Настоящее изобретение относится к кристаллической форме и усовершенствованным способам синтеза конкретного производного фталазинона, промежуточным соединениям, используемым в ходе указанного синтеза, фармацевтическим композициям на основе указанной кристаллической формы и применению указанной кристаллической формы.
Фермент млекопитающих PARP (Poly-(ADP-ribose)polymerase, поли-(АДФ-рибоза)-полимераза, мультидоменный белок с молекулярной массой 113 кДа) участвует в передаче сигнала о повреждении ДНК благодаря способности указанного фермента распознавать и быстро связываться с разрывами одинарной или двойной цепи ДНК (D'Amours, et al., Biochem. J., 342, 249-268 (1999)).
Некоторые наблюдения привели к заключению о том, что PARP принимает участие во множестве функций, связанных с ДНК, включая амплификацию генов, деление клеток, дифференцировку, апоптоз, эксцизионную репарацию оснований ДНК, а также оказывает влияние на длину теломера и стабильность хромосом (d'Adda di Fagagna, etal., Nature Gen., 23(1), 76-80 (1999)).
Исследования механизма, посредством которого PARP модулирует репарацию ДНК и другие процессы, показали, что PARP играет важную роль в образовании поли-АДФ-рибозных цепей в клеточном ядре (Althaus, F.R. And Richter, С., ADP-Ribosylation of Proteins: Enzymology and Biological Significance, Springer-Verlag, Berlin, 1987)).Связанная с ДНК активированная PARP использует НАД (никотинамид аденин динуклеотид) для синтеза поли-(АДФ-рибозы) во множестве ядерных целевых белков, включая топоизомеразу, гистоны и непосредственно PARP (Rhun, et al., Biochem. Biophys. Res. Commun., 245, 1-10 (1998)).
Поли-АДФ-рибозилирование также связывают с протеканием злокачественных превращений. Так, например, активность PARP является более высокой в изолированных ядрах SV40-трансформированных фибробластов, в то время как и лейкозные клетки, и раковые клетки толстой кишки проявляют более высокую ферментативную активность по сравнению с активностью эквивалентных нормальных лейкоцитов и слизистой оболочки толстой кишки (Miva, et al., Arch. Biochem. Biophys., 181, 313-321 (1977); Burzio, et al., Proc. Soc. Exp Biol. Med., 149, 933-938 (1975); и Hirai, et al., Cancer Res., 43, 3441-3446 (1983)).
Для установления функциональной роли поли-АДФ-рибозилирования в репарации ДНК использовали ряд низкомолекулярных ингибиторов PARP. В клетках, обработанных алкилирующими агентами, ингибирование PARP приводит к заметному увеличению количества разрывов в цепях ДНК и цитолизу (Durkacz, etal., Nature, 283, 593-596 (1980); Berger, N. A., Radiation Research, 101, 4-14(1985)).
Впоследствии было показано, что такие ингибиторы усиливают ответную реакцию на облучение за счет подавления репарации потенциально летальных повреждений (Ben-Hur, et al., British Journal of Cancer, 49 (Suppl. VI)), 34-42 (1984); Schlicker, et al., Int. J. Radiat. Bioi., 75, 91-100 (1999)). Имеются сообщения о том, что ингибиторы PARP являются эффективными для радиационной сенсибилизации гипоксических опухолевых клеток (US 5,032,617; US 5,215,738 и US 5,041,653).
Кроме того, животные с нокаутом PARP (PARP -/-) проявляют геномную нестабильность в ответ на действие алкилирующих агентов и γ-излучения (Wang, et al., Genes Dev., 9, 509-520 (1995); Menissier de Murcia, et al., Proc. Natl. Acad. Sci. USA, 94, 7303-7307 (1997)).
Показана также роль PARP в развитии некоторых сосудистых заболеваний, септического шока, ишемических повреждений и нейротоксичности (Cantoni et al., Biochim. Biophys. Acta, 1014, 1-7 (1989); Szabo et al., J. Clin. Invest, 100, 723-735 (1997)). В ходе исследований ингибиторов PARP было показано, что повреждение ДНК кислородными радикалами, приводящее к разрывам цепей ДНК, распознаваемых далее PARP, является основным фактором, способствующим развитию таких болезненных состояний (Cosi et al., J. Neurosci. Res., 39, 38-36 (1994); Cosi et al., Proc. Natl. Acad. Sci. U.S.A., 93, 4688-4692 (1996)). Позднее было показано, что PARP играет роль в патогенезе геморрагического шока (Liaudet et al., Proc. Nail. Acad. Sci. U.S.A., 97(3), 10203-10208 (2000)).
Кроме того, было показано, что эффективная ретровирусная инфекция клеток млекопитающих блокируется путем ингибирования активности PARP. При этом было продемонстрировано, что такое ингибирование рекомбинантных ретровирусных векторных инфекций происходит во множестве различных типов клеток (Gaken et al., J. Virology, 70(6), 39992-4000 (1996). В связи с этим были разработаны ингибиторы PARP, предназначенные для применения при лечении вирусных заболеваний и рака (WO 91/18591).
Кроме того, полагают, что ингибирование PARP задерживает появление характеристик старения в фибробластах человека (Rattan and dark, Biochem. Biophys. Res. Comm., 201(2), 665-672 (1994)). Это может быть связано с той ролью, которую PARP играет в контролировании функций теломеров (d'Adda di Fagagna, etal., Nature Gen., 23(1), 76-80 (1999)).
В публикации WO 2004/080976 предложен ряд производных фталазинона, описана их активность в отношении ингибирования PARP и последовательное применение указанных производных для лечения рака как в дополнение к лучевой или химиотерапии, так и в качестве самостоятельных агентов.
В WO 2005/053662 предложено применение ингибиторов PARP, в частности, производных фталазинона, в качестве ингибиторов эксцизионной репарации оснований (base excision repair, BER). Также предложено применение указанных ингибиторов для получения лекарственных препаратов для лечения раковых заболеваний, характеризующихся недостаточной активностью репарации двухнитевых разрывов ДНК, зависимой от гомологичной рекомбинации (Homologous Recombination, HR), в частности, раковых заболеваний, при которых имеет место фенотип с дефицитом генов BRCA1 и/или BRCA2.
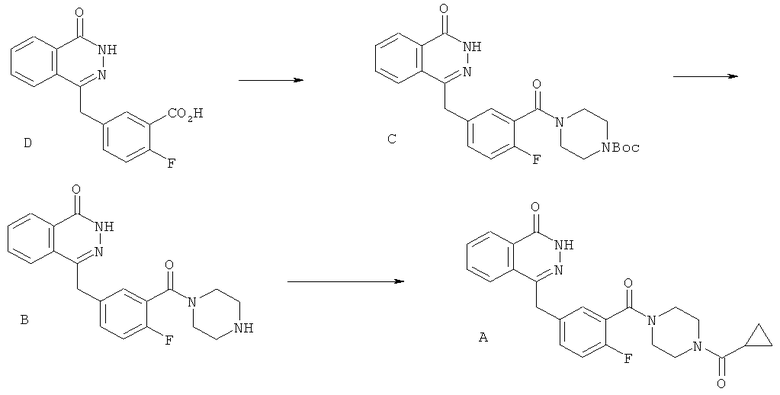
Особый интерес представляет описанный в WO 2004/080976 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-он (соединение А):
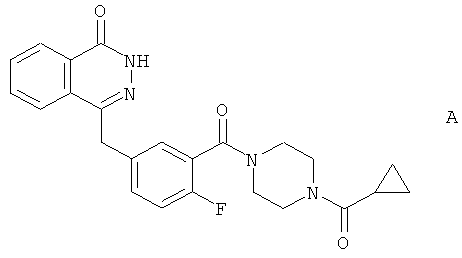
Согласно WO 2004/080976, соединение А синтезировали в числе других библиотечных соединений из 4-[4-фтор-3-(пиперазин-1-карбонил)-бензил]-2Н-фталазин-1-она (соединение В):
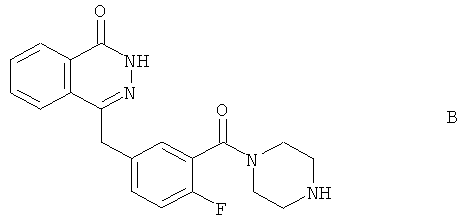
путем добавления циклопропанкарбонилхлорида:
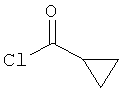
к раствору соединения (В) в дихлорметане с последующим добавлением основания Хюнига (N,N-диизопропилэтиламина). Эту реакцию проводили при перемешивании при температуре окружающей среды в течение 16 часов, а полученное соединение очищали с помощью препаративной ВЭЖХ.
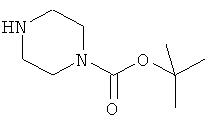
Производное (В) пиперазина получили путем удаления защитных групп из трет-бутилового эфира 4-[2-фтор-5-(4-оксо-3,4-дигидрофталазин-илметил)-бензоил]пиперазин-1-карбоновой кислоты (соединения С):
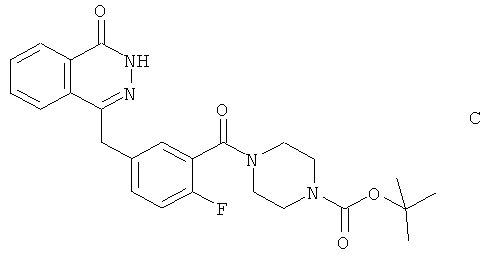
при помощи 6М HCl и этанола в течение 1 часа с последующим подщелачиванием аммиаком до рН 9 и экстракцией дихлорметаном.
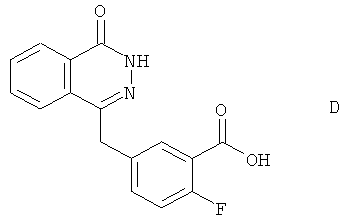
Пиперазиновое производное (С), содержащее защитную группу Boc, получили из 2-фтор-5-(4-оксо-3,4-дигидрофталазин-1-илметил)-бензойной кислоты (соединение D):
путем добавления трет-бутилового эфира пиперазин-1-карбоновой кислоты:
2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфата (HBTU) и N,N,-диизопропилметиламина в диметилацетамиде с последующим перемешиванием в течение 18 часов.
Некоторые формы соединения А могут иметь полезные свойства, например, с точки зрения их растворимости и/или стабильности и/или биодоступности и/или содержания в них примесей и/или фильтруемости и/или характеристик при сушке и/или пониженной гигроскопичности и/или простоты их обработки и/или измельчения и/или получения из них таблеток. Желательно также иметь усовершенствованный способ синтеза, подходящий для синтеза соединения А в достаточно крупных масштабах.
Соответственно, согласно первому аспекту в настоящем изобретении предложен 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-он (соединение А) в по существу кристаллической форме, в частности, в форме А.
Приведенный выше термин "в по существу кристаллической форме" означает, что по меньшей мере 50% по массе соединения А находится в кристаллической форме, предпочтительно - меньшей мере 70% по массе, 80% или 90%. В некоторых вариантах реализации по меньшей мере 95% по массе, 99% по массе или даже 99.5% по массе или более может находиться в кристаллической форме.
Кристаллическая форма А соединения А имеет рентгенограмму (λ=1.5418 Å), содержащую следующие характеристические пики:
Кристаллическая форма А соединения А может иметь также следующие дополнительные пики на рентгенограмме (λ=1.5418 Å):
Кристаллическую форму А соединения А может также характеризовать любая комбинация трех или более пиков из вышеуказанных 10 пиков.
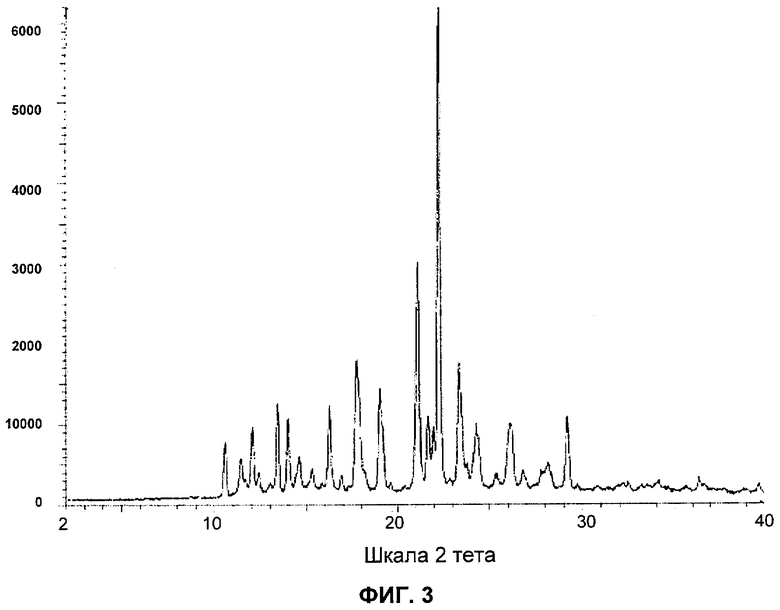
На фигуре 3 представлена репрезентативная порошковая рентгенограмма формы А соединения А.
Не желая быть связанными какой-либо конкретной теорией, полагают, что соединение А может легко образовывать структуру, в которой молекулы растворителя способны располагаться в кристаллической решетке. Такие сольваты, не обязательно являясь стехиометрическими по своей природе, могут состоять из одного чистого сольвата (например, метанолат соединения А и тетрагидрофуранат соединения А) или потенциально способны содержать компоненты нескольких растворителей (например, метанол и диэтиловый эфир). Молекулы растворителя обычно располагаются в карманах, образованных молекулами соединения А. При некоторых условиях размер указанных карманов может изменяться в достаточной степени для вмещения целого ряда растворителей, что приводит лишь к небольшому изменению общей структуры материала и, следовательно, лишь к небольшим сдвигам отражений на рентгенограмме.
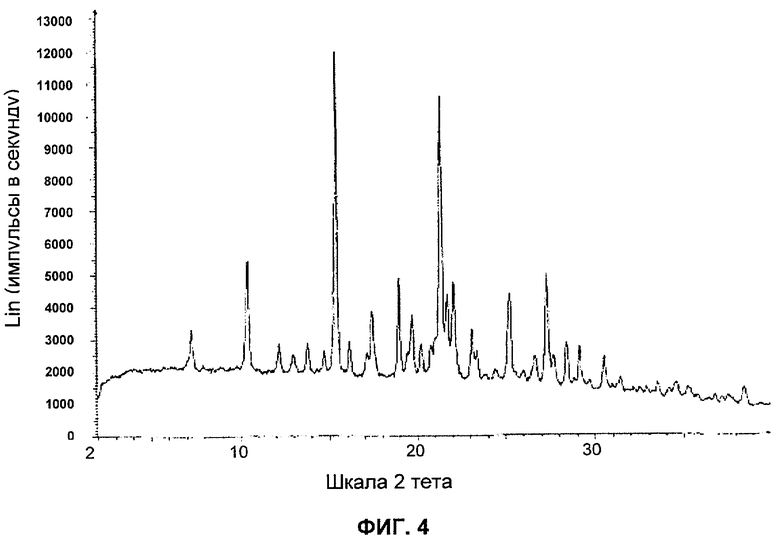
Сольваты, в том числе имеющие одинаковую общую структуру, были получены в ходе исследований созревания растворов и кристаллизации из дихлорметана, этилацетата, метанола, этанола, изопропанола, 2-бутанона, трет-бутилметилового эфира, толуола, тетрагидрофурана, воды, циклогексана, циклопропилметилкетона, 1,2-дихлорэтана, этилтрифторацетата, фторбензолгексафторизопропанола, метилнонафторбутилового эфира, 2-метил-1-пропанола, нитрометана, пропионитрила, трихлорэтилена, ααα-трифтортолуола, гептана, диоксана, ацетонитрила как в виде чистых растворителей, так и в сочетании с другим растворителем. Рентгенограмма наиболее распространенных сольватных структур представлена на фигуре 4 и обычно содержит пики с наибольшей интенсивностью в следующих положениях:
Следует понимать, что относительная интенсивность пиков, показанных на фигурах, может изменяться в зависимости от ориентации образца при испытании, а также от типа и установок применяемого прибора. Поэтому интенсивности на представленных рентгенограммах являются иллюстративными и не предназначены для сравнения абсолютных величин.
Форма А соединения А по существу не содержит растворителя. Термин "по существу не содержит растворителя", используемый в данном описании, относится к форме, содержащей лишь незначительное количество растворителя, например, к форме, общее содержание в которой любых растворителей составляет 0,5% по массе или менее. Общее содержание любого растворителя, включая воду, может составлять 0,25%, 0,1%, 0,05% или 0,025% по массе или менее.
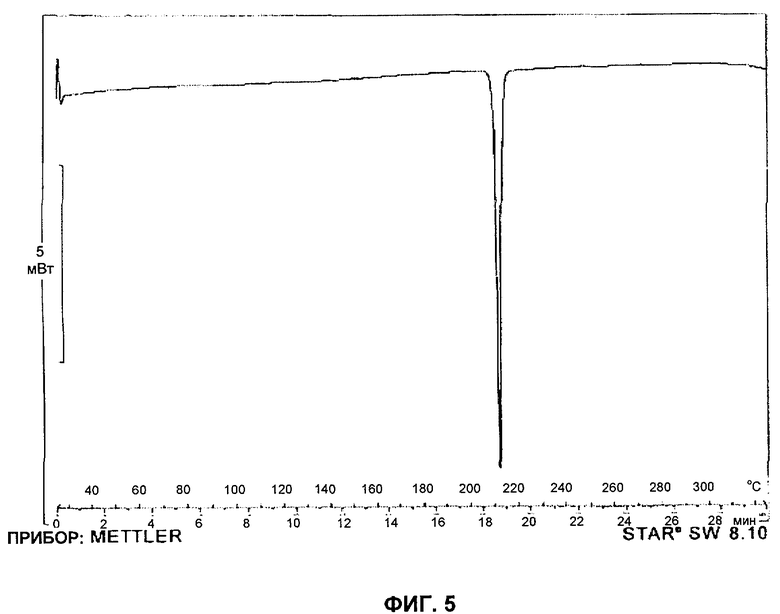
Форму А соединения А можно охарактеризовать также при помощи ДСК (дифференциальной сканирующей калориметрии). При нагревании от 25°С до 325°С со скоростью 10°С в минуту форма А соединения А начинает плавиться при 210.1°±1°С. Репрезентативная кривая ДСК для формы А соединения А представлена на фигуре 5.
Согласно второму аспекту в настоящем изобретении предложен способ получения 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она (соединения А) в виде кристаллической формы А, включающий кристаллизацию соединения А в растворителе с последующим вытеснением растворителя из кристаллической формы вытесняющим агентом. Указанный вытесняющий агент может представлять собой воду или смесь C1-2 спирта и воды.
Согласно первому варианту реализации указанный способ включает следующие стадии:
(1) кристаллизацию 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она (соединения А) из растворителя,
(2) обработку кристаллического соединения А этанолом, если исходный растворитель не является этанолом,
(3) обработку кристаллического соединения А водой для удаления захваченного этанола,
(4) сушку полученного продукта.
Растворитель, применяемый при начальной кристаллизации, может представлять собой, например, дихлорметан или ацетонитрил.
Способы получения формы А в общем случае могут включать замену растворителя. Установлено, что соединение А кристаллизуется таким образом, что в кристаллической решетке образуются каналы, которые могут захватывать растворитель, что усложняет удаление последнего.
Способ согласно первому варианту реализации изобретения подходит для применения, в частности, если растворитель, используемый для кристаллизации соединения А, представляет собой дихлорметан. Операцию замещения используемого в качестве растворителя дихлорметана на этанол можно провести путем перегонки раствора соединения А при атмосферном давлении в присутствии этанола. Замещение завершается, когда температура приближается к точке кипения этанола, например, по меньшей мере 73°С. В частности, указанное замещение можно произвести путем отгонки основной части дихлорметана с последующим добавлением некоторого объема этанола. Затем дистилляцию продолжают, заменяя порции дистиллята равными объемами этанола.
Кристаллизацию соединения А из этанола можно провести путем охлаждения раствора до температуры ниже 15°С, предпочтительно - ниже 10°С и более предпочтительно - примерно до 8°С. Затем кристаллы соединения А можно извлечь из раствора при помощи фильтрования.
Кристаллическое соединение А можно обработать водой, чтобы удалить захваченный этанол, путем суспендирования кристаллического материала в воде и кипячения с обратным холодильником в течение достаточного времени, например, в течение по меньшей мере трех часов, предпочтительно - в течение примерно четырех часов. Кристаллическое соединение А можно извлечь из водной суспензии при помощи фильтрования.
Сушку продукта, полученного на вышеуказанной стадии, легко осуществить, например, путем нагревания продукта в печи при температуре по меньшей мере 60°С, предпочтительно - примерно 70°С.
Согласно другому варианту реализации предложенный способ включает следующие операции:
(1) получение соединения А в виде кристаллической формы, содержащей растворитель,
(2) обработку соединения А в виде указанной кристаллической формы смесью воды и C1-2 спирта, если исходный растворитель, применяемый при синтезе соединения А в кристаллической форме, не является смесью воды и C1-2 спирта (т.е. метанола, этанола),
(3) сушку полученного продукта.
Затем полученный продукт можно обработать смесью воды и C1-2 спирта и высушить, получив соединение А в кристаллической форме А.
Соотношение воды и C1-2 спирта в смеси предпочтительно находится в пределах от 2:1 до 1:2 по объему, более предпочтительно - от 1.5:1 до 1:1.5 по объему. Особенно предпочтительная смесь содержит 1 часть воды на 1.2 части C1-2 спирта. Другая особенно предпочтительная смесь содержит 2 части воды на 1 часть C1-2 спирта. C1-2 спирт предпочтительно представляет собой этанол.
Соединение А в виде кристаллической формы можно получить путем кристаллизации соединения А из растворителя, как описано выше.
Обработку растворителем на операции (2) можно провести путем суспендирования соединения А в смеси воды и C1-2 спирта и нагревания до кипения с обратным холодильником при перемешивании. После этого можно охладить суспензию до температуры 55-65°С и отфильтровать, например, через пластинку из целита. Фильтровальную пластинку можно промыть смесью воды и C1-2 спирта до проведения перегонки при атмосферном давлении (обычно 1 атмосфера) или при более высоком давлении. Перегонку можно остановить, получив при этом суспензию, которую выдерживают при температуре окружающей среды перед последующим фильтрованием. Полученный осадок на фильтре можно промыть водой.
Сушку продукта, полученного на вышеуказанной стадии, легко осуществить, например, путем нагревания продукта в печи при температуре по меньшей мере 50°С, предпочтительно - примерно 60°С.
Последующую обработку можно произвести аналогично тому, как описано выше.
Согласно третьему варианту реализации указанный способ включает:
(1) суспендирование соединения А в смеси воды и C1-2 спирта в качестве растворителя,
(2) нагревание суспензии до кипения с обратным холодильником,
(3) охлаждение раствора и внесение затравки, представляющей собой соединение А в виде формы А,
(4) сушку полученного продукта.
Полученный продукт можно дополнительно обработать смесью воды и C1-2 спирта и высушить с получением соединения А в кристаллической форме А.
Соотношение воды и C1-2 спирта в смеси предпочтительно находится в пределах от 2:1 до 1:5 по объему, более предпочтительно - от 1:2 до 1:4 по объему. Особенно предпочтительная смесь содержит 1 часть воды на 3 части C1-2 спирта. C1-2 спирт предпочтительно представляет собой этанол.
Стадия (3) может включать охлаждение раствора до температуры 65-75°С (например, до 70°С) и фильтрование, например, через целитную пластинку. Фильтрующую пластинку можно промыть смесью воды и C1-2 спирта перед проведением перегонки (проводимой, например, при атмосферном давлении или более высоком давлении). Внесение затравки можно осуществить после охлаждения полученного фильтрата до температуры 40-50°С (например, 45°С). Полученную суспензию можно охладить до температуры окружающей среды (например, 20°С) в течение 2-3 часов (например, 2,5 часа) и выдержать при указанной температуре в течение времени, достаточного для протекания кристаллизации. Этот период времени может составлять от 12 до 24 часов и может составлять примерно 16 часов. По прошествии указанного времени можно добавить дополнительное количество воды. Воду можно добавлять медленно в объеме, примерно равном суммарному объему присутствующего растворителя (воды и C1-2 спирта), например, в течение 4-6 (в частности, 5) часов. После добавления воды суспензию можно выдержать при температуре окружающей среды, например, в течение 2 часов.
Затем суспензию можно отфильтровать, а полученный осадок на фильтре промыть смесью C1-2 спирта и воды (в соотношении от 1:3 до 1:2, например, 1:2.3).
Сушку продукта, полученного на вышеуказанной стадии, легко осуществить, например, путем нагревания продукта в печи под вакуумом при температуре от 40 до 60°С.
Согласно третьему аспекту в настоящем изобретении предложен способ синтеза соединения А из соединения В, включающий следующую стадию:
(1) контролируемое добавление предварительно смешанного раствора триэтиламина и циклопропанкарбонилхлорида в подходящем органическом растворителе (например, в дихлорметане, ДХМ) к раствору соединения В в том же самом органическом растворителе при поддержании температуры раствора равной менее 20°С.
Согласно некоторым вариантам реализации указанный способ включает дополнительно следующую стадию:
(2) перемешивание (например, с помощью мешалки) раствора со стадии (1) до завершения реакции при поддержании температуры раствора равной менее 20°С.
Указанное добавление на стадии (1) можно осуществлять по каплям.
Предложенный способ лучше поддается контролю по сравнению со способом, описанным в WO 2004/080976, что обеспечивает более региоселективное присоединение хлорангидрида. Известный из уровня техники способ, в меньшей степени поддающийся контролю, может приводить к присоединению хлорангидрида по атому азота или кислорода во фталазиноне, так же как и по заданному атому азота в пиперидине.
Указанный выше способ предпочтительно осуществляют в атмосфере азота.
Предпочтительно также поддерживать температуру раствора на стадии (2) в пределах от 10 до 15°С.
Продукт, полученный в результате указанного взаимодействия, предпочтительно по меньшей мере один раз промывают водой. Более предпочтительно указанная обработка продукта включает начальную и окончательную промывку водой, а также стадии промежуточной промывки разбавленной кислотой, например, 5% раствором лимонной кислоты, а затем - разбавленным основанием, например, 5% раствором карбоната натрия.
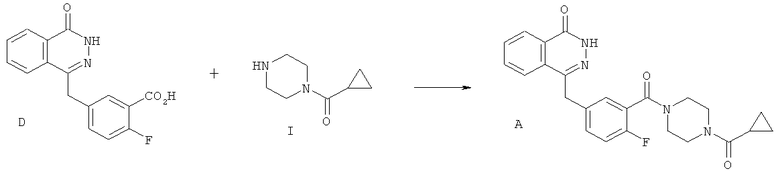
Согласно четвертому аспекту в настоящем изобретении предложен способ синтеза соединения А из соединения D, включающий взаимодействие соединения D с 1-(циклопропилкарбонил)пиперазином или его солью минеральной кислоты в присутствии агента, связывающего амид, и основания, например, амина (в том числе третичного амина, в частности, диизопропилэтиламина).
Указанная соль минеральной кислоты может представлять собой, например, солянокислую соль.
Добавление 1-(циклопропилкарбонил)пиперазина или его соли минеральной кислоты к соединению D можно осуществить в любом подходящем растворителе, например, в ацетонитриле. Агент, связывающий амид, предпочтительно представляет собой 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат (HBTU). Указанный агент предпочтительно добавляют к раствору, содержащему 1-(циклопропилкарбонил)пиперазин или соли минеральной кислоты указанного соединения, диизопропилэтиламин и соединения D, в течение некоторого периода времени, например, в течение 30 минут. Температуру полученного раствора можно поддерживать равной 25°С или ниже (или 20°С или ниже, например, 18°С). После проведения указанного добавления полученный раствор можно выдержать в течение некоторого периода времени. Предпочтительный температурный режим представляет собой выдержку раствора в течение 2 часов при комнатной температуре.
Образовавшееся соединение А можно извлечь из раствора путем охлаждения раствора до температуры ниже 10°С (или ниже 5°С, например, до 3°С) в течение некоторого периода времени (например, 1 часа) с последующим фильтрованием. Полученное соединение А можно промыть, например, холодным ацетонитрилом.
В WO 2004/080976 предложена следующая схема получения соединения D:
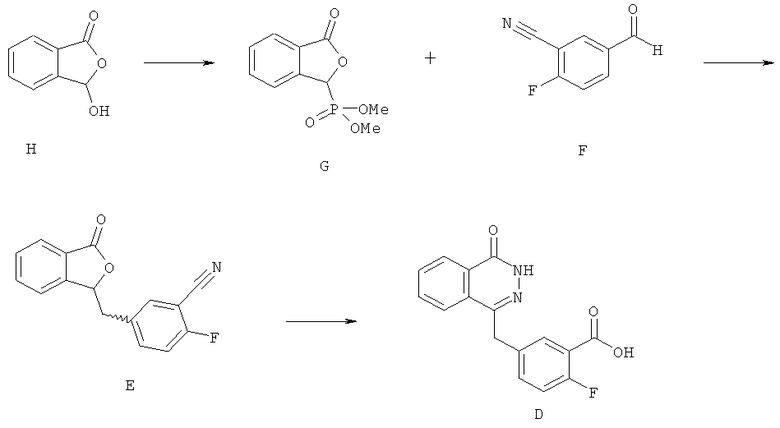
Диметилфосфит добавили по каплям в раствор метоксида натрия в метаноле при 0°С. Затем в реакционную смесь, представляющую собой суспензию в метаноле, порциями добавили 2-карбоксибензальдегид (Н), поддерживая температуру равной менее 5°С. Образовавшийся бледно-желтый раствор нагрели до 20°С в течение 1 часа. В реакционную смесь по каплям добавили метансульфокислоту и выпарили образовавшуюся белую суспензию под вакуумом. Полученный белый осадок погасили водой и экстрагировали хлороформом. Объединенные органические экстракты промыли водой, высушили над MgSO4 и выпарили под вакуумом, получив диметиловый эфир (3-оксо-1,3-дигидроизобензофуран-1-ил)фосфоновой кислоты (G) в виде белого осадка (выход: 95%). Этот продукт использовали на следующей стадии без дополнительной очистки.
К смеси диметилового эфира (3-оксо-1,3-дигидроизобензофуран-1-ил)фосфоновой кислоты (G) в тетрагидрофуране и 2-фтор-5-формилбензонитрила (F) в тетрагидрофуране по каплям в течение 25 минут добавили триэтиламин, поддерживая температуру равной менее 15°С. Затем реакционную смесь медленно нагрели до 20°С в течение 1 часа и сконцентрировали под вакуумом. Белый осадок суспендировали в воде в течение 30 минут, отфильтровали, промыли водой, гексаном и диэтиловым эфиром и высушили, получив 2-фтор-5-(3-оксо-3Н-изобензофуран-1-илиденметил)бензонитрил (Е) в виде смеси 50:50 изомеров Е и Z (выход: 96%).
К суспензии 2-фтор-5-(3-оксо-3Н-изобензофуран-1-илиденметил)бензонитрила (Е) в воде добавили водный раствор гидроксида натрия и нагрели реакционную смесь в атмосфере азота до 90°С в течение 30 минут. Затем реакционную смесь частично охладили до 70°С, добавили гидразин гидрат и перемешивали в течение 18 часов при 70°С. Далее реакционную смесь охладили до температуры окружающей среды и подкислили 2М HCl до рН 4. Смесь перемешали в течение 10 минут и отфильтровали. Полученный осадок промыли водой, гексаном, простым эфиром, этилацетатом и высушили, получив соединение D в виде светло-розового порошка (выход: 77%).
Желательно также иметь усовершенствованный способ синтеза соединения D.
В соответствии с пятым аспектом в настоящем изобретении предложен способ синтеза соединения D, включающий следующие стадии:
(а) синтез диэтил-(3-оксо-1,3-дигидро-2-бензофуран-1-ил)фосфоната (G') из 2-карбоксибензальдегида (Н),
(б) синтез 2-фтор-5-[(Е/Z)-(3-оксо-2-бензофуран-1(3Н)-илиден)метил]бензонитрила (Е) из диэтил-(3-оксо-1,3-дигидро-2-бензофуран-1-ил)фосфоната.
Предпочтительно не выделять соединение G' в ходе синтеза. Данный способ исключает применение натриевой соли диметилфосфита, которая является нестабильной (Pelchowicz, et al., J. Chem. Soc, 4348-4350 (1961)) в спиртовом растворе. Предпочтительно, чтобы стадия (а) проходила в 2-метилтетрагидрофуране, в котором натриевая соль диэтилфосфита является стабильной. Соль можно получить in situ путем добавления диэтилфосфита к охлажденному раствору трет-амилата натрия в 2-метилтетрагидрофуране. После реакции с натриевой солью диэтилфосфита можно провести реакцию с метансульфокислотой.
Стадию (б) можно проводить в 2-метилтетрагидрофуране с добавлением триэтиламина.
Способ синтеза соединения D может дополнительно включать следующие стадии:
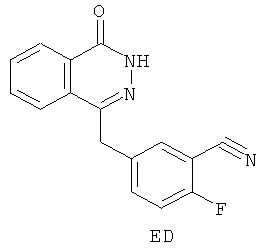
(в) синтез 2-фтор-5-[(4-оксо-3,4-дигидрофталазин-1-ил)метил]бензонитрила (ED):
из соединения Е путем взаимодействия с гидразин гидратом, и
(г) синтез соединения D из соединения ED путем взаимодействия с гидроксидом натрия.
Стадию (в) можно осуществить с использованием от 1.1 до 1.3 эквивалента гидразин гидрата в тетрагидрофуране с последующей нейтрализацией избытка гидразин гидрата уксусной кислотой.
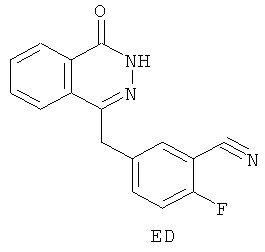
Согласно шестому аспекту в настоящем изобретении предложено соединение ED:
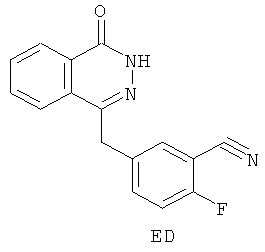
и его применение в синтезе соединения D.
Согласно следующему аспекту в настоящем изобретении предложена соль минеральной кислоты 1-(циклопропилкарбонил)пиперазина и способ ее синтеза путем реакции пиперазина с уксусной кислотой с последующим добавлением циклопропанкарбонилхлорида.
Согласно седьмому аспекту в настоящем изобретении предложена фармацевтическая композиция, содержащая соединение согласно первому аспекту изобретения и фармацевтически приемлемый носитель или разбавитель.
Согласно восьмому аспекту в настоящем изобретении предложено соединение согласно первому аспекту, подходящее для применения в способе лечения человека или животного.
Согласно девятому аспекту в настоящем изобретении предложено применение соединения согласно первому аспекту изобретения для получения лекарственного препарата для ингибирования активности PARP.
Согласно другим аспектам в изобретении предложено применение соединения согласно первому аспекту изобретения для получения лекарственного препарата, подходящего для лечения сосудистого заболевания, септического шока, ишемического повреждения, нейротоксичности, геморрагического шока, вирусной инфекции или заболеваний, которые поддаются облегчению путем ингибирования активности PARP.
Согласно еще одному аспекту в изобретении предложено применение соединения согласно первому аспекту изобретения для получения лекарственного препарата, подходящего для применения в качестве вспомогательного средства при лечении рака или для повышения восприимчивости раковых клеток по отношению к лечению ионизирующим излучением или химиотерапевтическими агентами.
Согласно другим аспектам в настоящем изобретении предложен способ лечения заболевания, поддающегося облегчению путем ингибирования активности PARP, включающий введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения согласно первому аспекту изобретения, предпочтительно - в форме фармацевтической композиции, и способ лечения рака, включающий введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения согласно первому аспекту изобретения, предпочтительно в форме фармацевтической композиции, одновременно или последовательно с ионизирующим излучением или химиотерапевтическими агентами.
Согласно другим аспектам настоящего изобретения указанные соединения можно использовать для получения лекарственного препарата для лечения ракового заболевания, характеризующегося нарушением пути репарации двухнитевого разрыва (ДНР) ДНК, зависимой от гомологичной рекомбинации (ГР), или для лечения пациента, страдающего раковым заболеванием, связанным с недостаточной активностью ГР-зависимой репарации ДНК, при этом указанный способ включает введение указанному пациенту терапевтически эффективного количества указанного соединения.
Путь ГР-зависимой репарации ДНР ДНК устраняет двухнитевые разрывы (ДНР) в ДНК посредством гомологичного механизма с восстанавлением непрерывности спирали ДНК (К.К.Khanna and S.P.Jackson, Nat. Genet. 27(3): 247-254 (2001)). Компоненты пути ГР-зависимой репарации ДНР ДНК включают, в частности, но без ограничения ATM (NM_000051), RAD51 (NM_002875), RAD51L1 (NM_002877), RAD51C (NM_002876), RAD51L3 (NM_002878), DMC1 (NM_007068), XRCC2 (NM_005431), XRCC3 (NM_005432), RAD52 (NM_002879), RAD54L (NM_003579), RAD54B (NM_012415), BRCA1 (NM_007295), BRCA2 (NM_000059), RAD50 (NM_005732), MRE11A (NM_005590) и NBS1 (NM_002485). Другие белки, участвующие в пути ГР-зависимой репарации ДНР ДНК, включают регуляторные факторы, в частности, EMSY (Hughes-Davies, et al., Cell, 115, pp 523-535). Компоненты ГР описаны также в работе Wood, et. at., Science, 291, 1284-1289 (2001).
Рак, связанный с нарушением ГР-зависимой репарации ДНР ДНК, может содержать или включать одну или несколько раковых клеток, которые имеют пониженную или нулевую способность к репарации ДНР ДНК посредством указанного пути по сравнению с нормальными клетками, т.е. активность пути ГР-зависимой репарации ДНР ДНК может быть пониженной или отсутствовать в одной или нескольких раковых клетках.
Активность одного или нескольких компонентов пути ГР-зависимой репарации ДНР ДНК может отсутствовать в одной или нескольких раковых клетках индивидуума, страдающего раковым заболеванием, связанным с недостаточной репарацией ДНР ДНК, зависимой от ГР. Компоненты пути ГР-зависимой репарации ДНР ДНК хорошо известны специалистам в данной области техники (см., например, Wood, et al., Science, 291, 1284-1289 (2001)) и включают вышеуказанные компоненты.
Согласно некоторым предпочтительным вариантам реализации изобретения раковые клетки могут иметь фенотип с дефектом BRCA1 и/или BRCA2, т.е. активность BRCA1 и/или BRCA2 в раковых клетках может быть уменьшена или отсутствовать. Раковые клетки такого фенотипа могут иметь нарушение по BRCA1 и/или BRCA2, т.е. экспрессия и/или активность BRCA1 и/или BRCA2 может быть уменьшена или отсутствовать в раковых клетках, например, вследствие мутации или полиморфизма кодирующей нуклеиновой кислоты или вследствие амплификации, мутации или полиморфизма в гене, кодирующем регуляторный фактор, например, в гене EMSY, который кодирует регуляторный фактор BRCA2 (Hughes-Davies, etal., Cell, 115, 523-535).
BRCA1 и BRCA2 известны как опухолеподавляющие гены, аллели дикого типа которых часто утрачиваются в опухолях гетерозиготных носителей (Jasin М., Опсодепе, 21(58), 8981-93 (2002); Tutt, et al., Trends Mol Med., 8(12), 571-6, (2002)). Связь мутаций BRCA1 и/или BRCA2 с раком молочной железы хорошо известна специалистам в данной области техники (Radice, P.J., Exp.Clin Cancer Res., 21 (3 Suppl), 9-12 (2002). Известно также, что амплификация гена EMSY, который кодирует фактор связывания BRCA2, связана с раком молочной железы и яичника.
Носители мутаций в BRCA1 и/или BRCA2 также подвержены риску развития рака яичника, простаты и поджелудочной железы.
Согласно некоторым предпочтительным вариантам реализации индивидуум является гетерозиготным по одной или нескольким вариациям, в частности, мутациям и полиморфизмам, в BRCA1 и/или BRCA2 или в их регуляторе. Способы определения вариации в BRCA1 и BRCA2 хорошо известны специалистам в данной области техники и описаны, например, в ЕР 699754, ЕР 705903, Neuhausen, S.L. and Ostrander, E.A., Genet. Test., 1, 75-83 (1992); Chappnis, P.O. and Foulkes, W.D., Cancer Treat Res, 107, 29-59 (2002); Janatova М., et al., Neoplasma, 50(4), 246-50 (2003); Jancarkova, N., Ceska Gynekol., 68(1), 11-6 (2003). Определение амплификации фактора связывания BRCA2 EMSY описано в работе Hughes-Davies, et al., Cell. 115, 523-535.
Мутации и полиморфизмы, связанные с раком, можно обнаружить на уровне нуклеиновых кислот путем определения присутствия вариантной последовательности нуклеиновых кислот или на уровне белка путем определения присутствия варианта (т.е. мутанта или аллельного варианта) полипептида.
Краткое описание чертежей
На Фиг.1 представлен ЯМР-спектр соединения А после обработки водой (пример 1).
На Фиг.2 представлена порошковая рентгенограмма соединения А после обработки водой (пример 1).
На Фиг.3 представлена репрезентативная порошковая рентгенограмма формы А соединения А.
На Фиг.4 представлена репрезентативная порошковая рентгенограмма сольватной формы соединения А.
На Фиг.5 представлена репрезентативная кривая ДСК формы А соединения А, полученная при нагревании от 25°С до 325°С со скоростью 10°С в минуту.
Применение
В настоящем изобретении предложено соединение А в виде формы А в качестве активного соединения, в частности, проявляющего активность в отношении ингибирования активности PARP.
Термин "активный", используемый в настоящем описании, относится к соединению, которое способно ингибировать активность PARP. В приведенных далее примерах описан анализ, который можно успешно использовать для определения способности соединения ингибировать PARP.
В настоящем изобретении также предложен способ ингибирования активности PARP в клетке, включающий приведение указанной клетки в контакт с эффективным количеством активного соединения, предпочтительно в виде фармацевтически приемлемой композиции. Такой способ можно осуществить in vitro или in vivo.
Так, например, образец клеток можно вырастить in vitro, привести активное соединение в контакт с указанными клетками и наблюдать влияние соединения на эти клетки. Примером такого "влияния" может служить величина репарации ДНК, произведенной за определенное время. Если активное соединение оказывает свое влияние на определенные клетки, это можно использовать в качестве прогностического или диагностического маркера эффективности соединения в способах лечения пациента, являющегося носителем клеток того же типа.
Термин "лечение", используемый в контексте лечения патологического состояния, в общем случае относится к лечению и терапии человека или животного (например, при применении в ветеринарии), при котором достигается некоторый желаемый терапевтический эффект, например, ингибирование прогресса патологического состояния, и включает снижение скорости прогрессирования, прекращение прогрессирования, облегчение патологического состояния и излечение патологического состояния. Кроме того, указанный термин включает лечение в качестве профилактической меры (т.е. профилактики).
Термин "вспомогательное средство", используемый в настоящем описании, относится к применению активного соединения в сочетании с известными терапевтическими средствами. Такие средства включают цитотоксические режимы применения лекарственных препаратов и/или ионизирующего излучения, которые используют при лечении различных типов рака. Известно, в частности, что указанные активные соединения усиливают действие ряда противораковых химиотерапевтических препаратов, включая топоизомеразный класс ядов (например, топотекан, иринотекан, рубитекан), большинство известных алкилирующих агентов (например, DTIC (дакарбазин), темозоламид) и препараты на основе платины (например, карбоплатин, цисплатин), используемые при лечении рака.
Активное соединение можно использовать также в качестве добавки к клеточной культуре для ингибирования PARP, например, для сенсибилизации клеток к действию известных химиотерапевтических агентов или ионизирующего излучения in vitro.
Кроме того, активное соединение можно использовать как часть анализа in vitro, например, для того, чтобы определить, насколько эффективно конкретное соединение для лечения кандидата-хозяина.
Введение
Активное соединение или фармацевтическая композиция, содержащая активное соединение, можно вводить субъекту любым подходящим способом, системно/периферически или по месту желаемого действия, в том числе, но без ограничения, перорально (например, путем проглатывания), топически (в том числе трансдермально, интраназально, окулярно, трансбуккально и сублингвально), пульмонарно (например, путем ингаляционной или инсуффляционной терапии при использовании, например, аэрозоля, в частности, через рот или нос), ректально, вагинально, парентерально, например, путем инъекции, в том числе подкожно, внутрикожно, внутримышечно, внутривенно, внутриартериально, внутрисердечно, подоболочечно, интраспинально, интракапсулярно, подкапсулярно, интраорбитально, интраперитонеально, интратрахеально, субкутикулярно, интраартикулярно, субарахноидально, интрастернально, а также путем имплантации депо, например, подкожно или внутримышечно.
Субъект может представлять собой эукариот, животное, позвоночное животное, млекопитающее, грызуна (например, морскую свинку, хомяка, крысу, мышь), представителя семейства мышиных (например, мышь), представителя семейства псовых (например, собаку), представителя семейства кошачьих (например, кошку), представителя семейства лошадиных (например, лошадь), примата, обезьяну (например, мартышкообразную или человекообразную), представителя семейства мартышкообразных (например, мартышку, бабуина), представителя семейства человекообразных (гориллу, шимпанзе, орангутанга, гиббона) или человека.
Составы
Активное соединение можно вводить отдельно, однако предпочтительно введение соединения в составе фармацевтической композиции (например, состава), содержащей активное соединение, как определено выше, вместе с одним или несколькими фармацевтически приемлемыми носителями, адъювантами, эксципиентами, разбавителями, наполнителями, буферами, стабилизаторами, консервантами, лубрикантами или другими веществами, хорошо известными специалистам в данной области техники, и, возможно с другими терапевтическими или профилактическими агентами.
Таким образом, в настоящем изобретении также предложены фармацевтические композиции, как определено выше, и способы получения фармацевтической композиции, включающие смешивание активного соединения, как определено выше, с одним или несколькими фармацевтически приемлемыми носителями, эксципиентами, буферами, адъювантами, стабилизаторами или другими веществами, как указано в настоящем описании, таким образом, чтобы активное соединение оставалось в виде кристаллической формы А.
Термин "фармацевтически приемлемый", используемый в данном описании, относится к соединениям, материалам, композициям и/или дозированным формам, которые на основании весомого медицинского заключения являются подходящими для применения в контакте с тканями субъекта (например, человека), не вызывая при этом излишней токсичности, раздражения, аллергической реакции или других проблем или осложнений, в соответствии с разумным отношением риска и ожидаемой пользы. Каждый из носителей, эксципиентов и т.д. также должен быть "приемлемым" в смысле совместимости с другими ингредиентами состава.
Подходящие носители, разбавители, эксципиенты и т.д. указаны в стандартной фармацевтической литературе. См., например, "Handbook of Pharmaceutical Additives", 2nd Edition (eds. M.Ash and I.Ash), 2001 (Synapse Information Resources, Inc., Endicott, New York, USA), "Remington's Pharmaceutical Sciences", 20th edition, pub. Lippincott, Williams & Wilkins, 2000 и "Handbook of Pharmaceutical Excipients", 2nd edition, 1994.
Составы согласно изобретению могут быть легко представлены в виде стандартных лекарственных форм и могут быть получены любыми способами, хорошо известными специалистам в области фармацевтики. Такие способы включают операцию объединения активного соединения с носителем, содержащим один или несколько вспомогательных ингредиентов. В общем случае составы получают путем равномерного и тщательного смешивания активного соединения с жидкими носителями или тонкоизмельченными твердыми носителями, или с теми и другими, а затем, в случае необходимости, формования продукта.
Составы могут иметь форму суспензий, таблеток, гранул, порошков, капсул, крахмальных облаток, пилюль или паст.
Составы, подходящие для перорального введения (например, путем проглатывания), могут быть получены в форме отдельных единиц, в частности, капсул, крахмальных облаток или таблеток, каждая из которых содержит заранее заданное количество активного соединения, порошка или гранул, в форме суспензии в водной или неводной жидкости или в форме пасты.
Таблетку можно изготовить известными способами, например, прессованием или формовкой, возможно, с добавлением одного или нескольких вспомогательных ингредиентов. Прессованные таблетки можно получить путем прессования на соответствующем оборудовании активного соединения в свободнотекучей форме, в частности, в форме порошка или гранул, возможно смешанного с одним или несколькими связующими (например, с повидоном, желатином, гуммиарабиком, сорбитом, трагакантом, гидроксипропилметилцеллюлозой), наполнителями или разбавителями (например, с лактозой, микрокристаллической целлюлозой, дикальций-фосфатом), лубрикантами (например, со стеаратом магния, тальком, диоксидом кремния), дезинтегрирующими агентами (например, с крахмалгликолятом натрия, поперечно-сшитым повидоном, поперечно-сшитой натрийкарбоксиметилцеллюлозой), поверхностно-активными или диспергирующими или смачивающими агентами (например, с лаурилсульфатом натрия) и консервантами (например, с метил-п-гидроксибензоатом, пропил-п-гидроксибензоатом, сорбиновой кислотой). Формованные таблетки можно получить путем формования в соответствующей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. На таблетки можно нанести покрытие или насечки, при этом состав таблеток может быть подобран таким образом, чтобы обеспечивать замедленное или регулируемое высвобождение применяемого активного соединения, например, с использованием гидроксипропилметилцеллюлозы в различных соотношениях, с обеспечением требуемого профиля высвобождения. На таблетки можно нанести энтеросолюбильное покрытие, чтобы обеспечить высвобождение в определенных частях кишечника, а не в желудке.
Капсула может содержать активное соединение в виде суспензии.
Составы, подходящие для топического введения (например, трансдермального, интраназального, окулярного, трансбуккального и сублингвального), можно получать в форме пасты.
Составы, подходящие для топического введения в глаз, включают также глазные капли, в которых активное соединение суспендировано в подходящем носителе, в частности, в водном растворителе, подходящем для растворения данного активного соединения.
Составы, подходящие для назального введения, в которых носитель является твердым веществом, в частности, крупнозернистым порошком с размером частиц в пределах примерно от 20 до примерно 500 микрон, вводят таким же образом, как нюхательный табак, т.е. путем быстрого вдыхания через носовой проход из контейнера с порошком, находящегося в непосредственной близости от носа.
Составы, подходящие для введения путем ингаляции, включают распыляемые аэрозоли, содержащиеся в контейнере под давлением, требующие применения подходящего пропеллента, в частности, дифхлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или других подходящих газов.
Дозировка
Следует понимать, что подходящая дозировка активного соединения и композиций, содержащих указанное активное соединение, может быть различной для каждого конкретного пациента. Определение оптимальной дозы в общем случае включает соотнесение терапевтической пользы и риска или вредных побочных эффектов лечения согласно настоящему изобретению. Выбранная дозировка зависит от множества факторов, включая, без ограничения, активность конкретного соединения, способ введения, время введения, скорость выведения соединения, длительность лечения, другие лекарственные препараты, соединения и/или материалы, применяемые в комбинации, возраст, пол, вес, патологическое состояние, общее состояние здоровья и анамнез конкретного пациента. Дозу соединения и способ введения в конечном итоге определяет лечащий врач, при этом в общем случае дозирование должно обеспечивать в месте действия локальные концентрации, обеспечивающие желаемый эффект, не вызывая значительных опасных или вредных побочных эффектов.
Введение in vivo можно осуществлять в виде однократной дозы, непрерывно или последовательно (например, дробными дозами через подходящие интервалы времени) на протяжении курса лечения. Способы определения наиболее эффективных способов введения и доз хорошо известны специалистам в данной области техники и изменяются в зависимости от конкретного состава, применяемого для лечения, цели лечения, целевой клетки, подвергаемой воздействию, и субъекта, подвергаемого лечению. Однократное или многократное введение можно осуществлять в соответствии с величиной дозы и схемой дозирования, выбранными лечащим врачом.
В общем случае подходящая доза активного соединения находится в пределах примерно от 10 мг до примерно 600 мг на м2 поверхности тела и вес субъекта в день.
Примеры
Пример 1: Синтез соединения А
Исходное вещество (D) синтезировали способом, описанным в WO 2004/080976.
Способы
Препаративная ВЭЖХ
Образцы очистили при помощи масс-направленной системы очистки Waters с использованием насоса Water 600 LC, колонки Waters Xterra C18 (5 мкм 19 мм × 50 мм) и масс-спектрометра Micromass ZQ в режиме определения положительных ионов при электрораспылительной ионизации. Подвижные фазы А (0,1% муравьиной кислоты в воде) и В (0,1% муравьиной кислоты в ацетонитриле) использовали с применением градиента от 5% В до 100% в течение 7 минут, выдержка в течение 3 минут, при расходе 20 мл/мин.
Аналитическая ВЭЖХ с масс-спектрометрией
Анализ методом ВЭЖХ провели с использованием насоса Spectra System P4000 и колонки Jones Genesis C18 (4 мкм, 50 мм × 4,6 мм). Подвижные фазы А (0,1% муравьиной кислоты в воде) и В (ацетонитрил) использовали с применением градиента от 5% В в течение 1 минуты с подъемом до 98% через 5 минут, выдержка в течение 3 минут, при скорости потока 2 мл/мин. Детектирование проводили при помощи детектора TSP UV 6000LP при 254 нм УФ и в диапазоне 210-600 нм с фотодиодной матрицей. Использовали масс-спектрометр Finnigan LCQ в режиме определения положительных ионов при электрораспылении.
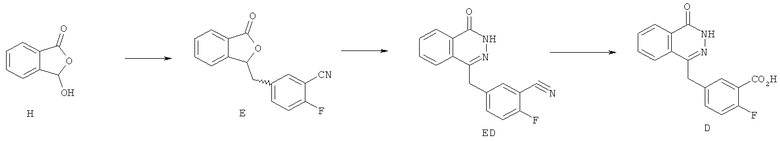
ЯМР
Спектры 1Н ЯМР записывали при помощи спектрометра Bruker DPX 300 при частоте 300 МГц. Химические сдвиги определяли в частях на миллион (ррт) по шкале δ по отношению к тетраметилсилановому внутреннему стандарту. Если не указано иное, все образцы растворяли в диметилсульфоксиде (ДМСО-d6).
(а) Сложный трет-бутиловый эфир 4-[2-фтор-5-(4-оксо-3,4-дигидрофталазин-илметил)-бензоил]пиперазин-1-карбоновой кислоты (соединение С)
В перемешанный раствор исходного вещества D (850 г) в диметилацетамиде (ДМА) (3561 мл) при температуре окружающей среды в атмосфере азота добавили одной порцией HBTU 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурониумгексафторфосфат) (1402 г). Затем добавили основание Хюнига (iPr2Net, 1096 мл), поддерживая температуру в пределах от 15 до 25°С, и раствор 1-Вос-пиперазина (637 г) в ДМА (1428 мл) также при поддержании температуры в пределах от 15 до 25°С.
Раствор перемешивали при температуре окружающей среды в течение 2 часов и отобрали образцы для определения полноты протекания реакции (методом ВЭЖХ). После завершения реакции раствор перелили в интенсивно перемешиваемую воду (17085 мл), поддерживая температуру в пределах от 15 до 25°С, затем отфильтровали осадок, промыли водой (2×7131 мл), гексаном (2×7131 мл) и метил-трет-бутиловым эфиром (МТБЭ) (2×3561 мл). Осадок высушили в течение ночи, а затем отобрали образцы для определения содержания воды и химической чистоты.
Затем эту реакцию повторили, см. таблицу:
(б) 4-[4-фтор-3-(пиперазин-1-карбонил)-бензил]-2Н-фталазин-1-он (соединение В)
В перемешанный раствор промышленных метилированных спиртов (ПМС) (2200 мл) и концентрированной HCl (4400 мл) при температуре окружающей среды в атмосфере азота порциями добавили соединение С (2780.2 г), при этом пенообразование регулировали скоростью добавления. Затем раствор перемешали при 15-25°С в течение 30 минут и отобрали образец для определения полноты протекания реакции (методом ВЭЖХ).
После завершения раствор выпарили, чтобы удалить ПМС, а водный раствор экстрагировали CH2Cl2 (2×3500 мл) и откорректировали рН>8 при помощи концентрированного аммиака. Полученную суспензию разбавили водой (10000 мл) и экстрагировали CH2Cl2 (4×3500 мл), промыли водой (2×2000 мл), осушили над MgSO4 (250 г) и выпарили. Сырой продукт суспендировали в CH2Cl2 (3500 мл) и добавили в МТБЭ (5000 мл). Образовавшуюся суспензию отфильтровали и высушили при 50°С в течение ночи, получив 611,0 г (выход 58,5%) вещества с чистотой 94,12%.
4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-он (соединение А)
В перемешанную суспензию соединения В (1290 г) в CH2Cl2 (15480 мл) в атмосфере азота добавили по каплям предварительно перемешанный раствор триэтиламина (470 мл) и циклопропанкарбонилхлорида (306 мл) в CH2Cl2 (1290 мл), поддерживая температуру ниже 20°С. Затем раствор перемешали при 10-15°С в течение 15 минут и отобрали образец для определения полноты протекания реакции. В реакционной смеси обнаружили всего 1,18% исходного вещества В, поэтому реакцию посчитали завершенной и продукт реакции подвергли следующей обработке.
Реакционную смесь промыли водой (7595 мл), 5% раствором лимонной кислоты (7595 мл), 5% раствором карбоната натрия (7595 мл) и снова водой (7595 мл). Органический слой осушили над сульфатом магния (500 г).
Затем отделили слой продукта, содержащий CH2Cl2, отфильтровали через целит и перенесли в сосуд объемом 25 л. Отогнали CH2Cl2 (8445 мл) при помощи перегонки и добавили этанол (10000 мл). Продолжили перегонку, заменяя каждые 4000 мл удаляемого дистиллята этанолом (4000 мл) до тех пор, пока температура головной фракции не достигла 73.7°С. Затем объем реакционной смеси уменьшили (до 7730 мл), при этом ее температура достигла 78.9°С, и выдержали реакционную смесь в течение ночи для охлаждения до 8°С. Затем отфильтровали образовавшийся осадок, промыли этанолом (1290 мл) и высушили в течение ночи при 70°С.
Выход = 1377.3 г (90%). Чистота согласно анализу способом ВЭЖХ (99.34% [площадь в %]). Содержание этанола - 0.01% и CH2Cl2 - 0.01% согласно анализу методом газовой хроматографии.
Спектр ЯМР 1H соединения А (ДМСО-d6) после обработки водой показан на фигуре 1.
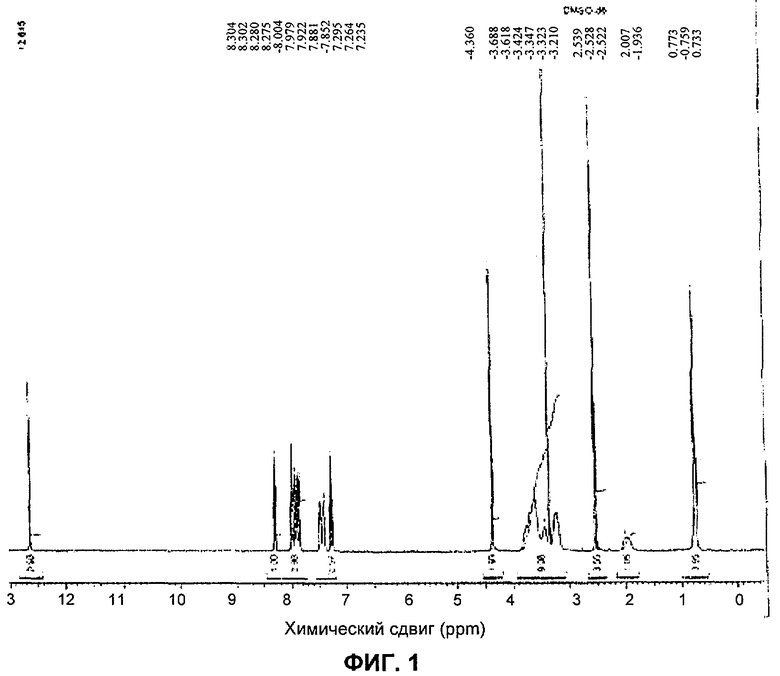
Порошковая рентгенограмма соединения А после обработки водой показана на фигуре 2, где указанное соединение представляет собой форму А.
Пример 2: Альтернативный способ синтеза соединения А с использованием 1-(циклопропилкарбонил)пиперазина
Способы (также для примеров 3 и 4)
ЯМР
Спектры ЯМР 1H регистрировали при помощи спектрометра DPX 400 при частоте 400 МГц. Химические сдвиги определяли в частях на миллион (ppm) по шкале δ по отношению к тетраметилсилановому внутреннему стандарту. Если не указано иное, все образцы растворяли в диметилсульфоксиде (ДМСС-d6).
Масс-спектры
Масс-спектры регистрировали при помощи масс-спектрометрической ионной ловушки Agilent XCT с использованием тандемной масс-спектрометрии (MS/MS) для подтверждения структуры. Прибор работал в режиме определения положительных ионов при электрораспылительной ионизации.
(а) 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-он (соединение А)
4-[2-фтор-5-(4-оксо-3,4-дигидрофталазин-1-ил)метил]-бензойную кислоту (D) (15.23 г, 51.07 ммоль) суспендировали при перемешивании в атмосфере азота в ацетонитриле (96 мл). Добавили диизопропилэтиламин (19.6 мл, 112,3 ммоль), а затем - 1-циклопропилкарбонилпиперазин (I) (9.45 г, 61.28 ммоль) и ацетонитрил (1 мл). Реакционную смесь охладили до 18°С. В течение 30 минут добавили о-бензотриазол-1-ил-тетраметилуронийгексафторфосфат (25.18 г, 66.39 ммоль) и перемешивали реакционную смесь в течение 2 часов при температуре окружающей среды. Реакционную смесь охладили до 3°С и выдержали при этой температуре в течение 1 часа, а затем отфильтровали. Отфильтрованный осадок промыли холодным (3°С) ацетонитрилом (20 мл), а затем высушили под вакуумом при максимальной температуре 40°С, получив титульное соединение в виде бледно-желтого порошка (20.21 г).
Масс-спектр: МН+435
ЯМР 1Н (400 МГц, ДМСО-d6) δ: 0.70 (m, 4Н), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
Пример 3: Альтернативный способ синтеза соединения А с использованием солянокислой соли 1-(циклопропилкарбонил)пиперазина
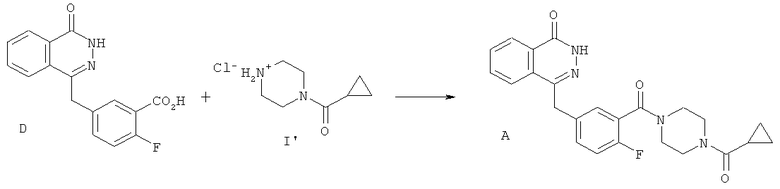
(а) Солянокислая соль 1-(циклопропилкарбонил)пиперазина (I')
В уксусную кислоту (700 мл) в течение 15 минут порциями добавили пиперазин (50.00 г, 0.581 моль) при перемешивании в атмосфере азота. Затем реакционную смесь нагрели до 40°С и выдержали при этой температуре до полного растворения. В течение 15 минут добавили циклопропанкарбонилхлорид (59.2 мл, 0.638 моль). Реакционную смесь перемешали при температуре окружающей среды в течение ночи. Затем реакционную смесь отфильтровали, а фильтрат подвергли перегонке при пониженном давлении до получения примерно 430 мл дистиллята. В реакционную смесь добавили толуол (550 мл) и продолжили перегонку при пониженном давлении до получения еще 400 мл дистиллята. Добавили еще одну порцию толуола (550 мл) и продолжили перегонку при пониженном давлении до получения 350 мл дистиллята. Полученную суспензию разбавили толуолом (200 мл) и перемешивали в течение ночи. Затем добавили толуол (500 мл), чтобы разбавить суспензию. Суспензию отфильтровали, промыли толуолом (100 мл) и высушили под вакуумом при 40°С, получив титульное соединение в виде серовато-белого порошка (86.78 г).
Масс-спектр: МН+155
ЯМР 1Н (400 МГц, D2O) δ: 0.92 (m, 4Н), 1.98 (m, 1Н), 3.29 (m, 2H), 3.38 (m, 2H), 3.84 (m, 2H), 4.08 (m, 2H).
(б) Соединение А
2-фтор-5-[(4-оксо-3,4-дигидрофталазин-1-ил)метил]бензойную кислоту (D) (0.95 г, 3.19 ммоль) суспендировали в ацетонитриле (4 мл) при перемешивании в атмосфере азота. Добавили 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурониумгексафторфосфат (HBTU) (1.45 г, 3.83 ммоль), а затем - соль HCl 1-циклопропилкарбонилпиперазина (I') (0.73 г, 3.83 ммоль). В течение 3 минут добавили диизопропилэтиламин (1.39 мл, 7,98 ммоль) и перемешивали реакционную смесь в течение ночи при температуре окружающей среды. Затем реакционную смесь охладили до 5°С, выдержали при этой температуре в течение 1 часа и отфильтровали. Отфильтрованный осадок промыли холодным (3°С) ацетонитрилом (2 мл), затем высушили под вакуумом при максимальной температуре 40°С, получив титульное соединение в виде бледно-желтого порошка (0.93 г).
(в) Рекристаллизация соединения А из водного метанола
Соединение А (9.40 г, 21.64 ммоль) с стадии (б) суспендировали в смеси воды (100 мл) и метанола (120 мл). Суспензию нагрели до обратного стекания при перемешивании. Образовавшийся мутный раствор охладили до 60°С и профильтровали через пластинку из гарболита. Фильтрующую пластинку промыли смесью воды (5 мл) и метанола (5 мл). Фильтрат подвергли дистилляции при атмосферном давлении до тех пор, пока не получили 115 мл дистиллята. Затем дистилляцию прекратили и выдержали образовавшуюся суспензию для охлаждения до температуры окружающей среды. Далее суспензию перемешали в течение ~18 часов и профильтровали. Отфильтрованный осадок промыли водой (20 мл), затем высушили в вакууме при максимальной температуре 60°С, получив титульное соединение в форме А в виде белого порошка (8.67 г).
Масс-спектр: МН+435
ЯМР 1Н (400 МГц, ДMCO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
(г) Рекристаллизация соединения А из водного этанола
Соединение А (9.40 г, 21.64 ммоль) с стадии (б) суспендировали в смеси воды (100 мл) и этанола (50 мл). Суспензию нагрели до обратного стекания при перемешивании. Образовавшийся мутный раствор охладили до 60°С и профильтровали через пластинку из гарболита. Фильтрующую пластинку промыли смесью воды (5 мл) и этанола (5 мл). Фильтрат подвергли дистилляции при атмосферном давлении до тех пор, пока не получили 53 мл дистиллята. Затем дистилляцию прекратили и выдержали образовавшуюся суспензию для охлаждения до температуры окружающей среды. Затем суспензию перемешали в течение ~18 часов и профильтровали. Отфильтрованный осадок промыли водой (20 мл), затем высушили в вакууме при максимальной температуре 60°С, получив титульное соединение в форме А в виде белого порошка (8.74 г).
Масс-спектр: МН+435
ЯМР 1Н (400 МГц. ДМСО-d6) δ: 0.70 (m, 4Н), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
Пример 4: Альтернативный способ синтеза соединения D
(а) 2-фтор-5-[(Е/Z)-(3-оксо-2-бензофуран-1(3Н)-илиден)метил]бензонитрил (E)
Трет-амилат натрия (99.00 г, 0.854 моль) и 2-метилтетрагидрофуран (960 мл) охладили до 2°С в атмосфере азота. По каплям добавили диэтилфосфит (110 мл, 0.855 моль), поддерживая температуру <5°С. Затем добавили 2-метилтетрагидрофуран (40 мл) для промывки линии, через которую осуществлялось добавление (line wash). Реакционную смесь перемешали при 2°С в течение 1 часа 40 минут. Добавили раствор 2-карбоксибензальдегида (Н) (80 г, 0.533 моль) в 2-метилтетрагидрофуране (200 мл), поддерживая температуру < 7°С во время добавления. После этого добавили для промывки линии, через которую осуществлялось добавление, 2-метилтетрагидрофуран (40 мл). Реакционную смесь нагрели до 20°С и выдержали при 20°С в течение 20 минут. В течение 1 часа 10 минут добавили метансульфоновую кислоту (7 мл, 0.101 моль), а затем - 2-метилтетрагидрофуран (40 мл). Реакционную смесь перемешали при 20°С в течение ночи. Добавили метансульфоновую кислоту (7 мл, 0.101 моль), а затем - 2-метилтетрагидрофуран (7 мл) и перемешали реакционную смесь при 20°с в течение следующих 4 часов. Добавили воду (400 мл) при температуре окружающей среды и полученную двухфазную смесь перемешали при температуре окружающей среды в течение 20 минут. Нижний водный слой удалили, а в органический слой добавили раствор бикарбоната калия (53.50 г, 0.534 моль) в воде (400 мл). Двухфазную смесь перемешали при температуре окружающей среды в течение 20 минут и затем удалили нижний водный слой. Органическую фракцию (раствор диэтил(3-оксо-1,3-дигидро-2-бензофуран-1-ил)фосфоната) сохраняли. В органическую фракцию добавили 2-фтор-5-формилбензонитрил (64 г, 0.429 моль) и перемешали смесь при 20°С. По каплям добавили триэтиламин (66 мл, 0.473 моль), а затем 2-метилтетрагидрофуран (7 мл). Реакционную смесь перемешали при 20°С в течение ночи, затем охладили до 5°С, отфильтровали, промыли промышленным метилированным спиртом (480 мл), а затем высушили в вакууме при максимальной температуре 40°С, получив титульное соединение (91.2 г).
Масс-спектр: МН+266
ЯМР 1Н (400 МГц, ДМСО-d6) δ: 6.89 (s, 1H, главный изомер), 6.94 (s, 1H, побочный изомер), 7.40 (dd, 1H, побочный изомер), 7.58 (t, 1H, оба изомера), 7.70 (t, 1H, оба изомера), 7.89, (t, 1H, оба изомера), 7.95 (d, 1H, оба изомера), 8.05 (d, 1H, оба изомера), 8.15 (m, 2H, главный изомер).
(б) 2-фтор-5-[(4-оксо-3,4-дигидрофталазин-1-ил)метил]бензонитрил (ED)
2-фтор-5-[(Е/Z)-(3-оксо-2-бензофуран-1(3Н)-илиден)метил]бензонитрил (Е) (20 г, 75.40 ммоль) и тетрагидрофуран (200 мл) перемешали при температуре окружающей среды в атмосфере азота в течение 30 минут. Добавили гидразин моногидрат (4.40 мл, 90.53 ммоль), а затем для промывки линии, через которую осуществлялось добавление, тетрагидрофуран (4 мл). Реакционную смесь перемешали при температуре окружающей среды в течение 1 часа 45 минут. Добавили уксусную кислоту (1.10 мл, 19.20 ммоль) и нагрели реакционную смесь до 60°С. Реакционную смесь выдержали при 60°С в течение ночи. Затем реакционную смесь охладили до 50°С и по каплям добавили воду (200 мл). Во время добавления поддерживали температуру равной 45°С. Реакционную смесь охладили до 20°С, отфильтровали, промыли смесью воды (30 мл) и тетрагидрофурана (30 мл), а затем высушили под вакуумом при максимальной температуре 40°С, получив титульное соединение (18.7 г).
Масс-спектр: МН+280
ЯМР 1Н (400 МГц, ДМСО-d6) δ: 4.38 (s, 4Н), 7.46 (t, 1H), 7.72 (m, 1H), 7.85 (dt, 1H), 7.92 (m, 2H), 7.99 (d, 1H), 8.27 (dd, 1H), 12.57 (s, 1H).
(в) 2-фтор-5-(4-оксо-3,4-дигидрофталазин-1-илметил)-бензойная кислота (D)
2-фтор-5-[(4-оксо-3,4-дигидрофталазин-1-ил)метил]бензонитрил (ED) (9.60 г, 34.37 ммоль) и воду (40 мл) перемешали при 20°С. Добавили 2М гидроксид натрия (36 мл, 72 ммоль), реакционную смесь нагрели до 90°С и выдержали при этой температуре в течение ночи. Затем охладили реакционную смесь до температуры окружающей среды и отфильтровали. Фильтровальную пластинку промыли водой (10 мл), и объединенный фильтрат добавили к 2М HCl (56 мл, 112.00 ммоль) при 60°С в течение 40 минут. Полученную суспензию охладили до 50°С, отфильтровали, промыли водой (57 мл) и высушили под вакуумом при максимальной температуре 60°С, получив титульное соединение (9.72 г).
Масс-спектр: МН+299
ЯМР 1H (400 МГц, ДМСО-d6) δ: 4.36 (s, 2H), 7.24 (dd, 1H), 7.59 (m, 1H), 7.84 (dt, 2H), 7.90 (dt, 1H), 7.98 (d, 1H), 8.27 (dd, 1H), 12.59 (s, 1H), 13.22 (br s, 1H).
Пример 5: Перекристаллизация соединения А из водного раствора этанола
4-[3-{[4-(циклопропанкарбонил)пиперазин-1-ил]карбонил}-4-фторбензил]фталазин-1(2Н)-он (соединение А) (20.00 г, 44.66 ммоль) суспендировали в смеси воды (50 мл) и этанола (150 мл). Суспензию нагревали до кипения с обратным холодильником при перемешивании. Образовавшийся раствор охладили до 70°С и отфильтровали. Фильтрующую пластинку промыли смесью воды (8 мл) и этанола (22 мл).
Фильтрат охладили до 45°С при перемешивании. Добавили 4-[3-{[4-(циклопропанкарбонил)пиперазин-1-ил]карбонил}-4-фторбензил] фталазин-1(2Н)-он (соединение А) в форме А (0.08 г) в качестве затравки. Полученную суспензию охладили до 20°С в течение 2.5 часов и перемешали при этой температуре в течение следующих 16 часов, чтобы обеспечить протекание кристаллизации. Добавили воду (200 мл) в течение 5 часов, поддерживая температуру равной 20°С. После добавления суспензию выдержали при 20°С в течение 2 часов.
Суспензию отфильтровали, отфильтрованный осадок промыли смесью этанола (24 мл) и воды (56 мл). Полученный осадок высушили под вакуумом при 40-60°С, получив титульное соединение (форму А) в виде серовато-белого порошка (18.1 г).
Способы получения данных, представленных на фигурах 3-5
Порошковая рентгенограмма - фигура 3 (соединение А в виде формы А)
Дифракцию рентгеновских лучей на порошке определяли при помощи дифрактометра Bruker D5000 (длина волны рентгеновских лучей 1.5418 Å, источник Сu, напряжение 40 кВ, термоэлектронная эмиссия 40 мА). Образцы сканировали в диапазоне 2-40° 2⊝ с шириной шага 0.02° и временем отсчета импульсов, составляющим 4 секунды.
Порошковая рентгенограмма - фигура 4 (соединение А в виде сольватированной формы)
Дифракцию рентгеновских лучей на сольватном семействе определяли при помощи дифрактометра Inel XRG-3000 (длина волны рентгеновских лучей 1.5418 Ǻ, источник Сu, напряжение 40 кВ, термоэлектронная эмиссия 30 мА). Образцы сканировали в диапазоне 2.5-40° 2⊝ с типичной шириной шага 0.03° и суммарным временем отсчета импульсов 300 секунд.
Дифференциальная сканирующая калориметрия (ДСК) - фигура 5
Кривые ДСК регистрировали при помощи прибора Mettler DSC820E с робототехнической системой TSO801RO. Типично менее 5 мг вещества, содержащегося в 40 мкл алюминиевой кювете с перфорированной крышкой, нагревали в пределах температуры от 25°С до 325°С при постоянной скорости нагревания 10°С в минуту. При этом использовали продувку газообразным азотом с расходом 100 мл в минуту.
Пример 6
Ингибирующее действие
Для оценки ингибирующего действия активного соединения использовали следующий анализ, который позволяет определять величину IC50.
PARP млекопитающего, выделенный из ядерного экстракта клеток HeLa, инкубировали с Z-буфером (25 мМ Hepes (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота) (Sigma), 12.5 мМ MgCl2 (Sigma), 1 мМ ДТТ (дитиотреитол) (Sigma), 10% глицерин (Sigma), 0.001% NP-40 (Sigma), pH 7.4) в планшете с 96 лунками FlashPlates (торговая марка) (NEN, UK) и добавили указанные ингибиторы с различными концентрациями. Все соединения разбавили ДМСО и получили конечную концентрацию образцов от 10 до 0.001 мкМ с конечной концентрацией ДМСО 1% в лунке. Общий объем образца составлял 40 мкл.
После 10 минут инкубирования при 30°С инициировали реакции, добавив 10 мкл реакционной смеси, содержащей НАД (никотинамид аденин динуклеотид) (5 мкМ), 3H-НАД и 30-мерные олигомеры двухнитевых ДНК. Получили отмеченные положительные и отрицательные реакционные лунки в сочетании с лунками с соединениями (неизвестные), чтобы рассчитать % ферментной активности. Затем планшеты встряхивали в течение 2 минут и инкубировали при 30°С в течение 45 минут.
После инкубации погасили реакцию, добавив в каждую лунку 50 мкл 30% уксусной кислоты. Затем планшеты встряхивали в течение 1 часа при температуре окружающей среды.
Планшеты перенесли в систему TopCount NXT (торговая марка) (Packard, UK) для подсчета сцинтилляций. Записанные значения представляли собой число импульсов в минуту (cpm) после 30-секундного подсчета для каждой лунки.
Затем рассчитали % ферментной активности для соединения, используя следующее уравнение:
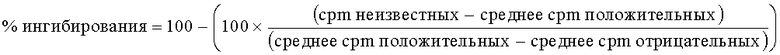
Рассчитали значения IC50 (концентрация, при которой ингибируется 50% ферментной активности) в пределах различных концентраций, обычно от 10 мкМ до 0.001 мкМ. Эти значения IC50 использовали для сравнения, чтобы определить повышенное потенцирование испытуемых соединений.
Соединение А имеет величину IC50, примерно равную 5 нМ.
Коэффициент потенцирования
Коэффициент потенцирования (PF50) для активного соединения рассчитывают как отношение IC50 роста контрольных клеток к IC50 роста клеток+ингибитор PARP. Кривые ингибирования роста как для контрольных, так и для обработанных соединением клеток показаны в присутствии алкилирующего агента - метилметансульфоната (ММС). Испытуемое соединение использовали с фиксированной концентрацией 0.2 микромоля. Концентрация ММС находилась в пределах от 0 до 10 мкг/мл.
Рост клеток оценивали при помощи анализа с сульфородамином В (SRB) (Skehan, P., et al., (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 82, 1107-1112). 2000 клеток HeLa поместили в каждую лунку микротитрационного планшета с 96 плоскодонными лунками объемом 100 мкл и инкубировали в течение 6 часов при 37°С. В лунки с клетками добавили чистую среду или среду, содержащую ингибитор PARP с конечной концентрацией 0.5, 1 или 5 мкМ. Клетки выращивали в течение 1 часа, а потом к необработанным клеткам или клеткам, обработанным ингибитором PARP, добавили ММС с определенным диапазоном концентраций (типично -0, 1, 2, 3, 5, 7 и 10 мкг/мл). Клетки, обработанные ингибитором PARP, использовали для определения ингибирования роста ингибитором PARP.
Клетки выдержали еще 16 часов, затем заменили среду и выращивали клетки в течение следующих 72 часов при 37°С. После этого среду удалили и закрепили клетки 100 мкл ледяной 10% (масса/объем) трихлоруксусной кислоты. Планшеты инкубировали при 4°С в течение 20 минут, а затем четыре раза промыли водой.
Каждую лунку клеток окрасили 100 мкл 0.4% (масса/объем) SRB в 1% уксусной кислоте в течение 20 минут с последующей четырехкратной промывкой 1% уксусной кислотой. Затем планшеты высушили в течение 2 часов при температуре окружающей среды. Краситель в окрашенных клетках солюбилюзировали, добавив в каждую лунку 100 мкл 10 мМ трис-основания. Планшеты осторожно встряхнули и выдержали при температуре окружающей среды в течение 30 минут, а затем измерили оптическую плотность при длине волны 564 нм при помощи микротитрационного планшет-ридера Microquant.
Соединение А имеет величину PF50 при 200 нМ, равную по меньшей мере 20.
| название | год | авторы | номер документа |
|---|---|---|---|
| 4-[3-(4-ЦИКЛОПРОПАНКАРБОНИЛ-ПИПЕРАЗИН-1-КАРБОНИЛ)-4-ФТОР-БЕНЗИЛ]-2Н-ФТАЛАЗИН-1-ОН | 2008 |
|
RU2487868C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2686314C1 |
| НОВЫЕ ФТАЛАЗИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2636585C2 |
| ДИГИДРОПИРИДОФТАЛАЗИНОНОВЫЕ ИНГИБИТОРЫ ПОЛИ(АДФ-РИБОЗА)ПОЛИМЕРАЗЫ | 2009 |
|
RU2514937C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПОЛИ(ADP-РИБОЗА) ПОЛИМЕРАЗЫ (PARP) | 2007 |
|
RU2472782C2 |
| ПИРИДАЗИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2008 |
|
RU2490265C2 |
| СОЕДИНЕНИЯ 8-ФТОРФТАЛАЗИН-1(2Н)-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ БРУТОНА | 2012 |
|
RU2622391C2 |
| ПРОИЗВОДНОЕ ФТАЛАЗИНОНКЕТОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2564527C2 |
| 4-ПИРИМИДИН- ИЛИ ПИРИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ ИНДОЛ-3-ИЛ-АЛКИЛПИПЕРАЗИНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2114111C1 |
| Производные бензимидазол-2-пиперазина, полезные в качестве ингибитора поли(АДФ-рибоза)-полимеразы (PARP) | 2014 |
|
RU2649002C2 |
Изобретение относится к 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-ону в виде кристаллической формы А, а также к способам его получения и фармацевтической композиции для ингибирования PARP на его основе. Технический результат - получена новая форма 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она, которая содержит меньше примесей и может использоваться для получения фармацевтических лекарственных форм большей степени чистоты. 9 н. и 3 з.п. ф-лы, 5 ил., 6 пр.
1. Соединение, представляющее собой 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-он в виде кристаллической формы А, порошковая рентгенограмма которого содержит следующие характеристические пики:
2. Соединение по п.1, отличающееся тем, что порошковая рентгенограмма указанного соединения содержит следующие характеристические пики:
3. Соединение по любому из пп.1 или 2, отличающееся тем, что температура начала плавления указанного соединения при анализе методом дифференциальной сканирующей калориметрии составляет 210,1°С±1°С при нагревании от 25°С до 325°С со скоростью 10°С в минуту.
4. Способ получения 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она (соединения А) в виде кристаллической формы А, включающий следующие стадии:
(1) кристаллизацию 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она из растворителя,
(2) обработку указанного кристаллического соединения А этанолом, если указанный исходный растворитель не является этанолом,
(3) обработку указанного кристаллического соединения А водой для удаления захваченного этанола,
(4) сушку полученного продукта.
5. Способ получения 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она (соединения А) в виде кристаллической формы А, включающий следующие стадии:
(1) кристаллизацию 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она из растворителя,
(2) нагревание указанного соединения в смеси с водой и С1-2 спиртом, если исходный растворитель, используемый при синтезе соединения А в виде указанной кристаллической формы, не является смесью воды и С1-2 спирта,
(3) перегонку указанной смеси при атмосферном давлении; и
(4) сушку полученного продукта.
6. Способ получения 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она (соединения А) в виде кристаллической формы А, включающий следующие стадии:
(1) суспендирование соединения А в смеси воды и C1-2 спирта в качестве растворителя,
(2) нагревание указанной суспензии до кипения с обратным холодильником,
(3) охлаждение указанного раствора и введение затравки, представляющей собой соединение А в виде формы А,
(4) сушку полученного продукта.
7. Способ синтеза 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она из 4-[4-фтор-3-(пиперазин-1-карбонил)-бензил]-2Н-фталазин-1-она, включающий следующую стадию:
(1) контролируемое добавление предварительно смешанного раствора триэтиламина и циклопропанкарбонилхлорида в органическом растворителе к раствору 4-[4-фтор-3-(пиперазин-1-карбонил)-бензил]-2Н-фталазин-1-она в этом же органическом растворителе при поддержании температуры указанного раствора равной менее 20°С.
8. Способ синтеза 4-[3-(4-циклопропанкарбонилпиперазин-1-карбонил)-4-фторбензил]-2Н-фталазин-1-она из 2-фтор-5-(4-оксо-3,4-дигидрофталазин-1-илметил)-бензойной кислоты, включающий взаимодействие 2-фтор-5-(4-оксо-3,4-дигидрофталазин-1-илметил)-бензойной кислоты с 1-(циклопропилкарбонил)пиперазином или его солью минеральной кислоты в растворителе в присутствии агента, связывающего амид, представляющего собой 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат (HBTU), и основанием, представляющим собой четвертичный амин при поддержании температуры при 25°С или менее.
9. Способ синтеза 2-фтор-5-(4-оксо-3,4-дигидрофталазин-1-илметил)-бензойной кислоты, включающий следующие стадии:
(а) синтез диэтил-(3-оксо-1,3-дигидро-2-бензофуран-1-ил)фосфоната из 2-карбоксибензальдегида в метилтетрагидрофуране по реакции с натриевой солью диэтилфосфита;
(б) синтез 2-фтор-5-[(Е/Z)-(3-оксо-2-бензофуран-1(3Н)-илиден)метил]бензонитрила из диэтил-(3-оксо-1,3-дигидро-2-бензофуран-1-ил)фосфоната в метилтетрагидрофуране по реакции с метансульфоновой кислотой.
10. Способ по п.9, отличающийся тем, что включает дополнительно следующие стадии:
(в) синтез 2-фтор-5-[(4-оксо-3,4-дигидрофталазин-1-ил)метил]бензонитрила (ED):
путем взаимодействия 2-фтор-5-[(Е/Z)-(3-оксо-2-бензофуран-1(3Н)-илидин)метил]бензонитрила с гидразин гидратом, и
(г) синтез 2-фтор-5-(4-оксо-3,4-дигидро-фталазин-1-илметил)-бензойной кислоты путем взаимодействия соединения ED с гидроксидом натрия.
11. Соединение, представляющее собой 2-фтор-5-[(4-оксо-3,4-дигидрофталазин-1-ил)метил]бензонитрил (ED):
12. Фармацевтическая композиция для ингибирования PARP, содержащая соединение по любому из пп.1-3 и фармацевтически приемлемый носитель или разбавитель.
| WO 2004080976 А1, 23.09.2004 | |||
| ПРИСПОСОБЛЕНИЕ ДЛЯ ТОЧНОЙ УСТАНОВКИ В ФОРМАХ МЕТАЛЛИЧЕСКИХ ЧАСТЕЙ ПРИ ЗАЛИВКЕ ИХ В ОТЛИВАЕМЫЕ ИЗДЕЛИЯ | 1926 |
|
SU6300A1 |

