Область техники, к которой относится изобретение
Настоящее изобретение относится к новым окисленным липидам и способам применения окисленных липидов для лечения или профилактики воспалений, ассоциированных с эндогенными окисленными липидами. Способы по настоящему изобретению можно применять для лечения или предотвращения воспалительных заболеваний и нарушений, таких как, например, атеросклероз и родственные нарушения, аутоиммунные заболевания или нарушения и пролиферативные заболевания или нарушения.
Сердечно-сосудистые заболевания представляют собой главный фактор риска для здоровья в промышленно развитых странах. Атеросклероз, наиболее распространенное сердечно-сосудистое заболевание, является основной причиной инфарктов, инсультов и гангрен конечностей и, как следствие этого, главной причиной смертности в США. Атеросклероз является комплексным заболеванием, обусловленным клетками многих типов и молекулярными факторами (для более детального ознакомления см. публикацию Ross, 1993, Nature 362:801-809). Процесс, возникающий в результате поражения эндотелиальных клеток и клеток гладких мышц (SMC) стенок артерии, включает образование отложений, состоящих из фиброзной, жировой и волокнистой ткани, или бляшек, предшествующих и сопутствующих воспалению. Дестабилизация бляшек может вызвать дальнейшие осложнения, такие как разрыв сосудов и тромбоз, которые возникают в результате воспалительной и фибропролиферативной реакции на многообразные формы повреждений. Например, считается, что сдвиговые напряжения являются причиной частого образования атеросклеротических бляшек в системе кровообращения на участках возникновения турбулентного потока крови, таких как разветвления сосудов и структуры неправильной формы.
Первым наблюдаемым явлением при образовании атеросклеротических бляшек является сцепление клеток зоны воспаления, таких как макрофаги, образуемые из моноцитов, с эндотелиальным слоем сосудов и проникновение в субэндотелиальное пространство. Повышенные уровни LDL в плазме вызывают отложение липидов на стенках сосудов, в результате которого расположенные рядом эндотелиальные клетки продуцируют окисленный липопротеин низкой плотности (LDL). Кроме того, захват липопротеина внеклеточным матриксом ведет к прогрессирующему окислению LDL липоксигеназами, активным кислородом, пероксинитритом и/или миелопероксидазой. Указанные окисленные LDL затем поглощаются в больших количествах моноцитами через фагоцитарные рецепторы, экспрессированные на их поверхностях.
Наполненные липидами моноциты и клетки гладких мышц (SMC), именуемые ксантомными клетками, являются главным компонентом жировых полосок. Взаимодействие между ксантомными клетками, эндотелиальными клетками и окружающими их клетками гладких мышц вызывает состояние хронического местного воспаления, которое в конечном счете ведет к активации эндотелиальных клеток, повышенному апоптозу макрофагов, пролиферации и миграции клеток гладких мышц и образованию фиброзных бляшек (Hajjar, D.P. and Haberland, M.E, J. Biol. Chem. 1997, Sep 12; 272(37):22975-78). В результате разрушения бляшек и образования тромбов происходит закупорка кровеносных сосудов, которая препятствует кровотоку и вызывает ишемию, состояние, характеризующееся недостатком кислорода в тканях вследствие недостаточной перфузии. Когда блокируется подача крови к сердцу по указанным артериям, у субъекта возникает “инфаркт”; когда происходит закупорка артерий головного мозга, у данного субъекта возникает инсульт. При сужении артерий, снабжающих кровью конечности, появляется сильная боль, ухудшается физическая подвижность и может возникнуть необходимость в ампутации.
Окисленный LDL участвует в патогенезе атеросклероза и атеротромбоза, оказывая воздействие на моноциты и клетки гладких мышц, вызывая апоптоз эндотелиальных клеток и нарушая баланс антикоагулянта в эндотелии. Окисленный LDL также ингибирует антиатерогенное HDL-ассоциированное разрушение окисленных фосфолипидов (Mertens, A and Holvoet, P., FASEB J. 2001 Oct; 15(12):2073-84). Данная ассоциация подтверждена многими исследованиями, демонстрирующими присутствие окисленного LDL в бляшках в разных животных моделях атерогенеза и замедление атерогенеза в результате ингибирования окисления фармакологическими и/или генетическими средствами (см., например, Witztum J. and Steinberg, D, Trends Cardiovasc Med 2001 Apr-May; 11(3-4):93-102 для ознакомления с текущей научной литературой). Действительно окисленный LDL и малондиальдегид (MDA)-модифицированный LDL недавно были предложены в качестве точных маркеров крови для определения 1-й и 2-й стадии коронарной болезни сердца (патент США № 6309888, выданный Holvoet et al., и патент США № 6255070, выданный Witztum et al.).
Снижение окисления и активности LDL легло в основу ряда предложенных клинических методов лечения и профилактики сердечно-сосудистых заболеваний. Букала и др. (Bucala, et al., патент США № 5869534) описали способы модуляции перокисления липидов в результате сокращения конечного продукта прогрессирующего гликозилирования, представляющего собой липид, характерный для образования ксантомных клеток вследствие возрастных изменений, заболевания и диабета. Танг и др. (Tang et al., at Incyte Pharmaceuticals, Inc., патент США № 5945308) описали способ идентификации и клинического применения рецептора окисленного LDL человека при лечении сердечно-сосудистых и аутоиммунных заболеваний, а также рака.
Атеросклероз и аутоиммунные заболевания
С учетом предполагаемой роли сильной воспалительной фибропролиферативной реакции в случае атеросклероза и ишемии многие ученые предприняли попытку определить аутоиммунный компонент поражения сосудов. В случае аутоиммунных заболеваний иммунная система узнает и атакует компоненты, которые обычно не являются антигенными (аутоантигены), помимо воздействия на проникающие в организм чужеродные антигены. Аутоиммунные заболевания классифицируются как заболевания, опосредуемые аутоантителами или клетками. Типичными аутоиммунными заболеваниями, опосредуемыми аутоантителами, являются миастения и идиопатическая тромботическая пурпура (ITP), и типичными клеткоопосредуемыми заболеваниями являются тиреоидит Хасимото и (юношеский) диабет типа I.
Признание того, что иммуноопосредуемые процессы преобладают в местах атеросклеротических поражений, стало возможным в результате исследования лимфоцитов и макрофагов на ранних стадиях, а именно жировых полосок. Было обнаружено, что указанные лимфоциты, которые включают преобладающую популяцию CD4+ клеток (остальные клетки являются CD8+ клетками), являются более многочисленными, чем макрофаги на ранних стадиях поражения при сравнении с более поздними стадиями поражения, когда данное соотношение меняется на обратное. При изучении полученных результатов возникают вопросы относительно того, отражает ли такое явление первичную иммунную сенсибилизацию на возможный антиген или альтернативно является простым эпифеноменом ранее индуцированного местного поражения тканей. Независимо от факторов, определяющих наполнение бляшки на начальной стадии образования указанными клетками зоны воспаления, они, по-видимому, характеризуют активированное состояние, определяемое сопутствующей экспрессией HLA-DR главного комплекса гистосовместимости класса II и рецептора интерлейкина (IL), а также общих лейкоцитарных антигенов (CD45R0) и интегрина очень позднего антигена 1 (VLA-1).
Воспалительная реакция, возникающая на ранних стадиях атеросклеротического поражения, может быть первичным инициирующим событием, вызывающим продуцирование разных цитокинов локальными клетками (то есть эндотелиальными клетками, макрофагами, клетками гладких мышц и клетками зоны воспаления), или может быть формой реакции защитной иммунной системы организма на опасный процесс. Некоторые цитокины, которые, как было установлено, более интенсивно продуцируются резидентными клетками, включают TNF-α, IL-1, IL-2, IL-6 IL-8, IFN-γ и хемоаттрактант моноцитов пептид-1 (МСР-1). Кроме того, установлено, что происходит сверхэкспрессия тромбоцитарного фактора роста (PDGF) и инсулиноподобного фактора роста (IGF) всеми клеточными компонентами в атеросклеротических бляшках, что, по-видимому, усиливает предварительную воспалительную реакцию в результате костимулирущего действия митогенного и хемотаксического факторов. Уемура и др. (Uyemura et al., Cross regulatory roles of IL-12 and IL-10 in atherosclerosis. J. Clin Invest. 1996, 97; 2130-2138) установили паттерн Т-клеточных цитокинов типа 1 в атеросклеротических поражениях человека, характеризующихся сильной экспрессией IFN-γ, а не мРНК IL-4 по сравнению с нормальными артериями. Кроме того, на пораженном участке была обнаружена сверхэкспрессия IL-12, фактора роста Т-клеток, продуцируемого главным образом активированными моноцитами и селективным индуктором паттерна цитокинов Th1, о чем свидетельствует большое количество его основной гетеродимерной формы мРНК р70 и р40 (его доминантный индуцибельный белок).
Аналогично явному свидетельству преобладания клеточной иммунной системы в атеросклеротической бляшке, имеются также многочисленные данные, подтверждающие участие локальной гуморальной иммунной системы. Так, в бляшках было обнаружено отложение иммуноглобулинов и комплементарных компонентов помимо повышенной экспрессии рецепторов C3b и C3Bi в резидентных макрофагах.
Ценные свидетельства участия иммуноопосредуемого воспаления в развитии атеросклероза были получены в результате исследования животных моделей. У мышей с нарушенной иммунологической реактивностью (с отсутствием МНС класса I) атеросклероз развивается быстрее по сравнению с иммунокомпетентными мышами. Кроме того, введение мышам C57BL/6 (Emeson E.E., Shen M.L., Accelerated atherosclerosis in hyperlipidemic C57BL/6 mice treated with cyclosporin A. Am. J. Pathol 1993; 142:1906-1915) и новозеландским белым кроликам (Roselaar S.E., Schonfeld G., Daugherty A. Enhanced development of atherosclerosis in cholesterol fed rabbits by suppression of cell mediated immunity. J. Clin. Invest. 1995; 96:1389-1394) циклоспорина А, сильнодействующего супрессора транскрипции IL-2, вызывало значительно более сильный атеросклероз при “нормальной” “нагрузке” липопротеина. Последние исследования могут пролить свет на возможную роль иммунной системы в противодействии самопроизвольному воспалительному процессу в атеросклеротической бляшке.
Атеросклероз не является классическим аутоиммунным заболеванием, хотя некоторые из его проявлений, таких как образование бляшек, закупоривающих кровеносные сосуды, можно отнести к аберрантной иммунологической реактивности. В классическом аутоиммунном заболевании очень часто можно выявить сенсибилизирущий аутоантиген, атакуемый иммунной системой и компонентами иммунной системы, которые узнают данный аутоантиген (гуморальными, то есть аутоантителами, или клеточными, то есть лимфоцитами). Кроме того, известно, что такое заболевание может быть вызвано у здоровых животных в результате пассивного переноса указанных компонентов иммунной системы, или в случае человека данное заболевание может передаваться от больной беременной женщины ее ребенку. Многие из указанных признаков отсутствуют в атеросклерозе. Кроме того, данное заболевание имеет общие факторы риска, такие как гипертензия, диабет, пониженная физическая активность, курение и другие, указанное заболевание поражает пожилых людей и характеризуется другим генетическим предпочтением по сравнению с классическими аутоиммунными заболеваниями.
Лечение аутоиммунного воспалительного заболевания может быть направлено на подавление или изменение общей и/или специфичной для болезни иммунной реактивности. Так, например, Аелло (Aiello, патенты США №№ 6034102 и 6114395) описал применение эстрогеноподобных соединений для лечения и профилактики атеросклероза и развития атеросклеротического поражения путем ингибирования рекрутинга клеток зоны воспаления. Аналогичным образом Медфорд и др. (Medford et al., патент США № 5846959) описали методы профилактики образования окисленной PUFA для лечения свердечно-сосудистых и других воспалительных заболеваний, обусловленных молекулой межклеточной адгезии VCAM-1. Кроме того, Фалб (Falb, патент США № 6156500) описал ряд сигнальных молекул и молекул межклеточной адгезии, присутствующих в большом количестве в атеросклеротической бляшке, в качестве потенциальных мишеней для противовоспалительной терапии.
Так как окисленный LDL несомненно участвует в патогенезе атеросклероза (см. выше), было исследовано влияние, оказываемое основными компонентами бляшки на аутоиммунитет в процессах развития атероматозного заболевания.
Иммунологическая реактивность на окисленный LDL. Известно, что окисленный LDL (Ox LDL) оказывает хемотаксическое действие на Т-клетки и моноциты. Известно также, что Ox LDL и его побочные продукты индуцируют экспрессию таких факторов как хемотаксический фактор моноцитов 1, секрецию колониестимулирующего фактора и активирующие свойства тромбоцитов, которые являются сильнодействующими стимуляторами роста.
Активное участие иммунной реакции клеток в атеросклерозе было обосновано Стеммом С. и др. (Stemme S., et al., Proc. Natl. Acad. Sci. USA 1995; 92:3893-97), которые выделили CD4+ в клонах бляшек, реагирующих на Ox LDL в качестве стимулов. Клоны, соответствующие Ox LDL (4 из 27), продуцировали в основном γ-интереферон, а не IL-4. Необходимо выяснить, является ли данная реакция способом контактирования вышеуказанных клонов Т-клеток с иммунной системой клетки при помощи сильного стимулирующего иммуногена (Ox LDL) или данная реакция служит средством борьбы с неактивным атеросклеротическим процессом.
Данные, относящиеся к участию гуморальных механизмов и их значению, являются гораздо более спорными. В одном недавно выполненном исследовании было сообщено о повышенных уровнях антител против MDA-LDL, являющегося метаболитом окисления LDL, у женщин, страдающих заболеванием сердца и/или диабетом (Dotevall, et al., Clin. Sci. 2001 Nov; 101(5):523-31). Другие исследователи продемонстрировали антитела, узнающие многие эпитопы в окисленном LDL, что свидетельствует об иммунной реакции на липид и компоненты аполипопротеина (Steinerova A., et al., Physiol Res. 2001; 50(2): 131-41) в случае атеросклероза и других заболеваний, таких как диабет, вазоренальный синдром, уремия, ревматическая атака и системная красная волчанка. В нескольких научных публикациях были отмечены повышенные уровни антител к Ox LDL по мере развития атеросклероза (определяемого по степени стеноза сонной артерии, тяжести заболевания периферических сосудов и т.д.). Недавно Шерер и др. (Sherer et al., Cardiology 2001; 95(1):20-4) продемонстрировали повышенные уровни антител к кардиолипину, бета 2GPI и OxLDL, при ишемической болезни сердца. Таким образом, по-видимому, существует согласие относительно присутствия антител к Ox LDL в форме иммунных комплексов в атеросклеротических бляшках, хотя действительная значимость данного открытия до сих пор не установлена.
Существует гипотеза, что антитела к Ox LDL играют активную роль в метаболизме липопротеина. Так, известно, что иммунные комплексы Ox LDL и соответствующих им антител более эффективно поглощаются макрофагами в суспензии по сравнению с Ox LDL. Из данного открытия нельзя сделать никаких выводов о патогенезе атеросклероза, так как до сих пор не получен ответ на вопрос, является ли ускоренное поглощение Ox LDL макрофагами благоприятным или вредным фактором.
Важные данные, касающиеся значимости гуморальной иммунной системы в процессе атерогенеза, ученые получают при исследовании животных моделей. Установлено, что гипериммунизация кроликов с отсутствием рецептора LDL гомологичным окисленным LDL вызывает продуцирование большого количества антител против Ox LDL, в результате чего происходит значительное уменьшение величины атеросклеротических поражений по сравнению с контрольной группой, которой вводили физиологический раствор с фосфатным буфером (PBS). Уменьшение образования бляшек было также достигнуто при иммунизации кроликов липосомами с большим содержанием холестерина при одновременном продуцировании антител против холестерина, причем данный эффект сопровождался 35% снижением уровней холестерина, содержащего липопротеин с очень низкой плотностью.
Таким образом, в лабораторных и клинических исследованиях была всесторонне продемонстрирована патогенная роль компонентов окисленного LDL и их значение в качестве аутоантигенов в случае атеросклероза и других заболеваний.
Опосредуемая слизистой оболочкой иммуномодуляция при лечении аутоиммунных заболеваний
В недавнем прошлом были разработаны новые методы и фармацевтические препараты, пригодные для лечения аутоиммунных заболеваний (и родственных опосредуемых Т-клетками воспалительных заболеваний, таких как отторжение аллотрансплантата и неврологические заболевания, обусловленные ретровирусами). Указанные методы лечения модулируют иммунную систему, вызывая толерантность к заболеванию, при пероральном введении или через слизистую оболочку, например, путем ингаляции, при использовании в качестве толерогенных аутоантигенов, антигенов-свидетелей или подавляющих болезнь фрагментов или аналогов аутоантигенов или антигенов-свидетелей. Такие методы лечения описаны, например, в патенте США № 5935577, выданном Вейнеру и др. Аутоантигены и антигены-свидетели рассмотрены ниже (для общего ознакомления с толерантностью, индуцируемой через слизистую оболочку, см. публикацию Nagler-Anderson, C., Crit. Rev. Immunol. 2000; 20(2):103-20). Установлено, что внутривенное введение аутоантигенов (и их фрагментов, содержащих иммунодоминантные эпитопные области их молекул) вызывает супрессию иммунной реакции под действием механизма, именуемого клональной толерантностью. Клональная толерантность дезактивирует только Т-клетки иммунной атаки, специфичные к определенному антигену, в результате чего происходит значительное снижение иммунной реакции на данный антиген. Таким образом, Т-клетки, стимулирущие аутоиммунную реакцию и являющиеся специфичными к аутоантигену с индуцированной толерантностью, больше не пролиферируют под воздействием данного антигена. Подобное уменьшение пролиферации снижает также иммунные реакции, определяющие симптомы аутоиммунного заболевания (такие как поражение нервной ткани, наблюдаемое в случае рассеянного склероза). Кроме того, существуют данные, что пероральное введение аутоантигенов (или иммунодоминантных фрагментов) в виде однократной дозы или в значительно больших количествах по сравнению с теми, которые запускают “активную супрессию”, могут также индуцировать толерантность вследствие анергии (или делеции клона).
В научной литературе описан также метод лечения на основании активной супрессии. Механизм активной супрессии отличается от механизма клональной толерантности. Указанный метод, всесторонне описанный в заявке на патент РСТ/US93/01705, включает пероральное введение или введение через слизистую оболочку антигенов, специфичных к ткани, подвергающейся аутоиммунному воздействию. Указанные антигены именуются “антигенами-свидетелями”. Такое лечение вызывает индукцию регуляторных (супрессорных) Т-клеток в лимфоидной ткани, ассоциированной с кишечником (GALT), лимфоидной ткани, ассоциированной с бронхами (BALT), или чаще всего в лимфоидной ткани, ассоциированной со слизистой оболочкой (MALT) (MALT включает GALT и BALT). Указанные регуляторные клетки выделяются в кровь или лимфатическую ткань и затем мигрируют в орган или ткань, пораженную аутоиммунным заболеванием, и подавляют аутоиммунную атаку на пораженный орган или ткань. Т-клетки, выявленные антигеном-свидетелем (которые узнают по крайней мере одну антигенную детерминанту антигена-свидетеля, используемого для их выявления), направленно воздействуют на очаг аутоиммунной атаки, где они опосредуют локальное выделение определенных иммуномодуляторных факторов и цитокинов, таких как трансформирующий β-фактор роста (TGF-β), интерлейкин-4 (IL-4) и/или интерлейкин-10 (IL-10). Из вышеуказанных факторов TGF-β является антиген-неспецифическим иммуносупрессивным фактором в связи с тем, что он подавляет иммунную атаку независимо от антигена, запускающего данную атаку (однако, поскольку толеризация антигеном-свидетелем, достигаемая при пероральном введении или при введении через слизистую оболочку, вызывает выделение TGF-β только в непосредственной близости от места аутоиммунной атаки, она не обеспечивает системную иммуносупрессию). IL-4 и IL-10 также являются антиген-неспецифическими иммунорегуляторными цитокинами. IL-4, в частности, усиливает реакцию Т-клеток-хелперов типа 2 (Th2), то есть воздействует на предшественники Т-клеток и вызывает их дифференцировку с образованием предпочтительно Th2-клеток за счет реакций Th1-клеток. IL-4 также косвенно ингибирует обострение, вызванное Th1-клетками. IL-10 является прямым ингибитором реакций Th1-клеток. После пероральной толеризации млекопитающих, страдающих аутоиммунным заболеванием, при помощи антигенов-свидетелей наблюдаются повышенные уровни TGF-β, IL-4 и IL-10 в очаге аутоиммунной атаки (Chen, Y. et al., Science, 265:1237-1240, 1994). Механизм подавления антигеном-свидетелем подтвержден фон Герретом и др. (von Herreth et al., J. Clin. Invest., 96:1324-1331, September 1996).
Пероральная иммуномодуляция, вызывающая пероральную толерантность, была эффективно применена в животных моделях при лечении воспалительного заболевания кишечника путем перорального введения пробиотических бактерий (Dunne, C., et al., Antonie Van Leeuwenhoek 1999 Jul-Nov; 76(1-4):279-92), аутоиммунного гломерулонефрита путем перорального введения гломерулярной базальной мембраны (Reynolds, J. et al., J. Am. Soc. Nephrol. 2001 Jan; 12(1):61-70), экспериментального аллергического энцефаломиелита (ЕАЕ, который равнозначен рассеянному склерозу или MS) путем перорального введения основного белка миелина (МВР), адъювантного и коллагенового артрита путем перорального введения субъекту соответственно коллагена и HSP-65. В Бостонской компании Autoimmune было проведено несколько экспериментов с участием людей по предотвращению диабета, рассеянного склероза, ревматоидного артрита и увеита. Результаты экспериментов с участием людей были менее впечатляющими по сравнению с экспериментами на животных, отличных от человека, однако, был достигнут некоторый успех по профилактике артрита.
Была также исследована иммуномодуляция путем индукции пероральной толерантности к аутоантигенам, обнаруженным в местах поражения атеросклеротическими бляшками. Исследование эпитопов, узнаваемых Т-клетками, и титров Ig в клинических и экспериментальных моделях атеросклероза позволили выявить три антигена-кандидата для подавления воспаления в атероматозных поражениях: окисленный LDL, вызываемый стрессом хитшоковый белок HSP 65 и кардиолипинсвязывающий бета-белок 2GP1. В заявке на патент США № 09/806400 на имя Шоенфельда и др. (поданной 30 сентября 1999 г.), которая полностью включена в данное описание изобретения, описано сокращение примерно на 30% атерогенеза в артериях у генетически предрасположенных трансгенных мышей с отсутствием рецептора LDL-RD, которым перорально вводили окисленный LDL человека. Однако указанное защитное действие достигалось в результате перорального введения неочищенного антигенного препарата, включающего центрифугированный, фильтрованный и очищенный LDL из сыворотки человека, которный подвергали продолжительному окислению Cu++ или малондиальдегидом (MDA). Хотя было достигнуто значительное ингибирование атерогенеза вследствие пероральной толерантности, не были идентифицированы специфические липидные антигены или иммуногенетические компоненты LDL. Другими возникшими препятствиями были неустойчивость, присущая неочищенному окисленному LDL in vivo вследствие ферментативной активности, поглощение окисленного LDL печенью, иммунные механизмы клетки и гетерогенность, свойственная разным донорам. Вполне вероятно, что устойчивый, лучше определенный аналог окисленного LDL обеспечит более эффективную иммуномодуляцию (например, в результате пероральной толерантности).
Индукция иммунной толерантности и последующее предотвращение или ингибирование аутоиммунных воспалительных процессов было продемонстрировано при введении супрессивных антигенов через слизистую оболочку кроме слизистой оболочки кишечника. Мембранная ткань вокруг глаз и слизистая оболочка носовой полости и кишечника подвержены многим инвазиям, а также воздействию аутоантигенов и обладают механизмами иммунореактивности. Так, Росси и др. (Rossi, et al., Scand J. Immunol 1999 Aug; 50(2):177-82) обнаружили, что введение глиадина через нос было таким же эффективным, как и внутривенное введение при ослаблении иммунной реакции на антиген в модели болезни органов брюшной полости у мышей. Аналогичным образом введение через нос антигена рецептора ацетилхолина было более эффективным, чем пероральное введение, для замедления и уменьшения мышечной слабости и пролиферации специфических лимфоцитов в модели миастении у мышей (Shi, F.D. et al., J. Immunol. 1999 May 15; 162(10):5757-63). Поэтому иммуногенные соединения, предназначенные для введения через слизистую оболочку, для внутривенного или внутрибрюшинного введения, должны быть оптимально адаптированы для способов введения через нос и другие мембраны.
Таким образом, существует насущная потребность в новых, хорошо определенных, синтетических производных окисленных фосфолипидов и родственных веществах, обладающих более высокой метаболической устойчивостью и эффективной иммуномодуляцией, вызываемой, например, в результате перорального, внутривенного, внутрибрюшинного введения и введения через слизистую оболочку.
Синтез окисленных фосфолипидов
Модифицированые фосфолипиды находят разное применение. Например, фосфолипиды, несущие активные соединения, растворяющиеся в липидах, могут быть введены в композиции, предназначенные для чрескожного и трансмембранного введения (патент США № 5985292, выданный Fournerou et al.), и производные фосфолипидов могут быть введены в липосомы и биовекторы, предназначенные для доставки лекарственных средств (см., например, патенты США № 6261597 и 6017513, выданные соответственно Куртцу, Бетбедеру и др., и патент США № 4614796). В патенте США № 5660855 описаны липидные конструкции холестерина, производного аминоманнозы, которые заключены в липосомы и пригодны для направленного воздействия на клетки или ткани гладких мышц. Указанные препараты уменьшают рестеноз артерий методами РТСА. Использование липосом для лечения атеросклероза далее описано в заявке WO 95/23592 Хоупа и Родригеза, которые предложили фармацевтические композиции из однослойных липосом, содержащих фосфолипиды. Липосомы, описанные в заявке WO 95/23592, позволяют оптимизировать приток холестерина из атеросклеротических бляшек и обычно являются неокисленными фосфолипидами.
Известно, что производные модифицированных фосфолипидов, имитирующие структуру фактора активации тромбоцитов (PAF), оказывают фармацевтически активное действие в случае разных нарушений и заболеваний, влияющих на такие функции, как проницаемость сосудов, кровяное давление, работа сердца и т.д. Существует предположение, что одна группа указанных производных может обладать противораковым действием (патент США № 4778912, выданный Inoue et al.). В патенте США № 4329302 описаны синтетические соединения фосфоглицеридов, а именно производные лизолецитина, которые опосредуют активацию тромбоцитов. Хотя соединения, описанные в патенте США № 4329302, представляют собой простой 1-О-алкиловый эфир или ацилфосфоглицериды 1-О-жирных кислот, установлено, что ацилирование короткой цепи лизолецитина позволяет получить соединения, активирующие тромбоциты, в отличие от ацилирования длинной цепи, и что простой 1-О-алкиловый эфир в биологическом отношении превосходит ацильные производные 1-О-жирной кислоты с точки зрения имитации PAF.
Влияние структуры разных фосфолипидов на их биологическую активность было исследовано Токумура и др. (Tokumura et al., Journal of Pharmacology and Experimental Therapeutics. July 1981, Vol. 219, No. 1) и описано в патенте США № 4827011, выданном Висснеру и др., применительно к гипертензии.
Другая группа эфирных производных модифицированных фосфолипидов описана в Китайском патенте № 642665, выданном Берхтольду. Было установлено, что указанные эфирные производные модифицированных фосфолипидов пригодны для использования в хроматографическом разделении, но могут оказывать некоторое физиологическое действие.
Окисление фосфолипидов происходит in vivo под действием свободных радикалов и в результате ферментативных реакций, часто возникающих в атеросклеротической бляшке. Получение окисленных фосфолипидов in vitro обычно включает простое химическое окисление естественного LDL или фосфолипидного компонента LDL. Исследователи, изучающие роль окисленного LDL, использовали, например, ионы железа и аскорбиновую кислоту (Itabe, H., et al., J. Biol. Chem. 1996; 271:33208-217), а также сульфат меди (George, J. et al., Atherosclerosis. 1998; 138:147-152; Ameli, S. et al., Arteriosclerosis Thromb Vasc. Biol. 1996; 16:1074-79) для получения молекул окисленного или умеренно окисленного фосфолипида, аналогичных молекулам, ассоциированным с компонентами бляшек. Было установлено, что аналогично полученные молекулы идентичны аутоантигенам, ассоциированным с атерогенезом (Watson A.D et al., J. Biol. Chem. 1997; 272:13597-607), и способны индуцировать защитную иммунную толерантность против атерогенеза (заявка на патент США № 09/806400 на имя Шоенфелд и др., поданная 30 сентября 1999 г.) у мышей. Аналогичным образом Койке (Koike, патент США № 5561052) описывает способ получения окисленных липидов и фосфолипидов с использованием сульфата меди и пероксид-дисмутазы для получения окисленной арахидоновой или линолевой кислоты и окисленного LDL для диагностических целей. Дэвис и др. (Davies et al., J. Biol. Chem. 2001, 276:16015) описывают использование окисленных фосфолипидов в качестве агонистов активированных пролифератором рецепторов пероксисомы.
1-Пальмитоил-2-(5-оксовалероил)-sn-глицеро-3-фосфохолин (POVPC, см. пример I для ознакомления с описанием двухмерной структуры) и его производные, такие как 1-пальмитоил-2-глутароил-sn-глицеро-3-фосфохолин (PGPC), являются типичными примерами окисленных этерифицированных фосфолипидов, которые были исследованы в связи с атерогенезом (см., например, публикации Boullier et al., J. Biol Chem. 2000, 275:9163; Subbanagounder et al., Circulation Research, 1999, pp. 311). Было также исследовано действие других структурных аналогов, относящихся к данному классу окисленных фосфолипидов (см., например, Subbanagounder et al., Arterioscler. Thromb. Nasc. Biol. 2000, pp. 2248; Leitinger et al., Proc. Nat. Ac. Sci. 1999, 96:12010).
Однако недостатком применения in vivo вышеуказанных окисленных фосфолипидов является узнавание, связывание и метаболизм активного компонента в организме, в результате чего важное значение приобретают соображения дозирования и устойчивости после введения.
Кроме того, применяемые методы окисления являются недостаточно специфичными, в результате чего образуются разные окисленные продукты, что делает необходимым дальнейшую очистку или использование неочищенных антигенных соединений. Данная проблема становится еще сложнее в случае естественного LDL, даже если он очищен.
Таким образом, существует общепризнанная потребность в новых синтетических окисленных фосфолипидах, усовершенствованных способах синтеза указанных фосфолипидов и их применении в качестве иммуномодуляторов при отсутствии вышеуказанных ограничений.
Сущность изобретения
Одним объектом настоящего изобретения является соединение общей формулы I
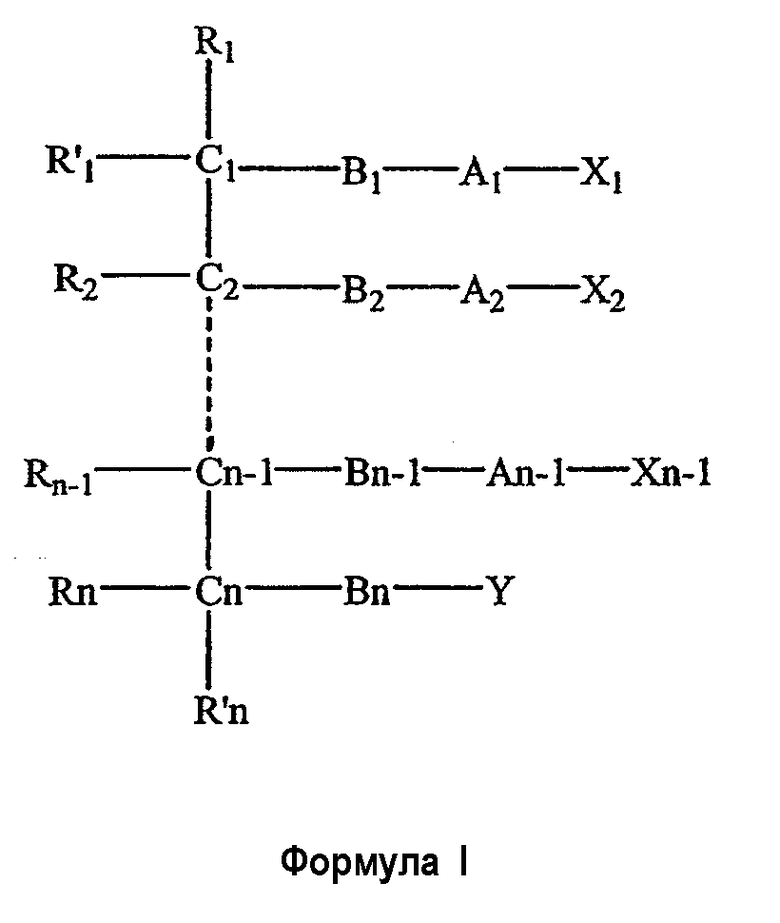
где
n является целым числом от 1 до 6, причем, если n=1, Cn, Bn, Rn, R'n и Y отсутствуют;
каждый из B1, B2, …Bn-1 и Bn независимо выбирают из группы, состоящей из кислорода, серы, азота, фосфора и кремния, причем каждый из указанного азота, фосфора и кремния замещен по крайней мере одним заместителем, выбранным из группы, состоящей из водорода, электронов неподеленной пары, алкила, галогена, циклоалкила, арила, гидрокси, тиогидрокси, алкокси, арилокси, тиоарилокси, тиоалкокси и оксо;
каждый из А1, А2, …An-1 и An независимо выбирают из группы, состоящей из CR''R''', C=O и С=S;
Y выбирают из группы, состоящей из водорода, алкила, арила, циклоалкила, карбокси, сахарида, фосфорной кислоты, фосфорилхолина, фосфорилэтаноламина, фосфорилсерина, фосфорилкардиолипина, фосфорилинозита, этилфосфохолина, фосфорилметанола, фосфорилэтанола, фосфорилпропанола, фосфорилбутанола, фосфорилэтаноламин-N-лактозы, фосфоэтаноламин-N-[метокси(пропиленгликоль)], фосфоинозит-4-фосфата, фосфоинозит-4,5-бифосфоната, пирофосфата, фосфоэтаноламиндиэтилентриаминпентаацетата, динитрофенилфосфоэтаноламина и фосфоглицерина; и
каждый из Х1, Х2, …Xn-1 независимо является насыщенным или ненасыщенным углеводородом общей формулы II
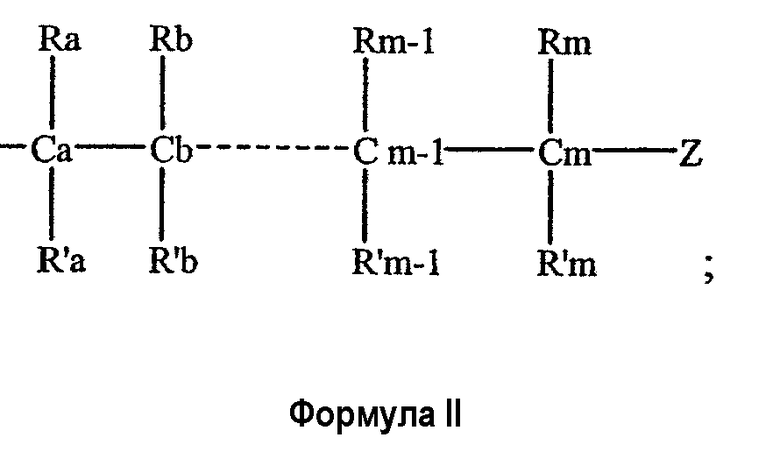
где
m является целым числом от 1 до 26; и
Z выбирают из группы, состоящей из
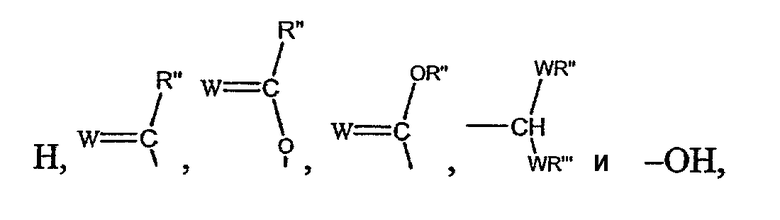
где
W выбирают из группы, состоящей из кислорода, серы, азота и фосфора, каждый из указанного азота и фосфора замещен по крайней мере одним заместителем, выбранным из группы, состоящей из водорода, электронов неподеленной пары, алкила, галогена, циклоалкила, арила, гидрокси, тиогидрокси, алкокси, арилокси, тиоарилокси, тиоалкокси и оксо; и
по крайней мере один из Х1, Х2, …Xn-1 Z не является водородом;
и где
каждый из R1, R'1, R2, …Rn-1, Rn, R'n, каждый из R'' и R''' и каждый из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm независимо выбирают из группы, состоящей из водорода, связи, алкила, алкенила, алкинила, циклоалкила, арила, гетероарила, гетероалициклической группы, галогена, тригалогенметила, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, фосфоната, фосфата, фосфинила, сульфонила, сульфинила, сульфонамида, амида, карбонила, тиокарбонила, С-карбокси, О-карбокси, С-карбамата, N-карбамат, С-тиокарбокси, S-тиокарбокси и амино, или альтернативно по крайней мере два из R1, R'1, R2, …Rn-1, Rn и R'n и/или по крайней мере два из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm образуют по крайней мере одно четырех-, пяти- или шестичленное ароматическое, гетероароматическое, алициклическое или гетероалициклическое кольцо; и
каждый из С1, С2, …Cn-1, Cn и каждый из Са, Cb, …Cm-1 и Cm является хиральным или нехиральным атомом углерода, каждый хиральный атом углерода имеет S-конфигурацию и/или R-конфигурацию;
его фармацевтически приемлемая соль, пролекарство, гидрат или сольват.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения по крайней мере один из А1, А2, … и An-1 означает CR''R''' и по крайней мере один из А1, А2, … и An-1 связан с Х1, Х2 … или Xn-1, который включает Z, не являющийся водородом.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения n равно 3 и по крайней мере один из А1 и А2 означает CR''R'''. Предпочтительно А2 означает CR''R''' и Х2 включает Z, не являющийся водородом. Далее предпочтительно оба А1 и А2 означают CR''R'''.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения Z выбирают из группы, состоящей из
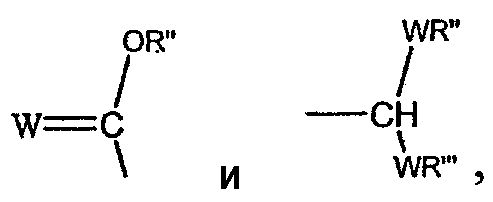
где W предпочтительно означает кислород и оба R'' и R''' независимо выбирают из группы, состоящей из водорода и алкила.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения n равно 1 и по крайней мере один из R1 и R'1 означает фосфат или фосфонат.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения n равно 5 или 6 и по крайней мере один из R1, R'1 и по крайней мере один из Rn и R'n образуют по крайней мере одно гетероалициклическое кольцо, например, моносахаридное кольцо.
Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая в качестве активного ингредиента описанное выше соединение и фармацевтически приемлемый носитель.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения фармацевтическая композиция упакована в упаковочный материал и идентифицирована оттиском, сделанным в упаковочном материале, или надписью, нанесенной на указанный упаковочный материал, как препарат, предназначенный для лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом, как это подробно описано ниже.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения фармацевтическая композиция далее содержит по крайней мере одно дополнительное соединение, пригодное для лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом, как это подробно описано ниже.
Другим объектом настоящего изобретения является способ лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом, который включает введение нуждающемуся субъекту терапевтически эффективного количества по крайней мере одного окисленного липида, позволяющего лечить или предотвратить у субъекта воспаление, ассоциированное с эндогенным окисленным липидом.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения окисленный липид выбирают из группы, состоящей из окисленного фосфолипида, фактора активации тромбоцитов, плазмалогена, замещенного или незамещенного углеводорода, содержащего 3-30 атомов углерода и концевой окисленной группы, окисленного сфинголипида, окисленного гликолипида, окисленного мембранного липида и любого их аналога или производного.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения окисленный липид имеет вышеуказанную общую формулу I.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения окисленный липид выбирают из группы, состоящей из 1-пальмитоил-2-азелаоил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-азелаоил-sn-глицеро-3-фосфохолина, 1-пальмитоил-2-глутароил-sn-глицеро-3-фосфохолина (PGPC), 1-пальмитоил-2-(5-оксовалероил)-sn-глицеро-3-фосфохолина (POVPC), 1-пальмитоил-2-(9-оксононаноил)-sn-глицеро-3-фосфохолина, 1-гексадецил-2-ацетоил-sn-глицеро-3-фосфохолина, 1-октадецил-2-ацетоил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-бутироил-sn-глицеро-3-фосфохолина, 1-октадецил-2-бутироил-sn-глицеро-3-фосфохолина, 1-пальмитоил-2-ацетоил-sn-глицеро-3-фосфохолина, 1-октадеценил-2-ацетоил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-(гомогаммалиноленоил)-sn-глицеро-3-фосфохолина, 1-гексадецил-2-арахидоноил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-эйкозапентаеноил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-докозагексаеноил-sn-глицеро-3-фосфохолина, 1-октадецил-2-метил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-бутеноил-sn-глицеро-3-фосфохолина, Lyso PAF C16, Lyso PAF C18, 1-O-1'-(Z)-гексадеценил-2-[12-[(7-нитро-2-1,3-бензоксадиазол-4-ил)амино]-додеканоил]-sn-глицеро-3-фосфохолина, 1-О-1'-(Z)-гексадеценил-2-олеоил-sn-глицеро-3-фосфохолина, 1-О-1'-(Z)-гексадеценил-2-арахидоноил-sn-глицеро-3-фосфохолина, 1-О-1'-(Z)-гексадеценил-2-докозагексаеноил-sn-глицеро-3-фосфохолина, 1-О-1'-(Z)-гексадеценил-2-олеоил-sn-глицеро-3-фосфоэтаноламина, 1-О-1'-(Z)-гексадеценил-2-арахидоноил-sn-глицеро-3-фосфоэтаноламина и 1-О-1'-(Z)-гексадеценил-2-докозагексаеноил-sn-глицеро-3-фосфоэтаноламина.
В соответствии с другими признаками предпочтительных вариантов осуществления изобретения указанный способ далее включает введение субъекту терапевтически эффективного количества по крайней мере одного дополнительного соединения, предназначенного для лечения или профилактики воспаления, ассоциированного с эндогенным окисленным LDL.
По крайней мере одно дополнительное соединение предпочтительно выбирают из группы, включающей ингибитор HMGCoA-редуктазы (статин), адъювант слизистой оболочки, кортикостероид, средство стероидной противовоспалительной терапии, средство нестероидной противовоспалительной терапии, аналгетик, фактор роста, токсин, HSP, бета-2-гликопротеин I, ингибитор белка, обеспечивающего перенос сложного холестерилового эфира (CETP), агонист пролиферирующего активированного рецептора периксосомы (PPAR), противоатеросклерозное средство, антипролиферативное средство, эзетимид, никотиновую кислоту, ингибитор сквалена, ApoE Milano и любое их производное и аналог.
Воспаление по настоящему изобретению ассоциировано с заболеваниями и нарушениями, такими как, например, идиопатические воспалительные заболевания или нарушения, хронические воспалительные заболевания или нарушения, острые воспалительные заболевания или нарушения, аутоиммунные заболевания или нарушения, инфекционные заболевания или нарушения, воспалительные злокачественные заболевания или нарушения, воспалительные заболевания или нарушения, связанные с трансплантацией, воспалительные дегенеративные заболевания или нарушения, заболевания или нарушения, обусловленные гиперчувствительностью, воспалительные сердечно-сосудистые заболевания или нарушения, воспалительные цереброваскулярные заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительные заболевания или нарушения желез, воспалительные желудочно-кишечные заболевания или нарушения, воспалительные кожные заболевания или нарушения, воспалительные заболевания или нарушения печени, воспалительные неврологические заболевания или нарушения, воспалительные заболевания или нарушения костно-мышечной системы, воспалительные заболевания или нарушения почек, воспалительные заболевания или нарушения репродуктивной системы, воспалительные системные заболевания или нарушения, воспалительные заболевания или нарушения соединительной ткани, воспалительные опухоли, некрозы, воспалительные заболевания или нарушения, связанные с имплантатом, воспалительные процессы, связанные со старением, заболевания или нарушения, обусловленные иммунодефицитом, пролиферативные заболевания и нарушения, а также воспалительные легочные заболевания или нарушения, как это подробно описано ниже.
Настоящее изобретение позволяет успешно преодолеть известные в настоящее время недостатки благодаря созданию новых синтетических окисленных липидов, устранить ограничения, связанные с известными в настоящее время синтетическими окисленными липидами и способами лечения или профилактики воспалений, ассоциированных с эндогенным окисленным липидом, при использовании синтетических окисленных липидов.
За исключением особо оговоренных случаев, все технические и научные термины, использованные в данном описании изобретения, имеют значения, обычно употребляемые специалистами в области, к которой относится данное изобретение. Ниже дано описание приемлемых способов и веществ, хотя при осуществлении на практике или проверке настоящего изобретения можно использовать способы и вещества, аналогичные или эквивалентные приведенным в данном описании изобретения. В случае возникновения противоречия необходимо обратиться к патенту с описанием изобретения, включающему определения терминов. Кроме того, вещества, способы и примеры приведены только для иллюстрации и не ограничивают объем изобретения.
Краткое описание чертежей
Настоящее изобретение описано только в виде примера со ссылкой на прилагаемые чертежи. При подробном рассмотрении чертежей необходимо подчеркнуть, что отдельные детали приведены только в качестве примера и для иллюстрации предпочтительных вариантов осуществления настоящего изобретения и представлены с учетом наиболее приемлемого и легко понимаемого описания принципов и концептуальных объектов данного изобретения. В данной связи следует отметить, что не ставилась цель более подробного описания структурных деталей изобретения, чем это необходимо для обоснованного понимания изобретения, при этом специалистам в данной области должно быть понятно из описания изобретения в сочетании с чертежами, каким образом можно осуществить на практике несколько вариантов данного изобретения.
На фиг.1 изображена блок-схема, на которой показан синтез простого эфира 2,5'-альдегид-лецитина, 1-гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолина (для D-ALLE) или 3-гексадецил-2-(5'-оксопентанил)-sn-глицеро-1-фосфохолина (для L-ALLE) (ALLE) способом синтеза по настоящему изобретению.
На фиг.2 изображена блок-схема, показывающая синтез POVPC по настоящему изобретению.
На фиг.3 изображена диаграмма, показывающая ингибирование начальной стадии атерогенеза у мышей с отсутствием apoE при помощи внутрибрюшинной иммунизации смешанными D- и L-изомерами ALLE. Мышей Apo-E KO в возрасте 5-7 недель иммунизировали 150 мкг/мышь смешанных D- или L-изомеров ALLE, связанных с очищенным производным белка туберкулина (ALLE L+D) (n=6), только очищенного производного белка туберкулина (PPD) (n=5) или не иммунизировали (контрольная группа) (n=7). Атерогенез выражен в виде площади атероматозных поражений в синусе аорты через 4,5 недели после 4-й иммунизации.
На фиг.4 изображена диаграмма, показывающая ингибирование начальной стадии атерогенеза у мышей Apo-E KO при пероральном введении ALLE. Мышам Apo-E KO в возрасте 6-7,5 недель перорально вводили смешанные D- и L-изомеры ALLE: 10 мкг/мышь (ALLE L+D 10 мкг) (n=11) или 1 мг/мышь (ALLE L+D 1 мг) (n=11) либо PBS (контрольная группа) (n=12) через день на протяжении 5 дней. Атерогенез выражен в виде площади атероматозных поражений в синусе аорты через 8 недель после последнего перорального введения.
На фиг.5 изображена диаграмма, показывающая ингибирование начальной стадии атерогенеза у мышей Apo-E KO при пероральном и назальном введении L-ALLE. Мышам Аро-Е КО в возрасте 7-10 недель перорально вводили 1 мг/мышь L-ALLE через день на протяжении 5 дней (OT L-ALLE) (n=11) или вводили в нос 10 мкг/мышь L-ALLE через день на протяжении 3 дней (NT L-ALLE) (n=11). Контрольным мышам перорально вводили такой же объем (0,2 мл) PBS (PBS ORAL) (n=12). Атерогенез выражен в виде площади атероматозных поражений в синусе аорты через 8 недель после последнего перорального или назального введения.
На фиг.6 изображена диаграмма, показывающая супрессию иммунной реактивности в отношении антигенов атеросклеротических бляшек, индуцируемую при пероральном введении синтетических окисленных фосфолипидов L-ALLE и POVPC. Мышам Аро-Е КО в возрасте 6 недель перорально вводили 1 мг/мышь L-ALLE (L-ALLE) (n=2) или POVPC (POVPC) (n=3) в 0,2 мл PBS либо только PBS (контрольная группа) (n=3) через день на протяжении 5 дней. Через одну неделю после последнего перорального введения мышей иммунизировали при помощи одной подкожной инъекции 50 мкг антигена окисленного LDL человека. Через 7 дней получали Т-клетки из лимфатического узла паховой области в соответствии с описанием, приведенным в нижеследующем разделе “Материалы и методы”, которые подвергали воздействию сенсибилизирующего антигена ox-LDL человека для анализа пролиферации in vitro. Пролиферация, являющаяся показателем иммунной реактивности, выражена в виде соотношения встраивания меченого тимидина в ДНК Т-клетки в присутствии и отсутствие антигена ox-LDL человека (индекс стимуляции, S.I.).
На фиг.7 изображена диаграмма, показывающая ингибирование развития поздней стадии атерогенеза у мышей Аро-Е КО при пероральном введении синтетических окисленных фосфолипидов D-ALLE, L-ALLE или POVPC. Мышам Аро-Е КО в возрасте 24,5 недель перорально вводили 1 мг/мышь L-ALLE (L-ALLE) (n=11), D-ALLE (D-ALLE) (n=9) или POVPC (POVPC) (n=10) через день на протяжении 5 дней с интервалами в 4 недели в течение 12 недель. Контрольным мышам перорально вводили такой же объем (0,2 мл) PBS (контрольная группа) (n=10) в соответствии с аналогичной схемой введения. Атерогенез выражен в виде площади атероматозных поражений в синусе аорты через 12 недель после первого перорального введения по сравнению с оценками поражения у неиммунизированных мышей в возрасте 24,5 недель до первого перорального введения (умерщвленных в 0-й период времени).
На фиг.8 изображен график, показывающий уменьшение содержания триглицерида в VLDL у мышей Аро-Е КО, индуцированное при пероральном введении синтетических окисленных фосфолипидов D-ALLE, L-ALLE или POVPC. Мышам Аро-Е КО в возрасте 24,5 недель перорально вводили 1 мг/мышь L-ALLE (треугольник) (n=11), D-ALLE (перевернутый треугольник) (n=9) или POVPC (квадрат) (n=10) через день на протяжении 5 дней с интервалами в 4 недели в течение 12 недель. Контрольным мышам перорально вводили такой же объем (0,2 мл) PBS (кружок) (n=10) в соответствии с аналогичной схемой введения. Содержание триглицерида (Tg, мг/мл) измеряли через 9 недель после времени t=0 ферментативным колориметрическим методом во фракциях VLDL после разделения собранных проб крови методом FPLC в соответствии с описанием, приведенным в нижеследующем разделе “Материалы и методы”.
На фиг.9 изображен график, показывающий уменьшение содержания холестерина в VLDL у мышей Аро-Е КО, индуцированное при пероральном введении синтетических окисленных фосфолипидов D-ALLE, L-ALLE или POVPC. Мышам Аро-Е КО в возрасте 24,5 недель перорально вводили 1 мг/мышь L-ALLE (треугольник) (n=11), D-ALLE (переверенутый треугольник) (n=9) или POVPC (квадрат) (n=10) через день на протяжении 5 дней с интервалами в 4 недели в течение 12 недель. Контрольным мышам перорально вводили такой же объем (0,2 мл) PBS (кружок) (n=10) в соответствии с аналогичной схемой введения. Содержание холестерина (холестерин, мг/мл) измеряли через 9 недель после времени t=0 ферментативным колориметрическим методом во фракциях VLDL после разделения собранных проб крови методом FPLC в соответствии с описанием, приведенным в нижеследующем разделе “Материалы и методы”.
На фиг.10 изображена двухмерная структура 1-гексадецил-2-(5'-карбоксибутил)-sn-глицеро-3-фосфохолина (CI-201, соединение VII), 1-гексадецил-2-(5',5'-диметоксипентилокси)-sn-глицеро-3-фосфохолина (соединение VIIIa) и 1-гексадецил-2-(5',5'-диэтоксипентилокси)-sn-глицеро-3-фосфохолина (соединение VIIIb).
На фиг.11 изображена диаграмма, показывающая ингибирование начальной стадии атерогенеза у мышей Аро-Е КО при пероральном введения CI-201. Мышам Аро-Е КО в возрасте 12 недель перорально вводили CI-201: 0,025 мг/мышь (n=14) или 0,2 мл PBS (контрольная группа) (n=15) каждый день на протяжении 8 недель (5 раз в неделю). Атеросклероз выражен в виде площади стероматозного поражения в синусе аорты через 11 недель после первого перорального введения.
На фиг.12 а-d представлены фотографии, показывающие уровни экспрессии цитокина в аорте мышей, которым вводили ALLE, CI-201, его этилацетальное производное (Et-ацеталь), его метилацетальное производное (Ме-ацеталь), oxLDL или PBS. В частности, на фигурах 12а и 12b показано увеличение уровня экспрессии IL-10 в аорте мышей, получавших ALLE, CI-201, Et-ацеталь, Ме-ацеталь и oxLDL, по сравнению с контрольными мышами (PBS) и снижение уровней экспрессии IFN-гамма в аортах мышей, которым вводили ALLE, CI-201, Ме-ацеталь и oxLDL, по сравнению с мышами, получавшими PBS; на фигурах 12с и 12d показано уменьшение экспрессии IL-12 у мышей, которым вводили ALLE, CI-201 и Et-ацеталь, по сравнению с группой, получавшей PBS. Мышам Аро-Е КО в возрасте 10-12 недель перорально вводили 1 мг/мышь/0,2 мл испытуемого антигена (ALLE, CI-201, Et-ацеталь, Ме-ацеталь) или 0,1 мг/мышь/0,2 мл oxLDL либо вводили 0,2 мл PBS. Пероральное введение производили 5 раз через день и экспрессию цитокина оценивали через 8 недель после последнего перорального введения.
На фиг.13 изображена столбчатая диаграмма, показывающая ослабление атерогенеза у мышей LDL-RD при пероральном введении oxLDL. Мышам LDL-RD перорально вводили PBS или 10, 100 и 1000 мкг/дозу oxLDL 5 раз через день. Атерогенез выражен в виде площади атероматозных поражений в синусе аорты через 5 недель после последнего перорального введения.
На фиг.14 а-b изображены столбчатые диаграммы, показывающие ингибирование атерогенеза у мышей Аро-Е КО при пероральном введения CI-201. Мышам Аро-Е КО перорально вводили PBS (контрольная группа) или 0,1, 1 и 10 мкг/дозу CI-201 в виде трех серий в начале каждого месяца, 5 раз через день в каждой серии. Атерогенез выражен в виде площади атероматозных поражений в синусе аорты через 12 недель после первого перорального введения. На фигуре 14а показана степень атеросклероза в каждой группе. На фигуре 14b показано сильное влияние низкой дозы CI-201 на атеросклероз по сравнению с “базовой” группой (умерщвленной в 0-й день) и контрольной группой.
На фиг.15 а-b изображены столбчатые диаграммы, показывающие повышение уровней IL-10 в сыворотке (фигура 15а) и предотвращение повышения SAA (фигура 15b) у мышей Аро-Е КО, иммунизированных CI-201. Мышам Аро-Е КО перорально вводили PBS (контрольная группа) или CI-201 5 раз через день. Сыворотку собирали в начале эксперимента, через 2 недели и 4 недели после первого перорального введения. Уровни маркеров оценивали в соответствии с описанием, приведенным в нижеследующем разделе “Материалы и методы”.
На фиг.16 а-b представлены фотографии (фигура 16а) и диаграмма (фигура 16b), на которых показаны уровни экспрессии цитокина в аорте мышей, получавших CI-201 или PBS. В частности, на фигурах 16а и 16b показано повышение уровня экспрессии IL-10 в аорте мышей, получавших CI-201, по сравнению с контрольными мышами (PBS), и уменьшение уровней экспрессии IFN-гамма в аортах мышей, получавших CI-201, по сравнению с мышами, которым вводили PBS. Мышам Аро-Е КО перорально вводили 1 мг/мышь CI-201 или 0,2 мл/мышь PBS 5 раз через день. Экспрессию противовоспалительного цитокина IL-10 и провоспалительного цитокина IFN-γ определяли через 8 недель после последнего перорального введения.
На фиг.17 представлены фотографии, показывающие результаты перорального введения CI-201, направленно воздействующего на аорту, мышам Аро-Е КО. Хотя введение CI-201, воздействующего на аорту, вызывало повышение уровня экспрессии IL-10 и снижение уровней экспрессии IFN-гамма по сравнению с введением PBS, не наблюдалось различий в экспрессии цитокина в селезенке и тонком кишечнике между группой, получавшей CI-201, и контрольной группой, получавшей PBS.
На фиг.18 изображена схема исследования для оценки ослабления адъювант-индуцированного артрита (AIA) у крыс, которым предварительно вводили CI-201.
На фиг.19 изображена столбчатая диаграмма, показывающая результат перорального введения CI-201 крысам, страдающим адъювант-индуцированным артритом (AIA), в виде оценки опухания лап. Крысам Lewis перорально вводили CI-201 или PBS (контрольная группа) 5 раз через день, после чего внутрикожно инъецировали суспензию туберкулезной микробактерии.
На фиг.20 изображена схема исследования для оценки ослабления адъювант-индуцированного артрита (AIA) у крыс, которым постоянно вводили CI-201.
На фиг.21 изображена столбчатая диаграмма, показывающая результат перорального введения CI-201 у крыс Lewis, страдающих AIA-индуцированным артритом, в виде оценки опухания лап. Крысам Lewis перорально вводили CI-201 или PBS (контрольная группа) 5 раз через день, после чего у них индуцировали артрит и постоянно перорально вводили CI-201 3 раза в неделю.
На фиг.22 изображены сравнительные графики, показывающие оценки артрита, контролируемые в процессе развития артрита у крыс, которым вводили CI-201 в разных концентрациях, по сравнению с крысами, которым вводили PBS.
На фиг.23 изображены сравнительные графики, показывающие процентное значение крыс с симптомами артрита после введения PBS (контрольная группа) и CI-201 в разных концентрациях.
На фиг.24 изображена столбчатая диаграмма, показывающая воздействие на раннюю стадию атерогенеза у мышей Аро-Е КО, индуцированное при пероральном введении предварительно окисленного соединения V. Самкам мышей Аро-Е КО в возрасте 8-10 недель перорально вводили соединение V: 5 мг/мышь (n=6), 1 мг/мышь (n=6), 0,2 мг/мышь (n=6) или PBS (контрольная группа) (n=7) через день на протяжении 5 дней. Атерогенез выражен в виде площади атероматозных поражений в синусе аорты через 8 недель после последнего перорального введения.
На фиг.25 изображена столбчатая диаграмма, показывающая воздействие на атерогенез у мышей Аро-Е КО, индуцированное при пероральном введении предварительно окисленного соединения V. Мышей Аро-Е КО в возрасте 23-26 недель умерщвляли в начале эксперимента (базовая группа B.L., n=10) либо вводили им PBS (контрольная группа, n=11) или 0,1 мкг/дозу соединения V (n=10) в виде трех серий в начале каждого месяца 5 раз через день в каждой серии. Атерогенез выражен в виде площади атероматозных поражений в синусе аорты через 12 недель после первого перорального введения.
Описание предпочтительных вариантов осуществления изобретения
Настоящее изобретение относится к композициям, содержащим окисленные липиды, и к способам их применения для лечения или профилактики воспаления, ассоциированного с эндогенными окисленными липидами. В частности, объектами настоящего изобретения являются (i) новые окисленные липиды; (ii) фармацевтические композиции, содержащие указанные липиды; (iii) способы применения новых окисленных липидов, а также других окисленных липидов для лечения или профилактики воспаления, ассоциированного с эндогенными окисленными липидами, и, как следствие этого, для лечения или профилактики заболеваний и нарушений, характеризующихся воспалением, которые включают, не ограничиваясь ими, атеросклероз, сердечно-сосудистые заболевания, цереброваскулярные заболевания, заболевания периферических сосудов, стеноз, рестеноз, стеноз в стенте, аутоиммунные заболевания или нарушения, воспалительные заболевания или нарушения, инфекционные заболевания или нарушения и пролиферативные заболевания или нарушения.
Принципы и способы осуществления настоящего изобретения могут быть лучше поняты со ссылкой на прилагаемые чертежи и описание изобретения.
Прежде чем перейти к подробному описанию по крайней мере одного варианта осуществления изобретения, необходимо отметить, что данное изобретение не ограничивается применением, подробно рассмотренным в нижеследующем описании или проиллюстрированным примерами. Данное изобретение включает другие варианты осуществления или может быть реализовано на практике разными способами. Кроме того, должно быть понятно, что фразеология и терминология, используемая в описании изобретения, служит только для описания и не ограничивает объем изобретения.
Экспериментальные и клинические данные свидетельствуют о прямом участии окисленного LDL (ox LDL) и компонентов LDL в этиологии повышенной воспалительной реакции в случае атеросклероза. Была продемонстрирована как клеточная, так и гуморальная иммунная реактивность в отношении окисленного LDL, связанного с бляшками, что позволяет предположить возникновение важного аутоиммунного компонента против окисленного LDL в процессе атерогенеза. Таким образом, против LDL, окисленного LDL и их компонентов были направлены многочисленные методы профилактики и лечения болезней сердца, сердечно-сосудистых заболеваний и заболеваний периферических сосудов.
В ранее выполненных исследованиях роли окисленного LDL и его компонентов в ослаблении иммунной реакции на эндогенный (например, связанный с бляшками) окисленный LDL использовали неочищенный антигенный препарат, содержащий центрифугированный, фильтрованный и очищенный LDL сыворотки человека, который подвергали длительному процессу окисления Cu++ или MDA, или синтезированные аналоги окисленного LDL. Так как фосфолипиды считаются активными компонентами LDL, исследования с использованием синтезированных аналогов окисленного LDL обычно включают применение окисленных фосфолипидов (например, POVPC и PGPC).
Хотя ранее считалось, что при пероральном введении окисленного LDL может быть достигнуто 30% уменьшение атерогенеза, что свидетельствует о защитном действии окисленного LDL, главным образом, вследствие пероральной толерантности, не были идентифицированы специфические липидные антигены или иммуногенные компоненты LDL. Другим препятствием, с которым ранее сталкивались исследователи, является неустойчивость, свойственная неочищенному окисленному LDL in vivo, вследствие ферментативной активности и поглощения окисленного LDL печенью и иммунными механизмами клеток. Такая неустойчивость свойственна также применению in vivo синтетических производных окисленного LDL, таких как POVPC и PGPC (описанных выше).
Поэтому до сих пор не была выявлена прямая корреляция между экзогенным окисленным LDL или его компонентами и эндогенным окисленным LDL в отношении иммуномодуляции. Ранее не были также обнаружены аналоги окисленного LDL, у которых отсутствует присущая им неустойчивость и другие ограничения, характерные для введения окисленного LDL, способного модулировать иммунную и/или воспалительную реакцию, ассоциированную с эндогенным окисленным LDL и другими эндогенными окисленными липидами.
В процессе создания настоящего изобретения было сделано предположение, что синтезированные окисленные липиды вообще и аналоги окисленного LDL, в частности, могут модулировать иммунную реактивность в отношении эндогенных окисленных липидов вообще и окисленного LDL, в частности, и, таким образом, могут быть использованы для лечения или профилактики многих заболеваний и нарушений, характеризующихся воспалением и/или измененной иммунной реакцией, таких как, например, атеросклероз и родственные заболевания или нарушения, а также других заболеваний и нарушений, ассоциированных с эндогенным окисленным липидом.
Воспаление, возникающее в процессе атерогенеза, часто ведет к осложнениям, таким как разрушение бляшек и тромбоз (Libby et al., Inflammation and atherosclerosis. Circulation 2002; 105:1135-1143).
Присутствие активированных Т-лимфоцитов в месте атеросклеротического поражения человека может указывать на их участие в возникновении и развитии заболевания (Ross R. Atherosclerosis-an inflammatory disease. NEJM. 1999; 340:115-126). Основной класс Т-лимфоцитов, CD4+, можно дифференцировать по линиям Th1 или Th2, которые функционально отличаются продуцированным цитокином: интерфероном (IFN)-γ, секретируемым клетками Th1, и интерлейкином (IL)-4, секретируемым клетками Th2. Главными индукторами клеток Th1 и Th2 являются соответственно IL-12 и IL-10 (Daugherty A. and Rateri DL., T lymphocytes in atherosclerosis the Yin-Yang of Th1 and Th2 Influence on lesion formation, Circ. Res. 2002; 90:1039-1040; Hansson G.K., Vaccination against atherosclerosis science or fiction. Circulation. 2002; 106:1599-1601).
Установлено, что Т-лимфоциты, выделенные из цельной крови субъектов, страдающих острым коронарным синдромом, или полученные из бляшек сонной артерии, специфически узнают Ox LDL и пролиферируют под воздействием Ox LDL (Hansson G.K. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc. Biol. 2001; 21:1876-90). Ox LDL и побочные продукты окисленных липидов (например, окисленные фосфолипиды) присутствуют в атеросклеротических бляшках (Witztum 2001, supra).
Следовательно, несмотря на то, что окислительная модификация LDL может быть необходимым условием быстрого накопления LDL в макрофагах и образования ксантомных клеток, она может также индуцировать иммуногенные эпитопы в молекуле LDL, вызывающие образование антител против Ox LDL.
Поэтому эпитопы окисленного LDL служат в качестве важных лигандов, опосредующих связывание и выведение поврежденных окислением частиц липопротеина и апоптозных клеток и индуцирующих естественную иммунную реакцию, вызывающую их удаление. С другой стороны, эпитопы окисленного LDL могут участвовать в иммунной активации, которая характеризует нарастающую атеросклеротическую бляшку.
С учетом вышеизложенного авторы настоящего изобретения пришли к выводу, что соединения, способные служить в качестве эпитопов окисленного LDL, могут модулировать иммунную реакцию, оказывая скорее благоприятное, чем вредное влияние на атерогенез. Другими словами, было установлено, что введение, предпочтительно пероральное, аналогов окисленного LDL, таких как окисленные фосфолипиды, вызывает толерантность к эндогенному окисленному LDL, образуемому в процессе атерогенеза и, таким образом, ослабляет воспалительную реакцию и замедляет развитие атерогенеза.
Недавно были опубликованы данные, подтверждающие возможность применения иммуномодуляции в качестве нового терапевтического подхода к лечению атеросклероза (Nicoletti et al., Induction of neonatal tolerance to oxidized lipoprotein reduces atherosclerosis in Apo E knockout (Apo-E KO) mice. Mol. Med.2000; 6(4):283-290). Было установлено, что внутрибрюшинная инъекция окисленного LDL новорожденным мышам (Аро-Е КО) вызывала Т-клеточную толерантность вследствие делеции клона, ослабление иммунной реакции на окисленный LDL и в результате этого пониженную восприимчивость к атеросклерозу.
В патогенезе атеросклероза, а также многих других заболеваний и нарушений участвует приобретенный и природный иммунитет. С учетом большого содержания липидов в месте поражения указанные липиды могут быть возможными мишенями для иммунной реакции, вызываемой атеросклерозом. Недавно было показано, что естественные Т-клетки-киллеры (NKT) могут узнавать липидные антигены, представляемые молекулами CD1. Молекулы CD1 представляют липидные антигены Т-клеткам в отличие от эволюционно родственных молекул главного комплекса гистосовместимости (МНС) класса I и II, которые показывают пептидные антигены. Однако подобно молекулам МНС класса I молекулы CD1 включают тяжелую цепь, связанную с легкой цепью β2-микроглобулина (β2М). Кристаллические структуры двух изоформ CD1, CD1b человека и CD1d мыши, характеризуются общей организацией доменов, напоминающей молекулы МНС класса I. В частности, антигенсвязывающий сайт в CD1 является гидрофобным, образующим каналы (CD1b) или карманы (CD1d мыши), в которых могут располагаться углеводородные цепи липидов. Узкое отверстие между α-спиралями позволяет отображать полярные части липида в области, доступной для узнавания рецепторами Т-клеток (TCR). Данная система облегчает связывание разных липидных молекул, присоединенных к разным полярным головным группам, создавая таким образом большой пул потенциальных CD1-представляемых антигенов (Zeng et al., Crystal structure of mouse CD1: an MHC-like fold with a large hydrophobic binding groove. Science 1997; 277:339-345).
Молекулы CD1 связывают антигены чужеродных липидов по мере того, как они обследуют эндосомные компартменты инфицированных антигенпредставляющих клеток. В отличие от Т-клеток, которые узнают CD1-ограниченные чужеродные липиды, CD1-ограниченные Т-клетки, взаимодействующие с аутоантигенами, функционируют в качестве “аутоэффекторов”, которые могут быть быстро стимулированы для выполнения хелперных и эффекторных функций при взаимодействии с CD1-экспрессирующими антиген-представляющими клетками. Функциональные отличия между субпопуляциями CD1-ограниченных Т-клеток и путями, по которым указанные клетки влияют на воспалительное и толерогенное действие дендритных клеток и активируют естественные клетки-киллеры и другие лимфоциты, позволяют определить, каким образом CD1-ограниченные Т-клетки регулируют антимикробные реакции, противоопухолевый иммунитет и баланс между толерантностью и аутоиммунитетом (Vincent et al., Understanding the function of CD1-restricted T cells. Nat. Immunol. 2003; 4:517-23).
Тупин и др. (Tupin et al., CD1d-dependent Activation of NKT Cells Aggravates Atherosclerosis. J. Exp. Med. 2004; 199:417-22) исследовали роль CD1d-ограниченных естественных Т-клеток-киллеров в атеросклерозе, используя мышей с отсутствием аполипопротеина Е (ароЕ(-/-) и мышей АроЕ(-/-), скрещенных с мышами CD1d(-/-), (CD1d(-/-)apoE(-/-)), у которых наблюдалось 25% уменьшение величины поражения по сравнению с мышами ароЕ(-/-). Введение альфа-галактозилцерамида, синтетического гликолипида, активирующего естественные Т-клетки-киллеры с помощью CD1d, вызывало 50% увеличение величины поражения у мышей ароЕ(-/-) и при этом не влияло на величину поражения у мышей ароЕ(-/-)CD1d(-/-). Полученные результаты показывают, что активация CD1d-ограниченных естественных Т-клеток-киллеров обостряет атеросклероз. Жоу и др. (Zhou et al., Editing of CD1d-bound lipid antigens by endosomal lipid transfer proteins. Science. 2004; 303:523-7) обнаружили, что мыши с отсутствием просапозина, предшественника семейства белков, переносящих эндосомные липиды (LTP), характеризуются специфическими дефектами CD1d-опосредуемого представления антигенов и у них отсутствуют естественные Т-клетки-киллеры Vα14. In vitro сапозины экстрагировали мономерные липиды из мембран и CD1, стимулируя таким образом загрузку и редактирование липидов в CD1. Временные комплексы CD1, липида и LTP предполагают создание модели “перетягивания”, в которой обмен липидов между CD1 и LTP происходит на основе их соответствующего сродства к липидам. LTP создают ранее неизвестную связь между липидным обменом и иммунитетом и, по-видимому, оказывают сильное влияние на спектр аутоантигенов, опухолевых антигенов и антигенов микробных липидов.
Активация макрофагов (М2) типа 2 является альтернативным путем, ведущим к классической активации макрофагов. Клетки М2 являются антиген-представляющими клетками (АРС), которые присутствуют в тонком слое кишечника в виде части ассоциированной с кишечником иммунной системы. Клетки М2 реагируют на экспрессию IL-10 вместо классической реакции макрофагов на цитокины Th1, как это описано ниже.
Активированные макрофаги используют в качестве антиген-представляющих клеток (АРС). Узнавание антигенов Т-клетками является главным событием, контролирующим приобретенную иммунную реакцию.
Классическим путем IFN-γ-зависимой активации макрофагом клетками Th1-типа является хорошо известный клеточный иммунитет. Активация макрофагов зависит от продуктов специфически активированных Т-клеток-хелперов, лимфоцитов Th1-типа и естественных клеток-киллеров, в частности, от IFN-γ и сети цитокинов, включающих IL-12 и IL-18, продуцируемых АРС. В последнее десятилетие получила признание концепция альтернативного пути активации макрофагов цитокинами Th2-типа IL-4 и IL-13 вместе с IL-10, которая, как считается, определяет особый фенотип макрофагов, совместимый с другой ролью в гуморальном иммунитете и восстановлении.
IL-4 и IL-13 усиливают экспрессию рецептора маннозы и молекул МНС класса II макрофагами, которые стимулируют эндоцитоз и представление антигенов.
Иммуноглобулины и иммунные комплексы могут связываться с активирующими и ингибирующими рецепторами для Fc и комплемента. Кроме того, лигирование Fc-рецептора оказывает значительное влияние на выделение цитокинов, таких как IL-12/IL-10 и IL-4, антиген-представляющими клетками и другими клетками врожденной и приобретенной иммунной системы (Gordon S. Alternative Activation of Macrophages, Nat. Rev. Immunol. 3:23-34; 2003).
Макрофаги, активированные вызывающими воспаление раздражителями (например, IFN-γ) и введенные в иммунные комплексы, оказывают совершенно противоположное действие вместо реакции на Th1: при повышенных уровнях IL-12 и средних уровнях IL-10 происходит значительное уменьшение уровней IL-12 и увеличение уровней IL-10. IL-10 оказывает иммуносупрессивное действие на макрофаги (Anderson, C.F. and Mosser, D.M. A Novel Phenotype for an Activated Macrophages: the Type 2 Activated Macrophage J. Leukoc. Biol.72:101-106; 2002). IL-10 воздействует на рецептор плазматической мембраны, отличающийся от рецепторов для IL-4 и IL-13, при этом отличается его влияние на экспрессию гена макрофага, включая более сильное ингибирование антиген-представляющих и эффекторных функций наряду с активацией выбранных генов или функций (Gordon S. Alternative Activation of Macrophages. Nat. Rev. Immunol. 3:23-34; 2003).
Следовательно, можно сделать вывод, что помимо воздействия на атеросклероз и другие болезни, которые непосредственно ассоциированы с окисленным LDL, иммуномодуляцию и противовоспалительное действие, вызываемое (синтетическими) аналогами окисленного LDL, можно использовать для лечения и профилактики других заболеваний и нарушений, прямо или косвенно ассоциированных с эндогенным окисленным LDL и другими окисленными липидами. Данный вывод был подтвержден несколькими исследованиями, целью которых была иммунотерапия аутоиммунных заболеваний человека, таких как ревматоидный артрит (RA), диабет типа I и рассеянный склероз, путем модуляции отдельных иммунных путей, участвующих в воспалении, или приобретения толерантности к разным антигенам (Bielekova et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167-1175; Kappos et al. Induction of a non-encephalitogenic type 2 Т helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. Nat. Med. 2000;6:1176-1182).
Таким образом, имеются многочисленные данные, которые подтверждают наличие взаимосвязи между липидами, воспалением и иммунной системой и указывают на прямую связь между ними.
Пытаясь усовершенствовать лечение воспалений, заболеваний и нарушений, ассоциированных с окисленными липидами, авторы настоящего изобретения создали новые синтетические окисленные фосфолипиды и структурно родственные соединения, у которых отсутствуют ограничения, характерные для окисленного LDL и других известных окисленных фосфолипидов и липидов (которые были указаны выше).
Как показано в приведенном ниже разделе “Примеры”, при реализации настоящего изоретения на практике было подтверждено, что пероральное введение и/или введение через слизистую оболочку новых аналогов окисленного LDL модулирует иммунную и/или воспалительную реакцию на эндогенный окисленный LDL, в результате чего происходит ослабление воспалительной реакции в воспалительных заболеваниях, таких как атеросклероз и ревматоидный артрит. Полученные результаты ясно показывают влияние экзогенных окисленных липидов на воспалительные и иммунные процессы с участием эндогенных окисленных липидов.
Таким образом, одним объектом настоящего изобретения являются новые соединения, имитирующие иммуномодулирующее действие, индуцируемое окисленным LDL, и/или воспаление, ассоциированное с окисленным LDL и/или другими окисленными липидами, при отсутствии ограничений, характерных для окисленного LDL и других окисленных липидов, благодаря чему они могут быть использованы для лечения воспалительных заболеваний и нарушений, вызываемых окисленными липидами, при пероральном введении или введении через слизистую оболочку.
Так как известно, что окисленные фосфолипиды являются активными компонентами ox LDL, и, кроме того, биологические мембраны обычно включают фосфолипиды, главным образом фосфоглицериды, соединения по настоящему изобретению имеют структуру на основе окисленных фосфолипидов вообще и окисленных фосфоглицеридов в частности.
Все соединения по настоящему изобретению имеют общую формулу I
где
n является целым числом от 1 до 6, причем, если n=1, Cn, Bn, Rn, R'n и Y отсутствуют;
каждый из B1, B2, …Bn-1 и Bn независимо выбирают из группы, состоящей из кислорода, серы, азота, фосфора и кремния, причем каждый из указанного азота, фосфора и кремния замещен по крайней мере одним заместителем, выбранным из группы, состоящей из водорода, электронов неподеленной пары, алкила, галогена, циклоалкила, арила, гидрокси, тиогидрокси, алкокси, арилокси, тиоарилокси, тиоалкокси и оксо;
каждый из А1, А2, …An-1 и An независимо выбирают из группы, состоящей из CR''R''', C=O и С=S;
Y выбирают из группы, состоящей из водорода, алкила, арила, циклоалкила, карбокси, сахарида, фосфорной кислоты, фосфорилхолина, фосфорилэтаноламина, фосфорилсерина, фосфорилкардиолипина, фосфорилинозита, этилфосфохолина, фосфорилметанола, фосфорилэтанола, фосфорилпропанола, фосфорилбутанола, фосфорилэтаноламин-N-лактозы, фосфоэтаноламин-N-[метокси(пропиленгликоль)], фосфоинозит-4-фосфата, фосфоинозит-4,5-бифосфоната, пирофосфата, фосфоэтаноламиндиэтилентриаминпентаацетата, динитрофенилфосфоэтаноламина и фосфоглицерина; и
каждый из Х1, Х2, …Xn-1 независимо является насыщенным или ненасыщенным углеводородом общей формулы II
где
m является целым числом от 1 до 26; и
Z выбирают из группы, состоящей из
где
W выбирают из группы, состоящей из кислорода, серы, азота и фосфора, причем каждый из указанного азота и фосфора замещен по крайней мере одним заместителем, выбранным из группы, состоящей из водорода, электронов неподеленной пары, алкила, галогена, циклоалкила, арила, гидрокси, тиогидрокси, алкокси, арилокси, тиоарилокси, тиоалкокси и оксо; и
по крайней мере один из Х1, Х2, …Xn-1 Z не является водородом;
и где
каждый из R1, R'1, R2, …Rn-1, Rn, R'n, каждый из R'' и R''' и каждый из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm независимо выбирают из группы, состоящей из водорода, связи, алкила, алкенила, алкинила, циклоалкила, арила, гетероарила, галогена, тригалогенметила, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, фосфоната, фосфата, фосфинила, сульфонила, сульфинила, сульфонамида, амида, карбонила, тиокарбонила, С-карбокси, О-карбокси, С-карбамата, N-карбамата, С-тиокарбокси, S-тиокарбокси и амино, или альтернативно по крайней мере два из R1, R'1, R2, …Rn-1, Rn и R'n и/или по крайней мере два из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm образуют по крайней мере одно четырех-, пяти- или шестичленное ароматическое, гетероароматическое, алициклическое или гетероалициклическое кольцо; и
каждый из С1, С2, …Cn-1, Cn и каждый из Са, Cb, …Cm-1 и Cm является хиральным или нехиральным атомом углерода, причем каждый хиральный атом углерода имеет S-конфигурацию и/или R-конфигурацию;
и включают фармацевтически приемлемую соль, пролекарство, гидрат или сольват.
Специалисту в данной области должно быть понятно, что возможность введения всех заместителей (например, R1-Rn, Ra-Rm, R'', R''') в указанные положения зависит от валентности и химической совместимости заместителя, замещаемого положения и других заместителей. Поэтому в объем настоящего изобретения входят все возможные заместителя для любого положения.
В используемом здесь значении термин “алкил” означает насыщенный алифатический углеводород, включающий группы с прямой и разветвленной цепью. Алкильная группа предпочтительно имеет 1-20 атомов углерода. Указанный числовой интервал, например, “1-20” означает, что группа, в данном случае алкильная группа, может содержать 1 атом углерода, 2 атома углерода, 3 атома углерода и т.д. до 20 атомов углерода включительно. Более предпочтительно алкил имеет цепь средней длины, включающую от 1 до 10 атомов углерода. Наиболее предпочтительно, за исключением особо оговоренных случаев, алкил является низшим алкилом, включающим 1-4 атома углерода. Алкильная группа может быть замещенной или незамещенной. В случае замещения замещающей группой может быть, например, циклоалкил, алкенил, алкинил, арил, гетероарил, гетероалициклическая группа, галоген, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, сульфинил, сульфонил, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, О-карбокси, сульфонамидо и амино в указанных здесь значениях.
“Циклоалкильная” группа означает группу с моноциклическим или конденсированным кольцом, состоящим из атомов углерода (то есть с кольцами, которые имеют общую смежную пару атомов углерода), в которой одно или несколько колец не имеют полностью конъюгированную систему пи-электронов. Неограничивающие примеры циклоалкильных групп включают циклопропан, цирклобутан, циклопентан, циклопентен, циклогексан, циклогексадиен, циклогептан, циклогептатриен и адамантан. Циклоалкильная группа может быть замещенной или незамещенной. В случае замещения замещающей группой может быть, например, алкил, алкенил, алкинил, арил, гетероарил, гетероалициклическая группа, галоген, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, сульфинил, сульфонил, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, О-карбокси, сульфонамидо и амино в указанных здесь значениях.
“Алкенильная” группа означает алкильную группу, которая содержит по крайней мере два атома углерода и по крайней мере одну углерод-углеродную двойную связь.
“Алкинильная” группа означает алкильную группу, которая содержит по крайней мере два атома углерода и по крайней мере одну углерод-углеродную тройную связь.
“Арильная” группа означает группу с моноциклическим или конденсированным кольцом, состоящим из атомов углерода (то есть с кольцами, которые имеют общую смежную пару атомов углерода), которая имеет полностью конъюгированную систему пи-электронов. Неограничивающие примеры арильных групп включают фенил, нафталенил и антраценил. Арильная группа может быть замещенной или незамещенной. В случае замещения замещающей группой может быть, например, алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, гетероалициклическая группа, галоген, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, сульфинил, сульфонил, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, О-карбокси, сульфонамидо и амино в указанных здесь значениях.
“Гетероарильная” группа означает группу с моноциклическим или конденсированным кольцом (то есть с кольцами, которые имеют общую смежную пару атомов), которая содержит в кольце один или несколько атомов, таких как, например, азот, кислород и сера, и дополнительно включает полностью конъюгированную систему пи-электронов. Неограничивающие примеры гетероарильных групп включают пиррол, фуран, тиофен, имидазол, оксазол, тиазол, пиразол, пиридин, пиримидин, хинолин, изохинолин и пурин. Гетероарильная группа может быть замещенной или незамещенной. В случае замещения замещающей группой может быть, например, алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, гетероалициклическая группа, галоген, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, сульфинил, сульфонил, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, О-карбокси, сульфонамидо и амино в указанных здесь значениях.
“Гетероалициклическая” группа означает группу с моноциклическим или конденсированным кольцом, которая содержит в кольце один или несколько атомов, таких как азот, кислород и сера. Указанные кольца могут также иметь одну или несколько двойных связей. Однако такие кольца не имеют полностью конъгированную систему пи-электронов. Гетероалициклическая группа может быть замещенной или незамещенной. В случае замещения замещающей группой могут быть, например, электроны неподеленной пары, алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, гетероалициклическая группа, галоген, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, сульфинил, сульфонил, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, О-карбокси, сульфонамидо и амино в указанных здесь значениях. Типичными примерами являются пиперидин, пиперазин, тетрагидрофуран, тетрагидропиран, морфолино и тому подобные.
“Гидроксильная” группа означает группу -ОН.
“Азид” означает группу -N=N.
“Алкоксильная” группа означает -О-алкильную и -О-циклоалкильную группу в указанных здесь значениях.
“Арилокси” группа означает -О-арильную и -О-гетероарильную группу в указанных здесь значениях.
“Тиогидроксильная” группа означает группу -SH.
“Тиоалкоксильная” группа означает -S-алкильную группу и -S-циклоалкильную группу в указанных здесь значениях.
“Тиоарилокси” группа означает -S-арильную и -S-гетероарильную группу в указанных здесь значениях.
“Карбонильная” группа означает группу -С(=О)-R, где R означает водород, алкил, алкенил, циклоалкил, арил, гетероарил (связанный атомом углерода в кольце) или гетероалициклическую группу (связанную атомом углерода в кольце) в указанных здесь значениях.
“Альдегидная” группа означает карбонильную группу, в которой R означает водород.
“Тиокарбонильная” группа означает группу -С(=S)-R, в которой R имеет указанные здесь значения.
“С-карбокси” группа означает группы -С(=О)-О-R, в которых R имеет указанные здесь значения.
“О-карбокси” группа означает группу RC(=O)-O-, в которой R имеет указанные здесь значения.
“Оксо” группа означает группу =О.
Группа “карбоновой кислоты” означает С-карбоксильную группу, в которой R означает водород.
“Галоген” означает фтор, хлор, бром или иод.
“Тригалогенметильная” группа означает группу -СХ3, в которой Х означает галоген в указанных здесь значениях.
“Сульфинильная” группа означает группу -S(=O)-R, в которой R имеет указанные здесь значения.
“Сульфонильная” группа означает группу -S(=O)2-R, в которой R имеет указанные здесь значения.
“S-сульфонамидо” группа означает группу -S(=O)2-NR2, в которой R имеет указанные здесь значения.
“N-сульфонамидо” группа означает группу RS(=O)2-NR, в которой R имеет указанные здесь значения.
“О-карбамильная” группа означает группу -ОС(=О)-NR2, в которой каждый из R имеют указанные здесь значения.
“N-карбамильная” группа означает группу ROC(=O)-NR-, в которой каждый из R имеют указанные здесь значения.
“О-тиокарбамильная” группа означает группу -OC(=S)-NR2, в которой R имеет указанные здесь значения.
“N-тиокарбамильная” группа означает группу ROC(=S)NR-, в которой каждый из R имеет указанные здесь значения.
“Амино” группа означает группу -NR2, в которой каждый из R имеют указанные здесь значения.
“C-амидо” группа означает группу -С(=O)-NR2, в которой каждый из R имеют указанные здесь значения.
“N-амидо” группа означает группу RC(=O)-NR-, в которой каждый из R имеют указанные здесь значения.
“Мочевина” означает группу -NRC(=O)-NR2, в которой каждый из R имеют указанные здесь значения.
“Гуанидино” группа означает группу -RNC(=N)-NR2, в которой каждый из R имеют указанные здесь значения.
“Гуанильная” группа означает группу R2NC(=N)-, в которой каждый из R имеют указанные здесь значения.
“Нитро” группа означает группу -NO2.
“Циано” группа означает группу -С≡N.
Термин “фосфонил” или “фосфонат” означает группу -Р(=О)(OR)2, в которой R имеет указанные выше значения. Термин “фосфат” означает группу -О-Р(=О)(OR)2, в которой каждый из R имеют указанные выше значения.
“Фосфорная кислота” означает фосфатную группу, в которой каждый из R означают водород.
Термин “фосфинил” означает группу -PR2, в которой каждый из R имеют указанные выше значения.
Термин “тиомочевина” означает группу -NR-C(=S)-NR-, в которой каждый из R имеют указанные выше значения.
Термин “сахарид” означает одно или несколько звеньев сахара, а именно звено сахара с открытой цепью или циклическое звено сахара (например, звенья на основе пиранозы или фуранозы), и включает любой моносахарид, дисахарид и олигосахарид за исключением особо оговоренных случаев.
Как видно из приведенной выше общей формулы I, соединения по настоящему изобретению имеют остов, состоящий из 1-6 атомов углерода, в котором по крайней мере один из указанных атомов углерода ковалентно присоединен к атому водорода, углеводородной группе (алкил, арил и т.д.), карбоксильной группе (например, ацил, карбоновая кислота и т.д.) или фосфорильной группе (которая именуется также фосфатной группой или просто фосфатом) и другие 1-5 атомов углерода остова ковалентно присоединены к углеводородным цепям (Х1-Xn-1) с помощью гетероатома (В1-Bn в вышеприведенной общей формуле I). Указанные углеводородные цепи могут включать насыщенные или ненасыщенные, замещенные или незамещенные цепи, необязательно прерываемые ароматическими, алициклическими, гетероалициклическими и/или гетероароматическими частями, приведенными в данном описании изобретения и показанными в общей формуле II, причем по крайней мере одна из указанных цепей заканчивается окисленной группой, определяемой выше как Z, не являющийся водородом.
В используемом здесь значении термин “углеводород” означает соединение, которое включает атомы водорода и атомы углерода, ковалентно связанные друг с другом. В насыщенном углеводороде все Са-Cm ковалентно присоединены к соседним атомам при помощи простой сигма-связи. В ненасыщенном углеводороде по крайней мере два соседних атома Са-Cm соединены друг с другом двойной связью или тройной связью.
Все углеводородные цепи по настоящему изобретению могут включать от 1 до 26 атомов углерода, более предпочтительно от 3 до 26 атомов углерода. Углеводородные цепи с концевой окисленной группой Z обычно имеют меньшую длину и предпочтительно включают от 3 до 10 атомов углерода, более предпочтительно от 3 до 6 атомов углерода, без учета атома углерода в окисленной группе.
LDL является липопротеином, состоящим из функционально разных частей (компонентов). Среди указанных частей находятся фосфолипиды, которые, как известно, играют важную роль в процессе воздействия окисленного LDL на заболевания, ассоциированные с образованием бляшек.
В используемом здесь значении термин “часть” или “компонент” означает главную часть функционально активной молекулы, которая связана с другой молекулой при сохранении своей активности. Фосфолипиды являются природными веществами, содержащими в конце цепи неполярную липидную группу и высокополярную фосфатидильную группу. Наиболее распространенными фосфолипидами в природе являются фосфоглицериды, которые состоят из глицеринового остова с присоединенными к нему ацильными частями жирных кислот. Фосфоглицериды, такие как ацилфосфоглицериды 1,2-О-жирных кислот, а также их окисленные модификации, такие как POVPC и PGPC, были использованы в исследованиях атерогенеза, как это было подробно описано выше.
В разных биологических процессах, таких как воспаление, помимо LDL, фосфолипидов и фосфоглицеридов, участвуют и другие липиды. Такие липиды включают, например, сфинголипиды, гликолипиды и другие мембранные липиды.
Вышеописанные соединения по настоящему изобретению созданы главным образом на основе структуры фосфоглицеридов, в соответствии с которой в предпочтительном варианте осуществления изобретения n в общей формуле I равно 3. Такие соединения определяются в данном описании изобретения как аналоги окисленного фосфоглицерида, в то время как все соединения по настоящему изобретению определяются как окисленные липиды и включают их аналоги и производные.
В используемом здесь значении термин “аналоги” означает соединения, которые в структурном отношении являются родственными целевой молекуле (например, окисленные фосфолипиды, окисленный LDL и т.д.) и поэтому могут обладать такой же биологической активностью.
Термин “производные” означает целевые молекулы, которые были химически модифицированы, но сохранили неизменной главную часть, например, целевые молекулы, которые были замещены дополнительными или разными заместителями, целевые молекулы, часть которых была окислена или гидролизована, и тому подобные.
С учетом неустойчивости, присущей ацильной части О-жирных кислот в естественных фосфоглицеридах, а также в других структурно родственных соединениях, которая возникает вследствие высокой подверженности быстрому гидролизу в биологических системах под действием фосфолипазы А1 (см., например, “A Textbook of Drug Design and Development”, Povl Krogsgaard-Larsen and Hans Bundgaard, eds., Harwood Academic Publishers, chapter 13, pages 478-480), соединения по настоящему изобретению были созданы с включением по крайней мере одной части простого эфира О-жирных кислот, как это показано в вышеприведенной общей формуле I, в которой, когда n равно 3, по крайней мере один из А1 и А2 предпочтительно является группой CR''R'''. Соединения, в которых один из А1 и А2 является группой CR''R''', определяются в данном описании изобретения как моноэтерифицированные аналоги фосфоглицерида, в то время как соединения, в которых каждый из А1 и А2 являются CR''R''', определяются как диэтерифицированные аналоги фосфоглицерида и характеризуются улучшенной устойчивостью in vivo, особенно по сравнению с известными в настоящее время синтетическими окисленными фосфоглицеридами (например, POVPC и PGPC).
Как показанно в приведенной выше общей формуле I, когда n равно 3, по крайней мере один из Х1 и Х2 является углеводородной цепью, которая заканчивается окисленной группой, при этом Z не является водородом. Однако, так как в естественных производных окисленного LDL цепь окисленного алкила обычно находится во втором положении и, как установлено, биологическая активность нескольких фосфолипидов непосредственно зависит от ее структуры (см. раздел “Уровень техники” для подробного ознакомления с данным вопросом), в другом предпочтительном варианте осуществления настоящего изобретения Х2 является углеводородной цепью, которая заканчивается окисленной группой.
Как далее показано в приведенной выше общей формуле II, окисленная группа может быть, например,
(альдегидом),

(карбоновой кислотой) и
(ацеталем), а также их производными, такими как, например, любое карбоксильное или тиокарбоксильное производное (например, сложный эфир карбоновой кислоты, в котором W означает кислород и R'' означает алкил, арил, циклоалкил и подобные группы), иминопроизводными (в которых W означает атом азота), амидопроизводными (в которых W означает кислород и R'' означает амин), фосфиновыми или фосфонатными производными и многими другими, которые были описаны выше.
Одним примером нового этерифицированного окисленного фосфоглицерида по настоящему изобретению является простой эфир 2,5'-альдегидлецитина (ALLE): 1-гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолин (D-ALLE), 3-гексадецил-2-(5'-оксопентанил)-sn-глицеро-1-фосфохолин (L-ALLE) и их рацемическая смесь, синтез и применение которых более подробно описаны в нижеследующем разделе “Примеры”.
Однако, так как известно, что альдегиды являются неустойчивыми соединениями, которые легко окисляются, предпочтительные примеры новых этерифицированных окисленных фосфоглицеридов по настоящему изобретению включают кислотное производное 1-гексадецил-2-(5'-карбоксибутил)-sn-глицеро-3-фосфохолина (также определяемое далее как IC-201) и его соответствующие ацетали 1-гексадецил-2-(5',5'-диметоксипентилокси)-sn-глицеро-3-фосфохолин и 1-гексадецил-2-(5',5'-диэтоксипентилокси)-sn-глицеро-3-фосфохолин (см. фигуру 10 для ознакомления с двухмерными структурными формулами), синтез и применение которых более подробно описаны в нижеследующем разделе “Примеры”.
Помимо вышеописанных окисленных липидов, выделенных из фосфоглицеридов, в объем настоящего изобретения входят также окисленные липиды, выделенные, например, из сфинголипидов. Аналоги в виде окисленных сфинголипидов по настоящему изобретению имеют вышеуказанную общую формулу I, в которой n равно 3, Y означает водород, В2 означает NH и А2 означает С=О, при этом углеводородная цепь с концевой окисленной группой присоединена к амиду, обозначенному Х2, или к С1.
Хотя окисленные фосфоглицериды выделены из глицерина, который является молекулой моносахарида, и окисленные сфинголипиды выделены из сфингозина, аминоспирта, можно предположить, что окисленные фосфолипиды, выделенные из других биологически широко распространенных структурных звеньев спирта, должны обладать таким же действием. Кроме того, так как не было выявлено корреляции между расстоянием расположения окисленной части и фосфатидильной части в окисленных фосфолипидах, считается, что окисленные липиды, выделенных из остова, включающего 4-6 атомов углерода, должны сохранять структурные характеристики, подобные характеристикам окисленных фосфоглицеридов и, таким образом, должны обладать такой же антигенностью и иммуномодулирующей активностью, благодаря чему они могут быть использованы аналогично производным окисленных фосфоглицеридов, рассмотренным в данном описании изобретения.
Предпочтительным примером такого структурного звена спирта является структурное звено моносахарида, такое как, например, глюкоза, эритрит и треит.
В другом предпочтительном варианте осуществления изобретения соединения по настоящему изобретению вклчают до 6 атомов углерода в цепи остова. Атомы углерода в цепи остова могут быть линейно присоединены друг к другу с образованием остова моносахарида с открытой цепью или альтернативно с образованием гетероалициклического остова моносахарида, а именно остова пиранозы или фуранозы, при этом в приведенной выше общей формуле один из R1 и R'1 ковалентно присоединен к одному из Rn или R'n простой эфирной связью (связь R-O-R).
Альтернативно соединения по настоящему изобретению могут содержать 4-6 атомов углерода в цепи остова, которые образуют несахаридное кольцо, а именно четырех-, пяти- или шестичленное углеродное или гетероалициклическое кольцо, при этом в приведенной выше общей формуле I один из R1 и R'1 ковалентно присоединен к одному из Rn или R'n разными связями (например, сигма-связью, π-связью, карбоксильной связью, простой эфирной связью, тиоэфирной связью и любой другой связью).
Как далее показано в приведенной выше общей формуле I, Y означает фосфорильную часть (например, фосфорилхолин, фосфорилэтаноламин и т.д.) или нефосфорильную часть (например, водород, ацил или алкил). Когда Y означает нефосфорильную часть, полученное соединение является не фосфолипидом, а соединением диглицерида, для n=3, или любым другим соединением, полученным из спирта, например соединением, выделенным из моносахарида.
Так как не сообщалось об какой-либо активности фосфорильной группы в отношении иммуномодулирующей активности окисленного LDL, можно предположить, что такие нефосфорильные соединения должны сохранить структурные характеристики, подобные вышеуказанным окисленным фосфолипидам, следовательно, должны обладать антигенностью и иммуномодулирующей активностью и поэтому могут быть использованы аналогично производным окисленного фосфолипида, рассмотренным в данном описании изобретения.
В одном варианте осуществления настоящего изобретения Y означает сахарид, как было указано выше, и, таким образом, данное соединение по настоящему изобретению является окисленным аналогом гликолипидов.
В другом варианте осуществления изобретения данное соединение является окисленным аналогом любого мембранного липида.
Предпочтительные структурные особенности, рассмотренные выше для окисленных фосфоглицеридов, относятся ко всем вышеописанным соединениям. Следовательно, в предпочтительном варианте настоящего изобретения по крайней мере один из А1, А2, … и An-1 является группой CR''R''', при этом соединение включает по крайней мере одно этерифицированную боковую цепь. С учетом неустойчивости О-ацильной боковой цепи желательно, чтобы по крайней мере одна из окисленных групп в Х1-Xn-1 была связана с такой этерифицированной боковой цепью.
Хотя естественные фосфолипиды и окисленные фосфолипиды обычно включают О-ацильные цепи, существует данные, свидетельствующие о том, что тиоловые производные окисленных фосфолипидов, которые включают, например, S-ацильные цепи, могут обладать такой же биологической активностью (см., например, публикации Reddy et al. Antitumor ether lipids: an improved synthesis of ilmofosine and an entioselective synthesis of an ilmofosine analog. Tetrahedron Letters. 1994;17:2679-2682; Batia and Hajdu. Stereospecific synthesis of ether and thioether phospholipids. The use of L-glyceric acid as a chiral phospholipids precursor. J. Org. Chem. 1988;53:5034-5039; Bosies et al. Lipids. 1987;22:947; Bosies et al. Ger. Offen. DE 3,906,952 [C.A. 1991, 114,102394w]; and Herrmann et al. NCI-EORTC Symposium on New Drugs in Cancer Therapy, Amsterdam, March 1992]. Так как тиолы характеризутся повышенной биологической устойчивостью, такие соединения могут быть весьма полезными.
В одном варианте осуществления настоящего изобретения по крайней мере один из В1-Bn означает серу, при этом по крайней мере одна из боковых цепей является тиолированной S-ацильной или S-алкильной цепью. В другом варианте осуществления изобретения по крайней мере один из Х1-Xn-1, включающий окисленную группу, связан с такой тиолированной боковой цепью.
Альтернативно каждый из В1-Bn могут представлять собой биологически совместимый гетероатом, не являющийся атомом кислорода и серы, такой как, например, азот, фосфор или кремний, как показано в приведенной выше общей формуле I.
Помимо структурных особенностей, рассмотренных в данном описании изобретения, соединения по настоящему изобретению могут быть далее замещены в любом положении, например, у любого атома углерода в боковой цепи или у любого атома углерода в остове. Хотя существует много возможных заместителей, описанных выше, которые входят в объем настоящего изобретения, предпочтительными заместителями являются, например, галоген и арил.
Хотя соединения по настоящему изобретению были созданы на основе окисленных фосфолипидов, таких как фосфоглицериды, авторы изобретения считают, что простая окисленная углеводородная цепь, которая необязательно присоединена к полярной группе, должна обладать такой же антигенностью и иммуномодулирущей активностью, что и вышеописанные аналоги окисленного фосфолипида.
Такая окисленная углеводродная цепь является общим признаком метаболитов арахидоновой кислоты. Арахидоновая кислота является полиненасыщенной жирной кислотой, имеющей 20 атомов углерода, которая образуется in vivo в результате ферментативного гидролиза фосфолипидов, содержащих указанную кислоту. Выделенная арахидоновая кислота окисляется в ряд важных физиологически активных веществ определенными липоксигеназами, и после целого ряда дополнительных ферментативных реакций указанные физиологически активные вещества метаболизируются в семейство классических простагландинов (PG), простациклинов (PGI2) и тромбоксанов (ТХ) А2, которые являются активными во многих биологических путях обмена. Все указанные метаболиты имеют общий признак, которым является цепь, состоящая из шести атомов углерода и заканчивающаяся окисляемой двойной связью.
Как было указано выше и будет далее продемонстрировано в разделе “Примеры”, важное значение имеет наличие окисленной группы в аналогах окисленного LDL, которые предназначены для имитации иммуномодуляции, индуцируемой ox LDL. Так, в сравнительных исследованиях, например, было показано, что соединение V, неокисленное соединение, соответствующее CI-201 (соединение VII), является неактивным, в то время как CI-201 является активным (см., например, примеры XIV и XV в нижеследующем разделе “Примеры”). Кроме того, на основании пути обмена арахидоновой кислоты можно предположить, что другие окисленные фосфолипиды проходят такой же путь обмена, в результате чего происходит выделение окисленной боковой цепи. Как будет описано ниже, окисленная боковая цепь предпочтительно включает от 3 до 7 атомов углерода и поэтому аналогична цепи, состоящей из шести атомов углерода, в метаболитах арахидоновой кислоты. Кроме того, вышеописанный механизм CD1, который предполагает участие липидов в иммунной системе, показывает, что гидрофильная головная группа, то есть головная группа углерод-С2 и/или углерод-С3 в CD1-d, наиболее вероятно является антигенной детерминантой, представляемой иммунной системе, так как она является частью, представляемой желобком CD1, который скрывает гидрофобную часть молекулы, указывая на определенную роль гидрофильного эпитопа в положении углерод-С2.
В окисленных фосфолипидах, таких как фосфоглицериды, окисленная боковая цепь присоединена к фосфоглицериновому остову. Однако, как было указано выше, фосфоглицериновый остов не выполняет какой-либо определенной роли.
В предпочтительном варианте осуществления настоящего изобретения n равно 1, при этом соединение по настоящему изобретению имеет простую углеводородную цепь с концевой окисленной группой. Хотя такая окисленная простая углеводородная цепь является неполярной, она может быть присоединена к полярной группе, такой как фосфорильная группа, при этом, как показано в приведенной выше общей формуле I, когда n равно 1, по крайней мере один из R1 и R'1 является фосфатной или фосфонатной группой. Альтернативно по крайней мере один из R1 и R'1 можно выбрать из других биологически совместимых полярных групп, таких как, например, пептиды, сахариды и тому подобные.
В зависимости от заместителей, все атомы углерода в вышеописанных соединениях, а именно С1-Cn и Са-Cm, могут быть хиральными или нехиральными. Любой хиральный атом углерода, присутствующий в соединениях по настоящему изобретению, может иметь R-конфигурацию, S-конфигурацию или быть рацемическим. Таким образом, в объем настоящего изобретения входят любые комбинации хиральных и рацемических атомов углерода, включая все возможные стереоизомеры, оптические изомеры, энантиомеры и аномеры. Как показано в нижеследующем разделе “Примеры”, соединения по настоящему изобретению могут быть синтезированы при сохранении конфигурации исходного вещества. Соединения по настоящему изобретению могут быть далее избирательно синтезированы с учетом стереохимии окисленной группы. В результате выбора соответствущих исходных веществ и соответствующих условий синтеза можно определить оптическую чистоту (например, включение хиральных и/или рацемических атомов углерода) и стереоизомеры полученных своединений. В случае получения рацемических смесей можно использовать известные методы для разделения оптических или стереоизомеров. Такие методы описаны, например, в публикации “Organic chemistry, fourth Edition by Paula Yurkanis Bruice, page 180-185 and page 214, Prentice Hall, Upper Sadde River, NJ 07458”.
В объем настоящего изобретения далее входят любые фармацевтически приемлемые соли, пролекарства, гидраты и сольваты вышеописанных соединений.
Термин “пролекарство” означает средство, которое превращается в активное соединение (активное исходное лекарственное вещество) in vivo. Пролекарства обычно используют для облегчения введения исходного лекарственного вещества. Пролекарства, например, могут быть биологически пригодны для перорального введения, в то время как исходное лекарственное вещество может быть не пригодно для такого введения. Пролекарство может также характеризоваться лучшей растворимостью по сравнению с исходным лекарственным веществом в фармацевтических композициях. Кроме того, пролекарства часто использут для пролонгированного высвобождения активного соеднения in vivo. Неограничивающим примером пролекарства может быть соединение по настоящему изобретению, имеющее одну или несколько частей карбоновой кислоты, которое вводят в виде сложного эфира (“пролекарство”). Такое пролекарство гидролизуется in vivo с образованием свободного соединения (исходное лекарственное вещество). Выбранный сложный эфир может влиять как на характеристики растворимости, так и на скорость гидролиза пролекарства.
Термин “фармацевтически приемлемая соль” означает заряженную разновидность исходного соединения и его противоион, который обычно используют для изменения характеристик растворимости исходного соединения и/или для уменьшения раздражения, причиняемого организму исходным соединением, без изменения биологической активности и свойств вводимого соединения. Неограничивающим примером фармацевтически приемлемой соли может быть карбоксилатный анион и катион, который включает, не ограничиваясь ими, аммоний, натрий, калий и тому подобные.
Термин “сольват” означает комплекс с переменной стехиометрией (например, ди-, три-, тетра-, пента-, гекса и так далее), который образуется растворенным веществом (соединением по настоящему изобретению) и растворителем, причем растворитель не должен влиять на биологическую активность растворенного вещества. Приемлемые растворители включают, например, этанол, уксусную кислоту и тому подобные.
Термин “гидрат” означает вышеуказанный сольват, в котором растворителем является вода.
Как будет подробно описано ниже, новые соединения по настоящему изобретению обладают высокой иммуномодулирующей активностью и поэтому могут быть использованы в разных терапевтических применениях. Использование указанных соединений в терапевтических применениях включает введение указанных соединений per se или в составе фармацевтической композиции, в которой такое соединение смешано с приемлемыми носителями или наполнителями.
Таким образом, другим объектом настоящего изобретения является фармацевтическая композиция, которая содержит в качестве активного ингредиента любые вышеописанные соединения общей формулы I и фармацевтически приемлемый носитель.
В используемом здесь значении термин “фармацевтическая композиция” означает препарат, содержащий один или несколько вышеописанных активных ингредиентов вместе с другими химическими компонентами, такими как физиологически приемлемые носители и наполнители. Целью создания фармацевтической композиции является облегчение введения соединения в организм субъекта.
Термин “активный ингредиент” означает соединения (например, ALLE, CI-201 и другие соединения, соответствующие приведенной выше общей формуле I), обладающие биологическим действием.
Термины “физиологически приемлемый носитель” и “фармацевтически приемлемый носитель”, которые имеют взаимозаменяемые значения, означают носитель или разбавитель, который не вызывает значительного раздражения организма и не ухудшает биологическую активность и свойства вводимого соединения. Адъювант входит в определение указанных терминов.
Термин “наполнитель” означает инертное вещество, добавляемое в фармацевтическую композицию для дальнейшего облегчения введения активного ингредиента. Неограничивающие примеры наполнителей включают карбонат кальция, фосфат кальция, разные сахара и крахмалы, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли.
Методы получения и введения лекарственных средств описаны в публикации “Remington's Pharmaceutical Sciences,” Mack Publishing Co., Easton, P.A., latest edition, которая включена в данное описание изобретения в качестве ссылки.
Приемлемые способы введения могут включать, например, пероральное и ректальное введение, введение через слизистую оболочку, особенно в нос, кишечное или парентеральное введение, включающее внутримышечные, подкожные и интрамедуллярные инъекции, а также подоболочечные, прямые интравентрикулярные, внутривенные, внутрибрюшинные, назальные или внутриглазные инъекции.
Альтернативно фармацевтическую композицию можно вводить не системно, а локально, например, в виде инъекции фармацевтической композиции непосредственно в пораженный участок ткани субъекта.
В предпочтительном варианте осуществления настоящего изобретения фармацевтические композиции предназначены для модуляции иммунной и/или воспалительной реакции при введении через слизистую оболочку.
В другом предпочтительном варианте осуществления настоящего изобретения фармацевтическая композиция предназначена для модуляции иммунной и/или воспалительной реакции при пероральном введении.
Фармацевтические композиции по настоящему изобретению предпочтительно предназначены для назального или внутрибрюшинного введения, как это подробно описано ниже.
Фармацевтические композиции по настоящему изобретению могут быть получены методами, хорошо известными в данной области, например путем смешивания, растворения, гранулирования, изготовления драже, растирания в порошок, эмульгирования, инкапсулирования, заключения в оболочку или лиофилизации.
Фармацевтические композиции, предназначенные для применения по настоящему изобретению, могут быть получены обычным способом с использованием одного или нескольких физиологически приемлемых носителей, включающих наполнители и вспомогательные вещества, которые облегчают обработку активных ингредентов в препаратах и могут быть использованы в фармацевтическом отношении. Конкретный препарат зависит от выбранного способа введения.
Для получения инъекционных препаратов активные ингредиенты фармацевтической композиции могут быть получены в водных растворах, предпочтительно в физиологически совместимых буферах, таких как раствор Хэнка, раствор Рингера или физологический раствор. Для введения через слизистую оболочку используют проникающие вещества, соответствующие преодолеваемому барьеру. Такие проникающие вещества хорошо известны в данной области.
Фармацевтическая композиция для перорального введения может быть получена в результате объединения активных соединений с фармацевтически приемлемыми носителями, хорошо известными в данной области. Такие носители позволяют получить фармацевтические композиции в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и тому подобного, которые предназначены для приема внутрь субъектом. Фармакологические препараты для перорального введения могут быть получены с использованием твердого наполнителя, необязательного измельчения полученной смеси и получения гранул из указанного смеси после добавления при желании приемлемых вспомогательных веществ для изготовления таблеток или сердцевины драже. Приемлемыми наполнителями являются, в частности, сахара, включающие лактозу, сахарозу, маннит или сорбит; целлюлозные препараты, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий-карбометилцеллюлоза; и/или физиологически приемлемые полимеры, такие как поливинилпирролидон (PVP). При желании могут быть добавлены вещества, улучшающие распадаемость, такие как перекрестно сшитый поливинилпирролидон, агар, альгиновая кислота или ее соль, такая как альгинат натрия.
На сердцевину драже наносят приемлемое покрытие. Для этой цели можно использовать концентрированные растворы сахара, которые могут необязательно содержать аравийскую камедь, тальк, поливинилпирролидон, гелеобразный карбополь, полиэтиленгликоль, диоксид титана, растворы лака и приемлемые органические растворители или смеси растворителей. В таблетки или покрытия драже для идентификации или характеристики разных комбинаций доз активных соединений могут быть добавлены красители или пигменты.
Фармацевтические композиции, которые могут быть использованы перорально, включают жесткие капсулы, изготовленные из желатина, а также мягкие герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Жесткие капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связывающими веществами, такими как крахмалы, смазывающими веществами, такими как тальк или стеарат магния, и необязательно со стабилизаторами. В мягких капсулах активные ингредиенты могут быть растворены или суспендированы в приемлемых липидах, таких как жирные кислоты, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. Все препараты для перорального введения должны быть в дозированной форме, пригодной для выбранного способа введения.
Композиции для трансбуккального введения могут быть в форме таблеток или лепешек, полученных обычным способом.
Лекарственные препараты для ингаляции через нос, содержащие активные ингредиенты по настоящему изобретению, обычно вводят в виде аэрозоля из баллона, находящегося под давлением, или ингалятора с использованием приемлемого пропеллента, такого как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан или диоксид углерода. В случае находящегося под давлением аэрозоля устройство может быть снабжено клапаном для подачи отмеренного количества. Капсулы и кассеты, изготовленные, например, из желатина, которые предназначены для использования в дозирующем устройстве, могут содержать порошкообразную смесь соединения и приемлемую порошкообразную основу, такую как лактоза или крахмал.
Фармацевтическая композиция по настоящему изобретению может быть предназначена для парентерального введения, например, в виде инъекции ударной дозы вещества или непрерывного влияния. Препараты для инъекций могут быть получены в дозированной лекарственной форме, например в ампулах или емкостях, рассчитанных на несколько доз, с необязательно добавленным консервантом. Указанные композиции могут представлять собой суспензии, растворы или эмульсии в мясляных или водных разбавителях и могут содержать образующие агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты.
Фармацевтические композиции для парентерального введения включают водные растворы активного препарата в водорастворимой форме. Кроме того, суспензии активных ингредиентов могут быть получены в виде инъекционных суспензий на масляной или водной основе. Приемлемые липофильные растворители или носители включают жирные кислоты, такие как кунжутное масло, или синтетические жирные кислоты, такие как этилолеат, триглицериды или липосомы. Водные инъекционные суспензии могут содержать вещества, повышающие вязкость суспензии, такие как натрий-карбоксиметилцеллюлоза, сорбит или декстран. Суспензия может необязательно содержать приемлемые стабилизаторы или агенты, повышающие растворимость активных ингредиентов, в результате чего могут быть получены высококонцентрированные растворы.
Альтернативно, активный ингредиент может находиться в порошкообразной форме, предназначенной для растворения непосредственно перед применением в приемлемом растворителе, таком как, например, стерильный апирогенный водный раствор.
Фармацевтическая композиция по настоящему изобретению может быть также получена в виде композиции для ректального введения, такой как суппозитории или удерживающая клизма, с использованием, например, обычных основ для суппозиториев, таких как масло какао и другие глицериды.
Фармацевтические композиции по настоящему изобретению включают композиции, в которых активные ингредиенты присутствуют в эффективном количестве, достаточном для достижения поставленной цели. В частности, терапевтически эффективное количество означает такое количество активных ингредиентов, которое позволяет эффективно предотвратить, устранить или ослабить симптомы заболевания (например, атеросклероза) или продлить жизнь субъекта, подвергаемого лечению.
Специалисты в данной области могут легко определить терапевтически эффективное количество, особенно с учетом приведенного подробного описания изобретения.
Для любого препарата, используемого при осуществлении способов по данному изобретению, терапевтически эффективное количество или дозу можно сначала определить, выполняя анализы in vitro и исследуя культуры клеток. Например, дозу можно определить при помощи животных моделей, позволяющих установить требуемую концентрацию или титр. Такую информацию можно использовать для более точного определения доз, пригодных для лечения человека.
Токсичность и терапевтическую эффективность активных ингредиентов, рассмотренных в данном описании изобретения, можно определить стандартными фармацевтическими методами in vitro с использованием культур клеток или экспериментальных животных. Данные, полученные в результате указанных анализов in vitro, исследования культур клеток и животных, можно использовать для определения диапазона доз, пригодных для лечения человека. Доза может изменяться в зависимости от используемой лекарственной формы и способа введения. Точный препарат, способ введения и доза могут быть выбраны лечащим врачом с учетом состояния нуждающегося субъекта (см., например, публикацию Fingl, et al., 1975, in “The Pharmacological Basis of Therapeutics”, Ch.1, p.1).
Величина дозы и интервалы между приемами лекарственного средства могут быть определены индивидуально для достижения в плазме или головном мозге уровней активого ингредиента, достаточных для индукции или подавления ангиогенеза (минимальная эффективная концентрация, МЕС). Минимальная эффективная концентрация может быть различной для каждого препарата, но может быть определена на основании данных, полученных in vitro. Дозы, необходимые для достижения минимальной эффективной концентрации, зависят от индивидуальных особенностей и способа введения. Для определения концентраций в плазме можно использовать известные методы обнаружения.
В зависимости от тяжести заболевания и реакции на проводимое лечение можно производить однократное или многократное введение дозы, при этом курс лечения может продолжаться от нескольких дней до нескольких недель, до полного выздоровления или ослабления симптомов заболевания.
Количество вводимой композиции, несомненно, зависит от состояния субъекта, нуждающегося в лечении, тяжести поражения, способа введения, решения лечащего врача и т.д.
Композиции по настоящему изобретению при желании могут находиться в упаковке или дозирущем устройстве, таком как набор, утвержденный Управлением по контролю за продуктами и лекарствами (FDA), который может включать одну или несколько дозированных лекарственных форм, содержащих активных ингредиент. Упаковка может представлять собой металлическую или пластиковую фольгу, такую как блистерная упаковка. К упаковке или дозирующему устройству может прилагаться инструкция по введению. Упаковка или дозирущее устройство может быть также снабжено уведомлением, соответствующим форме, предписанной правительственным агентством, регулирующим производство, применение или продажу фармацевтических препаратов, которое должно содержать информацию о санкционировании данным агентством формы композиций или пригодности для человека или животных. Таким уведомлением, например, может быть маркировка, утвержденная Управлением по контролю за продуктами и лекарствами США для прописанных лекарственных средств, или вкладыш о пригодности. Кроме того, могут быть получены композиции, содержащие препарат по настоящему изобретению вместе с совместимым фармацевтическим носителем, которые помещают в соответствующую емкость и маркируют с указанием заболевания, для лечения которого они предназначены, как будет подробно описано ниже.
Таким образом, в предпочтительном варианте осуществления настоящего изобретения фармацевтическую композицию упаковывают в упаковочный материал и указывают в печатном вкладыше или на упаковочном материале, что данная композиция предназначена для лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом. Список типичных заболеваний и нарушений, ассоциированных с таким воспалением, приведен ниже.
Как будет более подробно описано ниже, фармацевтическая композиция по настоящему изобретению может далее включать дополнительное соединение, пригодное для лечения или профилактики вышеуказанного воспаления.
Как подробно описано в нижеследующем разделе “Примеры”, новые соединения по настоящему изобретению позволяют эффективно модулировать иммунную реакцию и/или воспалительную реакцию, ассоциированную с эндогенным окисленным LDL, вызывая таким образом ослабление симптомов заболеваний, ассоциированных с эндогенным окисленным LDL. Полученные результаты ясно показывают, что (i) иммунная и/или воспалительная реакция на эндогенный окисленный LDL, в частности, и эндогенные окисленные липиды вообще может быть модулирована любым соединением, структурно родственным окисленному липиду; и (ii) соединения, способные модулировать иммунную и/или воспалительную реакцию на окисленные липиды, можно использовать для лечения или профилактики воспаления, ассоциированного с эндогенными окисленными липидами.
Таким образом, другим объектом настоящего изобретения является способ лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом. Способ по данному объекту настоящего изобретения осуществляют путем введения нуждающемуся субъекту терапевтически эффективного количества одного или нескольких окисленных липидов.
В используемом здесь значении термин “эндогенный окисленный липид” означает один или несколько окисленных липидов, которые присутствуют или образуются in vivo в результате воспалительных и других процессов, опосредуемых клетками или гуморальной системой.
Термин “способ” означает способы, средства, методы и процедуры, выполняемые для достижения поставленной цели, которые включают, не ограничиваясь ими, способы, средства, методы и процедуры, которые известны в данной области или могут быть легко разработаны на основе известных способов, средств, методов и процедур специалистами в химической, фармакологической, биологической, биохимической и медицинской областях.
В используемом здесь значении термин “лечение или профилактика” означает устранение, по существу ингибирование, замедление или реверсию развития заболевания, ослабление клинических симптомов заболевания или предотвращение возникновения клинических симптомов заболевания.
Субъектами, которым может быть назначено такое лечение, являются субъекты, страдающие заболеваниями или нарушениями, ассоциированными с воспалением, которые подробно описаны ниже. Предпочтительными субъектами по настоящему изобретению являются млекопитающие, такие как собаки, кошки, овцы, свиньи, лошади и крупный рогатый скот. Субъектами по настоящему изобретению предпочтительно являются люди.
Термин “окисленный липид” означает природное или предпочтительно синтезированное соединение, имеющее общие структурные признаки с природным липидом, окисленным липидом и любыми его компонентами, частями, аналогами и производными. Например, окисленный LDL состоит из нескольких функционально и структурно разных частей, и в определение данного термина входят любые синтезированные соединения, имеющие общие структурные признаки с любыми из указанных частей. В определение данного термина далее входит любое производное таких аналогов.
Типичные примеры окисленных липидов включают, не ограничиваясь ими, окисленные фосфолипиды, аналоги фактора активации тромбоцитов, аналоги плазмалогена, замещенные или незамещенные углеводороды, содержащие 3-30 атомов углерода и концевую окисленную группу, окисленные аналоги сфинголипидов, окисленные аналоги гликолипидов, окисленные аналоги мембранных липидов и любые их аналоги или производные.
Фосфолипиды вообще и фосфоглицериды, в частности, представляют собой хорошо известные липиды, которые являются также компонентами окисленного LDL. Фосфоглицериды являются производными фосфоглицерина, который содержит одну или несколько ацильных групп жирных кислот или ацильных групп, присоединенных к фосфоглицериновому остову.
Синтезированные окисленные фосфолипиды могут быть эффективно использованы при осуществлении способа по данному объекту настоящего изобретения. Типичные примеры известных синтетических окисленных фосфолипидов включают, не ограничиваясь ими, 1-пальмитоил-2-азелаоил-sn-глицеро-3-фосфохолин, 1-гексадецил-2-азелаоил-sn-глицеро-3-фосфохолин, 1-пальмитоил-2-глутароил-sn-глицеро-3-фосфохолин (PGPC), 1-пальмитоил-2-(5-оксовалероил)-sn-глицеро-3-фосфохолин (POVPC) и 1-пальмитоил-2-(9-оксононаноил)-sn-глицеро-3-фосфохолин.
Более предпочтительные примеры синтетических окисленных фосфолипидов включают вышеописанные соединения по настоящему изобретению, в которых n=1. Указанные соединения определены в данном описании изобретения как замещенные или незамещенные углеводороды, содержащие 3-30 атомов углерода и концевую окисленную группу.
Другие соединения, которые имеют структуру, родственную окисленным фосфоглицеридам, и поэтому могут быть эффективно использованы при осуществлении данного и других объектов настоящего изобретения, являются аналогами фактора активации тромбоцитов (PAF).
PAF представляют собой 1-алкил-2-ацетил-sn-глицеро-3-фосфохолины, естественные глицеролипиды, связанные простой эфирной связью. Алкильная цепь в положении sn-1 обычно является ненасыщенным алкилом, имеющим 16-18 атомов углерода. Некоторые хорошо известные аналоги PAF обычно характеризуются замещением ацильной части в положении sn-2 длинноцепной ацильной частью (например, ацилом жирной кислоты). Дополнительные аналоги PAF включают окислительную модификацию в положении sn-1 ненасыщенной О-алкильной цепи или в положении sn-2 ацильной цепи жирной кислоты.
Типичные примеры известных аналогов PSF, которые могут быть использованы в настоящем изобретении, включают, не ограничиваясь ими, 1-пальмитоил-2-(9-оксононаноил)-sn-глицеро-3-фосфохолин, 1-гексадецил-2-ацетоил-sn-глицеро-3-фосфохолин, 1-октадецил-2-ацетоил-sn-глицеро-3-фосфохолин, 1-гексадецил-2-бутироил-sn-глицеро-3-фосфохолин, 1-октадецил-2-бутироил-sn-глицеро-3-фосфохолин, 1-пальмитоил-2-ацетоил-sn-глицеро-3-фосфохолин, 1-октадеценил-2-ацетоил-sn-глицеро-3-фосфохолин, 1-гексадецил-2-(гомогаммалиноленоил)-sn-глицеро-3-фосфохолин, 1-гексадецил-2-арахидоноил-sn-глицеро-3-фосфохолин, 1-гексадецил-2-эйкозапентаеноил-sn-глицеро-3-фосфохолин, 1-гексадецил-2-докозагексаеноил-sn-глицеро-3-фосфохолин, 1-октадецил-2-метил-sn-глицеро-3-фосфохолин, 1-гексадецил-2-бутеноил-sn-глицеро-3-фосфохолин, Lyso PAF C16 и Lyso PAF C18. Однако в контексте настоящего изобретения могут быть также использованы любые другие аналоги PAF или их производные.
Дополнительные соединения, которые имеют структуру, родственную окисленным фосфоглицеридам, и поэтому могут быть эффективно использованы по данному и другим объектам настоящего изобретения, являются аналогами плазмалогена.
Плазмалогены представляют собой 1-алкил-2-ацетил-sn-глицеро-3-фосфатидил, естественные глицеролипиды, связанные простой эфирной связью, в которых алкильная цепь в положении sn-1 обычно является насыщенной. Некоторые хорошо известные аналоги плазмалогенов обычно характеризуются замещением ацильной части в положении sn-2 длинноцепной ацильной частью (например, ацил жирной кислоты) и далее включают окислительную модификацию в положении sn-1 или в положении sn-2.
Типичные примеры известных аналогов плазмалогена, которые могут быть использованы в контексте настоящего изобретения, включают, не ограничиваясь ими, 1-О-1'-(Z)-гексадеценил-2-[12-[(7-нитро-2-1,3-бензоксадиазол-4-ил)амино]додеканоил]-sn-глицеро-3-фосфохолин, 1-О-1'-(Z)-гексадеценил-2-олеоил-sn-глицеро-3-фосфохолин, 1-О-1'-(Z)-гексадеценил-1-арахидоноил-sn-глицеро-3-фосфохолин, 1-О-1'-(Z)-гексадеценил-2-докозагексаеноил-sn-глицеро-3-фосфохолин, 1-О-1'-(Z)-гексадеценил-2-олеоил-sn-глицеро-3-фосфоэтаноламин, 1-О-1'-(Z)-гексадеценил-2-арахидоноил-sn-глицеро-3-фосфоэтаноламин и 1-О-1'-(Z)-гексадеценил-2-докозагексаеноил-sn-глицеро-3-фосфоэтаноламин. Однако в контексте настоящего изобретения могут быть также использованы любые другие аналоги плазмалогенов или их производные.
В используемом здесь значении термин “воспаление, ассоциированное с эндогенным окисленным липидом” означает воспаление, которое ассоциировано с образованием или присутствием in vivo одного или нескольких окисленных липидов (таких как окисленный LDL, окисленные мембранные липиды и т.д.).
Воспаление является защитной реакцией организма на повреждение. Воспалительные реакции опосредованы несколькими цитокинами, к которым относятся IFN-γ и IL-10. IFN-γ участвует в патогенезе разных аутоиммунных и хронических воспалительных состояний. С другой стороны, IL-10 ингибирует продуцирование IFN-γ активированными иммунокомпетентными клетками, такими как ТН2 м М2, из чего следует, что данный цитокин (IL-10) является главным противовоспалительным “щитом”.
Сильное воспаление часто носит разрушительный характер и ведет к возникновению многочисленных заболеваний и нарушений. Как было подробно описано выше, сильная воспалительная реакция обычно ассоциирована с эпитопами окисленных липидов.
Как показано в нижеследующем разделе “Примеры”, модуляция иммунной реакции на окисленный LDL при помощи синтетических аналогов окисленного LDL оказывает противовоспалительное действие. Указанное противовоспалительное действие можно использовать для лечения или профилактики заболеваний или нарушений, обусловленных воспалением, которое ассоциировано с эндогенным окисленным LDL или любым другим эндогенным окисленным липидом. Такие заболевания и нарушения включают, например, заболевания или нарушения, обусловленные образованием бляшек, которые включают, не ограничиваясь ими, атеросклероз, атеросклеротические сердечно-сосудистые заболевания, цереброваскулярные заболевания, заболевания периферических сосудов, стеноз, рестеноз и стеноз в стенте, аутоиммунные заболевания или нарушения, нейродегенеративные заболевания или нарушения, пролиферативные заболевания или нарушения и процессы, связанные со старением.
Таким образом, типичные примеры заболеваний или нарушений, обусловленных воспалением, которое в свою очередь ассоциировано с эндогенными окисленными липидами, и поэтому пригодных для лечения способом по настоящему изобретению, включают, например, идиопатические воспалительные заболевания или нарушения, хронические воспалительные заболевания или нарушения, острые воспалительные заболевания или нарушения, аутоиммунные заболевания или нарушения, инфекционные заболевания или нарушения, воспалительные злокачественные заболевания или нарушения, воспалительные заболевания или нарушения, связанные с трансплантацией, воспалительные дегенеративные заболевания или нарушения, заболевания или нарушения, обусловленные гиперчувствительностью, воспалительные сердечно-сосудистые заболевания или нарушения, воспалительные цереброваскулярные заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительные заболевания или нарушения желез, воспалительные желудочно-кишечные заболевания или нарушения, воспалительные кожные заболевания или нарушения, воспалительные заболевания или нарушения печени, воспалительные неврологические заболевания или нарушения, воспалительные заболевания или нарушения костно-мышечной системы, воспалительные заболевания или нарушения почек, воспалительные заболевания или нарушения репродуктивной системы, воспалительные системные заболевания или нарушения, воспалительные заболевания или нарушения соединительной ткани, воспалительные опухоли, некрозы, воспалительные заболевания или нарушения, связанные с имплантатом, воспалительные процессы, связанные со старением, заболевания или нарушения, обусловленные иммунодефицитом, пролиферативные заболевания и нарушения, а также воспалительные легочные заболевания или нарушения, как это подробно описано ниже.
Неограничивающие примеры гиперчувствительности включают гиперчувствительность типа I, гиперчувствительность типа II, гиперчувствительность типа III, гиперчувствительность типа IV, гиперчувствительность немедленного типа, антителоопосредованную гиперчувствительность, гиперчувствительность, опосредованную иммунным комплексом, гиперчувствительность, опосредованную Т-лимфоцитами, гиперчувствительность замедленного типа, гиперчувствительность, опосредованную Т-лимфоцитами-хелперами, гиперчувствительность, опосредованную цитотоксическими Т-лимфоцитами, гиперчувствительность, опосредованную ТН1-лимфоцитами, и гиперчувствительность, опосредованную ТН2-лимфоцитами.
Неограничивающие примеры воспалительных сердечно-сосудистых заболеваний или нарушений включают обтурирующие заболевания или нарушения, атеросклероз, порок клапана сердца, стеноз, рестеноз, стеноз в стенте, инфаркт миокарда, коронарную болезнь сердца, острые коронарные синдромы, застойную сердечную недостаточность, стенокардию, ишемическую болезнь миокарда, тромбоз, грануломатоз Вегенера, артериит Такаясу, синдром Кавасаки, аутоиммунное заболевание или нарушение, направленное против фактора VIII, некротический васкулит мелких сосудов, микроскопическую ангиопатию, синдром Черга и Штрауса, олигоиммунный очаговый некротический гломерулонефрит, серповидный гломерулонефрит, антифосфолипидный синдром, антителоиндуцированную сердечную недостаточность, тромботическую пурпуру, аутоиммунную гемолитическую анемию, аутоиммунное заболевание сердца, болезнь Шагаса и аутоиммунитет против Т-лимфоцитов-хелперов.
Стеноз является обтурирующим заболеванием сосудистой сети, которое обычно возникает в результате образования атеросклеротических бляшек и повышенной активности тромбоцитов и поражает главным образом сосудистую систему сердца.
Рестеноз является прогрессирующей повторной обтурацией, которая часто возникает после удаления обтураций в стенозированной сосудистой системе. В тех случаях, когда раскрытое состояние сосуда может быть обеспечено механической поддержкой стента, может возникнуть стеноз в стенте, вызывающий повторную обтурацию восстановленного сосуда.
Неограничивающие примеры цереброваскулярных заболеваний или нарушений включают инсульт, воспаление сосудов головного мозга, внутримозговое кровоизлияние и позвоночно-артериальную недостаточность.
Неограничивающие примеры заболеваний и нарушений периферических сосудов включают гангрену, диабетический васкулит, ишемическую болезнь кишечника, тромбоз, диабетическую ретинопатию и диабетическую нефропатию.
Неограничивающие примеры аутоиммунных заболеваний и нарушений включают все заболевания, вызываемый иммунной реакцией, такой как аутоантитело- или клеточноопосредуемая иммунная реакция на аутоантиген и тому подобные. Типичными примерами являются хронический ревматоидный артрит, болезнь Стилла, системная красная волчанка, склеродерма, комплексное заболевание соединительной ткани, нодозный полиартериит, полимиозит/дерматомиозит, синдром Шегрена, синдром Бехчета, рассеянный склероз, аутоиммунный диабет, болезнь Хасимото, псориаз, первичная микседема, пернициозная анемия, миастения, хронический активный гепатит, аутоиммунная гемолитическая анемия, идиопатическая тромботическая пурпура, увеит, васкулит и гепарининдукцированная тромбоцитопения.
Неограничивающие примеры воспалительных заболеваний или нарушений желез включают заболевания или нарушения поджелудочной железы, диабет типа I, заболевания или нарушения щитовидной железы, болезнь Грейвса, тиреоидит, спонтанный аутоиммунный тиреоидит, тиреоидит Хасимото, идиопатическую микседему, аутоиммунное заболевание яичника, аутоиммунное мужское бесплодие, аутоиммунный простатит и аутоиммунный плюригландулярный синдром типа I.
Неограничивающие примеры воспалительных желудочно-кишечных заболеваний или нарушений включают колит, илеит, болезнь Крона, хроническое воспалительное заболевание тонкой кишки, синдром раздраженной толстой кишки, хроническое воспалительное заболевание толстой кишки, глютеновую болезнь, неспецифический язвенный колит, язву, кожную язву, пролежни, язву желудка, пептическую язву, щечную язву, носоглоточную язву, язву пищевода, язву двенадцатиперстной кишки и язву желудочно-кишечного тракта.
Неограничивающие примеры воспалителных кожных заболеваний или нарушений включают угри и аутоиммунное буллезное заболевание кожи.
Неограничивающие примеры воспалительных заболеваний или нарушений печени включают аутоиммунный гепатит, цирроз печени и билиарный цирроз печени.
Неограничивающие примеры воспалительных неврологических заболеваний или нарушений включают рассеянный склероз, болезнь Альцгеймера, болезнь Паркинсона, миастению, двигательную невропатию, синдром Гийена-Барре, аутоиммунную невропатию, миастенический синдром Ламберта-Итона, паранеопластическое неврологическое заболевание или нарушение, паранеопластическую мозжечковую атрофию, непаранеопластический синдром тугоподвижности, прогрессирующую мозжечковую атрофию, энцефалит Расмуссена, боковой амиотрофический склероз, хорею Сиденгама, синдром Жилль де ла Туретта, аутоиммунную полиэндокринопатию, невропатию, обусловленную нарушением иммунитета, приобретенную невромиотонию, множественный артрогрипоз, болезнь Гентингтона, слабоумие, обусловленное СПИДом, боковой амиотрофический склероз (AML), рассеянный склероз, инсульт, воспалительное заболевание сетчатки, воспалительное заболевание глаз, ретробульбарный неврит, губковидную энцефалопатию, мигрень, головную боль, гистаминовую головную боль и синдром тугоподвижности.
Неограничивающие примеры воспалительных заболеваний или нарушений соединительной ткани включают аутоиммунный миозит, первичный синдром Шегрена, аутоиммунное заболевание или нарушение гладких мышц, миозит, тендинит, воспаление связок, хондрит, воспаление суставов, синовиальное воспаление, синдром канала запястья, артрит, ревматоидный артрит, остеоартрит, анкилозирующий спондилоартрит, воспаление костной системы, аутоиммунное заболевание уха и аутоиммунное заболевание внутреннего уха.
Неограничивающие примеры воспалительных заболеваний или нарушений почек включают аутоиммунный интерстициальный нефрит и/или рак почки.
Неограничивающие примеры воспалительных заболеваний или нарушений репродуктивной системы включают повторную потерю плода, кисту яичника, заболевание или нарушение, связанное с менструальным циклом.
Неограничивающие примеры воспалительных системных заболеваний или нарушений включают системную красную волчанку, системный склероз, септический шок, синдром токсического шока и кахексию.
Неограничивающие примеры инфекционных заболеваний или нарушений включают хронические инфекционные заболевания или нарушения, подострые инфекционные заболевания или нарушения, острые инфекционные заболевания или нарушения, вирусные заболевания или нарушения, бактериальные заболевания или нарушения, заболевания или нарушения, вызванные простейшими, паразитарные заболевания или нарушения, микоз, микоплазменные заболевания или нарушения, гангрену, сепсис, заболевания или нарушения, вызванные насекомыми, грипп, туберкулез, малярию, синдром приобретенного иммунодефицита и тяжелое острое респираторное заболевание.
Неограничивающие примеры воспалительных заболеваний или нарушений, связанных с трансплантацией, включают отторжение трансплантата, хроническое отторжение трансплантата, подострое отторжение трансплантата, острое отторжение трансплантата, сверхострое отторжение трансплантата и реакцию “трансплантат против хозяина”. Типичные имплантаты включают протезный имплантат, грудной имплантат, силиконовый имплантат, зубной имплантат, имплантат полового члена, сердечный имплантат, искусственный сустав, устройство для восстановления перелома костей, костный имплантат, имплантат для доставки лекарственного средства, катетер, сердечный ритмоводитель, искусственное сердце, искусственный клапан сердца, имплантат, высвобождающий лекарственное средство, электрод и дыхательную трубку.
Неограничивающие примеры воспалительных опухолей включают злокачественную опухоль, доброкачественную опухоль, солидную опухоль, метастатическую опухоль и несолидную опухоль.
Неограничивающие примеры воспалительных легочных заболеваний и нарушений включают астму, аллергическую астму, эмфизему, хроническое обтурирующее заболевание легких, саркоидоз и бронхит.
Примером пролиферативного заболевания или нарушения является рак.
В научной литературе недавно было описано применение фосфолипидов и метаболитов фосфолипидов для лечения или профилактики заболеваний и синдромов, таких как, например, окислительный стресс в процессе старения (Onorato J.M., et al., Annal NY. Acad. Sci. 1998 Nov. 20; 854:277-90), ревматоидный артрит (RA) (Paimela L, et al., Ann. Rheum. Dis. 1996 Aug; 55(8):558-9), болезнь Стилла (Savolainen A, et al., 1995; 24(4):209-11), воспалительное заболевание толстой кишки (IBD) (Sawai T, et al., Pediatr Surg. Int. 2001 May; 17(4):269-74) и рак почки (Noguchi S, et al., Buiochem Biophys. Res. Commun. 1992 Jan 31; 182(2):544-50), что еще более подтвержает пригодность аналогов окисленного LDL для лечения или профилактики указанных заболеваний или нарушений.
В соответствии со способом по настоящему изобретению окисленные липиды можно вводить нуждающемуся субъекту разными способами, которые включают, например, пероральное и ректальное введение, введение через слизистую оболочку, особенно в нос, кишечное или парентеральное введение, включающее внутримышечные, подкожные и интрамедуллярные инъекции, а также подоболочечные, прямые интравентрикулярные, внутривенные, внутрибрюшинные, назальные или внутриглазные инъекции. Однако, как подробно описано в данном описании изобретения и далее показано в нижеследующем разделе “Примеры”, предпочтительными способами введения являются пероральный, через слизистую оболочку, назальный, внутрикожный (подкожный) и внутрибрюшинный способы введения.
В одном варианте осуществления изобретения внутрибрюшинно вводят 0,1-100 мг/кг окисленного липида в приемлемом носителе, который включает, не ограничиваясь ими, PBS или глицерин, один-три раза в неделю в соответствии со схемой постоянного или чередующегося введения.
В другом варианте осуществления изобретения в нос вводят 0,1-100 мг/кг окисленного липида в приемлемом носителе, который включает, не ограничиваясь ими, PBS или глицерин, один-три раза в неделю в соответствии со схемой постоянного или чередующегося введения.
В другом варианте осуществления изобретения подкожно вводят 0,1-100 мг/кг окисленного липида в приемлемом носителе, который включает, не ограничиваясь ими, PBS или глицерин, один-три раза в неделю в соответствии со схемой постоянного или чередующегося введения.
В другом варианте осуществления изобретения перорально вводят 0,1-100 мг/кг окисленного липида в приемлемом носителе, который включает, не ограничиваясь ими, PBS или глицерин, один-три раза в неделю в соответствии со схемой постоянного или чередующегося введения.
Способ по настоящему изобретению может далее включать введение вышеописанных композиций вместе с одним или несколькими дополнительными соединениями, предназначенными для лечения или профилактики вышеописанного воспаления, ассоциированного с эндогенным окисленным липидом.
Способ по настоящему изобретению может включать совместное введение, осуществляемое до, одновременно или после введения окисленных липидов, терапевтически эффективного количества одного или нескольких дополнительных соединений, при этом фармацевтическая композиция по настоящему изобретению может содержать помимо соединений по настоящему изобретению такие дополнительные соединения.
Типичные примеры дополнительных соединений, которые предназначены для лечения или профилактики воспаления, ассоциированного с вышеописанным эндогенным окисленным липидом, и поэтому пригодны для использования в данном варианте осуществления настоящего изобретения, включают, не ограничиваясь ими, ингибиторы HMGCoA-реуктазы (статины), адъюванты слизистой оболочки, кортикостероиды, средства стероидной противовоспалительной терапии, средства нестероидной противовоспалительной терапии, аналгетики, факторы роста, токсины, ингибиторы белка, обеспечивающего перенос сложного холестерилового эфира (CETP), агонисты пролиферирующего активированного рецептора пероксисомы (PPAR), противоатеросклерозные средства, антипролиферативные средства, эзетимид, никотиновую кислоту, ингибиторы сквалена, ApoE Milano, HSP, бета-2-гликопротеин I и любое их производное и аналог.
Ингибиторы HMGCoA-редуктазы (статины) являтся хорошо известными лекарственными средствами, которые эффективно снижают уровни LDL-холестерина в результате ингибирования фермента, регулирующего скорость продуцирования холестерина, и усиления выведения печенью LDL-холестерина, присутствующего в крови. Неограничивающие примеры обычно прописываемых статинов включают аторвастатин, флувастатин, ловастатин, правастатин и симвастатин.
Эзетимиб является первым средством в новом классе ингибиторов абсорбции холестерина, который эффективно и избирательно ингибирует поглощенние пищевого и билиарного холестерина у щеточной каемки кишечного эпителия, не влияя на поглощение триглицерида или жирорастворимых витаминов. Таким образом, эзетимиб уменьшает общее поступление холестерина в печень и индуцирует повышенную экспрессию рецепторов LDL, что способствует большему удалению LDL-C из плазмы.
Пероксисома является одномембранной органеллой, присутствующей почти во всех эукариотических клетках. Одним из наиболее важных метаболических процессов, опосредуемых пероксисомой, является β-окисление жирных кислот с длинными и очень длинными цепями. Пероксисома участвует также в синтезе желчной кислоты, холестерина и плазмалогена, метаболизме аминокислот и пурина.
Никотиновая кислота является известным агентом, снижающим уровни общего холестерина, LDL-холестерина и триглицерида и повышающим уровни HDL-холестерина. Существует три типа лекарственных средств на основе никотиновой кислоты: средства немедленного действия, отсроченного действия и пролонгированного действия. Никотиновая кислота или ниацин, водорастворимый витамин В, улучшает все липопротеины при введении в дозах, превышающих потребность в данном витамине.
Сквален, изопреноидное соединение, в структурном отношении подобное бета-каротину, является перомежуточным метаболитом в процессе синтеза холестерина. Организм человека поглощает примерно 60 процентов пищевого сквалена. Он переносится в сыворотке, будучи связанным с липопротеинами очень низкой плотности, и распределяется по всем тканям человека с достижением наибольшей концентрации в коже, где он является одним из основных компонентов поверхностных липидов кожи. Ингибиторы сквалена (например, монооксигеназа и синтаза) служат в качестве ингибиторов биосинтеза холестерина.
Агонисты пролиферирующего активированного рецептора (PPAR), например фибраты, являются членами надсемейства нуклеарных рецепторов, активируемыми жирными кислотами, которые играют важную роль в метаболизме липидов и глюкозы и применяются для лечения заболеваний обмена веществ, связанных с ожирением, таких как гиперлипемия, устойчивость к инсулину и ишемическая болезнь сердца. Фибраты обычно эффективно снижают повышенные уровни триглицеридов и холестерина в плазме и действуют в качестве агонистов PPAR. Наиболее выраженным действием фибратов является снижение концентрации липопротеинов с высоким содержанием триглицеридов (TRL) в плазме. Уровни LDL-холестерина (LDL-C) обычно снижаются у субъектов с повышенными исходными концентрациями в плазме, и уровни HDL-холестерина (HDL-C) обычно повышаются при низких исходных концентрациях в плазме. Неограничивающие примеры обычно прописываемых фибратов включают безафибрат, гемфиброзил и фенофибрат.
Ингибиторы белка, обеспечивающего перенос сложного холестерилового эфира, (CETP) играют важную роль в атерогенезе, уменьшая накопление сложного холестерилового эфира в макрофагах и стенках артерий, в результате чего сокращается образование ксантомных клеток и поглощение холестерина. Наиболее перспективным известным в настоящее время ингибитором СЕТР является авизимиб.
ApoA-I Milano, обычно используемый в виде рекомбинантного комплекса с фосфолипидом (ЕТС-216), вызывает значительную регрессию атеросклероза коронарной артерии.
Установлено, что совместное введение адъювантов слизистой оболочки имеет важное значение для предотвращения инвазии инфекционных агентов через поверхности слизистых оболочек. На начальных стадиях индукции иммунной реакции слизистой оболочки поглощение перорально или назально вводимых антигенов достигается благодаря уникальной совокупности антиген-отбирающих клеток, М-клеток, расположенных в фолликулоассоциированном эпителии (FAE) индуктивных сайтов. После успешного поглощения антигены немедленно процессируются и представляются подстилающими дендритными клетками (DC).
Неограничивающие примеры средств нестероидной противовоспалительной терапии включают оксикамы, такие как пироксикам, изоксикам, теноксикам, судоксикам и СР-14304; салицилаты, такие как аспирин, дизальцид, бенорилат, трилизат, сафаприн, солприн, дифлунизал и фендозал; производные уксусной кислоты, такие как диклофенак, фенклофенак, индометацин, сулиндак, толметин, изоксепак, фурофенак, тиопинак, зидометацин, ацеметацин, фентиазак, зомепирак, клинданак, оксепинак, фелбинак и кеторолак; фенаматы, такие как мефенаминовая, меклофенаминовая, флуфенаминовая, нифлуминовая и толфенаминовая кислоты; производные пропионовой кислоты, такие как ибупрофен, напроксен, беноксапрофен, флурбипрофен, кетопрофен, фенопрофен, фенбуфен, индопрофен, пирпрофен, карпрофен, оксапрозин, пранопрофен, миропрофен, тиоксапрофен, супрофен, альминопрофен и тиапрофеновая кислота; пиразолы, такие как фенилбутазон, оксифенбутазон, фепразон, азапропазон и триметазон.
Неограничивающие примеры средств стероидной противовоспалительной терапии включают, не ограничиваясь ими, кортикостероиды, такие как гидрокортизон, гидроксилтриамцинолон, альфа-метил-дексаметазон, фосфат дексаметазона, дипропионаты беклометазона, валерат клобетазола, дезонид, дезоксиметазон, ацетат дезоксикортикостерона, дексаметазон, дихлоризон, диацетат дифлоразона, валерат дифлукортолона, флуадренолон, ацетонид флуклоролона, флудрокортизон, пивалат флуметазона, ацетонид флуозинолона, флуоцинонид, сложные бутиловые эфиры флукортина, флуокортолон, ацетат флупреднидена (флупреднилидена), флурандренолон, гальцинонид, ацетат гидрокортизона, бутират гидрокортизона, метилпреднизолон, ацетонид триамцинолона, кортизон, кортодоксон, флуцетонид, флудрокортизон, диацетат дифторозона, флурадренолон, флудрокортизон, диацетат дифлурозона, ацетонид флурадренолона, медризон, амцинафел, амцинафид, бетаметазон и баланс его сложных эфиров, хлорпреднизон, ацетат хлорпреднизона, клокортелон, клесцинолон, дихлоризон, дифлурпреднат, флуклоронид, флунизолид, фторометалон, флуперолон, флупреднизолон, валерат гидрокортизона, циклопентилпропионат гидрокортизона, гидрокортамат, мепреднизон, параметазон, преднизолон, преднизон, дипропионат беклометазона, триамцинолон и их смеси.
Неограничивающие примеры аналгетиков (болеутоляющих средств) включают аспирин и другие салицилаты (такие как салицилат холина или магния), ибупрофен, кетопрофен, напроксен-натрий и ацетаминофен.
Факторы роста являются гормонами, выполнящими целый ряд функций, включающих регулирование образования молекул межклеточной адгезии, изменение пролиферации клеток, увеличение васкуляризации, повышение синтеза коллагена, регулирование костного метаболизма и изменение миграции клеток на данном участке. Неограничивающие примеры факторов роста включают инсулиноподобный фактор роста 1 (IGF-1), трансформирущий β-фактор роста (TGF-β), костный морфогенный белок (ВМР) и тому подобные.
Неограничивающие примеры токсинов включают холероген, который служит также в качестве адъюванта.
Неограничивающие примеры антипролиферативных средств включают алкилирущий агент, такой как азотистый иприт, этиленимин и метилмеламин, алкилсульфонат, нитрозомочевину и триазен; антиметаболит, такой как аналог фолиевой кислоты, аналог пиримидина и аналог пурина; природный продукт, такой как алкалоид барвинка, эпиподофиллотоксин, антибиотик, фермент, таксан и модификатор биологической реакции; разные агенты, такие как координационный комплекс платины, антрацендион, антрациклин, замещенная мочевина, производное метилгидразина или адренокортикальный супрессор; гормон или антагонист, такой как адренокортикостероид, прогестин, эстроген, антиэстроген, андроген, антиандроген или аналог гонадолиберина. Конкретные примеры химиотерапевтических средств включают, например, азотистый иприт, эпиподофиллотоксин, антибиотик, координационный комплекс платины, блеомицин, доксорубицин, паклитаксел, этопозид, циклофосфамид 4-ОН и цисплатин.
Семейстсо HSP состоит примерно из 25 белков, отличающихся молекулярной массой с высококонсервативными структурами. Почти у всех людей возникают клеточные и гуморальные иммунные реакции против микробного хитшокового белка 60 (HSP60). Из-за высокой степени антигенной гомологии, существующей между микробным (бактериальным или паразитарным) и человеческим HSP60, иммунитет в отношении микробов чреват опасностью перекрестной реактивности с HSP60 человека, экспрессируемым эндотелиальными клетками артерий в условиях стресса. Действительный аутоиммунитет против измененного аутологичного HSP60 может также запустить данный процесс (Wick et al., Atherosclerosis as an autoimmune disease: an update. TRENDS in Immunology. 2001; 22(12):665-669). HSP участвует в качестве аутоантигена-мишени в нескольких экспериментальных аутоиммунных заболеваниях (атрит, диабет типа I). Антитела против HSP65 и против HSP60 были явно ассоциированы с атероматозными поражениями у человека. Исследования, выполненные с использованием кроликов и мышей, показывают, что возникновение HSP65-индуцируемой иммунной реакцией в результате иммунизации рекомбинантным белком или препаратом Mycobacterium tuberculosis с высоким содержанием HSP65 усиливает атерогенез. Используя аутоиммунные процессы в отношении HSP65 в качестве возможного антигена-кандидата, который вызывает состояние иммунологической толерантности в результате “толеризации” слизистой оболочки, для блокировки указанных реакций, авторы настоящего изобретения обнаружили ослабление начальной стадии атеросклероза у мышей, которым перорально вводили HSP65, по сравнению с мышами, которым перорально вводили BSA или PBS (Harats et al., Oral tolerance with heat shock protein 65 attenuates mycobacterium tuberculosis induces and high fat diet driven atherosclerosis lesions. J. Am. Coll. Cardiol. 2002; 40:1333-1338); полученный результат был подтвержден Мароном, который показал, что назальная вакцинация HSP уменьшает воспалительный процесс, ассоциированный с атеросклерозом (Maron et al., Mucosal administration of heat shock protein-65 decreases atherosclerosis and inflammation in aortic arch of low density lipoprotein receptor-deficient mice. Circulation. 2002; 106:1708-1715).
Бета-2-гликопротеин I (бета2GPI) является фосфолипидсвязывающим белком, который служит в качестве мишени для протромботических антител против фосфолипида. Недавно было установлено, что данный белок вызывает иммуноопосредуемую реакцию и усиливает атеросклероз у мышей. β-Антитела к бета-2-GPI обладают способностью активировать моноциты и эндотелиальные клетки и могут индуцировать иммунную реакцию на бета2GPI у мышей с повышенным риском возникновения атеросклероза, ускоряя развитие данного заболевания. Когда бета2GPI-реактивный лимфатический узел и клетки селезенки переносили в организм мышей с отсутствием рецептора LDL, они стимулировали образование жировых полосок, подтверждая непосредственную проатерогенную роль бета2GPI-специфических лимфоцитов. Индукция иммунологической толерантности к бета2GPI в результате предварительного перорального введения антигена позволяла значительно уменьшить степень атеросклеротических поражений. Таким образом, бета2GPI является предполагаемым модулятором атеросклеротических бляшек, и поэтому, вероятно, может быть использован в качестве иммуномодулятора развития бляшек. Пероральное введение бета2GPI ингибирует реактивность клеток лимфатического узла в отношении бета2GPI у мышей, иммунизированных против белка человека. В клетках лимфатического узла бета2GPI-толерантных мышей, иммунизированных против бета2GPI, увеличивалось продуцирование IL-4 и IL-10 при инициировании соответствующим белком. Таким образом, пероральное введение бета2GPI является эффективным средством подавления атерогенеза у мышей (George et al. Suppression of early atherosclerosis in LDL-receptor deficient mice by oral tolerance with beta2-glycoprotein I. Cardiovasc. Res. 2004; 62(3):603-9).
Дополнительные цели, преимущества и отличительные признаки настоящего изобретения будут очевидны для специалистов в данной области при ознакомлении с нижеследующими примерами, не ограничивающими объем изобретения. Кроме того, все варианты осуществления и объекты настоящего изобретения, рассмотренные в описании изобретения и сформулированные в приведенной ниже формуле изобретения, находят экспериментальное подтверждение в нижеследующих примерах.
Примеры
Далее приведены примеры, которые вместе с описанием изобретения иллюстрируют данное изобретение, не ограничивая его объем.
Номенклатура, использованная в описании изобретения и лабораторных процедурах, относится к биохимическим и иммунологическим методам. Такие методы всесторонне описаны в научной литературе. См., например, публикации "Cell Biology: A Laboratory Handbook", Volumes I-III Cellis, J. E., ed. (1994); "Current Protocols in Immunology" Volumes I-III Coligan J. E., ed. (1994); Stites et al. (eds), "Basic and Clinical Immunology" (8th Edition), Appleton & Lange, Norwalk, CT (1994); Mishell and Shiigi (eds), "Selected Methods in Cellular Immunology", W. H. Freeman and Co., New York (1980); существующие иммуноанализы также всесторонне описаны в патентной и научной литературе; см., например, патенты США №№ 3791932, 3839153, 3850752, 3850578, 3853987, 3867517, 3879262, 3901654, 3935074, 3984533, 3996345, 4034074, 4098876, 4879219, 5011771 и 5281521 и публикацию “Methods in Enzymology” Vol. 1-317, Academic Press; Marshak et al., которые полностью включены в данное описание изобретения в качестве ссылки. В описание изобретения включены также другие общие ссылки. Считается, что указанные методы хорошо известны в данной области и представлены только для удобства читателя. Вся информация, содержащаяся в приведенных публикациях, включена в данное описание изобретения в качестве ссылки.
Материалы и общие методы
Животные
Мыши с отсутствием Аро-Е. Мыши с отсутствием аполипопротеина Е (Аро-Е КО), используемые в данных экспериментах, относятся к предрасположенной к атеросклерозу линии C57BL/6J-Apoetmlunc. У мышей, гомозиготных в отношении мутаций Apoetmlunc, наблюдается значительное повышение уровней общего холестерина в плазме, на которое не влияет ни возраст, ни пол. Жировые полоски в проксимальной аорте обнаружены в возрасте 3 месяцев. Поражения увеличиваются с возрастом и прогрессируют с образованием поражений, содержащих меньше липида, но больше удлиненных клеток, типичных для более поздней стадии предатеросклеротического поражения.
Создание линии. Мутантная линия Apoetmlunc была создана в лаборатории д-ра Нобуйо Маеда в Университете Северной Каролины в Чапел-хилл. Использовали линию клеток ES E14Tg2a, выделенных у 129 мышей. Используемая плазмида получила название pNMC109, и исходная линия была обозначена Т-89. Линия C57BL/6J была получена в результате десятикратного обратного скрещивания мышей с мутацией Apoetmlunc с мышами C57BL/6J (Plump et al., Severe hypercholesterolemia and atherosclerosis in apolipoprotein-E deficient mice created by homologous recombination in ES cells. Cell 1992; 71: 343-353; Zhang et al. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992; 258: 468-471).
Мышей содержали в лечебнице для животных Sheba (Tel-Hashomer, Israel) с 12-часовым циклом чередования света и темноты при 22-24°С и кормили лабораторным кормом с нормальным содержанием жира (лабораторный корм для грызунов Purina № 5001), включающим 0,027% холестерина и примерно 4,5% общего жира, воду давали по потребности.
Мыши LDL-RD. Мыши LDL-RD [LDLr<mlHer>LDL-/-(C57B/6 50% JSL 25% I129 25%)] в возрасте 8-12 недель были получены из лечебницы для животных Hadassah (Hadassah Hospital, Israel).
Крысы Lewis. Самцы крыс Lewis в возрасте 9-11 недель были предоставлены компанией Harlan laboratories (Израиль).
Иммунизация
I. Внутрибрюшинная иммунизации ALLE. Аналог простого эфира фосфолипида (ALLE D+L) объединяли с очищенным производным белка из туберкулина (PPD). Исходный раствор ALLE (D+L) растворяли в этаноле (99 мг/мл). 5 мг ALLE (D+L) (50,5 мкл из исходного раствора) разбавляли до 5 мг/мл 0,25 М раствором фосфатного буфера, рН 7,2, перемешивая при 0°С (на бане со льдом). 1,5 мг D- и L-ALLE в 300 мкл фосфатного буфера добавляли к 0,6 мг PPD, растворенного в 300 мкл фосфатного буфера. Гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (5 мг; Sigma, St Louis. MO), растворенный в 50 мкл воды, добавляли при перемешивании в течение 20 минут при 4°С. Остальные активные сайты блокировали 100 мкл 1 М раствора глицина. Объединенные соединения диализовали против физиологического раствора с фосфатным буфером (PBS), доведенного до 3 мл с помощью PBS, и хранили при 4°С. Иммунизацию 0,3 мл (150 мкг) антигена на одну мышь выполняли внутрибрюшинно 4 раза через 2 недели.
II. Подкожная иммунизация окисленным LDL человека. Окисленный LDL человека получали из собранной плазмы человека (плотность 1,019-1,063 г/мл) ультрацентрифугированием и окисляли Cu в течение ночи (для чего добавляли 15 мкл 1 мМ CuSO4 на каждый мл LDL, предварительно разведенного до концентрации, равной 1 мг/мл). Окисленный LDL диализовали против PBS и фильтровали. Для иммунизации окисленный LDL растворяли в PBS и смешивали с равными объемами неполного адъюванта Фрейнда. Иммунизацию выполняли в виде одной подкожной инъекции 50 мкг антигена/мышь в 0,2 мл объеме. Мышей иммунизировали один раз через один-три дня после последнего перорального введения и умерщвляли через 7-10 дней после иммунизации.
Определение уровня холестерина. После окончания эксперимента при помощи сердечной пункции получали 1-1,5 мл крови, к каждому образцу добавляли 1000 ед./мл гепарина и определяли уровни общего холестерина в плазме автоматизированным ферментативным методом (Boehringer Mannheim, Germany).
Анализ методом FPLC. Анализ содержания холестерина и липидов в липопротеинах методом жидкостной хроматографии белка быстрого разрешения выполняли в системе для FPLC (Pharmacia LKB FRAC-200, Pharmacia, Peapack, NJ) с колонкой, заполненной суперозой 6 HR 10/30 (Amersham Pharmacia Biotech, Inc. Peapack, NJ). В пробоотборной трубке для автоматического пробоотборника необходим минимальный объем образца, равный 300 мкл (кровь, собранную у 3 мышей, разбавляли в отношении 1:2 и фильтровали перед загрузкой) для полного заполнения 200 мкл петли для пробы. Собирали фракции 10-40, каждая из которых содержала 0,5 мл. 250 мкл образец из каждой фракции смешивали с только что приготовленным холестериновым или триглицеридным реагентом, инкубировали в течение 5 минут при 37°С и анализировали спектрофотометрическим методом при 500 нм.
Оценка атеросклероза. Количественное определение атеросклеротических поражений в виде жировых полосок производили путем вычисления величины поражения в синусе аорты в соответствии с описанием, приведенным в научной литературе (Paigen et al., Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 1987; 68:231-140), и вычисления величины поражения в аорте. После перфузии трис-буфером с EDTA в физиологическом растворе у животных удаляли сердце и аорту и осторожно счищали периферический жир. Верхний срез сердца погружали в среду ОСТ (10,24 мас./мас.% поливинилового спирта; 4,26 мас./мас.% полиэтиленгликоля; 85,50 мас./мас.% инертных ингредиентов) и замораживали. Анализировали каждый второй срез (толщиной 10 мкм) синуса аорты (400 мкм). Дистальную часть синуса аорты опознавали по трем створкам клапана, которые являются местом соединения аорты с сердцем. Срезы оценивали в отношении отложений жировых полосок после окрашивания масляным красным О. Площадь поражения на одном срезе оценивали по сетке (Rubin et al. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein A-I. Nature 1991; 353:265-267) путем подсчета неидентифицированных, пронумерованных образцов, выполняемого наблюдателем. Аорту рассекали от сердца и удаляли окружающие адвентициальные ткани. Аорту фиксировали и окрашивали сосуды суданом в соответствии с описанием, приведенным в научной литературе (Bauman and Mangold, J. Org. Chem. 31: 498, 1966).
Измерения и количественное определение атеросклеротических поражений в плазме. Уровни общего холестерина и общего триглицерида в плазме измеряли методом COBAS MIRA. У умерщвленных животных удаляли сердца и криосрезы корня аорты окрашивали масляным красным О. Площадь атеросклеротического поражения определяли при помощи компьютерного анализа (Image Pro Plus) и полученные результаты подтверждали исследованием под микроскопом.
Анализы пролиферации. Мышам перорально вводили ALLE, POVPC или PBS аналогично описанию метода оценки атеросклероза и иммунизировали через один день после последнего перорального введения окисленным LDL, полученным из вышеописанного очищенного LDL человека.
Пролиферацию определяли через восемь дней после иммунизации окисленным LDL следующим образом: селезенки или лимфатические узлы получали путем измельчения тканей на сите с размером ячеек 100 меш. (лимфатические узлы с выполненной иммунизацией и селезенки без иммунизации). Эритроциты лизировали холодной стерильной дважды дистиллированной водой (6 мл) в течение 30 секунд и добавляли 2 мл 3,5% NaCl. Добавляли неполную среду (10 мл), клетки центрифугировали в течение 7 минут со скоростью 1700 об/мин, вторично суспендировали в среде RPMI и производиди подсчет в гемоцитометре при разведении в отношении 1:20 (10 мкл клеток + 190 мкл трипанового синего). Пролиферацию измеряли путем введения [3H]-тимидина в ДНК трех одинаковых образцов 100 мкл эритроцитарной массы (2,5×106 клеток/мл) на 96-луночном титрационном микропланшете. Добавляли три одинаковых образца окисленного LDL (0-10 мкг/мл, 100 мкл/лунку), клетки инкубировали в течение 72 часов (37°С, 5% СО2 и влажность около 98%) и добавляли 10 мкл 3[H]-тимидина (0,5 мкCi/лунку). Клетки инкубировали еще один день, затем собирали, переносили в стекловолоконные фильтры при помощи харвестера (Brandel) и производили подсчет в β-счетчике (Lumitron). Для анализа цитокинов собирали супернатант без добавления 3[H]-тимидина и анализировали методом ELISA.
Отдельной группе мышей перорально вводили ALLE или PBS и иммунизировали окисленным LDL вышеописанным методом через один день после перорального введения последней дозы. Лимфатические узлы паховой области (полученные через 8 дней после иммунизации) удаляли у 3 мышей в каждой группе для исследования пролиферации. 1×106 клеток на мл инкубировали в виде трех одинаковых образцов в течение 72 часов в 0,2 мл культуральной среды в ячейках титрационного микропланшета в присутствии 10 мкг/мл окисленного LDL. Пролиферацию измеряли путем введения [3H]-тимидина в ДНК на протяжении последних 12 часов инкубации. Результаты были выражены в виде индекса стимуляции (S.I): отношение средней радиоактивности (число импульсов в минуту) антигена к средней фоновой радиоактивности (число импульсов в минуту), полученной при отсутствии антигена. Стандартное отклонение всегда было равно <10% среднего числа импульсов в минуту.
Оценка маркеров воспаления в сыворотке. Сыворотку разделяли центрифугированием и хранили при -70°С. Анализ маркеров воспаления выполняли методом ELISA (IL-10; R&D и SAA; BIOSOURCE).
Анализ методом RT-PCR. Аорты, селезенки и тонкий кишечник удаляли у иммунизированных и неиммунизированных мышей (в стерильных условиях) и замораживали в жидком азоте. Органы измельчали на сите и получали РНК с использованием набора Rneasy (QIAGEN). Оразцы РНК исследовали в спектрофотометре и нормализовали относительно β-актина. Обратную транскрипцию РНК в кДНК и полимеразную реакцию синтеза цепи (PCR) с использованием затравок выполняли при помощи набора для выполнения RT-PCR в одной пробирке Titan (ROCHE). Результаты были получены на 1% агарозном геле и документированы на пленке.
Статистический анализ. Анализ ANOVA по одному признаку использовали для сравнения независимых значений. В качестве статистически значимого значения была принята величина р<0,05.
Пример I
Синтез иммуномодулирующих антигенов, представляющих собой простой эфир 2,5'-альдегидлецитина (ALLE) и POVPC
ALLE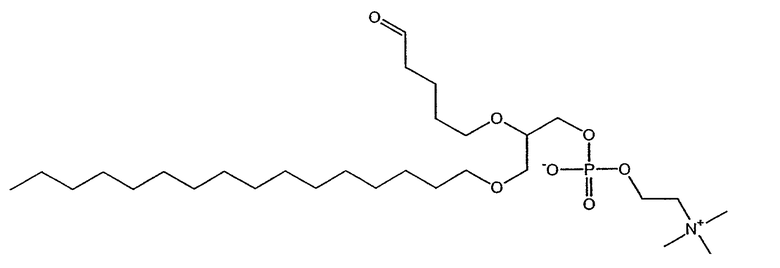
1-гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолин
POVPC
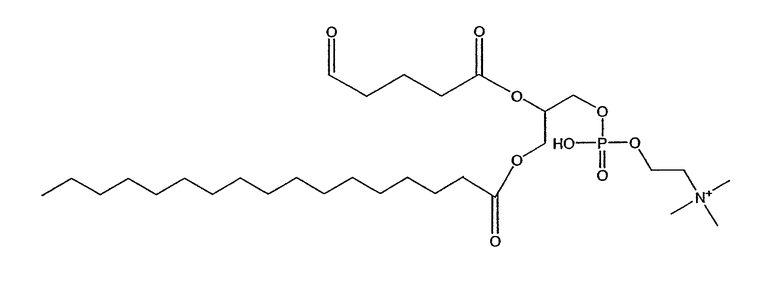
1-гексадеканоил-2-(5'-оксовалероил)-sn-3-фосфохолин
Синтез простого эфира 2,5'-альдегидлецитина (ALLE)
Простой эфир 2,5'-альдегидлецитина (ALLE) синтезировали в соответствии с модифицированными общими методами синтеза аналогов простого эфира лецитинов, описанными в публикациях Eibl H., et al., Ann. Chem. 709:226-230(1967), W.J. Baumann and H.K. Mangold, J. Org. Chem. 31498 (1996), E. Baer and Buchnea JBC. 230447 (1958), Halperin G. et al., Methods in Enzymology 129838-846 (1986). Нижеследующий способ относится к получению соединений, двухмерное изображение которых представлено на фигуре 1.
Простой эфир гексадецилглицерина. D-Ацетонглицерин (4 г) для синтеза L-ALLE или L-ацетонглицерин для синтеза D-ALLE, порошкообразный гидроксид калия (примерно 10 г) и гексадецилбромид (9,3 г) в бензоле (100 мл) премешивали и нагревали с обратным холодильником в течение 5 часов, удаляя при этом воду, образующуюся в результате азеотропной дистилляции (сравните публикации W.J. Baumann and H.K. Mangold, J. Org. Chem. 29:3055, 1964 и F. Paltauf, Monatsh. 99:1277, 1968). Объем растворителя постепенно уменьшали примерно до 20 мл, полученную смесь охлаждали до комнатной температуры и растворяли в простом эфире (100 мл). Полученный раствор промывали водой (2×50 мл) и удаляли растворитель при пониженном давлении. К остатку добавляли 100 мл смеси метанол:вода:концентрированная хлористоводородная кислота (90:10:5) и смесь нагревали с обратным холодильником в течение 10 минут. Продукт экстрагировали простым эфиром (200 мл) и последовательно промывали водой (50 мл), 10% раствором гидроксида натрия (20 мл) и снова водой (20 мл объемы) до нейтрального состояния. Растворитель удаляли при пониженном давлении и продукт (8,8 г) кристаллизовали из гексана, получая при этом 7,4 г чистого 1-гексадецил-глицерилового эфира (соединение I, фигура 1) для синтеза D-ALLE или 3-гексадецилглицерилового эфира для синтеза L-ALLE.
5-Гексенилметансульфонат. Смесь 5-гексен-1-ола (12 мл) и сухого пиридина (25 мл) охлаждали до температуры от -4°С до -10°С на ледяной бане с солью. По каплям добавляли метансульфонилхлорид (10 мл) в течение 60 минут и выдерживали смесь при 4°С в течение 48 часов. Добавляли лед (20 г), смесь оставляли выстаиваться в течение 30 минут и экстрагировали продукт простым эфиром (200 мл). Органическую фазу промывали водой (20 мл), 10% хлористоводородной кислотой, 10% раствором бикарбоната натрия (20 мл) и снова водой (20 мл). Растворитель выпаривали и неочищенный продукт хроматографировали на силикагеле 60 (100 г), используя смесь CHCl3:EtOAc (80:20) в качестве элюента, в результате чего было получено 14 г 5-гексенилметансульфоната.
1-Гексадецилокси-3-тритилокси-2-пропанол (для D-ALLE) или 3-гексадецилокси-1-тритилокси-2-пропанол (для L-ALLE) (соединение II). 1-Гексадецилоксиглицерин (для D-ALLE) или 3-гексадецилоксиглицерин (для L-ALLE) (7,9 г), трифенилхлорметан (8,4 г) и сухой пиридин (40 мл) нагревали при 100°С в течение 12 часов. Смесь охлаждали, добавляли 300 мл простого эфира и 150 мл охлаждаемой льдом воды и переносили реакционную смесь в делительную воронку. Органическую фазу последовательно промывали 50 мл воды со льдом, 1% раствором карбоната калия (до основного состояния) и 50 мл воды, затем сушили над безводным сульфатом натрия. Растворитель выпаривали, остаток растворяли в 150 мл теплого петролейного эфира и полученный раствор охлаждали при 4°С в течение ночи. Осадок отфильтровывали, фильтрат упаривали и остаток перекристиллизовывали из 20 мл этилацетата при -30°С, получая при этом 8,2 г 1-гексадецилокси-3-тритилокси-2-пропанола (для D-ALLE) или 3-гексадецилокси-1-тритилокси-2-пропанола (для L-ALLE) (соединение II, фигура 1), температура плавления 49°С.
1-Гексадецил-2-(5'-гексенил)глицериловый эфир (для D-ALLE) или 3-гексадецил-2-(5'-гексенил)глицериловый эфир (для L-ALLE) (соединение IV). 1-Гексадецилокси-3-тритилокси-2-пропанол (для D-ALLE) или 3-гексадецилокси-1-тритилокси-2-пропанол (для L-ALLE) (соединение II, фигура 1) (5,5 г) этерифицировали 5-гексенилметансульфоном, используя порошкообразный гидроксид калия в бензольном растворе аналогично вышеприведенному описанию. Неочищенный продукт 1-гексадецилокси-2-(5'-гексенилокси)-sn-3-тритилоксипропан (для D-ALLE) или 3-гексадецилокси-2-(5'-гексенилокси)-sn-3-тритилоксипропан (для L-ALLE) (соединение III, фигура 1) растворяли в 100 мл смеси метанол:вода:концентрированная хлористоводородная кислота (90:10:5) и реакционную смесь нагревали с обратным холодильником в течение 6 часов. Продукт экстрагировали простым эфиром, промывали водой и удаляли растворитель. Остаток растворяли в петролейном эфире (100 мл) и выдерживали при 4°С в течение ночи, в результате чего происходило осаждение большей части трифенилкарбинола. Продукт фильтровали и удаляли растворитель из фильтрата, после чего неочищенный продукт хроматографировали на силикагеле 60 (40 г), используя смесь хлороформ:петролейный эфир (1:1) в качестве элюента, в результате чего было получено 1,8 г чистого 1-гексадецил-2-(5'-гексенил)глицерилового эфира (для D-ALLE) или 3-гексадецил-2-(5'-гексенил)глицерилового эфира (для L-ALLE) (соединение IV, фигура 1).
1-Гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфохолин (для D-ALLE) или 3-гексадецил-2-(5'-гексенил)-sn-глицеро-1-фосфохолин (для L-ALLE) (соединение V). Нижеследующий способ является модификацией способа, описанного в публикации Eibl H., et al. Ann. Chem 709:226-230, 1967.
Раствор 1-гексадецил-2-гексенилглицерилового эфира (для D-ALLE) или 3-гексадецил-2-гексенилглицерилового эфира (для L-ALLE) (соединение IV, фигура 1) (2 г) в сухом хлороформе (15 мл) по каплям добавляли к перемешиваемому охлажденному раствору (от -4°С до -10°С) сухого триэтиламина (3 мл) и 2-бромэтилдихлорфосфата (1,25 мл, полученного описанным ниже способом) в сухом хлороформе (15 мл) в течение 1 часа. Смесь выдерживали при комнатной температуре в течение 6 часов и затем нагревали до 40°С в течение 12 часов. Полученный темно-коричневый раствор охлаждали до 0°С и добавляли 0,1 М раствор хлорида калия (15 мл). Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 60 минут. Добавляли метанол (25 мл) и хлороформ (50 мл), органическую фазу промывали 0,1 М раствором хлористоводородной кислоты (20 мл) и водой (20 мл). Растворитель выпаривали, неочищенный продукт растворяли в метаноле (15 мл), раствор переносили в пробирку для культуры и охлаждали на ледяной бане с солью. Добавляли холодный триметиламин (3 мл, -20°С) и герметично закупоривали пробирку. Смесь нагревали до 55°С в течение 12 часов, охлаждали до комнатной температуры и растворитель выпаривали в потоке азота. Остаток экстрагировали смесью хлороформ:метанол (2:1) (25 мл) и промывали 1 М раствором карбоната калия (10 мл) и водой (2×10 мл). Растворитель удаляли при пониженном давлении и неочищенные продукты хроматографировали на силикагеле 60 (20 г), используя смесь хлороформ:метанол (60:40), в результате чего было получено 1,5 г 1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфохолина (для D-ALLE) или 3-гексадецил-2-(5'-гексенил)-sn-глицеро-1-фосфохолина (для L-ALLE) (соединение V, фигура 1). Структура соединения V была подтверждена ЯМР и масс-спектрометрией.
1-Гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолин (для D-ALLE) или 3-гексадецил-2-(5'-оксопентанил)-sn-глицеро-1-фосфохолин (для L-ALLE) (соединение VI). Раствор соединения V (0,5 г) в муравьиной кислоте (15 мл) и 30% пероксиде водорода (3,5 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (50 мл) и экстрагировали смесью хлороформ:метанол (2:1) (5×50 мл). Растворитель выпаривали при пониженном давлении, остаток смешивали с метанолом (10 мл) и водой (4 мл) и перемешивали при комнатной температуре в течение 60 минут. Затем добавляли 80% фосфорную кислоту (2 мл) и метапериодат калия (0,8 г). Смесь выдерживали при комнатной температуре в течение ночи, разбавляли водой (50 мл) и экстрагировали смесью хлороформ:метанол (2:1) (50 мл). Органическую фазу промывали 10% раствором бисульфита натрия (10 мл) и водой (10 мл). Растворитель удаляли при пониженном давлении, неочищенный продукт хроматографировали на силикагеле 60 (10 г), используя смесь хлороформ:метанол (1:1) в качестве элюента, в результате чего было получено 249 мг 1-гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолина (для D-ALLE) или 3-гексадецил-2-(5'-оксопентанил)-sn-глицеро-1-фосфохолина (для L-ALLE) (соединение VI, фигура 1), имеющих значение Rf 0,15 (система для ТСХ, хлороформ:метанол:вода (60:40:8)) и характеризующихся положительной реакцией с динитрофенилгидразином. Химическая структура соединения VI была подтверждена ЯМР и масс-спектрометрией.
В соответствии с альтернативным способом этиленовую группу превращали в альдегидную группу озонированием и каталитическим гидрированием карбонатом кальция на палладии.
Получение 2-бромэтилдихлорфосфата. 2-Бромэтилдихлорфосфат получали, добавляя по каплям только что перегнанный 2-бромэтанол (0,5 М, полученный способом, описанным в публикации Gilman Org. Synth. 12:117, 1926) к охлаждаемому льдом раствору только что перегнанного оксихлорида фосфора (0,5 М) в сухом хлороформе в течение 1 часа, затем реакционную смесь нагревали с обратным холодильником в течение 5 часов и подвергали вакуумной перегонке (температура кипения 66-68°С под давлением 0,4-0,5 мм Hg). Реагент хранили до использования (-20°С) в атмосфере азота в небольших герметично закупоренных ампулах (Hansen W.H. et al. Lipids 17(6):453-459, 1982).
Синтез 1-гексадецил-2-(5'-карбоксибутил)-sn-глицеро-3-фосфохолина (CI-201). 1-Гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолин (соединение VI, полученное вышеописанным способом), 0,55 г (0,001 моль), растворяли в t-BuOH (30 мл). По каплям добавляли раствор NaClO2 (0,9 г, 0,01 моль) и NaH2PO4 (0,96 г, 0,07 моль) в 25 мл воды в течение 30 минут и перемешивали смесь при комнатной температуре в течение 3 часов. Реакционную смесь подкисляли до рН=3 концентрированной хлористоводородной кислотой и экстрагировали смесью хлороформ:метанол (2:1). Органическую фазу отделяли и растворитель выпаривали. Остаток очищали хроматографией на силикагеле, используя смесь хлороформ:метанол:вода (70:27:23), в результате чего был получен 1-гексадецил-2-(5'-карбоксибутил)-sn-глицеро-3-фосфохолин (0,42 г, выход 72%). Химическая структура (соединение VII, фигура 10) была подтверждена ЯМР и масс-спектрометрией.
Синтез 1-гексадецил-2-(5',5'-диметоксипентилокси)-sn-глицеро-3-фосфохолина. 1-Гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолин (соединение VI, полученное вышеописанным способом), 0,50 г (0,89 ммоль), растворяли в муравьиной кислоте (15 мл) и добавляли 30% пероксид водорода (3,5 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду (50 мл) и продукт экстрагировали смесью хлороформ:метанол (2:1) (2×50 мл). Органическую фазу промывали 10% водным раствором бикарбоната натрия (10 мл) и водой (10 мл) и растворитель удаляли при пониженном давлении. Остаток (0,4 г) растворяли в метаноле (10 мл), добавляли 10% водный раствор гидроксида натрия (4 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли 80% фосфорную кислоту (2 мл) и метапериодат калия (0,8 г) и продолжали перемешивать реакционную смесь в течение ночи. Добавляли смесь хлороформ:метанол (2:1) (50 мл), органическую фазу промывали 10% водным раствором бисульфита натрия (10 мл) и водой (10 мл) и растворитель удаляли в вакууме. Остаток (0,3 г) очищали хроматографией на силикагеле (10 г), используя смесь хлороформ:метанол (60:40-40:60) в качестве градиентного элюента, в результате чего был получен 1-гексадецил-2-(5',5'-диметоксипентилокси)-sn-глицеро-3-фосфохолин (0,25 г, выход 46%). Химическая структура (соединение VIIIa, фигура 10) была подтверждена ЯМР и масс-спектрометрией.
Синтез 1-гексадецил-2-(5',5'-диэтоксипентилокси)-sn-глицеро-3-фосфохолина. Неочищенный 1-гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолин (соединение VI, полученное вышеописанным способом), 50 мг (0,088 ммоль), растворяли в этаноле (10 мл) в атмосфере азота. Добавляли триэтилортоформиат (0,053 мл, 0,0476 г, 0,32 ммоль) и 3 капли концентрированной серной кислоты и реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем добавляли дихлорметан (75 мл) и реакционную смесь переносили в делительную воронку, последовательно промывали водой (75 мл), 2,5% водным раствором бикарбоната натрия (75 мл) и водой (75 мл) и сушили над безводным сульфатом натрия. Реакционную смесь фильтровали и растворитель удаляли в вакууме, получая при этом 50 мг неочищенного 1-гексадецил-2-(5',5'-диэтоксипентилокси)-sn-глицеро-3-фосфохолина. Структура была подтверждена ЯМР и масс-спектроскопией (соединение VIIIb, фигура 10).
Синтез 1-гексадеканоил-2-(5'-оксовалероил)-sn-3-глицерофосфохолина (POVPC). Смесь 1-гексадеканоил-sn-3-глицерофосфохолина (соединение I, фигура 2) (3 г), 5-гексеновой кислоты (1,2 мл), 1,3-дициклогексилкарбодиамида (DCC, 4,05 г) и N,N-диметиламинопиридина (DMP, 1,6 г) в дихлорметане (100 мл, только что перегнанного из пентоксида фосфора) тщательно перемешивали в течение 4 дней при комнатной температуре. Затем смесь хроматографировали на силикагеле 60 (40 г) и продукт, 1-гексадеканоил-2-(5'-гексеноил)-sn-3-глицерофосфохолин (2,8 г, соединение II, фигура 2), элюировали смесью хлороформ:метанол (25:75). Элюент растворяли в смеси 30% пероксид водорода:муравьиная кислота (4:15) и раствор перемешивали в течение ночи при комнатной температуре. Добавляли воду (50 мл), продукт экстрагировали смесью хлороформ:метанол (2:1) (100 мл) и органическую фазу промывали водой. Растворитель выпаривали при пониженном давлении, остаток растворяли в метаноле (15 мл) и 10% растворе аммиака (5 мл) и полученный раствор перемешивали при комнатной температуре в течение 6 часов. Неочищенный 1-гексадеканоил-2-(5',6'-дигидрокси)гексаноил-sn-3-глицерофосфохолин (соединение III, фигура 2) (структура была подтверждена ЯМР и масс-спектрометрией) подвергали дальнейшему взаимодействию без очистки. К раствору добавляли 80% фосфорную кислоту (3 мл) и метапериодат натрия (1 г), смесь перемешивали при комнатной температуре в течение ночи и экстрагировали смесью хлорофрм:метанол (2:1). Продукт очищали хроматографией на силикагеле 60 (20 г), используя смесь хлороформ:метанол (25:75) в качестве элюента. Было получено 850 мг 1-гексадеканоил-2-(5'-оксовалероил)-sn-3-глицерофосфохолина (POVPC, соединение IV, фигура 2), который характеризовался хроматографической подвижностью лецитина при выполнении ТСХ и положительным взаимодействием с динитрофенилгидразином. Структуру определяли методом ЯМР и масс-спектрометрии.
Альтернативно этиленовую группу превращали в альдегид озонированием и каталитическим гидрированием карбонатом кальция на палладии.
Пример II
Специфическое ингибирование атерогенеза у генетически предрасположенных мышей (Аро-Е КО) иммунизацией против L-ALLE + D-ALLE
Авторы настоящего изобретения установили, что иммунизация устойчивым этерифицированным синтетическим ALLE, являющимся компонентом LDL, позволяет уменьшить степень образования атеросклеротических бляшек у предрасположенных мышей. 19 самок мышей Аро Е/С 57 в возрасте 5-7 недель делили на 3 группы. В группе А (n=6) мышей иммунизировали внутрибрюшинно в соответствии с описанием, приведенным в разделе “Материалы и методы”, 150 мкг/мышь L-ALLE + D-ALLE один раз в 2 недели (0,3 мл/мышь), повторяя данную процедуру 4 раза. В группе В (n=6) мышей иммунизировали очищенным производным белка (PPD) токсина туберкулина один раз в 2 недели (0,3 мл/мышь). В группе С (n=7) мышей не иммунизировали. У мышей во всех трех группах брали пробы крови до иммунизации (время 0) и через одну неделю после второй иммунизации для определения антител против ох LDL, антител против ALLE и профиля липидов. Оценку атеросклероза производили вышеописанным методом через 4,5 недели после 4-й иммунизации. Мышей во всех группах взвешивали через каждые 2 недели на протяжении всего эксперимента. Все мыши получали нормальный корм, содержащий 4,5 мас.% жира (0,02% холестерина), и воду по потребности.
Ингибирование атерогенеза в результате иммунизации мышей Аро-Е КО при помощи ALLE
L-ALLE+D-ALLE 150 мкг/
мышь N=6
N=5
группа
без
иммунизации N=7
кие
данные
Как показано на фигуре 3, результаты, приведенные в таблице I, указывают на значительное уменьшение атероматозных поражений, измеренных в тканях сердца ALLE-иммунизированных мышей, по сравнению с PPD-иммунизированными мышами и неиммунизированными контрольными мышами. Не было обнаружено значительного влияния на другие измеренные показатели, такие как прирост массы, уровни триглицерида или холестерина в плазме или иммунокомпетентность, измеренная на основании уровней иммуносупрессивного цитокина TGF-β. Таким образом, иммунизация синтетическим ALLE, являющимся компонентом окисленного LDL (смесь рацемических форм D и L), в значительной степени (>50%) защищает генетически предрасположенных мышей Аро-Е КО от возникновения атеросклеротического поражения. Существенное, но менее значительное уменьшение бляшкообразования наблюдалось у мышей, иммунизированных PPD.
Пример III
Ингибирование атерогенеза у генетически предрасположенных мышей (Аро-Е КО) в результате перорального введения L-ALLE и D-ALLE
Внутрибрюшинная иммунизация сложноэфирными аналогами компонентов бляшкообразования обеспечила эффективное ингибирование атерогенеза у мышей Аро-Е КО (фигура 1). Была исследована возможность перорального введения L- и D-ALLE для подавления атерогенеза. 34 самца мышей Аро-Е КО в возрасте 8-10 недель делили на три группы. В группе А (n=11) мышам перорально вводили через желудочный зонд L-ALLE + D-ALLE, суспендированные в PBS, содержащем 5% этанола (1 мг/мышь), через день в течение 5 дней. В группе В (n=11) мышам перорально вводили 10 мкг/мышь L-ALLE+D-ALLE, суспендированных в PBS, содержащем 5% этанола, через день в течение 5 дней (0,2 мл/мышь). Мышам в группе С (n=12) вводили PBS (содержащий такой же объем этанола, что и в группах А+В). У мышей брали пробы крови до перорального введения (время 0) и в конце эксперимента (конец) для определения профиля липидов. Атеросклероз оценивали в синусе аорты вышеописанным методом через 8 недель после последнего перорального введения. Мышей взвешивали через каждые 2 недели на протяжении всего эксперимента. Все мыши получали нормальный корм, содержащий 4,5 мас.% жира (0,02% холестерина), и воду по потребности.
Ингибирование атерогенеза у мышей Аро-Е КО в результате перорального введения L-ALLE и D-ALLE
N=12
ALLE N=11
N=11
Как показано на фигуре 4, результаты, приведенные в таблице 2, указавают на значительное ослабление развития атеросклероза, измеренного в тканях мышей, которым перорально вводили низкие дозы (10 мкг-1 мг/мышь) ALLE, по сравнению с неиммунизированными контрольными мышами. Не было обнаружено значительного влияния на другие измеренные показатели, такие как прирост массы, уровни триглицерида или холестерина в плазме или иммунокомпетентность, измеренная на основании уровней иммуносупрессивного цитокина TGF-β. Таким образом, пероральное введения синтетического ALLE, являщегося компонентом окисленного LDL, в значительной степени (>50%) защищает генетически предрасположенных мышей Аро-Е КО от атеросклероза, причем такая защита сравнима с защитой, достигаемой при внутрибрюшинной иммунизации (см. фигуру 1).
Пример IV
Ингибирование атерогенеза у генетически предрасположенных мышей (Аро-Е КО) в результате индукции иммуномодуляции при пероральном и назальном введении L-ALLE
Механизмы иммуномодуляции, опосредуемой слизистой оболочкой, активно действуют в слизистой оболочке носа и кишечника. Поэтому сравнивали эффективность подавления атерогенеза у мышей Аро-Е КО при назальном и пероральном введеним L- и D-ALLE. 34 самца мышей Аро-Е КО в возрасте 7-10 недель делили на 3 группы. В группе А (n=11) мышам перорально вводили L-ALLE, суспендированный в PBS, содержащем 5% этанола (1 мг/мышь/0,2 мл), через день в течение 5 дней. В группе В (n=11) мышам вводили в нос способом, описанным в разделе “Материалы и методы”, 10 мкг/мышь/10 мкг L-ALLE, суспендированного в PBS, через день в течение 3 дней. Мышам в группе С (n=12) перорально и назально вводили PBS (содержащий такой же объем этанола, что и в группах А+В). У мышей брали пробы крови до перорального введения (время 0) и в конце эксперимента (конец) для определения профиля липидов. Атеросклероз оценивали в синусе аорты вышеописанным способом через 8 недель после последнего перорального введения. Мышей взвешивали через каждые 2 недели на протяжении всего эксперимента. Все мыши получали нормальный корм, содержащий 4,5 мас.% жира (0,02% холестерина), и воду по потребности.
Влияние перорального и назального введения L-ALLE на показатели обмена веществ и атерогенеза у мышей Аро-Е КО
1 мг
(N=11)
ALLE
10 мкг (N=11)
(N=12)
данные
Как показано на фигуре 5, результаты, приведенные в таблице 3, указывают на эффективное ингибирование атерогенеза, измеренного в тканях мышей, которым вводили в нос низкие дозы (10 мкг/мышь) ALLE, по сравнению с неимунизированными контрольными мышами. Назальное введение подобно пероральному введению не оказывало значительного влияния на другие измеренные показатели, такие как прирост массы, уровни триглицерида или холестерина в плазме. Таким образом, синтетический ALLE, являющийся компонентом окисленного LDL, в значительной степени (>50%) защищает генетически предрасположенных мышей Аро-Е КО от атеросклероза как при пероральном, так и назальном введении.
Пример V
Подавление специфической иммунной реакции против oxLDL у генетически предрасположенных мышей (Аро-Е КО) при пероральном введении L-ALLE или POVPV
Иммуномодуляция, индуцируемая в результате введения через слизистую оболочку окисленных аналогов LDL, может быть опосредована подавлением специфических иммунных реакций на антигены, связанные с бляшками. POVPC (1-гексадеканоил-2-(5'-оксовалероил)-sn-глицерофосфохолин) является неэфирным аналогом окисленного LDL, который в отличие от ALLE подвержен разрушению в печени и кишечнике. Пролиферацию лимфоцитов в ответ на пероральное введение POVPC и более устойчивого аналога ALLE измеряли у мышей Аро-Е КО. 8 самцов мышей Аро-Е КО в возрасте 6 недель делили на 3 группы. В группе А (n=2) мышам вышеописанным способом через желудочный зонд вводили 1 мг/мышь L-ALLE, суспендированного в 0,2 мл PBS, через день в течение 5 дней. В группе В (n=3) мышам вышеописанным способом перорально вводили 1 мг/мышь POVPC, суспендированного в 0,2 мл PBS, через день в течение 5 дней. Мышам в группе С (n=3, контрольная группа) перорально вводили 200 мкл PBS через день в течение 5 дней. Иммунную реактивность стимулировали путем иммунизации окисленным LDL человека способом, описанным в разделе “Материалы и методы”, через один день после последнего перорального введения. Через одну неделю после иммунизации у мышей удаляли лимфатические узлы для выполнения анализа пролиферации. Все мыши получали нормальный корм, содержащий 4,5 мас.% жира (0,02% холестерина), и воду по потребности.
Подавление иммунной реакции на ох-LDL человека у мышей Аро-Е КО в результате предварительного перорального введения синтетического окисленного LDL (ALLE и POVPC)
Как показано на фигуре 6, результаты, приведенные в таблице 4, указывают на значительное подавление иммунной реактивности в отношении антигена окисленного LDL человека, измеренное на основании ингибирования пролиферации в лимфатических узлах мышей Аро-Е КО. Лимфоциты мышей, которым перорально вводили ингибирующие атерогенез дозы (1 мг/мышь) ALLE или POVPC, характеризовались пониженным индексом стимуляции после иммунизации ox-LDL по сравнению с контрольными мышами (PBS). Так как индукция иммуномодуляции при пероральном введении подобно назальному введению не оказывала значительного влияния на другие измеренные показатели, такие как прирост массы, уровни триглицерида или холестерина в плазме или иммунокомпетентность (см. таблицы 1, 2 и 3), полученные результаты свидетельствуют о специфическом подавлении иммунной реактивности против ox-LDL. Таким образом, пероральное введение синтетического L-ALLE, являющегося компонентом окисленного LDL, является эффективным способом ослабления клеточной иммунной реакции на иммуногенные и атерогенные компоненты бляшки у генетически предрасположенных мышей Аро-Е КО. На фигуре 4 также показано подобное, но менее эффективное ингибирование пролиферации при пероральном введении менее устойчивого синтетического POVPC, являющегося компонентом LDL.
Пример VI
Ингибирование атерогенеза у генетически предрасположенных мышей (Аро-Е КО) в результате перорального введения D- и L-изомеров ALLE и POVPC
Так как было установлено, что пероральное введение ALLE и POVPC ингибирует начальную стадию атерогенеза и иммунную реактивность в отношении антигена LDL человека, связанного с бляшками, было произведено сравнение способности D- и L-изомеров эфирного аналога LDL и неэфирного аналога POVPC подавлять развитие атерогенеза у более старых мышей. Их влияние на триглицеридные и холестериновые фракции VLDL также контролировали методом FPLC. 57 самцов мышей Аро-Е КО в возрасте 24,5 недель делили на 5 групп. В группе А (n=11) мышам вышеописанным способом вводили через желудочный зонд 1 мг/мышь L-ALLE, суспендированного в 0,2 мл PBS, через день в течение 5 дней. В группе В (n=9) мышам вышеописанным способом перорально вводили 1 мг/мышь D-ALLE, суспендированного в 0,2 мл PBS, через день в течение 5 дней. В группе С (n=10) мышам вышеописанным способом через желудочный зонд вводили 1 мг/мышь POVPC, суспендированного в 0,2 мл PBS, через день в течение 5 дней. Контрольной группе D (n=10) перорально вводили PBS (содержащий такой же объем этанола, что и в группах А, В, С). Базовую группу умерщвляли в период времени = 0. Испытуемые антигены перорально вводили через каждые 4 недели (5 пероральных введений; через день), начиная с 24,5-недельного возраста, на протяжении 12 недель (3 серии пероральных введений).
У мышей брали пробы крови до перорального введения (время 0), после 2-й серии перорального введения и в конце эксперимента (конец) для определения профиля липидов, фракционирования липидов и уровней в плазме. Атеросклероз оценивали вышеописанным способом в синусе аорты и в аорте. У животных удаляли селезенки для выполнения анализа пролиферации через 12 недель после первого перорального введения. Массу регистрировали через каждые 2 недели на протяжении всего эксперимента. Все мыши получали нормальный корм, содержащий 4,5 мас.% жира (0,02% холестерина), и воду по потребности.
Ингибирование атерогенеза у мышей Аро-Е КО в результате перорального введения L-ALLE, D-ALLE и POVPC
кие данные
(% от
общей площади)
Как показано на фигуре 7, результаты, приведенные в таблице 5, указывают на эффективное ингибирование поздней стадии атерогенеза, измеренное в тканях более старых мышей после продолжительного перорального введения D- и L-изомеров ALLE и POVPC в дозе, равной 1 мг/мышь, по сравнению с контрольными мышами, которым перорально вводили PBS. Пероральное введение указанных соединений не оказывало значительного влияния на другие измеренные показатели, такие как прирост массы, уровни общего триглицерида или холестерина в плазме. Таким образом, синтетические D-, L-ALLE и POVPC, являющиеся компонентами окисленного LDL, обладают антиатерогенной активностью, почти полностью защищая от развития атерогенеза (по сравнению с оценками поражения в возрасте 24,5 недель) генетически предрасположенных мышей Аро-Е КО. Весьма удивительным является то, что ингибирование атерогенеза аналогами окисленного LDL сопровождается значительным снижением уровней холестерина VLDL и триглицеридов, измеренным методом FPLC (фигуры 8 и 9).
Пример VII
Ингибирование атерогенеза у генетически предрасположенных мышей (Аро-Е КО) в результате перорального введения CI-201
Была исследована способность подавлять атерогенез устойчивой формой этерифицированного фосфолипида, кислотным производным ALLE, CI-201. Самцов мышей Аро-Е КО в возрасте 12 недель делили на две группы. В группе А (n=14) мышам перорально вводили через желудочный зонд CI-201 (0,025 мг/дозу), суспендированный в PBS, каждый день в течение 8 недель (5 раз в неделю). Мышам в группе В (n=15) вводили PBS (контрольная группа). Атеросклероз оценивали вышеописанным способом. Все мыши получали нормальный корм, содержащий 4,5 мас.% жира (0,02% холестерина), и воду по потребности.
Как показано на фигуре 11, результаты свидетельствуют о значительном замедлении развития атеросклероза в тканях мышей, которым перорально вводили низкие дозы CI-201, по сравнению с неиммунизированными контрольными мышами (PBS). Поражение в синусе аорты в группе, получавшей CI-201, было равно 125192±19824 мкм2 и в контрольной группе (получавшей PBS) было равно 185400±20947 мкм2, что указывает на 33% (Р=0,051) уменьшение поражения в синусе аорты при пероральном введении низкой дозы CI-201. Экспрессия IL-10 (определенная методом RT-PCR) в аорте была на 40% выше в группе, получавшей CI-201, по сравнению с контрольной группой. Повышенные уровни экспрессии IL-10 в органе-мишени, аорте, подтверждают противовоспалительное действие, индуцируемое при пероральном введении CI-201. Таким образом, устойчивый синтетический CI-201 окисленного LDL вызывает иммуномодуляцию и оказывает противовоспалительное действие при пероральном введении.
Пример VIII
Экспрессия цитокинов в аорте мышей Аро-Е КО, иммунизированных окисленными фосфолипидами (ALLE, CI-201, Et-ацеталь, Ме-ацеталь и oxLDL)
Влияние ALLE, CI-201, его соответствующих ацетальных производных Et-ацеталь и Ме-ацеталь (соединения IIa и IIb, фигура 10) и oxLDL на экспрессию цитокинов в органе-мишени, аорте, определяли вышеописанным методом RT-PCR. Мышам Аро-Е КО перорально вводили 1 мг/мышь ALLE, 1 мг/мышь CI-201, 1 мг/мышь Et-ацеталя, 1 мг/мышь Ме-ацеталя, 0,1 мг/мыль oxLDL или 0,2 мл/мыль PBS. Пероральное введение производили 5 раз через день. Экспрессию противовоспалительного цитокина IL-10 и провоспалительных цитокинов IFN-γ и IL-12 определяли через 8 недель после последнего перорального введения.
Как показано на фигурах 12а и 12b, у мышей, которым вводили ALLE, CI-201, Et-ацеталь, Ме-ацеталь и oxLDL, наблюдались повышенные уровни экспрессии IL-10 по сравнению с контрольной группой, получавшей PBS. Как показано на фигурах 12с и 12d, наблюдалось противоположное воздействие на уровень экспрессии IFN-γ и IL-12. Пониженные уровни экспрессии IFN-γ были обнаружены у мышей, которым вводили ALLE, CI-201, Ме-ацеталь и oxLDL, и пониженные уровни IL-12 были обнаружены у мышей, которым вводили ALLE CI-201, Et-ацеталь и oxLDL.
Пример IX
Ингибирование атерогенеза у мышей LDL-RD в результате индукции иммуномодуляции при пероральном введении oxLDL
Для демонстрации того, что вышеописанные синтетические окисленные фосфолипиды вызывают такое же действие, что и окисленный LDL человека, была создана модель для оценки влияния oxLDL на развитие атеросклероза у мышей.
Мышей LDL-RD в возрасте 8-12 недель распределяли в разные группы по возрасту, массе тела и профилю липидов (холестерин и триглицерид). Каждой группе вводили oxLDL в увеличивающихся дозах (10, 100 или 1000 мкг/дозу, растворенный в PBS в общем объеме 0,2 мл PBS), альбумин (100 мкг/дозу, растворенный в PBS в общем объеме 0,2 мл PBS) или PBS (0,2 мл) 5 раз через день. Через день после последнего перорального введения мышам давали по потребности корм, способствующий развитию атеросклероза (“западный рацион”), и в течение пяти недель содержали с 12-часовым циклом чередования темноты и света.
Мышей умерщвляли через 6,5 недель после первого перорального введения и определяли площадь атеросклеротических бляшек в синусе аорты вышеописанным способом.
Влияние oxLDL на показатели обмена веществ приведены в нижеследующей таблице 6. Как показано в таблице 6, oxLDL, не воздействуя на массу тела или уровни холестерина, в дозе 100 мкг/дозу значительно (Р<0,05) снижал уровни триглицерида по сравнению с контрольными группами, которым вводили PBS и альбумин.
Влияние введения oxLDL на показатели обмена веществ у мышей LDL-RD
OxLDL
1000 мкг/дозу
OxLDL
100
мкг/дозу
Е
PBS
тичес-
кие
данные
(мг/дл)
(мг/дл)
Примечание: “масса” означает массу в граммах; “холестерин” означает уровень холестерина в плазме и “триглицерид” означает уровень триглицеридов в плазме, выраженный в мг/дл.
Замедление атерогенеза при пероральном введении OxLDL показано на фигуре 13 и в нижеследующей таблице 7. Как показано на фигуре 13, введение 100 мкг/дозу и 1000 мкг/дозу oxLDL значительно уменьшает (Р<0,001) площадь поражения в синусе аорты на 45% по сравнению с контрольными группами (которым вводили PBS или сывороточный альбумин человека (HAS)).
Влияние введения OxLDL на площадь атеросклеротического поражения в синусе аорты
10 мкг/дозу
тичес-
кие
данные
в синусе
аорты (мкм2)
Пример Х
Ингибирование атерогенеза у генетически предрасположенных мышей (Аро-Е КО) в результате перорального введения CI-201
В качестве модели предотвращения развития атерогенеза использовали мышей Аро-Е КО в возрасте 26-28 недель (АРО-Е -/-<tm1Unc> [C57B6J]). Мышей распределяли в разные группы по возрасту, массе тела и профилю липидов (холестерин и триглицерид). Одна группа, которую умерщвляли в начале эксперимента, служила в качестве “базовой” группы. Всем другим группам перорально вводили возрастающие дозы CI-201 (0,1, 1 или 10 мкг/дозу, растворнного в PBS, содержащем 0,05% этанола, в общем объеме 0,2 мл PBS). Контрольной группе вводили PBS (0,05% этанола, 0,2 мл).
Мышам вводили CI-201 или PBS в виде трех серий в начале каждого месяца, причем каждая серия включала 5 пероральных введений, выполняемых через день. Все мыши получали нормальный корм, содержащий 4,5 мас.% жира (0,02% холестерина), и воду по потребности, при этом мышей содержали с 12-часовым циклом чередования темноты и света.
Через 12 недель мышей умерщвляли и опредедяли площадь атеросклеротических бляшек в синусе аорты вышеописанным способом.
Влияние введения CI-201 на показатели обмена веществ приведены в нижеследующей таблице 8. Результаты показывают, что CI-201 не влияет на массу тела и профиль липидов у экспериментальных мышей.
Влияние введения CI-201 на показатели обмена веществ у мышей Аро-Е КО
ские данные
Примечание: “масса” означает массу в граммах; “холестерин” означает уровень холестерина в плазме и “триглицерид” означает уровень триглицеридов в плазме, выраженный в мг/дл.
Замедление атерогенеза при пероральном введении CI-201 показано на фигурах 14а-b и в нижеследущей таблице 9.
Влияние CI-201 на площадь атеросклеротического поражения в синусе аорты
1 мкг/дозу
PBS
данные
20505
20909
25772
21920
29248
Результаты, приведенные в таблице 9, показывают, что введение CI-201 полностью ингибирует развитие болезни, при этом площади поражения в синусе аорты мышей, которым вводили разные дозы CI-201, были аналогичны поражениям в базовой группе.
В отличие от этого у мышей, которым вводили PBS, наблюдалось 50% статистически значимое (р<0,01) увеличение атеросклеротического поражения в синусе аорты по сравнению с базовой группой (328491±21920 мкм2 в группе, которой вводили PBS, по сравнению с 218602±29248 мкм2 в базовой группе).
Как показано на фигуре 14а, все дозы CI-201 ингибировали развитие болезни, причем наиболее эффективной дозой была минимальная доза, равная 0,1 мкг/дозу. Как показано на фигуре 14b, в группе, которой вводили 0,1 мкг/дозу CI-201, наблюдалось 92% статистически значимое (Р<0,05) уменьшение атеросклеротического поражения в синусе аорты по сравнению с группой, которой вводили PBS (328491±21920 мкм2 в группе, получавшей PBS, по сравнению с 228000±25772 мкм2 в группе, получавшей CI-201).
Пример XI
Увеличение маркеров воспаления в сыворотке мышей Аро-Е КО, иммунизированных CI-201
С учетом значительного ингибирования развития атеросклероза при пероральном введении CI-201, которое, как было описано выше, не сопровождается индуцированным изменением массы тела или профиля липидов, исследовали влияние перорального введения CI-201 на уровень маркеров воспаления в сыворотке с целью изучения его механизма действия.
Как установлено в экспериментальных и клинических исследованиях, IL-10 является главным защитным цитокином роста и устойчивости бляшек. Например, Калигиури др. (Caligiuri et al., Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol. Med. 2003; 9(1-2):10-17) недавно сообщили, что величина поражения значительно увеличивается у мышей с отсутствием Аро-Е и IL-10 по сравнению с контрольными мышами, при этом повышается протеолитическая и прокоагулирующая активность, указывая на то, что IL-10 может уменьшать атеросклероз и улучшать устойчивость бляшек.
Другим маркером острого воспалительного состояния является сывороточный амилоид А (SAA), высокочувствительный маркер воспаления, количество которого может увеличиваться в 1000 раз во время воспаления. SAA так же, как и CRP (С-реактивный белок) синтезируется печенью под воздействием IL-1, IL-6 и TNF (Balke and Ridker, Novel clinical markers of vascular wall inflammation, Circ. Res. 2001; 89:763-771). Установлено, что SAA экспрессируется в месте атеросклеротического поражения клетками нескольких типов (Meek et al., Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proc. Natl. Acad. Sci. USA 1994; 91:3186-3190; Uhlar and Whitehead. Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem 1999; 265:501-523).
Воспаление инициируют прежде всего активированные моноциты крови и макрофаги ткани в месте нахождения раздражителя, вызывающего воспаление. Активированные макрофаги выделяют целый ряд первичных медиаторов воспаления, наиболее важными из которых являются члены семейств цитокинов IL-1 и TNF, которые запускают выделение целого ряда вторичных цитокинов и хемокинов (IL-6, IL-8 и МСР). Хемотаксическая активность указанных молекул направляет лейкоциты к месту воспаления, где они в свою очередь выделяют другие провоспалительные цитокины.
Мышам Аро-Е КО перорально вводили 0,1 мкг/мышь CI-201 или 0,2 мл/мышь pBS через день 5 раз. У мышей брали сыворотку до начала эксперимента (день 0), в конце эксперимента (две недели) и через две недели после окончания эксперимента (4 недели) и определяли уровень маркеров воспаления IL-10 и сывороточного амилоида А (SAA).
Полученные данные представлены на фигуре 15а (для уровней IL-10) и на фигуре 15b (для уровней SAA).
Как показано на фигуре 15а, в конце эксперимента (2 недели) наблюдалось значительное увеличение уровней IL-10 в сыворотке, и еще через 2 недели (4 недели) было отмечено снижение уровней. В контрольной группе, которой вводили PBS, на протяжении всего эксперимента не было отмечено изменений уровней IL-10 в сыворотке.
Как показано на фигуре 15b, уровни SAA в сыворотке значительно повысились в контрольной группе, при этом не неблюдалось изменения уровней SAA в сыворотке в группе, которой вводили CI-201.
Полученные результаты ясно показывают, что в результате повышения уровней IL-10 в сыворотке CI-201 индуцирует противовоспалительную реакцию, которая может подавить провоспалительную реакцию, о которой свидетельствуют повышенные уровни SAA. Системное воспаление, характеризующееся высокими уровнями SAA, может стимулировать дестабилизацию атеросклеротических бляшек помимо возможного прямого воздействия на атерогенез. Полученные результаты еще более подтверждают прямое воздействие CI-201 на воспалительные процессы.
Пример XII
Экспрессия цитокинов в разных органах мышей Аро-Е КО, иммунизированных CI-201
Влияние иммунизации CI-201 на эксперссию цитокина в органе-мишени, аорте, а также в селезенке, печени, почках и тонком кишечнике определяли с помощью вышеописанного метода RT-PCR. Мышам Аро-Е КО перорально вводили 1 мг/дозу CI-201 или 0,2 мл/мышь PBS через день 5 раз. Экспрессию противовоспалительного цитокина IL-10 и провоспалительного цитокина IFN-γ определяли через 8 недель после последнего перорального введения. Полученные данные приведены на фигурах 16а-b и на фигуре 17.
Как показано на фигурах 16а и 16b, у мышей, которым вводили CI-201, наблюдались повышенные уровни противовоспалительного цитокина IL-10 по сравнению с контрольной группой, которой вводили PBS, и противоположное действие, а именно пониженный уровень экспрессии, было характерено для провоспалительного цитокина IFN-γ в группе, которой вводили CI-201.
Усиление противовоспалительной реакции, о чем свидетельствуют повышенные уровни IL-10, сопровождающееся ослаблением провоспалительной реакции, о чем свидетельствуют пониженные уровни IFN-γ, еще больше подчеркивает роль иммуномодуляции, индуцированной CI-201, которая выражается в переключении реакции с Th1 на Th2 в аорте, а также ее противовоспалительное действие.
В то время как CI-201 усиливает противовоспалительную реакцию в органе-мишени, аорте, такой эффект отсутствует в других органах. Как показано на фигуре 17, не наблюдается различий в экспрессии цитокинов в селезенке и тонком кишечнике между группой, которой вводили CI-201, и контрольной группой, которой вводили PBS. Это позволяет предположить, что перорально вводимый антиген воздействует на пейеровы бляшки, находящиеся в указанном органе. В печени и почке также не наблюдалось изменения экспрессии цитокинов (данные не приведены).
Полученные выше результаты позволяют предположить, что аналоги окисленных фосфолипидов по настоящему изобретению ингибируют атеросклероз, активируя путь, затрагивающий как иммунную систему, так и место воспаления. Однако вполне возможно, что другие механизмы также участвуют в достижении наиболее сильного ингибирующего действия.
Пример XIII
Ингибирование ревматоидного артрита у крыс, страдающих адъювант-индуцированным артритом, в результате перорального введения CI-201
Ревматоидный артрит (RA) является тяжелым аутоиммунным заболеванием, характеризующимся хроническим воспалением и разрушением суставов. Адъювант-индуцированный артрит (AIA) является первой экспериментальной моделью артрита (Pearson. Development of arthritis, periarthritis and periostitis in rats given adjuvant. Proc. Soc. Exp. Biol. Med. 1956; 91:95-101; Pearson and Wood. Studies of polyarthritis and other lesions induced in rats by injection of mycobacterial adjuvant. I. general clinical and pathologic characteristics and some modifying factors. Arthritis Rheum 1959; 2: 440-459).
Морфологию поражений на начальной стадии AIA определяет клеточно-опосредованный иммунитет (CMI). За инфильтрацией лимфоцитов следует отек, отложение фибрина и некроз, сопровождающийся пролиферацией синовиоцитов и фибробластов и активацией остеобластов и остеокластов. Воспалительный инфильтрат в местах поражения суставов AIA содержит Т-клетки, активированные специфическими антигенами. Цитокины Th1, такие как IL-17, IFN-γ и TNF-α, экспрессированы на начальной стадии AIA вместе с цитокинами, характерными для активации макрофагов. На более поздней стадии заболевания повышаются уровни IL-4, IL-6 и JE (гомолог белка 1 хемоатрактанта моноцитов мыши) и TGF-β. Происходит локальное выделение протеолитических ферментов и/или свободных радикалов кислорода, которые вызывают постепенно разрушение коллагена типа II и IX, матрикса и со временем кости (Van Eden and Waksman. Immune regulation in adjuvant-induced arthritis. Possible implications for innovative therapeutic strategies in arthritis. Arthritis Rheum. 2003; 48(7):1788-1796).
Несколько попыток иммунотерапии аутоиммунных заболеваний человека, таких как ревматоидный артрит (RA), диабет типа I и рассеянный склероз, путем модуляции отдельных иммунных путей, участвующих в воспалении, или приобретения толерантности к разным антигенам, показали, что такой подход может быть перспективным (Bielekova et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167-1175; Kappos et al. Induction of a non-encephalitogenic type 2 Т helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. Nat. Med. 2000;6:1176-1182).
У многих субъектов ревматоидный артрит сопровождается сердечно-сосудистыми заболеваниями, причем именно эти заболевания являются главной причиной увеличения смертности. Считается, что аутоантитела против кардиолипина (CL) и модифицированных окислением липопротеинов низкой плотности (LDL, окисленный медью), в том числе LDL, модифицированного малондиальдегидом (MDA-LDL), могут служить для прогнозирования сердечно-сосудистых заболеваний. Установлено, что у субъектов, страдающих ревматоидным артритом, наблюдаются повышенные уровни аутоантител против окисленного медью липопротеина низкой плотности, модифицированных малондиальдегидом липопротеинов низкой плотности и кардиолипида (Cvetkovic et al. Increased levels of autoantibodies against copper-oxidized low density lipoprotein, malondialdehyde-modified low density lipoprotein and cardiolipin in patients with rheumatoid arthritis. Rheumatology. 2002; 41:988-995). Кроме того, существуют данные о наличии окисленного липопротеина низкой плотности в синовиальной жидкости субъектов, страдающих ревматоидным артритом (Dai et al. Evidence for oxidized low density lipoprotein in synovial fluid from rheumatoid arthritis patients. Free Radic. Res. 2000; 32(6): 479-486).
Так как было установлено, что CI-201 позволяет эффективно модулировать иммунную реакцию на Ox LDL и усиливать противовоспалительную реакцию, было исследовано его влияние на развитие артрита.
Самцам мышей Lewis в возрасте девяти недель перорально вводили разные дозы CI-201 (4 мг/кг или 0,4 мг/кг) или PBS через день 5 раз. Затем вызывали адъювант-индуцированный артрит при помощи внутрикожной инъекции 0,1 мл суспензии туберкулезной микробактерии. Тяжесть артрита контролировали путем измерения распухания лап и подвижности животных. Схема исследования показана на фигуре 18. Результаты приведены на фигуре 19.
Как показано на фигуре 19, предварительное введение более высокой дозы (4,0 мг/кг) CI-201 вызывало значительное уменьшение опухания лап у крыс по сравнению с контрольными крысами, которым вводили PBS.
В то время как крысы, которым вводили PBS, с трудом двигались при помощи только задних лап, подвижность крыс, которым предварительно вводили более высокую дозу CI-201, была близка к подвижности здоровых крыс.
Для оценки влияния постоянного введения CI-201 на состояние здоровья AIA-индуцированных крыс Lewis самцам крыс Lewis в возрасте 9 недель до индукции AIA с помощью внутрикожной инъекции 0,1 мл суспензии туберкулезной микробактерии через день 5 раз перорально вводили указанный препарат и продолжали постоянно вводить указанный препарат три раза в неделю в течение 30 дней. Схема исследования показана на фигуре 20. Результаты приведены на фигурах 21-23.
Как показано на фигурах 21-23, постоянное введение высокой дозы CI-201 значительно замедляло развитие артрита по всем исследуемым параметрам.
Полученные результаты ясно показывают, что помимо атеросклероза CI-201 может оказывать противовоспалительное действие на классическое воспалительное заболевание RA.
CD4+ Т-клетки-хелперы и макрофаги проникают через синовиальную мембрану (SM) в случае хронического, деструктивного ревматоидного артрита и, вероятно, играют главную роль в стимулировании и сохранении процесса заболевания. Из CD4+ Т-клеток можно выделить субпопуляцию Th1, характеризующуюся преобладающим продуцированием IFN-γ. Преобладание Th1-клеток провоспалительного типа является условием возникновения RA (Schmidt-Weber et al. Cytokine gene activation in synovial membrane, regional lymth nodes, and spleen during the course of rat adjuvant arthritis. Cell. Immunol. 1999; 195:53-65). Макрофаги также сильно активированы в воспалительном процессе, обусловленном ревматоидным артритом (RA), как локально, так и системно.
Сходство воспалительной реакции, возникающей при атеросклерозе и артрите, подтверждает предположение, что CI-201 индуцирует противовоспалительную реакцию в случае AIA, аналогичную реакции, описанной выше для атеросклероза.
Поэтому можно сделать вывод, что введение CI-201 вызывает повышение уровня IL-10 в модели AIA, в свою очередь IL-10 может подавлять провоспалительные цитокины, ослабляя таким образом течение заболевания, как это было продемонстрировано меньшим опуханием лап и лучшей подвижностью. Поэтому полученные результаты предполагают, что аналоги окисленных фосфолипидов по настоящему изобретению могут служить в качестве нового семейства терапевтических средств, предназначенных для лечения ревматоидного артрита, а также других аутоиммунных и/или воспалительных заболеваний.
Пример XIV
Пероральное введение предварительно окисленного соединения V генетически предрасположенных мышам (Аро-Е КО) - влияние окисленной группы на ингибирование атерогенеза
Влияние окисленной группы в ALLE и CI-201 исследовали путем сравнения влияния перорального введения ALLE и CI-201 на начальную стадию атерогенеза и последующее образование атеросклеротических бляшек с влиянием предварительно окисленного производного указанных веществ, а именно соединения V (1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфохолин, пример I).
25 самок мышей Аро-Е КО в возрасте 8-10 недель делили на 4 группы. Каждой группе перорально вводили 5 мг/мышь соединения V, суспендированного в 0,2 мл PBS (группа А, n=6), 1 мг/мышь соединения V”, суспендированного в 0,2 мл PBS (группа В, n=6), 0,2 мг/мышь соединения V, суспендированного в 0,2 мл PBS (группа С, n=6), и PBS (группа D, контрольная группа, n=7) через день в течение 5 дней. Через восемь недель после последнего перорального введения мышей умерщвляли. У мышей брали пробы крови до начала эксперимента (время 0) и в конце эксперимента (конец) для определения профиля липидов. Атеросклероз оценивали в сердце вышеописанным способом. Все мыши получали нормальный корм, содержащий 4,5 мас.% жира (0,02% холестерина), и воду по потребности.
Влияние введения соединения V на показатели обмена веществ и атерогенез показано в нижеследующей таблице 10. Влияние соединения V на атерогенез далее показано на фигуре 24.
Влияние предварительно окисленного производного соединения V на показатели обмена веществ и площадь атеросклеротического поражения в синусе аорты у мышей АроЕ КО
р)
Как показано на фигуре 24, в то время как пероральное введение окисленных соединений CI-201 и ALLE значительно ингибировало атерогенез у мышей Аро-Е КО, введение предварительно окисленного производного соединения V не влияло на атерогенез, что свидетельствует о важности наличия окисленной группы при лечении атерогенеза.
Пример XV
Пероральное введение предварительно окисленного соединения V генетически предрасположенным мышам (Аро-Е КО) - влияние окисленной группы на развитие атерогенеза
Далее исследовали возможность использования предварительно окисленного соединения V для предотвращения развития атерогенеза в вышеописанной модели с использованием мышей АроЕ КО. Мышей Аро-Е КО в возрасте 23-26 недель (АРО-Е -/-<tm1Unc>[C57B/6J]) распределяли в разные группы по возрасту, массе тела и профилю липидов (холестерин и триглицерид). Одна группа, которую умерщвляли в начале эксперимента, служила в качестве “базовой” группы (B.L., n=10). Второй группе вводили соединение V (0,1 мкг/дозу, n=10), растворенное в PBS, содержащем 0,05% этанола, в общем объеме 0,2 мл PBS. Контрольной группе вводили PBS (0,05% этанола, 0,2 мл) (n=11).
Мышам вводили соединение V или PBS в виде трех серий в начале каждого месяца, причем каждая серия включала 5 пероральных введений, выполняемых через день. Все мыши получали нормальный корм, содержащий 4,5 мас.% жира (0,02% холестерина), и воду по потребности, при этом мышей содержали с 12-часовым циклом чередования темноты и света.
Через 12 недель мышей умерщвляли и определяли профиль липидов и площадь атеросклеротических бляшек в синусе аорты вышеописанным способом.
Результаты, приведенные в нижеследущей таблице 11 и на фигуре 25, ясно показывают, что пероральное введение соединения V не влияло на развитие атерогенеза.
Влияние предварительно окисленного производного соединения V на показатели обмена веществ и площадь атеросклеротического поражения в синусе аорты у мышей АроЕ КО
0,1 мкг/дозу
Несмотря на то, что данное изобретение описано на основании конкретных вариантов его осществления, очевидно, что специалистам в данной области должны быть видны многие альтернативы, модификации и варианты настоящего изобретения. Поэтому настоящее изобретение относится ко всем таким альтернативам, модификациям и вариантам, которые входят в объем прилагаемой формулы изобретения.
Все публикации, патенты и заявки на патент, приведенные в описании изобретения, полностью включены в данное описание изобретения в качестве ссылки, как если бы все отдельные публикации, патенты или заявки на патент были специально включены в данное описание изобретения в качестве ссылки. Кроме того, цитирование и указание любых ссылок в данной заявке не должно рассматриваться как признание того, что такая ссылка характеризует известный уровень техники по отношению к настоящему изобретению.
Дополнительные представляющие интерес ссылки
(не указанные в тексте описания изобретения)
Wick G, Schett G, Amberger A, Kleindienst R, Xu Q. Is atherosclerosis an immunologically mediated disease? Immunol Today 1995; 16: 27-33.
Libby P, Hansson GK. Involvement of the immune system in human atherogenesis: current knowledge and unanswered questions. Lab Invest 1991; 64: 5-15.
Steinberg D, Parathasarathy S, Carew ТЕ, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med 1989; 320: 915-924.
Witztum J. The oxidation hypothesis of atherosclerosis. Lancet 1994; 344: 793-795.
Palinski W, Miller E, Witztum JL. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc Natl Acad Sci USA. 1995;92: 821-825.
George J, Afek A, Gilburd B, Levy Y, Levkovitz H, Shaish A, Goldberg I, Kopolovic Y, Wick G, Shoenfeld Y, Harats D. Hyperimmunization of ApoE deficient mice with homologous oxLDL suppresses early atherogenesis. Atherosclerosis. 1998; 138: 147-152.
Weiner H, Freidman A, Miller A. Oral tolerance: immunologic mechanisms and treatment of animal and human organ specific autoimmune diseases by oral administration of autoantigens. Annu Rev Immunol 1994; 12: 809-837.
Palinski W, Ord VA, Plump AS, Breslow JL, Steinberg D, Witztum JL. Apo-E-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation-specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler Thromb 1994; 14: 605-616
Roselaar SE, Kakkanathu PX, Daugherty A. Lymphocyte populations in atherosclerotic lesions of apo-E -/- and LDL receptor -/- mice; Decreasing density with disease progression. Arterioscler Thromb Vase Biol 1996; 16: 1013-1018.
Palinski W, Yla-Herttuala S, Rosenfeld ME, Butler SW, Socher SA, Parthasarathy S, Curtiss LK, Witztum JL. Antisera and monoclonal antibodies specific for epitopes generated during oxidative modification of low density lipoprotein. Arteriosclerosis 1990; 10: 325-335.
Ou Z., Ogamo A., Guo L., Konda Y., Harigaya Y. and Nakagawa Y. Anal. Biochem. 227: 289-294, 1995.
E. Baer and Buchnea JBC. 230,447,1958.
Quintana FJ, Carmi P, Mor F and Cohen R. Inhibition of adjuvant arthritis by a DNA vaccine encoding human heat shock protein 60. J. Immunol.2002; 169:3422-3428.
Cobelens PM, Heijnen CJ, Nieuwenhois EES et al. Treatment of adjuvant induced arthritis by oral administration of mycobacterial Hsp65 during disease. Arthritis Rheum. 2000;43(12):2694-2702.
Cobelens PM, Kavelaars A, Van Der Zee R et al. Dynamics of mycobacterial HSP65-induced T-cell cytokine expression during oral tolerance induction in adjuvant arthritis. Rheumatology. 2002;41:775-779.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОКИСЛЕННЫЕ ЛИПИДЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2004 |
|
RU2362567C2 |
| ОКИСЛЕННЫЕ СОЕДИНЕНИЯ ЛИПИДОВ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2532546C2 |
| СПОСОБ УЛУЧШЕНИЯ СТРУКТУРЫ И/ИЛИ ФУНКЦИЙ АРТЕРИОЛ | 2005 |
|
RU2414236C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ОКИСЛЕННЫХ ФОСФОЛИПИДОВ | 2005 |
|
RU2399626C2 |
| ЛИПИДНЫЕ НАНОЧАСТИЦЫ | 2021 |
|
RU2837542C1 |
| ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ОБЛЕГЧАТЬ ПО МЕНЬШЕЙ МЕРЕ ОДИН СИМПТОМ ВОСПАЛИТЕЛЬНОГО СОСТОЯНИЯ ПЕПТИД, СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ АТЕРОСКЛЕРОЗА С ИХ ПОМОЩЬЮ | 2005 |
|
RU2448977C2 |
| ПЭГИЛИРОВАННАЯ ЛИПИДНАЯ НАНОЧАСТИЦА С БИОАКТИВНЫМ ЛИПОФИЛЬНЫМ СОЕДИНЕНИЕМ | 2016 |
|
RU2737893C2 |
| КОМПОЗИЦИИ ДЛЯ НАРУЖНОГО ПРИМЕНЕНИЯ, СОДЕРЖАЩИЕ АДЕНОЗИЛКОБАЛАМИН, ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ КОЖИ | 2006 |
|
RU2379039C2 |
| ЛИПИДНОЕ СОЕДИНЕНИЕ И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2021 |
|
RU2825571C1 |
| НОСИТЕЛЬ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2008 |
|
RU2476204C2 |
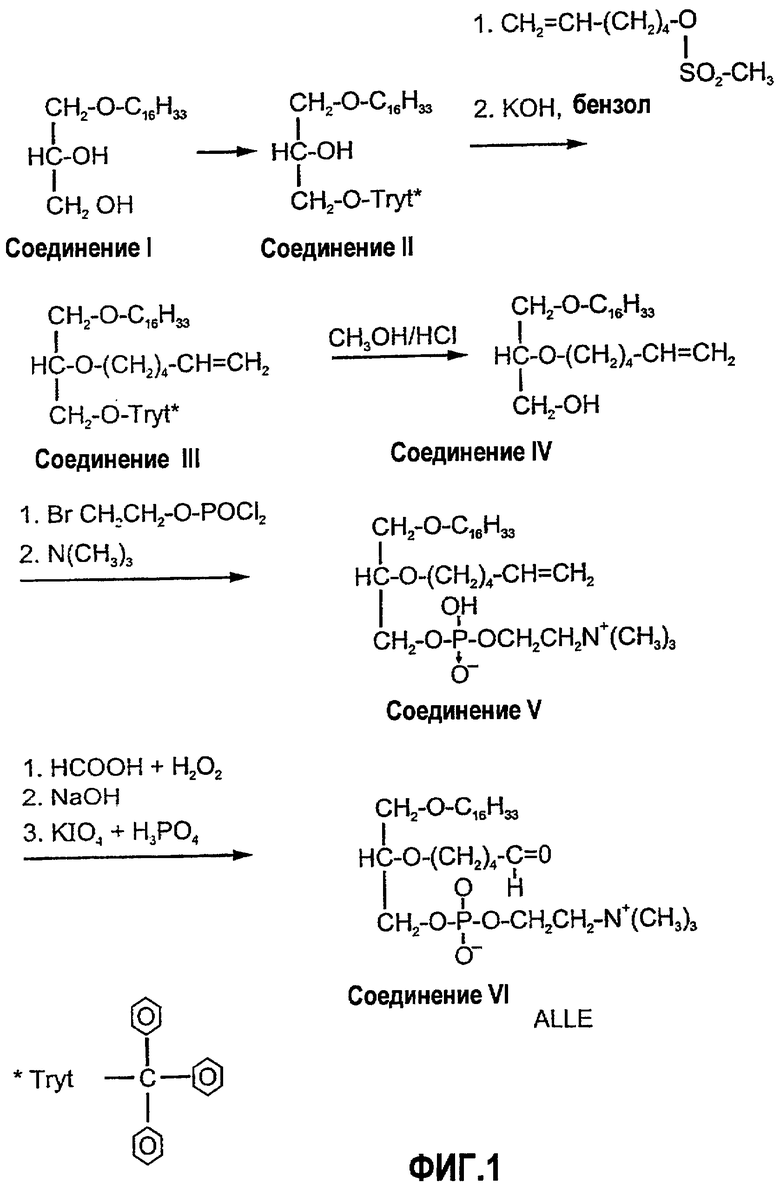
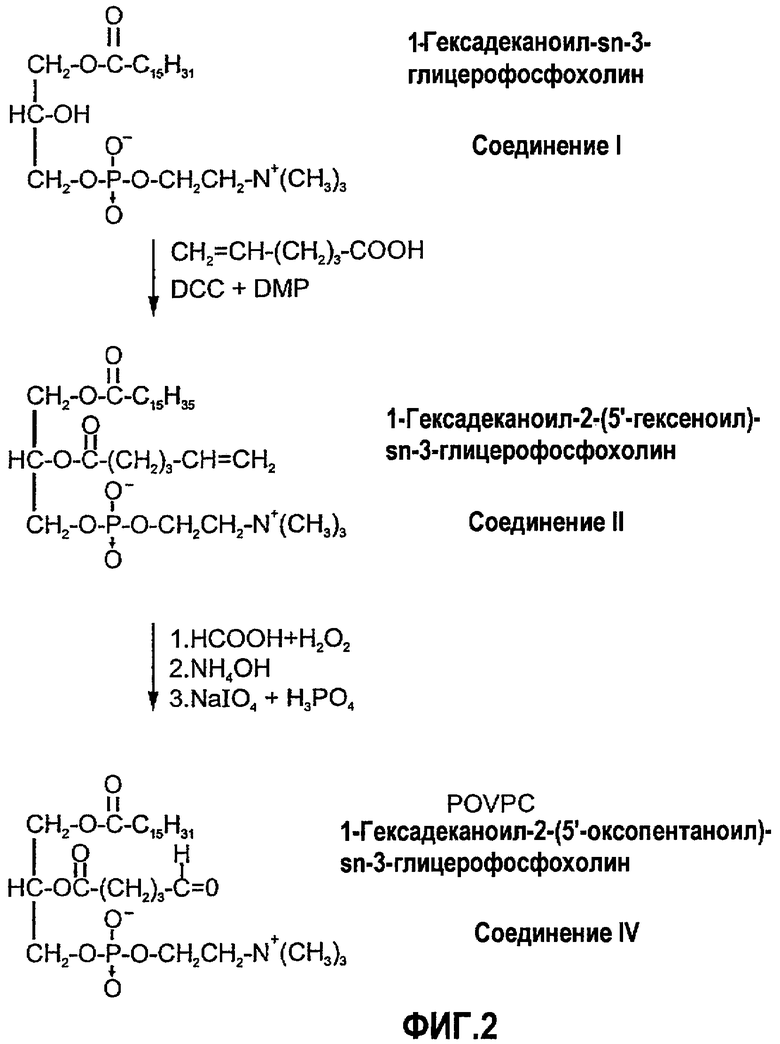
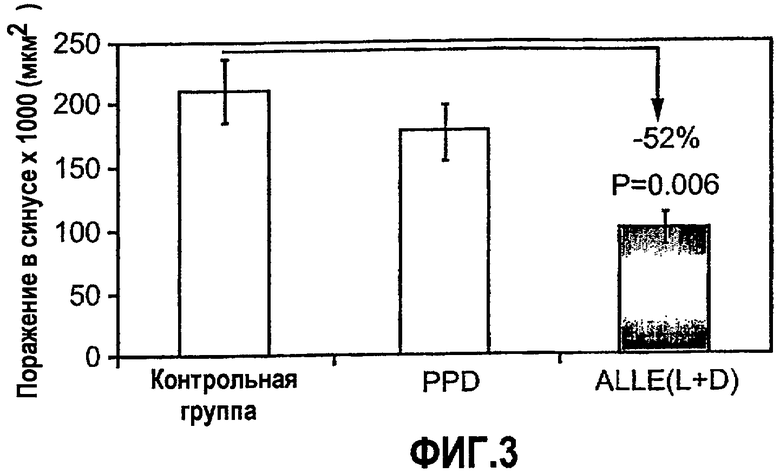
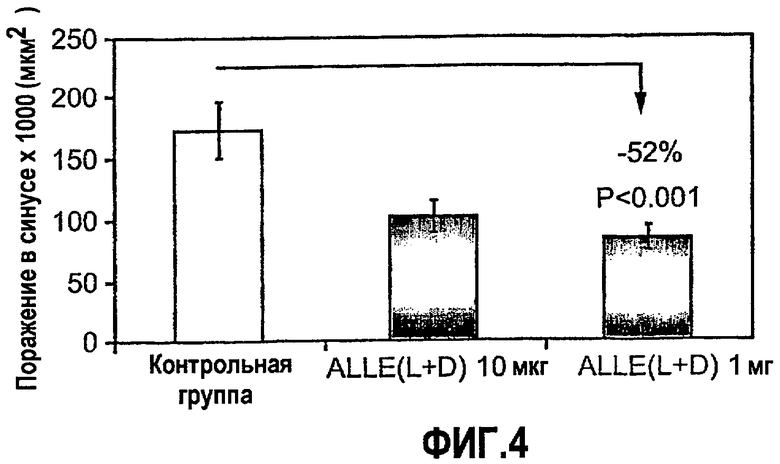
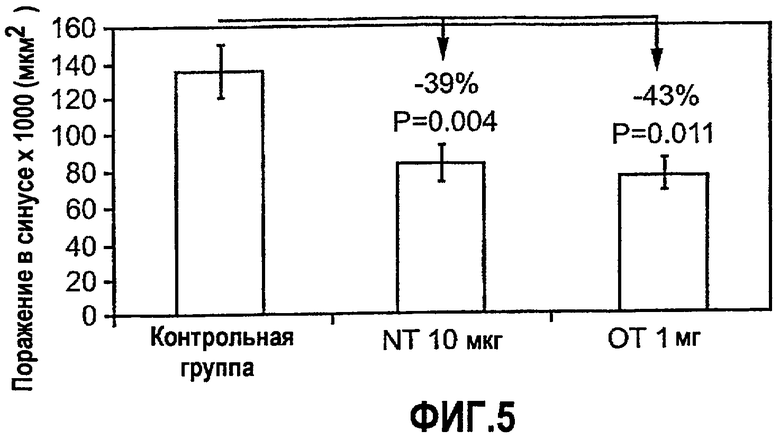
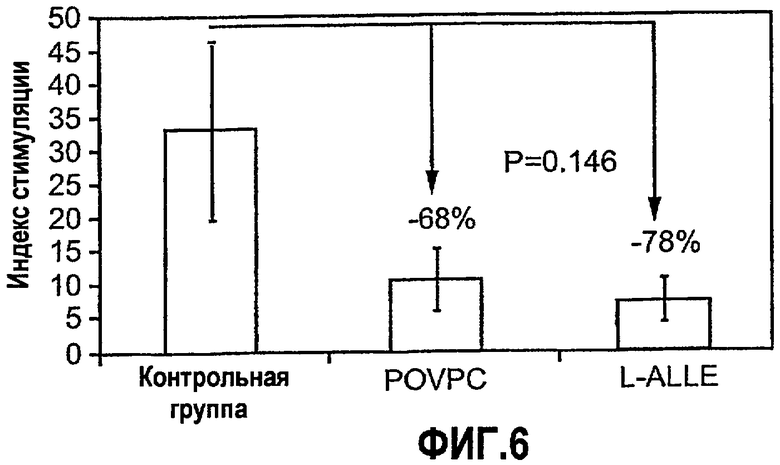
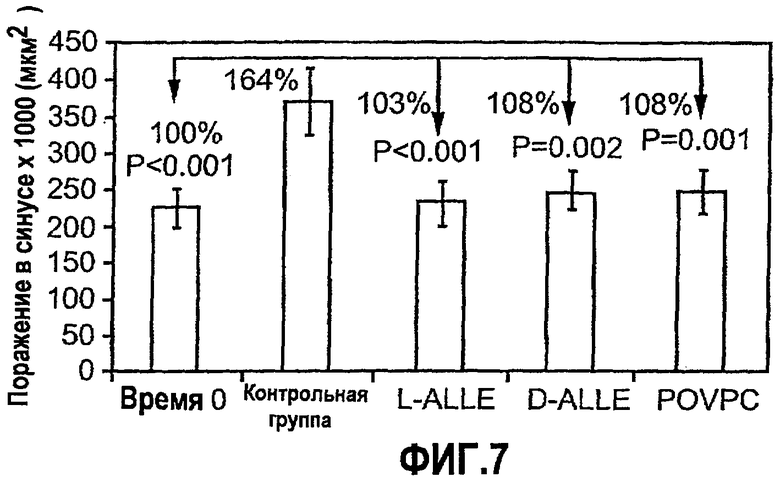
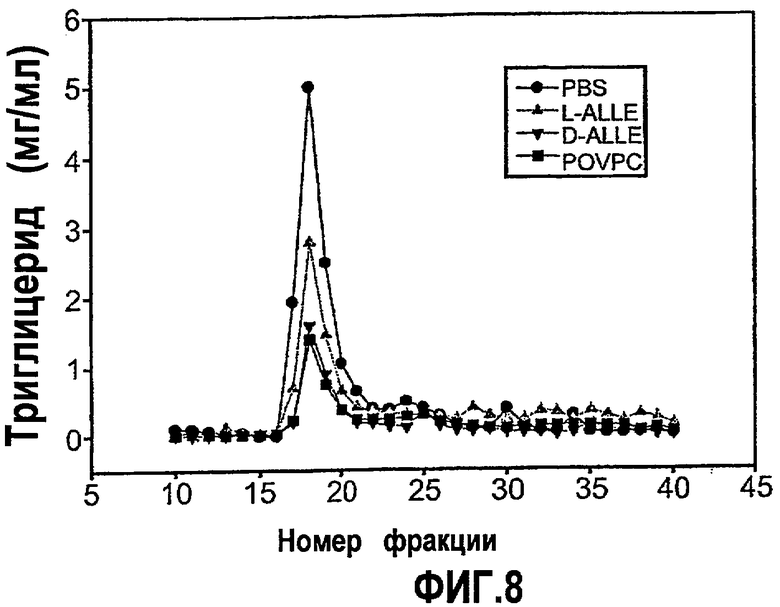
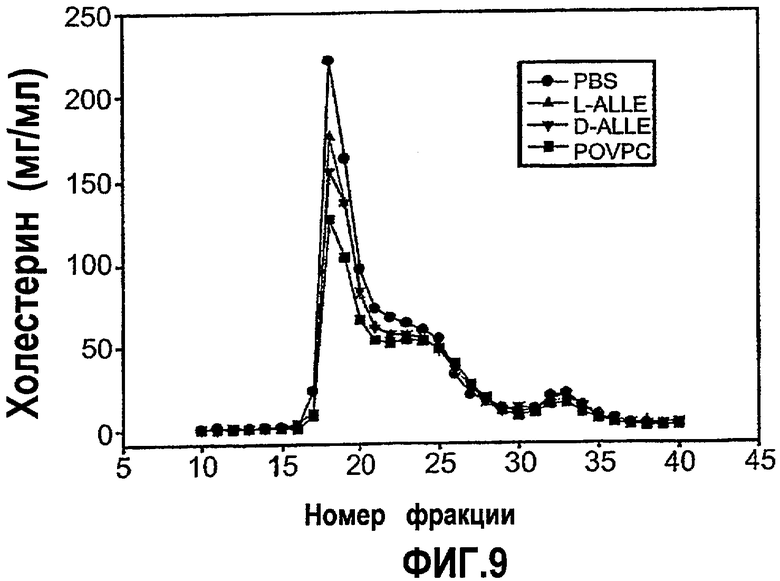
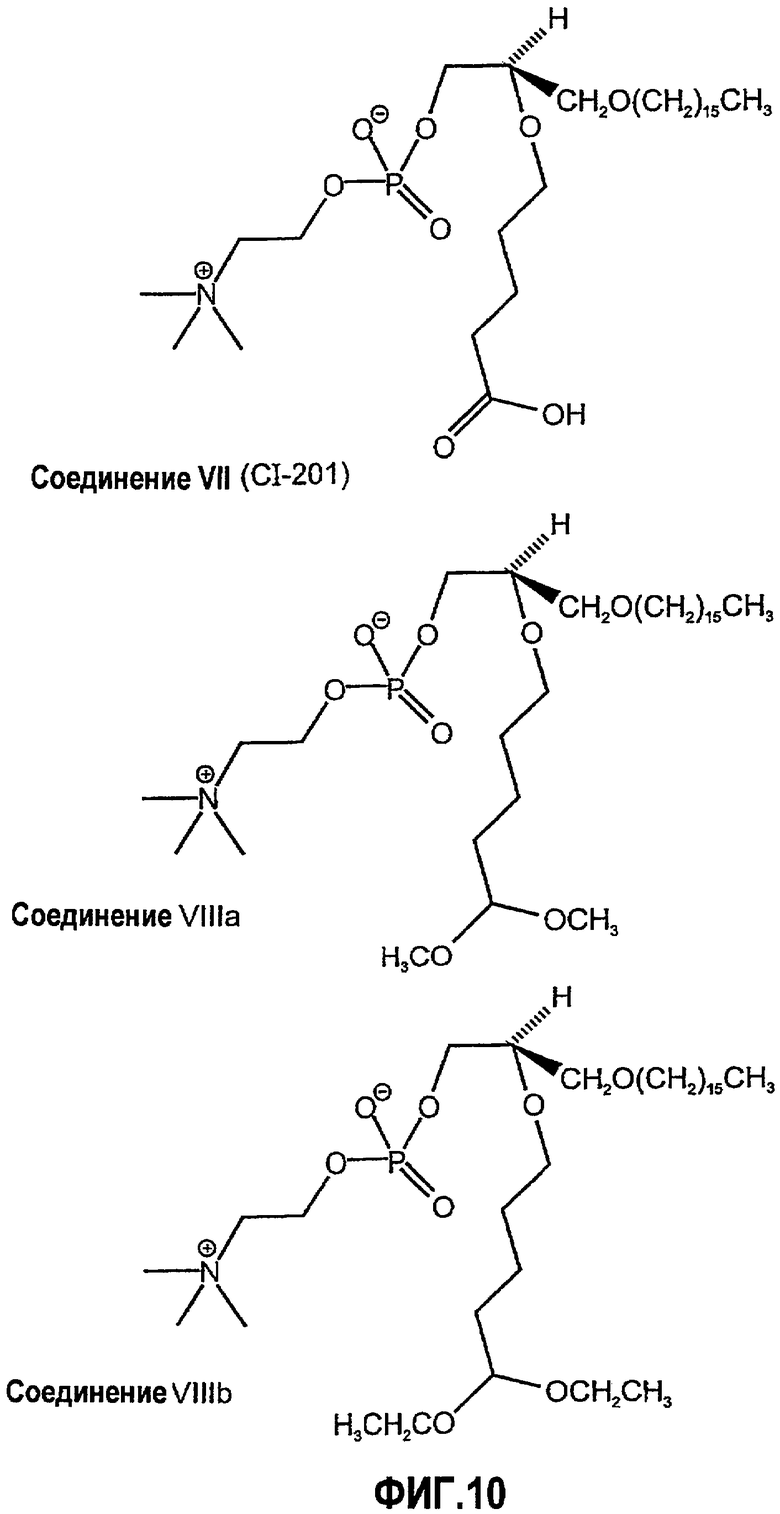
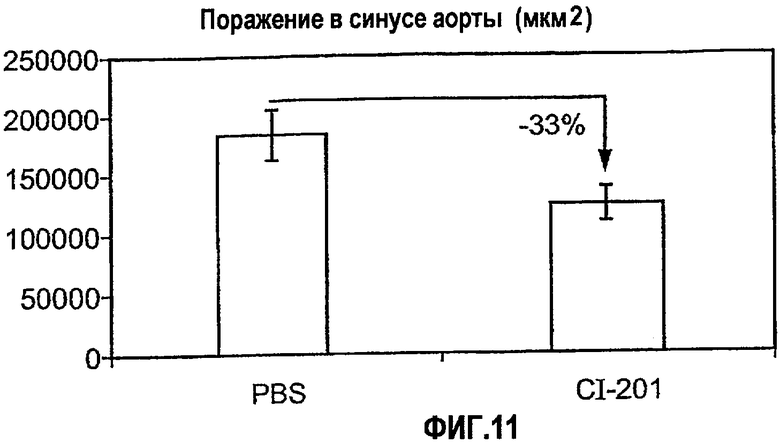
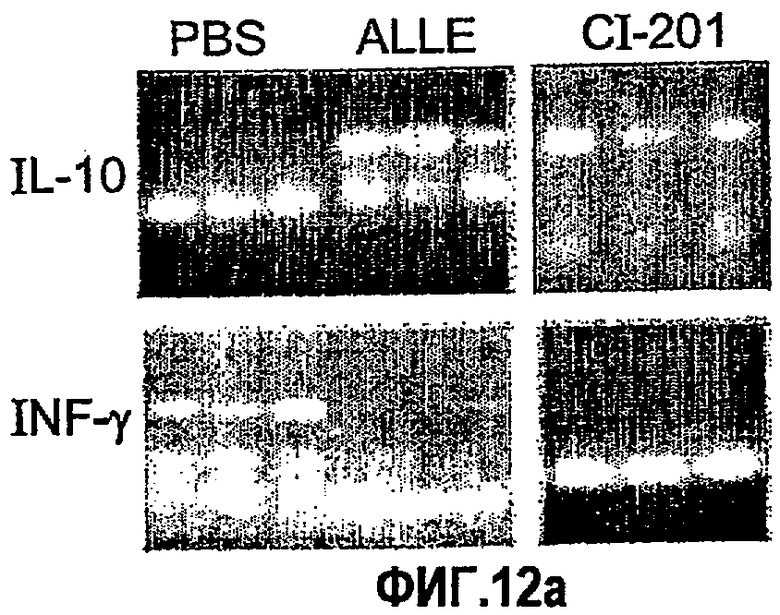
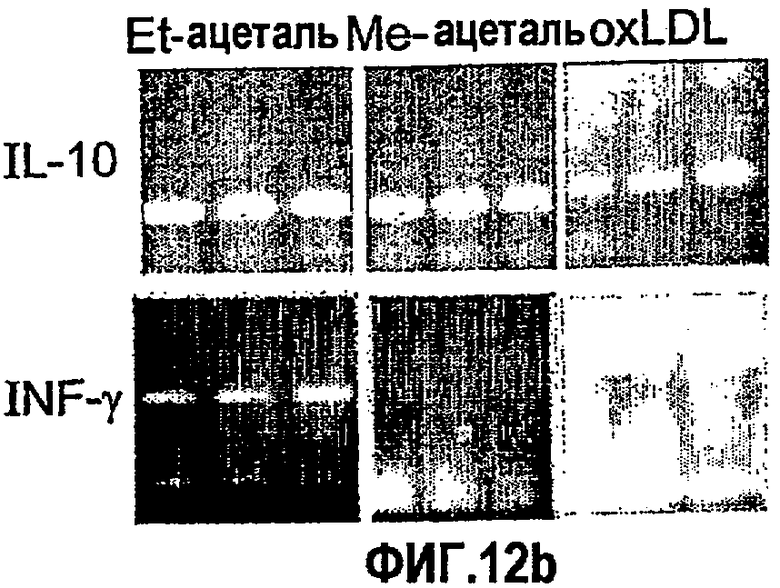
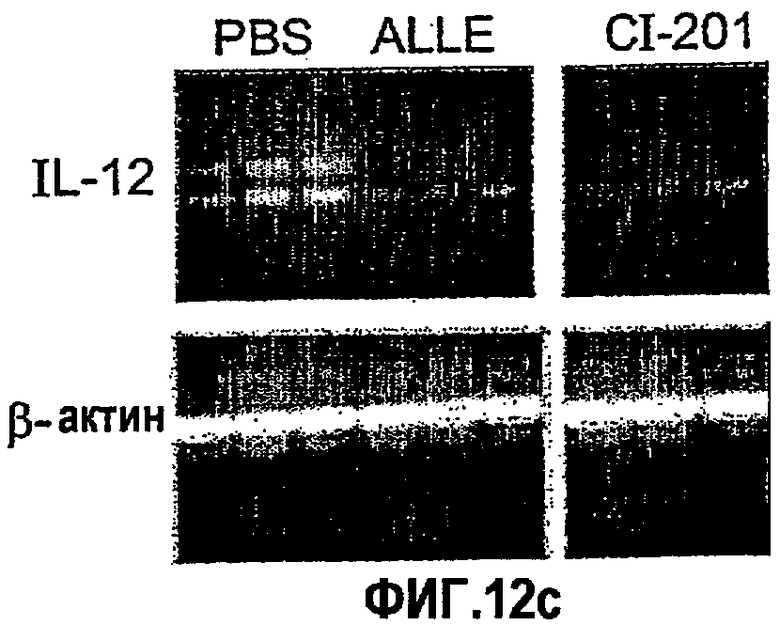
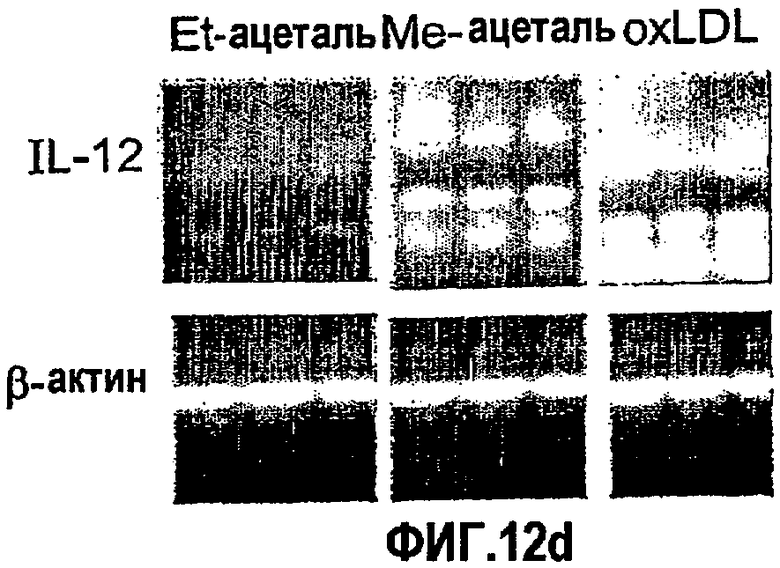
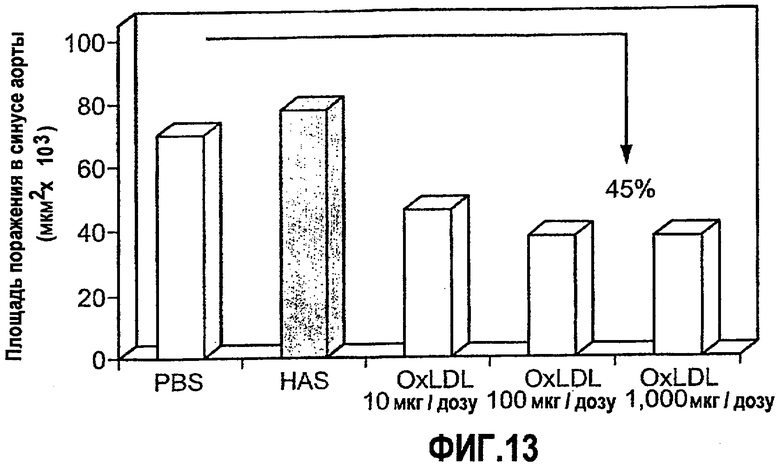
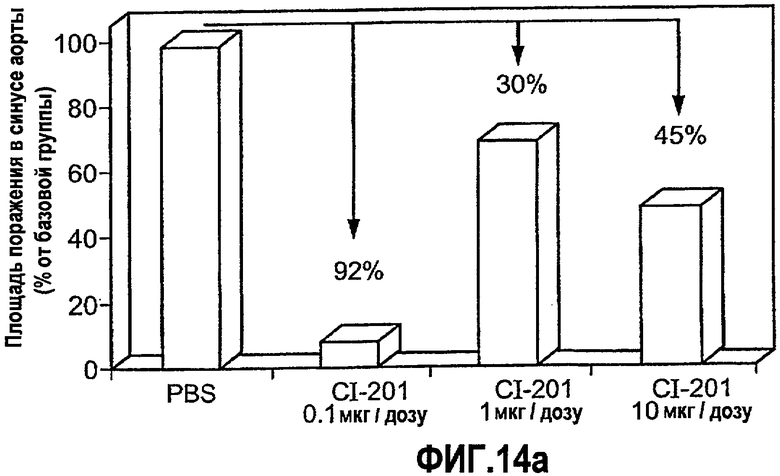
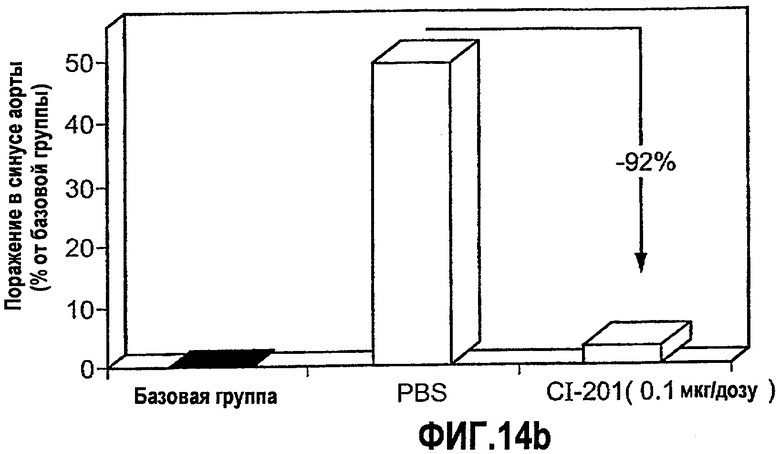
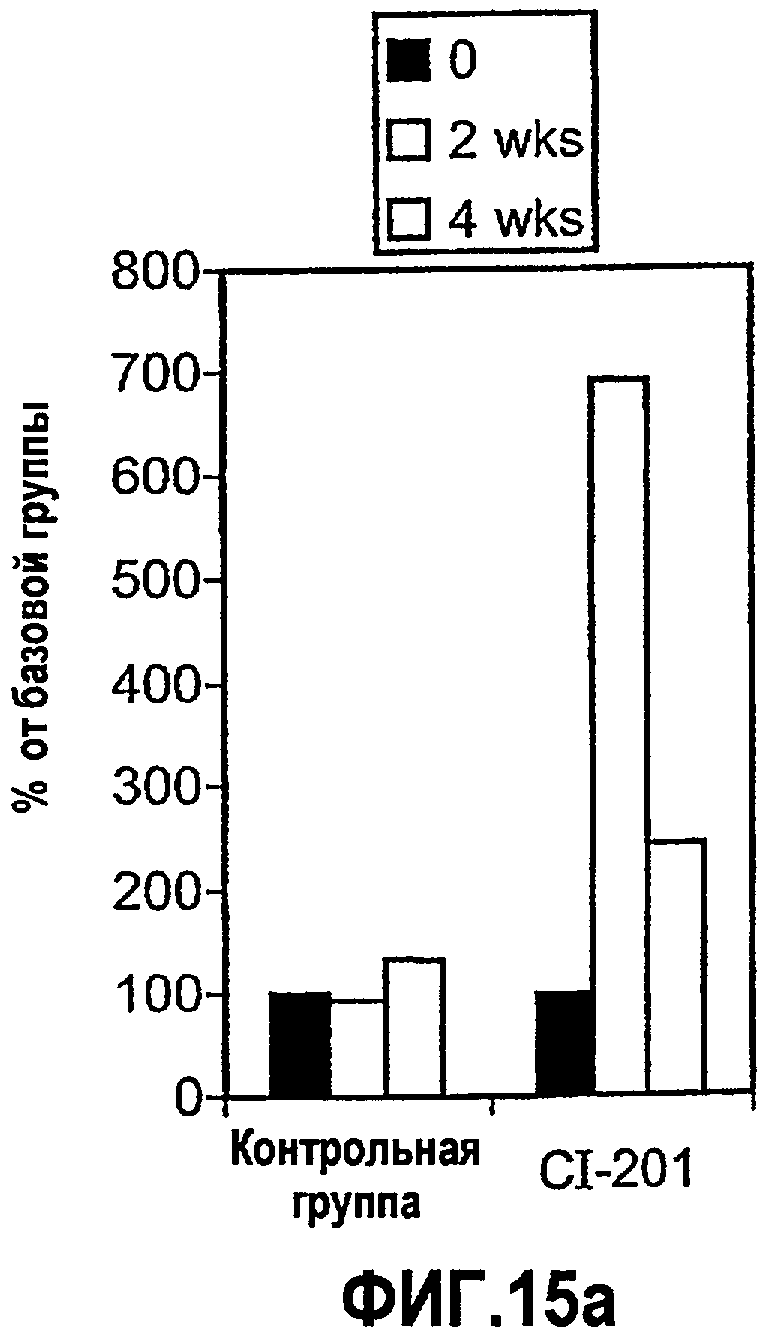
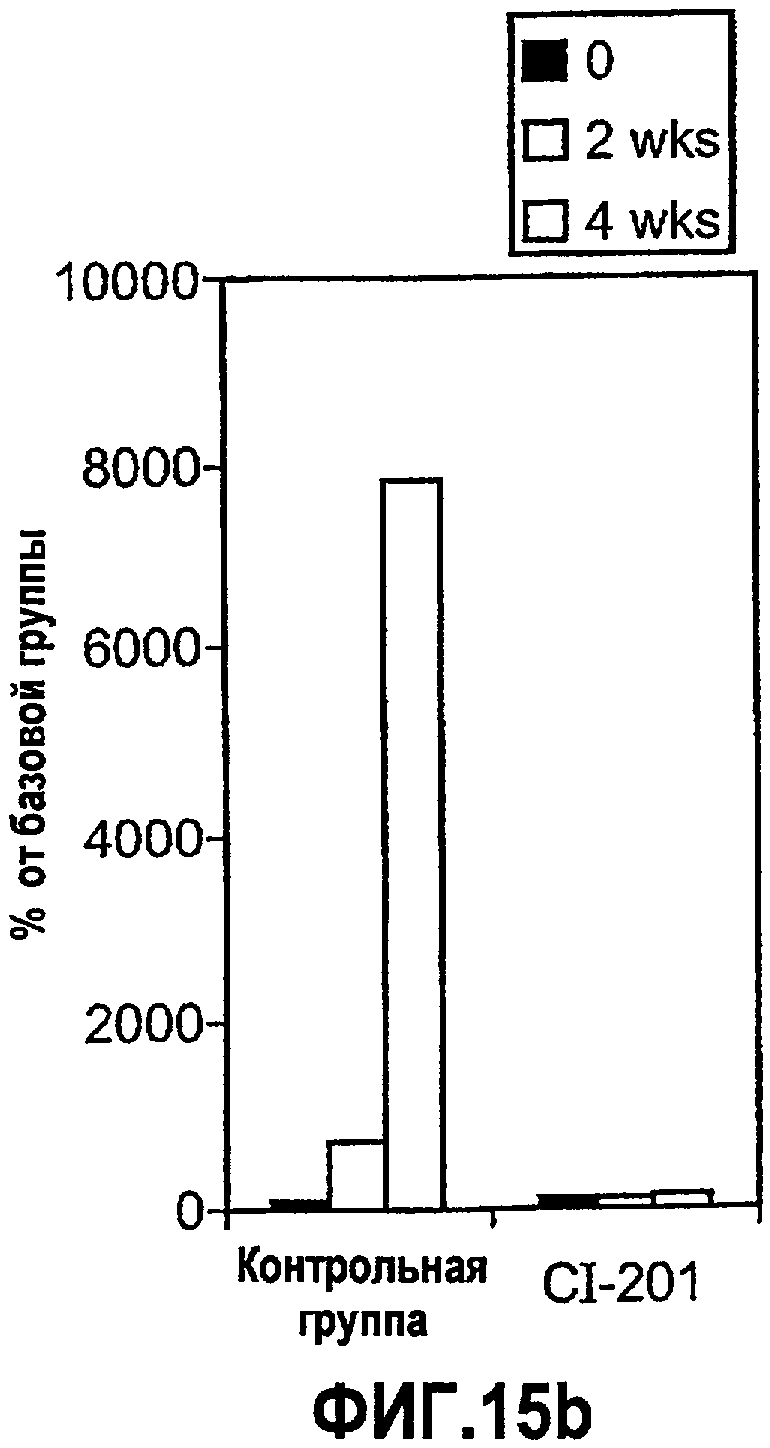
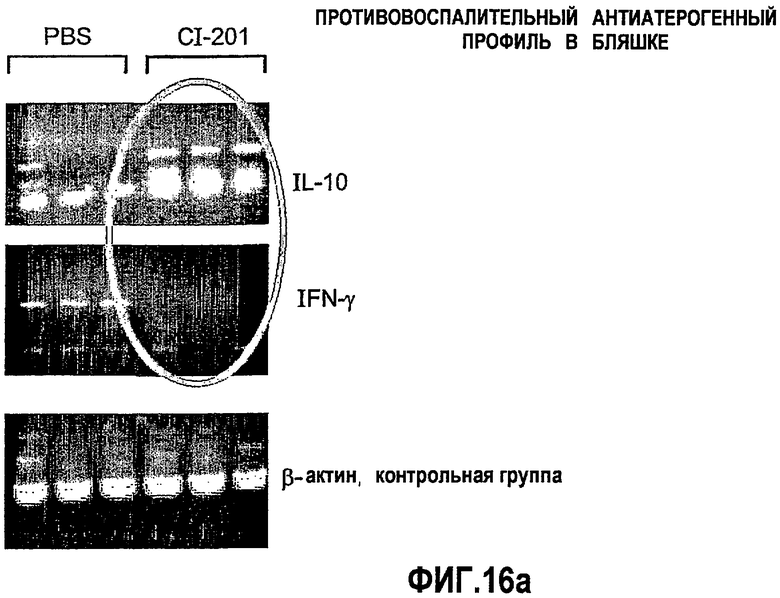
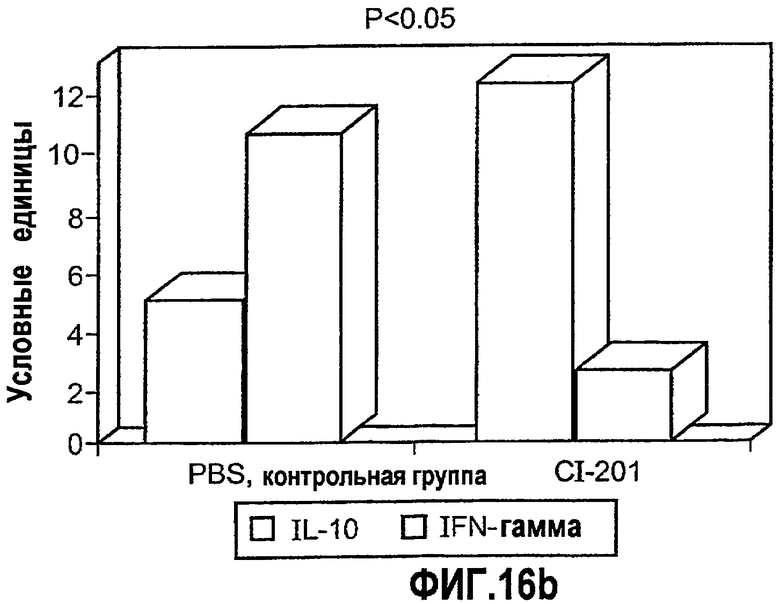
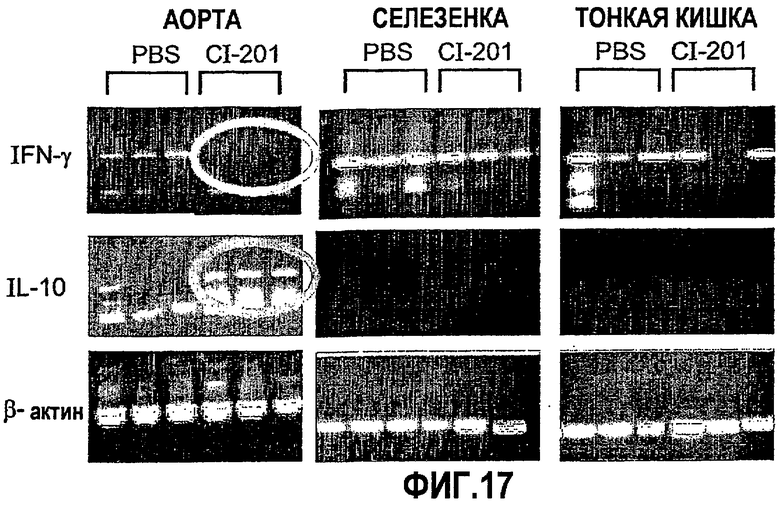
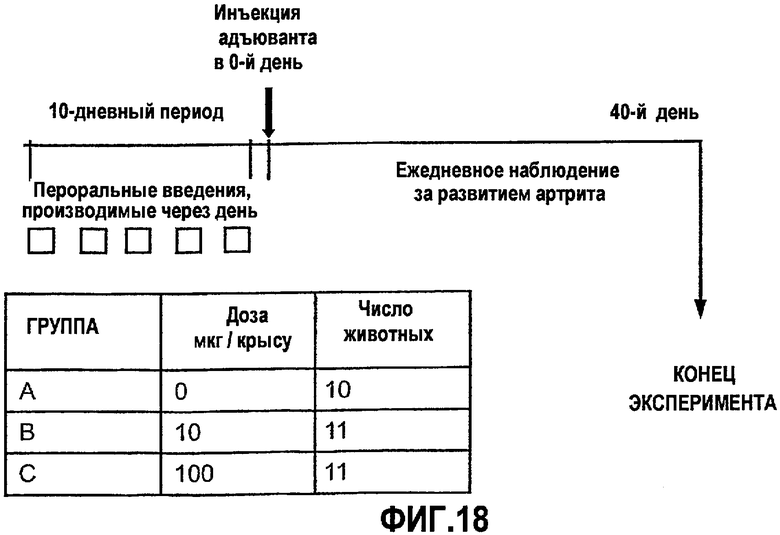

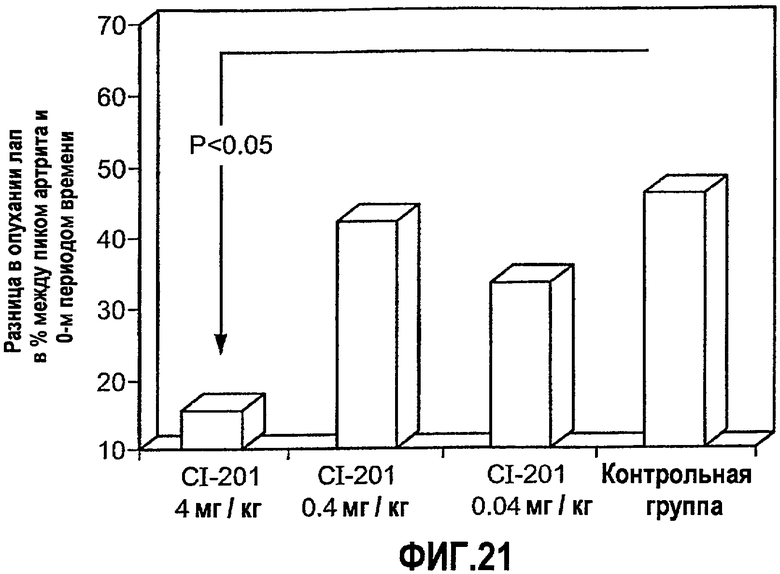
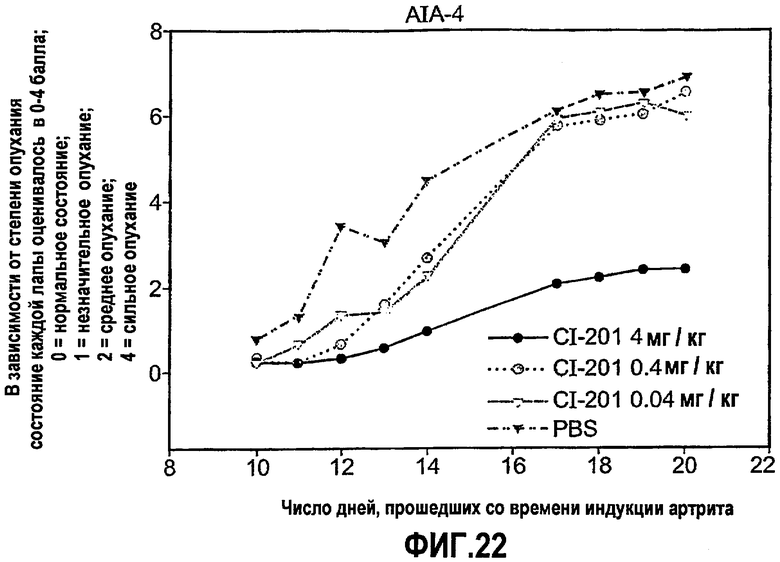
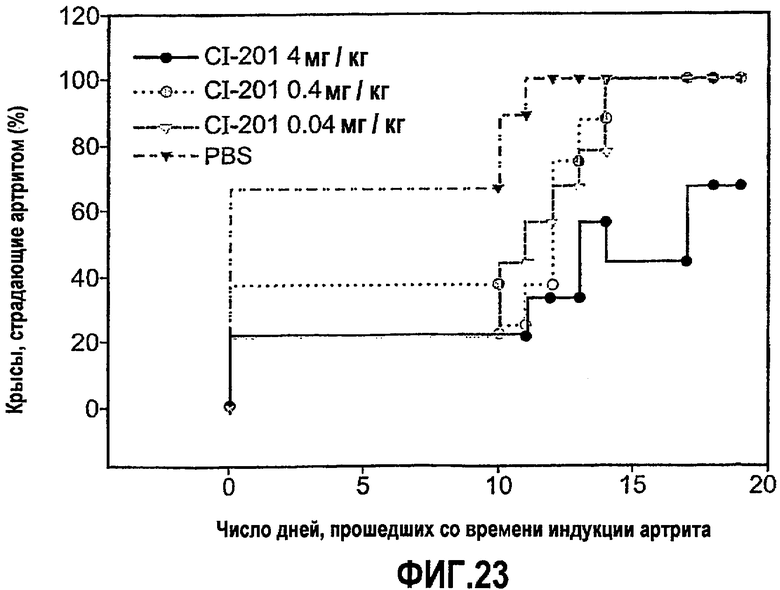
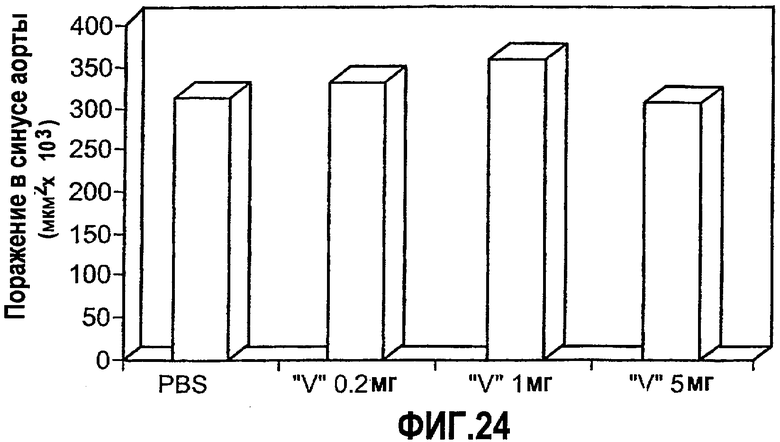
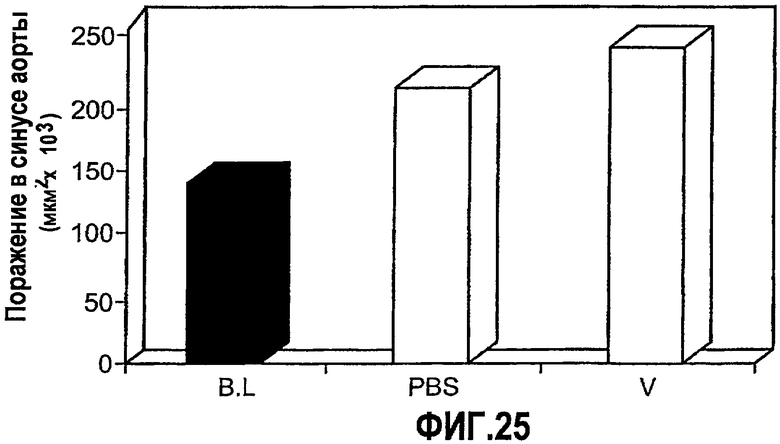
Настоящее изобретение относится к фармацевтическим композициям, включающим синтетические окисленные липиды, и способам применения окисленных липидов для лечения и профилактики воспаления, ассоциированного с эндогенным окисленным липидом. 6 н. и 33 з.п. ф-лы, 11 табл., 15 пр., 25 ил.
1. Фармацевтическая композиция, обладающая иммуномодулирующей активностью, содержащая в качестве активного ингредиента терапевтически эффективное количество соединения общей формулы I:
,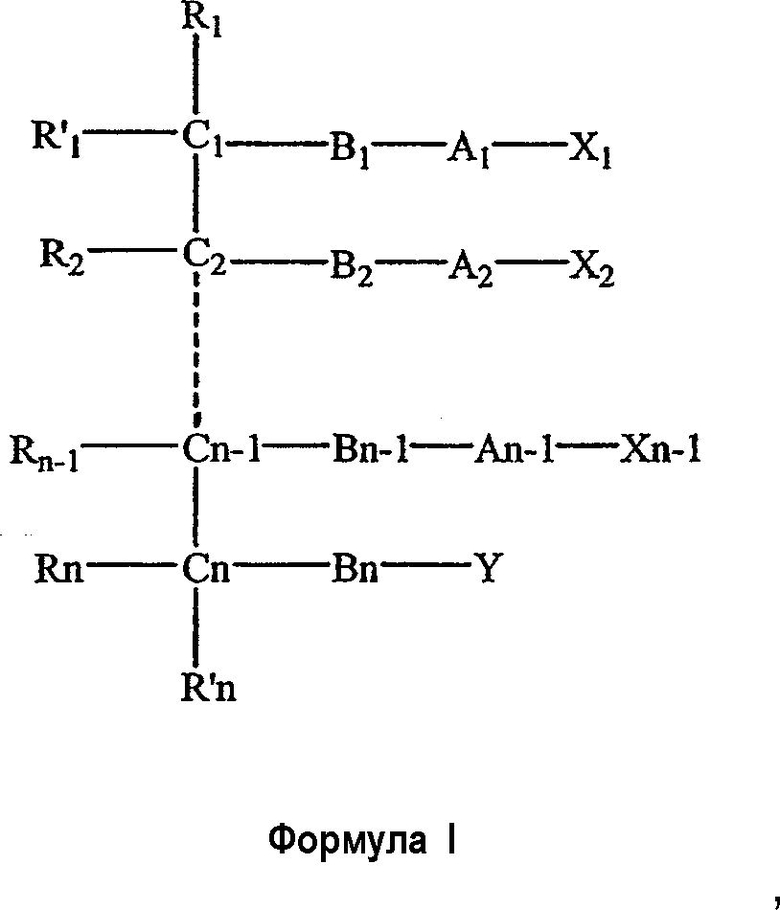
где n является целым числом 3;
В1 является кислородом или серой;
каждый из В2 и В3 является кислородом;
каждый из А1 и А2 является СН2;
Y выбирают из группы, состоящей из водорода, фосфорной кислоты, фосфорилхолина и фосфорилэтаноламина; и
каждый из X1 и Х2 независимо означает насыщенный или ненасыщенный углеводород общей формулы II:
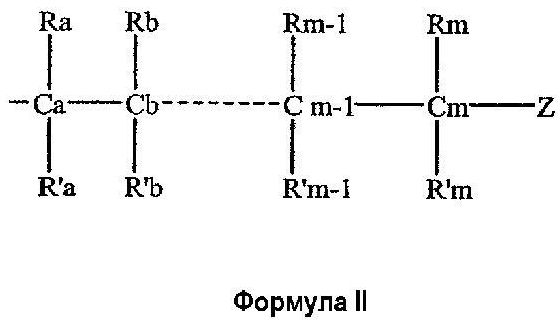
где m является целым числом от 2 до 19; и
Z выбирают из группы, состоящей из:
где W является кислородом; и
по меньшей мере один из X1 и Х2 включает Z, который не является
водородом; и
где каждый из R1, R2, R3, 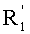 и
и 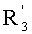 является водородом;
является водородом;
каждый из R'' и R''' независимо выбран из группы, состоящей из водорода и алкила;
каждый из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm независимо выбран из группы, состоящей из водорода, связи и гидроксигруппы; и
или его стереоизомера, оптического изомера, рацемической смеси, фармацевтически приемлемой соли,
и фармацевтически приемлемый носитель,
причем указанная фармацевтическая композиция упакована в упаковочный материал и идентифицирована оттиском, сделанным в указанном упаковочном материале или на упаковочном материале, как предназначенная для применения при лечении или предупреждения воспаления, связанного с эндогенным окисленным липидом, причем указанное воспаление связано с заболеванием или нарушением, выбранным из группы, состоящей из воспалительного желудочно-кишечного заболевания или нарушения, пролиферативного заболевания или нарушения и диабета.
2. Фармацевтическая композиция по п.1, где указанное воспалительное желудочно-кишечное заболевание или нарушение выбрано из группы, состоящей из колита, илеита, болезни Крона, хронического воспалительного заболевания тонкой кишки, синдрома воспаленной толстой кишки, хронического воспалительного заболевания толстой кишки, целиакии, язвенного колита, язвы, изъязвлений кожи, пролежней, язвы желудка, пептической язвы, язвы щеки, носоглоточной язвы, язвы пищевода, язвы двенадцатиперстной кишки и желудочно-кишечной язвы.
3. Фармацевтическая композиция по п.2, где указанное воспалительное желудочно-кишечное заболевание или нарушение выбрано из группы, состоящей из колита и хронического воспалительного заболевания толстого кишечника.
4. Фармацевтическая композиция по п.2, где указанное воспалительное желудочно-кишечное заболевание или нарушение выбрано из группы, состоящей из болезни Крона и язвенного колита.
5. Фармацевтическая композиция по п.1, где указанное пролиферативное заболевание или нарушение представляет собой злокачественную опухоль.
6. Фармацевтическая композиция по п.1, где указанное заболевание или нарушение представляет собой диабет.
7. Фармацевтическая композиция, обладающая иммуномодулирующей активностью, содержащая в качестве активного ингредиента терапевтически эффективное количество соединения общей формулы:
где n является целым числом 3;
В1 является кислородом или серой;
каждый из В2 и В3 является кислородом;
каждый из A1 и А2 является СН2;
Y выбирают из группы, состоящей из водорода, фосфорной кислоты, фосфорилхолина и фосфорилэтаноламина; и
каждый из X1 и Х2 независимо означает насыщенный или ненасыщенный углеводород общей формулы II:
где m является целым числом от 2 до 19; и
Z выбирают из группы, состоящей из:
где W является кислородом; и
по меньшей мере один из X1 и Х2 включает Z, который не является
водородом; и
где каждый из R1, R2, R3, 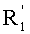 и
и 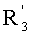 является водородом;
является водородом;
каждый из R'' и R''' независимо выбран из группы, состоящей из водорода и алкила;
каждый из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm независимо выбран из группы, состоящей из водорода, связи и гидроксигруппы;
или его стереоизомера, оптического изомера, рацемической смеси, фармацевтически приемлемой соли,
и фармацевтически приемлемый носитель,
причем указанная фармацевтическая композиция упакована в упаковочный материал и идентифицирована оттиском, сделанным в указанном упаковочном материале или на упаковочном материале, как предназначенная для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-6, интерлейкина-17, моноцитарного хемотактического белка-1 (МСР-1), моноцитарного хемотактического белка-3 (МСР-3) и фактора некроза опухоли-α (TNF-α).
8. Фармацевтическая композиция, обладающая иммуномодулирующей активностью, содержащая в качестве активного ингредиента терапевтически эффективное количество соединения общей формулы:
где n является целым числом 3;
B1 является кислородом или серой;
каждый из В2 и В3 является кислородом;
каждый из A1 и А2 является СН2;
Y выбирают из группы, состоящей из водорода, фосфорной кислоты, фосфорилхолина и фосфорилэтаноламина; и
каждый из Х1 и Х2 независимо означает насыщенный или ненасыщенный углеводород общей формулы II:
где m является целым числом от 2 до 19; и
Z выбирают из группы, состоящей из:
где W является кислородом; и
по меньшей мере один из X1 и Х2 включает Z, который не является
водородом; и
где каждый из R1, R2, R3, 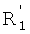 и
и 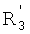 является водородом;
является водородом;
каждый из R'' и R''' независимо выбран из группы, состоящей из водорода и алкила;
каждый из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm независимо выбран из группы, состоящей из водорода, связи и гидроксигруппы; и
или его стереоизомера, оптического изомера, рацемической смеси,
фармацевтически приемлемой соли,
и фармацевтически приемлемый носитель,
причем указанная фармацевтическая композиция упакована в упаковочный материал и идентифицирована оттиском, сделанным в указанном упаковочном материале или на упаковочном материале, как предназначенная для применения при лечении заболевания или нарушения, при котором является благоприятным снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-6, интерлейкина-17, моноцитарного хемотактического белка-1 (МСР-1), моноцитарного хемотактического белка-3 (МСР-3) и фактора некроза опухоли-α (TNF-α).
9. Фармацевтическая композиция по любому из пп.1-8, содержащая соединение, где Х2 включает Z, не являющийся водородом.
10. Фармацевтическая композиция по п.9, где указанный Z выбран из
группы, состоящей из
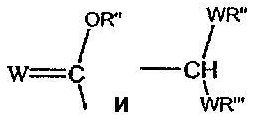
11. Фармацевтическая композиция по п.10, где Y является фосфорилхолином.
12. Фармацевтическая композиция по п.11, где В1 представляет собой кислород.
13. Фармацевтическая композиция по п.12, где Х2 является насыщенным углеводородом общей формулы II:
где m является целым числом 3;
Z является  , где W является кислородом; и
, где W является кислородом; и
каждый из R'', Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm является водородом.
14. Фармацевтическая композиция по п.13, где X1 является насыщенным углеводородом общей формулы II:
где m является целым числом от 11 до 15;
Z является Н; и
каждый из R'', Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm является водородом.
15. Фармацевтическая композиция по п.14, где указанное соединение выбрано из группы, состоящей из 1-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина, 1-додецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина и их фармацевтически приемлемой соли.
16. Фармацевтическая композиция по п.14, содержащая соединение формулы III:
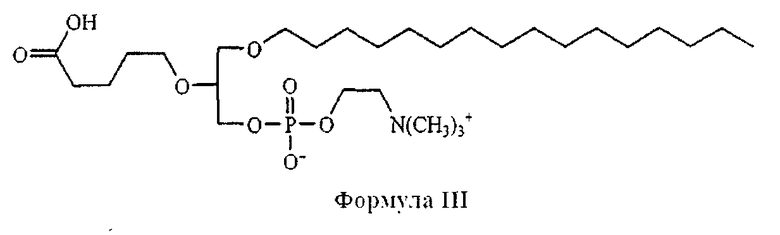
или его фармацевтически приемлемую соль.
17. Фармацевтическая композиция по любому из пп.1-8, содержащая соединение, выбранное из группы, состоящей из:
1-гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолина;
3-гексадецил-2-(5'-оксопентанил)-sn-глицеро-1-фосфохолина;
1-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина;
1-гексадецил-2-(6'-карбокси)гексанил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(6'-карбокси)гексанил-sn-глицеро-3-фосфоэтаноламина;
1-гексадецил-2-(3'-карбокси)пропил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(3'-карбокси)пропил-sn-глицеро-3-фосфоэтаноламина;
1-гексадецил-2-(4'-карбоксиметил)бутил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(5',5'-диэтоксипентил)-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(5',5'-диметоксипентил)-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(5',6'-дигидрокси)гексил-sn-глицеро-3-фосфохолина;
1-додецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфата;
1-гексадецил-2-(4'-карбокси)бутилглицерина;
1-(цис-9-гексадеценил)-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина;
1-(15'-карбокси)пентадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина;
1 -(15'-карбокси)пентадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина;
1-S-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина;
1-S-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина;
и их фармацевтически приемлемых солей.
18. Способ лечения или предупреждения воспаления, связанного с эндогенным окисленным липидом, причем указанное воспаление связано с заболеванием или нарушением, выбранным из группы, состоящей из воспалительного желудочно-кишечного заболевания или нарушения, пролиферативного заболевания или нарушения и диабета, причем способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества по меньшей мере одного окисленного липида общей формулы I:
где n является целым числом 3;
В1 является кислородом или серой;
каждый из В2 и В3 является кислородом;
каждый из А1 и А2 независимо выбирают из группы, состоящей из СН2 и C=О;
Y выбирают из группы, состоящей из водорода, фосфорной кислоты, фосфорилхолина и фосфорилэтаноламина; и
каждый из Х1 и Х2 независимо означает насыщенный или ненасыщенный углеводород общей формулы II:
где m является целым числом от 1 до 26; и
Z выбирают из группы, состоящей из:
где W является кислородом; и
по меньшей мере один из X1 и Х2 включает Z, не являющийся водородом; и
где каждый из R1, R2, R3, 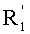 и
и 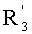 является водородом;
является водородом;
каждый из R'' и R''' независимо выбран из группы, состоящей из водорода и алкила;
каждый из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm независимо выбран из группы, состоящей из водорода, связи и гидроксигруппы;
или его стереоизомера, оптического изомера, рацемической смеси, фармацевтически приемлемой соли,
таким образом, осуществляя лечение или предупреждение у указанного субъекта воспалительного заболевания или нарушения, ассоциированного с эндогенным окисленным липидом.
19. Способ по п.18, где указанное воспалительное желудочно-кишечное заболевание или нарушение выбрано из группы, состоящей из колита, илеита, болезни Крона, хронического воспалительного заболевания тонкой кишки, синдрома воспаленной толстой кишки, хронического воспалительного заболевания толстой кишки, целиакии, язвенного колита, язвы, изъязвлений кожи, пролежней, язвы желудка, пептической язвы, язвы щеки, носоглоточной язвы, язвы пищевода, язвы двенадцатиперстной кишки и желудочно-кишечной язвы.
20. Способ по п.19, где указанное воспалительное желудочно-кишечное заболевание или нарушение выбрано из группы, состоящей из колита и хронического воспалительного заболевания толстого кишечника.
21. Способ по п.19, где указанное воспалительное желудочно-кишечное заболевание или нарушение выбрано из группы, состоящей из язвенного колита и болезни Крона.
22. Способ по п.18, где указанное пролиферативное заболевание или нарушение представляет собой злокачественную опухоль.
23. Способ по п.18, где указанное заболевание или нарушение представляет собой диабет.
24. Способ снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-6, интерлейкина-17, моноцитарного хемотактического белка-1 (МСР-1), моноцитарного хемотактического белка-3 (МСР-3) и фактора некроза опухоли-α (TNF-α) у субъекта, причем способ включает введение субъекту эффективного количества по меньшей мере одного окисленного липида общей формулы I:
где n является целым числом 3;
В1 является кислородом или серой;
каждый из В2 и В3 является кислородом;
каждый из А1 и А2 независимо выбирают из группы, состоящей из СН2 и С=O;
Y выбирают из группы, состоящей из водорода, фосфорной кислоты, фосфорилхолина и фосфорилэтаноламина; и
каждый из X1 и Х2 независимо означает насыщенный или ненасыщенный углеводород общей формулы II:
где m является целым числом от 1 до 26; и
Z выбирают из группы, состоящей из:
где W является кислородом; и
по меньшей мере один из X1 и Х2 включает Z, не являющийся водородом; и
где каждый из R1, R2, R3,
и 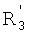 является водородом;
является водородом;
каждый из R'' и R''' независимо выбран из группы, состоящей из водорода и алкила;
каждый из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm независимо выбран из группы, состоящей из водорода, связи и гидроксигруппы;
или его стереоизомера, оптического изомера, рацемической смеси, фармацевтически приемлемой соли.
25. Способ лечения заболевания или нарушения, при котором является благоприятным снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-6, интерлейкина-17, моноцитарного хемотактического белка-1 (МСР-1), моноцитарного хемотактического белка-3 (МСР-3) и фактора некроза опухоли-α (TNF-α), включающий введение субъекту эффективного количества по меньшей мере одного окисленного липида общей формулы I:
где n является целым числом 3;
В1 является кислородом или серой;
каждый из В2 и В3 является кислородом;
каждый из А1 и А2 независимо выбирают из группы, состоящей из CH2 и С=O;
Y выбирают из группы, состоящей из водорода, фосфорной кислоты, фосфорилхолина и фосфорилэтаноламина; и
каждый из X1 и Х2 независимо означает насыщенный или ненасыщенный углеводород общей формулы II:
где m является целым числом от 1 до 26; и
Z выбирают из группы, состоящей из:
где W является кислородом; и
по меньшей мере один из X1 и Х2 включает Z, не являющийся водородом; и
где каждый из R1, R2, R3, 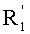 и
и 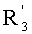 является водородом;
является водородом;
каждый из R'' и R''' независимо выбран из группы, состоящей из водорода и алкила;
каждый из Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm независимо выбран из группы, состоящей из водорода, связи и гидроксигруппы;
или его стереоизомера, оптического изомера, рацемической смеси, фармацевтически приемлемой соли.
26. Способ по любому из пп.18-25, где по меньшей мере один из А1 и А2 является СН2.
27. Способ по п.26, где А2 является СН2.
28. Способ по п.26, где А1 и А2 является СН2.
29. Способ по любому из пп.18-25, где Х2 включает Z, не являющийся водородом.
30. Способ по п.29, где указанный Z выбран из группы, состоящей из
31. Способ по п.30, где Y является фосфорилхолином.
32. Способ по п.31, где В1 является кислородом.
33. Способ по п.32, где Х2 является насыщенным углеводородом общей формулы II:
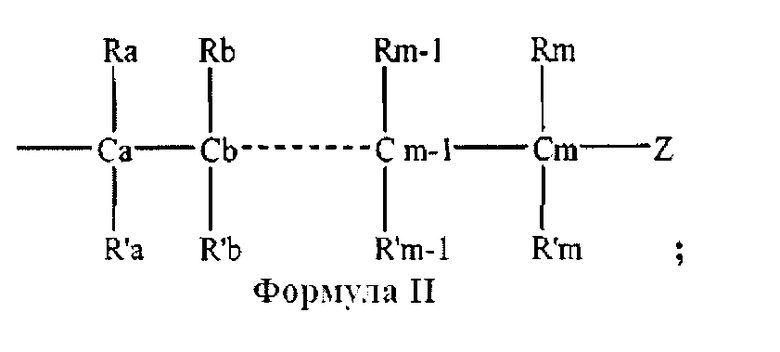
где m является целым числом 3;
Z является ,
где W является кислородом; и
каждый из R'', Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm является водородом.
34. Способ по п.33, где X1 является насыщенным углеводородом общей формулы II:
где m является целым числом от 11 до 15;
Z является Н; и
каждый из R'', Ra, R'a, Rb, R'b, …Rm-1, R'm-1, Rm и R'm является водородом.
35. Способ по п.34, где указанный окисленный липид выбран из группы, состоящей из 1-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина, 1-додецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина и их фармацевтически приемлемой соли.
36. Способ по п.34, где указанный окисленный липид представляет собой соединение формулы III:
или его фармацевтически приемлемую соль.
37. Способ по любому из пп.18-25, где указанный окисленный липид выбран из группы, состоящей из: 1-пальмитоил-2-азелаоил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-азелаоил-sn-глицеро-3-фосфохолина, 1-пальмитоил-2-глутароил-sn-глицеро-3-фосфохолина (PGPC), 1-пальмитоил-2-(5-оксовалероил)-sn-глицеро-3-фосфохолина (POVPC), 1-пальмитоил-2-(9-оксононаноил)-sn-глицеро-3-фосфохолина, 1-гексадецил-2-ацетоил-sn-глицеро-3-фосфохолина, 1-октадецил-2-ацетоил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-бутироил-sn-глицеро-3-фосфохолина, 1-октадецил-2-бутироил-sn-глицеро-3-фосфохолина, 1-пальмитоил-2-ацетоил-sn-глицеро-3-фосфохолина, 1-октадеценил-2-ацетоил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-(гомогаммалиноленоил)-sn-глицеро-3-фосфохолина, 1-гексадецил-2-арахидоноил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-эйкозапентаеноил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-докозагексаноил-sn-глицеро-3-фосфохолина, 1-октадецил-2-метил-sn-глицеро-3-фосфохолина, 1-гексадецил-2-бутеноил-sn-глицеро-3-фосфохолина, Lyso PAF С16, Lyso PAF С18, 1-О-1'-(Z)-гексадеценил-2-[12-[(7-нитро-2-1,3-бензоксадиазол-4-ил)амино]додеканоил]-sn-глицеро-3-фосфохолина, 1-О-1'-(Z)-гексадеценил-2-олеоил-sn-глицеро-3-фосфохолина, 1-О-1'-(Z)-гексадеценил-2-арахидоноил-sn-глицеро-3-фосфохолина, 1-O-1'-(Z)-гексадеценил-2-докозагексаеноил-sn-глицеро-3-фосфохолина, 1-O-1'-(Z)-гексадеценил-2-олеоил-sn-глицеро-3-фосфоэтаноламина, 1-O-1'-(Z)-гексадеценил-2-арахидоноил-sn-глицеро-3-фосфоэтаноламина, 1-О-1'-(Z)-гексадеценил-2-докозагексаеноил-sn-глицеро-3-фосфоэтаноламина, 1-S-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина и 1-S-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина.
38. Способ по п.18, который дополнительно включает введение указанному субъекту терапевтически эффективного количества по меньшей мере одного дополнительного соединения, способного осуществлять лечение или профилактику указанного воспаления, где указанным дополнительным соединением является ингибитор редуктазы HMGCoA (статин).
39. Способ по любому из пп.18-25, где указанный окисленный липид выбран из группы, состоящей из:
1-гексадецил-2-(5'-оксопентанил)-sn-глицеро-3-фосфохолина;
3-гексадецил-2-(5'-оксопентанил)-sn-глицеро-1-фосфохолина;
1-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина;
1-гексадецил-2-(6'-карбокси)гексанил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(6'-карбокси)гексанил-sn-глицеро-3-фосфоэтаноламина;
1-гексадецил-2-(3'-карбокси)пропил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(3'-карбокси)пропил-sn-глицеро-3-фосфоэтаноламина;
1-гексадецил-2-(4'-карбоксиметил)бутил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(5',5'-диэтоксипентил)-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(5',5'-диметоксипентил)-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(5',6'-дигидрокси)гексил-sn-глицеро-3-фосфохолина;
1-додецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина;
1-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфата;
1-гексадецил-2-(4'-карбокси)бутилглицерина;
1-(цис-9-гексадеценил)-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина;
1-(15'-карбокси)пентадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина;
1-(15'-карбокси)пентадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина;
1-S-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфохолина; 1-S-гексадецил-2-(4'-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина;
и их фармацевтически приемлемых солей.
| Способ получения производных глицерофосфолипидов | 1985 |
|
SU1400511A3 |
| US 5061626 А, 29.10.1991 | |||
| WATSON A D | |||
| еt al | |||
| “Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo." JOURNAL OF BIOLOGICAL CHEMISTRY, 1997, Vol: 272, Nr | |||
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |

