Изобретение относится к способу комбинированного биотехнологического и химического получения метионина. Изобретение относится прежде всего к ферментативному получению L-гомосерина с последующим его химическим превращением в L-метионин в одну или несколько стадий.
В настоящее время аминокислоту метионин повсеместно производят в больших объемах в промышленном масштабе, что свидетельствует о его важном коммерческом значении.
Метионин находит применение во многих областях, например используется в фармацевтических продуктах, а также продуктах, предназначенных для укрепления здоровья и поддержания хорошего физического состояния. Однако в первую очередь метионин используется в качестве добавки ко многим кормам для различных полезных животных и в таковом качестве может применяться и в рацемической форме, и в виде его чистых энантиомеров.
В промышленном масштабе метионин получают химическим путем по реакции Бухерера-Берга, представляющей собой разновидность реакции Штреккера. При этом исходные метилмеркаптопропионовый альдегид (получаемый из акролеина и метилмеркаптана), синильную кислоту, аммиак и диоксид углерода превращают в 5-(2-метилмеркаптоэтил)гидантоин (метионингидантоин), который затем путем щелочного гидролиза превращают в метионинат щелочного металла, из которого (метионината) в свою очередь затем путем нейтрализации кислотой, например серной кислотой или угольной кислотой, высвобождают метионин. Метионин можно получать и различными иными способами, например реакцией амидокарбонилирования, гидролизом протеинов или ферментацией.
С начала промышленного, крупномасштабного производства метионина существовала необходимость в разработке экономически эффективного и вместе с тем экологически безвредного процесса его получения.
Недостаток реакции Штреккера, равно как и реакции Бухерера-Берга состоит в необходимости применения токсичных соединений-предшественников, а именно: синильной кислоты и акролеина в качестве структурных звеньев с C1, соответственно С3. Синильную кислоту получают из метана и аммиака при высоких температурах. Акролеин получают частичным окислением пропена, получаемого в свою очередь из нефтяного сырья. Процесс получения метионина более подробно описан, например, в ЕР 1256571. Процесс получения акролеина более подробно описан, например, в ЕР 417723. Оба эти процесса связаны с высокими техническими затратами и значительными энергозатратами.
В связи с ростом в последние годы цены на нефть соответственно возрастала и стоимость акролеина, экономическая целесообразность использования которого поэтому в качестве структурного звена для синтеза метионина постоянно снижалась. Помимо этого токсичностью и физическими свойствами синильной кислоты и акролеина с учетом требований безопасности и охраны окружающей среды обусловлены соответствующие дополнительные затраты при их использовании в больших количествах.
При химическом синтезе метионин образуется в виде рацемической смеси D- и L-энантиомеров. Этот рацемат можно непосредственно использовать в качестве кормовой добавки, поскольку в условиях in vivo существует механизм превращения неприродного D-энантиомера в природный L-энантиомер. Однако такое превращение связано с потерей определенного количества метионина, а тем самым и с соответствующим снижением биоэффективности по сравнению с таковой при поступлении в организм чистого L-энантиомера в таком же количестве. Поэтому для достижения одного и того же эффекта требуется больше рацемического D,L-метионина, чем L-метионина.
По указанным выше причинам существовала необходимость в разработке экономически более эффективного и экологически менее вредного, а также надежного в осуществлении процесса получения метионина. При этом в первую очередь требовалось предложить пригодный для реализации в промышленном масштабе процесс получения метионина, обогащенного его L-энантиомером (L-метионина), особенно предпочтительно метионина в виде его L-энантиомера с максимально возможной степенью чистоты.
Недостаток существующих на сегодняшний день процессов получения L-метионина с помощью микроорганизмов, описанных, например, в WO 04/024933, состоит в сравнительно невысоком выходе целевого продукта. Причины этого обусловлены прежде всего проблемами, связанными со строгой организацией механизма регуляции микробиологического синтеза L-метионина, с выделением метионина из продуцирующих его клеток в ферментационный бульон, равно как и с энергоемкой реакционной стадией с участием восьми электронов при восстановлении сульфата до сероводорода. С другой стороны, ограниченной растворимостью метионина в воде, соответственно в водных ферментационных бульонах при высокой производительности биосинтеза метионина обусловлено его выпадение в осадок в ферментационной среде, что тем самым осложняет его очистку. Сложность и трудоемкость очистки метионина в свою очередь приводит к образованию значительных количеств отходов, утилизация которых сопряжена с существенными затратами.
В WO 05/059155 описан усовершенствованный способ выделения L-метионина из ферментационных бульонов. Однако подобное усовершенствование предполагает проведение сравнительно сложной последовательности стадий, включающей нагрев и растворение L-метионина в ферментационном бульоне, отфильтровывание биомассы при определенной температуре и дальнейшую обработку отфильтрованной метионинсодержащей биомассы, упаривание маточного раствора, охлаждение, кристаллизацию, отфильтровывание L-метионина от маточного раствора с последующими промывкой и сушкой L-метионина и возвращение маточных растворов в технологический процесс, а также образование двух разных потоков продукта, а именно: одного потока продукта с низкой и одного потока продукта с высокой концентрацией L-метионина. Однако подобное неизбежное образование метионина двоякого качества в свою очередь также приводит к значительному росту затрат и помимо этого нежелательно с маркетинговой точки зрения.
Указанными проблемами в конечном итоге обусловлен меньший общий выход L-метионина при его получении чисто ферментативным способом по сравнению, например, с выходом L-лизина при его получении также ферментативными способами, которые уже на протяжении многих лет успешно используются в технике, и/или обусловлено соответствующее увеличение затрат на ферментативное производство L-метионина.
Исходя из описанного выше уровня техники и присущих ему недостатков, в основу настоящего изобретения была положена задача прежде всего предложить способ получения метионина, который позволял бы устранить подробно рассмотренные выше недостатки известных из уровня техники способов. Такой способ должен исходя по возможности из другого доступного и получаемого ферментативным путем соединения-предшественника максимально простым путем и без использования вышеназванных опасных химикалиев обеспечивать получение L-, D-, соответственно D,L-метионина, предпочтительно, однако, L-метионина, и при этом прежде всего должен не иметь недостатков традиционных химических способов получения метионина, а также способов его прямого биотехнологического получения.
Еще одна задача изобретения состояла в разработке способа получения метионина по меньшей мере частично исходя из природного или воспроизводимого сырья.
Задача изобретения состояла также в том, чтобы разработать технически несложный в осуществлении способ получения L-метионина в приемлемых количествах и с требуемой чистотой.
Эти, а также другие, конкретно не указанные задачи, которые, однако, со всей очевидностью вытекают из контекста настоящего описания, решаются благодаря тому, что в качестве исходной используют иную, доступную и получаемую эффективнее ферментативным путем аминокислоту, из которой затем после соответствующего химического превращения без применения вышеназванных опасных химикалиев получают L-, D-, соответственно D,L-метионин, но прежде всего L-метионин. Тем самым удается избежать недостатков традиционных химических процессов получения метионина и недостатков традиционных процессов прямого ферментативного получения L-метионина. В качестве наиболее пригодной для использования согласно изобретению аминокислотой зарекомендовал себя гомосерин, который в отличие от метионина обладает высокой водорастворимостью и который можно получать также ферментативными способами.
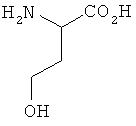
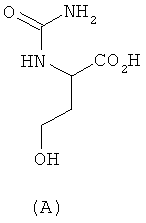
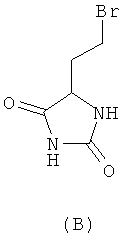
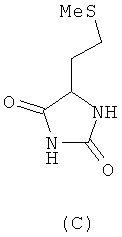
При синтезе метионина способом, который описан у Livak, Britton, VanderWeele и Murray ("Synthesis ofdi-methionine". Journal of the American Chemical Society 67, 1945, p. 2218-2220) и при осуществлении которого D,L-гомосерин образуется в качестве промежуточного продукта, исходят сначала из D,L-2-амино-4-бутиролактона, из которого через промежуточные D,L-гомосерин, N-карбамоилгомосерин, 4-(2-бромметил)гидантоин и 4-(2-метилтиоэтил)гидантоин в завершение получают D,L-метионин:
Дейтерированные производные гомосерина HO-CHD-CH2-CH(HNCOOtBu)COOtBu, соответственно H3CC6H4SO2O-CHD-CH2-CH(HNCOOtBu)COOtBu (где tBu обозначает трет-бутил) использовали согласно Son и Woodard ("Stereochemical mechanism of iodoacetic acid mediated decomposition of L-methionine to L-homoserine lactone", Journal of the American Chemical Society 111(4), 1989, p. 1363-1367) в качестве соединений-предшественников для получения дейтерированного соответственно в положении 4 L-гомосерина. Об использовании в процессе синтеза гомосерина этим способом недейтерированных соединений HO-CH2-CH2-CH(HNCOOtBu)COOtBu, соответственно Н3СС6Н4SO2O-СН2-СН2-СН(ННСOOtBu)COOtBu в указанной публикации ничего не говорится.
Представленные ниже соединения 3,6-ди(2-гидроксиэтил)-2,5-дикетопиперазин, 3,6-ди(2-хлорэтил)-2,5-дикетопиперазин, соответственно 3,6-ди(2-метилтиоэтил)-2,5-дикетопиперазин представляют собой химические промежуточные продукты, через которые согласно US 2397628 получают D,L-метионин исходя, однако, не из гомосерина, а из 2-ацетил-4-бутиролактона:
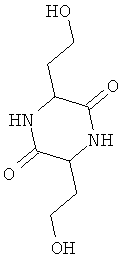
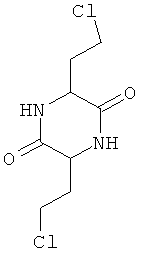
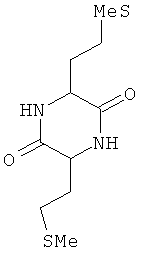
Помимо названных существуют и другие способы получения D,L-метионина, при осуществлении которых также исходят не из гомосерина, а, например, из 2-ацетил-4-бутиролактона, из которого сначала получают промежуточный 2-амино-4-бутиролактон или соответствующим образом защищенный 2-амино-4-бутиролактон, как это описано у Snyder, Andreen, John, Cannon и Peters ("Convenient synthesis of dl-methionine". Journal of the American Chemical Society 64, 1942, p.2082-2084).
При проведении синтеза способом, описанным у Plieninger ("Die Aufspaltung des γ-Butyrolactons und α-Amino-γ-butyrolactons mit Natriummethylmercaptid bzw. -selenid. Eine Synthese des Methionins", Chemische Berichte 83, 1950, cc.265-268), исходят из 2-амино-4-бутиролактона.
Представленные ниже соединения 3,6-ди(2-винил)-2,5-дикетопиперазин, соответственно 3,6-ди(2-бромэтил)-2,5-дикетопиперазин также представляют собой химические соединения-предшественники
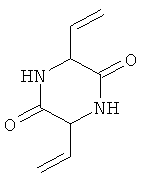
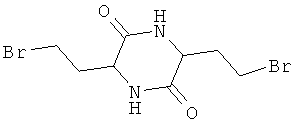
которые согласно Snyder и Chiddix ("Non-Markovnikov addition in reactions of 3,6-divinyl-2,5-diketopiperazme", Journal of the American Chemical Society 66, 1944, p.1002-1004) образуются в качестве промежуточных в процессе получения D,L-метионина. В этом случае гомосерин также не применяют.
Указанные выше задачи решаются прежде всего с помощью способа, заявленного в п.1 формулы изобретения. Различные предпочтительные варианты осуществления предлагаемого в изобретении способа представлены в зависимых пунктах формулы изобретения.
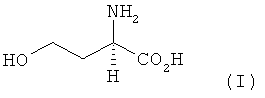
Предлагаемый в изобретении способ получения L-метионина, D-метионина или любой смеси L- и D-метионина, при осуществлении которого исходят из гомосерина и L-гомосерин, D-гомосерин или соответствующие смеси L- и D-гомосерина следующей формулы I
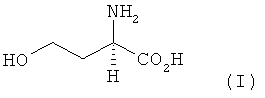
химическим путем превращают в метионин без образования при этом ни одного из таких промежуточных продуктов, как N-карбамоилгомосерин, 4-(2-бромэтил)гидантоин и 4-(2-метилтиоэтил)гидантоин (формулы А-С)
удается избежать недостатков вышеописанных чисто химических способов получения метионина или способов его прямого биотехнологического получения.
Этих недостатков удается избежать прежде всего в тех случаях, когда используемый L-гомосерин получен ферментативным путем. Общеизвестно, что L-гомосерин можно получать ферментативным путем с помощью микроорганизмов (микробиологический синтез), прежде всего бактерий семейства Enterobacteriaceae или коринеформных бактерий, с использованием при этом источников углерода, таких, например, как сахароза, глюкоза, фруктоза и глицерин либо их смеси, и традиционных источников азота, таких, например, как аммиак.
Примеры способов микробиологического получения L-гомосерина с использованием бактерий семейства Enterobacteriaceae, прежде всего бактерий Escherichia coli, можно найти в публикациях US 6303348, US 6887691, US 6960455 или ЕР 1217076 Al.
Примеры способов микробиологического получения L-гомосерина, где с использованием коринеформных бактерий, прежде всего бактерий Corynebacterium glutamicum, можно найти в публикациях US 3189526 или US 3598701.
Использование согласно изобретению полученного ферментативным путем L-гомосерина позволяет избежать необходимости в применении для получения метионина указанных выше относительно опасных исходных веществ - акролеина и синильной кислоты.
Вместе с тем может также оказаться целесообразным смешивать полученный ферментативным путем L-гомосерин с полученным классическим химическим способом рацемическим D,L-гомосерином и соответственно использовать полученную смесь D- и L-гомосерина для химического превращения с получением в результате соответствующих смесей D- и L-метионина. Использование подобного варианта может оказаться предпочтительным, в частности при необходимости утилизации D-/L-гомосерина в качестве отходов химических процессов его получения. Равным образом можно применять и чистый D-гомосерин. Использование этого варианта может оказаться предпочтительным при необходимости утилизации D-гомосерина в качестве отходов процесса расщепления рацемической смеси D-/L-гомосерина. Следует, однако, отметить, что использование чистого D-гомосерина предпочтительно, как правило, лишь при необходимости целенаправленного получения D-метионина.
Применение же только L-гомосерина, полученного ферментативным путем, позволяет напрямую получать L-метионин, а именно: путем проведения химических стадий предлагаемого в изобретении способа, которые не изменяют L-конфигурацию. Использование только L-гомосерина приводит в конечном итоге к получению чистого L-метионина, который непосредственно может применяться в фармацевтике и в пищевой промышленности и который при его использовании в кормах для животных характеризуется также более высокой биоэффективностью по сравнению с обычным D,L-метионином. Данный аспект предлагаемого в изобретении способа, как правило, имеет исключительно важное практическое значение.
В одном из предпочтительных вариантов осуществления предлагаемого в изобретении способа используют содержащий L-гомосерин твердый продукт, полученный из содержащего L-гомосерин ферментационного бульона путем удаления из него воды. Преимущество этого варианта состоит в возможности отделения образующихся в процессе ферментации побочных продуктов лишь на завершающей стадии очистки L-метионина и тем самым в экономии соответствующих затрат на очистку. Вместе с тем в некоторых случаях образующиеся при ферментации побочные продукты и/или примеси можно и не отделять от конечного продукта и оставлять в нем, если только они не препятствуют последующему химическому превращению или если их присутствие в конечном продукте даже желательно. Сказанное относится прежде всего к тем случаям, когда подобные побочные продукты и/или примеси сами обладают питательными свойствами, а L-метионин используется для получения добавок к кормам. Такие обладающие питательной ценностью соединения могут представлять собой, например, другие аминокислоты или белковые вещества.
В соответствии с этим объектом изобретения является также смешанный продукт, состоящий из L-метионина и образующихся при ферментативном получении L-гомосерина побочных продуктов и/или примесей.
Содержащий L-гомосерин ферментационный бульон целесообразно приготавливать путем культивирования выделяющего L-гомосерин микроорганизма в соответствующей питательной среде.
В качестве такого микроорганизма предпочтительно использовать бактерии, прежде всего бактерии рода Corynebacterium или Escherichia.
Помимо этого предпочтительно, чтобы концентрация L-гомосерина в ферментационном бульоне составляла 1 г/л.
При создании изобретения неожиданно было установлено, что химическое превращение L- и/или D-гомосерина можно проводить непосредственно взаимодействием с метилмеркаптаном (MeSH), при необходимости в присутствии кислотного катализатора. Существенное преимущество подобной возможности заключается в прямом получении конечного продукта, т.е. L-метионина, в одну единственную химическую стадию. Метилмеркаптан можно при этом использовать в большом избытке, а неизрасходованный метилмеркаптан можно затем легко отделять и возвращать в технологический процесс, поскольку в отличие от аминокислоты он при комнатной температуре представляет собой газообразное соединение.
MeSH предпочтительно при этом использовать в количестве от 1 до 100 мол. эквивалентов, более предпочтительно от 1 до 50 мол. эквивалентов.
Для ускорения реакции и повышения выхода продукта предпочтительно также использовать кислотный катализатор, выбранный из группы, включающей кислоты Бренстеда с показателем кислотности (рКа) не более 3.
К таким кислотам относятся, например, НСl, HBr, HI, H2SO4, (щелочной металл)НSO4, Н3РO4, (щелочной металл)Н2РO4, где под щелочным металлом подразумевается литий, натрий, калий, рубидий или цезий, полифосфорная кислота, С1-С12алкилсульфоновая кислота, С6-С10арилсульфоновая кислота, трифторметансульфоновая кислота, трифторуксусная кислота или сополимер тетрафторэтилена и перфтор-3,6-диоксо-4-метил-7-октенсульфоновой кислоты (нафион). Преимущество применения нафиона в качестве твердого катализатора заключается прежде всего в возможности его простого отделения от реакционной смеси по завершении реакции и возвращения в технологический процесс.
Равным образом может оказаться предпочтительным использование кислоты Льюиса в качестве катализатора. Из числа таких катализаторов можно назвать прежде всего по меньшей мере низкомолекулярную кислоту Льюиса, выбранную из группы, включающей АlСl3, ZnCl2, BF3·OEt2, SnCl2 и FеСl3.
В качестве предпочтительных для использования в указанных целях зарекомендовали себя также сильнокислотные ионообменные смолы, которые также можно регенерировать особо простым путем, прежде всего необязательно замещенная, например, сшитая дивинилбензолом, полистиролсульфокислотная смола.
Согласно изобретению могут использоваться также гетерогенные кислотные катализаторы из группы, включающей цеолит, монтмориллонит и WO3- и Сs2О-содержащий оксид алюминия. Среди названных оксидов алюминия предпочтительны таковые с содержанием WO3 в количестве от 5 до 15% и с содержанием Cs2O в количестве от 5 до 15%.
Реакцию целесообразно проводить в растворе и/или в суспензии в присутствии воды и/или органического растворителя. При проведении реакции в присутствии воды может оказаться целесообразным непосредственно исходить из содержащего L-гомосерин водного ферментационного раствора, из которого при необходимости удалены присутствующие в нем твердые вещества, поскольку таким путем удается избежать необходимости в проведении последующих стадий переработки. Вместе с тем, может также оказаться предпочтительным использовать и содержащий воду сырой L-гомосерин.
Так, в частности, согласно изобретению можно использовать воду и/или по меньшей мере один низкомолекулярный органический растворитель, выбранный из группы, включающей С3-С6кетоны, предпочтительно метилизобутилкетон (МИБК) или ацетон, С1-С4спирты с прямой либо разветвленной цепью, С4-С10карбоксилаты, предпочтительно этил- или бутилацетат, С3-С6карбоксамиды, предпочтительно диметилформамид (ДМФ) или диметилацетамид, ароматические соединения с С6-С10, предпочтительно толуол, и циклические карбонаты с С3-С7, предпочтительно этиленкарбонат, пропиленкарбонат или бутиленкарбонат. Кроме того, растворителем или по меньшей мере сорастворителем может также служить метилмеркаптан при его применении в соответствующем избытке.
В другом предпочтительном варианте осуществления изобретения предлагаемый в нем способ химического превращения L- и/или D-гомосерина в метионин может также заключаться в том, что на первой стадии путем введения уходящей группы Y по атому С4 гомосерина получают соединение формулы II
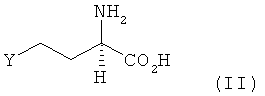
где Y обозначает галоген (хлор, бром или иод), сульфонилоксигруппу (n-толуолсульфонилоксигруппу [n-TsO], С6Н5SO3, Н3СSО3, Н5С2SО3 или СF3SО2), сульфат (ОSО3Н) или фосфат (ОРО3Н), и затем на второй стадии соединение формулы II взаимодействием с MeSH превращают в L-метионин, D-метионин либо в соответствующую смесь L- и D-метионина.
Уходящую группу Y, когда она представляет собой галоген, предпочтительно вводить на первой стадии соответственно взаимодействием гомосерина с РСl5, РСl3, ВВr3, РI3, РОСl3, SOCl2 или SOBr2.
Уходящую группу Y, когда она представляет собой сульфонилоксигруппу, предпочтительно вводить на первой стадии соответственно взаимодействием с хлорангидридом n-толуолсульфоновой кислоты (n-TsCl), С6Н5SO2Сl, Н3СSO2Сl, H5C2SO2Cl или СF3SO2Сl.
Для введения же уходящей группы Y, когда она представляет собой сульфат, на первой стадии обычно используют соответственно SO3, H2SO4 или олеум, а когда она представляет собой фосфат (ОРО3Н), - предпочтительно использовать полифосфорную кислоту.
Активация гомосерина введением соответствующей уходящей группы Y в положение 4 позволяет особо простым путем вводить на следующей стадии MeS-группу замещением уходящей группы Y.
Такую реакцию замещения предпочтительно проводить взаимодействием соединения формулы II с MeSH в присутствии оснóвного или кислотного катализатора.
В качестве оснóвных катализаторов для применения в указанных целях пригодны прежде всего NaOH, КОН, пиридин, триметиламин, триэтиламин или ацетат, карбонат, соответственно гидрокарбонат щелочных или щелочноземельных металлов, где под щелочными металлами подразумеваются литий, натрий, калий, рубидий или цезий, а под щелочноземельными металлами подразумеваются магний, кальций или барий.
Для применения в качестве кислотных катализаторов пригодны прежде всего НСl, HBr, HI, H2SO4, (щелочной металл)НSO4, Н3РO4, (щелочной металл)Н2РO4, где под щелочным металлом подразумевается литий, натрий, калий, рубидий или цезий, полифосфорная кислота, С1-С12алкилсульфоновая кислота, С6-С10арилсульфоновая кислота, трифторметансульфоновая кислота, трифторуксусная кислота или сополимер тетрафторэтилена и перфтор-3,6-диоксо-4-метил-7-октенсульфоновой кислоты (нафион).
Указанную реакцию предпочтительно проводить в присутствии органического растворителя и/или воды.
В качестве органического растворителя предпочтительно при этом использовать низкомолекулярный органический растворитель, выбранный из группы, включающей С3-С6кетоны, предпочтительно метилизобутилкетон (МИБК) или ацетон, С1-С4спирты с прямой либо разветвленной цепью, С4-С10карбоксилаты, предпочтительно этил- или бутилацетат, С3-С6карбоксамиды, предпочтительно ДМФ или диметилацетамид, ароматические соединения с С6-С10, предпочтительно толуол, и циклические карбонаты с С3-С7, предпочтительно этиленкарбонат, пропиленкарбонат или бутиленкарбонат.
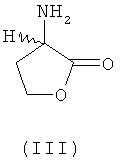
В еще одном предпочтительном варианте осуществления изобретения предлагаемый в нем способ химического превращения L- и/или D-гомосерина в метионин может также заключаться в том, что на первой стадии путем циклизации в присутствии кислотного катализатора получают соответствующий 2-амино-4-бутиролактон формулы III либо его соль (формула IV)
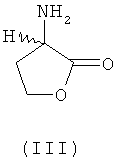
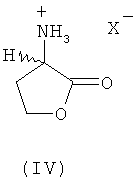
где Х обозначает Cl, Br, I, HSO4 (SO4)1/2, H2PO4, (HPO4)1/2, (PO4)1/3 или R'-SO3 (где R' представляет собой метил, этил, фенил, тозил), и затем этот лактон, соответственно его соль на второй стадии взаимодействием с MeSH превращают в L-метионин, D-метионин либо в соответствующую смесь L- и D-метионина. При этом прежде всего соль представляет собой стабильный промежуточный продукт, который можно направлять на промежуточное хранение или транспортировать, что является немаловажным преимуществом.
Для применения в качестве кислотного катализатора пригодны кислоты, выбранные из группы, включающей кислоты Бренстеда с рКа не более 3.
Предпочтительно при этом использовать в качестве кислотного катализатора НСl, HBr, HI, Н3SО4, (щелочной металл)HSO4, Н3PO4, (щелочной металл)Н2РO4, где под щелочным металлом подразумевается литий, натрий, калий, рубидий или цезий, полифосфорную кислоту, С1-С12алкилсульфоновую кислоту, С6-С10арилсульфоновую кислоту, трифторметансульфоновую кислоту, трифторуксусную кислоту или сополимер тетрафторэтилена и перфтор-3,6-диоксо-4-метил-7-октенсульфоновой кислоты (нафион).
Для применения в качестве кислотного катализатора в равной степени пригодны также сильнокислотные ионообменные смолы, а среди них - прежде всего необязательно замещенные, предпочтительно сшитые дивинилбензолом полистиролсульфокислотные смолы.
Согласно изобретению можно использовать также гетерогенные кислотные катализаторы из группы, включающей WO3- и Сs2О-содержащий оксид алюминия, цеолит и монтмориллонит. Среди названных оксидов алюминия предпочтительны таковые с содержанием WO3 от 5 до 15% и с содержанием Cs2O от 5 до 15%.
В качестве катализаторов можно использовать и кислоты Льюиса и прежде всего низкомолекулярные кислоты Льюиса из группы, включающей АlСl3, ZnCl2, ВF3·ОЕt2, SnCl2, FеСl3, которые широко доступны и недороги.
В еще одном предпочтительном варианте осуществления изобретения предлагаемый в нем способ химического превращения гомосерина в метионин может также заключаться в проведении следующих стадий:
a) L- и/или D-гомосерин подвергают N-ацилированию ацилирующим агентом до N-ацил-L- и/или -D-гомосерина формулы V
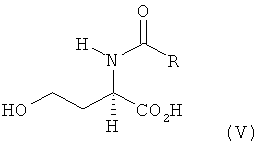
в которой R обозначает водород, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, фенил, моно-, ди- либо тригалогеналкил, где галогеном является F либо Сl, предпочтительно СF3 или ССl3, бензилоксикарбонил либо С1-С4алкилоксикарбонил, предпочтительно трет-бутилоксикарбонил или метилоксикарбонил,
б) полученный на стадии а) N-ацилгомосерин формулы V его взаимодействием с MeSH в присутствии оснóвного или кислотного катализатора превращают в N-ацилметионин формулы VI
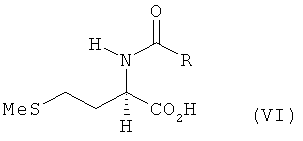
в) полученный на стадии б) N-ацил-L- и/или -D-метионин гидролизуют до соответствующего метионина.
В зависимости от конкретного выбора условий реакции либо на стадии а) первично образуется соответствующий O-ацилгомосерин, превращаемый затем в результате перегруппировки в N-ацилгомосерин формулы V, либо последний образуется на этой стадии непосредственно.
Для ацилирования на стадии а) предпочтительно использовать ацилирующий агент общей формулы R-CO-X1, где Х1 может представлять собой R1 COO, OR2 (где R2 обозначает метил или этил), Сl либо Br, a R и R1 могут иметь идентичные либо разные значения и обозначают водород, метил, этил, н-пропил, изопропил, н-бутил, втиор-бутил, трет-бутил, фенил, моно-, ди- либо тригалогеналкил, где галогеном является F либо Сl, предпочтительно СF3 или ССl3, бензилоксикарбонил либо С1-С4алкилоксикарбонил, предпочтительно трет-бутилоксикарбонил или метилоксикарбонил.
В качестве основного катализатора на стадии б) можно использовать NaOH, КОН, пиридин, триметиламин, триэтиламин или ацетат, карбонат, соответственно гидрокарбонат щелочных или щелочноземельных металлов, где под щелочными металлами подразумеваются литий, натрий, калий, рубидий или цезий, а под щелочноземельными металлами подразумеваются магний, кальций или барий.
Для применения в качестве кислотного катализатора на стадии б) пригодны прежде всего НСl, HBr, HI, H2SO4, (щелочной металл)HSO4, Н3РO4, (щелочной металл)Н2РO4, где под щелочным металлом подразумевается литий, натрий, калий, рубидий или цезий, полифосфорная кислота, С1-С12алкилсульфоновая кислота, С6-С10арилсульфоновая кислота, трифторметансульфоновая кислота, трифторуксусная кислота или сополимер тетрафторэтилена и перфтор-3,6-диоксо-4-метил-7-октенсульфоновой кислоты (нафион).
В еще одном предпочтительном варианте осуществления изобретения предлагаемый в нем способ химического превращения гомосерина в метионин может заключаться также в проведении следующих стадий:
a) L- и/или D-гомосерин подвергают N-ацилированию ацилирующим агентом до N-ацил-L- и/или -D-гомосерина формулы V
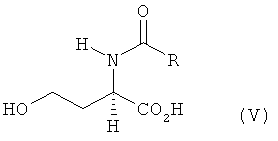
в которой R обозначает водород, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, фенил, моно-, ди- либо тригалогеналкил, где галогеном является F либо Сl, предпочтительно СF3 или ССl3, бензилоксикарбонил либо С1-С4алкилоксикарбонил, предпочтительно трет-бутилоксикарбонил или метилоксикарбонил,
б) полученное на стадии а) соединение формулы V путем введения уходящей группы Y по атому С4 переводят в соединение формулы VI
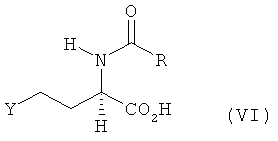
в которой Y обозначает галоген (хлор, бром или иод), сульфонилоксигруппу (n-TsO, С6Н5SO3, Н3СSО3 или Н5С2SO3), сульфат (OSO3Н) или фосфат (ОРО3Н),
в) полученное на стадии б) соединение формулы VI его взаимодействием с MeSH в присутствии оснóвного или кислотного катализатора превращают в N-ацил-L-метионин, N-ацил-D-метионин либо в соответствующую смесь N-ацил-L-и/или -D-метионина формулы VII
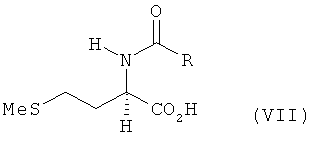
г) полученный на стадии в) N-ацил-L- и/или -D-метионин формулы VII гидролизуют до L- и/или D-метионина.
Соединение формулы V получают при этом в зависимости от конкретного выбора условий реакции либо путем перегруппировки первично образованного O-ацилгомосерина с получением N-ацилгомосерина, либо путем лактонизации in situ в сочетании с ацилированием с последующим размыканием кольца.
Для ацилирования на стадии а) предпочтительно использовать ацилирующий агент общей формулы R-CO-X1, где Х1 представляет собой R1COO, OR2 (где R2 обозначает метил или этил), Сl либо Br, a R и R1 могут иметь идентичные либо разные значения и обозначают водород, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, фенил, моно-, ди- либо тригалогеналкил, где галогеном является F либо Сl, предпочтительно СF3 или ССl3, бензилоксикарбонил либо С1-С4алкилоксикарбонил, предпочтительно трет-бутилоксикарбонил или метилоксикарбонил.
Уходящую группу Y, когда она представляет собой галоген, предпочтительно вводить на первой стадии соответственно взаимодействием гомосерина с РСl3, ВВr3, РI3, SOCl2 или SOBr2.
Уходящую группу Y, когда она представляет собой сульфонилоксигруппу, предпочтительно вводить на первой стадии соответственно взаимодействием с хлорангидридом n-толуолсульфоновой кислоты (n-TsCl), С6Н5SО2Сl, H3CSO2Cl, H5C2SO2Cl или СF3SO2Сl.
Для введения же уходящей группы Y, когда она представляет собой сульфат, на первой стадии обычно используют соответственно SO3, H2SO4 или олеум. Когда же уходящая группа Y представляет собой фосфат (ОРО3Н), то для ее введения на первой стадии предпочтительно использовать полифосфорную кислоту.
Активация N-ацилгомосерина введением соответствующей уходящей группы Y в положение 4 позволяет особо простым путем вводить на следующей стадии MeS-группу замещением уходящей группы Y.
Для применения в качестве оснóвных катализаторов на стадии в) пригодны прежде всего NaOH, КОН, пиридин, триметиламин, триэтиламин или ацетат, карбонат, соответственно гидрокарбонат щелочных или щелочноземельных металлов, где под щелочными металлами подразумеваются литий, натрий, калий, рубидий или цезий, а под щелочноземельными металлами подразумеваются магний, кальций или барий.
Для применения в качестве кислотных катализаторов на стадии в) пригодны прежде всего НСl, HBr, HI, Н2SO4, (щелочной металл)НSO4, Н3РO4, (щелочной металл)Н2РO4, где под щелочным металлом подразумевается литий, натрий, калий, рубидий или цезий, полифосфорная кислота, С1-С12алкилсульфоновая кислота, С6-С10арилсульфоновая кислота, трифторметансульфоновая кислота, трифторуксусная кислота или сополимер тетрафторэтилена и перфтор-3,6-диоксо-4-метил-7-октенсульфоновой кислоты (нафион).
В еще одном предпочтительном варианте осуществления изобретения предлагаемый в нем способ химического превращения L- и/или D-гомосерина в метионин может также заключаться в проведении следующих стадий:
а) L- и/или D-гомосерин подвергают N-ацилированию ацилирующим агентом и циклизации до N-ацил-L- и/или -D-гомосеринлактона формулы VIII
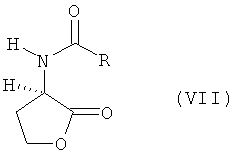
в которой R обозначает водород, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, фенил, моно-, ди- либо тригалогеналкил, где галогеном является F либо Сl, предпочтительно СF3 или ССl3, бензилоксикарбонил либо С1-С4алкилоксикарбонил, предпочтительно трет-бутилоксикарбонил или метилоксикарбонил, б) полученный на стадии а) N-ацилгомосеринлактон его взаимодействием с MeSH в присутствии основного или кислотного катализатора превращают в соответствующий N-ацилметионин формулы VII
в) полученный на стадии б) N-ацил-L- и/или -D-метионин гидролизуют при температуре выше 95°С до соответствующего метионина.
Для ацилирования на стадии а) предпочтительно использовать ацилирующий агент общей формулы R-CO-X1, где Х1 может представлять собой R1COO, OR2 (где R2 обозначает метил или этил), Сl либо Br, a R и R1 могут иметь идентичные либо разные значения и обозначают водород, метил, этил, н-пропил, изопропил, н-бутил, бтиор-бутил, трет-бутл, фенил, моно-, ди- либо тригалогеналкил, где галогеном является F либо Сl, предпочтительно СF3 или ССl3, бензилоксикарбонил либо С1-С4алкилоксикарбонил, предпочтительно трет-бутилоксикарбонил или метилоксикарбонил. N-ацелирование на стадии а) проводят при этом либо путем перегруппировки первично образованного O-ацилгомосерина до N-ацилгомосерина с последующей циклизацией, либо путем лактонизации in situ в сочетании с прямым N-ацилированием.
Помимо этого при ацилировании на стадии а) предпочтительно использовать в качестве растворителя карбоновую кислоту RCOOH или R1COOH, где R и R1 имеют указанные выше значения, при необходимости в присутствии еще одного сорастворителя, выбранного из группы, включающей С3-С6кетоны, предпочтительно метилизобутилкетон (МИБК) или ацетон, С4-С10карбоксилаты, предпочтительно этил- или бутилацетат, С3-С6карбоксамиды, предпочтительно ДМФ или диметилацетамид, ароматические соединения с С6-С10, предпочтительно толуол, и циклические карбонаты с С3-С7, предпочтительно этиленкарбонат, пропиленкарбонат или бутиленкарбонат.
В качестве основных катализаторов на стадии а) предпочтительно использовать производные пиридина, особенно предпочтительно диметиламинопиридин (ДМАП) или карбонилдиимидазол.
Реакцию на стадии а) предпочтительно при этом проводить при температуре в интервале от 20 до 100°С, прежде всего в интервале от 50 до 90°С.
В качестве оснóвного катализатора на стадии б) предпочтительно использовать при этом катализатор, выбранный из группы, включающий гидроксиды тетраалкиламмония с максимум 48 С-атомами, гидроксиды, карбонаты, гидрокарбонаты и ацетаты щелочных, соответственно щелочноземельных металлов, где под щелочными металлами подразумеваются литий, натрий, калий, рубидий или цезий, а под щелочноземельными металлами подразумеваются магний, кальций или барий, третичные амины с максимум 36 С-атомами и 1-4 N-атомами, тетра(С1-С4алкил)гуанидин, бициклические амины, предпочтительно ДБУ (1,8-диазабицикло[5.4.0]ундец-7-ен) и ТБД (1,5,7-триазабицикло[4.4.0]дец-5-ен), пиридин и сильнощелочные ионообменные смолы.
К числу других оснóвных катализаторов, которые предпочтительно использовать на стадии б), относятся триалкиламины общей формулы NR3R4R5, где R3, R4 и R5 могут иметь идентичные или разные значения и обозначают С1-С12алкильный остаток с прямой либо разветвленной цепью, предпочтительно метил, этил, н-пропил, изопропил, н-бутил или втор-бутил.
К наиболее предпочтительным оснóвным катализаторам относятся N(метил)3, N(метил)2(этил), N(метил)(этил)2, N(этил)3, N(н-пропил)3, N(этил)(изопропил)2 или N(н-бутил)3, а также диазабициклооктан (ДАБЦО), ДБУ, ТБД, гексаметилентетрамин, тетраметилэтилендиамин или тетраметилгуанидин.
Равным образом особенно предпочтительно использовать в качестве оснóвных катализаторов R3 R4 R5 R6 N-гидроксид, Li-, Na-, K-, Rb-, Cs-гидроксид, Mg-, Ca-, Ва-гидроксид, где R3, R4, R5 и R6 могут иметь идентичные или разные значения и обозначают С1-С12алкильный остаток с прямой либо разветвленной цепью, предпочтительно метил, этил, н-пропил, изопропил, н-бутил или втор-бутил.
Помимо вышеназванных в качестве наиболее предпочтительных основных катализаторов можно использовать также замещенные группой R7R8 NR9, сшитые полистирольные смолы, где R7, R8 и R9 могут иметь идентичные или разные значения и обозначают С1-С4алкильный остаток с прямой либо необязательно разветвленной цепью, предпочтительно метил, этил, н-пропил или н-бутил.
Для обеспечения быстрого протекания реакции на стадии б) и максимально возможной ее полноты целесообразно использовать 1-20 мол. эквивалентов, предпочтительно 1-10 мол. эквивалентов, основания в пересчете на эквивалент гидроксида, соответственно N-эквивалент.
При использовании же на стадии б) кислотного катализатора его предпочтительно выбирать из группы, включающей кислоты Бренстеда с показателем рКа менее 3 или кислоты Льюиса.
Предпочтительны при этом в качестве кислотных катализаторов НСl, НВr, HI, H2SO4, (щелочной металл)HSO4, Н3РO4, (щелочной металл)Н2РO4, где под щелочным металлом подразумевается литий, натрий, калий, рубидий или цезий, полифосфорная кислота, С1-С12алкилсульфоновая кислота, С6-С10арилсульфоновая кислота, трифторметансульфоновая кислота, трифторуксусная кислота или сополимер тетрафторэтилена и перфтор-3,6-диоксо-4-метил-7-октенсульфоновой кислоты (нафион).
В качестве кислотных катализаторов можно, однако, использовать также сильнокислотные ионообменные смолы, которые по завершении реакции можно легко отделять. Предпочтительны при этом необязательно замещенные, особенно предпочтительно сшитые дивинилбензолом полистиролсульфокислотные смолы.
Равным образом можно использовать и гетерогенные кислотные катализаторы из группы, включающей WO3- и Сs2О-содержащий оксид алюминия, цеолит и монтмориллонит. Среди названных оксидов алюминия предпочтительны таковые с содержанием WO3 в количестве от 5 до 15% и с содержанием Сs2О в количестве от 5 до 15%.
В равной степени предпочтительно использовать в качестве катализаторов и кислоты Льюиса. В качестве такой кислоты предпочтительна низкомолекулярная кислота Льюиса, выбранная из группы, включающей АlСl3, ZnCl2, BF3·OEt2, SnCl2, FеСl3.
Помимо этого реакцию на стадии б) предпочтительно проводить в растворе и/или суспензии в органическом растворителе.
В качестве растворителя при этом можно использовать воду и/или по меньшей мере один низкомолекулярный органический растворитель, выбранный из группы, включающей С3-С6кетоны, предпочтительно метилизобутилкетон (МИБК) или ацетон, С1-С4спирты с прямой либо разветвленной цепью, С4-С10карбоксилаты, предпочтительно этил- или бутилацетат, С3-С6карбоксамиды, предпочтительно ДМФ или диметилацетамид, ароматические соединения с С6-С10, предпочтительно толуол, и циклические карбонаты с С3-С7, предпочтительно этиленкарбонат, пропиленкарбонат или бутиленкарбонат.
Гидролиз на стадии в) можно проводить в водном растворе и/или суспензии.
Вместе с тем, однако, может также оказаться предпочтительным дополнительно использовать по меньшей мере один низкомолекулярный органический растворитель, выбранный из группы, включающей С3-С6кетоны, предпочтительно метилизобутилкетон (МИБК) или ацетон, С1-С4спирты с прямой либо разветвленной цепью, С4-С10карбоксилаты, предпочтительно этил-или бутилацетат, С3-С6карбоксамиды, предпочтительно ДМФ или диметилацетамид, ароматические соединения с С6-С10; предпочтительно толуол, и циклические карбонаты с С3-С7, предпочтительно этиленкарбонат, пропиленкарбонат или бутиленкарбонат.
Реакцию на стадии в) обычно проводят при температуре в интервале от 90 до 180°С, предпочтительно от 100 до 160°С, прежде всего от 120 до 150°С, наиболее предпочтительно от 130 до 140°С.
Для ускорения реакции гидролиза на стадии в) дополнительно можно работать в присутствии кислотного катализатора, основного катализатора или используемой в качестве катализатора кислоты Льюиса либо кислотного катализатора в сочетании с кислотой Льюиса.
Предлагаемый в изобретении способ комбинированного биотехнологического и химического получения метионина обладает целым рядом преимуществ по сравнению с соответствующим традиционным способом, прежде всего с учетом указанной выше потребности в экономически более эффективном и более надежном процессе, который к тому же должен обеспечивать получение L-метионина.
Во-первых, использование сахаров вместо пропена (соответственно акролеина) позволяет повысить экономическую эффективность производства метионина, учитывая, с одной стороны, современную стоимость сырья, а с другой стороны, достигнутую независимость от непрерывно растущих цен на сырую нефть.
Во-вторых, используемые сахара относятся к возобновляемым видам сырья, что позволяет в этом отношении внести ценный вклад в сбережение природных ресурсов. Помимо этого сахара существенно безопаснее образующихся в условиях промышленного производства промежуточных продуктов - акролеина и синильной кислоты, и поэтому замена этих видов сырья на сахара в качестве исходных веществ позволяет значительно снизить возможные риски, сопутствующие производственному процессу и тем самым повысить его безопасность и надежность.
В-третьих, использование ферментационной стадии, обеспечивающей энантиоселективное получение L-гомосерина, в сочетании с соответствующими, сравнительно мягкими химическими стадиями позволяет превращать L-гомосерин в L-метионин без рацемизации и получать таким путем метионин в виде его чистого L-энантиомера. Как указывалось выше, L-метионин обладает более высокой биодоступностью по сравнению с производимым в настоящее время D,L-метионином.
В-четвертых, получение метионина в виде его чистого L-энантиомера комбинированным способом описанного выше типа позволяет эффективно и без особых сложностей устранить рассмотренные выше проблемы, связанные с производством L-метионина исключительно биотехнологическим путем.
Ниже изобретение более подробно поясняется на примерах, которые, однако, не ограничивают его объем.
Прямое превращение L-гомосерина в L-метионин
Пример 1: Реакция в присутствии гетерогенного катализатора (7-10% WO3/7-10% Cs2O на Al2O3 в качестве носителя, выпускается фирмой Degussa)

L-гомосерин (полученный биотехнологическим путем) и тонкоизмельченный гетерогенный катализатор помещали в автоклав, после чего в него добавляли MeSH в виде жидкости. Затем автоклав нагревали до 140°С и выдерживали при этой температуре в течение 2,5 ч. После сброса давления и удаления MeSH промывали 20%-ным водным раствором NaOH. После фильтрации выход L-метионина по данным ЖХВР-анализа (жидкостная хроматография высокого разрешения) составил 3%.
Для сравнения: При проведении аналогичного эксперимента с использованием чистого Al2O3 в качестве носителя были обнаружены лишь следы метионина.
Пример 2: Реакция с изопропилтиолом (iPrSH) и с использованием кислоты/кислоты Льюиса (не подпадает под объем изобретения)
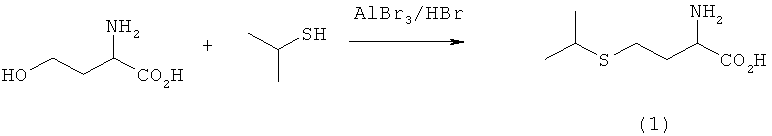
iPrSH (20 мл) медленно насыщали газообразным НВr. Затем добавляли L-гомосерин (10 ммолей) и смесь перемешивали в течение 10 мин. После этого добавляли АlСl3 (40 ммолей) и реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Далее реакцию в реакционной смеси прекращали добавлением смеси Н2О/НСl и подщелачивали NaOH. После отделения Аl(ОН)3 вакуум-фильтрацией раствор фильтрата концентрировали досуха и анализировали посредством ЖХВР. Выход соединения (1) составил 8,2%.
Активация L-гомосерина по атому С4 и превращение в L-метионин
Пример 3: Активация сульфатом с последующим нуклеофильным замещением при взаимодействии с NaSMe
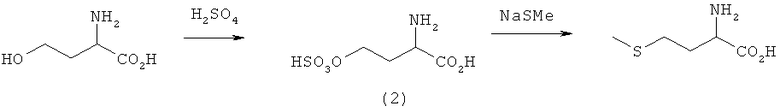
L-гомосерин (19,4 ммоля) при охлаждении смешивали с концентрированной H2SO4 (10 мл). Образовавшуюся реакционную смесь перемешивали в течение 30 мин до полного растворения гомосерина. Затем раствор оставляли стоять в течение 3 ч при комнатной температуре. После этого реакционный раствор добавляли к 800 мл охлажденного до -78°С диэтилового эфира, тщательно перемешивали и надосадочный раствор декантировали. Твердое вещество трижды промывали при -78°С диэтиловым эфиром порциями по 200 мл. После отделения бело-желтого твердого вещества вакуум-фильтрацией его в течение 2 ч сушили в вакууме, создаваемом масляным насосом. Выход сульфата (2) составил 88,0%.
Полученный сульфат (19 ммолей) растворяли в ДМСО (20 мл) и смешивали с NaSMe (50 ммолей). Этот реакционный раствор перемешивали при 80°С и через 90 мин анализировали посредством ЖХВР, согласно которой выход L-метионина составил 19,6%. В результате повторного эксперимента с использованием N-метилпирролидона (N-МП) в качестве растворителя выход L-метионина по истечении 10 мин составил 33,6%.
Циклизация L-гомосерина и его последующее превращение в L-метионин
Пример 4: Получение гидрохлорида 2-амино-4-бутиролактона
Активация путем образования лактона с последующим нуклеофильным замещением при взаимодействии с MeSH
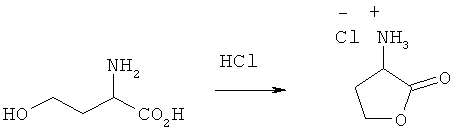
L-гомосерин (0,84 моля) смешивали с 600 мл концентрированной НС1 (6,1 моля). Полученный раствор перемешивали в течение примерно 15 мин до полного растворения всех компонентов, после чего в течение 1,5 ч под вакуумом удаляли воду. Остаток сушили. Выход гидрохлорида 2-амино-4-бутиролактона составил 99%.
Пример 5: Превращение гидрохлорида 2-амино-4-бутиролактона в L-метионин
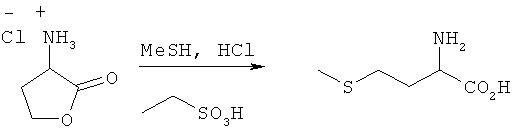
Гидрохлорид 2-амино-4-бутиролактона (22 ммоля) помещали в этансульфокислоте (0,2 моля), насыщенной НСl, в автоклав и к этой смеси добавляли MeSH (0,83 моля) в жидком виде. Затем автоклав закрывали и нагревали до 70°С с выдержкой при этой температуре в течение 5 ч. После сброса давления и охлаждения реакционный раствор анализировали посредством ЖХВР. Выход L-метионина составил 21%.
Пример 6: Превращение гидробромида 2-амино-4-бутиролактона в L-метионин
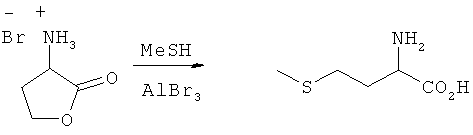
В автоклаве высокого давления к MeSH (50 мл) осторожно добавляли бромид алюминия (75 ммолей). Затем добавляли бромид аминолактона (продукт фирмы Aldrich) (25 ммолей). Автоклав встряхивали в течение 1 ч при комнатной температуре, а затем еще в течение 2 ч при 40°С. Далее автоклав охлаждали и сбрасывали в нем давление. После удаления MeSH остаток резко охлаждали водой и значение рН с помощью NaOH устанавливали на щелочное. Образовавшийся осадок удаляли путем фильтрации. Выход метионина составил 33%.
Пример 7: Превращение гидрохлорида 2-амино-4-бутиролактона в 2-амино-4-метилтиомасляную кислоту
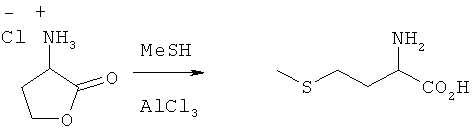
Хлорид аминолактона (10 ммолей), а также АlСl3 (30 ммолей) помещали в автоклав, после чего при перемешивании медленно смешивали с MeSH (30 мл). Затем смесь в течение 71 ч перемешивали при комнатной температуре. После прекращения реакции в реакционной смеси добавлением к ней воды посредством ЖХВР определяли выход 2-амино-4-метилтиомасляной кислоты, который составил 27%.
Пример 8: Превращение гидрохлорида 2-амино-4-бутиролактона в 2-амино-4-изопропилтиомасляную кислоту (не подпадает под объем изобретения)
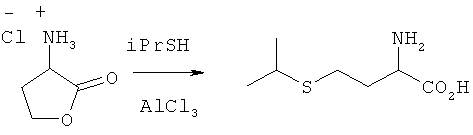
Изопропилтиол (iPrSH, 20 мл) смешивали при перемешивании с АlСl3 (30 ммолей). Затем добавляли хлорид аминолактона (10 ммолей) и смесь в течение 24 ч перемешивали при комнатной температуре. После прекращения реакции в реакционной смеси добавлением к ней воды посредством ЖХВР определяли выход 2-амино-4-изопропилтиомасляной кислоты, который составил 77%.
Пример 9: Превращение гидрохлорида 2-амино-4-бутиролактона в L-метионин
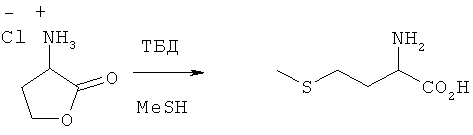
Гидрохлорид 2-амино-4-бутиролактона (70 ммолей) и ТБД (1,5,7-триазабицикло[4.4.0]дец-5-ен) (140 ммолей) помещали в автоклав, после чего добавляли жидкий MeSH. Закрытый автоклав нагревали до 70°C с выдержкой при этой температуре в течение 2,5 ч. Затем автоклав слегка охлаждали и сбрасывали в нем давление. Далее удаляли MeSH и остаток исследовали посредством ЖХВР. Выход L-метионина составил 21%.
Пример 10: Циклизация L-гомосерина и N-ацилирование до N-ацил-2-амино-4-бутиролактона с последующим его превращением в N-ацил-L-метионин (предшественник L-метионина)
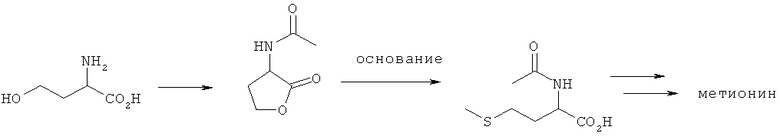
L-гомосерин (2 моля) суспендировали в 900 мл уксусного ангидрида и смешивали с диметиламинопиридином, который добавляли на кончике шпателя. Затем смесь медленно нагревали до 60°С. По истечении примерно 1 ч температура быстро возрастала до 100°С. Далее реакционную смесь в течение 90 мин перемешивали при 80°С и под вакуумом сушили досуха. Полученное желтое масло растворяли в изопропаноле (600 мл) и оставляли стоять на ночь при 0°С. Образовавшиеся кристаллы отфильтровывали, промывали холодным изопропанолом и в завершение сушили под вакуумом. Согласно данным ЖХВР-анализа выход N-ацетил-2-амино-4-бутиролактона составил 60%, а его чистота составила 99%.
Далее N-ацетил-2-амино-4-бутиролактон (1 экв.) его взаимодействием с различными основаниями в MeSH превращали в N-ацетилметионин. Затем смесь из N-ацетиламинолактона, соответствующего основания и MeSH (14 экв.) нагревали в закрытом автоклаве. После охлаждения, сброса давления и удаления MeSH образовавшийся маслянистый остаток исследовали посредством ЖХВР. Другие подробности проведенных опытов, равно как и достигнутый выход N-ацетил-L-метионина представлены ниже в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КЕТОКИСЛОТ И ИХ ПРОИЗВОДНЫХ | 2008 |
|
RU2536046C2 |
| ЗАМЕЩЕННЫЕ 2-МЕТИЛТИОЭТИЛОМ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ДОБАВОК К КОРМАМ | 2008 |
|
RU2516833C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛМЕРКАПТАНА ИЗ ДИАЛКИЛСУЛЬФИДОВ И ДИАЛКИЛПОЛИСУЛЬФИДОВ | 2008 |
|
RU2490255C2 |
| КЕТОМЕТИОНИНКЕТАЛИ И ИХ ПРОИЗВОДНЫЕ | 2007 |
|
RU2483062C2 |
| СИНТЕЗ И ПРИМЕНЕНИЕ 2-ОКСО-4-МЕТИЛТИОБУТАНОВОЙ КИСЛОТЫ, ЕЕ СОЛЕЙ И ПРОИЗВОДНЫХ | 2005 |
|
RU2385862C2 |
| СПОСОБ ПОЛУЧЕНИЯ БЛОКАТОРА АНГИОТЕНЗИНОВОГО РЕЦЕПТОРА | 2003 |
|
RU2412173C2 |
| ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ МЕТИОНИЛМЕТИОНИНА В КАЧЕСТВЕ ДОБАВКИ К КОРМАМ ДЛЯ РЫБ И РАКООБРАЗНЫХ | 2009 |
|
RU2599783C2 |
| ДИПЕПТИДЫ В КАЧЕСТВЕ КОРМОВЫХ ДОБАВОК | 2010 |
|
RU2536467C2 |
| Способ получения промежуточного соединения L-глюфосината и L-глюфосината | 2019 |
|
RU2799336C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2194048C2 |
Изобретение относится к новым способам получения L-метионина, D-метионина или любой смеси L- и D-метионина исходя из гомосерина и характеризуется тем, что L-гомосерин, D-гомосерин или смеси L- и D-гомосерина формулы
путем химического превращения переводят в метионин без образования при этом промежуточных N-карбамоилгомосерина, 4-(2-бромэтил)гидантоина и 4-(2-метилтиоэтил)гидантоина. Согласно первому из предлагаемых способов химическое превращение заключается в том, что на первой стадии путем циклизации в присутствии кислотного катализатора получает соответствующий 2-амино-4-бутиролактон формулы III либо его соль формулы IV
(значения Х приведены в п.1 формулы изобретения), и затем этот лактон или его соль на второй стадии взаимодействием с MeSH превращают в L-метионин, D-метионин либо в смесь L- и D-метионина. Согласно второму из предлагаемых способов химическое превращение состоит из стадий а)-в). На стадии а) гомосерин формулы (I) подвергают оснóвному каталитическому N-ацилированию ацилирующим агентом и циклизации до N-ацил-L- и/или -D-гомосеринлактона формулы 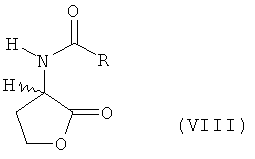
при температуре в интервале от 20 до 100°С, предпочтительно от 50 до 90°С, причем ацилирующий агент имеет общую формулу R-CO-X1 (значения R и X1 приведены в п.6 формулы изобретения). На стадии б) полученный на стадии а) N-ацилгомосеринлактон его взаимодействием с MeSH в присутствии основного катализатора превращают в соответствующий N-ацилметионин формулы
На стадии в) полученный на стадии б) N-ацилметионин гидролизуют при температуре выше 95°С до соответствующего метионина. Оснóвный катализатор, используемый на стадии б), представляет собой триалкиламин общей формулы NR3R4R5 (значения R3, R4 и R5 указаны в п.6 формулы изобретения) или ДАБЦО, ДБУ, ТБД, гексаметилентетрамин, тетраметилэтилендиамин или тетраметилгуанидин. При этом на стадии б) используют 1-20 мол. эквивалентов, предпочтительно 1-10 мол. эквивалентов, основания в пересчете на эквивалент гидроксида. 2 н. и 16 з.п. ф-лы, 1 табл., 10 пр.
1. Способ получения L-метионина, D-метионина или любой смеси L- и D-метионина исходя из гомосерина, характеризующийся тем, что L-гомосерин, D-гомосерин или соответствующие смеси L- и D-гомосерина формулы I
путем химического превращения переводят в метионин без образования при этом промежуточных N-карбамоилгомосерина, 4-(2-бромэтил)гидантоина и 4-(2-метилтиоэтил)гидантоина, отличающийся тем, что химическое превращение L- и/или D-гомосерина заключается в том, что на первой стадии путем циклизации в присутствии кислотного катализатора получают соответствующий 2-амино-4-бутиролактон формулы III либо его соль (формула IV)
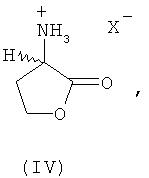
где Х обозначает Cl, Br, I, HSO4, (SO4)1/2, Н2РО4, (HPO4)1/2, (PO4)1/3 или R'-SO3 (где R' представляет собой метил, этил, фенил, тозил), и затем этот лактон, соответственно его соль на второй стадии взаимодействием с MeSH превращают в L-метионин, D-метионин либо в соответствующую смесь L- и D-метионина.
2. Способ по п.1, отличающийся тем, что на первой стадии циклизации используют кислотный катализатор, выбранный из группы, включающей кислоты Бренстеда с рКа не более 3.
3. Способ по п.2, отличающийся тем, что в качестве кислотного катализатора используют НСl, HBr, HI, H2SO4, (щелочной металл) НSO4, Н3РO4, (щелочной металл) Н2РO4, где под щелочным металлом подразумевается литий, натрий, калий, рубидий или цезий, полифосфорную кислоту, С1-С12алкилсульфоновую кислоту, С6-С10арилсульфоновую кислоту, трифторметансульфоновую кислоту, трифторуксусную кислоту или сополимер тетрафторэтилена и перфтор-3,6-диоксо-4-метил-7-октенсульфоновой кислоты (нафион).
4. Способ по п.1, отличающийся тем, что в качестве катализатора на второй стадии используют кислоту Льюиса.
5. Способ по п.4, отличающийся тем, что в качестве служащей катализатором кислоты Льюиса используют низкомолекулярную кислоту Льюиса, выбранную из группы, включающей АlСl3, ZnCl2, ВF3·ОЕt2, SnCl2, FeCl3.
6. Способ получения L-метионина, D-метионина или любой смеси L- и D-метионина исходя из гомосерина, характеризующийся тем, что L-гомосерин, D-гомосерин или соответствующие смеси L- и D-гомосерина формулы I
путем химического превращения переводят в метионин без образования при этом промежуточных N-карбамоилгомосерина, 4-(2-бромэтил)гидантоина и 4-(2-метилтиоэтил)гидантоина, отличающийся тем, что химическое превращение L- и/или D-гомосерина заключается в проведении следующих стадий:
а) подвергают основному каталитическому N-ацилированию ацилирующим агентом и циклизации до N-ацил-L- и/или -D-гомосеринлактона формулы VIII
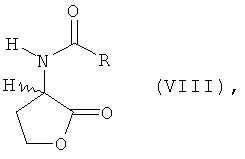
в которой R обозначает водород, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, фенил, моно-, ди- либо тригалогеналкил, где галогеном является F либо Сl, предпочтительно СF3 или ССl3, бензилоксикарбонил либо С1-С4алкилоксикарбонил, предпочтительно трет-бутилоксикарбонил или метилоксикарбонил,
при температуре в интервале от 20 до 100°С, предпочтительно от 50 до 90°С,
причем ацилирующий агент имеет общую формулу R-CO-X1, где X1 представляет собой R1COO, OR2 (где R2 обозначает метил или этил), Сl либо Вr, а R и R1 могут иметь идентичные либо разные значения и обозначают водород, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, фенил, моно-, ди- либо тригалогеналкил, где галогеном является F либо Сl, предпочтительно СF3 или ССl3,
б) полученный на стадии а) N-ацилгомосеринлактон его взаимодействием с MeSH в присутствии основного катализатора превращают в соответствующий N-ацилметионин формулы VII
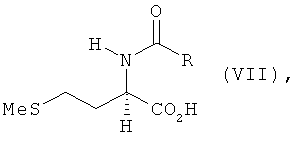
в) полученный на стадии б) N-ацилметионин гидролизуют при температуре выше 95°С до соответствующего метионина, в котором основный катализатор, используемый на стадии б), представляет собой триалкиламин общей формулы NR3R4R5, где R3, R4 и R5 могут иметь идентичные или разные значения и обозначают С1-С12алкильный остаток с прямой либо разветвленной цепью, предпочтительно метил, этил, н-пропил, изопропил, н-бутил или втор-бутил или в качестве основного катализатора используют ДАБЦО, ДБУ, ТБД, гексаметилентетрамин, тетраметилэтилендиамин или тетраметилгуанидин, и на стадии б) используют 1-20 мол. эквивалентов, предпочтительно 1-10 мол. эквивалентов, основания в пересчете на эквивалент гидроксида, соответственно N-эквивалент.
7. Способ по п.6, отличающийся тем, что на стадии а) в качестве катализатора используют производное пиридина, предпочтительно диметиламинопиридин (ДМАП) или карбонилдиимидазол.
8. Способ по п.6, отличающийся тем, что в качестве основного катализатора используют N(метил)3, N(метил)2(этил), N(метил)(этил)2, N(этил)3, N(н-пропил)3, N(этил)(изопропил)2 или N(н-бутил)3.
9. Способ по одному из пп.6, 7, отличающийся тем, что реакцию на стадии в) проводят при температуре в интервале от 100 до 160°С, прежде всего от 120 до 150°С, наиболее предпочтительно от 130 до 140°С.
10. Способ по одному из пп.6, 7, отличающийся тем, что реакцию на стадии в) дополнительно проводят в присутствии кислотного катализатора, основного катализатора или используемой в качестве катализатора кислоты Льюиса либо кислотного катализатора в сочетании с кислотой Льюиса.
11. Способ по одному из пп.1 или 6, отличающийся тем, что используют L-гомосерин, полученный ферментативным путем.
12. Способ по одному из пп.1 или 6, отличающийся тем, что используют только гомосерин с L-конфигурацией.
13. Способ по п.12, отличающийся тем, что используют L-гомосерин, полученный ферментативным путем.
14. Способ по одному из пп.12 или 13, отличающийся тем, что используют содержащий L-гомосерин твердый продукт, полученный из содержащего L-гомосерин ферментационного бульона путем удаления воды.
15. Способ по п.14, отличающийся тем, что содержащий L-гомосерин ферментационный бульон получен путем культивирования выделяющего L-гомосерин микроорганизма в соответствующей питательной среде.
16. Способ по п.15, отличающийся тем, что микроорганизм представляет собой бактерию.
17. Способ по п.16, отличающийся тем, что бактерия представляет собой бактерию рода Corynebacterium или Escherichia.
18. Способ по п.14, отличающийся тем, что концентрация L-гомосерина в ферментационном бульоне составляет по меньшей мере 1 г/л.
| J.E.LIVAK et al, Synthesis of dl-methionine, J | |||
| AM | |||
| CHEM | |||
| SOC., 1945, 67(12), 2218-2220 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ получения производных аминокислоты | 1985 |
|
SU1468411A3 |

