Изобретение относится к области химико-фармацевтической промышленности и медицины и касается нового соединения, которое может найти применение в качестве противоязвенного препарата.
Заболевания органов пищеварения всегда находились в центре внимания врачей по причине своей значительной распространенности. Из большого числа болезней данной группы особое социальное и медицинское значение имеет язвенная болезнь желудка и двенадцатиперстной кишки, поскольку по частоте она превышает всю другую патологию желудочно-кишечного тракта. Так, среди европейского населения уровень заболеваемости язвенной болезнью достигает 5% (от 0,1-0,3% в Западной Европе до 1,5-5% в России); в течение года при отсутствии лечения до 60% язв рецидивируют.
По объему продаж на мировом рынке в 2004 году противоязвенные препараты занимают второе место среди других терапевтических групп препаратов, их доля составляет 25,5% от всего объема продаваемых лекарств. Причем анализ результатов продаж за предыдущие годы показывает, что имеется очевидный рост годовых объемов продаж в среднем на 5%. Приведенные данные подготовлены Николь Грэй, специалистом журнала Pharmaceutical Executive и опубликованы на сайте www.pharmexec.com в статье "50 лучших фармацевтических компаний 2004 года".
На заболеваемость язвенной болезнью (ЯБ) влияют следующие факторы: социально-экономические и демографические (чаще болеют жители городов); чаще болеют жители развитых стран; чаще болеет население северных районов.
К факторам риска развития ЯБ относят: мужской пол; группу крови 0 (I); низкую кислотообразующую функцию желудка; курение; стресс.
Лечебные мероприятия при язвенной болезни направляют на снижение повреждающего воздействия желудочного сока в отношении окружающих тканей (антисекреторные препараты); повышение резистентности подвергающихся его воздействию тканей, в первую очередь слизистой желудка (гастропротекторные средства), и антимикробная терапия, направленная на уничтожение микроорганизмов Helicobacter Pylori (HP).
Основа лечения ЯБ - курсовая или поддерживающая терапия антисекреторными препаратами: ингибиторами протонного насоса, блокаторами Н2-рецепторов гистамина, антацидными препаратами, препаратами висмута (висмута субсалицилат/субцитрат), сукральфатом (комбинация гидроокиси алюминия и сахарного октасульфата). При этом некогда популярные препараты 80-х годов - блокаторы Н2-гистаминовых рецепторов - в значительной мере уступили место более эффективным антисекреторным средствам - ингибиторам протонной помпы (ИПП).
Блокаторы протонного насоса впервые были синтезированы в 1976 году. Ими оказались два вещества - тимопразол и омепразол. Тимопразол не нашел широкого применения, а омепразол получил широкое распространение. Именно он был официально рекомендован для клинического применения в 1988 году на Всемирном конгрессе гастроэнтерологов в Риме. Одной из причин такого признания омепразола стало подтверждение его высокой эффективности в уничтожении бактерии HP, открытие которой в 1983 году заставило пересмотреть взгляды на природу гастродуоденальных заболеваний. Значимость хеликобактерной инфекции приобрела огромное значение еще и потому, что эксперты ВОЗ, после анализа результатов многочисленных исследований, признали ее канцерогенной для человека. В связи с этим чрезвычайную актуальность приобретают вопросы эрадикации - уничтожения HP в слизистой оболочке желудка с целью обеспечить благоприятные условия для заживления язв.
Несмотря на всеобщее признание омепразола как первого эффективного представителя ИПП, поиски в направлении совершенствования ИПП продолжались. И в 1992 году специалисты японской фирмы Такеда синтезировали ИПП нового поколения - лансопразол. Спустя несколько лет в арсенале врачей всего мира появились ИПП следующих генераций: пантопразол, рабепразол, эзомепразол.
Таким образом, ИПП - наиболее часто используемые препараты для лечения язвы желудка и двенадцатиперстной кишки в настоящее время во всем мире.
ИПП используются для лечения ЯБ чуть больше 15 лет, поэтому еще не в полной мере известно о том, насколько они безопасны. Опасные побочные эффекты или представляющие угрозу для жизни пациента взаимодействия нового препарата с другими лекарственными средствами могут не быть выявлены до тех пор, пока не будет накоплен достаточный опыт применения - речь может идти о сотнях тысяч случаев использования. Однако уже и сейчас появляются сведения о негативном воздействии препаратов этого типа (в основном омепразола) на печень при применении у детей [1]. В клинике также отмечены такие побочные эффекты при применении ИПП, как диарея, тошнота, абдоминальная колика, сонливость, головная боль.
При длительном применении омепразола и других ингибиторов протонной помпы (так же как и при применении некоторых других антисекреторных препаратов) возможно стойкое и глубокое угнетение кислотной желудочной секреции, а в некоторых случаях - атрофии секреторных клеток желудка, что приводит к серьезным осложнениям (развитие новообразований типа гастриномы, аденокарциомы, обсеменение HP, кишечные инфекции, расстройство всасывания кишечником жиров, минералов и витаминов). Все указанные осложнения описаны в следующих работах: [2-12].
Наряду с этим, надо учитывать, что метаболизм ИПП происходит с участием цитохрома Р450 - оксидазной системы, поэтому при длительном применении ИПП может развиться толерантность к данным препаратам при истощении активности данной ферментной системы [13]. С этим, возможно, связан синдром отмены препарата - рецидивы заболевания отмечены в разные сроки (от 2 до 22 недель), в среднем 14 недель после отмены омепразола [14].
Наблюдаются и другие побочные и нежелательные эффекты препаратов этого класса, которые, в частности, указаны в инструкции по применению препаратов (головная боль, диарея, аллергические реакции, гинекомастия и др.).
Таким образом, ИПП являются эффективными препаратами, получившими широкое распространение для лечения ЯБ, однако необходимо учитывать их побочные эффекты и неблагоприятные последствия (наиболее опасные, вплоть до смертельных исходов - это инфицирование HP и развитие опухолей желудка), а также определенный процент случаев, когда они оказываются неэффективными при лечении. Поэтому поиск новых противоязвенных средств является важной и актуальной задачей.
Наиболее перспективным направлением в создании новых противоязвенных препаратов является получение препарата с гастропротекторными свойствами, так как в этом случае не происходит «жесткого» подавления желудочной секреции, а следовательно, снижается риск таких неблагоприятных событий, как обсеменение желудка HP и развитие онкологических процессов. В ряде случаев - при сохранении нормальной секреции соляной кислоты или пониженной - подавление желудочной секреции и не нужно. В этих случаях достаточно «защитить» слизистую оболочку желудка от раздражающего действия желудочного сока.
Препараты, отвечающие этим требованиям, созданы на основе простагландинов. Положительными свойствами простагландинов при лечении язвы желудка является их способность активировать протеинкиназу, которая, воздействуя на клеточные мембраны, может защищать слизистую оболочку желудка от агрессивного влияния кислоты и пепсина, а также стимуляция ими секреции слизи. С другой стороны, простагландины обладают антисекреторной активностью в отношении желудочного сока. Такое сочетание противоязвенных эффектов делает простагландины очень привлекательными в качестве лекарственных средств, однако опыт применения этих препаратов относительно невелик, только на уровне клинических испытаний [15, 16, 17].
Перспективным направлением является создание гастропротекторных препаратов - доноров оксида азота (NO). Оксид азота действует как многофункциональный гастропротекторный медиатор, влияющий на ряд аспектов физиологии желудочно-кишечного тракта (ЖКТ), включая секрецию слизи и бикарбоната, ток крови в стенках ЖКТ [18]. Кроме того, известны противомикробные свойства NO, в частности, в отношении бактерий HP [19]. Прямым антисекреторным действием доноры NO не обладают, но бикарбонат, секретируемый в желудке под действием доноров оксида азота, взаимодействует с соляной кислотой и таким образом нейтрализует ее.
В качестве противоязвенных средств используются также вещества, блокирующие секрецию соляной кислоты: это блокаторы H2-рецепторов (фамотидин, ранитидин, циметидин) и ингибиторы протонного насоса. Блокаторы Н2-рецепторов имеют ряд недостатков по сравнению с ингибиторами протонного насоса: во-первых, блокируется только сигнал, возникающий при связывании гистамина, во-вторых, их действие обратимо и они быстро элиминируются из кровеносного русла, поэтому пациент должен пить несколько таблеток в день. И, наконец, их применение приводит к усилению синтеза рецепторов для гистамина, в результате чего после отмены Н2-блокаторов наблюдается рикошетная вспышка секреции соляной кислоты.
Из ингибиторов протонного насоса первым на рынке в 80-е годы появился омепразол (фирма «Астра-Зенека»), который представляет собой замещенный бензимидазол. Ингибиторы протонного насоса, имеющиеся на рынке в настоящее время, также относятся к замещенным бензимидазолам. Это нексиум (очищенный S-изомер омепразола или эзомепразол, фирма «Астра-Зенека»), многочисленные генерики омепразола, париет (рабепразол, фирма «Янсен Силаг») и контролок (пантопразол, фирма «Бик Гульден», последний в России отсутствует). Все эти соединения представляют собой пролекарства. Они являются слабыми основаниями, которые при попадании в кислую среду париетальных канальцев связывают H+ и претерпевают внутримолекулярную перестройку, приводящую к их превращению в собственно лекарство, активный сульфенамид, взаимодействующий с SH-группами Н,К-АТФазы, экспонированными в полость секреторного канальца. Сульфенамиды образуют ковалентную связь с SH-группами, вследствие чего их действие необратимо [33]. Их эффект устраняется только после элиминации молекулы Н,К-АТФазы, модифицированной ингибитором, а время полужизни Н,К-АТФазы человека составляет около 40 часов. Именно по этой причине применение необратимых ингибиторов протонного насоса второго поколения, таких как париет и нексиум, обеспечивает очень высокую эффективность лечения кислотозависимых заболеваний. При лечении язвенной болезни этими ингибиторами в сочетании с антибиотиками, устраняющими инфекцию ИР, эффективность составляет более 90% [33].
Кроме необратимых ингибиторов протонного насоса известны также обратимые ингибиторы, лекарства на основе которых до настоящего времени не выпускаются. По-видимому, это связано с очень высокой эффективностью и безопасностью действия лекарств на основе необратимых ингибиторов протонного насоса.
Среди обратимых ингибиторов протонного насоса наиболее известен имидозопиридин SCH-28080, который ингибирует Н,К-АТФазу конкурентным по отношению к К+ способом с константой ингибирования (Кi) в ряду 0,2-0,24 мкМ [34]. Аналоги SCH-28080 взаимодействуют с фрагментом a-субъединицы Н,К-АТФазы длиной в 44 аминокислотных остатка, начинающимся с Leu-854 и заканчивающимся Arg-897. Этот фрагмент гомологичен подобному у Н,К-АТФазы, которую SCH-28080 также ингибирует, но с меньшим сродством. По-видимому, он представляет собой калий-связывающий центр этих родственных ферментов. Помимо SCH-28080 известен более специфичный ингибитор SK&F, который ингибирует только Н,К-АТФазу с 0,5 мкМ и конкурирует с SCH-28080 и К+, а также SK&F 96356, ингибирующий Н,К-АТФазу с Кi=0,07 мкМ, который также конкурирует с SCH-28080 и К+.
Препарат SCH 28080, имеющий формулу:

может быть указан в качестве наиболее близкого аналога [32]. Соединение является эффективным, однако возможно конкурирующее действие по отношению к ионам калия, что может привести к неблагоприятным побочным эффектам.
Задачей изобретения является поиск новых средств, обладающих эффективной противоязвенной активностью, но не имеющих побочных действий известных средств.
Задача решается синтезом нового соединения, представляющего собой производное пиридопиразиндионов. Согласно изобретению предлагается соединение - 9-(хинонил-2)-2-п-этоксифенилэтил-4,5,6,7,8,9,10,11-октагидропиридо[1,2-а]пиразиндион-1,4 формулы (I) и его фармацевтически приемлемые комплексные производные.

Синтез целевого соединения I
Исходным для синтеза является производное хромена (II), которое под действием хлорацетилхлорида превращено в соответствующее хлорацетильное производное (III).
Действие на последнее β (п-этоксифенил)этиламина привело к раскрытию пиранового цикла, образованию пиперазиндионового кольца. Последующее окисление гидрохинонового фрагмента в промежуточном IV привело к целевому соединению I. (Получение хромена II описано нами в работе: В.М.Любчанская, Л.М. Алексеева, С.А. Савина, А.С.Шашков, В.Г.Граник. Изв. АН, сер. хим., 2002, №10, стр.1736-1743.)

Получение III
К суспензии 4,23 г (19 ммоля) хромена II в 35 мл толуола добавляют при перемешивании 7,7 мл (95 ммоля) хлорацетилхлорида, кипятят 1 час, охлаждают, отфильтровывают 5,47 г III (98%), т.пл. 159-162°С (спирт). Спектр 1Н ЯМР (d6 ДМСО) δ, м.д.: 2,01 (кв. 2Н), 2,89 (т., 2Н), 3,71 (уш. с. 2Н), 4,38 (с. 2Н), 7,03 (м. 2Н), 7,26 (д. 1Н), 9,60 (с. 1Н).
Получение IV
К раствору 0, 88 г (3 ммоль) III в 6 мл ДМФА прибавляют 12 ммоля β (п-этоксифенил)этиламина, перемешивают 10 час при 20°С, разбавляют водой, отфильтровывают 1 г IV, выход 93%, т.пл. 169-171°С (ацетонитрил).
Получение I
К суспензии 1 ммоля IV в 10 мл хлороформа прибавляют раствор 0,14 г бикарбоната натрия в 2,5 мл воды и окисляющую смесь из 0,6 г K3[Fе(СN)6], 0,14 г NaHCO3 и 0,18 г поташа, 6 мл воды. Смесь перемешивают 1 час при 20°С, органический слой отделяют и упаривают, остаток растирают с эфиром, получают 85% I, т.пл. 137-140°С. Найдено: С 68.36, Н 6.12, N 6.57. C24H24N2O5. Вычислено: С 68.56, Н 5.75, N 6.66. Спектр 1Н ЯМР (ДМСО-d6, δ, м.д.): 1.32, 4.25 (тр., кв. 3Н, СН3СН2, J=7,5 Гц), 1.86 (к, 2Н, 7-СН2, J=7,5 Гц), 2.34 (уш.с., 2Н, 8-СН2), 2,68 и 3.40 (оба т., по 2Н, 2-СН2СН2, J=7,2 Гц), 4.04 (с., 2Н, 3-СН2), 6.68 (д., 1Н, J=2,8 Гц), 6.78 (к., 1Н, 4'-Н), 6.89 (к., 1Н, 5'-Н, J=8,4 Гц), 6,80-7,10 (AA'BB', 4H, С6Н4).
В качестве фармацевтически приемлемых комплексных соединений могут быть получены, например, соединения с циклодекстринами или глицирризиновой кислотой. Новые соединения обладают антисекреторной и гастропротекторной активностью.
Пример 1. Исследование противоязвенной активности соединения I изучали на следующих моделях:
1.1. Повреждения слизистой оболочки желудка у мышей, вызванные введением 0,6 N соляной кислоты и индометацина [26]
Нелинейных мышей-самцов массой 23-24 г лишали пищи в течение 24 часов, доступ к воде не ограничивали. Исследуемое соединение в дозах 25, 50 и 100 мг/кг вводили зондом в желудок за 1 ч, 3 ч и 6 ч до введения 0,6 N соляной кислоты (5 мл/кг) и индометацина (20 мг/кг), еще через 1 час животных забивали, извлекали желудки и подсчитывали число язв. В качестве препаратов сравнения применяли фамотидин в дозах 25 мг/кг и 50 мг/кг внутрижелудочно и омепразол в дозе 50 мг/кг внутрижелудочно.
1.2. Повреждения слизистой оболочки у мышей, вызванные введением абсолютного этанола [27]
Нелинейных мышей-самцов массой 20-23 г лишали пищи за 24 часа до опыта и за 18 часов до опыта ограничивали доступ к воде. Исследуемое соединение в дозах 25, 50 и 100 мг/кг вводили зондом в желудок за 1 ч, 3 ч и 6 ч до введения абсолютного этанола (0,3 мл), еще через час животных забивали, извлекали желудки и измеряли длину поврежденных участков слизистой оболочки в миллиметрах.
В опытах использовали контроль - животных, не получавших исследуемое соединение, а только по 0,3 мл физиологического раствора. В качестве препаратов сравнения применяли фамотидин в дозах 25 и 50 мг/кг внутрь желудка и омепразол в дозе 50 мг/кг внутрижелудочно.
1.3. Повреждения слизистой оболочки желудка у крыс, вызванные введением соединения 48/80 [28]
Исследуемое соединение в дозе 25 и 100 мг/кг вводили зондом в желудок за 1 ч и 6 ч до внутрибрюшинного введения соединения 48/80, развитие повреждений слизистой оболочки желудка наблюдали через 3 часа после введения соединения 48/80. Животных забивали, извлекали желудки и измеряли длину поврежденных участков слизистой оболочки в миллиметрах. В качестве препаратов сравнения применяли фамотидин в дозах 25 и 50 мг/кг внутрь и омепразол в дозе 50 мг/кг, оба внутрижелудочно.
1.4. Хроническая язва желудка, вызванная введением крысам 0,025 мл 20% уксусной кислоты [29]
Хроническую язву желудка у крыс вызывали введением 0,025 мл 20% уксусной кислоты под серозную оболочку желудка на границе фундального и антрального отделов. Через сутки после операции исследуемое соединение (в дозе 50 мг/кг) и препараты сравнения (омепразол - 50 мг/кг и фамотидин - 50 мг/кг) вводили зондом в желудок один раз в день в течение 21 дня. Каждые 7 дней по 6 животных из каждой группы забивали и определяли площадь язвенного дефекта желудка в мм2.
Пример 2. Исследование антисекреторной активности
2.1. Изучение базальной секреции у крыс под влиянием исследуемых веществ [30]
Исследование проводили на нелинейных крысах-самцах массой 180-200 г. Животных лишали пищи в течение 24 часов, воду не ограничивали. Под эфирным наркозом вскрывали брюшную полость и накладывали лигатуру на пилорический отдел желудка. Через 4 часа после операции исследовали желудочное содержимое; определяли объем секреции в расчете на 100 г массы тела животного, рН желудочного сока, свободную соляную кислоту и общую кислотность желудочного сока. Исследуемое соединение вводили внутрь в дозе 100 мг/кг в виде водной взвеси с добавлением твин-80 за 60 минут до перевязки пилоруса. Контрольным животным вводили дистиллированную воду с добавлением твин-80. В качестве препаратов сравнения применяли фамотидин в дозах 25 и 50 мг/кг внутрижелудочно и омепразол в дозе 50 мг/кг внутрижелудочно.
2.2 Изучение секреции желудка, стимулированной гистамином, пентагастрином и 1-аминопиреном
Исследование проводили на нелинейных крысах-самцах массой 180-200 г. Животных лишали пищи в течение 24 часов, воду не ограничивали. Перед наложением лигатур на пилорический отдел желудка крысам вводили подкожно гистамин (2,5 мг/кг), другой группе животных пентагастрин 10 мг/кг подкожно и третьей группе животных 1-аминопирен 1 мг/кг. Исследуемое соединение вводили внутрь при дозе 100 мг/кг в виде водной взвеси с добавлением твин-80 за 1 ч и 3 ч до перевязки пилоруса. Контрольным животным вводили дистиллированную воду с добавлением твин-80. В качестве препаратов сравнения применяли фамотидин в дозах 25 и 50 мг/кг внутрижелудочно и омепразол в дозе 50 мг/кг внутрижелудочно. Через 4 часа после операции исследовали желудочное содержимое; определяли объем секреции в расчете на 100 г массы тела животного, рН желудочного сока, свободную соляную кислоту и общую кислотность желудочного сока.
Пример 3. Исследование моторно-двигательной активности
Исследование пропульсивной активности кишечника [31]
Исследуемые соединения вводили нелинейным мышам-самцам массой 22-24 г зондом в желудок за 60 минут до введения активированного угля (0,3 мл 10% суспензии в желудок). Через 2 часа животных забивали с помощью СО2 и измеряли длину участка тонкого кишечника (в см), заполненного активированным углем. Контрольным животным вводили внутрижелудочно по 0,3 мл воды, препаратом сравнения был церукал в дозе 25 мг/кг внутрижелудочно.
Пример 4. Определение токсичности (ЛД50) исследуемых веществ при однократном введении [32]
Результаты исследования
1. Показано, что изученные производные пиридопиразиндиона обладают гастропротекторным действием в отношении острых язв желудка, вызванных введением соляной кислоты, индометацина, абсолютного этанрла, соединения 48/80; а также оказывают противоязвенное действие, способствуя заживлению язвенного дефекта на модели хронической уксусной язвы желудка. Их эффективность выше, чем у фамотидина, и равна омепразолу. По длительности эффекта изученные соединения также равны омепразолу и фамотидину, пролонгированным действием они не обладают.
2. При изучении антисекреторной активности показано, что омепразол блокирует базальную секрецию на 25% через 1 час после его введения и на 40% - через 3 часа; фамотидин: через 1 час - на 56% и через 3 часа - на 80%. Изучаемые новые соединения не подавляют базальную кислотную желудочную секрецию, что принципиально отличает их от известных противоязвенных средств (блокаторов H2-рецепторов, ингибиторов протонной помпы).
Таким образом, фамотидин сильно, а омепразол - умеренно блокируют базальную желудочную секрецию. Изученное новое соединение не подавляет базальный уровень желудочной секреции.
При изучении стимулированной секреции желудочного сока показано, что омепразол блокирует секрецию, стимулированную гистамином, на 70% через 1 час после его введения и на 80% - через 3 часа; стимулированную пентагастрином также в среднем на 90% и стимулированную 1-аминопиреном - на 90%. Фамотидин блокирует стимулированную гистамином секрецию на 90% через 1 час после его введения и на 95% - через 3 часа; стимулированную пентагастрином в среднем на 40% и стимулированную 1-аминопиреном - через 1 час - на 36% и через 3 часа - на 40%. Новое соединение блокирует секрецию, стимулированную гистамином, на 40% через 1 час после его введения и на 57% - через 3 часа; стимулированную пентагастрином также снижает в среднем на 54% и стимулированную 1-аминопиреном - на 50%.
Таким образом, омепразол блокирует секрецию желудочного сока, стимулированную всеми тремя соединениями (гистамином, пентагастрином, 1-аминопиреном), фамотидин блокирует только секрецию, стимулированную гистамином, и слабо - другими соединениями (пентагастрином и 1-аминопиреном), изученное соединение блокирует все три вида стимулированной секреции (так же как и омепразол), но более умерено.
3. При изучении моторно-двигательной активности желудка показано, что омепразол замедляет моторно-двигательную работу кишечника в среднем на 35%, фамотидин - не влияет на моторно-двигательную активность кишечника, новое исследованное соединение также не влияет на моторно-двигательную активность кишечника.
Таким образом, влияние на моторно-двигательную активность кишечника оказывает только омепразол. Данное свойство препарата, особенно при его длительном применении может отрицательно сказаться на основном противоязвенном эффекте, так как при замедлении моторно-двигательной активности желудка и кишечника содержимое желудка оказывает дополнительное раздражающее действие на язвенный дефект.
4. При изучении общей токсичности показано, что новое соединение и его производные малотоксичны, так же как и омепразол. При сравнении ЛД50 омепразола, фамотидина и изученных соединений показано, что новые соединения менее токсичны, по сравнению с фамотидином и омепразолом.
Пример 5. Проверка действия соединения I на ферментативную активность Н,K-АТФазы из слизистой оболочки желудка кролика, исследование обратимости этого действия и оценка эффекта этого соединения в присутствии ионов К+
Препарат микросом, обогащенный активностью Н,К-АТФазы, был получен по методу, предложенному R.A. Farley и L.D. Faller. Слизистую оболочку желудка получали из кролика, которого предварительно подвергали декапитации. Все операции проводили на льду. Желудок разрезали по линии наибольшей кривизны, промывали и соскребали со стенок желудка слизистую оболочку. Полученную слизистую замораживали и хранили при -80°С.
Для выделения препарата микросом, обогащенных Н,К-АТФазой, слизистую желудка размораживали и гомогенизировали в среде выделения (0,25 М сахароза, 5 мМ PIPES, 20 мМ ТРИС, рН 7,4) гомогенизатором типа Polytron (макс. обороты, 30 сек). Соотношение веса ткани:среда выделения - 1:20.
Далее гомогенат центрифугировали 10 мин при 10000g. Осадок отбрасывали, а супернатант центрифугировали 1 час при 100000g. Полученный осадок ресуспендировали в среде выделения (в минимальном объеме, необходимом для ресуспендирования). Суспензию наслаивали на ступенчатый градиент Фикола 400, приготовленный на среде выделения: 12% раствор Фикола, 4% раствор Фикола. Далее центрифугировали 180 мин при 100000g. После центрифугирования на границе 4% Фикола и 12% Фикола видна белая полоса, которая представляет собой фракцию микросом, обогащенную Н,K-АТФазой. Фракцию собирали, разделяли на аликвоты и хранили при -80°С.
Активность измеряли по нарастанию в среде неорганического фосфата, который образуется при гидролизе АТФ. Концентрацию неорганического фосфата измеряли по методу Ратбуна и Бетлах. Среда инкубации содержала: 30 мМ имидазола, рН 7,4, 3 мМ MgCl2, 130 мМ NaCl, 20 мМ KСl, 3 мМ АТР, 2 мМ уабаина, 0,1 мМ ЭГТА, 2,25 мкМ валиномицина, 5 мМ NaN3, 5 мкМ СССР (протонофор карбонил цианид м-хлорофенил гидразон).
В инкубационную пробу вносили среду инкубации и размороженную и гомогенизированную фракцию микросом. Пробу инкубировали при 37°С 20 мин и останавливали реакцию добавлением холодного стоп-раствора (3 М ацетат натрия, 3,7% формальдегид, рН 4,3).
Для оценки содержания фосфата, получаемого при энзиматическом гидролизе АТР, в пробу добавляли 2% раствор молибдата аммония и раствор хлорида олова (3 мг/мл). После развития синей окраски, через 15 мин, пробы колориметрировали при длине волны 650 нм.
Активность Н,K-АТФазы определяли как активность, ингибируемую 0,1 мМ SCH-28080. Подавляемая этим ингибитором активность Н,K-АТФазы составляла около 30%. Концентрация исследуемого соединения варьировали в диапазоне от 10-8 до 10-4 М.
Результаты исследования
Соединение I оказывает ингибирующее действие на активность Н,K-АТФазы из слизистой оболочки желудка кролика с I50=10-6 М. Эффект соединения I на Н,K-АТФазу обратим, он развивается во времени мгновенно и устраняется при разведении комплекса фермент-ингибитор.
Было исследовано, существует ли конкуренция между I и ионами калия. С этой целью исследована зависимость активности Н,K-АТФазы от концентрации KСl, а затем - ингибирующее действие I на этот фермент при насыщающих (20 мМ) и ненасыщающих (2 мМ) концентрациях KСl.
Установлено, что соединение I не конкурирует с ионами К+, напротив, эффект соединения I на активность Н,K-АТФазы из слизистой оболочки желудка кролика усиливался под действием ионов К+: значение I50 составило 10-5 М при 2 мМ КСl и 10-6 М при 20 мМ КСl.
Таким образом, производное пиридопиразиндиона (соединение I) обратимо ингибирует гидролитическую активность Н,K-АТФазы, значение I50 составляет 10-6 М при 20 мМ KСl; ингибиторная способность соединения I усиливается при повышении концентрации K+. Значение I50 снижается от 10-5 М при 2 мМ KСl до 10-6 М при 20 мМ KСl. Такое свойство соединения I дает преимущество при его использовании в живых системах.
Полученные результаты свидетельствуют, что соединение I является эффективным обратимым ингибитором Н,K-АТФазы. Однако в отличие от известного обратимого ингибитора SCH-28080 действие I не является конкурентным по отношению к калию. По-видимому, соединение I не связывается в калий-связывающем центре фермента. Более того, К+ усиливает ингибирующее действие соединения I. Это может оказаться важным вне зависимости от того, со стороны цитоплазмы или со стороны секреторного канальца действует ингибитор, так как концентрация К+ может достигать высоких значений и в секреторном канальце, который непосредственно связан с полостью желудка. Таким образом, соединение I является новым неизвестным ранее типом обратимых ингибиторов Н,K-АТФазы, действие которых не конкурентно по отношению к К+.
Пример 6. Исследование противоязвенной активности комплекса соединения I
Соединение I может образовывать комплексы с веществами, обладающими следующими свойствами:
- имеющими торообразную или иную сходную с ней структуру либо способность образовывать торообразные структуры, у которых внешняя поверхность гидрофильна, а внутренняя липофильна;
- имеющими сравнительно непрочные Ван-дер-Ваальсовы или водородные связи с соединением I.
Например, такими свойствами обладают циклодекстрины и глицирризиновая кислота.
1. Получение комплексных соединений
А) 5 г γ-циклодекстрина растворяют в 50 мл дистиллированной воды. В полученном растворе при температуре 35-40°С под действием ультразвука растворяют 250 мг соединения I. Воду удаляют лиофилизацией и остаток сушат в вакууме 1 мм рт.ст. при температуре 20°С. Получают 5,2-5,3 г комплексного соединения с содержанием основного вещества около 5%.
Об образовании комплекса свидетельствует смещение сигналов ароматических протонов в спектрах 1H ЯМР на 0,15-0,2 м.д.
Б) 7,5 г γ-циклодекстрина растворяют в 50 мл дистиллированной воды. В полученном растворе при температуре 35-40°С под действием ультразвука растворяют 500 мг соединения I. Воду удаляют лиофилизацией и остаток сушат в вакууме 1 мм. рт.ст. при температуре 20°С. Получают 7,9-8,1 г комплекса с содержанием основного вещества около 6,2%.
Об образовании комплекса свидетельствует смещение сигналов ароматических протонов в спектрах 1Н ЯМР на 0,1-0,2 м.д.
2. Исследование противоязвенной активности комплекса соединения I
Исследование противоязвенной активности соединения I (9-(хинонил-2)-2-п-этоксифенилэтил-4,5,6,7,8,9,10,11-октагидропиридо[1,2-а]пиразиндион-1,4) и его комплексных соединений изучали на модели повреждения слизистой оболочки желудка у мышей, вызванного введением 0,6 N соляной кислоты и индометацина.
Мышей-самцов массой 23-24 г лишали пищи в течение 24 часов, доступ к воде не ограничивали. Комплекс соединения I с циклодекстрином (ЦД) вводили в виде раствора 1,25 г комплекса в 10 мл дистиллированной воды. Раствор вводили зондом в желудок в объемах 0,1, 0,2 и 0,3 мл, что эквивалентно 25, 50 и 75 мг/кг соединения I соответственно. Комплекс соединения I с глицирризиновой кислотой (ГК) вводили в виде раствора 1,0 г комплекса в 10 мл дистиллированной воды. Раствор комплекса вводили зондом в желудок в объемах 0,1, 0,2 и 0,3 мл, что эквивалентно 25, 50 и 75 мг/кг соединения I соответственно. Соединение I без комплексообразователя также вводили в дозах 25, 50 и 75 мг/кг зондом в желудок. Во всех случаях исследуемые вещества вводили за 6, 3 и 1 час до введения 0,6 N соляной кислоты (5 мл/кг) и индометацина (20 мг/кг). Через 1 час после введения соляной кислоты и индометацина животных забивали, извлекали желудки и подсчитывали число язв. Результаты представлены в таблице 1.
Как видно из представленных данных, соединение I, его комплексы с циклодекстрином и глицирризиновой кислотой по изучаемому показателю достоверно не различались между собой по своему противоязвенному действию. Таким образом, комплексы соединения I обладают свойствами, аналогичными соединению I.




| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ЯЗВЕННОЙ БОЛЕЗНИ ЖЕЛУДКА И/ИЛИ 12-ПЕРСТНОЙ КИШКИ | 2010 |
|
RU2448702C2 |
| Средство, обладающее противоязвенной активностью | 1988 |
|
SU1831338A3 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ЯЗВЫ ЖЕЛУДКА И ДВЕНАДЦАТИПЕРСТНОЙ КИШКИ В ФОРМЕ ТВЕРДЫХ ЖЕЛАТИНОВЫХ КАПСУЛ, СОДЕРЖАЩАЯ РАБЕПРАЗОЛ ИЛИ ЕГО ПРОИЗВОДНЫЕ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2414889C2 |
| СПОСОБ ВЫБОРА ЛЕЧЕНИЯ ЯЗВЕННОЙ БОЛЕЗНИ ДВЕНАДЦАТИПЕРСТНОЙ КИШКИ | 2003 |
|
RU2252709C1 |
| КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ СЕКРЕЦИЮ КИСЛОТЫ В ЖЕЛУДКЕ | 2003 |
|
RU2340358C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ РАБЕПРАЗОЛ НАТРИЯ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2014 |
|
RU2554735C1 |
| 3-[3-(МОРФОЛИН-4-ИЛ)ПРОПИЛ]-2-[(2,2,3-ТРИМЕТИЛЦИКЛОПЕНТ-3-ЕН-1-ИЛ)МЕТИЛ]-1,3-ТИАЗОЛИДИН-4-ОН, ОБЛАДАЮЩИЙ ПРОТИВОЯЗВЕННОЙ И ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2017 |
|
RU2643669C1 |
| АННЕЛИРОВАННЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ ПРОТОНОВОГО НАСОСА ДЛЯ ЛЕЧЕНИЯ ЯЗВЫ | 2003 |
|
RU2336872C2 |
| РЕЖИМ ВВЕДЕНИЯ ИНГИБИТОРОВ Н, К-АТФазы | 1997 |
|
RU2203662C2 |
| СПОСОБ ПРЕДУПРЕЖДЕНИЯ РАЗВИТИЯ ЯЗВ СЛИЗИСТОЙ ОБОЛОЧКИ ЖЕЛУДКА, ВЫЗВАННЫХ НЕСТЕРОИДНЫМ ПРОТИВОВОСПАЛИТЕЛЬНЫМ ПРЕПАРАТОМ ДИКЛОФЕНАКОМ, В ЭКСПЕРИМЕНТЕ | 2024 |
|
RU2840889C1 |
Настоящее изобретение относится к области органической химии, а именно к соединению 9-(хинонил-2)-2-п-этоксифенилэтил-4,5,6,7,8,9,10,11-октагидропиридо[1,2-а]пиразин-1,4-диону формулы I или к его фармацевтически приемлемым комплексным производным. Также изобретение относится к применению соединения формулы I. Технический результат: получено новое производное октагидропиридо[1,2-а]пиразин-1,4-диона, которое может быть применимо в качестве противоязвенного средства. 2 н. и 2 з.п. ф-лы, 1 табл., 6 пр.
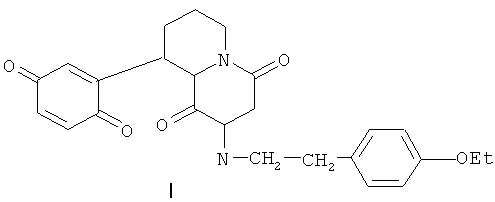
1. Соединение, представляющее собой 9-(хинонил-2)-2-п-этоксифенилэтил-4,5,6,7,8,9,10,11-октагидропиридо[1,2-а]пиразиндион-1,4 формулы (I)
или его фармацевтически приемлемое комплексное производное.
2. Соединение по п.1, обладающее обратимым ингибирующим действием в отношении Н,К-АТФазы.
3. Соединение по п.1, обладающее антисекреторной и гастропротекторной активностью.
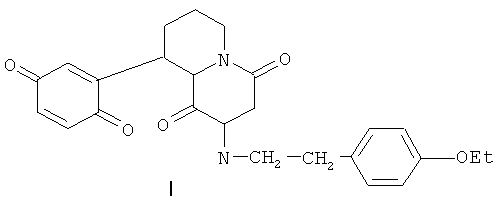
4. Применение соединения, представляющего собой 9-(хинонил-2)-2-п-этоксифенилэтил-4,5,6,7,8,9,10,11-октагидропиридо[1,2-а]пиразиндион-1,4 формулы (I)
или его фармацевтически приемлемого комплексного производного в качестве противоязвенного средства.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Устройство для защиты от помпажа компрессора | 1988 |
|
SU1663238A1 |
| СПОСОБ ПОЛУЧЕПИЯ 5-ОКСО-1Н,2,3,4,5,6,7,8,11-ОКТАГИДРО-ПИРИДО- | 0 |
|
SU241446A1 |

