Изобретение относится к области органической химии, а именно к способу получения п-ацетиламинофенола I (парацетамол), который обладает анальгезирующим действием и является составной частью ряда лекарственных препаратов.
Известны, защищенные патентами, способы получения п-ацетиламинофенола из различных производных фенола или анилина. Так, при нагревании п-аминофенилацетата в течение 5 часов при температуре 120°С образуется парацетамол с выходом 43% [1].
При нагревании 4-ацетоксиацетофенона в смеси с серой, водным аммиаком получен п-ацетиламинофенол с выходом 61% [2].
Одноступенчатое восстановление-ацетилирование п-нитрофенола осуществлено при взаимодействии его с тиоацетатом калия в присутствии тритона Х-405 [3]. При этом выход п-ацетиламинофенола составляет 80%.
К недостаткам указанных способов следует отнести использование дорогостоящих химических реагентов, низкие выходы целевого продукта, наличие большого количества отходов.
Анализ научно-технической литературы показал, что наиболее простым и технически доступным является синтез парацетамола I из п-нитрофенола III по схеме 1 (путь: II→III→IV→I).
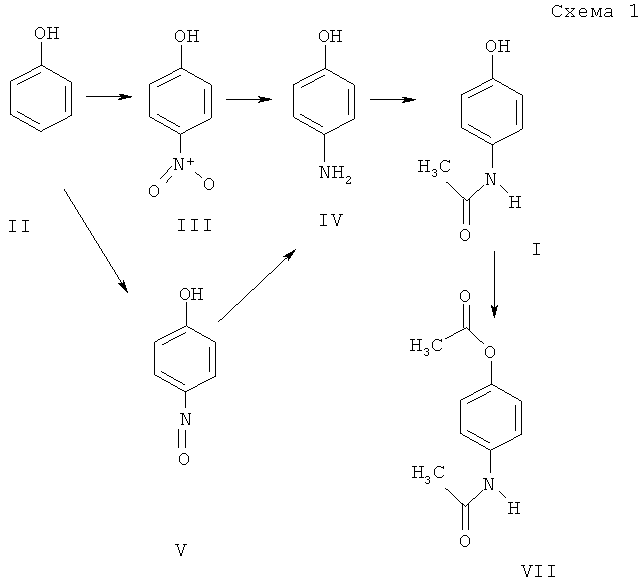
Однако существенным недостатком описанных в литературе превращений для получения парацетамола является использование в качестве исходного материала труднодоступного п-нитрофенола III, который получают нитрованием фенола II с выходом менее 30% [4]. Более доступным сырьем для синтеза парацетамола может быть п-нитрозофенол V, который получают нитрозированием фенола II с выходом более 80% [5, 6].
Наиболее близким к заявляемому способу по исходному сырью и его химическим превращениям является синтез парацетамола из п-нитрозофенола V (схема 1, путь: II→V→IV→I). В литературе описаны методики восстановления п-нитрозофенола V до п-аминофенола IV [7], который ацилируется при нагревании в уксусной кислоте до парацетамола I [8]. Однако восстановление нитрозофенола известно только реагентными методами, что сопряжено с необходимостью выделения и очистки промежуточного аминофенола IV и обуславливает появление большого количества токсичных отходов. Так в работе [7] описано восстановление нитрозофенола V в токе сероводорода с образованием при выделении и очистке п-аминофенола серусодержащих отходов.
В то же время в литературе отсутствуют данные о безотходных, экологически чистых, эффективных каталитических способах гидрирования нитрозофенола V.
Отсутствие технических решений по каталитическому гидрированию обусловлено высокой реакционной способностью п-нитрозофенола. который легко взаимодействует с образующимся в ходе гидрирования аминофенолом с образованием различных побочных соединений (например, диазосоединений).
Следует отметить, что поскольку процессы каталитического гидрирования протекают на поверхности раздела фаз, то скорость восстановления в значительной мере определяется эффективностью перемешивания реакционной среды. В то же время побочные процессы протекают во всем объеме реакционной массы и скорость их протекания может быть значительно выше реакции гидрирования.
Скорость гидрирования можно увеличивать повышением температуры реакции. Однако с увеличением температуры реакции ускоряются и побочные процессы с участием нитрозо- и аминопроизводных, протекающие во всем объеме реакционной массы.
Важнейшими параметрами, регулирующими процессы гидрирования, являются тип катализатора и применяемый растворитель. Варьирование этих параметров может изменить как механизм протекания каталитических процессов, так и повлиять на скорость побочных превращений.
Таким образом для предотвращения побочных реакций необходимо было найти такие условия, чтобы реакция гидрирования протекала быстрее, чем побочные процессы. При этом образующийся аминофенол целесообразно было бы защитить от участия в побочных процессах и провести процесс быстро, но при низких температурах.
Очевидно, что разработка практически реализуемого способа получения парацетамола гидрированием нитрозофенола является сложной задачей со многими неизвестными параметрами и далеко неочевидными перспективами ее решения.
Действительно в условиях лаборатории было установлено, что при гидрировании п-нитрозофенола в диоксане в присутствии уксусного ангидрида над катализатором Ni-Re реакция гидрирования через 2-2,5 часа прекращается, однако в качестве основного продукта из реакционной массы выделено производное диазобензола (схема 2, пример 1).

Строение диазобензола подтверждено данными ПМР-спектроскопии и масс-спектрометрии.
Так масса соединения, определенная масс-спектрометрически, соответствует расчетной. В ПМР спектре наблюдаются синглеты протонов метильных групп ацильных фрагментов и дублеты ароматических протонов.
С учетом всего вышеизложенного задачей настоящего изобретения являлось разработка простого, экологичного, технологичного способа получения парацетамола, основанного на доступном сырье.
Поставленная задача решается описываемым способом получения парацетамола I, который заключается в том, что п-нитрозофенол V перемешивают при температуре 20-50°С в растворе этилацетата в присутствии Pd/C-содержащего катализатора при давлении водорода 2.0-4.0 атм в течение 0.5-2 часов с последующим ацилированием образующегося п-аминофенола в реакторе для гидрирования.
Окончание реакции восстановления (полноту превращения исходного нитрозофенола) определяют по реакции Либермана [9] на отсутствие в реакционной смеси соединения, содержащего нитрозогруппу (пример 4).
Целевой продукт - парацетамол выделяют известными методами, в том числе фильтрацией от катализатора, отгонкой под вакуумом этилацетата, обработкой остатка водой и фильтрованием.
Идентичность образцов целевого продукта, полученного предлагаемым способом и ацилированием заведомого п-аминофенола, подтверждена совпадением температуры плавления соответствующих образцов и отсутствием температурной депрессии, для пробы смешения.
ПМР спектры сравниваемых образцов полностью идентичны.
Величина избытка используемого для ацилирования, образующегося аминофенола, уксусного ангидрида определялась экспериментально. Для полного эффективного связывания образующегося аминофенола достаточно добавления 2.0-кратного избытка уксусного ангидрида по отношению к исходному нитрозофенолу (один моль расходуется на связывание выделяющейся при восстановлении воды, а второй - для ацилирования образующегося аминофенола).
Наиболее оптимальным для эффективного проведения процесса гидрирования, позволяющим минимизировать побочные процессы и быстро провести гидрирование п-аминофенола, является применение в качестве катализатора Pd/C в среде этилацетата. Практически важным является и то, что использование этилацетата позволяет не только технологически просто осуществить процесс гидрирования и выделить целевой продукт, но легко регенерировать этот растворитель для многократного его использования.
Экспериментально установлено, что для полного превращения п-нитрозофенола в п-аминофенол при температуре 20-30°С достаточно 1.5-2 часового перемешивания реакционной массы. При повышении температуры реакции до 45-50°С время превращения существенно сокращается.
Описываемый способ получения парацетамола по сравнению с известными имеет следующие преимущества:
1. Предлагаемый способ основан на доступном отечественном сырье - нитрозофеноле.
2. Применение в качестве реакционной среды этилацетата в сочетании с катализатором Pd/C позволяет быстро в мягких условиях осуществить гидрирование п-нитрозофенола и избежать образования существенных количеств побочных продуктов. Применяемый для гидрирования растворитель - этилацетат может быть легко регенерирован.
3. Предлагаемый способ получения парацетамола позволяет осуществить в одном реакторе процессы гидрирования нитрозофенола и ацетилирование образующегося аминофенола с образованием целевого парацетамола.
4. Внедрение однореакторного процесса гидрирования-ацетилирования позволяет исключить стадии выделения и очистки промежуточного п-аминофенола, связанные с необходимостью очистки сточных вод, что уменьшает расход реагентов и существенно упрощает технологическую схему.
5. Предлагаемый метод контроля протекания реакции гидрирования позволяет точно контролировать время восстановления по чувствительной и простой аналитической реакции на нитрозогруппу, что исключает перерасход электроэнергии и положительно влияет на качество целевого продукта.
Пример 1. Гидрирование п-нитрозофенола на Ni-Re
0.8 г п-нитрозофенола, 0.8 г пасты Ni-Re промытой диоксаном и отжатого на фильтре Ni-Re, 50 мл диоксана, 3,5 мл уксусного ангидрида и 1 мл триэтиламина перемешивали в стальном реакторе при 45-45°С под давлением водорода 2.5-3.0 атм. Методом тонкослойной хроматографии было установлено, что полное превращение исходного происходит по истечении 2-х часов. Катализатор отфильтровывали. Растворитель отгоняли в вакууме до вязкого осадка. К остатку добавляли 3-5 мл ледяной воды и перемешивали в течение 1 часа. Образовавшийся осадок желтого цвета отфильтровывали. Полученный осадок перемешивали в 5 мл горячего (65-70°С) этанола и отфильтровывали. Получили 0.42 г желтого осадка с т.пл. 185-187°С. Масс-спектр: m/z 298 (30%). Спектр ЯМР 1Н, δ, м.д.: 2.33 (с, 6Н, 2×СОСН3), 7.33 (д, 4Н, J=8.6 Гц), 7.94 (д, 4Н, J=8.6 Гц).
Пример 2. Получение п-ацетиламинофенола в соответствии с оптимальными условиями описываемого способа
В металлический реактор емкостью 1 л, снабженный перемешивающим устройством, гильзой для термометра, манометром, газоподводящими трубками и рубашкой, подключенной к водяному термостату для нагрева и охлаждения реакционной массы, загружают 2.0 г (16.2 ммоль) п-нитрозофенола в 100 мл этилацетата, 0.1 г Pd/C (10%). Реакционную смесь перемешивают под давлением водорода 2.0-4.0 атм водорода при температуре 20-30°С в течение 1-2 часов. При отрицательной реакции отобранной пробы на наличие в смеси нитрозофенола (реакция Либермана на нитрозогруппу) в реакционную массу добавляют 3.3 г (32.4 ммоль) уксусного ангидрида и перемешивают еще 15 минут. Реакционную массу фильтруют от катализатора, растворитель отгоняют под вакуумом. Остаток разбавляют 10-15 мл холодной воды, охлаждают до 5-10°С и перемешивают при этой температуре 0.5-1.0 час. Выпавший осадок парацетамола отфильтровывают, промывают 10-15 мл ледяной воды. После высушивания получают 2.06 г (84% от теории). Т.пл. 169-171°С (Т. пл. лит. 170-172°С). Спектр ЯМР 1Н, δ, м.д.: 1.98 (с, 3Н, СН3), 6.62 (д, 2Н, J=4.5 Гц), 7.30 (д, 2Н, J=4.5 Гц), 8.86 (с, 1Н, NH), 9.47 (с, 1Н, ОН).
Для получения фармакопейного парацетамола перекристаллизовывают полученый продукт из 18-20 мл дистиллированной воды с добавлением 0.1 г активированного угля. Получают 1.65 г (67%) парацетамола, соответствующего требованиям Государственной фармакопеи. В случае отрицательного анализа кристаллов на цветность проводят дополнительную кристаллизацию с добавлением гидросульфита натрия для осветления из расчета 0.01 г на 1 г парацетамола. При упаривании под вакуумом маточника реакционной массы досуха и промывкой полученного твердого остатка в 5-7 мл 3-5% водного аммиака получают 0.25 г (8.4%) диацетильного производного VII. Т. пл. 135-137°С. Спектр ЯМР 1Н, δ, м.д.: 2.04 (с, 3Н, СОСН3), 2.25 (с, 3Н, СОСН3), 6.95 (д, 2Н, J=4.8 Гц), 7.57 (д, 2Н, J=4.8 Гц), 9.83 (с, 1Н, NH).
Пример 3. Получение п-ацетиламинофенола при 45-50°С
2.0 г (16.2 ммоль) п-нитрозофенола в 100 мл этилацетата, 0.1 г Pd/C (10%) перемешивают под давлением водорода 2.5-3.5 атм водорода при температуре 40-45°С в течение 0.5 часа. При отрицательной реакции отобранной пробы на наличие в смеси нитрозофенола (реакция Либермана на нитрозогруппу) в реакционную массу добавляют 3.3.г (32.4 ммоль) уксусного ангидрида и перемешивают еще 10 минут.
Выделение целевого продукта осуществляют аналогично описанному в примере 1. Выход целевого продукта в этом случае также составляет 80-85%.
Пример 4. Определение нитрозофенола в реакционной массе (реакция Либермана)
На часовое стекло пипеткой наносят 2 капли 20%-ного раствора резорцина в этаноле и 2 капли концентрированной серной кислоты. После перемешивания компонентов добавляют 1 каплю исследуемого раствора. Если в течение 1 минуты появляется буро-фиолетовое окрашивание, переходящее в красно-малиновое, это подтверждает наличие в испытуемом растворе нитрозопроизводного. Отсутствие окрашивания указывает на отсутствие в растворе нитрозопроизводного.
Предлагаемый способ предполагается использовать для синтеза парацетамола в качестве субстанции для получения лекарственных препаратов.
Источники информации
1. Патент US 5221769 (1993); (А1).
2. Патент US 5932075 (1999); (А1).
3. Патент US 2004/138509 (2004); (А1).
4. А.М.Беркенгейм. Практикум по синтетическим лекарствам и душистым веществам и фотореактивам. Москва-Ленинград, 1942, стр.82-83.
5. Препаративная органическая химия. М., Госхимиздат, 1959. стр.235.
6. С.С.Гитис, А.И.Глаз, А.В.Иванов. Практикум по органической химии. Москва. Высшая школа. 1991. стр.120.
7. Препаративная органическая химия. М., Госхимиздат, 1959. стр.517.
8. М.М.Кацнельсон, Приготовление синтетических фармацевтических препаратов. Издание 2. Под редакцией А.Е.Чичибабина. Гостехиздательство. М., 1923, стр.44.
9. Химическая энциклопедия. Научное издательство «Большая Российская энциклопедия». Москва 1992 г. Том 3, стр.275 (542).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПАРАЦЕТАМОЛА | 2014 |
|
RU2574733C1 |
| СПОСОБ ПОЛУЧЕНИЯ п-АЦЕТИЛАМИНОФЕНОЛА | 2012 |
|
RU2495865C1 |
| Способ получения пара-ацетиламинофенола | 2022 |
|
RU2800098C1 |
| СПОСОБ И УСТАНОВКА ПОЛУЧЕНИЯ ПАРАЦЕТАМОЛА ИЗ ПАРА-НИТРОЗОФЕНОЛА И/ИЛИ ПАРА-НИТРОФЕНОЛА В СРЕДЕ УКСУСНОЙ КИСЛОТЫ | 2023 |
|
RU2818763C1 |
| СПОСОБ И УСТАНОВКА ПОЛУЧЕНИЯ ПАРАЦЕТАМОЛА ИЗ ФЕНОЛА | 2023 |
|
RU2814270C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПАРАЦЕТАМОЛА | 2016 |
|
RU2668500C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПАРА-АМИНОФЕНОЛА И АЦЕТАТА ЦИНКА ПОСРЕДСТВОМ ВОССТАНОВЛЕНИЯ ПАРА-НИТРОЗОФЕНОЛА МЕТАЛЛИЧЕСКИМ ЦИНКОМ В СРЕДЕ УКСУСНОЙ КИСЛОТЫ | 2023 |
|
RU2798466C1 |
| Способ получения аминофенолов | 2022 |
|
RU2800099C1 |
| СПОСОБ ПОЛУЧЕНИЯ п-АМИНОФЕНОЛА И УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ п-АМИНОФЕНОЛА ЭТИМ СПОСОБОМ | 2023 |
|
RU2822065C1 |
| СПОСОБ И УСТАНОВКА ДЛЯ ПОЛУЧЕНИЯ ПАРА-АМИНОФЕНОЛА ИЗ ФЕНОЛА ПУТЁМ ПОСЛЕДОВАТЕЛЬНОГО НИТРОЗИРОВАНИЯ И ВОССТАНОВЛЕНИЯ СУЛЬФИДОМ АММОНИЯ | 2023 |
|
RU2801692C1 |
Предложен новый способ получения парацетамола, заключающийся в восстановлении п-нитрозофенола, проводимом в этилацетате в присутствии Pd/C-содержащего катализатора при давлении водорода 2,0-4,0 атм и температуре 20-50°С, последующем ацилировании образующегося п-аминофенола и выделении целевого продукта. Технический результат заключается в упрощении технологии и повышении выхода целевого продукта. 4 пр.
Способ получения п-ацетиламинофенола (парацетамола) формулы I
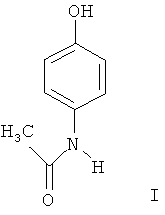
путем восстановления п-нитрозофенола, последующим ацилированием образующегося п-аминофенола и выделением целевого продукта отличающийся тем, что для упрощения технологии и повышения выхода продукта, процесс восстановления ведут в этилацетате в присутствии Pd/C-содержащего катализатора при давлении водорода 2,0-4,0 атм при температуре 20-50°С.
| US 5932075 A1, 03.08.1999 | |||
| US 5221769 A1, 22.07.1993 | |||
| Способ получения -аминофенолов | 1975 |
|
SU536167A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| СПОСОБ ПОЛУЧЕНИЯ ТАБЛЕТОК ПАРАЦЕТАМОЛА | 1992 |
|
RU2068689C1 |
| Jacquet, J | |||
| ПРИБОР ДЛЯ ЗАПИСИ И ВОСПРОИЗВЕДЕНИЯ ЗВУКОВ | 1923 |
|
SU1974A1 |
| Iqbal, Z | |||
| et al | |||
| "Different practical approaches to synthesire paracetamol" PAKISTAN JOURNAL OF SCIENCE | |||

