Настоящее изобретение относится к новым пептидам и их производным, которые потенциально обладают улучшенными терапевтическими качествами для лечения неопластических болезней по сравнению с Доластатином-10. Кроме того, в отличие от Доластатина-10, который должен быть выделен в лабораторных условиях из редких природных источников, соединения настоящего изобретения могут быть синтезированы традиционными способами, как это описано подробно ниже. Кроме того, Доластатин-10 нестабилен в кислой среде. Отмечено, что даже незначительные изменения в структуре могут вызвать полную потерю его активности (Biochemical Pharmacology, т. 40, N 8, 1859-64, 1990).
Соединения настоящего изобретения включают новые пептиды формулы I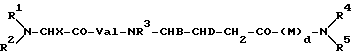
где R1 обозначает C1-C7-алкил;
R2 обозначает C1-C4-алкил;
X обозначает C1-C5 -алкил;
R3 обозначает низший алкил;
B обозначает C1-C5-алкил;
D обозначает C1-C5-алкокси;
M обозначает фенилаланил;
R4 обозначает водород;
R5 обозначает водород или бензил;
d равно 0 или 1,
или их соли с физиологически допустимыми кислотами
Настоящее изобретение также описывает способы получения соединений формулы I, фармакологические композиции, содержащие указанные соединения вместе с фармакологически приемлемым носителем и способы их использования для лечения рака у млекопитающих.
Соединения формулы 1 состоят предпочтительно из L-аминокислот или компонентов, полученных L-аминокислот, но они могут содержать одну или более D-аминокислот или компоненты, полученные из D-аминокислот.
Особенно пригодными физиологически приемлемыми кислотами являются: соляная кислота, лимонная кислота, винная кислота, молочная кислота, фосфорная кислота, метансульфоновая кислота, уксусная кислота, муравьиная кислота, малеиновая кислота, фумаровая кислота, яблочная кислота, янтарная кислота, малоновая кислота, серная кислота, L-глутаминовая кислота, L-аспарагиновая кислота, пировиноградная кислота, слизевая кислота, бензойная кислота, глюкуроновая кислота, щавелевая кислота, аскорбиновая кислота и ацетилглицин.
Новые соединения могут быть получены известными способами. Так, соединения могут быть собраны последовательно или путем связывания подходящих мелких фрагментов. При последовательном соединении, начиная с атома углерода в конце цепи, пептидную цепь удлиняют шаг за шагом каждый раз одной аминокислотой или элементом структуры. При связывании фрагментов возможно соединение между собой фрагментов различной длины и фрагментов, которые, в свою очередь, могут быть получены последовательной сборкой в структуру из аминокислот или строительных блоков.
Как при последовательном соединении, так и при связывании фрагментов, необходимо соединить структурные единицы с образованием амидной связи. Для этого пригодны ферментативные и химические способы.
Химические способы формирования амидной связи детально описаны у Muller, Methoden der organischen Chemie, т. XV/2, стр. 1-364, Thieme Verlag, Штутгарт, 1974; Stewart, Young, Solid Phase Peptide Synthesis, стр. 31-34, 71-82, Pierce Chemical Company, Рокфорд, 1984; Bodanszky, Klausner, Ondetti, Peptide Synthesis, стр. 85-128, John Wiley and Sons, Нью-Йорк, 1976 и в других известных работах по химии пептидов. Особенное предпочтение отдают азидному способу, симметричному и смешанному ангидридному способу, при которых in situ производятся или заранее формируются активные эфиры, использованию N-карбоксиангидридов аминокислот, защищенных уретаном, и образованию амидной связи с использованием связывающих реагентов, особенно дициклогексилкарбодиимида (ДЦК), диизопропилкарбодиимида (ДИК), 1-этоксикарбонил-2-этокси-1,2- дигидрохинолина (ЭЭДХ), 1-этил-3-(3-диметиламинопропил)- карбодиимидгидрохлорида (ЭДКИ), н-пропанфосфонового ангидрида (ПФА), N, N-бис(оксо-3-оксазолидинил)-амидофосфорилхлорида (БОФ-Х), бром-трис-пирролидинофосфонийгекса-фторфосфата (ПиБрФ), дифенилфосфорилазида (ДФФА), реагента Кастро (БОФ, ПиБоф), солей O-бензотриазолил-N, N, N',N'-тетраметилурония (ГБТУ), диэтилфосфорилцианида (ДЭФЦ), 2,5-дифенил-2,3-дигидро-3-оксо-4- гидрокситиофендиоксида (реагент Стеглиха; ГОТДО) и 1,1'-карбонилдиимидазола (КДИ). Связывающие реагенты могут быть применены отдельно или в комбинации с добавками, такими как N,N-диметил-4-аминопиридин (ДМАП), N-гидроксибензотриазол (ГОБт), N-гидроксибензотриазин (ГООБт), N-гидроксисукцинимид (ГОСу) или 2-гидроксипиридин.
Если обычно можно обойтись без защитных групп при ферментативном синтезе пептидов, при химическом синтезе обратимая защита реактивных групп, не включенных в образование амидной связи, необходима для обоих реагентов. Три общепринятых метода защитных групп предпочтительны для химического синтеза пептидов: методы с использованием бензилоксикарбонила (Z), трет.-бутоксикарбонила (Boc) и 9-фторенилметоксикарбонила (Fmoc). В каждом случае указана защитная группа на α-аминогруппе структурной единицы, удлиняющей цепь. Детальный обзор групп, защищающих аминокислоты, опубликован у  , Methoden der organischen Chemie, т. XV/1, стр. 20-906, Thieme Verlag, Штутгарт, 1974. Структурные единицы, используемые для соединения пептидной цепи, могут вступать в реакцию в растворе, в суспензии или с помощью метода, сходного с описанным Merrifield в J.Amer.Chem.Soc., 85, (1963), 2149. Особенно предпочтительными методами являются те, в которых пептиды соединяются последовательно или путем связывания фрагментов с использованием методов защитных групп Z, Boc или Fmoc, причем один из реагентов в указанном методе Merrifield связан с нерастворимым полимерным носителем (также называемым ниже полимером). Обычно это влечет за собой то, что пептид последовательно соединяется на полимерном носителе с использованием метода Boc или Fmoc защитной группы, растущая пептидная цепь ковалентно связывается атомом углерода в конце цепи с нерастворимыми частицами полимера (ср. фиг. 1 и 2). Этот процесс делает возможным отделение реагентов и побочных продуктов фильтрацией, что делает ненужной рекристаллизацию полупродукта.
, Methoden der organischen Chemie, т. XV/1, стр. 20-906, Thieme Verlag, Штутгарт, 1974. Структурные единицы, используемые для соединения пептидной цепи, могут вступать в реакцию в растворе, в суспензии или с помощью метода, сходного с описанным Merrifield в J.Amer.Chem.Soc., 85, (1963), 2149. Особенно предпочтительными методами являются те, в которых пептиды соединяются последовательно или путем связывания фрагментов с использованием методов защитных групп Z, Boc или Fmoc, причем один из реагентов в указанном методе Merrifield связан с нерастворимым полимерным носителем (также называемым ниже полимером). Обычно это влечет за собой то, что пептид последовательно соединяется на полимерном носителе с использованием метода Boc или Fmoc защитной группы, растущая пептидная цепь ковалентно связывается атомом углерода в конце цепи с нерастворимыми частицами полимера (ср. фиг. 1 и 2). Этот процесс делает возможным отделение реагентов и побочных продуктов фильтрацией, что делает ненужной рекристаллизацию полупродукта.
Защищенные аминокислоты или строительные блоки могут связываться с любыми пригодными полимерами, которые должны быть только растворимы в используемых растворителях и иметь стабильную физическую форму, которая облегчает фильтрацию. Полимер должен содержать функциональную группу, с которой первая защищенная аминокислота может быть прочно связана ковалентной связью. Для этой цели пригодно широкое разнообразие полимеров, в том числе целлюлоза, поливиниловый спирт, полиметакрилат, сульфированный полистирол, сополимер хлорметилированного стирола/дивинилбензола (полимер Меррифилда), 4-метилбензгидриламиновый полимер (МБГА-полимер), фенилацетамидометиловый полимер (Фам-полимер), пара-бензил-окси- бензил-спирт-полимер, бензгидриламиновый полимер (БГА-полимер), 4-(гидроксиметил)бензоилоксиметиловый полимер, полимер Breipohl и др. (Tetrahedron Letters, 28 (1987) 565; поставляемый BACHEM), 4-(2,4- диметоксифениламинометил)фенокси-полимер (поставляемый Novabiochem) или орто-хлортритиловый полимер (поставляемый Biohellas).
Пригодными для образования амидной связи в растворе являются все растворители, которые инертны в условиях реакции, особенно вода, N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), ацетонитрил, дихлорметан (ДХМ), 1,4-диоксан, тетрагидрофуран (ТГФ), N-метил-2-пирролидон (NMП) и смесь указанных растворителей. Синтез на полимерном носителе может проходить во всех инертных органических растворителях, в которых используемые производные аминокислот растворимы; однако предпочтительные растворители, кроме того, обладают свойством набухать в полимере, например ДМФ, ДХМ, NMП, ацетонитрил и ДМСО, и смеси этих растворителей. После того, как синтез закончен, соединение отщепляют от полимерного носителя. Условия, в которых возможно отщепление различных типов полимеров, описаны в литературе. Наиболее часто используются реакции расщепления, которые катализируются кислотой или палладием, особенно расщепление в жидком безводном фторводороде, в безводной трифторметан-сульфоновой кислоте, в разбавленной или концентрированной трифторуксусной кислоте, расщепление, катализируемое палладием, в тетрагидрофуране (ТГФ) или ТГФ-ДХМ смесях в присутствии слабого основания, такого, как морфолин, или расщепление в смесях уксусная кислота/дихлорметан/трифторэтанол. В зависимости от выбранных защитных групп они могут быть сохранены, а также отщеплены в условиях расщепления. Частичное нарушение защиты пептида может также иметь смысл, когда происходят реакции с образованием производных. Диалкилированные соединения с атомом азота на конце цепи могут быть получены либо путем связывания соответствующих N,N-диалкиламинокислот в растворе или на полимерном носителе, путем восстановительного алкилирования в растворе (например, NaCNBH3 в MeOH) или путем восстановительного алкилирования связанного с полимером соединения в ДМФ /1% уксусная кислота с NaCNBH3 и соответствующими альдегидами. Соединения с мостиками γ- или δ-лактама могут быть получены путем включения в пептидную цепь соответствующих структурных единиц дипептида, соединенных лактамовой мостиковой связью (R. Freidinger, J. Org. Chem. (1982) 104-109). Соединения с содержащими тиазол, оксазол, тиазолин или оксазолин дипептидными строительными блоками могут быть получены путем включения в пептидную цепь соответствующих дипептидных структурных единиц (U. Schmidt и др., Syntesis (1987), 233-236; P. Jouin и др., Tetrahedron Letters (1992), 2807-2810; P. Wipf и др., Tetrahedron Letters (1992), 907-910; W. R. Tully, J. Med. Chem. (1991), 2065; Synthesis(1987), 235; T.Shioiri и др. , J. Org. Chem. (1987), 1252- 1255; R. Pettit и др., J. Am.Chem. Som. (1989), 5463-65). Строительные блоки, имеющие структуру -NR3-CHB-CHD-CHE-CO- и - NR4-CHG-CHK-CHL-CO-, могут быть получены в соответствии с литературными данными путем реакции, например, соответствующих защищенных альдегидов аминокислоты с соответствующими алкилированными соединениями, например фосфонатами, фосфорными илидами, реагентом Эванса и т.д. (S. Shibuya и др., Heterocycles (1990), 1597-1600; М. Braun и др., Angew. Chem. (1987), 24-37; Angew. Chem. 1992, 104, N 10; T. Shioiri и др., Peptide Chemistry 1989, N. Yanaihara (Ed. ), 291-296; Pettit и др., J. Am. Chem. Soc. 1989, 111, 5463; T. Shioiri, Tetrahedron Letters 1991, 931-934; К. Koga, Tetrahedron Letters 1991, 2395-2398).
Соединения настоящего изобретения могут быть использованы для ингибирования или другого типа лечения твердых опухолей (например, опухолей легких, молочной железы, толстой кишки, предстательной железы, мочевого пузыря, прямой кишки, или опухоли эндометрия) или злокачественных гематологических заболеваний (например, лейкемии, лимфомы) путем назначения млекопитающим этих соединений в качестве лекарств. Это назначение может быть сделано любым способом, обычно применяемым для фармацевтических, предпочтительно онкологических агентов, в том числе оральным или парентеральным способами, в том числе подкожным, внутривенным, внутримышечным и внутрибрюшинным. Соединения могут быть назначены по отдельности или в виде фармацевтических композиций, содержащих соединение формулы (I) вместе с фармацевтически пригодным носителем, подходящим для желаемого способа применения. Такие фармацевтические композиции могут быть комбинированными продуктами, то есть содержать другие терапевтически активные ингредиенты.
Назначаемая для млекопитающего доза должна содержать эффективное ингибирующее опухоль количество ингредиента, которое будет зависеть от известных факторов, включающих биологическую активность конкретного применяемого соединения; способов назначения; возраста, состояния здоровья и веса реципиента; характера и степени проявления симптомов; частоты лечения; назначения других курсов лечения; и желаемого эффекта. Обычная ежедневная доза должна быть в пределах от 5 до 250 мг на килограмм веса при оральном применении и примерно от 1 до 100 мг на килограмм веса при парентеральном применении.
Подходящие формы дозировки содержат примерно от 10 до 500 мг активного ингредиента на единицу. Активный ингредиент должен, таким образом, составлять примерно 1-90 вес.% от общего веса композиции.
Новые соединения могут назначаться в обычных твердых или жидких фармацевтически применяемых формах, в том числе в виде непокрытых или покрытых (пленкой) таблеток, капсул, порошков, гранул, суппозиториев или растворов. Их получают общепринятыми способами. Для этой цели активные вещества могут быть соединены с обычными фармацевтическими вспомогательными средствами, такими, как, связующие для таблеток, наполнителями, консервантами, диспергаторами таблеток, регуляторами расхода, пластификаторами, смачивающими агентами, дисперсантами, эмульгаторами, растворителями, соединениями, способствующими выходу, антиокислителями и/или газами-пропеллентами (ср. Н. Sucker и др. : Pharmazeutische Technologie, Thieme-Verlag, Штутгарт, 1978). Применяемые формы, полученные таким способом, как правило содержат 1-90 вес. % активного вещества.
Следующие примеры служат для иллюстрации данного изобретения. Протеиногенные аминокислоты, упоминаемые в примерах, обозначены при помощи известного трехбуквенного кода. Другие сокращения: ТФА - трифторуксусная кислота, УК - уксусная кислота, Bu - бутил, Et - этил, Me - метил, Bzl - бензил.
А. Общие процедуры
I. Соединения, заявляемые в пункте I формулы изобретения, или синтезированы путем классического синтеза в растворе с использованием Z- и Boc-методологии, как описано выше, или при помощи стандартных методов синтеза в твердой фазе, ручным способом или с помощью полностью автоматической модели синтезатора 431А, поставляемого APPLIED BIOSYSTEMS. Использованы следующие различные циклы синтеза для Boc и Fmoc методов защитной группы:
а) Цикл синтеза для метода Вос-защитной группы
1. 30% трифторуксусная кислота в ДХМ 1х3 мин
2. 50% трифторуксусная кислота в ДХМ 1х1 мин
3. Промывка ДХМ 5х1 мин
4. 5% диизопропилэтиламин в ДХМ 1х1 мин
5. 5% диизопропилэтиламин в МП 1х1 мин
6. Промывка NMП 5х1 мин
7. Добавление преактивированной защищенной аминокислоты (активация с 1 эквивалентом ДЦК и 1 эквивалентом ГОБт NМП/ДХМ); Соединение пептида (1-я часть) 1х30 мин
8. Добавление ДМСО к реакционной смеси до тех пор, пока содержание ДМСО не достигнет 20 об.%
9. Соединение пептида (2-я часть) 1х16 мин
10. Добавление 3,8 эквивалентов диизопропилэтиламина к реакционной смеси
11. Соединение пептида (3-я часть) 1х7 мин
12. Промывка ДХМ 3х1 мин
13. Если превращение неполное, повторение соединения (вернуться к 5)
14. 10% уксусный ангидрид, 5% диизопропилэтиламин в ДХМ 1х2 мин
15. 10% уксусный ангидрид в ДХМ 1х4 мин
16. Промывка ДХМ 4х1 мин
17. Вернуться к 1.
БОФ-Х и ПиБрф использовали в качестве реагентов для соединения аминокислот следующих за N-метиламинокислотами или строительными блоками, несущими N-метилгруппу. Время реакции соответственно увеличили. При синтезе в растворе наиболее предпочтительно для этого типа связывания использование соответственно либо Boc-аминокислота-NKA (N-трет.-бутилоксикарбонил-аминокислота-N- карбоксиангидриды), или Z-аминокислота-NKA (N-бензилоксикарбонил- аминокислота-N-карбоксиангидриды).
б) Цикл синтеза для метода Fmoc защитной группы
1. Промывка ДМФ 1х1 мин
2. 20% пиперидин в ДМФ 1х4 мин
3. 20% пиперидин в ДМФ 1х16 мин
4. Промывка ДМФ 5х1 мин
5. Добавление преактивированной защищенной аминокислоты (активация 1 эквивалентом ТБТУ и 1,5 эквивалентом ДИПЭА в ДМФ); Соединение пептида 1х61 мин
6. Промывка ДМФ 3х1 мин
7. Если превращение неполное, повторить соединение (вернуться к 5)
8. 10% уксусный ангидрид в ДМФ 1х8 мин
9. Промывка ДМФ 3х1 мин
10. Вернуться к 2.
БОФ-Х и ПиБрф использовали в качестве реагентов для соединения аминокислот, следующих за N-метиламинокислотами или элементами структуры, несущих N-метил группу. Время реакции соответственно увеличили.
II. Редуктивное алкилирование N-содержащего конца цепи
У пептидного полимера, приготовленного по AIа или AIб разрушили защиту N-содержащего конца цепи (стадии 2-4 по AIб или 1-6 в AIа) и затем провели реакцию с 3-кратным молярным превышением альдегида или кетона в ДМФ/1% уксусной кислоте с добавлением 3 эквивалентов NaCNBH3. После завершения реакции (отрицательный тест Кайзера) полимер несколько раз промыли водой, изопропанолом, ДМФ и дихлорметаном.
III. Переработка пептидных полимеров, полученных по п.п. Iа и II
Пептидный полимер высушивали при пониженном давлении и переносили в реакционный сосуд прибора TEFLON HF (поставляемого PENINSULA). После добавления поглотителя, предпочтительно анизола (1 мл/г полимера), а для триптофан-содержащих пептидов - тиола, для того, чтобы отделить индольную формилгруппу, предпочтительно этандитиола (0,5 мл/г полимера), конденсировали во фтороводороде (10 мл/г полимера) при одновременном охлаждении с жидким N2. Смесь оставляли нагреваться до 0oC и перемешивали при этой температуре в течение 45 мин. Фторводород затем отделяли при пониженном давлении и остаток промывали этилацетатом для удаления остатков поглотителя. Соединение выделяли с помощью 30% уксусной кислоты и фильтрат лиофилизовали.
IV. Переработка пептидных полимеров, полученных по п.п. Iб и II
Пептидный полимер высушивали при пониженном давлении и затем подвергали одной из следующих процедур расщепления в зависимости от состава аминокислот (Wade, Tregear, Howard Florey Fmoc Workshop Manual, Melbourne, 1985).
Суспензию пептидного полимера в подходящей смеси ТФК перемешивали при комнатной температуре в течение определенного времени, и затем полимер отфильтровывали и промывали с ТФК и ДХМ. Фильтрат и смывы концентрировали и соединение осаждали путем добавления диэтилового эфира. После охлаждения в ледяной бане осадок отфильтровывали с помощью 30% уксусной кислоты и лиофилизовали.
V. Когда используют орто-хлоротритиловый полимер (поставляемый Biohellas), суспензию пептидного полимера в смеси уксусная кислота/трифторэтанол/дихлорметан (1:1:3) перемешивают при комнатной температуре в течение 1 часа. Полимер затем отфильтровывают отсасыванием и тщательно промывают расслаивающим раствором. Объединенные фильтраты концентрируют в вакууме и обрабатывают водой. Осажденный твердый осадок удаляют с помощью фильтрации или центрифугирования, промывают диэтиловым эфиром и высушивают при пониженном давлении.
VI. Очистка и исследование соединений
Очистку проводили с помощью гель-хроматографии (SEPHADEX G-10, G-15/10% HOAc, SEPHADEX LH20/MeOH) с или без последующей хроматографии при умеренном давлении (стационарная фаза: HD-SIL С- 18, 20-45 μ, 100  мобильная фаза: градиент с А = 0,1% ТФК/MeOH, В = 0,1% ТФК/H2О). Чистоту получившихся продуктов определяли с помощью аналитической жидкостной хроматографии высокого давления (стационарная фаза: 100 2,1 мм VYDAC С-18, 5 л, 300
мобильная фаза: градиент с А = 0,1% ТФК/MeOH, В = 0,1% ТФК/H2О). Чистоту получившихся продуктов определяли с помощью аналитической жидкостной хроматографии высокого давления (стационарная фаза: 100 2,1 мм VYDAC С-18, 5 л, 300  , мобильная фаза: градиент CH3CN/H2O, 0,1% уксусная кислота в качестве буфера, при 40oC). Исследование проводили с помощью масс-спектрометрии с бомбардировкой тяжелыми ядрами и 1H или 13C-спектроскопии.
, мобильная фаза: градиент CH3CN/H2O, 0,1% уксусная кислота в качестве буфера, при 40oC). Исследование проводили с помощью масс-спектрометрии с бомбардировкой тяжелыми ядрами и 1H или 13C-спектроскопии.
Б. Специальные процедуры
Пример 1
0,4 г Фенилаланинамидгидрохлорида (2 ммол) и 0,55 г Boc-N(CH3)-CH[CH(CH3)2] - CH(OCH3)-CH2-COOH (2 ммоля; синтезированного в соответствии с литературными данными: S.Shibuya и др., Heterocycles, т. 31) N 9, 1597-1600 (1990)) растворяли в ДМФ. После добавления 0,4 г ДЭФЦ (2,2 ммоля) и 1,4 мл диизопропиламина (ДИПА), реакционную смесь перемешивали в течение ночи при комнатной температуре, растворитель выпаривали при пониженном давлении и остаток улавливали с помощью этилацетата и тщательно промывали 5% водным раствором лимонной кислоты, водой, 5% NaHCO3 и раствором NaCl. Органический слой высушивали с помощью Na2SO4, отфильтровывали и упаривали досуха. Остаток (0,66 г) высушивали в вакууме, лишали защиты с помощью 5 мл ТФК/ДХМ (1:1) в течение 2 часов и упаривали досуха. Полученный таким образом лишенный защиты фрагмент растворяли в ДМФ и проводили реакцию с 0,44 г N- трет.-бутилоксикарбонил-валин-N-карбоксиангидридом (1,8 ммоля) при 45oC. После перемешивания в течение 5 часов растворитель выпаривали в вакууме, добавляли этилацетат и органический слой тщательно промывали с помощью 5% водной лимонной кислоты, 5% водного NaHCO3, воды и водного NaCl и высушивали с помощью Na2SO4. Растворитель выпаривали при пониженном давлении и остаток (0,7 г, 1,3 ммоля) обрабатывали 5 мл ТФК/ДХМ (1:1) в течение 2 часов. После испарения смеси растворителей и высушивания с помощью КОН остаток растворяли в ДМФ и 0,19 г N,N-диметилвалина (1,3 ммоля; синтезированного в соответствии с литературными данными: см., например, R.E.Bowmann, J.Chem.Soc.,1959, 1342) и добавляли 0,25 г ДЭФЦ (1,5 ммоля) и 0,7 мл ДИПЭА. Реакционную смесь перемешивали в течение ночи при комнатной температуре, упарили досуха и хроматографировали на колонке SEPHADEX LH-20. Фракции продукта собирали и выпаривали растворитель, получив 0,46 г соединения I, м. вес 561,7.
Пример 2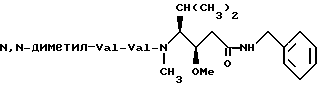
0,29 г Бензиламина (2 ммоля) и 0,55 г Boc-N (CH3-CH[CH(CH3)2]-CH(OCH3)-CH2-COOH (2 ммоля; синтезированного в соответствии с литературными данными: S. Shibuya и др. Heterocycles, т. 31, N 9, 1597-1600, (1990)) растворяли в ДМФ. После добавления 0,4 г ДЭФЦ (2,2 ммоля) и 1,4 мл ДИПЭА реакционную смесь перемешивали в течение ночи при комнатной температуре, растворитель выпаривали при пониженном давлении и остаток улавливали с помощью этилацетата и тщательно промывали 5% водным раствором лимонной кислоты, водой, 5% NaHCO3 и раствором NaCl. Органический слой высушивали с помощью Na2SO4, отфильтровывали и упаривали досуха. Остаток (0,51 г) высушивали в вакууме, лишали защиты с помощью 5 мл ТФК/ДХМ (1:1) в течение 2 часов и упаривали досуха. Полученный таким образом лишенный защиты фрагмент растворили в ДМФ и провели реакцию с 0,4 г N-трет.-бутилоксикарбонил-валин-N-карбоксиангидридом (1,6 ммоля) при 45oC. После перемешивания в течение 5 часов растворитель выпаривали в вакууме, добавляли этилацетат и органический слой тщательно промывали с помощью 5% водной лимонной кислоты, 5% водного NaHCO3, воды и водного NaCl и высушивали с помощью Na2SO3. Растворитель выпаривали при пониженном давлении и остаток (0,51 г, 1,1 ммоля) обрабатывали 5 мл ТФК/ДХМ (1: 1) в течение 2 часов. После испарения смеси растворителей и высушивания с помощью КОН остаток растворяли в ДМФ и 0,16 г N,N-диметилвалина (1,3 ммоля) и добавляли 0,23 г ДЭФЦ (1,4 ммоля) и 0,7 мл ДИПЭА. Реакционную смесь перемешивали в течение ночи при комнатной температуре, упаривали досуха и хроматографировали на колонке SEPHADEX LH-20. Фракции продукта собирали и выпаривали растворитель, получив 0,36 г соединения I (м. вес 432,6).
Остальные соединения могут быть получены в соответствии и примерами 1 и 2.
Соединения настоящего изобретения могут быть испытаны на противораковую активность обычными методами, включающими, например, методы, описанные ниже.
А. Методология in vitro
Цитотоксичность может быть измерена с использованием стандартной методологии для прикрепленных линий клеток, такой как, метод тетразолия для микрокультуры (ММТ). Детали этого метода были опубликованы (Alley, МС и др., Cancer Research, 48: 589-601, 1988). Экспоненциально растущие культуры клеток опухоли, такие, как НТ-29 карциномы толстой кишки или LX-1 опухоли легкого, использовали для получения микротитра высеиваемых на чашки культур. Клетки высеивали по 5000-20000 клеток на ячейку в 96-ячеистые планшеты (в 150 мкл среды) и выращивали в течение ночи при 37oC. Испытуемые соединения добавляли в 10-кратном разбавлении, варьируя от 10-4 до 10-10 моля. Затем клетки инкубировали в течение 48 часов. Для определения количества клеток, выживших в каждой ячейке, добавляли ММТ-краситель (50 мкл 3 мг/мл раствора 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолиумбромида в физиологическом растворе). Эту смесь инкубировали при 37oC в течение 5 часов, а затем в каждую ячейку добавляли по 50 мкл 25% SDS (натрийдодецилсульфат) при pH 2. После инкубации в течение ночи определяли меру поглощения света в каждой ячейке при 550 нм с помощью считывающего устройства ELISA. Вычисляют значения средних +/- SD (средних квадратичных отклонений) данных от дублирующих ячеек с использованием формулы % О/К (процент обработанных выживших клеток к контролю).
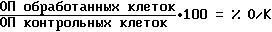
Концентрация испытываемого соединения, при которой при О/К дает 50%-ное ингибирование роста, обозначается как ИК50
Б. Методология in vivo
Соединения настоящего изобретения в дальнейшем могут быть исследованы любым из многих методов доклинических испытаний для определения активности in vivo, которыми определяют клиническую пригодность. Такие исследования проводят на лишенных волос мышах, которым трансплантировали ("ксенографировали") опухолевую ткань, предпочтительно человека, как это хорошо известно в данной области техники. Испытуемые соединения оценивают на их противоопухолевую эффективность по применению на несущих трансплантат (ксенографт) мышах.
Более конкретно, опухоли человека, которые были выращены на лишенных волос мышах без вилочковой железы, трансплантируют в новых животных-реципиентов, используя фрагменты опухоли, имеющие размеры около 50 мг. День трансплантации обозначают как день 0. Спустя 6-10 дней мышей обрабатывают исследуемыми соединениями с помощью внутривенной или внутрибрюшинной инъекции группами по 5-10 мышей для каждой дозы. Соединения давали ежедневно в течение 5 дней, 10 дней или 15 дней в дозах 10-100 мг/кг веса тела. Диаметры опухолей и веса тела измеряли дважды в неделю. Объемы опухоли вычисляют, используя диаметры, измеренные кронциркулем Вернье, по формуле:
(длина • ширина2)/2 = мг веса опухоли
Для каждой обработанной группы вычисляют средние значения веса опухоли и значения О/К определяют для каждой группы по сравнению с контрольными необработанными опухолями.
При определении цитотоксичности in vitro установлено, что пептид согласно примеру 1 имеет ИК50 = 1 • 10-7, а пептид согласно примеру 2 - 2 • 10-6
Соединения не обладают значительными токсическими побочными эффектами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПЕПТИДА ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2116312C1 |
| ИНГИБИТОРЫ ФАКТОРА ХА | 1995 |
|
RU2152954C1 |
| СИНТЕЗ ИНСУЛИНОТРОПНЫХ ПЕПТИДОВ | 2007 |
|
RU2448978C2 |
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 15 | 1998 |
|
RU2195462C2 |
| ТВЕРДОФАЗНЫЙ СИНТЕЗ ПЕПТИДОВ С ЗАЩИТНЫМИ ГРУППАМИ ВОС И FMOC | 2007 |
|
RU2439075C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ РЕГУЛИРОВАТЬ ПРОЛИФЕРАЦИЮ КЛЕТОК, И СПОСОБЫ ИНГИБИРОВАНИЯ КЛЕТОЧНОЙ ПРОЛИФЕРАЦИИ ЧЕЛОВЕКА И ЖИВОТНОГО (ВАРИАНТЫ) | 1989 |
|
RU2079509C1 |
| ГЛИКОЗИЛИРОВАННЫЙ ПОЛИПЕПТИД И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2012 |
|
RU2636456C2 |
| ПРОТИВООПУХОЛЕВЫЕ ПЕПТИДЫ | 1996 |
|
RU2182911C2 |
| ЦИКЛОПЕПТИДЫ ИЛИ ИХ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2129563C1 |
| НОВЫЙ МАРКЕР ДЛЯ ДИАГНОСТИКИ РАКА ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ | 2020 |
|
RU2802849C2 |

Пептиды общей формулы I, где R1 - C1-C7-алкил; R2 - C1-C4-алкил; X - C1-C5-алкил; R3 - низший алкил; B - C1-C5-алкил; D - C1-C5-алкокси; M - фениланилин, R4 - H; R5 - H или бензил; d = 0 или 1, или их соли обладают улучшенными терапевтическими качествами для лечения неопластических болезней по сравнению с Доластатином-10, но в отличие от него может быть синтезирован традиционными способами. 2 c.п. ф-лы, 2 табл. 

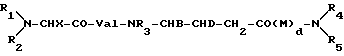
где R1 - C1 - C7-алкил;
R2 - C1 - C4-алкил;
X - C1 - C5-алкил;
R3 - низший алкил;
B - C1 - C5-алкил;
D - C1 - C5-алкокси;
M - фенилаланил;
R4 - водород;
R5 - водород или бензил;
d = 0 или 1,
или их соли с физиологически допустимыми кислотами.
| Способ получения пептидов | 1977 |
|
SU753358A3 |
| Способ получения полипептидов | 1977 |
|
SU904518A3 |
| Способ получения производных тетрапептидов или их солей | 1978 |
|
SU908246A3 |
| US 4816444 A, 1989 | |||
| US 4879278 A, 1989 | |||
| US 4978744 A, 1990 | |||
| РАБОЧИЙ ОРГАН ДЛЯ ЗАГЛАЖИВАНИЯ ПОВЕРХНОСТЕЙБЕТОННЫХ | 0 |
|
SU357332A1 |
| ПЕЧЬ ДЛЯ СЖИГАНИЯ МУСОРА | 0 |
|
SU222283A1 |
| Pettit G.R | |||
| et al | |||
| J | |||
| Amer | |||
| chem | |||
| Soc | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
