Настоящее изобретение относится к химическим способам приготовления определенных производных хиназолина или их фармацевтически приемлемым солям. Изобретение также относится к способу приготовления определенных промежуточных соединений, пригодных для приготовления производных хиназолина и к способам приготовления производных хиназолина, используя указанные промежуточные соединения.
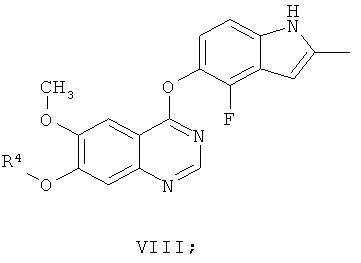
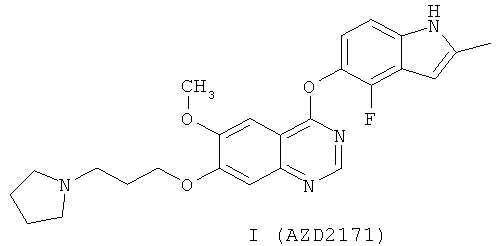
В частности, настоящее изобретение относится к химическим способам и промежуточным соединениям, пригодным для приготовления соединения 4-(4-фтор-2-метилиндол-1Н-5-илокси)-6-метокси-7-[3-(пирролидин-1-ил)пропокси]хиназолин. Это соединение подпадает под раскрытие WO 00/47212 и описано в Примере 240 этой заявки.
Соединение 4-(4-фтор-2-метилиндол-1Н-5-илокси)-6-метокси-7-[3-(пирролидин-1-ил)пропокси]хиназолин описано в настоящей заявке с помощью формулы I
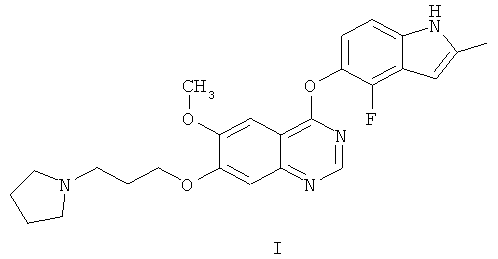
и как AZD2171, кодовый номер, под которым известно соединение.
Нормальный ангиогенез играет важную роль в разнообразных процессах, включая эмбриональное развитие, заживление ран и некоторых составляющих женской репродуктивной функции. Нежелательный или патологический ангиогенез связан с болезненными состояниями, включая диабетическую ретинопатию, псориаз, злокачественные новообразования, ревматоидный артрит, атерому, саркому Капоши и гемангиому (Fan и др., 1995, Trends Pharmacol. Sci. 16: 57-66; Folkman, 1995, Nature Medicine 1: 27-31). Полагают, что изменение проницаемости сосудов играет важную роль как в нормальных, так и в патологических физиологических процессах (Cullinan-Bove и др., 1993, Endocrinology 133: 829-837; Senger и др., 1993, Cancer and Metastasis Reviews, 12: 303-324). Были идентифицированы определенные полипептиды, которые в условиях in vitro обладают стимулирующим действием на рост эндотелиальных клеток, включая кислые и щелочные факторы роста фибробластов (aFGF и bFGF) и фактор роста эндотелия сосудов (VEGF). Активность фактора роста VEGF, посредством ограниченной экспрессии его рецепторов, в отличие от таковой у FGF, является относительно специфичной по отношению к клеткам эндотелия. Исследования, проведенные в последнее время, убедительно свидетельствуют о том, что VEGF является существенным стимулятором как нормального, так и патологического ангиогенеза (Jakeman и др., 1993, Endocrinology, 133: 848-859; Kolch и др., 1995, Breast Cancer Research and Treatment, 36: 139-155) и проницаемости сосудов (Connolly и др., 1989, J. Biol. Chem. 264: 20017-20024). Антагонистическое действие по отношению к VEGF путем блокирования VEGF при помощи антитела может приводить к ингибированию роста опухолей (Kim и др., 1993, Nature 362: 841-844).
Рецепторные тирозинкиназы (RTK) занимают важное место в передаче биохимических сигналов через плазматическую мембрану клеток. Как правило, эти трансмембранные молекулы включают внеклеточный лиганд-связывающий домен, связанный при помощи сегмента в плазматической мембране с внутриклеточным доменом тирозинкиназы. Связывание лиганда с рецептором приводит к стимулированию связанной с рецептором активности тирозинкиназы, что приводит к фосфорилированию остатков тирозина как в рецепторе, так и в других внутриклеточных молекулах. Эти изменения в фосфорилировании тирозина инициируют каскадный путь передачи сигналов, что приводит ко многим ответным реакциям клеток. К настоящему времени было идентифицировано по крайней мере девятнадцать различных подсемейств RTK на основании гомологии аминокислотных последовательностей. Одно из этих подсемейств в настоящее время состоит из fins-подобного рецептора тирозинкиназы, Flt-1 (также называется VEGFR-1), рецептора KDR, содержащего домен со вставкой киназы (который также называется VEGFR-2 или Flk-1) и другого fins-подобного рецептора тирозинкиназы, Flt-4. Было показано, что две эти родственные RTK, Flt-1 и KDR, связывают VEGF с высоким сродством (De Vries и др., 1992, Science 255: 989-991; Terman и др., 1992, Biochem Biophys. Res. Comm. 1992, 187: 1579-1586). Связывание VEGF с этими рецепторами, которые экспрессируются в гетерологичных клетках, ассоциировано с изменениями уровня фосфорилирования тирозина белков клетки и потоками кальция.
VEGF является ключевым стимулятором васкулогенеза и ангиогенеза. Этот цитокин вызывает фенотипическое образование отростков сосудов путем стимулирования пролиферации эндотелиальных клеток, экспрессии протеазы и миграции, и последующей организацией этих клеток с образованием капилляра (Keck и др., Science (Washington DC), 246: 1309-1312, 1989; Lamoreaux и др., Microvasc. Res., 55: 29-42,1998; Pepper и др., Enzyme Protein, 49: 138-162, 1996). Кроме того, VEGF вызывает значительное повышение проницаемости сосудов (Dvorak и др., Int. Arch. Allergy Immunol., 707: 233-235, 1995; Bates и др., Physiol. (Lond.), 533: 263-272, 2001), стимулируя формирование сверхпроницаемости, недоразвитой сосудистой сети, что является характерной особенностью патологического ангиогенеза.
Было показано, что активирование только KDR достаточно для активизации всех основных фенотипических ответных реакций на VEGF, включая пролиферацию эндотелиальных клеток, миграцию и выживание, и индуцирование проницаемости сосудов (Meyer и др., EMBO J., 18: 363-374, 1999; Zeng и др., J. Biol. Chem., 276: 32714-32719, 2001; Gille и др., J. Biol. Chem., 276: 3222-3230, 2001).
Ангиогенез и/или повышенная проницаемость сосудов присутствует при многих различных болезненных состояниях, включая злокачественное новообразование (включая лейкоз, множественную миелому и лимфому), диабет, псориаз, ревматоидный артрит, саркому Капоши, гемангиому, острую и хроническую нефропатии, атерому, артериальный рестеноз, аутоиммунные заболевания, астму, острое воспаление, чрезмерное образование рубцов и спаек, лимфатический отек, эндометриоз, дисфункциональное маточное кровотечение и заболевания глаз с пролиферацией сосудов сетчатки, включая дегенерацию желтого пятна, связанную со старением.
AZD2171 является эффективным ингибитором VEGF RTK и проявляет >800-5000-кратную селективность по сравнению с VEGFR-2, по сравнению с тирозинкиназой рецептора фактора роста эпидермиса, тирозинкиназой ErbB2 рецептора, тирозинкиназой ТЕК (Tie-2) рецептора и циклин-зависимой киназой-2. AZD2171 проявляет очень хорошую активность в условиях in vitro как (а) фермент и (б) в исследованиях HUVEC, которые описаны в WO 00/47212 (страницы 80-83). Значения AZD2171 IC50 для ингибирования выделенной KDR (VEGFR-2), Flt-1 (VEGFR-1) и Flt-4 (VEGFR-3) тирозинкиназной активности в ферментативном исследовании составляли <2 нМ, 5±2 нМ и ≤3 нМ соответственно. AZD2171 эффективно ингибирует VEGF-стимулированную пролиферацию клеток эндотелия (IC50 значение 0,4±0,2 нМ в исследовании HUVEC), но не ингибирует пролиферацию базальных клеток эндотелия существенно при >1250 раз больше концентрации (IC50 значение >500 нМ). Рост ксенотранслантированной опухоли Calu-6 на модели солидной опухоли в условиях in vivo, описанной в WO 00/47212 (страница 83), ингибируется на 49%**, 69%*** и 91%*** через 28 дней после ежедневного один раз в сутки перорального лечения с помощью 1, 5, 3 и 6 мг/кг/сутки AZD2171 соответственно (Р**<0,01, Р***<0,0001; односторонний t критерий). Было показано, что AZD2171 проявляет широкий спектр противоопухолевой активности на многих моделях после ежедневного один раз в сутки перорального лечения (Wedge и др., (2005) Cancer Research 65(10), 4389-4440).
В WO 02/12227 описаны несколько возможных путей получения индолокси бициклических соединений. Однако в WO 02/12227 специфически не раскрыт способ получения соединения формулы I.
В WO 00/47212 описан путь получения соединений формулы I (см. Пример 240). Этот путь получения соединения формулы I удовлетворительный для синтеза относительно небольших количеств соединения. Однако в этом пути задействован линейный, а не конвергентный синтез, для которого требуется применение многих стадий очистки и выделения большого количества промежуточных соединений. По существу, суммарный выход синтеза не является высоким. Существует потребность в более эффективном синтезе соединений формулы I, пригодном для получения больших количеств этого соединения. Также существует потребность в более эффективном синтезе промежуточных соединений, пригодных для синтеза соединений формулы I, для получения больших количеств этих промежуточных соединений.
Предпочтительно, новые синтезы должны минимизировать количество промежуточных соединений, которые необходимо выделять, и не должны использовать дорогие и трудоемкие методики очистки. Дополнительно, новые синтезы должны образовывать постоянно высококачественные соединения, в частности, для того, чтобы образовать высококачественное соединение формулы I для соответствия требованиям высокой чистоты для фармацевтического продукта. Новые синтезы также должны использовать методики и реагенты, которые могут безопасно использоваться на производственном предприятии и которые отвечают рекомендациям по охране окружающей среды.
В соответствии с настоящим изобретением, сейчас мы обеспечили улучшенные способы приготовления AZD2171, соединения формулы I.
В соответствии с настоящим изобретением, также обеспечиваются способы для приготовления ключевых промежуточных соединений, которые могут использоваться для приготовления AZD2171.
Новые способы являются благоприятными, поскольку они позволяют получить соединения высокого качества и с высоким выходом в большом количестве. Способы позволяют существенно уменьшить количество промежуточных соединений, которые следует выделить, и, в целом, являются более конвергентными, чем предыдущие пути. Такие изменения обеспечивают существенные преимущества относительно времени и затрат.
Для избегания неопределенности термин "AZD2171", как используется в настоящей заявке, относится к свободному основанию AZD2171, если специально не указано иначе.
Ключевым промежуточным соединением, которое может использоваться для получения AZD2171, является 2-метил-4-фтор-5-гидрокси-индол, соединение формулы II
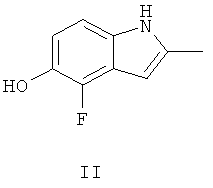
В Примере 237 WO 00/47212 описаны три пути приготовления соединения формулы II.
(i) В первом пути задействована реакция 2-фтор-4-нитроанизола с 4-хлорфеноксиацетонитрилом в диметилформамидном (ДМФА) растворителе в присутствии трет-бутоксида калия с последующим восстановлением с водородом, используя катализатор палладий на древесном угле, с получением смеси 4-фтор-5-метоксииндола и 6-фтор-5-метоксииндола. После защиты с азота индола с трет-бутоксикарбонилом смесь защищенных индолов подвергают реакции в тетрагидрофурановом (ТГФ) растворителе с трет-бутиллитием и метилйодидом, затем с трифторуксусной кислотой, получая смесь 6-фтор-5-метокси-2-метил-индола и 4-фтор-5-метокси-метилиндола. После очистки 4-фтор-5-метокси-метилиндол подвергают реакции с трибромидом бора в метиленхлориде, получая соединение формулы II, 4-фтор-5-гидрокси-2-метилиндол.
(ii) Во втором пути задействована реакция этилацетоацетата с 1,2,3-трифтор-4-нитробензолом в ТГФ в присутствии гидрида натрия с образованием 3-ацетилметил-1,2-дифтор-4-нитробензола. Затем 3-ацетилметил-1,2-дифтор-4-нитробензол подвергают реакции с триметилортоформиатом в метиленхлориде в присутствии монтмориллонита с образованием 1,2-дифтор-3-(2,2-диметоксипропил)-4-нитробензола. После этого 1,2-дифтор-3-(2,2-диметоксипропил)-4-нитробензол подвергают реакции с бензиловым спиртом в диметилацетамиде (DMA) в присутствии гидрида натрия с образованием 3-ацетилметил-1-бензилокси-2-фтор-4-нитробензола. Это соединение циклизируют и снимают защиту путем взаимодействия с 10% палладием на древесном угле в этаноле/уксусной кислоте в присутствии водорода, получая соединение формулы II, 4-фтор-5-гидрокси-2-метилиндол.
(iii) В третьем пути задействована реакция 1,2-дифтор-3-(2,2-диметоксипропил)-4-нитробензола с метилатом натрия в метаноле, получая 3-ацетилметил-2-фтор-1-метокси-4-нитробензол. Это соединение циклизируют и снимают защиту путем взаимодействия с трихлоридом титана в ацетоне в присутствии ацетата аммония, получая 4-фтор-5-метокси-2-метилиндол. Затем 4-фтор-5-метокси-метилиндол подвергают реакции с трибромидом бора в метиленхлориде, получая соединение формулы II, 4-фтор-5-гидрокси-2-метилиндол.
Пути, описанные в документах предшествующего уровня техники, для получения соединения формулы II достаточны для синтеза относительно небольших количеств соединения. Тем не менее, они все требуют выделения каждого из промежуточных соединений и, следовательно, предусматривают много стадий выделения и/или очистки. Это приводит к удовлетворительному суммарному выходу соединения формулы II, используемого в небольшом масштабе. Однако пути, описанные в документах предшествующего уровня техники, неприемлемы для применения в промышленном масштабе, поскольку они предусматривают много стадий выделения и/или очистки, которые не могут быть эффективно осуществлены в промышленном масштабе. В частности, пути, описанные в документах предшествующего уровня техники, непригодны для применения для приготовления фармацевтического продукта с высокой чистотой.
Следовательно, существует потребность в более эффективном синтезе соединения формулы II, подходящего для применения для получения бóльших количеств этого соединения. Предпочтительно, новые синтезы не должны предусматривать дорогостоящие и трудоемкие выделения и/или очистки. Таким образом, в новом синтезе должно быть уменьшено количество необходимых методик выделения и/или очистки, посредством этого уменьшена стоимость и время производства. Предпочтительно, в новом синтезе должно быть минимизировано количество растворителей, используемых при осуществлении метода, что улучшает экологические показатели и предоставляет возможность для восстановления растворителя. Предпочтительно, новый синтез также должен обеспечивать устойчивый и надежный способ выделения соединения формулы II и равным образом обеспечивать высокое качество соединения формулы II, например, для того, чтобы соответствовать нормативным требования для внедрения исходных веществ в получение фармацевтических продуктов.
В международной патентной заявке, номер публикации WO 2004/009542 описан альтернативный способ получения 2-метил-4-фтор-5-гидрокси-индола.
В соответствии с первым аспектом настоящего изобретения обеспечивается способ приготовления соединения формулы II из производного нитробензола формулы III
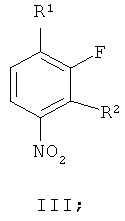
где R1 и R2 независимо выбирают из фтора, хлора, брома, йода и необязательно замещенного алкилсульфонилокси, такого как трифторокси или тозилокси,
где способ включает стадии:
(а) взаимодействие соединения формулы III со сложным эфиром формулы (IV)
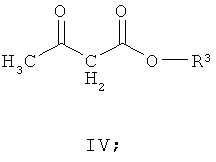
где R3 представляет собой подходящую этерифицирующую группу,
получая соединение формулы V
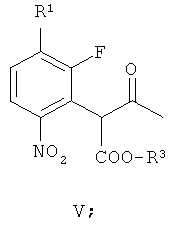
(б) взаимодействие соединения формулы V с гидроксильным ионом в присутствии соли арил-алкил аммония или соли тетра-алкил аммония, получая соединение формулы VI
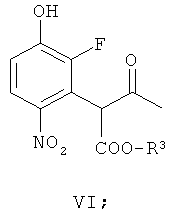
(в) взаимодействие соединения формулы VI, получая соединение формулы VII
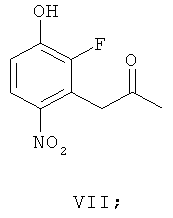
(г) восстановление соединения формулы VII, получая соединение формулы II. В одном варианте осуществления R1 и R2 независимо выбирают из фтора, хлора, брома и йода. В другом варианте осуществления R1 и R2 независимо выбирают из фтора, хлора и брома. В другом варианте осуществления R1 представляет собой фтор и R2 представляет собой бром. В другом варианте осуществления оба R1 и R2 представляют собой фтор, R3 представляет собой подходящую этерифицирующую группу, такую как необязательно замещенный C1-6алкил или необязательно замещенный бензил.
Квалифицированный специалист в данной области техники способен выбрать подходящие эстерифицирующие группы, которые не будут препятствовать способам этого варианта осуществления изобретения и будет позволять осуществить удаление сложноэфирной группы при осуществлении стадии способа (в).
В одном варианте осуществления R3 представляет собой C1-6алкил или бензил. В другом варианте осуществления R3 представляет собой С1-4алкил. В дальнейшем варианте осуществления R3 представляет собой С4алкил, подходящ трет-бутил.
Условия реакции для способа (а)
Реакцию способа (а) подходяще осуществляют в присутствии подходящего растворителя, такого как тетрагидрофуран или ацетонитрил, или в другом варианте осуществления подходящего неполярного растворителя, такого как толуол, триметилбензол или ксилол, в присутствии подходящего основания, такого как трет-бутоксид натрия или трет-пентоксид натрия. В другом варианте осуществления неполярный растворитель выбирают из толуола или триметилбензола.
Реакцию стадии (а) осуществляют при температуре в интервале, например, от 50 до 110°С, подходяще в интервале от 60 до 80°С, более подходяще в интервале от 65 до 75°С.
Соединение формулы IV подходяще может быть выбрано из метил 3-оксобутаноата, этил 3-оксобутаноата, пропил 3-оксобутаноата, бутил 3-оксобутаноата, втор-бутил 3-оксобутаноата и трет-бутил 3-оксобутаноата. Альтернативно соединение формулы IV может быть выбрано из метил 3-оксобутаноата, этил 3-оксобутаноата и трет-бутил 3-оксобутаноата. Подходяще можно использовать трет-бутил 3-оксобутаноат, так как для него требуется применение более мягких условий реакции в способе, что обладает преимуществом, поскольку реакции легче осуществить и при этом получают меньшее количество побочных продуктов.
Условия реакции для способа (б)
Реакцию способа (б) подходяще осуществляют в подходящем растворителе, таком как вода, или смешивающемся с водой растворителе, таком как тетрагидрофуран или ацетонитрил в присутствии подходящего основания, такого как гидроксид натрия, гидроксид калия или гидроксид лития.
Реакцию стадии (б) осуществляют при температуре в интервале, например, от 30 до 70°С, подходяще в интервале от 40 до 60°С, более подходяще в интервале от 45 до 55°С.
Примеры солей арил-алкил аммония включают Тритон В (гидроксид триметил бензил аммония), который является коммерчески доступным. Примеры солей тетра-алкил аммония включают хлорид тетра-бутил аммония и бромид тетра-бутил аммония.
Условия реакции для способа (в)
Реакцию способа (в) подходяще осуществляют в подходящем растворителе, таком как дихлорметан в присутствии кислоты, такой как трифторуксусной кислоты, или толуол в присутствии кислоты, такой как пара-толуол сульфоновой кислоты, уксусной кислоты, пропионовой кислоты или смесь уксусной кислоты и серной кислоты.
Если используют трифторуксусную кислоту в дихлорметане, то реакцию стадии (в) осуществляют при температуре в интервале, например, от 0 до 40°С, подходяще в интервале от 10 до 35°С, более подходяще в интервале от 20 до 30°С. Если используют толуол в присутствии пара-толуол сульфоновой кислоты, уксусной кислоты, пропионовой кислоты или смеси уксусной кислоты и серной кислоты, то реакцию стадии (в) осуществляют при температуре в интервале между 80°С и точкой кипения смеси растворитель/кислота. В одном варианте осуществления температура составляет 90°С.
Условия реакции для способа (г)
Квалифицированному специалисту в данной области техники хорошо известны различные способы, подходящие для восстановления соединения формулы VII.
Например, дитионит натрия или газообразный водород в присутствии подходящего катализатора, такого как палладий на угле. Если используют дитионит натрия, то реакцию способа (г) подходяще осуществляют в подходящем растворителе, таком как вода, или смешивающемся с водой растворителе, таком как тетрагидрофуран, ацетонитрил или спирт, например, метанол, этанол или изопропанол, благоприятно в присутствии подходящего основания, такого как карбонат калия или карбонат натрия. Более подробные сведения можно найти в Comprehensive Organic Transformations" под ред. Richard C.Larock, опубликованной John Wiley и Sons, 2-е изд., которая включена в настоящую заявку в качестве ссылки.
Реакцию стадии (г) осуществляют при температуре в интервале, например, от 0 до 50°С, подходяще в интервале от 10 до 40°С, более подходяще в интервале от 20 до 30°С.
Способ первого аспекта настоящего изобретения является благоприятным, так как он обеспечивает возможность получения соединения формулы II высокого качества и с высоким выходом в промышленном масштабе.
Стадии (а)-(в) необязательно можно осуществлять в виде непрерывного процесса без выделения и/или очистки промежуточных соединений формул V и VI. Это существенно уменьшает время и стоимость приготовления соединения формулы II в промышленном масштабе.
В одном аспекте способ приготовления соединения формулы II дополнительно может включать стадию (д) выделения и/или очистки соединения формулы II. Стадия (д) может включать любые подходящие стадии или методики для выделения желательного продукта, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники. Предпочтительные стадии, которые будут использоваться, будут обеспечивать продукт высокого качества и высокой чистоты.
Стадия (д), например, также может включать кристаллизацию, используя подходящую систему растворителей. Примером подходящей системы растворителей является система растворителей, содержащих растворенный продукт в дихлорметане и кристаллизацию путем добавления изогексана или изогептана, что обеспечивает получение соединение формулы II с высокой чистотой, обычно с чистотой больше 90%, подходяще больше 98%.
В другом аспекте изобретения обеспечивается соединение формулы VI, или его соль, или его защищенное производное. Примерами защищенных производных являются соединения, в которых гидрокси группа заменена C1-6алкокси или арилокси.
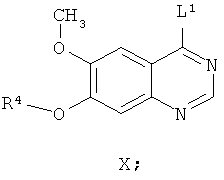
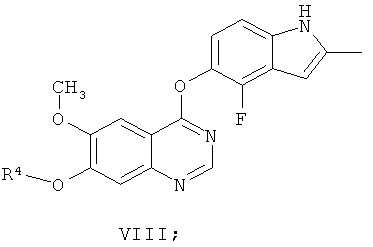
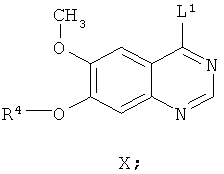
Другим ключевым промежуточным соединением, которое может использоваться для получения AZD2171, является соединение формулы VIII
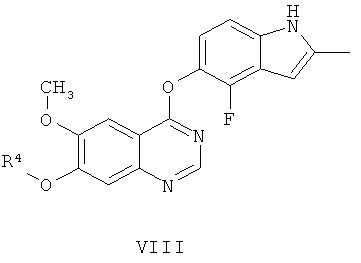
где R4 представляет собой защитную группу.
В примере 7 заявки WO 03/064413 описан путь приготовления соединения формулы VIII, где R4 представляет собой бензил. В этот путь задействована реакция свободного основания 7-бензилокси-4-хлор-6-метоксихиназолина с 4-фтор-5-гидрокси-2-метилиндолом и карбонатом калия в N-метил пирролидиноне в качестве растворителя, получая соединение формулы VIII. В примере 7 заявки WO 03/064413 указано, что 7-бензилокси-4-хлор-6-метоксихиназолин получают из 7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-она путем взаимодействия с тионилхлоридом в диметилформамиде в качестве растворителя.
Пути, описанные в документах предшествующего уровня техники, для получения соединения формулы VIII достаточны для синтеза относительно небольших количеств соединения. Однако для всех их необходимо выделение и/или очистка промежуточных соединений. Это приводит к удовлетворительному, но не высокому, суммарному выходу соединения формулы VIII.
Следовательно, существует потребность в более эффективном синтезе соединения формулы VIII, подходящем для получения больших количеств этого соединения. Предпочтительно, в новом синтезе не должны применяться дорогие и трудоемкие методики выделения и/или очистки. Следовательно, в новом синтезе должно быть уменьшено количество необходимых методик выделения и/или очистки, уменьшая таким образом стоимость и время приготовления. Новый способ также подходяще должен обеспечивать эффективное выделение соединения формулы VIII в кристаллической форме с высокой чистотой и выходом, где кристаллическая форма будет иметь хорошие фильтрационные характеристики.
В соответствии со вторым аспектом настоящего изобретения обеспечивается способ приготовления соединения формулы VIII
где R4 представляет собой защитную группу
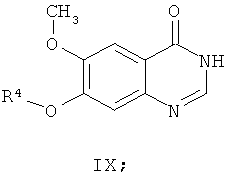
из соединения формулы IX
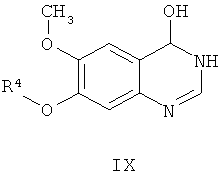
где способ включает стадии:
(е) взаимодействие соединения формулы IX со средством, используемым для получения производных, получая соединение формулы Х,
где L1 представляет собой уходящую группу; и
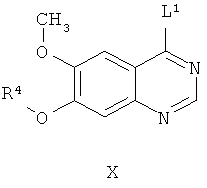
(ж) взаимодействие соединения формулы Х с соединением формулы II (2-метил-4-фтор-5-гидрокси-индол) необязательно in situ и необязательно в присутствии растворителя, используемого на стадии (е), получая соединение формулы VIII.
Термин 'защитная группа' относится к группам, которые легко удаляются в мягких кислотных условиях, нейтральных условиях или мягких щелочных условиях. Подходящими методами защиты являются методы, известные специалисту в данной области техники. Могут применяться общепринятые защитные группы в соответствии со стандартной практикой (для иллюстрации см. T.W.Green, Protective Groups in Organic Synthesis, John Wiley и Sons, 1991). Подходящими защитными группами на R4 являются бензил, замещенный бензил (например, С1-4алкоксибензил, ди-алкоксибензил, алкилбензил и ди-С1-4адлкибензил), трет-бутил, 1,1-диметил-1-этилметил, аллил, замещенный аллил (такой как С1-4алкилаллил) или метоксиэтоксиметил. В другом варианте осуществления R4 представляет собой бензил.
Для избежания неопределенности термин 'in situ' обозначает, что реакцию осуществляют без выделения продуктов с предыдущей стадии способа.
Способ второго аспекта изобретения является благоприятным, так как он обеспечивает возможность получения соединения формулы VIII с высокой чистотой и высоким выходом в промышленном масштабе.
Средство, используемое для получения производных, может включать любое подходящее средство для введения уходящей группы в 4-е положение соединения формулы IX. Примеры для L1 включают хлор, бром, йод и необязательно замещенный алкилсульфонил, такой как трифлил и тозил. Примеры средств, используемых для получения производных, включают хлорирующий реагент (такой как оксихлорид фосфора), бромирующий реагент (такой как оксибромид фосфора или смесь N-бромсукцинимида и три-изопропил фосфита) и йодирующий реагент.
Если средство, используемое для получения производных, представляет собой хлорирующий, бромирующий или йодирующий реагент, то стадия способа (е) может включать:
(е) взаимодействие соединения формулы IX с подходящим средством, используемым для получения производных в присутствии подходящего основания и подходящего растворителя, где реакцию осуществляют путем:
(е-1) добавления смеси соединения формулы IX и основания в растворителе к смеси средства, используемого для получения производных, в растворителе при температуре в интервале от 60 до 110°С в течение приблизительно 60 минут;
или
(е-2) добавления средства, используемого для получения производных, к смеси соединения формулы IX и основания в растворителе при температуре окружающей среды в течение приблизительно 15 минут и после этого нагревания реакционной смеси в течение приблизительно 90 минут до температуры в интервале от 70 до 90°С и перемешивания реакционной смеси при этой температуре приблизительно в течение 1 часа; или
(е-3) добавления средства, используемого для получения производных, к смеси соединения формулы IX и основания в растворителе при температуре в интервале от 60 до 110°С в течение приблизительно 15 минут.
Подходящий растворитель на стадии (е) выбирают из толуола, хлорбензола, 1,2-диметоксиэтана, ацетонитрила и анизола. В одном варианте осуществления растворитель представляет собой анизол или толуол. В другом варианте осуществления растворитель представляет собой анизол.
Подходящий растворитель на стадии (ж) выбирают из толуола, хлорбензола, 1,2-диметоксиэтана и анизола. В одном варианте осуществления растворитель представляет собой анизол или толуол. В другом варианте осуществления растворитель представляет собой анизол.
К растворителям может быть необходимо добавлять сорастворитель или сорастворители на стадии (е) и (ж), например, для растворения промежуточных соединений хлорбензилина или индола. Например, анизол необязательно может содержать ацетонитрил и N-метил пиролидинон и 1,2-диметоксиэтан может содержать N-метил пиролидинон.
В одном аспекте изобретения стадии (е) и (ж) осуществляют в толуоле в качестве растворителя.
В другом аспекте изобретения стадии (е) и (ж) осуществляют в анизоле в качестве растворителя.
Продукт со стадии (е) нет необходимости выделять перед осуществлением стадии (ж). Это позволяет осуществить способ в виде непрерывного способа без выделения и/или очистки промежуточного соединения формулы X. Это существенно уменьшает время и стоимость приготовления соединения формулы VIII в промышленном масштабе. Применение анизола в качестве растворителя реакции является благоприятным, поскольку этот растворитель сводит к минимуму образование побочных продуктов. Выбор растворителя также позволяет осуществить легкое и удобное выделение соединения формулы VIII. Например, если реакционную смесь охлаждают до температуры окружающей среды, то соединение формулы VIII обычно образует твердое вещество, где твердое вещество затем может быть собрано с помощью любого общепринятого метода.
Режим добавления реагентов на стадии (е) (то есть как описано на стадиях (е-1), (е-2) и (е-3)) является благоприятным, так как это сводит к минимуму образование побочных продуктов/примесей на этой стадии. Уменьшение образования побочных продуктов/примесей предоставляет возможность использования промежуточного соединения формулы X, полученного на стадии (е), на стадии (ж) без выделения и/или очистки. Уменьшение образования побочных продуктов/примесей на стадии (е) также предоставляет возможность правильной стереохимии реагентов на стадии (ж) способа и, следовательно, более эффективного осуществления реакции на этой стадии. В свою очередь, это обеспечивает высокий выход и высокую чистоту соединения формулы IX на стадии (ж).
Подходящий хлорирующий реагент для применения на стадии (е) представляет собой оксихлорид фосфора. Обычно, на стадии (е), применяют молярный избыток хлорирующего реагента по отношению к соединению формулы IX. Например, можно использовать молярный избыток в интервале от 1 до 2,0, подходяще в интервале от 1,2 до 1,4.
Подходящим основанием для применения на стадии (е) является основание, выбранное из триэтиламина и N,N-диизопропилэтиламина. В частности, основание представляет собой диизопроэтиламин. Добавление к реакционной смеси источника хлорида (такого как, например, гидрохлорид триэтиламина) может уменьшать образование побочных продуктов.
На стадии (е-1) реакцию осуществляют при температуре в интервале от 60 до 80°С, подходяще в интервале от 65 до 75°С, более подходяще в интервале от 70 до 75°С.
На стадии (е-2) добавление реагентов осуществляют при температуре окружающей среды. Под термином "температура окружающей среды" мы подразумеваем температуру в интервале от -10 до 30°С, в особенности температуру в интервале от 10 до 20°С, более предпочтительно температуру около 15°С. После этого реакционную смесь нагревали до температуры в интервале от 70 до 90°С, подходяще в интервале от 75 до 85°С, более подходяще в интервале от 80 до 85°С.
На стадии (е-3) реакцию осуществляют при температуре в интервале от 70 до 90°С, подходяще в интервале от 75 до 85°С, более подходяще в интервале от 80 до 85°С.
На стадии (е) термин "приблизительно" используется в выражениях "приблизительно 60 минут", "приблизительно 15 минут", "приблизительно 90 минут и "приблизительно 1 час" для указания того, что указанные промежутки времени не должны истолковываться как абсолютные значения, поскольку, что следует принять во внимание специалисту в данной области техники, периоды времени могут незначительно отличаться. Например, указанные периоды времени могут отличаться на ±50%, предпочтительно на ±15%, предпочтительно на ±10% от значений, указанных на стадии (е).
В одном аспекте изобретения, после осуществления стадии (е) способа, соединение формулы Х выделяют и/или очищают, например, перед хранением, транспортировкой и/или дальнейшей реакцией. Таким образом, в одном аспекте изобретения способ получения соединения формулы Х дополнительно включает стадию выделения соединения формулы X. Стадия может включать любые подходящие стадии или методики для выделения желательного продукта, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники. Предпочтительные стадии, которые будут использоваться, будут обеспечивать продукт высокого качества и высокой чистоты.
Реакцию стадии (ж) осуществляют при температуре в интервале от 60 до 85°С, подходяще в интервале от 65 до 80°С, более подходяще в интервале от 70 до 75°С.
В одном аспекте изобретения, после осуществления стадии (ж) способа, соединение формулы VIII выделяют и/или очищают, например, перед хранением, транспортировкой и/или дальнейшей реакцией. Таким образом, в одном аспекте изобретения способ получения соединения формулы VIII дополнительно включает стадию (з) выделения соединения формулы VIII. Стадия (з) может включать любые подходящие стадии или методики для выделения желательного продукта, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники. Предпочтительные стадии, которые будут использоваться, будут обеспечивать продукт высокого качества и высокой чистоты. Реакционная смесь может быть охлаждена до температуры окружающей среды, при этой температуре соединение формулы VIII обычно образует твердое вещество, и твердое вещество, образованное таким образом, может быть собрано с помощью любого подходящего метода, например, путем фильтрации.
Оба соединения, и соединение формулы IX, и исходное вещество - производное нитробензола формулы III являются коммерчески доступными или могут быть получены с помощью общепринятых методов. Например, соединение формулы IX может быть получено, как описано в примере 5 получение исходных веществ.
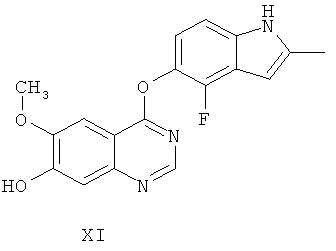
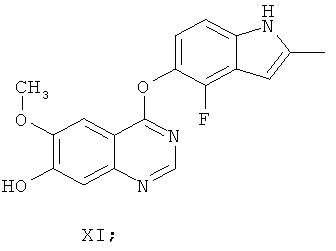
Другим ключевым промежуточным соединением, которое может применяться для получения AZD2171, является 7-гидрокси-4-(4-фтор-2-метилиндол-5-илокси)-6-метоксихиназолин, соединение формулы XI
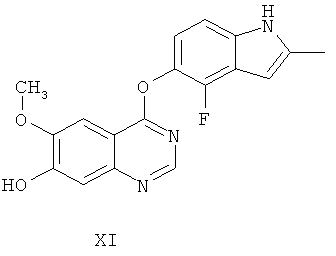
В примере 7 заявки WO 03/064413 описан путь приготовления соединения формулы XI. В этот путь задействована реакция 7-бензилокси-4-(2-метил-4-фториндол-5-илокси)-6-метоксихиназолина (соединение формулы VIII) с формиатом аммония в диметилформамиде, содержащем 10% палладий на угле, получая соединение формулы XI.
Этот путь, описанный в документах предшествующего уровня техники, для получения соединения формулы XI является достаточным для синтеза относительно небольших количеств соединения. Однако для него необходимо выделение и/или очистка промежуточных соединений. Это приводит к удовлетворительному, но не высокому, суммарному выходу соединения формулы XI.
Следовательно, существует потребность в более эффективном синтезе соединения формулы XI, подходящем для получения бóльших количеств этого соединения. Предпочтительно, в новом синтезе не должны применяться дорогие и трудоемкие методики очистки. Следовательно, в новом синтезе должно быть уменьшено количество необходимых методик выделения и/или очистки, уменьшая таким образом стоимость и время приготовления. Предпочтительно, в новом синтезе должно быть сведено к минимуму количество растворителей, используемых при осуществлении способа, что улучшает экологические показатели и предоставляет возможность для восстановления растворителя. Новый синтез также должен предоставить возможность эффективной кристаллизации соединения формулы XI в кристаллической форме с хорошими фильтрационными характеристиками и с высокой чистотой и выходом.
В соответствии с третьим аспектом настоящего изобретения обеспечивается способ приготовления 7-гидрокси-4-(4-фтор-2-метилиндол-5-илокси)-6-метоксихиназолина, соединение формулы XI
из соединения формулы IX
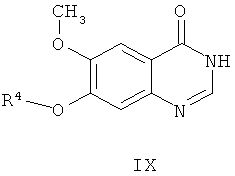
где R4 представляет собой защитную группу,
где способ включает стадии:
(е) взаимодействие соединения формулы IX со средством, используемым для получения производных, получая соединение формулы Х,
где L1 представляет собой уходящую группу
(ж) взаимодействие соединения формулы Х с соединением формулы II (2-метил-4-фтор-5-гидрокси-индол), необязательно in situ, необязательно в присутствии растворителя, используемого на стадии (е), получая соединение формулы VIII
(и) удаление R4 из соединения формулы VIII, получая соединение формулы XI или его соль,
и затем соединение формулы XI, полученное в форме свободного основания, может быть превращено в солевую форму и соединение формулы XI, полученное в форме соли, может быть превращено в форму свободного основания или в форму альтернативной соли, при необходимости.
Если средство, используемое для получения производных, представляет собой хлорирующий, бромирующий или йодирующий реагент, то стадия способа (е) может включать:
(е) взаимодействие соединения формулы IX с подходящим средством, используемым для получения производных в присутствии подходящего основания и подходящего растворителя, где реакцию осуществляют путем:
(е-1) добавления смеси соединения формулы IX и основания в растворителе к смеси средства, используемого для получения производных, в растворителе при температуре в интервале от 60 до 110°С в течение приблизительно 60 минут; или
(е-2) добавления средства, используемого для получения производных, к смеси соединения формулы IX и основания в растворителе при температуре окружающей среды в течение приблизительно 15 минут и после этого нагревания реакционной смеси в течение приблизительно 90 минут до температуры в интервале от 70 до 90°С и перемешивания реакционной смеси при этой температуре приблизительно в течение 1 часа; или
(е-3) добавления средства, используемого для получения производных, к смеси соединения формулы IX и основания в растворителе при температуре в интервале от 60 до 110°С в течение приблизительно 15 минут.
Способ согласно третьему аспекту изобретения является благоприятным, так как он обеспечивает возможность получения соединения формулы XI с высокой чистотой и высоким выходом в промышленном масштабе.
Условия реакции для способа (и)
Реакцию стадии (и) осуществляют при температуре в интервале от 20 до 60°С, более подходяще в интервале от 35 до 45°С.
В одном аспекте изобретения восстановление соединения формулы VIII осуществляют путем каталитического гидрирования, например, используя газообразный водород и подходящий катализатор, такой как палладий на угле.
В другом аспекте изобретения восстановление соединения формулы VIII осуществляют путем каталитической трансферной гидрогенизации, используя например, донор негазообразного водорода, такой как циклогексен или формиат аммония и подходящий катализатор, такой как палладий на угле.
Подходящие растворители для стадии (и) включают N-метилпирролидинон (NMP), диметилформамид или диметилацетамид.
В одном аспекте изобретения, после осуществления стадии (и) способа, соединение формулы XI выделяют и/или очищают. Можно использовать любые подходящие стадии или методики для выделения и/или очистки желательного продукта, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники. Предпочтительные стадии, которые будут использоваться, будут обеспечивать продукт высокого качества и высокой чистоты. Например, соединение формулы XI может быть выделено из NMP путем добавления антирастворителя, такого как вода, метанол, этанол, изопропанол, бутанол или ацетонитрил.
В другом варианте осуществления изобретения, после осуществления способа (и), соединение формулы XI используют in situ на следующей стадии в способе.
В соответствии с дальнейшим вариантом осуществления третьего аспекта изобретения обеспечивается способ приготовления соединения формулы XI из соединения формулы X, который включает стадии способа (ж) и (и) выше.
В соответствии с дальнейшим вариантом осуществления третьего аспекта изобретения обеспечивается способ приготовления 7-гидрокси-4-(4-фтор-2-метилиндол-5-илокси)-6-метоксихиназолина, соединение формулы XI
из соединения формулы IX
где R4 представляет собой защитную группу,
где способ включает стадии:
(е) взаимодействие соединения формулы IX со средством, используемым для получения производных, получая соединение формулы Х,
где L1 представляет собой уходящую группу,
(ж-1) взаимодействие соединения формулы Х с соединением формулы VII
необязательно in situ, необязательно в присутствии растворителя, используемого на стадии (е), получая соединение формулы XIV
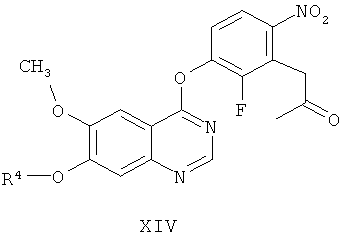
(и-1) восстановление соединения формулы XIV, получая соединение формулы XI
Условия реакции для способа (е)
Условия реакции для стадии (е) описаны выше.
Средство, используемое для получения производных, может содержать любое подходящее средство для введения уходящей группы в 4-е положение соединения формулы IX. Примерами для L1 являются хлор, бром, йод и необязательно замещенный алкилсульфонил, такой как трифлил и тозил. Примеры средств, используемых для получения производных, включают хлорирующий реагент (такой как оксихлорид фосфора), бромирующий реагент (такой как оксибромид фосфора или смесь N-бромсукцинимида и три-изопропил фосфита) и йодирующий реагент.
R4 представляет собой защитную группу, как определено выше.
Условия реакции для способа (ж-1)
Реакцию стадии (ж-1) осуществляют путем добавления раствора соли соединения формулы VII к раствору соединения формулы Х в растворителе, используемом на стадии (е-1). Соль соединения формулы VII может быть получена путем применения гидроксида лития, гидроксида калия, гидроксида натрия, карбоната лития, карбоната калия, карбоната натрия или карбоната цезия. Оптимально, используют гидроксид натрия. Соль образовывается при температуре от -20°С до +20°С, более подходяще в интервале от -10°С до 0°С. Раствор соли соединения формулы VII добавляли к раствору соединения формулы Х в растворителе, используемом на стадии (е-1) при температуре от 60°С до 100°С, подходяще в интервале 70-90°С.
Условия реакции для способа (и-1)
Реакцию стадии (и-1) осуществляют при температуре в интервале от 20 до 60°С, более подходяще в интервале от 35 до 45°С.
В одном аспекте изобретения восстановление соединения формулы XIV осуществляют путем каталитического гидрирования, например, используя газообразный водород и подходящий катализатор, такой как палладий на угле.
В другом аспекте изобретения восстановление соединения формулы XIV осуществляют путем каталитической трансферной гидрогенизации, используя например, донор негазообразного водорода, такой как циклогексен или формиат аммония, и подходящий катализатор, такой как палладий на угле.
Подходящие растворители для стадии (и) включают N-метилпирролидинон (NMP), диметилформамид или диметилацетамид.
В одном аспекте изобретения, после осуществления стадии (и-1) способа, соединение формулы XI выделяют и/или очищают. Можно использовать любые подходящие стадии или методики для выделения и/или очистки желательного продукта, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники. Предпочтительные стадии, которые будут использоваться, будут обеспечивать продукт высокого качества и высокой чистоты. Например, соединение формулы XI может быть выделено из NMP путем добавления антирастворителя, такого как вода, метанол, этанол, изопропанол, бутанол или ацетонитрил.
В другом варианте осуществления изобретения, после осуществления стадии (и-1), соединение формулы XI используют in situ на следующей стадии в способе.
В соответствии с дальнейшим вариантом осуществления третьего аспекта изобретения обеспечивается способ приготовления соединения формулы XI из соединения формулы X, который включает стадии способа (ж-1) и (и-1) выше.
В дальнейшем варианте осуществления изобретения обеспечивается соединение формулы XIV.
Дальнейший вариант осуществления третьего аспекта изобретения применим для приготовления различных кольцевых систем, замещенных 4-фтор-2-метилиндол-5-илокси. Таким образом, в соответствии с этим дальнейшим вариантом осуществления обеспечивается способ приготовления соединения формулы XI-1
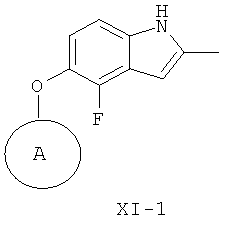
где А представляет собой подходящую кольцевую систему
из соединения формулы Х-1, где L1 представляет собой уходящую группу,
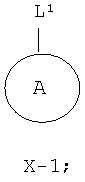
который включает:
(ж-2) взаимодействие соединения формулы Х-1 с соединением формулы VII
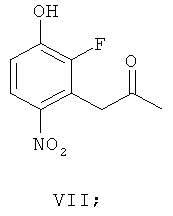
получая соединение формулы XIV-1
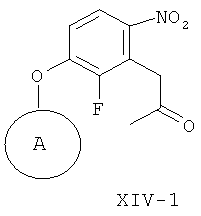
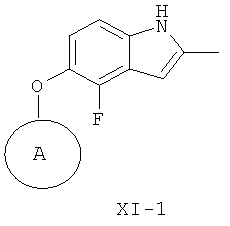
(и-2) восстановление соединения формулы XIV-1, получая соединение формулы XI-1
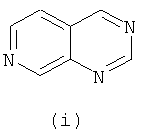
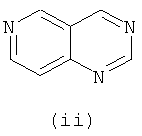
Подходящие кольцевые системы для кольца А представляют собой кольца, способные активироваться для предоставления возможности вытеснения активирующей группы ионом фенолата, то есть структурой формулы VII. Такие кольцевые системы включают:
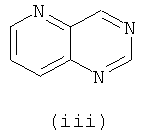
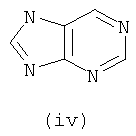
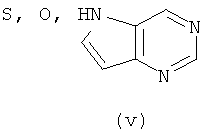
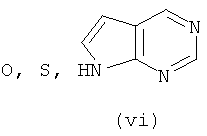
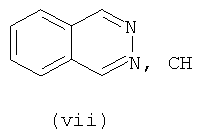
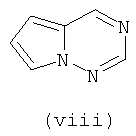
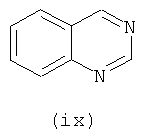
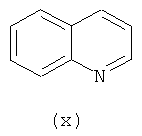
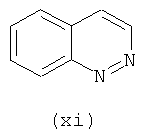
В одном варианте осуществления кольцо А выбирают из хиназолина, хинолина, циннолина и пирролотриазина. В другом варианте осуществления кольцо А представляет собой хиназолин. В другом варианте осуществления кольцо А представляет собой пирролотриазин. В другом варианте осуществления кольцо А представляет собой хиназолин и L1 находится в 4-м положении хиназолинового кольца. В другом варианте осуществления кольцо А представляет собой пирролотриазин и L1 находится в 4-м положении пирролотриазинового кольца.
Примерами для L1 являются хлор, бром, йод и необязательно замещенный алкилсульфонилокси или арилсульфонилокси, такой как трифлилокси и n-тозилокси.
Условия реакции для способа (ж-2)
Условия реакции для способа (ж-2) аналогичны условиям, описанным для способа (ж-1).
Условия реакции для способа (и-2)
Условия реакции для способа (и-2) аналогичны условиям, описанным для способа (и-1).
Для специалиста в данной области техники будет понятным, что кольцевая система А может быть замещена одной или несколькими группами. Такие группы могут не поддаваться влиянию способов в этом варианте осуществления изобретения или могут нуждаться в защите при осуществлении способов в этом варианте осуществления. Для квалифицированного специалиста в данной области техники должны быть известны стратегии защиты такой группы, такие как применение защитных групп, применение щадящих условий реакции, которые еще облегчают осуществление способов в вариантах осуществления и/или применение альтернативных катализаторов. Можно использовать общепринятые защитные группы в соответствии со стандартной практикой (для иллюстрации см. T.W.Green, Protective Groups in Organic Synthesis, John Wiley и Sons, 1991), как описано выше.
В одном аспекте изобретения, после осуществления стадии (и-2) способа, соединение формулы XI-1 выделяют и/или очищают. Можно использовать любые подходящие стадии или методики для выделения и/или очистки желательного продукта, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники. Предпочтительные стадии, которые будут использоваться, будут обеспечивать продукт высокого качества и высокой чистоты.
В другом варианте осуществления изобретения, после осуществления стадии (и-2), соединение формулы XI-1 используют in situ на следующей стадии в способе.
Дальнейший вариант осуществления обеспечивает способ приготовления соединения формулы XI-1 из соединения формулы Х-1, который включает стадии способа (ж-2) и (и-2) выше.
В дальнейшем варианте осуществления изобретения обеспечивается соединение формулы XIV-1.
Второстепенные варианты для вышеописанного способа также охватываются объемом изобретения. Примером является вариант, в котором соединение формулы VI
где R3 представляет собой подходящую этерифицирующую группу, как определено выше, например, C1-6алкил или бензил,
используют вместо соединения формулы VII в способе (ж-2). Таким образом, в соответствии с этим дальнейшим вариантом осуществления обеспечивается способ приготовления соединения формулы XI-1
из соединения формулы Х-1, где L1 представляет собой уходящую группу,
который включает
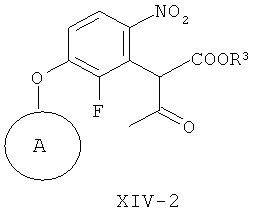
(ж-3) взаимодействие соединения формулы Х-1 с соединением формулы VI
получая соединение формулы XIV-2
(и-2) восстановление соединения формулы XIV-2, получая соединение формулы XI-2
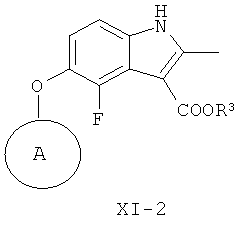
и (к-3) гидролиз соединения формулы XI-2, получая соединение формулы XI-1.
Примерами для L1 являются хлор, бром, йод и необязательно замещенный алкилсульфонил, такой как трифлил и тозил.
Условия реакции для способа (ж-3)
Условия реакции для способа (ж-3) аналогичны условиям, описанным для способа (ж-1).
Условия реакции для способа (и-3)
Условия реакции для способа (и-3) аналогичны условиям, описанным для способа (и-1).
Условия реакции для способа (к-3)
Реакцию способа (К-3) подходяще осуществляют в подходящем растворителе, таком как вода, или смешивающемся с водой растворителе, таком как тетрагидрофуран или ацетонитрил в присутствии подходящего основания, такого как гидроксид натрия, гидроксид калия или гидроксид лития.
Реакцию стадии (К-3) осуществляют при температуре в интервале, например, от 30 до 70°С, подходяще в интервале от 40 до 60°С, более подходяще в интервале от 45 до 55°С.
Примеры солей арил-алкил аммония включают Тритон В (гидроксид триметил бензил аммония), который является коммерчески доступным. Примеры солей тетра-алкил аммония включают хлорид тетра-бутил аммония и бромид тетра-бутил аммония.
В одном аспекте изобретения, после осуществления стадии (к-3) способа, соединение формулы XI-1 выделяют и/или очищают. Можно использовать любые подходящие стадии или методики для выделения и/или очистки желательного продукта, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники. Предпочтительные стадии, которые будут использоваться, будут обеспечивать продукт высокого качества и высокой чистоты.
В другом варианте осуществления изобретения, после осуществления стадии (к-3), соединение формулы XI-1 используют in situ на следующей стадии в способе.
Дальнейший вариант осуществления обеспечивает способ приготовления соединения формулы XI-1 из соединения формулы Х-1, который включает стадии способа (ж-3), (и-3) и (к-3) выше.
В дальнейшем варианте осуществления изобретения обеспечиваются соединения формулы XIV-2 и формулы XI-2.
В соответствии с четвертым аспектом настоящего изобретения обеспечивается способ приготовления 7-гидрокси-4-(4-фтор-2-метилиндол-5-илокси)-6-метоксихиназолина, соединение формулы XI
из соединения формулы IX
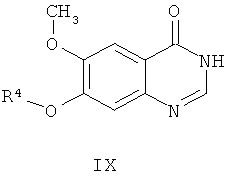
где R4 представляет собой защитную группу,
где способ включает стадии:
(е) взаимодействие соединения формулы IХ со средством, используемым для получения производных, получая соединение формулы Х,
где L1 представляет собой уходящую группу
(ж) взаимодействие соединения формулы Х с соединением формулы II (2-метил-4-фтор-5-гидрокси-индол) in situ в присутствии растворителя, используемого на стадии (е), получая соединение формулы VIII
(з) выделение соединения формулы VIII и
(к) удаление R4 из соединения формулы VIII, получая соединение формулы XI или его соль (например, его натриевую или калиевую соль),
и затем соединение формулы XI, полученное в форме свободной кислоты, может быть превращено в солевую форму и соединение формулы XI, полученное в форме соли, может быть превращено в свободную кислоту или в форму альтернативной соли, при необходимости.
Если средство, используемое для получения производных, представляет собой хлорирующий, бромирующий или йодирующий реагент, стадия способа (е) может включать:
(е) взаимодействие соединения формулы IX с подходящим средством, используемым для получения производных в присутствии подходящего основания и растворителя, выбранного из толуола и анизола, где реакцию осуществляют путем:
(е-1) добавления смеси соединения формулы IX и основания в растворителе к смеси средства, используемого для получения производных, в растворителе при температуре в интервале от 60 до 110°С в течение приблизительно 60 минут; или
(е-2) добавления средства, используемого для получения производных, к смеси соединения формулы IX и основания в растворителе при температуре окружающей среды в течение приблизительно 15 минут и после этого нагревания реакционной смеси в течение приблизительно 90 минут до температуры в интервале от 70 до 90°С и перемешивания реакционной смеси при этой температуре приблизительно в течение 1 часа; или
(е-3) добавления средства, используемого для получения производных, к смеси соединения формулы IX и основания в растворителе при температуре в интервале от 60 до 110°С в течение приблизительно 15 минут.
Способ согласно четвертому аспекту изобретения является благоприятным, так как он обеспечивает возможность получения соединения формулы XI с высокой чистотой и высоким выходом в промышленном масштабе.
В этом аспекте изобретения, после приготовления соединения формулы VIII на стадии (ж), соединение выделяют и, необязательно, очищают на стадии (з) способа. После этого выделенное соединение формулы VIII используют на стадии (к) для приготовления соединения формулы XI, либо непосредственно, либо после хранения в течение подходящего периода времени. Выделение соединения формулы VIII на стадии (з) является благоприятным, поскольку это предоставляет возможность широкого выбора способов удаления R4 группы из соединения формулы VIII на стадии (к), например, по сравнению с тем вариантом, когда эту стадию осуществляют in situ.
Условия реакции для способа (к)
Стадия (к) может включать любые подходящие стадии или методики для удаления R4, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники. Предпочтительные стадии, которые будут использоваться, будут обеспечивать продукт высокого качества и высокой чистоты. Например, на стадии (к), если R4 представляет собой бензильную группу, которая может быть удалена путем каталитического гидрирования, такого как газообразный водород, и подходящего катализатора, такого как палладий на угле. Применение каталитического гидрирования является благоприятным, поскольку оно обеспечивает высокоэффективный и щадящий метод удаления бензильной группы и поскольку он предоставляет возможность эффективного удаления побочных продуктов из потока отходов.
Реакцию стадии (к) можно осуществлять при любой температуре и в любом растворителе, подходящем для конкретного используемого метода удаления бензильной группы. Например, с N-метилпирролидиноном в качестве растворителя при температуре в интервале от 20 до 60°С.
В одном аспекте изобретения, после осуществления стадии (к) способа, соединение формулы XI выделяют и/или очищают. Можно использовать любые подходящие стадии или методики для выделения и/или очистки желательного продукта, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники. Предпочтительные стадии, которые будут использоваться, будут обеспечивать продукт высокого качества и высокой чистоты.
В соответствии с пятым аспектом изобретения обеспечивается способ приготовления AZD2171
из соединения формулы IX
где R4 представляет собой защитную группу.
где способ включает стадии превращения соединения формулы IX в соединение формулы XI
путем осуществления способа, как обсуждается выше для третьего или четвертого аспекта изобретения, и
(л) взаимодействие соединения формулы XI с соединением формулы XII или соединение формулы XIII
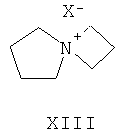
где L2 представляет собой уходящую группу и X- представляет собой подходящий противоион, такой как PF6 - (гексафторфосфат), хлорид, бромид или тетрафенилборат, в присутствии подходящего основания, получая соединение формулы I (AZD2171) или его соль.
В одном варианте осуществления соединение формулы XII обеспечивается в виде гидрохлоридной соли. В другом варианте осуществления соединение формулы XII обеспечивается в виде оксалатной соли.
В соответствии с дальнейшим вариантом осуществления изобретения обеспечивается оксалатная соль соединения формулы XII.
Примером для L2 являются хлор, бром, йод, мезилокси и тозилокси. В другом варианте осуществления примером для L2 являются хлор, бром и йод.
В одном варианте осуществления на стадии (л) соединение формулы XI подвергают реакции с соединением формулы XII. В другом варианте осуществления in step (л) соединение формулы XI подвергают реакции с соединением формулы XIII.
И после этого соединение формулы I, полученное в форме свободного основания, может быть превращено в солевую форму и соединение формулы I, полученное в форме соли, может быть превращено в форму свободного основания или в форму альтернативной соли, при необходимости.
Способ согласно пятому аспекту изобретения является благоприятным, так как он обеспечивает возможность получения соединения формулы I с высокой чистотой и высоким выходом в промышленном масштабе. Обычно способ согласно пятому аспекту настоящего изобретения осуществляют с выходом больше 80%. Способ согласно пятому аспекту изобретения также является благоприятным по меньшей мере с учетом аргументов, обсуждаемых выше для третьего и четвертого аспектов изобретения.
В одном варианте осуществления соединение формулы XI выделяют и/или очищают перед осуществлением стадии (л), например, используя любые подходящие стадии или методики, которые описаны в литературе и/или известны квалифицированному специалисту в данной области техники, как обсуждалось выше. В другом варианте осуществления соединение формулы XI подвергают реакции in-situ с соединением формулы XII или формулы XIII.
Условия реакции для способа (л)
Подходящее основание для применения на стадии (л) выбирают из карбоната натрия, карбоната калия, гидроксида натрия, гидроксида калия, трет-бутоксида калия и карбоната цезия.
Стадию (л) можно осуществлять в любом подходящем растворителе и при любой подходящей температуре.
Если основание, используемое на стадии (л), выбирают из карбоната натрия и карбоната калия, то подходящими растворителями являются, например, N-метилпирролидинон и N,N-диметилформамид. В этом аспекте стадию (л) обычно можно осуществлять при температуре в интервале от 60 до 105°С, подходяще в интервале от 80 до 100°С, подходяще в интервале от 75 до 85°С.
Способ согласно пятому аспекту изобретения является благоприятным, поскольку он предоставляет возможность получения AZD2171 с высокой чистотой и высоким выходом в промышленном масштабе. Обычно каждую из стадий способа согласно пятому аспекту настоящего изобретения осуществляют с выходом больше 80%.
Согласно шестому аспекту настоящего изобретения обеспечивается способ приготовления AZD2171 из соединения формулы IX
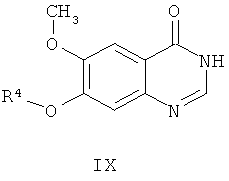
где R4 представляет собой защитную группу,
где способ включает стадии:
(е) взаимодействие соединения формулы IX со средством, используемым для получения производных, получая соединение формулы Х,
где L1 представляет собой уходящую группу,
(ж) взаимодействие соединения формулы Х с 2-метил-4-фтор-5-гидрокси-индолом, необязательно in situ, необязательно в присутствии растворителя, используемого на стадии (е), получая соединение формулы VIII
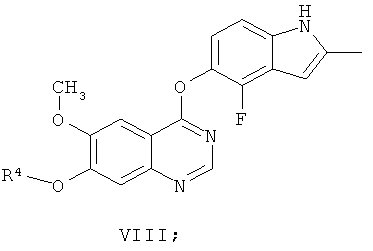
(и) удаление R4 из соединения формулы VIII, получая соединение формулы XI
(л) взаимодействие соединения формулы XI с соединением формулы XII или соединением формулы XIII
где L2 представляет собой уходящую группу и X- представляет собой подходящий противоион, такой как PF6 - (гексафторфосфат), хлорид или бромид,
в присутствии подходящего основания, получая соединение формулы I (AZD2171) или его соль;
затем AZD2171, полученное в форме свободного основания, при необходимости, может быть превращено в форму фармацевтически приемлемой соли.
Способ согласно шестому аспекту изобретения является благоприятным, поскольку он предоставляет возможность получения AZD2171 с высокой чистотой и высоким выходом в промышленном масштабе. Обычно каждую из стадий способа согласно седьмому аспекту настоящего изобретения осуществляют с выходом больше 80%.
Если средство, используемое для получения производных, представляет собой хлорирующий, бромирующий или йодирующий реагент, то стадия способа (е) может включать:
(е) взаимодействие соединения формулы IX с подходящим средством, используемым для получения производных в присутствии подходящего основания и растворителя, выбранного из толуола и анизола, где реакцию осуществляют путем:
(е-1) добавления смеси соединения формулы DC и основания в растворителе к смеси средства, используемого для получения производных, в растворителе при температуре в интервале от 60 до 110°С в течение приблизительно 60 минут; или
(е-2) добавления средства, используемого для получения производных, к смеси соединения формулы IX и основания в растворителе при температуре окружающей среды в течение приблизительно 15 минут и после этого нагревания реакционной смеси в течение приблизительно 90 минут до температуры в интервале от 70 до 90°С и перемешивания реакционной смеси при этой температуре приблизительно в течение 1 часа; или
(е-3) добавления средства, используемого для получения производных, к смеси соединения формулы IX и основания в растворителе при температуре в интервале от 60 до 110°С в течение приблизительно 15 минут.
Предпочтительные аспекты способа согласно шестому аспекту изобретения изложены выше для конкретных стадий, как описано во втором, третьем, четвертом и пятом аспектах настоящего изобретения. В частности, предпочтительные аспекты способа согласно шестому аспекту изобретения изложены выше, например, на конкретных стадиях третьей и пятой.
Подходяще, основание, используемое на стадии (л) способа шестого аспекта настоящего изобретения, представляет собой карбонат калия, а подходящий растворитель представляет собой N-метилпирролидинон.
Далее изобретение иллюстрируется следующими примерами, которые не ограничивают его объем, в которых, если специально не указано, иначе:
(i) упаривания осуществляют на роторном испарителе в вакууме и процедуры обработки осуществляют после удаления остатков твердых веществ, таких как осушители, путем фильтрации;
(ii) выходы представлены только с целью иллюстрации и необязательно, что могут быть достигнуты максимальные значения;
(iii) структуры конечных продуктов подтверждали с помощью ядерного (обычно протонного) магнитного резонанса (ЯМР) и масс-спектральных методик; значения химических сдвигов протонного магнитного резонанса измеряли на дельта-шкале и пики мультиплетности представляли следующим образом: s, синглет; d, дублет; t, триплет; m, мультиплет; br, широкий; q, квартет; quin, квинтет; все образцы анализировали на Bruker DPX 400 МГц при 3000 К в указанном растворителе, 16 сканирований, время повторения импульсов 10 секунд;
(iv) промежуточные соединения обычно не были полностью охарактеризованы, и их чистоту оценивали с помощью ЯМР-анализа;
(v) химические символы имеют их обычное значение; используются единицы и символы СИ; и
(vi) использованы следующие сокращения:
Пример 1
Получение 4-фтор-2-метил-1H-индол-5-ола (небольшой масштаб)
Получение трет-бутил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата (небольшой масштаб)
трет-Бутил ацетоацетат (3,852 г) добавляли к перемешиваемой смеси трет-пентоксида натрия (2,804 г) в толуоле (26 мл) при 40°С. Смесь нагревали до 70°С и добавляли 1,2,3-трифтор-4-нитробензол (2,00 г). Смесь поддерживали при 70°С в течение 3 часов. Смесь охлаждали до 25°С и 20% мас./мас. серной кислоты добавляли для доведения значения pH смеси до 1. Добавляли бикарбонат натрия для доведения значения pH смеси до pH 5. Добавляли воду (5 мл) и насыщенный хлорид натрия (5 мл). Нижний водный слой отбрасывали и органический слой последовательно промывали водой (7 мл), водой (7 мл), 2,3% мас./мас. водным раствором бикарбоната натрия (2,75 мл) и затем водой (6 мл). Органический слой перегоняли при пониженном давлении (50 мбар), получая раствор трет-бутил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата в толуоле.
Небольшое количество перегоняли в вакууме при 100°С, 0,4 мбар, получая очищенный образец трет-бутил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата.
Масс-спектр [М-Н]- 314.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,27 (s, 9 Н кето) 1,30 (s, 9 Н енол) 1,86 (s, 3 Н енол) 2,45 (s, 3 Н кето) 5,66 (s, 1 Н кето) 7,60 (td, J-9,38, 7.97 Гц. 1 Н кето) 7,75 (td, J=9,35, 8,03 Гц, 1 Н енол) 7,91 (dt, J=9,19, 2,36 Гц, 1 Н кето) 8,05 (ddd, J=9,24, 4,71, 1,89 Гц, 1 Н енол) 13,12 (d, J=0,65 Гц, 1 Н енол).
Получение трет-бутил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноата (небольшой масштаб)
Раствор трет-бутил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата (3 г, содержащий 2,38 г трет-бутил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата) разводили толуолом (5 мл) и экстрагировали 40% мас./мас. водным раствором Тритон В (3,28 мл). Затем к водному экстракту дополнительно добавляли 40% мас./мас. водного Тритон В (5,96 мл) и гидроксида натрия (2,053 г). Раствор нагревали при 50°С в течение 18 часов. Раствор охлаждали до 25°С и добавляли воду (8,45 мл), затем 20% мас./мас. серной кислоты (14,3 мл) и после этого дихлорметан (4 мл). Органический слой отделяли и водный слой дополнительно экстрагировали дихлорметаном (4 мл). Объединенные органические слои перегоняли при пониженном давлении, получая трет-бутил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноат 1,59 г.
Масс-спектр [М-Н]- 312.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,29 (s, 9 Н енол) 1,38 (s, 9 Н кето) 1,79 (s, 3 Н енол) 2,35 (s, 3 Н кето) 5,40 (s, 1 Н кето) 6,90 (t, J=9,05 Гц, 1 Н кето) 6,97 (t, J=9,00 Гц, 1 Н енол) 7,77 (d, J=9,05 Гц, 1 Н кето) 7,89 (dd, 7=9,16, 1,40 Гц, 1 Н енол) 13,04 (br.s., 1 Н енол).
Получение 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-она (небольшой масштаб)
К раствору трет-бутил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноата (7,89 г) в дихлорметане добавляли трифторуксусную кислоту (11,2 г) и смесь перемешивали при температуре окружающей среды в течение 20 часов. Смесь концентрировали, получая густую пасту, и добавляли толуол (14 мл) и смесь перегоняли на роторном испарителе (77 мбар, баня 40°С). Дополнительно добавляли толуол (14 мл) и смесь перегоняли. Добавляли гидроксид натрия (7,4% мас./мас., 20 мл) и нижний водный слой отделяли и промывали толуолом (18 мл). Водный слой разводили водой (20 мл) и нагревали до 40°С. Добавляли уксусную кислоту (13 мл), затем серную кислоту (20% мас./мас., 20 мл). Смесь охлаждали до 0°С и дополнительно добавляли воду (43 мл). Смесь охлаждали до -5°С и оставляли на ночь. Твердое вещество фильтровали и промывали водой (24 мл). Твердое вещество высушивали в вакуумном шкафу, получая 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-он (1,65 г, 30,7%).
Масс-спектр [М-Н]- 214.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 2,27 (s, 3 Н) 4,18 (d, J=1,62 Гц, 2 Н) 7,06 (t, J=9,00 Гц, 1 Н) 7,92 (dd, J=9,16, 1,72 Гц, 1Н) 11,43 (br.s., 1H).
Получение 4-фтор-2-метил-1H-индол-5-ола (небольшой масштаб)
1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-он (1 г) растворяли в растворе карбоната калия (1,3 г) в воде (13 мл). По каплям добавляли раствор дитионита натрия (5,24 г) в воде (12,3 мл). Смесь перемешивали в течение 1,5 часа, затем оставляли отстаиваться в течение ночи. Твердое вещество фильтровали и промывали водой (6 мл). Твердое вещество высушивали при 35°С в вакуумном сушильном шкафу, получая неочищенный 4-фтор-2-метил-1H-индол-5-ол (0,5 г, 64,5%).
Неочищенное твердое вещество (250 мг) растворяли в дихлорметане (6,75 мл) и фильтровали через подложку диоксида кремния (250 мг). Фильтровальную подложку промывали дихлорметаном (3,4 мл). Объединенные фильтраты перегоняли, удаляя 6 мл дистиллята. Затем концентрат по каплям добавляли к изогексану (4,25 мл) и смесь концентрировали, удаляя 3 мл дистиллята. Смесь охлаждали на ледяной бане и осадок фильтровали и промывали изогексаном (0,9 мл). Твердое вещество высушивали в вакуумном шкафу при 35°С, получая 4-фтор-2-метил-1H-индол-5-ол (180 мг, 72%)
Масс-спектр [М+Н]+ 166.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 2,33 (s, 3 Н) 6,02-6,05 (m, 1 H) 6,64 (t, J=8,41 Гц, 1 Н) 6,87 (d, J=8,51 Гц, 1 Н) 8,68 (s, 1 H) 10,81 (br.s., 1 H).
Пример 2
Получение 4-фтор-2-метил-1H-индол-5-ола (крупный масштаб)
Получение трет-бутил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата (крупный масштаб)
К толуолу (810 л) загружали трет-пентоксид натрия (91 кг, 2,3 экв.). Смесь нагревали до 40°С и добавляли трет-бутил ацетоацетат (124,3 кг, 2,2 экв.). Смесь нагревали до 70°С и добавляли трифторнитробензол (63,1 кг, 1,0 экв.). Температуру поддерживали при 70°С в течение 3 часов. Смесь охлаждали до 25°С и добавляли 20% мас./мас. серную кислоту для доведения pH до 5. Нижний водный слой отбрасывали и органический слой два раза промывали водой (2×227 кг воды), 2,3% мас./мас. бикарбоната натрия (192 кг) и затем водой (192 кг). Органический слой перегоняли в вакууме (50 мбар), удаляя 574 кг дистиллята, получая раствор трет-бутил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата в толуоле.
Получение трет-бутил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноата (крупный масштаб)
Раствор трет-бутил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата со стадии выше экстрагировали 40% мас./мас. водным Тритон В (164 кг, 1,10 экв.). К водной фазе дополнительно добавляли 40%мас./мас. Тритон В (298 кг, 2,0 экв.), после этого гидроксид натрия (97 кг, 6,8 экв.). Раствор нагревали при 50°С в течение 18 часов. Смесь охлаждали до 25°С и добавляли воду (392 кг), затем 20% мас./мас. серную кислоту (664 кг) и после этого дихлорметан (187 л). Дополнительно добавляли 20% мас./мас. серную кислоту до достижения значения рН смеси 5. Органический слой отделяли и водный слой дополнительно экстрагировали дихлорметаном (187 л). Объединенные органические слои перегоняли при пониженном давлении (500 мбар), удаляя 160 кг дистиллята, получая трет-бутил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноат в виде раствора в дихлорметане.
Получение 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-она (крупный масштаб)
К трет-бутил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноату в дихлорметане со стадий выше дополнительно добавляли дихлорметан (123 л), после этого трифторуксусную кислоту (158 кг), в то время как температуру поддерживали при 25°С. Реакционную смесь перемешивали в течение 20 часов. Смесь перегоняли, удаляя 283 кг дистиллята. Добавляли толуол (176 л) и смесь перегоняли, удаляя 208 кг дистиллята. Дополнительно добавляли толуол (176 л) и смесь перегоняли, удаляя 133 кг дистиллята. К остатку добавляли 7,4% мас./мас. водный гидроксид натрия (-705 кг) до достижения значения pH смеси 10,5. Водный слой отделяли и промывали толуолом (250 л). Водный слой разводили водой (250 л), нагревали до 40°С и добавляли уксусную кислоту (191,4 кг), понижая значение pH от 10,1 до 3,8. Затем значение pH доводили до 1 с помощью 20% мас./мас. серной кислоты (~315 кг). Смесь охлаждали до 0°С и затравливали с помощью 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-она. Дополнительно добавляли воду (600 кг) и твердое вещество выделяли путем фильтрации. Осадок на фильтре промывали водой (300 л). Продукт высушивали в вакууме (50 мбар) при 40°С. Выход: 56,0 кг, 74% на основе 1,2,3-трифтор-4-нитробензола.
Получение 4-фтор-2-метил-1H-индол-5-ола (крупный масштаб)
К раствору карбоната калия (79 кг) в воде (800 кг) добавляли 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-он (61 кг) и смесь перемешивали, получая раствор. К этому раствору при 25°С добавляли раствор дитионита натрия (298 кг) в воде (750 кг). Смесь поддерживали при 25°С в течение 2 часов. Продукт выделяли путем фильтрации, промывая осадок на фильтре водой (366 кг). Продукт высушивали при пониженном давлении (50 мбар) при 35°С. Выход: 34 кг, 72%.
Неочищенный 4-фтор-2-метил-1Н-индол-5-ол (33 кг) растворяли в дихлорметане (880 л) и фильтровали через диоксид кремния (33 кг). Фильтр промывали дихлорметаном (440 л). Объединенные фильтраты перегоняли, удаляя 835 л дистиллята. Этот концентрат быстро добавляли к изогексану (360 кг), получая в результате суспензию. Партию перегоняли, удаляя 436 л дистиллята. Партию охлаждали до 0°С, выдерживали в течение 1 часа и затем фильтровали. Осадок на фильтре промывали изогексаном (73 кг). Продукт высушивали при пониженном давлении (50 мбар) при 35°С. Выход: 31 кг, 68% на основе 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-она.
Пример 3
Получение 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-она из метил ацетоацетата
Получение метил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата
трет-Пентоксид натрия (13,41 г) добавляли к мезитилену (112 мл) и взвесь нагревали до 50°С, получая красно-коричневый раствор. К нему добавляли метил ацетоацетат (14,28 г). Смесь выделяла тепло до 75°С, образуя густую оранжевую/желтую взвесь. Температуру доводили до 70°С и добавляли 1,2,3-трифтор-4-нитробензол (10,0 г), получая оранжевый раствор. Смесь поддерживали приблизительно при 70°С в течение 3 часов, после этого позволяли охладиться до температуры окружающей среды и оставляли на ночь. К смеси добавляли соляную кислоту (6% мас./мас., 43 мл). Нижний водный слой отделяли и органический слой дополнительно два раза промывали соляной кислотой (6% мас./мас., 36 мл). Органический слой промывали водным раствором бикарбоната натрия (2,3% мас./мас., 15 мл) и затем водой (30 мл). Дополнительно загружали мезитилен (21 мл) и смесь упаривали на роторном испарителе (25 мбар, баня 40°С), получая 25 мл раствор.
Небольшое количество этого раствора (5 мл) концентрировали (68°С, 10 мбар), получая метил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноат (2,22 г) в виде желтого/коричневого масла.
Масс-спектр [М-Н]- 272.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,85 (s, 3 Н енол) 2,47 (s, 3 Н кето) 3,50 (s, 3 Н кето*) 3,63 (s, 3 Н енол*) 5,79 (s, 1 Н кето) 7,63 (td, J=9,38, 7,97 Гц, 1 Н кето) 7,78 (td, J=9,38, 7,97 Гц, 1 Н енол) 7,93 (ddd, J=9,21, 4,69, 1,94 Гц, 1 Н кето) 8,08 (ddd, J=9,27, 4,74, 1,94 Гц, 1 Н енол) 12,88 (s, 1 H енол).
* = взаимозаменяемо
Получение метил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноата
К раствору метил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата в мезитилене (11,01 г, 20 мл раствор) добавляли Тритон В (40% мас./мас., 50,54 г). Разделение было плохим, поэтому дополнительно добавляли мезитилен (8 мл). Нижний водный слой отделяли и к нему дополнительно добавляли Тритон В (40% мас./мас., 50,54 Г). Водную смесь поддерживали при 55°С в течение 16 часов. Смесь охлаждали до температуры окружающей среды и добавляли соляную кислоту (32% мас./мас., 19 мл) в течение 15 минут до достижения значения pH 5. Добавляли дихлорметан (24 мл) и нижний органический слой отделяли. Водный слой дополнительно экстрагировали дихлорметаном (24 мл) и объединенную органическую фазу подщелачивали путем добавления водного гидроксида натрия (8,5% мас./мас., 23 мл) до pH 10,5. Водную фазу дополнительно промывали изогексаном (23 мл), получая водный раствор метил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноата.
Небольшое количество упаривали насухо на роторном испарителе, получая образец для анализа.
Масс-спектр [М-Н]- 270.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,75 (s, 3 Н енол) 3,59 (s, 3 Н енол) 6,56 (t, J=9,27 Гц. 1 Н енол) 7,86 (dd, J=9,43, 1,24 Гц, 1 Н енол) 12,72 (br.s., 1 H енол).
Получение 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-она
Добавляли воду (17,7 мл) к уксусной кислоте (15,6 мл), после этого к серной кислоте (16,6 мл). К этому раствору добавляли водный раствор, содержащий метил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноат (8,195 г) и смесь нагревали при 90°С в течение 4 часов. Смесь охлаждали до 80°С и добавляли воду (24,6 мл). Смесь охлаждали до 40°С и добавляли затравку 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-он (2 мг). Смесь охлаждали до 0°С и оставляли перемешивать в течение ночи. Твердое вещество фильтровали, три раза промывали водой (15 мл) и высушивали в вакууме при 40°С, получая 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-он (320 мг).
Масс-спектр [М-Н]- 214.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,75 (s, 3 Н енол) 3,59 (s, 3 Н енол) 6,56 (t, J=9,27 Гц, 1 Н енол) 7,86 (dd, J=9,43, 1,24 Гц, 1 Н енол) 12,72 (br.s., 1 Н енол).
Пример 4
Получение 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-она из этил ацетоацетата
Получение этил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата
трет-пентоксид натрия (33,52 г) добавляли к мезитилену (280 мл) и смесь нагревали до 50°С. К суспензии добавляли этил ацетоацетат (39,61 г) в течение 10 минут. Реакционная смеса выделяла тепло приблизительно до 60°С. Густую взвесь нагревали до 70°С и добавляли 1,2,3-трифтор-4-нитробензол (25,0 г). Смесь выделяла тепло до 80°С. Смесь поддерживали при 70-80°С в течение 3 часов, после этого позволяли охладиться до температуры окружающей среды и оставляли на ночь. К смеси добавляли соляную кислоту (6% мас./мас., 100 мл) до достижения значения pH 1. Водный слой отделяли и отбрасывали и органический слой два раза промывали соляной кислотой 6% мас./мас., 90 мл), затем водным бикарбонатом натрия (2,3% мас./мас., 37,5 мл) и в завершение водой (75 мл). К органическому слою дополнительно добавляли мезитилен (53 мл) и смесь перегоняли на роторном испарителе (10 мбар, баня 60°С), получая раствор этил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноат в мезитилене (90 мл, содержащие 36,22 г этил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата).
Небольшой образец (10 мл) дополнительно упаривали на роторном испарителе (12 мбар, баня 60°С).
Масс-спектр [М-Н]- 284.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,03 (t, J=7,11 Гц, 3 Н кето*) 1,05 (t, J=7,11 Гц, 3 Н енол*) 1,86 (s, 3 Н енол) 2,47 (s, 3 Н кето) 3,89-4,22 (m, 2 Н кето + 2 Н енол) 5,76 (s, 1 Н кето) 7.62 (td, J=9,32, 7,97 Гц, 1 Н кето) 7,78 (td, J=9,35, 8,03 Гц, 1 Н енол) 7,93 (ddd, J=9,19, 4,71, 1,94 Гц, 1 Н кето) 8,08 (ddd, J=9,27, 4,69, 1,99 Гц, 1 Н енол) 13,00 (s, 1Н енол).
Получение этил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноата
Раствор этил 2-(2,3-дифтор-6-нитрофенил)-3-оксобутаноата (34,56 г) в мезитилене (общий объем 80 мл) экстрагировали Тритон В (40% мас./мас., 140,4 г). Водный слой дополнительно разводили Тритон В (40% мас./мас., 140,4 г), нагревали до 50°С и выдерживали в течение 16 часов. Смесь охлаждали до температуры окружающей среды и подкисляли до pH 5 путем добавления соляной кислоты (32% мас./мас., 74 мл). Смесь два раза экстрагировали дихлорметаном (69 мл) и объединенные органические слои экстрагировали водным гидроксидом натрия (8,5% мас./мас., 67 мл). Водный экстракт два раза промывали изогексаном (67,5 мл), получая водный раствор этил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноата (80 мл, содержащие 31,92 г этил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноата, обеспечивая 100% выход).
Небольшой образец упаривали насухо на роторном испарителе и анализировали.
Масс-спектр [М-Н]- 286.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 0,91 (t, J=7,06 Гц, 3 Н) 1,49 (s, 3 H) 3,58-3,89 (m, 2 H) 5,97 (t, J=9,32 Гц, 1 Н) 7,68 (d, J=10,34 Гц, 1 Н).
Получение 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-она
К смеси уксусной кислоты (57 мл) и воды (68 мл) добавляли серную кислоту (61 мл). Смесь нагревали до 97°С и добавляли водный раствор, содержащий этил 2-(2-фтор-3-гидрокси-6-нитрофенил)-3-оксобутаноат (31,6 г). Смесь нагревали при 97°С в течение 3,5 часов, затем охлаждали до 80°С и добавляли воду (95 мл). Смесь охлаждали до 40°С и затравливали с помощью 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-она (2 мг). Смесь охлаждали до 0°С и перемешивали в течение ночи. Смесь фильтровали и три раза промывали водой (58 мл). Продукт высушивали в вакуумном шкафу, получая 1-(2-фтор-3-гидрокси-6-нитрофенил)-пропан-2-он (8,42 г, 35,7%).
Масс-спектр [М-Н]- 212.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 2,27 (s, 3 Н) 4,18 (s, 2 H) 7,06 (t, J=8,94 Гц, 1 Н) 7,92 (dd, J=9,21, 1,67 Гц, 1 H) 11,44 (br.s., 1 H).
Пример 5
Получение 7-(бензилокси)-4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метоксихиназолина
7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-он (28 кг) и триэтиламин гидрохлорид (2,7 кг) добавляли к анизолу (244 кг). Добавляли N,N-диизопропилэтиламин (19,2 кг), затем линейно промывали анизолом (20 кг). Смесь охлаждали до 15°С и добавляли оксихлорид фосфора (19,8 кг) в течение 5 минут, затем линейно промывали анизолом (13,9 кг). После перемешивания в течение 15 минут смесь нагревали до 80°С в течение 90 минут и выдерживали при 80°С в течение 1 часа. Когда реакция завершалась, то партию переносили в новый реактор, затем линейно промывали анизолом (39,7 кг). Смесь охлаждали до 40°С и добавляли 16,19% мас./мас. водный раствор гидроксида натрия (132 кг) в течение 15 минут, позволяя температуре повыситься до 50°С, после этого линейно промывали водой (5 кг). Смесь поддерживали при 50°С в течение 30 минут, затем нагревали до 80°С. Партию фильтровали через фильтр Gaf, затем линейно промывали анизолом (10 кг). Нижний водный слой отделяли и верхний органический слой промывали при 80°С 20,9% мас./мас. водным раствором хлорида натрия (98,2 кг). Смесь перегоняли при пониженном давлении (80 мбар), получая остаточный объем 224 л. Добавляли 1-метил-2-пирролидинон (28,9) и смесь нагревали до 80°С, получая раствор 7-(бензилокси)-4-хлор-6-метоксихиназолина.
4-Фтор-2-метил-1H-индол-5-ол (17,2 кг) растворяли в ацетонитриле (85 кг). Раствор дегазировали путем выдерживания в вакууме (150 мбар) и затем повышали давление до 21,6 бар с помощью азота. Это осуществляли в течение четырех часов. Раствор охлаждали до 0°С и добавляли 16,19% мас./мас. водный раствор гидроксида натрия (25,7 кг) в течение 15 минут, поддерживая температуру при 0°С, затем линейно промывали ацетонитрилом (3 кг). Этот холодный раствор добавляли в течение 45 минут к раствору 7-(бензилокси)-4-хлор-6-метоксихиназолина при 80°С, затем линейно промывали ацетонитрилом (22 кг). Смесь поддерживали при 80°С в течение 4 часов, после этого два раза промывали водой (42 кг). Смесь охлаждали до 20°С и перегоняли при пониженном давлении (250 мбар) до достижения температуры партии 70°С. После этого вакуум доводили до 80 мбар и перегонку продолжали до остаточного объема 168 л. Партию охлаждали до 65°С и добавляли раствор воды (56 кг) в метаноле (133 кг), поддерживая температуру при 60-65°С. Смесь охлаждали до 20°С в течение 1 часа, затем выдерживали при 20°С в течение 2 часов. Продукт выделяли путем фильтрации, осадок промывали два раза метанолом (33 кг). Продукт высушивали с помощью азота при 40°С. Выход: 32,7 кг, 77%.
Масс-спектр [М+Н]+ 430.
1H ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 2,42 (s, 3 Н) 4,00 (s, 3 H) 5,36 (s, 2 H) 6,24 (s, 1 H) 6,99 (dd, J=8,57, 7,49 Гц, 1 Н) 7,16 (d, J=9,05 Гц, 1 Н) 7,35-7,41 (m, 1 H) 7,41-7,47 (m, 2 H) 7,51 (s, 1 H) 7,50-7,55 (m, 2 H) 7,63 (s, 1 H) 8,50 (s, 1 H) 11,33 (br.s., 1H).
Исходное вещество 7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-он получали следующим образом.
Смесь ванилиновой кислоты (200 г), ацетонитрила (600 мл) и N-этилдиизопропиламина (580 мл) нагревали в колбе с обратным холодильником. Затем добавляли бензил бромид (347 мл) в течение 3 часов. Реакционную смесь поддерживали в колбе с обратным холодильником в течение 15 часов. Добавляли триэтиламин (50 мл) и реакционную смесь поддерживали в колбе с обратным холодильником дополнительно в течение 30 минут. Добавляли ацетонитрил (400 мл) и реакционную смесь нагревали до 81°С. Добавляли воду (300 мл) и реакционную смесь охлаждали до 45°С. Реакционную смесь поддерживали при 45°С в течение 30 минут до осуществления кристаллизации. Затем реакционной смеси позволяли охладиться до 24°С и затем дополнительно охлаждали до 8°С и продукт (бензил 4-(бензилокси)-3-метоксибензоат) выделяли путем фильтрации. Твердое вещество промывали водой (3×500 мл) и после этого высушивали в вакууме при 45°С. Выход: 387 г, 93,4%.
Масс-спектр (М+H)+=349,2.
1Н ЯМР спектр (CDCl3) 3,9 (s, 3Н), 5,2 (s, 2Н), 5,3 (s, 2H), 6,9 (d, 1H), 7,2-7,4 (m, 10Н), 7,6-7,7 (m, 2H).
Бензил 4-(бензилокси)-3-метоксибензоат (78 г) смешивали с дихлорметаном (580 мл), водой (72 мл) и ледяной уксусной кислотой (288 мл). Смесь охлаждали до 10°С. Концентрированную серную кислоту (108 мл) добавляли контролируемым образом, поддерживая температуру реакционной смеси ниже 25°С. Затем добавляли концентрированную азотную кислоту (17,5 мл), поддерживая температуру реакционной смеси ниже 20°С. Затем реакционную смесь перемешивали при 20°С в течение 23 часов. Нижний водный слой удаляли и органический слой промывали водой (290 мл). Органический слой отделяли и перегоняли до 270 мл при атмосферном давлении. К реакционной смеси добавляли изопропанол (750 мл) при 45°С. Затем реакционную смесь нагревали до 40°С и перемешивали при этой температуре в течение 15 минут. Затем полученную суспензию охлаждали до 20°С, после этого до 5°С и поддерживали при этой температуре в течение одного часа. Продукт (бензил 4-(бензилокси)-5-метокси-2-нитробензоат) выделяли путем фильтрации, промывали изопропанолом (200 мл) и высушивали ниже чем 25°С. Выход: 78,4 г, 89,6%;
Масс-спектр (M+H)+=394,1.
1Н ЯМР спектр (CDCl3) 3,9 (s, 3Н), 5,2 (s, 2H), 5,3 (s, 2H), 7,1 (s, 1H), 7,3-7,4 (m, 10Н), 7,5 (s, 1H).
Бензил 4-(бензилокси)-5-метокси-2-нитробензоат (77 г) растворяли в ацетонитриле (882 мл). К раствору добавляли дитионит натрия (160,5 г) и температуру доводили до 25°С. Затем добавляли воду (588 мл), поддерживая температуру при 25°С. Значение pH поддерживали при 6, используя 8,8 М гидроксид натрия при восстановлении. После этого взвесь нагревали до 65°С и нижнюю водную фазу удаляли. Затем добавляли концентрированную соляную кислоту (35% мас./мас., 7,25 мл). Взвеси позволяли охладиться до 40°С и затем до 20°С. Добавляли раствор гидроксида натрия (47% мас./мас., 12,4 мл) и взвесь охлаждали до 0°С. Продукт (бензил 2-амино-4-(бензилокси)-5-метоксибензоат) выделяли путем фильтрации, промывали водой (2×196 мл) и затем высушивали при 40°С в вакууме. Выход: 66,2 г, 92,4%.
Масс-спектр (М+Н)+=364,1.
1Н ЯМР спектр (CDCl3) 3,8 (s, 3Н), 5,1 (s, 2Н), 5,3 (s, 2H), 6,2 (s, 1H), 7,3-7,4 (m, 10Н).
Бензил 2-амино-4-(бензилокси)-5-метоксибензоат (5,55 кг), формамидин ацетат (2,2 кг) и изобутанол (33,3 л) смешивали. Затем реакционную смесь нагревали до 97°С и перемешивали при этой температуре в течение 6 часов. Реакционную после этого смесь охлаждали до 25°С по меньшей мере в течение часа и затем перемешивали при этой температуре в течение 30 минут. Продукт (7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-он) выделяли путем фильтрации, промывали изобутанолом (6,1 л) и высушивали в вакуумном сушильном шкафу при температуре от 40 до 45°С. Выход: 4,25 кг, 98%.
Масс-спектр (М+H)+=283,1.
1Н ЯМР спектр (ДМСОd6) 3,9 (s, 3Н), 5,3 (s, 2H), 7,3 (s, 1H), 7,3-7,5 (m, 6H), 8,0 (s, 1H).
Пример 6
Получение 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-[3-(пирролидин-1-ил)пропокси]хиназолина
7-(Бензилокси)-4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метоксихиназолин(40 кг) растворяли в 1-метил-2-пирролидиноне (206 кг) при 40-45°С. Этот раствор загружали в инертный сосуд под давлением, содержащий катализатор 10% палладий на угле (0,235 кг приблизительно 50% воды влажн. катализатора). Осуществляли линейное промывание 1-метил-2-пирролидинона при 40-45°С (35 кг). Смесь гидрировали при 45°С и 3 бар и.д. в течение 3,5 часов. Реактор продували азотом и рециркулировали через фильтр Gaf, затем фильтровали через фильтр 1 мкм Pall в новый реактор. Осуществляли линейное промывание 1-метил-2-пирролидиноном (66 кг) через фильтры, получая раствор 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метоксихиназолин-7-ола. К этому раствору добавляли карбонат калия (10,3 кг) и смесь нагревали до 80°С.
71,5% мас./мас. водного раствора гидрохлорида 1-(3-хлорпропил)пирролидина (25 кг) разводили водой (14 л) и добавляли метил трет-бутиловый эфир (12 л). Перемешиваемую смесь подщелачивали до pH>11 путем добавления 46% мас./мас. водного раствора гидроксида натрия (6,5 л). Слои отделяли и нижний водный слой дополнительно экстрагировали метил трет-бутиловым эфиром (12 л). Объединенные органические слои промывали 20% мас./мас. водным раствором хлорида натрия (12,5 кг), получая раствор 1-(3-хлорпропил)пирролидина в метил трет-бутиловом эфире.
Раствор 1-(3-хлорпропил)пирролидина в метил трет-бутиловом эфире добавляли к горячему раствору 4-[(4-фтор-2-метил-1Н-индол-5-ил)окси]-6-метоксихиназолин-7-ола в 1-метил-2-пирролидиноне. Осуществляли линейное промывание метил трет-бутиловым эфиром (4 кг). Смесь поддерживали при 80°С в течение 3 часов, затем добавляли воду (348 кг) в течение 2 часов. Суспензию охлаждали до 60°С в течение 4 часов и выдерживали при 60°С в течение 12 часов. Продукт выделяли путем фильтрации и осадок промывали 1:1 мас./мас. 1-метил-2-пирролидинон/вода (64 кг), затем три раза водой (62 кг). Продукт высушивали с горячим азотом при 45°С. Выход: 34,8 кг при 100% мас./мас., 83%.
Масс-спектр [М+Н]+ 451.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,66-1,73 (m, 4 H) 1,95-2,03 (m, 2 H) 2,42 (s, 3 H) 2,43-2,48 (m, 4 H) 2,57 (t, J=7,11 Гц, 2 H) 4,00 (s, 3 H) 4,25 (t, J=6,41 Гц, 2 H) 6,24 (s, 1 H) 6,99 (dd, J=8,51, 7,44 Гц, 1 H) 7,16 (d, J=8,62 Гц, 1 H) 7,38 (s, 1 H) 7,60 (s, 1 H) 8,50 (s, 1 H) 11,33 (br. s., 1 H).
Водный раствор гидрохлорида 1-(3-хлорпропил)пирролидина готовили следующим образом.
Раствор 1-бром-3-хлорпропана (99,5 кг) в толуоле (237 кг) нагревали до 40-45°С. К этому раствору добавляли пирролидин (94,5 кг) в течение 1,5 часа, поддерживая температуру при 40-45°С. Осуществляли линейное промывание толуолом (37 кг) и реакцию поддерживали при 40-45°С дополнительно в течение четырех часов. Реакционную смесь охлаждали до 20-25°С и промывали водой (211 кг). Дополнительно добавляли воду (138 кг) и значение pH доводили до 8,8-9,0 путем добавления 34% мас./мас. соляной кислоты (4,7 кг). Водную фазу отделяли и отбрасывали. К органической фазе добавляли 34% мас./мас. соляную кислоту (61 кг) до достижения значения pH 0,5-1,0. Водную фазу отделяли и концентрировали в вакууме, поддерживая температуру <50°С до тех пор, пока содержание толуола не составляло <0,1%, получая водный раствор гидрохлорида 1-(3-хлорпропил)пирролидина 129,1 кг в концентрации 71,5% мас./мас., 79,9%.
Масс-спектр [М+Н]+ 148.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,79-2,06 (m, 4 H) 2,12-2,24 (m, 2 H) 2,90-3,03 (m, 2 H) 3,15-3,24 (m, 2 H) 3,44-3,56 (m, 2 H) 3,76 (t, J=6,41 Гц, 2 H) 11,33(br.s., 1H).
4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-[3-(пирролидин-1-ил)пропокси]хиназолин может быть приготовлен аналогично, используя раствор метил трет-бутилового эфира 1-(3-хлорпропил)пирролидина, полученный из 1-(3-хлорпропил)пирролидин оксалата.
1-(3-Хлорпропил)пирролидин оксалат (134,8 кг) суспендировали в воде (226 л) и добавляли метил трет-бутиловый эфир (83,4 кг). Перемешиваемую смесь подщелачивали до pH>11 путем добавления 49% мас./мас. водного раствора гидроксида калия (138,9 кг). Осуществляли линейное промывание водой (22,6 л). Нижний водный слой отделяли и переносили во второй реактор. Верхний органический слой переносили в бочки объемом 200 л. Осуществляли линейное промывание метил трет-бутиловым эфиром (16,7 кг). Водный слой повторно загружали в оригинальный реактор и дополнительно экстрагировали метил трет-бутиловым эфиром (83,4 кг). Нижний водный слой отбрасывали. Оригинальный органический слой перезагружали из бочек. Осуществляли линейное промывание метил трет-бутиловым эфиром (16,7 кг). Объединенные органические слои промывали 23% мас./мас. водным раствором хлорида калия (64,8 кг), получая раствор 1-(3-хлорпропил)пирролидина (83,9 кг) в метил трет-бутиловом эфире. После этого можно использовать требуемое количество раствора, если это является подходящим.
1-(3-Хлорпропил)пирролидин оксалат получали следующим образом.
К раствору 1-бром-3-хлорпропана (190 г) в толуоле (455 мл) при 40°С добавляли пирролидин (173 г), поддерживая температуру при 40-45°С. Затем осуществляли линейное промывание толуолом (40 мл). Смесь поддерживали при 40-45°С в течение 4 часов, затем охлаждали до 20°С и выдерживали в течение 6 часов. Смесь промывали водой (400 мл), затем дополнительно добавляли воду (265 мл) и значение pH доводили до 8,8-9,0 при экстрагировании с помощью 37% соляная кислота (9,5 мл). Водный слой отделяли.
Органический слой добавляли к раствору дигидрата щавелевой кислоты (129,31 г) в смеси изопропанола (1070 мл) и воды 109 мл) в течение 1 часа при 65-70°С. Смесь охлаждали до 55°С и поддерживали при этой температуре в течение 30 минут для инициации кристаллизации. После этого смесь охлаждали до 10°С в течение 1,5 часа и выдерживали при 10°С в течение часа перед фильтрованием. Твердое вещество промывали метил трет-бутиловым эфиром (400 мл) и в завершение метил трет-бутиловым эфиром (300 мл). Твердое вещество высушивали при 40°С в вакуумном сушильном шкафу, получая 1-(3-хлорпропил)пирролидин оксалат.
Выход 208,23 г, 72,5% теоретического.
Масс-спектр [М+Н]+ 148.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,85-1,97 (m, 4 Н) 2,06-2,16 (m, 2 H) 3,12-3,30 (m, 6 Н) 3,71 (t, J=6,41 Гц, 2 Н) 9.47 (br.s., 1 Н).
Пример 7
Получение малеатной соли 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолина
4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолин (28,4 кг в концентрации 100% мас./мас.) суспендировали в метаноле (284 л). Смесь дегазировали путем выдерживания в вакууме (-0,7 бар и.д.), затем вакуум ликвидировали с помощью азота. Это осуществляли три раза. Взвесь нагревали в колбе с обратным холодильником, получая прозрачный раствор. Раствор охлаждали до 60°С и рециркулировали через фильтр Gaf, затем фильтровали через фильтр 1 мкм Pall в кристаллизатор. Осуществляли линейное промывание метанолом (85 л), затем дегазировали, как описано выше.
Метанол (114 л) охлаждали до 0°С и дегазировали, как описано выше. Добавляли малеиновую кислоту (6,96 кг) и смесь перемешивали, получая прозрачный раствор, поддерживая в это время температуру при 0°С. Его переносили с помощью фильтра 1 мкм Pall в кристаллизатор, поддерживая температуру в кристаллизаторе при 55-60°С. Осуществляли линейное промывание метанолом (43 л). Температуру доводили до 55°С и добавляли микронизованную малеатную соль 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолина (форма А, 0,43 кг). Смесь поддерживали при 55°С в течение 3 часов, затем охлаждали до 40°С в течение 7 часов, затем охлаждали до -5°С в течение 6 часов. Суспензию поддерживали при -5°С в течение 20 часов. Продукт выделяли путем фильтрации, промывая осадок на фильтре метанолом (128 л). Продукт высушивали под горячим азотом при 50°С. Выход: 27,3 кг, 76%.
Масс-спектр [М+Н]+ 451.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,97 (br.s., 4 Н) 2,17-2,29 (m, 2 Н) 2,42 (s, 3 Н) 3,13 (br.s., 2 Н) 3,32-3,38 (m, 2 H) 3,60 (br.s., 2 H) 4,01 (s, 3 H) 4,32 (t, J=6,03 Гц, 2 Н) 6,01 (s, 2 H) 6,24 (s, 1 H) 6,98 (dd, J=8,51, 7,44 Гц, 1 Н) 7,16 (d, J=8,73 Гц, 1 H) 7,43 (s, 1 H) 7,64 (s, 1 H) 8,52 (s, 1 H) 9,36 (br.s., 2 H) 11,33 (br.s., 1 H).
Пример 8
Получение 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолина, используя соли 4-азониаспиро[3,4]октана
Получение хлорида 4-азониаспиро[3,4]октана
1-(3-Хлорпропил)пирролидин (1,142 г) растворяли в метаноле (38 мл) и раствор нагревали в колбе с обратным холодильником в течение ночи. Раствор упаривали насухо на роторном испарителе, получая 4-азониаспиро[3,4]октан хлорид в виде масла с примесями.
Выход 1,14 г.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,90-2,00 (m, 4H) 2,4-2,48 (m, 2H) 3,54-3,61 (m, 4H) 4,27 (t, J=8,1 Гц, 4H).
M+ 112.
Получение гексафторфосфата 4-азониаспиро[3,4]октана
Водный раствор хлорида 4-азониаспиро[3,4]октан получали путем добавления 1-(3-хлорпропил)пирролидина (128 мл) к воде (100 мл) при 94°С в течение 25 минут. Смесь перемешивали дополнительно в течение 5 минут, получая прозрачный раствор. Его охлаждали до 20°С и хранили в бутылке. Осуществляли линейные промывания водой (2×25 мл).
Добавляли раствор хлорида 4-азониаспиро[3,4]октана в воде (88 мл) в течение 10 минут к раствору гексафторфосфата натрия (50,3 г) в воде (100 мл), затем линейно промывали водой (12 мл). Дополнительно добавляли воду (50 мл) для перемешивания густой суспензии. Смесь нагревали до 90°С для перемешивания. Твердое вещество фильтровали и промывали водой (2×100 мл) и высушивали в вакуумном сушильном шкафу, получая гексафторфосфат 4-азониаспиро[3,4]октана (54,14 г, 70,3% теории).
ИК (KBr Диск) длина волны (см-1) 1450, 1348, 1323, 1100, 831, 557.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,89-2,01 (m, 4H) 2,40-2,50 (m, 2H) 3,53-3,61 (m, 4H) 4,27 (t, J=8,1 Гц, 4H).
Получение тетрафенилбората 4-азониаспиро[3,4]октана
К раствору тетрафенилбората натрия (1,90 г) в ацетоне (8 мл) при 20°С добавляли водный хлорид 4-азониаспиро[3,4]октана (1,64 мл, полученные, как описано выше).
Полученное твердое вещество фильтровали, промывали водой (3×10 мл) и высушивали в вакуумном сушильном шкафу, получая тетрафенилборат 4-азониаспиро[3,4]октана (2,17 г, 90% теории).
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,92 (m, 4H) 2,07-2,47 (m, 2H) 3,52-3,55 (m, 4H) 4,23 (t, J=8,1 Гц, 4H), 6,77-6,80 (m, 4H) 6,90-6,94 (m, 8H) 7,15-7,20 (m, 8H).
13С ЯМР спектр (100 МГц, ДМСО-d6) δ част. на млн. 14,12 (s, 1C) 20,71 (s, 1C) 62,01 (s, br, 2C) 121,45 (s, 8C) 125,21 (m, 8C) 135,5 (s, 4C) 163,3 (m, 4C).
Получение бромида 4-азониаспиро[3,4]октана
1,3-дибромпропан (287 мл) нагревали до 45°С и добавляли пирролидин (23,4 мл) в течение 4 часов. После этого смесь охлаждали до комнатной температуры и упаривали насухо на роторном испарителе. К оставшемуся маслу добавляли воду (200 мл) и небольшой органический нижний слой отделяли. Раствор промывали дихлорметаном (20 мл). Водный слой подщелачивали путем добавления карбоната калия (42,8 г) и экстрагировали дихлорметаном (2×80 мл). Объединенные органические экстракты высушивали над сульфатом магния и упаривали насухо на роторном испарителе. Остаток растирали в порошок путем добавления ацетона (30 мл) и перемешивали в течение ночи. Твердое вещество фильтровали, промывали ацетон (30 мл) и высушивали в вакуумном сушильном шкафу при 40°С.
Выход 3,06 г, 5,7%.
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 1,90-1,99 (m, 4H) 2,41-2,50 (m, 2H) 3,53-3,66 (m, 4H) 4,30 (t, J=8,1 Гц, 4H).
М+ 112.
Получение 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолина, используя хлорид 4-азониаспиро[3,4]октана
Хлорид 4-азониаспиро[3,4]октана (1,14 г) добавляли к смеси 4-[(4-фтор-2-метил-1Н-индол-5-ил)окси]-6-метоксихиназолин-7-ола (2,5 г), 1-метил-2-пирролидинона (25 мл), метил трет-бутилового эфира (1,8 мл) и карбоната калия (815 мг) при 80°С. После перемешивания при 80°С в течение 3 часов добавляли воду (27 мл) в течение 40 минут. Смеси позволяли охладиться до 20°С и оставляли на ночь. Смесь повторно нагревали до 60°С и фильтровали. Осадок на фильтре промывали 1:1 об./об. 1-метил-2-пирролидинон/вода (5 мл) и затем водой (4×5 мл). Твердое вещество высушивали в вакуумном шкафу, получая 4-[(4-фтор-2-метил-1Н-индол-5-ил)окси]-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолин.
Выход 2,29 г при 94,2% мас./мас., 65,0% теории.
Получение 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолина, используя бромид 4-азониаспиро[3,4]октана
К раствору 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метоксихиназолин-7-ола (2,5 г) в 1-метил-2-пирролидиноне (25 мл) и метил трет-бутиловом эфире (1,8 мл) добавляли карбонат калия (815 мг). Смесь дегазировали путем выдерживания в вакууме и разрежали с помощью азота. Это осуществляли пять раз. Смесь нагревали до 80°С и добавляли бромид 4-азониаспиро[3,4]октана (1,75 г). Смесь поддерживали при 80°С в течение 3,5 часов. К горячей смеси добавляли воду (27 мл) в течение 2 часов. Смесь охлаждали до 64°С и твердое вещество фильтровали. Твердое вещество промывали 1:1 об./об. 1-метил-2-пирролидинон/вода (5 мл) и затем водой (4×5 мл). Твердое вещество высушивали в вакуумном шкафу, получая 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолин.
Выход 2,75 г при 94,2% мас./мас., 78,0% теоретического.
Получение 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолина, используя гексафторфосфат 4-азониаспиро[3,4]октана
Реакцию осуществляли, как и для бромидной соли, но используя гексафторфосфат 4-азониаспиро[3,4]октана (1,99 г).
Выход 2,20 г при 94,2% мас./мас., 62,4% теоретического.
Пример 9
Получение 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метоксихиназолин-7-ола путем гидрирования 1-(3-{[7-(бензилокси)-6-метоксихиназолин-4-ил]окси}-2-фтор-6-нитрофенил)ацетона
Получение 1-(3-{[7-(бензилокси)-6-метоксихиназолин-4-ил]окси}-2-фтор-6-нитрофенил)ацетона
1-(2-Фтор-3-гидрокси-6-нитрофенил)-пропан-2-он (4,286 г) суспендировали в ацетонитриле (16 мл) и охлаждали до -5°С. К взвеси добавляли водный гидроксид натрия (0,93 мл 46,9% мас./мас. раствор). Осуществляли линейное промывание ацетонитрилом (16 мл).
Это охлажденный раствор добавляли к раствору 7-(бензилокси)-4-хлор-6-метоксихиназолина (4,26 г) в анизоле (28,8 мл) при 80°С в течение 15 минут. Осуществляли линейное промывание ацетонитрилом (16 мл). Смесь нагревали при 80°С в течение 21 часов. Мешалку останавливали и нижний водный слой отделяли. Верхний органический слой дополнительно промывали водой (6 мл) при 80°С. Партию охлаждали до 20°С и концентрировали при пониженном давлении (130 мбар), позволяя температуре партии достичь 100°С, собирая 24 мл дистиллята. Температуру партии доводили до 65°С и добавляли раствор воды (4 мл) в метаноле (28 мл). Смесь охлаждали до 20°С и продукт фильтровали. Осадок на фильтре промывали метанолом (2×12 мл) и высушивали в вакуумном сушильном шкафу в течение ночи, получая 1-(3-{[7-(бензилокси)-6-метоксихиназолин-4-ил]окси}-2-фтор-6-нитрофенил)ацетон (4,543 г, 67% теории).
1Н ЯМР спектр (400 МГц, ДМСО-d6) δ част. на млн. 2,29 (s, 3 Н) 4,00 (s, 3 H) 4,27 (s, 2 H) 5,37 (s, 2 H) 7,38 (m, 1 H) 7,44 (m, 2 H) 7,53 (m, 2 H) 7,56 (s, 1 H) 7,62 (s, 1 H) 7,77 (dd, J=9,1, 7,8 Гц, 1Н) 8,12 (dd, J=9,05, 0,81 Гц, 1 H) 8,59 (s, 1 H).
Масс-спектр [М+Н]+.
Гидрирование 1-(3-{[7-(бензилокси)-6-метоксихиназолин-4-ил]окси}-2-фтор-6-нитрофенил)ацетона с получением 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метоксихиназолин-7-ола
К раствору 1-(3-{[7-(бензилокси)-6-метоксихиназолин-4-ил]окси}-2-фтор-6-нитрофенил)ацетона (0,75 г) в 1-метил-2-пирролидиноне (7,5 мл) загружали катализатор 10% палладий на древесном угле (38,6 мг приблизительно 50% водн. влажн. пасты). Смесь нагревали до 50°С и гидрировали под давлением 4 бар в атмосфере водорода в течение 2,75 часа. При этом получали раствор, содержащий 28% площади пика 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метоксихиназолин-7-ола. ВЭЖХ время удерживания 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метоксихиназолин-7-ола 12,36 мин. Это было идентичным времени удерживания 4-[(4-фтор-2-метил-1H-индол-5-ил)окси]-6-метоксихиназолин-7-ола, приготовленного в примере 6.
ВЭЖХ метод: колонка Betabasic C18 3,5 мкм, 150 мм×4,6 мм внутр. диаметра. Температура 30°С. Вводимый объем 10 мкл. УФ 260 нм, ответ >0,1 мин. (2 с). Скорость потока 1,0 мл/мин. Сбор данных 17 мин. Время прогона 21 мин. Элюент А вода; Элюент В ацетонитрил; Элюент С 1% водная трифторуксусная кислота.
[М+Н]+ 340.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4-(4-БРОМ-2-ФТОРАНИЛИНО)-6-МЕТОКСИ-7(1-МЕТИЛПИПЕРИДИН-4-ИЛМЕТОКСИ)ХИНАЗОЛИНА(ZD6474) И СПОСОБЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2448102C2 |
| 4-ГЕТЕРОЦИКЛИЛ-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2137762C1 |
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ КАССЕТНЫХ ТРАНСПОРТЕРОВ | 2008 |
|
RU2512682C2 |
| ИНГИБИТОР EGFR И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2702631C2 |
| СПОСОБ ФТОРИРОВАНИЯ АНИЛИДНЫХ ПРОИЗВОДНЫХ И ФТОРИРОВАННЫЕ ПРОИЗВОДНЫЕ БЕНЗОТИАЗОЛА В КАЧЕСТВЕ IN VIVO ВИЗУАЛИЗИРУЮЩИХ АГЕНТОВ | 2006 |
|
RU2419601C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| Бензотиофены и родственные соединения в качестве агонистов STING | 2019 |
|
RU2806274C2 |
| ЗАМЕЩЕННЫЕ N-ФЕНИЛБИПИРРОЛИДИНКАРБОКСАМИДЫ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2008 |
|
RU2477719C2 |
| ПРОИЗВОДНЫЕ 5-АМИНОЦИКЛИЛМЕТИЛОКСАЗОЛИДИН-2-ОНА | 2008 |
|
RU2492169C2 |
Изобретение относится к способу получения соединения формулы II
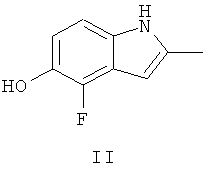
из соединения формулы III
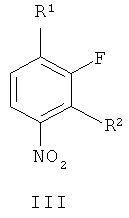 ;
;
где R1 и R2 независимо означают Cl, Br, F, I, взаимодействием его со сложным эфиром формулы (IV)
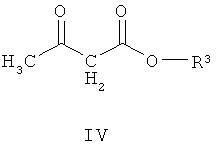 ;
;
где R3 означает С1-6алкил, с получением соединение формулы V
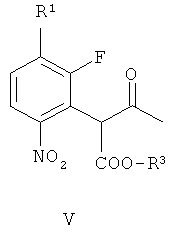
которое подвергают взаимодействию с гидроксильным ионом в присутствии соли арил-алкил аммония или соли тетра-алкил аммония, с получением соединение формулы VI
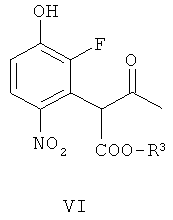
его последующей обработки с получением соединения формулы VII
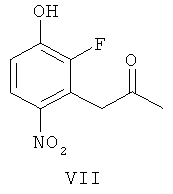
которое восстанавливают с получением соединения формулы II. 2 н. и 3 з.п. ф-лы, 9 пр.
1. Способ получения соединения формулы II
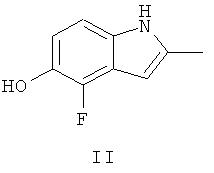
из соединения формулы III:
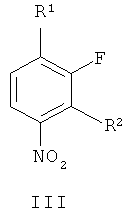 ;
;
где R1 и R2 независимо выбирают из хлора, брома, фтора и йода;
где способ включает стадии:
(а) взаимодействие соединения формулы III со сложным эфиром формулы (IV)
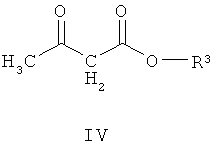 ;
;
где R3 представляет собой C1-6алкил;
получая соединение формулы V:
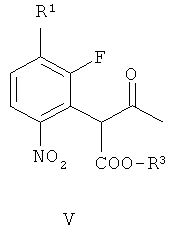 ;
;
(б) взаимодействие соединения формулы V с гидроксильным ионом в присутствии соли арил-алкил аммония или соли тетра-алкил аммония, получая соединение формулы VI
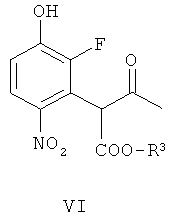 ;
;
(в) взаимодействие соединения формулы VI, получая соединение формулы VII;
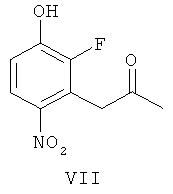 ;
;
(г) восстановление соединения формулы VII, получая соединение формулы II.
2. Способ по п.1, в котором R1 и R2 оба представляют собой фтор.
3. Способ по п.1, в котором стадию (а) осуществляют в растворителе, выбранном из толуола, триметилбензола и ксилола.
4. Способ по п.1, в котором Тритон В используют на стадии (б).
5. Соединение формулы VI:
как определено в п.1, или его соль.
| WO 00/47212 А, 17.08.2000 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ПРОИЗВОДНЫЕ АМИНОСПИРТОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2001 |
|
RU2233839C1 |

