Область техники
Настоящее изобретение относится к титаносиликатному материалу и способу его получения. А именно, настоящее изобретение относится к содержащему благородный металл титаносиликатному материалу и способу его получения.
Предпосылки изобретения
Титаносиликат представляет собой новый тип гетероатомного молекулярного сита, разработанный в начале 1980-х годов. В настоящее время синтетические титаносиликатные молекулярные сита включают TS-1 со структурой типа MFI, TS-2 со структурой типа MEL, Ti-MCM-22 со структурой типа MWW и TS-48 со структурой с относительно более крупными порами. Среди упомянутых титаносиликатных молекулярных сит титаносиликат TS-1, разработанный компанией Enichem, Италия, представляет собой новый титаносиликат, обладающий превосходной каталитической селективностью и окислительной способностью и получаемый путем введения переходного металла, титана, в каркас молекулярного сита со структурой ZSM-5. TS-1 обладает не только свойством каталитического окисления титана, но и функцией конфигурационной селективности и превосходной стабильностью молекулярных сит ZSM-5. В качестве катализатора этот титаносиликатный материал может быть использован для катализа окисления различных органических веществ, например, эпоксидирования олефинов, частичного окисления алканов, окисления спиртов, гидроксилирования фенолов, аммоксидирования циклонов и т.п. При окислении органических веществ с использованием молекулярных сит TS-1 в качестве окислителя может быть использован свободный от примесей пероксид водорода в низкой концентрации с тем, чтобы избежать использования сложной технологии и загрязнения окружающей среды в процессе окисления. Он также имеет такие преимущества, как рациональное использование энергии, экономичность, благоприятность для окружающей среды, несравнимые с обычной системой окисления, и более высокую селективность реакции. Таким образом, он имеет большие перспективы применения в промышленности. Титаносиликат в качестве катализатора селективного окисления органических веществ считается ключевым в области молекулярно-ситового катализа.
Н2О2 представляет собой общепризнанный экологически чистый окислитель, и единственным побочным продуктом его окисления является вода. Однако водный раствор Н2О2 сложно хранить и перевозить. Н2О2 очень неустойчив и разлагается под воздействием тепла, света, шероховатой поверхности, тяжелых металлов и других примесей. Кроме того, из-за его коррозионной активности должны предприниматься особые меры безопасности при его упаковке, хранении и транспортировке. Таким образом, этот химический продукт может быть эффективно использован только в случае, если Н2О2 используют на месте, либо процесс производства Н2О2 объединен с осуществляемым далее технологическим процессом, в котором используется Н2О2.
Н2О2 может быть синтезирован непосредственно из Н2 и О2, при этом степень использования исходных веществ достигает 100%. Таким образом, возлагаются надежды на использование Н2 и О2 для синтеза Н2О2 непосредственно на месте и последующее окисление органического материала с тем, чтобы посредством прямого использования Н2О2 решить проблемы стоимости и безопасности. Поскольку Pt, Pd, Au и т.д. являются эффективными компонентами для синтезирования Н2О2 из Н2 и О2, во многих патентных документах сообщалось об исследованиях по нанесению их на титаносиликатный материал с целью производства на месте Н2О2 для селективного окисления органических веществ. Например, Meiers R. и другие (J. Catal., 1998, 176:376-386) провели исследования по газофазному эпоксидированию пропилена с использованием в качестве катализатора Pt-Pd/TS-1. Кроме того, в US 6867312В1 и US 6884898В1 также описаны подобные исследования. Хотя упомянутый метод осуществляется в мягких реакционных условиях и имеет хорошую селективность (возможно, выше 95%), ему присущи недостатки - относительно более низкая активность катализатора, плохая стабильность катализатора и т.п. Таким образом, основные задачи исследований и совершенствования упомянутого метода заключаются в приготовлении и модификации соответствующих катализаторов с целью увеличения степени конверсии реакций и повышения устойчивости к дезактивации и регенерируемости катализатора.
Описание изобретения
Ввиду недостатков, свойственных нанесенным на титаносиликатный материал благородным металлам, таким как Pt, Pd, Au и т.п., с целью производства на месте Н2О2 для процесса реакции селективного окисления органических веществ, настоящим изобретением предлагается содержащий благородный металл титаносиликатный материал и способ его получения.
Содержащий благородный металл титаносиликатный материал, предложенный в настоящем изобретении, отличается тем, что упомянутый материал представлен оксидной формой xTiO2·100SiO2·yEOm·zE, где х составляет в диапазоне от 0,001 до 50,0; (y+z) составляет в диапазоне от 0,0001 до 20,0 и y/z<5; Е представляет собой один или более благородных металлов, выбранных из группы, состоящей из Ru, Rh, Pd, Re, Os, Ir, Pt, Ag и Au; m является числом, отвечающим степени окисления Е. Кристаллические зерна упомянутого материала обладают полой структурой или изогнутой (вогнуто-выпуклой) структурой.
В содержащем благородный металл титаносиликатном материале, предложенном в настоящем изобретении, х предпочтительно составляет в диапазоне от 0,005 до 25 или от 0,001 до 20, более предпочтительно - от 0,005 до 20; (y+z) предпочтительно составляет в диапазоне от 0,005 до 20 или от 0,001 до 10, более предпочтительно - от 0,005 до 10, наиболее предпочтительно - от 0,01 до 7; y/z предпочтительно составляет меньше 3, более предпочтительно - меньше 2, более предпочтительно - меньше 1, наиболее предпочтительно - от 0,01 до 0,8. Упомянутый благородный металл предпочтительно является одним или более металлом, выбранным из группы, состоящей из Pd, Pt, Ag и Au, более предпочтительно - Pd и/или Pt. Когда благородный металл - это два или более металла, выбранных из этой группы, упомянутая величина у представляет собой сумму величины у для каждого благородного металла; а упомянутая величина z является суммой величины z для каждого благородного металла. Например, когда благородный металл - это Pt и Pd, упомянутый материал представлен оксидной формой xTiO2·100SiO2·y1PtO·y2PdO·z1Pt·z2Pd, т.е. y=y1+y2; и z=z1+z2. Кристаллические зерна упомянутого материала полностью или частично обладают полой структурой, и полость полых кристаллических зерен упомянутого материала имеет радиальную протяженность 0,1-500 нм, предпочтительно, 0,5-300 нм. Адсорбционная способность по бензолу упомянутого материала, измеренная при условиях температуры 25ºС, Р/Р0=0,10 и времени адсорбции 1 ч, составляет по меньшей мере 25 мг/г, предпочтительно, по меньшей мере 35 мг/г. Между изотермой адсорбции и изотермой десорбции низкотемпературной адсорбции азота упомянутым материалом имеется петля гистерезиса. При относительном давлении Р/Р0 примерно 0,60 разность между адсорбционной способностью по азоту при десорбции и адсорбционной способностью по азоту при адсорбции составляет более 2% адсорбционной способности по азоту при адсорбции. Форма полости упомянутого материала может быть различной, не фиксированной формой, например, круглой, или прямоугольной, или неправильной многоугольной, или неправильной круглой, или комбинациями этих форм. Кристаллические зерна упомянутого материала представляют собой монокристаллические зерна или агрегатные кристаллические зерна, агрегированные из множества кристаллических зерен.
Кристаллические зерна материала, предложенного в настоящем изобретении, могут полностью или частично иметь полую структуру или изогнутую структуру.
Что касается материала, предложенного в настоящем изобретении, то полые кристаллические зерна являются выгодными для дисперсии молекул реагентов и продуктов, что повышает синергический эффект между благородным металлом и титаносиликатом; а благородные металлы обладают более высокой диспергируемостью. Кроме того, полая структура содержащего благородный металл титаносиликатного материала, предложенного в настоящем изобретении, обладает сильной способностью вмещать углеродистые отложения. По сравнению с уровнем техники (например, обычным методом пропитки носителя), селективность, каталитическая активность и устойчивость продукта реакции заметно увеличиваются в реакции окисления, например, реакции получения пропиленоксида путем эпоксидирования пропилена.
Кроме этого настоящим изобретением предлагается два способа получения вышеуказанного содержащего благородный металл титаносиликатного материала.
Один из способов, предложенных в настоящем изобретении, включает гомогенное смешивание титаносиликата, защитного средства, источника благородного металла, восстановителя, источника щелочи с водой, подачу данной смеси в реакционный сосуд для гидротермальной обработки, фильтрование, промывку, сушку с получением этого материала. Более конкретно, упомянутый способ включает стадии:
(1) гомогенное смешивание титаносиликата, защитного средства, источника благородного металла, восстановителя, источника щелочи с водой с получением смеси, обладающей соотношением титаносиликат (г):защитное средство (моль):источник щелочи (моль):восстановитель (моль):источник благородного металла (г, рассчитанный по индивидуальному веществу благородного металла):вода (моль) 100:(0,0-5,0):(0,005-5,0):(0,005-15,0):(0,005-10,0):(200-10000); и
(2) подачу смеси, полученной на стадии (1), в реакционный сосуд, реагирование при условиях гидротермальной обработки, выделение продукта с получением титаносиликатного материала.
В упомянутом первом способе получения смесь обладает предпочтительным молярным соотношением титаносиликат (г):защитное средство (моль):источник щелочи (моль):восстановитель (моль): источник благородного металла (г, рассчитанный по индивидуальному веществу благородного металла): вода 100:(0,005-1,0):(0,01-2,0):(0,01-10,0):(0,01-5,0):(500-5000).
В вышеуказанном первом способе получения титаносиликат на стадии (1) включает различные типы титаносиликатных молекулярных сит с разными структурами, например, TS-1, TS-2, Ti-BETA, Ti-MCM-22, Ti-MCM-41, Ti-ZSM-48, Ti-ZSM-12, Ti-MMM-1, Ti-SBA-15, Ti-MSU, Ti-MCM-48 и т.п., предпочтительно, TS-1.
В вышеуказанном первом способе получения защитное средство представляет собой полимер или поверхностно-активное вещество, причем этот полимер выбран из группы, состоящей из полипропилена, полиэтиленгликоля, полистирола, поливинилхлорида, полиэтилена и их производных; а поверхностно-активное вещество выбрано из группы, состоящей из анионогенных поверхностно-активных веществ, катионогенных поверхностно-активных веществ и неионогенных поверхностно-активных веществ.
Восстановитель на стадии (1) вышеупомянутого способа получения может быть выбран из группы, состоящей из гидразина, боргидрида и цитрата натрия, причем гидразин выбран из группы, состоящей из гидрата гидразина, гидрохлорида гидразина и сульфата гидразина; а упомянутый боргидрид выбран из группы, состоящей из боргидрида натрия и боргидрида калия.
Источник благородного металла на стадии (1) вышеупомянутого способа получения представляет собой неорганическое или органическое вещество упомянутого благородного металла, которое может быть выбрано из группы, состоящей из оксидов, галогенидов, карбонатов, нитратов, аммонийнитратов, солей хлористого аммония, гидроксидов и других комплексов благородного металла. Если взять в качестве примера палладий, источник палладия может представлять собой неорганический и/или органический источник палладия, причем неорганический источник палладия выбирают из группы, состоящей из оксида палладия, карбоната палладия, хлорида палладия, нитрата палладия, аммонийнитрата палладия, аммонийхлорида палладия, гидроксида палладия и других комплексов палладия; органический источник палладия выбирают из группы, состоящей из ацетата палладия и ацетилацетоната палладия.
Источник щелочи на стадии (1) вышеупомянутого способа получения представляет собой неорганический или органический источник щелочи, причем неорганический источник щелочи выбирают из группы, состоящей из аммиака, гидроксида натрия, гидроксида калия и гидроксида бария; а органический источник щелочи выбирают из группы, состоящей из карбамида, щелочных соединений четвертичного аммония, соединений алифатических аминов, соединений аминоспиртов и их смесей.
Щелочные соединения четвертичного аммония имеют общую формулу (R1)4NOH, где R1 обозначает алкил с 1-4 атомами углерода, предпочтительно, пропил.
Соединения алифатических аминов имеют общую формулу R2(NH2)n, где R2 обозначает алкил или алкилиден с 1-6 атомами углерода, и n равно 1 или 2. Соединения алифатических аминов выбирают из группы, состоящей из этиламина, н-бутиламина, бутандиамина и гександиамина.
Соединения аминоспиртов имеют общую формулу (HOR3)mNH(3-m), где R3 обозначает алкил с 1-4 атомами углерода, и m равно 1, 2 или 3. Соединения аминоспиртов выбирают из группы, состоящей из моноэтаноламина, диэтаноламина и триэтаноламина.
В вышеуказанном первом способе получения защитное средство может добавляться или не добавляться.
Условия гидротермальной обработки на стадии (2) вышеупомянутого способа получения относятся к гидротермальной обработке в течение 2-360 ч при температуре 80-200ºС и аутогенном давлении. Упомянутый процесс выделения хорошо известен специалистам в данной области, и в нем нет ничего особенного. Упомянутый процесс выделения, как правило, включает такие процессы, как промывка, сушка кристаллизованного продукта и т.п.
Второй способ получения, предложенный в настоящем изобретении, включает, в частности, следующие стадии:
(1) гомогенное смешивание источника титана, источника кремния, источника щелочи, защитного средства, источника благородного металла с водой с получением смеси, обладающей молярным соотношением источник кремния:источник титана:источник щелочи: источник благородного металла: защитное средство:вода 100:(0,005-50,0):(0,005-20,0):(0,005-10,0):(0,0001-5,0):(200-10000), где источник кремния рассчитывают как SiO2, источник титана рассчитывают как TiO2; а источник благородного металла рассчитывают как простое вещество; гидротермальную кристаллизацию смеси в течение по меньшей мере 2 ч при 120-200ºС, извлечение, фильтрование, сушку и прокаливание продукта с получением промежуточного кристаллического материала; и
(2) подачу промежуточного кристаллического материала, полученного на стадии (1), в оставшийся на стадии (1) фильтрат, добавление восстановителя в молярном соотношении 0,1-10 к источнику благородного металла, добавленному на стадии (1), гидротермальную обработку в течение 2-360 ч при 80-200ºС и аутогенном давлении, и выделение продукта с получением титаносиликатного материала по настоящему изобретению.
Во втором способе получения, предложенном в настоящем изобретении, стадия (2) может быть повторена один раз или, если нужно, многократно.
Во втором способе получения, предложенном в настоящем изобретении, смесь на стадии (1) обладает предпочтительным молярным соотношением источник кремния:источник титана:источник щелочи: источник благородного металла:защитное средство:вода 100:(0,01-10,0):(0,01-10,0):(0,01-5,0):(0,0005-1,0):(500-5000).
Во втором способе получения, предложенном в настоящем изобретении, источник кремния на стадии (1) выбирают из группы, состоящей из геля кремниевой кислоты (силикагеля), золя кремниевой кислоты и органического силиката, причем предпочтительным является органический силикат. Этот органический силикат имеет общую формулу (R4)4SiO4, где R4 обозначает алкил с 1-4 атомами углерода, предпочтительно, этил.
Во втором способе получения, предложенном в настоящем изобретении, источник титана представляет собой неорганическую соль титана или органический титанат, предпочтительно, органический титанат. Эту неорганическую соль титана выбирают из группы, состоящей из TiCl4, Ti(SO4)2 и TiOCl2. Органический титанат имеет общую формулу Ti(OR5)4, где R5 обозначает алкил с 1-6 атомами углерода, предпочтительно, алкил с 2-4 атомами углерода.
Во втором способе получения, предложенном в настоящем изобретении, источник щелочи на стадии (1) представляет собой щелочное соединение четвертичного аммония или смесь щелочного соединения четвертичного аммония, соединения алифатического амина и соединения аминоспирта. Щелочное соединение четвертичного аммония имеет общую формулу (R6)4NOH, где R6 обозначает алкил с 1-4 атомами углерода, предпочтительно, пропил. Соединение алифатического амина имеет общую формулу R7(NH2)n, где R7 обозначает алкил или алкилиден с 1-6 атомами углерода, и n равно 1 или 2. Соединение алифатического амина выбирают из группы, состоящей из этиламина, н-бутиламина, бутандиамина и гександиамина. Соединение аминоспирта имеет общую формулу (HOR8)mNH(3-m), где R8 обозначает алкил с 1-4 атомами углерода, и m равно 1, 2 или 3. Соединение аминоспирта выбирают из группы, состоящей из моноэтаноламина, диэтаноламина и триэтаноламина.
Во втором способе получения, предложенном в настоящем изобретении, защитное средство представляет собой полимер или поверхностно-активное вещество, причем полимер выбирают из группы, состоящей из полипропилена, полиэтиленгликоля, полистирола, поливинилхлорида, полиэтилена и их производных; а поверхностно-активное вещество выбирают из группы, состоящей из анионогенных поверхностно-активных веществ, катионогенных поверхностно-активных веществ и неионогенных поверхностно-активных веществ.
Источник благородного металла на стадии (1) вышеупомянутого второго способа получения представляет собой неорганическое или органическое вещество упомянутого благородного металла, которое может быть выбрано из группы, состоящей из оксидов, галогенидов, карбонатов, нитратов, аммонийнитратов, солей хлористого аммония, гидроксидов и других комплексов благородного металла. Если взять в качестве примера палладий, то источник палладия может представлять собой неорганический и/или органический источник палладия, причем неорганический источник палладия выбирают из группы, состоящей из оксида палладия, карбоната палладия, хлорида палладия, нитрата палладия, аммонийнитрата палладия, аммонийхлорида палладия, гидроксида палладия и других комплексов палладия; органический источник палладия выбирают из группы, состоящей из ацетата палладия и ацетилацетоната палладия.
Во втором способе получения, предложенном в настоящем изобретении, восстановитель на стадии (1) выбирают из группы, состоящей из гидроксиламина, гидразина, боргидрида и цитрата натрия, причем гидразин выбирают из группы, состоящей из гидрата гидразина, гидрохлорида гидразина и сульфата гидразина; а упомянутый боргидрид выбирают из группы, состоящей из боргидрида натрия и боргидрида калия.
По сравнению с уровнем техники каталитическая и окислительная активность и селективность продукта способов получения, предложенных в настоящем изобретении, явно повышены. При этом указанные способы обеспечивают более высокую каталитическую активность и стабильность (см. пример 12). Кроме того, поскольку полая или изогнутая структура, имеющаяся у кристаллических зерен материала, предложенного в настоящей заявке, является благоприятной для диспергирования молекул реагентов и продуктов, особенно, макромолекул (например, ароматических соединений), в ходе каталитической реакции, он особенно выгоден для каталитического окисления ароматических соединений, циклических соединений и т.п.
Краткое описание чертежей
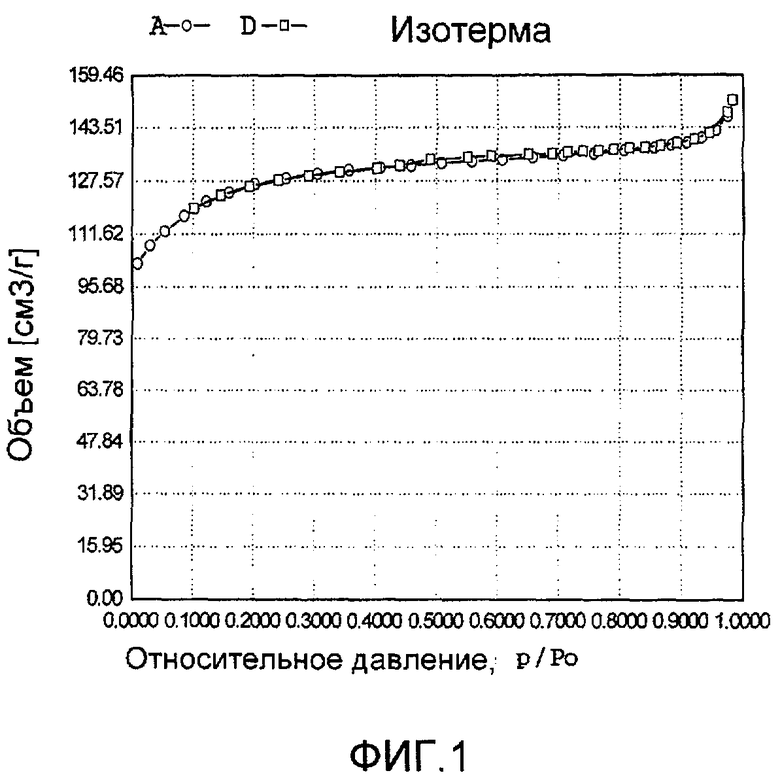
На фиг. 1 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота сравнительного образца DB-1 в Сравнительном Примере 1.
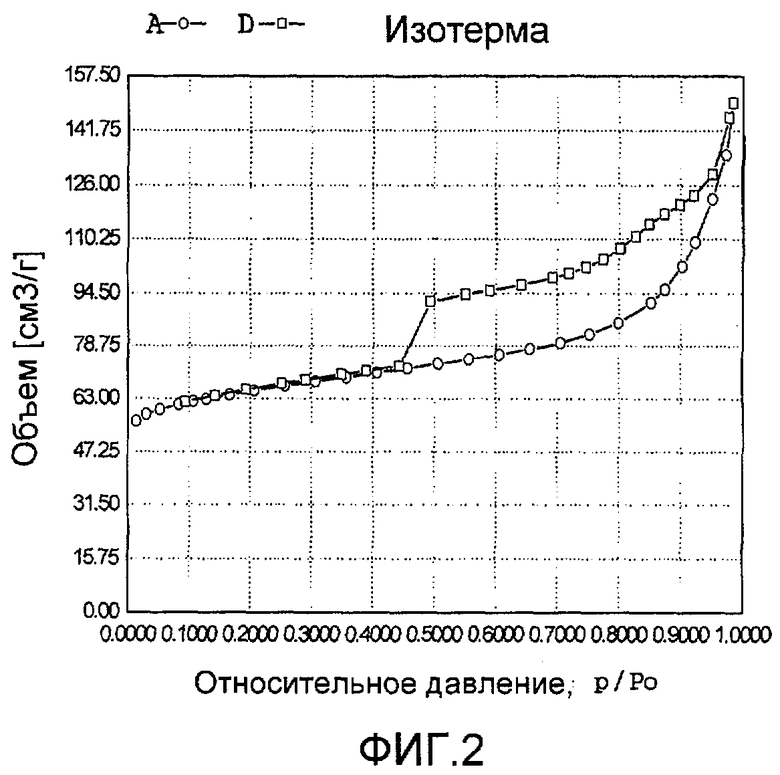
На фиг. 2 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца А в Примере 1.
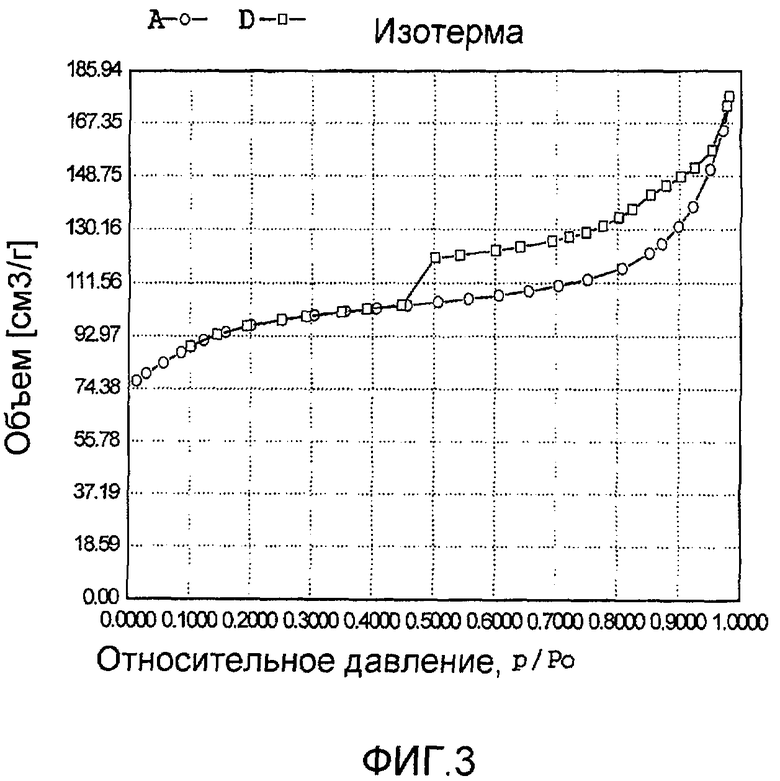
На фиг. 3 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца В в Примере 2.
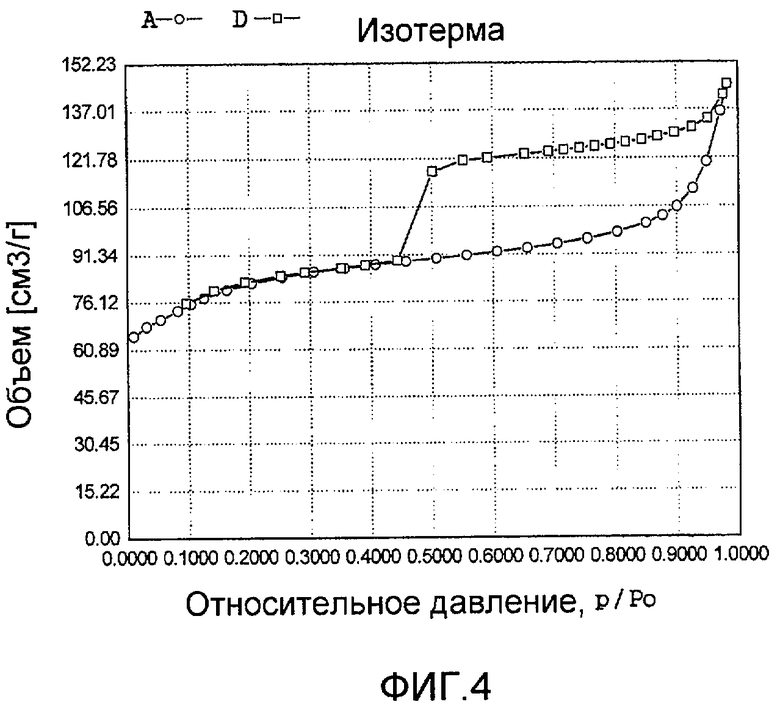
На фиг. 4 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца С в Примере 3.
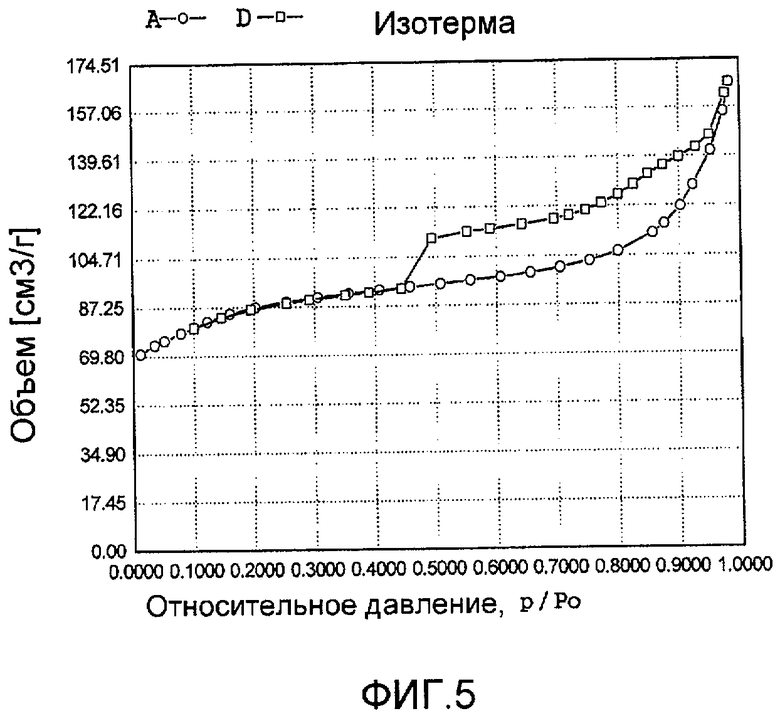
На фиг. 5 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца D в Примере 4.
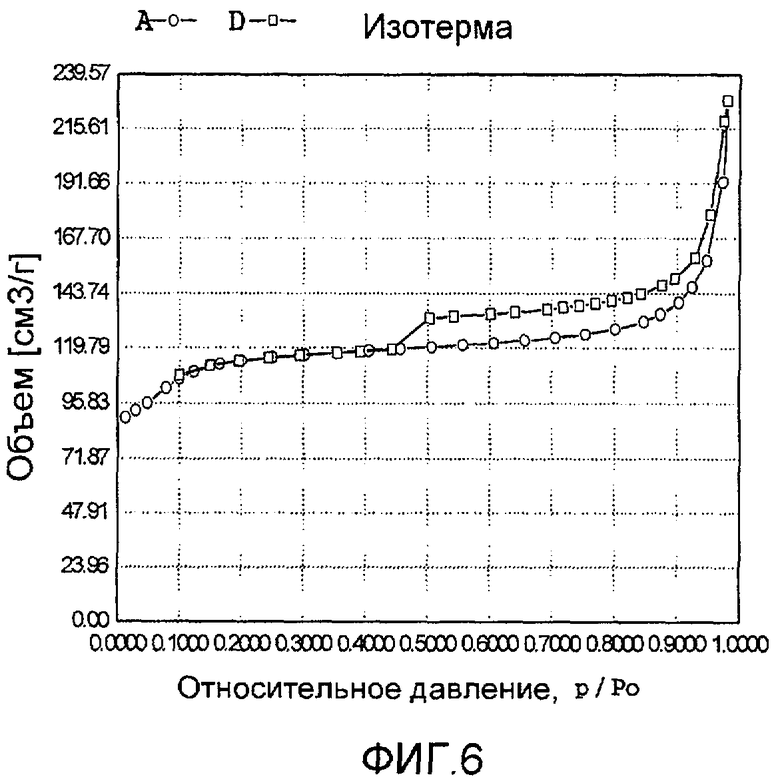
На фиг. 6 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца Е в Примере 5.
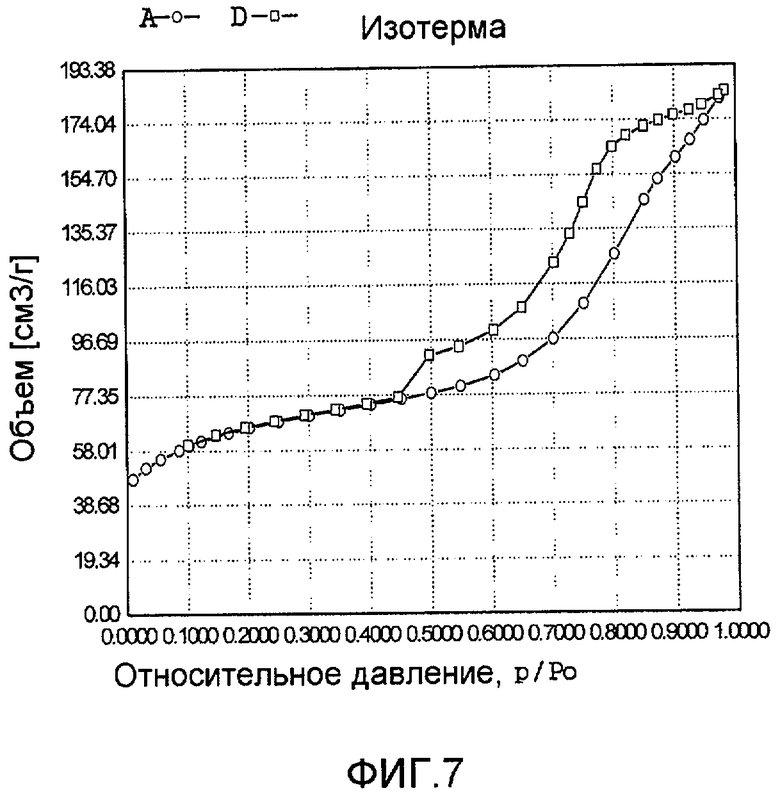
На фиг. 7 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца F в Примере 6.
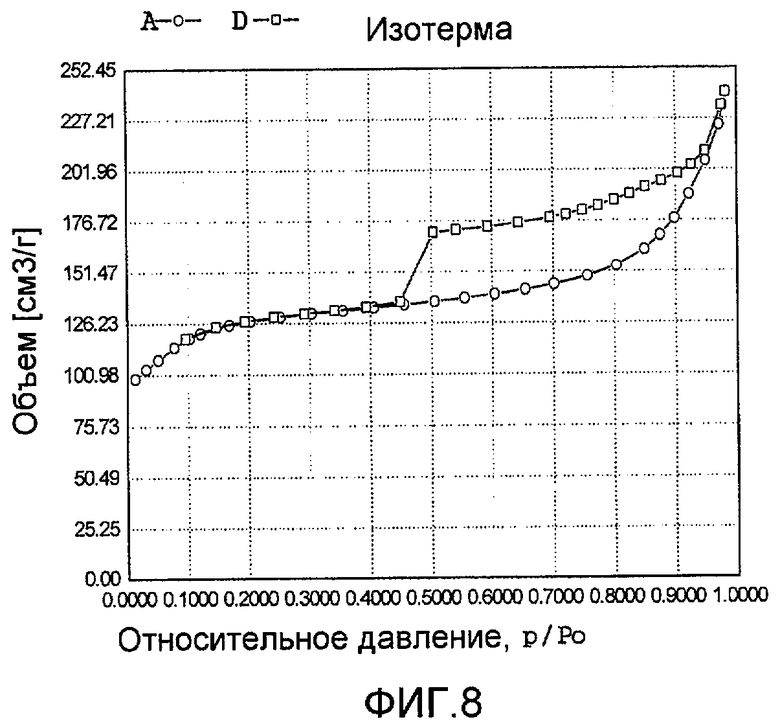
На фиг. 8 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца G в Примере 7.
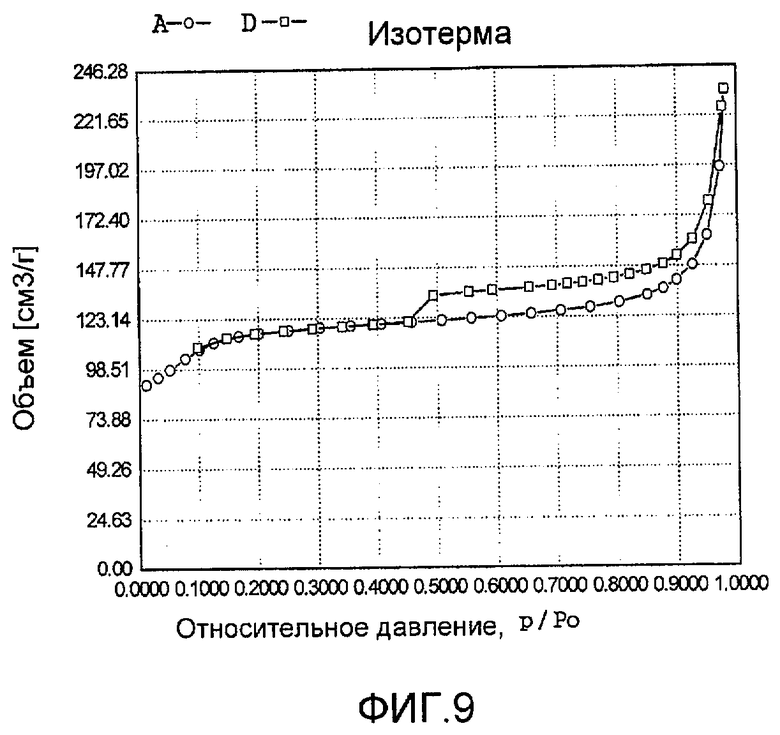
На фиг. 9 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца Н в Примере 8.
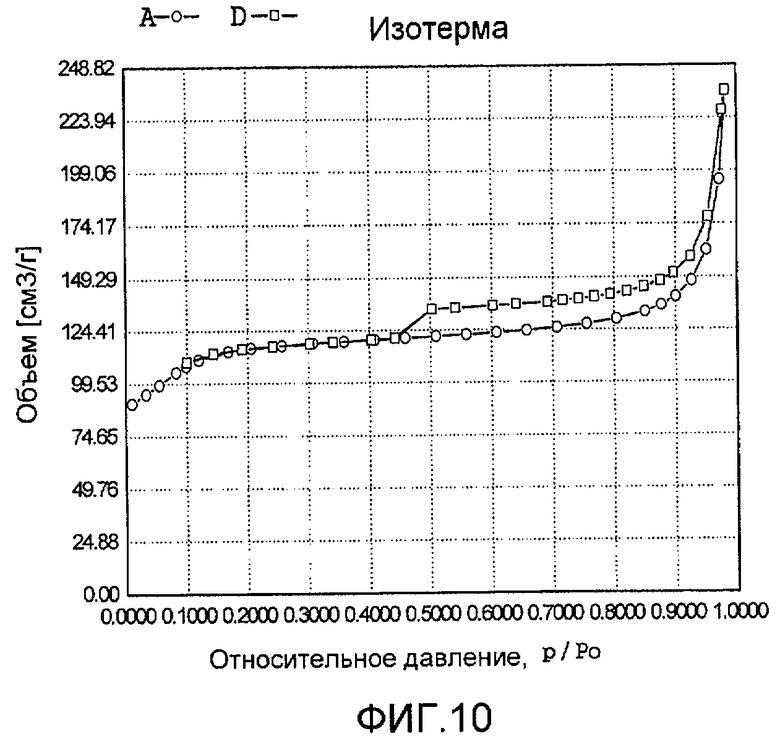
На фиг. 10 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца I в Примере 9.
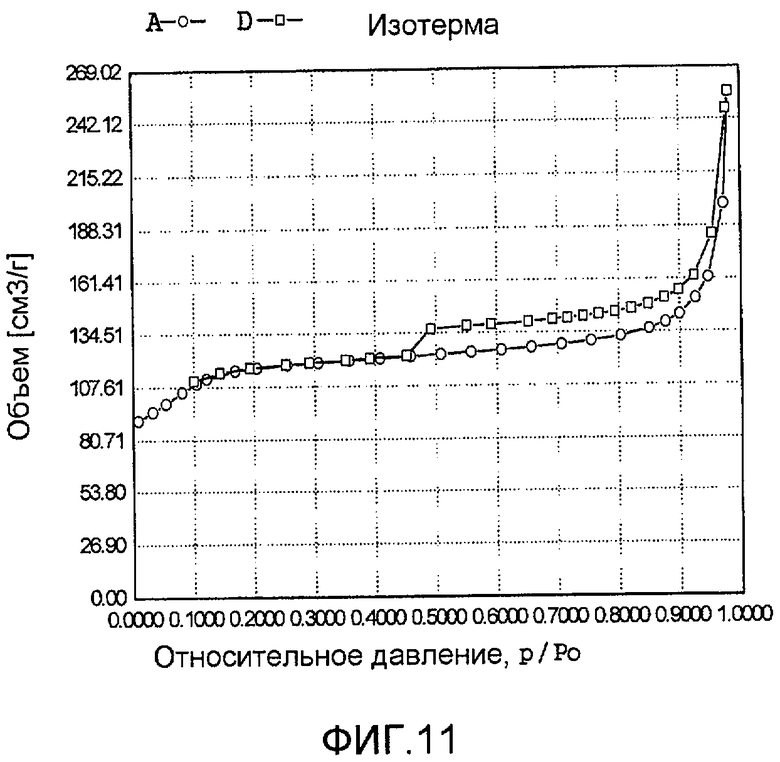
На фиг. 11 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца J в Примере 10.
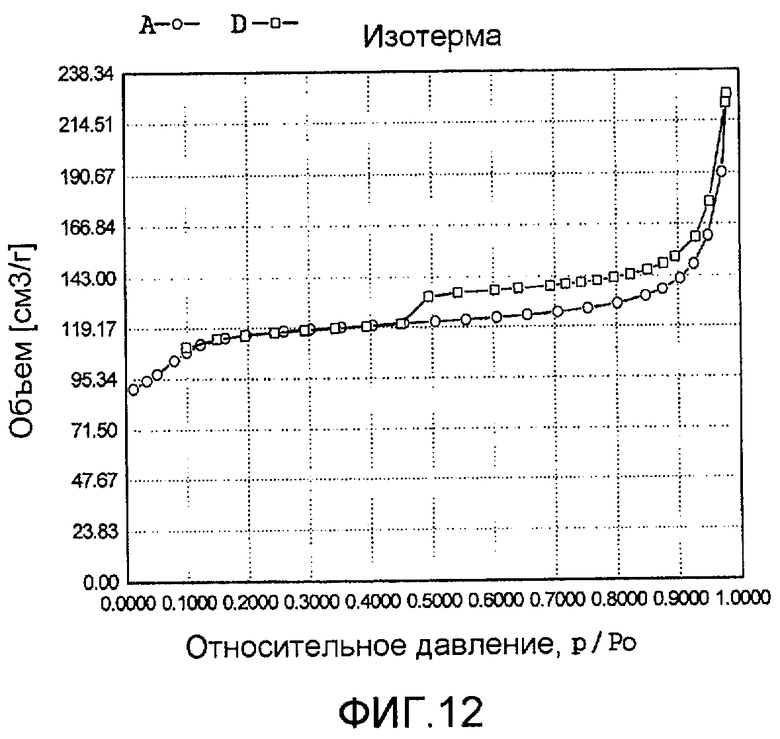
На фиг. 12 представлена адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота образца K в Примере 11.
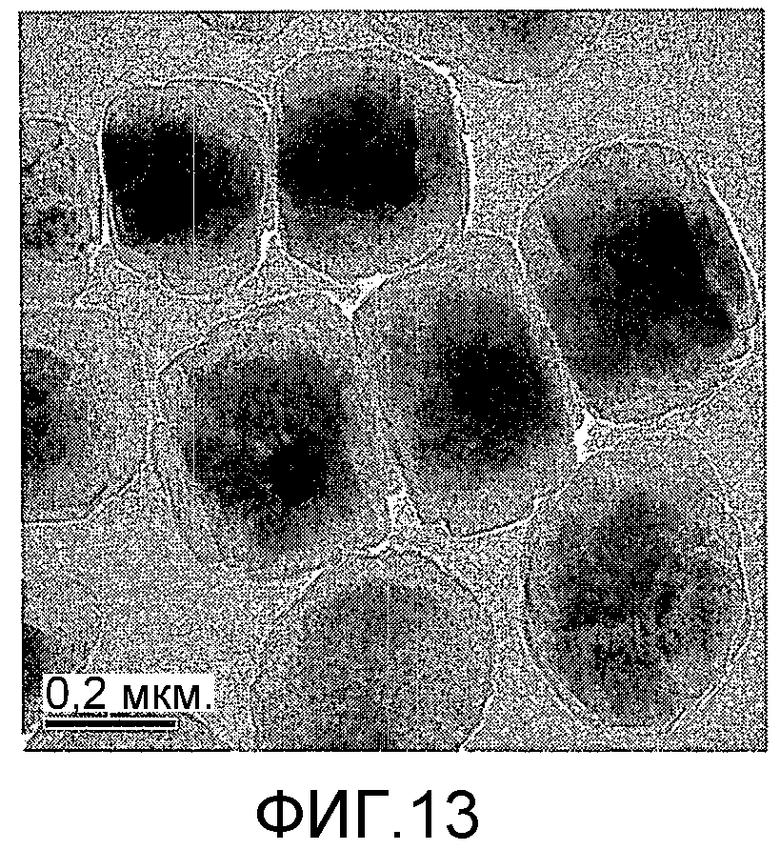
На фиг. 13 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение сравнительного образца DB-1 в Сравнительном Примере 1.
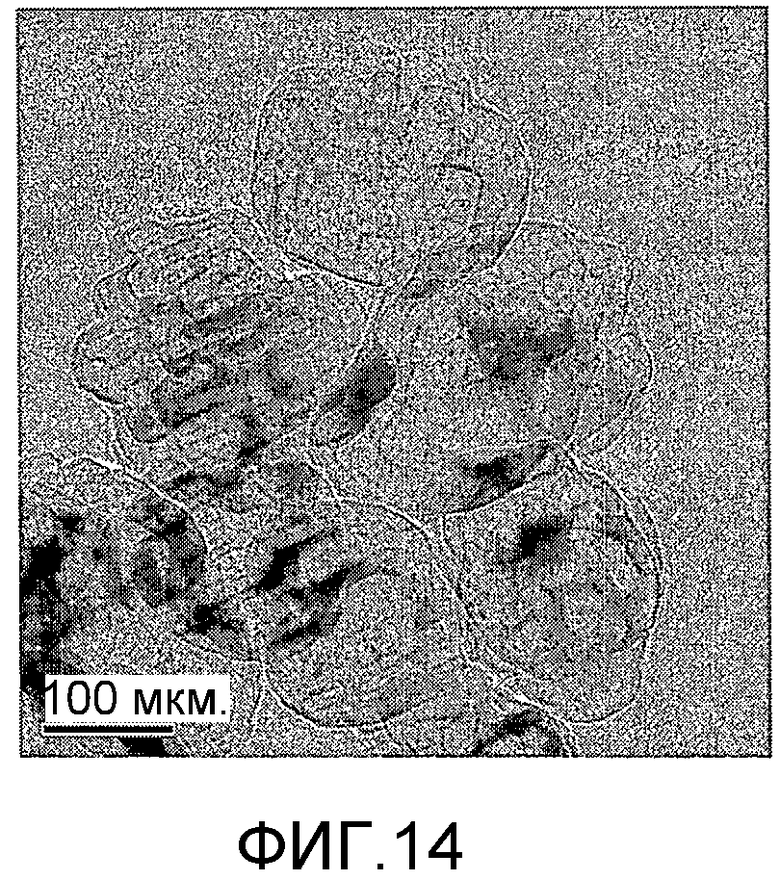
На фиг. 14 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца А в Примере 1.
На фиг. 15 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца В в Примере 2.
На фиг. 16 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца С в Примере 3.
На фиг. 17 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца D в Примере 4.
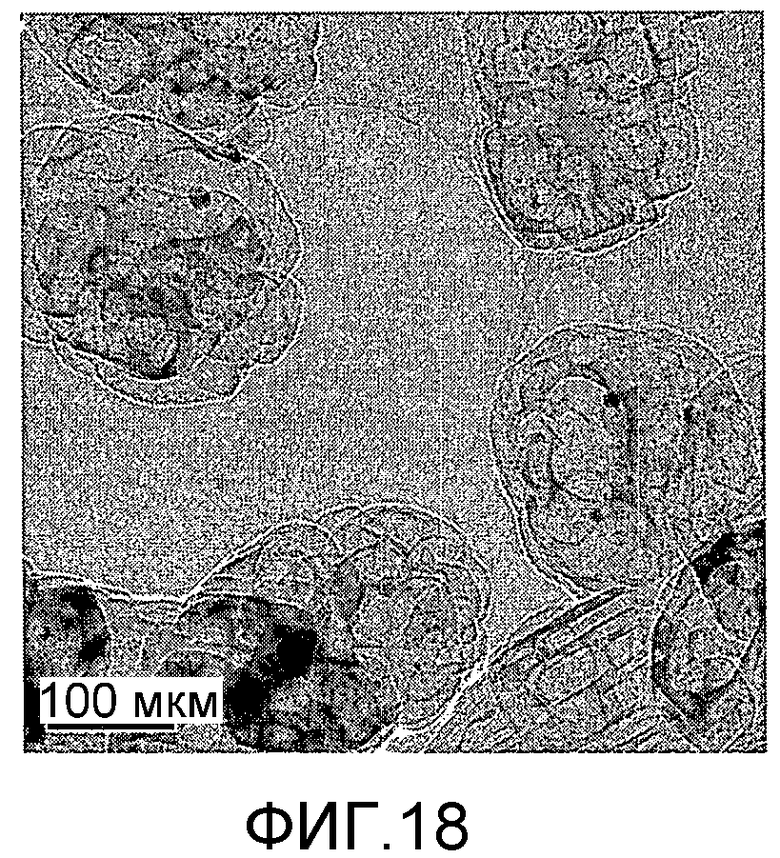
На фиг. 18 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца Е в Примере 5.
На фиг. 19 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца F в Примере 6.
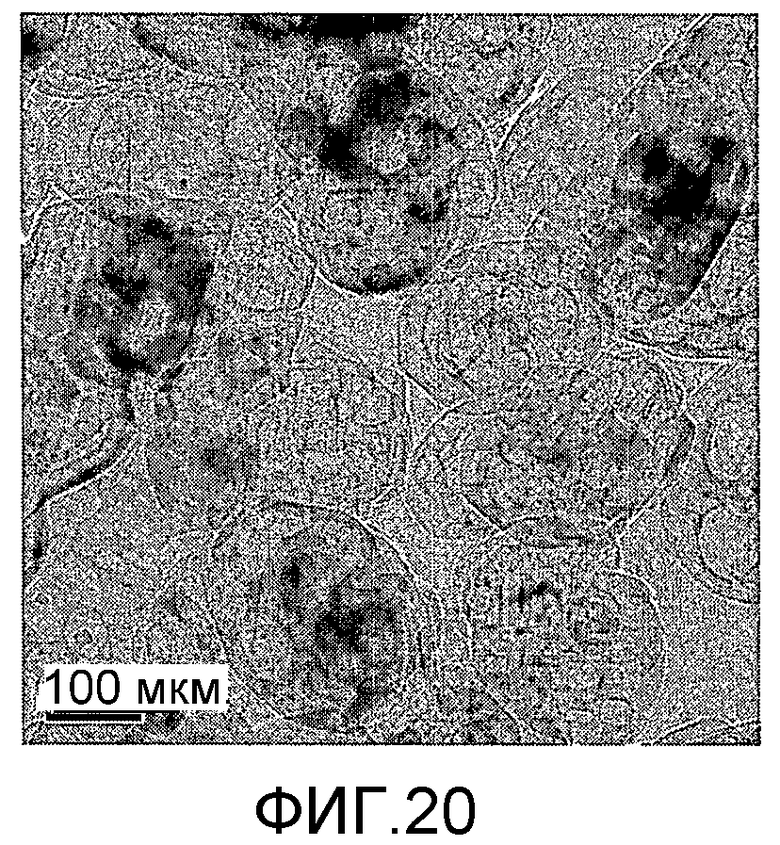
На фиг. 20 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца G в Примере 7.
На фиг. 21 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца Н в Примере 8.
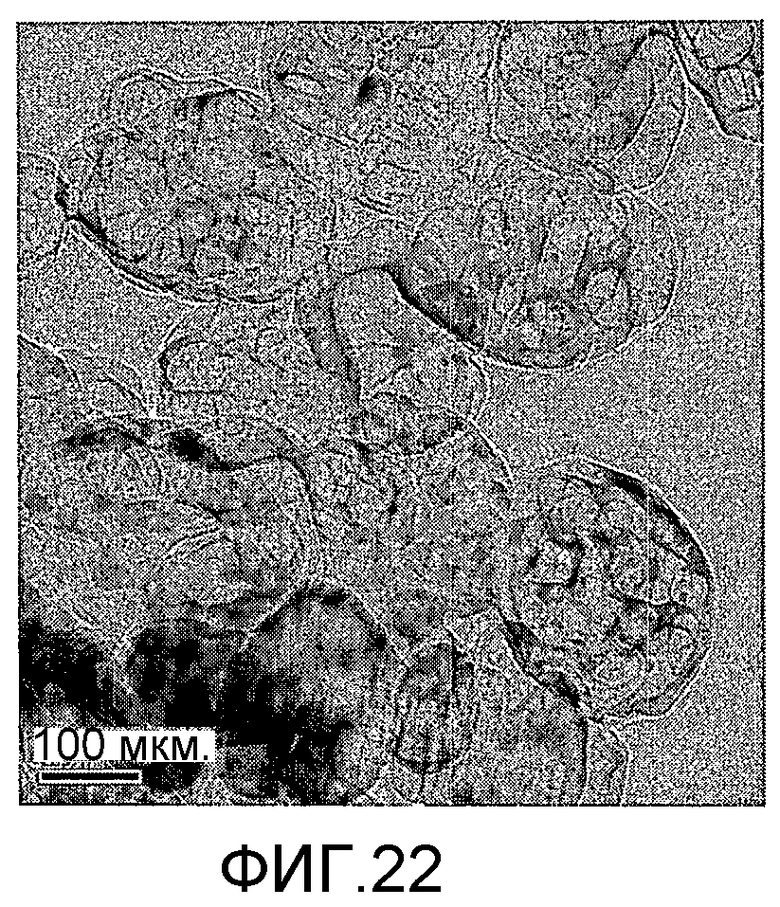
На фиг. 22 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца I в Примере 9.
На фиг. 23 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца J в Примере 10.
На фиг. 24 представлено полученное просвечивающим электронным микроскопом (ПЭМ) изображение образца K в Примере 11.
Варианты осуществления изобретения
Следующие ниже примеры обеспечивают дополнительное пояснение настоящего изобретения, но при этом не ограничивают настоящее изобретение.
Все реагенты, использованные в примерах, были серийно выпускаемыми продуктами химически чистых реагентов. Титаносиликат TS-1, использованный в сравнительных примерах и примерах, был получен в соответствии со способом, описанным в уровне техники в Zeolites, 1992, Vol.12, 943; использованный в них титаносиликат Ti-BETA был получен в соответствии со способом, описанным в уровне техники в J. Catal., 1994, Vol.145, 151; титаносиликат TS-2, использованный в сравнительных примерах и примерах, был получен в соответствии со способом, описанным в уровне техники в Appl. Catal., 1990, Vol.58, L1; использованный в них титаносиликат Ti-ZSM-48 был получен в соответствии со способом, описанным в уровне техники в J. Chem. Soc. Chem. Commun., 1994, 745; использованный в них титаносиликат Ti-ZSM-12 был получен в соответствии со способом, описанным в уровне техники в Zeolites, 1995, Vol.15, 236. Адсорбционно-десорбционная изотерма низкотемпературной адсорбции азота этих образцов была получена в соответствии со стандартным методом ASTM D4222-98 на приборе для измерения статической адсорбции азота (Static Nitrogen Adsorption Device) ASAP2405 компании Micromeritics, США. ПЭМ-Изображения образцов были получены при помощи просвечивающего электронного микроскопа (ПЭМ) Tecnai типа G2F20S-TWIN компании FEI, Голландия, при ускоряющем напряжении 20 кВ. Кроме того, у этих образов была измерена величина адсорбции бензола с помощью обычного процесса статической адсорбции.
Сравнительный Пример 1
Этот сравнительный пример иллюстрирует процесс обычного приготовления нанесенного на носитель катализатора палладий/титаносиликат.
20 г титаносиликата TS-1 и 20 мл раствора комплекса аммонийнитрата палладия с концентрацией 0,01 г/мл (в расчете на атом палладия) добавили к 20 мл деионизированной воды. Смесь перемешали до гомогенного состояния, подходящим образом герметизировали, пропитывали в течение 24 ч при температуре 40ºС, подвергли естественной сушке и восстановительной активации в течение 3 ч в атмосфере водорода при 150ºС с получением обычного нанесенного на носитель катализатора палладий/титаносиликат DB-1. При характеризации этот катализатор был представлен оксидной формой 6TiO2·100SiO2·0,7PdO·0,3Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 1) отсутствовала петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна твердотельная структура, а не полая или изогнутая структура (фиг. 13).
Пример 1
20 г титаносиликата TS-1, раствор комплекса аммонийнитрата палладия с концентрацией 0,01 г/мл (в расчете на атом палладия), подходящее количество гидрата гидразина и бромида цетилтриметиламмония добавили к водному раствору гидроксида тетрапропиламмония (с массовой концентрацией 10%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением титаносиликат (г):бромид цетилтриметиламмония (моль):гидроксид тетрапропиламмония (моль):гидрат гидразина (моль):комплекс амонийнитрата палладия (г, в расчете на палладий):вода (моль) 100:0,005:0,5:3,0:2,0:1000. Затем эту смесь подавали в герметичный реакционный сосуд из нержавеющей стали, подвергали гидротермальной обработке в течение 48 ч при 150ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 180ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала А по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 4TiO2·100SiO2·0,01PdO·0,09Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 2) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 14).
Пример 2
20 г титаносиликата Ti-ВЕТА, раствор комплекса хлорида палладия с концентрацией 0,01 г/мл (в расчете на атом палладия), подходящее количество гидрохлорида гидразина и полипропилена добавили к водному раствору гидроксида натрия (с массовой концентрацией 15%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением титаносиликат (г):полипропилен (моль):гидроксид натрия (моль):гидрохлорид гидразина (моль):хлорид палладия (г, в расчете на палладий):вода (моль) 100:0,9:1,8:0,15:0,1:4600. Затем эту смесь подавали в герметичный реакционный сосуд из нержавеющей стали, подвергали гидротермальной обработке в течение 24 ч при 180ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 110ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала В по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 8TiO2·100SiO2·0,006PdO·0,008Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 3) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 15).
Пример 3
Тетраэтилортосиликат, тетрабутилтитанат, ацетат палладия с концентрацией 0,01 г/мл (в расчете на атом палладия) и Tween 80 добавили к водному раствору гидроксида тетрапропиламмония и бутандиамина (с массовой концентрацией 10%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением источник кремния:источник титана:источник щелочи:источник палладия:защитное средство:вода 100:0,03:0,5:0,05:0,02:550, где источник кремния рассчитан как SiO2, источник титана рассчитан как TiO2; источник палладия рассчитан как Pd. Затем эту смесь подавали в герметичный реакционный сосуд, подвергали гидротермальной обработке в течение 120 ч при 120ºС и аутогенном давлении. Образовавшееся в результате вещество извлекли, отфильтровали, высушили и прокалили с получением промежуточного кристаллического материала. Указанный промежуточный кристаллический материал подавали в оставшийся фильтрат, затем добавили подходящее количество гидрата гидразина для гидротермальной обработки в течение 36 ч при 170ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 150ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала С по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 0,008TiO2·100SiO2·0,01PdO·0,2Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 4) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 16).
Пример 4
Золь кремниевой кислоты, тетрабутилтитанат, аммонийхлорид палладия с концентрацией 0,01 г/мл (в расчете на атом палладия) и додецилбензолсульфонат натрия добавили к водному раствору гидроксида тетрапропиламмония и бутандиамина (с массовой концентрацией 15%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением источник кремния:источник титана:источник щелочи:источник палладия:защитное средство:вода 100:2,0:5,2:2,0:0,5:2500, где источник кремния рассчитан как SiO2, источник титана рассчитан как TiO2; источник палладия рассчитан как Pd. Затем эту смесь подавали в герметичный реакционный сосуд из нержавеющей стали, подвергали гидротермальной обработке в течение 96 ч при 150ºС и аутогенном давлении. Образовавшееся в результате вещество извлекли, отфильтровали, высушили и прокалили с получением промежуточного кристаллического материала. Указанный промежуточный кристаллический материал подавали в оставшийся фильтрат, а затем добавили подходящее количество гидрохлорида гидразина для гидротермальной обработки в течение 48 ч при 120ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 120ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала D по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 19TiO2·100SiO2·0,5PdO·1,3Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 5) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 17).
Пример 5
20 г титаносиликата TS-2, ацетат палладия с концентрацией 0,01 г/мл (в расчете на атом палладия), подходящее количество боргидрида натрия и Tween 80 добавили к водному раствору бутандиамина (с массовой концентрацией 10%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением титаносиликат (г):Tween 80 (моль):бутандиамин (моль):боргидрид натрия (моль):ацетат палладия (г, в расчете на палладий):вода (моль) 100:0,1:0,02:0,05:0,03:520. Затем эту смесь подавали в герметичный реакционный сосуд из нержавеющей стали, подвергали гидротермальной обработке в течение 120 ч при 120ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 150ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала Е по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 0,1TiO2·100SiO2·0,66PdO·0,12Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 6) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 18).
Пример 6
20 г титаносиликата Ti-ZSM-48, аммонийхлорид палладия с концентрацией 0,01 г/мл (в расчете на атом палладия), подходящее количество сульфата гидразина и Pluronic P123 добавили к водному раствору гидроксида тетрапропиламмония (с массовой концентрацией 10%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением титаносиликат (г):Pluronic P123 (моль):гидроксид тетрапропиламмония (моль):сульфат гидразина (моль):аммонийхлорид палладия (г, в расчете на палладий):вода (моль) 100:0,5:0,1:8,5:4,8:2000. Затем эту смесь подавали в герметичный реакционный сосуд из нержавеющей стали, подвергали гидротермальной обработке в течение 240 ч при 90ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 120ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала F по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 0,04TiO2·100SiO2·3,6PdO·1,1Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 7) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 19).
Пример 7
Тетраэтилортосиликат, тетраэтилтитанат, ацетат палладия с концентрацией 0,01 г/мл (в расчете на атом палладия) и бромид цетилтриметиламмония добавили к водному раствору гидроксида тетрапропиламмония (с массовой концентрацией 13%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением источник кремния:источник титана:источник щелочи:источник палладия:защитное средство:вода 100:8,2:7,5:0,1:0,005:800, где источник кремния рассчитан как SiO2, источник титана рассчитан как TiO2; источник палладия рассчитан как Pd. Затем эту смесь подавали в герметичный реакционный сосуд, подвергали гидротермальной обработке в течение 96 ч при 160ºС и аутогенном давлении. Образовавшееся в результате вещество извлекли, отфильтровали, высушили и прокалили с получением промежуточного кристаллического материала. Указанный промежуточный кристаллический материал подавали в оставшийся фильтрат, а затем добавили подходящее количество гидрохлорида гидразина для гидротермальной обработки в течение 36 ч при 170ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 150ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала G по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 23TiO2·100SiO2·0,004PdO·0,8Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 8) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 20).
Пример 8
Тетраметилортосиликат, TiCl4, аммонийнитрат палладия с концентрацией 0,01 г/мл (в расчете на атом палладия) и полихлорвинил добавили к водному раствору гидроксида тетрапропиламмония (с массовой концентрацией 15%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением источник кремния:источник титана:источник щелочи:источник палладия:защитное средство:вода 100:5,0:0,02:4,5:0,9:4800, где источник кремния рассчитан как SiO2, источник титана рассчитан как TiO2; источник палладия рассчитан как Pd. Затем эту смесь подавали в герметичный реакционный сосуд, подвергали гидротермальной обработке в течение 96 ч при 150ºС и аутогенном давлении. Образовавшееся в результате вещество извлекли, отфильтровали, высушили и прокалили с получением промежуточного кристаллического материала. Указанный промежуточный кристаллический материал подавали в оставшийся фильтрат, а затем добавили подходящее количество боргидрида натрия для гидротермальной обработки в течение 48 ч при 120ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 120ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала Н по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 12TiO2·100SiO2·0,01PdO·6,4Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 9) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 21).
Пример 9
20 г титаносиликата Ti-ZSM-12, 10 мл раствора ацетата палладия в этаноле с концентрацией 0,01 г/мл (в расчете на атом палладия), подходящее количество цитрата натрия и полиэтиленгликоля добавили к водному раствору триэтаноламина (с массовой концентрацией 18%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением титаносиликат (г):полиэтиленгликоль (моль):триэтаноламин (моль):цитрат натрия (моль):ацетат палладия (г, в расчете на палладий):вода (моль) 100:0,01:1,2:0,05:1,0:1500. Затем эту смесь подавали в реакционный сосуд, подвергали гидротермальной обработке в течение 320 ч при 130ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 140ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала I по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 0,5TiO2·100SiO2·0,7PdO·1,3Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 10) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 22).
Пример 10
Тетраэтилортосиликат, тетрапропилтитанат, ацетат палладия с концентрацией 0,01 г/мл (в расчете на атом палладия) и бромид тетрадецилтриметиламмония добавили к водному раствору гидроксида тетрапропиламмония (с массовой концентрацией 13%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением источник кремния:источник титана:источник щелочи:источник палладия:защитное средство:вода 100:0,1:0,1:1,1:0,001:1500, где источник кремния рассчитан как SiO2, источник титана рассчитан как TiO2; источник палладия рассчитан как Pd. Затем эту смесь подавали в герметичный реакционный сосуд из нержавеющей стали, подвергали гидротермальной обработке в течение 72 ч при 160ºС и аутогенном давлении. Образовавшееся в результате вещество извлекли, отфильтровали, высушили и прокалили с получением промежуточного кристаллического материала. Указанный промежуточный кристаллический материал подавали в оставшийся фильтрат, а затем добавили подходящее количество гидрохлорида гидразина для гидротермальной обработки в течение 36 ч при 170ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 150ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала J по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 2TiO2·100SiO2·0,6PdO·3,3Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 11) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 23).
Пример 11
20 г титаносиликата TS-1, раствор комплекса аммонийнитрата палладия и аммонийнитрата платины с концентрацией 0,01 г/мл (в расчете на атом палладия или платины), гидрат гидразина и бромид цетилтриметиламмония добавили к водному раствору гидроксида тетрапропиламмония (с массовой концентрацией 14%), смешали и перемешали до гомогенного состояния с получением смеси, обладающей соотношением титаносиликат (г):бромид цетилтриметиламмония (моль):гидроксид тетрапропиламмония (моль):гидрат гидразина (моль):аммонийнитрат платины (г, в расчете на платину):аммонийнитрат палладия (г, в расчете на палладий):вода (моль) 100:0,1:1,2:2,0:0,8:1,2:1800. Затем эту смесь подавали в герметичный реакционный сосуд из нержавеющей стали, подвергали гидротермальной обработке в течение 72 ч при 180ºС и аутогенном давлении. Образовавшееся в результате вещество отфильтровали, промыли водой, подвергли естественной сушке и далее сушили при 180ºС в течение 3 ч с получением нового содержащего благородный металл титаносиликатного материала К по настоящему изобретению. При характеризации этот материал был представлен оксидной формой 4TiO2·100SiO2·0,3PdO·0,1PtO·0,9Pd·0,7Pt. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота (фиг. 12) имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура (фиг. 24).
Сравнительный Пример 2
Этот сравнительный пример иллюстрирует процесс обычного приготовления нанесенного на носитель катализатора палладий-платина/титаносиликат.
20 г титаносиликата TS-1 и по 10 мл растворов комплекса аммонийнитрата палладия и аммонийнитрата платины с концентрацией 0,01 г/мл (в расчете на атом палладия или платины) добавили к 20 мл деионизированной воды. Смесь перемешали до гомогенного состояния, надлежащим образом герметизировали, пропитывали в течение 24 ч при температуре 40ºС, подвергли естественной сушке и восстановительной активации в течение 3 ч в атмосфере водорода при 150ºС с получением обычного нанесенного на носитель катализатора палладий-платина/титаносиликат DB-2. При характеризации этот катализатор был представлен оксидной формой 6TiO2·100SiO2·0,8PdO·0,4PtO ·0,2Pd·0,5Pt. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота отсутствовала петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), полая структура не зафиксирована.
Сравнительный Пример 3
Этот сравнительный пример иллюстрирует процесс обычного приготовления нанесенного на носитель катализатора палладий/титаносиликат.
20 г титаносиликата, полученного в соответствии со способом, описанным в Примере 1 документа CN1132699C, и 20 мл раствора комплекса аммонийнитрата палладия с концентрацией 0,01 г/мл (в расчете на атом палладия) добавили к 20 мл деионизированной воды. Смесь перемешали до гомогенного состояния, надлежащим образом герметизировали, пропитывали в течение 24 ч при температуре 40ºС, подвергли естественной сушке и восстановительной активации в течение 3 ч в атмосфере водорода при 150ºС с получением нанесенного на носитель катализатора палладий-платина/титаносиликат DB-3. При характеризации этот катализатор был представлен оксидной формой 6TiO2·100SiO2·0,9PdO·0,1Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота имела место петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна полая структура.
Сравнительный Пример 4
Этот сравнительный пример иллюстрирует процесс обычного приготовления нанесенного на носитель катализатора палладий/титаносиликат.
20 г титаносиликата TS-2 и 15 мл раствора комплекса аммонийнитрата палладия с концентрацией 0,01 г/мл (в расчете на атом палладия) добавили к 30 мл деионизированной воды. Смесь перемешали до гомогенного состояния, надлежащим образом герметизировали, пропитывали в течение 24 ч при температуре 40ºС, подвергли естественной сушке и восстановительной активации в течение 1 ч в атмосфере водорода при 150ºС с получением обычного нанесенного на носитель катализатора палладий/титаносиликат DB-4. При характеризации этот катализатор был представлен оксидной формой 7TiO2·100SiO2·0,2PdO·0,6Pd. В его адсорбционно-десорбционной изотерме низкотемпературной адсорбции азота отсутствовала петля гистерезиса. На изображении, полученном просвечивающим электронным микроскопом (ПЭМ), видна твердотельная, а не полая структура.
Пример 12
Этот пример иллюстрирует влияние на реакцию получения пропиленоксида путем газофазного эпоксидирования пропилена образцов катализатора, полученных в примерах и сравнительных примерах настоящего изобретения, в присутствии водорода.
0,5 г каждого из образцов, полученных в описанных выше Примерах 1-11 и Сравнительных Примерах 1, 2, 3 и 4, соответственно добавили в реактор эпоксидирования, содержащий 80 мл метанола. В этот реактор подавали пропилен, кислород, водород и азот с образованием газообразной смеси пропилен-кислород-водород-азот (с молярным соотношением 1:1:1:7). Эпоксидирование этой смеси проводили при условиях температуры 60ºС, давления 1,0 МПа и объемного расхода пропилена 10 ч-1 с получением пропиленоксида (ПО).
Данные о степени конверсии пропилена и селективности ПО через 2 ч и 12 ч после начала реакции соответственно приведены в Таблицах 1 и 2.
Из Таблиц 1 и 2 видно, что активность материалов, предложенных в настоящем изобретении, заметно выше, чем материалов сравнительных образцов, а их селективность также в некоторой степени увеличена, что указывает на то, что их каталитическая активность при окислении и селективность явно улучшены по сравнению с уровнем техники, и при этом материалы по настоящему изобретению обладают лучшей стабильностью каталитической активности.
Данные по величине адсорбции бензола при условиях 25ºС, Р/Р0=0,10 и времени адсорбции 1 ч, а также радиальной протяженности полостей у образцов приведены в Таблице 3.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ГИДРООКИСЛЕНИЯ С ИСПОЛЬЗОВАНИЕМ КАТАЛИЗАТОРА, ПОЛУЧЕННОГО ИЗ КЛАСТЕРНОГО КОМПЛЕКСА ЗОЛОТА | 2007 |
|
RU2445159C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИМЕТИЛСУЛЬФОКСИДА | 2013 |
|
RU2649576C2 |
| СПОСОБ ПОЛУЧЕНИЯ МАТЕРИАЛА С МИКРОМЕЗОПОРИСТОЙ СТРУКТУРОЙ | 2005 |
|
RU2282587C1 |
| СПОСОБ ОЧИСТКИ БЕНЗИНА | 2017 |
|
RU2742646C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА, СОДЕРЖАЩЕГО ЗОЛОТО И ТИТАН | 2000 |
|
RU2232636C2 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОГО ТИТАНОСИЛИКАТА | 2014 |
|
RU2567314C1 |
| КАТАЛИЗАТОР ДЛЯ НЕЙТРАЛИЗАЦИИ ОТРАБОТАВШИХ ГАЗОВ ДИЗЕЛЬНЫХ ДВИГАТЕЛЕЙ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2259228C2 |
| СПОСОБ ОЧИСТКИ БЕНЗИНА | 2017 |
|
RU2754030C2 |
| Способ получения кристаллического титаносиликата | 2023 |
|
RU2825282C1 |
| СПОСОБ СКЕЛЕТНОЙ ИЗОМЕРИЗАЦИИ Н-БУТЕНОВ В ИЗОБУТИЛЕН | 2012 |
|
RU2475470C1 |







Изобретение относится к титаносиликатным материалам и способам их получения. Описан содержащий благородный металл титаносиликатный материал, являющийся катализатором, характеризующийся тем, что упомянутый материал представлен оксидной формой xTiO2·100SiO2·yEOm·zE, где x составляет в диапазоне от 0,001 до 50,0; (y+z) составляет в диапазоне от 0,0001 до 20,0 и y/z<5; E представляет собой один или более благородных металлов, выбранных из группы, состоящей из Ru, Rh, Pd, Re, Os, Ir, Pt, Ag и Au; m является числом, отвечающим степени окисления E; и кристаллические зерна упомянутого материала обладают полой структурой или изогнутой структурой. Описан способ получения указанного выше материала, включающий следующие стадии: (1) гомогенное смешивание титаносиликата, защитного средства, источника благородного металла, восстановителя, источника щелочи с водой с получением смеси, обладающей соотношением титаносиликат:защитное средство:источник щелочи:восстановитель:источник благородного металла:вода 100:(0,0-5,0):(0,005-5,0):(0,005-15,0):(0,005-10,0):(200-10000), где титаносиликат рассчитывают в граммах; защитное средство, источник щелочи, восстановитель и воду рассчитывают в молях; а источник благородного металла рассчитывают в граммах простого вещества благородного металла; и (2) подачу смеси, полученной на стадии (1), в реакционный сосуд, реагирование при условиях гидротермальной обработки, выделение продукта с получением титаносиликатного материала, причем упомянутые условия гидротермальной обработки относятся к гидротермальной обработке в течение 2-360 ч при температуре 80-200°С и аутогенном давлении. Описан способ получения указанного выше материала, включающий следующие стадии: (1) гомогенное смешивание источника титана, источника кремния, источника щелочи, защитного средства, источника благородного металла с водой с получением смеси, обладающей соотношением источник кремния:источник титана:источник щелочи:источник благородного металла:защитное средство:вода 100:(0,005-50,0):(0,005-20,0):(0,005-10,0):(0,0001-5,0):(200-10000), где источник кремния рассчитывают как SiO2, источник титана рассчитывают как TiO2; и источник благородного металла рассчитывают как простое вещество; гидротермальную кристаллизацию смеси в течение по меньшей мере 2 ч при 120-200°С и аутогенном давлении, извлечение, фильтрование, сушку и прокаливание продукта с получением промежуточного кристаллического материала; (2) подачу промежуточного кристаллического материала, полученного на стадии (1), в фильтрат, полученный после фильтрования на стадии (1), добавление восстановителя в молярном соотношении 0,1-10 к источнику благородного металла, добавленному на стадии (1), гидротермальную обработку в течение 2-360 ч при 80-200°С и аутогенном давлении, и выделение продукта с получением титаносиликатного материала. Технический результат - получен материал, характеризующийся увеличением селективности, каталитической активности в реакциях окисления. 3 н. и з.п. ф-лы; 3 табл.; 24 ил.; 12 пр.; 4 ср.пр.
1. Содержащий благородный металл титаносиликатный материал, являющийся катализатором, отличающийся тем, что упомянутый материал представлен оксидной формой xTiO2·100SiO2·yEOm·zE, где х составляет в диапазоне от 0,001 до 50,0; (y+z) составляет в диапазоне от 0,0001 до 20,0 и y/z<5; E представляет собой один или более благородных металлов, выбранных из группы, состоящей из Ru, Rh, Pd, Re, Os, Ir, Pt, Ag и Au; m является числом, отвечающим степени окисления E; и кристаллические зерна упомянутого материала обладают полой структурой или изогнутой структурой.
2. Титаносиликатный материал по п.1, отличающийся тем, что упомянутый благородный металл Е является одним или более благородными металлами, выбранными из группы, Pd, Pt, Ag и Au.
3. Титаносиликатный материал по п.2, отличающийся тем, что упомянутый благородный металл Е является Pd и/или Pt.
4. Титаносиликатный материал по п.3, отличающийся тем, что упомянутый благородный металл Е является Pd.
5. Титаносиликатный материал по п.1, отличающийся тем, что x составляет в диапазоне от 0,005 до 25,0; (y+z) составляет в диапазоне от 0,005 до 20,0 и y/z<3.
6. Титаносиликатный материал по п.1, отличающийся тем, что x составляет от 0,001 до 20,0; (y+z) составляет в диапазоне от 0,001 до 10,0 и y/z<2.
7. Титаносиликатный материал по п.1, отличающийся тем, что x составляет в диапазоне от 0,005 до 20,0; (y+z) составляет в диапазоне от 0,005 до 10,0 и y/z<1.
8. Титаносиликатный материал по п.1, отличающийся тем, что адсорбционная способность по бензолу упомянутого материала составляет по меньшей мере 25 мг/г, измеренная при условиях температуры 25°С, P/P0=0,10 и времени адсорбции 1 ч.
9. Титаносиликатный материал по п.1, отличающийся тем, что адсорбционная способность по бензолу упомянутого материала составляет по меньшей мере 35 мг/г, измеренная при условиях температуры 25°С, P/P0=0,10 и времени адсорбции 1 ч.
10. Титаносиликатный материал по п.1, отличающийся тем, что между изотермой адсорбции и изотермой десорбции низкотемпературной адсорбции азота упомянутым материалом имеется петля гистерезиса.
11. Титаносиликатный материал по п.1, отличающийся тем, что при относительном давлении Р/Р0 примерно 0,60 разность между адсорбционной способностью по азоту при десорбции и адсорбционной способностью по азоту при адсорбции составляет более 2% адсорбционной способности по азоту при адсорбции.
12. Титаносиликатный материал по п.1, отличающийся тем, что кристаллические зерна упомянутого материала полностью или частично имеют полую структуру или изогнутую структуру.
13. Титаносиликатный материал по п.12, отличающийся тем, что полость полых кристаллических зерен упомянутого материала имеет радиальную протяженность 0,1-500 нм.
14. Титаносиликатный материал по п.12, отличающийся тем, что полость полых кристаллических зерен упомянутого материала имеет радиальную протяженность 0,5-300 нм.
15. Титаносиликатный материал по п.12, отличающийся тем, что полость полых кристаллических зерен упомянутого материала имеет форму, выбранную из прямоугольной, круглой, неправильной круглой, неправильной многоугольной и их комбинаций.
16. Титаносиликатный материал по п.1, отличающийся тем, что кристаллические зерна упомянутого материала представляют собой монокристаллические зерна или агрегатные кристаллические зерна, агрегированные из множества кристаллических зерен.
17. Способ получения содержащего благородный металл титаносиликатного материала, являющегося катализатором, по п.1, отличающийся тем, что он включает следующие стадии:
(1) гомогенное смешивание титаносиликата, защитного средства, источника благородного металла, восстановителя, источника щелочи с водой с получением смеси, обладающей соотношением титаносиликат:защитное средство:источник щелочи:восстановитель:источник благородного металла:вода 100:(0,0-5,0):(0,005-5,0):(0,005-15,0):(0,005-10,0):(200-10000), где титаносиликат рассчитывают в граммах; защитное средство, источник щелочи, восстановитель и воду рассчитывают в молях; а источник благородного металла рассчитывают в граммах простого вещества благородного металла; и
(2) подачу смеси, полученной на стадии (1), в реакционный сосуд, реагирование при условиях гидротермальной обработки, выделение продукта с получением титаносиликатного материала, причем упомянутые условия гидротермальной обработки относятся к гидротермальной обработке в течение 2-360 ч при температуре 80-200°С и аутогенном давлении.
18. Способ получения по п.17, отличающийся тем, что титаносиликат на стадии (1) выбран из группы, состоящей из TS-1, TS-2, Ti-BETA, Ti-МСМ-41, Ti-ZSM-48, Ti-ZSM-12, Ti-MMM-1, Ti-MCM-22, Ti-SBA-15, Ti-MSU, Ti-MCM-48 и их смесей.
19. Способ получения по п.17, отличающийся тем, что титаносиликат на стадии (1) представляет собой TS-1.
20. Способ получения по п.17, отличающийся тем, что защитное средство на стадии (1) представляет собой полимер, подобранный из группы, состоящей из полипропилена, полиэтиленгликоля, полистирола, поливинилхлорида, полиэтилена и их производных или их смесей.
21. Способ получения по п.17, отличающийся тем, что защитное средство на стадии (1) представляет собой поверхностно-активное вещество, выбранное из группы, состоящей из анионогенных поверхностно-активных веществ, катионогенных поверхностно-активных веществ и неионогенных поверхностно-активных веществ.
22. Способ получения по п.17, отличающийся тем, что восстановитель на стадии (1) выбран из группы, состоящей из гидразина, боргидрида и цитрата натрия.
23. Способ получения по п.17, отличающийся тем, что источник благородного металла на стадии (1) выбран из группы, состоящей из оксидов, галогенидов, карбонатов, нитратов, аммонийнитратов, солей хлористого аммония, гидроксидов и других комплексов благородного металла.
24. Способ получения по п.17, отличающийся тем, что источник благородного металла на стадии (1) представляет собой источник палладия.
25. Способ получения по п.24, отличающийся тем, что источник палладия выбран из группы, состоящей из оксида палладия, карбоната палладия, хлорида палладия, нитрата палладия, аммонийнитрата палладия, аммонийхлорида палладия, гидроксида палладия и других комплексов палладия, или выбран из группы, состоящей из ацетата палладия и ацетилацетоната палладия.
26. Способ получения по п.17, отличающийся тем, что источник щелочи на стадии (1) выбран из группы, состоящей из аммиака, гидроксида натрия, гидроксида калия и гидроксида бария, или выбран из группы, состоящей из карбамида, щелочных соединений четвертичного аммония, соединений алифатических аминов, соединений аминоспиртов и их смесей.
27. Способ получения по п.26, отличающийся тем, что щелочные соединения четвертичного аммония имеют общую формулу (R1)4NOH, где R1 обозначает алкил с 1-4 атомами углерода.
28. Способ получения по п.27, отличающийся тем, что R1 обозначает пропил.
29. Способ получения по п.26, отличающийся тем, что соединения алифатических аминов имеют общую формулу R2(NH2)n, где R2 обозначает алкил или алкилиден с 1-6 атомами углерода, и n равно 1 или 2.
30. Способ получения по п.29, отличающийся тем, что соединения алифатических аминов выбраны из группы, состоящей из этиламина, н-бутиламина, бутандиамина и гександиамина.
31. Способ получения по п.26, отличающийся тем, что соединения аминоспиртов имеют общую формулу (HOR3)mNH(3-m), где R3 обозначает алкил с 1-4 атомами углерода, и m равно 1, 2 или 3.
32. Способ получения по п.31, отличающийся тем, что соединения аминоспиртов выбраны из группы, состоящей из моноэтаноламина, диэтаноламина и триэтаноламина.
33. Способ получения по п.17, отличающийся тем, что смесь на стадии (1) содержит титаносиликат:защитное средство:источник щелочи:восстановитель:источник благородного металла:воду в соотношении 100:(0,005-1,0):(0,01-2,0):(0,01-10,0):(0,01-5,0):(500-5000), где титаносиликатное молекулярное сито рассчитывают в граммах; защитное средство, источник щелочи, восстановитель и воду рассчитывают в молях; источник благородного металла рассчитывают в граммах простого вещества благородного металла.
34. Способ получения содержащего благородный металл титаносиликатного материала, являющегося катализатором, по п.1, отличающийся тем, что он включает следующие стадии:
(1) гомогенное смешивание источника титана, источника кремния, источника щелочи, защитного средства, источника благородного металла с водой с получением смеси, обладающей соотношением источник кремния:источник титана: источник щелочи:источник благородного металла:защитное средство:вода 100:(0,005-50,0):(0,005-20,0):(0,005-10,0):(0,0001-5,0):(200-10000), где источник кремния рассчитывают как SiO2, источник титана рассчитывают как TiO2; и источник благородного металла рассчитывают как простое вещество; гидротермальную кристаллизацию смеси в течение по меньшей мере 2 ч при 120-200°С и аутогенном давлении, извлечение, фильтрование, сушку и прокаливание продукта с получением промежуточного кристаллического материала;
(2) подачу промежуточного кристаллического материала, полученного на стадии (1), в фильтрат, полученный после фильтрования на стадии (1), добавление восстановителя в молярном соотношении 0,1-10 к источнику благородного металла, добавленному на стадии (1), гидротермальную обработку в течение 2-360 ч при 80-200°С и аутогенном давлении, и выделение продукта с получением титаносиликатного материала.
35. Способ получения по п.34, отличающийся тем, что источник кремния на стадии (1) выбран из группы, состоящей из геля кремниевой кислоты, золя кремниевой кислоты и органического силиката.
36. Способ получения по п.35, отличающийся тем, что органический силикат имеет общую формулу R4 4SiO4, где R4 обозначает алкил с 1-4 атомами углерода.
37. Способ получения по п.36, отличающийся тем, что R4 обозначает этил.
38. Способ получения по п.34, отличающийся тем, что источник титана представляет собой неорганическую соль титана или органический титанат.
39. Способ получения по п.38, отличающийся тем, что неорганическая соль титана выбрана из группы, состоящей из TiCl4, Ti(SO4)2 и TiOСl2.
40. Способ получения по п.38, отличающийся тем, что органический титанат имеет общую формулу Ti(OR5)4, где R5 обозначает алкил с 1-6 атомами углерода.
41. Способ получения по п.40, отличающийся тем, что R5 обозначает алкил с 2-4 атомами углерода.
42. Способ получения по п.34, отличающийся тем, что источник щелочи на стадии (1) представляет собой щелочное соединение четвертичного аммония или смесь щелочного соединения четвертичного аммония, соединения алифатического амина и соединения аминоспирта.
43. Способ получения по п.41, отличающийся тем, что щелочное соединение четвертичного аммония имеет общую формулу (R6)4NOH, где R6 обозначает алкил с 1-4 атомами углерода.
44. Способ получения по п.43, отличающийся тем, что R6 обозначает пропил.
45. Способ получения по п.42, отличающийся тем, что соединение алифатического амина имеет общую формулу R7(NH2)n, где R7 обозначает алкил или алкилиден с 1-6 атомами углерода, и n равно 1 или 2.
46. Способ получения по п.42, отличающийся тем, что соединение алифатического амина выбрано из группы, состоящей из этиламина, н-бутиламина, бутандиамина и гександиамина.
47. Способ получения по п.42, отличающийся тем, что соединение аминоспирта имеет общую формулу (HOR8)mNH(3-m), где R8 обозначает алкил с 1-4 атомами углерода, и m равно 1, 2 или 3.
48. Способ получения по п.42, отличающийся тем, что соединение аминоспирта выбрано из группы, состоящей из моноэтаноламина, диэтаноламина и триэтаноламина.
49. Способ получения по п.34, отличающийся тем, что защитное средство на стадии (1) представляет собой полимер или поверхностно-активное вещество, причем упомянутый полимер выбран из группы, состоящей из полипропилена, полиэтиленгликоля, полистирола, поливинилхлорида, полиэтилена и их производных и их смесей; а поверхностно-активное вещество выбрано из группы, состоящей из анионогенных поверхностно-активных веществ, катионогенных поверхностно-активных веществ и неионогенных поверхностно-активных веществ.
50. Способ получения по п.34, отличающийся тем, что источник благородного металла на стадии (1) представляет собой неорганическое или органическое вещество упомянутого благородного металла.
51. Способ получения по п.50, отличающийся тем, что источник благородного металла представляет собой неорганический или органический источник палладия.
52. Способ получения по п.51, отличающийся тем, что неорганический источник палладия выбран из группы, состоящей из оксида палладия, карбоната палладия, хлорида палладия, нитрата палладия, аммонийнитрата палладия, аммонийхлорида палладия, гидроксида палладия и других комплексов палладия; а упомянутый органический источник палладия выбран из группы, состоящей из ацетата палладия и ацетилацетоната палладия.
53. Способ получения по п.34, отличающийся тем, что смесь на стадии (1) обладает молярным соотношением источник кремния:источник титана:источник щелочи:источник палладия:защитное средство:вода 100:(0,01-10,0):(0,01-10,0):(0,01-5,0):(0,0005-1,0):(500-5000).
54. Способ получения по п.34, отличающийся тем, что восстановитель на стадии (2) выбран из группы, состоящей из гидроксиламина, гидразина, боргидрида, цитрата натрия и их смесей.
55. Способ получения по п.54, отличающийся тем, что гидразин выбран из группы, состоящей из гидрата гидразина, гидрохлорида гидразина и сульфата гидразина; а упомянутый боргидрид выбран из группы, состоящей из боргидрида натрия и боргидрида калия.
| WO 2000059632 A1, 12.10.2000 | |||
| Транспортное судно для плавания в ледовых условиях | 1980 |
|
SU906784A1 |
| WO 1994019277 A, 01.09.1994 | |||
| СПОСОБ ПОЛУЧЕНИЯ МАТЕРИАЛА С МИКРОМЕЗОПОРИСТОЙ СТРУКТУРОЙ | 2005 |
|
RU2282587C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРИДИНОВЫХ ОСНОВАНИЙ | 2000 |
|
RU2243217C2 |

