Изобретение относится к новым производным 1-азабициклононана, к способам их получения, к их применению в качестве фармацевтических средств и к содержащим их фармацевтическим композициям.
Первый вариант осуществления настоящего изобретения более предпочтительно относится к соединению формулы (I)
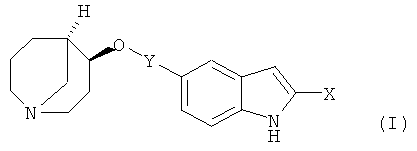
в которой
Х обозначает водород или гидроксигруппу и
Y обозначает одну из следующих групп:
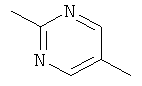
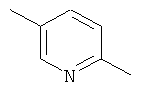
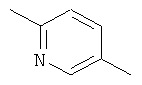
в форме свободного основания или соли присоединения с кислотой.
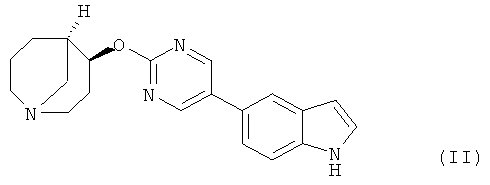
Предпочтительным соединением, предлагаемым в настоящем изобретении, является (4S,5R)-4-[5-(1Н-индол-5-ил)-пиримидин-2-илокси]-1-азабицикло[3.3.1]нонан, обладающий приведенной ниже формулой.
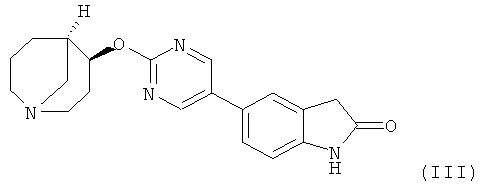
Другим предпочтительным соединением, предлагаемым в настоящем изобретении, является 5-{2-[(4S,5R)-(1-азабицикло[3.3.1]нон-4-ил)окси]-пиримидин-5-ил}-1,3-дигидроиндол-2-он, обладающий приведенной ниже формулой.
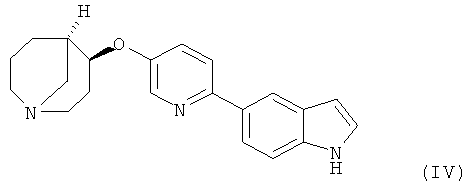
Другим предпочтительным соединением, предлагаемым в настоящем изобретении, является (4S,5R)-4-[6-(1Н-индол-5-ил)-пиридин-3-илокси]-1-азабицикло[3.3.1] нонан, обладающий приведенной ниже формулой.
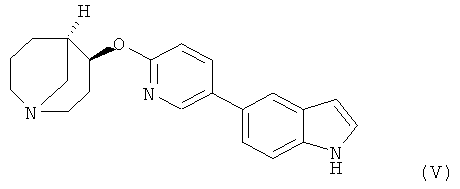
Другим предпочтительным соединением, предлагаемым в настоящем изобретении, является (4S,5R)-4-[5-(lH-индoл-5-ил)-пиpидин-2-илoкcи]-1-азабицикло[3.3.1]нонан, обладающий приведенной ниже формулой.
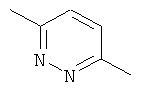
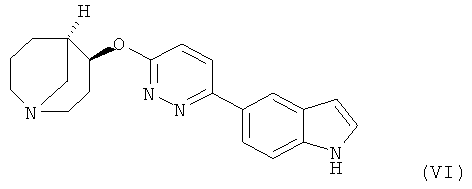
Другим предпочтительным соединением, предлагаемым в настоящем изобретении, является (4S,5R)-4-[6-(1Н-индол-5-ил)-пиридазин-3-илокси]-1-азабицикло[3.3.1]нонан, обладающий приведенной ниже формулой.
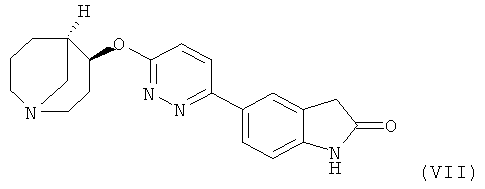
Другим предпочтительным соединением, предлагаемым в настоящем изобретении, является 5-{6-[(4S,5R)-(1-азабицикло[3.3.1]нон-4-ил)окси]-пиридазин-3-ил}-1,3-дигидроиндол-2-он, обладающий приведенной ниже формулой.
Соединения формулы (I) существуют в форме свободного основания или соли присоединения с кислотой. В настоящем описании, если не указано иное, выражение "соединения формулы (I)" следует понимать, как включающее соединения в любой форме, например, в форме свободного основания или соли присоединения с кислотой. Также включены соли, которые являются непригодными для использования в фармацевтике, но которые можно использовать, например, для выделения или очистки свободных соединений формулы (I), такие как пикраты или перхлораты. Для терапевтических целей используют только фармацевтически приемлемые соли или свободные соединения (если это является подходящим, то в форме фармацевтических препаратов) и поэтому они являются предпочтительными.
Соединения формулы (I) могут существовать в форме различных изомеров, например, кето-енольных таутомеров. В настоящем описании, если не указано иное, выражение "соединения формулы (I)" следует понимать, как включающее соединения в любой форме, например, в кето-форме или в енольной форме или в форме любой их смеси.
Если соединения, соли и т.п. указаны во множественном числе, это также означает одно соединение, соль и т.п.
Другой вариант осуществления настоящего изобретения также относится к способам получения соединений формулы (I).
Первый способ включает стадии
i) реакции соединения формулы (IX)
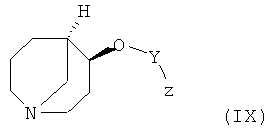
в которой Y является таким, как определено выше, и z обозначает отщепляющуюся группу, такую как Сl, Вr, I, тозилат с соединением формулы (X)
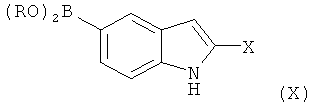
в которой R обозначает Н, C1-C4 алкил или оба RO совместно с атомом В, к которому они присоединены, образуют гетероциклический фрагмент, Х является таким, как определено выше, и
ii) извлечения полученного таким образом соединения формулы (I).
Второй способ включает стадии
i) реакции соединения формулы (XI)
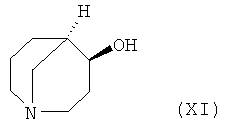
с соединением формулы XII
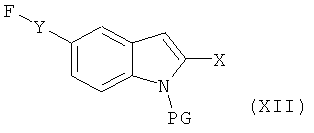
в которой Х и Y являются такими, как определено выше, и PG обозначает подходящую защитную группу,
ii) последующее удаление защитной группы из полученного таким образом соединения и
iii) извлечения полученного таким образом соединения формулы (I) в форме свободного основания или соли присоединения с кислотой.
Этот способ является особенно подходящим, если Y обозначает 3-пиридильный фрагмент.
Исходные вещества являются известными или могут быть получены по известным методикам. Синтез исходных веществ описан, например, в GB 123456, который включен в настоящее изобретение в качестве ссылки.
К описанным выше отдельным стадиям реакций могут относиться следующие положения:
а. Одну или большее количество функциональных групп, например, карбоксигруппу, гидроксигруппу, аминогруппу или меркаптогруппу в исходных веществах может понадобиться защитить. Использующиеся защитные группы могут уже содержаться в предшественниках и защищать соответствующие функциональные группы от нежелательных вторичных реакций, таких как ацилирование, образование простых эфиров, образование сложных эфиров, окисление, сольволиз и аналогичные реакции. Сами защитные группы отличаются тем, что они удаляются легко, т.е. без нежелательных вторичных реакций, обычно путем сольволиза, восстановления, фотолиза или при воздействии ферментов, например, при условиях, аналогичных физиологическим условиям, и тем, что они не содержатся в конечных продуктах. Специалист знает или легко может установить, какие защитные группы являются подходящими для реакций, указанных выше и ниже в настоящем изобретении. Защита таких функциональных групп такими защитными группами, сами защитные группы и реакции их удаления описаны, например, в стандартных справочных пособиях, таких как J.F.W.McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973, in T.W.Greene, "Protective Groups in Organic Synthesis", Wiley, New York 1981, in "The Peptidcs"; Volume 3 (editors: E.Gross and J.Meienhofer), Academic Press, London and New York 1981, in "Mcthoden der organischen Chemie" (Methods of organic chemistry), Houben Weyl, 4th edition, Volume 15/1, Gcorg Thieme Verlag, Stuttgart 1974, in H.-D.Jakubke and H.Jescheit, "Aminosaurcn, Peptide, Proteine" (Amino acids, peptides, proteins), Verlag Chemie, Weinheim, Deerfield Beach, and Basel 1982, and in Jochen Lchmann, "Chemie der Kohlenhydrate: Monosaccharide und Derivate" (Chemistry of carbohydrates: monosaccharides and derivatives), Georg Thieme Verlag, Stuttgart 1974.
b. Соли присоединения с кислотами можно получить из свободных оснований по известным методикам, и наоборот. Альтернативно можно использовать оптически чистые исходные вещества. Соли присоединения с кислотами, подходящие для применения в контексте настоящего изобретения, включают, например, гидрохлориды.
c. Смеси стереоизомеров, например, смеси диастереоизомеров, можно разделить на соответствующие изомеры по методикам, которые сами по себе известны. Например, смеси диастереоизомеров, можно разделить на отдельные диастереоизомеры с помощью фракционной кристаллизации, хроматографии, распределения между растворителями и с помощью аналогичных методик. Это разделение можно выполнить для исходных соединений или для самих соединений формулы I. Энантиомеры можно разделить путем образования солей диастереоизомеров, например, путем образования соли с энантиомерно чистой хиральной кислотой или с помощью хроматографии, например, ВЭЖХ, с использованием хроматографических субстратов с хиральными лигандами. Альтернативно можно использовать оптически чистые исходные вещества.
d. Разбавителями, подходящими для проведения описанных выше процедур, предпочтительно являются инертные органические растворители. Они, в частности, включают алифатические, алициклические и ароматические, необязательно галогенированные углеводороды, такие как, например, бензин, бензол, толуол, ксилол, хлорбензол, дихлорбензол, петролейный эфир, гексан, циклогексан, дихлорметан, хлороформ, тетрахлорид углерода; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, диоксан, тетрагидрофуран и диметиловый эфир этиленгликоля и диэтиловый эфир этиленгликоля; кетоны, такие как, ацетон, бутанон и метилизопропилкетон; нитрилы, такие как, ацетонитрил, пропионитрил и бутиронитрил; амиды, такие как, N,N-диметилформамид, N,N-диметилацетамид, N-метилформамидин, N-метилпирролидон и триамид гексаметилфосфорной кислоты; сложные эфиры, такие как, метилацетат и этилацетат; сульфоксиды, такие как, диметилсульфоксид, спирты, такие как, метанол, этанол н- и изопропанол, монометиловый эфир этиленгликоля, моноэтиловый эфир этиленгликоля, монометиловый эфир диэтиленгликоля, моноэтиловый эфир диэтиленгликоля. Кроме того, можно использовать смеси разбавителей. В зависимости от исходных веществ, условий проведения реакции и вспомогательных веществ могут быть подходящими вода или содержащие воду разбавители. Кроме того, в качестве разбавителя одновременно можно использовать исходное вещество.
e. Температуру проведения реакции можно менять в относительно широком диапазоне. Обычно способы осуществляют при температурах от 0 до 150°C, предпочтительно - от 10 до 120°C. Условия проведения реакций депротонирования можно менять в относительно широком диапазоне. Обычно способы осуществляют при температурах от -150 до +50°C, предпочтительно от -75 до 0°C.
f. Реакции обычно проводят при атмосферном давлении. Однако способы, предлагаемые в настоящем изобретении, также можно осуществлять при повышенном или пониженном давлении - обычно от 0,1 до 10 бар.
g. Исходные вещества обычно используются примерно в эквимолярных количествах. Однако также можно использовать относительно большой избыток одного из компонентов. Реакцию обычно проводят в подходящем разбавителе в присутствии вспомогательного вещества для проведения реакции и реакционную смесь обычно перемешивают при повышенной температуре в течение нескольких часов.
h. Обработку реакционных смесей в соответствии с указанными выше способами и очистку полученных таким образом соединений можно проводить по известным методикам (см. примеры получения).
Соединения, предлагаемые в настоящем изобретении, по данным исследований in vitro и на животных обладают ценными фармакологическими характеристиками и поэтому применимы в качестве фармацевтических средств.
Так, обнаружено, что соединения, предлагаемые в настоящем изобретении, являются холинергическими лигандами nAChR. Кроме того, предпочтительные соединения, предлагаемые в настоящем изобретении, проявляют селективную активность по отношению к α7-nAChR. В частности, может обнаружиться, что соединения, предлагаемые в настоящем изобретении, являются агонистами, частичными агонистами, антагонистами или аллостерическими модуляторами этого рецептора.
Благодаря своим фармакологическим профилям предполагается, что соединения, предлагаемые в настоящем изобретении, применимы для лечения самых различных заболеваний или патологических состояний, включая заболевания, связанные с ЦНС (центральная нервная система), заболевания, связанные с ПНС (периферическая нервная система), заболевания, связанные с воспалением, болью и симптомами отмены, вызванными токсикоманией. Заболевания или нарушения, связанные с ЦНС, включают генерализованные тревожные нарушения, нарушения познавательной способности, недостаточность и нарушение способности к обучению и памяти, болезнь Альцгеймера (БА), продромальную БА, слабое нарушение познавательной способности у пожилых (СНП), амнестическое СНП, нарушение памяти, связанное с возрастом, синдром дефицита внимания с гиперактивностью (СДВГ), болезнь Паркинсона, болезнь Гентингтона, БАС (боковой амиотрофический склероз), прионные нейродегенеративные нарушения, такие как болезнь Крейтцфельда-Якоба и болезнь куру, болезнь Туретта, психоз, депрессию и депрессивные нарушения, манию, маниакальную депрессию, шизофрению, недостаточность познавательной способности при шизофрении, обсессивно-компульсивные нарушения, панические расстройства, нарушения питания, нарколепсию, ноцицепцию, вызванное СПИД слабоумие, старческое слабоумие, слабое нарушение познавательной способности, связанное с возрастом, аутизм, дислексию, позднюю дискинезию, эпилепсию и судорожные синдромы, посттравматические стрессовые нарушения, временную аноксию, ложное слабоумие, предменструальный синдром, синдром поздней фазы лютеинизации, синдром хронической усталости и десинхроноз, развивающийся при полетах на реактивных самолетах. Кроме того, соединения, предлагаемые в настоящем изобретении, можно применять для лечения эндокринных нарушений, таких как тиреотоксикоз, фсохромоцитома, гипертензии и аритмий, а также стенокардии, гиперкинезии, преждевременной эякуляции и затруднения эрекции. В дополнение к этому соединения, предлагаемые в настоящем изобретении, можно применять для лечения воспалительных нарушений (Wang et al., Nature 2003, 421, 384; de Jonge et al., Nature Immunology 2005, 6, 844; Saeed et al., JEM 2005, 7, 1113), нарушений или патологических состояний, включая воспалительные кожные нарушения, ревматоидный артрит, послеоперационную кишечную непроходимость, болезнь Крона, воспалительной болезни кишечника, язвенного колита, сепсиса, фибромиалгии, панкреатита и диареи. Соединения, предлагаемые в настоящем изобретении, также можно применять для лечения симптомов отмены, вызванных прекращением употребления создающих зависимость веществ, таких как героин, кокаин, табак, никотин, опиоиды, бензодиазепины и алкоголь. Кроме того, соединения, предлагаемые в настоящем изобретении, можно применять для лечения боли, например, вызванной мигренью, послеоперационной боли, фантомной боли в ампутированных конечностях или боли, связанной с раком. Боль может представлять собой воспалительную или невропатическую боль, центральную боль, хроническую головную боль, боль, обусловленную диабетической невропатией, посттерапевтической невралгией или поражением периферического нерва.
Кроме того, дегенеративные глазные заболевания, которые можно лечить, включают глазные заболевания, которые прямо или косвенно могут включать дегенерацию клеток сетчатки, включая ишемические ретинопатии в целом, внутреннюю ишемическую невропатию зрительного нерва, все формы неврита зрительного нерва, возрастную дегенерацию желтого пятна (ВДП) в ее сухих формах (сухая ВДП) и мокрых формах (мокрая ВДП), диабетическую ретинопатию, кистевидный отек желтого пятна (КОЖ), отслоение сетчатки, пигментную дегенерацию сетчатки, дегенерацию желтого пятна Штаргардта, желточноформную дегенерацию желтого пятна Беста, врожденный амавроз Лебера и другие типы наследственной дегенерации сетчатки, патологическую миопию, ретролетальную фиброплазию и наследственную невропатию зрительного нерва Лебера.
Обнаружено, что воздействие комбинации, которая включает по меньшей мере один агонист никотинового альфа-7 рецептора и по меньшей мере одно соединение, выбранное из группы, включающей (а) обычные антипсихотические средства и (b) атипичные антипсихотические средства, при лечении психических нарушений больше аддитивного эффекта объединенных лекарственных средств. В частности, комбинации, раскрытые в настоящем изобретении, можно использовать для лечения шизофрении, которая является стойкой по отношению к монотерапии с использованием только одного из компонентов комбинации.
Поэтому настоящее изобретение относится к комбинации, такой как комбинированный препарат или фармацевтическая композиция, которая включает по меньшей мере один агонист никотинового альфа-7 рецептора и по меньшей мере одно соединение, выбранное из группы, включающей (а) обычные антипсихотические средства и (b) атипичные антипсихотические средства, в которой активные ингредиенты содержатся в каждом случае в свободной форме или в форме фармацевтически приемлемой соли, и необязательно по меньшей мере один фармацевтически приемлемый носитель, предназначенный для одновременного, раздельного или последовательного применения.
Термин "психические нарушения" при использовании в настоящем изобретении включает, но не ограничивается только ими, шизофрению, тревожные нарушения, депрессию и биполярные нарушения. Предпочтительно, если психическим нарушением, подвергающимся лечению комбинацией, раскрытой в настоящем изобретении, является шизофрения, более предпочтительно - шизофрения, которая является стойкой по отношению к монотерапии с использованием только одного из компонентов комбинации.
Термин "обычные антипсихотические средства" при использовании в настоящем изобретении включает, но не ограничивается только ими, галоперидол, флуфеназин, тиотиксен и флупентиксол.
Термин "атипичные антипсихотические средства" при использовании в настоящем изобретении включает, но не ограничивается только ими, клозарил, рисперидон, оланзапин, кветиапин, зипрасидон и арипипразол.
В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, применяют в качестве диагностических средств и/или лигандов для ПЭТ, например, для идентификации и локализации никотиновых рецепторов в различных тканях. Соответствующим образом меченные изотопами агенты, предлагаемые в настоящем изобретении, обладают ценными характеристиками при использовании в качестве агентов для мечения образцов для гистопатологических исследований, визуализирующих агентов и/или биомаркеров, далее называемых "маркерами" для селективного мечения nAChR. Агенты, предлагаемые в настоящем изобретении, более предпочтительно применять в качестве маркеров для альфа-7 nAChR рецепторов in vitro или in vivo. В частности, соединения, предлагаемые в настоящем изобретении, соответствующим образом меченные изотопами, применимы в качестве маркеров для ПЭТ. Такие маркеры для ПЭТ помечены одним или большим количеством атомов, выбранных из группы, включающей 11С, 13N, 15О, 18F.
Поэтому агенты, предлагаемые в настоящем изобретении, применимы, например, для определения степени занятости рецептора лекарственным средством, действующим на nAChR, или в диагностических целях при исследовании заболеваний, обусловленных дисбалансом или нарушением функции nAChR, и для мониторинга эффективности медикаментозного лечения таких заболеваний.
В соответствии с указанным выше настоящее изобретение относится к агенту, предлагаемому в настоящем изобретении, предназначенному для применения в качестве маркера для визуализации при нейроисследованиях.
В другом варианте осуществления настоящее изобретение относится к композиции для мечения структур головного мозга и периферической нервной системы, содержащих nAChR, in vivo и in vitro, включающей агент, предлагаемый в настоящем изобретении.
В еще одном варианте осуществления настоящее изобретение относится к способу мечения структур головного мозга и периферической нервной системы, содержащих nAChR, in vivo и in vitro, который включает взаимодействие ткани головного мозга с агентом, предлагаемым в настоящем изобретении.
Способ, предлагаемый в настоящем изобретении, может включать дополнительную стадию, предназначенную для установления того, осуществилось ли мечение целевой структуры агентом, предлагаемым в настоящем изобретении. Эту дополнительную стадию можно провести путем изучения целевой структуры с помощью позитронной эмиссионной томографии (ПЭТ) или однофотонной эмиссионной компьютерной томографии (ОФЭКТ), или любого другого устройства, позволяющего регистрировать радиоактивное излучение.
В частности, агенты, предлагаемые в настоящем изобретении, являются агонистами никотинового ацетилхолинового рецептора α7 (nAChR α7).
При функциональных анализах агенты, предлагаемые в настоящем изобретении, обнаруживают высокое сродство по отношению к α7 nAChR, о чем свидетельствуют указанные ниже исследования:
а. Функциональный анализ сродства к α7 nAChR проводят с использованием линии клеток гипофиза крыс, стабильно экспрессирующей α7 nAChR. Вкратце, методика состоит в следующем: Клетки GH3, рекомбинантно экспрессирующие nAChR α7, за 72 ч до эксперимента высевают в черные 96-луночные планшеты и инкубируют при 37°C в увлажненной атмосфере (5% CO2/95% воздуха). В день проведения эксперимента среду удаляют путем встряхивания планшетов и заменяют на 100 мкл среды для выращивания, содержащей чувствительный к кальцию флуоресцентный краситель, в присутствии 2,5 мМ пробеницида (Sigma). Клетки инкубируют при 37°C в увлажненной атмосфере (5% СO2/95% воздуха) в течение 1 ч. Планшеты встряхивают для удаления избытка Fluo-4, дважды промывают забуференным с помощью Hepes (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота) солевым раствором (в мМ: NaCl - 130, КСl - 5,4, CaCl2 - 2, MgSO4 - 0,8, NaH2PO4 - 0,9, глюкоза - 25, Hepes - 20, pH - 7,4; СРХ (сбалансированный солевой раствор Хенкса)) и в них помещают 100 мкл СРХ, при необходимости содержащего антагонист. Инкубирование в присутствии антагониста проводят в течение 3-5 мин. Затем планшеты помещают в визуализирующее устройство считывания планшетов и регистрируют сигнал флуоресценции. В этом анализе соединения, предлагаемые в настоящем изобретении, обладают значениями рЕС50, равными от примерно 5 до примерно 9. В этом исследовании предпочтительными являются частичные и активные агонисты.
b. Для оценки антагонистической активности соединений, предлагаемых в настоящем изобретении, по отношению к нейрональному nAChR α4β2 человека проводят аналогичный функциональный анализ с использованием линии эпителиальных клеток человека, стабильно экспрессирующей подтип α4β2 человека (Michelmore et al., Naunyn-Schmiedeberg's Arch. Pharmacol. (2002) 366, 235). При этом анализе предпочтительные соединения, предлагаемые в настоящем изобретении, обнаруживают селективность по отношению к подтипу α7 nAChR.
c. Для оценки антагонистической активности соединений, предлагаемых в настоящем изобретении, по отношению к "ганглиозному подтипу" (α3β4), никотиновому рецептору мышечного типа (α1β1γδ) и рецептору 5-НТ3, проводят функциональные анализы, аналогичные описанным выше в разделе а), с использованием линии эпителиальных клеток человека, стабильно экспрессирующей ганглиозный подтип человека, линии клеток, эндогенно экспрессирующей никотиновые рецепторы мышечного типа, или линии клеток, эндогенно экспрессирующей мышиный рецептор 5-НТ3 (Michelmore et al. Naunyn-Schmiedeberg's Arch. Pharmacol. (2002) 366, 235. Соединения, которые обнаруживают небольшую активность или не обнаруживают активности по отношению к α3β4 nAChR, к никотиновому рецептору мышечного подтипа, а также к рецептору 5-НТ3, являются особенно предпочтительными.
В экспериментах на мышах, характеризующихся недостаточной способностью нервной системы модулировать свою чувствительность по отношению к поступающим сигналам (мыши DBA/2), описанных в публикации S.Leonard et al., in Schizophrenia Bulletin 22, 431-445 (1996), соединения, предлагаемые в настоящем изобретении, значительно стимулируют эту способность при концентрациях, равных от примерно 10 до примерно 40 мкМ.
Можно показать, что соединения, предлагаемые в настоящем изобретении, улучшают внимание при исследовании внимания на грызунах (Robbins, J. Neuropsychiatry Clin. Neurosci. (2001) 13, 326-35), а именно в 5-позиционном исследовании времени реакции (5-CSRTT). При этом исследовании крысы должны смотреть на стенку, содержащую 5 отверстий. Если на одно из них направляется световой импульс, то крыса в течение 5 секунд должна коснуться носом этого отверстия и тогда в качестве награды она получает кусочек пищи, поступающий из кормушки, находящейся в противоположной стенке.
Соединения, предлагаемые в настоящем изобретении, также могут улучшать способность к обучению/память при исследовании группового поведения мышей и крыс (Ennaceur and Delacour, Behav. Brain Res. (1988) 31, 47-59).
Поэтому соединения, предлагаемые в настоящем изобретении, применимы для предупреждения и лечения (включая ослабление и предупреждение) различных нарушений, предпочтительно - указанных выше. Применимость агонистов α7 nAChR в случае нейродегенерации описана в литературе, например, в публикации Wang et al., J. Biol. Chem. 275, 5626-5632 (2000).
Разумеется, при лечении указанных выше и других нарушений подходящая доза соединения (активного ингредиента), предлагаемого в настоящем изобретении, будет меняться в зависимости, например, от субъекта, пути введения и характера и тяжести подвергающегося лечению патологического состояния, а также от относительной активности конкретного использующегося средства, предлагаемого в настоящем изобретении. Например, необходимое количество активного средства можно установить с помощью известных методик исследования in vitro и in vivo путем определения того, как долго концентрация активного средства в плазме крови сохраняется при значении, приемлемом для обеспечения лечебного эффекта. Обычно указывают, что для животных удовлетворительные результаты достигаются при суточных дозах, равных от примерно 0,01 до примерно 30,0 мг/кг при пероральном введении. Для людей назначаемая суточная доза находится в диапазоне от примерно 0,7 до примерно 1400 мг/сутки при пероральном введении, например, от примерно 50 до 200 мг (для человека массой 70 кг), которую обычно вводят однократно или в виде разделенных доз до 4 раз в сутки, или в форме с замедленным высвобождением средства. Дозированные формы для перорального введения предпочтительно включают от примерно 1,75 или 2,0 до примерно 700 или 1400 мг соединения, предлагаемого в настоящем изобретении, в смеси с соответствующим фармацевтически приемлемым разбавителем или носителем.
Фармацевтические композиции содержат, например, от примерно 0,1 до примерно 99,9%, предпочтительно - от примерно 20 до примерно 60% активного ингредиента (ингредиентов).
Примеры композиций, содержащих соединение, предлагаемое в настоящем изобретении, включают, например, твердую дисперсию, водный раствор, например, содержащий солюбилизирующий агент, микроэмульсию и суспензию, например, соли соединения формулы 1 или свободного основания соединения формулы I, в диапазоне от 0,1 до 1%, например, 0,5%. С помощью подходящей буферной добавки значение pH композиции может быть установлено в диапазоне, например, от 3,5 до 9,5, например, 4,5.
Соединения, предлагаемые в настоящем изобретении, также используются в качестве химикатов для научных исследований.
В случае применения в контексте настоящего изобретения соединение формулы I и/или его фармацевтически приемлемую соль можно вводить в виде одного активного средства или в комбинации с одним или большим количеством других активных средств формулы I и/или их фармацевтически приемлемых солей или, предпочтительно, других активных средств, обычно специально применяющихся для лечения нарушений, указанных в настоящем изобретении, или дополнительных других нарушений, любым обычным образом, например, перорально, например, в виде таблеток, капсул или назальных аэрозолей, или парентерально, например, в виде растворов или суспензий для инъекций. Такие другие активные средства, применяющиеся в таких комбинациях, предпочтительно выбраны из группы, включающей бензодиазепины, селективные ингибиторы повторного поглощения серотонина (СИПС), селективные ингибиторы повторного поглощения серотонина и норэпинефрина (СИПН), обычные антипсихотические средства, атипичные антипсихотические средства, буспирон, карбамазепин, окскарбазепин, габапентин и прегабалин.
СИПС, пригодный для применения в настоящем изобретении, предпочтительно выбран из группы, включающей флуоксетин, флувоксамин, сертралин, пароксетин, циталопрам и эсциталопрам. СИПН, пригодный для применения в настоящем изобретении, предпочтительно выбран из группы, включающей венлафаксин и дулоксетин. Термин "бензодиазепины" при использовании в настоящем изобретении включает, но не ограничивается только ими, клоназепам, диазепам и лоразепам. Термин "обычные антипсихотические средства" при использовании в настоящем изобретении включает, но не ограничивается только ими, галоперидол, флуфеназин, тиотиксен и флупентиксол. Термин "атипичные антипсихотические средства" при использовании в настоящем изобретении означает клозарил, рисперидон, оланзапин, кветиапин, зипразидон или арипипразол.
Буспирон можно вводить в виде свободного основания или соли, например, в виде его гидрохлорида, например, в той форме, в которой он имеется в продаже, например, под торговым названием буспар™ или беспар™. Его можно получить и вводить, например, как это описано в US 3717634. Флуоксетин можно вводить, например, в виде его гидрохлорида, в котором он имеется в продаже, например, под торговым названием прозак™. Его можно получить и вводить, например, как это описано в СА 2002182. Паркосетин ((3S,4R)-3-[(1,3-бензодиоксол-5-илокси)метил]-4-(4-фторфенил)пиперидин) можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием паксил™. Его можно получить и вводить, например, как это описано в US 3912743. Сертралин можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием золофт™. Его можно получить и вводить, например, как это описано в US 4536518. Клоназепам можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием антелепсин™. Диазепам можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием диазепам десинит™. Лоразепам можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием тавор™. Циталопрам можно вводить в виде свободного основания или соли, например, в виде его гидробромида, например, в той форме, в которой он имеется в продаже, например, под торговым названием ципрамил™. Эсциталопрам можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием ципралекс™. Его можно получить и вводить, например, как это описано в AU 623144. Венлафаксин можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием тревилор™. Дулоксетин можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием цимбалта™. Его можно получить и вводить, например, как это описано в СА 1302421. Карбамазепин можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием тегретал™ или тегретол™. Окскарбазепин можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием трилептал™. Окскарбазепин хорошо известен из литературы [см. например, Schuetz H. et al., Xenobiotica (GB), 16(8), 769-778 (1986)]. Габапентин можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием нейронтин™. Галоперидол можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием галоперидол СТАДА™. Флуфеназин можно вводить, например, в виде его дигидрохлорида, в котором он имеется в продаже, например, под торговым названием проликсин™. Тиотиксен можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием наван™. Его можно получить, например, как это описано в US 3310553. Флупентиксол можно вводить, например, в виде его дигидрохлорида, например, в той форме, в которой он имеется в продаже, например, под торговым названием эмергил™ или в виде его деканоата, например, в той форме, в которой он имеется в продаже, например, под торговым названием депиксол™. Его можно получить, например, как это описано в ВР 925538. Клозарил можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием лепонекс™. Его можно получить, например, как это описано в US 3539573. Рисперидон можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием риспердал™. Оланзапин можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием зипрекса™. Кветиапин можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием сероквел™. Зипразидон можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием геодон™. Его можно получить, например, как это описано в GB 281309. Арипипразол можно вводить, например, в той форме, в которой он имеется в продаже, например, под торговым названием абилифи™. Его можно получить, например, как это описано в US 5006528.
Структура активных ингредиентов, обозначаемых кодовыми номерами, родовыми или торговыми названиями, приведена в последнем издании стандартного справочника "The Merck Index" или в базах данных, например, Patents International (например, IMS World Publications). Соответствующее их содержание включено в настоящую заявку в качестве ссылки. Любой специалист в данной области техники вполне может идентифицировать активные агенты и на основании этих ссылок также может изготовить и исследовать фармацевтические показания и характеристики с помощью стандартных моделей, как in vitro, так и in vivo.
В случае комбинации фармацевтические композиции, предназначенные для раздельного введения компонентов комбинации и/или предназначенные для введения компонентов в фиксированной комбинации, т.е. одной галеновой композиции, включающей по меньшей мере 2 компонента комбинации, предлагаемые в настоящем изобретении, можно получить по методике, которая сама по себе известна, и они пригодны для энтерального, такого как пероральное, или ректального и парентерального введения млекопитающим, включая человека, и содержат терапевтически эффективное количество по меньшей мере одного фармакологически активного компонента комбинации по отдельности или в комбинации с одним или большим количеством фармацевтически приемлемых носителей, особенно подходящих для энтерального или парентерального введения. Если применяющиеся компоненты комбинации вводят в том виде, в котором они имеются в продаже, в виде отдельных лекарственных средств, то, если в настоящем изобретении не указано иное, для обеспечения описанного в настоящем изобретении благоприятного эффекта их дозировка и путь введения могут быть такими, как это указано в информации, приведенной в листке-вкладыше соответствующего имеющегося в продаже лекарственного средства.
Фармацевтическими препаратами для комбинированной терапии при энтеральном или парентеральном введении являются, например, препараты в виде разовых дозированных форм, таких как таблетки, покрытые сахаром, таблетки, капсулы или суппозитории, а также ампулы. Если не указано иное, то их готовят по методикам, которые сами по себе известны, например, с помощью обычных методик смешивания, гранулирования, нанесения покрытия из сахара, растворения или лиофилизации. Следует понимать, что разовое количество компонента комбинации, содержащееся в отдельной дозе каждой дозированной формы, само по себе не должно являться эффективным количеством, поскольку необходимое эффективное количество может быть обеспечено путем введения не только одной дозированной формы, но и двух или большего количества дозированных форм.
В частности, терапевтически эффективное количество каждого компонента комбинации можно вводить одновременно или последовательно и в любом порядке и компоненты можно вводить по отдельности (например, последовательно через постоянные или переменные промежутки времени) или в виде фиксированной комбинации. Например, способ лечения (включая ослабление) нарушения в контексте настоящего изобретения может включать (i) введение компонента комбинации (а) (соединения, предлагаемого в настоящем изобретении) в форме свободного основания или фармацевтически приемлемой соли и (ii) введение компонента комбинации (b) (например, другого соединения, предлагаемого в настоящем изобретении, или активного ингредиента, описываемого другой формулой) в форме свободного основания или фармацевтически приемлемой соли одновременно или последовательно в любом порядке, в количествах, которые совместно являются терапевтически эффективными, предпочтительно - в синергетически эффективных количествах, например, в виде суточных доз, содержащих количества, описанные в настоящем изобретении. Отдельные компоненты комбинации можно вводить по отдельности в разные моменты времени в течение курса лечения или одновременно в виде разделенных или разовых комбинированных форм. Кроме того, термин "введение" также включает применение пролекарства компонента комбинации, которое in vivo превращается в сам компонент комбинации. Поэтому следует понимать, что настоящее изобретение включает все такие режимы одновременного и/или поочередного лечения и термин "введение" следует интерпретировать соответствующим образом.
Эффективная использующаяся доза компонентов комбинации может меняться, например, в зависимости от конкретного применяющегося соединения или фармацевтической композиции, пути введения, подвергающегося лечению нарушения и/или тяжести подвергающегося лечению нарушения. Таким образом дозировочный режим выбирается в зависимости от множества факторов, включая путь введения, метаболизм соединения в почках и печени пациента. Врач, клиницист или ветеринар с общей подготовкой в данной области техники может легко определить и назначить эффективное количество одного из активных ингредиентов, необходимое для предупреждения, ослабления, противодействия или подавления нарушения. Оптимальная точность при установлении концентраций активных ингредиентов в диапазоне, в котором обеспечивается эффективность без проявления токсичности, требует выбора режима на основании кинетики поступления активного ингредиента в необходимые области организма.
В соответствии с указанным выше настоящее изобретение также относится к:
(1) Соединению формулы 1 и/или его соли, предназначенной для применения в целях диагностики или терапевтического лечения млекопитающего, предпочтительно - человека; предпочтительно - для применения в качестве агониста альфа-7 рецептора, например, для применения в целях лечения (включая ослабление) любого одного или большего количества нарушений, предпочтительно - любого одного или большего количества конкретных нарушений, указанных выше и ниже в настоящем изобретении.
(2) Фармацевтической композиции, включающей в качестве активного ингредиента соединение формулы 1 и/или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым разбавителем или носителем.
(2') Фармацевтической композиции, предназначенной для лечения или предупреждения нарушения, при лечении которого играет роль активация альфа-7 рецептора или он участвует и/или в котором проявляется активность альфа-7 рецептора, предпочтительно - любого одного или большего количества нарушений, указанных выше и ниже в настоящем изобретении, включающей соединение формулы 1 и/или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель.
(3) Способу лечения нарушения, предпочтительно - любого одного или большего количества конкретных нарушений, указанных выше в настоящем изобретении, у субъекта, нуждающегося в таком лечении, включающему введение фармацевтически эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли.
(3') Способу лечения или предупреждения нарушения, при лечении которого играет роль активация альфа-7 рецептора или он участвует и/или в котором проявляется активность альфа-7 рецептора, включающему введение нуждающемуся в нем млекопитающему терапевтически эффективного количества соединения формулы 1 и/или его фармацевтически приемлемой соли.
(4) Применению соединения формулы 1 и/или его фармацевтически приемлемой соли, для приготовления лекарственного средства, предназначенного для лечения или предупреждения заболевания или патологического состояния, при лечении которого играет роль активация альфа-7 рецептора или он участвует и/или в котором проявляется активность альфа-7 рецептора, предпочтительно - одного или большего количества нарушений, указанных выше.
(5) Способу, описанному выше, включающему совместное введение, например, одновременное или последовательное, терапевтически эффективного количества альфа-7 агониста формулы 1 и/или его фармацевтически приемлемой соли и второго фармацевтически активного соединения и/или его фармацевтически приемлемой соли, указанное второе фармацевтически активное соединение и/или его соль является особенно подходящим для применения при лечении любого одного или большего количества нарушений, указанных выше и ниже в настоящем изобретении.
(6) Комбинации, включающей терапевтически эффективное количество альфа-7 агониста формулы 1 и/или его фармацевтически приемлемой соли и второго фармацевтически активного соединения и/или его фармацевтически приемлемой соли, указанное второе фармацевтически активное соединение является особенно подходящим для применения при лечении любого одного или большего количества конкретных нарушений, указанных выше в настоящем изобретении.
Приведенные ниже примеры предназначены для иллюстрации настоящего изобретения без наложения ограничений на его объем. Используются следующие аббревиатуры:
Температуру измеряют в градусах Цельсия. Если не указано иное, то реакции проводят при комнатной температуре. Структуру конечных продуктов, промежуточных продуктов и исходных веществ подтверждают с помощью стандартных методик анализа, например, микроанализа, и спектроскопических характеристик (например, МС (масс-спектрометрия), ИК (инфракрасная спектроскопия), ЯМР (ядерный магнитный резонанс)).
Пример 1 (4S,5R)-4-[5-(1Н-Индол-5-ил)-пиримидин-2-илокси]-1-азабицикло[3.3.1]нонан
1-Азабицикло[3.3.1]нонан-4-он (11,92 г, 85,6 ммоля) растворяют в 160 мл МeОН и охлаждают до -10°C. Порциями прибавляют NaBH4 (1,69 г, 42,9 ммоля), так чтобы внутренняя температура не превышала 0°C. Реакционную смесь перемешивают при -10°C в течение 1 ч. Прибавляют воду и растворители выпаривают. Оставшееся твердое вещество растворяют в смеси МТБЭ/МеОН, фильтруют через Hyflo и фильтрат выпаривают и получают 19,28 г неочищенного продукта, который очищают с помощью хроматографии на оксиде алюминия (400 г, элюент: МТБЭ/МеОН от 95:5 до 80:20) и получают 10,96 г (91%) (4SR,5RS)-1-азабицикло[3.3.1]нонан-4-ола.
К уксусному ангидриду (150 мл) при охлаждении порциями прибавляют (4SR,5RS)-(1-азабицикло[3.3.1]нонан-4-ол (21.34 г, 151 ммоля). Реакционную смесь нагревают при 120°С в течение 2,5 ч, выпаривают и экстрагируют смесью ТГФ/насыщенный раствор K2CO3. Водный слой повторно экстрагируют с помощью ТГФ. Объединенные органические слои промывают рассолом, сушат над Na2SO4, фильтруют и выпаривают. Неочищенный продукт очищают с помощью перегонки из колбы в колбу (ВВ, 90°С) и получают 24,07 г (87%) 1-азабицикло[3.3.1]нон-4-илового эфира (4SR,5RS)-уксусной кислоты.
1-Азабицикло[3.3.1]нон-4-иловый эфир уксусной кислоты (120,0 г, 655 ммоля) растворяют в 200 мл абсолютного EtOH и 50 мл воды. Прибавляют L(+)-винную кислоту (98,3 г, 655 ммоля) и смесь кипятят с обратным холодильником в течение 1 мин. Смесь охлаждают до КТ, затем до 4°C. Осадок отфильтровывают, промывают с помощью EtOH и трижды перекристаллизовывают из смеси EtOH/H2O 4:1 и получают 41,16 г тартрата ([α]D KT=-14,72° (c=0,265, MeOH)), который после обработки насыщенным раствором Nа2СО3 дает 22,6 г (123 ммоля, 19%) 1-азабицикло[3.3.1]нон-4-илового эфира (4S,5R)-уксусной кислоты в виде свободного основания. Еще 10,7 г (32.1 ммоля, 5%) тартрата можно получить из маточных растворов с помощью еще 3 перекристаллизации из смеси EtOH/H2O 4:1.
1-Азабицикло[3.3.1]нон-4-иловый эфир уксусной кислоты (5,45 г, 29,7 ммоля) растворяют в 10% водном растворе NaOH и перемешивают в течение 1 ч при 50°C. После охлаждения до КТ смесь экстрагируют смесью ТГФ/рассол. Органический слой сушат над Na2SO4, фильтруют и фильтрат выпаривают и получают 3,83 г (91%) (4S,5R)-1-азабицикло[3.3.1]нонан-4-ола, который растворяют в 50 мл ТГФ и охлаждают до 0°C. По каплям прибавляют NHMDS (35 мл 1 М раствора в ТГФ), реакционную смесь перемешивают при КТ в течение 0,5 ч и затем прибавляют к предварительно охлажденному (-15°C) раствору 5-бром-2-хлорпиримидина (5,89 г, 30,5 ммоля) в ТГФ. Смесь перемешивают в течение 15 ч при КТ, затем разбавляют с помощью ТГФ и экстрагируют 1М водным раствором NaOH и рассолом. Водные слои дважды повторно экстрагируют с помощью ТГФ, объединенные органические слои сушат над Na2SO4, фильтруют и фильтрат выпаривают. Полученный неочищенный продукт (10,15 г) перекристаллизовывают из смеси CH3CN/MeOH и получают 5,21 г (65%) (4S,5R)-4-(5-бромпиримидин-2-илокси)-1-азабицикло[3.3.1]нонана.
5,19 г (17,4 ммоля) (4S,5R)-4-(5-Бромпиримидин-2-илокси)-1-азабицикло[3.3.1]нонана растворяют в 200 мл смеси толуол/ЕtOН 9:1. Прибавляют 5-индолилбороновую кислоту (3,47 г, 21,6 ммоля), Рd(РРh3)4 (1,04 г, 0,873 ммоля) и раствор Nа2СО3 (7,38 г, 69,6 ммоля) в 35 мл H2O. Реакционную смесь перемешивают при 90°С в течение 15 ч. После охлаждения до КТ смесь фильтруют через Hyflo, фильтрат экстрагируют водой и рассолом. Водные слои повторно экстрагируют с помощью AcOEt и объединенные органические слои сушат над Na2SO4, фильтруют и фильтрат выпаривают. Полученный неочищенный продукт очищают с помощью ФХ (255 г силикагеля, элюент - AcOEt/MeOH/Et3N 70:27:3) и перекристаллизовывают из EtOH и получают 3,87 г (67%) (4S,5R)-4-[5-(1Н-индол-5-ил)-пиримидин-2-илокси]-1-азабицикло[3.3.1]нонана. МС (ЭР+ (ионизация электрораспылением)): m/e=335 (МН+), т.пл. 195-199°C.
Получение предшественника, 1-азабицикло[3.3.1]нонан-4-она, проводят в соответствии с публикацией М.G.Kim et al., J. Med. Chem. (2003) 46, 2216.
Пример 2 (Изготовление мягких капсул)
5000 Капсул из мягкого желатина, каждая из которых в качестве активного ингредиента содержит 0,05 г одного из соединений формулы (I), указанных в предыдущих примерах, готовят следующим образом:
250 г Измельченного в порошок активного ингредиента суспендируют в 2 л Lauroglykol® (лаурат пропиленгликоля, Gattefossé S.A., Saint Priest, France) и размалывают в устройстве для мокрого размола с получением частиц размером примерно от 1 до 3 мкм. Затем с помощью машины для наполнения капсул порции смеси по 0,419 г помещают в капсулы из мягкого желатина.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-АЗАБИЦИКЛО[3.3.1]НОНАНОВ | 2005 |
|
RU2445313C2 |
| (1-АЗАБИЦИКЛО[3,3,1]НОН-4-ИЛ)-[5-(1Н-ИНДОЛ-5-ИЛ)-ГЕТЕРОАРИЛ]-АМИНЫ, КАК ХОЛИНЕРГИЧЕСКИЕ ЛИГАНДЫ nAChR, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ И НЕЙРОДЕГЕНЕРАТИВНЫХ НАРУШЕНИЙ | 2006 |
|
RU2440350C2 |
| 3-(ГЕТЕРОАРИЛОКСИ)-2-АЛКИЛ-1-АЗАБИЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ, КАК ЛИГАНДЫ АЛЬФА-7-nAChR (НИКОТИНОВЫХ АЦЕТИЛХОЛИНОВЫХ РЕЦЕПТОРОВ), ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 2005 |
|
RU2402551C2 |
| АЗАБИЦИКЛИЧЕСКИЕ АЛКАНОВЫЕ ПРОИЗВОДНЫЕ, ЗАМЕЩЕННЫЕ КОНДЕНСИРОВАННЫМ БИЦИКЛОГЕТЕРОЦИКЛОМ | 2007 |
|
RU2437884C2 |
| СЕЛЕКТИВНЫЕ К ПОДТИПУ РЕЦЕПТОРА АЗАБИЦИКЛОАЛКАНОВЫЕ ПРОИЗВОДНЫЕ | 2008 |
|
RU2417984C1 |
| МОНОГИДРАТ ПРОИЗВОДНОГО АЗААДАМАНТАНА | 2011 |
|
RU2579121C2 |
| АЗАБИЦИКЛОАЛКИЛЬНЫЕ ЭФИРЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АГОНИСТОВ АЛЬФА7-nAChR | 2003 |
|
RU2352569C2 |
| АМИДЫ ДИАЗАБИЦИКЛОАЛКАНОВ, СЕЛЕКТИВНЫЕ В ОТНОШЕНИИ АЦЕТИЛХОЛИНОВОГО ПОДТИПА НИКОТИНОВЫХ РЕЦЕПТОРОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 2007 |
|
RU2448969C2 |
| АМИДЫ ДИАЗАБИЦИКЛОАЛКАНОВ, СЕЛЕКТИВНЫЕ В ОТНОШЕНИИ АЦЕТИЛХОЛИНОВОГО ПОДТИПА НИКОТИНОВЫХ РЕЦЕПТОРОВ | 2007 |
|
RU2517693C2 |
| АЗААДАМАНТАНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2450002C2 |
Изобретение относится к соединениям в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой, выбранных из группы, включающей:
(4S,5R)-4-[5-(1Н-Индол-5-ил)-пиримидин-2-илокси]-1-азабицикло[3.3.1]нонан,
5-{2-[(4S,5R)-(1-Азабицикло[3.3.1]нон-4-ил)окси]-пиримидин-5-ил}-1,3-дигидроиндол-2-он,
(4S,5R)-4-[6-(1Н-Индол-5-ил)-пиридин-3-илокси]-1-азабицикло[3.3.1]нонан,
(4S,5R)-4-[5-(1Н-Индол-5-ил)-пиридин-2-илокси]-1-азабицикло[3.3.1]нонан,
(4S,5R)-4-[6-(1Н-Индол-5-ил)-пиридазин-3-илокси]-1-азабицикло[3.3.1]нонан и
5-{6-[(4S,5R)-(1-Азабицикло[3.3.1]нон-4-ил)окси]-пиридазин-3-ил}-1,3-дигидроиндол-2-он, которые обладают агонистической активностью в отношении nAChR α7, что позволяет использовать их в фармацевтических композициях и для приготовления лекарственного средства, предназначенного для предупреждения и лечения нарушения памяти. 4 н. и 7 з.п. ф-лы, 2 пр.
1. Соединение в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой, причем указанное соединение выбрано из группы, включающей:
(4S,5R)-4-[5-(1Н-индол-5-ил)-пиримидин-2-илокси]-1-азабицикло[3.3.1]нонан,
5-{2-[(4S,5R)-(1-азабицикло[3.3.1]нон-4-ил)окси]-пиримидин-5-ил}-1,3-дигидроиндол-2-он,
(4S,5R)-4-[6-(1Н-индол-5-ил)-пиридин-3-илокси]-1-азабицикло[3.3.1]нонан,
(4S,5R)-4-[5-(1Н-индол-5-ил)-пиридин-2-илокси]-1-азабицикло[3.3.1]нонан,
(4S,5R)-4-[6-(1Н-индол-5-ил)-пиридазин-3-илокси]-1-азабицикло[3.3.1]нонан и
5-{6-[(4S,5R)-(1-азабицикло[3.3.1]нон-4-ил)окси]-пиридазин-3-ил}-1,3-дигидроиндол-2-он.
2. Соединение в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой по п.1, представляющее собой (4S,5R)-4-[5-(1Н-индол-5-ил)-пиримидин-2-илокси]-1-азабицикло[3.3.1]нонан.
3. Соединение в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой по п.1, представляющее собой 5-{2-[(4S,5R)-(1-азабицикло[3.3.1]нон-4-ил)окси]-пиримидин-5-ил}-1,3-дигидроиндол-2-он.
4. Соединение в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой по п.1, представляющее собой (4S,5R)-4-[6-(1Н-индол-5-ил)-пиридин-3-илокси]-1-азабицикло[3.3.1]нонан.
5. Соединение в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой по п.1, представляющее собой (4S,5R)-4-[5-(1Н-индол-5-ил)-пиридин-2-илокси]-1-азабицикло[3.3.1]нонан.
6. Соединение в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой по п.1, представляющее собой (4S,5R)-4-[6-(1Н-индол-5-ил)-пиридазин-3-илокси]-1-азабицикло[3.3.1]нонан.
7. Соединение в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой по п.1, представляющее собой 5-{6-[(4S,5R)-(1-азабицикло[3.3.1]нон-4-ил)окси]-пиридазин-3-ил}-1,3-дигидроиндол-2-он.
8. Соединение по одному из пп.1-7 в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой, предназначенное для применения в качестве фармацевтического средства, обладающего агонистической активностью в отношении nAChR α7.
9. Фармацевтическая композиция, обладающая агонистической активностью в отношении nAChR α7, включающая соединение по одному из пп.1-7 в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой совместно с фармацевтическим носителем или разбавителем.
10. Применение соединения по одному из пп.1-7 в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой в качестве фармацевтического средства, предназначенного для предупреждения и лечения нарушения памяти.
11. Применение соединения по одному из пп.1-7 в форме свободного основания или фармацевтически приемлемой соли присоединения с кислотой для приготовления лекарственного средства, предназначенного для предупреждения, лечения и/или задержки прогрессирования нарушения памяти.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ получения производных 3-азабицикло 3,3,1 нонана,или их изомеров,или их фармацевтически приемлемых солей присоединения кислот (его варианты) | 1984 |
|
SU1395141A3 |

