Предлагаемое изобретение относится к биологически активным пептидам и белкам, способным ингибировать подвижность клеток, стимулированную хемокином МСР-1, а также к лекарственным средствам на их основе.
Известно, что при непосредственном участии хемотаксических цитокинов - хемокинов в организме осуществляется привлечение лейкоцитов в участки воспаления, их активация и дифференцировка. Кроме того, известно, что воспаление является неотъемлемым компонентом многих патологических состояний, таких как онкогенез, атеросклероз, аутоиммунные заболевания, болезнь Альцгеймера и др. [1]. Хемокины - это семейство небольших по массе основных белков (8-12 кДа), действие которых осуществляется через специальные хемокиновые рецепторы. Установлено, что в патогенезе атеросклероза ведущая роль принадлежит хемокину МСР-1 (monocyte chemoattractant protein-1), который экспрессируется макрофагами, гладкомышечными и эндотелиальными клетками стенки сосуда [2]. Ниже представлена аминокислотная последовательность МСР-1 человека:
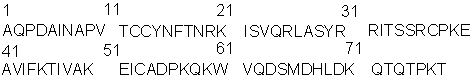
Присутствие в атеросклеротической бляшке моноцитов/макрофагов и Т-клеток свидетельствует о развитии локального воспалительного процесса в сосудистой стенке [3]. Значительная роль МСР-1 в развитии воспаления и, в частности, в атерогенезе [4] вызвала необходимость синтеза ингибиторов МСР-1.
Известны вещества непептидной (пиринидилимидазольной) природы, тормозящие миграцию моноцитарных клеток посредством блокирования некоторых звеньев внутриклеточной сигнализации [5], вследствие чего в отличие от пептидов препараты такого класса часто являются токсичными.
Известно [6], что пептид последовательности 51-62 МСР-1 (Н-EICADPKQKWVQ-OH) ингибирует миграцию моноцитов крови человека и клеток моноцитарной линии ТНР-1, стимулированных хемокинами, и не оказывает влияния на миграцию клеток, вызванную хемоаттрактантами нехимокиновой природы.
Известно, что аналог пептида последовательности 51-62 МСР-1 с заменой аланина на лейцин в положении 4 и валина на изолейцин в положении 11: H-EICLDPKQKWIQ-OH в 3-4 раза более активен, чем пептид 51-62 МСР-1, и согласно Reckless и Grainger [6] является лучшим кандидатом для проявления ингибирующей активности in vivo.
Известен также пептид следующей структуры [7]:
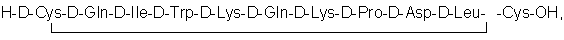 представляющий собой ретроэнантиоаналог пептида последовательности 51-62 МСР-1. Этот пептид в настоящее время является наиболее эффективным в качестве ингибитора миграции клеток in vitro и выбран в качестве прототипа заявляемого изобретения.
представляющий собой ретроэнантиоаналог пептида последовательности 51-62 МСР-1. Этот пептид в настоящее время является наиболее эффективным в качестве ингибитора миграции клеток in vitro и выбран в качестве прототипа заявляемого изобретения.
Недостатками прототипа являются сложность синтеза, обусловленная наличием дисульфидной связи в составе структуры пептида, что, как правило, приводит к уменьшению выхода целевого продукта за счет стадии циклизации, а также дороговизна синтеза (все аминокислоты, входящие в состав этого пептида D-конфигурации, что в несколько раз увеличивает стоимость исходных продуктов для синтеза этого пептида по сравнению с аналогичными пептидами, состоящими из остатков L-аминокислот).
Известно также, что С-концевая область хемокина МСР-1 не имеет функционального значения в стимуляции клеточной миграции [8, 9] и пептиды из этой области не конкурируют с меченым МСР-1 при взамодействии с рецепторами ТНР-1 клеток [10].
Задачей, стоящей перед авторами настоящего изобретения, явился поиск и синтез пептидов, ингибирующих МСР-1-стимулированную миграцию моноцитарных клеток, причем синтез таких пептидов должен быть значительно проще и дешевле по сравнению с синтезом пептида-прототипа. Актуальность такого исследования связана, в частности, с поиском соединений для покрытия стентов, используемых при коронарной ангиопластике у больных с ишемической болезнью сердца для предупреждения развития рестенозов (повторного сужения сосудов).
Поставленная задача решается путем синтеза пептида формулы: Н-DHLDKQTQTPKT-OH или его фармацевтически приемлемых солей. В структуре заявляемого пептида по сравнению с прототипом отсутствует дисульфидная связь, что всегда упрощает синтез и очистку целевого продукта. Заявляемый пептид содержит одинаковое с прототипом количество аминокислотных остатков - 12, но все они - L-конфигурации, благодаря чему затраты на синтез уменьшаются в несколько раз, поскольку D-аминокислиты гораздо дороже L-аминокислот.
Неочевидность изобретения обусловлена тем, что поставленная задача решается путем синтеза пептида последовательности 66-76 МСР-1 из С-концевой части молекулы МСР-1, хотя из литературы известно, что С-концевая область хемокина МСР-1 не имеет функционального значения в стимуляции клеточной миграции [8.9] и пептиды из этой области не конкурируют с меченым МСР-1 при взамодействии с рецепторами ТНР-1 клеток [10]. Таким образом, из литературных данных не следовало, что поиск пептидных ингибиторов МСР-1 стимулированной подвижности моноцитарых клеток из С-концевой части молекулы МСР-1 может привести к желаемым результатам. Заявляемый пептид нетоксичен, поскольку является фрагментом эндогенного белка МСР-1.
Следует отметить, что наряду с заявляемым пептидом подобной ингибирующей активностью могут обладать и его аналоги общей формулы: X1-DHLDKQTQTPKT-X2, или их фармацевтически приемлемые соли, или их эфиры, или амиды, где X1-H, низший ацил или X1 содержит один или несколько аминокислотных остатков, причем аминокислоты выбраны из алифатической, ароматической или гетероциклической групп, Х2-ОН, либо - NH2, или содержит один или несколько аминокислотных остатков, причем аминокислоты выбраны из алифатической, ароматической или гетероциклической групп. Так, в литературе [10] имеются данные о том, что амидирование фрагментов МСР-1 усиливает их активность.
В структуре заявляемого пептида по сравнению с прототипом отсутствует дисульфидная связь, что всегда упрощает синтез и очистку целевого продукта. Заявляемый пептид содержит одинаковое с прототипом количество аминокислотных остатков - 12, но все они - L-конфигурации, благодаря чему затраты на синтез пептида уменьшаются в несколько раз, поскольку D-аминокислоты гораздо дороже L-аминокислот.
Синтез заявляемого пептида осуществляется по стандартной технологии пептидного синтеза на твердой фазе [11] с применением Fmoc-(9-флуоренилметоксикарбонил)-технологии. В качестве нерастворимого носителя использовали сополимер стирола с 1% дивинилбензола с кислотолабильными якорными группами. Для блокирования функциональных групп боковых цепей аминокислот применяли следующие защиты: трет-бутильную для карбоксильных групп аспарагиновой и глутаминовой кислот и гидроксильных функций серина и треонина; трет-бутилоксикарбонильную (Boc) - защиту для ε-аминогруппы лизина; 2, 2, 5, 7, 8-пентаметилхроман-6-сульфонильную (Pmc) - защиту для гуанидиновой функции аргинина; третильную (Trt) - группу для карбоксамидной функции глутамина. Пептидную цепь наращивали по одной аминокислоте, начиная синтез с С-конца. Для создания амидных связей использовали N,N' - диизопропилкарбодиимид в присутствии 1- гидроксибензотриазола (DIC/HOBt-метод). Для заключительного деблокирования и отщепления пептидов от носителя применяли трифторуксусную кислоту со скэвенджерами. Сырой продукт твердофазного синтеза очищали с использованием препаративной ВЭЖХ.
Пример 1. Синтез пептида H-DHLDKQTQTPKT-OH. Пептид синтезировали по стандартной программе для однократной конденсации Fmoc-аминокислот. Согласно этой программе синтетический цикл включал 20-минутную активацию 1 ммоль присоединяемой аминокислоты в присутствии эквивалентных количеств DIC и НОВТ в NMP (N-метилпирролидоне), деблокирование α-аминогрупп 20% раствором пиперидина в NMP в течение 20 мин, конденсацию с 1 ммоль (4-кратным избытком) ацилирующего агента в NMP в течение 37 мин и все необходимые промежуточные промывки пептидилполимера.
Для заключительного деблокирования и отщепления пептида от носителя пептидилполимеры суспендировали в 10 мл деблокирующей смеси: трифторуксусная кислота (TFA) - фенол - триизобутилсилан (TIBS) - тиоанизол - 1,2-этандитиол (EDT) (8.25: 0.5: 0.5: 0.5: 0.25) и перемешивали при 20°С 2 ч. Смолу отфильтровывали, промывали TFA (2×1 мл), фильтрат упаривали до объема 3-5 мл и прибавляли 100 мл сухого эфира или этилацетата. Осадок отфильтровывали, промывали эфиром и этилацетатом, сушили.
Очистка пептида. Полученный сырой продукт растворяли в 5-10% уксусной кислоте (АсОН) и хроматографировали на колонке (25×250 мм) с Диасорбом-130Т C16, 10 мкм. Элюцию проводили градиентом концентрации (0.5% в минуту) СН3CN в 0.01М ацетате аммония, рН 4.5 со скоростью потока 12 мл/мин. Фракции, соответствующие целевому продукту, объединяли, упаривали, остаток растворяли в воде и лиофилизовали. Чистота полученного продукта - 96% по данным высокоэффективной жидкостной хроматографии (ВЭЖХ). Выход в расчете на стартовую аминокислоту составил 46%. Для C59P98N18O22 найдено: MALDI-MS (масс-спектрометрия с лазерной десорбцией): m/z [М+Н]+ 1411,7, вычислено [М+Н]+ 1411,5. Структура полученного соединения подтверждена спектром ядерного магнитного резонанса (1Н-ЯМР).
Тестирование синтезированного пептида проводилось по оценке его влияния на миграцию промоноцитарных клеток линии ТНР-1 и моноцитов периферической крови человека, стимулированную МСР-1.
Пример 2. Определение миграционной способности клеток. Миграционную способность клеток оценивали, используя камеру Бойдена [12]. В нижние ячейки камеры Бойдена помещали раствор МСР-1, 50-100 нг/мл, в верхние ячейки камеры вносили по 50 мкл суспензии клеток. Верхние и нижние ячейки разделяли поликарбонатной мембраной с размером пор 5 мкм (для оценки миграции моноцитов) и 8 мкм (для оценки миграции клеток ТНР-1). Камеру переносили в СО2-инкубатор на 1 час (при исследовании миграции моноцитов) или на 3 часа (при изучении миграции клеток ТНР-1). Непромигрировавшие клетки счищали, клетки, оставшиеся на фильтре, фиксировали в метаноле 5 мин и окрашивали красителем Гимза. Мембрану с прокрашенными клетками сканировали на сканере HP ScanJet 5300С и обсчитывали при помощи программы SigmaGel. Данные представляли в относительных единицах, представляющих отношение клеточной миграции в опыте к контрольной величине, полученной при оценке миграции клеток, прошедших сквозь фильтр в отсутствии хемоаттрактанта.
Пептид растворяли в бидистиллированной Н2О в концентрации 10 мМ и добавляли как в клеточную суспензию перед внесением в микрокамеру, так и в нижние ячейки микрокамеры. Конечная концентрация пептидов составляла 100 мкМ. Достоверность различий в данных по миграции клеток оценивали с помощью t-критерия Стьюдента.
Было показано, что заявляемый пептид ингибировал МСР-1-опосредованную миграцию клеток ТНР-1 на 30±5%, в то время как пептид-прототип ингибировал МСР-1-опосредованную миграцию клеток ТНР-1 на 15±5%. Было показано также, что заявляемый пептид ингибировал миграцию моноцитов в условиях модели in vitro на 21±5%, в то время как пептид-прототип ингибировал миграцию моноцитов в условиях модели in vitro на 32±7%. Эти данные свидетельствуют о сходном ингибирующем влиянии заявляемого пептида и прототипа. Полученные данные проиллюстрированы на приведенном чертеже.
Заявляемый пептид нетоксичен, поскольку является фрагментом эндогенного белка МСР-1.
СПИСОК ЛИТЕРАТУРЫ
1. Baggiolini M. //Nature. 1998. V.392. P. 565-568.
2. Libby P. //J. Intern. Med. 2000. V.247. P.349-358.
3. Ross R. // Am Heart J. 1999. V.138. P. 419-420.
4. Rollins B.L. // Blood. 1997. V.90. P.909-928.
5. Lee J.C. // Immunophatology. 2000. V. 47. P. 185-201.
6. Reckleess J., Grainger D. J. II Biochem. J. 1999. V. 340. P. 803-Gong J.-H., Clark-Levis I. // J Exp.Med. 1995. V 181. P. 631-640.
7. Reckleess J., Tatalick L. M., Grainger D. J. // Immunology. 2001. V. 103. P. 1-17.
8. Нап К.Н., Green S.R., Tangirala R.K., Tanaka S., Quenhenberger O. // J. Biol. Chem. 1999. V. 274. P. 32055-32062.
9. Jarnagin К., Grunherger D., Mulkins M., Wong В., Hemmerich S., Paavola C; Bloom A., Bhakta S., Diehl F., Freedman R., McCarley D., Polsky I., Ping-Tsou A., Kosaka A., Handel T.M. // Biochemistry. 1999. V.38. P. 16167-16177.
10. Steitz S.A.. Hasegava K., Chiang S.-L., Coob R.R., Castro MA, Lobl T.J\ Yamada M., Lazarides E., Cardarelli HP.M. //FEBS Letters. 1998. V.430. P. 158-164.
11. Neimark J. and Brian J. P.// Peptide Research. 1993. V/6. P 216.
12. Valente A.J., Rozek M.M., Schwartz C.J., Graves D.T. //Biochem. Biophys. Res. Commun. 1991. V. 176. P. 309-314.
13. Falk W., Goodwin R. H. Jr., Leonard E.J. // J. Immunol. Methods. 1980. V. 33. P.239-247.
| название | год | авторы | номер документа |
|---|---|---|---|
| Пептидное лекарственное средство | 2022 |
|
RU2827765C2 |
| ПЕПТИД ДЛЯ ИНГИБИРОВАНИЯ ФРАКТАЛКИН-СТИМУЛИРОВАННОЙ МИГРАЦИИ МОНОЦИТАРНЫХ КЛЕТОК | 2011 |
|
RU2461565C1 |
| Пептид, обладающий способностью ингибировать миграцию клеток, стимулированную белком TARC | 2016 |
|
RU2629198C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С ПРОЛОНГИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ ДОДЕКАПЕПТИДА ИНГРАМОН | 2022 |
|
RU2793124C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДОДЕКАПЕПТИДА И ТРИПЕПТИД ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2340626C1 |
| СИНТЕТИЧЕСКИЕ АНАЛОГИ IL-10 | 1997 |
|
RU2194054C2 |
| Антитела против эотаксина 2, которые распознают дополнительные связывающие CCR3 хемокины | 2015 |
|
RU2705255C2 |
| ИСПОЛЬЗОВАНИЕ МСР-1 ДЛЯ ИНДУКЦИИ ГОТОВНОСТИ К РАСКРЫТИЮ ШЕЙКИ МАТКИ | 1995 |
|
RU2207873C2 |
| АНТИТЕЛА К ЧЕЛОВЕЧЕСКОМУ МСР-1 | 2001 |
|
RU2314316C2 |
| АНТИТЕЛА ПРОТИВ МОНОЦИТАРНОГО ХЕМОАТТРАКТАНТНОГО БЕЛКА-1 (МСР-1) И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2339647C2 |
Изобретение относится к биологически активным пептидам, способным ингибировать подвижность клеток, стимулированную хемокином МСР-1. Предложен пептид формулы H-DHLDKQTQTPKT-OH и его фармацевтически приемлемые соли. Заявленный пептид может использоваться в кардиологии для покрытия стентов при ангиопластике с целью предупреждения рестенозов. 1 ил.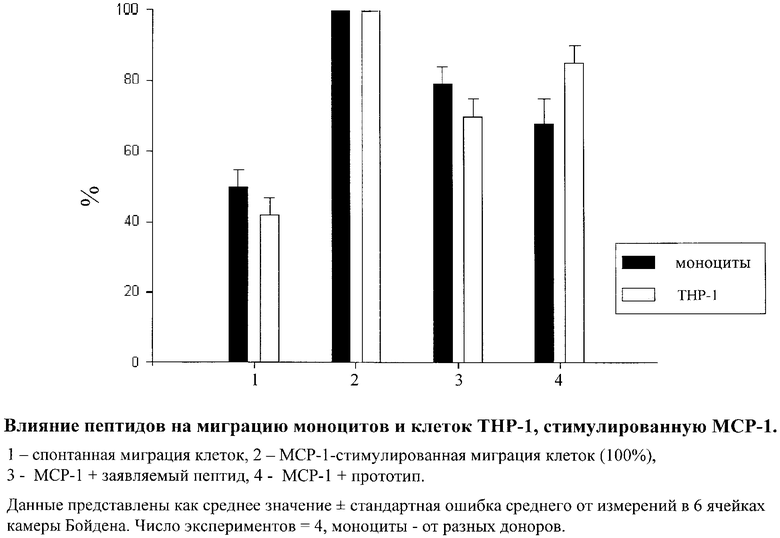
Пептид H-DHLDKQTQTPKT-OH и его фармацевтически приемлемые соли.
| ЦИТОМОДУЛИРУЮЩИЕ ЛИПОФИЛЬНЫЕ ПЕПТИДЫ ДЛЯ МОДУЛЯЦИИ АКТИВНОСТИ ИММУННОЙ СИСТЕМЫ И ПОДАВЛЕНИЯ ВОСПАЛЕНИЯ | 1998 |
|
RU2214417C2 |

