ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
По данной заявке испрашивается приоритет на основании ранее поданной заявки на выдачу патента Японии №210804/2007 (дата подачи 13 августа 2007 года). Полное описание заявки на выдачу патента Японии 210804/2007 включено в этот документ посредством ссылки.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения пирипиропеновых производных, применимых в качестве средств для борьбы с вредителями, и более конкретно относится к способу получения пирипиропеновых производных, которые содержат ацилоксигруппы в 1- и 11-положениях и гидроксильную группу в 7-положении.
Уровень техники
Пирипиропеновые производные, содержащие ацилокси в 1- и 11-положениях и гидроксил в 7-положении, представляют собой соединения, которые можно использовать для борьбы с вредителями, как описано в WO 2006/129714.
В WO 2006/129714 и в опубликованной заявке на выдачу патента Японии №259569/1996 раскрыт способ получения пирипиропеновых производных, содержащих ацилокси в 1- и 11-положениях и гидроксил в 7-положении. В соответствии со способом получения пирипиропеновые производные очищают или выделяют из множества продуктов, полученных путем неселективного гидролиза ацилокси с использованием 1,7,11-триацилоксисоединения в качестве исходного соединения. Однако этот способ получения страдает такими недостатками, как малый выход и непригодность для крупномасштабного синтеза.
Кроме того, в опубликованной заявке на выдачу патента Японии №259569/1996 описано использование сочетания защитных групп для синтеза пирипиропеновых производных; а в Journal of Antibiotics Vol. 49, No. 11, p. 1149, 1996, Bioorganic Medicinal Chemistry Letter Vol. 5, No. 22, p. 2683, 1995, опубликованной заявке на выдачу патента Японии №269065/1996 и WO 2008/013336 описан пример синтеза, в котором ацил вводят в 7-положение с использованием защитной группы. Однако в указанных документах не описан конкретный способ, в котором защитная группа используется для получения пирипиропеновых производных, содержащих ацилокси в 1- и 11-положениях и гидроксил в 7-положении.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения обнаружили, что пирипиропеновые производные, содержащие ацилокси в 1- и 11-положениях и гидроксильную группу в 7-положении, могут быть получены с высоким выходом из пирипиропена A (опубликованная заявка на выдачу патента Японии №259569/1996; Bioorganic Medicinal Chemistry Letter Vol. 5, No. 22, p. 2683, 1995; и WO 2004/060065), полученного в виде встречающегося в природе вещества, с использованием подходящей защитной группы. Настоящее изобретение было осуществлено на основании данного открытия.
Соответственно, целью настоящего изобретения является предоставление способа получения пирипиропеновых производных, применимых в качестве средств для борьбы с вредителями, и соединений в качестве промежуточных продуктов для получения пирипиропеновых производных.
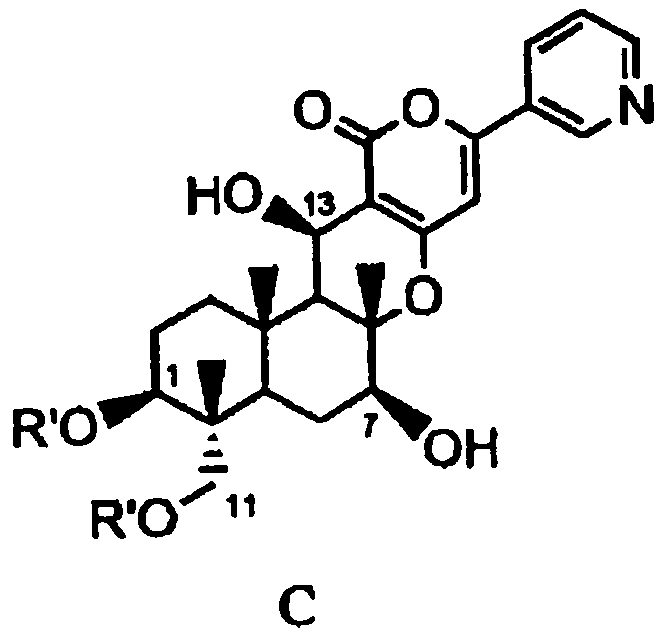
В соответствии с первым аспектом настоящее изобретение относится к способу получения соединения C, представленного формулой C:
[Химическая формула 1]
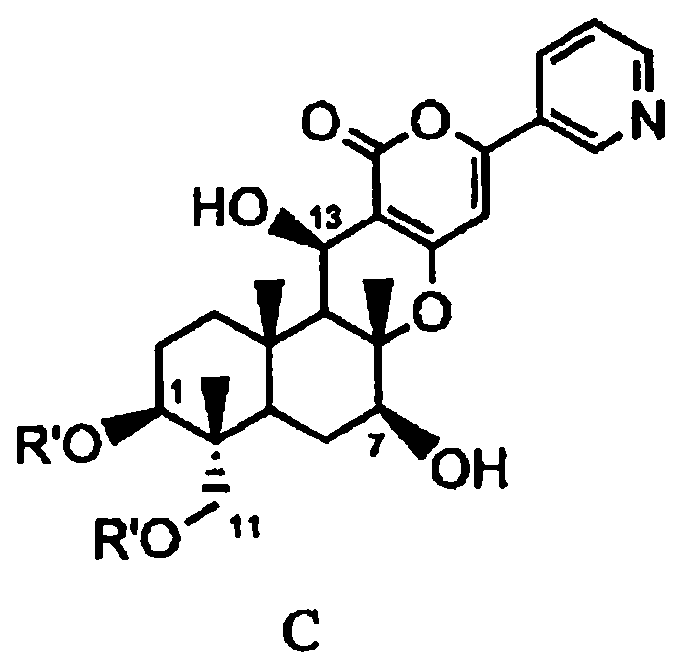 ,
,
в которой R' представляет собой линейный, разветвленный или циклический C2-6алкилкарбонил, при условии, что если алкильный фрагмент в алкилкарбонильной группе является разветвленным или циклическим, то R' представляет собой C3-6алкилкарбонил, причем способ включает стадии:
(a1) гидролиза ацетила в 7-положении соединения A1, представленного формулой A1:
[Химическая формула 2]
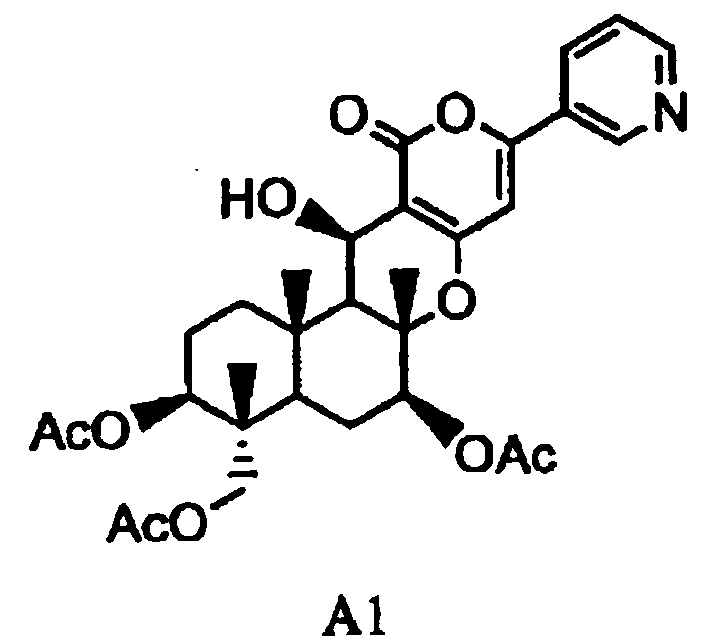 ,
,
в которой Ac представляет собой ацетил,
основанием для селективного дезацилирования соединения A1, затем защиты гидроксила в 7-положении с получением соединения B1, представленного формулой B1:
[Химическая формула 3]
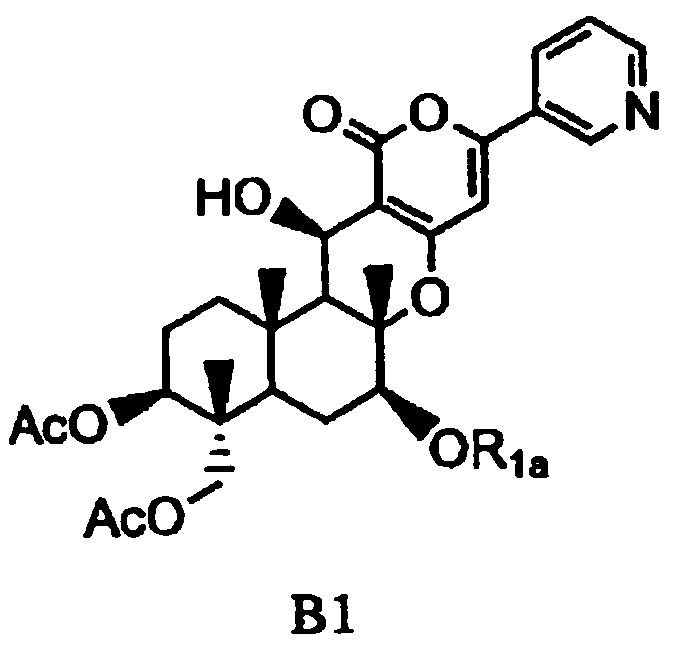 ,
,
в которой
значение Ac определено выше,
R1a представляет собой необязательно замещенный линейный C2-4алкилкарбонил; необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил; необязательно замещенный атомом галогена C1-6алкилокси-C1-6алкил; необязательно замещенный атомом галогена C1-6алкилтио-C1-6алкил; необязательно замещенный атомом галогена линейный, разветвленный или циклический C1-4алкил, при условии, что если алкил в C1-4алкильной группе является разветвленным или циклическим, то алкильная группа представляет собой C3-4алкил; необязательно замещенный атомом галогена C2-6алкенил; необязательно замещенный атомом галогена C2-6алкинил; необязательно замещенный бензил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу,
где в R1a необязательно присутствующий на алкилкарбониле заместитель выбирают из группы, состоящей из атомов галогена, C1-4алкилокси, C1-4галогеналкилокси, C1-4алкилкарбонила, C1-4галогеналкилкарбонила, C1-4алкилкарбонилокси и C1-4галогеналкилкарбонилокси, а необязательно присутствующий на гетероциклической группе и бензиле заместитель выбирают из группы, состоящей из атомов галогена, C1-4алкила, C1-4алкилокси, C1-4галогеналкилокси, C1-4алкилтио, C1-4галогеналкила, C1-4алкилкарбонила, C1-4галогеналкилкарбонила, C1-4алкилкарбонилокси, C1-4галогеналкилкарбонилокси, нитро и циано,
затем гидролиза ацетила в 1- и 11-положениях соединения B1 основанием для дезацилирования соединения B1 с получением тем самым соединения Fa, представленного формулой Fa:
[Химическая формула 4]
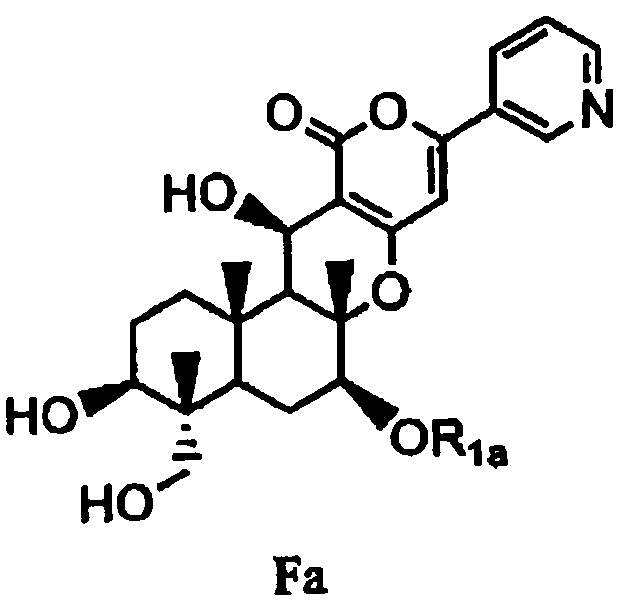 ,
,
в которой значение R1a определено выше, или
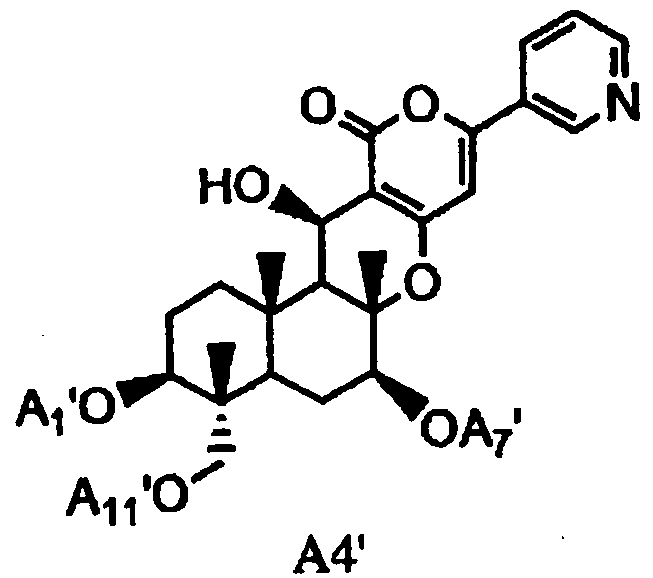
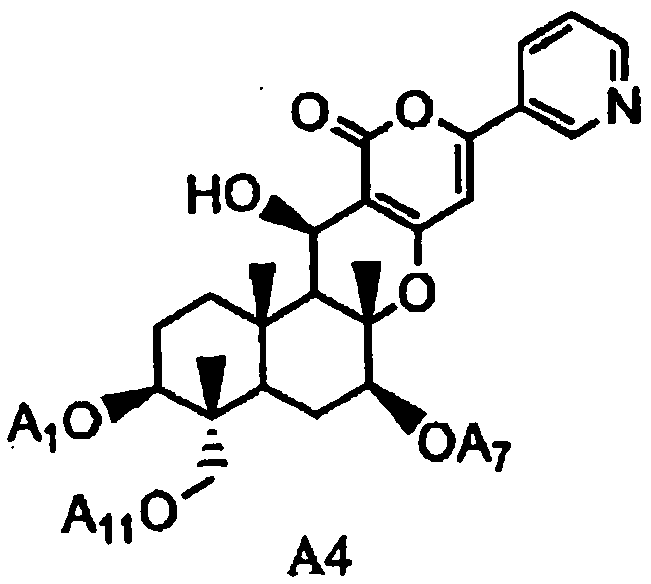
(a2) гидролиза ацила в 1-, 7- и 11-положениях соединения A1 или соединения A4', представленного формулой A4':
[Химическая формула 5]
 ,
,
в которой A1', A7' и A11', которые могут быть одинаковыми или разными, представляют собой ацетил или пропионил, при условии, что A1', A7' и A11' одновременно не представляют собой ацетил,
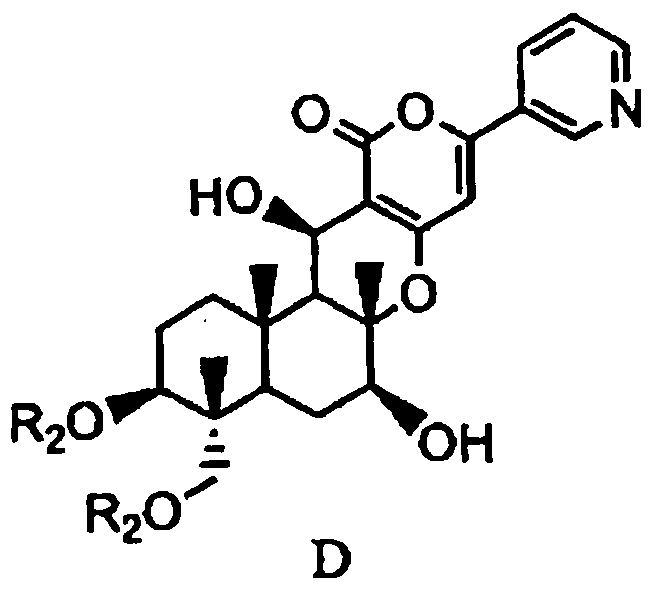
основанием для дезацилирования соединения A1 или A4', и затем защиты гидроксила в 1- и 11-положениях с получением соединения D, представленного формулой D:
[Химическая формула 6]
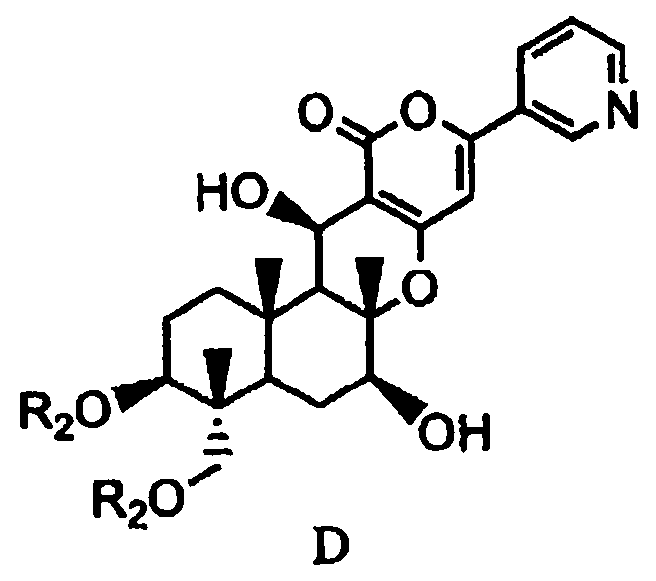 ,
,
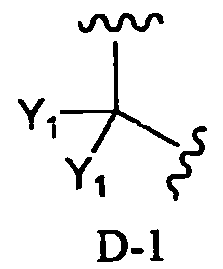
в которой два R2 образуют вместе группу, выбранную из групп, представленных формулами D-1, D-2, D-3 и D-4:
[Химическая формула 7]
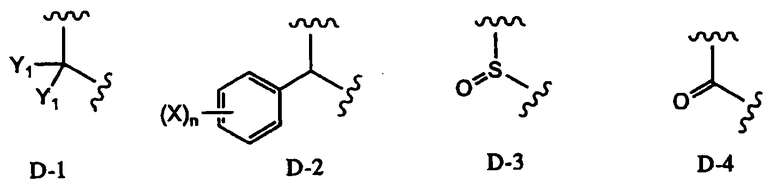 ,
,
в которых Y1 представляет собой атом водорода или C1-4алкил; заместители X, которые могут быть одинаковыми или разными, представляют собой атом водорода, C1-4алкокси или нитро; и n равно 0-5,
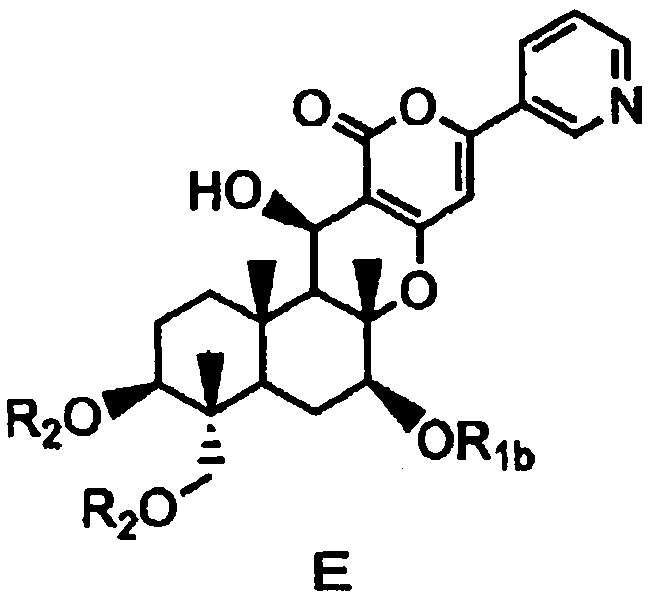
затем защиты гидроксила в 7-положении соединения D с получением соединения E, представленного формулой E:
[Химическая формула 8]
 ,
,
в которой
R1b представляет собой формил; необязательно замещенный линейный C1-4алкилкарбонил; необязательно замещенный бензил; необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил; необязательно замещенный атомом галогена C1-6алкилокси-C1-6алкил; необязательно замещенный атомом галогена C1-6алкилтио-C1-6алкил; необязательно замещенный атомом галогена линейный, разветвленный или циклический C1-4алкил, при условии, что если алкил в C1-4алкильной группе является разветвленным или циклическим, то алкильная группа представляет собой C3-4алкил; необязательно замещенный атомом галогена C2-6алкенил; необязательно замещенный атомом галогена C2-6алкинил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу,
где в R1b необязательно присутствующий на алкилкарбониле заместитель выбирают из группы, состоящей из атомов галогена, C1-4алкилокси, C1-4галогеналкилокси, C1-4алкилкарбонила, C1-4галогеналкилкарбонила, C1-4алкилкарбонилокси и C1-4галогеналкилкарбонилокси, а необязательно присутствующий на гетероциклической группе и бензиле заместитель выбирают из группы, состоящей из атомов галогена, C1-4алкила, C1-4алкилокси, C1-4галогеналкилокси, C1-4алкилтио, C1-4галогеналкила, C1-4алкилкарбонила, C1-4галогеналкилкарбонила, C1-4алкилкарбонилокси, C1-4галогеналкилкарбонилокси, нитро и циано, и
значение R2 определено выше,
с последующим удалением защитных групп в 1- и 11-положениях соединения E с получением соединения Fb, представленного формулой Fb:
[Химическая формула 9]
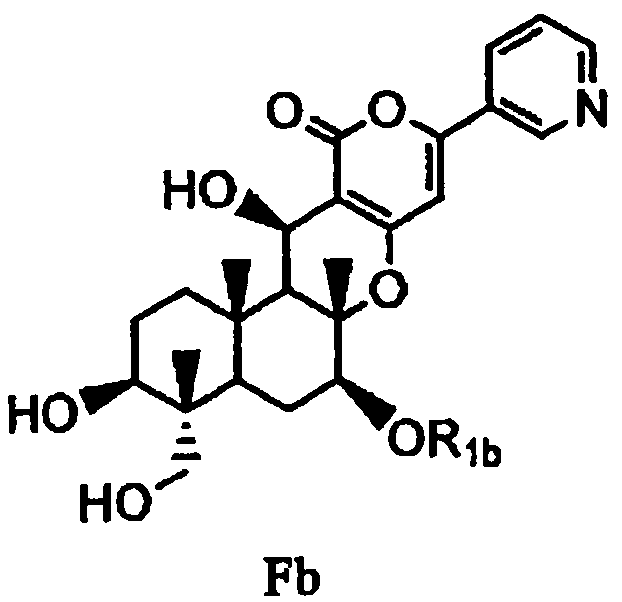 ,
,
в которой значение R1b определено выше, и
(b) ацилирования гидроксила в 1- и 11-положениях соединения Fa или Fb ацилирующим агентом, соответствующим предполагаемой R', с получением соединения B2a или B2b, представленного формулой B2a или B2b:
[Химическая формула 10]
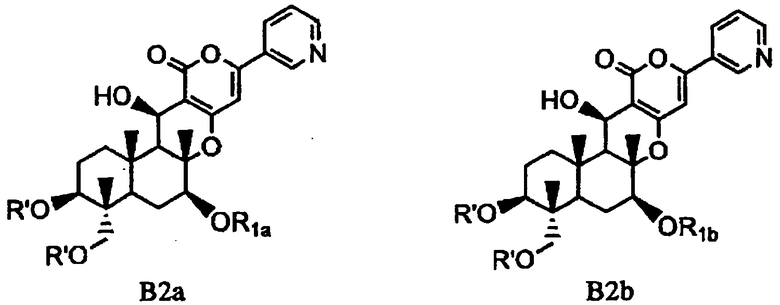 ,
,
в которых значения R1a, R1b и R' определены выше,
а затем удаления защитной группы в 7-положении соединения B2a или соединения B2b.
В соответствии со вторым аспектом настоящее изобретение относится к способу получения соединения B2a, представленного формулой B2a:
[Химическая формула 11]
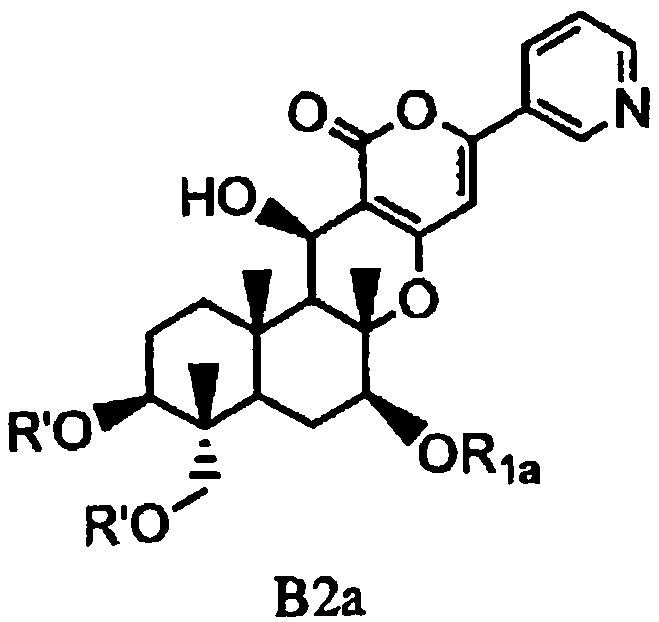 ,
,
в которой значение R1a определено выше, и R' представляет собой циклический C3-6алкилкарбонил, причем способ включает:
гидролиз ацетила в 7-положении соединения A1, описанного выше, основанием для селективного дезацилирования соединения A1, затем защиту гидроксила в 7-положении с получением соединения B1, представленного формулой B1:
[Химическая формула 12]
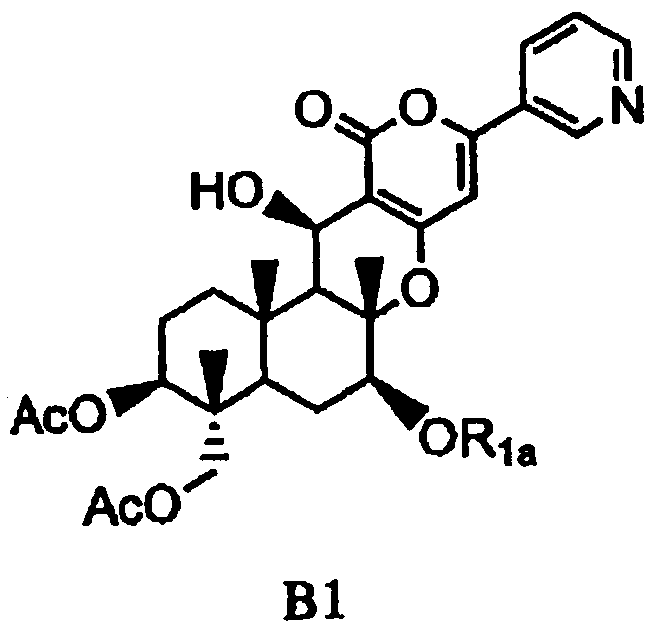 ,
,
в которой значения Ac и R1a определены выше,
затем гидролиз ацетила в 1- и 11-положениях соединения B1 основанием для дезацилирования соединения B1, с получением тем самым соединения Fa, представленного формулой Fa:
[Химическая формула 13]
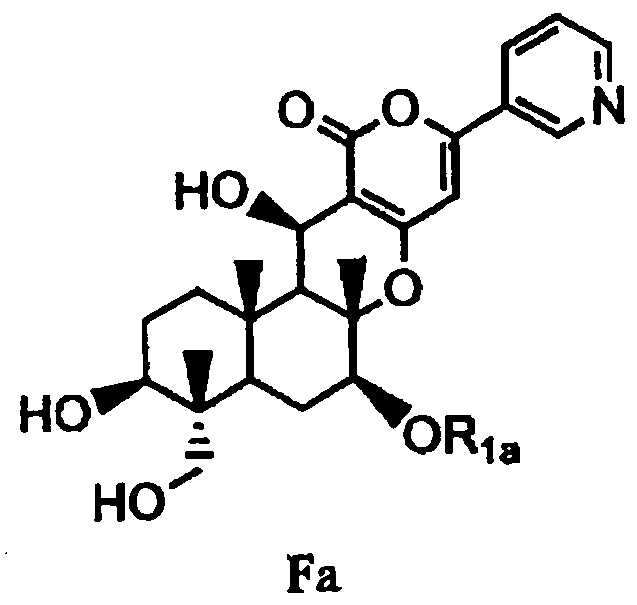 ,
,
в которой значение R1a определено выше,
а затем ацилирование гидроксила в 1- и 11-положениях соединения Fa ацилирующим агентом, соответствующим предполагаемой R'.
В соответствии с третьим аспектом настоящее изобретение относится к способу получения соединения B2b, представленного описанной выше формулой B2b, в которой R' представляет собой циклический C3-6алкилкарбонил, причем способ включает:
гидролиз ацила в 1-, 7- и 11-положениях соединения A4, представленного формулой A4:
[Химическая формула 14]
 ,
,
в которой A1, A7 и A11, которые могут быть одинаковыми или разными, представляют собой ацетил или пропионил,
основанием для дезацилирования соединения A4, затем защиту гидроксила в 1- и 11-положениях с получением соединения D, затем защиту гидроксила в 7-положении соединения D с получением описанного выше соединения E, с последующим удалением защитной группы в 1- и 11-положениях соединения E с получением описанного выше соединения Fb, а затем ацилированием гидроксила в 1- и 11-положениях соединения Fb ацилирующим агентом, соответствующим R'.
В соответствии с четвертым аспектом настоящее изобретение относится к способу получения соединения C, представленного формулой C, в которой R' представляет собой циклический C3-6алкилкарбонил. Способ включает ацилирование гидроксила в 1- и 11-положениях описанного выше соединения Fb ацилирующим агентом, соответствующим R', с получением соединения B2b, а затем удаление защитной группы в 7-положении соединения B2b.
В соответствии с пятым аспектом настоящее изобретение относится к способу получения описанного выше соединения C. Способ включает гидролиз ацила в 1-, 7- и 11-положениях соединения A4 основанием для дезацилирования соединения A4, затем защиту гидроксила в 1- и 11-положениях с получением соединения D, затем защиту гидроксила в 7-положении соединения D с получением соединения E, с последующим удалением защитных групп в 1- и 11-положениях соединения E с получением соединения Fb, а затем ацилированием гидроксила в 1- и 11-положениях соединения Fb ацилирующим агентом, соответствующим R', с получением соединения B2b, а затем удаление защитной группы в 7-положении соединения B2b.
В соответствии с шестым аспектом настоящее изобретение относится к соединению, которое содержит ацилокси в 1- и 11-положениях и гидроксил в 7-положении и применимо в качестве промежуточного продукта для получения пирипиропеновых производных. Соединение представлено формулой B2b:
[Химическая формула 15]
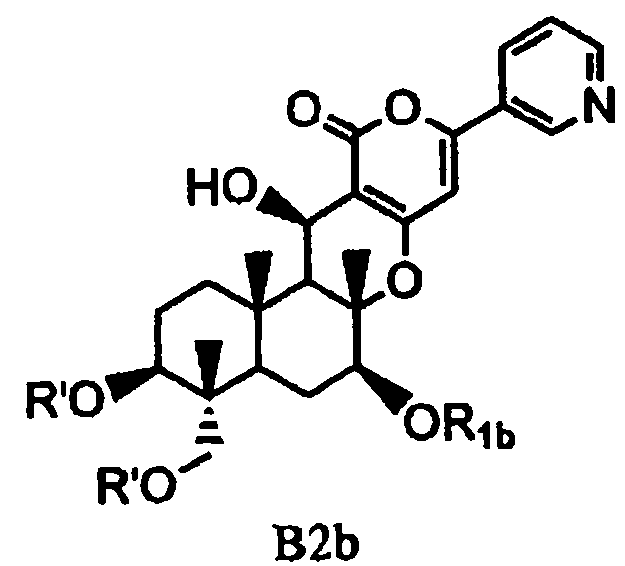 ,
,
в которой значение R1b определено выше; и R' представляет собой циклический C3-6алкилкарбонил.
Настоящее изобретение позволяет получать с высоким выходом пирипиропеновые производные, которые содержат ацилоксигруппы в 1- и 11-положениях и гидроксильную группу в 7-положении и которые применимы в качестве средств для борьбы с вредителями.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Используемый в этом документе термин «галоген» означает фтор, хлор, бром или йод.
Термины «алкил», «алкенил» или «алкинил», используемые в этом тексте в качестве заместителя или части заместителя, означают алкил, алкенил или алкинил линейного, разветвленного или циклического типа, или сочетанного типа, если иное не определено особо.
Обозначение «Ca-b», используемое в этом документе, применительно к заместителю означает, что число атомов углерода, содержащееся в заместителе, составляет от a до b. Кроме того, «Ca-b» в термине «Ca-bалкилкарбонил» означает, что число атомов углерода в алкильном фрагменте, за исключением атомов углерода в карбонильном фрагменте, составляет от a до b.
Используемый в этом документе термин «галогеналкил» означает алкил, замещенный, по крайней мере, одним атомом галогена. По аналогии, термины «галогеналкилокси», «галогеналкилкарбонил» и «галогеналкилкарбонилокси», соответственно, означают алкилокси, замещенную, по крайней мере, одним атомом галогена, алкилкарбонил, замещенный, по крайней мере, одним атомом галогена, и алкилкарбонилокси, замещенный, по крайней мере, одним атомом галогена.
Конкретные примеры линейной, разветвленной или циклической C2-6алкилкарбонильной группы, представленной R', в которой если алкильный фрагмент в C2-6алкилкарбонильной группе является разветвленным или циклическим, то алкильный фрагмент представляет собой C3-6алкилкарбонил, включают циклопропанкарбонил и пропионил. Алкилкарбонильная группа предпочтительно представляет собой циклический C3-6алкилкарбонил, более предпочтительно циклопропанкарбонил.
Конкретные примеры группы -SiR3R4R5, в которой каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил, представленные R1a и R1b, включают триметилсилил, триэтилсилил, триизопропилсилил, трет-бутилдиметилсилил и трет-бутилдифенилсилил. Группа -SiR3R4R5 является необязательно замещенной, и такие заместители включают атомы галогена. В группе -SiR3R4R5 все R3, R4 и R5 предпочтительно представляют собой линейный или разветвленный C1-6алкил, то есть группа -SiR3R4R5 предпочтительно представляет собой алкилсилил, более предпочтительно трет-бутилдиметилсилил.
Конкретные примеры линейной, разветвленной или циклической C1-4алкильной группы, представленной R1a и R1b, в которой если алкил в C1-4алкильной группе является разветвленным или циклическим, то алкильная группа представляет собой C3-4алкил, включают метил, этил, пропил, изопропил, циклопропил, н-бутил, изобутил и трет-бутил. Алкильная группа является необязательно замещенной, и такие заместители включают атомы галогена.
Конкретные примеры C2-6алкенильной группы, представленной R1a и R1b, включают винил, (1- или 2-)пропенил, (1-, 2- или 3-)бутенил, (1-, 2-, 3- или 4-)пентенил и (1-, 2-, 3-, 4- или 5-)гексенил. Алкенильная группа является необязательно замещенной, и такие заместители включают атомы галогена.
Конкретные примеры C2-6алкинильной группы, представленной R1a и R1b, включают этинил, (1- или 2-)пропинил, (1-, 2- или 3-)бутинил, (1-, 2-, 3- или 4-)пентинил и (1-, 2-, 3-, 4- или 5-)гексинил. Алкинильная группа является необязательно замещенной, и такие заместители включают атомы галогена.
Конкретные примеры насыщенной или ненасыщенной 5- или 6-членной гетероциклической группы, представленной R1a и R1b, включают тетрагидропиранил, тетрагидротиопиранил, тетрагидрофуранил и тетрагидротиофуранил. Гетероциклическая группа является необязательно замещенной, и такие заместители включают атомы галогена, C1-4алкил, C1-4алкилокси, C1-4галогеналкилокси, C1-4алкилтио, C1-4галогеналкил, C1-4алкилкарбонил, C1-4галогеналкилкарбонил, C1-4алкилкарбонилокси, C1-4галогеналкилкарбонилокси, нитро и циано. Гетероциклическая группа предпочтительно представляет собой тетрагидропиранил.
Конкретные примеры линейной C2-4алкилкарбонильной группы, представленной R1a, включают пропионил, пропилкарбонил и н-бутилкарбонил. Алкилкарбонильная группа является необязательно замещенной, и такие заместители включают атомы галогена, C1-4алкилокси, C1-4галогеналкилокси, C1-4алкилкарбонил, C1-4галогеналкилкарбонил, C1-4алкилкарбонилокси и C1-4галогеналкилкарбонилокси.
Конкретные примеры линейной C1-4алкилкарбонильной группы, представленной R1b, включают ацетил, пропионил, пропилкарбонил и н-бутилкарбонил. Алкилкарбонильная группа является необязательно замещенной, и такие заместители включают атомы галогена, C1-4алкилокси, C1-4галогеналкилокси, C1-4алкилкарбонил, C1-4галогеналкилкарбонил, C1-4алкилкарбонилокси и C1-4галогеналкилкарбонилокси.
C1-6алкилокси-C1-6алкильная группа, представленная R1a и R1b, является необязательно замещенной, и такие заместители включают атомы галогена.
C1-6алкилтио-C1-6алкильная группа, представленная R1a и R1b, является необязательно замещенной, и такие заместители включают атомы галогена.
Бензильная группа, представленная R1a и R1b, является необязательно замещенной, и такие заместители включают атомы галогена, C1-4алкил, C1-4алкилокси, C1-4галогеналкилокси, C1-4алкилтио, C1-4галогеналкил, C1-4алкилкарбонил, C1-4галогеналкилкарбонил, C1-4алкилкарбонилокси, C1-4галогеналкилкарбонилокси, нитро и циано.
Предпочтительно R1a представляет собой необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу, более предпочтительно группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил; или насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу, еще более предпочтительно группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил; или тетрагидропиранил, наиболее предпочтительно трет-бутилдиметилсилил или тетрагидропиранил.
Предпочтительно R1b представляет собой ацетил, хлорацетил, необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу, или необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил, более предпочтительно ацетил, хлорацетил или необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил, еще более предпочтительно ацетил, хлорацетил или трет-бутилдиметилсилил, наиболее предпочтительно ацетил или хлорацетил.
Заместитель, образованный путем объединения вместе двух заместителей R2, предпочтительно представляет собой группу, представленную формулой D-1 или D-2:
[Химическая формула 16]
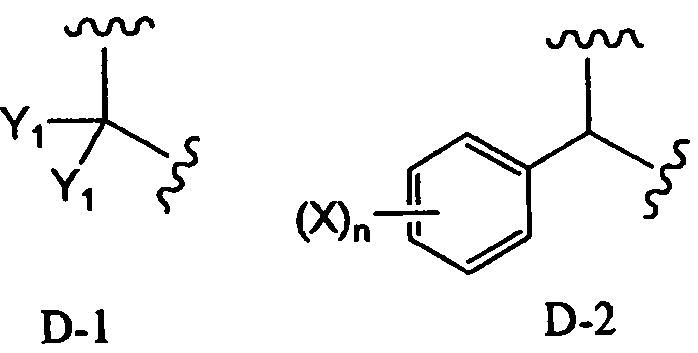 ,
,
в которой Y1 представляет собой атом водорода или C1-4алкил; заместители X, которые могут быть одинаковыми или разными, представляют собой атом водорода, C1-4алкокси или нитро; и n равно 0-5, более предпочтительно изопропилиден, бензилиден или пара-метоксибензилиден. В соответствии с другим вариантом осуществления заместитель, образованный путем объединения вместе двух заместителей R2, предпочтительно представляет собой D-1, более предпочтительно изопропилиден.
Предпочтительно каждый A1, A7 и A11 представляют собой ацетил.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения, в способе в соответствии с первым аспектом настоящего изобретения или в способе в соответствии с пятым аспектом настоящего изобретения R' представляет собой циклический C3-6алкилкарбонил.
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения, в способе в соответствии с первым аспектом настоящего изобретения R1a представляет собой необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу.
В соответствии с еще одним предпочтительным вариантом осуществления настоящего изобретения, в способе в соответствии с первым аспектом настоящего изобретения или в способе в соответствии с третьим аспектом настоящего изобретения R1b представляет собой ацетил, хлорацетил или необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил.
В соответствии с дополнительным предпочтительным вариантом осуществления настоящего изобретения, в способе в соответствии с первым аспектом настоящего изобретения или в способе в соответствии с третьим аспектом настоящего изобретения два заместителя R2 образуют вместе группу, представленную формулой D-1 или D-2:
[Химическая формула 17]
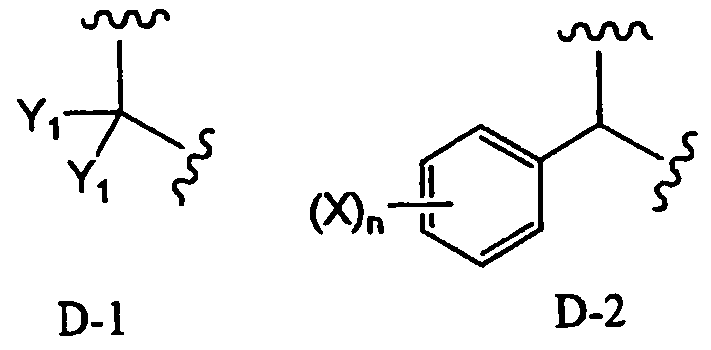 ,
,
в которой Y1 представляет собой атом водорода или C1-4алкил; заместители X, которые могут быть одинаковыми или разными, представляют собой атом водорода, C1-4алкокси или нитро; и n равно 0-5.
В соответствии с еще одним дополнительным предпочтительным вариантом осуществления настоящего изобретения, в способе в соответствии со вторым аспектом настоящего изобретения R1a представляет собой необязательно замещенный линейный C2-4алкилкарбонил; необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу.
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения, в способе в соответствии с шестым аспектом настоящего изобретения R1b представляет собой ацетил, хлорацетил, необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил, или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу; и R' представляет собой циклический C3-6алкилкарбонил.
В соответствии с еще одним другим предпочтительным вариантом осуществления настоящего изобретения, в способе R' в формулах B2a, B2b и C представляет собой пропионил или циклопропанкарбонил.
В соответствии с дополнительным предпочтительным вариантом осуществления настоящего изобретения, в способе предполагаемое соединение получают через соединение Fa, где R1a в формуле B1, Fa или B2a представляет собой необязательно замещенный линейный или разветвленный алкилсилил или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу.
В соответствии с еще одним дополнительным предпочтительным вариантом осуществления настоящего изобретения, в способе предполагаемое соединение получают через соединение Fb, где R1b в формуле E, Fb или B2b представляет собой ацетил, хлорацетил или необязательно замещенный линейный или разветвленный алкилсилил.
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения, в способе предполагаемое соединение получают через соединение Fb, где R2 в формуле D или E представляет собой группу, представленную формулой D-3.
В соответствии с более предпочтительным вариантом осуществления настоящего изобретения, предполагаемое соединение получают через соединения D, E, Fb и B2b, где R2 в формуле D или E представляет собой группу, представленную формулой D-3; R1b в формуле E, Fb или B2b представляет собой ацетил, хлорацетил или необязательно замещенный линейный или разветвленный алкилсилил; и R' в формулах B2b и C представляет собой циклопропанкарбонил.
В соответствии с другим аспектом настоящее изобретение относится к способу получения соединения C, представленного формулой C:
[Химическая формула 18]
 ,
,
в которой R' представляет собой циклический C3-6алкилкарбонил, причем способ включает:
гидролиз ацила в 1-, 7- и 11-положениях соединения A4, представленного формулой A4:
[Химическая формула 19]
 ,
,
в которой A1, A7 и A11, которые могут быть одинаковыми или разными, представляют собой ацетил или пропионил,
основанием для дезацилирования соединения A4, а затем защиту гидроксила в 1- и 11-положениях с получением соединения D, представленного формулой D:
[Химическая формула 20]
 ,
,
в которой два R2 образуют вместе группу, представленную формулой D-1:
[Химическая формула 21]
 ,
,
в которой Y1 представляет собой атом водорода или C1-4алкил,
затем защиту гидроксила в 7-положении соединения D с получением соединения E, представленного формулой E:
[Химическая формула 22]
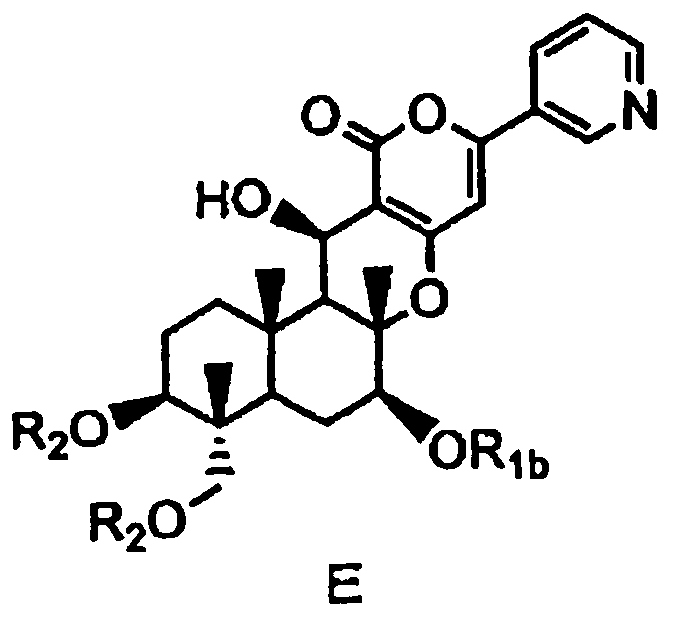 ,
,
в которой R1b представляет собой ацетил или хлорацетил, и значение R2 определено выше,
затем удаление защитных групп в 1- и 11-положениях соединения E с получением соединения Fb, представленного формулой Fb:
[Химическая формула 23]
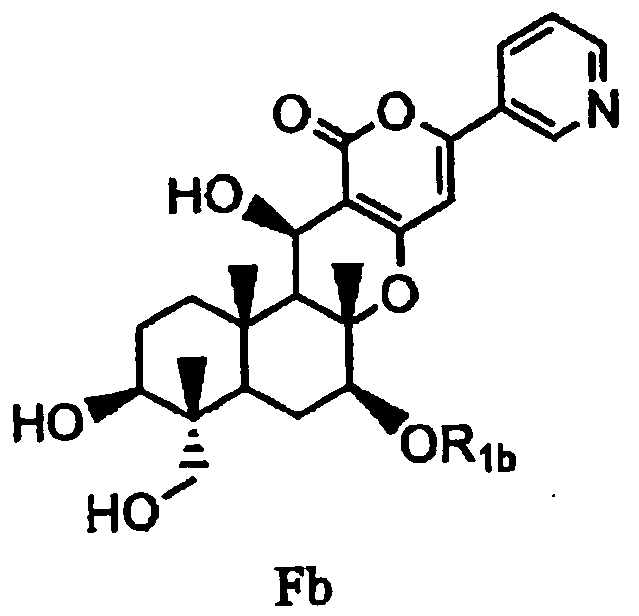 ,
,
затем ацилирование гидроксила в 1- и 11-положениях соединения Fb ацилирующим агентом, соответствующим R', с получением соединения B2b, представленного формулой B2b:
[Химическая формула 24]
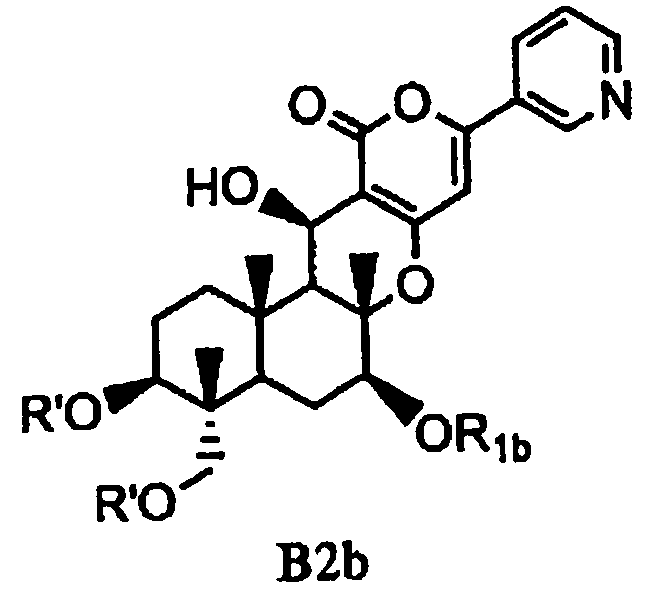 ,
,
в которой значения R1b и R' определены выше,
а затем удаление защитной группы в 7-положении соединения B2b.
Настоящее изобретение может быть подробно описано в соответствии со следующей схемой.
[Химическая формула 25]
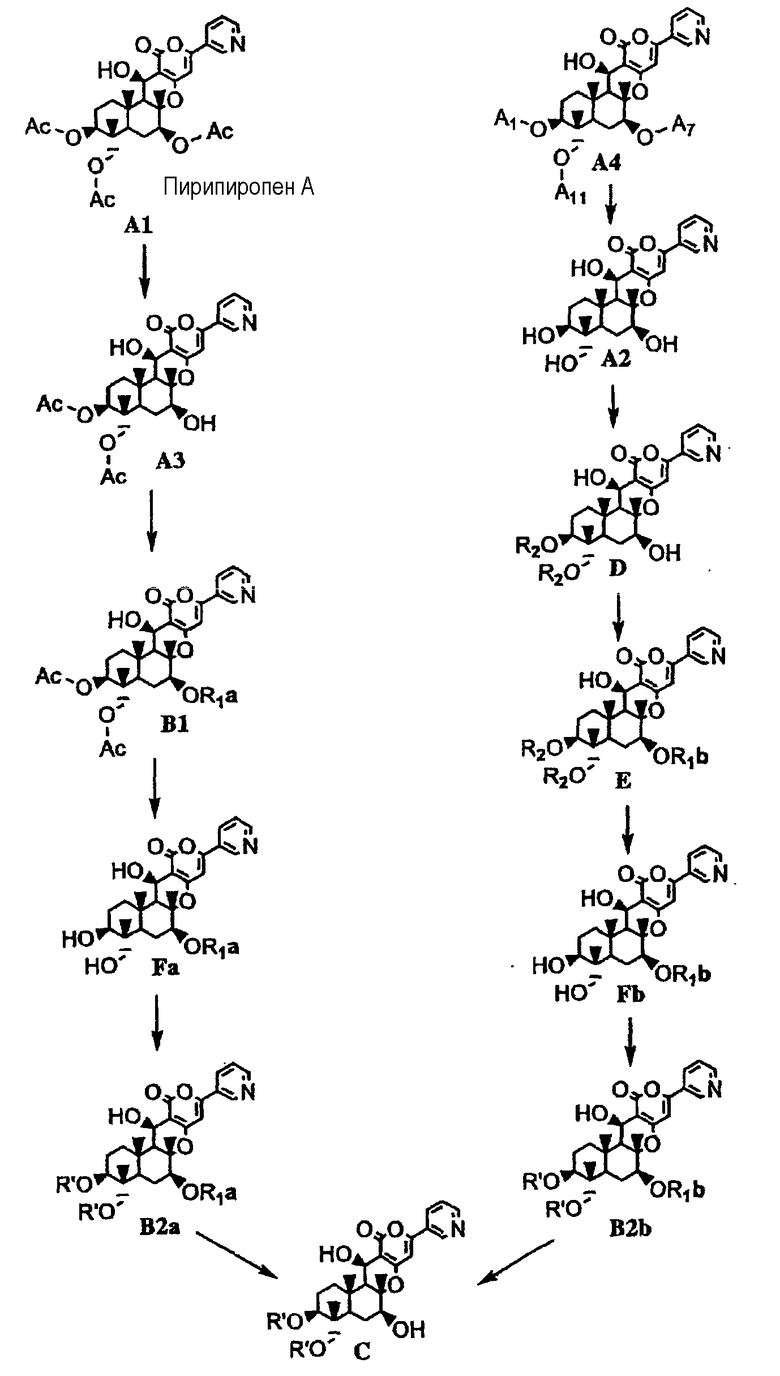
В этой схеме значения Ac, R1a, R1b, A1, A7, A11 и R2 определены выше; R' представляет собой линейный, разветвленный или циклический C2-6алкилкарбонил, где если алкильный фрагмент в C2-6алкилкарбонильной группе является разветвленным или циклическим, то алкильный фрагмент представляет собой C3-6алкилкарбонил.
Продукт каждой стадии может использоваться на последующей стадии без дополнительной обработки.
1-1: Получение соединения A3 из соединения A1
Соединение A1 может быть получено способами, описанными, например, в опубликованной заявке на выдачу патента Японии №184158/1994, WO 2004/060065, опубликованной заявке на выдачу патента Японии №259569/1996 или Bioorganic Medicinal Chemistry Letter Vol. 5, No. 22, p. 2683.
Растворители, используемые на стадии получения соединения A3 из соединения A1, включают спиртовые растворители, содержащие 1-4 атома углерода, такие как метанол, эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан, апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид и ацетонитрил, галоидзамещенные растворители, такие как дихлорметан и хлороформ, и воду, и смешанные растворители, составленные из двух или более из указанных растворителей.
Основания, используемые в этом документе, включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития и гидроксид бария, щелочные металлы, такие как метоксид натрия, этоксид натрия и трет-бутоксид калия, алкоксиды щелочноземельных металлов, или органические основания, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гидразин и гуанидин. Предпочтительными являются 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, метоксид натрия и трет-бутоксид калия. Особенно предпочтительными являются 1,8-диазабицикло[5.4.0]ундец-7-ен и трет-бутоксид калия.
Количество используемого основания предпочтительно составляет от 0,01 до 1,2 эквивалентов относительно количества соединения A1. Температура реакции предпочтительно составляет от -20°C до 50°C. Продолжительность реакции предпочтительно составляет от 0,5 ч до 7 суток.
1-2: Получение соединения B1 из соединения A3
На стадии получения соединения B1 из соединения A3 гидроксил в 7-положении может быть защищен с использованием галогенида R1a, представленного R1a-Hal, где Hal представляет собой атом галогена, ангидрида кислоты R1a или смешанного ангидрида кислоты R1a, соответствующего предполагаемой R1a, или 3,4-дигидропирана в присутствии основания, в присутствии кислоты или в отсутствие основания и кислоты, или с использованием конденсирующего агента, такого как дициклогексилкарбодиимид, 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид, карбонилдиимидазол, дипиридила дисульфид, диимидазоила дисульфид, 1,3,5-трихлорбензоила хлорид, 1,3,5-трихлорбензоила ангидрид, PyBop или PyBrop.
Эта стадия может осуществляться в отсутствие или в присутствии растворителя. Используемые в этом документе растворители включают кетоновые растворители, такие как ацетон и диэтилкетон, эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир и тетрагидрофуран, сложноэфирные растворители, такие как этилацетат и бутилацетат, апротонные полярные органические растворители, такие как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и ацетонитрил, полярные органические растворители, такие как пиридин, галоидзамещенные углеводородные растворители, такие как дихлорметан и хлороформ, или ароматические углеводородные растворители, такие как толуол, и смешанные растворители, составленные из двух или более из указанных растворителей.
Используемые в этом документе основания включают, например, карбонат натрия, карбонат калия, гидрид натрия, трет-бутоксид калия, метоксид натрия, этоксид натрия, пиридин, диметиламинопиридин, имидазол, 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, триэтиламин или диизопропилэтиламин.
Используемые в этом документе кислоты включают, например, пара-толуолсульфоновую кислоту, пара-толуолсульфоновой кислоты моногидрат, пиридиния пара-толуолсульфонат, 10-камфорсульфоновую кислоту, соляную кислоту или серную кислоту.
Температура реакции предпочтительно составляет от -20°C до 50°C. Продолжительность реакции предпочтительно составляет от 0,5 ч до 4 суток.
1-3: Получение соединения Fa из соединения B1
Растворители, используемые на стадии получения соединения Fa из соединения B1, включают спиртовые растворители, содержащие 1-4 атома углерода, такие как метанол, эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан, апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид и ацетонитрил, галоидзамещенные растворители, такие как дихлорметан и хлороформ, или воду, и смешанные растворители, составленные из двух или более из указанных растворителей.
Используемые в этом документе основания включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития и гидроксид бария, щелочноземельные металлы, такие как метоксид натрия, этоксид натрия и трет-бутоксид калия, алкоксиды щелочноземельных металлов, или органические основания, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гидразин и гуанидин. Предпочтительными являются 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия и гидроксид калия. Особенно предпочтительным является карбонат калия.
Количество используемого основания предпочтительно составляет от 0,01 до 10 эквивалентов относительно количества соединения B1. Температура реакции предпочтительно составляет от -20°C до 50°C. Продолжительность реакции предпочтительно составляет от 0,5 до 48 ч.
2-1: Получение соединения A2 из соединения A4
Соединение A4 и соединение A4' могут являться встречающимися в природе веществами, полученными способами, описанными, например, в опубликованной заявке на выдачу патента Японии №184158/1994, WO 94/09147 и опубликованной заявке на выдачу патента Японии №239385/1996. В качестве альтернативы, например, производные получают способом, описанным, например, в опубликованной заявке на выдачу патента Японии №259569/1996.
Растворители, используемые на стадии получения соединения A2 из соединения A4, включают спиртовые растворители, содержащие 1-4 атома углерода, такие как метанол, эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан, апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид и ацетонитрил, галоидзамещенные растворители, такие как дихлорметан и хлороформ, или воду, и смешанные растворители, составленные из двух или более из указанных растворителей.
Используемые в этом документе основания включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития и гидроксид бария, щелочноземельные металлы, такие как метоксид натрия, этоксид натрия и трет-бутоксид калия, алкоксиды щелочноземельных металлов, или органические основания, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гидразин и гуанидин. Предпочтительными являются 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия и гидроксид калия. Особенно предпочтительным является карбонат калия.
Количество используемого основания предпочтительно составляет от 0,01 до 10 эквивалентов относительно количества соединения A4. Температура реакции предпочтительно составляет от -20°C до 50°C. Продолжительность реакции предпочтительно составляет от 0,5 до 48 ч.
В соответствии с этим этапом соединение A2 может быть по аналогии получено из соединения A1 или соединения A4' (соединения, аналогичные соединению A4, за исключением того, что A1, A7 и A11 в соединении A4 представляют собой A1', A7' и A11', соответственно).
2-2: Получение соединения E через соединение D из соединения A2
Растворители, используемые на стадии получения соединения D из соединения A2, включают кетоновые растворители, такие как ацетон и диэтилкетон, эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, диоксан и тетрагидрофуран, сложноэфирные растворители, такие как этилацетат и бутилацетат, апротонные полярные органические растворители, такие как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и ацетонитрил, галоидзамещенные углеводородные растворители, такие как дихлорметан и хлороформ, или ароматические углеводородные растворители, такие как толуол, и смешанные растворители, составленные из двух или более из указанных растворителей.
Гидроксил 1- и 11-положениях может быть замещен с использованием, например, диметоксипропана, ацетона, необязательно замещенного бензальдегида или его диметилацетальной формы, 2-метоксипропена, 2-этоксипропена, фосгена, трифосгена, трихлорацетилхлорида, пара-нитробензилоксикарбонилхлорида или карбонилдиимидазола, соответствующего предполагаемой R2. Кроме того, предпочтительно используют кислотный катализатор, такой как пара-толуолсульфоновая кислота, пара-толуолсульфоновой кислоты моногидрат, пиридиния пара-толуолсульфонат, 10-камфорсульфоновая кислота, фтороводород, соляная кислота, бромоводород, серная кислота, йод, хлорид железа, хлорид олова, хлорид цинка, хлорид алюминия, триметилхлорсилан, триметилсилилтрифлат или 2,3-дихлор-5,6-дициано-1,4-бензохинон (предпочтительно пиридиния пара-толуолсульфонат и пара-толуолсульфоновая кислота), в количестве от 0,001 до 20 эквивалентов, более предпочтительно от 0,01 до 5 эквивалентов, еще более предпочтительно от 0,01 до 0,04 эквивалента относительно соединения A2.
Температура реакции предпочтительно составляет от -20°C до 50°C, более предпочтительно от комнатной температуры до 50°C. Продолжительность реакции предпочтительно составляет от 0,5 до 48 ч.
Более конкретно, например, соединения D и E, которые представляют собой соединения, в которых R2 представляет собой D-1, могут быть получены путем проведения реакции с использованием реагента для введения защитных групп, такого как диметоксипропан, 2-метоксипропен или 2-этоксипропен, в присутствии кислотного катализатора, такого как пара-толуолсульфоновая кислота, пара-толуолсульфоновой кислоты моногидрат, пиридиния пара-толуолсульфонат или 10-камфорсульфоновая кислота, в количестве от 0,001 до 20 эквивалентов, предпочтительно от 0,01 до 5 эквивалентов, более предпочтительно от 0,01 до 0,04 эквивалента относительно соединения A2, или путем проведения реакции в ацетоне с использованием пара-толуолсульфоновой кислоты, пиридиния пара-толуолсульфоната, серной кислоты или сульфата меди в количестве от 0,001 до 20 эквивалентов относительно соединения A2.
Соединения D и E, которые представляют собой соединения, в которых R2 представляет собой D-2, могут быть получены путем проведения реакции с использованием необязательно замещенного бензальдегида или его диметилацетальной формы в присутствии кислотного катализатора, такого как пара-толуолсульфоновая кислота, пара-толуолсульфоновой кислоты моногидрат, пиридиния пара-толуолсульфонат или хлорид цинка, в количестве от 0,001 до 20 эквивалентов относительно соединения A2.
Далее, стадия получения соединения E из соединения D может осуществляться в отсутствие или в присутствии растворителя. Используемые в этом документе растворители включают кетоновые растворители, такие как ацетон, диэтилкетон, эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, диоксан и тетрагидрофуран, сложноэфирные растворители, такие как этилацетат и бутилацетат, апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид и ацетонитрил, полярные органические растворители, такие как пиридин, галоидзамещенные углеводородные растворители, такие как дихлорметан и хлороформ, или ароматические углеводородные растворители, такие как толуол, и смешанные растворители, составленные из двух или более из указанных растворителей.
Защитная группа, соответствующая R1b, может быть введена по гидроксилу в 7-положении с использованием галогенида R1b, представленного R1b-Hal, R1bOH, R1bCl, (R1b)2O, смешанного ангидрида кислоты R1b или 3,4-дигидропирана в присутствии основания, в присутствии кислоты, или в отсутствие основания или кислоты, или с использованием конденсирующего агента, такого как дициклогексилкарбодиимид, 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид, карбонилдиимидазол, дипиридила дисульфид, диимидазоила дисульфид, 1,3,5-трихлорбензоила хлорид, 1,3,5-трихлорбензоила ангидрид, PyBop или PyBrop.
Используемые в этом документе основания включают, например, карбонат натрия, карбонат калия, гидрид натрия, трет-бутоксид калия, метоксид натрия, этоксид натрия, пиридин, диметиламинопиридин, имидазол, 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, триэтиламин или диизопропилэтиламин.
Используемые в этом документе кислоты включают, например, пара-толуолсульфоновую кислоту, пара-толуолсульфоновой кислоты моногидрат, пиридиния пара-толуолсульфонат, 10-камфорсульфоновую кислоту, соляную кислоту или серную кислоту.
Температура реакции предпочтительно составляет от -20°C до 50°C. Продолжительность реакции предпочтительно составляет от 0,5 ч до 7 суток.
Более конкретно, например, соединение, в котором R1b представляет собой линейный C1-4алкилкарбонил, необязательно замещенный атомом галогена, например, ацетил или хлорацетил, может быть получено путем проведения при температуре от -20°C до 50°C реакции с использованием R1bCl или (R1b)2O в количестве от 1 до 20 эквивалентов относительно соединения D и пиридина, диметиламинопиридина или триэтиламина в качестве основания в количестве от 0,1 до 20 эквивалентов относительно соединения D, в отсутствие растворителя или в тетрагидрофуране, дихлорметане, N,N-диметилформамиде или пиридине или в смешанном растворителе, составленном из двух или более из указанных растворителей.
Соединение, в котором R1b представляет собой необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил, может быть получено путем проведения при температуре от -20°C до 50°C реакции с использованием галогенида R1b в количестве от 1 до 10 эквивалентов относительно соединения D и имидазола в качестве основания в количестве от 1 до 10 эквивалентов относительно соединения D в дихлорметане, хлороформе, N,N-диметилформамиде, N,N-диметилацетамиде, или в смешанном растворителе, составленном из двух или более из указанных растворителей.
2-3: Получение соединения Fb из соединения E
Растворители, используемые на стадии получения соединения Fb из соединения E, включают спиртовые растворители, содержащие 1-4 атома углерода, такие как метанол, кетоновые растворители, такие как ацетон и диэтилкетон, эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан, апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид и ацетонитрил, галоидзамещенные растворители, такие как дихлорметан и хлороформ, или воду, и смешанные растворители, составленные из двух или более из указанных растворителей.
При снятии защитной группы с фрагмента R2 может использоваться органическая кислота, такая как уксусная кислота, трифторуксусная кислота, трифторуксусный ангидрид, фтороводород, соляная кислота, бромоводород, серная кислота, пара-толуолсульфоновая кислота, пара-толуолсульфоновой кислоты моногидрат, пиридиния пара-толуолсульфонат или 10-камфорсульфоновая кислота, или катализатор гидрирования, такой как хлорид бора, бромид магния, динитроцинк, хлорид висмута, хлорид церия, хлорид железа, хлорид олова, хлорид цинка, хлорид алюминия, палладий на угле или гидроксид палладия, в количестве от 0,01 до 20 эквивалентов относительно соединения E, в зависимости от типа защитной группы.
Температура реакции предпочтительно составляет от -20°C до 50°C. Продолжительность реакции предпочтительно составляет от 0,5 до 48 ч.
Более конкретно, например, если R2 в соединении E представляет собой D-1, то соединение Fb может быть получено путем проведения реакции с использованием 0,01-20 эквивалентов (относительно соединения E) соляной кислоты, уксусной кислоты, пара-толуолсульфоновой кислоты, пара-толуолсульфоновой кислоты моногидрата, пиридиния пара-толуолсульфоната, динитроцинка или хлорида висмута (предпочтительно соляной кислоты, уксусной кислоты или пиридиния пара-толуолсульфоната) в воде, метаноле, тетрагидрофуране, дихлорметане, хлороформе, N,N-диметилформамиде, ацетонитриле или уксусной кислоте или в смешанном растворителе, составленном из двух или более из указанных растворителей, при температуре от -20°C до 50°C, предпочтительно при температуре от комнатной до 40°C.
Если R2 представляет собой D-2, то соединение Fb может быть получено путем проведения реакции с использованием 0,01-20 эквивалентов (относительно соединения E) 10-камфорсульфоновой кислоты в воде, метаноле, тетрагидрофуране или хлороформе или в смешанном растворителе, составленном из двух или более из указанных растворителей, при температуре от -20°C до 50°C, предпочтительно при температуре от комнатной до 40°C.
3: Получение соединения B2a из соединения Fa и получение соединения B2b из соединения Fb
Стадия получения соединения B2a из соединения Fa и стадия получения соединения B2b из соединения Fb могут быть осуществлены в отсутствие или в присутствии растворителя. Используемые в этом документе растворители включают кетоновые растворители, такие как ацетон и диэтилкетон, эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир и тетрагидрофуран, сложноэфирные растворители, такие как этилацетат и бутилацетат, апротонные полярные органические растворители, такие как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и ацетонитрил, галоидзамещенные углеводородные растворители, такие как дихлорметан и хлороформ, или ароматические углеводородные растворители, такие как толуол, и смешанные растворители, составленные из двух или более из указанных растворителей.
Группа R' может быть введена в 1- и 11-положение с использованием R'OH, R'Cl, (R')2O или смешанного ангидрида кислоты, соответствующего предполагаемой R', в присутствии или в отсутствие основания или с использованием конденсирующего агента, такого как дициклогексилкарбодиимид, 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид, карбонилдиимидазол, дипиридила дисульфид, диимидазоила дисульфид, 1,3,5-трихлорбензоила хлорид, 1,3,5-трихлорбензоила ангидрид, PyBop или PyBrop.
Используемые в этом документе основания включают, например, карбонат натрия, карбонат калия, гидрид натрия, трет-бутоксид калия, метоксид натрия, этоксид натрия, пиридин, 4-диметиламинопиридин, имидазол, 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, триэтиламин и диизопропилэтиламин (предпочтительно пиридин).
Температура реакции предпочтительно составляет от -20°C до 50°C, более предпочтительно от 0°C до 30°C. Продолжительность реакции предпочтительно составляет от 0,5 до 48 ч.
4: Получение соединения C из соединения B2a или соединения B2b
Растворители, используемые на стадии получения соединения C из соединения B2a или B2b, включают спиртовые растворители, содержащие 1-4 атома углерода, такие как метанол, эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан, апротонные полярные органические растворители, такие как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и ацетонитрил, галоидзамещенные углеводородные растворители, такие как дихлорметан и хлороформ, ароматические углеводородные растворители, такие как толуол, или воду, и смешанные растворители, составленные из двух или более из указанных растворителей.
Снятие защитной группы с R1b в соединении B2b может быть осуществлено в зависимости от типа защитной группы. Например, если R1b представляет собой формил, ацетил или хлорацетил, то в качестве основания может использоваться, например, неорганические основание, такое как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития или гидроксид бария, щелочной металл, такой как метоксид натрия, этоксид натрия или трет-бутоксид калия, алкоксид щелочноземельного металла, органическое основание, такое как 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гидразин или гуанидин, предпочтительно метоксид натрия, гидроксид натрия, карбонат натрия или гидрокарбонат натрия, в количестве от 0,01 до 10 эквивалентов, предпочтительно в количестве от 0,1 до 2 эквивалентов относительно соединения B2b.
Если R1b представляет собой необязательно замещенный атомом галогена C1-6алкилокси-C1-6алкил, необязательно замещенный атомом галогена C1-6алкилтио-C1-6алкил, необязательно замещенный атомом галогена линейный, разветвленный или циклический C1-4алкил, необязательно замещенный атомом галогена C2-6алкенил, необязательно замещенный атомом галогена C2-6алкинил, необязательно замещенное насыщенное или ненасыщенное 5- или 6-членное гетероциклическое кольцо, необязательно замещенный бензил или необязательно замещенную атомом галогена группу -SiR3R4R5, то может использоваться, например, органическая кислота, такая как пара-толуолсульфоновая кислота, пара-толуолсульфоновой кислоты моногидрат, пиридиния пара-толуолсульфонат, фтороводород/пиридин, тригидрофторид/триэтиламин, уксусная кислота, хлорид уксусной кислоты, трифторуксусная кислота, трифторуксусный ангидрид, фтороводород, соляная кислота, бромоводород, серная кислота, тиофенол или 10-камфорсульфоновая кислота, катализатор гидрирования, такой как хлорид бора, бромид бора, бромид магния, хлорид церия, хлорид меди, сульфат меди, хлорид лития, хлорид железа, хлорид олова, хлорид цинка, бромид цинка, хлорид алюминия, хлорид титана, палладий на угле, гидроксид палладия или хлорид палладия, триметилхлорсилан, триметилйодсилан, триметилсилилтрифлат или 2,3-дихлор-5,6-дициано-1,4-бензохинон, в количестве от 0,1 до 10 эквивалентов относительно соединения B2b.
Температура реакции предпочтительно составляет от -20°C до 50°C, более предпочтительно от 0°C до комнатной температуры. Продолжительность реакции предпочтительно составляет от 0,5 ч до 7 суток.
В соответствии с этой стадией соединение C может быть по аналогии получено из соединения B2a.
ПРИМЕРЫ
Настоящее изобретение дополнительно проиллюстрировано последующими примерами, которые не предназначены для ограничения настоящего изобретения.
Пример 1
Синтез 7-дезацетилпирипиропена A
Пирипиропен A (30 мг) растворяли в 80% водном растворе метанола (2 мл). К раствору добавляли 1,8-диазабицикло[5.4.0]-ундец-7-ен (9 мг), и перемешивали смесь при комнатной температуре в течение 1,5 ч. К реакционному раствору добавляли уксусную кислоту для остановки реакции. Затем растворитель удаляли путем выпаривания в условиях пониженного давления, к остатку добавляли воду, и экстрагировали смесь этилацетатом. Этилацетатный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-дезацетилпирипиропена A. Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, ацетон/гексан=1/1) с получением 7-дезацетилпирипиропена A (17 мг, выход 61%). Результаты измерений методами МС(ИЭР) и 1H-ЯМР показали, что соединение представляет собой соединение PR-7, описанное в опубликованной заявке на выдачу патента Японии №259569/1996.
Пример 2
Синтез 7-O-трет-бутилдиметилсилил-7-дезацетилпирипиропена A
7-Дезацетилпирипиропен A (30 мг), синтезированный способом, описанным в примере 1, растворяли в N,N-диметилформамиде (5 мл), и добавляли к раствору имидазол (113 мг) и трет-бутилдиметилхлорсилан (250 мг). Смесь перемешивали при комнатной температуре в течение 24 ч. Затем реакционный раствор вливали в воду, и экстрагировали смесь этилацетатом. Этилацетатный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния, и удаляли растворитель путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-трет-бутилдиметилсилил-7-дезацетилпирипиропена A (470 мг).
МС(ИЭР): m/z 656 (M+H)+;
1H-ЯМР (CDCl3): δ 0,11 (3H, с), 0,16 (3H, с), 0,90 (3H, с), 0,96 (9H, с), 1,30-1,38 (1H, м), 1,32-1,37 (1H, м), 1,41 (3H, с), 1,60 (3H, с), 1,61-1,69 (2H, м), 1,77-1,92 (1H, м), 2,05 (6H, с), 2,15 (1H, м), 2,89 (1H, д, J=2,4 Гц), 3,64-3,70 (2H, м), 3,73 (1H, д, J=11,6 Гц), 3,83 (1H, д, J=11,6 Гц), 4,78 (1H, дд, J=4,8, 11,2 Гц), 4,99 (1H, м), 6,36 (1H, с), 7,42 (1H, дд, J=4,8, 8,0 Гц), 8,11 (1H, д, J=8,0 Гц), 8,70 (1H, д, J=4,4 Гц), 9,00 (1H, д, J=2,0 Гц).
Пример 3
Синтез 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетилпирипиропена A
7-O-трет-бутилдиметилсилил-7-дезацетилпирипиропен A (470 мг), полученный в примере 2, растворяли в 88% водном растворе метанола (40 мл). К раствору добавляли карбонат калия (307 мг), и перемешивали смесь при комнатной температуре в течение 19,5 ч. Растворитель удаляли путем выпаривания в условиях пониженного давления. К остатку добавляли воду и этилацетат. Оставшееся нерастворенным твердое вещество собирали путем фильтрации с получением 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетилпирипиропена A (65 мг). Маточный раствор экстрагировали этилацетатом. Затем этилацетатный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель удаляли путем выпаривания в условиях пониженного давления с получением 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетилпирипиропена A (235 мг). Таким образом, получали всего 300 мг (выход в две стадии из соединения примера 2: 95%) 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетилпирипиропена A.
МС(ИЭР): m/z 572 (M+H)+;
1H-ЯМР (CD3OD): δ 0,08 (3H, с), 0,13 (3H, с), 0,64 (3H, с), 0,90 (9H, с), 1,19 (1H, дт, J=3,6, 12,8 Гц), 1,31 (3H, с), 1,33-1,36 (2H, м), 1,48 (1H, т, J=12,0 Гц), 1,53 (3H, с), 1,62-1,80 (3H, м), 1,99-2,03 (1H, м), 3,16 (1H, д, J=10,8 Гц), 3,44 (1H, д, J=10,8 Гц), 3,56 (1H, дд, J=4,8, 11,6 Гц), 3,76 (1H, дд, J=5,2, 11,2 Гц), 4,86 (1H, д, J=3,2 Гц), 6,47 (1H, с), 7,47 (1H, ддд, J=0,8, 4,8, 8,0 Гц), 8,17 (1H, дт, J=2,0, 8,4 Гц), 8,55 (1H, дд, J=2,0, 4,8 Гц), 8,91 (1H, дд, J=0,8, 2,4 Гц).
Пример 4
Синтез 7-O-трет-бутилдиметилсилил-1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A
7-O-трет-бутилдиметилсилил-1,7,11-тридезацетилпирипиропен A (57 мг), синтезированный способом, описанным в примере 3, растворяли в N,N-диметилформамиде (2 мл). К раствору при 0°C добавляли пиридин (0,5 мл), перемешивали смесь при этой температуре в течение 30 мин, и добавляли к ней циклопропанкарбонилхлорид (62 мг). Смесь перемешивали при этой температуре в течение 3 ч, затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия, и удаляли растворитель путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-трет-бутилдиметилсилил-1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (87 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=30/1) с получением 7-O-трет-бутилдиметилсилил-1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (67 мг, выход 95%).
МС(ИЭР): m/z 708 (M+H)+;
1H-ЯМР (CDCl3): δ 0,11 (3H, с), 0,15 (3H, с), 0,85-0,88 (4H, м), 0,91 (3H, с), 0,96 (9H, с), 0,92-1,01 (4H, м), 1,25-1,36 (1H, м), 1,42 (3H, с), 1,45-1,47 (1H, м), 1,53-1,65 (5H, м), 1,58 (3H, с), 1,80-1,93 (2H, м), 2,12-2,16 (1H, м), 2,81 (1H, д, J=2,0 Гц), 3,65 (1H, д, J=12,0 Гц), 3,70 (1H, м), 3,91 (1H, д, J=11,6 Гц), 4,81 (1H, дд, J=4,8, 11,6 Гц), 4,98 (1H, м), 6,36 (1H, с), 7,41 (1H, дд, J=4,8, 8,0 Гц), 8,10 (1H, дт, J=2,0, 8,4 Гц), 8,69 (1H, дд, J=1,6, 4,8 Гц), 9,00 (1H, д, J=2,0 Гц).
Пример 5
Синтез 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A
7-O-трет-бутилдиметилсилил-1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропен A (100 мг), синтезированный способом, описанным в примере 4, растворяли в тетрагидрофуране (1,5 мл). К раствору при 0°C добавляли пиридин (0,6 мл) и фтороводород/пиридин (0,9 мл). Смесь перемешивали при этой температуре в течение 4 суток, затем добавляли к ней водный раствор гидрокарбоната натрия, и экстрагировали смесь хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (92 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=20/1) с получением 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (79 мг, выход 95%).
МС(ИЭР): m/z 594 (M+H)+;
1H-ЯМР (CDCl3): δ 0,85-0,88 (4H, м), 0,92 (3H, с), 0,96-1,01 (4H, м), 1,35 (1H, дт, J=4,0, 12,6 Гц), 1,42 (3H, с), 1,45-1,50 (2H, м), 1,56-1,63 (3H, м), 1,66 (3H, с), 1,79-1,93 (3H, м), 2,14 (1H, м), 2,17 (1H, д, J=3,6 Гц), 2,85 (1H, д, J=2,0 Гц), 3,74 (1H, д, J=12,0 Гц), 3,78-3,82 (1H, м), 3,86 (1H, д, J=11,6 Гц), 4,82 (1H, дд, J=5,2, 11,6 Гц), 4,99 (1H, м), 6,52 (1H, с), 7,42 (1H, дд, J=4,8, 8,0 Гц), 8,11 (1H, дт, J=1,9, 8,1 Гц), 8,70 (1H, дд, J=1,6, 4,8 Гц), 9,00 (1H, д, J=2,0 Гц).
Пример 6
Синтез 7-дезацетил-7-O-тетрагидропиранилпирипиропена A
7-Дезацетилпирипиропен A (500 мг), полученный способом, описанным в примере 1, растворяли в дихлорметане (10 мл), и добавляли к раствору 3,4-дигидропиран (372 мг) и пиридиния пара-толуолсульфонат (348 мг). Смесь перемешивали при комнатной температуре в течение 73,5 ч. Затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-дезацетил-7-O-тетрагидропиранилпирипиропена A (742 мг). Результаты измерений методами МС(ИЭР) и 1H-ЯМР показали, что соединение представляет собой соединение PR-44, описанное в опубликованной заявке на выдачу патента Японии №259569/1996.
Пример 7
Синтез 1,7,11-тридезацетил-7-O-тетрагидропиранилпирипиропена A
7-Дезацетил-7-O-тетрагидропиранилпирипиропен A (742 мг), полученный в примере 6, растворяли в 66% водном растворе метанола (9 мл). К раствору добавляли карбонат калия (511 мг), и перемешивали смесь при комнатной температуре в течение 4 ч. К смеси добавляли воду, и собирали оставшееся нерастворенным твердое вещество путем фильтрации с получением 1,7,11-тридезацетил-7-O-тетрагидропиранилпирипиропена A (453 мг, выход в две стадии из соединения примера 6: 90%).
МС(ИЭР): m/z 542 (M+H)+.
Пример 8
Синтез 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетил-7-O-тетрагидропиранилпирипиропена A
1,7,11-Тридезацетил-7-O-тетрагидропиранилпирипиропен A (450 мг), синтезированный способом, описанным в примере 7, растворяли в N,N-диметилформамиде (6 мл). К раствору при 0°C добавляли пиридин (3 мл), и перемешивали смесь при этой температуре в течение 10 мин. К смеси добавляли циклопропанкарбонилхлорид (525 мг). Смесь перемешивали при этой температуре в течение 1 ч. Затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния, и удаляли растворитель путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетил-7-O-тетрагидропиранилпирипиропена A (800 мг).
МС(ИЭР): m/z 678 (M+H)+.
Пример 9
Синтез 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A
1,11-O-дициклопропанкарбонил-1,7,11-тридезацетил-7-O-тетрагидропиранилпирипиропен A (800 мг), полученный в примере 8, растворяли в метаноле (8 мл), и при 0°C к раствору добавляли пара-толуолсульфоновой кислоты моногидрат (142 мг). Смесь перемешивали при этой температуре в течение 21,5 ч. Затем к ней добавляли водный раствор гидрокарбоната натрия. Метанол удаляли путем выпаривания в условиях пониженного давления, и экстрагировали остаток этилацетатом. Этилацетатный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (570 мг). Неочищенный продукт очищали методом хроматографии на силикагеле (Mega Bond Elut (Varian), ацетон/гексан=3/5) с получением 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (346 мг, выход в две стадии из соединения примера 8: 70%). Данные МС(ИЭР) и 1H-ЯМР соединения соответствуют таковым для соединения примера 5.
Пример 10
Синтез 1,7,11-тридезацетилпирипиропена A
Пирипиропен A (1 г) растворяли в 66% водном растворе метанола (15 мл). К раствору добавляли карбонат калия (355 мг), и перемешивали смесь при комнатной температуре в течение 20 ч. Растворитель удаляли путем выпаривания в условиях пониженного давления. К смеси добавляли этилацетат и воду, и собирали оставшиеся нерастворенными кристаллы путем фильтрации с получением 1,7,11-тридезацетилпирипиропена A (737 мг, выход 94%). Результаты измерений методами МС(ИЭР) и 1H-ЯМР показали, что соединение представляет собой соединение PR-3, описанное в опубликованной заявке на выдачу патента Японии №259569/1996.
Пример 11
Синтез 1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропена A
1,7,11-Тридезацетилпирипиропен A (200 мг), синтезированный способом, описанным в примере 10, растворяли в N,N-диметилформамиде (2 мл). К раствору добавляли ацетондиметилацеталь (456 мг) и пиридиния пара-толуолсульфонат (550 мг). Смесь перемешивали при комнатной температуре в течение 25,5 ч. Затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропена A. Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=10/1) с получением 1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропена A (171 мг, выход 79%). Результаты измерений методами МС(ИЭР) и 1H-ЯМР показали, что соединение представляет собой соединение PR-16, описанное в опубликованной заявке на выдачу патента Японии №269065/1996.
Пример 12
Синтез 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропена A
1,11-Тридезацетил-1,11-O-изопропилиденпирипиропен A (168 мг), синтезированный способом, описанным в примере 11, растворяли в N,N-диметилформамиде (2 мл). К раствору добавляли имидазол (92 мг) и трет-бутилдиметилхлорсилан (204 мг). Смесь перемешивали при комнатной температуре в течение 22 ч, затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропена A (193 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=20/1) с получением 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропена A (187 мг, выход 90%).
МС(ИЭР): m/z 612 (M+H)+;
1H-ЯМР (CDCl3): δ 0,11 (3H, с), 0,16 (3H, с), 0,96 (9H, с), 1,03 (1H, м), 1,10 (3H, с), 1,33 (1H, дт, J=3,6, 12,8 Гц), 1,40 (3H, с), 1,43 (3H, с), 1,44 (3H, с), 1,39-1,44 (1H, м), 1,55-1,58 (2H, м), 1,58 (3H, с), 1,64 (1H, кв, J=12,0 Гц), 1,81 (1H, дкв, J=3,6, 12,8 Гц), 2,20 (1H, дт, J=3,2, 12,8 Гц), 2,81 (1H, д, J=1,6 Гц), 3,42 (1H, д, J=10,8 Гц), 3,51 (1H, д, J=10,4 Гц), 3,50-3,53 (1H, м), 3,72 (1H, дд, J=4,8, 11,2 Гц), 4,97 (1H, м), 6,35 (1H, с), 7,41 (1H, дд, J=4,8, 8,0 Гц), 8,10 (1H, дт, J=1,6, 8,0 Гц), 8,69 (1H, дд, J=1,6, 4,8 Гц), 9,00 (1H, д, J=2,0 Гц).
Пример 13
Синтез 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетилпирипиропена A
7-O-трет-бутилдиметилсилил-1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропен A (116 мг), синтезированный способом, описанным в примере 12, растворяли в тетрагидрофуране (1 мл), и при 0°C к раствору добавляли 63% уксусную кислоту (4 мл). Смесь перемешивали при комнатной температуре в течение 24 ч. К смеси добавляли водный раствор гидрокарбоната натрия и экстрагировали ее хлороформом. Хлороформный слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетилпирипиропена A (101 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=10/1) с получением 7-O-трет-бутилдиметилсилил-1,7,11-тридезацетилпирипиропена A (91 мг, выход 84%).
МС(ИЭР): m/z 572 (M+H)+;
1H-ЯМР (CD3OD): δ 0,08 (3H, с), 0,13 (3H, с), 0,64 (3H, с), 0,90 (9H, с), 1,19 (1H, дт, J=3,6, 12,8 Гц), 1,31 (3H, с), 1,33-1,36 (2H, м), 1,48 (1H, т, J=12,0 Гц), 1,53 (3H, с), 1,62-1,80 (3H, м), 1,99-2,03 (1H, м), 3,16 (1H, д, J=10,8 Гц), 3,44 (1H, д, J=10,8 Гц), 3,56 (1H, дд, J=4,8, 11,6 Гц), 3,76 (1H, дд, J=5,2, 11,2 Гц), 4,86 (1H, д, J=3,2 Гц), 6,47 (1H, с), 7,47 (1H, ддд, J=0,8, 4,8, 8,0 Гц), 8,17 (1H, дт, J=2,0, 8,4 Гц), 8,55 (1H, дд, J=2,0, 4,8 Гц), 8,91 (1H, дд, J=0,8, 2,4 Гц).
Пример 14
Синтез 1,11-дидезацетил-1,11-O-изопропилиденпирипиропена A
1,7,11-Тридезацетил-1,11-O-изопропилиденпирипиропен A (100 мг), синтезированный способом, описанным в примере 11, растворяли в дихлорметане (2 мл). К раствору добавляли триэтиламин (61 мг), 4-диметиламинопиридин (7 мг) и уксусный ангидрид (26 мг). Смесь перемешивали при комнатной температуре в течение 4 ч. Затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия, и удаляли растворитель путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-дидезацетил-1,11-O-изопропилиденпирипиропена A (120 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=20/1) с получением 1,11-дидезацетил-1,11-O-изопропилиденпирипиропена A (103 мг, выход 95%). Результаты измерений методами МС(ИЭР) и 1H-ЯМР показали, что соединение представляет собой соединение PR-43, описанное в опубликованной заявке на выдачу патента Японии №269065/1996.
Пример 15
Синтез 1,11-дидезацетилпирипиропена A
1,11-Дидезацетил-1,11-O-изопропилиденпирипиропен A (99 мг), синтезированный способом, описанным в примере 14, растворяли в тетрагидрофуране (1,2 мл) и метаноле (2,4 мл), и добавляли к раствору пиридиния пара-толуолсульфонат (185 мг). Смесь перемешивали при комнатной температуре в течение 30 ч, затем добавляли к ней триэтиламин, и удаляли растворитель путем выпаривания в условиях пониженного давления. К остатку добавляли хлороформ и воду, и экстрагировали смесь хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия, и удаляли растворитель путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-дидезацетилпирипиропена A (85 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=60/1) с получением 1,11-дидезацетилпирипиропена A (64 мг, выход 72%). Результаты измерений методами МС(ИЭР) и 1H-ЯМР показали, что соединение представляет собой соединение PR-5, описанное в опубликованной заявке на выдачу патента Японии №259569/1996.
Пример 15a
Синтез 1,11-дидезацетилпирипиропена A
1,7,11-Тридезацетилпирипиропен A (9,67 г) суспендировали в N,N-диметилформамиде (48 мл). К суспензии добавляли ацетондиметилацеталь (6,61 г) и пара-толуолсульфоновой кислоты моногидрат (0,08 г), и перемешивали смесь при 38-41°C в течение 4 ч. К смеси добавляли 4-диметиламинопиридин (0,08 г) и удаляли растворитель путем выпаривания в условиях пониженного давления в течение 1,5 ч. Остаток охлаждали до 0°C. К охлажденному раствору добавляли триэтиламин (2,57 г) и уксусный ангидрид (2,37 г), и перемешивали смесь при этой температуре в течение 16 ч. Затем к реакционной смеси добавляли воду (96 г), и корректировали значение pH смеси до 7,17 путем добавления 5% соляной кислоты. Выпавший в осадок светло-желтый порошок собирали путем фильтрации и дважды промывали водой (20 г). Полученный тем самым неочищенный продукт суспендировали в метаноле (48 мл), добавляли к суспензии 15% соляную кислоту (4,7 г), и перемешивали смесь при 25-27°C в течение 2 ч. К смеси добавляли воду (33 мл), и фильтровали нерастворимые вещества, после чего корректировали значение pH до 4,41 путем добавления 5% водного раствора гидроксида натрия. Затем к смеси добавляли воду (31 мл). Выпавший в осадок светло-желтый порошок собирали путем фильтрации и дважды промывали 30% водным раствором метанола (20 мл). Промытый порошок сушили при 40°C в течение 23 ч с получением 8,62 г 1,11-дидезацетилпирипиропена A. Результаты измерений методом 1H-ЯМР показали, что данные 1H-ЯМР согласуются с таковыми для соединения, полученного в примере 15.
Пример 16
Синтез 1,11-O-дициклопропанкарбонил-1,11-дидезацетилпирипиропена A
1,11-Дидезацетилпирипиропен A (61 мг), синтезированный способом, описанным в примере 15, растворяли в N,N-диметилформамиде (1,2 мл). К раствору при 0°C добавляли пиридин (0,3 мл), перемешивали смесь при этой температуре в течение 10 мин, добавляли к смеси циклопропанкарбонилхлорид (77 мг) и перемешивали ее при этой температуре в течение 1,5 ч. Затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-O-дициклопропанкарбонил-1,11-дидезацетилпирипиропена A (97 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=20/1) с получением 1,11-O-дициклопропанкарбонил-1,11-дидезацетилпирипиропена A (73 мг, выход 93%).
МС(ИЭР): m/z 636 (M+H)+;
1H-ЯМР (CDCl3): δ 0,84-0,89 (4H, м), 0,89 (3H, с), 0,90-1,06 (4H, м), 1,37 (1H, дт, J=3,8, 13,2 Гц), 1,45 (3H, с), 1,53 (1H, д, J=4,0 Гц), 1,55-1,67 (4H, м), 1,70 (3H, с), 1,79-1,87 (2H, м), 1,89-1,94 (2H, м), 2,14-2,18 (1H, м), 2,16 (3H, с), 2,97 (1H, д, J=2,0 Гц), 3,77 (2H, с), 4,81 (1H, дд, J=4,8, 11,7 Гц), 5,00 (1H, м), 5,02 (1H, дд, J=5,0, 11,4 Гц), 6,46 (1H, с), 7,40 (1H, дд, J=4,9, 8,0 Гц), 8,09 (1H, дт, J=1,9, 8,1 Гц), 8,68 (1H, дд, J=1,6, 4,8 Гц), 9,00 (1H, д, J=2,0 Гц).
Пример 16a
Синтез 1,11-O-дициклопропанкарбонил-1,11-дидезацетилпирипиропена A
1,11-Дидезацетилпирипиропен A (25,76 г) суспендировали в этилацетате (130 мл), и добавляли к суспензии пиридин (15,84 г). Смесь охлаждали до 10-15°C. По каплям к смеси добавляли циклопропанкарбонилхлорид (15,70 г), и перемешивали смесь при 25-30°C в течение 3 ч. Реакционный раствор снова охлаждали до 10-15°C, и по каплям добавляли к нему воду (50 мл). Значение pH смеси корректировали до 2,59 путем добавления 5н соляной кислоты, после чего проводили разделение. Органический слой промывали 5% водным бикарбонатом натрия (50 мл) и 10% солевым раствором (50 мл) в указанном порядке. Полученный тем самым этилацетатный раствор удаляли путем выпаривания в условиях пониженного давления, а затем замещали метанолом для корректировки объема жидкости до 130 мл. По каплям к раствору добавляли воду (130 мл). Выпавший в осадок светло-желтый порошок собирали путем фильтрации, дважды промывали 50% водным раствором метанола (40 мл) и сушили при 40°C в течение 23 ч с получением 30,80 г 1,11-O-дициклопропанкарбонил-1,11-дидезацетилпирипиропена A. Результаты измерений методом 1H-ЯМР показали, что данные 1H-ЯМР согласуются с таковыми для соединения, полученного в примере 16.
Пример 17
Синтез 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A
1,11-O-дициклопропанкарбонил-1,11-дидезацетилпирипиропен A (67 мг), синтезированный способом, описанным в примере 16, растворяли в 95% водном растворе метанола (0,07 мл). К раствору при 0°C добавляли карбонат натрия (22 мг). Смесь перемешивали при этой температуре в течение 4 суток. Затем к смеси добавляли уксусную кислоту. Метанол удаляли путем выпаривания в условиях пониженного давления, и экстрагировали остаток хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (74 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=10/1) с получением 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (47 мг, выход 76%). Данные МС(ИЭР) и 1H-ЯМР для соединения согласуются с таковыми для соединения, полученного в примере 5.
Пример 17a
Синтез 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A
1,11-O-дициклопропанкарбонил-1,11-дидезацетилпирипиропен A (30,00 г) суспендировали в смешанной жидкости, состоящей из воды (20 мл) и метанола (190 мл), и охлаждали смесь до 0-5°C. К смеси добавляли 1M раствор метоксида натрия в метаноле (4,49 мл), и перемешивали смесь при этой температуре в течение 23 ч. К реакционному раствору добавляли 1,2% соляную кислоту (20 мл), и фильтровали смесь через 0,5-мкм фильтр, а затем корректировали объем жидкости приблизительно до 90 мл путем выпаривания в условиях пониженного давления. К остатку добавляли смешанную жидкость (метанол/вода=2/1, 120 мл) для корректировки объема жидкости приблизительно до 150 мл. Затем к остатку добавляли смешанную жидкость (метанол/вода=2/1, 120 мл) для корректировки объема жидкости приблизительно до 180 мл. Смесь перемешивали при комнатной температуре в течение 1 ч, затем охлаждали до 5°C и перемешивали в течение 17 ч. Выпавший в осадок светло-желтый порошок собирали путем фильтрации, дважды промывали 30% водным раствором метанола (50 мл) и сушили при 40°C в течение 22 ч с получением 23,82 г 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A. Результаты измерений методом 1H-ЯМР показали, что данные 1H-ЯМР согласуются с таковыми для соединения, полученного в примере 17.
Пример 18
Синтез 7-O-хлорацетил-1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропена A
1,7,11-Тридезацетил-1,11-O-изопропилиденпирипиропен A (100 мг), синтезированный способом, описанным в примере 11, растворяли в тетрагидрофуране (2 мл) и триэтиламине (61 мг), и добавляли к раствору хлоруксусный ангидрид (103 мг). Смесь перемешивали при комнатной температуре в течение 3,5 ч, затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-хлорацетил-1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропена A (118 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=20/1) с получением 7-O-хлорацетил-1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропена A (80 мг, выход 70%).
МС(ИЭР): m/z 574 (M+H)+;
1H-ЯМР (CDCl3): δ 1,11 (3H, с), 1,16 (1H, дд, J=2,4, 12,6 Гц), 1,33-1,41 (1H, м), 1,44 (3H, с), 1,45 (3H, с), 1,52 (1H, д, J=4,0 Гц), 1,58-1,65 (1H, м), 1,62 (3H, с), 1,70 (3H, с), 1,66-1,75 (2H, м), 1,77-1,86 (1H, м), 2,22 (1H, м), 2,90 (1H, д, J=2,0 Гц), 3,48 (2H, с), 3,54 (1H, дд, J=3,6, 12,0 Гц), 4,19 (2H, д, J=4,0 Гц), 5,00 (1H, м), 5,09 (1H, дд, J=5,6, 11,6 Гц), 6,45 (1H, с), 7,41 (1H, дд, J=4,8, 8,0 Гц), 8,10 (1H, дт, J=1,6, 8,0 Гц), 8,70 (1H, дд, J=1,6, 4,8 Гц), 9,02 (1H, д, J=1,6 Гц).
Пример 19
Синтез 7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A
7-O-хлорацетил-1,7,11-тридезацетил-1,11-O-изопропилиденпирипиропен A (35 мг), синтезированный способом, описанным в примере 18, растворяли в тетрагидрофуране (0,6 мл) и метаноле (1,2 мл), и добавляли к раствору пиридиния пара-толуолсульфонат (61 мг). Смесь перемешивали при комнатной температуре в течение 31 ч, затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A (30 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=10/1) с получением 7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A (24 мг, выход 74%).
МС(ИЭР): m/z 534 (M+H)+;
1H-ЯМР (CD3OD): δ 0,74 (3H, с), 1,32 (1H, м), 1,44 (3H, с), 1,54 (2H, м), 1,69-1,75 (2H, м), 1,75 (3H, с), 1,79-1,86 (1H, м), 1,91-1,94 (1H, м), 2,12 (1H, м), 3,26 (1H, д, J=11,6 Гц), 3,52 (1H, д, J=10,8 Гц), 3,67 (1H, дд, J=5,2, 11,6 Гц), 4,33 (2H, д, J=2,4 Гц), 4,98 (1H, м), 5,15 (1H, дд, J=5,2, 11,6 Гц), 6,79 (1H, с), 7,55 (1H, дд, J=4,8, 8,0 Гц), 8,28 (1H, дт, J=2,4, 8,0 Гц), 8,62 (1H, дд, J=1,6, 4,8 Гц), 9,02 (1H, д, J=2,4 Гц).
Пример 20
Синтез 7-O-хлорацетил-1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A
7-O-хлорацетил-1,7,11-тридезацетилпирипиропен A (21 мг), синтезированный способом, описанным в примере 19, растворяли в N,N-диметилформамиде (1,2 мл) и пиридине (0,3 мл), и при 0°C добавляли к раствору циклопропанкарбонилхлорид (25 мг). Смесь перемешивали при этой температуре в течение 2,5 ч. Затем реакционный раствор вливали в воду, и экстрагировали смесь хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-хлорацетил-1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (37 мг). Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=30/1) с получением 7-O-хлорацетил-1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (16 мг, выход 58%).
МС(ИЭР): m/z 670 (M+H)+;
1H-ЯМР (CDCl3): δ 0,85-0,90 (4H, м), 0,91 (3H, с), 0,96-1,08 (4H, м), 1,38 (1H, дт, J=4,0, 12,6 Гц), 1,45 (3H, с), 1,54-1,67 (5H, м), 1,72 (3H, с), 1,81-1,95 (3H, м), 2,17 (1H, м), 2,89 (1H, д, J=1,6 Гц), 3,78 (2H, с), 4,17 (2H, д, J=2,8 Гц), 4,82 (1H, дд, J=4,8, 11,6 Гц), 5,01 (1H, м), 5,09 (1H, дд, J=5,2, 11,6 Гц), 6,45 (1H, с), 7,41 (1H, дд, J=4,8, 8,0 Гц), 8,10 (1H, дт, J=1,6, 8,0 Гц), 8,69 (1H, дд, J=1,6, 4,8 Гц), 9,02 (1H, д, J=1,6 Гц).
Пример 21
Синтез 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A
7-O-хлорацетил-1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропен A (14 мг), синтезированный способом, описанным в примере 20, растворяли в 95% водном растворе метанола (1,4 мл). Затем к раствору добавляли гидрокарбонат натрия (1,9 мг). Смесь перемешивали при комнатной температуре в течение 3 ч. К смеси добавляли уксусную кислоту, а затем удаляли метанол путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A. Неочищенный продукт очищали методом препаративной тонкослойной колоночной хроматографии (силикагель Merck 60F254 0,5 мм, хлороформ/метанол=10/1) с получением 1,11-O-дициклопропанкарбонил-1,7,11-тридезацетилпирипиропена A (12 мг, выход 94%). Данные МС(ИЭР) и 1H-ЯМР для соединения согласуются с таковыми для соединения, полученного в примере 5.
Пример 22
Синтез 1,11-O-бензилиден-1,7,11-тридезацетилпирипиропена A
1,7,11-Тридезацетилпирипиропен A (1,0 г), синтезированный способом, описанным в примере 10, растворяли в N,N-диметилформамиде (10 мл). К раствору добавляли пиридиния пара-толуолсульфонат (2,75 г) и бензальдегида диметилацеталь (3,3 г). Смесь перемешивали при комнатной температуре в течение 5 ч. Затем реакционный раствор вливали в воду, и экстрагировали смесь этилацетатом. Этилацетатный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия, и удаляли растворитель путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-O-бензилиден-1,7,11-тридезацетилпирипиропена A. Неочищенный продукт очищали методом хроматографии на силикагеле (Mega Bond Elut (Varian), ацетон/хлороформ=1/10) с получением 1,11-O-бензилиден-1,7,11-тридезацетилпирипиропена A (887 мг, выход 74%). Результаты измерений методами МС(ИЭР) и 1H-ЯМР показали, что соединение представляет собой соединение PR-93, описанное в опубликованной заявке на выдачу патента Японии №269065/1996.
Пример 23
Синтез 1,11-O-бензилиден-7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A
1,11-O-бензилиден-1,7,11-тридезацетилпирипиропен A (1,0 г), синтезированный способом, описанным в примере 22, растворяли в пиридине (2,5 мл). К раствору при 0°C добавляли хлоруксусный ангидрид (206 мг). Смесь перемешивали при этой температуре в течение 1,5 ч, а затем реакционный раствор вливали в воду. Смесь экстрагировали этилацетатом. Этилацетатный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 1,11-O-бензилиден-7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A. Неочищенный продукт очищали методом хроматографии на силикагеле (Mega Bond Elut (Varian), ацетон/гексан=1/100) с получением 1,11-O-бензилиден-7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A (359 мг, выход 72%).
МС(ИЭР): m/z 622 (M+H)+;
1H-ЯМР (CDCl3): δ 1,19-1,22 (1H, м), 1,25 (3H, с), 1,41 (1H, м), 1,48 (3H, с), 1,53-1,56 (1H, м), 1,70 (3H, с), 1,70-1,84 (3H, м), 1,95 (1H, м), 2,27 (1H, м), 2,88 (1H, д, J=1,6 Гц), 3,49 (1H, д, J=10,4 Гц), 3,50-3,53 (1H, м), 3,89 (1H, д, J=10,4 Гц), 4,20 (2H, д, J=3,2 Гц), 5,02 (1H, м), 5,12 (1H, дд, J=5,2, 11,6 Гц), 5,54 (1H, с), 6,46 (1H, с), 7,33-7,43 (4H, м), 7,51 (2H, дд, J=1,6, 8,0 Гц), 8,11 (1H, дт, J=2,0, 8,0 Гц), 8,70 (1H, дд, J=1,6, 4,8 Гц), 9,02 (1H, д, J=2,0 Гц).
Пример 24
Синтез 7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A
1,11-O-бензилиден-7-O-хлорацетил-1,7,11-тридезацетилпирипиропен A (10 мг), синтезированный способом, описанным в примере 23, растворяли в хлороформе (1 мл) и метаноле (9 мл), и добавляли к раствору 10-камфорсульфоновую кислоту (3 мг). Смесь перемешивали при комнатной температуре в течение 5 суток. Затем реакционный раствор вливали в воду, и экстрагировали смесь хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A. Неочищенный продукт очищали методом хроматографии на силикагеле (Mega Bond Elut (Varian), ацетон/хлороформ=1/10) с получением 7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A (8 мг, выход 100%). Данные МС(ИЭР) и 1H-ЯМР для соединения согласуются с таковыми для соединения, полученного в примере 19.
Пример 25
Синтез 1,7,11-тридезацетил-1,11-O-пара-метоксибензилиденпирипиропена A
1,7,11-Тридезацетилпирипиропен A (1,0 г), синтезированный способом, описанным в примере 10, растворяли в N,N-диметилформамиде (22 мл). К раствору добавляли пиридиния пара-толуолсульфонат (2,76 г) и пара-метоксибензальдегида диметилацеталь (0,4 г). Смесь перемешивали при комнатной температуре в течение 4 ч. Затем реакционный раствор вливали в воду, и экстрагировали смесь этилацетатом. Этилацетатный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 1,7,11-тридезацетил-1,11-O-p-метоксибензилиденпирипиропена A. Неочищенный продукт очищали методом хроматографии на силикагеле (Mega Bond Elut (Varian), ацетон/хлороформ=1/10) с получением 1,7,11-тридезацетил-1,11-O-пара-метоксибензилиденпирипиропена A (520 мг, выход 41%). Результаты измерений методами МС(ИЭР) и 1H-ЯМР показали, что соединение представляет собой соединение PR-124, описанное в опубликованной заявке на выдачу патента Японии №269065/1996.
Пример 26
Синтез 7-O-хлорацетил-1,7,11-тридезацетил-1,11-O-пара-метоксибензилиденпирипиропена A
1,7,11-Тридезацетил-1,11-O-пара-метоксибензилиденпирипиропен A (100 мг), синтезированный способом, описанным в примере 25, растворяли в тетрагидрофуране (2 мл). К раствору добавляли триэтиламин (50 мг) и хлоруксусный ангидрид (60 мг). Смесь перемешивали при комнатной температуре в течение 2,5 ч, затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-хлорацетил-1,7,11-тридезацетил-1,11-O-пара-метоксибензилиденпирипиропена A. Неочищенный продукт перекристаллизовывали из метанола и этилацетата, а затем очищали перекристаллизованный продукт методом хроматографии на силикагеле (Mega Bond Elut (Varian), ацетон/хлороформ=1/30) с получением 7-O-хлорацетил-1,7,11-тридезацетил-1,11-O-пара-метоксибензилиденпирипиропена A (83 мг, выход 75%).
МС(ИЭР): m/z 652 (M+H)+;
1H-ЯМР (CDCl3): δ 1,20 (1H, м), 1,24 (3H, с), 1,40 (1H, м), 1,47 (3H, с), 1,54 (1H, д, J=3,6 Гц), 1,72 (3H, с), 1,66-1,80 (3H, м), 1,93 (1H, м), 2,26 (1H, м), 2,87 (1H, д, J=2,0 Гц), 3,47 (1H, д, J=10,0 Гц), 3,47-3,51 (1H, м), 3,80 (3H, с), 3,87 (1H, д, J=10,4 Гц), 4,20 (2H, д, J=3,2 Гц), 5,01 (1H, м), 5,12 (1H, дд, J=5,6, 11,6 Гц), 5,50 (1H, с), 6,46 (1H, с), 6,90 (2H, д, J=8,8 Гц), 7,41 (1H, дд, J=4,8, 12,0 Гц), 7,43 (2H, д, J=8,8 Гц), 8,10 (1H, дт, J=2,0, 8,4 Гц), 8,70 (1H, дд, J=2,4, 4,8 Гц), 9,02 (1H, д, J=2,0 Гц).
Пример 27
Синтез 7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A
7-O-хлорацетил-1,7,11-тридезацетил-1,11-O-пара-метоксибензилиденпирипиропен A (30 мг), синтезированный способом, описанным в примере 26, растворяли в хлороформе (5 мл) и метаноле (1 мл), и добавляли к раствору 10-камфорсульфоновую кислоту (3 мг). Смесь перемешивали при комнатной температуре в течение 5 суток, затем реакционный раствор вливали в воду и экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Затем растворитель удаляли путем выпаривания в условиях пониженного давления с получением неочищенного 7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A. Неочищенный продукт очищали методом хроматографии на силикагеле (Mega Bond Elut (Varian), ацетон/хлороформ=1:5) с получением 7-O-хлорацетил-1,7,11-тридезацетилпирипиропена A (14 мг, выход 69%). Данные МС(ИЭР) и 1H-ЯМР для соединения согласуются с таковыми для соединения, полученного в примере 19.
| название | год | авторы | номер документа |
|---|---|---|---|
| 1,5,6-ЗАМЕЩЕННЫЕ 2-ОКСО-3-ЦИАНО-1,6А-ДИАЗАТЕТРАГИДРОФЛУОРАНТЕНЫ | 2006 |
|
RU2389730C2 |
| КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2389731C2 |
| СРЕДСТВА БОРЬБЫ С СЕЛЬСКОХОЗЯЙСТВЕННЫМИ ВРЕДИТЕЛЯМИ | 2006 |
|
RU2405310C2 |
| ПУТИ ПРИМЕНЕНИЯ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2020 |
|
RU2795503C2 |
| аЭКЗО-АЗАСПИРО-ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2018 |
|
RU2795096C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИММУНОМОДУЛЯТОРЫ В КАЧЕСТВЕ ИНГИБИТОРА КОНТРОЛЬНЫХ ТОЧЕК ИММУННОГО ОТВЕТА PDL1 | 2020 |
|
RU2824127C2 |
| БЕНЗОКСАЗИНИЛ-АМИДОЦИКЛОПЕНТИЛ-ГЕТЕРОЦИКЛИЧЕСКИЕ МОДУЛЯТОРЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2301802C2 |
| 6,7,8,9-ЗАМЕЩЕННЫЕ 1-ФЕНИЛ-1,5-ДИГИДРОПИРИДО (3,2-b) ИНДОЛ-2-ОНЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТИИНФЕКЦИОННЫХ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2005 |
|
RU2377243C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2654857C2 |
Изобретение относится к способу получения соединения С, которое представлено следующей формулой С:
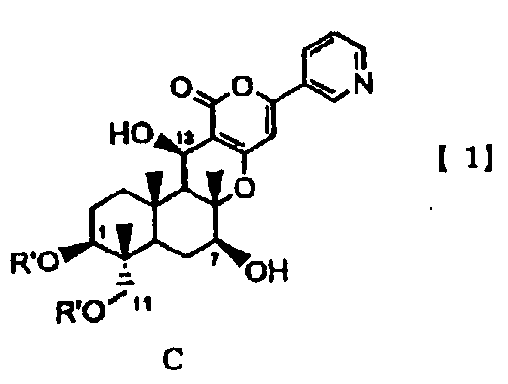
где R' представляет собой линейную, разветвленную или циклическую группу С2-6алкилкарбонильную группу. В этом способе в качестве защитной группы для гидроксигруппы в 7-положении используют R1b, где R1b представляет собой формильную группу, необязательно замещенную линейную С1-4алкилкарбонильную группу, необязательно замещенную бензильную группу, группу -SiR3R4R5, где R3, R4 и R5 независимо представляют собой линейную или разветвленную С1-6алкильную группу или фенильную группу, которая может быть замещена атомом галогена, С1-6алкилокси-С1-6алкильную группу, которая может быть замещена атомом галогена, С1-6алкилтио-С1-6алкильную группу, которая может быть замещена атомом галогена, линейную, разветвленную или циклическую С1-4алкильную группу, которая может быть замещена атомом галогена (в случае разветвленной или циклической она представляет собой С3-4алкильную группу), С2-6алкенильную группу, которая может быть замещена атомом галогена, С2-6алкинильную группу, которая может быть замещена атомом галогена, или необязательно замещенную насыщенную или ненасыщенную 5-членную или 6-членную гетероциклическую группу. Этот способ делает возможным получение с высоким выходом пирипиропенового производного, которая содержит ацилоксигруппу в 1-положении и в 11-положении и гидроксигруппу в 7-положении. Такое пирипиропеновое производное применимо в качестве средства для борьбы с вредителями. Изобретение также включает способы получения промежуточных соединений В2а и B2b и и само соединение формулы 2Bb. 6 н. и 9 з.п. ф-лы, 26 пр.
1. Способ получения соединения С, представленного формулой С:
[Химическая формула 1]
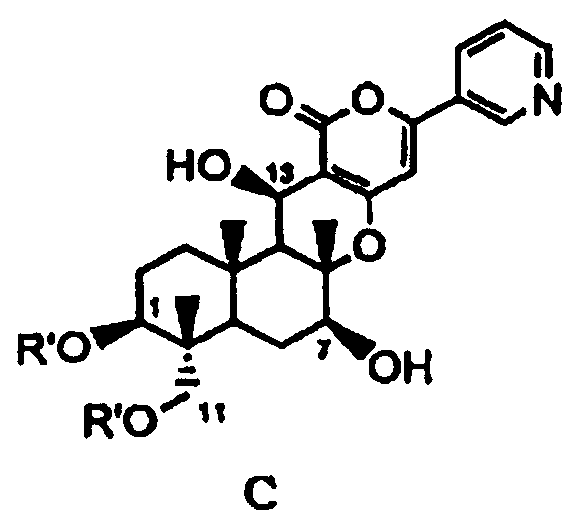
в которой R' представляет собой линейный, разветвленный или циклический С2-6алкилкарбонил, при условии, что если алкильный фрагмент в алкилкарбонильной группе является разветвленным или циклическим, то R' представляет собой С3-6алкилкарбонил, включающий стадии:
(a1) гидролиза ацетила в 7-положении соединения А1, представленного формулой А1:
[Химическая формула 2]
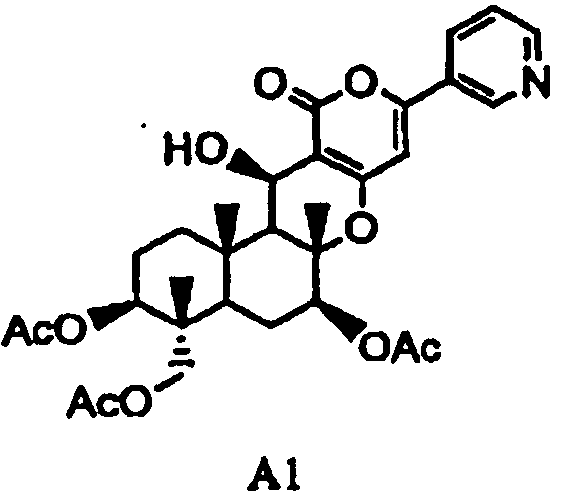
в которой Ас представляет собой ацетил,
основанием для селективного дезацилирования соединения А1, затем защиты гидроксила в 7-положении с получением соединения В1, представленного формулой В1:
[Химическая формула 3]
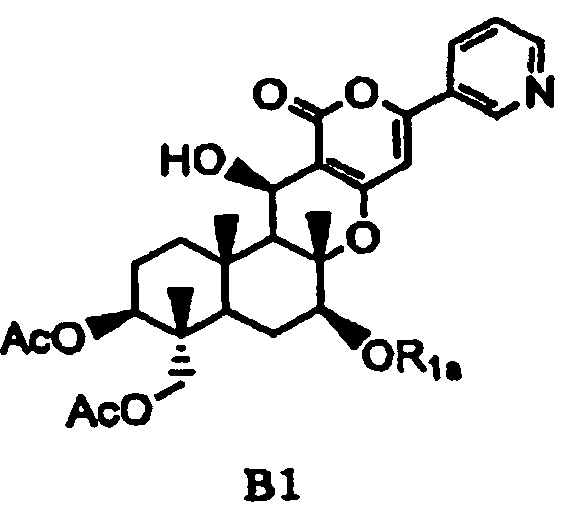
в которой значение Ас определено выше,
R1a представляет собой необязательно замещенный линейный С2-4алкилкарбонил; необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный С1-6алкил или фенил; необязательно замещенный атомом галогена С1-6алкилокси-С1-6алкил; необязательно замещенный атомом галогена С1-6алкилтио-С1-6алкил; необязательно замещенный атомом галогена линейный, разветвленный или циклический С1-4алкил, при условии, что если алкил в С1-4алкильной группе является разветвленным или циклическим, то алкильная группа представляет собой С3-4алкил; необязательно замещенный атомом галогена С2-6алкенил; необязательно замещенный атомом галогена С2-6алкинил; необязательно замещенный, бензил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу,
где в R1a необязательно присутствующий на алкилкарбониле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкилокси, C1-4галогеналкилокси, С1-4алкилкарбонила, С1-4галогеналкилкарбонила, C1-4алкилкарбонилокси и С1-4галогеналкилкарбонилокси, а необязательно присутствующий на гетероциклической группе и бензиле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкила, C1-4алкилокси, С1-4галогеналкилокси, С1-4алкилтио, С1-4галогеналкила, C1-4алкилкарбонила, С1-4галогеналкилкарбонила, С1-4алкилкарбонилокси, C1-4галогеналкилкарбонилокси, нитро и циано,
затем гидролиза ацетила в 1- и 11-положениях соединения В1 основанием для дезацилирования соединения В1 с получением тем самым соединения Fa, представленного формулой Fa:
[Химическая формула 4]
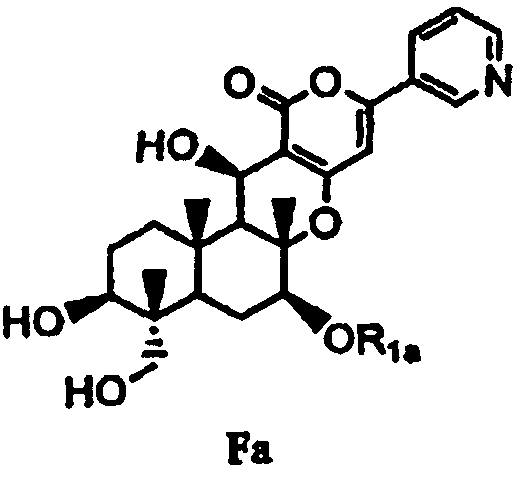
в которой значение R1a определено выше, или
(а2) гидролиза ацила в 1-, 7-и 11-положениях соединения А1 или соединения А4', представленного формулой А4':
[Химическая формула 5]
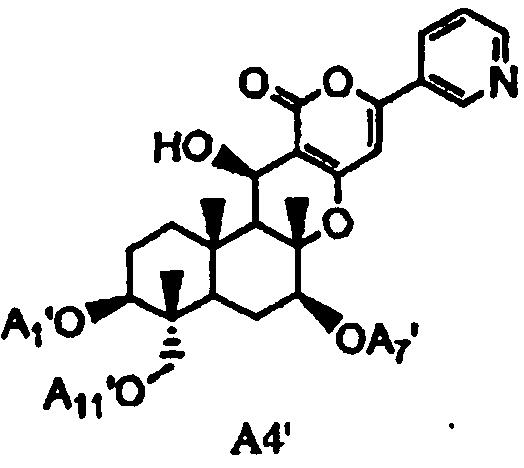
в которой 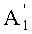
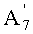
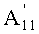
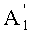
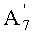
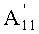
[Химическая формула 6]
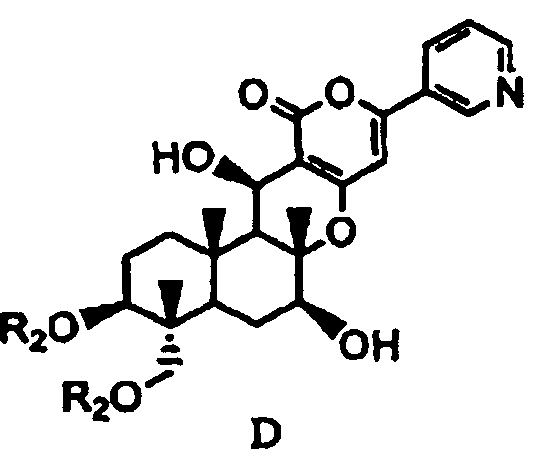
в которой два R2 образуют вместе группу, выбранную из групп, представленных формулами D-1, D-2, D-3 и D-4:
[Химическая формула 7]
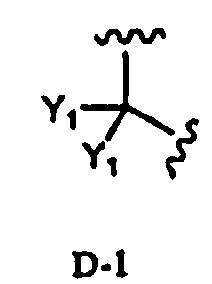
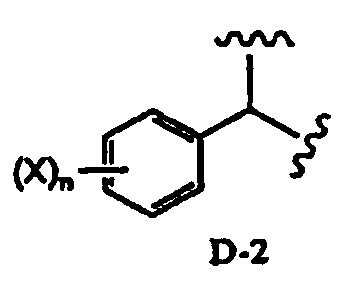
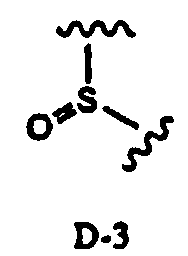
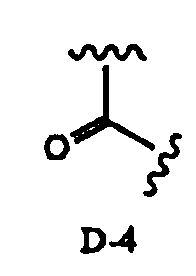
в которых Y1 представляет собой атом водорода или С1-4алкил;
заместители X, которые могут быть одинаковыми или разными, представляют собой атом водорода, С1-4алкокси или нитро; и n равно 0-5, затем защиты гидроксила в 7-положении соединения D с получением соединения Е, представленного формулой Е;
[Химическая формула 8]
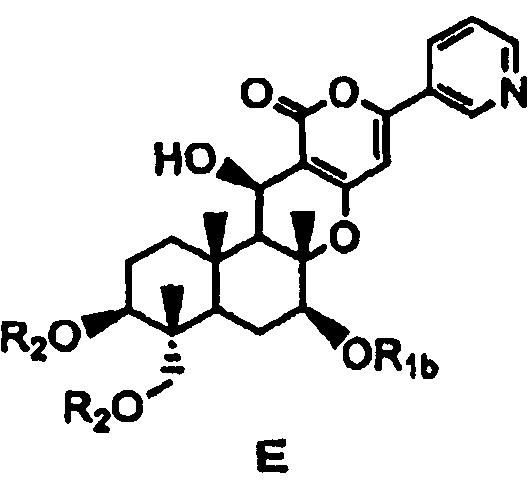
в которой R1b представляет собой формил; необязательно замещенный линейный C1-4алкилкарбонил; необязательно замещенный бензил;
необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил; необязательно замещенный атомом галогена C1-6алкилокси-С1-6алкил; необязательно замещенный атомом галогена C1-6алкилтио-С1-6алкил; необязательно замещенный атомом галогена линейный, разветвленный или циклический С1-4алкил, при условии, что если алкил в С1-4алкильной группе является разветвленным или циклическим, то алкильная группа представляет собой С3-4алкил; необязательно замещенный атомом галогена С2-6алкенил; необязательно замещенный атомом галогена С2-6алкинил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу,
где в R1b необязательно присутствующий на алкилкарбониле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкилокси, C1-4галогеналкилокси, С1-4алкилкарбонила, С1-4галогеналкилкарбонила, C1-4алкилкарбонилокси и С1-4галогеналкилкарбонилокси, а необязательно присутствующий на гетероциклической группе и бензиле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкила, C1-4алкилокси, С1-4галогеналкилокси, С1-4алкилтио, С1-4галогеналкила, C1-4алкилкарбонила, С1-4галогеналкилкарбонила, С1-4алкилкарбонилокси, C1-4галогеналкилкарбонилокси, нитро и циано, и
значение R2 определено выше,
затем удаления защитных групп в 1- и 11-положениях соединения Е с получением соединения Fb, представленного формулой Fb:
[Химическая формула 9]
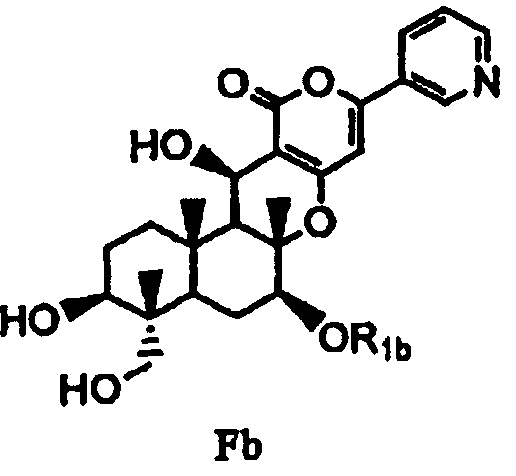
в которой значение R1b определено выше, и
(b) ацилирования гидроксила в 1- и 11-положениях соединения Fa или Fb ацилирующим агентом, соответствующим предполагаемой R', с получением соединения В2а или B2b, представленного формулой В2а или B2b:
[Химическая формула 10]
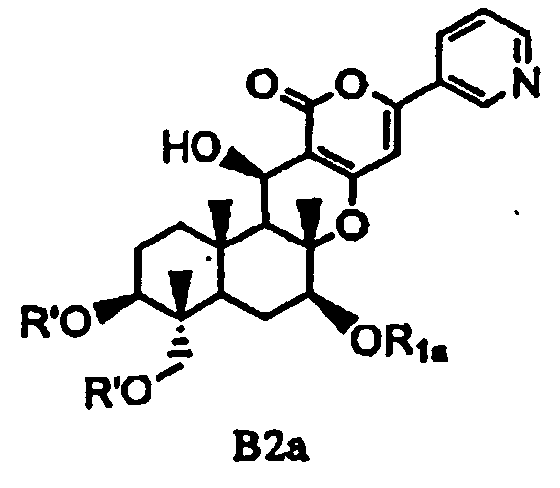
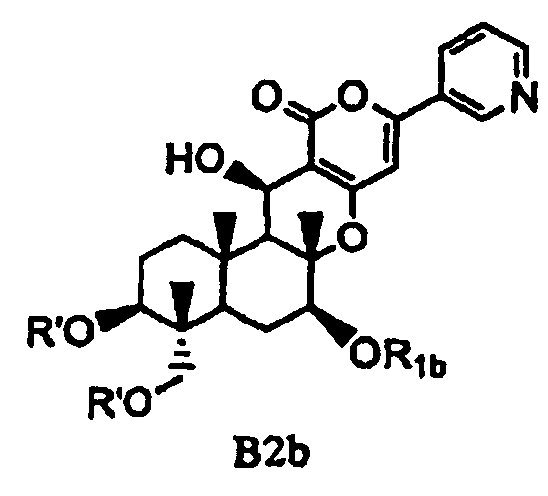
в которых значения R1a, R1b и R' определены выше, а затем удаления защитной группы в 7-положении соединения В2а или соединения B2b.
2. Способ по п.1, где R' представляет собой циклический С3-6алкилкарбонил.
3. Способ по п.1, где R1a представляет собой необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный С1-6алкил или фенил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу.
4. Способ по п.1, где R1b представляет собой ацетил; хлорацетил; или необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил.
5. Способ по п.1, где два R2 образуют вместе группу, представленную формулой D-1 или D-2:
[Химическая формула 11]
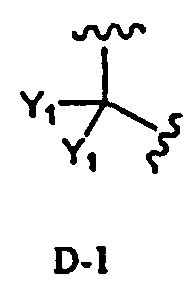
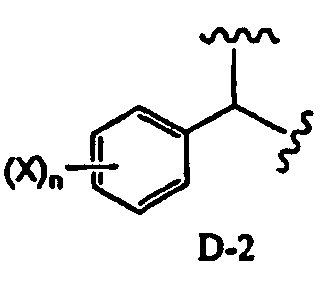
в которой Y1 представляет собой атом водорода или С1-4алкил; заместители X, которые могут быть одинаковыми или разными, представляют собой атом водорода, С1-4алкокси или нитро; и n равно 0-5.
6. Способ получения соединения В2а, представленного формулой В2а:
[Химическая формула 12]
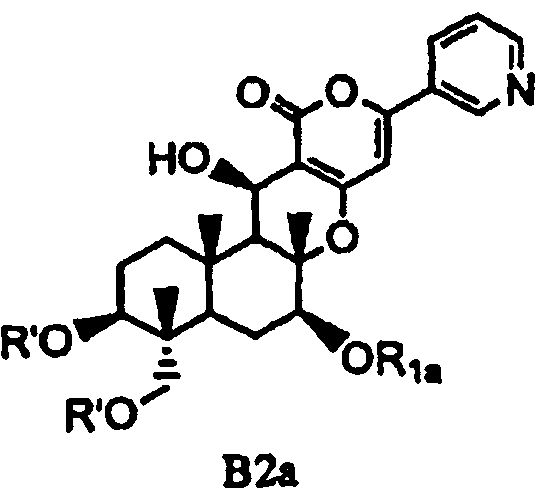
в которой R1a представляет собой необязательно замещенный линейный С2-4алкилкарбонил; необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный С1-6алкил или фенил; необязательно замещенный атомом галогена С1-6алкилокси-С1-6алкил; необязательно замещенный атомом галогена С1-6алкилтио-С1-6алкил; необязательно замещенный атомом галогена линейный, разветвленный или циклический С1-4алкил, при условии, что если алкил в С1-4алкильной группе является разветвленным или циклическим, то алкильная группа представляет собой С3-4алкил; необязательно замещенный атомом галогена С2-6алкенил; необязательно замещенный атомом галогена С3-6алкинил; необязательно замещенный бензил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу,
где в R1a необязательно присутствующий на алкилкарбониле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкилокси, C1-4галогеналкилокси, С1-4алкилкарбонила, С1-4галогеналкилкарбонила, C1-4алкилкарбонилокси и С1-4галогеналкилкарбонилокси, и необязательно присутствующий на гетероциклической группе и бензиле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкила, C1-4алкилокси, С1-4галогеналкилокси, С1-4алкилтио, С1-4галогеналкила, С1-4алкилкарбонила, С1-4галогеналкилкарбонила, С1-4алкилкарбонилокси, С1-4галогеналкилкарбонилокси, нитро и циано, и
R' представляет собой циклический С3-6алкилкарбонил, включающий:
гидролиз ацетила в 7-положении соединения А1, представленного формулой А1:
[Химическая формула 13]
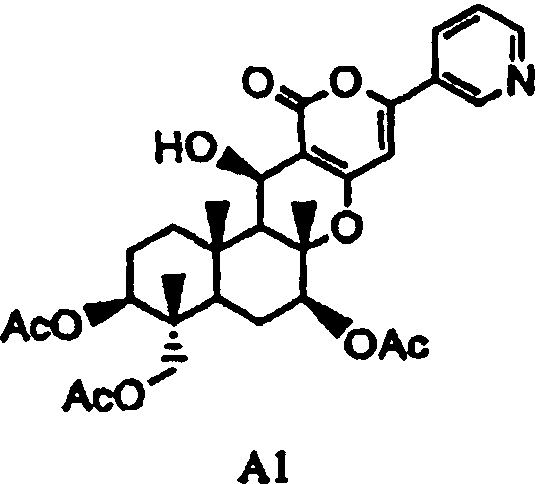
в которой Ас представляет собой ацетил,
основанием для селективного дезацилирования соединения А1, а затем защиту гидроксила в 7-положении с получением соединения В1, представленного формулой В1;
[Химическая формула 14]
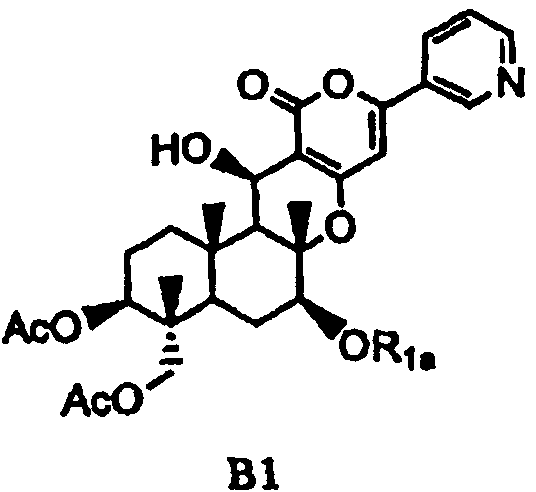
где значения Ас и R1a определены выше,
затем гидролиз в 1- и 11-положениях соединения В1 основанием для дезацилирования соединения В1 с получением тем самым соединения Fa, представленного формулой Fa:
[Химическая формула 15]
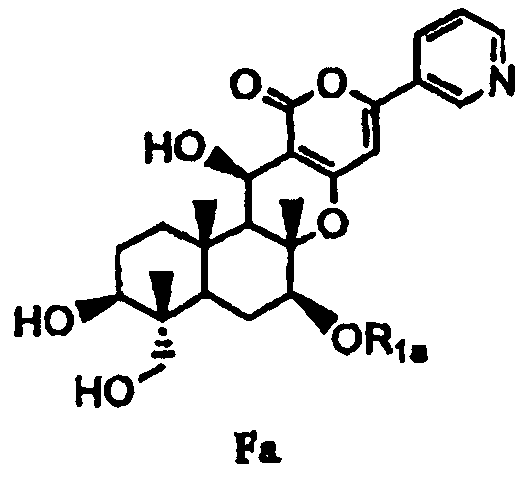
где значение R1a определено выше,
а затем ацилирование гидроксила в 1- и 11-положениях соединения Fa ацилирующим агентом, соответствующим предполагаемой R'.
7. Способ по п.6, где R1a представляет собой необязательно замещенный линейный С2-4алкилкарбонил; необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный С1-6алкил или фенил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу.
8. Способ получения соединения B2b, представленного формулой B2b:
[Химическая формула 16]
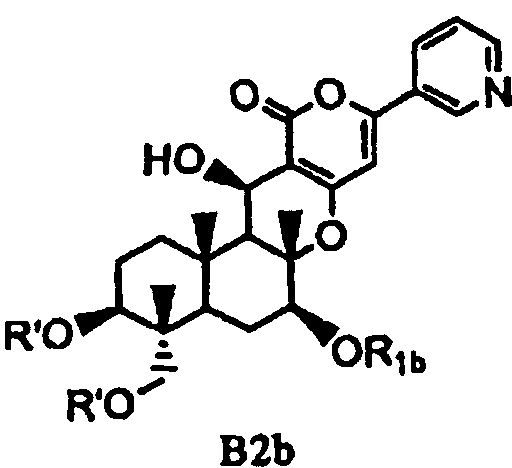
в которой R1b представляет собой формил; необязательно замещенный линейный C1-4алкилкарбонил; необязательно замещенный бензил;
необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный С1-4алкил или фенил; необязательно замещенный атомом галогена C1-6алкилокси-С1-6алкил; необязательно замещенный атомом галогена C1-6алкилтио-С1-6алкил; необязательно замещенный атомом галогена линейный, разветвленный или циклический С1-4алкил, при условии, что если алкил в С1-4алкильной группе является разветвленным или циклическим, то алкильная группа представляет собой С3-4алкил; необязательно замещенный атомом галогена С2-6алкенил; необязательно замещенный атомом галогена С3-6алкинил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу,
где в R1b необязательно присутствующий на алкилкарбониле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкилокси, C1-4галогеналкилокси, С1-4алкилкарбонила, С1-4галогеналкилкарбонила, C1-4алкилкарбонилокси и С1-4галогеналкилкарбонилокси, и необязательно присутствующий на гетероциклической группе и бензиле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкила, C1-4алкилокси, С1-4галогеналкилокси, С1-4алкилтио, С1-4галогеналкила, C1-4алкилкарбонила, С1-4галогеналкилкарбонила, С1-4алкилкарбонилокси, C1-4галогеналкилкарбонилокси, нитро и циано, и
R' представляет собой циклический С3-6алкилкарбонил, включающий:
гидролиз ацила в 1-, 7- и 11-положениях соединения А4, представленного формулой А4:
[Химическая формула 17]
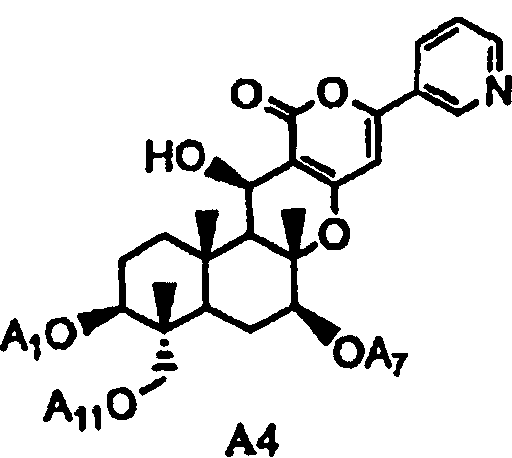
в которой A1, A7 и А11, которые могут быть одинаковыми или разными, представляют собой ацетил или пропионил,
основанием для дезацилирования соединения А4, а затем защиту гидроксила в 1-и 11-положениях с получением соединения D, представленного формулой D:
[Химическая формула 18]
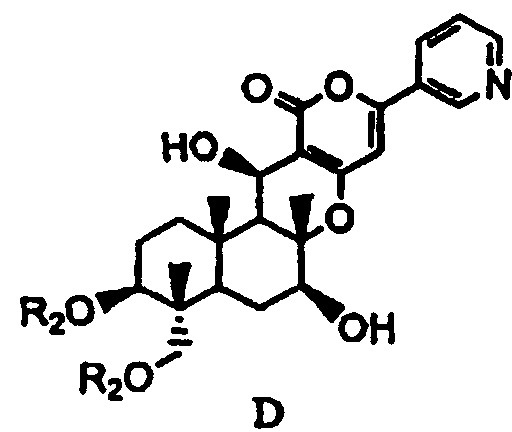
в которой два R2 образуют вместе группу, выбранную из групп, представленных формулами D-1, D-2, D-3 и D-4:
[Химическая формула 19]
в которых Y1 представляет собой атом водорода или С1-4алкил;
заместители X, которые могут быть одинаковыми или разными, представляют собой атом водорода, С1-4алкокси или нитро; и n равно 0-5, затем защиту гидроксила в 7-положении соединения D с получением соединения Е, представленного формулой Е;
[Химическая формула 20]
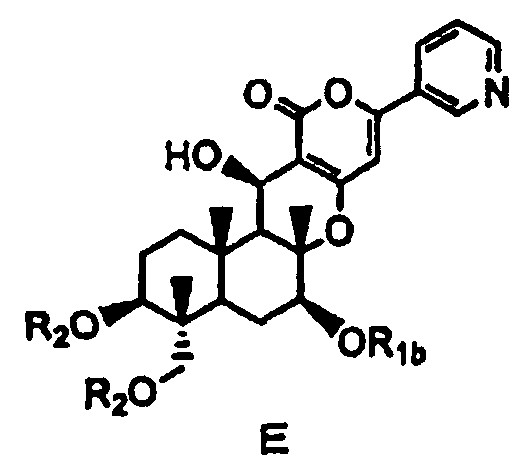
в которой значения R1b и R2 определены выше,
затем удаление защитной группы в 1- и 11-положениях соединения Е с получением соединения Fb, представленного формулой Fb:
[Химическая формула 21]
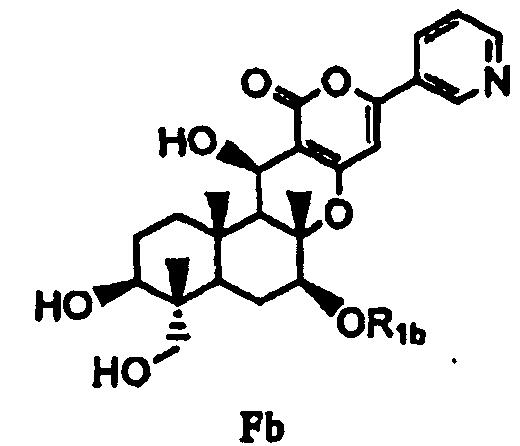
в которой значение R1b определено выше,
а затем ацилирование гидроксила в 1- и 11-положениях соединения Fb ацилирующим агентом, соответствующим R'.
9. Способ по п.8, где R1b представляет собой ацетил;
хлорацетил; или необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный С1-6алкил или фенил.
10. Способ по п.8, где два R2 образуют вместе группу, представленную формулой D-1 или D-2:
[Химическая формула 22]
в которой Y1 представляет собой атом водорода или С1-4алкил; заместители X, которые могут быть одинаковыми или разными, представляют собой атом водорода, С1-4алкокси или нитро; и n равно 0-5.
11. Способ получения соединения С, представленного формулой С:
[Химическая формула 23]
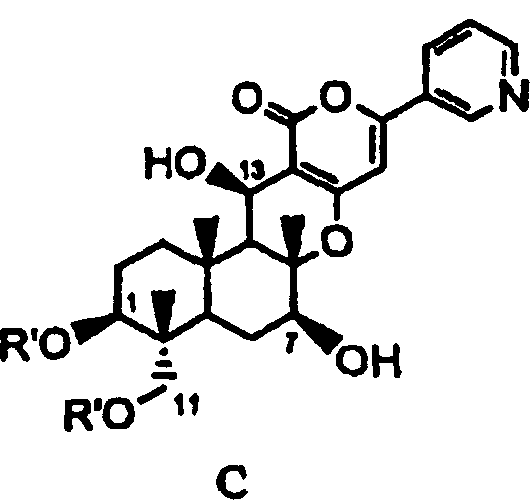
в которой R' представляет собой циклический С3-6алкилкарбонил, включающий:
ацилирование гидроксила в 1- и 11-положениях соединения Fb, представленного формулой Fb:
[Химическая формула 24]
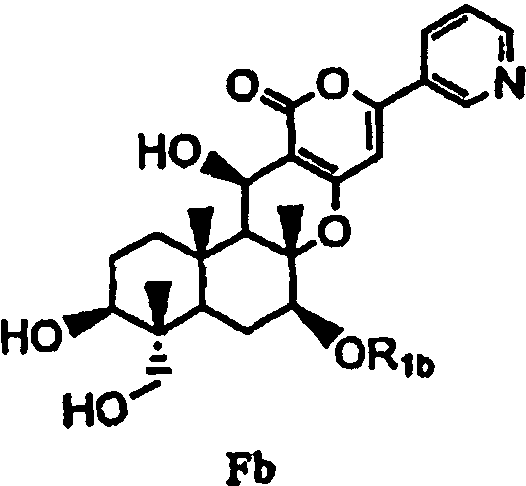
в которой R1b представляет собой формил; необязательно замещенный линейный C1-4алкилкарбонил; необязательно замещенный бензил; необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный С1-6алкил или фенил; необязательно замещенный атомом галогена С1-6алкилокси-С1-6алкил; необязательно замещенный атомом галогена C1-6алкилтио-С1-6алкил; необязательно замещенный атомом галогена линейный, разветвленный или циклический С1-4алкил, при условии, что если алкил в С1-4алкильной группе является разветвленным или циклическим, то алкильная группа представляет собой С3-4алкил; необязательно замещенный атомом галогена С2-6алкенил; необязательно замещенный атомом галогена С2-6алкинил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу,
где в R1b необязательно присутствующий на алкилкарбониле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкилокси, C1-4галогеналкилокси, С1-4алкилкарбонила, С1-4галогеналкилкарбонила, C1-4алкилкарбонилокси и С1-4галогеналкилкарбонилокси, и необязательно присутствующий на гетероциклической группе и бензиле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкила, C1-4алкилокси, С1-4галогеналкилокси, С1-4алкилтио, С1-4галогеналкила, C1-4алкилкарбонила, С1-4галогеналкилкарбонила, С1-4алкилкарбонилокси, C1-4галогеналкилкарбонилокси, нитро и циано,
ацилирующим агентом, соответствующим R', с получением соединения B2b, представленного формулой B2b:
[Химическая формула 25]
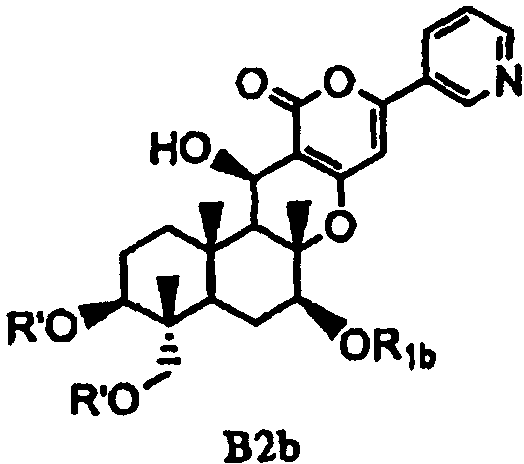
в которой значения R1b и R' определены выше,
а затем удаление защитной группы в 7-положении соединения B2b.
12. Способ получения соединения С, представленного формулой С:
[Химическая формула 26]
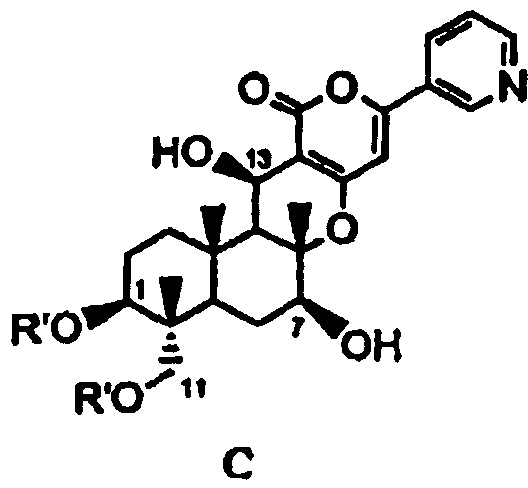
в которой R' представляет собой линейный, разветвленный или циклический С2-6алкилкарбонил, при условии, что если алкильный фрагмент в С2-6алкилкарбонильной группе является разветвленным или циклическим, то алкильный фрагмент представляет собой С3-6алкилкарбонил, включающий:
гидролиз ацила в 1-, 7- и 11-положениях соединения А4, представленного формулой А4:
[Химическая формула 27]
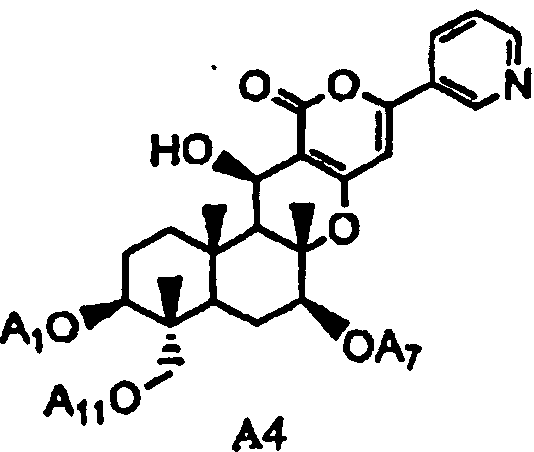
в которой A1, А7 и А11, которые могут быть одинаковыми или разными, представляют собой ацетил или пропионил,
основанием для дезацилирования соединения А4, затем защиту гидроксила в 1- и 11-положениях с получением соединения D, представленного формулой D:
[Химическая формула 28]
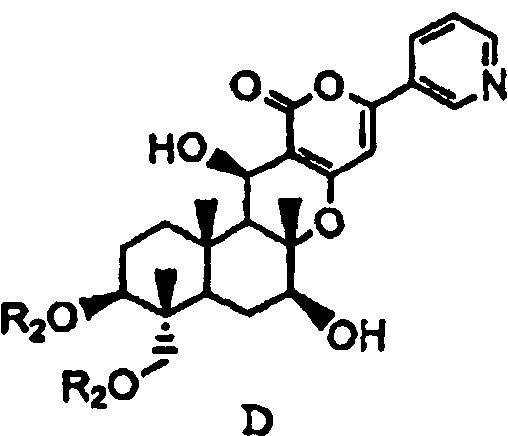
в которой два R2 образуют вместе группу, выбранную из групп, представленных формулами D-1, D-2, D-3 и D-4:
[Химическая формула 29]
в которых Y1 представляет собой атом водорода или С1-4алкил;
заместители X, которые могут быть одинаковыми или разными, представляют собой атом водорода, С1-4алкокси или нитро; и n равно 0-5, затем защиту гидроксила в 7-положении соединения D с получением соединения Е, представленного формулой Е:
[Химическая формула 30]
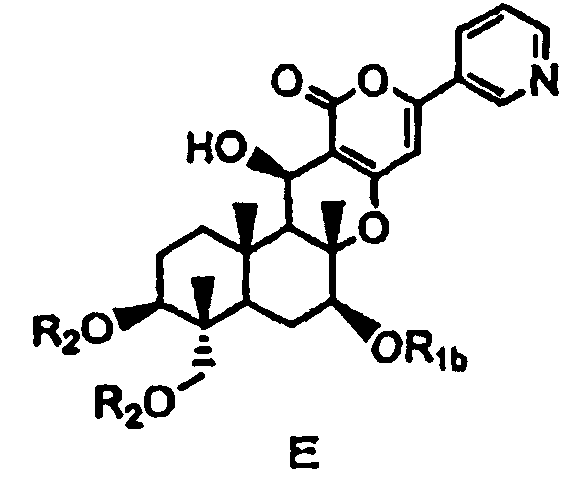
в которой R1b представляет собой формил; необязательно замещенный линейный C1-4алкилкарбонил; необязательно замещенный бензил; необязательно замещенную атомом галогена группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный С1-6алкил или фенил; необязательно замещенный атомом галогена С1-6алкилокси-С1-6алкил; необязательно замещенный атомом галогена С1-6алкилтио-С1-6алкил; необязательно замещенный атомом галогена линейный, разветвленный или циклический С1-4алкил, при условии, что если алкил в С1-4алкильной группе является разветвленным или циклическим, то алкильная группа представляет собой С3-4алкил; необязательно замещенный атомом галогена С2-6алкенил; необязательно замещенный атомом галогена С2-6алкинил; или необязательно замещенную насыщенную или ненасыщенную 5- или 6-членную гетероциклическую группу,
где в R1b необязательно присутствующий на алкилкарбониле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкилокси, С1-4галогеналкилокси, С1-4алкилкарбонила, С1-4галогеналкилкарбонила, C1-4алкилкарбонилокси и С1-4галогеналкилкарбонилокси, и необязательно присутствующий на гетероциклической группе и бензиле заместитель выбирают из группы, состоящей из атомов галогена, С1-4алкила, С1-4алкилокси, С1-4галогеналкилокси, С1-4алкилтио, С1-4галогеналкила, C1-4алкилкарбонила, С1-4галогеналкилкарбонила, С1-4алкилкарбонилокси, C1-4галогеналкилкарбонилокси, нитро и циано, и
значение R2 определено выше,
затем удаление защитных групп в 1- и 11-положениях соединения Е с получением соединения Fb, представленного формулой Fb:
[Химическая формула 31]
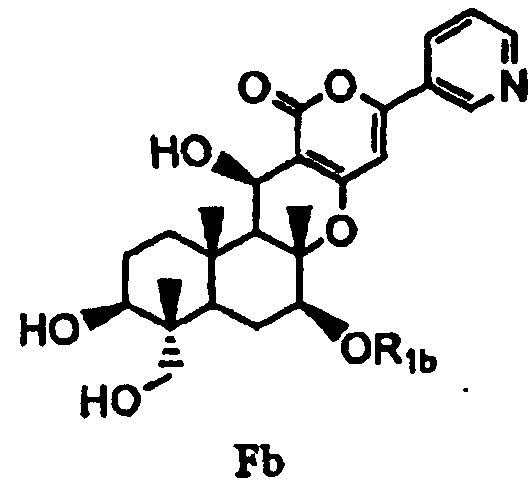
в которой значение R1b определено выше,
затем ацилирование гидроксила в 1- и 11-положениях соединения Fb ацилирующим агентом, соответствующим R', с получением соединения B2b, представленного формулой B2b:
[Химическая формула 32]
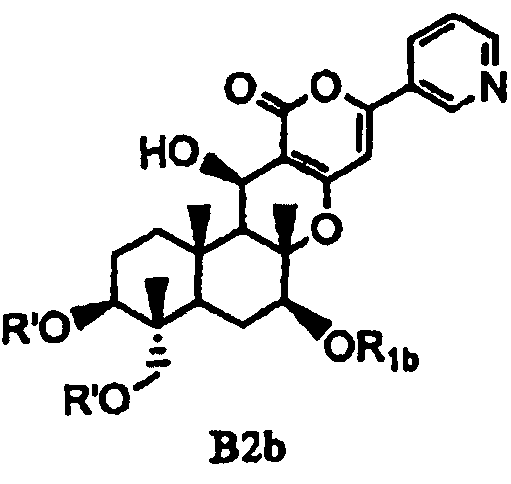
в которой значения R1b и R' определены выше,
а затем удаление защитной группы в 7-положении соединения B2b.
13. Способ по п.12, где R' представляет собой циклический С3-6алкилкарбонил.
14. Соединение, представленное формулой B2b:
[Химическая формула 33]
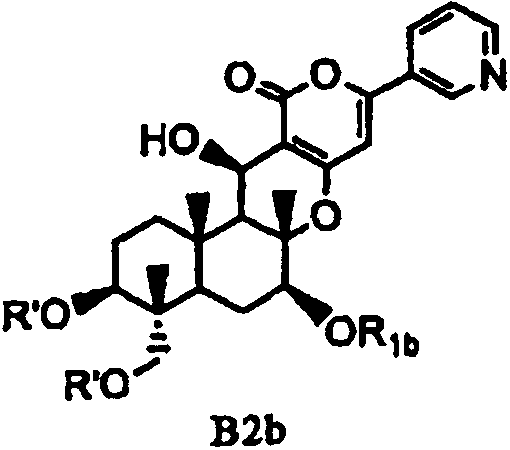
в которой R1b представляет собой линейный С1-4алкилкарбонил, необязательно замещенный атомом галогена; группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный C1-6алкил или фенил; тетрагидропиранильную группу, и R' представляет собой циклический С3-6алкилкарбонил.
15. Соединение по п.14, где R1b представляет собой ацетил; хлорацетил; группу -SiR3R4R5, где каждый R3, R4 и R5 независимо представляет собой линейный или разветвленный С1-6алкил или фенил; или тетрагидропиранильную группу, и R' представляет собой циклический C2-6алкилкарбонил.
| WO 2006129714 A1, 07.12.2006 | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Obata R et al, J | |||
| Antidiotics, 1996, v.49, no.11, p.1149-1156 | |||
| Obata R et al, Bioorganic & Medicinal Chemistry Letters, 1995, v.5, no.22, p.2683-2688 | |||
| Способ получения циклопропанкарбоксилатов | 1972 |
|
SU527133A3 |

