной форме, так и в связанной неактивной форме, определили путем взятия образцов венозной крови через 0, 5, 15, 30, 45 минут и 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 14, 16 и 24 часа после активации капсулы или введения дозы. Образцы крови собирали в пробирки, содержащие гепарин лития, центрифугировали в течение 30 минут при 1500g, 15 минут при 4°С. Плазму исследовали на наличие фенилэфрина в его связанной и несвязанной формах. Количество связанного фенилэфрина вычислили с помощью вычитания оцененного количества несвязанного фенилэфрина из оцененного количества всего фенилэфрина.
Значения концентрации в плазме всего, так же как и несвязанного фенилэфрина, подробно изложенные в Таблице 11, приведенной ниже, проиллюстрированы на Фиг.6. В частности, Фиг.6А показывает полулогарифмический график, и Фиг.6В показывает линейный график. Хотя, представленные как на Фиг.6А, так и на Фиг.6В графики для несвязанного фенилэфрина, как правило, накладываются на графики для общего фенилэфрина и поэтому их трудно различить. Сходство между общим и связанным фенилэфрином очевидно из Фиг.6С, на котором нанесены значения Сmax для общего, несвязанного и связанного фенилэфрина, доведенного до уровня дозы 1 мг, после доставки 10 мг и 30 мг фенилэфрина в ободочную кишку. Как показано на Фиг.6А, график для несвязанного фенилэфрина, доставленного Sudafed РЕ™, быстро достигает максимума и затем быстро спускается вниз до уровня ниже 0.1 нг/мл в течение первых 4 часов после введения дозы. Напротив, графики для несвязанного фенилэфрина, доставленного в ободочную кишку, как для 10 мг дозы, так и для 30 мг дозы, достигают максимума и спускаются вниз более плавно и объективно являются более постоянными в течение продолжительного периода времени.
Данные были проанализированы, применяя стандартный фармакокинетический способ анализа, для следующих параметров: Cmax, максимальная наблюдаемая концентрация в плазме; Tmax, время достижения Cmax; Tlag, момент, когда лекарственное средство стало поддаваться количественному определению в плазме; AUC0-24, площадь под кривой «концентрация-время» в диапазоне доз или активация к моменту 24 часов после принятия внутрь; AUC0-∞, площадь под кривой «концентрация-время» в диапазоне доз или активация к моменту времени, равному бесконечности; t1/2, конечный элиминационный период полувыведения. Относительная биодоступность, показанная в таблице как Frel окончательный, показывает биодоступность фенилэфрина введенного в ободочную кишку по отношению к фенилэфрину, введенному в виде пероральной лекарственной формы немедленного высвобождения. Результаты показаны в Таблицах 11, 12 и 13. Кроме того, профили концентрации в плазме общего, так же как и несвязанного фенилэфрина, представлены графически на Фиг.6.
Концентрация в плазме лекарственной формы немедленного высвобождения, содержащей фенилэфрин, быстро достигает максимума через около 1.5 часа после перорального введения и затем падает до половины максимальной концентрации за 2.5 часа (смотрите Таблицы 11, 12 и 13). Для сравнения фенилэфрин, доставленный в ободочную кишку и абсорбировавшийся из ободочной кишки, достигает его максимальной концентрации через около 6 часов (10 мг) или 8 часов (30 мг), медленно уменьшаясь в концентрации с достижением половины максимальной концентрации за около 12 часов (10 мг).
Кроме того, когда введение осуществляли в виде формы немедленного высвобождения, активная несвязанная форма составляла только очень маленькую часть концентрации фенилэфрина в плазме, который быстро достигал его максимальной концентрации в плазме и удалялся. Процентное содержание активного несвязанного фенилэфрина по сравнению с общим фенилэфрином, который присутствовал в плазме по прошествии времени, может быть определено путем деления AUC0-∞ для несвязанного фенилэфрина на AUC0-∞ для всего фенилэфрина. Когда 10 мг фенилэфрина было введено в форме немедленного высвобождения, активная несвязанная форма фенилэфрина составляла не более чем около 0.5% от всей концентрации фенилэфрина, обнаруживаемой в плазме со временем. Для сравнения, когда 10 мг фенилэфрина было введено путем высвобождения в ободочной кишке, активная несвязанная форма фенилэфрина составляла около 2.2% от всей концентрации фенилэфрина, обнаруживаемой в плазме со временем. Около 5% всего фенилэфрина было доступно в активной форме при введении 30 мг фенилэфрина путем высвобождения в ободочной кишке.
Введение таблетки с немедленным высвобождением, Sudafed РЕ™, приводит к быстрой абсорбции фенилэфрина. Значение Tmax находилось в пределах от 0.25 до 0.75 часов после введения дозы, и значение вылечены Cmax составляло 0.704±0.369 нг/мл. Величина AUC0-24, показывающая общее количество лекарственного средства, находящегося в плазме за время от момента введения дозы и до 24 часов после введения дозы, для несвязанного фенилэфрина составляла 0.561±0.165 (нг*час/мл) и терминальный период полураспада находился в пределах от 0.323 до 3.52 часов. Эти данные для несвязанного фенилэфрина хорошо согласуются с опубликованной информацией по фармакокинетике фенилэфрина.
Доставка 10 мг гидрохлорида фенилэфрина в ободочную кишку привела к уменьшенному Cmax и пролонгированному Tmax для несвязанного фенилэфрина по сравнению с лекарственной формой немедленного высвобождения. Однако величина AUC0-24 для несвязанного фенилэфрина была выше, чем эта величина для Sudafed РЕ™. Значение Tmax изменялось и находилось в пределах от 0.25 до 3 часов. Значение величины Cmax составляло 0.400±0.454 нг/мл, и значение величины AUC0-24 составляло 1.58±0.915 (нг*час/мл). Терминальный период полураспада находился в пределах от 2.91 до 13.5 часов, что больше, чем значения, сообщенные для таблетки с немедленным высвобождением. Данные подтверждают, что абсорбция в ободочной кишке была стадией, ограничивающей скорость элиминации, и что конечная фаза, таким образом, показывает продолжительную абсорбцию.
Так же как доставка 10 мг гидрохлорида фенилэфрина в ободочную кишку, доставка 30 мг гидрохлорида фенилэфрина приводит к пролонгированной абсорбции с пролонгированным Tmax и уменьшенным Cmax для несвязанного фенилэфрина. Tmax находился в пределах от 0.75 до 6.03 часов, и значение вылечены Cmax составляло 0.468±0.174 нг/мл. Значение величины AUC0-24 составляло 5.17±1.56 (нг*час/мл), и значение относительной биодоступности составляло 333±140% (в пределах от 213 до 544%) по сравнению с биодоступностью, измеренной для перорального введения фенилэфрина в форме с немедленным высвобождением. Терминальный период полураспада находился в пределах от 2.98 до 18.9 часов.
Результаты неожиданно показали, что абсорбция фенилэфрина в ободочной кишке демонстрирует более желательную стабильную концентрацию в плазме, по сравнению с пероральным введением фенилэфрина в форме немедленного высвобождения, и что более высокая часть фенилэфрина остается в несвязанной активной форме. При абсорбции в ободочной кишке, однако, отсутствует первоначальный всплеск, который дает пероральное введение формы с немедленным высвобождением.
Пример 2. Примерные лекарственные формы A-G были сделаны и профили растворения были определены.
Профили растворения примерных лекарственных форм A-G (подробно изложены в Таблице 2) были исследованы в течение периода времени 14 часов, применяя прибор для определения растворения USPI, установленный на 75 об/мин. Изучение растворения проводили при 37°С±0.5°С с 900 мл воды, буферизованной с помощью 0.5 мМ фосфатного буфера с рН 7.4. Через каждый интервал времени образец раствора анализировали с помощью ЖХВД (жидкостная хроматография высокого давления) при 215 нм для того, чтобы определить процент растворенного фенилэфрина. Результаты растворения, которые представляют собой среднее значение за 3 измерения, показаны в Таблице 15. Профиль растворения так же представлен на Фиг.3.
Пример 3. Примерная лекарственная форма, подробно изложенная в Таблице 9, была сделана и профили растворения были определены.
Профиль высвобождения фенилэфрина из таблетки, соответствующей примеру, подробно описанному в Таблице 9, изучали в течение периода времени 12 часов, применяя прибор для определения растворения USP II, стартующий при 50 оборотах в минуту. Изучение растворения проводили с помощью 750 мл имитированной жидкой желудочной среды TS USP (без ферментов) в течение первого часа. Через час значение рН среды подняли до рН 6.8 засчет добавления 250 мл 2.0М раствора трифосфата натрия, и изучение растворения проводили в течение дополнительных 11 часов. Через каждый интервал времени образец раствора анализировали для определения процентного содержания растворенного фенилэфрина. Фиг.4 приводит данные графическим образом.
Пример 4. Примерная лекарственная форма, подробно изложенная в Таблице 10, была сделана и профили растворения были определены.
Профиль высвобождения фенилэфрина из двухслойной таблетки, соответствующей примерной лекарственной форме I, подробно изложенной в Таблице 10, изучали в течение периода времени, равного 12 часам. Профиль высвобождения фенилэфрина из таблетки, соответствующей примеру, изучали за период времени, равный 12 часам, применяя прибор для определения растворения USP II, стартующий при 50 оборотах в минуту. Изучение растворения проводили с помощью 750 мл имитированной жидкой желудочной среды TS USP (без ферментов) в течение первого часа. Через час значение рН среды подняли до рН 6.8 засчет добавления 250 мл 2.0М раствора трифосфата натрия, и изучение растворения проводили в течение дополнительных 11 часов. Через каждый интервал времени образец раствора анализировали для определения процентного содержания растворенного фенилэфрина. Фиг.5 иллюстрирует профиль растворения, на основании среднего по 6 измерениям.
Пример 5. Фармакокинетическая модель абсорбции фенилэфрина в желудочно-кишечном тракте.
Для того чтобы помочь понять, как доза, скорость высвобождения и область абсорбции в желудочно-кишечном тракте влияют на уровни концентрации в плазме, разработали фармакокинетическую модель, путем введения избирательного поглощения вдоль желудочно-кишечного тракта и кинетики высвобождения лекарственного средства как входной функции, с целью прогнозирования концентраций несвязанного фенилэфрина в плазме. Предпочтительные интервалы для части фенилэфрина, доставляемой путем немедленного высвобождения (IR) в верхние отделы желудочно-кишечного тракта, путем пролонгированного высвобождения (SR) в верхние отделы желудочно-кишечного тракта, и/или путем доставки в ободочную кишку для обоих вариантов дозы, то есть как для 30 мг дозы, так и для 60 мг дозы фенилэфрина (РЕ), подытожены в Таблице 16.
Результаты симуляций для вариантов 10 мг дозы фенилэфрина с немедленным высвобождением, так же как и 30 мг дозы фенилэфрина, перекрывающих диапазоны, перечисленные в Таблице 16, представлены в Таблице 17, так же как и графически на Фиг.7. На основании результатов этих симуляций предпочтительный вариант выполнения изобретения для 30 мг дозы фенилэфрина обеспечивает следующий профиль доставки: около 7.5 мг фенилэфрина, доставляемого в форме с немедленным высвобождением в верхние отделы желудочно-кишечного тракта, и оставшаяся часть фенилэфрина, доставляемая в форме с пролонгированным высвобождением в верхние отделы желудочно-кишечного тракта и/или ободочную кишку (например, около 17.5 мг фенилэфрина в верхние отделы желудочно-кишечного тракта и около 5 мг фенилэфрина, доставленного в ободочную кишку).
b: Ссылкой является 10 мг таблетка Sudafed РЕ, принимаемая внутрь каждые 4 часа в течение 12 часов
Результаты симуляций для вариантов 10 мг дозы фенилэфрина с немедленным высвобождением, так же как и 60 мг дозы фенилэфрина, перекрывающих диапазоны, перечисленные в Таблице 16, представлены в Таблице 18, а так же графически на Фиг.8. На основании результатов этих симуляций предпочтительный вариант выполнения изобретения для 60 мг дозы фенилэфрина обеспечивает следующий профиль доставки: около 10 мг фенилэфрина, доставляемого в форме с немедленным высвобождением в верхние отделы желудочно-кишечного тракта, и оставшаяся часть фенилэфрина, доставляемая в форме с пролонгированным высвобождением в верхние отделы желудочно-кишечного тракта и/или ободочную кишку (например, около 20 мг фенилэфрина в верхние отделы желудочно-кишечного тракта и около 30 мг фенилэфрина, доставляемого в ободочную кишку).
b: Ссылкой является 10 мг таблетка Sudafed РЕ, принимаемая внутрь каждые 4 часа в течение 24 часов
Для вариантов выполнения изобретения, описанных здесь, ожидают, что композиции, содержащие фенилэфрин или его фармацевтически приемлемую соль в описанном ядре, слое (слоях) и покрытие, показывают профили концентрации в плазме, такие, как показано на Фиг.7 и Фиг.8. Поэтому данный, специально изучивший это описание изобретения специалист в данной области техники может осуществить на практике это изобретение, без чрезмерного экспериментирования, с достижением желаемого профиля дозы.
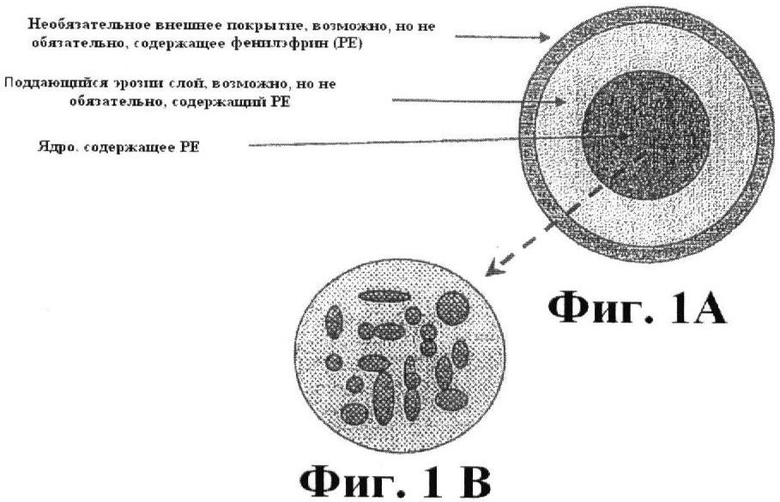
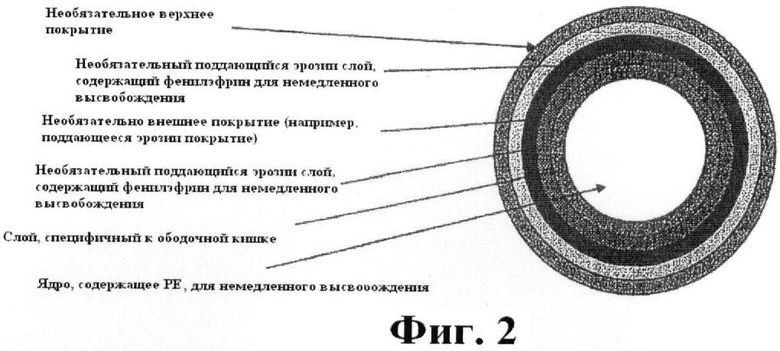
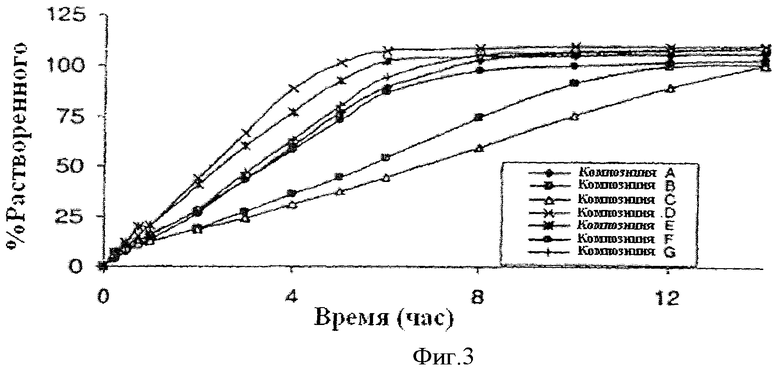
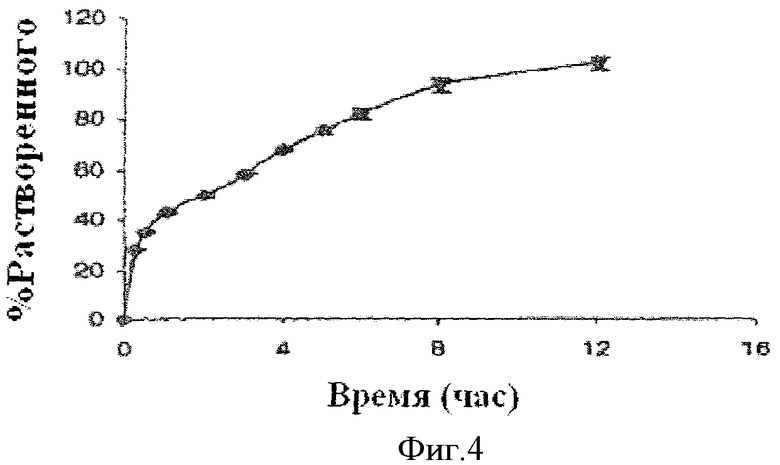
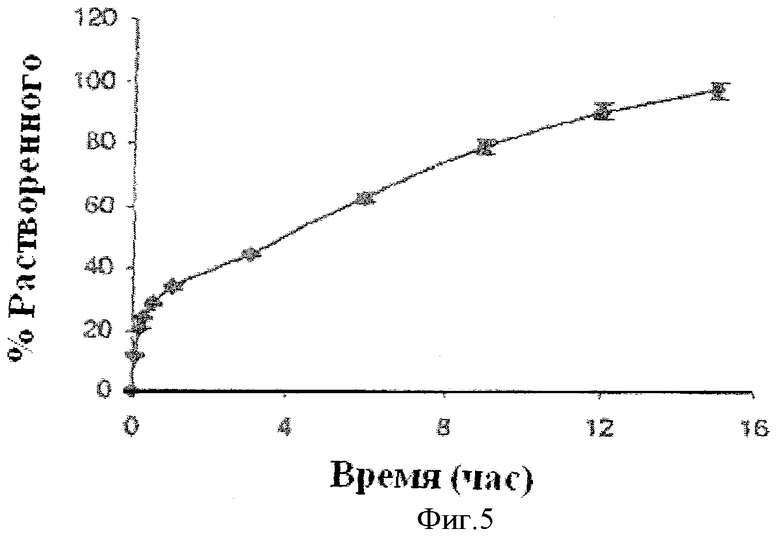
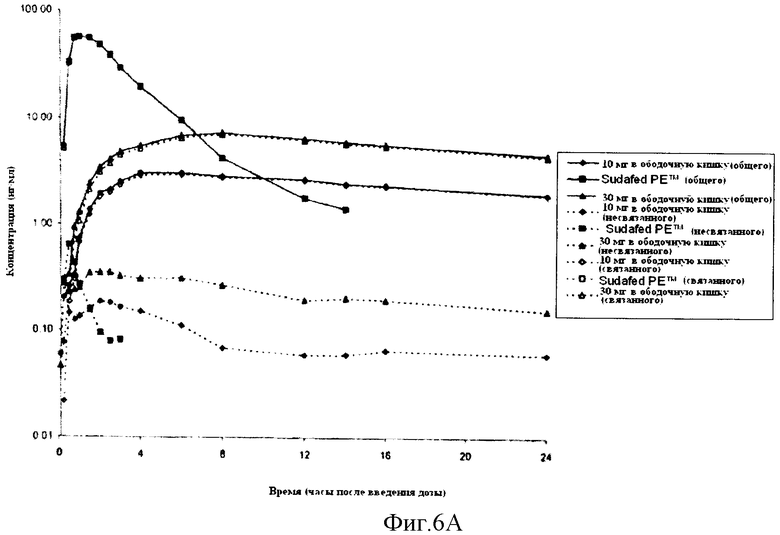
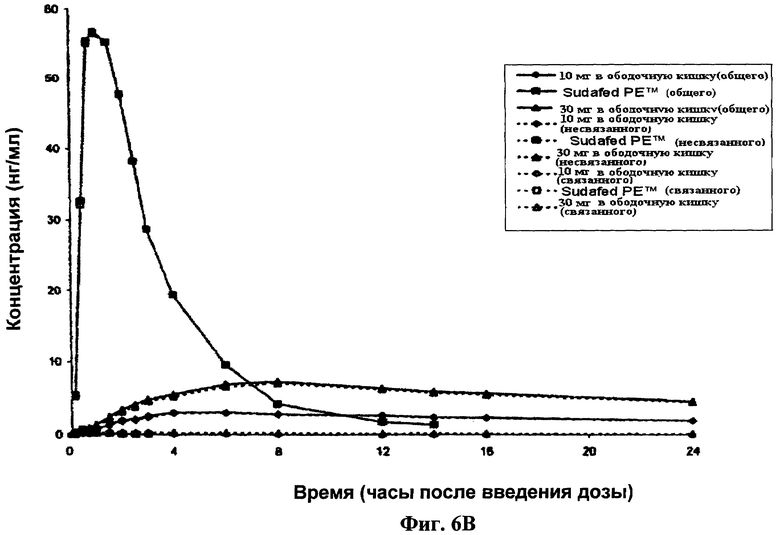
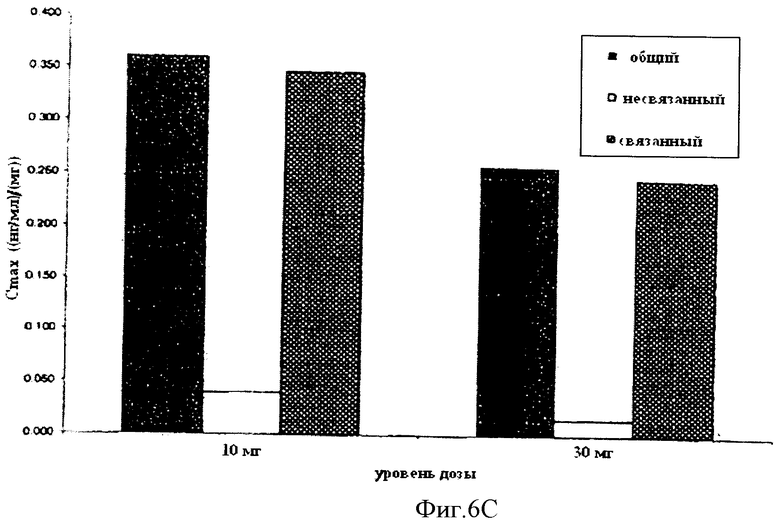
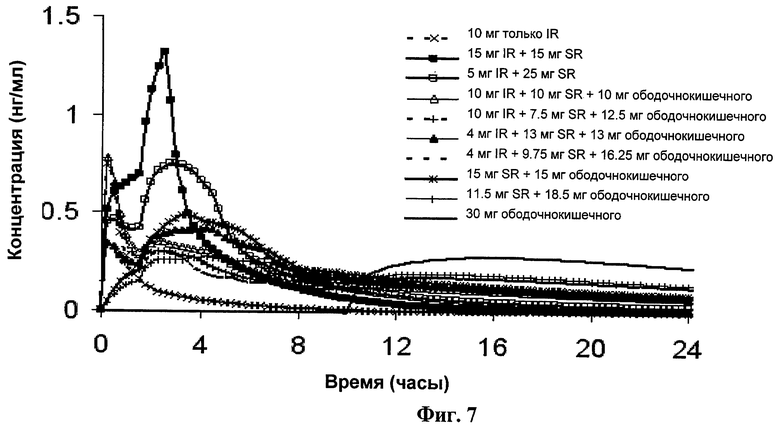
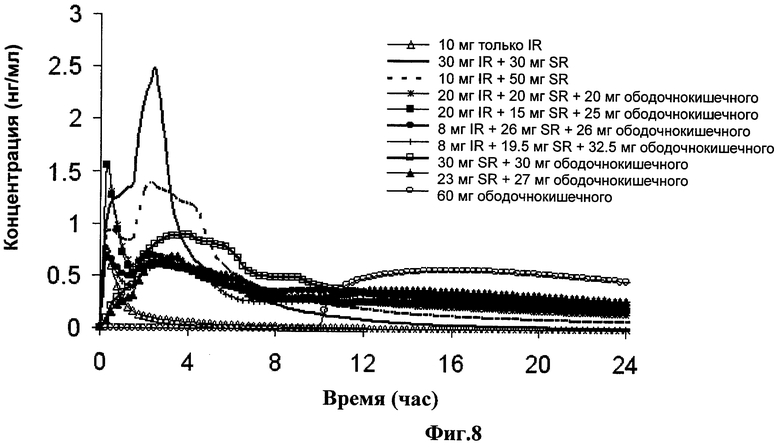
Изобретение раскрывает фармацевтическую композицию, подходящую для перорального введения. Фармацевтическая композиция содержит ядро, содержащее полимерную матрицу контролированного высвобождения, включающую гидроксипропилцеллюлозу и натриевую соль карбоксиметилцеллюлозы, и фенилэфрин или его фармацевтически приемлемую соль, и поддающийся эрозии слой, необязательно, содержащий фенилэфрин или его фармацевтически приемлемую соль. При приеме субъектом композиции внутрь полимерная матрица контролированного высвобождения из указанных компонентов обеспечивает нулевой или близкий к нулю порядок высвобождения фенилэфрина или его фармацевтически приемлемой соли, и по меньшей мере, часть фенилэфрина абсорбируется в ободочной кишке субъекта. Фармацевтическая композиция может, кроме того, содержать один или более дополнительных терапевтически активных агентов, выбранных из группы, состоящей из антигистамина, анальгетика, жаропонижающего средства и нестероидного противовоспалительного средства. Изобретение обеспечивает улучшенную биодоступность фенилэфрина и приводит к продолжительным концентрациям в плазме активного фенилэфрина при однократном болюсном введении препарата. 2 н. и 83 з.п. ф-лы, 8 ил., 18 табл.
1. Фармацевтическая композиция, подходящая для перорального введения, где композиция содержит:
a) ядро, содержащее полимерную матрицу контролированного высвобождения, включающую гидроксипропилцеллюлозу и натриевую соль карбоксиметилцеллюлозы, и фенилэфрин или его фармацевтически приемлемую соль; и
b) поддающийся эрозии слой, необязательно, содержащий фенилэфрин или его фармацевтически приемлемую соль;
где при приеме субъектом композиции внутрь полимерная матрица контролированного высвобождения обеспечивает нулевой или близкий к нулю порядок высвобождения фенилэфрина или его фармацевтически приемлемой соли и, по меньшей мере, часть фенилэфрина абсорбируется в ободочной кишке субъекта.
2. Фармацевтическая композиция по п.1, где по меньшей мере 5 мас.% фенилэфрина или его фармацевтически приемлемой соли абсорбируется в ободочной кишке субъекта.
3. Фармацевтическая композиция по п.1, где ядро содержит одну или более гранул, содержащих фенилэфрин или его фармацевтически приемлемую соль.
4. Фармацевтическая композиция по п.3, где одна или более гранул имеют поддающееся эрозии покрытие гранулы на поверхности вышеупомянутой гранулы (гранул).
5. Фармацевтическая композиция по п.4, где поддающееся эрозии покрытие гранулы содержит рН-чувствительный полимер или полимер, специфичный к ободочной кишке.
6. Фармацевтическая композиция по п.1, где ядро дополнительно содержит вещество, способствующее всасыванию.
7. Фармацевтическая композиция по п.1, где ядро обнажается посредством разрушения поддающегося эрозии слоя, по меньшей мере, через 5 ч после принятия субъектом.
8. Фармацевтическая композиция по п.7, где ядро обнажается посредством разрушения поддающегося эрозии слоя, по меньшей мере, через 8 ч после принятия субъектом.
9. Фармацевтическая композиция по п.1, где поддающийся эрозии слой содержит смесь гидроксипропилметилцеллюлозы, поливинилового спирта или привитого сополимера поливинилового спирта и полиэтиленгликоля и карбоксиметилцеллюлозы или натриевой соли карбоксиметилцеллюлозы.
10. Фармацевтическая композиция по п.9, где поддающийся эрозии слой дополнительно содержит микрокристаллическую целлюлозу.
11. Фармацевтическая композиция по п.1, где поддающийся эрозии слой частично или полностью инкапсулирует ядро.
12. Фармацевтическая композиция по п.1, дополнительно содержащая внешнее покрытие.
13. Фармацевтическая композиция по п.12, где внешнее покрытие содержит фенилэфрин или его фармацевтически приемлемую соль.
14. Фармацевтическая композиция по п.12, где внешним покрытием является внешняя оболочка.
15. Фармацевтическая композиция по п.12, где внешняя оболочка содержит фенилэфрин или его фармацевтически приемлемую соль и поливиниловый спирт.
16. Фармацевтическая композиция по п.12, где внешняя оболочка содержит фенилэфрин или его фармацевтически приемлемую соль и привитой сополимер поливинилового спирта и полиэтиленгликоля.
17. Фармацевтическая композиция по п.12, где внешняя оболочка содержит 1-2% (в/в) фенилэфрина или его фармацевтически приемлемой соли; 6-10% (в/в) полимера, основанного на поливиниловом спирте; и необязательно 0.9-1% (в/в) лоратадина.
18. Композиция по п.1, дополнительно содержащая один или более дополнительных терапевтических агентов.
19. Композиция по п.3, дополнительно содержащая один или более дополнительных терапевтических агентов.
20. Композиция по п.12, дополнительно содержащая один или более дополнительных терапевтических агентов.
21. Композиция по п.20, где один или более дополнительных терапевтических агентов находятся в ядре, поддающемся эрозии слое, внешней оболочке или любой комбинации двух или более из них.
22. Фармацевтическая композиция по п.1, где поддающийся эрозии слой представляет собой слой, специфичный к ободочной кишке;
причем при приеме субъектом композиции внутрь слой, специфичный к ободочной кишке, разрушается посредством действия специфичного для ободочной кишки условия в организме субъекта.
23. Фармацевтическая композиция по п.22, где специфичным для ободочной кишки условием является специфичный для ободочной кишки фермент.
24. Фармацевтическая композиция по п.23, где слой, специфичный к ободочной кишке, содержит один или более полимеров, выбранных из одного или более членов группы, состоящей из: N,N-диметилакриламида, N-трет-бутилакриламида, акриловой кислоты, поперечно сшитой с азоароматическими соединениями, хитозана, шеллака, горохового крахмала, амилозы, смеси этилцеллюлозы и амилозы и комбинации пектина, хитозана и гидроксипропилметилцеллюлозы.
25. Фармацевтическая композиция по п.22, где слой, специфичный к ободочной кишке, инкапсулирует ядро.
26. Композиция по п.22, дополнительно содержащая дополнительный поддающийся эрозии слой, необязательно содержащий фенилэфрин или его фармацевтически приемлемую соль.
27. Композиция по п.22 или 26, дополнительно содержащая внешнее покрытие.
28. Фармацевтическая композиция по п.27, где внешним покрытием является энтеральное покрытие.
29. Композиция по п.26, где поддающийся эрозии слой инкапсулирует слой, специфичный к ободочной кишке.
30. Фармацевтическая композиция по п.1, где поддающийся эрозии слой представляет собой рН-чувствительный слой, причем рН-чувствительный слой разрушается до обнажения ядра, по меньшей мере, через 5 ч после принятия субъектом композиции внутрь и причем рН-чувствительный слой полностью разрушается при рН в ободочной кишке.
31. Композиция по п.30, где значение рН составляет от около 5.5 до около 7.0.
32. Композиция по п.31, где значение рН составляет от около 5.7 до около 6.8.
33. Композиция по п.30, дополнительно содержащая дополнительный поддающийся эрозии слой, инкапсулирующий рН-чувствительный слой и ядро, причем дополнительный поддающийся эрозии слой необязательно содержит фенилэфрин или его фармацевтически приемлемую соль.
34. Композиция по п.30 или 33, дополнительно содержащая внешнее покрытие.
35. Фармацевтическая композиция по п.34, где внешним покрытием является энтеральное покрытие.
36. Композиция по п.30, где рН-чувствительный слой инкапсулирует ядро.
37. Фармацевтическая композиция по п.1, или 22, или 30, дополнительно содержащая верхнее покрытие.
38. Композиция по п.1, или 3, или 22 или 30, дополнительно содержащая один или более дополнительных терапевтических агентов.
39. Композиция по п.38, где один или более дополнительных терапевтических агентов выбираются из одного или более членов группы, состоящей из антигистамина, анальгетика, жаропонижающего средства и нестероидного противовоспалительного средства.
40. Композиция по п.38, где один или более дополнительный(ые) терапевтический(ие) агент(ы) находится(ятся) в составе формы немедленного высвобождения.
41. Композиция по п.38, где одним или более дополнительным терапевтическим агентом является лоратадин или деслоратадин в форме немедленного высвобождения.
42. Композиция по п.18, где один или более дополнительный(ые) терапевтический(ие) агент(ы) находится(ятся) в составе поддающегося эрозии слоя.
43. Композиция по п.19, где один или более дополнительный(ые) терапевтический(ие) агент(ы) находится(ятся) в составе поддающегося эрозии слоя.
44. Композиция по п.20, где один или более дополнительный(ые) терапевтический(ие) агент(ы) находится(ятся) в составе поддающегося эрозии слоя.
45. Композиция по п.21, где один или более дополнительный(ые) терапевтический(ие) агент(ы) находится(ятся) в составе поддающегося эрозии слоя.
46. Композиция по п.38, где композиция разработана в виде капсулы или таблетки.
47. Композиция по п.38, где один или более дополнительный(ые) терапевтический(ие) агент(ы) находится(ятся) в составе формы пролонгированного высвобождения.
48. Фармацевтическая композиция по любому из пп.1-47, подходящая для перорального введения, для доставки фенилэфрина или его фармацевтически приемлемой соли в однократной дозе в организм субъекта, нуждающегося в этом, причем неметаболизированный фенилэфрин обнаруживается в плазме вышеупомянутого субъекта в течение, по меньшей мере, 5 ч после принятия субъектом композиции внутрь.
49. Фармацевтическая композиция по п.48, где неметаболизированный фенилэфрин обнаруживается в плазме вышеупомянутого субъекта в течение, по меньшей мере, 6 ч после принятия субъектом композиции внутрь.
50. Фармацевтическая композиция по п.48, где неметаболизированный фенилэфрин обнаруживается в плазме вышеупомянутого субъекта в течение, по меньшей мере, 8 ч после принятия субъектом композиции внутрь.
51. Фармацевтическая композиция по п.48, где неметаболизированный фенилэфрин обнаруживается в плазме вышеупомянутого субъекта в течение, по меньшей мере, 12 ч после принятия субъектом композиции внутрь.
52. Фармацевтическая композиция по п.48, где неметаболизированный фенилэфрин обнаруживается в плазме вышеупомянутого субъекта в течение, по меньшей мере, 16 ч после принятия субъектом композиции внутрь.
53. Фармацевтическая композиция по п.48, где неметаболизированный фенилэфрин обнаруживается в плазме вышеупомянутого субъекта в течение, по меньшей мере, 20 ч после принятия субъектом композиции внутрь.
54. Фармацевтическая композиция по п.48, где неметаболизированный фенилэфрин обнаруживается в плазме вышеупомянутого субъекта в течение, по меньшей мере, 24 ч после принятия субъектом композиции внутрь.
55. Фармацевтическая композиция по п.48, где композиция разработана для доставки, по меньшей мере, части фенилэфрина или его фармацевтически приемлемой соли в ободочную кишку субъекта.
56. Фармацевтическая композиция по п.48, где композиция разработана для доставки, по меньшей мере, части фенилэфрина или его фармацевтически приемлемой соли путем пролонгированного высвобождения в верхних отделах желудочно-кишечного тракта субъекта.
57. Фармацевтическая композиция по п.48, где композиция разработана для доставки, по меньшей мере, части фенилэфрина или его фармацевтически приемлемой соли путем немедленного высвобождения в верхних отделах желудочно-кишечного тракта субъекта.
58. Фармацевтическая композиция по любому из пп.1-47, подходящая для перорального введения, для доставки фенилэфрина или его фармацевтически приемлемой соли, в однократной дозе, в организм субъекта, нуждающегося в этом, причем неметаболизированный фенилэфрин обнаруживается в плазме вышеупомянутого субъекта на момент около 5 ч, или около 6 ч, или около 8 ч, или около 12 ч, или около 16 ч, или около 20 ч или около 24 ч после принятия субъектом композиции внутрь.
59. Фармацевтическая композиция по п.52, где композиция разработана для доставки, по меньшей мере, части фенилэфрина или его фармацевтически приемлемой соли в ободочную кишку субъекта.
60. Фармацевтическая композиция по п.1, где ядро дополнительно содержит один или более из следующих компонентов: микрокристаллическая целлюлоза и стеарат магния.
61. Фармацевтическая композиция по п.60, где поддающийся эрозии слой дополнительно содержит один или более полимеров, основанных на поливиниловом спирте.
62. Фармацевтическая композиция по п.61, где поддающийся эрозии слой дополнительно содержит лоратадин или деслоратадин.
63. Фармацевтическая композиция по п.1, где
- ядро содержит следующие компоненты: около 4-5% (в/в) фенилэфрина или его фармацевтически приемлемой соли, около 4-5% (в/в) гидроксипропилцеллюлозы 300-600 сП при 10%, около 4-5% (в/в) гидроксипропилцеллюлозы 150-300 сП при 2%, около 16-17% (в/в) натриевой соли карбоксиметилцеллюлозы, около 59-61% (в/в) микрокристаллической целлюлозы и около 0.9-1% (в/в) стеарата магния; и
- поддающийся эрозии слой содержит следующие компоненты: около 1-2% (в/в) фенилэфрина или его фармацевтически приемлемой соли и около 6-10% (в/в) полимера, основанного на поливиниловом спирте.
64. Фармацевтическая композиция по п.63, где полимером, основанным на поливиниловом спирте, является привитой сополимер поливинилового спирта и полиэтиленгликоля.
65. Фармацевтическая композиция по п.63, где поддающийся эрозии слой дополнительно содержит около 0.9-1% (в/в) лоратадина или деслоратадина.
66. Фармацевтическая композиция по п.1, где ядро содержит следующие компоненты: около 22.5 мг фенилэфрина или его фармацевтически приемлемой соли, около 25 мг гидроксипропилцеллюлозы 300-600 сП при 10%, около 25 мг гидроксипропилцеллюлозы 150-300 сП при 2%, около 90 мг натриевой соли карбоксиметилцеллюлозы, около 332.5 мг микрокристаллической целлюлозы и около 5 мг стеарата магния.
67. Фармацевтическая композиция по п.1, где поддающийся эрозии слой содержит следующие компоненты: около 7.5 мг фенилэфрина или его фармацевтически приемлемой соли и около 36-54 мг полимера, основанного на поливиниловом спирте.
68. Фармацевтическая композиция по п.67, где полимером, основанным на поливиниловом спирте, является привитой сополимер поливинилового спирта и полиэтиленгликоля.
69. Фармацевтическая композиция по п.67, где поддающийся эрозии слой дополнительно содержит около 5 мг лоратадина или деслоратадина.
70. Применение композиции по любому из пп.1-69 для приготовления лекарственного средства для лечения симптомов простуды, гриппа, аллергии или неаллергического ринита у субъекта, нуждающегося в этом.
71. Применение по п.70, где лекарственное средство находится в форме для введения каждые 8, 12 или 24 ч.
72. Применение по п.70 или 71, где лекарственное средство находится в форме, подходящей для перорального введения фенилэфрина или его фармацевтически приемлемой соли субъекту, причем лекарственное средство доставляет, по меньшей мере, часть фенилэфрина в ободочную кишку субъекта, и где максимальная концентрация несвязанного фенилэфрина в плазме субъекта достигается в промежутке от около 5 до около 24 ч после введения.
73. Применение по п.72, где максимальная концентрация несвязанного фенилэфрина в плазме субъекта достигается в промежутке от около 6 до около 12 ч после введения.
74. Применение по п.70 или 71, где лекарственное средство находится в форме, подходящей для перорального введения фенилэфрина или его фармацевтически приемлемой соли субъекту, причем, по меньшей мере, часть фенилэфрина абсорбируется из ободочной кишки, таким образом поддерживая концентрацию несвязанного фенилэфрина в плазме субъекта на уровне, по меньшей мере, 0.1 нг/мл на момент около 6 ч после введения композиции.
75. Применение по п.74, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 0.5 нг/мл на момент около 6 ч после введения композиции.
76. Применение по п.75, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 1 нг/мл на момент около 6 ч после введения композиции.
77. Применение по п.76, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 2.5 нг/мл на момент около 6 ч после введения композиции.
78. Применение по п.74, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 0.1 нг/мл на момент около 12 ч после введения композиции.
79. Применение по п.78, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 0.5 нг/мл на момент около 12 ч после введения композиции.
80. Применение по п.79, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 1 нг/мл на момент около 12 ч после введения композиции.
81. Применение по п.80, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 2.5 нг/мл на момент около 12 ч после введения композиции.
82. Применение по п.74, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 0.1 нг/мл на момент около 24 ч после введения композиции.
83. Применение по п.82, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 0.5 нг/мл на момент около 24 ч после введения композиции.
84. Применение по п.83, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 1 нг/мл на момент около 24 ч после введения композиции.
85. Применение по п.84, где концентрация несвязанного фенилэфрина составляет, по меньшей мере, 2.5 нг/мл на момент около 24 ч после введения композиции.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| US 3558768 A, 26.01.1971. | |||

