Настоящее изобретение относится к кристаллической форме и фармацевтическим составам, а также к применению кристаллической формы.
Фермент млекопитающих поли-(АДФ-рибоза)-полимераза (PARP) (113 кДа мультидоменный белок) является частью системы передачи сигналов, связанных с повреждением ДНК, благодаря своей способности распознавать и быстро связываться с однонитевыми или двунитевыми разрывами ДНК (D'Amours, et al., Biochem. J., 342, 249-268 (1999)).
Ряд наблюдений позволил прийти к заключению, что PARP принимает участие в реализации разнообразных функций, связанных с ДНК, включая амплификацию гена, деление клетки, дифференцировку, апоптоз, эксцизионную репарацию ДНК, а также оказывает влияние на длину теломер и стабильность хромосомы (d'Adda di Fagagna, et al., Nature Gen., 23(1), 76-80 (1999)).
В ходе исследований механизма модуляции ферментом PARP репарации ДНК и других процессов, была выявлена его важность для формирования цепей поли-(АДФ-рибоза) в ядре клетки (Althaus, F.R. and Richter, С., ADP-Ribosylation of Proteins: Enzymology and Biological Significance, Springer-Verlag, Berlin (1987)). Связанная с ДНК, активированная PARP использует никотинамид-аденин-динуклеотид (NAD) для синтеза поли-(АДФ-рибозы) в различных целевых ядерных белках, включая топоизомеразу, гистоны и саму PARP (Rhun, et al., Biochem. Biophys. Res. Commun., 245, 1-10 (1998)).
Поли-АДФ-рибозилирование также связывают со злокачественной трансформацией. Например, активность PARP является более высокой в изолированных ядрах фибробластов, трансформированных SV40, а как лейкозные клетки и клетки опухоли ободочной кишки демонстрируют более высокую ферментативную активность, чем эквивалентные нормальные лейкоциты и слизистая оболочка ободочной кишки (Miwa, et al., Arch. Biochem. Biophys., 181, 313-321 (1977); Burzio, et al., Proc. Soc. Exp. Biol. Med., 149, 933-938 (1975); and Hirai, et al., Cancer Res., 43, 3441-3446 (1983)). Для выяснения функциональной роли поли-АДФ-рибозилирования в репарации ДНК использовали ряд низкомолекулярных ингибиторов PARP. В клетках, обработанных алкилирующими агентами, ингибирование PARP приводит к явному росту разрывов нити ДНК и лизису клеток (Durkacz, et al., Nature, 283, 593-596 (1980); Berger, N.A., Radiation Research, 101, 4-14 (1985)).
Позднее было показано, что такие ингибиторы усиливают эффект ответа на облучение посредством подавления репарации потенциально летального повреждения (Ben-Hur, et al., British Journal of Cancer, 49 (Suppl. VI), 34-42 (1984); Schlicker, et al., Int. J. Radiat. Biol., 75, 91-100 (1999)). Сообщалось, что ингибиторы PARP эффективны для чувствительных к облучению, гипоксических опухолевых клеток (US 5,032,617; US 5,215,738 и US 5,041,653).
Кроме того, животные с нокаутом по PARP (PARP-I-) демонстрируют нестабильность генома в ответ на алкилирующие агенты и γ-облучение (Wang, et al., Genes Dev., 9, 509-520 (1995); Menissier de Murcia, et al., Proc. Natl. Acad. Sci. USA, 94, 7303-7307 (1997)).
Также была продемонстрирована роль PARP в некоторых сосудистых заболеваниях, септическом шоке, ишемическом повреждении и нейротоксичности (Cantoni, et al., Biochim. Biophys. Acta, 1014, 1-7 (1989); Szabo, et al., J. Clin. Invest., 100, 723-735 (1997)). Как было показано в исследованиях ингибиторов PARP, повреждение ДНК кислородным радикалом, приводящее к разрывам нитей ДНК, которые впоследствии распознаются PARP, является главным фактором, способствующим развитию таких болезненных состояний (Cosi, et al., J. Neurosci. Res., 39, 38-46 (1994); Said, et al., Proc. Natl. Acad. Sci. U.S.A., 93, 4688-4692 (1996)). Совсем недавно было продемонстрировано, что PARP участвует в патогенезе геморрагического шока (Liaudet, et al., Proc. Natl. Acad. Sci. U.S.A., 97(3), 10203-10208 (2000)).
Также было продемонстрировано, что ингибирование активности PARP приводит к блокированию эффективной ретровирусной инфекции клеток млекопитающих. Было показано, что такое ингибирование рекомбинантных ретровирусных векторных инфекций встречается в разнообразных различных типах клеток (Gaken, et al., J. Virology, 70(6), 3992-4000 (1996)). Были разработаны ингибиторы PARP для использования в противовирусной терапии и при лечении рака (WO 91/18591).
Кроме того, было сделано предположение о том, что ингибирование PARP задерживает начало процесса старения в фибробластах человека (Rattan and dark, Biochem. Biophys. Res. Comm., 201(2), 665-672 (1994)). Это может быть связано с ролью, которую PARP играет в контроле функции теломер (d'Adda di Fagagna, et al., Nature Gen., 23(1), 76-80 (1999)).
В публикации WO 2004/080976 описан ряд производных фталазинона, их активность в ингибировании PARP и основанное на указанной активности использование в лечении рака в качестве дополнения к радиотерапии или химиотерапии или в качестве отдельного средства.
В публикации WO 2005/053662 описано применение ингибиторов PARP, в частности, производных фталазинона в качестве ингибиторов основной эксцизионной репарации (BER). Описано применение этих ингибиторов в производстве медикаментов для лечения злокачественных опухолей с дефицитом репарации двунитевых разрывов ДНК на основе гомологичной рекомбинации (HR), в частности, описано лечение злокачественных опухолей, имеющих дефицитный фенотип BRCA1 и/или BRCA2.
4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-он (соединение А), раскрытый в WO 2004/080976:
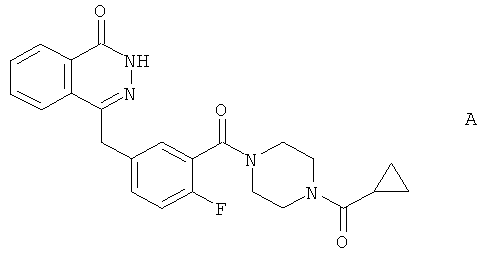
представляет особый интерес.
Кристаллическая форма соединения А (Форма А) описана в одновременно рассматриваемых заявках, которые испрашивают приоритет по заявке US 60/829,694, поданной 17 октября 2006 г., под названием «Phthalazinone Derivative» («Производное фталазинона»), включая US 11/873,671 и WO 2008/047082.
Конкретные формы соединения А могут обладать полезными свойствами, например, в отношении растворимости и/или стабильности и/или биодоступности и/или состава примесей и/или фильтрационных свойств и/или характеристик осушки и/или отсутствия гигроскопичности, и/или они могут обеспечивать более простое обращение и/или тонкое измельчение и/или таблетирование.
Соответственно, первый аспект настоящего изобретения обеспечивает 4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-он (соединение А) по существу в виде кристаллической Формы L.
Использованный выше термин «По существу в виде кристаллической Формы L» означает, что минимум 50% по весу соединения А представлено в виде Формы L, предпочтительно минимум 70%, 80% или 90% по весу. В некоторых случаях кристаллическая форма может составлять минимум 95% по весу, 99% или даже 99,5% по весу или более.
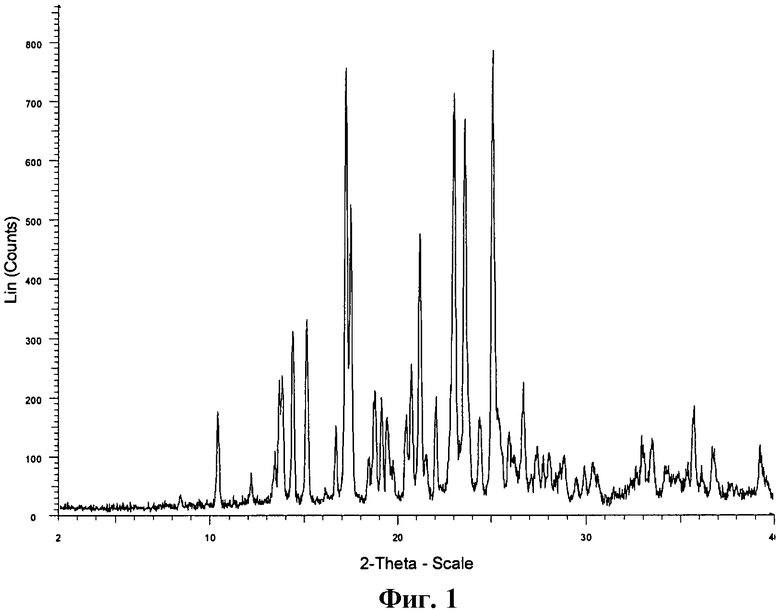
Соединение А в виде кристаллической Формы L имеет рентгеновскую дифрактограмму (λ=1.5418Е) со следующими характерными пиками:
Рентгеновская дифрактограмма Соединения А в виде кристаллической Формы L также может включать следующие дополнительные пики (λ=1.5418Е):
Соединение А в виде кристаллической Формы L также может характеризоваться любой комбинацией трех или более пиков, выбранных из списка 8 пиков выше.
Типичная порошковая рентгеновская дифрактограмма (XRD) соединения А в Форме L показана на Фиг.1.
Форма L соединения А по существу не содержит растворителя. Формулировка «в основном не содержит растворителя» в настоящем описании относится к форме, содержащей только незначительные количества какого-либо растворителя, например, к форме с общим количеством какого-либо растворителя 0,5% по весу или менее. Общее количество какого-либо растворителя может быть 0.25%, 0,1%, 0,05% или 0,025% по весу или менее.
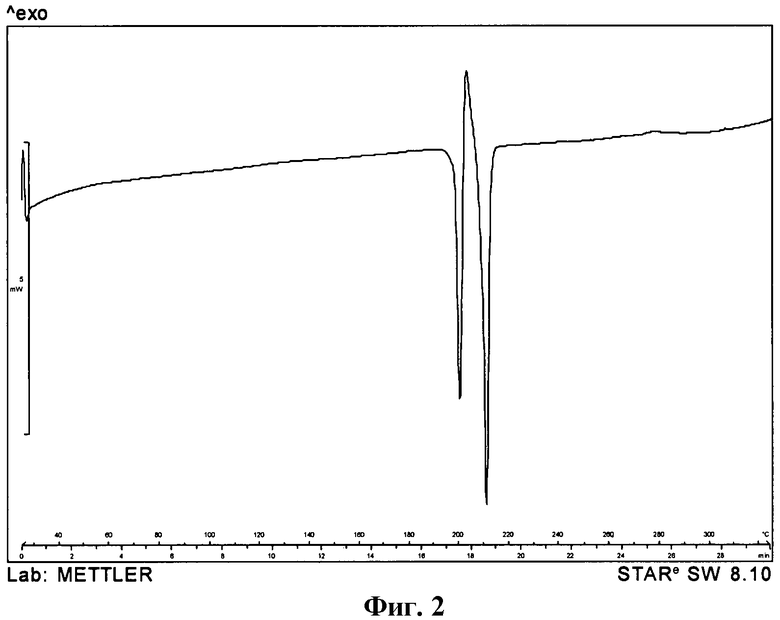
Форма L соединения А также может быть охарактеризована с использованием дифференциальной сканирующей калориметрии (DSC). Начало процесса плавления формы L соединения А при нагревании от 25°С до 325°С со скоростью 10°С в минуту будет соответствовать 198,5°С±1°С. Типичная кривая дифференциальной сканирующей калориметрии (DSC) соединения А в виде Формы L показана на Фиг.2. Вторая эндотерма соответствует плавлению Формы А, в которую превращается расплавленная Форма L.
Второй аспект настоящего изобретения обеспечивает способ получения 4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-она (соединение А) в виде кристаллической Формы L из соединения А, представленного в виде кристаллической Формы А.
Указанный способ включает суспендирование соединения А в виде Формы А или смеси Форм А и L в органическом растворителе, который может содержать до 30% воды в объемном отношении. В некоторых случаях смесь может не содержать Формы L или содержать 99%, 90% или 80% Формы А. В других случаях количество Формы L может достигать 50, 70 или даже 80% смеси. В некоторых случаях органический растворитель может не содержать воду. В других случаях органический растворитель может содержать до 25 или 20% воды. Растворитель можно выбрать из растворителя, который обеспечивает достаточную растворимость для превращения. В некоторых случаях растворитель выбирают из группы, включающей метанол, этанол, ацетон, ацетонитрил и нитрометан.
Суспендирование может быть осуществлено при температуре до точки кипения растворителя, но обычно его осуществляют при более низкой температуре. В некоторых случаях суспендирование осуществляют при температуре в диапазоне между 20°С и 50°С или даже 20°С и 40°С или 20°С и 30°С. При более низких температурах время суспендирования может быть увеличено. Как правило, суспендирование выполняют в течение, по меньшей мере, 3 часов, 6 часов или даже 24 часов. В некоторых случаях суспендирование осуществляют в течение минимум 3 дней, 4 дней, недели или даже 3 недель.
Третий аспект настоящего изобретения обеспечивает фармацевтический состав, включающий соединение согласно первому аспекту и фармацевтически приемлемый носитель или разбавитель.
Четвертый аспект настоящего изобретения обеспечивает соединение согласно первому аспекту для применения в лечении человека или животных.
Пятый аспект настоящего изобретения обеспечивает применение соединения согласно первому аспекту изобретения в изготовлении лекарственного средства для ингибирования активности PARP.
Другие аспекты изобретения обеспечивают применение соединения согласно первому аспекту настоящего изобретения в изготовлении лекарственного средства для лечения следующих заболеваний: заболевания сосудов; септический шок; ишемическое поражение; нейротоксичность; геморрагический шок; вирусная инфекция или заболевания, выраженность которых может быть снижена путем ингибирования активности PARP.
Другой аспект изобретения обеспечивает применение соединения согласно первому аспекту настоящего изобретения в изготовлении лекарственного средства для использования в качестве вспомогательного средства в терапии рака или для для повышения чувствительности раковых клеток к обработке ионизирующим излучением или химиотерапевтическими средствами.
Другие дальнейшие аспекты изобретения предусматривают лечение заболеваний путем ингибирования PARP, включающее введение субъекту, нуждающемуся в лечении, терапевтически эффективного количества соединения согласно первому аспекту предпочтительно в форме фармацевтического состава, и лечение рака, включающее введение субъекту, нуждающемуся в лечении, терапевтически эффективного количества соединения согласно первому аспекту в комбинации, предпочтительно в форме фармацевтического состава, одновременно или последовательно с ионизирующим облучением или химиотерапевтическими средствами.
В дальнейших аспектах настоящего изобретения соединения можно применять в изготовлении лекарственного средства для лечения рака, характеризующегося дефицитом репарации двухнитевых разрывов ДНК, по механизму гомологичной рекомбинации (HR), или для лечения пациента, больного раком, характеризующимся дефицитом репарации двухнитевых разрывов ДНК, по механизму гомологичной рекомбинации (HR), включающего введение в организм данного пациента терапевтически эффективного количества соединения.
Путь репарации двухнитевых разрывов ДНК, зависимый от гомологичной рекомбинации (HR), устраняет двухнитевые разрывы (DSB) в ДНК посредством воссоздания непрерывной спирали ДНК по механизмам гомологии (K.K.Khanna and S.P.Jackson, Nat. Genet. 27(3): 247-254 (2001)). Компоненты пути репарации двухнитевых разрывов ДНК, по механизму гомологичной рекомбинации (HR), включают, но не ограничиваются, ATM (NM_000051), RAD51 (NM_002875), RAD51L1 (NM_002877), RAD51C (NM_002876), RAD51L3 (NM_002878), DMC1 (NM_007068), XRCC2 (NM_005431), XRCC3 (NM_005432), RAD52 (NM_002879), RAD54L (NM_003579), RAD54B (NM_012415), BRCA1 (NM_007295), BRCA2 (NM_000059), RAD50 (NM_005732), MRE11A (NM_005590) и NBS1 (NM_002485). Другие белки, участвующие в репарации двухнитевых разрывов ДНК на основе гомологичной рекомбинации (HR), включают регуляторные факторы, такие как EMSY (Hughes-Davies, et al., Cell, 115, pp.523-535). Компоненты HR также описаны в Wood, et al., Science, 291, 1284-1289 (2001).
Рак с дефицитом репарации двухнитевых разрывов ДНК, по механизму гомологичной рекомбинации (HR), может включать или состоять из одной или более раковых клеток, у которых снижена или отсутствует способность к репарации двухнитевых разрывов ДНК по такому пути в сравнении с нормальными клетамик, то есть активность пути репарации двухнитевых разрывов ДНК, по механизму гомологичной рекомбинации (HR), может быть снижена или отсутствовать в одной или большем количестве раковых клеток.
Активность одного или более компонентов пути репарации двухнитевых разрывов ДНК, по механизму гомологичной рекомбинации (HR), может отсутствовать в одной или большем количестве раковых клеток пациента, больного раком, характеризующимся дефицитом репарации двухнитевых разрывов ДНК, по механизму гомологичной рекомбинации (HR). Компоненты пути репарации двухнитевых разрывов ДНК, по механизму гомологичной рекомбинации (HR), хорошо описаны в специализированной литературе (см. например, Wood, et al., Science, 291, 1284-1289 (2001)) и включают упомянутые выше компоненты.
В некоторых предпочтительных случаях раковые клетки могут иметь дефицитный фенотип BRCA1 и/или BRCA2, то есть в таких раковых клетках активность BRCA1 и/или BRCA2 снижена или отсутствует. Раковые клетки с таким фенотипом могут быть дефицитными по BRCA1 и/или BRCA2, то есть в раковых клетках экспрессия и/или активность BRCA1 и/или BRCA2 могут быть снижены или отсутствовать, например, в результате мутации или полиморфизма в кодирующей нуклеиновой кислоте или в результате амплификации, мутации или полиморфизма в гене, кодирующем регуляторный фактор, например, ген EMSY, который кодирует регуляторный фактор BRCA2 (Hughes-Davies, et al., Cell, 115, 523-535).
BRCA1 и BRCA2 являются известными супрессорами опухолей, аллели дикого типа которых часто отсутствуют в опухолях гетерозиготных носителей (Jasin М., Oncogene, 21(58), 8981-93 (2002); Tutt, et al., Trends Mol Med., 8(12), 571-6, (2002)). Связь мутаций BRCA1 и/или BRCA2 с раком молочной железы хорошо описана в специализированной литературе (Radice, P.J., Exp Clin Cancer Res., 21 (3 Suppl), 9-12 (2002)). Также известна связь между амплификацей гена EMSY, который кодирует фактор связывания BRCA2, и раком груди и яичников.
Носители мутаций в BRCA1 и/или BRCA2 также подвержены повышенному риску развития рака яичника, простаты и поджелудочной железы.
В некоторых предпочтительных случаях пациент является гетерозиготным по одному или более изменениям, таких как мутации и полиморфизм в BRCA1 и/или BRCA2 или какой-либо регулирующей последовательности этих генов. Детектирование изменений в BRCA1 и BRCA2 хорошо известно специалистам в данной области и описано, например, в ЕР 699754, ЕР 705903, Neuhausen, S.L. and Ostrander, E.A., Genet. Test, 1, 75-83 (1992); Chappnis, P.O. and Foulkes, W.D., Cancer Treat Res, 107, 29-59 (2002); Janatova М., et al., Neoplasma, 50(4), 246-50 (2003); Jancarkova, N., Ceska Gynekol., 68(1), 11-6 (2003)). Выявление амплификации фактора связывания BRCA2 EMSY описано в Hughes-Davies, et al., Cell, 115, 523-535.
Мутации и полиморфизм, ассоциированные с раком, могут быть обнаружены на уровне нуклеиновой кислоты посредством определения присутствия измененной последовательности нуклеиновой кислоты или на уровне белка посредством определения присутствия измененного полипептида (то есть мутантного или аллельного варианта).
Краткое описание рисунков
На Фиг.1 показана типичная дифрактограмма, полученная в результате анализа соединения А в виде Формы L методом рентгеновской дифракции в порошке (XRD).
На Фиг.2 показана типичная кривая дифференциальной сканирующей калориметрии (DSC) соединения А в виде Формы L, полученная посредством нагрева от 25°С до 325°С со скоростью 10°С в минуту.
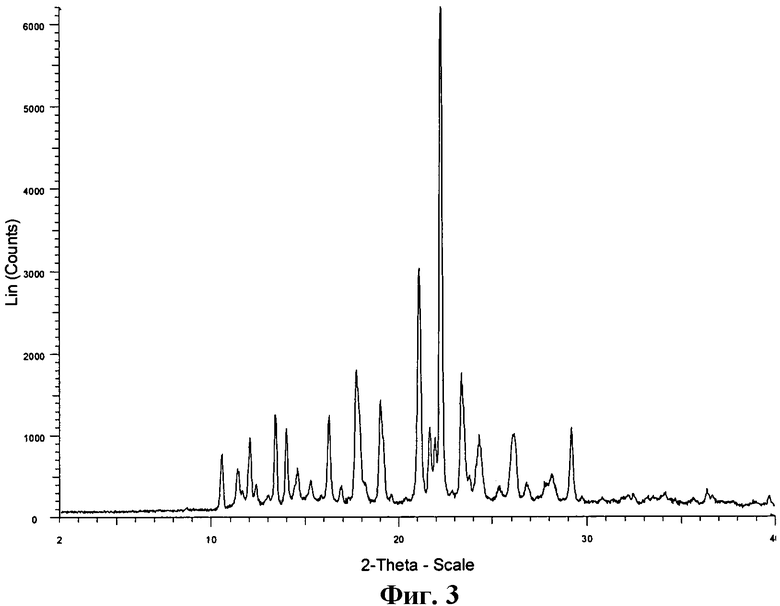
На Фиг.3 показана типичная дифрактограмма, полученная в результате анализа соединения А в виде Формы А методом рентгеновской дифракции в порошке (XRD).
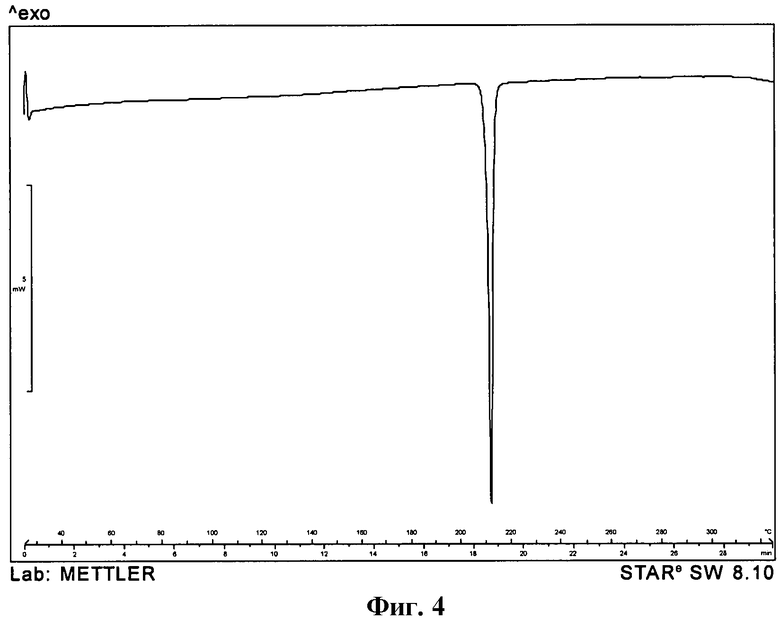
На Фиг.4 показана типичная кривая дифференциальной сканирующей калориметрии (DSC) соединения А в виде Формы А, полученная посредством нагрева от 25°С до 325°С со скоростью 10°С в минуту.
Применение
Согласно настоящему изобретению предложено соединение А в Форме L в виде активного соединения/в частности, способного к ингибированию активности PARP.
Термин «активное» в настоящем описании относится к соединению, обладающему способностью к ингибированию активности PARP. Один из видов анализа, который удобно использовать для оценки ингибирования PARP соединением, описан в примерах ниже.
Далее настоящее изобретение обеспечивает способ ингибирования активности PARP в клетке, включающий осуществление контакта данной клетки с эффективным количеством активного соединения, предпочтительно в форме фармацевтически приемлемого состава. Такой способ может быть реализован in vitro или in vivo.
Например, можно вырастить образец клеток in vitro, привести активное соединение в контакт с данными клетками и осуществлять наблюдение влияния соединения на эти клетки. В качестве иллюстрации «влияния» (эффекта) может быть определен объем репарации ДНК, произведенной в определенное время. В случае обнаружения влияния активного соединения на клетки, это можно использовать в качестве прогностического или диагностического маркера эффективности соединения в способах лечения пациента, несущего клетки того же клеточного типа.
Термин «лечение» в настоящем описании применительно к лечению состояния обычно относится к лечению и терапии либо человека, либо животного (например, при применении в ветеринарии), при котором достигают некоторого желаемого терапевтического эффекта, такого как подавление прогрессирования состояния, и включает снижение скорости прогрессирования (развития), остановку развития, снижение выраженности состояния и излечивание состояния. Также данный термин включает лечение как профилактическую меру (то есть профилактика).
Термин «вспомогательное средство» в настоящем описании относится к применению активного соединения совместно с известными терапевтическими средствами. Такие средства включают цитотоксические режимы лекарственных средств и/или ионизирующего облучения, как они используются при лечении различных типов рака. В частности, активные соединения, как известно, усиливают действие ряда химиотерапевтических средств лечения рака, которые охватывают класс топоизомераз ядов (например, топотекан, иринотекан, рубитекан), большинство известных алкилирующих агентов (например, DTIC, темозоламид) и препараты на основе платины (например, карбоплатин, цисплатин), используемые в лечении рака.
Активное соединение можно также применять в качестве добавки к клеточной культуре для ингибирования PARP, например, с целью повышения чувствительности клеток к известным химиотерапевтическим средствам или лечению ионизирующим облучением in vitro.
Активное соединение можно также применять в тесте in vitro, например, с целью определения, получит ли предполагаемый реципиент пользу от лечения рассматриваемым соединением.
Введение
Активное соединение или фармацевтический состав, содержащий активное соединение, можно вводить субъекту любым удобным путем введения, системно/периферически или в место желаемого действия, в том числе включая, в частности, но без ограничения, пероральное (например, путем проглатывания); топическое (в том числе, например, трансдермальное, интраназальное, введение в глаз, буккальное и сублингвальное); через легкие (например, при помощи ингаляционной или инсуффляционной терапии, с использованием, в частности, аэрозоля, например, через рот или нос); ректальное; вагинальное; парентеральное, например, путем инъекции, в том числе подкожной, интрадермальной, внутримышечной, внутривенной, внутриартериальной, интракардиальной, интратекальной, интраспинальной, интракапсулярной, субкапсулярной, в глазницу, интраперитонеальной, интратрахеальной, субкутикулярной, внутрисоставной, субарахноидальной и интрастернальной; с путем имплантирования депо, например, подкожно или внутримышечно.
Субъект может представлять собой эукариотический организм, животное, позвоночное животное, млекопитающее, грызуна (например, морская свинка, хомяк, крыса, мышь), животное семейства мышиных (например, мышь), представителя семейства псовых (например, собака), представителя семейства кошачьих (например, домашняя кошка), представителя семейства лошадиных (например, лошадь), примата, обезьяну (например, животное семейства мартышковых или человекообразных обезьян), животное семейства мартышковых (например, мармозетка, бабуин), животное семейства человекообразных обезьян (например, горилла, шимпанзе, орангутанг, гиббон) или человека.
Лекарственные формы
Активное соединение можно вводить отдельно, тем не менее, в предпочтительном случае оно имеет форму фармацевтического состава (например, лекарственная форма), содержащего активное соединение, определенное выше, совместно с одним или несколькими фармацевтически приемлемыми носителями, адъювантами, эксципиентами, разбавителями, наполнителями, буферами, стабилизаторами, консервантами, лубрикантами или другими материалами, хорошо известными специалистам в данной области, и, возможно, с другими терапевтическими или профилактическими агентами.
Таким образом, согласно настоящему изобретению предложены также фармацевтические составы, согласно приведенному выше определению, и способы получения фармацевтического состава, которые включают смешивание активного соединения, определенного выше, с одним или более фармацевтически приемлемыми носителями, эксципиентами, буферами, адъювантами, стабилизаторами или другими материалами согласно данному описанию так, чтобы активное соединение оставалось в виде кристаллической Формы L.
Термин «фармацевтически приемлемый» в данном описании относится к соединениям, материалам, составам и/или лекарственным формам, которые, согласно обоснованному медицинскому заключению, являются пригодными для применения в контакте с тканями субъекта (например, человека), не вызывая чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмерно с разумным отношением между выгодой и риском. При этом каждый носитель, эксципиент и т.д. также должен быть «приемлемым» в смысле совместимости с другими ингредиентами лекарственного средства.
Пригодные носители, эксципиенты и т.п. описаны в стандартной фармацевтической литературе. См., например, "Handbook of Pharmaceutical Additives", 2nd Edition (eds. M.Ash and I.Ash), 2001 (Synapse Information Resources, Inc., Endicott, New York, USA), "Remington's Pharmaceutical Sciences", 20th edition, pub. Lippincott, Williams & Wilkins, 2000; and "Handbook of Pharmaceutical Excipients", 2nd edition, 1994.
Лекарственные средства могут быть удобно представлены в дозированной лекарственной форме и получены способами, которые хорошо известны специалистам в области фармацевтики. Такие способы включают операцию объединения активного соединения с носителем, который образован одним или несколькими вспомогательными ингредиентами. В общем случае лекарственные средства получают путем равномерного и тщательного перемешивания активного соединения с жидкими носителями или тонко диспергированными твердыми носителями или обоими видами носителей, а затем в случае необходимости формуют продукт.
Лекарственные формы могут представлять собой суспензии, таблетки, гранулы, порошки, капсулы, облатки, пилюли или пасты.
Лекарственные формы, пригодные для перорального введения (например, путем проглатывания), могут быть представлены в форме раздельных элементов, в частности, капсул, облаток или таблеток, каждая из которых содержит предварительно определенную дозу активного соединения, в форме порошка или гранул, в форме суспензии в водной или неводной жидкости или в форме пасты.
Таблетка может быть изготовлена традиционными способами, например, прессованием или формовкой, возможно, с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки могут быть получены путем прессования в соответствующем аппарате активного соединения в сыпучей форме, в частности, порошка или гранул, возможно, смешанных с одним или более связующими (например, с повидоном, желатином, гуммиарабиком, сорбитом, трагакантом, гидроксипропилметилцеллюлозой); наполнителями или разбавителями (например, лактозой, микрокристаллической целлюлозой, гидрофосфатом кальция); лубрикантами (например, стеаратом магния, тальком, кремнеземом); разрыхлителями (например, натриевой солью гликолята крахмала, сшитым повидоном, сшитой натриевой карбоксиметилцеллюлозой); поверхностно-активными или диспергирующими или смачивающими агентами (например, лаурилсульфатом натрия); и консервантами (например, метил-р-гидроксибензоатом, пропил-р-гидроксибензоатом, сорбиновой кислотой). Формованные таблетки могут быть получены путем формования под давлением в соответствующем аппарате смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки могут иметь покрытие или рифление и могут быть приготовлены таким образом, чтобы обеспечивать медленное или регулируемое высвобождение активного соединения за счет использования, например, гидроксипропилметилцеллюлозы в различных пропорциях для получения желаемого профиля высвобождения. На таблетки можно нанести энтеросолюбильное покрытие для обеспечения высвобождения в частях кишечника, отличных от желудка.
Капсула может содержать активное соединение в виде суспензии.
Лекарственные средства, пригодные для топического введения (например, трансдермально, интраназально, через глаза, буккально и сублингвально) могут быть изготовлены в форме пасты.
Лекарственные формы, пригодные для топического введения в глаз, включают также глазные капли, в которых активное соединение суспендировано в пригодном носителе, в частности, в водном растворителе для активного соединения.
Лекарственные формы, пригодные для назального введения, в которых носитель является твердым, содержат крупный порошок с размером частиц, например, в пределах от примерно 20 до примерно 500 микрон, которые вводятся при вдыхании, т.е. путем быстрой ингаляции через носовой проход из контейнера с порошком, удерживаемого вблизи носа.
Лекарственные формы, пригодные для введения путем ингаляции, включают средства, представляющие собой аэрозольный спрей, подаваемый из находящегося под давлением контейнера под действием соответствующего газа-вытеснителя, в частности, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или других пригодных газов.
Дозировка
Следует понимать, что соответствующие дозировки активного соединения и составов, содержащих активное соединение, могут быть различными для различных пациентов. Определение оптимальной дозировки обычно включает подбор соотношения между уровнем терапевтической пользы и соответствующим риском или вредными побочными эффектами лечения согласно настоящему изобретению. Выбор уровня дозировки зависит от различных факторов, которые включают, в частности, но без ограничения, активность конкретного соединения, способ введения, время введения, скорость выведения соединения из организма, длительность лечения, другие лекарственные препараты, соединения и/или материалы, используемые в комбинации, а также возраст, пол, вес, тяжесть заболевания, общее состояние здоровья и историю болезни пациента. Дозу соединения и способ введения в конечном итоге определяет лечащий врач, однако в общем случае величина дозы должна создавать локальную концентрацию в месте воздействия, которая обеспечивает желаемый эффект, и при этом не вызывает существенных вредных или опасных побочных эффектов.
Введение in vivo можно осуществлять одной дозой, непрерывно или с перерывами (например, раздельными дозами через соответствующие интервалы времени) в течение курса лечения. Способы определения наиболее эффективных средств и доз введения хорошо известны специалистам в данной области и варьируют в зависимости от лекарственного средства, используемого для лечения, цели лечения, клетки-мишени, подвергаемой воздействию, и субъекта, получающего лечение. Уровень и схема дозирования, а также режим однократного и многократного введения выбирает лечащий врач.
В общем случае пригодная доза активного соединения находится в пределах примерно от 10 мг до примерно 600 мг на м2 площади тела субъекта в день.
Примеры
Общие методы
Порошковая рентгенодифрактометрия (XRD)
Порошковую рентгенодифрактометрию осуществляли при помощи дифрактометра Bruker D5000 (длина волны источника рентгеновского излучения 1.5418 Е Cu, напряжение 40 кВ, термоэлектронная эмиссия 40 мА). Образцы сканировали в диапазоне от 2-40° 2θ с шагом 0,02° и 1 сек. счетом.
Дифференциальная сканирующая калориметрия (DSC)
Дифференциальную сканирующую калориметрию (DSC) выполняли с использованием Mettler DSC820E с роботизированной системой TSO801RO. Обычно, менее 5 мг материала, находящегося в 40 мкл алюминиевой кювете с перфорированной крышкой, нагревали по диапазону температур от 25°С до 325°С при постоянной скорости нагревания 10°С в минуту. Для продувки использовали газообразный азот при скорости потока 100 мл в минуту.
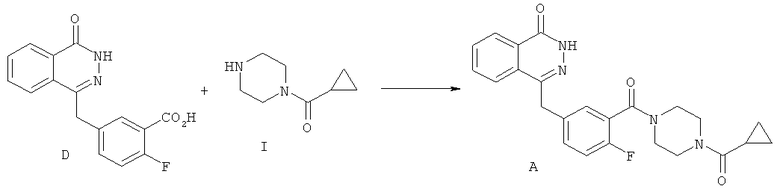
Получение Соединения А в виде Формы А
Ядерный магнитный резонанс (NMR)
1Н-ЯМР спектры регистрировали с использованием спектрометра Bruker DPX 400 при 400 МГц. Химические сдвиги регистрировали в миллионных долях (ppm) на 5-шкале относительно внутреннего стандарта тетраметилсилана. Если не указано иначе, все образцы растворяли в ДМСО-d6.
Масс-спектрогры
Масс-спектры регистрировали на масс-спектрометре с ионной ловушкой AgilentXCT методом тандемной масс-спектрометрии (MS/MS). Данный анализ использовали для подтверждения структуры. Прибор использовали в режиме электрораспыления положительных ионов.
(а) 4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-он (Соединение А)
Суспендировали 2-фтор-5-[(4-оксо-3,4-дигидрофталазин-1-ил) метил] бензойную кислоту (D) (15,23 г, 51,07 ммоль) с перемешиванием в атмосфере азота в ацетонитриле (96 мл). Добавили диизопропилэтиламин (19,6 мл, 112,3 ммоль) с последующим добавлением 1-циклопропилкарбонилпиреразина (л) (9,45 г, 61,28 ммоль) и ацетонитрила (1 мл). Охладили реакционную смесь до 18°С. Добавляли О-бензотриазол-1-ил-тетраметилуроний гексафторфосфат (25,18 г, 66,39 ммоль) в течение 30 минут и перемешивали реакционную смесь в течение 2 часов при комнатной температуре. Охладили реакционную смесь до 3°С и выдержали при этой температуре в течение 1 часа, а затем осуществили фильтрацию. Промыли осадок на фильтре холодным (3°С) ацетонитрилом (20 мл), после чего высушили в вакууме при температуре до 40°С с получением указанного соединения в форме твердого вещества бледно-желтого цвета (20,21 г).
Масс-спектр: МН+435
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
(b) Рекристаллизация соединения А из этанола
Суспендировали 4-(3-{[4-(циклопропилкарбонил)пиперазин-1-ил]карбонил}-4-фторбензил)фталазин-1(2Н)-он (соединение А) (20,00 г, 44,66 ммоль) в смеси воды (50 мл) и этанола (150 мл). Нагрели суспензию для дефлегмирования с перемешиванием. Затем охладили полученный раствор до 70°С и профильтровали. Промыли фильтр смесью воды (8 мл) и этанола (22 мл).
Охладили фильтрат до 45°С с перемешиванием. Добавили 4-(3-{[4-(циклопропилкарбонил)пиперазин-1-ил]карбонил}-4-фторбензил)фталазин-1(2Н)-он (соединение А) в Форме А (0,08 г), что обеспечило образование зародышей кристаллизации в смеси. Охлаждали полученную суспензию до 20°С в течение 2,5 часов и перемешивали ее при этой температуре в течение последующих 16 часов для осуществления кристаллизации. Добавляли воду (200 мл) в течение 5 часов, поддерживая температуру на уровне 20°С. В конце добавления выдерживали суспензию при температуре 20°С в течение 2 часов.
Профильтровали суспензию и промыли осадок на фильтре смесью этанола (24 мл) и воды (56 мл). Выгрузили и высушили отделенное твердое вещество под вакуумом при температуре 40-60°С для получения соединения А в виде Формы А - желтоватое твердое вещество (18,1 г).
На Фиг.3 показана типичная дифрактограмма, полученная методом рентгеновской дифрактометрии (XRD) соединения А в виде Формы А, а на Фиг.4 показана типичная кривая дифференциальной сканирующей калориметрии (DSC) соединения А в виде Формы А, полученной посредством нагрева от 25°С до 325°С со скоростью 10°С в минуту. Рентгеновскую дифрактограмму (XRD) получили, как изложено выше, но с 4 сек счетом.
Дифрактограмма Соединения А в виде кристаллической Формы А (λ=1.5418Е) содержит следующие характерные пики:
Дифрактограмма Соединения А в виде кристаллической Формы А также может содержать следующие дополнительные пики на дифракционной рентгенограмме (λ=1.5418Е):
Соединение А в виде кристаллической Формы А также может характеризоваться любой комбинацией трех или более пиков, выбранных из списка 10 пиков выше.
Начало процесса плавления Формы А соединения А при нагревании от 25°С до 325°С со скоростью 10°С в минуту будет соответствовать 210,1°С±1°С по DSC.
Пример 1
23 мг соединения А в виде Формы А взвесили во флакон. К этому твердому веществу добавили 0,25 мл раствора, изготовленного из 8,5 мл метанола и 1,5 мл воды (то есть 15% воды в объемном отношении). Полученную суспензию нагрели до 70°С, профильтровали и позволили ей медленно охладиться до температуры окружающей среды в закрытом флаконе. Достигнутая в результате кристаллизация привела к образованию твердого вещества, которое высушили в вакуумной печи. Продукт - соединение А в виде Формы L.
Пример 2
В реакционную пробирку добавили приблизительно 50 мг соединения А в виде Формы А и приблизительно 1 мл этанола/воды (80:20 в объемном отношении). Суспендировали смесь при температуре 40°С в течение 3 недель. Профильтровали образец и высушили его на фильтре в течение 10 минут, а затем сушили на лабораторном столе в течение ночи. Продукт-соединение А в виде Формы L.
Пример 3
Аналогично Примеру 2, суспендировали соединение А в виде Формы А в смеси этанола/воды (80:20 в объемном отношении) в течение 3 недель при температуре 60°С с получением соединения А в виде Формы L.
Пример 4
В реакционную пробирку добавили приблизительно 30 мг соединения А в виде Формы А и приблизительно 1 мл этанола/воды (80:20 в объемном отношении) с образованием насыщенной суспензии Формы А. К этой насыщенной суспензии добавили 30 мг соединения А в виде Формы L. Суспендировали смесь при температуре 25°С в течение 3 дней. Профильтровали образец и высушили его на фильтре в течение 10 минут, а затем сушили на лабораторном столе в течение всей ночи. Продукт - соединение А в виде Формы L, без Формы А.
Пример 5
Повторили Пример 4, но с использованием следующих растворителей для суспендирования смеси Формы А и Формы L, в этих экспериментов суспендировали образцы в течение 4 дней.
В каждом случае продукт представлял собой соединение А в виде кристаллической Формы L, без Формы А.
Пример 6
Ингибирующее действие
Для оценки ингибирующего действия активного соединения использовали следующий анализ для определения значения IC50.
Инкубировали поли-(АДФ-рибоза)-полимеразу (PARP) млекопитающего, выделенную из экстракта ядер клеткок Hela, с Z-буфером (25 мМ Hepes (Sigma); 12,5 мМ MgCl2 (Sigma); 50 мМ KCl (Sigma); 1 мМ DTT (Sigma); 10% Glycerol (Sigma) 0,001% NP-40 (Sigma); pH 7.4) в 96 ячейке FlashPlates (ТОРГОВАЯ МАРКА) (NEN, Великобритания) и добавили различные концентрации указанных ингибиторов. Все соединения разбавили в ДМСО и получили конечные концентрации для анализа в диапазоне от 10 до 0,01 мкМ, причем конечная концентрация ДМСО доставляла 1% на лунку. Общий объем исследуемой смеси составлял 40 мкл на лунку.
После 10 мин инкубации при температуре 30°С инициировали реакции добавлением 10 мкл реакционной смеси, содержащей никотинамид-аденин-динуклеотид (NAD) (5 мкМ), 3H-NAD и двухнитевые ДНК-олигонуклеотиды длиной 30 нуклеотидов. Выделенные лунки для положительной и отрицательной реакции обрабатывали одновременно с лунками, содержащими соединение (неизвестные), для расчета активности фермента в %. Затем встряхивали планшеты в течение 2 минут и инкубировали при температуре 30°С в течение 45 минут.
После инкубации гасили реакции добавлением 50 мкл 30%-ной уксусной кислоты в каждую лунку. Затем встряхивали планшеты в течение в течение 1 часа при комнатной температуре.
Перенесли планшеты на TopCount NXT (ТОРГОВАЯ МАРКА) (Packard, Великобритания) для регистрации сцинтилляции. Зарегистрированные значения представляют собой число импульсов в минуту (cpm) после 30 сек подсчета для каждой лунки.
Затем рассчитывали активность фермента в % для соединения, используя следующее уравнение:
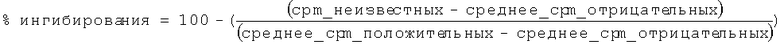
Рассчитали значения IC50 (концентрация, которое обеспечивает ингибирование 50% активности фермента) в диапазоне различных концентраций, обычно от 10 мкМ до 0,001 мкМ. Эти значения IC50 используют в качестве сравнительных величин для определения повышенной эффективности соединений.
Соединение А имеет значение IC50, равное приблизительно 5 нМ.
Коэффициент потенцирования
Коэффициент потенцирования (PF50) для активного соединения рассчитывают как отношение IC50 роста контрольной клетки к IC50 роста клетки + ингибитор PARP. Кривые подавления роста для контрольной клетки и обработанной соединением клетки даны в присутствии алкилирующего агента метилметансульфоната (MMS). Испытательное соединение использовалось при установленной концентрации 0,2 микромоль. Концентрации MMS были в диапазоне от 0 до 10 мкг/мл.
Оценку роста клетки осуществляли, используя анализ с сульфородамином В (SRB) (Skehan, P., et al., (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 82, 1107-1112.). 2000 клеток HeLa посеяли в каждой лунке плоскодонного микротитрационного планшета с 96 ячейками в объеме 100 мкл и культивировали в течение 6 часов при температуре 37°С. Среду заменяли либо на простую среду, либо на среду, содержащую ингибитор PARP в конечной концентрации 0,5,1 или 5 мкМ. Клеткам позволили расти в течение еще одного часа, после чего добавляли MMS в диапазоне концентраций (обычно 0, 1, 2, 3, 5, 7 и 10 мкг/мл) к необработанным клеткам или клеткам, обработанным ингибитором PARP. Для оценки подавления роста под действием ингибитора PARP использовали клетки, обработанные только ингибитором PARP.
Клетки оставили на еще 16 часов, после чего заменяли среду и позволяли клеткам расти в течение еще 72 часов при температуре 37°С. Затем среду удалили и зафиксировали клетки с помощью 100 мкл ледяной 10% (в отношении веса к объему) трихлоруксусной кислоты. Планшеты культивировали при температуре 4°С в течение 20 минут, а затем промыли четыре раза водой. Затем окрасили каждую лунку с клетками с помощью 100 мкл 0,4% (в отношении веса к объему) SRB в 1%-ной уксусной кислоте в течение 20 минут, после чего четыре раза промыли 1%-ной уксусной кислотой. Затем планшеты сушили в течение 2 часов при комнатной температуре. Солюбилизировали красящее вещество из окрашенных клеток добавлением 100 мкл 10 мМ Tris Base в каждую лунку. Осторожно встряхивали планшеты и оставили при комнатной температуре на 30 минут, после чего измеряли оптическую плотность при 564 нМ на спектрофотометре Microquant для считывания титрационных микропланшетов.
Соединение А при 200 нМ имеет значение PF50, равное 20 мин.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИМОРФНАЯ ФОРМА 4-[3-(4-ЦИКЛОПРОПАНКАРБОНИЛПИПЕРАЗИН-1-КАРБОНИЛ)-4-ФТОРБЕНЗИЛ]-2Н-ФТАЛАЗИН-1-ОНА | 2007 |
|
RU2465270C2 |
| ИНГИБИТОРЫ РЕПАРАЦИИ ПОВРЕЖДЕНИЯ ДНК ДЛЯ ЛЕЧЕНИЯ РАКА | 2004 |
|
RU2413515C2 |
| ИНГИБИТОРЫ РЕПАРАЦИИ ПОВРЕЖДЕНИЙ ДНК ДЛЯ ЛЕЧЕНИЯ РАКА | 2017 |
|
RU2755865C2 |
| СПОСОБЫ НА ОСНОВЕ ДЕТЕКТИРОВАНИЯ ОЧАГОВ RAD51 В ОПУХОЛЕВЫХ КЛЕТКАХ | 2018 |
|
RU2825699C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ SNS-595 ДЛЯ ЛЕЧЕНИЯ СУБЪЕКТОВ С ОНКОЛОГИЧЕСКИМИ ЗАБОЛЕВАНИЯМИ, ИМЕЮЩИХ СНИЖЕННУЮ АКТИВНОСТЬ BRCA2 | 2010 |
|
RU2558835C2 |
| ЛЕЧЕНИЕ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ С ПОМОЩЬЮ СОЕДИНЕНИЯ 4-ИОД-3-НИТРОБЕНЗАМИД В КОМБИНАЦИИ С ПРОТИВООПУХОЛЕВЫМИ СРЕДСТВАМИ | 2008 |
|
RU2480211C2 |
| ТРИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПАРП | 2004 |
|
RU2404183C2 |
| ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ 2-{4-[(3S)-ПИПЕРИДИН-3-ИЛ]ФЕНИЛ}-2Н-ИНДАЗОЛ-7-КАРБОКСАМИДА | 2009 |
|
RU2495035C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2686314C1 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ТЕТРАЦИКЛИЧЕСКИМИ АНАЛОГАМИ ХИНОЛОНА ДЛЯ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2752506C2 |
Изобретение относится к 4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-ону в виде кристаллической Формы L, имеющей характеристические пики на порошковой рентгеновской дифрактограмме, приведенные в формуле изобретения, способам получения Формы L, фармацевтическому составу, содержащему Форму L, и вариантам применения Формы L и составов, содержащих Форму L. Технический результат - получение новой кристаллической формы названного выше соединения, которое обладает ингибирующей активностью поли-(АДФ)полимеразы (PARP). Форма L не содержит примесей растворителя, что позволяет более точно определять дозировку активного соединения при лечении пациента. 8 н. и 9 з.п. ф-лы, 4 ил., 6 пр.
1. 4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-он в виде кристаллической Формы L, имеющей следующие характеристические пики на порошковой рентгеновской дифрактограмме (XRD):
2. Соединение по п.1, дифрактограмма которого, полученная методом порошковой рентгеновской дифрактометрии (XRD), содержит следующие характеристические пики (XRD):
3. Соединение по любому из пп.1-2, характеризующееся тем, что в исследовании методом дифференциальной сканирующей калориметрии (DSC) при нагреве от 25°С до 325°С со скоростью 10°С температура начала плавления составляет 198,5±1°С.
4. Способ получения 4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-она в виде кристаллической Формы L, имеющей следующие характеристические пики на порошковой рентгеновской дифрактограмме (XRD):
из 4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-она в виде кристаллической Формы А, имеющей следующие характеристические пики на порошковой рентгеновской дифрактограмме (XRD):
включающий суспендирование 4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-она в виде Формы А в органическом растворителе,
выбранном из группы, включающей метиловый спирт, этиловый спирт, ацетон, ацетонитрил, нитрометан, который возможно содержит до 30% об./об. воды.
5. Способ получения 4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-она в виде кристаллической Формы L, имеющей следующие характеристические пики на порошковой рентгеновской дифрактограмме (XRD):
включающий суспендирование4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-она в виде формы А, имеющей следующие характеристические пики на порошковой рентгеновской дифрактограмме (XRD):
и формы L, полученной по п.4, в органическом растворителе, выбранном из группы, включающей метиловый спирт, этиловый спирт, ацетон, ацетонитрил, нитрометан, который возможно содержит до 30% об./об. воды.
6. Фармацевтический состав, ингибирующий активность поли-(АДФ-рибозы)-полимеразы (PARP), включающий соединение по любому из пп.1-3 и фармацевтически приемлемый носитель или разбавитель.
7. Применение соединения по любому из пп.1-3 в изготовлении лекарственного средства для ингибирования активности PARP.
8. Применение соединения по любому из пп.1-3 в изготовлении лекарственного средства для лечения следующих заболеваний: заболевания сосудов; септический шок; ишемическое поражение; нейротоксичность; геморрагический шок; вирусная инфекция; или заболеваний, выраженность которых может быть снижена путем ингибирования активности PARP.
9. Применение соединения по любому из пп.1-3 в изготовлении лекарственного средства для использования в качестве вспомогательного средства в терапии рака или для повышения чувствительности раковых клеток к обработке ионизирующим излучением или химиотерапевтическими средствами.
10. Применение соединения по любому из пп.1-3 в изготовлении лекарственного средства для использования в лечении у пациента рака, характеризующегося дефицитом пути репарации двухнитевых разрывов ДНК, по механизму гомологичной рекомбинации (HR).
11. Применение по п.10, отличающееся тем, что рак включает одну или более раковых клеток, у которых снижена или отсутствует способность к репарации двухнитевых разрывов ДНК по механизму гомологичной рекомбинации (HR) по сравнению с нормальными клетками.
12. Применение по п.11, отличающееся тем, что раковые клетки имеют фенотип дефицита BRCA1 или BRCA2.
13. Применение по п.12, отличающееся тем, что раковые клетки характеризуются дефицитом BRCA1 или BRCA2.
14. Применение по п.10, отличающееся тем, что пациент является гетерозиготным по мутации в гене, кодирующем один из компонентов пути репарации двухнитевых разрывов ДНК по механизму гомологичной рекомбинации (HR).
15. Применение по п.14, отличающееся тем, что пациент является гетерозиготным по мутации в BRCA1 и/или BRCA2.
16. Применение по любому из пп.9-15, отличающееся тем, что указанный рак представляет собой рак молочной железы, яичника, поджелудочной железы или простаты.
17. Применение по любому из пп.9-15, отличающееся тем, что указанное лечение включает применение ионизирующего излучения или химиотерапевтического средства.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| ПРИСПОСОБЛЕНИЕ ДЛЯ ТОЧНОЙ УСТАНОВКИ В ФОРМАХ МЕТАЛЛИЧЕСКИХ ЧАСТЕЙ ПРИ ЗАЛИВКЕ ИХ В ОТЛИВАЕМЫЕ ИЗДЕЛИЯ | 1926 |
|
SU6300A1 |
| ТИХОНОВ А.И | |||
| и др | |||
| Биофармация | |||
| - Харьков: издательство НФаУ "Золотые страницы", 2003, с.22-23. | |||

