Изобретение относится к области химической технологии в пищевой промышленности, а именно к усовершенствованию способа переработки сивушных масел по ГОСТ 17071-91 производства этилового спирта на спиртоводочных комбинатах.
Сивушные масла являются побочным продуктом процесса производства этилового спирта и до настоящего времени подвергаются в основном лишь утилизации с целью освобождения аппаратуры спиртоводочных комбинатов.
Известны методы утилизации сивушных масел путем сжигания в топках в составе мазута (Климовский Д.Н., Смирнов В.Н. «Технология спирта» М. 1961) или путем использования в качестве сырья для выделения изоамилового спирта перегонкой на ректификационной установке (Патент РФ№2109724 от 23.05.2001, №2138476 от 25.06.98, №2196763 от 21.05.2001), а также отделением спиртов С5 и выше от воды методом экстракции углекислым газом в закритической области и ректификации (патент США №5284983 от 08.02.1994).
Прототипом изобретения служит патент РФ №2138476 от 25.06.98 (Гареев и др.) «Способ переработки сивушного масла». По данному способу процесс проводят на установке, состоящей из куба-испарителя, ректификационной колонны, оборудованной холодильником, флегмоделителем и сепаратором дистиллята, путем отгонки головной фракции сивушного масла до начала расслоения компонентов в сепараторе, азеотропной сушкой содержимого куба-испарителя и перегонкой оставшейся массы с выделением технического изоамилового спирта или продукта высшего качества.
Недостатком способа-прототипа является то, что осушенным получается лишь один продукт - изоамиловый спирт, а остальные компоненты (этиловый, пропиловые, бутиловые спирты и др.) - обводненными, что затрудняет или делает невозможным их квалифицированное использование. Известная рекомендация утилизировать их в смеси с топочным мазутом является нерациональной, поскольку в топочном мазуте эти компоненты также нерастворимы и качество мазута от этого ухудшается.
Задачей, на решение которой направлено заявляемое изобретение, является устранение указанного недостатка, а именно создание такой технологии переработки сивушных масел, которая позволила бы на 100% квалифицированно использовать все содержащиеся в них компоненты.
Поставленная задача решается тем, что в способе переработки сивушных масел спиртоводочных комбинатов путем их перегонки, в отличие от прототипа предварительно обезвоживают сивушные масла перегонкой с низкокипящим углеводородным растворителем, образующим с водой азеотропную смесь с последующим ее фракционированием на товарные продукты.
Поставленная задача решается также тем, что в качестве низкокипящего углеводородного растворителя используется н-гексан в количестве 0,03-0,07 м3 на 1 м3 сивушных масел.
Кроме того, для решения поставленной задачи после обезвоживания сивушных масел осуществляют методом четкой ректификации выделение изоамилового спирта чистотой более 99%.
При анализе патентной и научно-технической литературы авторами не обнаружено подобного заявляемому способа переработки сивушных масел, что позволяет сделать вывод о соответствии заявляемого способа критерию «новизна». Вместе с тем данное решение не является очевидным для среднего специалиста в данной области, что позволяет сделать вывод о соответствии критерию «изобретательский уровень». Действительно, ранее н-гексан применялся лишь для азеотропной отгонки воды из смесей ароматических углеводородов и нефетепродуктов, в которых вода практически не растворяется или растворяется ограниченно. Растворимость воды в бензоле - 0,05% (26°С), а в изоамиловом спирте на порядок выше - 0,1% (26°С), поэтому предположить, что н-гексан будет способен высушивать сивушные спирты, было невозможно. Однако мы впервые показали, что н-гексан способен в условиях азеотропной отгонки высушивать сивушные спирты.
На чертеже изображена общая схема установки переработки сивушных масел, работающая в периодическом режиме.
Установка состоит из емкости сырья (1) и емкости для вспомогательного углеводородного растворителя (н-гексан) (2), ректификационной колонны (3) с кубом (4), конденсатора-холодильника дистиллята (5), емкости-сепаратора дистиллята (6), флегмоделителя (7), дополнительной емкости (8), емкостей для сбора водного (9), углеводородных слоев, выделяемых последовательно фракций после осушки сырья из сепаратора (10, 11, 12) и емкости для кубового остатка (13).
Процесс осуществляется следующим образом: куб (4) объемом 2 м3 колонны, оборудованный подогревателем, заполняется сырьем (1 м3) и 0,05 м3 углеводородного растворителя в дополнительную емкость 8, затем последовательно фракции спиртов С2-С4 в емкость (10), изоамиловый спирт в емкость (11) и более высококипящие фракции в емкость (12).
Кубовый остаток после охлаждения и сливается в емкость (13).
Регенерированный углеводородный растворитель из дополнительной емкости (8) может быть использован при последующих разгонках вместо свежего, фракции дистиллята из емкостей (10, 11, 12) могут быть использованы в парфюмерной, лакокрасочной промышленности и как смесевой компонент в автомобильных бензинах. Кубовый остаток из емкости (13) используется как котельное топливо.
Установка переработки сивушных масел может быть спроектирована для работы и в непрерывном режиме.
Благодаря предложенному способу появляется возможность получать дополнительные товарные продукты, которые в зависимости от пределов вскипания могут быть использованы в лакокрасочной, парфюмерной промышленности, в производстве товаров народного потребления, а также как смесевой компонент автомобильных бензинов, полностью совместимый с ними и повышающий октановое число.
Пример 1
В куб колонны 4 объемом 2 м3, оборудованный подогревателем, залили 1 м3 сырья и 0,02 м3 н-гексана. Включили подачу охлаждающей воды в конденсатор и теплоносителя в обогреватель куба. Нагрели содержимое куба до начала прохождения паров через ректификационную колонну 3 (60°С), откуда пары с температурой 52°С поступают в конденсатор-холодильник 5, конденсат стекает в сепаратор 6, где расслаивается на водную и углеводородную фазы. Водная фаза накапливается в сепараторе, а углеводородная поступает в колонну 3 на орошение. Флегмоделитель 7 при этом работает только на колонну. Избыток водной фазы из сепаратора 6 отводим в емкость 9. Ректификацию начинаем при достижении температуры 68°С.
Водную фазу из сепаратора 6 после этого полностью сливаем в емкость 9, а углеводородную фазу на проход подаем в флегмоделитель 7, устанавливаем режим орошения для разделения компонентов. Вначале отбираем углеводородный растворитель в дополнительную емкость 8, затем последовательно фракции спиртов С2-С4 в емкость (10), изоамиловый спирт в емкость (11) и более высококипящие фракции в емкость (12). Кубовый остаток после охлаждения сливаем в емкость (13).
Анализ чистоты продукта проводился сравнением со стандартным образцом конечного продукта методом газожидкостной хроматографии на приборе Chrom-5 (длина колонки 1,2 м, неподвижная фаза - силикон SE-30 (5%), на Chromaton N-AW-DMCS (0.16-0.20 мм); рабочая температура 50-300°С, газ-носитель - гелий.
В данном примере чистота конечного продукта составила 82%.
Пример 2
В куб колонны 4 объемом 2 м3, оборудованный подогревателем, залили 1 м3 сырья и 0,03 м3 н-гексана. Включили подачу охлаждающей воды в конденсатор и теплоносителя в обогреватель куба. Нагрели содержимое куба до начала прохождения паров через ректификационную колонну 3 (60°С), откуда пары с температурой 52°С поступают в конденсатор-холодильник 5, конденсат стекает в сепаратор 6, где расслаивается на водную и углеводородную фазы. Водная фаза накапливается в сепараторе, а углеводородная поступает в колонну 3 на орошение. Флегмоделитель 7 при этом работает только на колонну. Избыток водной фазы из сепаратора 6 отводим в емкость 9. Ректификацию начинаем при достижении температуры 68°С.
Водную фазу из сепаратора 6 после этого полностью сливаем в емкость 9, а углеводородную фазу на проход подаем в флегмоделитель 7, устанавливаем режим орошения для разделения компонентов. Вначале отбираем углеводородный растворитель в дополнительную емкость 8, затем последовательно фракции спиртов С2-С4 в емкость (10), изоамиловый спирт в емкость (11) и более высококипящие фракции в емкость (12). Кубовый остаток после охлаждения сливаем в емкость (13).
Анализ чистоты продукта проводился сравнением со стандартным образцом конечного продукта методом газожидкостной хроматографии на приборе Chrom-5 (длина колонки 1,2 м, неподвижная фаза - силикон SE-30 (5%), на Chromaton N-AW-DMCS (0.16-0.20 мм); рабочая температура 50-300°С, газ-носитель - гелий.
В данном примере чистота конечного продукта составила 92%.
Пример 3
В куб колонны 4 объемом 2 м3, оборудованный подогревателем, залили 1 м3 сырья и 0,05 м3 н-гексана. Включили подачу охлаждающей воды в конденсатор и теплоносителя в обогреватель куба. Нагрели содержимое куба до начала прохождения паров через ректификационную колонну 3 (60°С), откуда пары с температурой 52°С поступают в конденсатор-холодильник 5, конденсат стекает в сепаратор 6, где расслаивается на водную и углеводородную фазы. Водная фаза накапливается в сепараторе, а углеводородная поступает в колонну 3 на орошение. Флегмоделитель 7 при этом работает только на колонну. Избыток водной фазы из сепаратора 6 отводим в емкость 9. Ректификацию начинаем при достижении температуры 68°С.
Водную фазу из сепаратора 6 после этого полностью сливаем в емкость 9, а углеводородную фазу на проход подаем в флегмоделитель 7, устанавливаем режим орошения для разделения компонентов. Вначале отбираем углеводородный растворитель в дополнительную емкость 8, затем последовательно фракции спиртов С2-С4 в емкость (10), изоамиловый спирт в емкость (11) и более высококипящие фракции в емкость (12). Кубовый остаток после охлаждения сливаем в емкость (13).
Анализ чистоты продукта проводился сравнением со стандартным образцом конечного продукта методом газожидкостной хроматографии на приборе Chrom-5 (длина колонки 1,2 м, неподвижная фаза - силикон SE-30 (5%), на Chromaton N-AW-DMCS (0.16-0.20 мм); рабочая температура 50-300°С, газ-носитель - гелий.
В данном примере чистота конечного продукта составила 99%.
Пример 4
В куб колонны 4 объемом 2 м3, оборудованный подогревателем, залили 1 м3 сырья и 0,07 м3 н-гексана. Включили подачу охлаждающей воды в конденсатор и теплоносителя в обогреватель куба. Нагрели содержимое куба до начала прохождения паров через ректификационную колонну 3 (60°С), откуда пары с температурой 52°С поступают в конденсатор-холодильник 5, конденсат стекает в сепаратор 6, где расслаивается на водную и углеводородную фазы. Водная фаза накапливается в сепараторе, а углеводородная поступает в колонну 3 на орошение. Флегмоделитель 7 при этом работает только на колонну. Избыток водной фазы из сепаратора 6 отводим в емкость 9. Ректификацию начинаем при достижении температуры 68°С.
Водную фазу из сепаратора 6 после этого полностью сливаем в емкость 9, а углеводородную фазу на проход подаем в флегмоделитель 7, устанавливаем режим орошения для разделения компонентов. Вначале отбираем углеводородный растворитель в дополнительную емкость 8, затем последовательно фракции спиртов С2-C4 в емкость (10), изоамиловый спирт в емкость (11) и более высококипящие фракции в емкость (12). Кубовый остаток после охлаждения сливаем в емкость (13).
Анализ чистоты продукта проводился сравнением со стандартным образцом конечного продукта методом газожидкостной хроматографии на приборе Chrom-5 (длина колонки 1,2 м, неподвижная фаза - силикон SE-30 (5%), на Chromaton N-AW-DMCS (0.16-0.20 мм); рабочая температура 50-300°С, газ-носитель - гелий.
В данном примере чистота конечного продукта составила 99%.
Пример 5
В куб колонны 4 объемом 2 м3, оборудованный подогревателем, залили 1 м3 сырья и 0,08 м3 н-гексана. Включили подачу охлаждающей воды в конденсатор и теплоносителя в обогреватель куба. Нагрели содержимое куба до начала прохождения паров через ректификационную колонну 3 (60°С), откуда пары с температурой 52°С поступают в конденсатор-холодильник 5, конденсат стекает в сепаратор 6, где расслаивается на водную и углеводородную фазы. Водная фаза накапливается в сепараторе, а углеводородная поступает в колонну 3 на орошение. Флегмоделитель 7 при этом работает только на колонну. Избыток водной фазы из сепаратора 6 отводим в емкость 9. Ректификацию начинаем при достижении температуры 68°С.
Водную фазу из сепаратора 6 после этого полностью сливаем в емкость 9, а углеводородную фазу на проход подаем в флегмоделитель 7, устанавливаем режим орошения для разделения компонентов. Вначале отбираем углеводородный растворитель в дополнительную емкость 8, затем последовательно фракции спиртов С2-С4 в емкость (10), изоамиловый спирт в емкость (11) и более высококипящие фракции в емкость (12). Кубовый остаток после охлаждения сливаем в емкость (13).
Анализ чистоты продукта проводился сравнением со стандартным образцом конечного продукта методом газожидкостной хроматографии на приборе Chrom-5 (длина колонки 1,2 м, неподвижная фаза - силикон SE-30 (5%), на Chromaton N-AW-DMCS (0.16-0.20 мм); рабочая температура 50-300°С, газ-носитель - гелий.
В данном примере чистота конечного продукта составила 99%.
Дальнейшее повышение расхода н-гексана нецелесообразно, так как это не ведет к повышению чистоты конечного продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРОИЗВОДСТВА СПИРТА ЭТИЛОВОГО РЕКТИФИКОВАННОГО "АЛЬФА" | 2007 |
|
RU2366711C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОАМИЛОВОГО СПИРТА (ВАРИАНТЫ) | 2001 |
|
RU2196763C1 |
| СПОСОБ ПЕРЕРАБОТКИ ПРОПЕНСОДЕРЖАЩЕЙ УГЛЕВОДОРОДНОЙ СМЕСИ | 2004 |
|
RU2264379C1 |
| СПОСОБ ПЕРЕРАБОТКИ СИВУШНОГО МАСЛА | 1998 |
|
RU2138476C1 |
| НИТРОРАСТВОРИТЕЛЬ, РАСТВОРИТЕЛЬ ОБЕЗЖИРИВАЮЩИЙ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2004 |
|
RU2270184C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ИЗОАМИЛОВОГО СПИРТА | 1996 |
|
RU2109724C1 |
| Способ выделения пропионитрила из продуктов гидроформилирования акрилонитрила | 1978 |
|
SU883019A1 |
| УНИВЕРСАЛЬНАЯ УСТАНОВКА ДЛЯ ОЧИСТКИ РЕКТИФИКАЦИЕЙ РАСТВОРИТЕЛЕЙ, ОТНОСЯЩИХСЯ К ОСНОВНЫМ КЛАССАМ ОРГАНИЧЕСКИХ РАСТВОРИТЕЛЕЙ, И СПОСОБЫ ОЧИСТКИ РЕКТИФИКАЦИЕЙ НА НЕЙ АЦЕТОНА, ИЗОПРОПИЛОВОГО СПИРТА, БЕНЗОЛА, ТОЛУОЛА, Н-БУТАНОЛА, ИЗОБУТАНОЛА, ЭТИЛАЦЕТАТА, Н-БУТИЛАЦЕТАТА И ЦИКЛОГЕКСАНА | 2004 |
|
RU2264840C1 |
| СПОСОБ РЕКТИФИКАЦИИ СПИРТОВОЙ БРАЖКИ | 1993 |
|
RU2106176C1 |
| СПОСОБ ПОЛУЧЕНИЯ РЕКТИФИКОВАННОГО СПИРТА | 2009 |
|
RU2421522C1 |
Изобретение относится к способу переработки сивушных масел спиртоводочных комбинатов путем их перегонки. Способ характеризуется тем, что осуществляют предварительное обезвоживание сивушных масел перегонкой с низкокипящим углеводородным растворителем, образующим с водой азеотропную смесь, с последующим ее фракционированием на товарные продукты, причем в качестве низкокипящего углеводородного растворителя используется н-гексан в количестве 0,03-0,07 м3 на 1 м3 сивушных масел. Предлагаемый способ предоставляет технологию переработки сивушных масел, которая позволила бы на 100% квалифицированно использовать все содержащиеся в них компоненты. 1 з.п. ф-лы, 5 пр., 1 ил.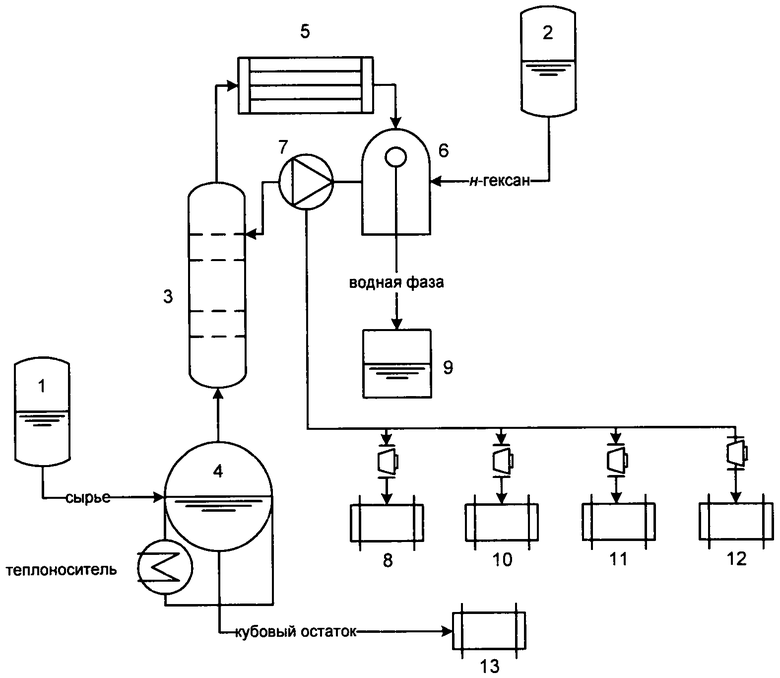
1. Способ переработки сивушных масел спиртоводочных комбинатов путем их перегонки, отличающийся тем, что осуществляют предварительное обезвоживание сивушных масел перегонкой с низкокипящим углеводородным растворителем, образующим с водой азеотропную смесь, с последующим ее фракционированием на товарные продукты, причем в качестве низкокипящего углеводородного растворителя используется н-гексан в количестве 0,03-0,07 м3 на 1 м3 сивушных масел.
2. Способ по п.1, отличающийся тем, что после обезвоживания сивушных масел осуществляют методом четкой ректификации выделение изоамилового спирта чистотой более 99%.
| Способ обезвоживания сивушного масла | 1951 |
|
SU94565A2 |
| Способ обезвоживания сивушного масла | 1950 |
|
SU90409A1 |
| СПОСОБ ПЕРЕРАБОТКИ СИВУШНОГО МАСЛА | 1998 |
|
RU2138476C1 |
| СПОСОБ ПЕРЕРАБОТКИ СИВУШНОГО МАСЛА | 2001 |
|
RU2183617C2 |

