ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к каталитической системе для применения при олигомеризации, такой как тримеризация или тетрамеризация этилена, и, более конкретно, к способу повышения активности и селективности олигомеризации этилена с применением каталитической системы, которая содержит соединения переходного металла или предшественник соединения переходного металла, промотор и лиганд, имеющий определенную стереоизомерную структуру.
УРОВЕНЬ ТЕХНИКИ
1-Гексен и 1-октен являются важными коммерческими исходными веществами, которые широко применяют в качестве мономеров или сомономеров в способах полимеризации для получения линейного полиэтилена низкой плотности, их получают очисткой продуктов олигомеризации этилена. Однако реакции олигомеризации этилена, проводимые до сих пор, являются неэффективными в том, что 1-гексен и 1-октен получают вместе со значительными количествами бутена, высших олигомеров и полиэтилена. При такой прежней технологии олигомеризации этилена выход требуемого продукта ограничен, поскольку обычно получают диапазон α-олефинов, соответствующий распределению продуктов Schulze-Flory или Poisson.
За последнее время исследования сфокусировались либо на получении 1-гексена селективной тримеризацией этилена посредством катализа переходным металлом, либо на получении 1-октена селективной тетрамеризацией этилена, и наиболее известными катализаторами переходных металлов для применения при тримеризации или тетрамеризации этилена являются катализаторы на основе хрома.
В публикации международной патентной заявки WO 02/04119 описан катализатор на основе хрома для тримеризации этилена, который содержит лиганд формулы (R1)(R2)X-Y-X(R3)(R4), в которой Х представляет собой атом фосфора, мышьяк или сурьма, Y представляет собой связывающую группу, такую как -N(R5)- и по меньшей мере один из R1, R2, R3 и R4 имеет полярный заместитель или электронодонорный заместитель.
В другой публикации описано применение (о-этилфенил)2PN(Ме)Р(о-этилфенил)2, лиганда, который обнаруживает каталитическую активность при получении 1-гексена в каталитических условиях и не имеет полярный заместитель по меньшей мере у одного из R1, R2, R3 и R4 (Antea Carter et al., Chem. Commun., 2002, p.858-859).
Кроме того, в выложенной публикации патента Кореи №2006-0002741 описано, что превосходную активность и селективность тримеризации этилена можно достичь на практике посредством применения лиганда PNP, содержащего неполярный заместитель в ортоположении фенильного кольца, присоединенного к атому фосфора, например (о-этилфенил)2PN(Ме)Р(о-этилфенил)2.
Между тем, в публикации международной патентной заявки WO 04/056479 описана повышенная селективность в способе получения 1-октена тетрамеризацией этилена с применением катализатора на основе хрома, содержащего лиганд PNP, не имеющий заместитель у фенильного кольца, присоединенного к атому фосфора. В данной патентной публикации примеры гетероатомного лиганда, который применяют в катализаторе для тетрамеризации этилена, включают в себя (фенил)2PN(изопропил)Р(фенил)2 и т.д.
В этой предшествующей публикации описано, что катализатор на основе хрома, содержащий гетероатомный лиганд, имеющий в качестве гетероатомов как атомы азота, так и фосфора, без каких-либо полярных заместителей на углеводородных или гетероуглеводородных группах у атома фосфора, можно применять для селективной тетрамеризации этилена для получения 1-октена, часто с селективностью свыше 70 масс.%.
Однако в предыдущих публикациях не указывают конкретный пример структуры гетероатомсодержащего лиганда, который может тетрамеризовать этилен при высокой селективности для получения 1-октена или тримеризовать этилен при высокой селективности для получения 1-гексена. Кроме того, в этих публикациях предлагают только структуру каркаса PNP-типа, такую как (R1)(R2)P-(R5)N-P(R3)(R4), которая является лигандом, имеющим селективность для получения 1-октена приблизительно 70 масс.%. Кроме того, возможные заместители для гетероатомных лигандов также ограничены.
Кроме того, у предыдущих лигандов с каркасом PNP-типа, содержащих гетероатомы, имеются проблемы в том, что их активность в реакциях получения 1-октена или 1-гексена изменяются с течением времени, и скорость реакции значительно снижается.
ОПИСАНИЕ
[Техническая проблема]
Заявителем проведены эксперименты по олигомеризации этилена при различных изменениях не только структуры между атомами Р и Р, но также заместителей R1, R2, R3 и R4 на атомах Р для преодоления проблем стабильности катализатора в предшествующих технологиях. В результате этого заявителем было обнаружено, что при применении изобретательского катализатора на основе хрома, имеющего лиганд с каркасом P-C-C-P, не содержащий азот, 1-гексен или 1-октен можно получить тримеризацией или тетрамеризацией этилена с высокой селективностью, и, кроме того, активность катализатора является стабильной на протяжении времени, достаточного для того, чтобы скорость реакции можно поддерживать постоянной. Кроме того, автором изобретения было обнаружено, что когда структуры, соседние с атомами углерода между двумя атомами фосфора в изобретательском лиганде, имеющем структуру каркаса P-C-C-P, изменяются стереоскопически, активность и селективность тримеризации и тетрамеризации может значительно повышаться. На основании этих открытий было завершено настоящее изобретение.
То есть задачей настоящего изобретения является предоставление каталитической системы с переходным металлом, которая содержит лиганд P-C-C-P, имеющий определенную стерическую структуру, и поэтому может иметь стабильную каталитическую активность, таким образом непрерывно поддерживая постоянную скорость реакции.
[Техническое разрешение задачи]
Для достижения указанной выше цели настоящее изобретение предоставляет каталитическую систему для селективной олигомеризации этилена, которая содержит соединения переходного металла или предшественник соединения переходного металла, промотор и лиганд, представленный одной из следующих формул 1-4:
[Формула 1]
[Формула 2]
[Формула 3]
[Формула 4]
где каждый из R1, R2, R3 и R4 независимо представляет собой углеводородный, замещенный углеводородный, гетероуглеводородный или замещенный гетероуглеводородный радикал, каждый из R5 и R6 независимо представляет собой углеводородный или замещенный углеводородный радикал и А представляет собой углеводородный, замещенный углеводородный, гетероуглеводородный или замещенный гетероуглеводородный радикал.
[Благоприятные эффекты]
Согласно настоящему изобретению, когда олигомеризацию этилена проводят с применением каталитической системы на основе хрома, либо содержащей линейный лиганд, имеющий (S,S)- или (R,R)-изомерную структуру P-C-C-P, которая является асимметричной относительно плоскости симметрии, либо содержащей транс-циклический лиганд, имеющий структуру (S,S)- или (R,R)-изомера P-C-C-P, активность и селективность тримеризации или тетрамеризации можно значительно увеличить согласно направлению расположения и структуре заместителей, соседних с атомами углерода между двумя атомами фосфора, и таким образом 1-гексен или 1-октен можно получить с высокой селективностью.
[Лучший вариант осуществления изобретения]
Далее настоящее изобретение будет описано более подробно.
Настоящее изобретение относится к каталитической системе на основе хрома для селективной олигомеризации этилена, которая содержит соединения переходного металла или предшественник соединения переходного металла, промотор и лиганд с каркасом P-C-C-P, обладающий стереоспецифичностью. С применением каталитической системой изобретения можно получать 1-гексен или 1-октен при высокой его активности и высокой селективности и поддержании стабильной реакционной активности. Согласно настоящему изобретению, активность и селективность тримеризации и тетрамеризации можно значительно повысить стерическим расположением структур, соседних с атомами углерода между двумя атомами фосфора, в частности в соединении с каркасом P-C-C-P, для применения в качестве лиганда.
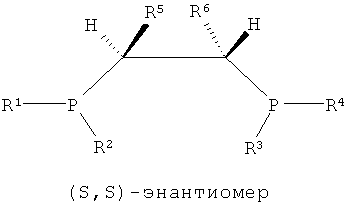
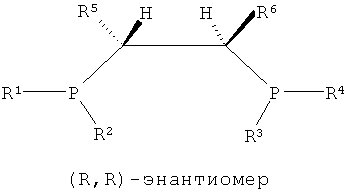
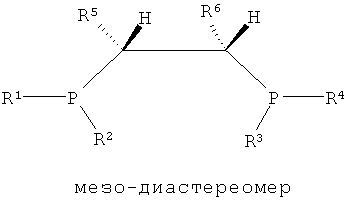
Как показано в формулах 1, 2 и 5, в структуре лиганда PCH(R5)CH(R6)P могут существовать три стереоизомера согласно ориентациям, в которых заместители R5 и R6, соседние с соответствующими атомами углерода, связываются с атомом углерода. То есть можно считать, что каждый из атомов углерода, присутствующих в структуре каркаса P-C-C-P, является хиральным атомом углерода, поскольку четыре заместителя, соседние с ним, являются полностью разными, и в этом случае два направления расположения для каждого атома углерода могут существовать согласно направлению, в котором заместители присоединяются к атомам углерода. Согласно направлению расположения заместителей, заместители на соответствующих хиральных атомах углерода можно разделить на заместители с (R)- и (S)-конфигурациями системой Кана-Ингольда-Прега.
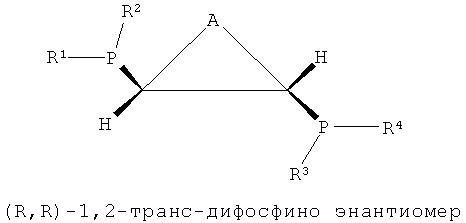
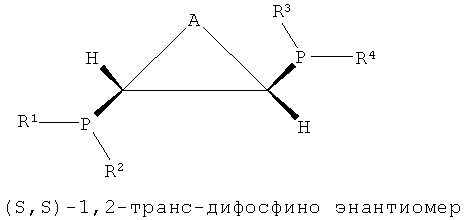
В структуре каркаса формулы I, приведенной ниже, заместители на первом атоме углерода имеют (S)-конфигурацию, поскольку три заместителя расположены в направлении против часовой стрелки согласно их приоритету, когда заместитель атом водорода, имеющий самый низший приоритет, направлен в сторону от наблюдателя. Кроме того, можно увидеть, что заместители на правом атоме углерода имеют (S)-конфигурацию, когда водород с самым низким приоритетом направлен в сторону от наблюдателя. Видно, что показанная ниже структура формулы 2 является (R,R)-изомером, когда ее рассматривают таким же образом, как указано выше. Структура показанной ниже формулы 5 является (R,S)-конфигурацией и мезо-изомером, поскольку два атома углерода являются хиральными. Таким же образом, в приведенной ниже циклической структуре формулы 6, когда два соседних дифосфиновых соединения имеют (R,S)-конфигурацию, дифосфиновые соединения направлены в одну и ту же сторону и являются цис-изомерами.
[Формула 1]
[Формула 2]
[Формула 5]
[Формула 6]
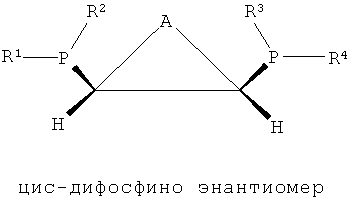
где каждый из R1, R2, R3 и R4 независимо представляет собой углеводородный, замещенный углеводородный, гетероуглеводородный или замещенный гетероуглеводородный радикал, каждый из R5 и R6 независимо представляет собой углеводородный или замещенный углеводородный радикал и А представляет собой углеводородный, замещенный углеводородный, гетероуглеводородный или замещенный гетероуглеводородный радикал.
В настоящем изобретении было подтверждено, что хиральные (S,S)- или (R,R)-изомеры обнаруживали весьма хорошую селективность и активность при тримеризации или тетрамеризации этилена по сравнению с ахиральными мезо-изомерами. Подобным же образом, было отмечено, что лигандное соединение, частично состоящее из ахиральной транс-циклической структуры P-C-C-P, как показано на формулах 3 и 4, обнаруживали весьма хорошую селективность и активность при тримеризации или тетрамеризации этилена по сравнению с хиральной цис-циклической структурой.
[Формула 3]

[Формула 4]
где каждый из R1, R2, R3 и R4 независимо представляет собой углеводородный, замещенный углеводородный, гетероуглеводородный или замещенный гетероуглеводородный радикал и А представляет собой углеводородный, замещенный углеводородный, гетероуглеводородный или замещенный гетероуглеводородный радикал.
То есть настоящее изобретение относится к каталитической системе, содержащей стереоизомерное соединение с каркасом P-C-C-P, которая имеет значительно повышенную селективность и активность при олигомеризации этилена и в то же время поддерживает стабильную реакционную активность, и более конкретно, к каталитической системе, содержащей лиганд, имеющий хиральную (S,S)- или (R,R)-изомерную структуру P-C-C-P.
В дальнейшем настоящее изобретение будет описано более подробно.
Каталитическая система для селективной олигомеризации этилена согласно настоящему изобретению содержит соединения переходного металла или предшествиник соединения переходного металла, промотор и стереоизомерный лиганд, имеющий структуру каркаса P-C-C-P, такая каталитическая система может иметь высокую активность и высокую селективность при олигомеризации этилена и может поддерживать стабильную реакционную активность. Более конкретно, настоящее изобретение относится к каталитической системе, содержащей лиганд, имеющий (S,S)- или (R,R)-изомерную структуру P-C-C-P, которая является стерически асимметричной относительно плоскости симметрии, как показано в следующих формулах 1-4:
[Формула 1]
[Формула 2]
[Формула 3]
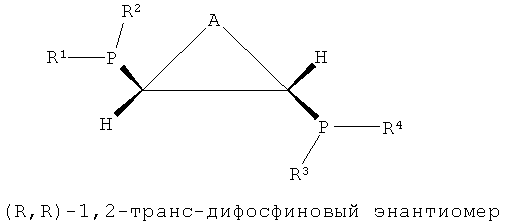
[Формула 4]
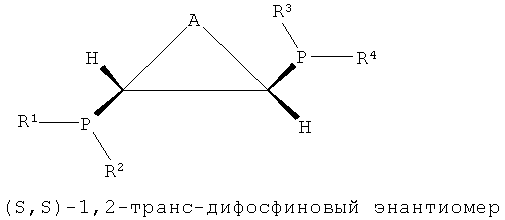
где каждый из R1, R2, R3 и R4 независимо представляет собой углеводородный, замещенный углеводородный, гетероуглеводородный или замещенный гетероуглеводородный радикал и А представляет собой углеводородный, замещенный углеводородный, гетероуглеводородный или замещенный гетероуглеводородный радикал.
В формулах 1-4 подходящие примеры R1, R2, R3 и R4 могут включать в себя фенил, бензил, нафтил, антраценил, мезитил, ксенил, метил, этил, этиленил, пропил, пропенил, пропинил, бутил, циклогексил, 4-метилциклогексил, 4-этилциклогексил, 4-изопропилциклогексил, толил, ксилил, 4-метилфенил, 4-этилфенил, 4-изопропилфенил, 4-трет-бутилфенил, 4-метоксифенил, 4-изопропоксифенил, кумил, метокси, этокси, фенокси, толилокси, диметиламино, тиометил, триметилсилил, диметилгидразин, 2-метилциклогексил, 2-этилциклогексил, 2-изопропилциклогексил, о-метилфенил, о-этилфенил, о-изопропилфенил, о-трет-бутилфенил, о-метоксифенил, о-изопропоксифенил, бифенил, нафтил и антраценил и каждый из них может быть выбран независимо.
Более предпочтительно, каждый из R1, R2, R3 и R4 может быть независимо выбран из группы, состоящей из фенила, толила, бифенила, нафтила, циклогексила, 4-метилфенила, 4-этилфенила, 4-изопропилфенила, 4-трет-бутилфенила, 4-метоксифенила, 4-изопропоксифенила, 2-метилциклогексила, 2-этилциклогексила, 2-изопропилциклогексила, о-метилфенила, о-этилфенила, о-изопропилфенила, о-трет-бутилфенила, о-метоксифенила и о-изопропоксифенила.
Каждый из R5 и R6 может быть независимо выбран из группы, состоящей из углеводородных групп и замещенных углеводородных групп. Более конкретно, их можно выбрать из группы, состоящей из алкила, арилокси, галогена, нитро, алкоксикарбонила, карбонилокси, алкокси, аминокарбонила, карбониламино, диалкиламино, их производных, и арила, замещенного любым заместителем.
Группа А может быть углеводородным, замещенным углеводородным, гетероуглеводородным или замещенным гетероуглеводородным радикалом и более конкретно, может быть выбрана из группы, состоящей из С2-С10алкилена, алкокси, алкоксикарбонила, карбонилокси, аминокарбонила, карбониламино, алкиламино и их производных.
При олигомеризации этилена согласно настоящему изобретению лигандом с каркасом P-C-C-P для стабильного поддержания селективности в отношении получения 1-гексена или 1-октена и реакционной активности может быть (S,S)- или (R,R)-изомерный линейный лиганд или транс-циклический лиганд. Кроме того, можно применять смесь двух изомеров, то есть лиганд, состоящий из многократной связи (S,S)- или (R,R)-(R1)(R2)P-(R5)CHCH(R6)-P(R3)(R4).
Примеры стереоизомерного лиганда, состоящего из каркаса P-C-C-P, для поддержания активности, селективности и стабильной активности при селективной олигомеризации этилена согласно настоящему изобретению, включают в себя (S,S)- или (R,R)-(фенил)2Р-СН(метил)СН(метил)-Р(фенил)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(метил)СН(метил)-Р(4-метоксифенил)2, (S,S)- или (R,R)-(4-метилфенил)2Р-СН(метил)СН(метил)-Р(4-метилфенил)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(метил)СН(метил)-Р(фенил)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(этил)СН(метил)-Р(4-этилфенил)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(этил)СН(метил)-Р(фенил)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(этил)СН(этил)-Р(4-этилфенил)2, (S,S)- или (R,R)-(фенил)2Р-СН(этил)СН(этил)-Р(фенил)2, (S,S)- или (R,R)-(фенил)2Р-СН(изопропил)СН(метил)-Р(фенил)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(изопропил)СН(метил)-Р(4-метоксифенил)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(изопропил)СН(метил)-Р(4-этилфенил)2, (S,S)- или (R,R)-(фенил)2Р-СН(н-пропил)СН(метил)-Р(фенил)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(н-пропил)СН(метил)-Р(4-метоксифенил)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(н-пропил)СН(метил)-Р(4-этилфенил)2, (S,S)- или (R,R)-(фенил)2Р-СН(изопропил)СН(этил)-Р(фенил)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(изопропил)СН(этил)-Р(4-метоксифенил)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(изопропил)СН(этил)-Р(4-этилфенил)2, (S,S)- или (R,R)-транс-1,2-ди-(Р(фенил)3)циклогексан, (S,S)- или (R,R)-транс-1,2-ди-(Р(4-метоксифенил)3)циклогексан, (S,S)- или (R,R)-транс-1,2-ди-(Р(4-этилфенил)2)циклогексан, (S,S)- или (R,R)-транс-1,2-ди-(Р(фенил)2)циклопентан, (S,S)-или (R,R)-транс-1,2-ди-(Р(4-метоксифенил)2)циклопентан, (S,S)- или (R,R)-1,2-ди-(Р(4-этилфенил)2)циклопентан, (S,S)- или (R,R)-3,4-ди-(Р(фенил)2)пиррол, (S,S)- или (R,R)-3,4-ди-(Р(4-метоксифенил)2)пиррол, (S,S)- или (R,R)-транс-3,4-ди-(Р(4-этилфенил)2)пиррол, (S,S)- или (R,R)-транс-3,4-ди-(Р(4-этилфенил)2)имидазол, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(диметиламин)СН(диметиламин)-Р(4-этилфенил)2, (S,S)- или (R,R)-(3-метоксифенил)2Р-СН(метил)СН(метил)-Р(3-метоксифенил)2, (S,S)- или (R,R)-(4-этоксифенил)2Р-СН(метил)СН(метил)-Р(о-этоксифенил)2, ((S,S)- или (R,R)-4-диметиламинфенил)2Р-СН(метил)СН(метил)Р(4-диметиламинфенил)2, (S,S)- или (R,R)-(4-этилциклогексил)2РСН(метил)СН(метил)Р(4-этилциклогексил)2, (S,S)- или (R,R)-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенил)2, (S,S)- или (R,R)-(2-изопропилфенил)2РСН(метил)СН(метил)Р(2-изопропилфенил)2, (S,S)- или (R,R)-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенил)2, (S,S)- или (R,R)-(2-этилфенил)2РСН(метил)СН(метил)Р(фенил)2, (S,S)- или (R,R)-(2-этилфенил)2РСН(этил)СН(метил)Р(2-этилфенил)2, (S,S)- или (R,R)-(2-этилфенил)2РСН(этил)СН(этил)Р(2-этилфенил)2, (S,S)- или (R,R)-(2-этилфенил)2РСН(изопропил)СН(метил)Р(2-этилфенил)2, (S,S)- или (R,R)-(2-этилфенил)2РСН(н-пропил)СН(метил)Р(2-этилфенил)2, (S,S)- или (R,R)-(2-этилфенил)2РСН(изопропил)СН(этил)Р(2-этилфенил)2, (S,S)- или (R,R)-транс-1,2-ди-(Р(2-этилфенил)2)циклогексан, (S,S)- или (R,R)-транс-1,2-ди-(Р(2-этилфенил)2)циклопентан, (S,S)- или (R,R)-транс-3,4-ди-(Р(2-этилфенил)2)пиррол, (S,S)- или (R,R)-транс-3,4-ди-(Р(2-этилфенил)2)имидазол, (S,S)- или (R,R)-(2-этилфенил)2РСН(диметиламин)СН(диметиламин)Р(2-этилфенил)2, (S,S)- или (R,R)-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенил)2, (S,S)- или (R,R)-(2-этоксифенил)2РСН(метил)СН(метил)Р(2-этоксифенил)2, (S,S)- или (R,R)-(2-диметиламинофенил)2РСН(метил)СН(метил)Р(2-диметиламинфенил)2, (S,S)- или (R,R)-(2-этилциклогексил)2РСН(метил)СН(метил)Р(2-этилциклогексил)2, (S,S)- или (R,R)-(фенил)2Р-СН(фенил)СН(фенил)-Р(фенил)2, (1S,2S)- или (1R,2R)-транс-бис(дифенилфосфино)циклогексана, и (1S,2S)- или (1R,2R)- транс-бис(ди(4-метоксифенил)фосфино)циклогексана, но объем настоящего изобретения не ограничивается ими, и лиганды согласно настоящему изобретению можно получить с применением различных способов, известных специалистам в данной области.
Стереоизомерная структура каркаса типа P-C-C-P лиганда согласно настоящему изобретению отличается от предыдущего гетеролиганда (R)nPN(R')P(R)m, и гетероатомами в структуре каркаса лиганда согласно настоящему изобретению являются только атомы фосфора (Р). То есть лиганд для применения в каталитической системе согласно настоящему изобретению содержит углерод-углеродную структуру каркаса без какого-либо атома азота между двумя атомами фосфора. Согласно настоящему изобретению пространственная структура лиганда подходящим образом регулируется в отношении направления расположения заместителей, присоединенных к атомам углерода, и поэтому каталитическая система согласно настоящему изобретению может проявлять превосходную каталитическую активность, может достигать селективности при получении 1-гексена или селективности при получении 1-октена выше чем 70 масс.%, и может поддерживать стабильную реакционную активность.
Для получения гексена или октена с высокой селективностью каталитическую систему с лигандом согласно настоящему изобретению можно получить посредством способа, содержащего стадию смешивания соединения переходного металла и активирующего агента в любом порядке.
Способ получения изобретательской каталитической системы согласно настоящему изобретению может содержать стадию получения координационного комплекса лиганда из соединения переходного металла и стереоизомерного лиганда, имеющего структуру каркаса P-C-C-P. Этот способ содержит либо стадию добавления к реакционной смеси координационного комплекса, полученного из лиганда с каркасом P-C-C-P и соединения переходного металла, либо стадию добавления в реактор лиганда с каркасом P-C-C-P и соединения переходного металла так, чтобы образовался координационный комплекс лиганда с каркасом P-C-C-P in situ.
Получение координационного комплекса лиганда с каркасом P-C-C-P in situ означает получение комплекса в среде, в которой имеет место каталитическая реакция. Чтобы получить координационный комплекс in situ, соединение переходного металла и лиганд с каркасом P-C-C-P предпочтительно добавляют так, чтобы отношение металл:лиганд обычно было приблизительно 0,01:1-100:1, предпочтительно приблизительно 0,1:1-10:1 и более предпочтительно 0,5:1-2:1.
Переходным металлом может быть любой такой металл, выбранный из группы, состоящей из хрома, молибдена, вольфрама, титана, тантала, ванадия и циркония. Предпочтительным является хром.
Когда соединение переходного металла, катализирующее олигомеризацию этилена согласно настоящему изобретению, смешивают с лигандом с каркасом P-C-C-P и промотором, оно может быть простой неорганической или органической солью или координационным или металлорганическим соединением. Этим соединением предпочтительно является хром или предшественник хрома. Хром или предшественник хрома предпочтительно выбирают из группы, состоящей из ацетилацетоната хрома(III), трис(тетрагидрофуран)трихлорхрома и 2-этилгексаноата хрома(III).
Кроме того, лиганд с каркасом P-C-C-P может быть присоединен к полимерной цепи, так что образовавшийся координационный комплекс лиганда со структурой каркаса P-C-C-P становится нерастворимой при температуре, выше комнатной температуры. Кроме того, лиганд с каркасом P-C-C-P или соединение переходного металла могут быть связаны с основой и фиксированы на основе, такой как диоксид кремния, силикагель, полисилоксан или оксид алюминия.
Промотором для применения в изобретательском способе может быть любое соединение, которое образует активный катализатор, когда его смешивают с лигандом с каркасом P-C-C-P и соединением переходного металла. Активатор можно также применять в смеси. Соединения, подходящие для применения в качестве активатора, включают в себя алюминийорганические соединения, борорганические соединения и органические соли.
Алюминийорганические соединения, подходящие для применения в качестве активатора в каталитической системе согласно настоящему изобретению, включают в себя такие соединения, как AIR3, где каждый из радикалов R независимо представляет собой С1-С12алкил, кислородсодержащий алкил или галогенид, и LiAIH4.
Примеры такого промотора включают в себя триметилалюминий (ТМА), триэтилалюминий (TEA), триизобутилалюминий (TIBA), три-н-октилалюминий, метилалюминийдихлорид, этилалюминийдихлорид, диметилалюминийхлорид, диэтилалюминийхлоридизопропоксид, этилалюминийсесквихлорид, метилалюминийсесквихлорид и алюминоксан.
В данной области хорошо известно, что алюминоксан является олигомерным соединением, которое можно обычно получить смешиванием воды и алкилалюминиевого соединения, например триметилалюминия. Полученное алюминоксановое олигомерное соединение может быть линейным соединением, циклическим соединением, соединением включения или их смесью.
Подходящие борорганические соединения включают в себя бороксин, NaBH4, триэтилборан, трифенилборан, комплексы трифенилборана и аммиака, трибутилборат, триизопропилборат, трис(пентафторфенил)боран, тритилтетра(пентафторфенил)борат, тетра(пентафторфенил)борат диметилфениламмония, тетра(пентафторфенил)борат диэтилфениламмония, тетра(пентафторфенил)борат этилдифениламмония и тетра(пентафторфенил)борат этилдифениламмония. Эти борорганические соединения можно применять в смеси с алюминийорганическими соединениями.
Кроме того, в качестве промоторов алюминоксан можно выбрать из алкилалюминоксанов, например метилалюминоксана (МАО) и этилалюминоксана (ЕАО), а также модифицированных алкилалюминоксанов, например модифицированного метилалюминоксана (ММАО). Модифицированный метилалюминоксан (Akzo Nobel) содержит помимо метильной группы разветвленную алкильную группу, такую как изобутильная группа, или н-октильную группу.
Промотором предпочтительно является метилалюминоксан (МАО) или этилалюминоксан (ЕАО).
Соединение переходного металла хрома и алюминоксан можно смешивать так, чтобы отношение алюминий:металл было приблизительно 1:1-10000:1 и предпочтительно приблизительно 1:1-1000:1.
Отдельные компоненты каталитической системы, описанной в контексте, можно добавлять для смешивания одновременно или последовательно в любом порядке в присутствии или в отсутствие растворителя с получением активного катализатора. Смешивание компонентов катализатора можно проводить при температуре между -20°С и 250°С. Во время смешивания компонентов катализатора присутствие олефина обычно имеет защитное действие, что таким образом обеспечивает повышенную каталитическую эффективность. Более предпочтительно, когда смешивание компонентов катализатора проводят при температуре, составляющей от 20°С до 100°С.
Продукт реакции, описанный в настоящем изобретении, то есть олигомер этилена, можно получить с применением каталитической системы изобретения и каталитического способа в присутствии или в отсутствие инертного растворителя посредством реакции в гомогенной жидкости, реакции в суспензии, когда каталитическая система частично или полностью является нерастворимой, реакции в двухфазной системе жидкость/жидкость, реакции во всем объеме, при которой олефин действует в качестве основной среды, или газофазной реакции.
Так, способ получения согласно настоящему изобретению можно проводить в инертном растворителе. То есть можно применять любой инертный растворитель, который не реагирует с каталитическим соединением или с активатором. Эти инертные растворители могут включать в себя насыщенные алифатические и ненасыщенные алифатические и ароматические углеводороды и галогензамещенный углеводород. Типичные растворители включают в себя бензол, толуол, ксилол, кумол, гептан, циклогексан, метилциклогексан, метилциклопентан, н-гексан, 1-гексен, 1-октен и тому подобное, но объем настоящего изобретения не ограничивается ими.
Реакцию олигомеризации согласно настоящему изобретению можно проводить при температуре от -20 до 250°С, предпочтительно от 15 до 130°С и более предпочтительно от 30 до 70°С.
Кроме того, способ согласно настоящему изобретению можно проводить при давлении от атмосферного давления до 5×107 Па (500 бар), предпочтительно 1-7×106 Па (10-70 бар) и более предпочтительно 3-5×106 Па (30-50 бар).
В варианте осуществления настоящего изобретения координационный комплекс стереоизомерного лиганда с каркасом P-C-C-P и условия реакции выбирают так, чтобы выход 1-гексена из этилена был больше чем 50 масс.%, и более предпочтительно, больше чем 70 масс.%. В контексте выход означает число граммов 1-гексена, образованных на 100 г образованного продукта реакции.
В другом варианте осуществления настоящего изобретения координационный комплекс стереоизомерного лиганда с каркасом P-C-C-P и условия реакции выбирают так, чтобы выход 1-октена из этилена был больше чем 50 масс.%, и более предпочтительно, больше чем 70 масс.%. В контексте выход означает число граммов 1-октена, образованных на 100 г образованного продукта реакции.
В зависимости от лиганда с каркасом P-C-C-P и условий реакции способ олигомеризации согласно настоящему изобретению может дать помимо 1-гексена или 1-октена различные количества 1-бутена, 1-гексена, метилциклопентана, метиленциклопентана, пропилциклопентана и ряд высших олигомеров и полиэтиленов.
Способ согласно настоящему изобретению можно проводить на установке, содержащей реактор любого типа. Примеры этого реактора включают в себя реактор периодического действия, реактор полупериодического действия и реактор непрерывного действия, но объем настоящего изобретения не ограничивается ими. Установка может содержать комбинацию реактора, впускного отверстия для введения олефинов и каталитической системы в реактор, линию для выгрузки продукта олигомеризации из реактора и по меньшей мере один сепаратор для разделения продукта олигомеризации, в котором каталитическая система может содержать соединение переходного металла, промотор и координацонный комплекс лиганда P-C-C-P, описываемый в контексте.
Согласно настоящему изобретению, 1-гексен или 1-октен можно получить при высокой активности и высокой селективности олигомеризацией этилена с применением каталитической системы для олигомеризации этилена согласно настоящему изобретению.
[Способ изобретения]
Далее настоящее изобретение будет описано более подробно обращением к нижеследующим примерам получения и примерам, но объем настоящего изобретения не ограничивается этими примерами.
[Примеры]
Пример 1 получения катализатора: получение лиганда (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенил)2
Получение катализатора проводили, как описано в В.Bosnich et al., J. Am. Chem. Soc. 99(19) (1977).
Ди-п-толуолсульфонат (2R,3R)-бутандиола получали из (2R,3R)-бутандиола. Этот способ получения проводили, как описано в R.В.Mitra et al., J. Am. Chem. Soc 84(1962). 100 мл (1,24 моль) сухого пиридина помещали в 1-литровую колбу на бане со смесью лед-вода и смешивали с 100 г (0,525 моль) п-толуолсульфонилхлорида и затем медленно, по каплям, добавляли 22 мл (0,245 моль) (2R,3R)-бутандиола. После повышения температуры до комнатной температуры на протяжении 20 минут полутвердую смесь выдерживали при комнатной температуре на протяжении ночи. К смеси добавляли избыточное количество льда в виде кусочков и смесь тщательно встряхивали так, чтобы не образовывалась сплошная масса. После медленного отделения кристаллического порошка его перемешивали вместе с кусочками льда в течение 2 часов и к смеси при интенсивном перемешивании добавляли измельченные кусочки льда и 70 мл концентрированной хлористоводородной кислоты. Экстрагированную суспензию фильтровали, осадок полностью промывали водой и сушили, таким образом получая 85 г (86,3%) ди-п-толуолсульфоната (2R,3R)-бутандиола (т.пл. 62-64°С).
В 1-литровую трехгорлую круглодонную колбу, снабженную 250-миллилитровой капельной воронкой, парциальным конденсатором горячего орошения и вводом для азота, загружали 95 г перекристаллизованного трифенола и 300 мл сухого тетрагидрофурана. К раствору при 25°С в атмосфере азота и при перемешивании добавляли 5,0 г тонких кусочков лития. В растворе сразу образовывался LiPPh2, и раствор изменял цвет в темно-красно-желтый при выделении большого количества тепла. Температуру раствора медленно повышали до 55°С на протяжении 1 часа и раствор перемешивали в течение 2 часов при охлаждении снова до 25°С. Образованный фениллитий разлагали добавлением по каплям 33 г перегнанного и очищенного трет-бутилхлорида на протяжении 45 минут. Прозрачный красно-желтый раствор кипятили в течение 5 минут и затем снова охлаждали до -4°С.
К охлажденному раствору по каплям на протяжении 1 часа по каплям добавляли 35 г вышеполученного ди-п-толуолсульфоната (2R,3R)-бутандиола, растворенного в 100 мл сухого ТГФ. Раствор медленно нагревали до комнатной температуры и затем перемешивали в течение 30 минут. К нему добавляли 30 мл продутой азотом воды и ТГФ удаляли дистилляцией при пониженном давлении, таким образом получая бесцветный продукт типа масла. Продукт два раза экстрагировали 150 мл эфира и затем сушили Na2SO4. Эфирный экстракт фильтровали с применением раствора 15 г гексагидрата перхлората никеля в 50 мл этанола в атмосфере азота. Na2SO4, оставшийся в фильтре, тщательно промывали эфиром и затем эфирный раствор добавляли к раствору соединения никеля. Красно-коричневый продукт типа масла, который содержал желтые кристаллы, был [Ni((SS)-хирафос)2](ClO4)2. Смесь масла и кристаллов добавляли к 15 г тиоцианата натрия (NaNCS), растворенного в 50 мл горячего этанола, и раствор интенсивно перемешивали в течение нескольких часов до образования однородного желто-коричневого твердого вещества, [Ni((SS)-хирафос)2NCS]NCS. Твердый продукт полностью промывали этанолом и затем промывали эфиром.
15 г комплекса никеля суспендировали в 150 мл этанола в атмосфере азота и суспензию нагревали с перемешиванием. К суспензии быстро добавляли раствор 4 г цианида натрия (NaCN). Комплекс никеля медленно растворяли с получением прозрачного красного раствора [Ni((SS)-хирафос)2CN3]-, который затем превращался в мутный раствор бежевого цвета. Горячий раствор перемешивали до образования желтой суспензии. Суспензию охлаждали и твердое вещество промывали два раза 25 мл воды и быстро охлаждали охлажденным льдом этанолом. Содержащее примеси твердое вещество бежевого цвета сушили при 25°С, добавляли к 125 мл кипящего безводного этанола и затем фильтровали через фритту. Фильтрование через фритту проводили при комнатной температуре в течение 12 часов и в результате этого фильтрат полностью удалялся и оставалось только бесцветное блестящее твердое вещество. Твердое вещество перекристаллизовывали из 60 мл безводного этанола, получая таким образом 5,5 г полностью бесцветного, чистого соединения (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенил)2.
Пример 1: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 и МАО
300 мл реактор из нержавеющей стали промывали азотом в вакууме и затем в него добавляли 100 мл циклогексана и МАО (4,0 ммоль AI). Затем температуру повышали до 45°С. В 50 мл контейнер Шленка в ящике с перчатками 3,5 мг (0,010 ммоль) Cr(III)(ацетилацетоната)3 в 10 мл толуола смешивали с 4,3 мг (0,010 ммоль) соединения (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора. Смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. В реактор загружали этилен до 3×106 Па (30 бар) и смесь перемешивали со скоростью 600 об/мин. Спустя 30 минут подачу этилена в реактор прекращали, перемешивание заканчивали для окончания реакции и реактор охлаждали до температуры ниже 10°С.
После выгрузки избыточного этилена из реактора к жидкости в реакторе добавляли этанол, содержащий 10 об.% хлористоводородной кислоты. Для анализа жидкости ГХ-FID в качестве внутреннего стандарта добавляли нонан. Небольшое количество образца органического слоя сушили над безводным сульфатом магния и затем анализировали ГХ-FID. Оставшийся органический слой фильтровали для отделения твердых парафиновых/полимерных продуктов. Эти твердые продукты сушили в сушильном шкафу при 100°С на протяжении ночи и взвешивали, получая таким образом 1,3 г полиэтилена. Анализ ГХ показал, что общая масса реакционной смеси была 38,2 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 2: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 и МАО
300 мл реактор из нержавеющей стали, который был таким же, как применяли в примере 1, промывали азотом в вакууме и затем в него добавляли 100 мл циклогексана и МАО (2,0 ммоль Al). Затем температуру в реакторе повышали до 45°С. В 50 мл контейнер Шленка в ящике с перчатками 0,7 мг (0,002 ммоль) Cr(III)(ацетилацетоната)3 в 10 мл толуола смешивали с 0,86 мг (0,002 ммоль) соединения (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора. Смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. В реактор загружали этилен до 3×106 Па (30 бар) и смесь перемешивали со скоростью 600 об/мин. Спустя 30 минут подачу этилена в реактор прекращали, перемешивание останавливали для окончания реакции и реактор охлаждали до температуры ниже 10°С.
После выгрузки избыточного этилена из реактора к жидкости в реакторе добавляли этанол, содержащий 10 об.% хлористоводородной кислоты. Для анализа жидкости ГХ-FID в качестве внутреннего стандарта добавляли нонан. Небольшое количество образца органического слоя сушили над безводным сульфатом магния и затем анализировали ГХ-FID. Оставшийся органический слой фильтровали для отделения твердых парафиновых/полимерных продуктов. Эти твердые продукты сушили в сушильном шкафу при 100°С на протяжении ночи и взвешивали. Анализ ГХ показал, что общая масса реакционной смеси была 18,0 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 3: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 и МАО
300 мл реактор из нержавеющей стали, который был таким же, как применяли в примере 1, промывали азотом в вакууме и затем в него добавляли 100 мл циклогексана и МАО (2,0 ммоль Al). Затем температуру в реакторе повышали до 45°С. В 50 мл контейнер Шленка в ящике с перчатками 3,75 мг (0,01 ммоль) CrCl3(тетрагидрофурана)3 в 10 мл толуола смешивали с 4,3 мг (0,01 ммоль) соединения (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора. Смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. В реактор загружали этилен до 3,0×106 Па (30 бар) и смесь перемешивали со скоростью 600 об/мин. Спустя 30 минут подачу этилена в реактор прекращали, перемешивание останавливали, чтобы таким образом остановить реакцию и реактор охлаждали до температуры ниже 10°С.
После выгрузки избыточного этилена из реактора к жидкости в реакторе добавляли этанол, содержащий 10 об.% хлористоводородной кислоты. Для анализа жидкости ГХ-FID в качестве внутреннего стандарта добавляли нонан. Небольшое количество образца органического слоя сушили над безводным сульфатом магния и затем анализировали ГХ-FID. Оставшийся органический слой фильтровали для отделения твердых парафиновых/полимерных продуктов. Эти твердые продукты сушили в сушильном шкафу при 100°С на протяжении ночи. Анализ ГХ показал, что общая масса реакционной смеси была 30,5 г, Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 4: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 и МАО
300 мл реактор из нержавеющей стали, который был таким же, как применяли в примере 1, промывали азотом в вакууме и затем в него добавляли 100 мл циклогексана и МАО (2,0 ммоль Al). Затем температуру в реакторе повышали до 45°С. В 50 мл контейнер Шленка в ящике с перчатками 4,0 мг (0,01 ммоль) Cr(этилгексаноата)3 в 10 мл толуола смешивали с 4,3 мг (0,01 ммоль) соединения (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора. Смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. В реактор загружали этилен до 3,0×106 Па (30 бар) и смесь перемешивали со скоростью 600 об/мин. Спустя 30 минут подачу этилена в реактор прекращали, перемешивание заканчивали для остановки реакции и реактор охлаждали до температуры ниже 10°С.
После выгрузки избыточного этилена из реактора к жидкости в реакторе добавляли этанол, содержащий 10 об.% хлористоводородной кислоты. Для анализа жидкости ГХ-FID в качестве внутреннего стандарта добавляли нонан. Небольшое количество образца органического слоя сушили над безводным сульфатом магния и затем анализировали FX-FID. Оставшийся органический слой фильтровали для отделения твердых парафиновых/полимерных продуктов. Эти твердые продукты сушили в сушильном шкафу при 100°С на протяжении ночи. Анализ ГХ показал, что масса реакционной смеси была 30,0 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 5: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 и МАО
300 мл реактор из нержавеющей стали примера 1 промывали азотом в вакууме и затем в него добавляли 100 мл циклогексана и МАО (2,0 ммоль Al). Затем температуру в реакторе повышали до 45°С. В 50 мл контейнер Шленка в ящике с перчатками 0,8 мг (0,002 ммоль) Cr(этилгексаноата)3 в 10 мл толуола смешивали с 0,86 мг (0,002 ммоль) соединения (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора. Смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. В реактор загружали этилен до 3,0×106 Па (30 бар) и смесь перемешивали со скоростью 600 об/мин. Спустя 30 минут подачу этилена в реактор прекращали, перемешивание прекращали, чтобы таким образом остановить реакцию и реактор охлаждали до температуры ниже 10°С.
После выгрузки избыточного этилена из реактора к жидкости в реакторе добавляли этанол, содержащий 10 об.% хлористоводородной кислоты. Для анализа жидкости ГХ-FID в качестве внутреннего стандарта добавляли нонан. Небольшое количество образца органического слоя сушили над безводным сульфатом магния и затем анализировали ГХ-FID. Оставшийся органический слой фильтровали для отделения твердых парафиновых/полимерных продуктов. Эти твердые продукты сушили в сушильном шкафу при 100°С. Анализ ГХ показал, что общая масса реакционной смеси была 11,2 г, Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 2 получения катализатора: получение лиганда (R,R)-(фенил)2РСН(метил)СН(метил)Р(фенил)2
Катализатор получали таким же способом, как в примере 1 получения катализатора, за исключением того, что в качестве исходного вещества реакции применяли (2S,3S)-бутандиол вместо (2R,3R)-бутандиола. Получали 5,1 г полностью бесцветного, чистого (R,R)-(фенил)2РСН(метил)СН(метил)Р(фенил)2.
Пример 6: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (R,R)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (R,R)-(фенил)2РСН(метил)СН(метил)Р(фенил)2 применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса продуктов реакции была 43,2 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 7: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (R,R)-(фенил)2РСН(метил)CH(метил)Р(фенила)2 и МАО
Повторяли способ примера 3, за исключением того, что (R,R)-(фенил)2РСН(метил)СН(метил)Р(фенил)2 применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 25,3 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 8: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (R,R)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 и МАО
Повторяли способ примера 4, за исключением того, что (R,R)-(фенил)2РСН(метил)СН(метил)Р(фенил)2 применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 40,9 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 3 получения катализатора: получение лиганда (S,S)-(4-метоксифенил)2РСН(метил)CH(метил)Р(4-метоксифенил)2
Получение катализатора проводили, как описано в В.Bosnich et al., J. Am. Chem. Soc. 99(19) (1977).
Получение ди-п-толуолсульфоната (2R,3R)-бутандиола из (2R,3R)-бутандиола проводили согласно способу примера 1 получения катализатора.
Получение три-(4-метоксифенил)фосфина проводили следующим образом. 91,1 г (3,75 моль) кусочков магния медленно добавляли к 95 мл (0,75 моль) 4-броманизола в 2 литрах ТГФ. После проведения интенсивной реакции реакционную смесь нагревали при кипячении с обратным холодильником в течение 2 часов, получая при этом реактив Гриньяра. Реактив Гриньяра добавляли по каплям к раствору 17,5 мл (0,2 моль) PCl3 в 2 литрах ТГФ при -78°С на протяжении 2 часов с перемешиванием. После завершения добавления по каплям баню сухой лед/ацетон удаляли и реакционную смесь нагревали до комнатной температуры. Реакционную смесь перемешивали на протяжении ночи и растворитель удаляли в вакууме. Фосфиновый продукт применяли в последующей стадии без выделения.
В 1-литровую трехгорлую колбу, снабженную 250-миллилитровой капельной воронкой, парциальным конденсатором горячего орошения и вводом для азота, загружали 70 г перекристаллизованного три-(4-метоксифенола) и 300 мл сухого тетрагидрофурана. К раствору при 25°С в атмосфере азота и при перемешивании добавляли 2,8 г тонких кусочков лития. В растворе сразу образовывался LiP(4-OMe-Ph)2 и раствор изменял цвет в темно-красно-желтый при выделении большого количества тепла. Температуру раствора медленно повышали до 55°С на протяжении 1 часа и раствор перемешивали в течение 2 часов при охлаждении снова до 25°С.Образованный метоксифениллитий разлагали добавлением по каплям 18,5 г перегнанного и очищенного трет-бутилхлорида на протяжении 45 минут. Прозрачный красно-желтый раствор кипятили в течение 5 минут и затем снова охлаждали до -4°С.
К охлажденному раствору на протяжении 1 часа по каплям добавляли 19,6 г вышеполученного ди-п-толуолсульфоната (2R,3R)-бутандиола, растворенного в 100 мл сухого ТГФ. Раствор медленно нагревали до комнатной температуры и затем перемешивали в течение 30 минут. К нему добавляли 30 мл продутой азотом воды и ТГФ удаляли дистилляцией при пониженном давлении, таким образом получая бесцветный продукт типа масла. Продукт два раза экстрагировали 150 мл эфира и затем сушили Na2SO4. Эфирный экстракт фильтровали и обрабатывали раствором 8,4 г гексагидрата перхлората никеля в 50 мл этанола в атмосфере азота. Na2SO4, оставшийся в фильтре, тщательно промывали эфиром и затем эфирный раствор добавляли к раствору соединения никеля. Красно-коричневый продукт типа масла, который часто содержал желтые кристаллы, был [Ni((2S,3S)-бис(ди-п-метоксифенил)фосфорбутан)2](ClO4)2. Смесь масла и кристаллов добавляли к 8,4 г тиоцианата натрия (NaNCS), растворенного в 50 мл горячего этанола, и раствор интенсивно перемешивали в течение нескольких часов до образования однородного желто-коричневого твердого вещества, [Ni((2S,3S)-бис(ди-п-метоксифенил)фосфорбутан)2NCS]NCS. Твердый продукт полностью промывали этанолом и затем промывали эфиром.
17 г комплекса никеля суспендировали в 150 мл этанола в атмосфере азота и нагревали с перемешиванием. К суспензии быстро добавляли раствор 4 г цианида натрия (NaCN). Комплекс никеля медленно растворяли с получением светло-красного раствора [Ni((2S,3S)-бис(ди-п-метоксифенил)фосфорбутан)2CN3]-, который затем превращался в мутный раствор бежевого цвета. Горячий раствор перемешивали до образования желтой суспензии. Суспензию охлаждали и твердое вещество промывали два раза 25 мл воды и быстро охлаждали охлажденным льдом этанолом. Содержащее примеси твердое вещество бежевого цвета сушили при 25°С, добавляли 125 мл кипящего безводного этанола и затем фильтровали через фритту. Фильтрование через фритту проводили при комнатной температуре в течение 12 часов и в результате этого фильтрат полностью удалялся и оставалось только бесцветное блестящее твердое вещество. Твердое вещество перекристаллизовывали из 60 мл безводного этанола, получая таким образом 6,2 г полностью бесцветного, чистого (S,S)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2.
Пример 9: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (S,S)-(4-метоксифенил)2Р-СН(метил)СН(метил)Р(4-метоксифенила)2 и МАО
Повторяли способ примера 3, за исключением того, что (S,S)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученный в примере 3 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса продуктов реакции была 22,3 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 10: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (S,S)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2 и МАО
Повторяли способ примера 3, за исключением того, что (S,S)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученный в примере 3 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 12,8 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 11: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (S,S)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2 и МАО
Повторяли способ примера 4, за исключением того, что (S,S)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученный в примере 3 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 24,1 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 4 получения катализатора: получение лиганда (R,R)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2
Повторяли способ примера 3 получения катализатора, за исключением того, что (2S,3S)-бутандиол применяли в качестве исходного вещества вместо (2R,3R)-бутандиола. Получали 6,2 г полностью бесцветного чистого (R,R)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2.
Пример 12: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (R,R)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2 и МАО
Повторяли способ примера 9, за исключением того, что (R,R)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученный в примере 4 получения катализатора, применяли вместо (S,S)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2 в качестве лиганда. В результате этого общая масса продуктов реакции была 25,7 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 13: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (R,R)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2 и МАО
Повторяли способ примера 10, за исключением того, что (R,R)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученный в примере 4 получения катализатора, применяли в качестве лиганда вместо (S,S)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2. В результате этого общая масса продуктов реакции была 10,3 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 14: тетрамеризация этилена с применением Cr(2-этилгексаноата)3 (R,R)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2 и МАО
Повторяли способ примера 11, за исключением того, что (R,R)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученный в примере 4 получения катализатора, применяли в качестве лиганда вместо (S,S)-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2. В результате этого общая масса продуктов реакции была 27,5 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 5 получения катализатора: получение лиганда (S,S)-(2-метоксифенил)2РСН(метил)CH(метил)Р(2-метоксифенил)2
Получение катализатора проводили, как описано в В.Bosnich et al., J. Am. Chem. Soc. 99(19) (1977).
Ди-п-толуолсульфонат (2R,3R)-бутандиола получали из (2R,3R)-бутандиола. Этот способ получения проводили, как описано в R.В.Mitra et al., J. Am. Chem. Soc 84 (1962). 100 мл (1,24 моль) сухого пиридина помещали в 1-литровую колбу на бане со смесью лед-вода и смешивали с 100 г (0,525 моль) п-толуолсульфонилхлорида и затем медленно, по каплям, добавляли 22 мл (0,245 моль) (2R,3R)-бутандиола. После повышения температуры раствора до комнатной температуры на протяжении 20 минут полутвердую смесь выдерживали при комнатной температуре на протяжении ночи. К смеси добавляли избыточное количество льда в виде кусочков и смесь тщательно встряхивали так, чтобы не образовывалась сплошная масса. После медленного отделения кристаллического порошка его перемешивали вместе с кусочками льда в течение 2 часов и к смеси при интенсивном перемешивании добавляли измельченные кусочки льда и 70 мл концентрированной хлористоводородной кислоты. Экстрагированную суспензию фильтровали, осадок полностью промывали водой и сушили, таким образом получая 85 г (86,3%) ди-п-толуолсульфоната (2R,3R)-бутандиола (т.пл. 62-64°С).
Получение три-(2-метоксифенил)фосфина проводили следующим образом. 91,1 г (3,75 моль) кусочков магния медленно добавляли к 95 мл (0,75 моль) 2-броманизола в 2 литрах ТГФ. После интенсивной реакции реакционную смесь нагревали при кипячении с обратным холодильником в течение 2 часов, получая при этом реактив Гриньяра. Реактив Гриньяра добавляли по каплям к раствору 17,5 мл (0,2 моль) PCl3 в 2 литрах ТГФ при -78°С на протяжении 2 часов с перемешиванием. После завершения добавления по каплям баню сухой лед/ацетон удаляли и реакционную смесь нагревали до комнатной температуры. Реакционную смесь перемешивали на протяжении ночи и растворитель удаляли в вакууме. Фосфиновый продукт применяли в следующей стадии без выделения.
В 1-литровую трехгорлую круглодонную колбу, снабженную 250-миллилитровой капельной воронкой, парциальным конденсатором горячего орошения и вводом для азота, загружали 70 г перекристаллизованного три-(2-метоксифенола) и 300 мл сухого тетрагидрофурана. К раствору при 25°С в атмосфере азота и при перемешивании добавляли 2,8 г тонких кусочков лития. В растворе сразу образовывался LiP(4-OMe-Ph)2 и раствор изменял цвет в темно-красно-желтый при выделении большого количества тепла. Температуру раствора медленно повышали до 55°С на протяжении 1 часа и раствор перемешивали в течение 2 часов при охлаждении его снова до 25°С. Образованный 2-метоксифениллитий разлагали добавлением по каплям 18,5 г перегнанного и очищенного трет-бутилхлорида на протяжении 45 минут. Прозрачный красно-желтый раствор кипятили в течение 5 минут и затем снова охлаждали до -4°С.
К охлажденному раствору по каплям на протяжении 1 часа добавляли 19,6 г вышеполученного ди-п-толуолсульфоната (2R,3R)-бутандиола, растворенного в 100 мл сухого ТГФ. Раствор медленно нагревали до комнатной температуры и затем перемешивали в течение 30 минут. К нему добавляли 30 мл продутой азотом воды и ТГФ удаляли дистилляцией при пониженном давлении, таким образом получая бесцветный продукт типа масла. Продукт два раза экстрагировали 150 мл эфира и затем сушили Na2SO4. Эфирный экстракт фильтровали и обрабатывали раствором 8,4 г гексагидрата перхлората никеля в 50 мл этанола в атмосфере азота. Na2SO4, оставшийся в фильтре, тщательно промывали эфиром и затем эфирный раствор добавляли к раствору соединения никеля. Красно-коричневый продукт типа масла, который содержал желтые кристаллы, был соединением [Ni((2S,3S)-бис(ди-п-метоксифенил)фосфорбутан)2](ClO4)2. Смесь масла и кристаллов добавляли к 8,4 г тиоцианата натрия (NaNCS), растворенного в 50 мл горячего этанола, и раствор интенсивно перемешивали в течение нескольких часов до образования однородного желто-коричневого твердого вещества, [Ni((2S,3S)-бис(ди-п-метоксифенил)фосфорбутан)2NCS]NCS. Твердый продукт полностью промывали этанолом и затем промывали эфиром.
17 г комплекса никеля суспендировали в 150 мл этанола в атмосфере азота и нагревали с перемешиванием. К суспензии быстро добавляли раствор 4 г цианида натрия (NaCN). Комплекс никеля медленно растворяли с получением светло-красного раствора [Ni((2S,3S)-бис(ди-п-метоксифенил)фосфорбутан)2CN3]-, который затем превращался в мутный раствор бежевого цвета. Горячий раствор перемешивали до образования желтой суспензии. Суспензию охлаждали и твердое вещество промывали два раза 25 мл воды и быстро охлаждали охлажденным льдом этанолом. Содержащее примеси твердое вещество бежевого цвета сушили при 25°С, добавляли к 125 мл кипящего безводного этанола и затем фильтровали через фритту. Фильтрование через фритту проводили при комнатной температуре в течение 12 часов и в результате этого фильтрат был полностью удален и оставалось только бесцветное блестящее твердое вещество. Твердое вещество перекристаллизовывали из 60 мл безводного этанола, получая таким образом 6,8 г полностью бесцветного, чистого (S,S)-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенил)2.
Пример 6 получения катализатора: получение лиганда (R,R)-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенил)2
Повторяли способ примера 5 получения катализатора, за исключением того, что (2S,3S)-бутандиол применяли в качестве исходного вещества вместо (2R,3R)-бутандиола. Получали 5,3 г полностью бесцветного чистого (R,R)-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенила)2.
Пример 7 получения катализатора: получение лиганда (S,S)-(2-этилфенил)2РСН(метил)CH(метил)Р(2-этилфенил)2
Повторяли способ примера 5 получения катализатора, за исключением того, что 2-бензилбромид применяли для получения три-(2-этилфенил)фосфина. Получали 5,7 г полностью бесцветного чистого (S,S)-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенила)2.
Пример 8 получения катализатора: получение лиганда (R,R)-(2-этилфенил)2РСН(метил)CH(метил)Р(2-этилфенил)2
Повторяли способ примера 5 получения катализатора, за исключением того, что (2S,3S)-бутандиол применяли в качестве исходного соединения реакции вместе (2R,3R)-бутандиола и 2-бензилбромид применяли для получения три-(2-этилфенил)фосфина. Получали 4,6 г полностью бесцветного чистого (R,R)-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенила)2.
Пример 9 получения катализатора: получение лиганда (S,S)-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2
Повторяли способ примера 1 получения катализатора, за исключением того, что (1R,2R)-1,2-дифенилэтандиол применяли в качестве исходного соединения. Получали 3,3 г полностью бесцветного чистого (1S,2S)-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2.
Пример 15: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (S,S)-(фенил)РСН(фенил)СН(фенил)Р(фенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (S,S)-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2, полученный в примере 9 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 15,7 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 16: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (S,S)-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2 и МАО
Повторяли способ примера 3, за исключением того, что (S,S)-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2, полученный в примере 9 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 10,1 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 17: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (S,S)-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2 и МАО
Повторяли способ примера 4, за исключением того, что (S,S)-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2, полученный в примере 3 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 21,5 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 10 получения катализатора: получение лиганда (R,R)-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2
Повторяли способ примера 1 получения катализатора, за исключением того, что (1S,2S)-1,2-дифенилэтандиол применяли в качестве исходного соединения. Получали 1,5 г полностью бесцветного чистого (1R,2R)-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2.
Пример 18: тримеризация этилена с применением Cr(III)(ацетилацетоната)3, (R,R)-(фенил)РСН(фенил)СН(фенил)Р(фенила)2 и МАО
Повторяли способ примера 15, за исключением того, что (R,R)-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2, полученный в примере 10 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 16,3 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 19: тримеризация этилена с применением CrCl3(тетрагидрофурана)3, (R,R)-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2 и МАО
Повторяли способ примера 16, за исключением того, что (R,R)-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2, полученный в примере 10 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 9,2 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 20: тримеризация этилена с применением Cr(2-этилгексаноата)3, (R,R)-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2 и МАО
Повторяли способ примера 17, за исключением того, что (R,R)-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2, полученный в примере 10 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 6,5 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 11 получения катализатора: получение лиганда (1S,2S)-транс-бис(дифенилфосфино)циклогексана
Повторяли способ примера 1 получения катализатора, за исключением того, что (1R,2R)-транс-циклогександиол применяли в качестве исходного соединения вместо (2R,3R)-бутандиола. Получали 3,6 г полностью бесцветного чистого (1S,2S)-транс-бис(дифенилфосфино)циклогексана.
Пример 21: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (1S,2S)-транс-бис(дифенилфосфино)циклогексана и МАО
Повторяли способ примера 1, за исключением того, что (1S,2S)-транс-бис(дифенилфосфино)циклогексан, полученный в примере 11 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 77,5 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 22: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (1S,2S)-транс-бис(дифенилфосфино)циклогексана и МАО
Повторяли способ примера 3, за исключением того, что (1S,2S)-транс-бис(дифенилфосфино)циклогексан, полученный в примере 11 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 52,3 г. Распределение продуктов этого примера суммировано ниже в таблице 1.
Пример 23: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (1S,2S)-транс-бис(дифенилфосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что (1S,2S)-транс-бис(дифенилфосфино)циклогексан, полученный в примере 11 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 74,9 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 12 получения катализатора: получение лиганда (1Р,2Р)-транс-бис(дифенилфосфино)циклогексана
Повторяли способ примера 1 получения катализатора, за исключением того, что (1S,2S)-транс-циклогександиол применяли в качестве исходного соединения вместо (2R,3R)-бутандиола. Получали 3,9 г полностью бесцветного чистого (1R,2R)-транс-бис(дифенилфосфино)циклогексана.
Пример 24: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (1R,2R)-транс-бис(дифенилфосфино)циклогексана и МАО
Повторяли способ примера 1, за исключением того, что (1R,2R)-транс-бис(дифенилфосфино)циклогексан, полученный в примере 12 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 83,5 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 25: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (1R,2R)-транс-бис(дифенилфосфино)циклогексана и МАО
Повторяли способ примера 3, за исключением того, что (1R,2R)-транс-бис(дифенилфосфино)циклогексан, полученный в примере 12 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 56,4 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 26: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (1R,2R)-транс-бис(дифенилфосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что (1R,2R)-транс-бис(дифенилфосфино)циклогексан, полученный в примере 12 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 75,6 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 13 получения катализатора: получение лиганда (1S,2S)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана
Повторяли способ примера 3 получения катализатора, за исключением того, что (1R,2R)-транс-циклогександиол применяли в качестве исходного соединения вместо (2R,3R)-бутандиола. Получали 3,8 г полностью бесцветного чистого (1S,2S)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана.
Пример 27: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (1S,2S)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 1, за исключением того, что (1S,2S)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексан, полученный в примере 13 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 124 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 28: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (1S,2S)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 3, за исключением того, что (1S,2S)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексан, полученный в примере 13 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 82,7 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 29: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (1S,2S)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что (1S,2S)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексан, полученный в примере 13 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 110,6 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 14 получения катализатора: получение лиганда (1R,2R)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана
Повторяли способ примера 3 получения катализатора, за исключением того, что (1S,2S)-транс-циклогександиол применяли в качестве исходного соединения вместо (2R,3R)-бутандиола. Получали 3,9 г полностью бесцветного чистого (1R,2R)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана.
Пример 30: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (1R,2R)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 1, за исключением того, что (1R,2R)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексан, полученный в примере 14 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 123,8 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 31: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (1R,2R)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 3, за исключением того, что (1R,2R)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексан, полученный в примере 14 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 90,2 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 32: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (1R,2R)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что (1R,2R)-транс-бис(ди-(4-метоксифенил)фосфино)циклогексан, полученный в примере 14 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 134 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 15 получения катализатора: получение лиганда (S,S)-(4-метилфенил)2РСН(метил)CH(метил)Р(4-метилфенил)2
Повторяли способ примера 3 получения катализатора, за исключением того, что 4-толилбромид применяли для получения три-(4-метилфенил)фосфина. Получали 3,9 г полностью бесцветного чистого (S,S)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2.
Пример 33: тетрамеризация этилена с применением Cr(III)ацетилацетоната)3, (S,S)-(4-метилфенил)2РСН(метил)CH(метил)Р(4-метилфенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (S,S)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2, полученный в примере 15 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 55,9 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 34: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (S,S)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2 и МАО
Повторяли способ примера 3, за исключением того, что (S,S)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2, полученный в примере 15 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 24,8 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 35: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (S,S)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2 и МАО
Повторяли способ примера 4, за исключением того, что (S,S)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2, полученный в примере 15 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 42,1 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 16 получения катализатора: получение лиганда (R,R)-(4-метилфенил)2РСН(метил)СН(метил)Р(-4-метилфенил)2
Повторяли способ примера 15 получения катализатора, за исключением того, что (2S,3S)-бутандиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 4,5 г полностью бесцветного чистого (R,R)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2.
Пример 36: тетрамеризация этилена с применением Cr(III)ацетилацетоната)3, (R,R)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2 и МАО
Повторяли способ примера 33, за исключением того, что (R,R)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2, полученный в примере 16 получения катализатора, применяли вместо (S,S)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 50,4 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 37 тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, (R,R)-(4-метилфенил)2РСН(метил)CH(метил)Р(4-метилфенила)2 и МАО
Повторяли способ примера 34, за исключением того, что (R,R)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2, полученный в примере 16 получения катализатора, применяли в качестве лиганда вместо (S,S)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2. В результате этого общая масса продуктов реакции была 22,1 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 38: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, (R,R)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2 и МАО
Повторяли способ примера 35, за исключением того, что (R,R)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2, полученный в примере 16 получения катализатора, применяли в качестве лиганда вместо (S,S)-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2. В результате этого общая масса продуктов реакции была 46,5 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 17 получения катализатора: получение лиганда (1S,2S)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана
Повторяли способ примера 5 получения катализатора, за исключением того, что (1R,2R)-транс-циклогександиол применяли в качестве исходного соединения вместо (2R,3R)-бутандиола. Получали 3,3 г полностью бесцветного чистого (1S,2S)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана.
Пример 39: тримеризация этилена с применением Cr(III)(ацетилацетоната)3, (1S,2S)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 1, за исключением того, что (1S,2S)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексан, полученный в примере 17 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 63,4 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 40: тримеризация этилена с применением CrCl3(тетрагидрофурана)3, (1S,2S)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 3, за исключением того, что (1S,2S)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексан, полученный в примере 17 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 26,8 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 41: тримеризация этилена с применением Cr(2-этилгексаноата)3, (1S,2S)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что (1S,2S)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексан, полученный в примере 17 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 43,4 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 18 получения катализатора: получение лиганда (1R,2R)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана
Повторяли способ примера 5 получения катализатора, за исключением того, что (1S,2S)-транс-циклогександиол применяли в качестве исходного соединения вместо (2R,3R)-бутандиола. Получали 2,5 г полностью бесцветного чистого (1R,2R)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана.
Пример 42: тримеризация этилена с применением Cr(III)(ацетилацетоната)3, (1R,2R)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 1, за исключением того, что (1R,2R)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексан, полученный в примере 18 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 75,4 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 43: тримеризация этилена с применением CrCl3(тетрагидрофурана)3, (1R,2R)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 3, за исключением того, что (1R,2R)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексан, полученный в примере 18 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 20,4 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 44: тримеризация этилена с применением Cr(2-этилгексаноата)3, (1R,2R)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что (1R,2R)-транс-бис(ди-(2-метоксифенил)фосфино)циклогексан, полученный в примере 18 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса продуктов реакции была 38,2 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 19 получения катализатора: получение лиганда (1S,2S)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана
Повторяли способ примера 5 получения катализатора, за исключением того, что (1R,2R)-транс-циклогександиол применяли в качестве исходного соединения вместо (2R,3R)-бутандиола и 2-бензилбромид применяли для получения три-(2-этилфенил)фосфина. Получали 4,1 г полностью бесцветного чистого (1S,2S)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана.
Пример 45: тримеризации этилена с применением Cr(III)(ацетилацетоната)3, (1S,2S)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана и МАО
Повторяли способ примера 1, за исключением того, что (1S,2S)-транс-бис(ди-(2-этилфенил)фосфино)циклогексан, полученный в примере 19 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 43,2 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 46: тримеризация этилена с применением CrCl3(тетрагидрофурана)3, (1S,2S)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана и МАО
Повторяли способ примера 3, за исключением того, что (1S,2S)-транс-бис(ди-(2-этилфенил)фосфино)циклогексан, полученный в примере 19 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 16,3 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 47: тримеризация этилена с применением Cr(2-этилгексаноата)3, (1S,2S)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что (1S,2S)-транс-бис(ди-(2-этилфенил)фосфино)циклогексан, полученный в примере 19 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 28,3 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 20 получения катализатора: получение лиганда (1R,2R)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана
Повторяли способ примера 5 получения катализатора, за исключением того, что (1S,2S)-транс-циклогександиол применяли в качестве исходного соединения вместо (2R,3R)-бутандиола и 2-бензилбромид применяли для получения три-(2-этилфенил)фосфина. Получали 2,9 г полностью бесцветного чистого (1R,2R)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана.
Пример 48: тримеризации этилена с применением Cr(III)(ацетилацетоната)3, (1R,2R)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана и МАО
Повторяли способ примера 1, за исключением того, что (1R,2R)-транс-бис(ди-(2-этилфенил)фосфино)циклогексан, полученный в примере 20 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 50,5 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 49: тримеризация этилена с применением CrCl3(тетрагидрофурана)3, (1R,2R)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана и МАО
Повторяли способ примера 3, за исключением того, что (1R,2R)-транс-бис(ди-(2-этилфенил)фосфино)циклогексан, полученный в примере 20 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 21,3 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 50: тримеризация этилена с применением Cr(2-этилгексаноата)3, (1R.2R)-транс-бис(ди-(2-этилфенил)фосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что (1R,2R)-транс-бис(ди-(2-этилфенил)фосфино)циклогексан, полученный в примере 20 получения катализатора, применяли в качестве лиганда вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2. В результате этого общая масса полученных продуктов реакции была 23,4 г. Распределение продуктов этого примера суммировано ниже в таблице 2.
Пример 21 получения катализатора: получение лиганда (S,S)-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенил)2
Повторяли способ примера 3 получения катализатора, за исключением того, что 3-толилбромид применяли для получения три-(2-метилфенил)фосфина.
Получали 3,6 г полностью бесцветного чистого (S,S)-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенила)2.
Пример 22 получения катализатора: получение лиганда (R,R)-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенил)2
Повторяли способ примера 21 получения катализатора, за исключением того, что (2S,3S)-бутандиол применяли в качестве исходного соединения вместо (2R,3R)-бутандиола. Получали 4,0 г полностью бесцветного чистого (R,R)-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенила)2.
Сравнительный пример 1 получения катализатора: получение лиганда мезо-(фенил)2РСН(метил)CH(метил)Р(фенил)2
Повторяли способ примера 1 получения катализатора, за исключением того, что мезо-бутандиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 5,7 г полностью бесцветного чистого мезо-(фенил)2РСН(метил)СН(метил)Р(фенила)2.
Сравнительный пример 1: тетрамеризация этилена с применением Cr(III)ацетилацетоната)3, лиганда мезо-(фенил)2РСН(метил)СН(метил)Р(фенила)2 и МАО
Повторяли способ примера 1, за исключением того, что мезо-(фенил)2РСН(метил)СН(метил)Р(фенил)2, полученный в сравнительном примере 1 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 7,3 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 2: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, лиганда мезо-(фенил)2РСН(метил)СН(метил)Р(фенил)2 и МАО
Повторяли способ примера 3, за исключением того, что мезо-(фенил)2РСН(метил)СН(метил)Р(фенил)2, полученный в сравнительном примере 1 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 4,3 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 3: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, лиганда мезо-(фенил)2РСН(метил)СН(метил)Р(фенил)2 и МАО
Повторяли способ примера 4, за исключением того, что мезо-(фенил)2РСН(метил)СН(метил)Р(фенил)2, полученный в сравнительном примере 1 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила) в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 6,8 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 2 получения катализатора: получение лиганда мезо-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2
Повторяли способ примера 3 получения катализатора, за исключением того, что мезо-бутандиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 3,3 г полностью бесцветного чистого мезо-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2.
Сравнительный пример 4: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, мезо-(4-метоксифенил)2РСН(метил)CH(метил)Р(4-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что мезо-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученный в сравнительном примере 2 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 2,3 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 5: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, мезо-(4-метоксифенил)2РСН(метил)CH(метил)Р(4-метоксифенила)2 и МАО
Повторяли способ примера 3, за исключением того, что мезо-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученный в сравнительном примере 2 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 3,5 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 6: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, мезо-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенила)2 и МАО
Повторяли способ примера 4, за исключением того, что мезо-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученный в сравнительном примере 2 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 3,9 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 3 получения катализатора: получение лиганда мезо-(2-метоксифенил)2РСН(метил)CH(метил)Р(2-метоксифенил)2
Повторяли способ примера 5 получения катализатора, за исключением того, что мезо-бутандиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 4,5 г полностью бесцветного чистого мезо-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенила)2.
Сравнительный пример 7: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, мезо-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что мезо-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенил)2, полученный в сравнительном примере 3 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 5,2 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 8: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, мезо-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенила)2 и МАО
Повторяли способ примера 3, за исключением того, что мезо-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенил)2, полученный в сравнительном примере 3 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 6,5 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 9: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, мезо-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенила)2 и МАО
Повторяли способ примера 4, за исключением того, что мезо-(2-метоксифенил)2РСН(метил)СН(метил)Р(2-метоксифенил)2, полученный в сравнительном примере 3 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 4,8 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 4 получения катализатора: получение лиганда мезо-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенил)2
Повторяли способ примера 5 получения катализатора, за исключением того, что мезо-бутандиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола и 2-бензилбромид применяли для получения три-(2-этилфенил)фосфина. Получали 4,4 г полностью бесцветного чистого мезо-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенила)2.
Сравнительный пример 10: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, мезо-(2-этилфенил)2РСН(метил)CH(метил)Р(2-этилфенила)2 и МАО
Повторяли способ примера 1, за исключением того, что мезо-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенил)2, полученный в сравнительном примере 4 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 10,5 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 11: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, мезо-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенила)2 и МАО
Повторяли способ примера 3, за исключением того, что мезо-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенил)2, полученный в сравнительном примере 4 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 5,3 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 12: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, мезо-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенила)2 и МАО
Повторяли способ примера 4, за исключением того, что мезо-(2-этилфенил)2РСН(метил)СН(метил)Р(2-этилфенил)2, полученный в сравнительном примере 4 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 6,2 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 5 получения катализатора: получение мезо-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2
Повторяли способ примера 1 получения катализатора, за исключением того, что (1R,2S)-1,2-дифенилэтандиол применяли в качестве исходного соединения реакции. Получали 2,6 г полностью бесцветного чистого мезо-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2.
Сравнительный пример 13: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, мезо-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2 и МАО
Повторяли способ примера 1, за исключением того, что мезо-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2, полученный в сравнительном примере 5 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 8,5 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 14: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, мезо-(фенил)2РСН(фенил)СН(фенил)Р(фенила)2 и МАО
Повторяли способ примера 3, за исключением того, что мезо-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2, полученный в сравнительном примере 5 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 3,2 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 15: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, лиганда мезо-(фенил)2РСН(фенил)CH(фенил)Р(фенила)2 и МАО
Повторяли способ примера 4, за исключением того, что мезо-(фенил)2РСН(фенил)СН(фенил)Р(фенил)2, полученный в сравнительном примере 5 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СК(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 3,8 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 6 получения катализатора: получение лиганда цис-1,2-бис(дифенилфосфино)циклогексана
Повторяли способ примера 1 получения катализатора, за исключением того, что цис-1,2-циклогександиол применяли в качестве исходного соединения реакции. Получали 4,3 г полностью бесцветного чистого цис-1,2-бис(дифенилфосфино)циклогексана.
Сравнительный пример 16: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, лиганда цис-1,2-бис(дифенилфосфино)циклогексан и МАО
Повторяли способ примера 1, за исключением того, что цис-1,2-бис(дифенилфосфино)циклогексан, полученный в сравнительном примере 6 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 15,4 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 17: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, лиганда цис-1,2-бис(дифенилфосфино)циклогексан и МАО
Повторяли способ примера 3, за исключением того, что цис-1,2-бис(дифенилфосфино)циклогексан, полученный в сравнительном примере 6 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 7 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 18: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, цис-1,2-бис(дифенилфосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что цис-1,2-бис(дифенилфосфино)циклогексан, полученный в сравнительном примере 6 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 10,8 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 7 получения катализатора: получение лиганда цис-1,2-бис(ди-(4-метоксифенил)фосфино)циклогексана
Повторяли способ примера 3 получения катализатора, за исключением того, что цис-1,2-циклогександиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 3,0 г полностью бесцветного чистого цис-1,2-бис(ди-(4-метоксифенил)фосфино)циклогексана.
Сравнительный пример 19: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, лиганда цис-1,2-бис(ди-(4-метоксифенил)фосфино)циклогексан и МАО
Повторяли способ примера 1, за исключением того, что цис-1,2-бис(ди-(4-метоксифенил)фосфино)циклогексан, полученный в сравнительном примере 7 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 3,9 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 20: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, лиганда цис-1,2-бис(ди-(4-метоксифенил)фосфино)циклогексан и МАО
Повторяли способ примера 3, за исключением того, что цис-1,2-бис(ди-(4-метоксифенил)фосфино)циклогексан, полученный в сравнительном примере 7 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 2,4 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 21: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, лиганда цис-1,2-бис(ди-(4-метоксифенил)фосфино)циклогексан и МАО
Повторяли способ примера 4, за исключением того, что цис-1,2-бис(ди-(4-метоксифенил)фосфино)циклогексан, полученный в сравнительном примере 7 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 2,8 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 8 получения катализатора: получение лиганда цис-1,2-бис(ди-(2-метоксифенил)фосфино)циклогексана
Повторяли способ примера 5 получения катализатора, за исключением того, что цис-1,2-циклогександиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 3,9 г полностью бесцветного чистого цис-1,2-бис(ди-(2-метоксифенил)фосфино)циклогексана.
Сравнительный пример 22: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, цис-1,2-бис(ди-(2-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 1, за исключением того, что цис-1,2-бис(ди-(2-метоксифенил)фосфино)циклогексан, полученный в сравнительном примере 8 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 7,1 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 23: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, цис-1,2-бис(ди-(2-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 3, за исключением того, что цис-1,2-бис(ди-(2-метоксифенил)фосфино)циклогексан, полученный в сравнительном примере 8 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 2,9 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 24: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, цис-1,2-бис(ди-(2-метоксифенил)фосфино)циклогексана и МАО
Повторяли способ примера 4, за исключением того, что цис-1,2-бис(ди-(2-метоксифенил)фосфино)циклогексан, полученный в сравнительном примере 8 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 4,7 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 9 получения катализатора: получение лиганда цис-1,2-бис(ди-(2-этилфенил)фосфино)циклогексан
Повторяли способ примера 5 получения катализатора, за исключением того, что цис-1,2-циклогександиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола и 2-бензилбромид применяли для получения три-(2-этилфенил)фосфина. Получали 3,0 г полностью бесцветного чистого цис-1,2-бис(ди-(2-этилфенил)фосфино)циклогексана.
Сравнительный пример 25: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, лиганда цис-1,2-бис(ди-(2-этилфенил)фосфино)циклогексан и МАО
Повторяли способ примера 1, за исключением того, что цис-1,2-бис(ди-(2-этилфенил)фосфино)циклогексан, полученный в сравнительном примере 9 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 7,9 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 26: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, лиганда цис-1,2-бис(ди-(2-этилфенил)фосфино)циклогексан и МАО
Повторяли способ примера 3, за исключением того, что цис-1,2-бис(ди-(2-этилфенил)фосфино)циклогексан, полученный в сравнительном примере 9 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 6,3 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 27: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, лиганда цис-1,2-бис(ди-(2-этилфенил)фосфино)циклогексан и МАО
Повторяли способ примера 4, за исключением того, что цис-1,2-бис(ди-(2-этилфенил)фосфино)циклогексан, полученный в сравнительном примере 9 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 3,1 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 10 получения катализатора: получение лиганда мезо-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2
Повторяли способ примера 15 получения катализатора, за исключением того, что мезо-бутандиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 4,5 г полностью бесцветного чистого мезо-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2.
Сравнительный пример 28: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, мезо-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)3 и МАО
Повторяли способ примера 1, за исключением того, что мезо-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2, полученный в сравнительном примере 10 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 12,3 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 29: тетрамеризация этилена с применением CrCl3(тетрагидрофурана)3, мезо-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2 и МАО
Повторяли способ примера 3, за исключением того, что мезо-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2, полученный в сравнительном примере 10 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 4,6 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 30: тетрамеризация этилена с применением Cr(2-этилгексаноата)3, мезо-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенила)2 и МАО
Повторяли способ примера 4, за исключением того, что мезо-(4-метилфенил)2РСН(метил)СН(метил)Р(4-метилфенил)2, полученный в сравнительном примере 10 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 6,3 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 11 получения катализатора: получение лиганда мезо-(2-метилфенил)2РСН(метил)CH(метил)Р(2-метилфенил)2
Повторяли способ примера 21 получения катализатора, за исключением того, что мезо-бутандиол применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 3,6 г полностью бесцветного чистого мезо-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенила)2.
Сравнительный пример 31: тримеризация этилена с применением Cr(III)(ацетилацетоната)3, мезо-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенила)2 и МАО
Повторяли способ примера 1, за исключением того, что мезо-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенил)2, полученный в сравнительном примере 11 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 2,5 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 32: тримеризация этилена с применением CrCl3(тетрагидрофурана)3, мезо-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенила)2 и МАО
Повторяли способ примера 3, за исключением того, что мезо-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенил)2, полученный в сравнительном примере 11 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 1,5 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 33: тримеризация этилена с применением Cr(2-этилгексаноата)3, мезо-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилсренила)2 и МАО
Повторяли способ примера 4, за исключением того, что мезо-(2-метилфенил)2РСН(метил)СН(метил)Р(2-метилфенил)2, полученный в сравнительном примере 11 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 1,6 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 12 получения катализатора: получение лиганда (фенил)2РСН2СН2Р(фенил)2
Согласно способу, описанному в R. N. Salvatore et al., Tetrahedron Letters 44 (2003), лиганд получали предоставлением возможности дифенилфосфину реагировать с 2 молярными эквивалентами дибромалкана в присутствии диметилформамида (ДМФА) и гидроксида цезия. В частности, 360 мг (2,14 ммоль) моногидратированного гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, содержащего 1,0 г активированного порошка молекулярного сита, имеющего размер пор 4Å, и смесь перемешивали в атмосфере азота. К смеси добавляли 0,38 мл (2,14 ммоль) диметилфосфина и смесь перемешивали при комнатной температуре в течение 1 часа с получением раствора темно-оранжевого цвета. К раствору по каплям добавляли 0,11 мл (1,29 ммоль) 1,2-дибромэтана, раствор становился белым. После предоставления раствору смеси возможности реагировать при комнатной температуре в течение 36 часов, к продукту реакции добавляли 60 мл дистиллированной воды и экстрагировали три раза 60 мл DMC. Органический слой промывали три раза дистиллированной водой и сушили безводным сульфатом натрия. Затем растворитель удаляли в вакууме и остаток перекристаллизовывали в растворителе бензола, получая таким образом 333 мг (выход 78%) чувствительных к действию воздуха белых кристаллов (333 мг, выход 78%).
Сравнительный пример 34: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (фенил)2РСН2СН2Р(фенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (фенил)2PCH2CH2P(фенил)2, полученный в сравнительном примере 12 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 5,5 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 35: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (фенил)2PCH2CH2P(фенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (фенил)2РСН2СН2Р(фенил)2, полученный в сравнительном примере 12 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда и давлением реакции было 4,5×106 Па (45 бар), В результате этого общая масса полученных продуктов реакции была 17,9 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 13 получения катализатора: получение лиганда (4-метоксифенил)2РСН2СН2Р(4-метоксифенил)2
Повторяли способ примера 3 получения катализатора, за исключением того, что этиленгликоль применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 4,1 г полностью бесцветного чистого (4-метоксифенил)2РСН2СН2Р(4-метоксифенила)2.
Сравнительный пример 36: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (4-метоксифенил)2PCH2CH2P(4-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (4-метоксифенил)2РСН2СН2Р(4-метоксифенил)2, полученный в сравнительном примере 13 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 6,3 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 37: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (4-метоксифенил)2РСН2СН2Р(4-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (4-метоксифенил)2РСН2СН2Р(4-метоксифенил)2, полученный в сравнительном примере 13 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда и давлением реакции было 4,5×106 Па (45 бар). В результате этого общая масса полученных продуктов реакции была 12,9 г. Распределение продуктов этого примера суммировано ниже в таблице 3.
Сравнительный пример 14 получения катализатора: получение лиганда (2-метоксифенил)2РСН2СН2Р(2-метоксифенил)2
Повторяли способ примера 5 получения катализатора, за исключением того, что этиленгликоль применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 3,5 г полностью бесцветного чистого (2-метоксифенил)2РСН2СН2Р(2-метоксифенила)2.
Сравнительный пример 38: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (2-метоксифенил)2РСН2СН2Р(2-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (2-метоксифенил)2РСН2СН2Р(2-метоксифенил)2, полученный в сравнительном примере 14 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого не был получен никакой продукт реакции.
Сравнительный пример 39: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (2-метоксифенил)2РСН2СН2Р(2-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (2-метоксифенил)2РСН2СН2Р(2-метоксифенил)2, полученный в сравнительном примере 14 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда и давлением реакции было 4,5×106 Па (45 бар). В результате этого не был получен никакой продукт реакции.
Сравнительный пример 15 получения катализатора: получение лиганда (2-зтмлфенил)2PCH2CH2P(2-этилфенил)2.
Повторяли способ примера 5 получения катализатора, за исключением того, что этиленгликоль применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола и 2-бензилбромид применяли для получения три-(2-этилфенил)фосфина. Получали 2,9 г полностью бесцветного чистого (2-этилфенил)2РСН2СН2Р(2-этилфенила)2.
Сравнительный пример 40: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (2-этилфенил)2РСН2СН2Р(2-этилфенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (2-этилфенил)2РСН2СН2Р(2-этилфенил)2, полученный в сравнительном примере 15 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого не был получен никакой продукт реакции.
Сравнительный пример 41: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (2-этилфенил)2РСН2СН2Р(2-этилфенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (2-этилфенил)2РСН2СН2Р(2-этилфенил)2, полученный в сравнительном примере 15 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда и давлением реакции было 4,5×106 Па (45 бар). В результате этого не был получен никакой продукт реакции.
Сравнительный пример 16 получения катализатора: получение лиганда (фенил)2РСН2Р(фенил)2
Повторяли способ сравнительного примера 10 получения катализатора, за исключением того, что дибромметан применяли в качестве исходного соединения реакции вместо 1,2-дибромэтана. Получали 390 мг полностью бесцветного чистого (фенил)2РСН2Р(фенила)2.
Сравнительный пример 42: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (фенил)2РСН2Р(фенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (фенил)2РСН2Р(фенил)2, полученный в сравнительном примере 16 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого общая масса полученных продуктов реакции была 1,2 г. Распределение продуктов этого примера суммировано ниже в таблице 4.
Сравнительный пример 43: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (фенил)2РСН2Р(фенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (фенил)2РСН2Р(фенил)2, полученный в сравнительном примере 16 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда и давлением реакции было 4,5×106 Па (45 бар). В результате этого общая масса полученных продуктов реакции была 1,7 г. Распределение продуктов этого примера суммировано ниже в таблице 4.
Сравнительный пример 17 получения катализатора: получение лиганда (4-метоксифенил)2РСН2Р(4-метоксифенил)2
Повторяли способ примера 3 получения катализатора, за исключением того, что дииодметан применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 4,7 г полностью бесцветного чистого (4-метоксифенил)2РСН2Р(4-метоксифенила)2.
Сравнительный пример 44: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (4-метоксифенил)2РСН2Р(4-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (4-метоксифенил)2РСН2Р(4-метоксифенил)2, полученный в сравнительном примере 17 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого не был получен никакой продукт реакции.
Сравнительный пример 45: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (4-метоксифенил)2РСН2Р(4-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (4-метоксифенил)2РСН2Р(4-метоксифенил)2, полученный в сравнительном примере 17 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда и давлением реакции было 4,5×106 Па (45 бар). В результате этого не был получен никакой продукт реакции.
Сравнительный пример 18 получения катализатора: получение лиганда (2-метоксифенил)2РСН2Р(2-метоксифенил)2
Повторяли способ примера 5 получения катализатора, за исключением того, что дииодметан применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 1,6 г полностью бесцветного чистого (2-метоксифенил)2РСН2Р(2-метоксифенила)2.
Сравнительный пример 46: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (2-метоксифенил)2РСН2Р(2-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (2-метоксифенил)2РСН2Р(2-метоксифенил)2, полученный в сравнительном примере 18 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого не был получен никакой продукт реакции.
Сравнительный пример 47: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (2-метоксифенил)2РСН2Р(2-метоксифенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (2-метоксифенил)2РСН2Р(2-метоксифенил)2, полученный в сравнительном примере 18 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда и давлением реакции было 4,5×106 Па (45 бар). В результате этого не был получен никакой продукт реакции.
Сравнительный пример 19 получения катализатора: получение лиганда (2-этилфенил)2РСН2Р(2-этилфенил)2
Повторяли способ примера 7 получения катализатора, за исключением того, что дииодметан применяли в качестве исходного соединения реакции вместо (2R,3R)-бутандиола. Получали 5,5 г полностью бесцветного чистого (2-этилфенил)2РСН2Р(2-этилфенила)2.
Сравнительный пример 48: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (2-этилфенил)2РСН2Р(2-этилфенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (2-этилфенил)2РСН2Р(2-этилфенил)2, полученный в сравнительном примере 19 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда. В результате этого не был получен никакой продукт реакции.
Сравнительный пример 49: тетрамеризация этилена с применением Cr(III)(ацетилацетоната)3, (2-этилфенил)2РСН2Р(2-этилфенила)2 и МАО
Повторяли способ примера 1, за исключением того, что (2-этилфенил)2РСН2Р(2-этилфенил)2, полученный в сравнительном примере 19 получения катализатора, применяли вместо (S,S)-(фенил)2РСН(метил)СН(метил)Р(фенила)2 в качестве лиганда и давлением реакции было 4,5×108 Па (45 бар). В результате этого не был получен никакой продукт реакции.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВЫСОКОАКТИВНЫЙ И ВЫСОКОСЕЛЕКТИВНЫЙ КАТАЛИЗАТОР ОЛИГОМЕРИЗАЦИИ ЭТИЛЕНА И СПОСОБ ПОЛУЧЕНИЯ ГЕКСЕНА ИЛИ ОКТЕНА С ПРИМЕНЕНИЕМ ДАННОГО КАТАЛИЗАТОРА | 2010 |
|
RU2541528C2 |
| КАТАЛИТИЧЕСКИЕ СИСТЕМЫ ДЛЯ ТЕТРАМЕРИЗАЦИИ ЭТИЛЕНА И СПОСОБ ПОЛУЧЕНИЯ 1-ОКТЕНА С ИХ ПРИМЕНЕНИЕМ | 2008 |
|
RU2461423C2 |
| Галогенсодержащее соединение и его применение, состав катализатора и способы олигомеризации, тримеризации и тетрамеризации этилена | 2019 |
|
RU2802019C2 |
| ГАЛОГЕНСОДЕРЖАЩЕЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛИГАНДА В КАТАЛИЗАТОРЕ ОЛИГОМЕРИЗАЦИИ ЭТИЛЕНА | 2019 |
|
RU2802018C2 |
| СИСТЕМА АРИЛФЕНОКСИКАТАЛИЗАТОРА ДЛЯ ПОЛУЧЕНИЯ ГОМОПОЛИМЕРА ЭТИЛЕНА ИЛИ СОПОЛИМЕРОВ ЭТИЛЕНА И АЛЬФА-ОЛЕФИНОВ | 2005 |
|
RU2374267C2 |
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ИСПОЛЬЗУЕМЫХПРИ ПОЛУЧЕНИИИНГИБИТОРОВ NEP | 2011 |
|
RU2573824C2 |
| ДИФОСФИНЫ И МЕТАЛЛОКОМПЛЕКСЫ | 2006 |
|
RU2408600C2 |
| КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ОЛИГОМЕРИЗАЦИИ ЭТИЛЕНА | 2008 |
|
RU2467797C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА И ЕГО АНАЛОГОВ | 2013 |
|
RU2619928C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПИОНОВОЙ КИСЛОТЫ | 2010 |
|
RU2544989C2 |
Каталитическая система для селективной олигомеризации этилена, которая содержит лиганд с каркасом P-C-C-P формулы 1, 2, 3 или 4, соединение хрома или молибдена или вольфрама, и промотор. Описаны также способ получения 1-гексена селективной тримеризацией этилена и способ получения 1-октена селективной тетрамеризацией этилена с применением указанной каталитической системы. Активность катализатора является стабильной на протяжении времени, достаточного для того, чтобы скорость реакции можно было поддерживать постоянной. 3 н. и 4 з.п. ф-лы, 4 табл., 50 пр.
[Формула 1]
[Формула 2]
[Формула 3]
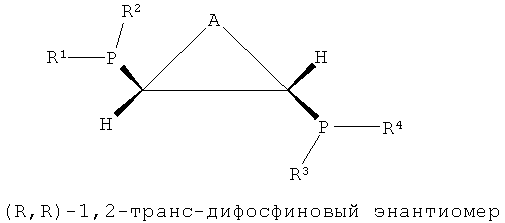
[Формула 4]
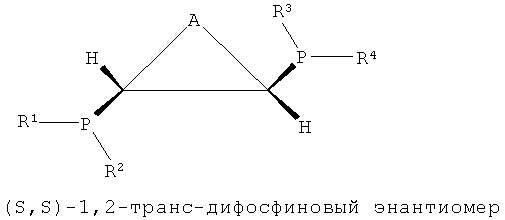
1. Каталитическая система для селективной олигомеризации этилена, которая содержит хром, или молибден, или вольфрам, или предшественник хрома, или молибдена, или вольфрама, промотор и лиганд, представленный одной из следующих формул 1-4:
[Формула 1]
[Формула 2]
[Формула 3]
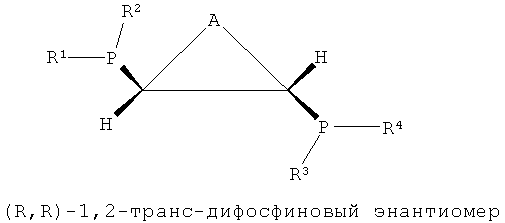
[Формула 4]
где каждый из R1, R2, R3 и R4 независимо выбран из группы, состоящей из фенила, бензила, нафтила, антраценила, 4-метилфенила, 4-этилфенила, 4-изопропилфенила, 4-трет-бутилфенила, 4-метоксифенила, 4-изопропоксифенила, о-метилфенила, о-этилфенила, о-изопропилфенила, о-трет-бутилфенила, о-метоксифенила, о-изопропоксифенила, бифенила, каждый из R5 и R6 независимо выбран из алкила или арила и А выбран из группы, состоящей из алкилена.
2. Каталитическая система по п.1, где лиганд является стерически асимметричным относительно плоскости симметрии.
3. Каталитическая система по п.1, где лиганд выбран из группы, состоящей из (S,S)- или (R,R)-(фенил)2Р-СН(метил)СН(метил)-Р(фенила)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(метил)СН(метил)-Р(4-метоксифенила)2, (S,S)- или (R,R)-(4-метилфенил)2Р-СН(метил)СН(метил)-Р(4-метилфенила)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(метил)СН(метил)-Р(фенила)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(этил)СН(метил)-Р(4-этилфенила)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(этил)СН(метил)-Р(фенила)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(этил)СН(этил)-Р(4-этилфенила)2, (S,S)- или (R,R)-(фенил)2Р-СН(этил)СН(этил)-Р(фенила)2, (S,S)-или (R,R)-(фенил)2Р-СН(изопропил)СН(метил)-Р(фенила)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(изопропил)СН(метил)-Р(4-метоксифенила)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(изопропил)СН(метил)-Р(4-этилфенила)2, (S,S)- или (R,R)-(фенил)2Р-СН(н-пропил)СН(метил)-Р(фенила)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(н-пропил)СН(метил)-Р(4-метоксифенила)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(н-пропил)СН(метил)-Р(4-этилфенила)2, (S,S)- или (R,R)-(фенил)2Р-СН(изопропил)СН(этил)-Р(фенила)2, (S,S)- или (R,R)-(4-метоксифенил)2Р-СН(изопропил)СН(этил)-Р(4-метоксифенила)2, (S,S)- или (R,R)-(4-этилфенил)2Р-СН(изопропил)СН(этил)-Р(4-этилфенила)2, (S,S)- или (R,R)-транс-1,2-ди-(Р(фенил)2)циклогексана, (S,S)- или (R,R)-транс-1,2-ди-(Р(4-метоксифенил)2)циклогексана, (S,S)- или (Р,Р)-транс-1,2-ди-(Р(4-этилфенил)2)циклогексана, (S,S) или (Р,Р)-транс-1,2-ди-(Р(фенил)2)циклопентана, (S,S)- или (R,R)-транс-1,2-ди-(Р(4-метоксифенил)2)циклопентана, (S,S)- или (R,R)-1,2-ди-(Р(4-этилфенил)2)циклопентана, (S,S)- или (R,R)-(фенил)2Р-СН(фенил)СН(фенил)-Р(фенил)2, (1S,2S)- или (1R,2R)-транс-бис(дифенилфосфино)циклогексана, и (1S,2S)- или (1R,2R)-транс-бис(ди(4-метоксифенил)фосфино)циклогексана.
4. Каталитическая система по пп.1-3, где указанный хром или предшественник хрома выбран из группы, состоящей из ацетилацетоната хрома(III), трис(тетрагидрофуран)трихлорхрома и 2-этилгексаноата хрома(III).
5. Каталитическая система по пп.1-3, где промотором является метилалюминоксан (МАО) или этилалюминоксан (ЕАО).
6. Способ получения 1-гексена селективной тримеризацией этилена с применением каталитической системы по любому из пп.1-3.
7. Способ получения 1-октена селективной тетрамеризацией этилена с применением каталитической системы по любому из пп.1-3.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ТРИМЕРИЗАЦИЯ И ОЛИГОМЕРИЗАЦИЯ ОЛЕФИНОВ С ИСПОЛЬЗОВАНИЕМ КАТАЛИЗАТОРА, ВКЛЮЧАЮЩЕГО ИСТОЧНИК ХРОМА, МОЛИБДЕНА ИЛИ ВОЛЬФРАМА И ЛИГАНД, СОДЕРЖАЩИЙ ПО МЕНЬШЕЙ МЕРЕ ОДИН АТОМ ФОСФОРА, МЫШЬЯКА ИЛИ СУРЬМЫ, СВЯЗАННЫЙ С ПО МЕНЬШЕЙ МЕРЕ ОДНОЙ (ГЕТЕРО)УГЛЕВОДОРОДНОЙ ГРУППОЙ | 2001 |
|
RU2299096C2 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

