Изобретение относится к биодеградируемым полимерным носителям для доставки противоопухолевых лекарственных средств, в частности, соединений класса таксанов, таких как паклитаксел, доцетаксел, антрациклиновых антибиотиков, таких как доксорубицин, эпирубицин и других.
Современная медицина имеет в своем распоряжении целый ряд эффективных противоопухолевых лекарственных средств, однако, их широкое применение в клинической практике может быть ограничено некоторыми негативными факторами, в частности, такими, как высокая токсичность, низкая растворимость в воде.
Одним из возможных путей решения указанной проблемы является связывание противоопухолевых лекарственных средств с полимерными носителями. Как установлено исследованиями, полимеры, имеющие высокую молекулярную массу, не способны легко диффундировать через нормальные капилляры и гломерулярный эндотелий, что предотвращает интоксикацию нормальной ткани. С другой стороны установлено, что злокачественные опухоли часто имеют нарушенный капиллярный эндотелий и более высокую проницаемость, чем сосудистая сеть нормальной ткани. Благодаря указанным факторам конъюгат полимер-лекарственное средство может селективно просачиваться из кровеносных сосудов в опухоли, приводя к накоплению в опухоли активного терапевтического средства. Кроме того, растворимые в воде полимеры могут обеспечивать солюбилизацию коньюгированных с ними нерастворимых лекарственных средств.
В настоящее время разработаны различные синтетические и природные полимеры, способные выполнять функцию носителей противоопухолевых лекарственных средств с обеспечением их опухоль - специфической доставки.
Так, в частности, известны полимерные носители для/противоопухолевых лекарственных средств класса таксанов, представляющие собой полиэтиленгликоль или его производные [RU 2002109594], гиалуроновую кислоту или ее производные [RU 2384593].
Указанные полимерные носители не являются биологически деградируемыми соединениями, вследствие чего могут накапливаться в организме, вызывая тем самым нежелательные последствия.
Известны полимерные носители на основе природных аминокислот, достоинством которых является биосовместимость и биодеградируемость.
Так, в частности, известен полимерный носитель для противоопухолевых лекарственных средств [EA 002400], представляющий собой полиаспарагиновую или полиглутаминовую кислоту или сополимер указанных аминокислот. Рассматриваемые полимерные носители имеют молекулярный вес от 5000 до 100000 Да, хорошо растворимы в воде, легко разрушаются лизосомными ферментами, стабильны в плазме, содержат достаточное количество функциональных групп для присоединения лекарственного средства
В рассматриваемом изобретении связь полимерного носителя с противоопухолевым лекарственным средством осуществляется с участием карбоксильных групп полимера и гидроксильных групп лекарственного средства, при этом образуются трудно разрушаемые сложноэфирные связи, расщепление которых происходит в условиях высокой щелочности среды. Это затрудняет высвобождение лекарственного средства в физиологических средах организма человека, pH которых лежат в области значений, близких к нейтральным или слабокислым.
Задачей заявляемого изобретения является создание нового биодеградируемого полимерного носителя для доставки противоопухолевого лекарственного средства.
По первому варианту изобретения биодеградируемый полимерный носитель для доставки противоопухолевого лекарственного средства представляет собой полимер, макромолекула которого имеет линейную конфигурацию цепи, и состоит из звеньев общей формулы
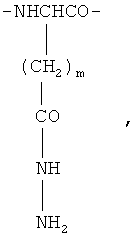
где m=1 или 2.
По второму варианту изобретения биодеградируемый полимерный носитель для доставки противоопухолевого лекарственного средства представляет собой полимер, макромолекула которого имеет линейную конфигурацию полимерной цепи и состоит из звеньев общей формулы
где m=1 или 2,
при этом полимерная цепь содержит С - концевую группу, представляющую собой (С6-С18) алкиламин или трет-бутилкарбазат.
Предлагаемый полимерный носитель по первому и второму вариантам изобретения представляет собой линейный полимер, структурными звеньями которого являются фрагменты β-гидразида аспарагиновой кислоты (поли-(β-гидразид аспарагиновой кислоты)) и/или фрагменты γ-гидразида глутаминовой кислоты (поли-(γ-гидразид глутаминовой кислоты)).
За счет того, что полимерная цепь заявляемого носителя по первому и второму вариантам изобретения включает производные природных водорастворимых полиаминокислот, обеспечивается его биодеградируемость, биосовместимость и возможность солюбилизации нерастворимых в воде коньюгированных с ним лекарственных средств.
Особенностью предлагаемого полимерного носителя по первому и второму вариантам является наличие в его макромолекуле ацил-гидразидных группировок, CONHNH2, расположенных в боковых цепях ее звеньев.
За счет наличия указанных ацил-гидразидных группировок обеспечивается химическое или физическое связывание заявляемого полимерного носителя по первому и второму вариантам изобретения с лекарственным средством.
Химическое связывание полимерного носителя по первому и второму вариантам изобретения может осуществляться с лекарственным средством, в составе молекул которого имеются альдегидные или кетонные группы, в частности, с соединениями класса таксанов или антрациклиновыми антибиотиками. При этом ацил-гидразидные группировки полимерного носителя и альдегидные или кетонные группы лекарственного средства образуют хемодеградируемые альдиминные или кетиминные связи.
Указанные альдиминная или кетиминная связи разрушаются при значениях pH среды около 7, что облегчает высвобождение лекарственного средства из полимерного носителя в физиологических средах организма человека, характеризующихся значением pH, близким к нейтральному или слабокислому.
Физическое связывание полимерного носителя по первому и второму вариантам изобретения может осуществляться с лекарственным средством после его включения в гидрофобное ядро полимерного носителя и формирования полимерных сеток в результате внутримолекулярных подшивок ацил-гидразидных группировок полимерных цепей.
В полимерном носителе по первому варианту изобретения на С-конце полимерной цепи содержится группа, химическая природа которой определяется выбранным для реакции полимеризации инициатором. В частности, это может быть OH-группа, изопропиламин (NНСН(СН3)2) и др.
В полимерном носителе по второму варианту изобретения С-концевая группа превращена в липофильный фрагмент, представляющий собой (С6-C18) алкиламин, в частности, октадециламин (CH4(CH2)16CH2NH2), или трет-бутилкарбазат
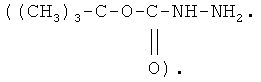
Модификация полимерного носителя по С-концу каким-либо из вышеуказанных липофильных фрагментов позволяет в случае физического связывания полимерного носителя с лекарственным средством обеспечить более медленное высвобождение последнего (депо-эффект).
Таким образом, техническим результатом, достигаемым при реализации заявляемого изобретения по первому и второму его вариантам, является создание нового полимерного биодеградируемого носителя, способного к химическому или физическому связыванию с противоопухолевым лекарственным средством с образованием легкоразрушаемых связей в физиологических средах организма человека.
Количество звеньев (n) в цепи макромолекулы полимерного носителя по первому и второму вариантам изобретения лежит в пределах от 100 до 2000.
Молекулярная масса (MM) полимерного носителя по первому и второму вариантам изобретения составляет величину от 10000 до 200000 Да.
Полимерный носитель по первому варианту изобретения получают, в частности, по следующей схеме:
Получение исходных мономеров: N - карбоксиангидридов сложных эфиров аспарагиновой и/или глутаминовой кислоты, выбранных из группы: β-метил-аспартата, β-бензил-аспартата, γ-метил-глутамата, γ-бензил-глутамата по известной методике [см., например, W. Daly, D. Poche. Tetrahedron Lett., 1988, V.29, №46, Р5859].
Синтез полиаспарагиновой или полиглутаминовой кислоты, имеющей на конце боковой цепи сложноэфирные защитные группы (-O-бензил- или -O-метил-), который осуществляют по известной методике [W. Daly, D. Poche. Tetrahedron Lett., 1988, V.29, №46, Р5859].
Обработка полученного полимера, в ходе которой сложноэфирные защитные группы (-O-бензил- или -O-метил-) превращаются в ацил-гидразидные группировки. В частности, указанную обработку осуществляют путем добавления 50-100 кратного избытка гидразин-гидрата или безводного гидразина к раствору соответствующего полимера в метаноле, этаноле или другом инертном растворителе при температуре от 35 до 50°C. Продолжительность указанной реакции составляет от 1 до 15 суток. По окончании реакции полимер высаживают в диэтиловый эфир, а затем промывают и сушат.
Полимерный носитель по второму варианту изобретения получают по той же схеме, что и для первого варианта, при этом при проведении синтеза полиаспарагиновой или полиглутаминовой кислоты для обеспечения модификации полимерного носителя по С-концу каким-либо из указанных выше химических соединений в качестве инициатора реакции полимеризации используют соответствующее химическое соединение.
Возможность реализации первого и второго вариантов изобретения показана в примерах конкретного выполнения.
Пример 1 (первый вариант изобретения).
Получали поли-(γ-гидразид глутаминовой кислоты), имеющий на С-конце изопропиламин (NHCH(СН3)2).
В качестве исходного мономера использовали N-карбоксиангидрид γ - бензил-глутамата, который получали следующим образом.
К суспензии, содержащей 8 г γ-бензилового эфира глутаминновой кислоты в 200 мл диоксана, нагретого до 50°C, прибавляли 3,3 г (1/3 эквивалент) трифосгена. Периодически реакционную смесь продували азотом для удаления HCl. Через 3 ч диоксан отгоняли на вакуумном роторе, полученную массу растворяли в этилацетате, а затем высаживали в петролейный эфир. Перекристаллизацию проводили из смеси этилацетата с петролейным эфиром несколько раз, чтобы избавиться от примесей хлористого водорода. Чистоту N-карбоксиангидрида γ-бензил глутамата оценивали с помощью тонкослойной хроматографии (ТСХ) в системе бензол - ацетон, взятых в отношении 1:1.
Выход N-карбоксиангидрида γ-бензил-глутамата составил 5,3 г. Тпл составила 95-97°C, что является близким к теоретическому значению Тпл.
Далее осуществляли полимеризацию N-карбоксиангидрида γ-бензил- глутамата следующим образом.
К раствору, содержащему 0,9 г (0,0034 моля) N-карбоксиангидрида γ-бензил-глутамата в 22,5 мл диоксана, добавляли в качестве инициатора полимеризации изопропиламин в количестве 5,9 мкл (0,0068 ммоля). Соотношение мономер: инициатор было равно 50:1. Реакционную смесь выдерживали пять дней при комнатной температуре, а затем полученный поли- (γ-бензил-глутамат) осаждали в диэтиловый эфир, промывали несколько раз диэтиловым эфиром и сушили на воздухе.
Выход указанного полимера составил 0,7 г.
Полученный полимер обрабатывали раствором гидразин-гидрата в метаноле при температуре 45°С, при этом гидразин-гидрат брали в 70-кратном избытке по отношению к полимеру.
Время реакции составило трое суток. Затем полимер высаживали в диэтиловый эфир, промывали и сушили.
Снимали ИК-спектр полученного полимера. Присутствия в полимере сложноэфирных групп не обнаружено, что свидетельствовало о том, что гидразинолиз прошел полностью.
Выход полученного поли- (γ-гидразида глутаминовой кислоты) составил 0,7 г.
ММ указанного полимера, определенная с использованием ГПХ, составила 140000 Да.
n=1400.
Пример 2 (второй вариант изобретения)
Получали поли- (β-гидразид аспарагиновой кислоты), модифицированный по С-концу октадециламином.
В качестве исходного мономера использовали N-карбоксиангидрид (3-метил-аспартата, который получали следующим образом.
К суспензии, содержащей 4,5 г (0,0245 моль) (β-метилового эфира аспарагиновой кислоты в 100 мл диоксана, нагретого до 50°C, прибавляли 2,43 г (1/3 эквивалент) трифосгена. Периодически реакционную смесь продували азотом для удаления остатков HCl. Через 4 ч по окончания реакции смесь фильтровали от не прореагировавшего хлоргидрата β-метиласпартата, и раствор концентрировали на вакуумном испарителе. Полученную массу растворяли в этилацетате, а затем высаживали в петролейный эфир. Перекристаллизацию проводили несколько раз, чтобы избавиться от примесей хлористого водорода. Чистоту N-карбоксиангидрида β-метил-аспартата оценивали с помощью ТСХ анализа, проводимого в системе бензол - ацетон, взятых в соотношении 1:1.
Выход N-карбоксиангидрида β-метил-аспартата составил 1,5 г. Тпл составила 60-61°C, что является близким к теоретическому значению Тпл.
Далее осуществляли полимеризацию N-карбоксиангидрида β-метил-аспартата следующим образом.
3,2 г (0,0185 моль) N-карбоксиангидрида β-метил-аспартата растворяли в 80 мл диоксана (4% раствор), к раствору добавляли в качестве инициатора полимеризации октадециламин в количестве 125 мг (0,462 ммоль). Соотношение мономер: инициатор было равно 40:1. Реакционную смесь выдерживали 14 дней при комнатной температуре, а затем полученный поли-(β-метил-аспартат) осаждали в диэтиловый эфир, промывали несколько раз диэтиловым эфиром и сушили на воздухе.
Выход указанного полимера составил 1,9 г.
Полученный полимер обрабатывали раствором гидразин-гидрата в метаноле при температуре 45°C, при этом гидразин-гидрат брали в 80-кратном избытке по отношению к полимеру.
Время реакции составило трое суток. Затем полимер высаживали в диэтиловый эфир, промывали и сушили.
Снимали ИК спектр полученного полимера. Присутствия в полимере сложноэфирных групп не обнаружено, что свидетельствовало о том, что гидразинолиз прошел полностью.
Выход полученного поли- (β-гидразида аспарагиновой кислоты), содержащего на С-конце октадециламин, составил 1,8 г.
ММ указанного полимера, определенная с использованием ГПХ, составила 160000 Да.
n=1600.
Пример 3 (второй вариант изобретения).
Получали поли- (β-гидразид аспарагиновой кислоты), модифицированный по С-концу третбутилкарбазатом.
В качестве исходного мономера использовали N-карбоксиангидрид β- метил-аспартата, полученный по примеру 2.
Осуществляли реакцию полимеризации N-карбоксиангидрида β-метил-аспартата, как описано в примере 2, но с использованием в качестве инициатора полимеризации трет-бутилкарбазата.
Выход полимера составил 1,9 г.
Полученный полимер обрабатывали раствором гидразин-гидрата в метаноле при температуре 45°С, при этом гидразин-гидрат брали в 50-кратном избытке по отношению к полимеру.
Время реакции составило трое суток. Затем полимер высаживали в диэтиловый эфир, промывали и сушили.
Снимали ИК-спектр полученного полимера. Присутствия в полимере сложноэфирных групп не обнаружено, что свидетельствовало о том, что гидразинолиз прошел полностью.
Выход полученного поли-(β-гидразида аспарагиновой кислоты), модифицированного по С-концу трет-бутилкарбазатом, составил 1,5 г.
ММ указанного полимера, определенная с использованием ГПХ, составила 160000 Да.
n=1600.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИОДЕГРАДИРУЕМЫЙ ПОЛИМЕРНЫЙ НОСИТЕЛЬ ДЛЯ ДОСТАВКИ ПРОТИВООПУХОЛЕВОГО ЛЕКАРСТВЕННОГО СРЕДСТВА | 2012 |
|
RU2500428C1 |
| Способ получения блок-сополимеров и инициатор-2,2 -ди(амидоаминоуксусной кислоты)дифенилдисульфид для осуществления способа | 1977 |
|
SU664970A1 |
| ГЛИКОПОЛИСИАЛИРОВАНИЕ БЕЛКОВ, НЕ ЯВЛЯЮЩИХСЯ БЕЛКАМИ СВЕРТЫВАНИЯ КРОВИ | 2014 |
|
RU2662807C2 |
| ПЕПТИДНЫЙ СОПОЛИМЕР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2009 |
|
RU2402576C1 |
| КОНЪЮГАТЫ ПОЛИГЛУТАМАТ-АМИНОКИСЛОТА И СПОСОБЫ | 2006 |
|
RU2472812C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ГИДРАЗИДОВ | 2008 |
|
RU2484849C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ БЛОК-СОПОЛИМЕР, ВКЛЮЧАЮЩИЙ СОЕДИНЕНИЕ БОРОНОВОЙ КИСЛОТЫ | 2012 |
|
RU2569847C2 |
| АНАЛОГ ДОЛОСТАТИНА | 1993 |
|
RU2132334C1 |
| КОНЪЮГАТ ГЛИКОПРОТЕИНА, ОБЛАДАЮЩЕГО АКТИВНОСТЬЮ ЭРИТРОПОЭТИНА, С ПРОИЗВОДНЫМИ N-ОКСИДА ПОЛИ-1,4-ЭТИЛЕНПИПЕРАЗИНА (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ КОНЪЮГАТА | 2013 |
|
RU2556378C2 |
| ГЛИКОПОЛИСИАЛИРОВАНИЕ БЕЛКОВ, НЕ ЯВЛЯЮЩИХСЯ БЕЛКАМИ СВЕРТЫВАНИЯ КРОВИ | 2010 |
|
RU2533619C2 |
Изобретение относится к химико-фармацевтической промышленности и представляет собой биодеградируемый полимерный носитель для доставки противоопухолевого лекарственного средства, макромолекула которого имеет линейную конфигурацию цепи и состоит из звеньев общей формулы
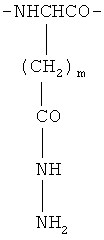
где m=1 или 2, при этом молекулярная масса полимерного носителя составляет величину от 10000 до 200000 Да. Изобретение обеспечивает создание нового полимерного биодеградируемого носителя, способного к химическому или физическому связыванию с противоопухолевым лекарственным средством с образованием легкоразрушаемых связей в физиологических средах организма человека. 2 н.п. ф-лы, 3 пр.
1. Биодеградируемый полимерный носитель для доставки противоопухолевого лекарственного средства, макромолекула которого имеет линейную конфигурацию полимерной цепи и состоит из звеньев общей формулы
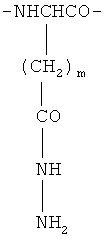
где m=1 или 2,
при этом молекулярная масса полимерного носителя составляет величину от 10000 до 200000 Да.
2. Биодеградируемый полимерный носитель для доставки противоопухолевого лекарственного средства, макромолекула которого имеет линейную конфигурацию полимерной цепи и состоит из звеньев общей формулы
где m=1 или 2,
при этом полимерная цепь содержит C-концевую группу, представляющую собой (C6-C18) алкиламин или трет-бутил карбазат, а молекулярная масса полимерного носителя составляет величину от 10000 до 200000 Да.
| Reyhanen Astaneh, Hamid Reza Moghimi, Mohammad Erfan, Hamid Mobedi / FORMULATION OF AN INJECTABLE IMPLANT FOR PEPTIDE DELIVERY AND MECHANISTIC STUDY OF THE EFFECT OF POLYMER MOLECULAR WEIGHT ON ITS RELEASE BEHAVIOR / DARU, 2006, V.14, No.2, p.65-70 | |||
| МОЛЕКУЛЯРНЫЙ КОМПЛЕКС ДЛЯ ТРАНСФЕКЦИИ КЛЕТОК МЛЕКОПИТАЮЩИХ, СОДЕРЖАЩИЙ ПЛАЗМИДНУЮ ДНК, МОДИФИЦИРОВАННЫЙ ПОЛИЭТИЛЕНИМИН И МОЛЕКУЛЫ-ЛИГАНДЫ | 2005 |
|
RU2303064C2 |
| US 20080253969 A1, 16.10.2008 | |||
| WO 2012040524 A1, 29.03.2012 | |||
| ПОЛИМЕРНЫЙ КОНЪЮГАТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2130462C1 |
| US 6441025 В2, 27.08.2002 | |||
| US 7744861 B2, 29.06.2010. | |||

